

Abstract
BACKGROUND:
Individuals with tuberous sclerosis complex (TSC) are at increased risk of epilepsy. Early seizure control improves developmental outcomes, making identifying at-risk patients critically important. Despite several identified risk factors, it remains difficult to predict. The purpose of the study was to confirm previously identified risk factors for epilepsy in patients with TSC and evaluate the combined risk prediction of these factors.
METHODS:
The study group (N=333) were participants with TSC enrolled in the TSC Autism Center of Excellence Research Network and UT TSC Biobank. The outcome was defined as having an epilepsy diagnosis. Potential risk factors included sex, TSC genotype, and tuber presence. Logistic regression was used to calculate the odds ratio, 95% confidence interval (CI), and p-value for the association between each variable and epilepsy. A clinical risk prediction model incorporating all risk factors was built. Area under the curve (AUC) was calculated to characterize the full model’s ability to discriminate individuals with TSC with and without epilepsy.
RESULTS:
The strongest risk for epilepsy was presence of tubers (95% CI: 2.39-10.89). Individuals with pathogenic TSC2 variants were 3-times more likely (95% CI: 1.55-6.36) to develop seizures compared to those with TSC from other causes. The combination of risk factors resulted in an AUC of 0.73.
CONCLUSIONS:
Simple characteristics of TSC patients can be combined to successfully predict epilepsy risk. A risk assessment model that incorporates sex, TSC genotype, protective TSC2 missense variant, and tuber presence correctly predicts epilepsy in 73% of TSC patients.
Keywords: tuberous sclerosis complex (TSC), epilepsy, seizures, risk prediction model, genotype, risk factors
Introduction
Tuberous sclerosis complex (TSC) is a genetic syndrome that affects approximately 1 in 6,000 individuals. It is caused by pathogenic variants in TSC1 and TSC2, predisposing the individual to benign tumor formation as well as neurological problems including seizures, intellectual disability, autism, and behavioral problems. Notably, the highest morbidity and mortality stems from neurological issues (Northrup et al., 2018), thus predicting which patients will develop these phenotypes carries significant prognostic value.
One of the most severe neurologic issues is epilepsy, which affects 70-90% of individuals with TSC (Asato and Hardan, 2004; Wong and Khong, 2006). A significant relationship between early-age-at-seizure-onset and poor developmental outcomes has been established (Capal et al., 2017; Jeste et al., 2016; Jansen et al., 2008; Gomez et al., 1982). Furthermore, several studies demonstrated that early control of epilepsy improves developmental outcomes (Cusmai et al., 2011; Jóźwiak et al. 2011, Bombardieri et al., 2010). Therefore, establishing early risk-prediction models for seizures is paramount in TSC clinical care (Davis et al, 2019; Wu, et al., 2016; Muzykewicz, et al., 2009), as tailoring treatment to surveil and intervene would optimize outcomes for patients with TSC.
Features implicated in seizure development among patients with TSC include: central nervous system lesions and white matter abnormalities (Curatolo, 2015; Im et al., 2015; Peters et al., 2012; Jansen et al., 2008), male sex (Au et al., 2007; Sancak et al., 2005), and TSC genotype. TSC2 pathogenic variants, in general, are associated with increased risk of infantile spasms and more severe general phenotype (Sancak et al., 2005; Dabora et al., 2002); however, individuals with certain TSC2 missense variants are less likely to develop epilepsy compared to patients with TSC from other causes (Fox et al., 2017; Farach et al., 2016; Ekong et al., 2016; van Eeghen et al., 2013; Wentink et al., 2012; Jansen et al., 2006; Mayer et al., 2004; O’Connor et al., 2003; Khare et al., 2001). Interictal epileptiform discharges found on electroencephalography (EEG) are useful in predicting impending epilepsy but are not early risk predictors (Wu et al., 2019; Wu et al., 2016).
In spite of these findings, providing patients with accurate and specific prognostic information remains a challenge. As the previously identified risk factors are not strong enough to accurately predict epilepsy themselves, a potential solution would be to combine the efforts of previous studies by incorporating identified individual risk factors into a risk prediction model to determine if the combination produces a more accurate risk prediction. Risk prediction models have been utilized in other single gene disorders (O’Mahony et al., 2013; Armstrong et al., 2009; Cassidy et al., 2008; Eagle et al., 2004) and provide an important approach to assessing outcome by identifying at-risk individuals, facilitating the design and planning of clinical trials, informing surveillance strategies, fostering the development of benefit-risk indices, and enabling estimates of the prevalence and cost of disease. The purpose of this study was to create a weighted risk prediction model for epilepsy in patients with TSC by testing cumulative clinical risk in a well-phenotyped and genotyped cohort of patients.
Materials and Methods
Study population:
The study was performed using participant data from two sources: the TSC Autism Center of Excellence Research Network (TACERN) study (NCT 01780441) (N=119) and UT TSC biobank (N=214). Participants were included if they had a clinical diagnosis of definite TSC, had undergone genetic testing for TSC1 and TSC2, and had reliable demographic and phenotypic information including sex, seizure history, and presence or absence of tubers. We also limited our study subjects to unrelated individuals.
TSC Genotyping:
Genotyping for the patients was performed prior to the study, either clinically or as part of the research protocol. Participants who had genotyping as part of the TACERN trial or UT Biobank study had Sanger sequencing of TSC1 and TSC2 with reflex to multiplex ligation-dependent probe amplification (MLPA), if Sanger sequencing was negative. All variants were classified using American College of Medical Genetics standards and guidelines (Richards et al., 2015) and the TSC1 or TSC2 Leiden Open Variation Databases (https://databases.lovd.nl/shared/genes/TSC1; http://chromium.lovd.nl/LOVD2/TSC/home.php?select_db=TSC2). Those who had benign, likely benign, variants of uncertain significance, or no variants were labeled “no mutation identified (NMI).” Those with pathogenic or likely pathogenic variants who met clinical criteria for TSC were considered positive for TSC1 or TSC2 disease-causing mutation. Known pathogenic variants considered protective against seizures included TSC2 missense variants in exons 23-33 (van Eeghen et al., 2013) along with the following TSC2 missense variants: p.Arg622Trp; p.Arg905Gln; p.Ser1036Pro; p.Arg1200Trp; p.Gln1503Pro; p.Gly1579Ser; p.Arg1713His (Khare et al 2001, O’Connor et al 2003, Mayer et al 2004, Jansen et al 2006, Wentink et al 2012, Farach et al 2017, Fox et al 2017).
Phenotypic Data:
Presence of seizures for the TACERN participants was determined by study clinicians based on history, EEG, and a daily seizure diary. Presence of tubers in TACERN participants was determined based on radiologist report of brain MRIs obtained at or after 24 months. Presence of seizures and/or tubers in UT TSC Biobank participants was based on both self-reported and clinical information. Participants with two or more unprovoked seizures were designated as having epilepsy. Participants were determined not to have epilepsy if they were at least twelve months old with no history of seizure. This timepoint was chosen based on a study demonstrating that the onset of epilepsy occurs by twelve months in the majority of patients with TSC (Curatolo et al., 2012).
Statistical Analysis:
A clinical risk prediction model was developed based on factors previously identified as associated with risk of seizure among individuals with TSC. Risk factors included sex (male, female), TSC pathogenic variant (TSC1, TSC2, NMI), protective missense variant in TSC2 (present, absent), and tuber formation (present, absent). Logistic regression was used to determine the association between each risk factor and seizure. Specifically, an unadjusted odds ratio (OR), 95% confidence interval (CI), and p-value was determined for each clinical factor. Next, a clinical risk prediction model was built that incorporated all risk factors to estimate the adjusted OR, 95% CI, and p-value for each risk factor. Finally, the area under the curve (AUC) was calculated to characterize the full model’s ability to discriminate individuals with TSC with and without seizure. A successful epilepsy risk prediction model was defined a priori to have AUC ≥ 0.70 (Hosmer and Lemeshow, 2000). All statistical analyses were conducted in R version 5.3.2.
Results
Three hundred thirty-three TSC participants had the necessary clinical and demographic information to be included in the full epilepsy risk prediction model, 261 of whom had epilepsy, while 72 did not have epilepsy (Table 1). Individuals with epilepsy were a median age of 36 months at last assessment and ranging up to 43 years of age. Individuals without epilepsy were a median age of 48 months at last assessment and ranging up to 57 years of age. Per exclusion criteria, none were younger than 12 months old. Nine participants had epilepsy despite having a variant that is associated with a lower likelihood of seizure (missense variants within exons 23-33: n=6; Arg905Gln: n=2; Arg1200Trp: n=1). The protective variants in the participants without epilepsy included: Arg622Trp (n=1); Arg1200Trp (n=3); and missense variants within exons 23-33 (n=3).
Table 1.
Characteristics of Participants
| With Epilepsy (n=261) | Without Epilepsy (n=72) | P-value* | |
|---|---|---|---|
| Sex, n (%) | 0.99 | ||
| Female | 133 (51.0) | 36 (50.0) | |
| Male | 128 (49.0) | 36 (50.0) | |
| Pathogenic variant, n (%) | <0.001 | ||
| TSC1 | 31 (11.9) | 20 (27.8) | |
| TSC2 NMI |
189 (72.4) 41 (15.7) |
33 (45.8) 19 (26.4) |
|
| Protective variant, n (%) | 0.06 | ||
| No | 252 (96.6) | 65 (90.3) | |
| Yes | 9 (3.4) | 7 (9.7) | |
| Tubers, n (%) | <0.001 | ||
| No | 17 (6.5) | 20 (27.8) | |
| Yes | 244 (93.5) | 52 (72.2) |
P-values are derived from the Chi-square test with continuity correction. NMI-no mutation identified, n-number of patients, %-percent of patients within the group.
The strongest risk for epilepsy was identified among participants with tubers, who had more than 5-times the adjusted odds of epilepsy than individuals with no tubers (adjusted OR: 5.1, 95% CI: 2.4-10.9; Table 2). Individuals with pathogenic TSC2 variants had 3.1-times the adjusted odds of epilepsy compared to individuals without known mutations (95% CI: 1.6-6.4), whereas individuals with pathogenic TSC1 variants had similar adjusted odds of epilepsy (adjusted OR: 0.7, 95% CI: 0.3-1.6). The epilepsy-protective association of specific missense variants in TSC2 were confirmed (adjusted OR: 0.3, 95% CI: 0.1-0.8). Male sex was not identified as a risk factor for seizure in our study population (adjusted OR: 0.9, 95% CI: 0.5-1.6). The model incorporating all previously reported risk factors for seizure in TSC patients resulted in an area under the curve of 0.73 (Figure 1).
Table 2.
Epilepsy clinical risk prediction model for individuals with tuberous sclerosis
| Risk predictors | Adjusted odds ratio (95% CI) | P-value |
|---|---|---|
| Sex (male) | 0.88 (0.50, 1.55) | 0.6 |
| TSC Genotype | ||
| NMI | Ref | |
| TSC1 | 0.71 (0.32, 1.62) | 0.4 |
| TSC2 | 3.14 (1.55, 6.36) | 0.002 |
| Protective variant | 0.25 (0.08, 0.78) | 0.017 |
| Tubers | 5.10 (2.39, 10.89) | <0.001 |
P-values are from the Wald test for each logistic regression coefficient. AUC- area under the receiver operating curve, CI- confidence interval, NMI-no mutation identified, Ref- reference
Figure 1.
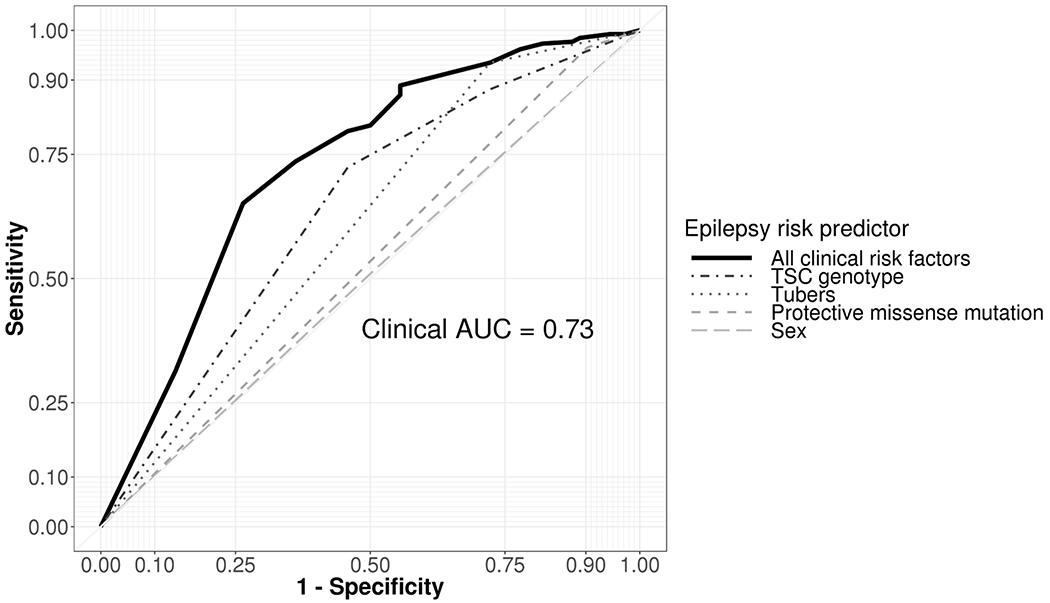
Receiver operating curve for the ability of sex, TSC genotype, protective missense variant, and tuber formation to individually and simultaneously predict the presence of epilepsy among individuals with TSC. The area under the curve (AUC) indicates that the model will indicate greater likelihood of epilepsy among 73% of randomly selected patients with TSC and epilepsy than a randomly chosen individual with TSC without epilepsy. Combination of risk factors has increased accuracy over individual risk predictors.
Discussion
In one of the first assessment to generate and evaluate a clinical risk prediction model for epilepsy among those with TSC, we found the combined risk prediction of sex, TSC genotype, protective TSC2 missense variants, and tuber presence offers improved epilepsy risk prediction beyond each risk factor individually. Notably, the epilepsy risk prediction model is based on factors that can easily be assessed upon specialist referral at or around the time of TSC diagnosis, so incorporation into the model would not necessitate extra testing. The individual risk factors, while helpful, do not accurately predict epilepsy alone. However, by including the known risk factors together in one model, clinicians are armed with more reliable and accurate information regarding risk of epilepsy in their patient. It is interesting that even with all of the factors incorporated, the risk prediction model can only predict with 73% reliability, demonstrating there is much to be learned about the pathogenesis of epilepsy in TSC. Still, the information is valuable as it may be used to both inform prognosis and guide treatment. For instance, due to the high risk of epilepsy in patients with TSC, current management recommendations include getting a baseline EEG (Kruger et al., 2012) to evaluate for unrecognized or sub-clinical seizures. EEGs are also useful as interictal epileptiform discharges serve as a biomarker for impending seizures, with seizure onset occurring an average of four months later (Wu et al., 2016; Wu et al., 2019). However, if seizure onset has not yet occurred and is not impending, the EEG is likely to be normal. For patients predicted to be at high risk for epilepsy, clinicians may choose to do closer surveillance with additional follow up EEGs as early identification and control of seizures is crucial to improving developmental outcomes (Cusmai et al., 2011; Jóźwiak et al. 2011, Bombardieri et al., 2010).
We specifically focused on risk factors that were previously reported and/or confirmed to be associated with epilepsy (Fox et al., 2017; Farach et al., 2016; Ekong et al., 2016; Curatolo, 2015; Im et al., 2015; van Eeghen et al., 2013; Peters et al., 2012; Wentink et al., 2012; Jansen et al., 2008; Au et al., 2007; Jansen et al., 2006; Sancak et al., 2005; Mayer et al., 2004; O’Connor et al., 2003; Dabora et al., 2002; Khare et al., 2001). As evidence regarding correlation between tuber count and/or location, and epilepsy, specifically, is weak and overall studies are conflicting (Doherty et al., 2005; Wong and Khong, 2006; Kassiri et al., 2011), the specifics of tubers, such as count and location were not included. Instead, the dichotomaous approach of presence or absence of tubers, which has better evidence (Curatolo, 2015; Im et al., 2015; Peters et al., 2012; Jansen et al., 2008) was used. Because the risk factors incorporated were proven risk factors, we did not divide our population into training and validation sets. In fact, our cohort reflected these previously described associations. For instance, presence of tubers and a TSC2 genotype that was not one of the protective missense variants reliably predicted seizures in our cohort. Interestingly, the strongest risk appeared to be presence of tubers. Tubers are present in about 90% of individuals with TSC (Krueger et al., 2012), so while the information is useful, it is not very specific. Males are previously reported to have more severe TSC phenotype (Au et al., 2007; Sancak et al., 2005); however, in our cohort, males did not appear to have increased risk for seizure, specifically. Notably, nine patients with a protective variant still had epilepsy and more patients with protective variants had epilepsy than did not. While those variants demonstrate statistical significance for protecting against epilepsy in our cohort, it is apparent they are not fully protective.
Our study should be considered in the light of certain strengths and limitations. First, we had a relatively large sample size and a subpopulation with very detailed phenotypic information. However, risks of specific aspects of epilepsy such as onset, type, and severity could not be ascertained as the information was not available for the majority of individuals in the study. Specifics of tubers, such as number and location, were not included in analysis as overall the correlation with epilepsy is unproven (Doherty et al., 2005; Wong and Khong, 2006; Kassiri et al., 2011). Information was self-reported for some of the participants and thus could be inaccurate. Finally, while we did not divide our sample into training validation sets, we: 1) selected factors that have been demonstrated to be associated with epilepsy among those with TSC; and 2) observed associations that were consistent with previous reports. Therefore, we believe our model accurately reflects the ability of epilepsy risk factors to simultaneously predict risk of epilepsy in patients with TSC.
This is the first model to predict manifestations of TSC and sets the groundwork for creation of more models in future studies to predict epilepsy onset, type, and severity; as well as other neurological features of TSC. While the current model has acceptable discrimination, it also demonstrates that there is much more to be learned about risks and pathogenesis of epilepsy in TSC. A future goal is to strengthen the model by incorporating genetic modifiers.
Conclusion
A successful epilepsy risk prediction model for patients with TSC can be made by incorporating known risks including sex, TSC genotype, protective variants, and presence of tubers. The model increases the utility of previously identified risk factors and may be strengthened if other risk factors are discovered, such as modifier genes.
HIGHLIGHTS.
Predicting which patients with TSC will develop epilepsy is crucial to early identification and intervention but remains difficult despite known clinical risk and protective factors
A risk assessment model that incorporates known risk factors for epilepsy including sex, TSC genotype, protective TSC2 missense variant, and tuber presence, correctly predicts risk of epilepsy in individuals with TSC better than individual factors alone
The strongest risk factors for epilepsy in those with TSC in the studied cohort was the presence of tubers and TSC2 pathogenic variant
This model accurately predicts epilepsy in 73% of individuals with TSC and may be strengthened with the discovery and incorporation of additional risk factors, such as modifier genes
Acknowledgements:
We are sincerely indebted to the generosity of the families and patients in TSC clinics across the United States who contributed their time and effort to this study. We would also like to thank the Tuberous Sclerosis Alliance for their continued support in TSC research.
Funding:
This work was supported by Autism Center of Excellence Network [1U01NS082320-01], the Developmental Synaptopathies Consortium [1U54NS092090-01], and the Department of Defense [W81XWH1810537]. The Developmental Synaptopathies Consortium [U54NS092090] is part of the Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), National Center for Advancing Translational Sciences (NCATS). Research reported in this publication was supported by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health (NINDS), Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD), National Institute of Mental Health (NIMH) and National Center for Advancing Translational Sciences (NCATS).
**Members of the Tuberous Sclerosis Autism Center of Excellence Research Network (TACERN): Principal Investigators: Sahin, M1; Krueger, D2; Bebin, M3; Wu, JY4; Northrup, H5; Co-Investigators: Warfield, S1; Peters, J1; Scherrer, B1; Goyal, M3; Project managers: Filip-Dhima, R1; Dies, K1; Bruns, S2; Neuropsychological assessment team: Hanson, E1; Bing, N2; Kent, B2; O’Kelley, S3; Williams, M E9; Pearson, D5; Data Coordinating Center at UAB: Cutter, G6; TS Alliance: Roberds, S7; Autism Speaks: Murray DS8; Affiliations for TACERN: 1Boston Children’s Hospital, Boston, MA, 2Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, 3University of Alabama at Birmingham, Birmingham, AL, 4University of California, Los Angeles, Los Angeles, CA, 5McGovern Medical School, University of Texas Health Science Center at Houston, Houston, TX, 6University of Alabama at Birmingham, Data Coordinating Center, Birmingham, AL, 7Tuberous Sclerosis Alliance, 8Autism Speaks, 9Keck School of Medicine of USC, University of Southern California, Children’s Hospital Los Angeles, Los Angeles, CA
References:
- 1.Armstrong TS, Cao Y, Scheurer ME, Vera-Bolaños E, Manning R, Okcu MF, Bondy M, Zhou R, Gilbert MR. Risk analysis of severe myelotoxicity with temozolomide: the effects of clinical and genetic factors. Neuro-oncology. 2009. Dec 1;11(6):825–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asato MR, Hardan AY. 2004. Neuropsychiatric problems in tuberous sclerosis complex. J Child Neurol 19:241–249. [DOI] [PubMed] [Google Scholar]
- 3.Au KS, Williams AT, Roach ES, Batchelor L, Sparagana SP, Delgado MR, Wheless JW, Baumgartner JE, Roa BB, Wilson CM, Smith-Knuppel TK. Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. Genetics in Medicine. 2007. Feb 1;9(2):88–100. [DOI] [PubMed] [Google Scholar]
- 4.Bombardieri R, Pinci M, Moavero R, Cerminara C, Curatolo P. Early control of seizures improves long-term outcome in children with tuberous sclerosis complex. European Journal of Paediatric Neurology. 2010. Mar 31;14(2):146–9. [DOI] [PubMed] [Google Scholar]
- 5.Cassidy A, Myles JP, van Tongeren M, Page RD, Liloglou T, Duffy SW, Field JK. The LLP risk model: an individual risk prediction model for lung cancer. British journal of cancer. 2008. Jan;98(2):270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Capal JK, Bernardino-Cuesta B, Horn PS, Murray D, Byars AW, Bing NM, Kent B, Pearson DA, Sahin M, Krueger DA, TACERN Study Group. Influence of seizures on early development in tuberous sclerosis complex. Epilepsy & Behavior. 2017. May 1;70:245–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Curatolo P Mechanistic target of rapamycin (mTOR) in tuberous sclerosis complex-associated epilepsy. Pediatric neurology. 2015. Mar 31;52(3):281–9. [DOI] [PubMed] [Google Scholar]
- 8.Curatolo P, Jóźwiak S, Nabbout R. Management of epilepsy associated with tuberous sclerosis complex (TSC): clinical recommendations. european journal of paediatric neurology. 2012. Nov 30;16(6):582–6. [DOI] [PubMed] [Google Scholar]
- 9.Cusmai R, Moavero R, Bombardieri R, Vigevano F, Curatolo P. Long-term neurological outcome in children with early-onset epilepsy associated with tuberous sclerosis. Epilepsy & Behavior. 2011. Dec 31;22(4):735–9. [DOI] [PubMed] [Google Scholar]
- 10.Dabora SL, Roberts P, Nieto A, Perez R, Jozwiak S, Franz D, Bissler J, Thiele EA, Sims K, Kwiatkowski DJ. Association between a high-expressing interferon-γ allele and a lower frequency of kidney angiomyolipomas in TSC2 patients. The American Journal of Human Genetics. 2002. Oct 31;71(4):750–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis PE, Kapur K, Filip-Dhima R, Trowbridge SK, Little E, Wilson A, Leuchter A, Bebin EM, Krueger D, Northrup H, Wu JY. Increased electroencephalography connectivity precedes epileptic spasm onset in infants with tuberous sclerosis complex. Epilepsia. 2019. Aug;60(8):1721–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doherty C, Goh S, Young Poussaint T, Erdag N, Thiele EA. Prognostic significance of tuber count and location in tuberous sclerosis complex. Journal of child neurology. 2005. Oct;20(10):837–41. [DOI] [PubMed] [Google Scholar]
- 13.Eagle KA, Lim MJ, Dabbous OH, Pieper KS, Goldberg RJ, Van de Werf F, Goodman SG, Granger CB, Steg PG, Gore JM, Budaj A. A validated prediction model for all forms of acute coronary syndrome: estimating the risk of 6-month postdischarge death in an international registry. Jama. 2004. Jun 9;291(22):2727–33. [DOI] [PubMed] [Google Scholar]
- 14.Ekong R, Nellist M, Hoogeveen-Westerveld M, Wentink M, Panzer J, Sparagana S, Emmett W, Dawson NL, Malinge MC, Nabbout R, Carbonara C. Variants within TSC2 exons 25 and 31 are very unlikely to cause clinically diagnosable tuberous sclerosis. Human mutation. 2016. Apr 1;37(4):364–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farach LS, Gibson WT, Sparagana SP, Nellist M, Stumpel CT, Hietala M, Friedman E, Pearson DA, Creighton SP, Wagemans A, Segel, Ben-Shalom E, Au KS, Northrup HR TSC2 c. 1864C> T variant associated with mild cases of tuberous sclerosis complex. American Journal of Medical Genetics Part A. 2017. Mar 1;173(3):771–5. [DOI] [PubMed] [Google Scholar]
- 16.Fox J, Ben-Shachar S, Uliel S, Svirsky R, Saitsu H, Matsumoto N, Fattal-Valevski A. Rare familial TSC2 gene mutation associated with atypical phenotype presentation of Tuberous Sclerosis Complex. Am J Med Genet A. 2017;173:744–8. [DOI] [PubMed] [Google Scholar]
- 17.Gomez MR, Kuntz NL, Westmoreland BF. Tuberous sclerosis, early onset of seizures, and mental subnormality Study of discordant homozygous twins. Neurology. 1982. Jun 1;32(6):604-604. [DOI] [PubMed] [Google Scholar]
- 18.Dw Hosmer, Lemeshow S. Applied Logistic Regression, 2nd Edition. Chapter 5. John Wiley and Sons, New York, NY, 2000. Pages 160–164. [Google Scholar]
- 19.Im K, Ahtam B, Haehn D, Peters JM, Warfield SK, Sahin M, Grant PE. Altered structural brain networks in tuberous sclerosis complex. Cerebral Cortex. 2015. Mar 5:bhv026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jansen AC, Sancak O, D’Agostino MD, Badhwar A, Roberts P, Gobbi G, Wilkinson R, Melanson D, Tampieri D, Koenekoop R, Gans M. Unusually mild tuberous sclerosis phenotype is associated with TSC2 R905Q mutation. Annals of neurology. 2006. Nov 1;60(5):528–39. [DOI] [PubMed] [Google Scholar]
- 21.Jansen FE, Vincken KL, Algra A, Anbeek P, Braams O, Nellist M, Zonnenberg BA, Jennekens-Schinkel A, van den Ouweland A, Halley D, Van Huffelen AC. Cognitive impairment in tuberous sclerosis complex is a multifactorial condition. Neurology. 2008. Mar 18;70(12):916–23. [DOI] [PubMed] [Google Scholar]
- 22.Jeste S, Varcin K, Helleman G, Gulsrud A, Sahin M, Wu J, Kasari C, Nelson C. Symptom Profiles of Autism Spectrum Disorder in Tuberous Sclerosis Complex (S10. 001). Neurology. 2016. Apr 5;86(16 Supplement):S10–001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jóźwiak S, Kotulska K, Domańska-Pakieła D, Łojszczyk B, Syczewska M, Chmielewski D, Dunin-Wąsowicz D, Kmieć T, Szymkiewicz-Dangel J, Kornacka M, Kawalec W. Antiepileptic treatment before the onset of seizures reduces epilepsy severity and risk of mental retardation in infants with tuberous sclerosis complex. European Journal of Paediatric Neurology. 2011. Sep 30;15(5):424–31. [DOI] [PubMed] [Google Scholar]
- 24.Kassiri J, Snyder TJ, Bhargava R, Wheatley BM, Sinclair DB. Cortical tubers, cognition, and epilepsy in tuberous sclerosis. Pediatric neurology. 2011. May 1;44(5):328–32. [DOI] [PubMed] [Google Scholar]
- 25.Khare L, Strizheva GD, Bailey JN, Au KS, Northrup H, Smith M, Smalley SL, Henske EP. A novel missense mutation in the GTPase activating protein homology region of TSC2 in two large families with tuberous sclerosis complex. Journal of medical genetics. 2001. May 1;38(5):347–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krueger DA, Northrup H, International Tuberous Sclerosis Complex Consensus Group. 2013.Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. 49:255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mayer K, Goedbloed M, van Zijl K, Nellist M, Rott HD. Characterization of a novel TSC2 missense mutation in the GAP related domain associated with minimal clinical manifestations of tuberous sclerosis. J Med Genet. 2004;41:e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muzykewicz DA, Costello DJ, Halpern EF, Thiele EA. Infantile spasms in tuberous sclerosis complex: prognostic utility of EEG. Epilepsia. 2009. Feb 1;50(2):290–6. [DOI] [PubMed] [Google Scholar]
- 29.Northrup H, Koenig MK, Pearson DA, Au KS. Tuberous Sclerosis Complex. 1999 [Updated 2018]. GeneReviews® Available from: https://www.ncbi.nlm.nih.gov/books/NBK1220/ [Google Scholar]
- 30.O’Connor SE, Kwiatkowski DJ, Roberts PS, Wollmann RL, Huttenlocher PR. A family with seizures and minor features of tuberous sclerosis and a novel TSC2 mutation. Neurology. 2003. Aug 12;61(3):409–12. [DOI] [PubMed] [Google Scholar]
- 31.O’Mahony C, Jichi F, Pavlou M, Monserrat L, Anastasakis A, Rapezzi C, Biagini E, Gimeno JR, Limongelli G, McKenna WJ, Omar RZ. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). European heart journal. 2013. Oct 14;35(30):2010–20. [DOI] [PubMed] [Google Scholar]
- 32.Peters JM, Sahin M, Vogel-Farley VK, Jeste SS, Nelson CA, Gregas MC, Prabhu SP, Scherrer B, Warfield SK. Loss of white matter microstructural integrity is associated with adverse neurological outcome in tuberous sclerosis complex. Academic radiology. 2012. Jan 31;19(1):17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. 2015. Genetics in medicine. 17(5):405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sancak O, Nellist M, Goedbloed M, Elfferich P, Wouters C, Maat-Kievit A, Zonnenberg B, Verhoef S, Halley D, van den Ouweland A. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype–phenotype correlations and comparison of diagnostic DNA techniques in tuberous sclerosis complex. European Journal of Human Genetics. 2005. Jun 1;13(6):731–41. [DOI] [PubMed] [Google Scholar]
- 35.van Eeghen AM, Nellist M, van Eeghen EE, Thiele EA. Central TSC2 missense mutations are associated with a reduced risk of infantile spasms. Epilepsy research. 2013. Jan 31;103(1):83–7. [DOI] [PubMed] [Google Scholar]
- 36.Wentink M, Nellist M, Hoogeveen-Westerveld M, Zonnenberg B, van der Kolk D, van Essen T, Park SM, Woods G, Cohn-Hokke P, Brussel W, Smeets E. Functional characterization of the TSC2 c. 3598C> T (p. R1200W) missense mutation that co-segregates with tuberous sclerosis complex in mildly affected kindreds. Clinical genetics. 2012. May 1;81(5):453–61. [DOI] [PubMed] [Google Scholar]
- 37.Wong V, Khong PL. 2006. Tuberous sclerosis complex: Correlation of MRI findings with comorbidities. J Child Neurol 21:99–105. [DOI] [PubMed] [Google Scholar]
- 38.Wu JY, Peters JM, Goyal M, Krueger D, Sahin M, Northrup H, Au KS, Cutter G, Bebin EM. Clinical electroencephalographic biomarker for impending epilepsy in asymptomatic tuberous sclerosis complex infants. Pediatric neurology. 2016. Jan 31;54:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu JY, Goyal M, Peters JM, Krueger D, Sahin M, Northrup H, Au KS, O’Kelley S, Williams M, Pearson DA, Hanson E. Scalp EEG spikes predict impending epilepsy in TSC infants: A longitudinal observational study. Epilepsia. 2019. Dec;60(12):2428–36. [DOI] [PMC free article] [PubMed] [Google Scholar]