

Abstract
Background:
A familial predisposition to sudden and/or arrhythmic death (SAD) in the setting of coronary artery disease (CAD) exists; however, the genetic basis is poorly understood.
Objective:
To determine whether a genome-wide polygenic score for CAD (GPSCAD) might have utility in SAD risk stratification in CAD patients without severe systolic dysfunction.
Methods:
A previously validated GPSCAD was generated utilizing genome-wide genotyping in 4698 PREDETERMINE participants of European ancestry with CAD and left ventricular ejection fraction (LVEF)>30–35%. The population was dichotomized according to top GPSCAD decile as defined by the general population, and absolute, proportional, and relative risks for SAD and non-SAD were estimated using competing risk analyses.
Results:
Over a median follow-up of 8.0 years, participants in the top GPSCAD decile were at elevated absolute SAD risk (8.0%; 95% CI, 5.1–12.4 versus 4.8%; 95% CI,3.3–7.0; p=0.005) and proportional SAD risk (29% vs 16%; p=0.0003) compared to the remainder. After controlling for LVEF, clinical factors, and ECG parameters, the top GPSCAD decile was associated with SAD (sHR1.77; 95% CI, 1.23–2.54; p=0.002) but not non-SAD (sHR 1.00, 95% CI 0.80–1.25; p=0.98) (p for Δ =0.003). The addition of the top GPSCAD decile to the multivariable model significantly improved net reclassification indexes (continuous NRI 14.0%, p=0.024 and categorical NRI 6.6%, p=0.005) but not the C-Index (difference in C-Index 0.007; p=0.143).
Conclusion:
Among CAD patients without severe systolic dysfunction, high GPSCAD specifically predicted SAD and enriched for both absolute and proportional SAD risk, identifying a population who might benefit from defibrillator therapy.
Keywords: polygenic risk score, coronary artery disease, sudden death
Condensed Abstract
Among coronary artery disease (CAD) patients without severe systolic dysfunction, the top decile of a genome-wide polygenic score for CAD (GPSCAD) identified a subpopulation at elevated absolute sudden and/or arrhythmic death (SAD) risk and in whom the proportion of deaths due to SAD was elevated compared to the remainder of the cohort. After controlling for ejection fraction, clinical factors, and ECG parameters, the top GPSCAD decile was associated with SAD but not non-SAD. These data in aggregate suggest that the GPSCAD might be useful in identifying a population who might benefit from defibrillator therapy.
Introduction
Sudden cardiac death remains a major international public health problem, responsible for 15%−20% of all deaths1. Coronary artery disease (CAD) is the most common substrate underlying sudden death2. While implantable cardioverter-defibrillator (ICD) improves survival in CHD patients with symptomatic heart failure and severe systolic dysfunction, more than 70% of sudden deaths occur in patients who do not meet this current paradigm for sudden death prevention3. In order to reduce the global burden of sudden death, it will be critical to improve risk stratification in this population. To do so, readily accessible, pragmatic screening tools that specifically predict sudden and/or arrhythmic death (SAD) are needed. Genetic markers, which can be easily measured at scale and at relatively low cost, represent one potential feasible option for SAD risk stratification in this broad population.
Since CAD underlies most SAD and there is a familial predisposition to SAD in the setting of CAD4–6, it is possible that both conditions share a common genetic basis. In support of this hypothesis, several studies have reported that variants that associate with CAD also associate with SAD risk in the general population7–9. Although these data have important mechanistic implications, the clinical impact is limited due to the low absolute risk of SAD in the general population10 and the lack of clarity as to whether these CAD variants in aggregate specifically predict SAD as opposed to other causes of death. To meaningfully advance SAD risk stratification, genetic markers will need to identify populations at clinically actionable levels of absolute SAD risk in whom SAD accounts for a substantial proportion of total mortality1.
In the present study, we investigate the utility of a validated genome-wide polygenic score (GPSCAD)11, 12 for SAD risk stratification in an intermediate-risk population with established CAD but without severe systolic disfunction and/or indication for a primary prevention ICD. The GPSCAD is comprised of over 6 million common variants and has been previously demonstrated to predict incident CAD11, 12 and accounts for up to 22% of CAD heritability13 in populations of European descent. We assessed the absolute and relative associations of the GPSCAD with SAD risk and other competing forms of mortality over 8 years of follow-up. These data were then used to explore whether the GPSCAD, in combination with standard clinical and ECG markers, might be able to identify a subset of the population at both high absolute and proportional SAD risk for inclusion in future trials of ICD therapy within this population.
Methods
Study Cohort
The PREDETERMINE study (ClinicalTrials.gov identifier: NCT01114269) is an ongoing multicenter, prospective cohort study of 5764 patients with CAD, as documented by significant stenoses on coronary angiography (>50% Left Main or >70% major epicardial vessel), prior coronary revascularization, or documented history of myocardial infarction (MI), who did not qualify for primary prevention ICD based upon having a left ventricular function (LVEF) more than 35% or an LVEF of 30% to 35% with New York Heart Association (NYHA) class I. Patients were also required to have either a clinical history of MI or LVEF < 50% for inclusion in the study. Exclusion criteria included history of heart transplant, cardiac arrest not associated with MI, current or planned ICD, or life expectancy less than 6 months1. Patients with non-European ancestry were excluded (n= 788) from this analysis because the GPSCAD was derived and validated in persons with European decent and the number of SAD events (n=21) in this subpopulation precluded stratified analyses. The present analysis includes 4698 who successfully underwent genotyping and were of European ancestry. This study was approved by the Mass General Brigham Institutional Review Board (protocol #: 2007P000840).
Assessment of GPSCAD
The methods for generating the GPSCAD used in this study has been previously published (Supplemental methods)11. Genotyping of the PREDETERMINE participants was performed using the Illumina GSAv1 genotyping array. Of the total n= 5488 participants who underwent genotyping (European and non-European ancestry), 97% were retained after quality control procedures that removed low-quality samples due to discordance between reported and genetically inferred sex. The overall call rate was >98%. Analysis was restricted to individual with European ancestry confirmed by projecting onto the 1000 Genomes population by computing principal components using EIGENSOFT software package14. Genetic variants not in Hardy-Weinberg equilibrium (P<1×10−15) were removed. Imputation of genotype dose for additional variants was performed using the Haplotype Reference consortium reference panel15. The previously reported GPSCAD was computed using all 6630150 variants with imputation score >0.3. Each participant had a raw GPSCAD score generated by multiplying the genotype dosage for each risk-increasing allele by its respective weight and the summing across all variants in the score using PLINK2 software16. We used a reference distribution from 1000 Genomes to create ancestry-adjusted scores, using the residual from a linear regression model that uses principal components to predict score.
Endpoint Ascertainment and Definitions
After enrolment, all participants in the PREDETERMINE study were followed centrally by the Clinical Coordinating Center at the Brigham and Women’s Hospital through questionnaires inquiring about cardiac arrest, ICD implantation, and other cardiovascular endpoints by mail or telephone, contact with postal authorities, obituary searches and serial searches of the National Death Index for names of non-respondents every 6 months. Endpoints occurring in a hospital setting were confirmed by medical records. For out-of-hospital deaths, standardized detailed interviews were conducted with family members and potential witnesses. Endpoints were confirmed from emergency medical service reports, emergency department and other medical records, autopsies and witness reports of the circumstances surrounding the death.
The primary endpoint was sudden and/or arrhythmic death (SAD). All deaths were classified according to timing (sudden vs non-sudden) and mechanism (arrhythmic vs non-arrhythmic)17. Sudden death was defined as an unexpected death due to cardiac or unknown causes that occurred within one hour of symptom onset or within 24 hours of being last witnessed to be symptom free. Arrhythmic death was defined as deaths preceded by an abrupt spontaneous collapse of circulation without antecedent circulatory or neurological impairment. Deaths before which the pulse gradually disappeared and/or those preceded by circulatory or neurologic impairment were considered non-arrhythmic deaths and were excluded from the SAD endpoint regardless of timing. Out-of-hospital cardiac arrests due to ventricular fibrillation (VF) successfully resuscitated with external electrical defibrillation were considered aborted arrhythmic deaths and included in the primary SAD endpoint. Deaths not characterized as SAD are termed “non-SAD”. Deaths were also classified as cardiac, non-cardiac, or due to an unknown cause (Supplemental methods).
For patients who received an ICD over the course of study follow-up, a secondary endpoint comprised of ICD therapies for ventricular tachycardia (VT)/VF >200 bpm was adjudicated using stored electrograms (Supplemental methods)18.
Statistical Analysis
For the main analyses, participants contributed person-time from enrollment to date of death, out-of-hospital cardiac arrest, ICD implant, last contact date, or February 21, 2021, whichever came first. A priori, the population was dichotomized into two groups, GPSCAD top decile and the rest of the cohort based upon prior studies demonstrating a population threshold of risk. Absolute rates of SAD and non-SAD, and the resultant proportion of deaths due to SAD, were estimated for the top GPSCAD decile versus the remainder of the population using cumulative incidence function curves accounting for competing risk of the alternative endpoint. Relative risks of SAD and non-SAD in the top GPSCAD decile versus the rest of the population were estimated from subdistribution hazard ratios (sHR) derived from competing risk Fine-Gray models. Multivariable models were adjusted for (i) age and sex, and (ii) age, sex, diabetes, hypertension, body mass index, atrial fibrillation, NYHA class, LVEF , smoking history, lipid lowering drugs, diuretics, parental history of SAD (maternal or paternal history of SAD), and a previously validated ECG score associated with SAD in this population.19 The ECG score assigned points as follows: contiguous Q wave: 1 point, QRS duration (<_80: 0 points, 81–110: 1 point, >110: 2 points), left ventricular hypertrophy (LVH): 1 point, prolonged JTc: 1 point. Scores were stratified into low (0–1 points), moderate (2 points), and high (≥ 3 points) risk. The continuous association between GPSCAD and modes of death was also examined using penalized spline basis Cox models controlling for the same variables. To examine whether the top GPSCAD decile was differentially associated with SAD versus non-SAD deaths, cause specific hazard models with likelihood ratio comparisons were performed20.
In a secondary analysis, patients were not censored at the time of ICD implant, and relative risks of a combined endpoint of SAD and ICD therapy (VT/VF > 200 bpm) and non-SAD were estimated using sub-distribution hazard ratios derived using Fine-Gray models as described above.
To examine whether the GPSCAD improved SAD risk prediction, the top decile of GPSCAD variable was added to the above multivariable model including clinical risk factors and ECG score, and the change in the C-index, integrated discrimination improvement (IDI), continuous net reclassification improvement (NRI), and categorical NRI based upon 8-year probabilities of SAD (0–<2%, 2-<8%,8-≤15, >15%) were calculated. These SAD cut-points were selected to be consistent with those used in a prior study in this cohort that reported improvements in SAD reclassification with the ECG score over 5-years of follow-up.19. Non-parametric bootstrapping with 1000 samples were used to build the confidence intervals for NRI and IDI. The adjusted C-Index (corrected for optimism) with parametric confidence intervals were based on bootstrapping with 100 samples. Cumulative incidences of SAD and non-SAD at 8 years stratified by quartiles of the baseline multivariable model were also assessed.
These data on absolute and proportional risk were used to explore the potential impact ICD therapy might have on overall mortality among patients in the top decile of GPSCAD as previously described1. These analyses assumed that ICD therapy would result in a 60% reduction in SAD with no reduction in non-SAD mortality in this population as observed in clinical trials of patients with severe systolic dysfunction21, 22. The percent reduction in total mortality and number needed to treat (NNT) were estimated based on the observed 8-year rates of SAD and total mortality in this cohort. The sample sizes required to achieve 80% power to detect these differences was then computed along with corresponding confidence intervals for the NNT using standard errors based on the observed rates and computed sample size. Statistical analyses were performed using SAS version 9.4 and R version 3.6.3. A two-tailed P< 0.05 was considered to indicate statistical significance.
Results
Baseline characteristics
Among the 4698 PRE-DETERMINE study participants with European ancestry, 646 (13.8%) were in the top general population-based GPSCAD decile. Compared to the rest of the cohort, participants in the top GPSCAD decile were younger (63 years, IQR 54–70 years vs 66 years, IQR 58–73 years), more likely to be female (27% vs 23%), have more severe CAD, a history of coronary artery bypass surgery and parental history of SAD, and less likely to have impaired renal function23 (Table 1). Over a median follow-up of 8.05 years (25th, 75th percentiles: 6.1, 9.05), there were 176 SAD events (22 cardiac arrest and 154 sudden cardiac death) and 833 non-SAD events.
Table 1.
Baseline Characteristics of Study Cohort Stratified by Polygenic Score for Coronary Artery Disease.
| Baseline Characteristics | Top Decile GPSCAD (n=646) |
Rest of Cohort (n=4052) |
P-value |
|---|---|---|---|
| Age, median (IQR) | 63.0 (54.0, 70.0) | 66.0 (58.0, 73.0) | <0.001 |
| Male Gender, n (%) | 471 (72.9) | 3122 (77.0) | 0.021 |
| Hypertension, n (%) | 488 (75.5) | 3041 (75.0) | 0. 788 |
| History of MI, n (%) | 597 (92.4) | 3664 (90.4) | 0. 106 |
| BMI, median (IQR) | 29.8 (26.3, 33.6) | 29.3 (26.1, 33.2) | 0.076 |
| Smoking Status, n (%) | 0. 283 | ||
| Never | 230 (35.6) | 1316 (32.5) | |
| Former | 325 (50.3) | 2153 (53.1) | |
| Current | 91 (14.1) | 582 (14.4) | |
| CAD Severity, n (%) | 0.020 | ||
| 0 Vessel/1 Vessel | 231 (36.9) | 1681 (42.7) | |
| 2 Vessel | 191 (30.5) | 1123 (28.5) | |
| 3 Vessel/Left main | 204 (32.6) | 1130 (28.7) | |
| History of Revascularization, n (%) | 603 (93.3) | 3752 (92.6) | 0. 498 |
| PCI | 505 (78.2) | 3243 (80.0) | 0. 274 |
| CABG | 247 (38.2) | 1316 (32.5) | 0. 004 |
| Family History SAD, n (%) | 191 (29.6) | 1031 (25.4) | 0. 027 |
| Diabetes mellitus, n (%) | 183 (28.3) | 1242 (30.7) | 00.233 |
| History of AF, n (%) | 90 (13.9) | 606 (15.0) | 0. 495 |
| Ejection Fraction, median (IQR) | 52.0 (45.0, 60.0) | 50.5 (45.0, 60.0) | 0.527 |
| Ejection Fraction | 0.837 | ||
| ≥ 60% | 191 (29.6) | 1127 (27.8) | |
| 50–59% | 223 (34.5) | 1431 (35.3) | |
| 40–49% | 191 (29.6) | 1228 (30.3) | |
| 30–39% | 41 (6.3) | 266 (6.6) | |
| NYHA Class, n (%) | 0. 272 | ||
| Class I | 532 (82.45) | 3236 (79.9) | |
| Class II | 88 (13.6) | 652 (16.1) | |
| Class III/IV | 25 (3.9) | 152 (3.8) | |
| ECG Score, n (%) | 0.416 | ||
| Low | 366 (56.7) | 2333 (57.6) | |
| Median | 188 (29.1) | 1217 (30.0) | |
| High | 92 (14.2) | 502 (12.4) | |
| eGFR, ml/min/1.73 m2 | 81.73 [66.83, 95.56] | 75.99 [59.07, 91.55] | <0.001 |
| eGFR class | <0.001 | ||
| <60 ml/min/1.73 m2 | 111 (17.2) | 1058 (26.2) | |
| Medications, n (%) | |||
| Aspirin | 578 (89.5) | 3561 (87.9) | 0. 246 |
| Beta-blocker | 536 (83.0) | 3325 (82.1) | 0. 573 |
| Lipid-lowering agent | 613 (94.9) | 3769 (93.0) | 0. 077 |
| ACE inhibitor/ARB | 433 (67.0) | 2778 (68.6) | 0. 437 |
| Aldosterone Antagonist | 9 (1.4) | 51 (1.3) | 0. 777 |
| Diuretic | 182 (28.2) | 1268 (31.3) | 0. 111 |
Abbreviations: CAD = coronary artery disease, PCI = percutaneous coronary intervention, CABG = coronary bypass artery graft, SAD=sudden and/or arrhythmic death, NYHA = New York Heart Association, ECG = electrocardiogram, eGFR = estimated glomerular filtration rate, ACE = angiotensin-converting enzyme, ARB = angiotensin receptor blocker
eGFR was calculated as follows: eGFR-EPIcr-cys_R = 135 X min(Scr/k,1)α X max(Scr/k,1)−0.544 X min(Scys/0.8,1)−0.323 X max(Scys/0.8,1)−0.778 X 0.9961age X 0.963 [if female]); where k=0.7 for female and 0.9 for male and α = 0.219 for female and −0.144 for male.
Absolute and Proportional Risk of SAD:
The unadjusted cumulative incidence of SAD over the course of the study, accounting for competing deaths, was significantly greater in the top decile of GPSCAD at 8.0% (95% CI,5.1–12.4) compared to the rest of the cohort [4.8% (95% CI,3.3–7.0), p for Δ =0.005, Central Illustration]. In contrast, the unadjusted cumulative incidence of non-SAD, accounting for competing deaths, was significantly lower in the top GPSCAD decile (22.0%; 95% CI, 16.3–29.7),] as compared to the rest of the cohort (Central Illustration, 28.9 % (95% CI, 22.5–37.2; p for Δ =0.005). Due to these differential associations with SAD and non-SAD, the proportion of deaths due to SAD was greater in the top GPSCAD decile versus the remainder of the cohort (29% versus 16%, p for proportion = 0.0003, Central Illustration).
Central Illustration.
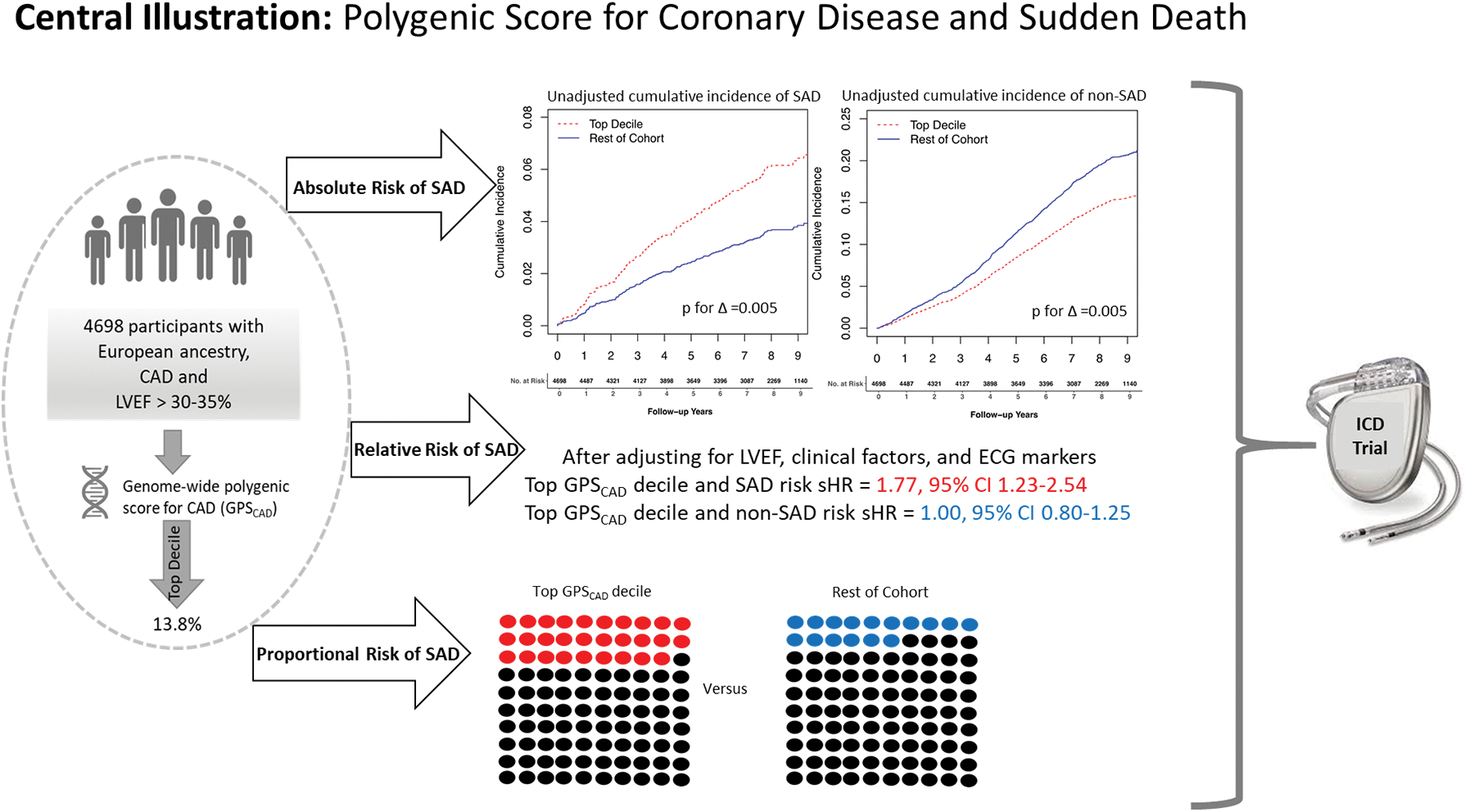
Unadjusted cumulative incidence of SAD and non-SAD. The red line represents top GPSCAD decile and the blue line represents the rest of the cohort. In panel A, the unadjusted cumulative incidence of SAD over the study period, accounting for competing deaths, was significantly greater in the top GPSCAD decile compared to the rest of the cohort. In panel B, the unadjusted cumulative incidence of non-SAD, accounting for competing deaths, was significantly lower in the top GPSCAD decile as compared to the rest of the cohort. GPSCAD decile = genome-wide polygenic score for coronary artery disease, non-SAD = non-sudden and non-arrhythmic death, SAD = sudden and/or arrhythmic death.
Relative Risks for SAD and Competing Causes of Death:
In age- and sex-adjusted Fine-Gray models, participants in the top GPSCAD decile were at 1.76-fold (95% CI, 1.23–2.53; p=0.002, Table 2, Central Illustration) increased risk for SAD compared to the remainder of the cohort. After controlling for LVEF, clinical factors including parental history of SAD, and ECG score, the association between the top GPSCAD decile and SAD was not materially altered (sHR 1.77, 95% CI, 1.23–2.54; p=0.002) and further control for CAD severity and renal function also did not alter the association with SAD (sHR1.81, 95% CI, 1.25–2.62; p=0.002). Results were also similar if ICD therapies for VT/VF were included in the SAD outcome (sHR 1.69, 95% CI, 1.20–2.38; p=0.003).
Table 2.
Age-Sex and Multivariable-Adjusted Association of Polygenic Score for Coronary Artery Disease and Sudden Death.
| GPSCAD | |||
|---|---|---|---|
| < Top Decile | Top Decile | P value | |
| SAD vs non-SAD | |||
| SAD events (n=176) | n=139 | n=37 | |
| Age- and Sex-Adjusted sHR (95% CI) |
1.00 (referent) |
1.76 (1.23–2.53) |
0.002 |
| Multivariable† sHR (95% CI) | 1.00 (referent) |
1.77 (1.23–2.54) |
0.002 |
| Non-SAD events (n=833) | n=744 | n=89 | |
| Age- and Sex-Adjusted sHR(95% CI) |
1.00 (referent) |
0.98 (0.79–1.22) |
0.86 |
| Multivariable† sHR (95% CI) | 1.00 (referent) |
1.00 (0.80–1.25) |
0.98 |
| Cardiac vs Non-Cardiac Death | |||
| Cardiac deaths (n= 320) | n=264 | n=56 | |
| Age- and Sex-Adjusted sHR(95% CI) | 1.00 (referent) |
1.56 (1.17–2.08) |
0.003 |
| Multivariable† sHR (95% CI) | 1.00 (referent) |
1.53 (1.15–2.04) |
0.004 |
| Non-cardiac deaths (n= 624) | N=558 | n=66 | |
| Age- and Sex-Adjusted sHR(95% CI) |
1.00 (referent) |
0.94 (0.73–1.22) |
0.65 |
| Multivariable† sHR (95% CI) | 1.00 (referent) |
0.97 (0.74–1.27) |
0.83 |
SAD: sudden arrhythmic death; non-SAD: non sudden arrhythmic death; sHR: subdistribution hazard ratio; CI: confidence interval *95% confidence intervals shown in parentheses.
Multivariable models adjusted with updated values for age, sex, diabetes, hypertension, body mass index, atrial fibrillation, New York Heart Association class, ejection fraction, smoking history, lipid lowering drugs, diuretics, family history of sudden arrhythmic death and ECG score comprised of contiguous Q wave, QRS duration, left ventricular hypertrophy and prolonged JTc).
In contrast, there was no association between the top GPSCAD decile and non-SAD in age-sex adjusted and multivariable models (multivariable sHR 1.00, 95% CI 0.80–1.25; Table 2, Central Illustration). Even when non-SAD deaths were limited to those due to cardiac causes, there remained no association between the top GPSCAD decile and non-SAD (sHR 1.25, 95% 0.76–2.04). Despite the lack of an association with non-SAD cardiac deaths, the association of the top GPSCAD decile with SAD translated into an elevated risk of cardiac death (sHR 1.53, 95% CI 1.15–2.04, Table 2). There was no association between the top GPSCAD decile and non-cardiac deaths (Table 2). In multivariable-adjusted cause specific models, the difference in the association between the top GPSCAD decile and SAD versus non-SAD was statistically significant (adjusted sHRs= 1.76, 95% CI 1.22–2.54 versus 1.01, 95% CI 0.81–1.27 respectively; p for difference = 0.015).
In secondary analyses, the continuous association of the ancestry-adjusted GPSCAD score with SAD was examined according to the distribution in the PREDETERMINE Cohort. The distribution of the GPSCAD score was shifted toward higher scores in this CAD population compared to participants of European ancestry in 1000 Genomes, but with a significant amount of overlap (Supplemental Figure 1). The 90th percentile of the adjusted GPSCAD scores were 1.21 for the 1000 Genomes, 1.44 for SAD controls and 1.88 for SAD cases in PREDETERMINE (Supplemental Figure 1). In multivariable adjusted spline models, the continuous association between the GPSCAD and SAD was non-linear with significant SAD risk elevations observed above a GPSCAD of 1.46, corresponding to the 89 percentile of the GPSCAD within the PREDETERMINE cohort (p value for non-linearity = 0.011, Figure 1).
Figure 1.
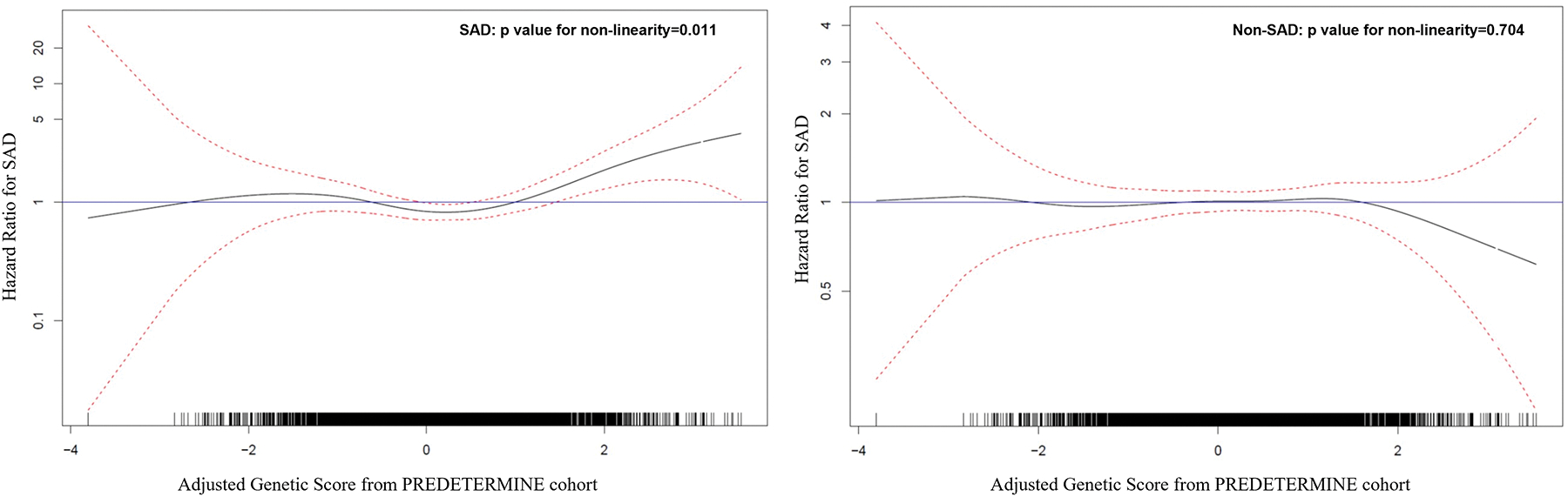
Continuous association between GPSCAD and modes of death. Results of the association between continuous GPSCAD and SAD is shown on the left and non-SAD on the right. The distribution of scores The black line represents the hazard ratio and the red dotted lines represent the 95% confidence interval. The multivariable-adjusted spline model demonstrate a non-linear association with significant SAD risk elevation above a GPSCAD score of 1.46. This was not seen for non-SAD risk. Please refer to the Statistical Analysis section for definitions of variables adjusted for in the model. GPSCAD decile = genome-wide polygenic score for coronary artery disease, non-SAD = non-sudden and non-arrhythmic death, SAD = sudden and/or arrhythmic death.
Improvements in SAD Risk Prediction (Table 3)
Table 3.
Discrimination and Reclassification Indices with Addition of Polygenic Score for Coronary Artery Disease.
| C-Index* | C-Index Differences | IDI (95% CI) | Continuous NRI (95% CI) | Categorical NRI (95% CI) | |
|---|---|---|---|---|---|
| Clinical Model + ECG | 0.704 (0.675, 0.733) | 1 [Reference] | 1 [Reference] | 1 [Reference] | 1 [Reference] |
| Clinical Model + ECG + top Decile GPS CAD | 0.712 (0.676, 0.740) | 0.007 (‒0.003, 0.017), p=0.143 | 0.003 (0.001, 0.006), p=0.004 | 0.140 (0.018, 0.258), p=0.024 | 0.066 (0.021,0.113) p=0.005 |
Clinical Model included age, sex, hypertension, diabetes mellitus, atrial fibrillation, left ventricular ejection fraction, NYHA functional class, smoking status, BMI, lipid lowering medication use, diuretics, and family history of SAD. The GPSCAD model included percentile of genetic risk score. The ECG + GPSCAD model included ECG score in addition to percentile of genetic risk score. IDI, integrated discrimination index. NRI, net reclassification index, Categorical NRI 8-year SAD strata (0–2%, 2%−8%, 8%−15%, >15%).
Adjusted C-index (corrected for optimism) with parametric confidence intervals based on bootstrapping with 100 samples.
To assess whether incorporating information on the top decile of GPSCAD improves measures of SAD risk prediction, we compared the baseline multivariable model including LVEF, clinical, and ECG factors to another which also included the top decile of GPSCAD. The unadjusted c-index for the model including the GPSCAD was 0.741, and after bootstrap adjustment for optimism, the C-Index was 0.712 (0.676–0.740). The adjusted c-index was greater, but not significantly different, than that generated from the model including clinical factors and ECG alone (0.704; 0.675–0.733; p difference =0.14). However, the addition of top GPSCAD decile to the baseline multivariable model significantly improved reclassification indices [continuous NRI 14.0% (1.8%−25.8%), p=0.024 and categorical NRI 6.6% (2.1%−11.3%), p=0.005, Table 3] and discrimination as assessed by the integrated discrimination improvement (IDI), 0.003 (0.001–0.006), p=0.004 ( Table 3). To further examine the incremental value of GPSCAD as compared to the model including clinical markers and ECG score, we reexamined 8-year rates of SAD according to GPSCAD stratified by quartiles of baseline model (Figure 2). The top GPSCAD decile enhanced the absolute rates of SAD, particularly among those deemed at intermediate and high risk by clinical risk markers and ECG (quartile 2, 3 and 4).
Figure 2.
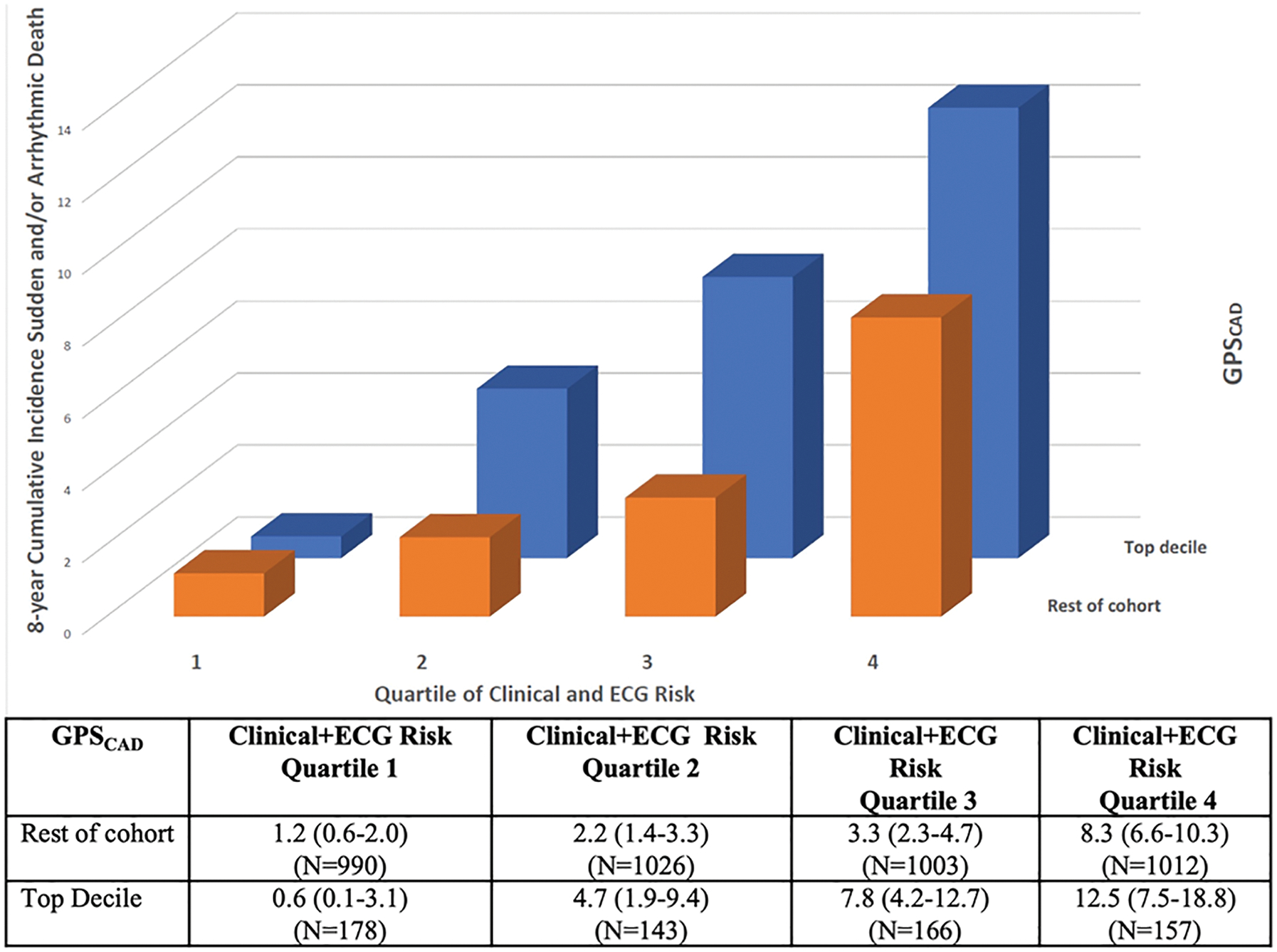
SAD rates according to GPSCAD and quartiles of clinical risk. The orange bars represent the 8 -year cumulative incidence of SAD across quartiles of a model including clinical markers and ECG score and the blue bars represent the incremental value of top GPSCAD decile when added to this baseline model. The top GPSCAD decile enhanced absolute rates of SAD, particularly for those deemed at intermediate and high SAD risk by the baseline model. GPSCAD decile = genome-wide polygenic score for coronary artery disease, SAD = sudden and/or arrhythmic death.
Potential Implications for ICD efficacy and Clinical Trial Design
In exploratory analyes, these data on absolute and proportional risk were used to estimate the theoretical efficacy of ICD therapy as measured by the number needed to treat (NNT) and percent mortality reduction in high-risk subgroups as defined by the top GPSCAD decile. If we assume that ICD therapy would reduce the risk of SAD by 60%, the participants in the top GPSCAD decile would be projected to have a ~17% relative risk reduction in all-cause mortality with ICD therapy and a NNT to save one life of 27, requiring a randomized trial of 3600 to detect that benefit over 8 years with 80% power and yielding a hypothetical CI for the NNT of 16–91. If the population was further risk stratified using a validated ECG score19, those in the top category of ECG score and GPSCAD decile would be estimated to have a ~24% relative risk reduction in all-cause mortality with ICD therapy and the projected NNT to save one life would be reduced to 11, requiring a randomized controlled trial of only 864 participants to demonstrate this mortality risk reduction and yielding a 95% CI of 7–37 for the NNT. If the latter trial followup was shortened to to 5 years, the estimated risk reduction would again be ~24% and the trial would require 1388 participants for 80% power.
Discussion
In this large, contemporary cohort of CAD patients without severe LV dysfunction, patients in the top GPSCAD decile were at 77% increased risk for SAD after accounting for LVEF, clinical factors, and ECG parameters. In contrast, there was no association between top GPSCAD decile and non-SAD, even when limited to cardiac causes, suggesting that the GPSCAD specifically predicts SAD and not other forms of cardiac mortality in patients with CAD. The top GPSCAD decile not only identified a subset of patients with clinically meaningful absolute risk, with a SAD event rate of 8.0% over the study period, but also identified a group where the proportion of deaths due to SAD was greater (29%) compared to the remainder of the cohort (16%). Among those in the highest quartile of clinical risk, the absolute SAD rate was further elevated in the top GPSCAD decile at 12.5% at 8-years.
Coronary artery disease, with and without MI, represents the single most prevalent cause of sudden cardiac death (SCD) and family history of SCD is a strong predictor of SCD4, 5, 24. Two prior case-control studies found that a history of SCD among a first-degree relative is an independent risk factor for experiencing ventricular fibrillation4 or SCD5 in the setting of an acute MI. Furthermore, SCD as a first manifestation of an acute coronary event appears to cluster in certain families4, 5. These data support the hypothesis that there is an important genetic component to SAD risk in CAD. Although early genome-wide association studies reported novel common variants associated with SAD risk25, 26; these results have not been replicated8. However, several studies have found that common variants associated with CAD are associated with elevations in SAD in general population samples7–9. These associations are relatively modest (relative risk estimates of ~1.3) and are thought to be reflective of underlying undiagnosed CAD within the population.
Our study builds on this previous literature and provides several novel findings. First, using a recently validated genome-wide polygenic score for developing CAD11, we demonstrate a strong association between the top GPSCAD decile and SAD in the presence of established CAD independent of parental history of SAD and other clinical risk markers. Thus, the observed association between common CAD associated genetic variants and SAD is not entirely explained by undiagnosed CAD and/or associated risk factors and entends beyond familial predisposition for SAD. Second, as compared to prior studies, we focused on an intermediate risk population where SAD rates are ~10-fold higher than in the general population1; and thus, the relative risk elevations observed in the top GPSCAD decile translated into clinically actionable absolute SAD rates. Third, we show that patients in the top GPSCAD decile were at specific increased risk of SAD without elevations in competing risk of other modes of death such that the proportional risk of SAD was also elevated. These latter novel findings provide support for the concept that SAD risk can be specifically identifed in context of other competing modes of death and have important implications for selecting patient populations that might derive a mortality benefit from the ICD18, 27.
Since the ICD only impacts deaths due to ventricular arrhythmia and has no impact on other causes of death, markers that specifically elevate SAD risk are needed to improve selection of patients that may derive a mortality benefit from the ICD1, 28. Apart from risk stratification with invasive electrophysiology studies in high risk patients29, very few, if any, individual risk markers have been documented to specifically identify increased SAD risk. The current risk stratification tools of LVEF <30–35% and symptomatic HF do not discriminate between SAD and non-SAD causes of cardiac death21, 22, 30; thus, mortality remains high in traditionally defined ‘high-risk’ patients after ICD placement31. To our knowledge, the GPSCAD represents one of the first risk stratification measures documented to be solely associated with SAD risk that can be relatively easily applied to broad populations. In exploratory analyses, we demonstrate how the GPSCAD in concert with the ECG might be used to design an achievable randomized controlled trial demonstrating meaningful mortality reductions and potentially clinically relevant numbers needed to treat to save one life with an ICD.
It is important to note, that not all measures of SAD risk prediction were improved with the addition of GPSCAD to a conventional risk model that included the ECG score. Indices of reclassification and some measures of discrimination were significantly improved; however, the c-Index was not. The NRI, which reflects discrimination and calibration within risk categories, may provide a more sensitive measure of model fit than the c-index which is based solely on rank.32
These data also raise important mechanistic questions. Since non-cardiac causes of death comprise the majority of non-SADs in this and other populations of patients without severe systolic dysfunction1, 33, the lack of an association of the GPSCAD with the overall incidence of non-SAD was not unexpected. However, the biological explanation for why the GPSCAD only predicts SAD and not other forms of cardiac mortality is less clear. It is feasible that those in the highest decile of GPSCAD may have a greater burden of rupture-prone atherosclerotic plaques that are more likely to rapidly transition from stable CAD to an unstable pathophysiology i.e., acute ischemia or acute MI resulting in SAD34, 35. Thus, an increased predisposition toward the occurrence of a severe intermediary acute coronary event may underlie the association of the top GPSCAD decile with SAD. Alternatively, there may also be pleotrophic effects on adverse myocardial scar and/or ion channel remodeling which might increase the predisposition toward fatal arrhythmias among patients with CHD. In support of the latter hypothesis, the relationship between the GPSCAD and arrhythmic events was similar when ICD therapies for VT/VF were included in the endpoint.
Strengths and Limitations
Our study has several strengths including large sample size of well-phenotyped patients with genotyping, stringent adjudication of modes of death, and explicit incorporation of competing risk. However, there are limitations that require further discussion. First, our analysis was performed among those participants in PREDETERMINE with European ancestry because the genome-wide polygenic risk score was derived and tested in a population with this genetic ancestry11 and the numbers of events in non-European ethnicities precluded a rigorous stratified analysis. Since ancestry has a significant influence over allele frequencies, linkage disequilibrium patterns, and effect sizes of common polymorphisms36, the predictive power of GPSCAD score and SAD needs further investigation in diverse populations. Second, although common variants for CAD have been found to predict SAD risk in other populations, these findings within this intermediate-risk CAD population would benefit from further validation before implemented in clinical practice. Third, given the small sample size and lack of adequate replication datasets, the discovery of specific variants associated with SAD and assessments of proportion of heritability were beyond the scope of the current manuscript. Fourth, although all efforts were made to ascertain medical records and circumstances surrounding deaths, and rigorous and widely accepted methods of SAD adjudication were utilized17, 37, the possibility of misclassification of mode of death cannot be excluded. Finally, the estimates for NNT in our exploratory modeling analysis are based on single observations and estimates of ICD benefit in patients with moderate-severe systolic dysfunction22; thus these estimates may be optimistic. The NNT and mortality reduction associated with ICD therapy in this population can only be determined through future randomized trials.
Conclusions
Among a large population with established CAD without severe systolic dysfunction, a polygenic risk score for CAD was strongly and specifically associated with increased risk of SAD. There was no association with other modes of death. The addition of top GPSCAD decile improved SAD risk stratification beyond contributions from LVEF, and clinical markers including parental history of SAD. These findings in aggregate suggest that the GPSCAD is a promising method to improve SAD risk stratification in CAD patients who do not currently qualify for an ICD and warrants further study.
Supplementary Material
Clinical Compentencies.
Competency in Medical Knowledge:
Most sudden and/or arrhythmic death (SAD) occur among patients who do not meet current guideline indications for implantable cardioverter defibrillator therapy. Identifying screening tools for SAD risk in this broader population are needed.
Translational Outlook 1:
GPSCAD score, in combination with other SAD risk markers, provides a promising risk stratification tool for the identification of patients who stand to benefit the most from sudden death preventive therapies but requires further investigation.
Translational Outlook 2:
Randomized clinical trials are needed to demonstrate mortality reductions and numbers needed to treat to save one life with an implantable cardioverter defibrillator using this comprehensive risk stratification strategy.
Funding:
PREDETERMINE was supported by research grants from the National Heart, Lung, and Blood Institute R01HL091069, St. Jude Medical Inc, and St Jude Medical Foundation (to CMA). Genetic analysis was supported by grants 1K08HG010155 and 1U01HG011719 (to A.V.K.) from the National Human Genome Research Institute.
Abbreviations
- ICD
implantable cardioverter-defibrillator
- SAD
sudden and/or arrhythmic death
- CAD
coronary artery disease
- GPS
genome-wide polygenic score
- LVEF
left ventricular ejection fraction
- MI
myocardial infarction
- VT
ventricular tachycardia
- VF
ventricular fibrillation
- sHR
subdistribution hazard ratios
- ECG
electrocardiogram
- IDI
integrated discrimination improvement
- NRI
net reclassification improvement
- NNT
number needed to treat
Footnotes
Disclosures: A.V.K. is an employee and holds equity in Verve Therapeutics; has served as a scientific advisor to Amgen, Maze Therapeutics, Navitor Pharmaceuticals, Sarepta Therapeutics, Novartis, Silence Therapeutics, Korro Bio, Veritas International, Color Health, Third Rock Ventures, Illumina, Foresite Labs, and Columbia University (NIH); received speaking fees from Illumina, MedGenome, Amgen, and the Novartis Institute for Biomedical Research; received a sponsored research agreement from IBM Research, and is listed as a co-inventor on a patent application for use of imaging data in assessing body fat distribution and associated cardiometabolic risk. The remaining authors have nothing to disclose.
References:
- 1.Chatterjee NA, Moorthy MV, Pester J, et al. Sudden Death in Patients With Coronary Heart Disease Without Severe Systolic Dysfunction. JAMA Cardiol. 2018;3:591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Myerburg RJ and Junttila MJ. Sudden cardiac death caused by coronary heart disease. Circulation. 2012;125:1043–52. [DOI] [PubMed] [Google Scholar]
- 3.Stecker EC, Vickers C, Waltz J, et al. Population-based analysis of sudden cardiac death with and without left ventricular systolic dysfunction: two-year findings from the Oregon Sudden Unexpected Death Study. J Am Coll Cardiol. 2006;47:1161–6. [DOI] [PubMed] [Google Scholar]
- 4.Dekker LR, Bezzina CR, Henriques JP, et al. Familial sudden death is an important risk factor for primary ventricular fibrillation: a case-control study in acute myocardial infarction patients. Circulation. 2006;114:1140–5. [DOI] [PubMed] [Google Scholar]
- 5.Kaikkonen KS, Kortelainen ML, Linna E and Huikuri HV. Family history and the risk of sudden cardiac death as a manifestation of an acute coronary event. Circulation. 2006;114:1462–7. [DOI] [PubMed] [Google Scholar]
- 6.Jabbari R, Engstrom T, Glinge C, et al. Incidence and risk factors of ventricular fibrillation before primary angioplasty in patients with first ST-elevation myocardial infarction: a nationwide study in Denmark. J Am Heart Assoc. 2015;4:e001399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newton-Cheh C, Cook NR, VanDenburgh M, Rimm EB, Ridker PM and Albert CM. A common variant at 9p21 is associated with sudden and arrhythmic cardiac death. Circulation. 2009;120:2062–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ashar FN, Mitchell RN, Albert CM, et al. A comprehensive evaluation of the genetic architecture of sudden cardiac arrest. Eur Heart J. 2018;39:3961–3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hernesniemi JA, Lyytikainen LP, Oksala N, et al. Predicting sudden cardiac death using common genetic risk variants for coronary artery disease. Eur Heart J. 2015;36:1669–75. [DOI] [PubMed] [Google Scholar]
- 10.Deo R and Albert CM. Epidemiology and genetics of sudden cardiac death. Circulation. 2012;125:620–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khera AV, Chaffin M, Aragam KG, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50:1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hindy G, Aragam KG, Ng K, et al. Genome-Wide Polygenic Score, Clinical Risk Factors, and Long-Term Trajectories of Coronary Artery Disease. Arterioscler Thromb Vasc Biol. 2020;40:2738–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nikpay M, Goel A, Won HH, et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47:1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA and Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9. [DOI] [PubMed] [Google Scholar]
- 15.McCarthy S, Das S, Kretzschmar W, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48:1279–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM and Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hinkle LE Jr. and Thaler HT. Clinical classification of cardiac deaths. Circulation. 1982;65:457–64. [DOI] [PubMed] [Google Scholar]
- 18.Younis A, Goldberger JJ, Kutyifa V, et al. Predicted benefit of an implantable cardioverter-defibrillator: the MADIT-ICD benefit score. Eur Heart J. 2021;42:1676–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chatterjee NA, Tikkanen JT, Panicker GK, et al. Simple electrocardiographic measures improve sudden arrhythmic death prediction in coronary disease. Eur Heart J. 2020;41:1988–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lunn M and McNeil D. Applying Cox regression to competing risks. Biometrics. 1995;51:524–32. [PubMed] [Google Scholar]
- 21.Greenberg H, Case RB, Moss AJ, et al. Analysis of mortality events in the Multicenter Automatic Defibrillator Implantation Trial (MADIT-II). J Am Coll Cardiol. 2004;43:1459–65. [DOI] [PubMed] [Google Scholar]
- 22.Packer DL, Prutkin JM, Hellkamp AS, et al. Impact of implantable cardioverter-defibrillator, amiodarone, and placebo on the mode of death in stable patients with heart failure: analysis from the sudden cardiac death in heart failure trial. Circulation. 2009;120:2170–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Delgado C, Baweja M, Crews DC, et al. A Unifying Approach for GFR Estimation: Recommendations of the NKF-ASN Task Force on Reassessing the Inclusion of Race in Diagnosing Kidney Disease. J Am Soc Nephrol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friedlander Y, Siscovick DS, Weinmann S, et al. Family history as a risk factor for primary cardiac arrest. Circulation. 1998;97:155–60. [DOI] [PubMed] [Google Scholar]
- 25.Arking DE, Junttila MJ, Goyette P, et al. Identification of a sudden cardiac death susceptibility locus at 2q24.2 through genome-wide association in European ancestry individuals. PLoS Genet. 2011;7:e1002158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bezzina CR, Pazoki R, et al. Genome-wide association study identifies a susceptibility locus at 21q21 for ventricular fibrillation in acute myocardial infarction. Nat Genet. 2010;42:688–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kristensen SL, Levy WC, Shadman R, et al. Risk Models for Prediction of Implantable Cardioverter-Defibrillator Benefit: Insights From the DANISH Trial. JACC Heart Fail. 2019;7:717–724. [DOI] [PubMed] [Google Scholar]
- 28.Solomon SD and Chatterjee NA. Who Benefits From Implantable Cardioverter-Defibrillators?: Integrating Absolute, Proportional, and Competing Risk. J Am Coll Cardiol. 2017;69:2619–2621. [DOI] [PubMed] [Google Scholar]
- 29.Buxton AE, Lee KL, Hafley GE, et al. Relation of ejection fraction and inducible ventricular tachycardia to mode of death in patients with coronary artery disease: an analysis of patients enrolled in the multicenter unsustained tachycardia trial. Circulation. 2002;106:2466–72. [DOI] [PubMed] [Google Scholar]
- 30.Solomon SD, Zelenkofske S, McMurray JJ, et al. Sudden death in patients with myocardial infarction and left ventricular dysfunction, heart failure, or both. N Engl J Med. 2005;352:2581–8. [DOI] [PubMed] [Google Scholar]
- 31.Merchant FM, Levy WC and Kramer DB. Time to Shock the System: Moving Beyond the Current Paradigm for Primary Prevention Implantable Cardioverter Defibrillator Use. J Am Heart Assoc. 2020;9:e015139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cook NR. Use and misuse of the receiver operating characteristic curve in risk prediction. Circulation. 2007;115:928–35. [DOI] [PubMed] [Google Scholar]
- 33.Vaduganathan M, Claggett BL, Chatterjee NA, et al. Sudden Death in Heart Failure With Preserved Ejection Fraction: A Competing Risks Analysis From the TOPCAT Trial. JACC Heart Fail. 2018;6:653–661. [DOI] [PubMed] [Google Scholar]
- 34.Ye S, Willeit J, Kronenberg F, Xu Q and Kiechl S. Association of Genetic Variation on Chromosome 9p21 With Susceptibility and Progression of Atherosclerosis: A Population-Based, Prospective Study. J Am Coll Cardiol. 2008;52:378–384. [DOI] [PubMed] [Google Scholar]
- 35.Harismendy O, Notani D, Song X, et al. 9p21 DNA variants associated with coronary artery disease impair interferon-gamma signalling response. Nature. 2011;470:264–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin AR, Gignoux CR, Walters RK, et al. Human Demographic History Impacts Genetic Risk Prediction across Diverse Populations. Am J Hum Genet. 2017;100:635–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fishman GI, Chugh SS, Dimarco JP, et al. Sudden cardiac death prediction and prevention: report from a National Heart, Lung, and Blood Institute and Heart Rhythm Society Workshop. Circulation. 2010;122:2335–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.