

Abstract
A robust, catalytic enantioselective method to construct challenging, biologically relevant, tertiary ether stereocenters has been developed. The process capitalizes on readily accessible bis(oxazoline) ligands to control the facial selectivity of the addition of copper acetylides to benzopyrylium triflates, reactive species generated in situ. Up to 99% enantiomeric excesses are achieved with a broad substrate scope. Using density functional theory (DFT) calculations, the origin of the experimentally observed enantiocontrol was attributed to additional non-covalent interactions observed in the transition state leading to the major enantiomer, such as π-stacking. The resultant substrates have direct applications in the synthesis of naturally occurring bioactive chromanones and tetrahydroxanthones.
Keywords: Asymmetric Catalysis, Tertiary Ether, Methodology, Chromanone, Alkynylation
Graphical Abstract

Phomoxanthone A (1, Scheme 1) is a naturally occurring dimeric tetrahydroxanthone with an enticing biological profile, including significant activity toward both cisplatin sensitive and resistant cancer cells.1 The impressive biological properties of phomoxanthone A have spurred investigations into its possible mode of action. A 2018 study from Bohler and coworkers found that 1 causes rapid disintegration of the inner mitochondrial membrane through a unique mode of action.1b Despite the recent studies, the full details of the biomolecular target(s) and mechanism of action of phomoxanthone A remain elusive.1c–d
Scheme 1.
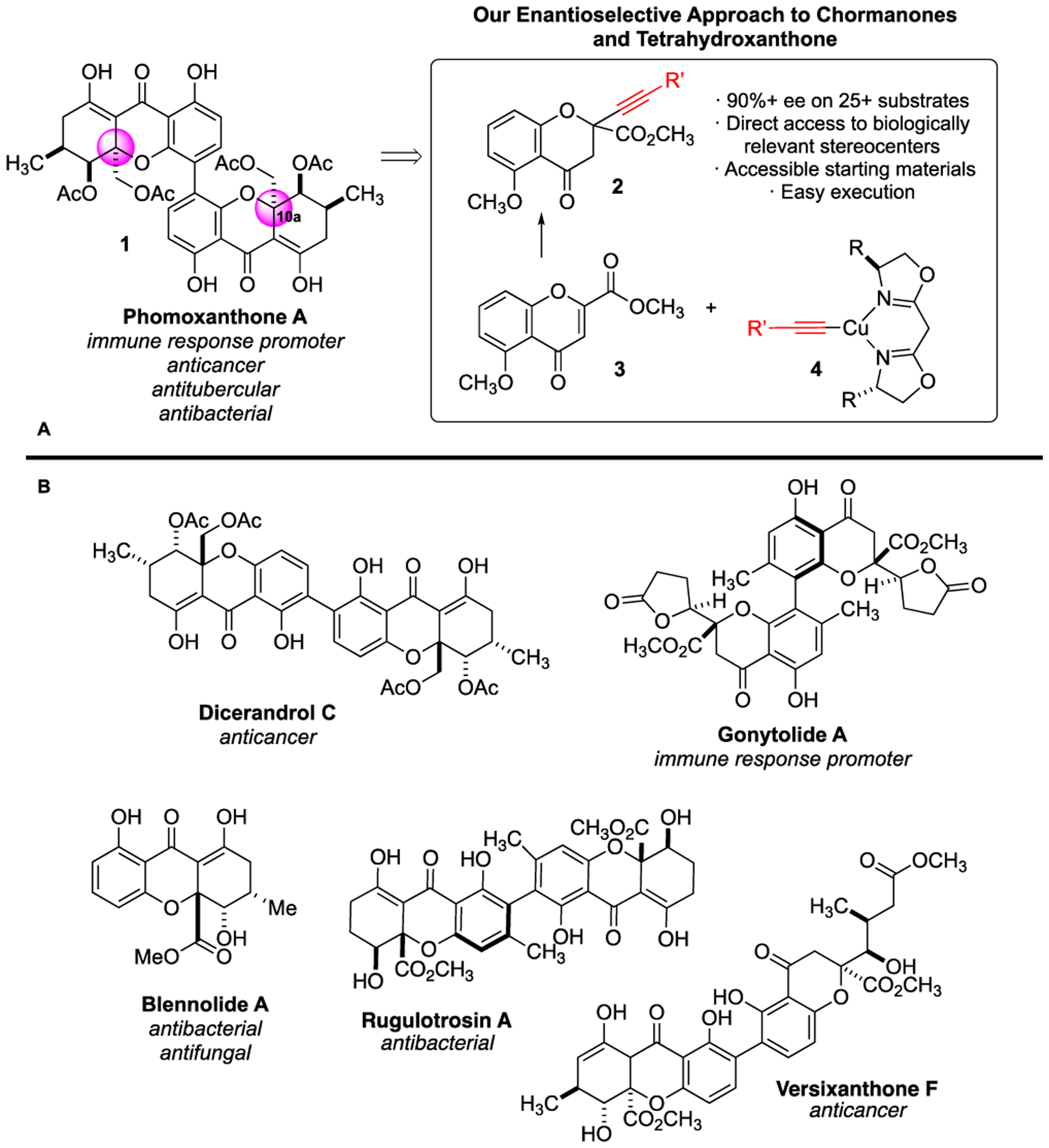
(A) Phomoxanthone A and our enantioselective approach toward its synthesis. (B) A selection of naturally occurring chromanones and tetrahydroxanthones.
The limited understanding of the mechanisms of action of phomoxanthone A may be due, in part, to the synthetic challenge it poses. Phomoxanthone A has not yet been made by chemical synthesis. Originally isolated by Isaka and coworkers in 2001 from a strain of the phomopsis fungus that grew as an endophyte on teak trees in northern Thailand, phomoxanthone A possesses a synthetically demanding scaffold.2 The 10a-stereocenter may be the most challenging aspect of 1 (highlighted in pink, Scheme 1A).3 As a chiral tertiary ether,4 position 10a is a sterically encumbered center with limited options in the literature for enantiocontrolled construction. Further, 10a is a stereocenter that is sensitive to racemization under both overly acidic and basic reaction conditions.
Although phomoxanthone A has not yet been synthesized, it belongs to a family of structurally related, secondary metabolites (Scheme 1B).5 Several of these species, such as rugulotrosin A6 and dicerandrol C,7 have attracted a synthetic following and routes have been established that enable their construction. The two most popular methods to construct the 10a stereocenter of the dimeric tetrahydroxanthones include (1) Porco’s racemic siloxyfuran addition, followed by resolution,8 and (2) Tietze’s intramolecular Wacker approach9 (Scheme 2A and 2B, respectively). While both routes were brilliantly executed by the Porco and Tietze groups, they are limited to substrates only accommodated by the respective resolution and Wacker technologies required.
Scheme 2.

Approaches toward the enantioselective construction of the 10a stereocenter of select chromanones/tetrahydroxanthones.
We envisioned developing a route to establish the 10a stereocenter of naturally inspired, dimeric chromanones and tetrahydroxanthones that is efficient, easily executed, robust to a broad substrate scope, and applicable to library construction. Ultimately, we hypothesized that we could utilize this methodology to synthesize a library of analogues of phomoxanthone A for structure-activity relationship studies and biomolecular target identification. Herein, we report the success of our copper bis(oxazoline)-catalyzed alkynylation of substituted chromenones (3) giving rise to chromanones 2 in excellent (up to 99%) enantiomeric excess (Scheme 1). The extensive substrate scope and transition-state models for this process are described herein.
As shown in Scheme 2, the enantioselective construction of the 10a tertiary ether stereocenter in 7 was inspired by our earlier discoveries: alkyl and aryl copper acetylides under the influence of bis(oxazoline) ligation add to benzopyrylium triflates (R=H) with excellent levels of enantiocontrol (>90% ee, Scheme 2D).10 While in the midst of our own reaction development, Aponick separately advanced the addition of copper acetylides to benzopyrylium triflates in the presence of StackPhos ligands (Scheme 2C).11 After identification of this copper acetylide route, we immediately invested heavily in applying this technology to the construction of the tertiary ether stereocenter in 7 (R = CO2CH3). Our first attempt applied the benzyl bis(oxazoline) ligand 8 (Scheme 2) toward the enantioselective construction of 7a. However, given the steric demands of this center, we achieved a promising, albeit mediocre, enantiomeric excess of 34% (entry 1, Table 1). The reaction system required significant optimization – mainly focused on the investigation of 30+ bis(oxazoline) ligands12 in order to reach excellent levels of enantiocontrol. A few of the key findings that were established en route to our final conditions are listed in Table 1.13 Some traction was gained in the improvement in yield and enantiomeric excess by switching from ligand 8 to t-butyl bis(oxazoline) ligand 9 (Scheme 2), giving rise to 7a in 87% yield and 38% ee (entry 2). Further optimization found that chlorobenzene improved the enantiomeric excess, especially when the reaction was conducted at low temperatures (entries 3–5). Specifically, a − 71% ee of 7a was achieved when the reaction was conducted with ligand 9 at −78 to −35 °C (entry 5). Finally, excellent levels of enantiocontrol, with opposite stereochemistry relative to ligand 9, were reached when the reaction was conducted with indanyl bis(oxazoline) ligand 10 (68% yield, 90% ee, entry 7).
Table 1.
Optimization of the Alkynylation Reactiona
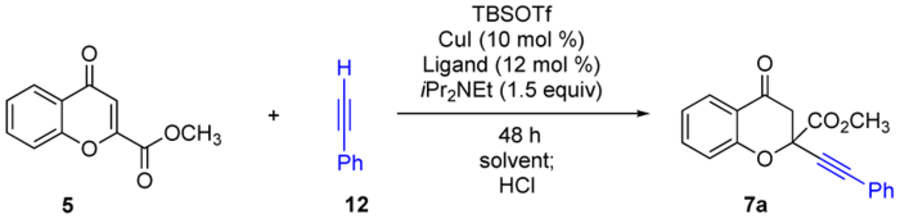
| |||||
|---|---|---|---|---|---|
| entry | ligand | solvent | temp. (°C) | yield (%) | ee (%) |
| 1 | 8 | toluene | 0 | 29 | 34 |
| 2 | 9 | toluene | 0 | 87 | −38 |
| 3 | 9 | PhCl | 0 | 69 | −42 |
| 4 | 9 | PhCl | −78 °C to −28 °C | 87 | −66 |
| 5 | 9 | PhCl | −78 °C to −35 °C | 78 | −71 |
| 6 | 10 | PhCl | −78 °C to −28 °C | 71 | 88 |
| 7 | 10 | PhCl | −78 °C to −35 °C | 68 | 90 |
See supporting information for extensive details on the reaction optimization and associated experimental procedures.
With a set of highly enantioselective reaction conditions identified, attention was turned toward an exploration of the substrate scope. We first examined the influence of the chromenone ester on enantiomeric excess (Table 2). A wide range of esters were tolerated in the reaction. The methyl ester of 5 gave rise to 7a in 68% yield and 90% ee, while the ethyl ester afforded 7b in 53% yield and 89% ee. The isopropyl and tert-butyl esters gave rise to 7c and 7d in 91% ee and 71% ee, respectively, while the benzyl ester afforded 7e in 83% ee. The trichloroethyl ester gave rise to 7f in 92% ee. Although the trichloroethyl ester was slightly higher in terms of enantiomeric excess, we continued with the methyl ester for the rest of our screening as it was both higher yielding and easier to prepare than the trichloroethyl ester.
Table 2.
Ester and Alkyne Substrate Studiesa

|
Isloated yields. See supporting information for detailed experimental procedures.
Next, we tested the influence of the alkyne on the yield and stereoselectivity of the reaction (Table 2). A variety of substituted phenyl acetylenes were readily accepted as reaction partners in the process, giving rise to excellent levels of enantiocontrol in all cases. For example, methyl groups at the 4-, 3-, or 2- position on the phenyl ring gave rise to their respective products 7g, 7h, and 7i in 90–92% ee. 4-Methoxyphenyl acetylene gave rise to 7j in 90% ee. Halogen substituents performed equally well; for instance, 4-Cl and 4-Br phenyl acetylene afforded 7k and 7l in 91% ee and 90% ee, respectively. 4-Trifluoromethylphenyl acetylene generated 7m in nearly 90% ee. Acetylenes containing 1-naphthyl and 2-naphthyl substituents were also well tolerated in the process.
Moving beyond phenyl acetylenes, the influence of terminal alkynes possessing heterocyclic and aliphatic substituents were studied in the reaction of chromenone 5 and alkyne 12 to yield 7 (Table 2). While the yields were reduced, thiophene and furan substituents on the alkyne were well tolerated and the corresponding products 7q-7s were isolated in excellent enantiocontrol (91–93% ee). Cyclopropyl acetylene was well tolerated, giving rise to 7t in 61% yield and 89% ee. Cyclohexyl acetylene offered a decent performance, yielding 7u with 81% ee. tert-Butyl acetylene gave rise to 7v in 89% yield and 91% ee with ligand 11 while cyclohexenyl acetylene gave rise to 7w in 66% yield and 86% ee.
The scope of the reaction was then investigated with respect to the chromenone (5, Table 3). Immediately we studied the reaction of the most biologically relevant 5-methoxy chromenone 5x with phenyl acetylene and were delighted to find that 7x was produced in 64% yield and 93% ee. A methoxy group was also well tolerated in the 7-position to generate 7y in 75% yield and 99% ee. Alkyl substituents were also accommodated; for instance, 7-methyl chromenone 5z afforded 7z in 67% yield and 82% ee. Halogens were well tolerated as substituents at the 7- and 8-positions; specifically, 7aa, 7ab, and 7ac were produced in 85% ee, 95% ee, and 93% ee, respectively. A 7-trifluoromethyl substituent gave rise to 7ad in 95% ee. In fact, a wide array of substituents in the 7-position afforded the products with excellent levels of enantiocontrol. For example, allyl, phenyl, alkynyl, and alkenyl substituents gave rise to products 7ae-7ah in 97–99% ee. The cyclopropyl alkynyl group was also well tolerated in the 5- and 6- positions, affording 7ai and 7aj in 81% ee and 98% ee, respectively. Recrystallization can be used to improve the enantiomeric excess of 7 when desired. For example, after recrystallization, 7n was isolated in 99% ee and used for x-ray crystallographic analysis (vide infra).
Table 3.
Chromanone Substrate Studiesa

|
Isolated yields. See supporting information for extenvise details on the substrate scope study, including additional substrates that are not shown on this table.
The combination of the generation of a biologically relevant, sterically congested stereocenter and impressive scope of the reaction prompted us to explore in-silico modeling of the enantio-determining step. First, using X-ray crystallographic analysis, we were able to collect evidence of the absolute configuration of the new stereocenter in 7n to be R when the reaction is conducted with the indanyl ligand 10 (Figure 1). From this one example, the rest of the substrates were reasoned to also favor the R enantiomer as the major product by analogy.
Figure 1.

Evidence of the Absolute Stereochemistry of 7na
a) The anisotropic displacement parameters are drawn at 50% probability level.
A possible reaction pathway is hypothesized in Scheme 3.14 The ligand coordinates to CuI giving rise to complex 13. The reaction of 13 with the alkyne and Hunig’s base generates the copper acetylide 14. The benzopyrylium ion 15,15 formed in situ upon exposure of 5 to TBSOTf, reacts with the copper acetylide in the proposed enantio-determining step to afford 16.16 The loss of the silyl group upon treatment with HCl then yields the final product 7.
Scheme 3.

Plausible Reaction Pathway
The proposed enantio-determining step was studied computationally with the B3LYP/6–31+G(d) (SDD for Cu)17 level of theory for the reaction phenyl acetylene with 5a under the influence of ligand 10. Scheme 4 depicts the transition states leading to the R enantiomer (the major enantiomer observed experimentally) and S enantiomer (the minor enantiomer observed experimentally). The transition state leading to observed major R enantiomer plausibly benefits from additional non-covalent interactions, such as π-stacking between the benzopyrylium ion and the indanyl side chain of the ligand (a length of 3.45Å was found between the ligand H and the aromatic ring). The existence of additional non-covalent interactions in the R transition versus the S transition state was confirmed in a non-covalent interaction plot analysis of the two transition states (Scheme 4 B, green regions = non-covalent interactions determined).18 The computationally predicted enantiomeric excess of 92% ee is similar to the experimentally observed 90% ee for 7a. The transition states for the formation of 7y were also determined using the same level and theory and, again, the calculated enantiomeric excess (85% ee) matched reasonably well with the experimen tally determined value of 99% ee, although there appear to be some electronic effects caused by the −OCH3 substituent that are not fully taken into account in our computational model thus causing a mild difference in experimental v. computational enantiomeric excess.19
Scheme 4.

(A) DFT Transition States Calculated with B3LYP/6–31+G(d) (SDD for Cu) Level of Theory in the Gas Phase at −35°C. (B) Non-Covalent Interaction Plots for Transition States
Early on in our studies it was observed that tert-butyl bis(oxazoline) ligand 9 affords the product 7a with opposite enantioselectivity when compared to the outcome of the alkynylation reaction with ligand 10 (see Table 1). To better understand the reason behind the switch in facial selectivity between the two ligands, the DFT transition states were calculated for the alkynylation reaction of 5a to generate 7a under the influence of 9 (Scheme 5). A study of the transition state structures suggests that the benzopyrylium ion is farther away from the ligand backbone (the closest distance found was 3.91Å in the transition state leading to the S enantiomer) with fewer non-covalent interaction present than what is observed with ligand 10 (see supporting information for non-covalent interaction plots). There may be a steric component that is driving the observed outcome of the alkynylation of 5a in the presence of ligand 9. Specifically, in the calculated transition state of the major enantiomer, the tert-butyldimethylsilyl group is oriented away from the tert-butyl side chain on the ligand ultimately generating a transition state leading to S-7a. Therefore, while ligand 9 may rely at least partially on steric hindrance for enantiocontrol, ligand 10 appears to involve a network of supportive non-covalent interactions between the ligand and benzopyrylium ion to control the facial selectivity of the alkyne addition reaction.
Scheme 5.

DFT Transition States Calculated with B3LYP/6–31+G(d) (SDD for Cu) Level of Theory in the Gas Phase at −35 °C of Alkynylation with Ligand 9.
Due to the variability in enantiomeric excess observed in the alkynylation of different chromenone esters (7a-f, Table 2), it was hypothesized that the size of the ester substituent may influence the stereochemical outcome of the reactions. The correlation of the steric parameters to the log of enantiomeric ratio is possible and can lend insight into the plausible role of steric bulk on reaction stereoselectivity.20 The Charton values of several esters explored in the reaction were plotted against their respective log(er) (Chart 1), and demonstrated a linear correlation between the Charton value of the ester and the enantioselectivity of the alkynylation reaction. Specifically, smaller ester substituents (e.g., methyl, ethyl) led to higher enantiomeric ratios.
Chart 1.

Plot of Enantiomeric Ratio versus Charton Values in the Enantioselective Alkynylation of Benzopyrylium Triflate Esters
Given our interest in applying this synthetic tactic toward the construction of biologically relevant compounds, we initiated preliminary studies to probe manipulations that would be tolerated by the newly prepared, enantio-enriched chromanone core (Scheme 6). The reduction of the alkyne was achieved upon treatment of 7a with Pd/C and H2 to give rise to 17 in an unoptimized 65% yield and 90% ee. The Schmidt rearrangement of 17 was accomplished under the influence of NaN3 and H2SO4, producing 18, a desirable 1,2-amino alcohol precursor, in 80% yield and 90% ee. The reduction of both the ketone and alkyne functional groups of 7a is achieved upon long exposure to Pd/C and H2. Under these conditions, chroman 19 was isolated in 81% yield and 90% ee from 7a. The ester can then be converted to the methyl group, giving rise to motifs found in naturally occurring molecules such as vitamin E. Chromane 20 was isolated in 77% yield and 90% ee over two steps from 19.
Scheme 6.

Select Synthetic Manipulations Tolerated by Enantioenriched Chromanone 7a.
In conclusion, a highly enantioselective route to sterically hindered and biologically relevant tertiary ether stereocenters has been developed. The methodology benefits from a broad substrate scope and employs readily available indanyl bis(oxazoline) ligands to control the stereoselective addition of copper acetylides to benzopyrylium triflates. The proposed transition state leading to the major enantiomer may benefit from additional noncovalent interactions, such as π-stacking interactions between the ligand and the benzopyrylium ion. Further DFT studies will aim on Cu ligand design to take advantage of favorable non-covalent interactions in the proposed enantio-determining step. Ongoing efforts are dedicated toward taking advantage of this robust reaction system to synthesize bioactive targets, such as naturally occurring dimeric chromanones and tetrahydroxanthones, that contain this challenging tertiary ether stereocenter.
Supplementary Material
ACKNOWLEDGMENT
The National Institutes of Health are gratefully acknowledged for funding these studies (1R35GM124804-01). C.M.H. acknowledges generous computational resources as provided by the Ohio Supercomputer Center.
Funding Sources
NIH NIGMS (1R35GM124804-01).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental Details (PDF)
Computational Details (PDF)
Conflict of Interest
A.E.M. is a cofounder of rTherapies, LLC.
REFERENCES
- (1).a) Rönsberg D; Debbab A; Mándi A; Vasylyeva V; Böhler P; Stork B; Engelke L; Hamcher A; Sawadogo R; Diederich M; Wray V; Lin W; Kassack MU; Janiak C; Scheu S; Wesselborg S; Kurtán T; Aly AH; Proksch P Pro-Apoptotic and Immunostimulatory Tetrahydroxanthone Dimers from the Endophytic Fungus Phomopsis longicolla. J. Org. Chem 2013, 78, 12409–12425; [DOI] [PubMed] [Google Scholar]; b) Böhler P; Stuhldreier F; Anand R; Kondadi A; Schlutermann D; Berleth N; Deitersen J; Wallot-Hieke N; Wu W; Frank M; Niemann H; Wesbuer E; Barbian A; Luyten T; Parys J; Weidtkamp-Peters S; Borchardt A; Reichert A; Pena-Blano A; Garcia-Saez A; Itskanov S; van der Bliek A; Proksch P; Wesselborg S; Stork B The Mycotoxin Phomoxanthone A Disturbs the Form and Function of the Inner Mitochondrial Membrane. Cell Death & Disease 2018, 9, 286–303; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang C; Engelke L; Bickel D; Hamacher A; Frank M; Proksch P; Gohlke H; Kassack MU The Tetrahydroxanthone-dimer Phomoxanthone A is a Strong Inducer of Apoptosis in Cis-platin-resistant Solid Cancer Cells. Bioorg. Med. Chem 2019, 27, 115044–115056; [DOI] [PubMed] [Google Scholar]; d) Yang R; Dong Q; Zu H; Gao X; Zhao Z; Qin J; Chen C; Luo D Identification of Phomoxanthone A and B as Protein Tyrosine Phosphate Inhibitors. ACS Omega 2020, 5, 25927–25935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Isaka M; Jaturapat A; Rukserre K; Damwisetkanjana K; Tanticharoen M; Thebtaranonth Y Phomoxanthones A and B, Novel Xanthone Dimers from the Endophytic Fungus Phomopsis Species. J. Nat. Prod 2001, 64, 1015–1018 [DOI] [PubMed] [Google Scholar]
- 3).For a review, see:; Nibbs AE Scheidt KA Asymmetric Methods for the Synthesis of Flavanones, Chromanones, and Azaflavanones. Eur. J. Org. Chem 2012, 449– 462; [DOI] [PMC free article] [PubMed] [Google Scholar]; for select examples of enantioselective 2-alkylchromanone synthesis, see:; a) Rao AV; Gaitonde AS; Prakash SP; Rao SP A Concise Synthesis of Chiral 2-Methyl Chromanon-4-ones: Stereoselective Build-up of the Chromanol Moiety of Anti-HIV Agent Calanolide. Tetrahedron Lett. 1994, 35, 6347 – 6350; [Google Scholar]; b) Kawasaki M; Kakuda H; Goto M; Kawabata S; Kometani T Asymmetric Synthesis of 2-Substituted Chroman-4-ones using Lipase-Catalyzed Kinetic Resolutions. Tetrahedron: Asymmetry 2003, 14, 1529 – 1534; [Google Scholar]; c) Biddle MM; Lin M;. Scheidt KA Catalytic Enantioselective Synthesis of Flavanones and Chromanones. J. Am. Chem. Soc 2007, 129, 3830 – 3831; [DOI] [PubMed] [Google Scholar]; d) Boekl H; Mackert R; Muramann C; Schweickert N US66646136B1, 2013; [Google Scholar]; e) Termath AO; Sebode H; Schlundt W; Stemmler RT; Netscher T; Bonrath W; Schmalz H-G Total Synthesis of (R,R,R)-α-Tocopherol through Asymmetric Cu-Catalyzed 1,4-Addition. Chem. Eur. J 2014, 20, 12051 – 12055; [DOI] [PubMed] [Google Scholar]; f) Brown MK; Degrado SJ; Hoveyda AH Highly Enantioselective Cu-Catalyzed Conjugate Additions of Dialkylzinc Reagents to Unsaturated Furanones and Pyranones: Preparation of Air-Stable and Catalytically Active Cu-Peptide Complexes. Angew. Chem. Int. Ed 2005, 44, 5306– 5310; Angew. Chem. 2005, 117, 5440 – 5444; [DOI] [PubMed] [Google Scholar]; g) Vila C; Hornillos V; Fananas-Mastral M; Feringa BL Catalytic Asymmetric Conjugate Addition of Grignard Reagents to Chromones. Chem. Commun 2013, 49, 5933– 5935; [DOI] [PubMed] [Google Scholar]; h) Trost BM; Gnanamani E; Kalnmals CA; Hung C-I; Tracy JS Direct and Enantio- and Diastereoselective Vinylogous Addition of Butenolides to Chromones Catalyzed by Zn-ProPhenol. J. Am. Chem. Soc 2019, 141, 1489–1493. [DOI] [PubMed] [Google Scholar]
- 4).For a recent review on the importance of chiral tertiary ethers and their limited synthetic accessibility, see:; Liu Y-L; Lin X-T Recent Advances in Catalytic Asymmetric Synthesis of Tertiary Alcohols via Nucleophilic Addition to Ketones. Adv. Synth. Catal 2019, 361, 876–918. [Google Scholar]; For select reports demonstrating the importance of constructing tertiary ether stereocenters from chromanones, see:; (a) Baek D; Ryu H; Ryu J; Lee J; Stoltz B; Hong S Catalytic Enantioselective Synthesis of Tetrasubstituted Chromanones via Palladium Catalyzed Asymmetric Conjugate Arylation using Chiral Pyridine-Dihydroisoquinoline Ligands. Chiral Chem. Sci 2020, 11, 4602–4607; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gerten A; Stanley L Palladium-Catalyzed Conjugate Additional of Arylboronic Acids to 2-Substituted Chromones in Aqueous Media. Tetrahedron Lett. 2016, 57, 5460–5463. [Google Scholar]
- 5).For recent reviews on dimeric chromanones and tetrahydroxanthones, see:; a) Wezeman T; Brase S; Masters K Xanthone Dimers: A Compound Family which is both Common and Privileged. Nat. Prod. Rep 2015, 32, 6–28; [DOI] [PubMed] [Google Scholar]; b) Masters K; Brase S Xanthones from Fungi, Lichens, and Bacteria: The Natural Products and their Synthesis. Chem. Rev 2012, 112, 3717–3776. [DOI] [PubMed] [Google Scholar]; For isolation and biological activities of gontyolides, see:; Kikuchi H; Isobe M; Sekiya M; Abe Y; Hoshikawa T; Ueda K; Kurata S; Katou Y; Oshima Y Structures of the Dimeric and Monomeric Chromanones, Gonytolides A-C, Isolated from the Fungus Gonytrichum sp. and Their Promoting Activities of the Innate Immune Responses. Org. Lett 2011, 13, 4624–4627. [DOI] [PubMed] [Google Scholar]; For isolation and biological activities of the blennolides, see:; Zhang W; Krohn K; Zia-Ullah; Flörke U; Pescitelli G; Di Bari L; Antus S; Kurtán T; Rheinheimer J; Draeger S; Schulz B New Mono- and Dimeric Members of the Secalonic Acid Family: Blennolides A-G Isolated from the Fungus Blennoria sp. Chem. Eur. J 2008, 14, 4913–4923. [DOI] [PubMed] [Google Scholar]; For isolation and biological data on the dicerandrols, see:; Wagenaar MM; Clardy J Dicerandrols, New Antibiotic and Cytotoxic Dimers Produced by the Fungus Phomposis longicolla Isolated from an Endagered Mint. J. Nat. Prod 2001, 64, 1006–1009. [DOI] [PubMed] [Google Scholar]; For isolation and biological data on the versixanthones, see:; Wu G; Yu G; Kurtan T; Mandi A; Peng J; Mo X; Liu M; Li H; Sun X; Li J; Zhu T; Gu Q; Li D Versixanthones A-F, Cytotoxic Xanthone-Chromanone Dimers from the Marine-Derived Fungus Aspergillus versicolor HDN1009. J. Nat. Prod 2015, 78, 2691–2698. [DOI] [PubMed] [Google Scholar]; For isolation and biological data on the rugulotrosins, see:; Stewart M; Capon RJ; White JM; Lacey E; Tennant S; Gill J; Shaddock M Rugulotrosins A and B: Two New Antibacterial Metabolites from an Australian Isolate of a Penicillium sp. J. Nat. Prod 2004, 67, 728–730. [DOI] [PubMed] [Google Scholar]
- 6).a) Qin T; Skraba-Joiner S; Khalil Z; Johnson R; Capon R; Porco JA Atropselective Synthesis of (−) and (+) Rugulotrosin A Utilizing Point-to-Axial Chirality Transfer. Nature Chemistry 2015, 7, 234–240; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen J; Li Y; Xiao Z; He H; Gau S Asymmetric Synthesis of Rugulotrosin A. Org. Lett 2020, 22, 1485–1489. [DOI] [PubMed] [Google Scholar]
- 7).Ganapathy D; Reiner JR; Valdomir G; Senthilkumar S; Tietze LF Enantioselective Total Synthesis and Structure Confirmation of the Natural Dimeric Tetrahydroxanthenone Diceradrol C. Chem. Eur. J 2017, 23, 2299–2302. [DOI] [PubMed] [Google Scholar]
- 8).a) Qin T; Iwata T; Ransom TT; Beutler JA; Porco JA Syntheses of Dimeric Tetrahydroxanthones with Varied Linkages: Investigations of “Shapeshifting” Properties. J. Am. Chem. Soc 2015, 137, 15225–15233; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Qin T; Porco JA Total Syntheses of Secalonic Acids A and D. Angew. Chem. Int. Ed 2014, 53, 3107–3110; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Qin T; Johnson R; Porco J Vinylogous Addition of Siloxyfurans to Benzopyryliums: A Concise Approach to the Tetrahydroxanthone Natural Products. J. Am. Chem. Soc 2011, 133, 1714–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9).Tietze LF; Ma L; Reiner JR; Jackenkroll S; Heidemann S Enantioselective Total Synthesis of (−)-Blennolide A. Chem. Eur. J 2013, 19, 8610–8614 [DOI] [PubMed] [Google Scholar]
- 10).Guan Y; Attard J; Mattson AE Copper Bis(oxazoline)-Catalyzed Enantioselective Alkynylation of Benzopyrylium Ions. Chem. Eur. J 2020, 26,1742–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).DeRatt LG; Pappoppula M; Aponick A A Facile Enantioselective Alkynylation of Chromones. Angew. Chem. Int. Ed 2019, 58, 8416–8420. [DOI] [PubMed] [Google Scholar]
- 12).For a review on bis(oxazoline) ligands in asymmetric catalysis, see:; Desimoni G; Faita G; Jorgensen HA C2-Symmetric Chiral Bis(Oxazoline) Ligands in Asymmetric Catalysis. Chem. Rev 2006, 106, 3561–3651. [DOI] [PubMed] [Google Scholar]
- 13). See supporting information for extensive details on the reaction optimization.
- 14).For related reactions that support this hypothesized pathway, see:; a) Srinivas H,D; Maity P; Yap GP; Watson M Enantioselective Copper-Catalyzed Alkynylation of Benzopyranyl Oxocarbenium Ions. J. Org. Chem 2015, 80, 4003–4016; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhong K; Shan C; Zhu L; Zhang T; Liu F; Shen B; Lan Y; Bai R Theroetical Study of the Addition of Cu-Carbenes to Acetylenes to Form Chiral Allenes. J. Am. Chem. Soc 2019. 141, 5772–5780. [DOI] [PubMed] [Google Scholar]
- 15).For select work on the generation and reactions of benzopyrylium triflates, see:; a) Iwasaki H, Kume T, Yamamoto Y, Akiba K, Reaction of K 4-t-Butyldimethylsiloxy-1-benzopyrylium Salt with Enol Silyl Ethers and Active Methylenes. Tetrahedron Lett. 1987, 28, 6355 – 6358; [Google Scholar]; b) Stubbing LA, Li FF, Furkert DP, V. Caprio E; Brimble MA Access to 2-Alkyl Chromanones via Conjugate Addition Approach. Tetrahedron 2012, 68, 6948–6956; [Google Scholar]; c) Liu J; Li Z; Tong P; Xie Z; Zhang Y; Li Y TMSI-Promoted Vinylogous Michael Addition of Siloxyfuran to 2-Substituted Chromones: A General Approach for the Total Synthesis of Chromanone Lactone Natural Products. J. Org. Chem 2015, 80, 1632 – 1643. [DOI] [PubMed] [Google Scholar]
- 16). See supporting information for the specific details of the computational approach.
- 17).For citations on the level of theory, see:; a) Becke AD Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys 1993, 98, 5648–5652; [Google Scholar]; b) Stephens PJ; Devlin FJ; Chabalowski CF; Frisch MJ Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem 1994, 98, 11623–11627; [Google Scholar]; c) Kim K; Jordan KD Comparison of Density Functional and MP2 Calculations on the Water Monomer and Dimer. J. Phys. Chem 1994, 98, 10089–10094; [Google Scholar]; d) Nicklass A; Dolg M; Stoll H; Preuss H; Nicklass A; Dolg M; Stoll H; Preuss H Ab Initio Energy-Adjusted Pseudopotentials for the Noble Gases Ne through Xe : Calculation of Atomic Dipole and Quadrupole Polarizabilities. J. Chem. Phys 1995, 102, 8942–8952; [Google Scholar]; e) Hariharan PC; Pople JA The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theor. Chim. Acta 1973, 28, 213–222; [Google Scholar]; f) Ditchfield R; Hehre WJ; Pople JA Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys 1971, 54, 724–728; [Google Scholar]; g) Gordon MS The Isomers of Silacyclopropane. Chem. Phys. Lett 1980, 76, 163–168. [Google Scholar]; h) Hariharan PC; Pople JA Accuracy of AHn Equilibrium Geometries by Single Determinant Molecular Orbital Theory. Mol. Phys 1974, 27, 209–214; [Google Scholar]; i) Hehre WJ; Ditchfield K; Pople JA Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys 1972, 56, 2257–2261. [Google Scholar]
- 18).Contreras-Garcia J; Johnson ER; Keinan S; Chaudret R; Piquemal J-P; Bertan D; Yang W NCIPLOT: A Program for Plotting Noncovlaent Interaction Reagions J. Chem. Theory Comput 2011, 7, 625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19).Schneebeli ST; Hall ML; Breslow R; Friesner R J. Am. Chem. Soc Quantitative DFT Modeling of the Enantiomeric Excess for Dioxirane-Catalyze Epoxidations. 2009, 131, 3965–3973. [DOI] [PMC free article] [PubMed] [Google Scholar]; See supporting information for additional details of the computational work.
- 20).Sigman MS; Miller JJ Examination of the Role of Taft-Type Steric Parameters in Asymmetric Catalysis. J. Org. Chem 2009, 74, 7633–7643. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.