

Synopsis:
Many microbes, toxins, autoimmune, and neoplastic diseases may cause liver inflammation; however, five main viruses whose main pathogenesis is liver disease are referred to as hepatitis A, B, C, D, and E viruses. These viruses cause a significant burden of global illness. With the exception of hepatitis A virus, all may cause chronic infection potentially leading to cirrhosis and hepatocellular carcinoma. Excellent serologic and nucleic acid detection methods are available for determining the precise etiology, and in some cases, the duration of infection. Diagnostics are critical for identifying those needing treatment and for monitoring the treatment success.
Keywords: viral hepatitis, viral diagnostics, hepatitis A, hepatitis B, hepatitis C, hepatitis D, hepatitis E
INTRODUCTION
The word hepatitis is derived from “itis” meaning inflammation and “hepar”, the Greek word for liver. Many conditions including alcohol ingestion and medications may cause liver inflammation. Although many viral and bacterial infections cause hepatitis (e.g., cytomegalovirus, Epstein Barr virus, influenza, yellow fever virus), five viruses primarily infect the liver and are named after the clinical disease, hepatitis A through E. A sixth, hepatitis G virus was initially thought to cause hepatitis, but subsequent studies did not confirm this hypothesis.1 This virus will not be discussed in this review.
Symptoms of hepatitis are not specific to a single hepatitis virus, and thus clinical presentation does not distinguish between different viral etiologies. Hepatitis virus infection may be mild or asymptomatic. In symptomatic cases, acute hepatitis is associated with flu-like illness, fever, fatigue, loss of appetite, abdominal pain, nausea, vomiting, jaundice, dark urine, clay colored stools, and rarely, fulminant hepatitis. Chronic hepatitis is frequently asymptomatic or mildly symptomatic. Over time, chronic viral hepatitis may lead to persistent inflammation, leading to fibrosis with resultant cirrhosis, liver failure, and hepatocellular carcinoma.
The transmission mode for hepatitis A and E is primarily through contaminated water or food (fecal-oral), though transfusion-related hepatitis A virus (HAV) has occurred. Hepatitis B and C are transmitted through sexual and parenteral exposure, and vertically from mother to child. Hepatitis D is only transmitted with hepatitis B virus (HBV) as described below.
Microbial diagnostic testing is increasingly utilizing molecular testing for nucleic acid testing (NAT; e.g. using the polymerase chain reaction [PCR]) or microbial proteins using mass spectrometry due to their more sensitive and specific detection of pathogens. However, in viral hepatitis, serologic tests remain the mainstay of diagnosis. In some circumstances serology requires concurrent NAT methods. Understanding hepatitis transmission modes, natural history and viral kinetics, and the limitations of testing is required for choosing appropriate diagnostic tests for viral hepatitis.
HEPATITIS A VIRUS
Hepatitis A virus (HAV) is a positive sense, single strand RNA virus classified as a member of the Hepatovirus genus within the family Picornaviridae. Viral RNA is directly translated into a single polyprotein cleaved by viral proteases into structural and nonstructural protein products. Although there are seven HAV genotypes, there is a single serotype. HAV is transmitted by ingesting contaminated food or water, and it enters the bloodstream through the oropharynx or intestinal epithelium. It is delivered to the liver where it replicates in hepatocytes and Kupffer cells. Although classically thought of as a non-enveloped virus, recent data show that a pseudo-enveloped particle is released from cells into the bloodstream or into the biliary tree. Virus released into the biliary tree is transported to the gastrointestinal tract for excretion, and the pseudo-envelope is removed from the particles by bile acids.2 The excreted non-enveloped particles are highly resistant to environmental stress and may remain infectious for prolonged periods of time.
Most HAV infections are asymptomatic in young children; however, HAV more commonly causes disease in older children and adults. The average time from exposure to clinical illness is 4 weeks (range 2 to 6 weeks), and fecal viral RNA levels rapidly fall at the time of clinical illness (Fig. 1).3 Anti-HAV antibodies and HAV-specific cytotoxic T cells are detected at the time of illness. Taken together, these findings suggest that HAV pathogenesis is immunologically mediated. Illness usually lasts less than 1 month; however, relapsing hepatitis, and rarely fulminant hepatitis A occur. A variety of extrahepatic complications are described, including arthritis and vasculitis. Following infection, there is no residual liver disease, and anti-HAV is protective against reinfection.
Figure 1.
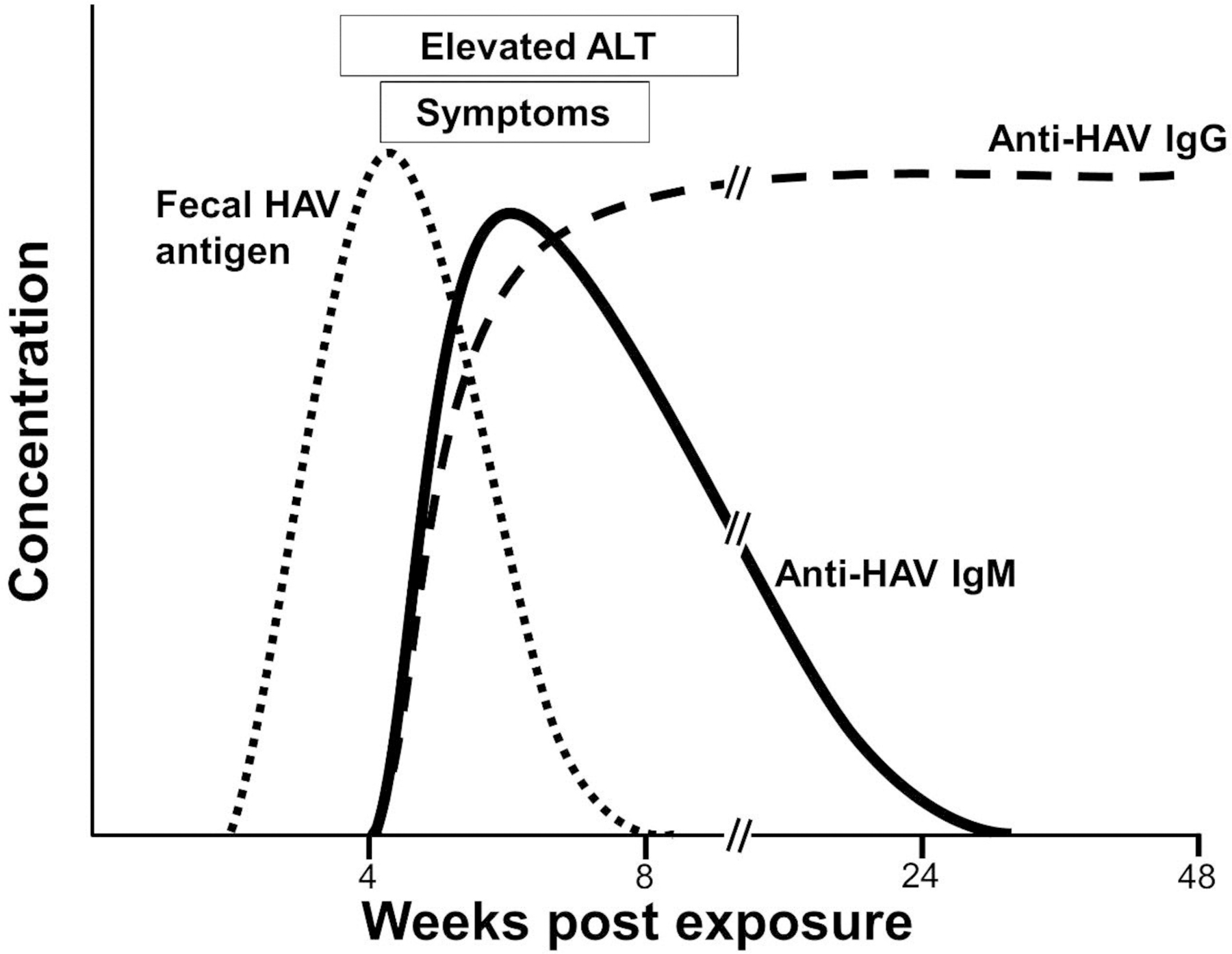
Schematic of the kinetics of viral, serologic, and clinical findings in hepatitis A virus infection.
DIAGNOSTIC EVALUATION
HAV viremia begins prior to illness and virus excretion into the stool via the biliary tree peaks around the time of maximal liver enzyme elevations (Fig. 1). Anti-HAV antibodies are detected at or shortly after elevation of ALT. Initially, anti-HAV IgM isotype antibodies are detected, and these typically fall below detection limits within 6 months post infection. Before IgM levels fall, anti-HAV IgG is detected. Anti-HAV IgG is protective and persists for life. Measurement of “total” anti-HAV antibodies identifies both IgG and IgM isotypes, and if present, there is lifelong protection against reinfection. Total antibodies will not distinguish acute versus chronic infection. IgM specific antibody testing is required, and if positive, testing is diagnostic of acute infection. Since HAV is an acute self-limited disease, and IgM antibodies are present prior to and during infection, there is no clinical indication for detecting virus by either antigen or RNA detection methods. As a result, no commercially available HAV antigen detection or NAT assays are available.
HEPATITIS B VIRUS
Hepatitis B virus (HBV) is an enveloped, partially double-stranded DNA virus within the family Hepadnaviridae. HBV is classified into 10 genotypes, A to J.4 Although genotypes have distinct geographic distributions and different rates of disease progression,4,5 clinical use of genotype determination requires further study.6 The infectious HBV virion is comprised of a lipoprotein envelope (hepatitis B surface antigen or HBsAg), the viral core protein or antigen (HBcAg) which assembles into the capsid encapsidating the partially double stranded, circular DNA genome (Figure 2A). The particle also carries the RNA dependent, DNA polymerase (RdDp or reverse transcriptase). DNA replication utilizes an RNA intermediate, explaining why some HIV antiviral drugs are active against HBV. The core protein precursor (pre-Core) contains a 28-amino acid region at the N-terminus. A truncated HBcAg containing the pre-core region is called the hepatitis B e antigen (HBeAg). This can be secreted by infected cells and is a marker of HBV replication and infectivity.7 HBsAg is released into the bloodstream at high concentrations in large spherical or tubular shaped particles (Fig 2B, C). Specific patterns of viral antigens and antibodies appear during acute and chronic infection. Therefore, understanding the different HBV antigens is critical for interpreting diagnostic tests for HBV infection.
Figure 2.
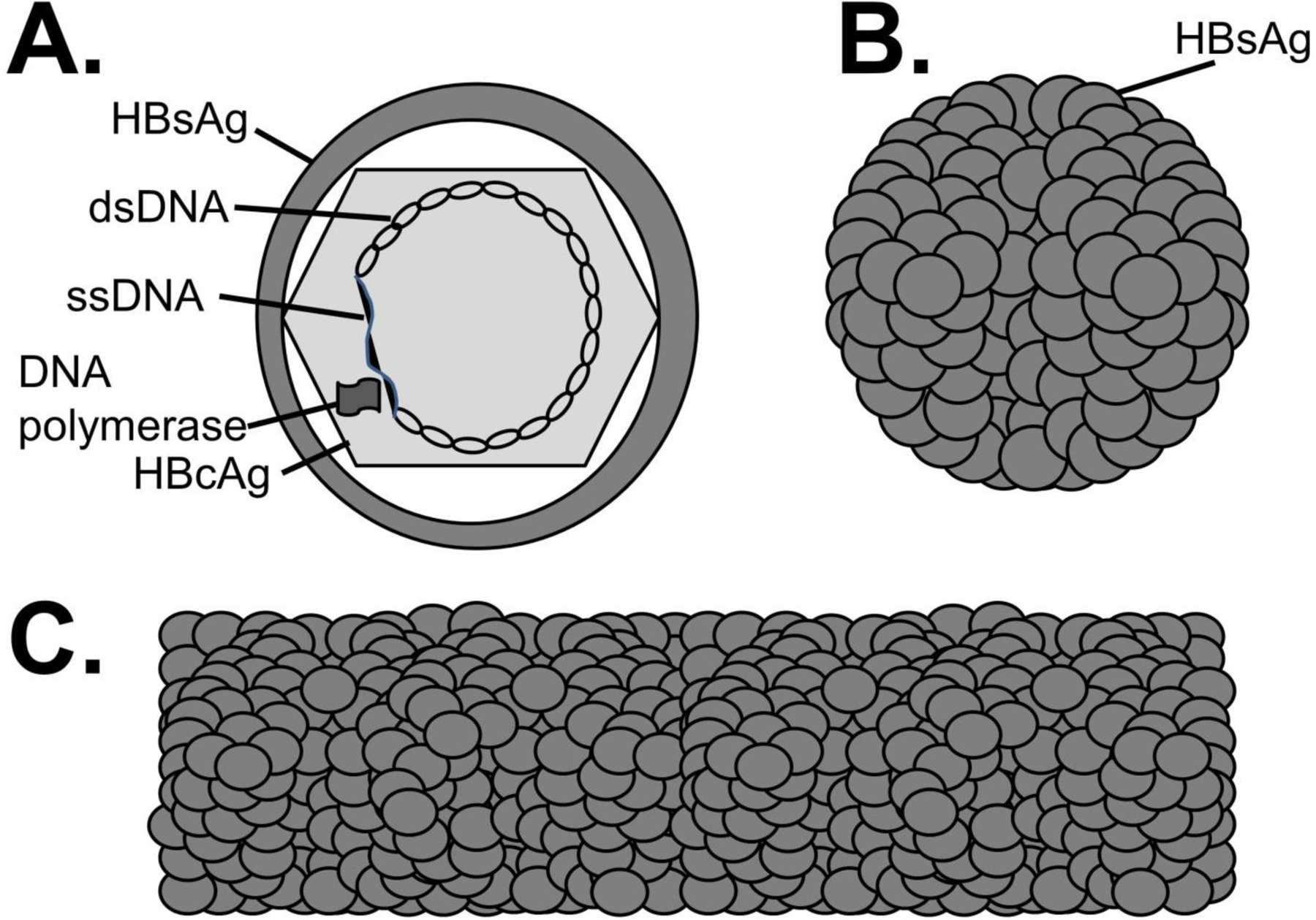
HBV virus particles. A. The infectious (Dane) particle contains surface antigen (HBsAg), the partially double stranded (ds) DNA genome (ss = single strand), the viral RNA dependent, DNA polymerase, and the core antigen (HBcAg). HBsAg is released into serum as spherical particles (B) or tubular structures (C). These particles do not carry viral DNA, HBcAg or the viral polymerase.
NATURAL HISTORY
HBV primarily infects hepatocytes although viral DNA is also detected in peripheral blood mononuclear cells. Viral DNA is detected in serum using NAT methods within 2 to 5 days after acquisition.8,9 DNA detection is highly sensitive, and thus a reliable marker of active HBV replication. HBsAg is detected using serologic methods and is detected 1 to 2 weeks after HBV DNA detection.10 HBsAg detection indicates active HBV replication. Since viral replication can be determined serologically, HBV DNA is usually not tested until HBsAg is detected.
Fewer than 15% of patients with HBV infection have clinical hepatitis, and less than half of these developed jaundice.11 Clinical illness correlates with the age and immunological maturity, and newborns and children rarely having significant clinical disease. Overall, approximately 95% of immunocompetent people have self-limited HBV infection, most recovering completely. The remaining individuals develop chronic infection. The rate of viremia clearance is inversely related to the magnitude of clinical illness; thus chronic infection is increased in newborns and immune compromised individuals who rarely have overt hepatitis.11,12 Vertical transmission from HBV-infected mothers to their children results in chronic infection in approximately 90% of cases.13
Symptomatic patients may have a pre-icteric or prodromal period followed by clinical hepatitis that typically occurs 11–24 weeks following exposure.8,9 Peak liver enzyme elevations occur after development of HBV-specific immune responses, supporting an immune-mediated component of HBV liver pathogenesis. In those with clinical illness, symptoms generally improve by the time jaundice develops, typically 2 to 6 weeks after peak serum HBV DNA levels. In acute, self-limited infection, HBV DNA and HBsAg levels generally fall below detection limits in the first 3–4 months post-infection.8
Serologic Evaluation of Infection
HBV core antigen and antibody
The first antibodies to appear are directed against the core protein (anti-HBc; Fig. 3). The first anti-HBc antibodies are IgM isotype, and these switch to the IgG isotype over 3 to 6 months. Once anti-HBc IgG antibodies develop, they are detected for life in most individuals, and measurement of “total” anti-HBc (detecting IgG and IgM anti-HBc isotypes) is the best marker for documenting prior HBV infection regardless of other serologic results. Total anti-HBc does not indicate the timing of infection, and anti-HBc IgM measurement is needed to determine if the infection is recent (Fig. 3 and 4).
Figure 3.
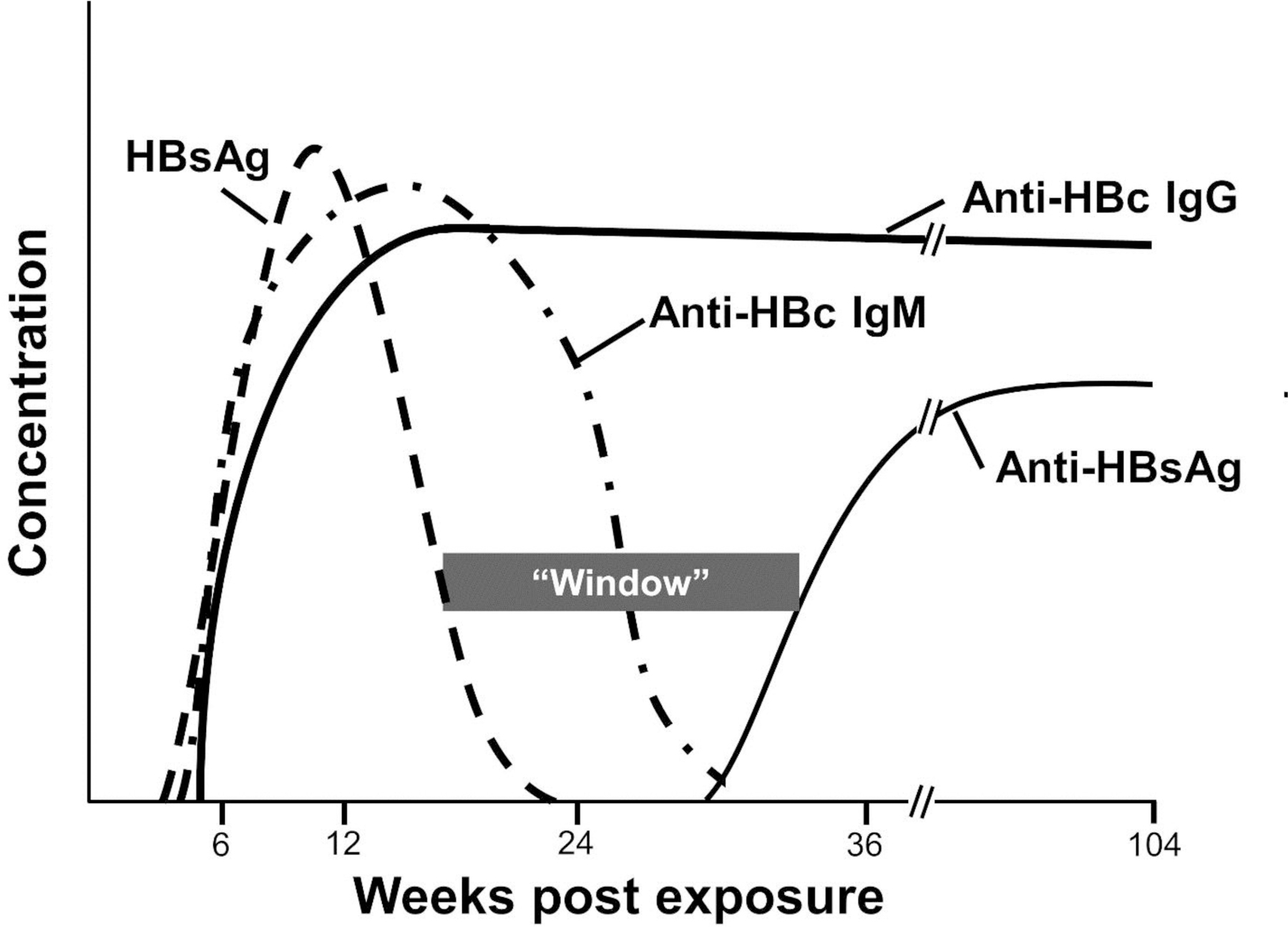
Viral and serologic findings in acute hepatitis B virus infection. HBsAg = hepatitis B virus surface antigen. Anti-HBsAg = anti-hepatitis B surface antibodies. Anti-HBc = anti-hepatitis B core antibodies. “Window” is time between between positive surface antigen and antibody that will not be detected unless anti-HBc antibodies are measured.
Figure 4.
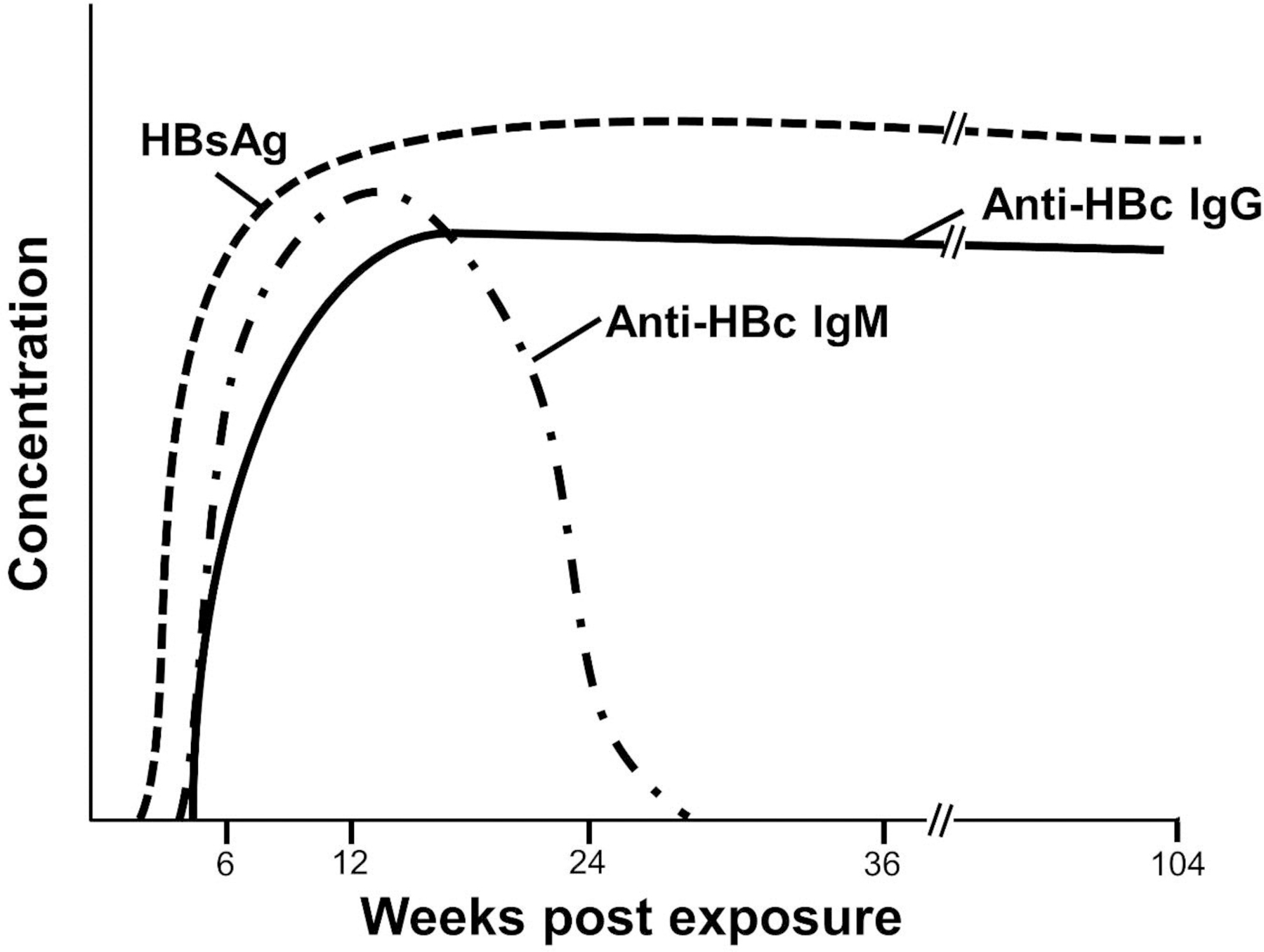
Viral and serologic findings in chronic hepatitis B virus infection. HBsAg = hepatitis B virus surface antigen. Anti-HBc = anti-hepatitis B core antibodies.
HBV surface antigen and antibody
In acute self-limited infection, antibodies to HBsAg are detected after HBsAg clearance and appearance of anti-HBc IgG. Anti-HBs is generally detected within 6 months after infection and is a marker of protection against re-infection.14 Current HBV vaccines are comprised of recombinant (non-infectious) HBsAg particles that elicit anti-HBs (Fig. 2B, C), thus vaccinated individuals have anti-HBs antibodies detected in serum.14 The presence or absence of total anti-HBc will differentiate prior infection from vaccination in the HBsAb positive person, as vaccination does not elicit anti-HBc.13 Since there is a period of time (window) when HBsAg and anti-HBs are both negative during acute infection, screening for HBV infection should include a total anti-HBc test (Fig. 3).
Although the patterns in figures 3 and 4 are highly consistent, one serological pattern occurs that is difficult to reconcile with clinical decision making. Specifically, HBV DNA without detectable anti-HBc occurs in immunocompromised people with acute hepatitis (13.8%), in HBV reactivation (41.4%) and chronic hepatitis B (44.8%)15–18. Approximately half of these patients develop anti-HBc over time (often at low levels), or HBc is detected using different anti-HBc assays.18 No mutations have been identified in core protein sequences in these individuals, thus this scenario appears to represent an immune recognition defect18. Therefore, it is not advisable to rely on a single anti-HBc negative result to exclude HBV infection in immunocompromised hosts and repeat anti-HBc testing or using different serologic testing methods should be considered.18
HBV e Antigen and Antibody
HBeAg is a variant of the HBcAg and is released into the circulation shortly after infection. Development of anti-HBe and loss of HBeAg during acute self-limited infection is a predictor of clearance. In chronic infection, development of anti-HBe occurs late in infection, usually after several years. Seroconversion to anti-HBe indicates a favorable outcome marking transition from high replication rates to low replication rates associated with inactive hepatitis B. However, some individuals with anti-HBe demonstrate active liver pathology. Mutations in the pre-core amino acid sequence have been identified, suggesting that mutation resulted in seroconversion to anti-HBe.19,20
Chronic HBV Infection
Chronic HBV infection is defined as detection of HBsAg on at least 2 separate occasions measured at least 6 months apart.21,22 Host cellular immune responses to virus-infected hepatocytes are thought responsible for hepatic inflammation causing liver injury. These contribute to the development of cirrhosis and hepatocellular carcinoma in chronic HBV infection. There are 4 phases in chronic HBV which are important in deciding when to treat HBV. These are summarized as:
Immunotolerance. Asymptomatic patients with positive HBsAg, HBeAg, and normal liver enzymes. High HBV DNA (> 20,000 IU/mL).
HBeAg-positive immunoactive disease. Individual may or may not have clinical liver disease. Positive HBsAg and HBeAg, positive or negative anti-HBe, elevated liver enzymes (> 2 times the upper limit of normal ALT), and high HBV DNA (> 20,000 IU/mL).
HBeAg-negative, inactive disease (inactive chronic HBV or low replicative infection). Positive HBsAg, negative HBeAg (positive anti-HBe), normal ALT, low HBV DNA (< 2,000 IU/mL). There may be fibrosis from previous inflammation.
HBeAg-negative immunoreactive disease. Negative HBsAg, HBeAg (positive anti-HBe), elevated liver enzymes (> 2 times upper limits of normal ALT), intermediate to high HBV DNA (> 2,000 IU/mL).
Although these phases of HBV pathogenesis do not have unique clinical presentations, disease is generally asymptomatic in the immunotolerance and HBeAg-negative inactive disease phase, but more active in the HBeAg-positive immunoactive and HBeAg-negative immunoreactive disease stages. Since either HBeAg-positive immunoactive disease and HBeAg-negative immunoreactivation disease may progress to liver failure, initiation of treatment is recommended in these situations. HBV DNA levels may fluctuate, though they are persistently elevated (> 20,000 IU/ml) in individuals with detectable HBeAg. HBV DNA can differentiate inactive carriers from patients with HBeAg-negative chronic hepatitis B23. Inactive chronic hepatitis B patients typically have HBV DNA < 2,000 IU/mL while those with immune active hepatitis B have HBV DNA > 20,000 IU/mL.
DIAGNOSTIC TESTING
Virus detection
HBsAg and HBeAg detection
HBV antigens are detected using solid-phase immunoassays. HBsAg particles or HBeAg protein is captured to the solid-phase with a monoclonal or polyclonal sera and a labeled secondary antibody to the specific antigen is used for detection. These assays use microparticles and are automated. Current detection methods use enzymatic, chemiluminescence, or fluorescence polarization methods to detect the antigens.24 HBsAg assays detect a minimum of 0.7 ng/mL of HBsAg, with newer tests detection limits down to 0.13 ng/mL.25
There are concerns that some assays cannot detect HBsAg variants with mutations in the major antigenic region that result in conformation changes. Many HBsAg immunoassays use antibodies directed against the main antigenic determinant (“a” determinant). Mutation in this region may account for the false-negative results by some assays.26–28 Thus, acute infection should always include screening for anti-HBc or HBV DNA.29 HBsAg quantitation is not needed in patients with chronic hepatitis B, although quantitative HBsAg has been used by some in monitoring patients receiving interferon-based therapies.22
Nucleic Acid Amplification Testing (NAT)
HBV DNA PCR
Quantitative HBV DNA testing is essential for determining the need for HBV treatment and for evaluating treatment response.22 Highly sensitivity NAT assays are important for diagnosis of HBeAg-negative chronic HBV and occult HBV, where DNA concentration may be quite low.30 Current HBV DNA quantification methods utilize real-time PCR. This has excellent analytical performance, including a low limit of detection and a broad linear range.30 However, characteristics vary among different commercial platforms. The World Health Organization (WHO) has generated an HBV DNA standard and results should be provided in international units (IU).31 Nevertheless, quantitative results may vary and the best practice for following DNA levels is to use the same assay from the same laboratory whenever possible.30
Serology
Anti-HBc, anti-HBs, and anti-HBe
Commercial detection of anti-HBc, anti-HBs, and anti-HBe antibodies utilizes enzyme linked immunosorbent assay (ELISA) methodologies. Several versions of each test are available using different detection methods and instrumentation. The two most commonly used methods employ a competitive approach (anti-HBc and anti-HBe) or a solid-phase, sandwich type approach (anti-HBs).14,32
Genotype testing
Currently, commercial testing of HBV genotypes is not recommended for clinical care with the exception of testing before interferon-based therapy, or when knowledge of the HBV genotype may aid risk stratification of disease progression.21,22
Resistance testing
Since transmission of resistance mutations is rare in North America, resistance testing is not recommended in treatment-naïve patients prior to starting therapy.33 Antiviral resistance may be useful for patients with past treatment experience, those with persistent or virological breakthrough while on antiviral therapy as defined by a 10-fold increase in serum HBV DNA from their nadir during treatment.22 Resistance is determined by evaluating specific sequence variation within the polymerase gene, and identifying polymorphisms demonstrated to correlate with antiviral resistance in vitro or in vivo. Current methods include restriction fragment length polymorphism, hybridization, and sequencing methodologies, and testing usually requires HBV DNA concentrations > 1,000 IU/mL.
LABORATORY USE IN MONITORING
Baseline studies
Patients with chronic HBV infection should be evaluated to determine the phase of infection as described above. ALT, HBV DNA, and HBeAg should be measured, and liver fibrosis quantified to allow prediction of long-term outcomes and inform treatment decisions. In patients receiving treatment, HBV DNA levels are the primary method to determine treatment response. Therefore, serial testing of hepatic function, quantitative HBV DNA, HBeAg, anti-HBe, as well as evaluation of liver fibrosis are needed to guide treatment decisions.21,22 In addition to HBV testing, laboratory monitoring during antiviral therapy should include measurement of HBsAg, HBeAg, anti-HBs, anti-HBe, and CBC, renal and hepatic function every 3–6 months. These tests will potentially identify progression of liver disease, extrahepatic manifestations of chronic HBV, and treatment toxicities.21 A summary of serologic and NAT results useful in different HBV clinical situations is shown in Table 1.
Table 1.
Diagnostic test patterns in acute and chronic hepatitis B virus infection
| HBsAg | Anti-HBs | Anti-HBc IgM | Total Anti-HBc | HBV DNA | Interpretation |
|---|---|---|---|---|---|
| + | − | + | +/− | + | Acute hepatitis B |
| − | + | − | + | − | Past infection |
| − | + | − | − | − | Vaccination |
| − | − | − | + | +/− | Previous infection, occult hepatitis B |
HEPATITIS C VIRUS
Hepatitis C Virus (HCV) is a positive-sense, single-stranded RNA virus. Its structure, genomic organization, and replication cycle support classification as a member of the family Flaviviridae, but unique enough to classify in a separate genus, Hepacivirus.1 The RNA genome is translated into a polyprotein that is cleaved by cellular and viral proteases into structural and nonstructural proteins. Due to extensive genetic diversity, there are 7 major genotypes and 67 HCV subtypes globally.34 Genotype distribution varies by geographic location,34 and globally, genotype 1 is a predominant genotype, followed by the genotype 3, then genotype 4, respectively.35
NATURAL HISTORY
Acute HCV refers to the first 6 months following acquisition of infection, regardless of symptoms.36,37 Infection is usually asymptomatic despite abnormal liver function test results.38,39 In symptomatic patients, most present with nonspecific flu-like symptoms.36 A minority of infected individuals develop symptoms of acute viral hepatitis including jaundice, anorexia, and abdominal discomfort.36 Although HCV infection may cause life-long infection, 15% to 30% of infections resolve spontaneously.40,41 These patients lose detectable HCV RNA, and in symptomatic patients, this typically occurs within 3 to 4 months following the onset of symptoms.42 Several variables are associated with HCV clearance including polymorphisms in the interferon-lambda −2 gene (IL28B), gender, race, age, and a variety of host immune markers (HLA-B, HLA-C, KIR).43,44 The timing of viral clearance influences the decision to consider therapy for acute hepatitis.45
Unfortunately, the majority of patients have persistent viremia. Chronic HCV infection is defined as lasting more than 6 months. Chronic infection is usually asymptomatic and may progress slowly and silently to chronic liver disease.41 The major liver disease caused by HCV is fibrosis and severe fibrosis (cirrhosis) develops in 20% to 30% of chronic HCV infections.41 Although cirrhosis can develop rapidly, it typically develops over 20 to 30 years. Several factors increase the risk of cirrhosis including alcohol use, male gender, age of acquisition, and immune suppression (HIV).41,46 HCV-infected people with cirrhosis are also at increased risk of hepatocellular carcinoma. Several extrahepatic manifestations of HCV are also recognized, including arthritis, keratoconjunctivitis sicca, lichen planus, glomerulonephritis, porphyria cutanea tarda, and type II cryoglobulinemia.41 Up to 80% of HCV infected people have detectable rheumatoid factor in serum; half of these also have cryoglobulins detected.47
HCV KINETICS
HCV RNA is not detected in serum during the first 1 to 2 weeks following transmission48, yet HCV RNA is the earliest marker of infection.38 Early in infection, HCV RNA levels range widely (from 2,500 to > 1 million IU/mL). Viremia precedes ALT and bilirubin elevations. The HCV RNA pattern early in infection often shows a peak, followed by a reduction in concentration. In patients who spontaneous clear infection, HCV RNA continues to rapidly fall until viremia is not detectable.49,50 An alternative pattern is sometimes seen, where low-level viremia (< 120 copies/mL) may precede an increase and plateau of HCV viral load (VL).38,51 The variability emphasizes the need to repeat HCV RNA testing in situations where the suspicion of early HCV is high. The majority of people with chronic HCV infection demonstrate fluctuating or high HCV RNA levels.40,50 HCV RNA levels frequently stabilize somewhat, and although there is a large range of concentrations between individuals, average HCV RNA levels are more than one million genome copies/mL plasma.52
Seroconversion is generally slow, ranging from 34 to 70 days in HIV-negative blood product recipients and intravenous drug users.53,54 Antibodies are rarely detected before liver enzymes peak or return to normal values. Thus, infection may be missed if liver enzymes alone were used as the indication for HCV testing.38 The delay is even greater in HIV-infected individuals, with median seroconversion ranging from 91 to 158 days.55 Antibody negative, HCV RNA positive infection is uncommon, but occurs in both HIV-infected and uninfected individuals.56,57 Although the reasons for variation in viremia and host immune response patterns are not clear, they likely are influenced by several factors including the route of entry, the nature of inoculum, the frequency of exposure, and viral-mediated interference with host immune responses.58,59
DIAGNOSTIC TESTING
Diagnostic testing for hepatitis C virus infection relies primarily upon the detection of antibodies by serologic testing and the direct detection of viral RNA by nucleic acid detection testing (NAT). Both HCV serology and NAT are validated, and FDA approved using serum or plasma as the specimen source. There is also a licensed serologic assay performed using oral fluid.60
Detection of virus
Antigen Detection
Detection and quantification of core antigen in serum or plasma utilizes enzyme-linked immunosorbent assay (EIA). This is used extensively in Europe and available globally, particularly in resource-limited settings due to target stability, simple instrumentation and cost.50 Detection of HCV core antigen usually correlates well with HCV RNA detection and thus may serve as a surrogate marker for viral replication.61,62 However, the assay has inferior sensitivity and specificity compared to HCV RNA testing when HCV RNA values are below 20,000 IU/mL. Therefore, this method is not commonly used in the U.S.61,62
Nucleic Acid Detection Testing (NAT)
HCV can be quantified using target amplification techniques (transcription-mediated amplification [TMA], or reverse transcription real time PCR [RT-real time-PCR]) methods with very low limits of detection, ranging from 1.0 – 1.7 log10 IU/mL. Quantitative methods are preferred to qualitative methods for establishing baseline HCV RNA concentration prior to treatment, and for following response during and following therapy. The American Association for the Study of Liver Diseases (AASLD) recommends use of FDA-approved, highly sensitive HCV VL with a limit of detection of less than 25 IU/mL.63
In the past, quantitative units of various assays did not report the same concentration of HCV RNA in clinical samples. The WHO established an international calibration standard for HCV RNA and defined international units/mL (IU/mL) to allow comparison of HCV RNA levels in clinical samples between laboratories and assays. Nevertheless, some problems remain due to intrinsic variability between instruments and laboratories including precision, reproducibility, and accuracy.64 It is important to remember that, since the methods rely upon logarithmic amplification of viral RNA, viral load changes less than 0.5 log10 IU/mL (e.g. 3-fold) may reflect differences in laboratory performance.65
Serology
Available HCV serologic assays in the United States include the second- and third-generation Enzyme Immunoassay (EIA). No single HCV antigen consistently elicits antibodies in humans, thus the assays use multiple HCV antigens. The first-generation HCV assay used a region of the non-structural protein NS4 (c100–3) to detect anti-HCV antibodies (See figure 5). This was refined in second generation tests to use regions of NS4 (regions termed C200, HC-31), NS3 (protease; c33c), and core (c22–3) proteins. This was further enhanced in third generation tests and the core antigen and NS4 antigens were changed (c22p, a peptide containing a major epitope residing in the core protein between amino acids 10 to 35), NS4 HC-31 to NS4 5–1-1p, and the NS5 protein was added. Second- and third-generation EIA tests have increased sensitivity and specificity and identify early seroconversion and atypical seroconversion better than first generation tests.66
Figure 5.
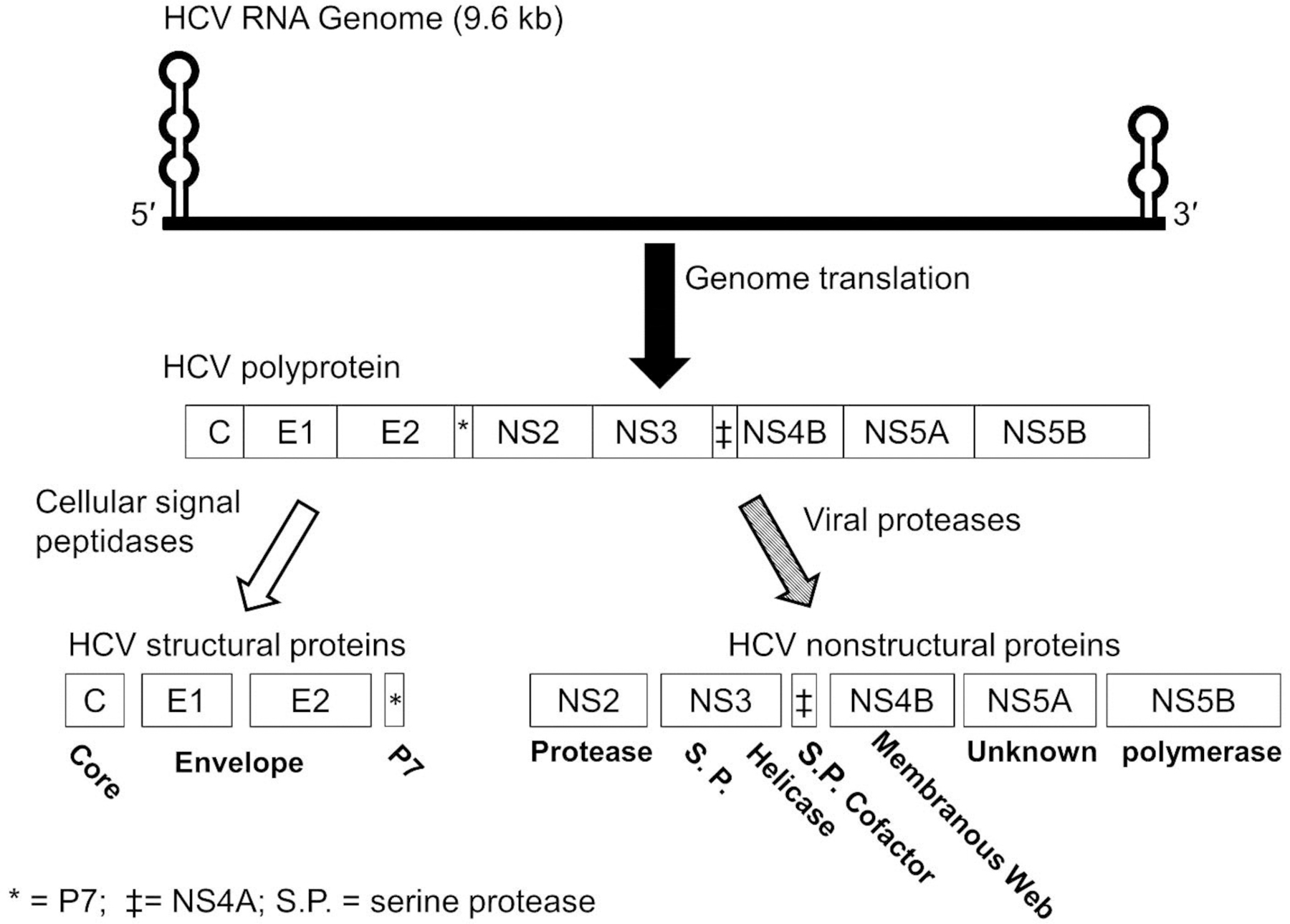
Hepatitis C virus genome organization (top), polyprotein structure (middle), and processing (bottom). The 5′ and 3′ untranslated regions contain highly structured, stem-loop regions.
Although second-generation assays are still available, they may yield false-negative results, and alternative testing with third-generation EIA or NAT should be considered in patients with negative EIA 2.0 results, particularly in those with a high index of suspicion.
In addition to the standard EIA tests, there is an FDA-approved, rapid, highly accurate point-of-care test available. It detects antibody using an indirect immunoassay technique using a nitrocellulose membrane coated with core, NS3, and NS4 antigens. The clinical performance is comparable to current laboratory-developed EIA methods, and this test may prove useful in addressing the problem of under-diagnosis of HCV.60 IgM testing has no utility in acute hepatitis C as it may is detected at the same time as with IgG and may persist for up to a year after HCV acquisition.67
Genotype and resistance testing
As noted above, there are 7 HCV genotypes. Determining the HCV genotype is useful for determining the best treatment regimen. This was particularly true in the era of interferon-based (IFN) treatments. In addition, many direct activing anti-HCV (DAA) therapies are genotype or subtype specific.68 HCV genotypes are determined using Sanger sequencing, next-generation sequencing (deep sequencing) or hybridization methods, and can utilize relatively short regions of the 5’ untranslated (5’UTR) genome sequence from samples and aligning these with reference sequences.69
Due to the error-prone HCV RNA dependent, RNA polymerase (RdRp), HCV amino acid polymorphisms naturally occur throughout the genome coding region that may be associated with resistance or reduced susceptibility to a member or even an entire class of DAAs. These sequence differences may or may not confer resistance to a specific drug in the class (protease inhibitor, polymerase inhibitor, NS5A inhibitor, etc.). In addition, resistance mutations may be selected during DAA treatment. The field of DAA HCV therapy is rapidly evolving, and there are increasing options for pan-genotypic inhibitors. Thus, the use of genotype and resistance testing is constantly changing. The AASLD provides and updates recommendation guidelines.63
DIAGNOSTIC APPROACH FOR HEPATITIS C
Currently, there are no validated methods to distinguish acute and chronic HCV infection.70 Infection duration estimation relies upon prior diagnostic testing results and an understanding of the mode of transmission, natural history, viral kinetics, and limitations of different laboratory test methodologies. Since seroconversion may be delayed for many weeks following exposure, diagnosis of HCV during acute infection requires HCV RNA detection.45 Further, HCV RNA may be negative or very low titer early in infection, thus repeat RNA testing is warranted two to four weeks following a negative test in cases where there is a high index of suspicion.50
Detection of HCV RNA without anti-HCV is strongly indicative of acute hepatitis in immunocompetent patients, particularly when it is followed by seroconversion.50 In clinical practice, this sequence of events is uncommonly detected. Most individuals with chronic HCV infection have positive RNA and antibody results, though infection occasionally does not appear to elicit detectable antibodies.50 Therefore, diagnosis is these patients should rely on HCV RNA. Table 2 provides a summary of diagnostic tests and interpretations.
Table 2.
Summary of laboratory result and interpretation
| Anti-HCV Ab | HCV RNA | Interpretation |
|---|---|---|
| Negative | Detectable | • Acute hepatitis C • Chronic hepatitis C in immunocompromised or exceptional cases |
| Detected | Undetectable | • Spontaneous resolved* • Treated infection |
| Detected | Detected | • Chronic infection • Acute infection (clinical history of exposure may help to distinguish) |
| Negative | Undetectable | • No evidence of hepatitis C infection** |
may require repeat HCV RNA testing to ensure the clearance
may require repeat HCV RNA testing if high clinical suspicious of early infection within 1–2 weeks of acquisition is suspected.
LABORATORY MONITORING
From AASLD guideline (www.HCVGuidance.org on March 19, 2018)63
Baseline laboratories
The HCV genotype should be determined in patients with chronic HCV infection, as it contributes to treatment options and prognosis. Resistance testing is also needed in some situations. Currently it is recommended for some anti-NS5A DAA therapies as a baseline in genotypes 1a and 3. However, this is an evolving field, and consultation with AASLD guidelines is highly recommended prior to treating HCV infection.63 Other laboratory studies needed prior to initiating HCV DAA therapy include CBC, electrolytes, renal function, liver enzymes, PT/INR, serologic evaluation for HIV and HBV, and an assessment of hepatic fibrosis. Methods to assess fibrosis include liver biopsy, imaging, and noninvasive markers (i.e. fibroscan).71
During therapy, CBC, creatinine, and liver enzymes are recommended 4 weeks after starting treatment and as clinically indicated for drug-related adverse effects. Quantitative HCV RNA testing is recommended 4 weeks after starting and 12 weeks after completing therapy. Testing may be considered at the end of treatment, and 24 weeks or longer following the completion of therapy.63
HEPATITIS D VIRUS
Hepatitis D virus (HDV) is a defective RNA virus. Since HDV requires the HBV lipid envelope (HBsAg, Fig. 2) to assemble into viral particles that are capable of infecting new cells, it is incapable of reproducing unless there is HBV co-infection.72 HDV is a member of the Deltavirus genus and there are 8 genotypes (genotype 1 to 8) which have distinct and specific geographical distribution.73
HDV can be co-transmitted with HBV to a person who does not have HBV infection (Figure 6). Alternatively, HDV may superinfect an individual with existing HBV infection (Figure 7).74 Clinically, infection with HDV is indistinguishable from the other viral hepatitis viruses, though on average hepatitis is more severe.75 In acute co-infection, HDV clears if HBV clears, thus approximately 95% of these infections resolve. HDV appears to be more aggressive, leading to more rapid cirrhosis and hepatocellular disease in chronically HBV-infected individuals superinfected with HDV.76 HDV becomes chronic in 70–90% of superinfections.75 HDV transmission risks are the same as HBV; though infections are highest in injecting drug users, people exposed to blood or blood products, and individuals from Mediterranean, sub-Sahara Africa, Middle East, the northern part of South America, and Central and Northern Asia.77 Guidelines recommend screening for HDV in immigrants from regions with high HDV endemicity, HBV-infected individuals with unexplained high ALT, and in those with uncertainty regarding HBV treatment initiation.22
Figure 6.

Schematic of the kinetics of viral and serologic findings in hepatitis D virus co-infection with HBV in a self-limited HBV infection. Since hepatitis B surface antigen (HBsAg) serves as the surface envelope protein for HDV, it must be present when HDV RNA is detected. Hepatitis delta virus antigen = HDV Ag, HBV serology as in Figure 3.
Figure 7.
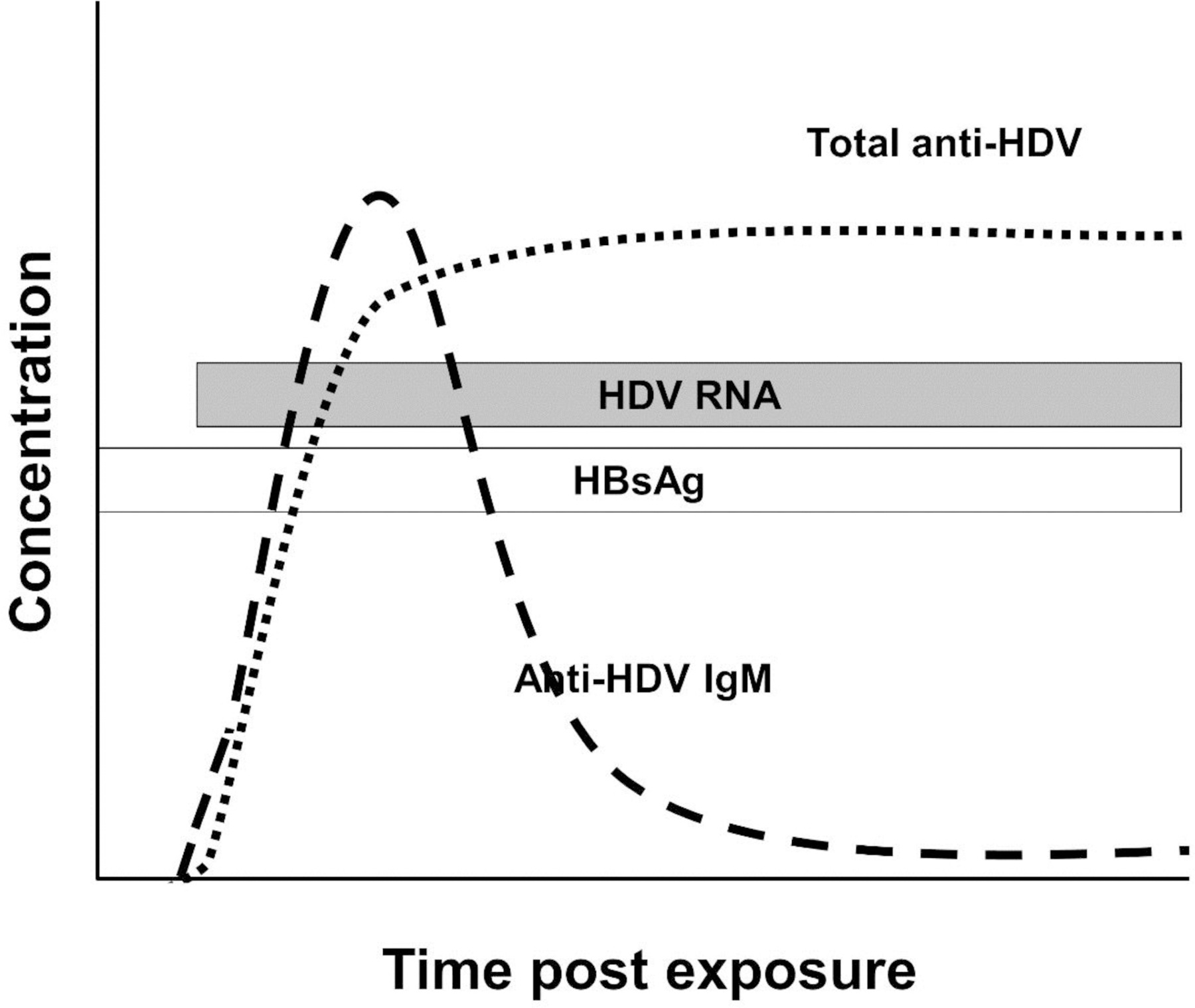
Schematic of the kinetics of viral and serologic findings in hepatitis D virus superinfection of a person with chronic HBV infection. Since hepatitis B surface antigen (HBsAg) serves as the surface envelope protein for HDV, it must be present when HDV RNA is detected. HBV serology as in Figure 4.
DIAGNOSTIC TESTING
Virus detection:
Hepatitis D virus antigen (HDAg)
Detection of HDAg is an indicator of acute infection.78 It appears early, but is very short-lived. Serum HDAg can be detected by either ELISA or radioimmunoassay (RIA), but is less sensitive than measuring HDV RNA.
HDV RNA
Quantitative HDV RNA represents the gold standard for diagnosis of HDV infection, and is useful for monitoring response to treatment, especially to assess a sustained virologic response (SVR) which is associated with cure.79,80 Available assays may not detect HDV RNA, and if they do, the measured RNA levels may be dramatically lower than actual levels, especially when measuring the African HDV genotypes (HDV-5 to HDV-8). This is attributed to sequence diversity causing primer mismatches, and potentially to the complex secondary structure of genomic RNA.81 New instruments are under development to improve performance characteristics regardless of the genotype.82 The WHO has developed an international standard HDV RNA preparation (WHO-HDV-IS) to serve as a quality control.83 Further, a commercially available automated quantitative real-time PCR method is available in reference laboratories in the United States.
Anti-HDV antibodies
Serological testing for HDV infection utilizes anti-HDV IgM antibody detection (anti-HD IgM). Anti-HD IgM is detected during the window period between HDAg and development of anti-HDV IgG (Figure 6). Anti-HD IgM is indicative of chronic infection when present at high titer.78 It rapidly declines in patients with self-limited infection. In contrast, anti-HD IgM persists in patients with chronic infection.84,85 Falling anti-HD IgM predicts resolution of chronic HDV infection. This may occur spontaneously, or be induced by anti-HBV therapy. Interestingly, anti-HD IgM rises in response to HDV-induced liver damage,86 and may be useful if HDV RNA is negative in the face of clinical features suggesting HDV-related liver disease, given the poor sensitivity of HDV RNA assays.87 Anti-HD IgG appears several weeks after anti-HD IgM and may persist for life regardless of clinical outcome or clearance of infection.85
The AASLD recommends screening by measuring anti-HDV antibodies (IgM and IgG). If either of these is positive, HDV-RNA testing is indicated to diagnose active infection.22
LABORATORY USE IN MONITORING
Baseline laboratories
Evaluation for treatment follows the same approach used for HBV, and includes liver enzymes, HBV DNA, and fibrosis evaluation.22 Anti-HBc IgM can be used to determine the status of acute co-infection (anti-HBc IgM positive) versus HDV infection of a chronic HBV carrier (anti-HBc IgM negative). This will provide some insight into the potential of developing chronic HDV infection.
There are no HDV specific antiviral therapies, but treating HBV effectively treats HDV. IFN-based therapies may have HDV effects. Monitoring HDV RNA during HBV therapy provides no predictive benefit. If an IFN-based therapy is used, HDV RNA should be monitored up to 24 weeks after completing treatment to determine if HDV clears regardless of HBV. Unfortunately, though IFN is the drug of choice for HDV, treatment success rates are < 60%.22,79,88
HEPATITIS E VIRUS
Hepatits E virus is a non-enveloped RNA virus classified within the Hepeviridae family. There are at least 4 genotypes, and genotypes 1 and 2 are found exclusively in humans, while genotypes 3 and 4 are zoonoses found in humans and other animals. There are at least 2 distinct epidemiological patterns. HEV1 and 2 are associated with large sporadic and epidemic outbreaks in developing countries. HEV is transmitted by the fecal-oral route, usually via contaminated water. Autochthonous cases of sporadic hepatitis in the developed world are associated with HEV3 and HEV4 infection. These are thought to be transmitted zoonotically by ingesting undercooked animal products (swine, deer, and unidentified sources) in addition to travelers visiting endemic regions.89–91 HEV should be considered in cases of unexplained acute hepatitis regardless of travel history.91 Although it was thought that HEV only caused acute, self-limited infection, it appears that HEV may cause chronic hepatitis resulting in rapidly progressing cirrhosis in immunocompromised hosts, including patients receiving kidney transplantation and those with HIV-1 infection.92–94
DIAGNOSTIC TESTING
Although there are no FDA approved tests at present, many commercial and reference laboratories have quantitative HEV RNA and serology for HEV available.
Detection of virus
HEV RNA
HEV RNA is detected using NAT methodologies, though no serologic method is available for HEV antigen. In patients with acute HEV infection, viremia peaks during the incubation period and early symptomatic phase (Figure 8).95,96 HEV fecal excretion is short lived and HEV RNA is not generally detected in serum or feces following biochemical hepatitis.95 In immunocompromised patients where HEV RNA persists for ≥ 3 months, it appears unlikely that spontaneous viral clearance will occur.97 Recently, the WHO developed an international standard RNA preparation for genotype 3a to facilitate accurate quantification of HEV RNA between laboratories.
Figure 8.
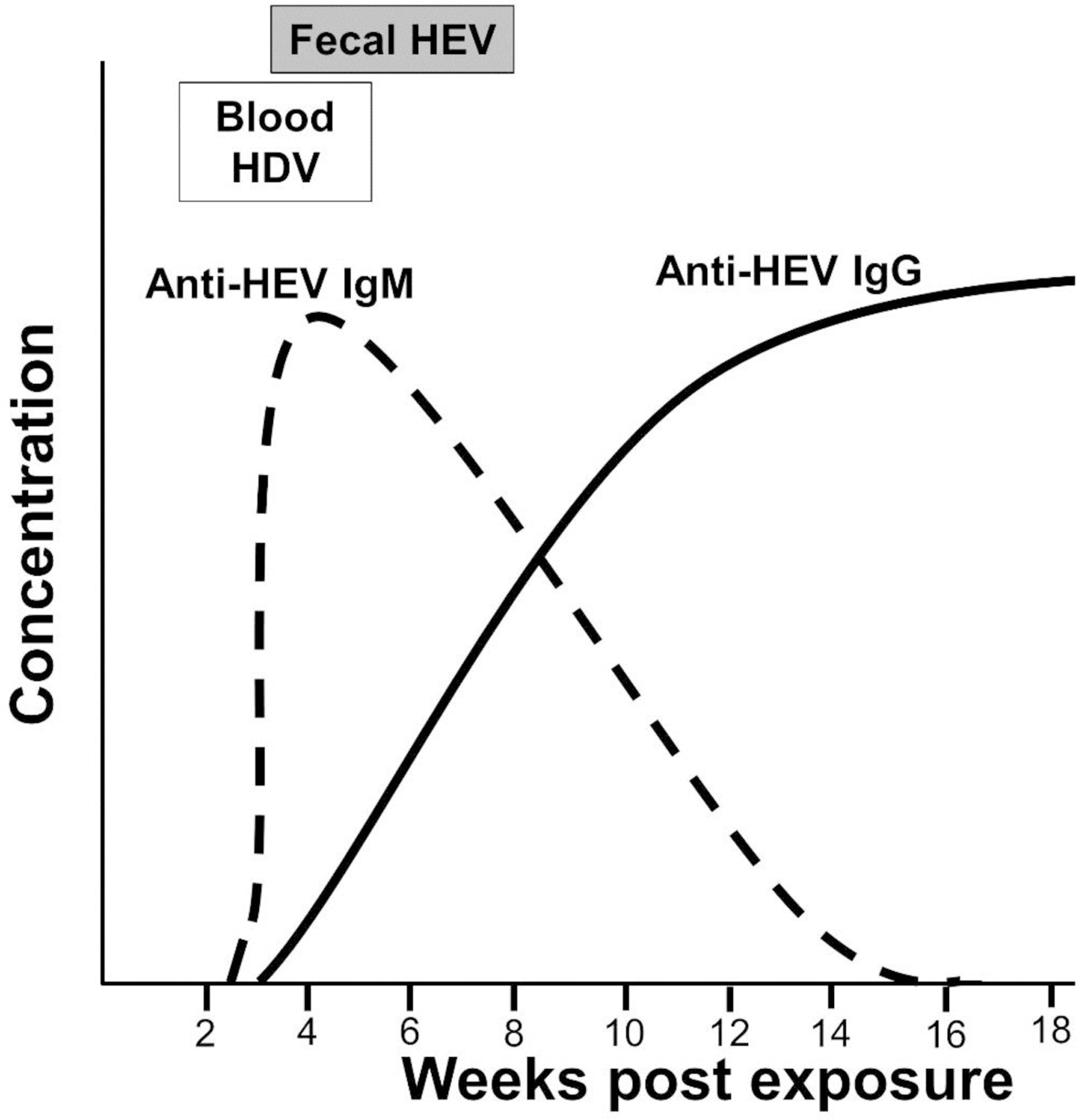
Viral and serologic findings in acute hepatitis E virus infection.
Diagnostic Testing
Anti-HEV IgM generally peaks before clinical illness, although levels remain high for approximately 8 weeks before rapidly falling. In general, anti-HEV IgM levels are below the level of detection by 32 weeks post illness.96 However, the sensitivity of the assay is highly variable for different genotypes, and validated assays having the best performance characteristics are recommended.98
Anti-HEV IgG is often present in patients at the time of acute hepatitis. Anti-HEV IgG levels peak approximately 4 weeks after symptom onset and remain at high levels for more than a year.96
Diagnosis of acute HEV is based on the detection of anti-HEV IgM and HEV RNA.96 Screening employs total and IgM-specific anti-HEV. If positive, HEV RNA should be measured. For individuals with HEV RNA detected, repeat testing is recommended. Chronic HEV infection is defined as having HEV RNA detected for a minimum of 3 months duration.97
Exceptions to the rule
Chronic infection with HEV is rare, but several reports describe persistent genotype 3 HEV viremia in immunocompromised hosts with rapidly progressive liver disease.93,94 Further confirmation of these reports in different geographic regions will be important for understanding the impact of this entity. Chronic HEV viremia without active hepatitis has been seen in individuals with HIV infection.92 Since anti-HEV IgM and/or IgG may not be elicited in patients with severe immunosuppression, the diagnosis of HEV may be challenging.99 Therefore, in immunocompromised patients a combination of IgM and IgG serology and NAT detection of viral RNA should be performed. Additional testing through an independent secondary source, such as the CDC, which offers both ELISA for HEV antibody, as well as fecal and serum HEV NAT should also be considered.100
Key points:
Viral hepatitis may be caused by many viruses, although five viruses are named for their primary manifestation of causing hepatic infection (hepatitis A, B, C, and E).
Although clinical presentation of viral hepatitis is insufficient to determine etiology, precise diagnosis of acute hepatitis A and B is feasible by serologic methods.
Although definitive characterization of infection duration is not possible for hepatitis C, D, and E, diagnosis of ongoing infection is possible using serology and nucleic acid amplification methods
Viral hepatitis diagnostic testing is critical for treatment initiation and/or monitoring treatment response in hepatitis B, C, and D.
Footnotes
Disclosure: There are no relevant commercial relationships to disclose. Grant support: Department of Veterans Affairs Merit Review Grants BX000207 (JTS), and NIAID R56AI126493
References
- 1.Stapleton JT, Foung S, Muerhoff AS, Bukh J, Simmonds P. The GB viruses: a review and proposed classification of GBV-A, GBV-C (HGV), and GBV-D in genus Pegivirus within the family Flaviviridae. J Gen Virol 2011;92(Pt 2):233–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hirai-Yuki A, Hensley L, Whitmire JK, Lemon SM. Biliary Secretion of Quasi-Enveloped Human Hepatitis A Virus. mBio 2016;7(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown EA, Stapleton JT. Hepatitis A In: Murray PR, Baron EJ, Jorgensen JH, Pfaller MA, Yolken RH, editors. Manual of Clinical Microbiology, 8th edition. Washington, DC: ASM Press; 2003.p. 1452–1463. [Google Scholar]
- 4.Sunbul M Hepatitis B virus genotypes: global distribution and clinical importance. World journal of gastroenterology 2014;20(18):5427–5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chotiyaputta W, Lok AS. Hepatitis B virus variants. Nature reviews Gastroenterology & hepatology 2009;6(8):453–462. [DOI] [PubMed] [Google Scholar]
- 6.Liang TJ. Hepatitis B: the virus and disease. Hepatology (Baltimore, Md) 2009;49(5 Suppl):S13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liang TJ, Ghany M. Hepatitis B e Antigen--the dangerous endgame of hepatitis B. N Engl J Med 2002;347(3):208–210. [DOI] [PubMed] [Google Scholar]
- 8.Whalley SA, Murray JM, Brown D, et al. Kinetics of acute hepatitis B virus infection in humans. The Journal of experimental medicine 2001;193(7):847–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pawlotsky JM. Hepatitis B virus (HBV) DNA assays (methods and practical use) and viral kinetics. Journal of hepatology 2003;39 Suppl 1:S31–35. [DOI] [PubMed] [Google Scholar]
- 10.Zaaijer HL, Vrielink H, Koot M. Early detection of hepatitis B surface antigen and detection of HBsAg mutants: a comparison of five assays. Vox sanguinis 2001;81(4):219–221. [DOI] [PubMed] [Google Scholar]
- 11.McMahon BJ, Alward WL, Hall DB, et al. Acute hepatitis B virus infection: relation of age to the clinical expression of disease and subsequent development of the carrier state. J Infect Dis 1985;151(4):599–603. [DOI] [PubMed] [Google Scholar]
- 12.Pol S Management of HBV in immunocompromised patients. Liver international : official journal of the International Association for the Study of the Liver 2013;33 Suppl 1:182–187. [DOI] [PubMed] [Google Scholar]
- 13.Bauer T, Sprinzl M, Protzer U. Immune control of hepatitis B virus. Digestive diseases (Basel, Switzerland) 2011;29(4):423–433. [DOI] [PubMed] [Google Scholar]
- 14.Cavalieri SJ, Hrabovsky S, Jorgensen T. Comparison of DiaSorin and Bio-Rad test kits for the detection of hepatitis B virus total core and surface antibodies on the Bio-Rad Evolis. American journal of clinical pathology 2010;133(1):110–113. [DOI] [PubMed] [Google Scholar]
- 15.Awerkiew S, Daumer M, Reiser M, et al. Reactivation of an occult hepatitis B virus escape mutant in an anti-HBs positive, anti-HBc negative lymphoma patient. Journal of clinical virology : the official publication of the Pan American Society for Clinical Virology 2007;38(1):83–86. [DOI] [PubMed] [Google Scholar]
- 16.Avettand-Fenoel V, Thabut D, Katlama C, Poynard T, Thibault V. Immune suppression as the etiology of failure to detect anti-HBc antibodies in patients with chronic hepatitis B virus infection. J Clin Microbiol 2006;44(6):2250–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feeney SA, McCaughey C, Watt AP, et al. Reactivation of occult hepatitis B virus infection following cytotoxic lymphoma therapy in an anti-HBc negative patient. Journal of medical virology 2013;85(4):597–601. [DOI] [PubMed] [Google Scholar]
- 18.Anastasiou OE, Widera M, Verheyen J, et al. Clinical course and core variability in HBV infected patients without detectable anti-HBc antibodies. Journal of clinical virology : the official publication of the Pan American Society for Clinical Virology 2017;93:46–52. [DOI] [PubMed] [Google Scholar]
- 19.Grandjacques C, Pradat P, Stuyver L, et al. Rapid detection of genotypes and mutations in the pre-core promoter and the pre-core region of hepatitis B virus genome: correlation with viral persistence and disease severity. Journal of hepatology 2000;33(3):430–439. [DOI] [PubMed] [Google Scholar]
- 20.Brunetto MR, Giarin MM, Oliveri F, et al. Wild-type and e antigen-minus hepatitis B viruses and course of chronic hepatitis. Proceedings of the National Academy of Sciences of the United States of America 1991;88(10):4186–4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang LSY, Covert E, Wilson E, Kottilil S. Chronic Hepatitis B Infection: A Review. Jama 2018;319(17):1802–1813. [DOI] [PubMed] [Google Scholar]
- 22.Terrault NA, Lok ASF, McMahon BJ, et al. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology (Baltimore, Md) 2018;67(4):1560–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chu CJ, Hussain M, Lok AS. Quantitative serum HBV DNA levels during different stages of chronic hepatitis B infection. Hepatology (Baltimore, Md) 2002;36(6):1408–1415. [DOI] [PubMed] [Google Scholar]
- 24.Weber B, Bayer A, Kirch P, Schluter V, Schlieper D, Melchior W. Improved detection of hepatitis B virus surface antigen by a new rapid automated assay. J Clin Microbiol 1999;37(8):2639–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biswas R, Tabor E, Hsia CC, et al. Comparative sensitivity of HBV NATs and HBsAg assays for detection of acute HBV infection. Transfusion 2003;43(6):788–798. [DOI] [PubMed] [Google Scholar]
- 26.Jongerius JM, Wester M, Cuypers HT, et al. New hepatitis B virus mutant form in a blood donor that is undetectable in several hepatitis B surface antigen screening assays. Transfusion 1998;38(1):56–59. [DOI] [PubMed] [Google Scholar]
- 27.Louisirirotchanakul S, Kanoksinsombat C, Theamboonlert A, Puthavatana P, Wasi C, Poovorawan Y. Mutation of the “a” determinant of HBsAg with discordant HBsAg diagnostic kits. Viral immunology 2004;17(3):440–444. [DOI] [PubMed] [Google Scholar]
- 28.Gerlich WH, Bremer C, Saniewski M, et al. Occult hepatitis B virus infection: detection and significance. Digestive diseases (Basel, Switzerland) 2010;28(1):116–125. [DOI] [PubMed] [Google Scholar]
- 29.Allain JP, Mihaljevic I, Gonzalez-Fraile MI, et al. Infectivity of blood products from donors with occult hepatitis B virus infection. Transfusion 2013;53(7):1405–1415. [DOI] [PubMed] [Google Scholar]
- 30.Valsamakis A Molecular testing in the diagnosis and management of chronic hepatitis B. Clin Microbiol Rev 2007;20(3):426–439, table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saldanha J, Gerlich W, Lelie N, Dawson P, Heermann K, Heath A. An international collaborative study to establish a World Health Organization international standard for hepatitis B virus DNA nucleic acid amplification techniques. Vox sanguinis 2001;80(1):63–71. [DOI] [PubMed] [Google Scholar]
- 32.Huzly D, Schenk T, Jilg W, Neumann-Haefelin D. Comparison of nine commercially available assays for quantification of antibody response to hepatitis B virus surface antigen. J Clin Microbiol 2008;46(4):1298–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lok AS, Ganova-Raeva L, Cloonan Y, et al. Prevalence of hepatitis B antiviral drug resistance variants in North American patients with chronic hepatitis B not receiving antiviral treatment. Journal of viral hepatitis 2017;24(11):1032–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith DB, Bukh J, Kuiken C, et al. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology (Baltimore, Md) 2014;59(1):318–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collaborators POH. Global prevalence and genotype distribution of hepatitis C virus infection in 2015: a modelling study. The lancet Gastroenterology & hepatology 2017;2(3):161–176. [DOI] [PubMed] [Google Scholar]
- 36.Westbrook RH, Dusheiko G. Natural history of hepatitis C. Journal of hepatology 2014;61(1 Suppl):S58–68. [DOI] [PubMed] [Google Scholar]
- 37.Webster DP, Klenerman P, Dusheiko GM. Hepatitis C. Lancet (London, England) 2015;385(9973):1124–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cox AL, Netski DM, Mosbruger T, et al. Prospective evaluation of community-acquired acute-phase hepatitis C virus infection. Clin Infect Dis 2005;40(7):951–958. [DOI] [PubMed] [Google Scholar]
- 39.Recommendations for prevention and control of hepatitis C virus (HCV) infection and HCV-related chronic disease. Centers for Disease Control and Prevention. MMWR Recommendations and reports : Morbidity and mortality weekly report Recommendations and reports 1998;47(Rr-19):1–39. [PubMed] [Google Scholar]
- 40.Thomson EC, Smith JA, Klenerman P. The natural history of early hepatitis C virus evolution; lessons from a global outbreak in human immunodeficiency virus-1-infected individuals. The Journal of general virology 2011;92(Pt 10):2227–2236. [DOI] [PMC free article] [PubMed]
- 41.Hoofnagle JH. Hepatitis C: the clinical spectrum of disease. Hepatology (Baltimore, Md) 1997;26(3 Suppl 1):15s–20s. [DOI] [PubMed] [Google Scholar]
- 42.Gerlach JT, Diepolder HM, Zachoval R, et al. Acute hepatitis C: high rate of both spontaneous and treatment-induced viral clearance. Gastroenterology 2003;125(1):80–88. [DOI] [PubMed] [Google Scholar]
- 43.Frias M, Rivero-Juarez A, Rodriguez-Cano D, et al. HLA-B, HLA-C and KIR improve the predictive value of IFNL3 for Hepatitis C spontaneous clearance. Scientific reports 2018;8(1):659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beinhardt S, Payer BA, Datz C, et al. A diagnostic score for the prediction of spontaneous resolution of acute hepatitis C virus infection. Journal of hepatology 2013;59(5):972–977. [DOI] [PubMed] [Google Scholar]
- 45.Mondelli MU, Cerino A, Cividini A. Acute hepatitis C: diagnosis and management. Journal of hepatology 2005;42 Suppl(1):S108–114. [DOI] [PubMed] [Google Scholar]
- 46.Marcellin P, Asselah T, Boyer N. Fibrosis and disease progression in hepatitis C. Hepatology (Baltimore, Md) 2002;36(5 Suppl 1):S47–56. [DOI] [PubMed] [Google Scholar]
- 47.Schmidt WN, Stapleton JT, LaBrecque DR, et al. Hepatitis C virus (HCV) infection and cryoglobulinemia: analysis of whole blood and plasma HCV-RNA concentrations and correlation with liver histology. Hepatology (Baltimore, Md) 2000;31(3):737–744. [DOI] [PubMed] [Google Scholar]
- 48.Farci P, Alter HJ, Wong D, et al. A long-term study of hepatitis C virus replication in non-A, non-B hepatitis. N Engl J Med 1991;325(2):98–104. [DOI] [PubMed] [Google Scholar]
- 49.Thimme R, Bukh J, Spangenberg HC, et al. Viral and immunological determinants of hepatitis C virus clearance, persistence, and disease. Proceedings of the National Academy of Sciences of the United States of America 2002;99(24):15661–15668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pawlotsky JM. Use and interpretation of virological tests for hepatitis C. Hepatology (Baltimore, Md) 2002;36(5 Suppl 1):S65–73. [DOI] [PubMed] [Google Scholar]
- 51.Glynn SA, Wright DJ, Kleinman SH, et al. Dynamics of viremia in early hepatitis C virus infection. Transfusion 2005;45(6):994–1002. [DOI] [PubMed] [Google Scholar]
- 52.Schijman A, Colina R, Mukomolov S, et al. Comparison of hepatitis C viral loads in patients with or without coinfection with different genotypes. Clinical and diagnostic laboratory immunology 2004;11(2):433–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Busch MP. Insights into the epidemiology, natural history and pathogenesis of hepatitis C virus infection from studies of infected donors and blood product recipients. Transfusion clinique et biologique : journal de la Societe francaise de transfusion sanguine 2001;8(3):200–206. [DOI] [PubMed] [Google Scholar]
- 54.Netski DM, Mosbruger T, Depla E, et al. Humoral immune response in acute hepatitis C virus infection. Clin Infect Dis 2005;41(5):667–675. [DOI] [PubMed] [Google Scholar]
- 55.Thomson EC, Nastouli E, Main J, et al. Delayed anti-HCV antibody response in HIV-positive men acutely infected with HCV. AIDS (London, England) 2009;23(1):89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schmidt WN, Wu P, Cederna J, Mitros FA, LaBrecque DR, Stapleton JT. Surreptitious hepatitis C virus (HCV) infection detected in the majority of patients with cryptogenic chronic hepatitis and negative HCV antibody tests. J Infect Dis 1997;176(1):27–33. [DOI] [PubMed] [Google Scholar]
- 57.George SL, Gebhardt J, Klinzman D, et al. Hepatitis C virus viremia in HIV-infected individuals with negative HCV antibody tests. Journal of acquired immune deficiency syndromes (1999) 2002;31(2):154–162. [DOI] [PubMed] [Google Scholar]
- 58.Rehermann B, Nascimbeni M. Immunology of hepatitis B virus and hepatitis C virus infection. Nature reviews Immunology 2005;5(3):215–229. [DOI] [PubMed] [Google Scholar]
- 59.Bhattarai N, McLinden JH, Xiang J, et al. Hepatitis C virus infection inhibits a Src-kinase regulatory phosphatase and reduces T cell activation in vivo. PLoS pathogens 2017;13(2):e1006232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee SR, Kardos KW, Schiff E, et al. Evaluation of a new, rapid test for detecting HCV infection, suitable for use with blood or oral fluid. Journal of virological methods 2011;172(1–2):27–31. [DOI] [PubMed] [Google Scholar]
- 61.Ross RS, Viazov S, Salloum S, Hilgard P, Gerken G, Roggendorf M. Analytical performance characteristics and clinical utility of a novel assay for total hepatitis C virus core antigen quantification. J Clin Microbiol 2010;48(4):1161–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tedder RS, Tuke P, Wallis N, Wright M, Nicholson L, Grant PR. Therapy-induced clearance of HCV core antigen from plasma predicts an end of treatment viral response. Journal of viral hepatitis 2013;20(1):65–71. [DOI] [PubMed] [Google Scholar]
- 63.America TAAftSoLDatIDSo. HCV Guidance: Recommendations for Testing, Managing, and Treating Hepatitis C Available at: https://www.hcvguidelines.org/ Accessed May 11, 2018. 2017.
- 64.Pawlotsky JM, Bouvier-Alias M, Hezode C, Darthuy F, Remire J, Dhumeaux D. Standardization of hepatitis C virus RNA quantification. Hepatology (Baltimore, Md) 2000;32(3):654–659. [DOI] [PubMed] [Google Scholar]
- 65.Pawlotsky JM. Measuring hepatitis C viremia in clinical samples: can we trust the assays? Hepatology (Baltimore, Md) 1997;26(1):1–4. [DOI] [PubMed] [Google Scholar]
- 66.Tobler LH, Stramer SL, Lee SR, et al. Impact of HCV 3.0 EIA relative to HCV 2.0 EIA on blood-donor screening. Transfusion 2003;43(10):1452–1459. [DOI] [PubMed] [Google Scholar]
- 67.Quiroga JA, Campillo ML, Catillo I, Bartolome J, Porres JC, Carreno V. IgM antibody to hepatitis C virus in acute and chronic hepatitis C. Hepatology (Baltimore, Md) 1991;14(1):38–43. [DOI] [PubMed] [Google Scholar]
- 68.Schmidt WN, Nelson DR, Pawlotsky JM, Sherman KE, Thomas DL, Chung RT. Direct-acting antiviral agents and the path to interferon independence. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association 2014;12(5):728–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Simmonds P Viral heterogeneity of the hepatitis C virus. Journal of hepatology 1999;31 Suppl 1:54–60. [DOI] [PubMed] [Google Scholar]
- 70.Orland JR, Wright TL, Cooper S. Acute hepatitis C. Hepatology (Baltimore, Md) 2001;33(2):321–327. [DOI] [PubMed] [Google Scholar]
- 71.Papastergiou V, Tsochatzis E, Burroughs AK. Non-invasive assessment of liver fibrosis. Annals of gastroenterology 2012;25(3):218–231. [PMC free article] [PubMed] [Google Scholar]
- 72.Wang CJ, Chen PJ, Wu JC, Patel D, Chen DS. Small-form hepatitis B surface antigen is sufficient to help in the assembly of hepatitis delta virus-like particles. Journal of virology 1991;65(12):6630–6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Le Gal F, Gault E, Ripault MP, et al. Eighth major clade for hepatitis delta virus. Emerg Infect Dis 2006;12(9):1447–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chatzinoff M, Friedman LS. Delta agent hepatitis. Infectious disease clinics of North America 1987;1(3):529–545. [PubMed] [Google Scholar]
- 75.Yurdaydin C, Idilman R, Bozkaya H, Bozdayi AM. Natural history and treatment of chronic delta hepatitis. Journal of viral hepatitis 2010;17(11):749–756. [DOI] [PubMed] [Google Scholar]
- 76.Romeo R, Del Ninno E, Rumi M, et al. A 28-year study of the course of hepatitis Delta infection: a risk factor for cirrhosis and hepatocellular carcinoma. Gastroenterology 2009;136(5):1629–1638. [DOI] [PubMed] [Google Scholar]
- 77.Pascarella S, Negro F. Hepatitis D virus: an update. Liver international : official journal of the International Association for the Study of the Liver 2011;31(1):7–21. [DOI] [PubMed] [Google Scholar]
- 78.Shattock AG, Morris MC. Evaluation of commercial enzyme immunoassays for detection of hepatitis delta antigen and anti-hepatitis delta virus (HDV) and immunoglobulin M anti-HDV antibodies. J Clin Microbiol 1991;29(9):1873–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Castelnau C, Le Gal F, Ripault MP, et al. Efficacy of peginterferon alpha-2b in chronic hepatitis delta: relevance of quantitative RT-PCR for follow-up. Hepatology (Baltimore, Md) 2006;44(3):728–735. [DOI] [PubMed] [Google Scholar]
- 80.Mederacke I, Bremer B, Heidrich B, et al. Establishment of a novel quantitative hepatitis D virus (HDV) RNA assay using the Cobas TaqMan platform to study HDV RNA kinetics. J Clin Microbiol 2010;48(6):2022–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Le Gal F, Brichler S, Sahli R, Chevret S, Gordien E. First international external quality assessment for hepatitis delta virus RNA quantification in plasma. Hepatology (Baltimore, Md). 2016;64(5):1483–1494. [DOI] [PubMed] [Google Scholar]
- 82.Le Gal F, Dziri S, Gerber A, et al. Performance Characteristics of a New Consensus Commercial Kit for Hepatitis D Virus RNA Viral Load Quantification. J Clin Microbiol 2017;55(2):431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chudy M, Hanschmann K-M, Bozdayi M, Kreß J, Nubling C, Group. CS. Collaborative study to establish a World Health Organization international standard for hepatitis D virus RNA for nucleic acid amplification technique (NAT)-based assays. Document WHO/BS/20132227 World Health Organization, Geneva, Switzerland 2013.
- 84.Smedile A, Lavarini C, Crivelli O, Raimondo G, Fassone M, Rizzetto M. Radioimmunoassay detection of IgM antibodies to the HBV-associated delta (delta) antigen:” clinical significance in delta infection. Journal of medical virology 1982;9(2):131–138. [DOI] [PubMed] [Google Scholar]
- 85.Aragona M, Macagno S, Caredda F, et al. Serological response to the hepatitis delta virus in hepatitis D. Lancet (London, England) 1987;1(8531):478–480. [DOI] [PubMed] [Google Scholar]
- 86.Borghesio E, Rosina F, Smedile A, et al. Serum immunoglobulin M antibody to hepatitis D as a surrogate marker of hepatitis D in interferon-treated patients and in patients who underwent liver transplantation. Hepatology (Baltimore, Md) 1998;27(3):873–876. [DOI] [PubMed] [Google Scholar]
- 87.Hughes SA, Wedemeyer H, Harrison PM. Hepatitis delta virus. Lancet (London, England) 2011;378(9785):73–85. [DOI] [PubMed] [Google Scholar]
- 88.Wedemeyer H, Yurdaydin C, Dalekos GN, et al. Peginterferon plus adefovir versus either drug alone for hepatitis delta. N Engl J Med 2011;364(4):322–331. [DOI] [PubMed] [Google Scholar]
- 89.Schlauder GG, Dawson GJ, Erker JC, et al. The sequence and phylogenetic analysis of a novel hepatitis E virus isolated from a patient with acute hepatitis reported in the United States. The Journal of general virology 1998;79 ( Pt 3):447–456. [DOI] [PubMed] [Google Scholar]
- 90.Kamar N, Dalton HR, Abravanel F, Izopet J. Hepatitis E virus infection. Clin Microbiol Rev 2014;27(1):116–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dalton HR, Bendall R, Ijaz S, Banks M. Hepatitis E: an emerging infection in developed countries. The Lancet Infectious diseases 2008;8(11):698–709. [DOI] [PubMed] [Google Scholar]
- 92.Dalton HR, Bendall RP, Keane FE, Tedder RS, Ijaz S. Persistent carriage of hepatitis E virus in patients with HIV infection. N Engl J Med 2009;361(10):1025–1027. [DOI] [PubMed] [Google Scholar]
- 93.Gerolami R, Moal V, Picard C, Colson P. Hepatitis E virus as an emerging cause of chronic liver disease in organ transplant recipients. Journal of hepatology 2009;50(3):622–624. [DOI] [PubMed] [Google Scholar]
- 94.Kamar N, Mansuy JM, Cointault O, et al. Hepatitis E virus-related cirrhosis in kidney- and kidney-pancreas-transplant recipients. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2008;8(8):1744–1748. [DOI] [PubMed] [Google Scholar]
- 95.Aggarwal R, Kini D, Sofat S, Naik SR, Krawczynski K. Duration of viraemia and faecal viral excretion in acute hepatitis E. Lancet (London, England) 2000;356(9235):1081–1082. [DOI] [PubMed] [Google Scholar]
- 96.Huang S, Zhang X, Jiang H, et al. Profile of acute infectious markers in sporadic hepatitis E. PloS one 2010;5(10):e13560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kamar N, Rostaing L, Legrand-Abravanel F, Izopet J. How should hepatitis E virus infection be defined in organ-transplant recipients? American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2013;13(7):1935–1936. [DOI] [PubMed] [Google Scholar]
- 98.Drobeniuc J, Meng J, Reuter G, et al. Serologic assays specific to immunoglobulin M antibodies against hepatitis E virus: pangenotypic evaluation of performances. Clin Infect Dis 2010;51(3):e24–27. [DOI] [PubMed] [Google Scholar]
- 99.Yoo N, Bernstein J, Caldwell C, et al. Hepatitis E virus infection in a liver transplant recipient: delayed diagnosis due to variable performance of serologic assays. Transplant infectious disease : an official journal of the Transplantation Society 2013;15(4):E166–168. [DOI] [PubMed] [Google Scholar]
- 100.Sue PK, Pisanic N, Heaney CD, et al. Variability of hepatitis E serologic assays in a pediatric liver transplant recipient: challenges to diagnosing hepatitis E virus infection in the United States. Transplant infectious disease : an official journal of the Transplantation Society 2015;17(2):284–288. [DOI] [PMC free article] [PubMed] [Google Scholar]