

Abstract
X-linked Charcot-Marie-Tooth disease type 1 (CMT1X) is a demyelinating neuropathy resulting from loss-of-function mutations affecting the GJB1/connexin 32 (Cx32) gene. We previously showed functional and morphological improvement in Gjb1-null mice following AAV9-mediated delivery of human Cx32 driven by the myelin protein zero (Mpz) promoter in Schwann cells. However, CMT1X mutants may interfere with virally delivered wild-type (WT) Cx32. To confirm the efficacy of this vector also in the presence of CMT1X mutants, we delivered AAV9-Mpz-GJB1 by lumbar intrathecal injection in R75W/Gjb1-null and N175D/Gjb1-null transgenic lines expressing Golgi-retained mutations, before and after the onset of the neuropathy. Widespread expression of virally delivered Cx32 was demonstrated in both genotypes. Re-establishment of WT Cx32 function resulted in improved muscle strength and increased sciatic nerve motor conduction velocities in all treated groups from both mutant lines when treated before as well as after the onset of the neuropathy. Furthermore, morphological analysis showed improvement of myelination and reduction of inflammation in lumbar motor roots and peripheral nerves. In conclusion, this study provides proof of principle for a clinically translatable gene therapy approach to treat CMT1X before and after the onset of the neuropathy, even in the presence of endogenously expressed Golgi-retained Cx32 mutants.
Keywords: X-linked Charcot-Marie-Tooth, gene therapy, AAV9, Golgi-retained mutations, demyelination, R75W, N175D, Connexin 32
Graphical abstract
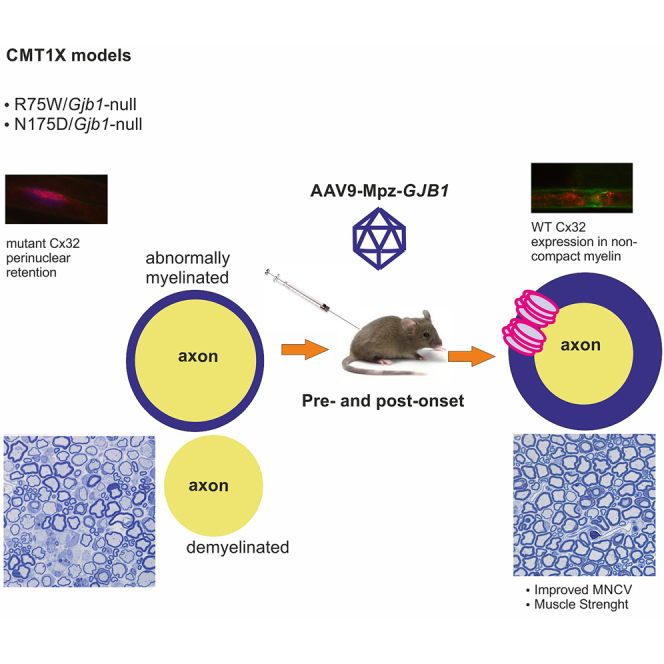
Kagiava and colleagues describe Schwann cell-targeted delivery of AAV9 following intrathecal injection in order to provide proof of principle for a clinically translatable gene therapy approach to treat CMT1X before and after the onset of the neuropathy, even in the presence of endogenously expressed Golgi-retained Cx32 mutants.
Introduction
X-linked Charcot-Marie-Tooth (CMT1X) is the second most common form of Charcot-Marie-Tooth (CMT) inherited neuropathy, affecting 7%–15% of all CMT patients. CMT1X affects mainly men with onset at the age of 5–20 years, while women usually present milder symptoms at a later stage.1,2,3,4,5,6 The disease is characterized by slowly progressive weakness and atrophy of distal limb muscles, loss of reflexes, and sensory loss.7,8,9 Nerve conduction velocities are typically reduced in affected men but may be normal in heterozygous women, while reduction in compound muscle action potential (CMAP) amplitude resulting from axonal degeneration and fiber loss is seen in both genders.10,11 Mutations in the GJB1 gene, which encodes the gap junction (GJ) protein connexin 32 (Cx32), are the cause of CMT1X neuropathy.12,13,14 Cx32 is located at the non-compact myelin at paranodal loops and Schmidt-Lanterman incisures of myelinating Schwann cells. Here, it forms GJ channels that traverse the myelin sheath and are responsible for the homeostasis and axon-glial signaling.15,16,17
There are over 400 mutations reported so far in CMT1X patients (https://neuropathybrowser.zuchnerlab.net/#/) affecting all domains of Cx32, even the non-coding regions.18,19 Many CMT1X mutations expressed in HeLa cells, including missense and in-frame mutations such as the R75W, N175D, and T55I, cause intracellular retention in the Golgi or the endoplasmic reticulum (ER), failure to form GJ channels, and finally degradation of the mutant Cx32 by the proteasomes or the lysosomes.20,21,22,23,24,25,26 However, several of them can still form GJ-like plaques and in certain cases even retain their ability to form functional GJ-plaques.27,28 In vivo data of the expression of Cx32 mutations show similarities to the data obtained in HeLa cells with no signs of toxic effects but rather loss-of-function mechanisms.25,29 These experimental data replicate the results of clinical studies showing that, despite the fact CMT1X patients express different types of mutations, they present a similar phenotype and disease progression.30
Despite the lack of gain-of-function effects on CMT1X mutations derived from hemizygous state, in vitro studies showed that some CMT1X mutants can have dominant-negative effects when co-expressed with wild-type (WT) Cx32. Co-expression mostly of Golgi-retained mutants such as R75W and N175D, but not of ER-retained mutants such as T55I, with WT Cx32 resulted in impaired function of WT Cx3216,22,23 and even caused intracellular retention of WT Cx32.22 In vivo data further demonstrated that ER-retained mutants do not cause any nerve pathology when expressed on a WT background, while expression of Golgi-retained mutants resulted in a mild nerve pathology on a WT background, indicating that they have dominant-negative effects on co-expressed WT Cx32 protein.29,31
In our previous studies we demonstrated therapeutic benefit in different CMT1X mouse models by single lumbar intrathecal or intra-sciatic injection of viral vectors expressing the human Cx32 32,33,34,35,36. Delivery of a lentiviral or adeno-associated viral (AAV) vector carrying human WT Cx32 resulted in the partial rescue of demyelinating neuropathy in Gjb1-null mice before and after the onset of the neuropathy.32,35 Although the results of these studies were encouraging, the use of the Gjb1-null mouse model does not fully replicate the clinical circumstances since most CMT1X patients express mutated forms of Cx32 rather than not expressing the protein. In order to proceed with a more translatable approach, we used mouse models expressing ER-retained (T55I) or Golgi-retained (R75W and N175D) mutants in a Gjb1-null background and delivered the WT Cx32 using a lentiviral vector. Our data showed that expression of Cx32 rescued the demyelinating neuropathy only in the model expressing the ER-retained but not in models expressing the Golgi-retained mutants.33 The limited ability of lentiviral vector to rescue the demyelinating neuropathy in Golgi-retained mutants indicated that higher expression levels of WT Cx32 might be needed in order to overcome the dominant-negative effects of mutant Cx32. The use of AAV vectors that remain episomal and are not under the control of the host genome transcription regulation in general provides higher expression levels and can effectively cross the blood-brain barrier (BBB).37,38,39,40,41,42 We previously showed that the AAV9 serotype, which is already used for the treatment of other neurological disorders such as spinal muscular atrophy (SMA), can provide strong expression of WT Cx32, resulting in the partial rescue of demyelinating neuropathy in the Gjb1-null model before and after the onset of the neuropathy.32 Thus, it may offer an alternative to treat all CMT1X mutations, including the Golgi-retained ones.
Based on these results, we used AAV9 to deliver the human WT Cx32 for the treatment of the demyelinating neuropathy in two CMT1X mouse models expressing Golgi-retained mutants, the R75W and the N175D on a Gjb1-null background. The vector was delivered by lumbar intrathecal injection before and after the onset of the neuropathy in order also to test the efficacy when nerve pathology is already established. Our results show that the use of AAV9 provides widespread expression of Cx32, which can rescue the demyelinating neuropathy in both Golgi-retained mouse models as indicated by functional and morphological assessments. Furthermore, our approach was effective at both early and late time points, although early intervention was more effective.
Results
Cx32 expression following intrathecal AAV9-Mpz.GJB1 injection in CMT1X mutant mice
In order to confirm the baseline transgene expression in the CMT1X mutant lines, we first used immunoblot analysis to detect EGFP and Cx32 levels in lumbar roots and sciatic nerve lysates from untreated R75W/Gjb1-null and N175D/Gjb1-null mice. These experiments confirmed the presence of both proteins in the peripheral nervous system (PNS) tissue lysates from mutant lines showing that expression levels of both proteins appeared higher in the N175D/Gjb1-null line, although not significantly compared to the R75W/Gjb1-null line (Figure S1).
In order to access expression of the virally delivered Cx32 in the two CMT1X transgenic lines, we immunostained for Cx32 and Caspr, a paranodal marker, in lumbar roots and sciatic nerves 6 weeks following intrathecal injection of the AAV9 vector carrying the GJB1 gene. These experiments confirmed the expression of Cx32 in both lumbar roots (Figures 1C and 1D) and sciatic nerves (Figures 1E and 1F) of injected mice at paranodal non-compact myelin areas where Cx32 GJ channels are normally formed, despite the presence of the mutant protein in the perinuclear cytoplasm of Schwann cells (Figures 1A–1F). Cx32 was also detected in the perinuclear cytoplasm of Schwann cells of non-injected mice, but we did not detect any at the paranodal areas of these mice (Figures 1A and 1B). Quantification of Cx32-immunoreactive paranodal areas among all paranodal areas visualized in each tissue based on Caspr paranodal labeling showed that expression rates (percentage of paranodal myelin areas that were immunoreactive for Cx32) were similar between the two mutants in both lumbar roots and sciatic nerves (Figures 1G and 1H). Expression rates of Cx32 in lumbar roots of R75W/Gjb1-null mice reached 50.3% ± 4.66% (n = 6 mice) and in N175D/Gjb1 null 58.1% ± 4.69% (n = 6 mice; p > 0.05). Expression rates in sciatic nerves reached 53.6% ± 3.23% in R75W/Gjb1 null mice (n = 10 nerves; n = 5 mice) and 55.1% ± 5.19% in N175D/Gjb1 null (n = 10 nerves; n = 5 mice; p > 0.05).
Figure 1.

Cx32 expression in Schwann cells of PNS tissues following intrathecal delivery of AAV9-Mpz.GJB1 in R75W/Gjb1-null and N175D/Gjb1-null mice
In paranodal myelin areas labeled with Caspr (green) (white arrows in A–F), in contrast to non-injected mice with absence of Cx32 expression (A and B), the virally delivered human WT Cx32 (red) is detected in lumbar roots (C and D) and sciatic nerves (E and F) of AAV9-Mpz.GJB1 injected mice (red arrows; C–F) 6 weeks post injection. Mutant Cx32 (red; asterisks; A–F) was detected in the perinuclear cytoplasm area of myelinating Schwann cells both in the tissues from non-injected (A and B) as well as in the tissues of AAV9-Mpz.GJB1-injected mice (C–F). Paranodal areas are shown in separate and merged channels under the overview images. Quantification of Cx32 immunoreactive paranodal areas showed similar expression rates of the virally delivered WT Cx32 in lumbar roots (G) and sciatic nerve teased fibers (H) of AAV9-Mpz.GJB1 R75W/Gjb1 null and N175D/Gjb1 null mice. Values represent mean±SEM. Scale bar, 50 μm.
Improvement of motor performance in pre-onset-treated R75W/Gjb1-null and N175D/Gjb1-null mice
Grip strength analysis was performed at baseline before the initiation of the experiment at 2 months of age and then at 4, 6, and 8 months of age at 2, 4, and 6 months post injection, respectively (Figure 2A). At baseline, at the age of 2 months, both groups in both genotypes did not differ (Figures 2B and 2G). The force generated by the hindlimbs of the mock group of the R75W/Gjb1-null mice reached 70.25 ± 3.19 g (n = 26), while the full group reached 76.86 ± 4.19 g (n = 26; p > 0.05). For the N175D/Gjb1-null, the mock group reached 72.55 ± 3.55 g (n = 24) and the full reached 69.61 ± 3.87 g (n = 24; p > 0.05). At 4 months of age, 2 months post treatment, we observed an improvement in the R75W/Gjb1-null-treated compared to the mock group (Figure 2C). The force reached 69.30 ± 3.37g (n = 25) in the mock group compared to 93.02 ± 3.09 g in the treated group (n = 26; p < 0.0001; Figure 2C). For the N175D/Gjb1-null groups, force reached 67.47 ± 4.15 g in the mock group (n = 24) compared to 90.22 ± 3.69 g in the treated group (n = 24; p = 0.002; Figure 2H). At 6 months of age, 4 months post treatment, we observed an improvement in both R75W/Gjb1-null and N175D/Gjb1-null fully treated mice compared to the respective mock groups (Figures 2D and 2I). R75W/Gjb1-null mock-treated mice reached 69.33 ± 2.52 g (n = 25), while treated mice reached 102.75 ± 4.29 g (n = 26; p < 0.0001; Figure 2D). Likewise, N175/Gjb1-null mock-treated mice reached 56.76 ± 2.98 g (n = 24) compared to 85.44 ± 3.66 g in treated mice (n = 23; p < 0.0001; Figure 2I). Finally, at 8 months of age, 6 months post treatment, fully treated mice of both lines showed improvement in the force generated by the hindlimbs. The R75W/Gjb1-null mock-treated group reached 69.36 ± 3.49 g (n = 25) compared to 105.98 ± 5.30 g in treated animals (n = 26; p < 0.0001; Figure 2E). N175D/Gjb1-null mock-treated groups reached 65.15 ± 3.27 g (n = 24) compared to 100.95 ± 4.18 g in treated mice (n = 23; p < 0.0001; Figure 2J). Over time, improvement was evident in treated mice of both lines examined, as indicated in Figures 2F and 2K, whereas mock-treated mice either remained stable (Figure 2F), as in the case of R75W/Gjb1-null, or showed decreasing strength, as in the case of the N175D/Gjb1-null line (Figure 2K).
Figure 2.

Analysis of functional outcomes in R75W/Gjb1-null and N175D/Gjb1-null mice injected pre-onset with AAV9-Mpz.GJB1 compared to AAV9-Mpz.Egfp (mock)
Diagram showing the pre-onset treatment trial design with timing of functional and morphological analysis (A). Results of foot grip analysis of R75W/Gjb1-null (B–F) and N175D/Gjb1-null mice (G–K) comparing AAV9-Mpz.GJB1-treated (GJB1) and mock-treated mice, as indicated. While there were no differences between the treatment groups before the injection (B and G), 2 months post-injection grip strength improved in treated mice of both genotypes compared to the mock groups (C and H). Improved performance continued at 4 months post injection (6 months old) (D and I) as well as at 6 months post injection (8 months of age) in treated mice of both mutant lines compared to mock groups (E and J). Longitudinal analysis within each group showed improved motor performance over time in treated R75W/Gjb1-null and N175D/Gjb1-null mutants while mock groups either remained stable or decreased (F and K). Sciatic nerve motor conduction velocity (MNCV) also showed significant improvement 6 months after treatment compared to the mock groups in both R75W/Gjb1-null-treated (L) and N175D/Gjb1-null-treated (N) mice. The amplitude of the compound muscle action potential (CMAP) was also evaluated in both genotypes with no differences observed between treated and mock groups (M and O). Values represent mean±SEM. Statistical analysis was performed using Mann-Whitney test (B–E, G–J, and L–O) or one-way ANOVA (F and K).
Although foot grip analysis proved to be the most sensitive test for the CMT1X mouse models, we also performed rotarod analysis. Motor balance and coordination were determined according to our previous studies using an accelerating rotarod apparatus.32 As with the foot grip analysis, mice were tested before the initiation of the experiment at 2 months of age and at 4, 6, and 8 months of age. Training of animals consisted of three trials per day with 15-min intervals for resting between trials for three consecutive days. Mice were placed on the rod with the speed gradually increasing from 4 to 40 rotations per minute (rpm). Testing was performed on the fourth day using two different speeds, 20 and 32 rpm. Latency (seconds) to fall was calculated for each speed.
At baseline, at the age of 2 months, both groups in both genotypes did not differ, as indicated in Figures S2A and S2I. At 4 months of age, 2 months post treatment, we observed an improvement in the R75W/Gjb1-null fully treated mice in both speeds examined (Figures S2B and S2F). In contrast, the N175D/Gjb1-null groups did not show any differences in both speeds examined at the same time point (Figures S2J and S2N). At 4 months post injection, similar results were obtained in both speeds examined. Fully treated R75W/Gjb1-null mice remained on the rotarod for a longer time compared to the mock-treated mice (Figures S2C and S2G), while N175D/Gjb1-null showed an improvement only at low speed (Figures S2K and S2O). Finally, 6 months post treatment we observed improved performance in R75W/Gjb1-null at both speeds (Figures S2D and S2H), while in the N175D/Gjb1-null we observed improved performance only at the low speed (Figures S2L and S2P).
Improved nerve conduction velocities in pre-onset-treated R75W/Gjb1-null and N175D/Gjb1-null mice
Motor nerve conduction velocity (MNCV) was measured in both mutant lines 6 months after pre-onset treatment in vivo from bilateral sciatic nerves following stimulation in anesthetized animals at the sciatic notch and distally at the knee. MNCV was improved in both R75W/Gjb1-null- and N175D/Gjb1-null-treated mice compared to their mock littermates. R75W/Gjb1-null mock-treated mice reached 27.52 ± 0.72 m/s (n = 11), while fully treated mice reached 38.39 ± 0.89 m/s (n = 11; p < 0.0001; Figure 2L). Likewise, N175D/Gjb1-null mock-treated mice reached 29.62 ± 0.50 m/s (n = 11), while treated mice reached 39.10 ± 0.98 m/s (n = 12; p < 0.0001; Figure 2N). We further measured the CMAP amplitude in both mutants examined. We did not observe any differences in the CMAP amplitude in either the R75W/Gjb1-null or N175D/Gjb1-null mice, as indicated in Figures 2M and 2O, respectively.
Neurofilament light concentrations in pre-onset-treated R75W/Gjb1-null and N175D/Gjb1-null mice
Further to behavioral and electrophysiological analysis, we measured the concentration of neurofilament light (NF-L) in plasma samples of untreated mice to determine the concentration of NF-L at different time points and then to examine whether NF-L could be a treatment-responsive biomarker in the CMT1X mouse models used in this study. For this, we used plasma samples from mock and fully treated R75W and N175D/Gjb1-null mice. In the R75W/Gjb1-null group, we observed an increase in the concentration of NF-L from 2 to 4 months of age, followed by a decrease at 6 and 8 months (p < 0.05; Figure S4A). In contrast, N175D/Gjb1-null mice showed a constant increase of NF-L concentration over time and up to 10 months of age (Figure S4B). However, when we compared the mock to the fully treated groups in the treatment trials, we did not observe any significant differences between the two groups in either of the two genotypes examined after pre-onset (Figures S4C and S4D) or post-onset treatment (Figures S4E and S4F).
Improved nerve pathology in pre-onset-treated R75W/Gjb1-null and N175D/Gjb1-null mice
We compared the pathological changes between treated and mock-treated 8-month-old R75W/Gjb1-null and N175D/Gjb1-null mice in lumbar ventral roots, in the femoral motor nerve, and in the mid-sciatic nerve. Improved pathology was observed in all tissues of R75W/Gjb1-null fully treated mice examined. Both abnormally myelinated fibers and foamy macrophages were decreased in treated mice compared to the mock group. In anterior lumbar roots (Figures 3A, 3B, 3E, and 3F), the ratio of abnormally myelinated fibers reached 0.21 ± 0.02 (n = 15) in mock-treated mice compared to 0.07 ± 0.01 in treated mice (n = 16; p < 0.0001; Figure 3I). Likewise, the numbers of foamy macrophages counted per 1,000 myelinated fibers were decreased in fully treated mice, reaching 3.65 ± 0.62 (n = 16) compared to 10.04 ± 0.91 in the mock group (n = 15; p < 0.0001; Figure 3J). The femoral motor nerves showed similar results (Figures 3M, 3N, 3Q, and 3R): the ratio of abnormally myelinated fibers decreased to 0.14 ± .0.01 (n = 12) in treated compared to mock-treated mice (0.27 ± 0.01; n = 11; p < 0.0001; Figure 3U), while foamy macrophages were decreased in treated (4.22 ± 0.47; n = 12) compared to the mock-treated mice (8.53 ± 0.53; n = 11; p < 0.0001; Figure 3V). Finally, improvement was also observed in both values of abnormally myelinated fibers and foamy macrophages in sciatic nerves (Figures S5A, S5B, S5E, and S5F), where abnormally myelinated fibers reached 0.04 ± 0.01 (n = 12) in treated and 0.09 ± 0.00 (n = 11; p < 0.0001; Figure S5I) in mock-treated mice. At the same time, the numbers of the foamy macrophages decreased in treated mice (2.73 ± 0.29; n = 12) compared to mock-treated mice (5.59 ± 0.42; n = 11; p < 0.0001; Figure S5J).
Figure 3.

Morphological analysis of anterior lumbar roots and femoral motor nerves of pre-onset-treated R75W/Gjb1-null and N175D/Gjb1-null mice at 8 months of age (6 months after treatment)
Representative images of semithin sections of R75W/Gjb1-null (A, B, E, and F) and N175D/Gjb1-null (C, D, G, and H) anterior motor lumbar spinal roots attached to the spinal cord, at low and higher magnification, as indicated. AAV9-Mpz.GJB1-injected mice show improved root myelination compared to mock-treated littermates with fewer demyelinated (∗) and remyelinated (r) fibers, as well as reduced numbers of foamy macrophages (arrows in E and G). Improved root pathology is confirmed by the morphometric analysis results, showing decreased ratios of abnormally myelinated fibers and reduced numbers of macrophages per 1,000 fibers in treated mice except for the macrophage reduction in N175D/Gjb1-null roots not reaching statistical significance (I–L). Semithin sections of femoral motor nerves of R75W/Gjb1-null (M, N, Q, and R) and N175D/Gjb1-null (O, P, S, and T) at lower and higher magnification as indicated, and corresponding morphometric analysis results (U–X) from mock- and full-vector-treated mice as indicated. AAV9-Mpz.GJB1-injected mice show improved myelination in femoral nerves compared to mock-treated littermates with fewer demyelinated and remyelinated fibers as well as reduction in the numbers of foamy macrophages, confirmed by quantification. Values represent mean±SEM. Statistical analysis was performed using the Mann-Whitney test.
The N175D/Gjb1-null early-treatment trial showed similar results to R75W/Gjb1-null mice. In anterior lumbar roots (Figures 3C, 3D, 3G, and 3H), the ratio of abnormally myelinated fibers reached 0.25 ± 0.01 (n = 11) in mock compared to 0.15 ± 0.02 in treated mice (n = 11; p = 0.0002; Figure 3K), while the numbers of foamy macrophages showed a trend for decrease in treated (9.18 ± 1.66; n = 11) compared to the mock-treated mice (13.35 ± 1.21; n = 11; p > 0.05; Figure 3L). Likewise, in the femoral motor nerves (Figures 3O, 3P, 3S, and 3T), the ratio of abnormally myelinated fibers decreased to 0.16 ± .0.01 (n = 11) in the fully treated compared to mock-treated mice (0.26 ± 0.01; n = 11; p < 0.0001; Figure 3W), and foamy macrophages were decreased in treated (3.89 ± 0.44; n = 11) compared to mock-treated littermates (6.85 ± 0.65; n = 11; p = 0.0052; Figure 3X). Finally, improvement was also observed in the sciatic nerves (Figures S5C, S5D, S5G, and S5H) where abnormally myelinated fibers reached 0.04 ± 0.00 (n = 11) in treated mice and 0.10 ± 0.01 (n = 11; p < 0.0001; Figure S5K) in mock-treated mice. Furthermore, foamy macrophages were decreased in fully treated (4.11 ± 0.25; n = 11) compared to mock-treated littermates (6.70 ± 0.43; n = 11; p < 0.0001; Figure S5L).
Improvement of functional performance in post-onset-treated R75W/Gjb1-null and N175D/Gjb1-null mice
Following the overall positive response to pre-onset treatment demonstrated in both the R75W and N175D/Gjb1-null mice CMT1X transgenic lines, we further evaluated whether later treatment given well after the onset of functional and pathological abnormalities could also provide a therapeutic benefit. Behavioral analysis including grip strength and rotarod testing was carried out as described for the pre-onset groups at baseline at 6 months of age and every 2 months until the end of the study at the age of 10 months. Grip strength evaluation showed that at baseline, at the age of 6 months, both groups of both genotypes had similar strength (Figures 4B and 4F). The force generated by the hindlimbs of the mock group of the R75W/Gjb1-null mice reached 89.17 ± 4.64 g (n = 38), while the full group reached 78.88 ± 3.17 g (n = 41; p > 0.05). For the N175D/Gjb1-null, the mock group reached 67.09 ± 2.53 g (n = 32) and the full reached 65.33 ± 2.84 g (n = 38; p > 0.05). At 8 months of age, 2 months post treatment, we observed an improvement in both genotypes in fully treated mice compared to the mock groups (Figures 4C and 4G). In the R75W/Gjb1-null line, the force of the mock group reached 76.85 ± 4.26 g (n = 38) compared to 111.34 ± 4.11 g in the treated group (n = 41; p < 0.0001; Figure 4C). In the N175D/Gjb1-null line, the mock group reached 77.25 ± 13.82 g (n = 32) compared to 84.71 ± 4.72 g in the treated group (n = 38; p = 0.002; Figure 4G). Finally, at 10 months of age, 4 months post treatment, we observed again a significant improvement of grip strength in both R75W/Gjb1-null- and N175D/Gjb1-null-treated mice compared to the respective mock groups (Figures 4D and 4H). R75W/Gjb1-null mice reached a strength of 89.23 ± 5.42 g in the mock (n = 38) compared to 125.04 ± 5.42 g in the treatment group (n = 41; p < 0.0001; Figure 4D). Likewise, N175D/Gjb1-null mice reached a force of 63.25 ± 3.85 g in the mock (n = 32) compared to 80.01 ± 4.32 g in the treated group (n = 38; p = 0.0081; Figure 4H). Improved function was evident from baseline to the end of the experiment in both lines examined, as indicated in Figures 4E and 4I, with increased force from 6 to 10 months of age in treated mice compared to the mock groups, which showed either stable force (Figure 4E), as in the case of R75W/Gjb1-null, or decreasing force, as in the case of N175D/Gjb1-null (Figure 4I).
Figure 4.

Analysis of functional outcomes in R75W/Gjb1-null and N175D/Gjb1-null mice treated post onset with AAV9-Mpz.GJB1 treated compared to AAV9-Mpz.Egfp-treated (mock) littermates
Diagram showing the post-onset treatment trial design with timing of functional and morphological analysis (A). Results of foot-grip analysis of R75W/Gjb1-null (B–E) and N175D/Gjb1-null mice (F–I) comparing AAV9-Mpz.GJB1-treated (GJB1) and mock-treated mice, as indicated. There were no differences between the treatment groups before the injection (B and F), but, 2 months post injection, grip strength improved in treated mice of both genotypes compared to the mock groups (C and G). Improved performance continued at 4 months post injection (10 months of age) in treated mice of both CMT1X lines compared to controls (D and H). Longitudinal analysis within each group showed improved motor performance over time in treated mice of both genotypes, while mock groups did not improve (E and I). Sciatic nerve motor conduction velocity (MNCV) also showed significant improvement 4 months after treatment compared to the mock groups in both R75W/Gjb1-null-treated (J) and N175D/Gjb1-null-treated (L) mice. CMAP amplitudes showed no significant differences between treated and mock groups (K and M). Values represent mean±SEM. Statistical analysis was performed using Mann-Whitney or one-way ANOVA.
Motor balance and coordination were further evaluated by rotarod testing at 6, 8, and 10 months of age, as described above for the pre-onset group. At baseline, at the age of 6 months, both groups in both genotypes showed similar performance at both speeds examined (Figures S3A, S3D, S3G, and S3J). However, late-treated animals from both transgenic lines failed to show any improvement in rotarod performance compared to the respective mock groups 2 and 4 months post injection (Figure S3).
Improved nerve conduction velocities in post-onset-treated R75W/Gjb1-null and N175D/Gjb1-null mice
We also conducted electrophysiological analysis of sciatic nerve function as in the early-treatment groups. Electrophysiology was conducted at 10 months of age, 4 months post injection. MNCV was improved in 10-month-old treated mice from both the R75W/Gjb1-null and N175D/Gjb1-null transgenic lines compared to their mock-treated littermates. Sciatic MNCV in 10-month-old R75W/Gjb1-null mice was on average 29.43 ± 0.75 m/s in the mock (n = 16) compared to 38.60 ± 1.39 m/s in the treated group (n = 16; p < 0.0001; Figure 4J). Likewise, in N175D/Gjb1-null mice, MNCV reached 29.46 ± 0.83 m/s in the mock group (n = 13) compared to 41.37 ± 0.91 m/s in the treatment group (n = 19; p < 0.0001; Figure 4L). Sciatic CMAP amplitudes were also measured but showed no changes between the mock and fully treated mice in either of the two genotypes (Figures 4K and 4M).
Improved nerve pathology in post-onset-treated R75W/Gjb1-null and N175D/Gjb1-null mice
Similar to the pre-onset groups, we performed detailed morphological analysis in anterior lumbar roots, mid-sciatic nerves, and femoral motor nerves of post-onset-treated compared to mock-treated 10-month-old R75W/Gjb1-null and N175D/Gjb1-null mice. Improved pathology was observed in all tissues examined in R75W/Gjb1-null mice treated after onset at the age of 6 months compared to mock-treated littermates. Both abnormally myelinated fibers and foamy macrophages were decreased in treated mice compared to the mock-treated mice. In anterior lumbar roots (Figures 5A, 5B, 5E, and 5F), the ratio of abnormally myelinated fibers reached 0.26 ± 0.02 (n = 12) in mock compared to 0.13 ± 0.03 in treated mice (n = 12; p = 0.0011; Figure 5I). Likewise, the numbers of foamy macrophages were decreased in treated mice (7.38 ± 0.62; n = 12) compared to the mock group (14.56 ± 1.09; n = 12; p = 0.0003; Figure 5J). Similar results were obtained in femoral motor nerves (Figures 5M, 5N, 5Q, and 5R), in which the ratio of abnormally myelinated fibers decreased to 0.23 ± .0.02 (n = 12) in treated mice compared to the mock group (0.32 ± 0.01; n = 12; p = 0.0111; Figure 5U) and foamy macrophages were decreased in treated (5.41 ± 0.87; n = 12) compared to the mock-treated animals (8.76 ± 0.55; n = 12; p = 0.0073; Figure 5V). Finally, improvement was observed also in the sciatic nerves (Figures S5M, S5N, S5Q, and S5R), where abnormally myelinated fibers reached 0.05 ± 0.01 (n = 11) in treated and 0.10 ± 0.01 in mock-treated mice (n = 12; p = 0.001; Figure S5U) and foamy macrophages were decreased in treated mice (3.54 ± 0.51; n = 11) compared to the mock group (6.79 ± 0.46; n = 12; p = 0.0013; Figure S5V).
Figure 5.

Morphological analysis of anterior lumbar roots and femoral motor nerves of 10-month-old R75W/Gjb1-null and N175D/Gjb1-null mice 4 months following post-onset intrathecal delivery of AAV9-Mpz.GJB1 or AAV9-Mpz.Egfp vectors
Representative images of semithin sections of R75W/Gjb1-null (A, B, E, and F) and N175D/Gjb1-null (C, D, G, and H) anterior motor lumbar spinal roots attached to the spinal cord, at low and higher magnification, as indicated. AAV9-Mpz.GJB1 injected mice show improved root myelination compared to mock-treated littermates with fewer demyelinated (∗) and remyelinated (r) fibers, as well as reduced numbers of foamy macrophages (arrows in E and G). Improved root pathology is confirmed by the morphometric analysis results showing decreased ratios of abnormally myelinated fibers and reduced numbers of macrophages per 1,000 fibers (I–L). Semithin sections of femoral motor nerves of R75W/Gjb1-null (M, N, Q, and R) and N175D/Gjb1-null (O, P, S, and T) at lower and higher magnification as indicated. The corresponding morphometric analysis results (U–X) from mock- and full-vector-treated mice confirm in AAV9-Mpz.GJB1 injected CMT1X mice of both genotypes improved myelination in femoral nerves compared to mock-treated littermates with fewer demyelinated and remyelinated fibers as well as reduction in the numbers of foamy macrophages. Values represent mean±SEM. Statistical analysis was performed using the Mann-Whitney test.
In N175D/Gjb1-null mice the post-onset treatment trial showed similar positive outcomes as in R75W/Gjb1-null mice. In the anterior lumbar roots (Figures 5C, 5D, 5G, and 5H), the ratio of abnormally myelinated fibers reached 0.32 ± 0.03 (n = 13) in mock compared to 0.16 ± 0.02 in treated mice (n = 12; p = 0.0012; Figure 5K), while the numbers of foamy macrophages decreased to 8.81 ± 1.74 in treated (n = 12) compared to 14.79 ± 1.20 in mock-treated animals (n = 13; p = 0.008; Figure 5L). Likewise, in femoral motor nerves (Figures 5O, 5P, 5S, and 5T) of fully treated N175D/Gjb1-null mice, the ratio of abnormally myelinated fibers decreased to 0.21 ± .0.02 (n = 12) compared to 0.31 ± 0.01 in the mock group (n = 13; p = 0.0001; Figure 5W) and foamy macrophages were decreased to 5.21 ± 0.42 in treated mice (n = 12) compared to the mock group (9.27 ± 0.71; n = 13; p < 0.0001; Figure 5X). Finally, in the mid-sciatic nerves (Figures S5O, S5P, S5S, and S5T), abnormally myelinated fibers were decreased to 0.06 ± 0.01 (n = 10) in treated compared to 0.10 ± 0.00 in mock-treated mice (n = 10; p = 0.0003; Figure S5W) and foamy macrophages were decreased to 4.55 ± 0.53 in treated (n = 10) compared to the mock group (7.59 ± 0.48; n = 10; p = 0.0015; Figure S5X).
We further performed axonal profiling using semithin sections of femoral motor nerves to assess axon loss and degeneration. This analysis showed no significant differences in axon diameter distribution between mock and fully treated mice of either mutant line (Figures S6A and S6B). Furthermore, total axon numbers did not significantly differ between the two groups in either mutant line: in the R75W/Gjb1-null mice mock group, 563.3 ± 39.57 (n = 6 nerves; three mice), and fully treated mice, 600.3 ± 9.06 (n = 6 nerves; three mice, p > 0.05); in N175D/Gjb1-null mice mock group, 588.3 ± 13.2 (n = 6 nerves; three mice), and fully treated mice, 613.7 ± 34.28 (n = 6 nerves; three mice, p > 0.05) (Figures S6C and S6D).
Discussion
Using a well-characterized AAV serotype, AAV9, we studied its efficacy to treat demyelinating peripheral neuropathy in two CMT1X transgenic mouse models expressing Golgi-retained mutant Cx32, the R75W/Gjb1-null, and N175D/Gjb1-null. Our results show widespread expression of virally delivered Cx32 in non-compact myelin areas throughout the PNS, resulting in improved motor performance and nerve pathology in both genotypes, following either early pre-onset or delayed post-onset treatment. Thus, in addition to previously demonstrated efficacy of AAV9-delivered gene replacement therapy in Gjb1-null mice,32 this study provides proof of concept for this treatment approach also in two clinically relevant models of CMT1X. Importantly, therapeutic benefit occurs despite the previously demonstrated interfering effects of Golgi-retained mutants such as R75W and N175D on co-expressed WT Cx32,22,29,31 supporting the clinical relevance and translatability of this treatment for CMT1X patients.
As in our previous studies, we delivered by lumbar intrathecal injection an AAV9 vector carrying the GJB1 gene driven by the Schwann cell-specific Mpz promoter in order to restrict expression in Schwann cells, the key cell responsible for the development of the peripheral neuropathy in CMT1X through a cell-autonomous mechanism.33,34,43 Here, we used two Golgi-retained mutant mouse models expressing the R75W or the N175D GJB1 mutation on a Gjb1-null background. Although both mutant models expressed similar levels of EGFP as expected, the N175D/Gjb1-null model showed a trend of higher expression levels of Cx32 levels in both roots and nerves. Variable Cx32 protein levels in the two models may result from different effects of random insertion into the genome during transgenic line generation, as well as the degree of degradation that retained Cx32 mutants undergo in proteasomes and lysosomes.21 These different baseline levels of mutant Cx32 in the two lines may explain minor variations in therapeutic outcomes, including slightly less significant response in function and morphology of the N175D//Gjb1-null line. Nevertheless, the AAV9-based approach overcomes any interfering effects, providing a clear therapeutic benefit in both CMT1X models. It should be noted that the levels of Cx32 mutants and the possible interfering effects with co-expressed WT Cx32 are likely overestimated in our transgenic models, because expression is driven by the 2',3'-cyclic nucleotide phosphodiesterase (CNP) promoter,29 and not by the Gjb1 promoter itself. This is also in accordance with the result of recently published R75W knockin mouse model in which no Cx32 expression could be detected, likely due to lower expression levels under Gjb1 promoter and rapid degradation of the mutant.44
Here, we present for the first time electrophysiological data obtained in transgenic R75W/Gjb1-null and N175D/Gjb1-null mice showing MNCV values that are similar to those reported in Gjb1-null32,45,46 and in R75W knockin44 mouse models. This shows that either complete deletion or the presence of a Cx32 mutant results in similar functional abnormalities and degree of MNCV slowing. This loss-of-function mechanism is attributed to defects in the formation or electrochemical properties of GJ channels formed by the R75W, R75P, or R75Q mutants.47 Furthermore, although mock-injected animals received the vector carrying the Egfp gene with Schwann cell-specific expression, we did not observe any differences in their functional outcomes compared to untreated Gjb1-null mice, in keeping with our previous studies.32,45,46,48 In this study, re-establishment of WT Cx32 expression resulted in improved MNCVs in treated animals, but the values did not reach the expected WT values, similar to what we have already shown in Gjb1-null treated mice.32 This may be due to the fact that the viral expression of Cx32 does not reach WT levels but also because our gene replacement only reaches a certain percentage of myelinating Schwann cells in PNS tissues, as previously quantified.32
Furthermore, our data from this and previous studies show that MNCV is a sensitive and treatment-responsive functional outcome measure for therapies in different models of demyelinating CMT neuropathies, including Gjb1-null,32,48 Sh3tc2−/−,49 and C61-heterozygous50 mice. Functional improvement after treatment was further confirmed by the results of foot grip analysis, which has proved to be the most reliable behavioral test for CMT1X mouse models. This is not surprising given the predominantly motor-fiber pathology in this model.51 Thus, the combination of electrophysiological and behavioral analysis can provide an accurate assessment of functional alterations in CMT models, as also demonstrated in previous studies either at the baseline or following treatment.32,44,48,50,52
Based on our previous results showing that lentiviral vector did not express Cx32 at high enough levels to overcome the interfering effects of Golgi-retained mutants,33 we proceeded with a use of a more potent vector, the AAV9, which is known to provide higher expression levels compared to lentiviral vectors due to its mostly episomal persistence escaping the host genome regulatory control that limits expression of integrated lentiviral vectors38,39 and its capacity to diffuse more efficiently in the tissues.40,41,42 The significant therapeutic benefit shown in this study achieved by AAV9 gene replacement in both Golgi-retained mutant models in contrast to failed efficacy of the previously tested lentiviral approach in the same models33 confirms the AAV advantage. This further indicates that higher expression levels of Cx32 can overcome possible interfering effects of certain Cx32 mutants, likely occurring during hemichannel formation in the Golgi.53,54
The advantage of our approach is that it can be applied for all CMT1X patients regardless of the type and cellular effects of the causative GJB1 mutations. Alternatively, more personalized therapies including combined silencing of the mutant gene and addition of the normal gene, or gene editing, could be proposed.55,56,57 However, this study demonstrates that gene addition is efficient enough without prior silencing of mutant Cx32 expression and can achieve an easier and more translatable approach for the treatment of CMT1X. It can be applied to all types of mutations, including Golgi- or ER-retained mutants, the latter previously shown to be rapidly degraded and to have no interfering effects with co-expressed WT Cx32.22
In our previous studies, we used NF-L as a biomarker for both progression and response to treatment in different demyelinating neuropathy models and showed an amelioration with at least early treatment.32,49,50 In particular, the Gjb1-null mouse model showed an elevation of NF-L concentration up to 4 months of age followed by a significant reduction until 12 months of age (our unpublished data). Here, we obtained similar results for the longitudinal study in the R75W/Gjb1-null mouse model with an increase at 4 months of age followed by a significant reduction by 8 months, although remaining at much higher levels compared to baseline. This is in accordance with what is already shown for CMT1X patients, where elevated concentrations of NF-L are observed followed by a significant reduction after a 6-year interval.58,59 This was not the case in the N175D/Gjb1-null mouse model showing a continuous increase up to 8 months of age that might be attributed to more active pathology in this mouse model compared to the R75W/Gjb1-null. Interestingly, N175D/Gjb1-null mice already showed significantly higher NF-L concentration at 2 months of age (baseline) compared to the R75W mutants (p < 0.0001), a stage prior to the development of the neuropathy. In contrast to our previous results, where we showed reduced NF-L concentration after treatment, this was not the case in this study, in which no differences were observed between mock and fully treated CMT1X mice, despite behavioral, electrophysiological, and morphological improvements. Thus, we find in these mouse models a lack of correlation between the severity of pathology and NF-L plasma levels. This is in accordance with studies in patients where no correlation was found between the weighted Charcor-Marie-Tooth Examination Scores (CMTES) and Charcot-Marie-Tooth Neuropathy Scores (CMTNS) and NF-L plasma concentrations, suggesting that there are some limitations in the use of NF-L as a possible biomarker for CMTs.58,59
CMT1X is a slowly progressive neuropathy with myelin pathology already present in childhood, accompanied by progressive axonal degeneration.60 Thus, it is essential to demonstrate that any treatment would be beneficial also after the onset of PNS pathology. Although we have already shown in the Gjb1-null mouse model that early intervention is more efficient than post-onset treatment,43 we also wanted to evaluate this delayed treatment approach in CMT1X mutant mouse models expressing different GJB1 mutations that more faithfully reproduce the molecular-genetic background of CMT1X patients. Our results show improvement in functional and morphological outcomes in both transgenic lines, although without reaching the WT levels, similar to the results obtained when treating Gjb1-null mice,32 when treated not only before but also well after the onset of the neuropathy. Although the outcomes of MNCV and foot grip strength were similar in pre- and post-onset treatment groups (p > 0.05), our rotarod data and morphological analysis results do show a lower response after late compared to early intervention. In particular, the rotarod analysis showed that there was an improvement in pre-onset-treated R75W/Gjb1-null mice at both speeds examined, while there was no improvement following post-onset treatment. N175D/Gjb1-null mice also showed a trend for improved motor performance at the low speed examined only after pre-onset treatment. Although some morphological improvements were also less pronounced in post-onset treatment groups, they remained significant in all tissues, confirming that they are more sensitive to measure therapeutic response. Nevertheless, morphological evaluation has not included more distal nerves or neuromuscular junction innervation,34 which may contribute to motor function and are more affected by progressive axonal degeneration. Overall, both our previous post-onset treatment results43 and the current results indicate that therapeutic intervention should be initiated as early as possible in order to achieve a higher therapeutic benefit in CMT1X.
Finally, our data show that the therapeutic effects of AAV9-mediated gene replacement in the PNS of CMT1X mutant mice can persist for at least up to 6 months following a single injection. This is an indication that AAV9 can be stably expressed at high levels even 6 months after transduction of the highly differentiated and post-mitotic myelinating Schwann cells, offering sufficient exogenous WT Cx32 production to overcome the effects of the co-expressed Cx32 mutant proteins even in older mice up to 10 months of age. Similar long-lasting therapeutic effect has been demonstrated when using AAV9-mediated gene silencing in Schwann cells in a model of CMT1A, with stable silencing effects and therapeutic benefit on myelination for at least 8 months after a single intrathecal injection.50 Moreover, intravenous injection of AAV9 in mice results in stable expression for up to 9 months.61
In conclusion, this study shows that GJB1 gene replacement by lumbar intrathecal delivery of AAV9 in two representative mouse models of CMT1X-expressing Golgi-retained Cx32 mutant can improve functional and morphological outcomes that are characteristic of this demyelinating neuropathy. Furthermore, AAV-mediated expression of WT Cx32 can overcome possible interfering effects of Golgi-retained mutants both before as well as after the onset of the neuropathy. This study provides a proof of concept for a translatable gene therapy approach to treat CMT1X patients irrespective of their underlying mutations. Further evaluation of the scale-up potential and safety of this approach in larger animals is warranted in other to advance clinical translation.
Materials and methods
Cloning and production of AAV vectors
We generated constructs for AAV-vector-mediated gene delivery designed to provide Schwann cell-specific expression of either the reporter gene EGFP (pAAV-Mpz.Egfp, mock vector) or Cx32 (pAAV-Mpz.GJB1, full, therapeutic vector), both under the rat 1.2-kb Mpz promoter shown to drive expression specifically in Schwann cells as already described in our previous studies.35,62
The pAAV-Mpz.Egfp and pAAV-Mpz.GJB1 plasmids were cross-packaged into AAV9 capsid. For AAV9 vector production, HEK293T cells are grown in 10-cm tissue culture dishes in IMDM supplemented with 10% FCS. Two hours prior to transfection, the medium is changed. When cells have reached approximately 70%–80% confluency, they are transfected using polyethylenimine (PEI; branched, molecular weight 25,000; Sigma) with a three-plasmid mix of pAAV, pRepCap, and pXX6 plasmid (provided by Dr. Jude Samulsky, University of North Carolina at Chapel Hill) at a ratio of 1:2:1, and the medium is changed 16 h after transfection. As it was shown previously63 that the cell medium contains significant amounts of virions, both the cells and the cell supernatant are collected 48 h later for vector purification. Cells are pelleted by centrifugation at 500 × g for 10 min and are subsequently re-dissolved in lysis buffer (0.5% sodium deoxycholate, 150 mM NaCl, 20 mM Tris, 50 U/mL Benzonase; Sigma-Aldrich, Munich, Germany) and incubated at 37°C for 1 h. The lysate is then clarified by centrifugation at 3,000 × g for 10 min and filtered in parallel with the supernatant through 0.45-μm Millipore filters (Millipore, San Salvador, El Salvador). Next, RNAse A and a protease inhibitor cocktail is added to both the lysate and the supernatant and incubated for 2 h at 37°C followed by clarification at 3,000 × g for 15 min. The preparations are then combined with a precipitation mix (PEG/NaCl) at a ratio of 3:1, which is incubated at 4°C overnight. The mixture is centrifuged at 3,000 × g for 30 min and the aqueous supernatant discarded. The virion-containing pellet is then resuspended in 3 mL of pellet suspension buffer (250 mM NaCl solution), which is clarified by centrifugation at 10,000 × g for 10 min at 4°C. The virus-containing aqueous layer is then transferred to Beckmann UltraClear SW41 tube and is centrifuged at 149,000 × g for 3 h. The aqueous layer is then discarded, and the viral pellet resuspended in 200 μL of pellet suspension buffer. The vector genome copy number is determined by the Picogreen method.64
Experimental animals
In this study, we used 2- and 6-month-old mice expressing two different CMT1X mutations on a Gjb1-null background, modeling the genotype of male CMT1X patients expressing a single mutated GJB1 allele. Gjb1-null mice (C57BL/6_129) were originally generated by Prof. Klaus Willecke, University of Bonn, Germany. In these mice, the neor gene was inserted in frame into the Exon 2 of GJB1 gene, which contains the open reading frame (ORF).65 We used the previously described and phenotypically characterized CMT1X transgenic models R75W/Gjb1-null29 and N175D/Gjb1-null mice,33 both generated in our transgenic mouse facility on C57BL/6 background, weighing 20–25 g. The R75W and N175D mutants are known to be retained in the Golgi.22,29 Mice were kept in a specific-pathogen-free animal facility, housed in open-top system cages. Wood bedding for laboratory mice, dried by high-temperature treatment, sieved, de-dusted, and of high absorbency, was used and mice were housed up to five in each cage. Standard mouse diet, certificate, for reproduction, weaning, and growth, and tap potable water, filtered and UV sterilized, were administered to the mice. Mice were kept in 12-h dark/12-h light cycle at a temperature of 22°C. Both male and female mice were used in our experiments and showed no (sex-related) differences in their behavioral performance or nerve pathology. All experimental procedures in this study were conducted in accordance with animal care protocols approved by the Cyprus Government’s Chief Veterinary Officer (project license CY/EXP/PR.L3/2017) according to national law, which is harmonized with EU guidelines (EC Directive 86/609/EEC).
Study design
For Cx32 expression analysis, we used R75W/Gjb1-null and N175D/Gjb1-null mice injected with the pAAV-Mpz.GJB1 (full) vector. For the treatment trial experiments, mice were treated at the age of 2 months for the early treatment and at 6 months of age for the late treatment (Figures 2A and 4A). Littermate mice were randomized to receive either AAV9-Mpz.GJB1 (full) treatment or AAV9-Mpz.Egfp (mock treatment, control group) and were assigned a coding number for further identification. Two-month-old mice were evaluated before the treatment and again at the age of 4 and 6 months, while 6-month-old mice were evaluated before the treatment and at the age of 8 and 10 months. Mice were evaluated by behavioral testing by an examiner blinded to the treatment condition and used at the age of 6 months for the early treatment and 10 months for the late treatment for electrophysiology or for quantitative morphometric analysis of semithin sections. Analysis of physiological and morphological results was also performed blinded to the treatment condition. For the early-treatment study, we used 26 R75W/Gjb1-null mice injected with the mock vector (15 for morphological outcomes and 11 for electrophysiological analysis) and 27 injected with the full vector (16 for morphological outcomes and 11 for electrophysiological analysis) as well as 22 N175D/Gjb1-null mice injected with the mock vector (11 for morphological outcomes and 11 for electrophysiological analysis) and 23 injected with the full vector (11 used for morphological outcomes and 12 for electrophysiological analysis). Finally, for the late treatment, we used 28 R75W/Gjb1-null mice injected with the mock vector (12 for morphological outcomes and 16 for electrophysiological analysis) and 28 injected with the full vector (12 for morphological outcomes and 16 for electrophysiological analysis), as well as 26 N175D/Gjb1-null mice injected with the mock vector (13 for morphological outcomes and 13 for electrophysiological analysis) and 31 injected with the full vector (12 for morphological outcomes and 19 for electrophysiological analysis). For the treatment trials, we used at least 10 mice for both electrophysiological and morphological analysis in order to account for the expected variability between mice and obtain adequate data for statistical analysis. All values obtained were included in the analysis without excluding any outliers. Foot grip tests were performed three times and the mean value was used.
Intrathecal vector delivery
We delivered the AAV vectors by a single lumbar intrathecal injection as previously described.35,66 Briefly, a small skin incision was made along the lower lumbar spine level of anesthetized mice to visualize the spine, and the AAV vector was delivered into the L5-L6 intervertebral space. A 50-μL Hamilton syringe (Hamilton, Giarmata, Romania) connected to a 26-gauge needle was used to inject 20 μL of AAV stock containing an estimated total of 2 × 1011 vector genomes (vg), at a maximum rate of 5 μL/min. A flick of the tail was considered indicative of successful intrathecal administration.
Immunofluorescence staining
For immunostaining, mice were anesthetized with avertin according to institutionally approved protocols, and then transcardially perfused with normal saline followed by fresh 4% paraformaldehyde in 0.1 M PBS buffer. The lumbar-sacral spinal cords with spinal roots attached, as well as the bilateral sciatic and femoral motor nerves, were dissected and post-fixed in 4% PFA, the spinal cord for 2 h and sciatic and femoral nerves for 30 min. Spinal roots were frozen for cryosections, while sciatic and femoral nerves were isolated and teased into fibers under a stereoscope. Teased fibers or sections were permeabilized in cold acetone and incubated at RT with a blocking solution of 5% BSA (Sigma-Aldrich, Munich, Germany) containing 0.5% Triton X (Sigma-Aldrich, Munich, Germany) for 1 h. Primary antibodies used were: mouse monoclonal antibody against contactin-associated protein (Caspr, 1:50; gift of Dr Elior Peles, Weizmann Institute of Science) and rabbit antisera against Cx32 (1:50; Invitrogen, Waltham, MA USA) all diluted in blocking solution and incubated overnight at 4°C. Slides were then washed in PBS and incubated with mouse cross-affinity fluorescein-conjugated (1:1,000; Invitrogen, Waltham, MA USA) and rabbit cross-affinity purified rhodamine-conjugated (1:500; Jackson ImmunoResearch, West Grove, USA) secondary antibodies for 1 h at RT. Cell nuclei were visualized with DAPI (1 μg/mL; Sigma, Munich, Germany). Slides were mounted with fluorescent mounting medium and images photographed under a fluorescence microscope with a digital camera using a fluorescence microscope (Nikon Eclipse Ni; Tokyo, Japan) with digital camera (DS-Qi2) using NIS-Elements software.
For quantification of viral Cx32 expression, we counted in lumbar root and sciatic nerve sections double stained for Cx32 and Caspr in n = 6 mice per genotype the number of Cx32-immunoreactive paranodal areas, expressed as a percentage of all paranodal areas visualized in each tissue section based on Caspr paranodal labeling.
Immunoblot analysis
Fresh sciatic and femoral nerves and lumbar spinal roots were collected from R75W/Gjb1-null and N175D/Gjb1-null mice and lysed in ice-cold RIPA buffer (10 mM sodium phosphate, pH 7.0, 150 mM NaCl, 2 mM EDTA, 50 mM sodium fluoride, 1% Nonidet P-40, 1% sodium deoxycholate, and 0.1% SDS, all from Sigma-Aldrich, Munich, Germany) containing a mixture of protease inhibitors (Roche, Sigma-Aldrich, Munich, Germany). Proteins (150 μg) from the lysates were fractionated by 12% SDS/PAGE and then transferred to a Hybond-C Extra membrane (GE Healthcare Life Sciences, Logan, USA) using a semidry transfer unit. Nonspecific sites on the membrane were blocked with 5% non-fat milk in PBS with Tween 20 (PBS-T) for 1 h at room temperature. Immunoblots were incubated with rabbit antisera against EGFP (1:1,000; Abcam, Cambridge, UK) or Cx32 (clone 918, 1:3,000)67 and mouse β-tubulin (1:4,000; Developmental Studies Hybridoma Bank, Iowa City, USA) at 4°C overnight. After washing, the immunoblots were incubated with an anti-mouse or anti-rabbit HRP-conjugated secondary antiserum (Jackson ImmunoResearch, diluted 1:3,000, West Grove, USA) in 5% milk-PBST for 1 h. The bound antibody was visualized by an enhanced chemiluminescence system (GE Healthcare Life Sciences, Logan, USA).
Behavioral analysis
Mice included in the treatment trial were subjected to grip strength and rotarod testing in order to access motor performance following delivery of the therapeutic or the mock AAV9 vector.
Grip strength testing
To measure grip strength, mice were held by the tail and lowered toward the apparatus (Ugo Basile, Varese, Italy) until they grabbed the grid with the hind paws. Mice were gently pulled back until they released the grid. Measurements of the force in grams were indicated on the equipment. Each session consisted of three consecutive trials, and measurements were averaged. Hindlimb force was compared between AAV9.Mpz-GJB1 and AAV9.Mpz-Egfp-treated mice.
Rotarod testing
Motor balance and coordination were determined as described previously68 using an accelerating rotarod apparatus (Ugo Basile, Varese, Italy). Training of animals consisted of three trials per day with 15-min rest period between trials, for three consecutive days. The mice were placed on the rod and the speed was gradually increased from 4 to 40 rpm. The trial lasted until the mouse fell from the rod or after the mouse remained on the rod for 600 s and was then removed. Testing was performed on the fourth day using two different speeds, 20 and 32 rpm. Latency to fall was calculated for each speed. Each mouse was placed on the rotarod three times at each speed used, and three different values were obtained for each speed. Mean values were used for each mouse at the two different speeds.
Electrophysiological analysis
MNCV
MNCV was measured in vivo using published methods46 from bilateral sciatic nerves following stimulation in anesthetized animals at the using two stimulation sites, one near the sciatic notch and one distally at the knee, via bipolar electrodes with supramaximal square-wave pulses (5 V) of 0.05 ms. The latencies of the CMAPs were recorded by a bipolar electrode inserted between digits 2 and 3 of the hind paw and measured from the stimulus artifact to the onset of the negative M-wave deflection. MNCV was calculated by dividing the distance between the stimulating and recording electrodes by the result of subtracting distal from proximal latency.
NF-L plasma level evaluation
To further access the potential effect of the treatment on neuroaxonal degeneration, we measured the plasma concentration of the axonal injury biomarker NF-L69 in untreated R75W/Gjb1-null and N175D/Gjb1-null mice at the ages of 2, 4, 6, and 8 months, as well as from mice carrying both mutations treated either with the therapeutic or with the mock vector, at the ages of 8 or 10 months for early or late treatment, respectively. Blood was collected prior to sacrificing the animals using standard methods.70 Blood samples were processed within 1 h. Blood was collected into EDTA-containing tubes and centrifuged at 20°C at 3,500 rpm for 10 min. Plasma was aliquoted and stored at −80°C until testing. Plasma NF-L concentration was measured at University College London (UCL) using a commercially available NF-Light kit on a single-molecule array (Simoa) HD-X instrument (Quanterix, Billerica, MA).71,72 All samples were measured in duplicate on one occasion using one batch of reagents. Intra-assay coefficients of variation were <15.
Morphometric analysis of myelination in lumbar roots and peripheral nerves
Mice were transcardially perfused with 2.5% glutaraldehyde in 0.1 M PBS buffer. The lumbar spinal cord with multiple spinal roots attached, as well as the femoral and sciatic nerves, were dissected and fixed overnight at 4°C, then osmicated, dehydrated, and embedded in Araldite resin (all purchased from Agar Scientific, Essex, UK). Transverse semithin sections (1 μm) of the lumbar spinal cord with roots and the middle portion of the femoral motor and sciatic nerves were obtained and stained with alkaline toluidine blue (Sigma-Aldrich, Munich, Germany). Sections were visualized with 10×, 20×, and 40× objective lenses and captured using a Nikon Eclipse Ni microscope (Tokyo, Japan) with digital camera (DS-Fi3) using NIS-Elements software. Images of whole root or transverse nerve sections were obtained at 100–200× final magnification, and a series of partially overlapping fields covering the entire cross-sectional area of the roots or the nerves were captured at 400× final magnification.
These images were used to examine the degree of abnormal myelination in all treatment groups as described previously.29,36,73 In brief, all demyelinated, remyelinated, and normally myelinated axons were counted in the entire root or nerve cross section using the following criteria: axons larger than 1 μm without a myelin sheath were considered demyelinated; axons with myelin sheaths <10% of the axonal diameter and/or axons surrounded by “onion bulbs” (i.e., circumferentially arranged Schwann cell processes and extracellular matrix) were considered remyelinated; all other myelinated axons were considered normally myelinated.
In addition, we counted the number of foamy macrophages present within the entire cross section of each root or nerve, as an indication of inflammation. Macrophages were identified in semithin sections at 400× magnification as cells laden with myelin debris, devoid of a basement membrane, and extending small, microvilli-like processes, as described previously.45,74 The macrophage count was calculated as the ratio per 1,000 myelinated fibers to account for size differences between different spinal roots and nerves. All pathological analyses were performed blinded to the treatment condition of each mouse. Finally, using semithin sections of femoral motor nerves from both full and mock treatment groups, total axons and the axonal diameter for axon profiling analysis were measured using the Image Pro Plus software (version 6.0; Media Cybernetics) using a custom-made macro.
Statistical analysis
Behavioral testing results, electrophysiological results, NF-L concentrations in the treatment trial groups, and morphological analysis data obtained from mock-treated and fully treated groups were compared using the Mann-Whitney U test (significance level for all comparisons, p < 0.05). NF-L levels at the timeline experiments were compared using one-way ANOVA with Tukey’s post-test. Tests were performed using GraphPad Instat3 software or GraphPad Prism 8 (GraphPad, San Diego, USA).
Data and code availability
Data are available upon request from the corresponding author.
Acknowledgments
This work was funded by the Muscular Dystrophy Association (MDA) and Charcot-Marie-Tooth Association (CMTA) (grant MDA 603003 to K.A.K.), as well as the UK Dementia Research Institute at UCL (UKDRI-1003 to H.Z. and A.H.).
Author contributions
A.K. conducted the experiments, acquired data, analyzed data, and wrote the manuscript. I.S. conducted cloning and mouse PCR screening. A.H. and H.Z. performed analysis of plasma NF-L levels. A.B. designed and produced vectors. K.A.K. designed research studies, analyzed data, and wrote the manuscript. All authors critically reviewed and approved the final manuscript.
Declaration of interests
H.Z. has served on scientific advisory boards and/or as a consultant for Abbvie, Acumen, Alector, Alzinova, ALZPath, Annexon, Apellis, Artery Therapeutics, AZTherapies, CogRx, Denali, Eisai, Nervgen, Novo Nordisk, Optoceutics, Passage Bio, Pinteon Therapeutics, Prothena, Red Abbey Labs, reMYND, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics, and Wave; has given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure, Biogen, and Roche; and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program (outside submitted work).
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2023.07.011.
Contributor Information
Alexia Kagiava, Email: kleopa@cing.ac.cy.
Kleopas A. Kleopa, Email: alexiak@cing.ac.cy.
Supplemental information
References
- 1.Jerath N.U., Gutmann L., Reddy C.G., Shy M.E. Charcot-marie-tooth disease type 1X in women: Electrodiagnostic findings. Muscle Nerve. 2016;54:728–732. doi: 10.1002/mus.25077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kleopa K.A., Scherer S.S. Molecular genetics of X-linked Charcot-Marie-Tooth disease. NeuroMolecular Med. 2006;8:107–122. doi: 10.1385/nmm:8:1-2:107. [DOI] [PubMed] [Google Scholar]
- 3.Liang C., Howells J., Kennerson M., Nicholson G.A., Burke D., Ng K. Axonal excitability in X-linked dominant Charcot Marie Tooth disease. Clin. Neurophysiol. 2014;125:1261–1269. doi: 10.1016/j.clinph.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 4.Lu Y.Y., Lyu H., Jin S.Q., Zuo Y.H., Liu J., Wang Z.X., Zhang W., Yuan Y. Clinical and Genetic Features of Chinese X-linked Charcot-Marie-Tooth Type 1 Disease. Chin. Med. J. 2017;130:1049–1054. doi: 10.4103/0366-6999.204925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martikainen M.H., Majamaa K. Novel GJB1 mutation causing adult-onset Charcot-Marie-Tooth disease in a female patient. Neuromuscul. Disord. 2013;23:899–901. doi: 10.1016/j.nmd.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 6.Saporta A.S.D., Sottile S.L., Miller L.J., Feely S.M.E., Siskind C.E., Shy M.E. Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann. Neurol. 2011;69:22–33. doi: 10.1002/ana.22166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Birouk N., LeGuern E., Maisonobe T., Rouger H., Gouider R., Tardieu S., Gugenheim M., Routon M.C., Léger J.M., Agid Y., et al. X-linked Charcot-Marie-Tooth disease with connexin 32 mutations: clinical and electrophysiologic study. Neurology. 1998;50:1074–1082. doi: 10.1212/wnl.50.4.1074. [DOI] [PubMed] [Google Scholar]
- 8.Dubourg O., Tardieu S., Birouk N., Gouider R., Léger J.M., Maisonobe T., Brice A., Bouche P., LeGuern E. Clinical, electrophysiological and molecular genetic characteristics of 93 patients with X-linked Charcot-Marie-Tooth disease. Brain. 2001;124:1958–1967. doi: 10.1093/brain/124.10.1958. [DOI] [PubMed] [Google Scholar]
- 9.Hahn A.F., Brown W.F., Koopman W.J., Feasby T.E. X-linked dominant hereditary motor and sensory neuropathy. Brain. 1990;113:1511–1525. doi: 10.1093/brain/113.5.1511. [DOI] [PubMed] [Google Scholar]
- 10.Record C.J., Skorupinska M., Laura M., Rossor A.M., Pareyson D., Pisciotta C., Feely S.M.E., Lloyd T.E., Horvath R., Sadjadi R., Herrmann D.N., et al. Genetic analysis and natural history of Charcot-Marie-Tooth disease CMTX1 due to GJB1 variants. Brain. 2023:awad187. doi: 10.1093/brain/awad187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siskind C.E., Murphy S.M., Ovens R., Polke J., Reilly M.M., Shy M.E. Phenotype expression in women with CMT1X. J. Peripher. Nerv. Syst. 2011;16:102–107. doi: 10.1111/j.1529-8027.2011.00332.x. [DOI] [PubMed] [Google Scholar]
- 12.Kleopa K.A., Abrams C.K., Scherer S.S. How do mutations in GJB1 cause X-linked Charcot-Marie-Tooth disease? Brain Res. 2012;1487:198–205. doi: 10.1016/j.brainres.2012.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kleopa K.A., Zamba-Papanicolaou E., Alevra X., Nicolaou P., Georgiou D.M., Hadjisavvas A., Kyriakides T., Christodoulou K. Phenotypic and cellular expression of two novel connexin32 mutations causing CMT1X. Neurology. 2006;66:396–402. doi: 10.1212/01.wnl.0000196479.93722.59. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y., Yin F. A Review of X-linked Charcot-Marie-Tooth Disease. J. Child Neurol. 2016;31:761–772. doi: 10.1177/0883073815604227. [DOI] [PubMed] [Google Scholar]
- 15.Balice-Gordon R.J., Bone L.J., Scherer S.S. Functional gap junctions in the schwann cell myelin sheath. J. Cell Biol. 1998;142:1095–1104. doi: 10.1083/jcb.142.4.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bruzzone R., White T.W., Paul D.L. Connections with connexins: the molecular basis of direct intercellular signaling. Eur. J. Biochem. 1996;238:1–27. doi: 10.1111/j.1432-1033.1996.0001q.x. [DOI] [PubMed] [Google Scholar]
- 17.Scherer S.S., Deschênes S.M., Xu Y.T., Grinspan J.B., Fischbeck K.H., Paul D.L. Connexin32 is a myelin-related protein in the PNS and CNS. J. Neurosci. 1995;15:8281–8294. doi: 10.1523/JNEUROSCI.15-12-08281.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murphy S.M., Polke J., Manji H., Blake J., Reiniger L., Sweeney M., Houlden H., Brandner S., Reilly M.M. A novel mutation in the nerve-specific 5'UTR of the GJB1 gene causes X-linked Charcot-Marie-Tooth disease. J. Peripher. Nerv. Syst. 2011;16:65–70. doi: 10.1111/j.1529-8027.2011.00321.x. [DOI] [PubMed] [Google Scholar]
- 19.Tomaselli P.J., Rossor A.M., Horga A., Jaunmuktane Z., Carr A., Saveri P., Piscosquito G., Pareyson D., Laura M., Blake J.C., et al. Mutations in noncoding regions of GJB1 are a major cause of X-linked CMT. Neurology. 2017;88:1445–1453. doi: 10.1212/WNL.0000000000003819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deschênes S.M., Walcott J.L., Wexler T.L., Scherer S.S., Fischbeck K.H. Altered trafficking of mutant connexin32. J. Neurosci. 1997;17:9077–9084. doi: 10.1523/JNEUROSCI.17-23-09077.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kleopa K.A., Yum S.W., Scherer S.S. Cellular mechanisms of connexin32 mutations associated with CNS manifestations. J. Neurosci. Res. 2002;68:522–534. doi: 10.1002/jnr.10255. [DOI] [PubMed] [Google Scholar]
- 22.Kyriakoudi S., Sargiannidou I., Kagiava A., Olympiou M., Kleopa K.A. Golgi-retained Cx32 mutants interfere with gene addition therapy for CMT1X. Hum. Mol. Genet. 2017;26:1622–1633. doi: 10.1093/hmg/ddx064. [DOI] [PubMed] [Google Scholar]
- 23.Omori Y., Mesnil M., Yamasaki H. Connexin 32 mutations from X-linked Charcot-Marie-Tooth disease patients: functional defects and dominant negative effects. Mol. Biol. Cell. 1996;7:907–916. doi: 10.1091/mbc.7.6.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoshimura T., Satake M., Ohnishi A., Tsutsumi Y., Fujikura Y. Mutations of connexin32 in Charcot-Marie-Tooth disease type X interfere with cell-to-cell communication but not cell proliferation and myelin-specific gene expression. J. Neurosci. Res. 1998;51:154–161. doi: 10.1002/(SICI)1097-4547(19980115)51:2<154::AID-JNR4>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 25.Yum S.W., Kleopa K.A., Shumas S., Scherer S.S. Diverse trafficking abnormalities of connexin32 mutants causing CMTX. Neurobiol. Dis. 2002;11:43–52. doi: 10.1006/nbdi.2002.0545. [DOI] [PubMed] [Google Scholar]
- 26.VanSlyke J.K., Deschenes S.M., Musil L.S. Intracellular transport, assembly, and degradation of wild-type and disease-linked mutant gap junction proteins. Mol. Biol. Cell. 2000;11:1933–1946. doi: 10.1091/mbc.11.6.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Castro C., Gómez-Hernandez J.M., Silander K., Barrio L.C. Altered formation of hemichannels and gap junction channels caused by C-terminal connexin-32 mutations. J. Neurosci. 1999;19:3752–3760. doi: 10.1523/JNEUROSCI.19-10-03752.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ressot C., Gomès D., Dautigny A., Pham-Dinh D., Bruzzone R. Connexin32 mutations associated with X-linked Charcot-Marie-Tooth disease show two distinct behaviors: loss of function and altered gating properties. J. Neurosci. 1998;18:4063–4075. doi: 10.1523/JNEUROSCI.18-11-04063.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sargiannidou I., Vavlitou N., Aristodemou S., Hadjisavvas A., Kyriacou K., Scherer S.S., Kleopa K.A. Connexin32 mutations cause loss of function in Schwann cells and oligodendrocytes leading to PNS and CNS myelination defects. J. Neurosci. 2009;29:4736–4749. doi: 10.1523/JNEUROSCI.0325-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shy M.E., Siskind C., Swan E.R., Krajewski K.M., Doherty T., Fuerst D.R., Ainsworth P.J., Lewis R.A., Scherer S.S., Hahn A.F. CMT1X phenotypes represent loss of GJB1 gene function. Neurology. 2007;68:849–855. doi: 10.1212/01.wnl.0000256709.08271.4d. [DOI] [PubMed] [Google Scholar]
- 31.Jeng L.J.B., Balice-Gordon R.J., Messing A., Fischbeck K.H., Scherer S.S. The effects of a dominant connexin32 mutant in myelinating Schwann cells. Mol. Cell. Neurosci. 2006;32:283–298. doi: 10.1016/j.mcn.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 32.Kagiava A., Karaiskos C., Richter J., Tryfonos C., Jennings M.J., Heslegrave A.J., Sargiannidou I., Stavrou M., Zetterberg H., Reilly M.M., et al. AAV9-mediated Schwann cell-targeted gene therapy rescues a model of demyelinating neuropathy. Gene Ther. 2021;28:659–675. doi: 10.1038/s41434-021-00250-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kagiava A., Karaiskos C., Richter J., Tryfonos C., Lapathitis G., Sargiannidou I., Christodoulou C., Kleopa K.A. Intrathecal gene therapy in mouse models expressing CMT1X mutations. Hum. Mol. Genet. 2018;27:1460–1473. doi: 10.1093/hmg/ddy056. [DOI] [PubMed] [Google Scholar]
- 34.Kagiava A., Richter J., Tryfonos C., Karaiskos C., Heslegrave A.J., Sargiannidou I., Rossor A.M., Zetterberg H., Reilly M.M., Christodoulou C., Kleopa K.A. Gene replacement therapy after neuropathy onset provides therapeutic benefit in a model of CMT1X. Hum. Mol. Genet. 2019;28:3528–3542. doi: 10.1093/hmg/ddz199. [DOI] [PubMed] [Google Scholar]
- 35.Kagiava A., Sargiannidou I., Theophilidis G., Karaiskos C., Richter J., Bashiardes S., Schiza N., Nearchou M., Christodoulou C., Scherer S.S., Kleopa K.A. Intrathecal gene therapy rescues a model of demyelinating peripheral neuropathy. Proc. Natl. Acad. Sci. USA. 2016;113:E2421–E2429. doi: 10.1073/pnas.1522202113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sargiannidou I., Kagiava A., Bashiardes S., Richter J., Christodoulou C., Scherer S.S., Kleopa K.A. Intraneural GJB1 gene delivery improves nerve pathology in a model of X-linked Charcot-Marie-Tooth disease. Ann. Neurol. 2015;78:303–316. doi: 10.1002/ana.24441. [DOI] [PubMed] [Google Scholar]
- 37.Blits B., Derks S., Twisk J., Ehlert E., Prins J., Verhaagen J. Adeno-associated viral vector (AAV)-mediated gene transfer in the red nucleus of the adult rat brain: comparative analysis of the transduction properties of seven AAV serotypes and lentiviral vectors. J. Neurosci. Methods. 2010;185:257–263. doi: 10.1016/j.jneumeth.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 38.Hutson T.H., Verhaagen J., Yáñez-Muñoz R.J., Moon L.D.F. Corticospinal tract transduction: a comparison of seven adeno-associated viral vector serotypes and a non-integrating lentiviral vector. Gene Ther. 2012;19:49–60. doi: 10.1038/gt.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mason M.R.J., Ehlert E.M.E., Eggers R., Pool C.W., Hermening S., Huseinovic A., Timmermans E., Blits B., Verhaagen J. Comparison of AAV serotypes for gene delivery to dorsal root ganglion neurons. Mol. Ther. 2010;18:715–724. doi: 10.1038/mt.2010.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bey K., Deniaud J., Dubreil L., Joussemet B., Cristini J., Ciron C., Hordeaux J., Le Boulc'h M., Marche K., Maquigneau M., et al. Intra-CSF AAV9 and AAVrh10 Administration in Nonhuman Primates: Promising Routes and Vectors for Which Neurological Diseases? Mol. Ther. Methods Clin. Dev. 2020;17:771–784. doi: 10.1016/j.omtm.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duque S., Joussemet B., Riviere C., Marais T., Dubreil L., Douar A.M., Fyfe J., Moullier P., Colle M.A., Barkats M. Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Mol. Ther. 2009;17:1187–1196. doi: 10.1038/mt.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Foust K.D., Nurre E., Montgomery C.L., Hernandez A., Chan C.M., Kaspar B.K. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kagiava A., Richter J., Tryfonos C., Leal-Julià M., Sargiannidou I., Christodoulou C., Bosch A., Kleopa K.A. Efficacy of AAV serotypes to target Schwann cells after intrathecal and intravenous delivery. Sci. Rep. 2021;11:23358. doi: 10.1038/s41598-021-02694-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abrams C.K., Lancaster E., Li J.J., Dungan G., Gong D., Scherer S.S., Freidin M.M. Knock-in mouse models for CMTX1 show a loss of function phenotype in the peripheral nervous system. Exp. Neurol. 2023;360:114277. doi: 10.1016/j.expneurol.2022.114277. [DOI] [PubMed] [Google Scholar]
- 45.Groh J., Heinl K., Kohl B., Wessig C., Greeske J., Fischer S., Martini R. Attenuation of MCP-1/CCL2 expression ameliorates neuropathy in a mouse model for Charcot-Marie-Tooth 1X. Hum. Mol. Genet. 2010;19:3530–3543. doi: 10.1093/hmg/ddq269. [DOI] [PubMed] [Google Scholar]
- 46.Zielasek J., Martini R., Toyka K.V. Functional abnormalities in P0-deficient mice resemble human hereditary neuropathies linked to P0 gene mutations. Muscle Nerve. 1996;19:946–952. doi: 10.1002/(SICI)1097-4598(199608)19:8<946::AID-MUS2>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 47.Abrams C.K., Islam M., Mahmoud R., Kwon T., Bargiello T.A., Freidin M.M. Functional requirement for a highly conserved charged residue at position 75 in the gap junction protein connexin 32. J. Biol. Chem. 2013;288:3609–3619. doi: 10.1074/jbc.M112.392670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ozes B., Myers M., Moss K., McKinney J., Ridgley A., Chen L., Bai S., Abrams C.K., Freidin M.M., Mendell J.R., Sahenk Z. AAV1.NT-3 gene therapy for X-linked Charcot-Marie-Tooth neuropathy type 1. Gene Ther. 2022;29:127–137. doi: 10.1038/s41434-021-00231-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schiza N., Georgiou E., Kagiava A., Médard J.J., Richter J., Tryfonos C., Sargiannidou I., Heslegrave A.J., Rossor A.M., Zetterberg H., et al. Gene replacement therapy in a model of Charcot-Marie-Tooth 4C neuropathy. Brain. 2019;142:1227–1241. doi: 10.1093/brain/awz064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stavrou M., Kagiava A., Choudury S.G., Jennings M.J., Wallace L.M., Fowler A.M., Heslegrave A., Richter J., Tryfonos C., Christodoulou C., et al. A translatable RNAi-driven gene therapy silences PMP22/Pmp22 genes and improves neuropathy in CMT1A mice. J. Clin. Invest. 2022;132:e159814. doi: 10.1172/JCI159814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scherer S.S., Xu Y.T., Nelles E., Fischbeck K., Willecke K., Bone L.J. Connexin32-null mice develop demyelinating peripheral neuropathy. Glia. 1998;24:8–20. doi: 10.1002/(sici)1098-1136(199809)24:1<8::aid-glia2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 52.Stavropoulos F., Sargiannidou I., Potamiti L., Kagiava A., Panayiotidis M.I., Bae J.H., Yeom S.C., Lee J.Y., Kleopa K.A. Aberrant Mitochondrial Dynamics and Exacerbated Response to Neuroinflammation in a Novel Mouse Model of CMT2A. Int. J. Mol. Sci. 2021;22:11569. doi: 10.3390/ijms222111569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Falk M.M. Biosynthesis and structural composition of gap junction intercellular membrane channels. Eur. J. Cell Biol. 2000;79:564–574. doi: 10.1078/0171-9335-00080. [DOI] [PubMed] [Google Scholar]
- 54.Maza J., Das Sarma J., Koval M. Defining a minimal motif required to prevent connexin oligomerization in the endoplasmic reticulum. J. Biol. Chem. 2005;280:21115–21121. doi: 10.1074/jbc.M412612200. [DOI] [PubMed] [Google Scholar]
- 55.Drouet V., Ruiz M., Zala D., Feyeux M., Auregan G., Cambon K., Troquier L., Carpentier J., Aubert S., Merienne N., et al. Allele-specific silencing of mutant huntingtin in rodent brain and human stem cells. PLoS One. 2014;9:e99341. doi: 10.1371/journal.pone.0099341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Southwell A.L., Skotte N.H., Kordasiewicz H.B., Østergaard M.E., Watt A.T., Carroll J.B., Doty C.N., Villanueva E.B., Petoukhov E., Vaid K., et al. In vivo evaluation of candidate allele-specific mutant huntingtin gene silencing antisense oligonucleotides. Mol. Ther. 2014;22:2093–2106. doi: 10.1038/mt.2014.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Trochet D., Prudhon B., Beuvin M., Peccate C., Lorain S., Julien L., Benkhelifa-Ziyyat S., Rabai A., Mamchaoui K., Ferry A., et al. Allele-specific silencing therapy for Dynamin 2-related dominant centronuclear myopathy. EMBO Mol. Med. 2018;10:239–253. doi: 10.15252/emmm.201707988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Millere E., Rots D., Simrén J., Ashton N.J., Kupats E., Micule I., Priedite V., Kurjane N., Blennow K., Gailite L., et al. Plasma neurofilament light chain as a potential biomarker in Charcot-Marie-Tooth disease. Eur. J. Neurol. 2021;28:974–981. doi: 10.1111/ene.14689. [DOI] [PubMed] [Google Scholar]
- 59.Rossor A.M., Kapoor M., Wellington H., Spaulding E., Sleigh J.N., Burgess R.W., Laura M., Zetterberg H., Bacha A., Wu X., et al. A longitudinal and cross-sectional study of plasma neurofilament light chain concentration in Charcot-Marie-Tooth disease. J. Peripher. Nerv. Syst. 2022;27:50–57. doi: 10.1111/jns.12477. [DOI] [PubMed] [Google Scholar]
- 60.Senderek J., Hermanns B., Bergmann C., Boroojerdi B., Bajbouj M., Hungs M., Ramaekers V.T., Quasthoff S., Karch D., Schröder J.M. X-linked dominant Charcot-Marie-Tooth neuropathy: clinical, electrophysiological, and morphological phenotype in four families with different connexin32 mutations(1) J. Neurol. Sci. 1999;167:90–101. doi: 10.1016/s0022-510x(99)00146-x. [DOI] [PubMed] [Google Scholar]
- 61.Zincarelli C., Soltys S., Rengo G., Rabinowitz J.E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 62.Scherer S.S., Xu Y.T., Messing A., Willecke K., Fischbeck K.H., Jeng L.J.B. Transgenic expression of human connexin32 in myelinating Schwann cells prevents demyelination in connexin32-null mice. J. Neurosci. 2005;25:1550–1559. doi: 10.1523/JNEUROSCI.3082-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Arden E., Metzger J.M. Inexpensive, serotype-independent protocol for native and bioengineered recombinant adeno-associated virus purification. J. Biol. Methods. 2016;3:e38. doi: 10.14440/jbm.2016.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Piedra J., Ontiveros M., Miravet S., Penalva C., Monfar M., Chillon M. Development of a rapid, robust, and universal picogreen-based method to titer adeno-associated vectors. Hum. Gene Ther. Methods. 2015;26:35–42. doi: 10.1089/hgtb.2014.120. [DOI] [PubMed] [Google Scholar]
- 65.Nelles E., Bützler C., Jung D., Temme A., Gabriel H.D., Dahl U., Traub O., Stümpel F., Jungermann K., Zielasek J., et al. Defective propagation of signals generated by sympathetic nerve stimulation in the liver of connexin32-deficient mice. Proc. Natl. Acad. Sci. USA. 1996;93:9565–9570. doi: 10.1073/pnas.93.18.9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kagiava A., Kleopa K.A. Intrathecal Delivery of Viral Vectors for Gene Therapy. Methods Mol. Biol. 2018;1791:277–285. doi: 10.1007/978-1-4939-7862-5_22. [DOI] [PubMed] [Google Scholar]
- 67.Ahn M., Lee J., Gustafsson A., Enriquez A., Lancaster E., Sul J.Y., Haydon P.G., Paul D.L., Huang Y., Abrams C.K., Scherer S.S. Cx29 and Cx32, two connexins expressed by myelinating glia, do not interact and are functionally distinct. J. Neurosci. Res. 2008;86:992–1006. doi: 10.1002/jnr.21561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Savvaki M., Theodorakis K., Zoupi L., Stamatakis A., Tivodar S., Kyriacou K., Stylianopoulou F., Karagogeos D. The expression of TAG-1 in glial cells is sufficient for the formation of the juxtaparanodal complex and the phenotypic rescue of tag-1 homozygous mutants in the CNS. J. Neurosci. 2010;30:13943–13954. doi: 10.1523/JNEUROSCI.2574-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gafson A.R., Barthélemy N.R., Bomont P., Carare R.O., Durham H.D., Julien J.P., Kuhle J., Leppert D., Nixon R.A., Weller R.O., et al. Neurofilaments: neurobiological foundations for biomarker applications. Brain. 2020;143:1975–1998. doi: 10.1093/brain/awaa098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Parasuraman S., Raveendran R., Kesavan R. Blood sample collection in small laboratory animals. J. Pharmacol. Pharmacother. 2010;1:87–93. doi: 10.4103/0976-500X.72350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rohrer J.D., Woollacott I.O.C., Dick K.M., Brotherhood E., Gordon E., Fellows A., Toombs J., Druyeh R., Cardoso M.J., Ourselin S., et al. Serum neurofilament light chain protein is a measure of disease intensity in frontotemporal dementia. Neurology. 2016;87:1329–1336. doi: 10.1212/WNL.0000000000003154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sandelius Å., Zetterberg H., Blennow K., Adiutori R., Malaspina A., Laura M., Reilly M.M., Rossor A.M. Plasma neurofilament light chain concentration in the inherited peripheral neuropathies. Neurology. 2018;90:e518–e524. doi: 10.1212/WNL.0000000000004932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vavlitou N., Sargiannidou I., Markoullis K., Kyriacou K., Scherer S.S., Kleopa K.A. Axonal pathology precedes demyelination in a mouse model of X-linked demyelinating/type I Charcot-Marie Tooth neuropathy. J. Neuropathol. Exp. Neurol. 2010;69:945–958. doi: 10.1097/NEN.0b013e3181efa658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kobsar I., Berghoff M., Samsam M., Wessig C., Mäurer M., Toyka K.V., Martini R. Preserved myelin integrity and reduced axonopathy in connexin32-deficient mice lacking the recombination activating gene-1. Brain. 2003;126:804–813. doi: 10.1093/brain/awg072. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available upon request from the corresponding author.