

Abstract
The mammalian neuromuscular junction (NMJ) comprises a presynaptic terminal, a postsynaptic receptor region on the muscle fiber (endplate), and the perisynaptic (terminal) Schwann cell. As with any synapse, the purpose of the NMJ is to transmit signals from the nervous system to muscle fibers. This neural control of muscle fibers is organized as motor units, which display distinct structural and functional phenotypes including differences in pre- and postsynaptic elements of NMJs. Motor units vary considerably in the frequency of their activation (both motor neuron discharge rate and duration/duty cycle), force generation, and susceptibility to fatigue. For earlier and more frequently recruited motor units, the structure and function of the activated NMJs must have high fidelity to ensure consistent activation and continued contractile response to sustain vital motor behaviors (e.g., breathing and postural balance). Similarly, for higher force less frequent behaviors (e.g., coughing and jumping), the structure and function of recruited NMJs must ensure short-term reliable activation but not activation sustained for a prolonged period in which fatigue may occur. The NMJ is highly plastic, changing structurally and functionally throughout the life span from embryonic development to old age. The NMJ also changes under pathological conditions including acute and chronic disease. Such neuroplasticity often varies across motor unit types.
Introduction
The structure and function of the neuromuscular junction (NMJ) provide insight into neural communication. The term “synapse” was introduced by Sir Charles Sherrington (1897) to describe the point of contact between a neuron and its target cell. For motor neurons and their target cells (skeletal muscle fibers), the synapse is the NMJ. This article focuses primarily on the mammalian NMJ, but we include important original observations on the NMJ in frog muscle (e.g., the research of Bernard Katz from the 1950s to 1970s), as well as information that is only available based on research in other nonmammalian species (e.g., seminal works by Oppenheim and Landmesser on the development of the NMJ in chick hindlimb muscle).
Using a microscope, Robert Hooke was the first to observe a living cell in 1665, which he described in his book Micrographia (98). In 1839, the concept that living organisms (both animals and plants) are composed of cells (the Cell Theory) was independently proposed by the German botanist Matthias Schleiden (410) and the German physiologist Theodor Schwann (411). Due to limitations in microscopy, it was initially unclear if the cell theory could be applied to the nervous system. However, with the introduction of the silver impregnation technique by Camillo Golgi (Golgi stain) in 1873 (167), it became possible to visualize neurons as single cells. However, Golgi continued to promote the Reticular Theory that the nervous system is a continuous interconnected network, or a reticulum, originally proposed by Gerlach in 1871 (162, 441). Based on the Cell Theory, an opposing view had emerged in the 1880s that the nervous system comprised discrete individual cells. In support, the neuroanatomist Santiago Ramón y Cajal used the Golgi staining technique to provide exquisite drawings of neural cells (154). Based on the work of Golgi and Cajal, the German anatomist Heinrich von Waldeyer-Hartz introduced the term “neuron” in 1891 to describe neural cells. The Neuron Theory (alternatively Neuron Doctrine) had important implications with respect to communication within the nervous system. In the Neuron Doctrine, a neuron communicates via synapses with other cells, for example, at the NMJ. Curiously, both Cajal and Golgi won the 1906 Nobel Prize for their work, despite their opposing views on the elemental structure of the nervous system.
During this period, the English neurologist William Gowers coined the term motor neuron (171) to describe the neurons involved in direct motor control of skeletal muscle fibers. The endpoints of motor neuron axons innervating muscle fibers are termed presynaptic terminals. The presynaptic terminal is specialized for neurotransmitter release, and the postsynaptic receptor region responds to the released neurotransmitters via selective receptors. The specialized postsynaptic region on skeletal muscle fibers is called the endplate, a term introduced by the German anatomist Wilhelm Krause as “motorische endplatte” (231). Cholinergic receptors (AChRs) at the endplate of skeletal muscle fibers are ionotropic receptors, which directly gate a cation channel in response to the binding of acetylcholine (ACh). Thus, in mammals, ACh induces depolarization of the postsynaptic membrane when it binds to the AChR.
In 1925, Edward Liddell and Sir Charles Sherrington defined a motor unit as a single motor neuron and all the muscle fibers it innervates (259). Functional differences exist across motor unit types, and in this article, we highlight these differences as an organizing principle for NMJ physiology. Different motor unit types vary in their overall activity and their frequency of activation. There must be fidelity of neuromuscular transmission at the NMJ to ensure appropriate activation and mechanical responses by motor unit muscle fibers. For example, within the diaphragm muscle (DIAm), some motor units involved in breathing are very active with a duty cycle of 30% to 50%, depending on the species. In contrast, other motor units are recruited only infrequently for higher force expulsive behaviors such as sneezing, coughing, parturition, defecation, and micturition (138, 426).
Motor Units
Motor units are the final common pathway for the neural control of muscle force and contraction and therefore movement. Once excited, an action potential transmitted via neurotransmitters across the NMJ to produce an action potential in all the muscle fibers within the unit that are then activated in an all-or-none fashion. The mechanical properties of a motor unit depend on the collective mechanical properties of the muscle fibers. Accordingly, the motor unit force will depend on the average force generated per fiber and the number of muscle fibers within the motor unit (innervation ratio). The central nervous system controls force generation by motor unit recruitment and modulation of the frequency of motor neuron action potentials.
Motor unit recruitment
Motor unit recruitment (i.e., initiation of motor neuron axonal action potentials in response to sufficient excitatory input) is dependent on the intrinsic electrophysiological properties of motor neurons. Accordingly, smaller motor neurons comprising more fatigue-resistant motor units are recruited before larger motor neurons that comprise stronger but more fatigable motor units. In his membrane theory, Julius Bernstein (1902) applied thermodynamic principles underlying the Nernst equation to describe the resting membrane potential in nerve axons. In his theory, depolarization during an action potential reflected an abrupt decrease in membrane resistance (increase in membrane conductance). With identification of the structure of the bipolar lipid cell membrane, it was recognized that the dielectric properties of the cell membrane established the ability of the cell membrane to store charge along the intra- and extracellular surfaces of the lipid bilayer, thereby forming a capacitor (417). Accordingly, cell membrane capacitance depends on surface area of the cell. The membrane surface area also relates to input resistance such that smaller motor neurons have higher . The relation between membrane potential and input current is described by Ohm’s Law as . However, it must be noted that Ohm’s Law represents a static state, whereas the dynamic nature of motor neuron depolarization and recruitment requires a change in over time , which represents a change in the charge stored on the membrane as driven by synaptic current ; Eq. (1)]. Smaller motor neurons have less surface area and thus a lower . For a given amount of , there is a greater in smaller compared with larger motor neurons making them more excitable (126, 507, 508). Whether explained through intrinsic differences in or , smaller motor neurons have a greater change in in response to a given (Figure 1). Gasser and Grundfest (1939) found that the size of a motor neuron is approximately proportional to the diameter of its motor axon and thus, action potential conduction velocity (155). Building on their work, Elwood Henneman introduced the size principle (1957), one of the leading concepts of motor unit physiology (193, 499). He observed that smaller motor neurons with slower conduction velocities were consistently recruited before larger motor neurons with faster conduction velocities. The size principle has been largely upheld under a variety of conditions with reversal of motor unit recruitment order occurring of the time. The occurrence of recruitment order reversal is more common during more dynamic tasks likely reflecting changes in the balance of excitatory and inhibitory synaptic input (67).
Figure 1.
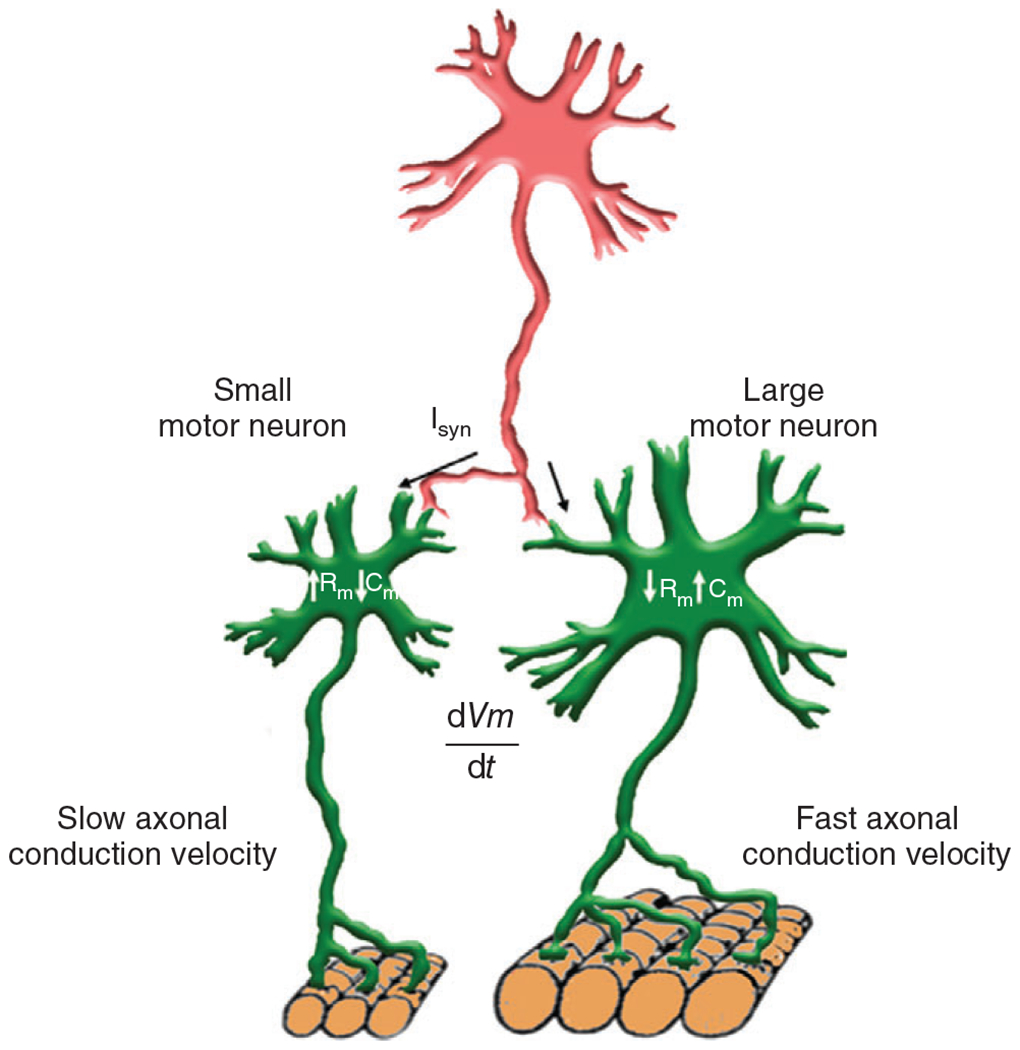
For a given level of glutamatergic input current (Isyn), the change in membrane potential will be greater for smaller motor neurons due to their lower membrane capacitance (size; ) and higher input resistance compared to larger motor neurons. Thus, smaller motor neurons will reach the threshold for action potential generation sooner (i.e., earlier recruitment) than larger motor neurons. Smaller motor neurons also have smaller axons with slower action potential propagation velocities compared to larger motor neurons. These relationships constitute the Henneman’s size principle for motor unit recruitment.
| (1) |
Distribution of motor neuron size
Generally, motor neurons innervating a skeletal muscle range in size (e.g., surface area), which underlies the selective recruitment of motor units during different motor behaviors. For example, there is an approximately five-fold range in surface areas of phrenic motor neurons in rodents (55, 137, 279, 351, 355). The distribution of phrenic motor neuron somal and total surface areas is generally Gaussian (Figure 2). Assuming a uniform innervation ratio of approximately 100 DIAm fibers innervated per phrenic motor neuron (144), the phrenic motor neuron pool can be functionally segregated into tertiles corresponding to motor unit types with the upper tertile of somal volumes likely innervating more fatigable motor units (see below). Importantly, the morphology of the motor axon and presynaptic terminal at NMJs varies proportionately with motor neuron surface area.
Figure 2.
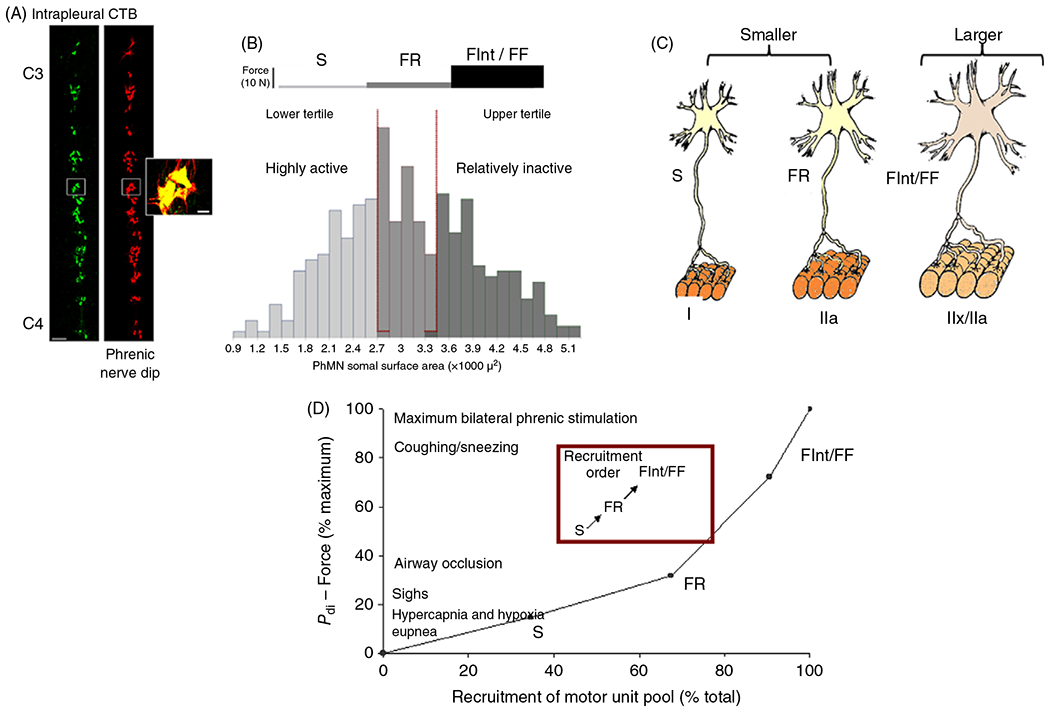
(A) Imaging of the phrenic motor neuron pool from rats acquired by confocal microscopy after labeling with tetramethylrhodaminedextran (nerve-dip) or cholera toxin B (CTB) injected into the intrapleural space. Reused, with permission, from Mantilla CB, et al., 2009/ELSEVIER (279). (B) The distribution of phrenic motor neuron somal surface areas divided into tertiles. (C) Smaller motor neurons comprise slow (S; type I muscle fibers) and fast, fatigue-resistant (FR; type IIa muscle fibers) motor units, whereas larger motor neurons comprise fast fatigue intermediate (FInt; type IIx muscle fibers) and fast fatigable (FF; type IIb muscle fibers). (D) In the rat diaphragm muscle (DIAm), a model predicting motor unit recruitment during different motor behaviors was developed based on (i) a recruitment order dependent on motor neuron size (S first FF last), (ii) the forces generated by each motor unit type, and (iii) the relative proportion of each motor unit type. The model predictions were compared to the transdiaphragmatic pressure (Pdi) or force generated by the DIAm (273). Based on this model, the lower Pdi generated during quiet breathing (eupnea) and hypercapnia/hypoxia stimulated breathing required the recruitment of only fatigue-resistant and motor units. In contrast, with more forceful (higher Pdi) DIAm efforts (e.g., coughing/sneezing and voiding) require recruitment of the entire phrenic motor neuron pool, including more fatigable FInt and FF motor units.
Motor unit/muscle fiber type classification
Muscle fiber types are classified based on the expression of different myosin heavy chain (MyHC) isoforms (407, 408) as determined by immunoreactivity to specific MyHC isoform antibodies. This classification of muscle fiber types also conforms with the mechanical and fatigue properties of fibers. In adult mammalian muscles, four isoforms can be identified via immunohistochemistry: , expressed in type I (slow, fatigue-resistant) fibers; expressed in type IIa (fast, fatigue-resistant) fibers; and/or expressed in type IIx/IIb (fast, fatigable) fibers (Figure 3). While and isoforms are uniquely expressed in type I and IIa muscle fibers of healthy mammals, respectively, and isoforms are typically co-expressed in muscle fibers in varying proportions related to their fatigability (431). For this reason, these fibers are more appropriately classified as type IIx/IIb.
Figure 3.
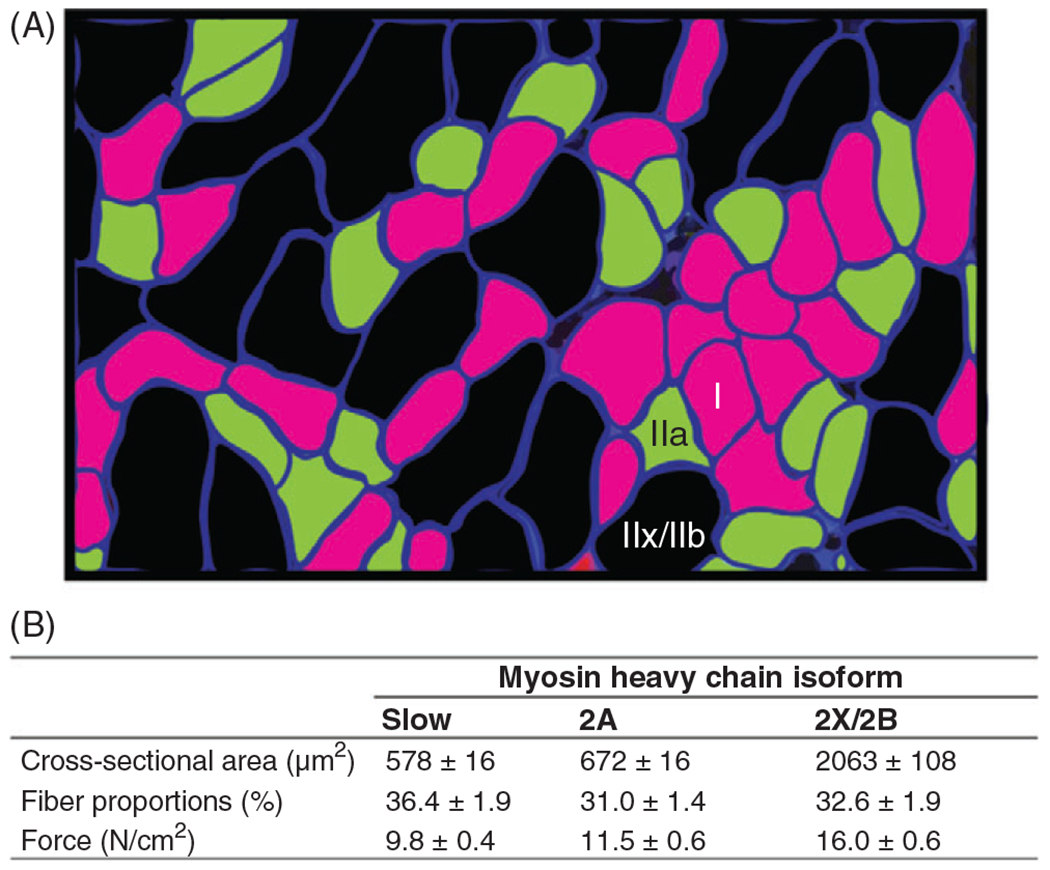
(A) Fiber-type classification in the rat diaphragm muscle (DIAm) is based on immunoreactivity to primary antibodies for myosin heavy chain (MyHC) isoforms (pseudo-colored in this example). Reused, with permission, from Mantilla CB, et al., 2010/ELSEVIER (273). (B) Composite table based on results from a number of studies displaying mean±SD of muscle fiber cross-sectional area, the proportion of fiber types within the DIAm, and specific force (273).
Motor units comprise many muscle fibers, with innervation ratios (number of muscle fibers per motor neuron) ranging from as few as 5 to (495). Glycogen depletion is one technique to identify the muscle fibers innervated by a single motor neuron. In this technique, a motor axon is repeatedly stimulated to deplete glycogen stores and thus identify innervated muscle fibers (104, 107, 145, 213, 235, 427). Using glycogen depletion, Burke et al. showed that the classification of motor unit types in the cat medial gastrocnemius muscle based on mechanical and fatigue properties corresponded with the histochemical classification of muscle fibers comprising the motor unit (65). Motor units are classified as either fast or slow based on the time to peak twitch force. This also corresponds with their velocity of shortening and force generation. Motor units are further classified as fatigue resistant or fatigable based on their susceptibility to force loss during repeated stimulation. Accordingly, slow (type S) motor units comprise type I fibers, fast, fatigue-resistant (type FR) motor units comprise type IIa fibers, fast, fatigue-intermediate (FInt) motor units comprise fibers co-expressing and fast, fatigable (FF) motor units comprise fibers co-expressing (Figure 2). Importantly, the range of motor behaviors accomplished by a given muscle depends on the motor unit composition and the appropriate recruitment of motor units. For example, in the DIAm, breathing requires the recruitment of fatigue-resistant type and FR motor units, while higher force expulsive behaviors require the recruitment of more fatigable type FInt and FF motor units (138, 426). Thus, the properties of the NMJ must match these marked differences in activation history.
Frequency modulation of motor units
The force generated by a motor unit depends on the contractile properties (contractile protein composition) of muscle fibers as well as the frequency of activation by the motor neuron. Muscle fibers produce force through a process of excitation-contraction coupling (see below). Neuromuscular transmission at the NMJ triggers excitation via an increase in cytosolic concentration leading to cross-bridge recruitment and muscle contraction (Figure 4).
Figure 4.
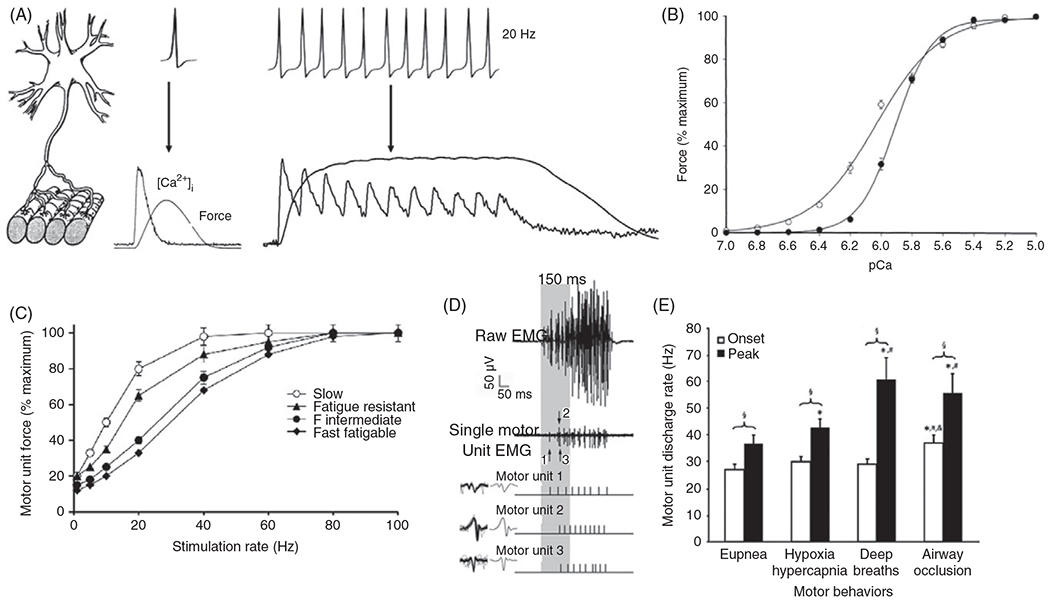
(A) An incoming motor neuron action potential is transduced to muscle fiber action potential, intracellular concentration , and force response in a 1:1 ratio. At lower stimulus intensities, the and force responses do not summate, but as the frequency of activation increases, both the and force responses summate until they fuse. (B) In permeabilized muscle fibers, the relationship between and muscle force exhibits a sigmoidal relation that is shifted left in type I fibers compared with type II fibers (159). (C) The relationship between cat diaphragm muscle (DIAm) motor unit force and phrenic nerve stimulation frequency also displays a sigmoidal curve, which is shifted leftward for slow motor units as compared to fast motor units. Fast fatigue intermediate (FInt) and fast fatigable (FF) motor units display the most rightward shifted force-frequency response curves (144, 425). (D) Single motor unit action potentials or compound summated action potentials in the rat diaphragm muscle (DIAm) were recorded using electromyographic (EMG) electrodes. Single motor unit action potentials were identified by their constant waveform. In this example, the discharge profiles of three single motor units in the rat DIAm were discriminated. (E) Once recruited, the discharge rate of DIAm motor units increases as inspiratory efforts proceed. As inspiratory drive increases, the difference between onset and peak motor unit discharge rate increases, reflected frequency modulation of force generation to accomplish different behaviors (415).
The force response of a muscle fiber to a single axonal action potential is the twitch force (Figure 4A). As the discharge rate increases, the force signals summate in time resulting in the fusion of twitch responses. There is a sigmoidal relation between motor unit force and the frequency of activation induced by electrical stimulation of motor axons (Figure 4C). At lower frequencies, there is high fidelity between axonal action potentials and the muscle fiber response (131, 213). The sigmoidal relation between the force response and the frequency of activation (force/frequency response) is due to differences in sensitivity and cross-bridge formation (excitation-contraction coupling). The binding affinity of troponin for is higher in type II DIAm fibers, which shifts their force- response to the left compared with type I fibers (Figure 4B) (158, 428). Accordingly, the concentration that produces of maximum force is higher in type II DIAm fibers compared with type I. The sensitivity of force generation is indexed by . The differences in force-frequency response of different fiber types are explained by essential differences in sensitivity and the force- response (Figure 4C).
The range of motor unit discharge rates across behaviors can be measured in vivo with electromyography using intramuscular or subcutaneous electrodes (Figure 4D) (38). In the rat DIAm, motor units display frequency coding with progressively increasing discharge rates during activation (e.g., lower onset discharge rate compared with peak discharge rates for higher forces; Figures 4D and 4E). The onset discharge rate of DIAm motor units during lower force ventilatory behaviors (e.g., eupnea, hypoxia-hypercapnia, and sighs) is fairly consistent at approximately 28 Hz (112, 273, 415, 470). During inspiratory efforts, the discharge rate of DIAm motor units progressively increases to approximately during eupnea, to approximately during hypoxia-hypercapnia, and to approximately during sighs. This range of discharge rates corresponds to the steepest portion of the force-frequency response curve of slow and FR motor units (Figure 4C). During higher force behaviors of the DIAm, including airway occlusion ( maximum force), the onset discharge rate of motor units is higher at approximately with a peak discharge rate is approximately (Figure 4E). Thus, across all motor behaviors of the DIAm, the range of motor unit discharge rates corresponds with the steepest portion of the sigmoidal force-frequency response curve (143).
Motor units are recruited to accomplish different DIAm motor behaviors that vary considerably in their duty cycles. Differences in motor unit activation (discharge rate and duty cycle) may drive structural and functional changes at the NMJ. Type and FR DIAm motor units consisting of type I and IIa muscle fibers are recruited to maintain breathing across the lifespan (Figure 4B) with duty cycles of approximately but at lower discharge rates (273). More fatigable DIAm motor units consisting of type IIx/IIb muscle fibers are recruited relatively infrequently for higher force and velocity expulsive behaviors (duty cycles ), but at higher discharge rates [Figure 6; (138, 225, 226).
Figure 6.
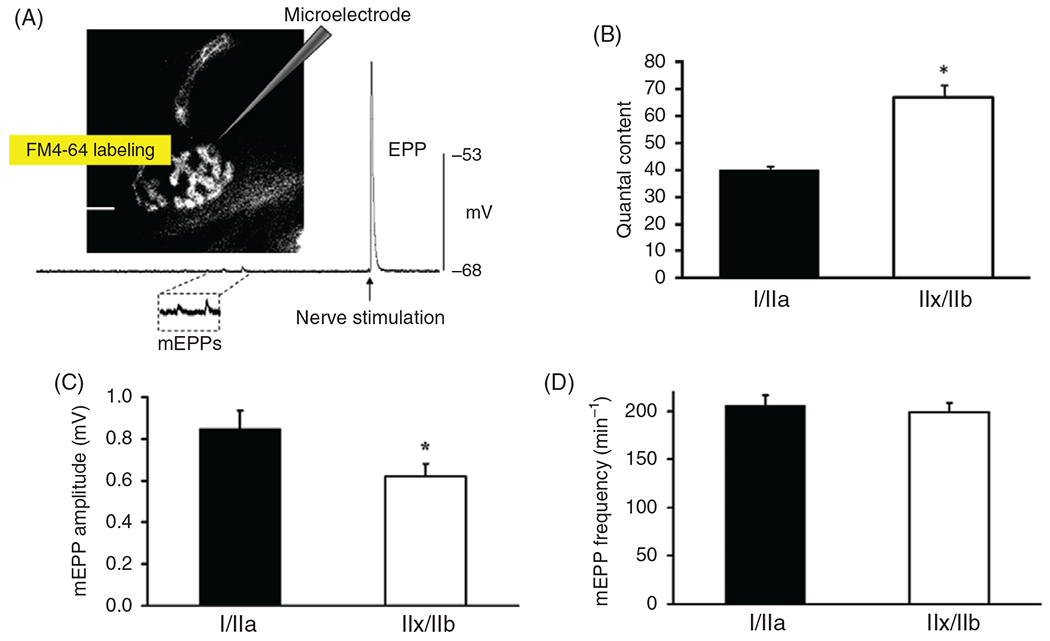
(A) Postsynaptic responses to spontaneous or evoked synaptic vesicle release can be measured electrophysiologically. In this example, the response to the spontaneous exocytosis of ACh was observed as miniature endplate potentials (mEPPs) and more concerted release as evoked endplate potentials (EPPs) were recorded using an intracellular microelectrode in the muscle fiber. The microelectrode (glass micropipette) was inserted into a rat diaphragm muscle (DIAm) fiber near the neuromuscular junction identified by labeling the presynaptic terminal using FM4-64. (B) Quantal content (QC determined as the ratio of EPP amplitude to mEPP amplitude) was significantly greater in type IIx/IIb DIAm fibers compared with type I and IIa fibers (*P < 0.01) (389). (C) In rat DIAm, the average mEPP amplitude recorded in type IIx/IIb fibers was significantly smaller than that in type I and IIa fibers (*P < 0.05) (389). (D) The frequency of spontaneous mEPPs was comparable among type I, IIa, and IIx/IIb fibers in the rat DIAm (389).
Neuromuscular Junctions
The NMJ comprises a presynaptic terminal, the postsynaptic endplate, and the terminal Schwann cell (Figure 5). The presynaptic terminal comprises structures and mechanisms for the mobilization, release, and reuptake of synaptic vesicles as well as support for local metabolic demands. The synaptic cleft ( across) is the space between the pre- and postsynaptic membranes and contains a basal lamina layer that is continuous with that of the muscle fibers and local Schwann cells. The mammalian postsynaptic endplate consists of junctional folds with AChRs clustered at the peaks of folds (124, 504) and voltage-gated channels in the troughs (127). The terminal Schwann cell plays a fundamental supporting role in the development and maintenance of the NMJ (123). ACh released at the presynaptic terminal induces cation conductance and depolarization at AChRs, which opens voltage-gated channels, generating an action potential that propagates along the muscle fiber membrane.
Figure 5.
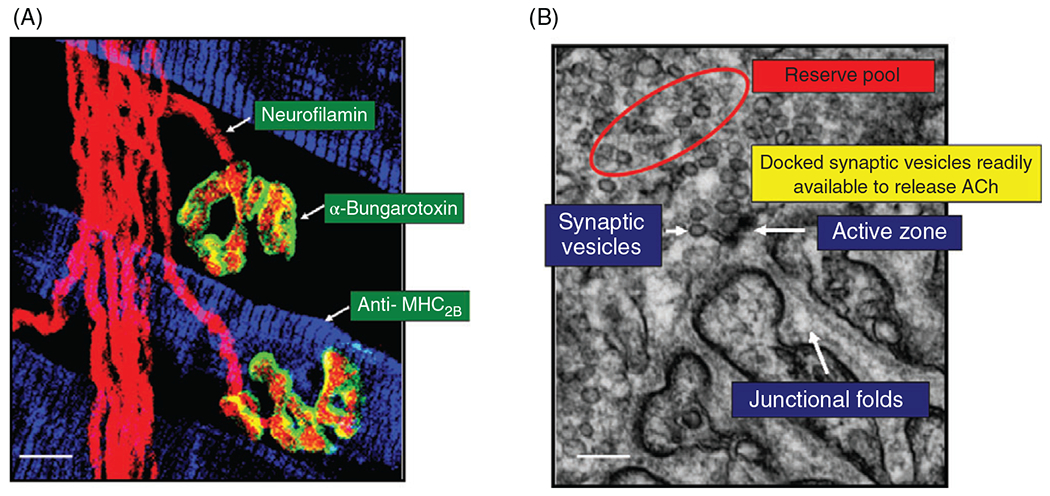
(A) Pre- and postsynaptic elements of neuromuscular junctions (NMJs) on rat diaphragm muscle (DIAm) fibers can be visualized by confocal microscopy. Phrenic motor axon and presynaptic terminals were labeled by fluorescence immunostaining using neurofilamin antibody (red). Motor endplates were labeled using fluorescently tagged -bungarotoxin (green), which binds to cholinergic receptors. DIAm fiber types were distinguished by immunofluorescence using antibodies specific to different myosin heavy chain isoforms. In this example, immunoreactivity for anti- was used. The isoform is co-expressed with in rat DIAm fibers (159) so they are classified as type IIx/IIb. Accordingly, the two NMJs shown in this image are on type IIx/IIb DIAm fibers. (B) Electron micrographic (EM) image of a NMJ on a type rat DIAm fiber in which it is possible to clearly distinguish the presynaptic terminal containing an active zone, synaptic vesicles, and the motor endplate containing junctional folds. Synaptic vesicles docked near the active zone hypothetically form a readily releasable pool, whereas non-fused synaptic vesicles form a reserve pool. It is assumed that the availability of synaptic vesicles to fuse with the presynaptic terminal membrane near the active zone depends on the distance from the active zones.
Presynaptic terminals
Motor axons traverse varying distances originating from somatotopically organized segments within the spinal cord in mammals (17, 18) and ending as the presynaptic terminal at specialized regions along muscle fibers, the endplate (433). The presynaptic terminal of the motor neuron axon primarily subserves signaling mechanisms for the transduction of membrane depolarization and the release of ACh through synaptic vesicles, that is, the impetus for excitation-contraction coupling. Presynaptic terminals also provide structures that enable retrograde communication from muscle fibers back to the presynaptic terminal and up toward the input region of the motor neurons. Presynaptic terminals undergo remodeling throughout the lifespan, from the embryo to old age.
Synaptic vesicles and synaptic vesicle release
From the 1950s to the 1980s, Bernard Katz and his colleagues provided seminal observations in NMJs of frog hindlimb muscles characterizing the role of synaptic vesicle release at the presynaptic terminal in neuromuscular transmission. These studies leveraged intracellular electrophysiological recordings using micropipette electrodes (261, 311), which they modified to measure depolarization of the postsynaptic membrane of NMJs in frog muscle fibers (118). Coincident with the advances provided by electrophysiological techniques, the electron microscope (EM) provided visualization of the presynaptic terminal. Using EM, De Robertis and Bennett observed that the presynaptic terminals of frog sympathetic ganglia and earthworm nerve cord neuropil contained many spherical electron translucent/transparent vesicles, which they termed synaptic vesicles (92). These investigators concluded that these synaptic vesicles were related to the particulate or granular fractions reported by others to contain ACh and catecholamines. In 1956, Palay (338) proposed that the vesicles observed by EM were the structural source of the small amplitude, subthreshold postsynaptic depolarizations that had been previously observed (103, 118, 119) (Figure 6), which Fatt and Katz termed miniature end-plate potentials (mEPPs) (119). Accordingly, the physiologically based hypothesis of quantal transmitter release had a structural correlate. Subsequently, support for the concept that synaptic vesicles are the structural unit of neurotransmitter release (quanta) came from studies using freeze-fracture EM, which visualized the fusion and release events of a synaptic vesicle at the presynaptic terminal in the frog cutaneous pectoris muscle (196).
The quantal theory of synaptic vesicle release was based on observations of the relationship between the average amplitude of spontaneous mEPPs compared to the amplitude of evoked endplate potentials (EPPs) (93, 199, 283, 454). Importantly, it was shown that the amplitude of EPPs was a multiple of the mean amplitude of mEPPs (93). Katz and colleagues recognized that and EPPs were not action potentials but followed passive cable properties such that the amplitude of the mEPP and EPP decayed depending on the distance from the endplate region (determined by a length constant).
Consistent with the passive properties of synaptic events at the NMJ, Katz demonstrated that mEPP amplitude varied with fiber size in the frog digitorum longus muscle (223). In agreement, in the rat DIAm, it was shown that the mean amplitude of mEPPs is greater in type I and IIa fibers compared with type IIx/IIb fibers (113, 389) (Figure 7), reflecting the smaller fiber cross-sectional area of type I and IIa fibers, which affects their intrinsic electrophysiological properties (389). The mean amplitude of evoked EPPs is similar across fiber types in the rat DIAm (113, 389) (Figure 7). Quantal content is calculated as the ratio of EPP to mean mEPP amplitudes, and thus reflects the number of synaptic vesicles (i.e., mEPPs) contributing to an EPP (93). The evoked QC of NMJs at type IIx/IIb DIAm fibers is greater than that at type I and IIa fibers (113, 389).
Figure 7.
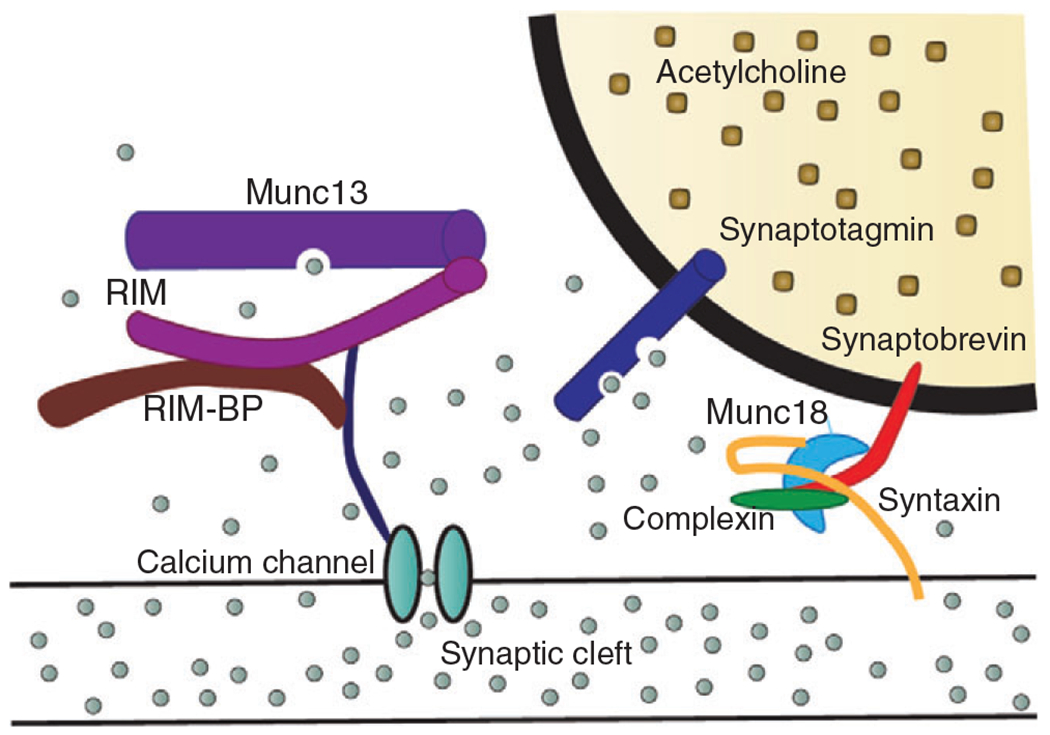
The SNARE protein complex mediates the docking and fusion of synaptic vesicles to the presynaptic terminal membrane. In response to a nerve action potential and depolarization of the presynaptic terminal, this process is initiated by the influx of (represented by the grey circles) via voltage-gated channels. The elevated intracellular concentration leads to increased binding to synaptotagmin, which then triggers the signaling cascade responsible for synaptic vesicle fusion to the presynaptic terminal membrane near active zones. The SNARE signaling cascade involves synaptobrevin, mammalian uncoordinated-18 (Munc18), complexin, and syntaxin working together to fuse the synaptic vesicle membrane to the presynaptic terminal membrane. channels are also tethered close to the synaptic vesicle by Munc13, rab3-interacting molecules (RIM), and RIM binding protein (RIM-BP).
While Fatt and Katz were the first to describe the distribution of mEPP amplitude and frequency at frog NMJs, Gage and Hubbard (1965) extended and confirmed similar findings in several different species including the mammalian NMJ. The frequency of mEPPs is related to the probability of spontaneous release and thus likely reflects the probability of synaptic vesicle fusion to the presynaptic terminal membrane and subsequent release (53, 287, 389). At rest, frequency is similar across different fiber types in the rat DIAm (113, 389). A small yet thorough body of research has examined whether mEPP frequency truly represents a spontaneous, random process (stochastic Poisson distribution). Cohen et al. tested the Poisson distribution of mEPP frequency from 11 different datasets in the sartorius muscle from a frog using 5 different goodness of fit tests (81). Additionally, they tested the independence of the interval between mEPPs using both autocorrelation analysis and a comparison of the unsmoothed power spectrums given a Poisson expectation. They concluded that the frequency of mEPPs was not accurately described by a true Poisson distribution. Segal et al. explored the distribution of frequency through Campbell’s theorem, which calculates the expected value and variance of a random point process but does not inherently require it to be a Poisson distribution, allowing for skewed distribution (414). This approach accurately predicted the distribution of frequencies except at high frequencies. Conclusions about the nature of frequency are partly obscured by observations of a nonquantal release of ACh (leakage) (222) and limited by damage caused by micropipettes, effects of any added stimulating agents, redistribution of ions (i.e., ) in isolated preparations, accumulation of in the synaptic cleft, and high rates of leakage (222, 414). Furthermore, Poisson distributions follow assumptions of pure independence such that any two events cannot occur at the same time, and the occurrence of one event will not affect the probability that a second event will occur (nondeterministic). Given the 3-D nature of NMJs and the singular recording micropipette within the muscle fiber, these assumptions seem too limited to accurately describe the release dynamics at NMJs.
Synaptic vesicles are relatively spherical with a lipid bilayer membrane interposed with protein structures to transport neurotransmitters and ions across the synaptic vesicle membrane (94, 180), trafficking proteins, and docking proteins. The neurotransmitter protein complex exchanges neurotransmitter for either protons ( 2 protons: 1 neurotransmitter) or through a process that acidifies the synaptic vesicle (44). Each synaptic vesicle at presynaptic terminals of motor neurons has been estimated to contain approximately 5,000 to molecules in the cutaneous pectoris muscle of frogs and the external oblique muscle of snakes (239). Trafficking proteins mediate the intracellular movement of the synaptic vesicles including axonal transport, synaptic vesicle pool formation, and docking in the active zone. Whereas the docking proteins allow fusion with the presynaptic terminal membrane and release of via exocytosis.
Some synaptic vesicles in the presynaptic terminal are docked to active zones, identified by electron-dense regions on the presynaptic terminal membrane. The synaptic vesicles docked to active zones are thus hypothesized to be readily available to release ACh upon stimulation of the presynaptic terminal (termed the “readily releasable pool”). This readily releasable pool of synaptic vesicles is distinguished structurally by their location in proximity to active zones, which are structurally aligned with the AChR dense region of the junctional folds at the endplate (Figure 6). The size of the readily releasable pool of synaptic vesicles is also defined functionally by the initial amplitude of the evoked EPP (QC—see below). Notably, while at higher stimulation frequencies, quantal estimates suggest total vesicular exocytosis at active zones, at low stimulation frequencies, exocytosis of the readily releasable pool is smaller. This suggests that not all active zones participate in exocytosis during stimulations in the physiological range (394). The presynaptic terminal also appears to contain other pools of synaptic vesicles that are not readily releasable (375). These synaptic vesicles are located throughout the presynaptic terminal and can dock with the presynaptic terminal membrane only when a site becomes available (94, 389). It is thought that synaptic vesicles that are closer to active zones, but not docked, constitute an “immediately available” pool (374), but the functional definition of this pool is not as well defined. Structurally, the number of synaptic vesicles in the immediately available pool can be arbitrarily determined based on distance (e.g., 200 nm) from the active zone. Although not clearly defined, the synaptic vesicles in the immediately available pool can be functionally determined by changes in QC during repetitive stimulation (see below). Other synaptic vesicles are bound within the presynaptic terminal and must be freed before they are available for docking and release, thus comprising a reserve pool. The number of docked synaptic vesicles can be counted to estimate the size of the readily releasable vesicle pool based on location, and QC can be determined electrophysiologically (see below). In the rat DIAm, the number of docked synaptic vesicles per active zone is not different across motor unit types. However, presynaptic terminals at type IIx/IIb DIAm fibers have a greater surface area with more active zones and thus, a larger readily releasable pool of synaptic vesicles (389). The number of synaptic vesicles in the structurally defined immediately available pool can only be estimated as the mean number of vesicles contained within an arbitrarily defined distance (e.g., 200 nm) from presynaptic terminal active zones (389, 409). Those synaptic vesicles outside of this arbitrary range constitute a reserve pool. With larger presynaptic terminals, the size of both the immediately available and reserve pools of synaptic vesicles is larger at type IIx/IIb DIAm fibers.
Mechanisms of synaptic vesicle release
Synaptic vesicle release involves a complex process requiring a cascade of intracellular mechanisms to mobilize, dock, fuse, and release ACh collectively termed the SNARE (soluble -ethylmaleimide-sensitive factor-associated protein receptor) complex. The fusion of synaptic vesicles and release of ACh occurs at active zones, which consist of bands of electron-dense “active zone material”—aggregates of structural macromolecules (ribbons, T-bars, beams, etc.) bound to the membrane-with the precise ordering of the structures that bind synaptic vesicles (307, 450, 484). This complex docks and primes synaptic vesicles for exocytosis. Synaptotagmin allows for -evoked synaptic vesicle fusion and exocytosis at the active zones. Close association of postsynaptic receptors to active zones is ensured via transsynaptic cell-adhesion molecules. Active zones comprise a set of core constituent proteins consisting of rab3-interacting molecule (RIM), RIM-binding protein (RIM-BP), mammalian uncoordinated-13 (Munc13), -liprin, and a protein rich in the amino acids glutamine, leucine, lysine, and serine (ELKS) (218). RIM, RIM-BP, and Munc13 are discussed below in relation to their role in neurotransmitter release via the SNARE complex. The proteins -liprin and ELKS are cytoskeletal and scaffolding proteins involved in active zone assembly (262, 447). In addition, ELKS are implicated in the regulation of influx within the presynaptic terminal (262). Active zones are situated immediately across the synaptic cleft from the region of the postsynaptic membrane that contains the highest density of AChRs. Based on studies in rat and mouse limb muscles and DIAm, there are several hundred (ranging from to 900) active zones per presynaptic terminal (74). Proteins comprising the SNARE complex are localized directly across the synaptic cleft from the region of the endplate containing AChRs (i.e., within the active zones) of frog NMJs.
In an elegant series of studies in NMJs at frog sartorius muscle, Katz and Miledi clearly demonstrated that synaptic vesicle release at the presynaptic terminal requires influx and a rise in (219–221). The rise in at the presynaptic terminal triggers synaptic vesicle fusion and exocytosis at the active zones, thereby releasing into the synaptic cleft. The presence of triggers synaptic vesicle docking in <100 μs (396), which some take as evidence that the SNARE complex is at least partially formed and possibly primed prior to synaptic vesicle docking (449). Voltage-gated channels at the presynaptic terminal consist of an subunit (pore-forming) and two auxiliary subunits (β and ). The subunit consists of four homologous domains, each with six transmembrane segments. Segments 1 through 4 are sensitive to changes in and segments 5 and 6 comprise the channel pore (496). Voltage-gated channels are tethered to the presynaptic terminal membrane and the SNARE complex by the proteins RIM, RIM-BP, and Munc13, which also play roles in docking the synaptic vesicles (Figure 7) (449). Voltage-gated channels are tethered less than away from the cellular membrane-bound SNARE within the active zones of frog NMJs (382, 383).
The SNARE complex provides the structure necessary to overcome the electrostatic and hydration repulsive forces between the membranes of the synaptic vesicle and the presynaptic terminal. Synaptic vesicle fusion and release of ACh require the coordination and interaction of multiple SNARE proteins (300, 419). Mutual negative charges on the exposed proteins will prevent fusion by electrostatic repulsion, and binds to sensitive proteins it provides ares of reversed polarity (47, 339). The density of SNARE proteins determines fusion kinetics via the extent of liposome fusion (210).
There are two lines of evidence for the existence of the SNARE complex in the presynaptic terminal of mammals: (i) studies using botulinum toxin, which destabilizes or prevents the formation of SNARE, provide a useful tool to study the SNARE complex in mammalian NMJs (487), and (ii) histochemical techniques using fluorescently labeled proteins to directly study the presence of and changes in key SNARE complex proteins. The proteins associated with the docking and release of synaptic vesicles directly form the SNARE complex and provide additional tethering between the SNARE complex and the membranes (Figure 8). The core SNARE proteins are synaptobrevin, synaptosomal-associated protein, 25 kDa (SNAP25), and syntaxin. Synaptobrevin is a highly prevalent transmembrane synaptic vesicle protein (~70/vesicle), whereas SNAP25 and syntaxin are associated with the cell membrane and provide an ionic coupled link with neighboring SNARE complexes (292). Synaptotagmin, complexin, Munc18, and Munc13 are regulatory proteins related to the SNARE complexes (Figure 8). Synaptotagmin is evolutionarily conserved and is the primary detector. In response to a rise in , synaptotagmin, as well as complexin, bind to syntaxin (34, 60). Complexin mediates SNARE oligomerization and organizes SNARE complexes into zig-zag patterns (240, 466). If complexin is absent, there is a suppression of fast, synchronous exocytosis and conversely an increase in spontaneous exocytosis (367). Finally, Munc18 interacts with syntaxin, which triggers a conformational change that allows it to bind with synaptobrevin (453). The absence of Munc18 entirely prevents synaptic vesicle fusion and leads to neuron degeneration (189). The formed SNARE complex undergoes a progressive zippering-like effect destabilizing the hydrophilic surfaces of the lipid bilayer and allowing the opening of a fusion pore (184). Finally, synaptophysin is another highly prevalent protein in the synaptic vesicle membrane but its functional role is poorly understood. Some evidence suggests that synaptophysin acts as a negative regulator of neurotransmitter release by modifying the probability of release (362). Differences in synaptophysin may explain fiber type differences in the probability of synaptic vesicle release (see below).
Figure 8.
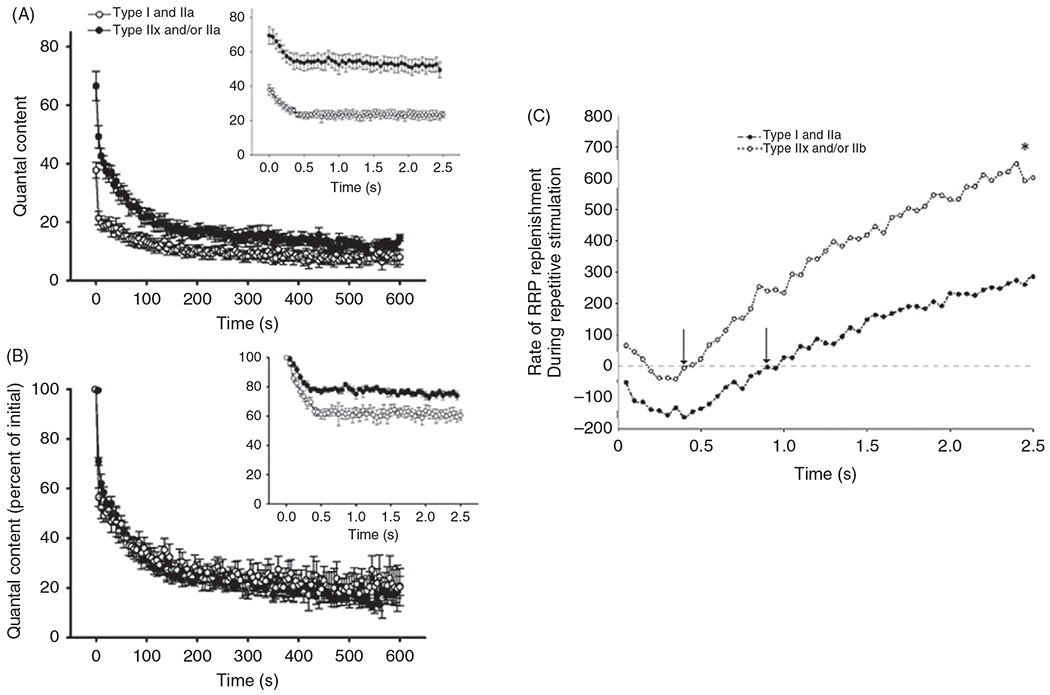
(A) The average decline in quantal content (QC) over a 10-min period of continuous 20-Hz stimulation at type I or IIa vs type IIx/IIb fibers in the rat diaphragm muscle (DIAm). At both fiber types, there was a rapid early decline in QC (1st 100 s) followed by a slower (late) decline (beyond 300 s). Inset: Shows the immediate decline in QC occurring within the first . (B) The relative change in QC (normalized to the initial QC) is similar across type I or IIa and type IIx/IIb DIAm fibers in both the early and late phases of decline. However, there was a fiber type difference in the immediate decline in ; inset), with a greater relative change at type I or IIa fibers compared to that at type fibers . (C) The rate of synaptic vesicle replenishment from the reserve pool during repetitive phrenic nerve stimulation (20 or pulse duration) was estimated based on the difference between the predicted depletion of the readily- releasable pool of synaptic vesicles and the actual QC measurements at DIAm fibers. A higher rate of synaptic vesicle replenishment was observed at type IIx/IIb fibers compared to type I and IIa fibers . Figure used with permission from (389).
Synaptic vesicle recycling
The presynaptic terminal membrane is in a constant state of turnover, which serves to release and reform synaptic vesicles, as well as provide a mechanism for retrograde transport of signaling molecules taken up through endocytosis (442). Importantly for synaptic transmission, synaptic vesicle recycling is required to replenish the readily releasable pool of synaptic vesicles to sustain quantal release (370, 377, 448) after approximately the first s of repetitive stimulation at type IIx/IIb fibers and after approximately 0.5 s at type I and IIa fibers in the rat DIAm (389) (Figure 8). The recycling of synaptic vesicles can be visualized using styryl dyes such as FM4-64 (272) (Figure 9). After synaptic vesicle fusion with the presynaptic terminal membrane, ACh is released via exocytosis thereby exposing the synaptic vesicle membrane to styryl dyes in the extracellular fluid. Styryl dyes such as FM4-64, introduced by William J. Betz in 1992 and named after Fei Mao (40), bind to the entire presynaptic terminal membrane, including the fused synaptic vesicle membranes. When the synaptic vesicle is recycled via endocytosis, the FM4-64 labeled membrane is internalized in the presynaptic terminal (37, 40, 366). After washing out the extracellular FM4-64, the recycled and internalized synaptic vesicles can be visualized using confocal fluorescence microscopy (Figure 10). The recycling of synaptic vesicles is faster in presynaptic terminals of type I fibers (366). By imaging FM4-64 uptake, the extent of synaptic vesicle recycling following stimulation at 10 Hz was reported to be greater in type I and IIa DIAm fibers compared with type IIx/IIb fibers. With repetitive stimulation at 50 Hz, the extent of FM4-64 “destaining” was greater in type I and IIa DIAm fibers compared to type IIx/IIb fibers, again indicating an increased rate of synaptic vesicle recycling (272). Two weeks of phrenic motor neuron and DIAm inactivity imposed by cervical spinal cord hemisection at results in an increase in the rate of synaptic vesicle recycling across all fiber types. In contrast, blocking action potential propagation along the phrenic nerve for two weeks using tetrodotoxin (TTX) markedly disrupted synaptic vesicle recycling. These results indicate that the synaptic vesicle pools at presynaptic terminals display considerable plasticity that can affect the efficacy of neuromuscular transmission.
Figure 9.
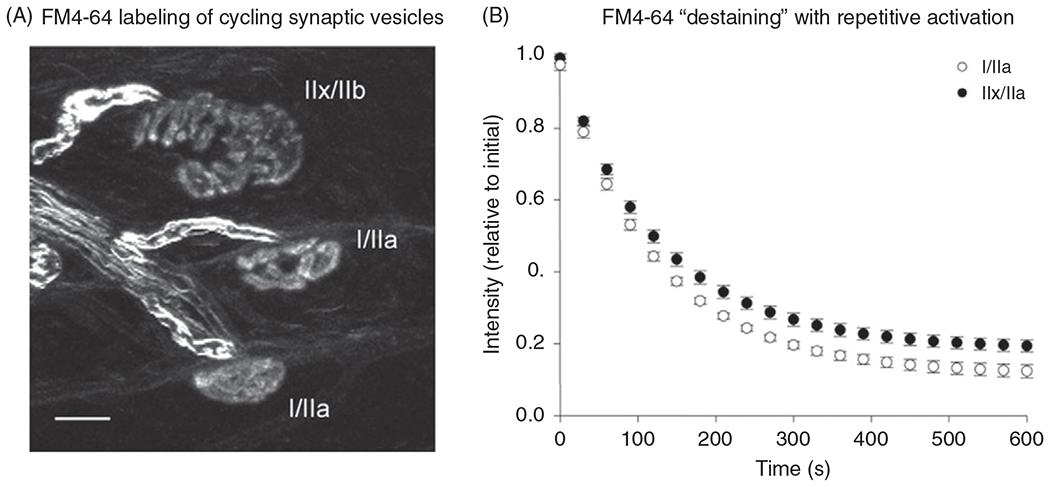
(A) Confocal imaging of presynaptic terminals at three different rat diaphragm muscle (DIAm) fibers labeled by the uptake of the styryl dye FM4-64. (B) After loading the terminals with FM4-64, the dye was washed from the bath and the terminals were destained by repetitive phrenic nerve stimulation at 10 Hz (0.5 ms supramaximal pulses with a 67% duty cycle for a 20-min period) compared with presynaptic terminals at NMJs on type IIx/IIb fibers. Values are mean±standard error for single-exponential fitted curves.
Figure 10.
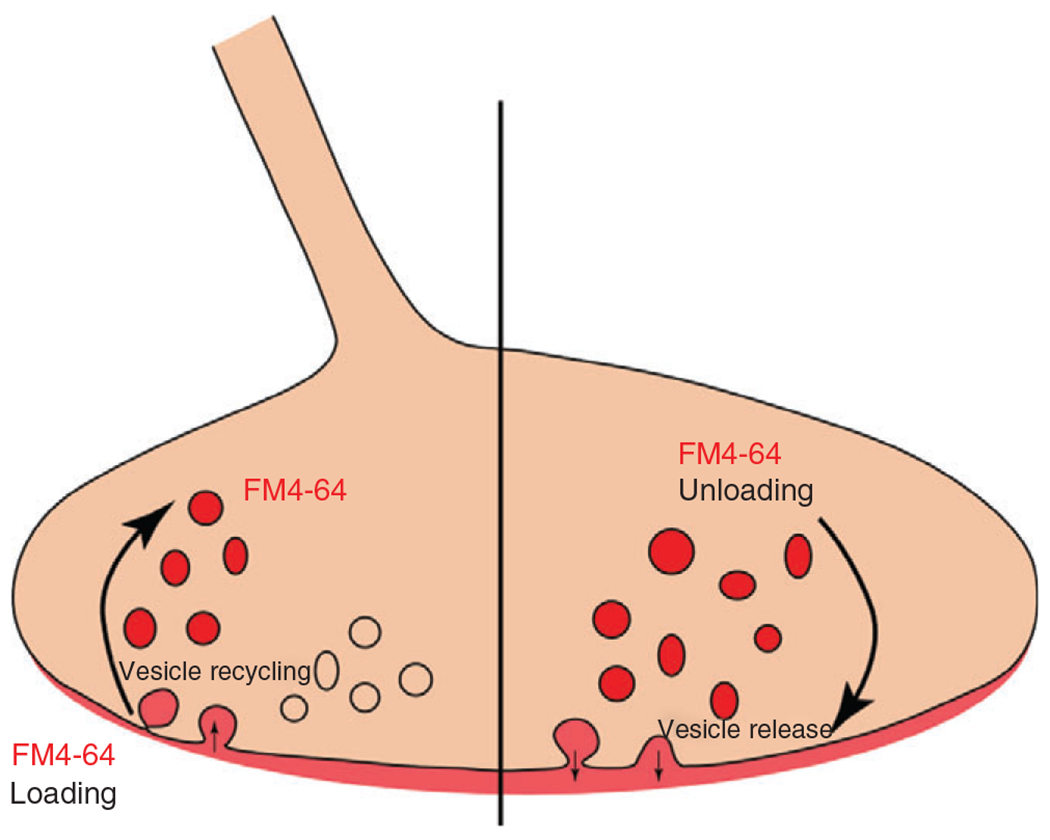
FM4-64 is applied extracellularly and is taken up into the presynaptic terminal through synaptic vesicle endocytosis. Confocal imaging during this initial process provides information on synaptic vesicle recycling. After loading the presynaptic terminal with FM4-64, and rinsing the remaining extracellular FM4-64, synaptic vesicle binding and release can be assessed by the decline in FM4-64 fluorescence.
The fastest proposed mechanism for synaptic vesicle endocytosis is a process termed “kiss-and-run” whereby the synaptic vesicle fuses, the pore opens, and is immediately closed again (376). While a fairly straightforward hypothesis, the ‘kiss-and-run’ mechanism of synaptic vesicle fusion and ACh release is the most difficult to verify due to the speed of the ‘kiss-and-run’ actions and limitations in approaches to directly study it (442).
Synaptic vesicle uptake occurs in a frequency-dependent manner (168). The nontoxic subunit of cholera toxin B (CTB), which binds to gangliosides on the motor neuron membrane can be used to label internalized membrane after endocytosis. When CTB is conjugated with a fluorescent marker, the internalized CTB can be imaged using confocal microscopy, which has been used to identify the uptake of lipid rafts. In unstimulated motor neurons or under conditions of low-frequency stimulation, endocytosis (lipid raft uptake) is not different across rat DIAm fiber types. However, at higher frequencies of stimulation, endocytosis increases (168). In general, motor units comprising type IIx/IIb fibers discharge at higher frequencies compared with motor units comprising type I and IIa (416, 426). Therefore, higher rates of lipid raft uptake through bulk endocytosis occur in larger motor neurons that innervate type IIx/IIb fibers.
Neurotrophic and myotrophic anterograde/retrograde communication
In addition to synaptic vesicles, there are other vesicles that are important in communication between muscle fibers and motor neurons via endocytosis and axonal transport. These other vesicles transport molecules that do not easily diffuse in the hydrophilic extracellular matrix (268) and can carry proteins, lipids, DNA, mRNA, and micro RNA that are involved in transmembrane transport, intracellular signaling pathways, cell dynamics, and metabolism (387). Clathrin is a large protein that lines the inner surface of the cell membrane and assists in the budding process to create endocytosed vesicles (340). Clathrin-mediated endocytosis is part of the process of recycling synaptic vesicles in which de novo vesicles are filled with ACh via acetylcholine transferase (AChT) and returned to the reserve pool synaptic vesicles (252). Clathrin-mediated endocytosis also mediates the internalizing of signaling molecules, that are bound to the presynaptic terminal membrane. Clathrin-mediated endocytosis is not directly affected by discharge rate (19, 195). Bulk endocytosis occurs through a process of lipid raft uptake, which accounts for synaptic vesicle recycling at higher frequencies (168). Bulk endocytosis is not molecule specific and requires postendocytic sorting mechanisms (442).
Retrograde signaling pathways from the presynaptic terminal back toward the motor neuron soma mediate myotrophic influences at the motor neuron as well as provide communication regarding the efficacy of neuromuscular transmission. Retrograde communication is mediated via the axonal transport of endocytosed vesicles. Direct evidence for the importance of retrograde signaling pathways at the NMJ predominantly come from research using drosophila; however, studies in mice that have manipulated protein expression in the muscle and observed changes in the motor neuron provide evidence for retrograde communication across the NMJ. For example, signaling cascades involving the secretion of Wnt glycoproteins are important mediators of NMJ development and maturation. Recently, researchers used mice with a muscle-specific -catenin (a downstream protein in the Wnt canonical pathway) knockout and found a decrease in presynaptic sensitivity at the motor neuron presynaptic terminal (258).
Retrograde signaling by brain-derived neurotrophic factor (BDNF) via its high affinity tropomyosin receptor kinase B (TrkB) receptor may also play an important role in maintaining the integrity of the presynaptic terminal. The motor neuron produces BDNF, which is released in synaptic vesicles after which it binds to its high affinity TrkB receptor. Subsequently, the BDNF/TrkB complex is internalized by endocytosis and then retrogradely transported to the motor neuron soma where it mediates signaling via phosphorylation of the transcription factor cAMP response element-binding protein (CREB). Endocytosis of BDNF/TrkB has a distinct advantage in the presence of cargo-binding adaptors that enable the specificity of molecule uptake (230, 476, 510).
Energetic demands of the presynaptic terminal
Repeated activation of the presynaptic terminals at NMJs has an energetic cost that is met by local ATP production by mitochondria. The volume density of mitochondria for a given surface area of a neuron predicts the ATP requirements for maintaining the membrane potential (492). The presynaptic terminal consumes energy (ATP hydrolysis) via the -ATPase pump to counter ion flow from action potentials and leakage currents to restore and maintain the resting membrane potential. ATP requirements for the -ATPase pump have been estimated based on recordings of conductance during an action potential. The rate of ATP consumption was estimated as a function of the current produced by the -ATPase pump, and Faraday’s constant (F; the charge per 1 mole of electrons). The ATP consumption rate was assumed to be directly proportional to the probability that an action potential would occur (187, 250). In the rat DIAm, phrenic motor neurons innervating type I and IIa fibers are highly active being recruited to sustain breathing with a duty cycle of to 50% (138, 426). Thus, it is likely that presynaptic terminals of NMJs at type I and IIa DIAm fibers exhibit higher mitochondrial volume densities compared to type IIx/IIb fibers to support the higher energetic demands at these NMJs.
The main isoform of the -ATPase pump in neuronal projections, and presumably the presynaptic compartment of the NMJ, is the subunit (51, 506), which appears to be optimized for high-intensity neuronal firing (16, 46, 85). Indeed, in motor neurons innervating type FF motor units (type IIx/IIb muscle fibers), which exhibit higher discharge rates upon recruitment, the expression of the subunit has been reported to be greater than that found in motor neurons innervating type S (type I muscle fibers) motor units (390). However, the utility of such selectivity is somewhat opaque, as it was also suggested that subunit expression was selective to gamma motor neurons (105). Like many molecular-based classifications of motor neurons, the fidelity of labeling in both motor neurons and their presynaptic terminals at the NMJ is difficult to disambiguate and the reproducibility in mixed motor units has not been established (280). Regardless, at type-identified presynaptic terminals, it remains to be seen whether -ATPase pump isoforms are specific to fiber type. Despite these caveats, in diseases such as Alzheimer’s (324), Parkinson’s (421), and importantly amyotrophic lateral sclerosis (ALS)—where marked presynaptic terminal/NMJ dysfunction occurs, -ATPase pump function is inhibited (106, 390), which may reflect mitochondrial dysfunction.
ATP is also hydrolyzed to provide the energy required to disassemble the SNARE complex and recycle synaptic vesicles (187, 482). Synaptic vesicles are filled with neurotransmitter via proton exchange, and an ATPase pump maintains the vesicular proton concentration (406). The SNARE complex must be disassembled and recycled to maintain the synaptic vesicle pool. A single molecule of ATP is hydrolyzed to power a “spring-loaded” protein unfolding for a single SNARE complex (395). As with ATP hydrolysis for the -ATPase pump to maintain the presynaptic terminal membrane potential, the ATP requirements for maintaining the synaptic vesicle pool are also dependent on motor neuron activity and thus the release of synaptic vesicles. In addition, there are motor unit/muscle fiber type differences in QC that would affect the energetic requirements of the SNARE complex.
Mitochondria within the presynaptic terminal serve two functions. First, mitochondrial oxidative phosphorylation provides ATP for local energy demands at the presynaptic terminal (30, 31, 479). The ATP demand depends on motor neuron discharge rate, as well as the presynaptic terminal surface area and the size of the synaptic vesicle pool. Second, as would be expected, with increases in stimulation frequency, also rises (88). If mitochondria are depolarized prior to stimulation, the rise in is much greater. Therefore, mitochondrial ATP production serves to buffer by direct influx into mitochondria via the mitochondrial uniporter (MCU) as well as sequestration via the sarcoplasmic-endoplasmic (SERCA) pump. A rise in is coupled to an increase in mitochondrial , which stimulates the tricarboxylic acid (TCA) cycle and leads to an increase in ATP production (99, 160)—excitation-energy coupling. Interestingly, mitochondrial volume density within the presynaptic terminal increases with continuous electrical stimulation, at least in Drosophila (468). Whether this adaptive response is also present in mammals has yet to be determined.
Mitochondrial sequestration of can either reduce or increase the probability of synaptic vesicle release by affecting (89). However, cell death can occur if is not tightly regulated across both the cytosolic and mitochondrial membranes. For example, if is not regulated and thus increases, a general increase in mitochondrial permeability occurs, including substances to which mitochondria are normally impervious. This increase in mitochondrial permeability can lead to an increase in internal mitochondrial pressure resulting in mitochondrial swelling and eventual rupture. Mitochondrial rupture results in the release of cytochrome triggering apoptotic signals within the cell and in some cases neuronal degeneration (318).
Amyotrophic lateral sclerosis reflects motor-unit specific neuronal degeneration
Degeneration and loss of motor neurons are pathognomonic for ALS, with muscle weakness and death by respiratory complications usually occurring within approximately 2 to 3 years of diagnosis (129, 473). Although only a minor fraction of ALS patients have a known genetic cause (the basis for rodent mutant models), there are no observed differences between pathology and prognosis between genetic and sporadic ALS (156). The common pathophysiological disturbances of motor neurons reflect synaptic dysfunction and dysmorphisms that precede cell death and symptoms (9, 72, 313, 384, 444). Across motor units, larger motor neurons comprising type FInt and FF motor units (type IIx/IIb fibers) are more vulnerable, with smaller motor neurons comprising type S and FR motor units (type I and IIa fibers) surviving relatively unscathed (100, 128, 135, 136, 190, 191, 227, 418).
Morphological derangements of the presynaptic terminal are often an early observation in rodent models of ALS employing a variety of genetic mutations and include frank denervation and alterations of distal axons and may exhibit sex differences (5, 286). These alterations include a reduction in the size of the synaptic vesicle pool, reduced colocalization of synapse-stabilizing proteins (such as dystrophin and rapsyn), and increases in distal axon arbors (i.e., collateral sprouting) (70, 77, 285, 313). Similar disturbances, including denervation, are also present in human ALS patients (62).
Although many of the principles expounded upon in this article apply to all mammals in general, there are some well-established differences in human NMJ structure and function compared to those of other mammals. Overall, human NMJs are amongst the smallest that exist in mammals, in both absolute terms and relative to size (in terms of both body mass and muscle fiber diameter), with a “nummular” (coin-shaped) appearance compared to rodents, cats, and dogs with morphologies more akin to “pretzel” shapes (49, 216, 434). One caveat to this is that the fiber-type dependence of NMJ size in many of these species, including humans remains uncharacterized or poorly documented. Despite the smaller cross-sectional areas of human NMJs, their surface area increases around 8-fold, due to the deepened folds of human NMJs compared to smaller mammalian counterparts (1, 435). This greater surface area effectively acts as an amplifier of acetylcholine postsynaptic responses, perhaps to compensate for the lower QC for a given stimulus frequency of humans compared to smaller mammals (86, 166, 435, 493, 494).
Despite motor unit-specific analyses being relatively uncommon, there are suggestions that morphological changes are selective to vulnerable FInt and FF motor units (type IIx/IIb muscle fibers) (148). However, the morphology of both presynaptic terminal and endplate is unlikely to be a useful surrogate for more sensitive assessments as denervation of endplates is seen in a remarkable number of normal, healthy human NMJs (43, 505) and can be misleading regarding the fidelity of signal transduction (130, 139, 488). Due to these confounding factors, simple estimates of synaptic invasion have gone out of vogue as, without the inclusion of a functional assessment, they appear to have no bearing in relation to the fidelity of neuromuscular transmission.
Functional neuromuscular transmission deficits, including QC and mEPP frequency abnormalities, and decreased facilitation of neuromuscular transmission are readily apparent in rodent models of ALS (9, 72, 310, 384) and may explain some of the EMG phenomena observed in clinical cohorts (91, 192, 217). The deficits in QC and mEPP frequency are more apparent in vulnerable FInt and FF motor units (type IIx/IIb muscle fibers) (9, 417). There is much debate regarding the timing of motor axon degeneration compared with motor neuron pathophysiology (483). Impaired axonal transport (480) and diminished trophic signaling (489, 490) undoubtedly contribute to a steady escalation of motor unit pathology (451, 490) concomitant or immediately following motor neuron loss and denervation (313, 444). In addition, marked loss of Schwann cell function is characteristic of ALS. Healthy Schwann cells, which respond to motor neuron discharge patterns, increased ACh release among other physiological changes to presynaptic terminal structure and function (9, 15, 285, 465, 471), contribute to neuromuscular impairment in ALS in a motor unit-specific manner (471).
Overall, in ALS, motor axon presynaptic terminals undergo remarkable remodeling in the attempt to ameliorate the effects of motor neuron loss and denervation on neuromuscular transmission fidelity. Indeed, these processes may be engaged in response to aberrant axonal signaling prior to denervation (285, 286). It should also be noted that studies show remarkably few muscle defects in ALS, aside from denervation-induced atrophy (75), providing an ideal substrate for recovery provided motor unit innervation can be effectively restored and motor neuron loss blunted (7, 474). Looking forward, interventions focused on preserving presynaptic terminal contact with the endplate in ALS may also benefit retrograde neurotrophic support derived from muscle-associated factors. Indeed, efforts to promote AChR clustering in ALS models using muscle-specific kinase (MuSK) promoting antibodies lead to the preservation of NMJ innervation, NMJ function, the preservation of motor neurons (prior to endstage), and gross muscle strength (69, 343). Efforts aimed at enhancing docking protein 7 (DOK7), which activates MuSK (203, 325) achieves similar ends, with improvements in NMJ innervation, ameliorates muscle atrophy, and increases life span, but not motor neuron survival (298). Much effort remains to characterize how these attempts to improve NMJ outcomes with ALS relate to the known vulnerabilities of FInt and FF-type motor units.
Postsynaptic endplates
The endplate consists of junctional folds in vertebrates, the peaks of which are paired tightly to the active zones of the presynaptic terminal. The local depolarization at the endplate is electrotonically conducted to the voltage-gated channels in the troughs of the junctional folds. The shape of the junctional folds at endplates function to facilitate the endplate potential (284) by guiding the depolarizing current toward voltage-gated channels that are densely populated in troughs of the junctional folds (68, 127). The folded shape is maintained by the basal lamina. This is supported by evidence that dissolution of the basal lamina alters the shape and depth of the folds. The junctional folds in the membrane change the membrane and axial resistance, both dependent on geometry, and therefore the length constant (an electrical constant that describes the exponential decay in passive depolarization over a distance). The length constant is greater for the folded area of the membrane compared with the unfolded membrane (392). This spatial arrangement plays an integral role in producing a muscle fiber action potential (115) to facilitate the excitation-contraction coupling process that results in force production (176, 401).
Slow (type I fibers) and fast fatigue-resistant (type IIa fibers) motor units in the rat DIAm have smaller and simpler endplate structures (e.g., of the surface area compared with more fatigable fast (type IIx/IIb fibers) motor units), which both reflects and supports their typically lower discharge rates and smaller muscle fibers (131, 168, 353). Endplates at type IIx/IIb fibers are larger, more complex, and more prone to the effects of remodeling. However, this does not appear to be directly generalizable across different muscles. Comparisons of endplate area across muscles in mice showed that NMJs in the soleus (predominantly slow) are smaller than NMJs in the extensor digitorum longus (predominantly fast) but bigger than NMJs in the extensor digitorum communis (25).
Nicotinic acetylcholine receptors
In 1905, John Langley was the first to introduce the concept that receptors are responsible for the cellular responses to applied substances (Receptor Theory), and he specifically described the responses of muscle fibers to the application of nicotine and curare (248). Nearly 30 years later, Henry Dale and Otto Loewi won the Nobel Prize for their shared work on the chemical transmission at neuronal synapses. Notably, Dale spent approximately 20 years working with ACh and in 1929 confirmed the presence of ACh in mammals. In 1934, he introduced the terms cholinergic and adrenergic nerve fibers to describe the substances released by these neurons. An interesting division in the concept of neurological signal transmission occurred with the emerging research from Dale and researchers such as John Eccles, colloquially known as “soup vs spark.” While Dale and colleagues proposed that neural signals were transmitted between cells via chemical substances, Eccles and others firmly held that synapses relied on electrical impulses transmitted to the target cell. In 1951, Eccles “came to accept it unreservedly by what Sir Henry regards as the scientific equivalent of a religious conversion (458).” Curiously, it was not until 1955 that David Nachmansohn theorized that there was a protein acting as the receptor for ACh, and the AChR was finally biochemically characterized in 1970 by Changeux and colleagues (73).
Nicotinic AChRs are pentameric, ionotropic receptors ( subunits) activating nonselective cation channels, which produce relatively large synchronous depolarizing currents across the peaks of the junctional folds at the endplate (257). The ACh-binding site at the nicotinic AChR lies at the junction of the subunit and either the (mature endplate) or (developing endplate) subunit (170). The density of AChRs across mammalian species (bat, mouse, rat) is surprisingly consistent with approximately (3) and was first described by Salpeter & Eldefrawi in 1973 (399). The EEP resulting from AChR activation is typically greater than the voltage threshold required to generate an action potential via activation of voltage-dependent channels (i.e., safety factor). The safety factor is affected by the number of AChRs available and the amount of ACh released (i.e., QC). A safety factor is required to ensure the fidelity of generating propagating muscle fiber action potentials in response to neural signals (493). The AChR-mediated depolarizing current is greater in type IIx/IIb fibers compared with type I and type IIa fibers due to differences in QC and the number of postsynaptic AChRs (391).
While mEPPs and EPPs are the results of the synchronous activation of many AChRs, the depolarization events represent the summation of current responses from each activated AChR channel. It is estimated that a single AChR has a 70% to 80% probability of opening in response to neurotransmitter release from the presynaptic terminal (245). The probability that an AChR will open partially depends on the concentration of ACh available to bind to the receptor (14, 224). High concentrations or repeated activation by ACh lead to desensitization of the AChR, which is measured as increased silent periods, where despite sufficient presence of ACh, the receptor channel does not open (97, 224). Based on data with currents elicited by ejecting ACh from a glass pipette, AChR channels open at a fast rate (246) (Figure 11), which is consistent with the steep rise observed during mEPPs. The initial depolarization resulting from AChR currents is sustained, directed, and amplified by the junctional folds toward the voltage-gated channels to trigger a muscle fiber action potential.
Figure 11.
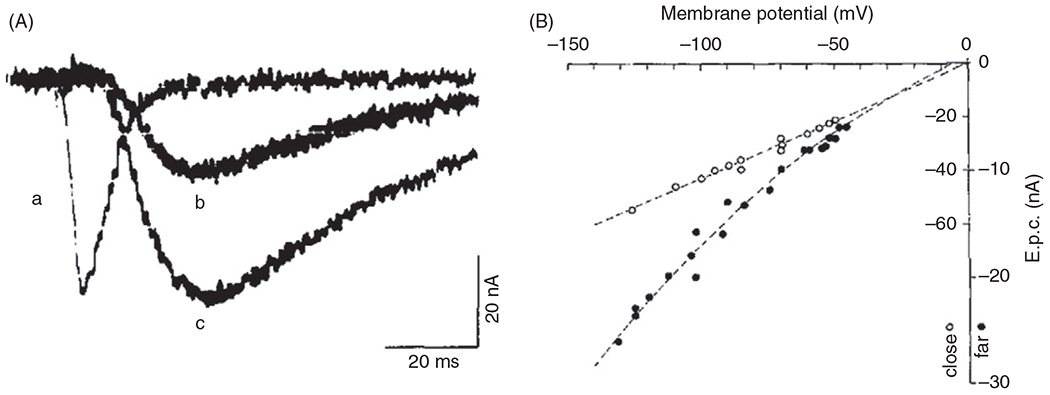
(A) Current traces from acetyl choline receptors (AChRs) at motor endplates of frog NMJs: (a) currents measured immediately above the AChR clusters and currents measured a short distance from the junctional folds at weaker (b) and stronger (c) stimulus pulses. Figures reproduced with permissions from (269). (B) The current-voltage (I-V) relationship for junctional (open) and extrajunctional (filled) AChRs at motor endplates of frog NMJs recorded using the patch-clamp technique. The slope of I-V relationship represents the conductance of the AChR channel. The reversal potential was indicating the AChR at frog NMJs is a non-selective cation channel.
Myasthenia gravis reflects postsynaptic pathophysiology
Myasthenia gravis is caused by the production of autoantibodies to either the AChR itself or to endplate AChR-clustering proteins MuSK, Agrin, LDL receptor-related protein 4 (LRP4), MuSK, Dok7, and Rapsyn (Figure 12), resulting in a breakdown of neuromuscular transmission and symptoms of muscle weakness and increased fatigue (164, 197, 198). In some forms, this auto-immune attack of AChRs or AChR-associated proteins is congenital (110). By contrast to aging and ALS, myasthenia gravis has effects spanning all muscle fiber types (79, 80, 282). Structural NMJ changes in myasthenia gravis include reduced number of AChRs (117, 163, 302, 303, 359), loss of presynaptic innervation of endplates (83, 359), and the derangement of postjunctional folds (108). Functional NMJ changes in myasthenia gravis include reduced safety factor (393), and a reduction of QC (EPP amplitude) (111, 255, 316, 371) despite increased QC (86, 299, 347, 348).
Figure 12.
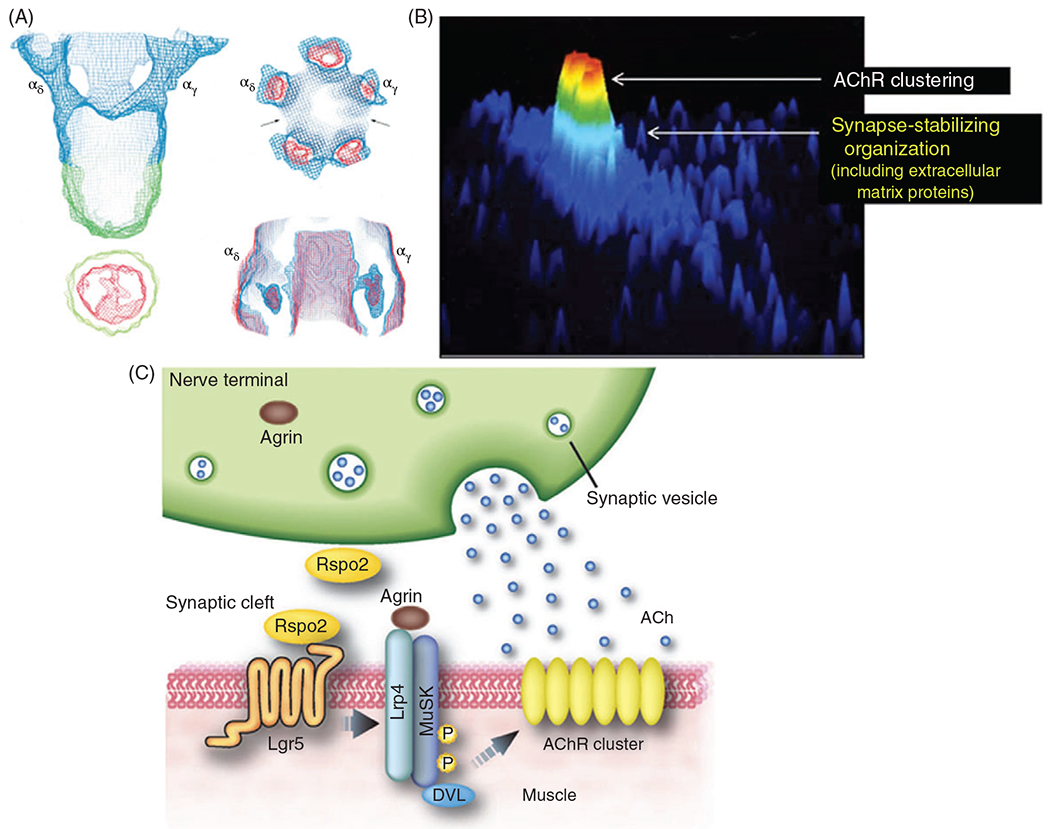
(A) Averaged images from electron microscopy (EM) were used to reconstruct the nicotinic acetylcholine receptor (AChR) at 4.6 A resolution. The top right panel is the intracellular perspective, and the bottom right is extracellular (297). (B) AChRs are more densely clustered, typically at the peak of the junctional folds (red denotes greater density) compared with the troughs. Although this image is of a cultured rat myotube, which does not consistently exhibit junctional folds. Reproduced with copyright permissions from (452). (C) Agrin is released from the presynaptic cleft and binds to muscle-specific kinase (MUSK) receptors on the muscle fiber. The binding of agrin to MuSK triggers an intracellular cascade that clusters the AChRs. Reproduced with copyright permissions from (308).
In relation to morbidity and mortality in myasthenia gravis, the prime mover is the health of the DIAm, with the major contributors to mortality being respiratory-tract infections such as pneumonia and influenza being major contributors to death (8, 294, 336, 346). Indeed, for patients with MuSKassociated myasthenia gravis, a clear respiratory muscle involvement occurs (242, 312, 337). A variety of treatments are effective for myasthenia gravis, with combined approaches aimed at increasing neuromuscular transmission (e.g., cholinesterase inhibitors) and immunomodulators, to reduce auto-immune activity (346, 422).
Efforts to understand the molecular underpinning of myasthenia gravis have involved a variety of animal model paradigms, including immuno-sensitization to the AChR and MuSK, which replicate the weakness, impaired neuromuscular transmission, and endplate fragmentation apparent in human clinical cases (59, 82, 83, 108, 109, 301, 346, 372). The muscle specificity of symptoms has been associated with developmental timelines of endplate formation, with those muscles that swiftly form synapses after initial in utero motor neuron to skeletal muscle contact being more sensitive to experimentally induced myasthenia gravis (358, 500). In the rodent DIAm, neuromuscular transmission failure is readily apparent (475), along with diminished mEPPs (66), a reduction in AChRs (293), and denervation (244). Some findings at human and rat endplates have suggested a compensatory increase in QC (347).
A key component of future efforts will be to ensure that as individuals are successfully treated for myasthenia gravis age, their adjunctive therapy for confounding factors should be considered. For example, vigilance regarding potential indications of respiratory neuromotor deficits occurring with sarcopenia (125), cardiovascular illness (363, 486), or age-associated infection risks (i.e., COVID-19) (305, 397, 440) are essential.
Terminal Schwann cells
Nonmyelinating terminal Schwann cells cap the NMJ and are a significant component of the NMJ. Louis-Antoine Ranvier first described terminal Schwann cells in 1878, but it has only been in the last approximately 30 years that their importance to the NMJ has been thoroughly explored. Terminal Schwann cells influence growth and stabilization of the NMJ, respond to motor neuron activity, and support NMJ remodeling. There is a moderate-to-strong linear relation between the number of terminal Schwann cells per NMJ (range of ) and the size of the endplate (263). The number of terminal Schwann cells present at the NMJ changes with the size of the NMJ through developmental stages. Terminal Schwann cells proliferate during development (12), axonal degeneration due to injury or disease (78, 400, 501), and axonal regeneration (11, 266, 503).
Terminal Schwann cells influence growth and stabilization of the NMJ
Terminal Schwann cells guide outgrowing motor axons, provide support to maintain established NMJs, and are key to the transition from the premature polysynaptic innervation to mature monosynaptic innervation of muscle fibers (Figure 13). Both nerves and Schwann cell precursors develop from neural crest cells. Schwann cell precursors then form immature Schwann cells at E15-E17 in rats and E13-E15 in mice (208, 209, 296). The transition from Schwann cell precursors to immature Schwann cells is synchronized with vascularization of the nerves and the developing perineurium. Motor neurons will still innervate muscles in frog models where Schwann cells have been ablated indicating that terminal Schwann cell development is not necessary for the outgrowth of motor neurons (364). Neurite outgrowth closely follows Schwann cell precursor outgrowth in tadpoles, leading some researchers to propose that Schwann cells guide nerve outgrowth (194). There is supporting evidence in mice and rats showing that after the application of neuregulin1 (NRG1), which causes Schwann cell sprouting, nerve processes extend along the Schwann cell sprouts (188, 469).
Figure 13.
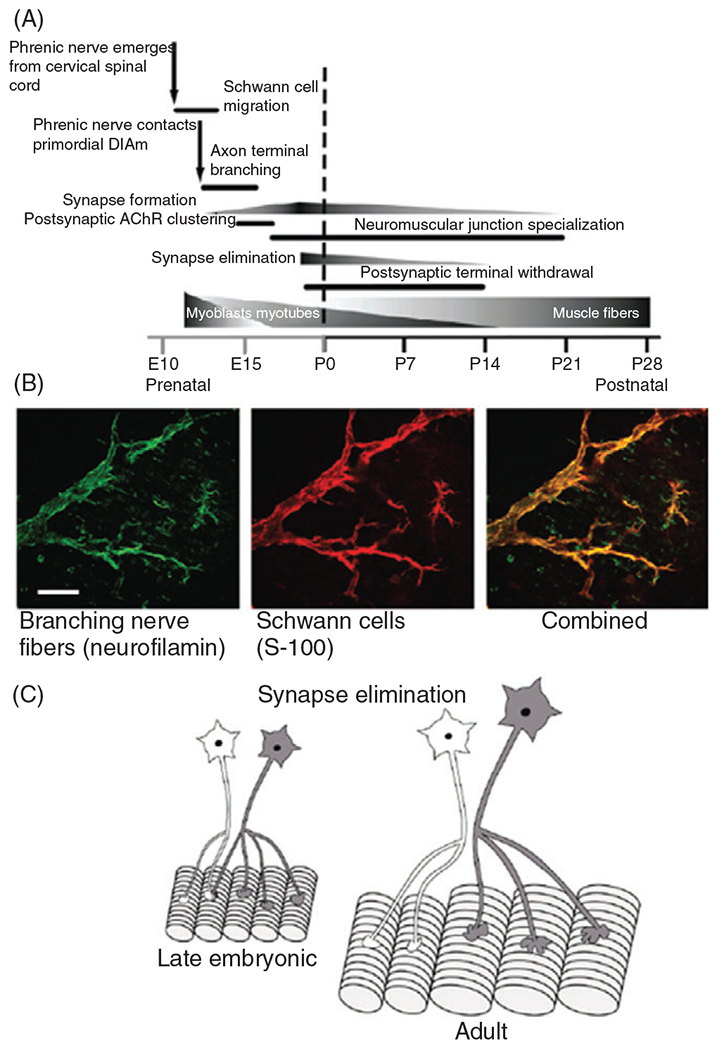
(A) Perinatal developmental timeline of the development of phrenic motor neurons and their innervation of muscle fibers in the rat diaphragm muscle (DIAm). (B) Photomicrographs of motor axons and presynaptic terminals labeled using an anti-body specific for neurofilamin (green) and Schwann cells labeled using an anti-body for S-100. The merged image shows the close association of motor axons to Schwann cells. (C) During late embryonic and early postnatal development, DIAm fibers are innervated by more than one phrenic motor neuron (i.e., polyneuronal innervation). Subsequently, polyneuronal innervation disappears via synapse elimination leaving each muscle fiber innervated by only one motor axon.
Terminal Schwann cells are important for motor neuron survival and health after outgrowth and contact with the muscle fibers. If Schwann cell precursors are absent, such as through a mutant mouse model, most motor neurons die by E18 (373). In mouse models where immature Schwann cells are ablated, motor nerves display the aberrant formation of nerve bundles on outgrowing axons (fasciculation), synaptic growth is stunted, and subsequently, a substantial portion of the nerve terminals retract (71). NRG1 release by motor neurons and ErbB receptor activity on terminal Schwann cells has been implicated as key to this motor neuron and terminal Schwann cell mutually dependent survival during development.
Finally, terminal Schwann cells mediate the transition from polysynaptic innervation to monosynaptic innervation of muscle fibers by motor neurons. Curiously, around P2 in mice, terminal Schwann cells begin to attack the NMJs along muscle fibers. The terminal Schwann cells do not determine which NMJ remains but begin to compete for direct contact with the AChR dense surfaces on muscle fibers to phagocytose displaced motor neuron terminals (438). The motor neuron that is successfully capable of maintaining the greatest contact with the endplate during this competition phase appears to remain as the single surviving presynaptic terminal. The most successful neuron is likely to be the most active neuron. Terminal Schwann cells randomly phagocytose presynaptic terminals during the transition from polysynaptic innervation to monosynaptic innervation at NMJs and the “axosomes”—fragments of the terminal containing intact organelles—are consumed by the terminal Schwann cells and integrated into the cytoplasm (42). The process is at least partially modulated by neuron-derived type III NRG1 during its peak in postnatal development (254) as manipulating the type II NRG1 signal also affects the rate of synaptic pruning. The high level of NRG1 triggers the terminal Schwann cells to intrude into the synaptic cleft.
Terminal Schwann cells and neuromuscular transmission in adults
Terminal Schwann cells sense NMJ activity through muscarinic AChRs as well as receptors for ATP, adenosine (A1 receptor), and substance P (52, 161, 380, 386). In 1992, Jahromi and colleagues as well as Reist and Smith found that terminal Schwann cells respond with an increase in after neurotransmitter release from the presynaptic terminal in the cutaneous pectoris muscle of frogs (204, 368). This increase in in the terminal Schwann cell is dependent on the type of stimulation. During continuous high-frequency stimulation, the increased evoked in Schwann cells (204, 368, 386) leads to synaptic potentiation (381). During repeated trains of high-frequency stimulation of the motor nerve, oscillatory responses occur, and synaptic depression is observed in the mouse soleus muscle (465). When terminal Schwann cells are ablated at frog NMJs, their electrophysiological and force-producing capabilities are significantly altered. After 1 week, EPP amplitude and mean QC are both decreased. The depression after high-frequency continuous stimulation and paired-pulse facilitation is reduced. Finally, twitch tension via nerve stimulation is significantly weaker (364).
Neuromuscular Transmission
The raison d’être of neuromuscular transmission is to faithfully propagate neural signals from the motor neuron to skeletal muscle fibers. To do this, ACh released at the presynaptic terminal induces cation conductance and depolarization at AChRs, which opens adjacent voltage-gated channels generating an action potential that propagates along the muscle fiber membrane. The cascade initiated by muscle fiber action potentials that leads to muscle fiber contraction is known as excitation-contraction coupling. The effectiveness of neuromuscular transmission governs the initiation of excitation-contraction coupling and thus motor system mechanical output.
Excitation-contraction coupling
Muscle fiber depolarization due to an action potential is passively transmitted down transverse tubules (t-tubules), where it activates voltage-sensitive dihydropyridine (DHP) receptors. The DHP receptors are fused with ryanodine receptor (RyR) channels in the sarcoplasmic reticulum (SR) that open to release into the cytosolic space. The increase increases binding to troponin C on the actin filament that interacts with troponin I to expose binding sites for myosin heads. The binding of myosin to actin (cross-bridge formation) underlies force production and contraction (176, 401).
Efficacy of neuromuscular transmission
Functionally, the purpose of the NMJ is effective neuromuscular transmission. The measure of the efficacy of neuromuscular transmission is the safety factor or safety margin, defined as the ratio or difference, respectively, between the amplitude of the evoked EPP and the threshold for generation of a muscle fiber action potential (493). In the rat DIAm, the safety factor at type IIx/IIb fibers during a single evoked response is higher compared with type I and IIa fibers. The increased safety factor in type IIx/IIb fibers is in line with the greater QC and the higher density of voltage-gated channels at the endplate in IIx/IIb muscle fibers (295, 389). However, with repetitive stimulation, EPP amplitude (QC) decreases to a greater extent in type IIx/IIb fibers, compromising their safety factor, and potentially leading to neuromuscular transmission failure.
The efficacy of neuromuscular transmission declines as the duration of motor neuron activity increases. The amplitude of EPPs (QC) decreases over time if the nerve is continuously stimulated, and this is likely due to a decrease in both probabilities of release and number of synaptic vesicles available to release (389) (Figure 9). In the rat DIAm, the absolute level of decline is not different but when normalized to initial QC the decline in QC is greater in the first few s of stimulation in type I and IIa fibers compared to IIx/IIb fibers. Based on estimates of the number of synaptic vesicles in the readily releasable pool at presynaptic terminals, it was determined that the initial decline in QC at type IIx/IIb fibers could be solely attributed to the depletion of the readily releasable pool (389). However, for presynaptic terminals at type I and IIa DIAm fibers, the decline in QC was much faster than would be predicted by a depletion of the readily releasable pool, which was interpreted as a decline in probability of synaptic vesicle release (Figure 14).
Figure 14.
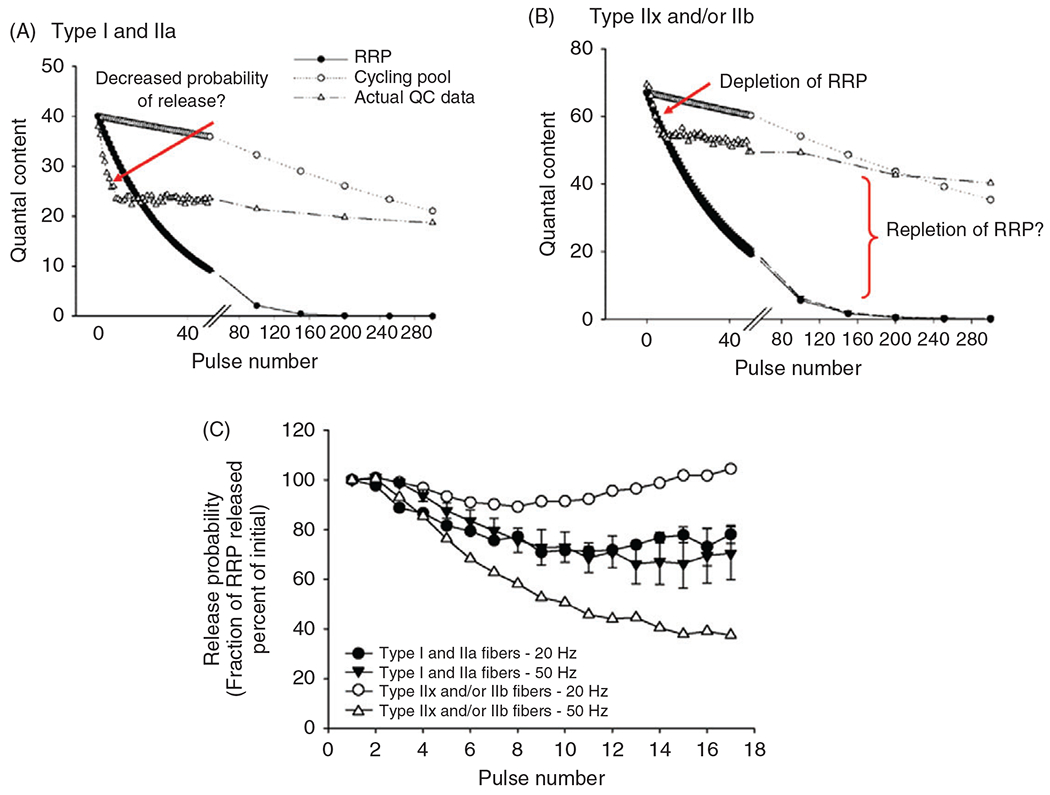
In the rat diaphragm muscle (DIAm), quantal content (QC) decreases with repeated phrenic nerve stimulation (at 20 or pulse duration) in all fiber types (A. type I and IIa; B. type IIx/IIb) (389). A model of the replenishment of the readily releasable pool (RRP) of synaptic vesicles was developed to explain the initial decline of QC and the subsequent leveling of QC. For type I/IIa fibers, the initial decline in QC is steeper than can be explained by the depletion of the RRP, so at least part of this depletion was attributed to a decrease in the probability of synaptic vesicle release. For type IIx/IIb fibers the initial decline in QC was parallel to the depletion of the RRP, which suggests that the depletion is causal. All fiber types exhibit an ability to continuously release synaptic vesicles despite a continual decline in the RRP, therefore the sustained release is due to the repletion of synaptic vesicles through synaptic vesicle recycling. The probability of release and repletion of the RRP is difficult to directly measure so these remain suggested mechanisms, hence the question mark in the figure. The probability of release remains relatively unchanged at low compared to high frequencies for type I and IIa fibers. In contrast, higher frequencies substantially decrease the probability of release at type IIx/IIb DIAm fibers.
In the frog, Del Castillo and Katz consistently observed neuromuscular transmission failure at the presynaptic terminal, reflected by the absence of a postsynaptic response despite sufficient nerve stimulation. These basic observations were accentuated by exposure to a low /high medium (93). Neuromuscular transmission failure can also result from a failure of the nerve action potential to propagate down all branches of the motor neuron (branch-point failure), a failure to release sufficient synaptic vesicles from the presynaptic terminal, or a failure of the motor endplate to respond to ACh release (e.g., reduced AChR sensitivity; Figure 15). Branch-point failure is frequency dependent and contributes more to neuromuscular transmission failure in the developing neuromuscular system, likely due to polyneuronal innervation, but plays less of a role in mature motor axons (142). With 2-min stimulation of the phrenic nerve at 10 Hz, the incidence of axonal propagation failure is approximately 4% in rats during the first postnatal week but is not observed at all in adult rats (Figure 16). By contrast, at 75 Hz the incidence of phrenic axonal propagation failure rate after 2-min is approximately 66% during the first postnatal week and approximately 26% in adult rats (Figure 16). There are two proposed mechanisms behind branch-point failure, relating to either: (i) transmembrane concentration or (ii) axonal geometry (2, 179, 429). As the membrane branches, membrane capacitance decreases while membrane resistance increases in the smaller branches. More importantly, axonal resistance increases reducing the length constant and decreasing the probability of action potential propagation (445, 446). Fatigable motor units typically have higher innervation ratios with each motor neuron having more axonal branches and are therefore more likely to fail. This was tested in the rat DIAm using the technique of glycogen depletion in which repetitive stimulation depletes muscle fiber glycogen. If axonal propagation fails to activate muscle fibers, this is reflected by reduced glycogen depletion. It was observed that the incidence of axonal propagation failure (lack of glycogen depletion) was more evident in type IIx/IIb DIAm fibers and increased with stimulation rate (213). At both high and low frequencies, type IIx/IIb fibers were found to be approximately 30% more susceptible to neuromuscular transmission failure than type I and IIa fibers despite the greater safety factor and QC exhibited by the type IIx/IIb fibers (213).
Figure 15.
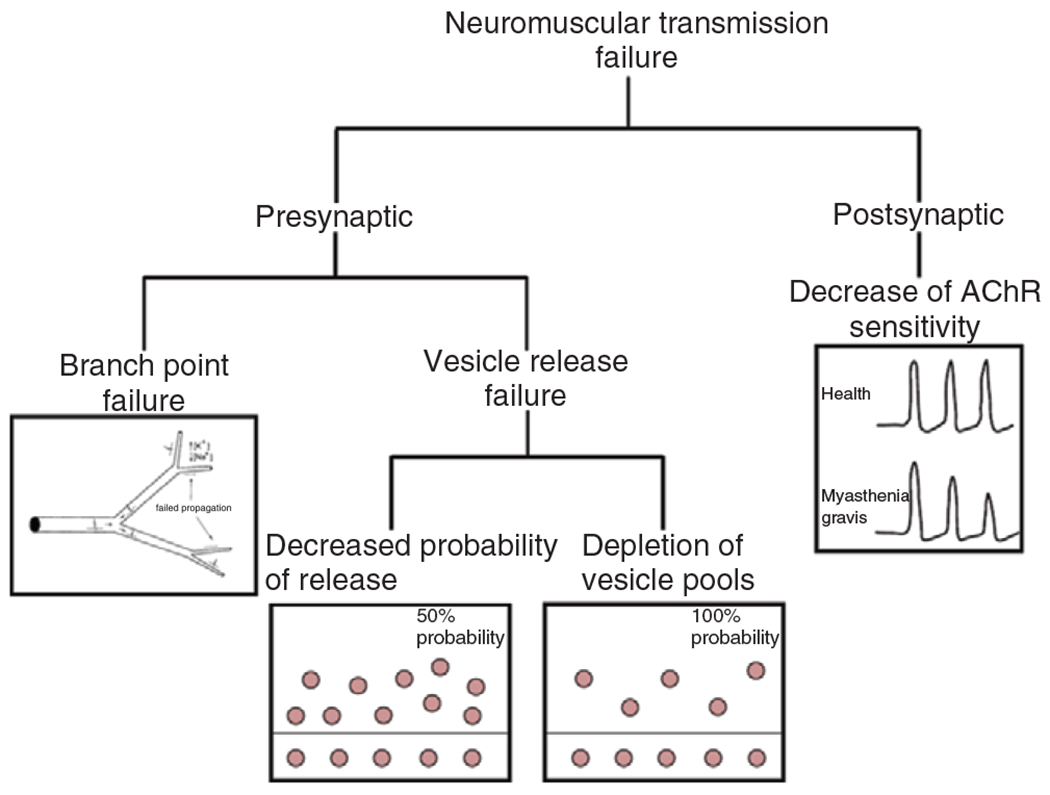
Neuromuscular transmission failure (NMTF) can occur due to failures at both the presynaptic and postsynaptic components of the neuromuscular junction (NMJ). At the presynaptic component, NMTF can be subdivided into branch point failure and a failure to release synaptic vesicles, which can be caused by either a decrease in the probability of release or depletion of the synaptic vesicle pools.
Figure 16.
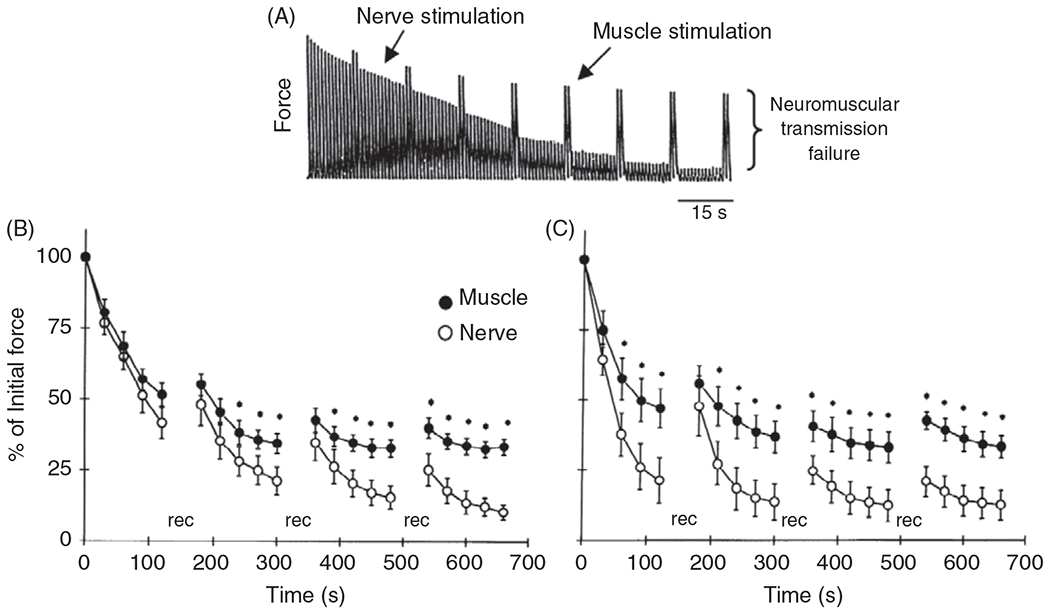
(A) One approach to test neuromuscular transmission failure (NMTF) is to repeatedly stimulate motor axons and compared the evoked forces to those evoked by intermittent direct muscle stimulation. NMTF is quantified as the difference between the amplitude of the forces evoked by direct muscle vs nerve stimulation. In the rat diaphragm muscle (DIAm), the extent of NMTF is dependent on the frequency of phrenic nerve stimulation such that at lower frequencies (B. 10 Hz) there is less divergence in the forces evoked by direct muscle versus nerve stimulation (i.e., less NMTF) than at higher frequencies (C. 75 Hz) (213).
At higher frequencies (40, 75, and 80 Hz), neuromuscular transmission failure could be due to both a failure to release synaptic vesicles or AChR desensitization. Synaptic vesicle release can fail due to a decrease in either the available pool of readily releasable synaptic vesicles or the probability of synaptic vesicle release. With repetitive stimulation, the probability of synaptic vesicle release decreases ((389); Figure 15). The fidelity in the propagation of action potentials along the axon may also relate to the level of myelin surrounding the axon, with deficits in myelination increasing the likelihood of axonal propagation failure (22, 149, 150, 412). In the continued presence of ACh, AChRs desensitize resulting in a reduced depolarizing current (224). There is no evidence that AChR differs across fiber types but the presence of ACh in the synaptic cleft increases with repeated stimulation at higher frequencies. Therefore, AChR desensitization is unlikely to explain the differences in neuromuscular transmission failure across fiber types.
A common paradigm to test neuromuscular transmission failure is to compare the force response evoked by nerve stimulation to that produced by direct muscle stimulation (bypassing the nerve) (Figure 16) (4, 28, 238). In previous studies using this technique, we reported a number of conditions that affect neuromuscular transmission failure at the DIAm in adult rats and mice, including aging (131, 175, 178), cardiopulmonary bypass (114), testosterone (45), corticosteroids (430), and hypothyroidism (350).
As mentioned above, we found that the neonatal rat DIAm is far more susceptible to neuromuscular transmission failure during repetitive activation than the adult (33, 122, 142, 357, 365). Neuromuscular transmission failure in the embryonic and early postnatal DIAm may be due in part, to the more extensive of phrenic axon branching before the elimination of polyneuronal innervation. Failure of action potential propagation at axonal branch points has been well established (Table 1) (142, 234, 236). Additionally, myelination of the motor axons develops over the first several weeks postnatally in mice, which strongly influences both intrinsic electrophysiological properties and action potential propagation (432). It is also possible that there are developmental differences in QC due to fewer synaptic vesicles in the readily releasable pool and differences in SNARE proteins that mediate fusion and exocytosis of synaptic vesicles. In the rat DIAm during the early postnatal period, the frequency and amplitude of spontaneous mEPPs are reduced and the amplitude of evoked EPPs is also reduced as compared to the adult DIAm of mice and rats (142, 304). In addition, neonates have smaller mEPP and EPP amplitudes, either due to fewer synaptic vesicles or fewer receptors (122). Both mEPP amplitude and safety factor are reduced in mouse genetic models with reduced neuregulin expression (402). Heterozygous mice with disruptions in neuregulin show a 50% reduction in AChR but also potentially compensate for fewer receptors through an increase in QC.
Table 1.
Incidence of Axonal Propagation Failure and Extent of Decrement in EPP Amplitude During Repetitive Stimulation at Different Ages (142)
| Stimulation rate (pps) |
||||||||
|---|---|---|---|---|---|---|---|---|
| 10 | 20 | 40 | 75 | 10 | 20 | 40 | 75 | |
|
|
|
|||||||
| Age | Propagation failure rate (%) | Decrement in EPP amplitude (%) | ||||||
| 1 week | 4.4±1.7a | 14.5±3.5a | 40.5±3.8a | 65.6±2.2 | 52.2±10.8 | 70.0±8.2 | 87.2±7.7a | 91.3±4.4 |
| 3 week | 0±0 | 2.0±1.0 | 16.7±4.2b | 37.5±4.8b | 63.7±9.7 | 61.2±5.2 | 78.5±3.1 | 77.4±2.8 |
| Adults | 0±0 | 2.5±0.5 | 4.0±2.4 | 26.0±5.1 | 61.3±7.8 | 62.8±6.2 | 73.2±2.7 | 75.1±1.3 |
Values are mean±SD. Propagation failure rate calculated as the proportion of absent evoked EPPs in the first 10 pulses of each train.
Significantly different from older animals (P < 0.01).
Significantly different from adults (P < 0.05).
Importantly, increased neuromuscular transmission failure occurs with motor unit expansion, whether associated with distal NMJ factors (axon terminals, synaptic cleft, or synaptic vesicles), more proximal disturbances such as branch-point failures, or both. If motor unit expansion occurs during perinatal development, NMJs per se, remain morphologically sound (130), despite marked motor neuron loss (54, 55). In this scenario, increased neuromuscular transmission failure is likely due to increased innervation ratios in the absence of denervation/reinnervation and pre- or postsynaptic NMJ defects (130, 139). Conversely, in scenarios such as age-associated sarcopenia, where motor neuron loss occurs, increased neuromuscular transmission failure (131) is likely associated with motor unit expansion (increased innervation ratio) thereby increasing branch point failures due to spared motor neurons re-innervating denervated fibers (137) and the frank degeneration of pre- or postsynaptic NMJ compartments (131, 354).
Energy requirements of neuromuscular transmission
The consumption of ATP at the muscle fiber relates to countering active and resting membrane currents -ATPase pump), cross-bridge cycling mechanics, and re-sequestering of following activation. Sadly, despite the abundance of information about -ATPase pump, and SERCA function in skeletal muscle, including the DIAm, there is a paucity of knowledge regarding the endplate of the NMJ. It is surprising that although many of the conditions that afflict the -ATPase pump, and sarcoplasmic reticulum transport ATPase (SERCA pump) function have an NMJ phenotype, the -ATPase pump, and SERCA at the NMJ have not been the focus of the investigation to date. Similar to their role in other cells, including the presynaptic component of the NMJ (see above), the maintenance of the postsynaptic membrane potential depends on ATP hydrolysis via the -ATPase pump. In skeletal muscle, a suite of -ATPase pump isoforms exist, with fatigue-resistant type I and type IIa fibers having increased expression of and isoforms and reduced expression of isoform compared with type IIx/IIb fibers (146, 202, 462). In a variety of conditions, including aging (511), muscular dystrophy (233), hypoxia, and chronic obstructive pulmonary disease (172, 291), the isoform-specific expression of the -ATPase pump is altered. Notably, the -ATPase pump does not appear to be concentrated at the NMJ endplate (10), although the fiber type dependence of this remains unexplored.
ATP hydrolysis is also necessary to re-sequester into the SR via the SERCA pump, and as SERCA facilitates relaxation its function is associated with muscle fatigue. In skeletal muscle, the predominant isoform is SERCA2A in type I fibers and SERCA1 in type II fibers (265, 306, 497). In DIAm, increased SERCA2A and/or decreased SERCA1 is correlated with improvements in fatigue resistance, while decreased SERCA2A and/or increased SERCA1 isoform expression results in decreased endurance (13, 212). Impairment of SERCA has been shown to occur in circumstances ranging from aging (50, 361, 461), hyperthyroidism (183), immobilization (398, 455), hypoxia and chronic obstructive pulmonary disease (335, 420), ALS (360), and muscular dystrophy (169, 289).
Following a single depolarizing action potential, or a single contraction, the magnitude of ATP consumption is likely to be greater in an individual type IIx/IIb fibers compared with type I and IIa fibers. However, allowing for the major differences in activation history across fiber types in the DIAm, ATP requirements for maintaining membrane potential and sequestering are much greater in type 1 and IIa fibers than in type IIx/IIb fibers. Thus, the cumulative rate of ATP consumption across a given time period (as opposed to a unitary single activation) would be much greater in I and IIa DIAm fibers.
Plasticity of NMJs Across the Age Spectrum
Across the aging spectrum, there are major changes in the structure and function of NMJs that affect neuromuscular transmission. During embryonic development, the arrival of motor axons coincides with the formation of muscle fibers from myoblasts and myotubes (myogenesis). During this embryonic and subsequent early postnatal period in mammals, muscle fibers can be influenced by more than one motor neuron (polyneuronal innervation) (Figure 13). In addition, during the perinatal period, there is a marked culling (loss) of motor neurons and elimination of synapses until the adult motor unit is established (Figure 13). On the other end of the aging spectrum, motor neurons are also lost (similar to ALS) and muscle fibers are denervated as a result. Reinnervation of denervated muscle fibers by surviving motor neurons expands motor unit innervation ratio. The mechanisms underlying NMJ plasticity during perinatal development and aging are not fully understood but have been intensely studied over the years.
Myogenesis and innervation coincide during development
In the embryonic rodent DIAm, the phrenic nerve arrives around embryonic day 11 (E11 in mice) and 13 (E13 in rats; Figure 13) (6), at a time when myogenesis is incomplete and the DIAm consists of myoblasts and myotubes. Myogenesis consists of two major phases: a determination phase, when myoblasts emerge from mesodermal progenitor cells, and a terminal differentiation phase, when myoblasts fuse to form myotubes followed by the formation of muscle fibers that express contractile proteins structurally organized in sarcomeres (173). The timing of myogenesis is influenced by the expression of muscle regulatory factors (MRFs), including myogenic differentiation factor 1 (MyoD) (90), myogenin (56), myogenic factor-5 (Myf-5) (57), and myogenic regulatory factor-4 (MRF4) (369). These MRFs are also involved in regulating MyHC isoform expression and the formation of adult muscle fiber types (200). The process of myogenesis and formation of adult fiber types is not complete until the emergence of and isoforms in type IIx/IIb fibers. In the rodent DIAm, type IIx/IIb fibers do not emerge until after the second postnatal week, and even then, type IIx/IIb fibers display differential growth into adulthood. Thus, the period between E11-E13 and postnatal day 14 (P14) represents a critical time for the organization of motor units that are essential for the diversity of muscle functions.
In myogenesis, myoblast fusion forms multinucleated myotubes and is essential for the subsequent formation of muscle fibers with expression of contractile proteins organized into sarcomeres. It is important to note that nerve outgrowth and arrival at the muscle are coincident with the critical period of contractile protein organization during myogenesis (275). However, the terminal differentiation of myoblasts and the formation of myotubes and muscle fibers are not dependent on innervation. Using rat DIAm injected with -botulinum toxin at E14, Harris concluded that primary myotubes can develop absent from any support by the nervous system; however, secondary myotube development depends on innervation (185). The transition from primary to secondary myotubes is a staggered process and innervation of the burgeoning secondary myotubes can occur through the transfer of a nerve terminal from a more densely populated primary myotube (102). Developing myofibrils must contract during the development or generation of secondary myotube ceases (185). Myotube and muscle fiber formation are primarily influenced by mechanical properties that affect protein expression in the myoblast cell membrane (e.g., a1-integrin), the cytoskeleton (e.g., actin and desmin), the basal lamina (e.g., muscle cell adhesion molecule—M-CAM), and the extracellular matrix (e.g., fibronectins, laminins, M-cadherin, and neural cell adhesion molecule—N-CAM) (228, 232). Thus, the terminal differentiation of myoblasts and their subsequent fusion to form multinucleated myotubes and muscle fibers involves complex processes.
During late embryonic and early postnatal development, the expression of MyHC isoforms in muscle fibers undergoes substantial transition with the early expression of and isoforms together with embryonic and neonatal isoforms (275). During this time, the size of motor neurons innervating muscle fibers is relatively homogeneous (132). After the second postnatal week in rodents, expression of and isoforms emerges, even in the absence of innervation (424). Singular expression of MyHC isoforms does not occur in rat DIAm fibers until the second postnatal week, and the final adult proportions of fiber types do not appear until the fourth postnatal week (157). However, after the second postnatal week in the rat DIAm, there is an approximately 2- to 3-fold growth of type IIx/IIb fibers compared with type I and IIa fibers (214, 349). With the postnatal emergence in type IIx/IIb DIAm fibers, there is a corresponding increase in specific force (force per cross-sectional area) and velocity of shortening (157). If the DIAm is denervated before the second postnatal week, the differential growth of type IIx/IIb fibers is blunted, suggesting a trophic influence of innervation. The period of postnatal growth of type IIx/IIb fibers in the rat DIAm also corresponds with an increase in size of the phrenic motor neurons (351, 356).
Polyneuronal innervation and synapse elimination during perinatal development
During embryonic and early postnatal development, each muscle fiber can be innervated by more than a single motor neuron—polyneuronal innervation (206, 320, 491) (Figure 13). Early observations regarding polyneuronal innervation were based on intracellular electrophysiological recordings of EPPs, where EPPs were complex and consisted of 2 to 4 “units” distinguishable by time and amplitude (365). Upon arrival of the phrenic nerve in the rat DIAm at E13 in rats, motor axons branch extensively but only start to form synapses after about 24 h (36, 173, 186). The reason for the short delay in branching and initial synapse formation is unclear, but it may be that cervical motor neurons are not electrically excitable and capable of generating and transmitting action potentials until E14 (256). In the DIAm, electrical stimulation of the phrenic nerve evokes an AChR-dependent postsynaptic response by E14 (182). The pattern of intramuscular axonal branching establishes a somatotopic organization in which phrenic motor neurons from higher cervical segments innervate the sternal region and more ventral aspects of the costal and crural regions (145, 249).
Emerging motor axons are accompanied by Schwann cells, which may be involved in the guidance and outgrowth of axons (123). Both Schwann cells and nerve terminals display coated vesicles suggesting that there may be the vesicular release of trophic factors (see below). Among the regulatory processes that may be involved are those associated with properties of the extracellular matrix, the release of trophic factors, and/or chemotactic substances. Insight into axonal outgrowth and branching derives from in vitro studies in rats and zebrafish, which suggest that the perineural sheath functions as a scaffold for targeting motor axons toward muscle fibers (237, 270, 463). Unfortunately, exploring these processes under in vivo conditions is difficult.
Trophic influences derived from both motor neurons (neurotrophic) and maturing muscle fibers (myotrophic) appear to be important in matching innervation with muscle fiber development (140, 141, 327, 334, 477, 498). During development, a complex interplay between programmed motor neuron cell death and the development of muscle fibers and their nascent innervations occurs in the third week of gestation (26, 309, 317, 457). Spontaneous motor unit activity has been proposed to regulate motor neuron survival and innervations in two ways: first, by the activity-dependent production of trophic factors (326), and second, by the regulation of neuromuscular synapse number, in line with Hebbian concepts (247). Experimentally, when in utero motor neuron pools have reduced activity (27, 87), excessive axonal branching ensues and an increased profligacy of nascent presynaptic terminals is observed (27, 87, 140, 141, 328, 329, 332, 333, 345). Conversely, in cases with increased motor neuron activity (27, 133, 134, 331), axonal branching and the number of NMJs are markedly restricted (27, 140, 141, 331).
In cases where neurotrophins may bind to multiple receptors, for example, BDNF binding to its high affinity TrKB receptor as well as the lower affinity p75NTR receptor, the overall signaling effect of BDNF depends on the concentration of BDNF and the relative expression of each receptor (319) (Figure 17). Notably, for BDNF, receptor-dependent responses involve the potentiation of canonical trophic effects via TrKB or the triggering of apoptosis by p75NTR (23, 32, 319, 341). There is a striking decline in the abundance of p75NTR receptors in motor neurons during embryonic development corresponding to the period of programmed cell death of motor neurons (101, 151–153). At birth and thereafter the expression of the p75NTR receptor in motor neurons is at a very low level. The developmental importance of the interplay between BDNF and p75NTR receptors is underscored by p75NTR knock-out mice, which exhibit delays in presynaptic terminal maturation and neurotransmission efficacy (207, 342). Interestingly, p75NTR receptors are expressed again in adult motor neurons as a result of injury (axotomy) and diseases afflicting motor neuron axons, such as ALS (215, 413). It remains to be determined if the adult expression of p75NTR contributes to the phenotype of developmental disorders of motor units (e.g., early-onset spasticity), other scenarios where motor neuron denervation occurs (e.g., DIAm sarcopenia), or if there is a motor unit type-dependence to p75NTR expression during pathological conditions.
Figure 17.
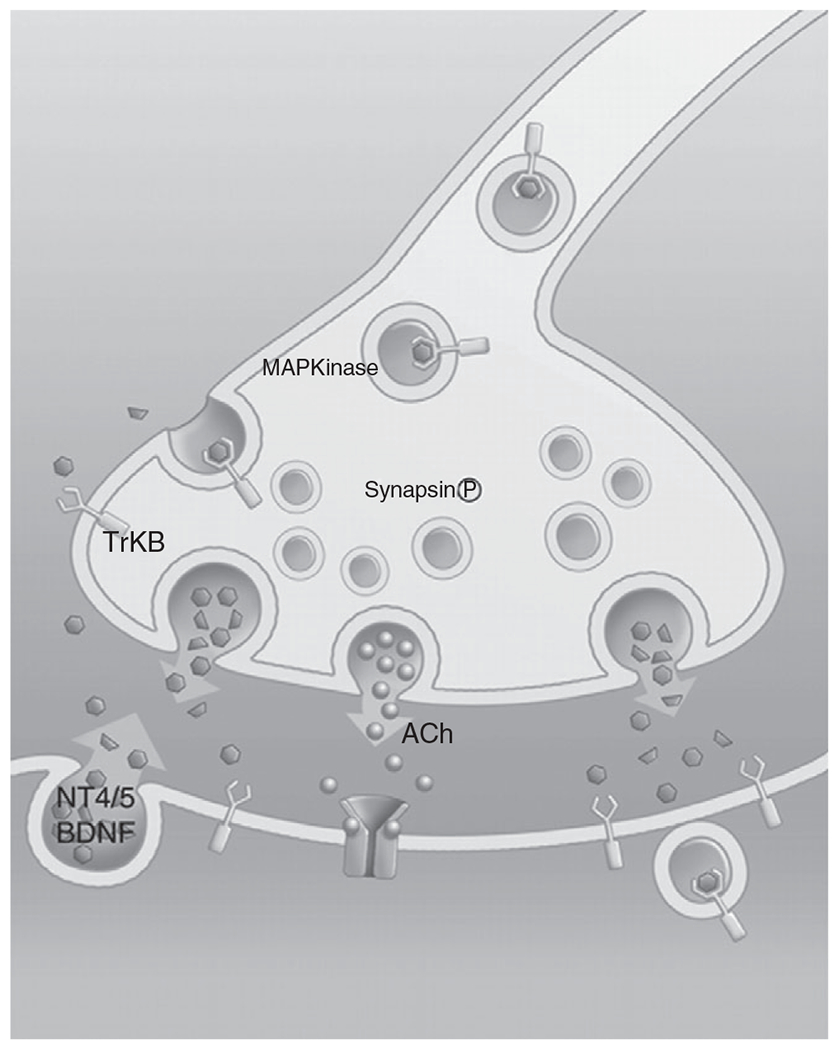
Neurotrophin 4 (NT-4) and brain-derived neurotrophic factor (BDNF) are predominantly produced and released by motor neurons, although there is some evidence suggesting a myogenic pathway for these neurotrophins. Both NT-4 and BDNF bind to a high affinity tropomyosin kinase B (TrkB) receptor located both pre- and postsynaptically. Some evidence suggests that TrkB and BDNF in the bound state are endocytosed and transported retrogradely via the axon to provide signaling to the soma. Republished, with permission, from Mantilla CB and Sieck GC, 2003 (274).
Initially, each myotube/myofiber can be contacted by multiple motor neurons (polyneuronal innervation). Subsequently, polyneuronal innervation disappears through the process of motor nerve terminal withdrawal and pruning of axonal branches, rather than complete degeneration of motor neurons, which in the mouse and rat DIAm is completed by about the second postnatal week (35, 36, 61, 344, 403). The rate of synapse elimination is initially rapid but slows until only one remains (459). The process of synapse elimination is not fully understood, but it has been suggested that competition among motor neurons for target cell innervation depends on activity and/or the fidelity of neuromuscular transmission (Hebbian competition) (29, 38, 39, 41, 63, 64, 120, 121, 206, 344, 365, 385). Accordingly, nerve terminals of more active motor neurons might persist, while those of less active motor neurons are eliminated. Synapse elimination is affected by imposed increases or decreases in neuromuscular transmission such that reducing activity also reduces the rate of synapse elimination and patterned high-frequency stimulation accelerates synapse elimination (321, 460, 464). However, the simple theory of activation does not support the fact that in adults, motor units with the largest innervation ratio (i.e., number of muscle fibers innervated by a single motor neuron) are often those that are least active (e.g., type IIx/IIb fibers comprising fast-fatigable motor units) (48, 144, 423). It is likely that the activity patterns of motor neurons change markedly during postnatal development with the growth of motor neurons and changes in intrinsic electrophysiological properties dictating excitability. For example, myelination of motor neurons, which has profound effects on electrophysiological properties, is not complete until the second to third postnatal week in mice (432). The most dramatic period of motor neuron growth occurs after synapse elimination is complete (i.e., after the second postnatal week in rats) (349). Another possibility is that the preservation of a presynaptic terminal depends on the fidelity of neuromuscular transmission. It has been shown that the incidence of neuromuscular transmission failure is much higher in the neonatal rat DIAm as compared with the adult (142). Therefore, presynaptic terminals from those phrenic motor neurons that fail to elicit a postsynaptic response are eliminated consistent with Hebbian competition. However, what underlies the precise matching of motor neurons and their presynaptic terminals with muscle fiber types is still unknown. The pattern and fidelity of activation may affect the secretion of synaptotrophins or synaptotoxins that contribute to the maintenance or elimination of synapses (403). Another possibility is that muscle fibers use intracellular signals called synaptomedins to maintain contact with effective nerve terminals, which may or may not be dependent on activity (21). At present, none of these possibilities can be excluded, and it is likely that a combination of mechanisms is responsible for the final pattern of motor unit innervation. However, since the properties of motor neurons and muscle fibers are closely linked, it is likely that the mechanisms underlying synapse elimination are also linked to muscle fiber lineage. Ultimately, the motor unit composition may also be affected by ongoing postnatal myogenesis and the formation of new myotubes and muscle fibers, which occurs until the third postnatal week in the rat DIAm (349). Thus, the final innervation ratio of motor units is not established in the rat DIAm until about postnatal day 28.
In rats, respiratory movements have been observed at E17 (174), which indicates that functional synapses and neuromuscular transmission are present at least within a few days after initial innervation. In the embryonic DIAm, we observed that transient cytosolic and contractile responses were evoked by electrical stimulation of the phrenic nerve as early as E12.5 in mice and E14 in rats. Thus, neuromuscular synapses are functional soon after the arrival of the presynaptic terminal. Importantly, the cytosolic and contractile responses were blocked by d-tubocurarine and -bungarotoxin indicating dependence on ACh release and activation of AChRs. Yet, in the embryonic rat DIAm just after initial innervation, there is no apparent postsynaptic endplate with limited clustering of AChRs until E17. The weak evoked contractile response in DIAm myotubes/myofibers is consistent with the emergence of sarcomeric organization of contractile proteins that are necessary for a mechanical response. Thus, phrenic nerve activity can elicit a mechanical response even before well-defined endplates are present. These primitive mechanical responses may be important in the maturation of myotubes into DIAm fibers (i.e., sarcomeric organization of contractile proteins and their alignment in myofibers).
Motor neuron loss in old age—Denervation and reinnervation
Neural plasticity refers to the ability of the nervous system to adapt to new and repeated experiences. While work in synaptic plasticity began in the field of psychology with Donald Hebb in the 1940s, his observations that repeated stimulation of a presynaptic neuron could facilitate the response in the postsynaptic neuron have had profound impact within the realm of neurophysiology (478). Plasticity is important during development as it reinforces and maintains useful synapses, ensuring a parsimonious neural network. During adulthood, plasticity allows the NMJ to adapt to environmental changes and stressors. Remodeling of the NMJ in response to degeneration, whether due to injury, disease, or aging, can restore or maintain neuromuscular function.
Aging is associated with a loss of motor neurons resulting in the denervation of muscle fibers (137, 467). We have found that age-related loss of phrenic motor neurons was restricted to larger motor neurons that innervate type IIx/IIb DIAm fibers in rats (137). The loss of motor neurons mandates NMJ reinnervation of surviving muscle fibers (131, 354). With reinnervation, there is increased axonal branching and de novo regeneration of presynaptic terminals at these fibers. There is greater fragmentation of presynaptic terminals associated with reinnervation in both rodent and human studies (84, 243, 322, 354, 379, 485), which is associated with spatial uncoupling between the active zone and receptor location across the synaptic cleft (131, 253, 354, 378). There have been similar reports of an age-related increase in complexity of nerve terminal arborization at type IIx/IIb fibers (353, 354). The age-related increase in the complexity of presynaptic nerve terminals results in an increase in presynaptic terminal surface area with aging, which may not improve neuromuscular transmission (354). Unfortunately, age-related changes in the ultrastructure of presynaptic terminals have not been fully characterized. However, the increased fragmentation of the NMJ in DIAm of older mice was not associated with any change in QC but was associated with a greater endplate current amplitude after repeated stimulation at ≤5 Hz (488). Similarly, the number of active zones is decreased with aging in presynaptic terminals of motor neurons innervating the sternomastoid muscle in mice (74). It is unknown whether there are age-related changes in SNARE protein mediated vesicle release and/or synaptic vesicle recycling. Such changes may compromise QC and the fidelity of neuromuscular transmission in old age. Indeed, increased DIAm neuromuscular transmission failure in older animals has been reported (131, 175).
Synaptic plasticity is modulated through neurotrophic signaling pathways. Neurotrophins are released by both neurons (neurogenic) and muscle fibers (myogenic; Figure 13) (509). Two of the most researched neurotrophins are BDNF and NT-4. Both BDNF and NT-4 enhance neuromuscular transmission during repetitive stimulations (278). Evidence suggests that both BDNF and NT-4 act through the high-affinity TrkB receptor. Indeed, inhibition of TrkB kinase reverses the facilitation of neuromuscular transmission in the presence of BDNF and NT-4. In aging, inhibition of the TrkB receptor leads to reduced overlap of presynaptic terminals and endplate regions and increases the proportion of denervated NMJs by approximately 20% (177).
Glial-derived neurotrophic factor (GDNF) also has profound effects on motor neurons. GDNF knockout mice exhibit 20% to 30% fewer motor neurons as well as altered axonal branching patterns (181). In contrast, overexpression can lead to increased motor neuron survival during development and extensive axonal sprouting patterns in mice (315, 330). Finally, neural growth factor (NGF) can augment evoked potentials, depolarize postsynaptic resting membrane potential, and limit the effects of AChR blockers.
Evidence of myotrophic signaling is provided by studies using nerve-muscle co-cultures from cloned cells and frog NMJs. Research with nerve-muscle co-cultures observed clustering of synaptic vesicles and synaptic vesicle-associated antigens closest to muscle tissue but not when the neurites were grown near other types of tissue suggesting a muscle-specific effect (264). There is also evidence that when motor neurons reinnervate muscle fibers, they do so at preexisting junctional folds as observed in the cutaneous pectoris of frogs (405), which provides muscle-derived signaling at the endplate. Myotrophic substances such as MyoD, myogenin, and MRF-4 have been implicated in the determination of fiber type (274). However, both MyoD and myogenin are also affected by innervation, thus blurring the distinction between myotrophic and neurotrophic influences (201).
Aggregation of AChRs at postsynaptic endplates during development
The progression in expression and aggregation of AChRs is concurrent with the outgrowth of motor axons and presynaptic contact with myotubes/muscle fibers, around E16 in mice (288). In rat DIAm, mEPPs and EPPs are measured as early as E14 (95), providing evidence for functional synaptic transmission at the NMJ. In the mouse DIAm, aggregation and clustering of AChRs are apparent at approximately E13. Results from studies on chick embryos have found that AChR aggregation occurs within a few hours of contact with motor axon terminals (147). Postnatally, the density and clustering of AChRs at endplates change from a plaque-to-pretzel morphology in mammals (281). The process of aggregation of AChRs has received considerable attention and critically involves agrin secretion by the nerve terminal (Figure 16). Agrin is a nerve-derived glycoprotein that binds to MuSK receptors on the muscle fiber membrane and activates a rapsyn-mediated clustering of AChR (58, 241, 275, 314, 404). Activation through intracellular pathways allows a single agrin molecule to cluster many AChRs (290). MuSK phosphorylation appears to be critical for AChR clustering, as clustering is absent in MuSK double mutant mice and in mice treated with an siRNA designed to block MuSK transcription (229). The expression of MuSK receptors varies along the length of muscle fibers, being most abundant in their central regions where MuSK can undergo autoactivation (267), leading to AChR clustering in the absence of neural influence. In neighboring sections of the muscle fiber, where MuSK is not as richly expressed, agrin released by motor axons can also lead to AChR clustering (260). Importantly, nerve-derived agrin release by presynaptic terminals and MuSK expression in muscle fibers appear to coordinate the formation of pre- and postsynaptic components of the NMJ and account for the apparent “targeting” of motor axons to “preformed” AChR clusters (481, 502), as well as the induction of new clusters by nerve-derived influences (211). Agrin is important not only for the development of the endplate but also for the development of mechanisms underlying excitation-contraction coupling in myotubes including the expression of ryanodine receptors and voltage-gated L-type channels (24, 456).
Another important neurotrophic influence is that of neuregulin, which is a member of the larger epidermal growth factor family (116, 402, 472, 512). Neuregulin mediates its influence through ErbB receptors to activate tyrosine kinases in maturing muscle fibers (251). Neuregulin is both a nerve-derived and muscle-derived trophic influence for myotube-to-muscle fiber differentiation during embryonic and early postnatal development, but the interactive roles of different trophic factor signaling pathways (e.g., agrin vs. neuregulin) in the emerging NMJ remains to be determined.
Following AChR aggregation, synaptic folds from and complex endplate structures differentiate with characteristics of adult NMJs in the rat DIAm. At birth, AChRs appear in irregular patches at an endplate that more closely resembles an oval or “spoon” (281). Within a few days, the endplate begins to form gutters (junctional folds), and AChRs not located at the peaks of junctional folds are degraded. Previously, we demonstrated that in the adult rat DIAm, endplate morphology varies across different fiber types, being far more complex in type IIx/IIb fibers compared with type I and IIa fibers (271, 352). This is consistent with work in other mammalian muscles (323, 443). By contrast, postsynaptic motor endplates in the fetal and neonatal DIAm are much smaller and far less complex compared with adults. The progression of endplate growth with development tightly matches muscle fiber growth (20). There is very little information regarding the mechanisms responsible for the development of fiber type-specific differences in endplate morphology (276). Importantly, type IIx/IIb fibers in the rodent DIAm do not emerge until after the second postnatal week (277). Furthermore, type IIx/IIb DIAm fibers display faster postnatal growth compared with the other fiber types beginning at the third postnatal week into adulthood (277). With an increase in motor endplate complexity, there is an increase in surface area with a greater number of AChRs to elicit an endplate potential. It is likely that the mechanisms underlying the growth of type IIx/IIb fibers and the increasing complexity of motor endplates are linked.
Changes related to aging in postsynaptic endplates
At the other end of the lifespan, there are also significant changes at the endplate that predominantly affect type IIx/IIb fibers (354). It is possible that trophic influences that affect normal remodeling are altered with aging (131, 354). It should be pointed out that an increase in the size of endplates does not necessarily correlate with improved synaptic efficacy. A possible reflection of the instability of NMJ remodeling may be the increased expression of neural cell adhesion molecule (NCAM) that occurs with aging (439). NCAM is involved in pre- and postsynaptic communication at the NMJ (76). Curiously, despite the increased prevalence with age, the NCAM response after denervation is suppressed with old age, and therefore, researchers have suggested that an impaired ability to appropriately modulate NCAM may be responsible for failed reinnervation potential (not all muscle fibers are successfully reinnervated) (165). In addition, there is an age-related gradual decrease in the number of AChRs at the motor endplate and increased extra-junctional receptors in the sternomastoid and DIAm of aged rats (84, 436, 437). In addition, the endplate increases in size and complexity on type IIx/IIb fibers but not on type I or IIa fibers in aged rats and mice (354, 388). Endplate growth is also paired with AChR disaggregation (205). Additionally, the voltage-gated sodium channels exhibit slower activation and inactivation kinetics and there is an increase in extrajunctional current (96). Disordering of the laminar structures of the NMJ also impairs neuromuscular transmission of the DIAm, which is exacerbated in aging (253). The effects of motor unit type on disaggregation and changes in voltage-gated channel physiology have not been explored.
Conclusion
The NMJ is the sole interface between the motor control centers (the motor neuron pools and their associated presynaptic circuits) and skeletal muscle fibers. As such, the NMJ is a potential Achilles’ heel for the uninterrupted and orderly activation of motor units. Many of the redundancies of the NMJ structure and function attempt to lessen the likelihood of signal transduction failure (see the sections on the readily releasable pool, vesicular recycling, and the safety factor). When these mechanisms fail, or if the motor neuron innervating muscle fibers dies and reinnervation and remodeling are impaired or incomplete, a suite of NMJ dysfunctions may occur. Often, these pathophysiological changes are specific to a subset of motor unit types, with selective vulnerability of FInt and FF motor units, as evidenced in aging and ALS. By contrast, myasthenia gravis is not selective, with pathology across multiple motor unit types.
Throughout this article, we have highlighted the plasticity and remodeling relation between the presynaptic and postsynaptic elements of motor units as an organizing principle of the NMJ. These relations extend beyond structural maintenance and include the coordination of neuro- and myotrophic factors, the maintenance of vesicular availability to demand for release, and the balance of ATP availability to consumption. Key advances in the field will likely be related to the motor unit type-specifics of these phenomena, the importance of which has recently been unveiled in concert with disease-associated studies (e.g., Schwann cells and the tripartite synapse in ALS). Such gains in understanding may also serve as a template for efforts focused on the central synapses within the CNS.
We conclude that with the ever-increasing array of experimental tools at our disposal, the molecular underpinnings of state-dependent NMJ functions (e.g., the paradoxical effects of BNDF and NMJ autophagy via the p75NTR receptor) will be unveiled. These discoveries will hopefully usher in a new era of NMJ therapeutics for a variety of developmental conditions and degenerative diseases.
Didactic Synopsis.
Major teaching points
The information contained in this article will help with teaching NMJ physiology using different motor unit/muscle fiber types as an organizing principle.
A detailed section on motor unit physiology provides insight into the unique mechanical and fatigue properties displayed by different muscle fiber types and how they are activated via neuromuscular transmission.
The NMJ comprises the presynaptic axon terminal, the postsynaptic receptor region at the muscle fiber, and the perisynaptic (terminal) Schwann cell that provides support and is essential for maintenance of the NMJ.
References
- 1.Ackermann F, Waites CL, Garner CC. Presynaptic active zones in invertebrates and vertebrates. EMBO Rep 16: 923–938, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adelman WJ, Palti Y, Senft JP. Potassium ion accumulation in a periaxonal space and its effect on the measurement of membrane potassium ion conductance. J Membr Biol 13: 387–410, 1973. [DOI] [PubMed] [Google Scholar]
- 3.Albuquerque EX, Barnard EA, Porter CW, Warnick JE. The density of acetylcholine receptors and their sensitivity in the postsynaptic membrane of muscle endplates. Proc Natl Acad Sci U S A 71: 2818–2822, 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aldrich TK, Shander A, Chaudhry I, Nagashima H. Fatigue of isolated rat diaphragm: Role of impaired neuromuscular transmission. J Appl Physiol 61 (3): 1077–1083, 1986. [DOI] [PubMed] [Google Scholar]
- 5.Alhindi A, Boehm I, Chaytow H. Small junction, big problems: Neuromuscular junction pathology in mouse models of amyotrophic lateral sclerosis (ALS). J Anat, 2021. DOI: 10.1111/joa.13463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allan DW, Greer JJ. Embryogenesis of the phrenic nerve and diaphragm in the fetal rat. J Comp Neurol 382: 459–468, 1997. [DOI] [PubMed] [Google Scholar]
- 7.Allodi I, Comley L, Nichterwitz S, Nizzardo M, Simone C, Benitez JA, Cao M, Corti S, Hedlund E. Differential neuronal vulnerability identifies IGF-2 as a protective factor in ALS. Sci Rep 6: 25960, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alshekhlee A, Miles JD, Katirji B, Preston DC, Kaminski HJ. Incidence and mortality rates of myasthenia gravis and myasthenic crisis in US hospitals. Neurology 72: 1548–1554, 2009. [DOI] [PubMed] [Google Scholar]
- 9.Arbour D, Tremblay E, Martineau E, Julien JP, Robitaille R. Early and persistent abnormal decoding by glial cells at the neuromuscular junction in an ALS model. J Neurosci 35: 688–706, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ariyasu RG, Deerinck TJ, Levinson SR, Ellisman MH. Distribution of (Na+ + K+)ATPase and sodium channels in skeletal muscle and electroplax. J Neurocytol 16: 511–522, 1987. [DOI] [PubMed] [Google Scholar]
- 11.Arthur-Farraj PJ, Latouche M, Wilton DK, Quintes S, Chabrol E, Banerjee A, Woodhoo A, Jenkins B, Rahman M, Turmaine M, Wicher GK, Mitter R, Greensmith L, Behrens A, Raivich G, Mirsky R, Jessen KR. c-Jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron 75: 633–647, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Asbury AK. Schwann cell proliferation in developing mouse sciatic nerve. A radioautographic study. J Cell Biol 34: 735–743, 1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aubier M, Viires N. Calcium ATPase and respiratory muscle function. Eur Respir J 11: 758–766, 1998. [PubMed] [Google Scholar]
- 14.Auerbach A, Akk G. Desensitization of mouse nicotinic acetylcholine receptor channels. A two-gate mechanism. J Gen Physiol 112: 181–197, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Auld DS, Robitaille R. Perisynaptic Schwann cells at the neuromuscular junction: Nerve- and activity-dependent contributions to synaptic efficacy, plasticity, and reinnervation. Neuroscientist 9: 144–157, 2003. [DOI] [PubMed] [Google Scholar]
- 16.Azarias G, Kruusmagi M, Connor S, Akkuratov EE, Liu XL, Lyons D, Brismar H, Broberger C, Aperia A. A specific and essential role for Na, K-ATPase alpha3 in neurons co-expressing alpha1 and alpha3. J Biol Chem 288: 2734–2743, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bacskai T, Fu Y, Sengul G, Rusznak Z, Paxinos G, Watson C. Musculotopic organization of the motor neurons supplying forelimb and shoulder girdle muscles in the mouse. Brain Struct Funct 218: 221–238, 2013. [DOI] [PubMed] [Google Scholar]
- 18.Bacskai T, Rusznak Z, Paxinos G, Watson C. Musculotopic organization of the motor neurons supplying the mouse hindlimb muscles: A quantitative study using Fluoro-Gold retrograde tracing. Brain Struct Funct 219: 303–321, 2014. [DOI] [PubMed] [Google Scholar]
- 19.Balaji J, Ryan TA. Single-vesicle imaging reveals that synaptic vesicle exocytosis and endocytosis are coupled by a single stochastic mode. Proc Natl Acad Sci U S A 104: 20576–20581, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balice-Gordon RJ, Lichtman JW. In vivo visualization of the growth of pre- and postsynaptic elements of neuromuscular junctions in the mouse. J Neurosci 10: 894–908, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balice-Gordon RJ, Lichtman JW. In vivo observations of pre- and postsynaptic changes during the transition from multiple to single innervation at developing neuromuscular junctions. J Neurosci 13: 834–855, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baloh RH, Strickland A, Ryu E, Le N, Fahrner T, Yang M, Nagarajan R, Milbrandt J. Congenital hypomyelinating neuropathy with lethal conduction failure in mice carrying the Egr2 I268N mutation. J Neurosci 29: 2312–2321, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R, Kohn J, Causing CG, Miller FD. The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol 140: 911–923, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bandi E, Jevsek M, Mars T, Jurdana M, Formaggio E, Sciancalepore M, Fumagalli G, Grubic Z, Ruzzier F, Lorenzon P. Neural agrin controls maturation of the excitation-contraction coupling mechanism in human myotubes developing in vitro. Am J Physiol Cell Physiol 294: C66–C73, 2008. [DOI] [PubMed] [Google Scholar]
- 25.Banker BQ, Kelly SS, Robbins N. Neuromuscular transmission and correlative morphology in young and old mice. J Physiol 339: 355–377, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Banks GB, Choy PT, Lavidis NA, Noakes PG. Neuromuscular synapses mediate motor axon branching and motoneuron survival during the embryonic period of programmed cell death. Dev Biol 257: 71–84, 2003. [DOI] [PubMed] [Google Scholar]
- 27.Banks GB, Kanjhan R, Wiese S, Kneussel M, Wong LM, O’Sullivan G, Sendtner M, Bellingham MC, Betz H, Noakes PG. Glycinergic and GABAergic synaptic activity differentially regulate motoneuron survival and skeletal muscle innervation. [Erratum appears in J Neurosci. 2005 Mar 16; 25 (11): 3018-21]. J Neurosci 25: 1249–1259, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baraka A. Nerve and muscle stimulation of the rat isolated phrenic nerve- diaphragm preparation. Anesth Analg 53: 594–596, 1974. [PubMed] [Google Scholar]
- 29.Barber MJ, Lichtman JW. Activity-driven synapse elimination leads paradoxically to domination by inactive neurons. J Neurosci 19: 9975–9985, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barrett EF, Barrett JN, David G. Mitochondria in motor nerve terminals: Function in health and in mutant superoxide dismutase 1 mouse models of familial ALS. J Bioenerg Biomembr 43: 581–586, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barrett EF, Barrett JN, David G. Dysfunctional mitochondrial Ca(2+) handling in mutant SOD1 mouse models of fALS: Integration of findings from motor neuron somata and motor terminals. Front Cell Neurosci 8: 184, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barrett GL. The p75 neurotrophin receptor and neuronal apoptosis. Prog Neurobiol 61: 205–229, 2000. [DOI] [PubMed] [Google Scholar]
- 33.Bazzy AR, Donnelly DF. Failure to generate action potentials in new-born diaphragms following nerve stimulation. Brain Res 600: 349–352, 1993. [DOI] [PubMed] [Google Scholar]
- 34.Bennett MK, Calakos N, Scheller RH. Syntaxin: A synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science 257: 255–259, 1992. [DOI] [PubMed] [Google Scholar]
- 35.Bennett MR, Lavidis NA. Segmental motor projections to rat muscles during the loss of polyneuronal innervation. Dev Brain Res 13: 1–7, 1984. [DOI] [PubMed] [Google Scholar]
- 36.Bennett MR, Pettigrew AG. The formation of synapses in striated muscle during development. J Physiol 241: 515–545, 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Betz WJ, Bewick GS, Ridge RM. Intracellular movements of fluorescently labeled synaptic vesicles in frog motor nerve terminals during nerve stimulation. Neuron 9: 805–813, 1992. [DOI] [PubMed] [Google Scholar]
- 38.Betz WJ, Caldwell JH, Ribchester RR. The size of motor units during post-natal development of rat lumbrical muscle. J Physiol 297: 463–478, 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Betz WJ, Caldwell JH, Ribchester RR. The effects of partial denervation at birth on the development of muscle fibres and motor units in rat lumbrical muscle. J Physiol 303: 265–279, 1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Betz WJ, Mao F, Bewick GS. Activity-dependent fluorescent staining and destaining of living vertebrate motor nerve terminals. J Neurosci 12: 363–375, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Betz WJ, Ribchester RR, Ridge RM. Competitive mechanisms underlying synapse elimination in the lumbrical muscle of the rat. J Neurobiol 21: 1–17, 1990. [DOI] [PubMed] [Google Scholar]
- 42.Bishop DL, Misgeld T, Walsh MK, Gan WB, Lichtman JW. Axon branch removal at developing synapses by axosome shedding. Neuron 44: 651–661, 2004. [DOI] [PubMed] [Google Scholar]
- 43.Bjornskov EK, Norris FH Jr, Mower-Kuby J. Quantitative axon terminal and end-plate morphology in amyotrophic lateral sclerosis. Arch Neurol 41: 527–530, 1984. [DOI] [PubMed] [Google Scholar]
- 44.Blakely RD, Edwards RH. Vesicular and plasma membrane transporters for neurotransmitters. Cold Spring Harb Perspect Biol 4: a005595, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blanco CE, Zhan WZ, Fang YH, Sieck GC. Exogenous testosterone treatment decreases diaphragm neuromuscular transmission failure in male rats. J Appl Physiol 90: 850–856, 2001. [DOI] [PubMed] [Google Scholar]
- 46.Blanco G, Mercer RW. Isozymes of the Na-K-ATPase: Heterogeneity in structure, diversity in function. Am J Phys 275: F633–F650, 1998. [DOI] [PubMed] [Google Scholar]
- 47.Bockmann RA, Grubmuller H. Multistep binding of divalent cations to phospholipid bilayers: A molecular dynamics study. Angew Chem Int Ed Engl 43: 1021–1024, 2004. [DOI] [PubMed] [Google Scholar]
- 48.Bodine SC, Roy RR, Eldred E, Edgerton VR. Maximal force as a function of anatomical features of motor units in the cat tibialis anterior. J Neurophysiol 57 (6): 1730–1745, 1987. [DOI] [PubMed] [Google Scholar]
- 49.Boehm I, Alhindi A, Leite AS, Logie C, Gibbs A, Murray O, Farrukh R, Pirie R, Proudfoot C, Clutton R, Wishart TM, Jones RA, Gillingwater TH. Comparative anatomy of the mammalian neuromuscular junction. J Anat 237: 827–836, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boncompagni S, Protasi F, Franzini-Armstrong C. Sequential stages in the age-dependent gradual formation and accumulation of tubular aggregates in fast twitch muscle fibers: SERCA and calsequestrin involvement. Age (Dordr) 34: 27–41, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bottger P, Tracz Z, Heuck A, Nissen P, Romero-Ramos M, Lykke-Hartmann K. Distribution of Na/K-ATPase alpha 3 isoform, a sodium-potassium P-type pump associated with rapid-onset of dystonia parkinsonism (RDP) in the adult mouse brain. J Comp Neurol 519: 376–404, 2011. [DOI] [PubMed] [Google Scholar]
- 52.Bourque MJ, Robitaille R. Endogenous peptidergic modulation of perisynaptic Schwann cells at the frog neuromuscular junction. J Physiol 512 (Pt 1): 197–209, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boyd IA, Martin AR. Spontaneous subthreshold activity at mammalian neural muscular junctions. J Physiol 132: 61–73, 1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brandenburg JE, Fogarty MJ, Brown AD, Sieck GC. Phrenic motor neuron loss in an animal model of early onset hypertonia. J Neurophysiol 123: 1682–1690, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brandenburg JE, Gransee HM, Fogarty MJ, Sieck GC. Differences in lumbar motor neuron pruning in an animal model of early onset spasticity. J Neurophysiol 120: 601–609, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Braun T, Bober E, Buschhausen-Denker G, Kohtz S, Grzeschik KH, Arnold HH, Kotz S. Differential expression of myogenic determination genes in muscle cells: Possible autoactivation by the Myf gene products. EMBO J 8: 3617–3625, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Braun T, Buschhausen-Denker G, Bober E, Tannich E, Arnold HH. A novel human muscle factor related to but distinct from MyoD1 induces myogenic conversion in 10T1/2 fibroblasts. EMBO J 8: 701–709, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brockhausen J, Cole RN, Gervasio OL, Ngo ST, Noakes PG, Phillips WD. Neural agrin increases postsynaptic ACh receptor packing by elevating rapsyn protein at the mouse neuromuscular synapse. Dev Neurobiol 68: 1153–1169, 2008. [DOI] [PubMed] [Google Scholar]
- 59.Brody IA, Engel WK. Denervation of muscle in myasthenia gravis. Report of a patient with myasthenia gravis for 47 years and histochemical signs of denervation. Arch Neurol 11: 350–354, 1964. [DOI] [PubMed] [Google Scholar]
- 60.Brose N, Petrenko AG, Sudhof TC, Jahn R. Synaptotagmin: A calcium sensor on the synaptic vesicle surface. Science 256: 1021–1025, 1992. [DOI] [PubMed] [Google Scholar]
- 61.Brown MC, Jansen JKS, Van Essen D. Polyneuronal innervation of skeletal muscle in new-born rats and its elimination during maturation. J Physiol 261: 387–422, 1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bruneteau G, Bauche S, Gonzalez de Aguilar JL, Brochier G, Mandjee N, Tanguy ML, Hussain G, Behin A, Khiami F, Sariali E, Hell-Remy C, Salachas F, Pradat PF, Lacomblez L, Nicole S, Fontaine B, Fardeau M, Loeffler JP, Meininger V, Fournier E, Koenig J, Hantai D. Endplate denervation correlates with Nogo-A muscle expression in amyotrophic lateral sclerosis patients. Ann Clin Transl Neurol 2: 362–372, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Buffelli M, Busetto G, Cangiano L, Cangiano A. Perinatal switch from synchronous to asynchronous activity of motoneurons: Link with synapse elimination. Proc Natl Acad Sci U S A 99: 13200–13205, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Buffelli M, Tognana E, Cangiano A, Busetto G. Activity-dependent vs. neurotrophic modulation of acetylcholine receptor expression: Evidence from rat soleus and extensor digitorum longus muscles confirms the exclusive role of activity. Eur J Neurosci 47: 1474–1481, 2018. [DOI] [PubMed] [Google Scholar]
- 65.Burke RE, Levine DN, Tsairis P, Zajac FE 3rd. Physiological types and histochemical profiles in motor units of the cat gastrocnemius. J Physiol 234: 723–748, 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Burres SA, Crayton JW, Gomez CM, Richman DP. Myasthenia induced by monoclonal anti-acetylcholine receptor antibodies: Clinical and electrophysiological aspects. Ann Neurol 9: 563–568, 1981. [DOI] [PubMed] [Google Scholar]
- 67.Butler JE, McKenzie DK, Gandevia SC. Discharge properties and recruitment of human diaphragmatic motor units during voluntary inspiratory tasks. J Physiol 518 (Pt 3): 907–920, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Caldwell JH. Clustering of sodium channels at the neuromuscular junction. Microsc Res Tech 49: 84–89, 2000. [DOI] [PubMed] [Google Scholar]
- 69.Cantor S, Zhang W, Delestree N, Remedio L, Mentis GZ, Burden SJ. Preserving neuromuscular synapses in ALS by stimulating MuSK with a therapeutic agonist antibody. elife 7: e34375, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cappello V, Vezzoli E, Righi M, Fossati M, Mariotti R, Crespi A, Patruno M, Bentivoglio M, Pietrini G, Francolini M. Analysis of neuromuscular junctions and effects of anabolic steroid administration in the SOD1G93A mouse model of ALS. Mol Cell Neurosci 51: 12–21, 2012. [DOI] [PubMed] [Google Scholar]
- 71.Ceci ML, Mardones-Krsulovic C, Sanchez M, Valdivia LE, Allende ML. Axon-Schwann cell interactions during peripheral nerve regeneration in zebrafish larvae. Neural Dev 9: 22, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chand KK, Lee KM, Lee JD, Qiu H, Willis EF, Lavidis NA, Hilliard MA, Noakes PG. Defects in synaptic transmission at the neuromuscular junction precede motor deficits in a TDP-43(Q331K) transgenic mouse model of amyotrophic lateral sclerosis. FASEB J 32: 2676–2689, 2018. [DOI] [PubMed] [Google Scholar]
- 73.Changeux JP, Kasai M, Lee CY. Use of a snake venom toxin to characterize the cholinergic receptor protein. Proc Natl Acad Sci U S A 67: 1241–1247, 1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen J, Mizushige T, Nishimune H. Active zone density is conserved during synaptic growth but impaired in aged mice. J Comp Neurol 520: 434–452, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cheng AJ, Allodi I, Chaillou T, Schlittler M, Ivarsson N, Lanner JT, Thams S, Hedlund E, Andersson DC. Intact single muscle fibres from SOD1(G93A) amyotrophic lateral sclerosis mice display preserved specific force, fatigue resistance and training-like adaptations. J Physiol 597: 3133–3146, 2019. [DOI] [PubMed] [Google Scholar]
- 76.Chipman PH, Schachner M, Rafuse VF. Presynaptic NCAM is required for motor neurons to functionally expand their peripheral field of innervation in partially denervated muscles. J Neurosci 34: 10497–10510, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Clark JA, Southam KA, Blizzard CA, King AE, Dickson TC. Axonal degeneration, distal collateral branching and neuromuscular junction architecture alterations occur prior to symptom onset in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. J Chem Neuroanat 76: 35–47, 2016. [DOI] [PubMed] [Google Scholar]
- 78.Clemence A, Mirsky R, Jessen KR. Non-myelin-forming Schwann cells proliferate rapidly during Wallerian degeneration in the rat sciatic nerve. J Neurocytol 18: 185–192, 1989. [DOI] [PubMed] [Google Scholar]
- 79.Coers C, Telerman-Toppet N. Morphological and histochemical changes of motor units in myasthenia. Ann N Y Acad Sci 274: 6–19, 1976. [DOI] [PubMed] [Google Scholar]
- 80.Coers C, Telerman-Toppet N, Gerard JM, Szliwowski H, Bethlem J, van Wijngaarden GK. Changes in motor innervation and histochemical pattern of muscle fibers in some congenital myopathies. Neurology 26: 1046–1053, 1976. [DOI] [PubMed] [Google Scholar]
- 81.Cohen I, Kita H, Van Der Kloot W. The Intervals Between Miniature End-plate Potentials in the Frog are Unlikely to be independently or exponentially distributed. J Physiol 236: 327–339, 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cole RN, Ghazanfari N, Ngo ST, Gervasio OL, Reddel SW, Phillips WD. Patient autoantibodies deplete postsynaptic muscle-specific kinase leading to disassembly of the ACh receptor scaffold and myasthenia gravis in mice. J Physiol 588: 3217–3229, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cole RN, Reddel SW, Gervasio OL, Phillips WD. Anti-MuSK patient antibodies disrupt the mouse neuromuscular junction. Ann Neurol 63: 782–789, 2008. [DOI] [PubMed] [Google Scholar]
- 84.Courtney J, Steinbach JH. Age changes in neuromucular junction morphology and acetylcholine receptor distribution on rat skeletal muscle fibres. J Physiol 320: 435–447, 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Crambert G, Hasler U, Beggah AT, Yu C, Modyanov NN, Horisberger JD, Lelievre L, Geering K. Transport and pharmacological properties of nine different human Na, K-ATPase isozymes. J Biol Chem 275: 1976–1986, 2000. [DOI] [PubMed] [Google Scholar]
- 86.Cull-Candy SG, Miledi R, Trautmann A, Uchitel OD. On the release of transmitter at normal, myasthenia gravis and myasthenic syndrome affected human end-plates. J Physiol 299: 621–638, 1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dahm LM, Landmesser LT. The regulation of synaptogenesis during normal development and following activity blockade. J Neurosci 11: 238–255, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.David G, Barrett EF. Stimulation-evoked increases in cytosolic [Ca(2+)] in mouse motor nerve terminals are limited by mitochondrial uptake and are temperature-dependent. J Neurosci 20: 7290–7296, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.David G, Barrett EF. Mitochondrial Ca2+ uptake prevents desynchronization of quantal release and minimizes depletion during repetitive stimulation of mouse motor nerve terminals. J Physiol 548: 425–438, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Davis RL, Weintraub H, Lassar B. Expression of a single transfected cDNA converts fibrobalsts to myoblasts. Cell 51: 987–1000, 1987. [DOI] [PubMed] [Google Scholar]
- 91.de Carvalho M, Swash M. Nerve conduction studies in amyotrophic lateral sclerosis. Muscle Nerve 23: 344–352, 2000. [DOI] [PubMed] [Google Scholar]
- 92.De Robertis ED, Bennett HS. Some features of the submicroscopic morphology of synapses in frog and earthworm. J Biophys Biochem Cytol 1: 47–58, 1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.del Castillo J, Katz B. Quantal components of the end-plate potential. J Physiol 124: 560–573, 1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Denker A, Rizzoli SO. Synaptic vesicle pools: An update. Front Synaptic Neurosci 2: 135, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dennis MJ, Ziskind-Conhaim L, Harris AJ. Development of neuromuscular junctions in rat embryos. Dev Biol 81: 266–279, 1981. [DOI] [PubMed] [Google Scholar]
- 96.Desaphy JF, De Luca A, Imbrici P, Conte CD. Modification by ageing of the tetrodotoxin-sensitive sodium channels in rat skeletal muscle fibres. Biochim Biophys Acta 1373: 37–46, 1998. [DOI] [PubMed] [Google Scholar]
- 97.Dionne VE, Leibowitz MD. Acetylcholine receptor kinetics. A description from single-channel currents at snake neuromuscular junctions. Biophys J 39: 253–261, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Donaldson IM. Robert Hooke’s Micrographia of 1665 and 1667. J R Coll Physicians Edinb 40: 374–376, 2010. [DOI] [PubMed] [Google Scholar]
- 99.Duchen MR. Ca(2+)-dependent changes in the mitochondrial energetics in single dissociated mouse sensory neurons. Biochem J 283 (Pt 1): 41–50, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dukkipati SS, Garrett TL, Elbasiouny SM. The vulnerability of spinal motoneurons and soma size plasticity in a mouse model of amyotrophic lateral sclerosis. J Physiol 596: 1723–1745, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dupuis L, Pehar M, Cassina P, Rene F, Castellanos R, Rouaux C, Gandelman M, Dimou L, Schwab ME, Loeffler JP, Barbeito L, Gonzalez de Aguilar JL. Nogo receptor antagonizes p75NTR-dependent motor neuron death. Proc Natl Acad Sci U S A 105: 740–745, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Duxson MJ, Ross JJ, Harris AJ. Transfer of differentiated synaptic terminals from primary myotubes to new-formed muscle cells during embryonic development in the rat. Neurosci Lett 71: 147–152, 1986. [DOI] [PubMed] [Google Scholar]
- 103.Eccles JCK B; Kuffler SW Nature of the “endplate potential” in curarized muscle. J Neurophysiol 4: 362–387, 1941. [Google Scholar]
- 104.Edstrom L, Kugelberg E. Histochemical composition, distribution of fibres and fatiguability of single motor units. Anterior tibial muscle of the rat. J Neurol Neurosurg Psychiatry 31: 424–433, 1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Edwards IJ, Bruce G, Lawrenson C, Howe L, Clapcote SJ, Deuchars SA, Deuchars J. Na+/K+ ATPase alpha1 and alpha3 isoforms are differentially expressed in alpha- and gamma-motoneurons. J Neurosci 33: 9913–9919, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ellis DZ, Rabe J, Sweadner KJ. Global loss of Na, K-ATPase and its nitric oxide-mediated regulation in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci 23: 43–51, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Enad JG, Fournier M, Sieck GC. Oxidative capacity and capillary density of diaphragm motor units. J Appl Physiol 67: 620–627, 1989. [DOI] [PubMed] [Google Scholar]
- 108.Engel AG, Lindstrom JM, Lambert EH, Lennon VA. Ultrastructural localization of the acetylcholine receptor in myasthenia gravis and in its experimental autoimmune model. Neurology 27: 307–315, 1977. [DOI] [PubMed] [Google Scholar]
- 109.Engel AG, Nagel A, Walls TJ, Harper CM, Waisburg HA. Congenital myasthenic syndromes: I. Deficiency and short open-time of the acetylcholine receptor. Muscle Nerve 16: 1284–1292, 1993. [DOI] [PubMed] [Google Scholar]
- 110.Engel AG, Shen XM, Selcen D, Sine SM. Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. Lancet Neurol 14: 461, 2015. [DOI] [PubMed] [Google Scholar]
- 111.Engel AG, Tsujihata M, Lambert EH, Lindstrom JM, Lennon VA. Experimental autoimmune myasthenia gravis: A sequential and quantitative study of the neuromuscular junction ultrastructure and electrophysiologic correlations. J Neuropathol Exp Neurol 35: 569–587, 1976. [DOI] [PubMed] [Google Scholar]
- 112.Enoka RM, Robinson GA, Kossev AR. A stable, selective electrode for recording single motor-unit potentials in humans. Exp Neurol 99: 761–764, 1988. [DOI] [PubMed] [Google Scholar]
- 113.Ermilov LG, Mantilla CB, Rowley KL, Sieck GC. Safety factor for neuromuscular transmission at type-identified diaphragm fibers. Muscle Nerve 35: 800–803, 2007. [DOI] [PubMed] [Google Scholar]
- 114.Ermilov LG, Pulido JN, Atchison FW, Zhan WZ, Ereth MH, Sieck GC, Mantilla CB. Impairment of diaphragm muscle force and neuromuscular transmission after normothermic cardiopulmonary bypass: Effect of low dose inhaled CO. Am J Physiol Regul Integr Comp Physiol 298: R784–R789, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Eshed-Eisenbach Y, Peles E. The clustering of voltage-gated sodium channels in various excitable membranes. Dev Neurobiol 81 (5): 427–437, 2019. DOI: 10.1002/dneu.22728. [DOI] [PubMed] [Google Scholar]
- 116.Falls DL, Rosen KM, Corfas G, Lane WS, Fischbach GD. ARIA, a protein that stimulates acetylcholine receptor synthesis, is a member of the neu ligand family. Cell 72: 801–815, 1993. [DOI] [PubMed] [Google Scholar]
- 117.Fambrough DM, Drachman DB, Satyamurti S. Neuromuscular junction in myasthenia gravis: Decreased acetylcholine receptors. Science 182: 293–295, 1973. [DOI] [PubMed] [Google Scholar]
- 118.Fatt P, Katz B. An analysis of the end-plate potential recorded with an intra-cellular electrode. J Physiol 115: 320–370, 1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fatt P, Katz B. Spontaneous subthreshold activity at motor nerve endings. J Physiol 117: 109–128, 1952. [PMC free article] [PubMed] [Google Scholar]
- 120.Favero M, Busetto G, Cangiano A. Spike timing plays a key role in synapse elimination at the neuromuscular junction. Proc Natl Acad Sci U S A 109: E1667–E1675, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Favero M, Massella O, Cangiano A, Buffelli M. On the mechanism of action of muscle fibre activity in synapse competition and elimination at the mammalian neuromuscular junction. Eur J Neurosci 29: 2327–2334, 2009. [DOI] [PubMed] [Google Scholar]
- 122.Feldman JD, Bazzy AR, Cummins TR, Haddad GG. Developmental changes in neuromuscular transmission in the rat diaphragm. J Appl Physiol 71: 280–286, 1991. [DOI] [PubMed] [Google Scholar]
- 123.Feng Z, Ko CP. The role of glial cells in the formation and maintenance of the neuromuscular junction. Ann N Y Acad Sci 1132: 19–28, 2008. [DOI] [PubMed] [Google Scholar]
- 124.Fertuck HC, Salpeter MM. Localization of acetylcholine receptor by 125I-labeled alpha-bungarotoxin binding at mouse motor endplates. Proc Natl Acad Sci U S A 71: 1376–1378, 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fish LA, Fallon JR. Multiple MuSK signaling pathways and the aging neuromuscular junction. Neurosci Lett 731: 135014, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fleshman JW,Munson JB, Sypert GW, Friedman WA. Rheobase, input resistance, and motor-unit type in medial gastrocnemius motoneurons in the cat. J Neurophysiol 46: 1326–1338, 1981. [DOI] [PubMed] [Google Scholar]
- 127.Flucher BE, Daniels MP. Distribution of Na+ channels and ankyrin in neuromuscular junctions is complementary to that of acetylcholine receptors and the 43 kd protein. Neuron 3: 163–175, 1989. [DOI] [PubMed] [Google Scholar]
- 128.Fogarty MJ. The bigger they are the harder they fall: Size-dependent vulnerability of motor neurons in amyotrophic lateral sclerosis. J Physiol 596: 2471–2472, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Fogarty MJ. Driven to decay: Excitability and synaptic abnormalities in amyotrophic lateral sclerosis. Brain Res Bull 140: 318–333, 2018. [DOI] [PubMed] [Google Scholar]
- 130.Fogarty MJ, Brandenburg JE, Sieck GC. Diaphragm neuromuscular transmission failure in a mouse model of an early-onset neuromotor disorder. J Appl Physiol (1985) 130 (3): 708–720, 2021. DOI: 10.1152/japplphysiol.00864.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Fogarty MJ, Gonzalez Porras MA, Mantilla CB, Sieck GC. Diaphragm neuromuscular transmission failure in aged rats. J Neurophysiol 122: 93–104, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fogarty MJ, Hammond LA, Kanjhan R, Bellingham MC, Noakes PG. A method for the three-dimensional reconstruction of Neurobiotin-filled neurons and the location of their synaptic inputs. Front Neural Circuits 7: 153, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Fogarty MJ, Kanjhan R, Bellingham MC, Noakes PG. Glycinergic neurotransmission: A potent regulator of embryonic motor neuron dendritic morphology and synaptic plasticity. J Neurosci 36: 80–87, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Fogarty MJ, Kanjhan R, Yanagawa Y, Noakes PG, Bellingham MC. Alterations in hypoglossal motor neurons due to GAD67 and VGAT deficiency in mice. Exp Neurol 289: 117–127, 2017. [DOI] [PubMed] [Google Scholar]
- 135.Fogarty MJ, Mu EWH, Lavidis NA, Noakes PG, Bellingham MC. Size-dependent dendritic maladaptations of hypoglossal motor neurons in SOD1(G93A) mice. Anat Rec (Hoboken) 304 (7): 1562–1581, 2021. DOI: 10.1002/ar.24542. [DOI] [PubMed] [Google Scholar]
- 136.Fogarty MJ, Mu EWH, Lavidis NA, Noakes PG, Bellingham MC. Size-dependent vulnerability of lumbar motor neuron dendritic degeneration in SOD1(G93A) mice. Anat Rec (Hoboken) 303: 1455–1471, 2020. [DOI] [PubMed] [Google Scholar]
- 137.Fogarty MJ, Omar TS, Zhan WZ, Mantilla CB, Sieck GC. Phrenic motor neuron loss in aged rats. J Neurophysiol 119: 1852–1862, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fogarty MJ, Sieck GC. Evolution and functional differentiation of the diaphragm muscle of mammals. Compr Physiol 9: 715–766, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fogarty MJ, Sieck GC, Brandenburg JE. Impaired neuromuscular transmission of the tibialis anterior in a rodent model of hypertonia. J Neurophysiol 123: 1864–1869, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fogarty MJ, Smallcombe KL, Yanagawa Y, Obata K, Bellingham MC, Noakes PG. Genetic deficiency of GABA differentially regulates respiratory and non-respiratory motor neuron development. PLoS One 8: e56257, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Fogarty MJ, Yanagawa Y, Obata K, Bellingham MC, Noakes PG. Genetic absence of the vesicular inhibitory amino acid transporter differentially regulates respiratory and locomotor motor neuron development. Brain Struct Funct 220: 525–540, 2015. [DOI] [PubMed] [Google Scholar]
- 142.Fournier M, Alula M, Sieck GC. Neuromuscular transmission failure during postnatal development. Neurosci Lett 125: 34–36, 1991. [DOI] [PubMed] [Google Scholar]
- 143.Fournier M, Sieck GC. Physiological properties of diaphragm motor units. Neurosci Abstr 10: 789, 1984. [Google Scholar]
- 144.Fournier M, Sieck GC. Mechanical properties of muscle units in the cat diaphragm. J Neurophysiol 59: 1055–1066, 1988. [DOI] [PubMed] [Google Scholar]
- 145.Fournier M, Sieck GC. Somatotopy in the segmental innervation of the cat diaphragm. J Appl Physiol 64: 291–298, 1988. [DOI] [PubMed] [Google Scholar]
- 146.Fowles JR, Green HJ, Ouyang J. Na+-K+-ATPase in rat skeletal muscle: Content, isoform, and activity characteristics. J Appl Physiol 96: 316, 2004-326, 1985. [DOI] [PubMed] [Google Scholar]
- 147.Frank E, Fischbach GD. Early events in neuromuscular junction formation in vitro. J Cell Biol 83: 143–158, 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci 20: 2534–2542, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Fukudome T, Shibuya N, Yoshimura T, Eguchi K. Short-term effects of prednisolone on neuromuscular transmission in the isolated mdx mouse diaphragm. Tohoku J Exp Med 192: 211–217, 2000. [DOI] [PubMed] [Google Scholar]
- 150.Gale AN, Gomez S, Duchen LW. Changes produced by a hypomyelinating neuropathy in muscle and its innervation. Morphological and physiological studies in the Trembler mouse. Brain 105: 373–393, 1982. [DOI] [PubMed] [Google Scholar]
- 151.Garcia N, Tomas M, Santafe MM, Besalduch N, Lanuza MA, Tomas J. The interaction between tropomyosin-related kinase B receptors and presynaptic muscarinic receptors modulates transmitter release in adult rodent motor nerve terminals. J Neurosci 30: 16514–16522, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Garcia N, Tomas M, Santafe MM, Lanuza MA, Besalduch N, Tomas J. Localization of brain-derived neurotrophic factor, neurotrophin-4, tropomyosin-related kinase b receptor, and p75 NTR receptor by high-resolution immunohistochemistry on the adult mouse neuromuscular junction. J Peripher Nerv Syst 15: 40–49, 2010. [DOI] [PubMed] [Google Scholar]
- 153.Garcia N, Tomas M, Santafe MM, Lanuza MA, Besalduch N, Tomas J. Blocking p75 (NTR) receptors alters polyinnervationz of neuromuscular synapses during development. J Neurosci Res 89: 1331–1341, 2011. [DOI] [PubMed] [Google Scholar]
- 154.Garcia-Lopez P, Garcia-Marin V, Freire M. The histological slides and drawings of cajal. Front Neuroanat 4: 9, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gasser HS, Grundfest H. Axon diameters in relation to the spike dimensions and the conduction velocity in mammalian A fibers. Am J Phys 127: 393–414, 1939. [Google Scholar]
- 156.Geevasinga N, Menon P, Ozdinler PH, Kiernan MC, Vucic S. Pathophysiological and diagnostic implications of cortical dysfunction in ALS. Nat Rev Neurol 12: 651–661, 2016. [DOI] [PubMed] [Google Scholar]
- 157.Geiger PC, Bailey JP, Mantilla CB, Zhan WZ, Sieck GC. Mechanisms underlying myosin heavy chain expression during development of the rat diaphragm muscle. J Appl Physiol 101: 1546–1555, 2006. [DOI] [PubMed] [Google Scholar]
- 158.Geiger PC, Cody MJ, Macken RL, Sieck GC. Maximum specific force depends on myosin heavy chain content in rat diaphragm muscle fibers. J Appl Physiol 89: 695–703, 2000. [DOI] [PubMed] [Google Scholar]
- 159.Geiger PC, Cody MJ, Sieck GC. Force-calcium relationship depends on myosin heavy chain and troponin isoforms in rat diaphragm muscle fibers. J Appl Physiol 87: 1894–1900, 1999. [DOI] [PubMed] [Google Scholar]
- 160.Gellerich FN, Gizatullina Z, Arandarcikaite O, Jerzembek D, Vielhaber S, Seppet E, Striggow F. Extramitochondrial Ca2+ in the nanomolar range regulates glutamate-dependent oxidative phosphorylation on demand. PLoS One 4: e8181, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Georgiou J, Robitaille R, Charlton MP. Muscarinic control of cytoskeleton in perisynaptic glia. J Neurosci 19: 3836–3846, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Gerlach J. Von den Ruckenmarke. Leipzig: Engelmann, 1871. [Google Scholar]
- 163.Ghazanfari N, Linsao EL, Trajanovska S, Morsch M, Gregorevic P, Liang SX, Reddel SW, Phillips WD. Forced expression of muscle specific kinase slows postsynaptic acetylcholine receptor loss in a mouse model of MuSK myasthenia gravis. Physiol Rep 3: e12658, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Gilhus NE, Tzartos S, Evoli A, Palace J, Burns TM, Verschuuren J. Myasthenia gravis. Nat Rev Dis Primers 5: 30, 2019. [DOI] [PubMed] [Google Scholar]
- 165.Gillon A, Sheard P. Elderly mouse skeletal muscle fibres have a diminished capacity to upregulate NCAM production in response to denervation. Biogerontology 16: 811–823, 2015. [DOI] [PubMed] [Google Scholar]
- 166.Glavinovic MI. Change of statistical parameters of transmitter release during various kinetic tests in unparalysed voltage-clamped rat diaphragm. J Physiol 290: 481–497, 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Golgi C. Sulla sostanza grigia del cervello. Gazetta Medica Italiana 33: 244–246, 1873. [Google Scholar]
- 168.Gonzalez Porras MA, Fogarty MJ, Gransee HM, Sieck GC, Mantilla CB. Frequency-dependent lipid raft uptake at rat diaphragm muscle axon terminals. Muscle Nerve 59: 611–618, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Goonasekera SA, Lam CK, Millay DP, Sargent MA, Hajjar RJ, Kranias EG, Molkentin JD. Mitigation of muscular dystrophy in mice by SERCA overexpression in skeletal muscle. J Clin Invest 121: 1044–1052, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Gotti C, Clementi F, Fornari A, Gaimarri A, Guiducci S, Manfredi I, Moretti M, Pedrazzi P, Pucci L, Zoli M. Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem Pharmacol 78: 703–711, 2009. [DOI] [PubMed] [Google Scholar]
- 171.Gowers WR. A Manual of Diseases of the Nervous System. P. Blakiston, Son & Company, 1898. [Google Scholar]
- 172.Green HJ, Duhamel TA, Holloway GP, Moule JW, Ouyang J, Ranney D, Tupling AR. Muscle Na+-K+-ATPase response during 16 h of heavy intermittent cycle exercise. Am J Physiol Endocrinol Metab 293: E523–E530, 2007. [DOI] [PubMed] [Google Scholar]
- 173.Greer JJ, Allan DW, Martin-Caraballo M, Lemke RP. An overview of phrenic nerve and diaphragm muscle development in the perinatal rat. J Appl Physiol 86: 779–786, 1999. [DOI] [PubMed] [Google Scholar]
- 174.Greer JJ, Smith JC, Feldman JL. Respiratory and locomotor patterns generated in the fetal rat brain stem-spinal cord in vitro. J Neurophysiol 67: 996–999, 1992. [DOI] [PubMed] [Google Scholar]
- 175.Greising SM, Ermilov LG, Sieck GC, Mantilla CB. Ageing and neurotrophic signalling effects on diaphragm neuromuscular function. J Physiol 593: 431–440, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Greising SM, Gransee HM, Mantilla CB, Sieck GC. Systems biology of skeletal muscle: Fiber type as an organizing principle. Wiley Interdiscip Rev Syst Biol Med 4: 457–473, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Greising SM, Stowe JM, Sieck GC, Mantilla CB. Role of TrkB kinase activity in aging diaphragm neuromuscular junctions. Exp Gerontol 72: 184–191, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Greising SM, Vasdev AK, Zhan WZ, Sieck GC, Mantilla CB. Chronic TrkB agonist treatment in old age does not mitigate diaphragm neuromuscular dysfunction. Physiol Rep 5: e13103, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Grossman Y, Parnas I, Spira ME. Differential conduction block in branches of a bifurcating axon. J Physiol 295: 283–305, 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Gundersen CB. The structure of the synaptic vesicle-plasma membrane interface constrains SNARE models of rapid, synchronous exocytosis at nerve terminals. Front Mol Neurosci 10: 48, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Haase G, Dessaud E, Garces A, de Bovis B, Birling M, Filippi P, Schmalbruch H, Arber S, deLapeyriere O. GDNF acts through PEA3 to regulate cell body positioning and muscle innervation of specific motor neuron pools. Neuron 35: 893–905, 2002. [DOI] [PubMed] [Google Scholar]
- 182.Hall ZW, Sanes JR. Synaptic structure and development: The neuromuscular junction. Cell 72 (Suppl): 99–121, 1993. [DOI] [PubMed] [Google Scholar]
- 183.Hamalainen N, Pette D. Myosin and SERCA isoform expression in denervated slow-twitch muscle of euthyroid and hyperthyroid rabbits. J Muscle Res Cell Motil 22: 453–457, 2001. [DOI] [PubMed] [Google Scholar]
- 184.Hanson PI, Roth R, Morisaki H, Jahn R, Heuser JE. Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick-freeze/deep-etch electron microscopy. Cell 90: 523–535, 1997. [DOI] [PubMed] [Google Scholar]
- 185.Harris AJ. Embryonic growth and innervation of rat skeletal muscles. I neural regulation of muscle fibre numbers. Philos Trans R Soc Lond Ser B Biol Sci 293: 257–277, 1981. [DOI] [PubMed] [Google Scholar]
- 186.Harris AJ. Embryonic growth and innervation of rat skeletal muscles. III. Neural regulation of junctional and extra-junctional acetylcholine receptor clusters. Philos Trans R Soc Lond Ser B Biol Sci 293: 287–314, 1981. [DOI] [PubMed] [Google Scholar]
- 187.Harris JJ, Jolivet R, Attwell D. Synaptic energy use and supply. Neuron 75: 762–777, 2012. [DOI] [PubMed] [Google Scholar]
- 188.Hayworth CR, Moody SE, Chodosh LA, Krieg P, Rimer M, Thompson WJ. Induction of neuregulin signaling in mouse schwann cells in vivo mimics responses to denervation. J Neurosci 26: 6873–6884, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Heeroma JH, Roelandse M, Wierda K, van Aerde KI, Toonen RF, Hensbroek RA, Brussaard A, Matus A, Verhage M. Trophic support delays but does not prevent cell-intrinsic degeneration of neurons deficient for munc18-1. Eur J Neurosci 20: 623–634, 2004. [DOI] [PubMed] [Google Scholar]
- 190.Hegedus J, Putman CT, Gordon T. Time course of preferential motor unit loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 28: 154–164, 2007. [DOI] [PubMed] [Google Scholar]
- 191.Hegedus J, Putman CT, Tyreman N, Gordon T. Preferential motor unit loss in the SOD1 G93A transgenic mouse model of amyotrophic lateral sclerosis. J Physiol 586: 3337–3351, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Henderson R, Baumann F, Hutchinson N, McCombe P. CMAP decrement in ALS. Muscle Nerve 39: 555–556, 2009. [DOI] [PubMed] [Google Scholar]
- 193.Henneman E, Somjen G, Carpenter DO. Functional significance of cell size in spinal motoneurons. J Neurophysiol 28: 560–580, 1965. [DOI] [PubMed] [Google Scholar]
- 194.Herrera AA, Qiang H, Ko CP. The role of perisynaptic Schwann cells in development of neuromuscular junctions in the frog (Xenopus laevis). J Neurobiol 45: 237–254, 2000. [DOI] [PubMed] [Google Scholar]
- 195.Heuser JE, Reese TS. Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J Cell Biol 57: 315–344, 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Heuser JE, Reese TS, Dennis MJ, Jan Y, Jan L, Evans L. Synaptic vesicle exocytosis captured by quick freezing and correlated with quantal transmitter release. J Cell Biol 81: 275–300, 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Higuchi O, Hamuro J, Motomura M, Yamanashi Y. Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann Neurol 69: 418–422, 2011. [DOI] [PubMed] [Google Scholar]
- 198.Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, Vincent A. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med 7: 365–368, 2001. [DOI] [PubMed] [Google Scholar]
- 199.Hounsgaard J Motor neurons. Compr Physiol 7: 463–484, 2017. [DOI] [PubMed] [Google Scholar]
- 200.Hughes SM, Koishi K, Rudnicki M, Maggs AM. MyoD protein is differentially accumulated in fast and slow skeletal muscle fibres and required for normal fibre type balance in rodents. Mech Dev 61: 151–163, 1997. [DOI] [PubMed] [Google Scholar]
- 201.Hughes SM, Taylor JM, Tapscott SJ, Gurley CM, Carter WJ, Peterson CA. Selective accumulation ofMyoD andmyogeninmRNAs in fast and slow adult skeletal muscle is controlled by innervation and hormones. Development 118: 1137–1147, 1993. [DOI] [PubMed] [Google Scholar]
- 202.Hundal HS, Marette A, Ramlal T, Liu Z, Klip A. Expression of beta subunit isoforms of the Na+, K(+)-ATPase is muscle type-specific. FEBS Lett 328: 253–258, 1993. [DOI] [PubMed] [Google Scholar]
- 203.Inoue A, Setoguchi K, Matsubara Y, Okada K, Sato N, Iwakura Y, Higuchi O, Yamanashi Y. Dok-7 activates the muscle receptor kinase MuSK and shapes synapse formation. Sci Signal 2: ra7, 2009. [DOI] [PubMed] [Google Scholar]
- 204.Jahromi BS, Robitaille R, Charlton MP. Transmitter release increases intracellular calcium in perisynaptic Schwann cells in situ. Neuron 8: 1069–1077, 1992. [DOI] [PubMed] [Google Scholar]
- 205.Jang YC, Van Remmen H. Age-associated alterations of the neuromuscular junction. Exp Gerontol 46: 193–198, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Jansen JKS, Fladby T. The perinatal organization of the innervation of skeletal muscle in mammals. Prog Neurobiol 34: 39–90, 1990. [DOI] [PubMed] [Google Scholar]
- 207.Je HS, Yang F, Ji Y, Potluri S, Fu XQ, Luo ZG, Nagappan G, Chan JP, Hempstead B, Son YJ, Lu B. ProBDNF and mature BDNF as punishment and reward signals for synapse elimination at mouse neuromuscular junctions. J Neurosci 33: 9957–9962, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Jessen KR, Mirsky R. Embryonic Schwann cell development: The biology of Schwann cell precursors and early Schwann cells. J Anat 191 (Pt 4): 501–505, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Jessen KR, Mirsky R. Origin and early development of Schwann cells. Microsc Res Tech 41: 393–402, 1998. [DOI] [PubMed] [Google Scholar]
- 210.Ji H, Coleman J, Yang R, Melia TJ, Rothman JE, Tareste D. Protein determinants of SNARE-mediated lipid mixing. Biophys J 99: 553–560, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Jo SA, Zhu X, Marchionni MA, Burden SJ. Neuregulins are concentrated at nerve-muscle synapses and activateACh-receptor gene expression. Nature 373: 158–161, 1995. [DOI] [PubMed] [Google Scholar]
- 212.Johansson C, Lunde PK, Gothe S, Lannergren J,Westerblad H. Isometric force and endurance in skeletal muscle of mice devoid of all known thyroid hormone receptors. J Physiol 547: 789–796, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Johnson BD, Sieck GC. Differential susceptibility of diaphragm muscle fibers to neuromuscular transmission failure. J Appl Physiol 75: 341–348, 1993. [DOI] [PubMed] [Google Scholar]
- 214.Johnson BD, Wilson LE, Zhan WZ, Watchko JF, Daood MJ, Sieck GC. Contractile properties of the developing diaphragm correlate with myosin heavy chain phenotype. J Appl Physiol 77: 481–487, 1994. [DOI] [PubMed] [Google Scholar]
- 215.Johnson H, Hokfelt T, Ulfhake B. Expression of p75(NTR), trkB and trkC in nonmanipulated and axotomized motoneurons of aged rats. Brain Res Mol Brain Res 69: 21–34, 1999. [DOI] [PubMed] [Google Scholar]
- 216.Jones RA, Harrison C, Eaton SL, Llavero Hurtado M, Graham LC, Alkhammash L, Oladiran OA, Gale A, Lamont DJ, Simpson H, Simmen MW, Soeller C, Wishart TM, Gillingwater TH. Cellular and molecular anatomy of the human neuromuscular junction. Cell Rep 21: 2348–2356, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Joyce NC, Carter GT. Electrodiagnosis in persons with amyotrophic lateral sclerosis. PM R 5: S89–S95, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Kaeser PS, Sudhof TC. RIM function in short- and long-term synaptic plasticity. Biochem Soc Trans 33: 1345–1349, 2005. [DOI] [PubMed] [Google Scholar]
- 219.Katz B, Miledi R. Propagation of electric activity in motor nerve terminals. Proc R Soc Lond 161: 453–482, 1965. [DOI] [PubMed] [Google Scholar]
- 220.Katz B, Miledi R. The release of acetylcholine from nerve endings by graded electric pulses. Proc R Soc Lond B Biol Sci 167: 23–38, 1967. [DOI] [PubMed] [Google Scholar]
- 221.Katz B, Miledi R. The timing of calcium action during neuromuscular transmission. J Physiol 189: 535–544, 1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Katz B, Miledi R. Transmitter leakage from motor nerve endings. Proc R Soc Lond B Biol Sci 196: 59–72, 1977. [DOI] [PubMed] [Google Scholar]
- 223.Katz B, Thesleff S. On the factors which determine the amplitude of the ‘miniature end-plate potential’. J Physiol 137: 267–278, 1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Katz B, Thesleff S. A study of the desensitization produced by acetylcholine at the motor end-plate. J Physiol 138: 63–80, 1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Khurram OU, Fogarty MJ, Rana S, Vang P, Sieck GC, Mantilla CB. Diaphragm muscle function following mid-cervical contusion injury in rats. J Appl Physiol (1985) 126 (1): 221–230, 2018. DOI: 10.1152/japplphysiol.00481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Khurram OU, Fogarty MJ, Sarrafian TL, Bhatt A, Mantilla CB, Sieck GC. Impact of aging on diaphragm muscle function in male and female Fischer 344 rats. Physiol Rep 6: e13786, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Kiernan JA, Hudson AJ. Changes in sizes of cortical and lower motor neurons in amyotrophic lateral sclerosis. Brain 114 (Pt 2): 843–853, 1991. [DOI] [PubMed] [Google Scholar]
- 228.Knudsen KA. Cell adhesion molecules in myogenesis. Curr Opin Cell Biol 2: 902–906, 1990. [DOI] [PubMed] [Google Scholar]
- 229.Kong XC, Barzaghi P, Ruegg MA. Inhibition of synapse assembly in mammalian muscle in vivo by RNA interference. EMBO Rep 5: 183–188, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Kononenko NL, Diril MK, Puchkov D, Kintscher M, Koo SJ, Pfuhl G, Winter Y,Wienisch M, Klingauf J, Breustedt J, Schmitz D, Maritzen T, Haucke V. Compromised fidelity of endocytic synaptic vesicle protein sorting in the absence of stonin 2. Proc Natl Acad Sci U S A 110: E526–E535, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Krause W Die Entladungshypothese und die motorischen Endplatten. Arch Mikrosk Anat 13: 170–179, 1877. [Google Scholar]
- 232.Krauss RS, Cole F, Gaio U, Takaesu G, Zhang W, Kang JS. Close encounters: Regulation of vertebrate skeletal myogenesis by cell-cell contact. J Cell Sci 118: 2355–2362, 2005. [DOI] [PubMed] [Google Scholar]
- 233.Kravtsova VV, Bouzinova EV, Chibalin AV, Matchkov VV, Krivoi II. Isoform-specific Na, K-ATPase and membrane cholesterol remodeling in motor endplates in distinct mouse models of myodystrophy. Am J Physiol Cell Physiol 318: C1030–C1041, 2020. [DOI] [PubMed] [Google Scholar]
- 234.Krnjevic K, Miledi R. Failure of neuromuscular propagation in rats. J Physiol 140: 440–461, 1958. [PMC free article] [PubMed] [Google Scholar]
- 235.Krnjevic K, Miledi R. Motor units in the rat diaphragm. J Physiol 140: 427–439, 1958. [PMC free article] [PubMed] [Google Scholar]
- 236.Krnjevic K, Miledi R. Presynaptic failure of neuromuscular propagation in rats. J Physiol Lond 149: 1–22, 1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Kucenas S, Takada N, Park HC, Woodruff E, Broadie K, Appel B. CNS-derived glia ensheath peripheral nerves and mediate motor root development. Nat Neurosci 11: 143–151, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Kuei JH, Shadmehr R, Sieck GC. Relative contribution of neurotransmission failure to diaphragm fatigue. J Appl Physiol 68: 174–180, 1990. [DOI] [PubMed] [Google Scholar]
- 239.Kuffler SW, Yoshikami D. The number of transmitter molecules in a quantum: An estimate from iontophoretic application of acetylcholine at the neuromuscular synapse. J Physiol 251: 465–482, 1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Kummel D, Krishnakumar SS, Radoff DT, Li F, Giraudo CG, Pincet F, Rothman JE, Reinisch KM. Complexin cross-links prefusion SNAREs into a zigzag array. Nat Struct Mol Biol 18: 927–933, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Kummer TT, Misgeld T, Sanes JR. Assembly of the postsynaptic membrane at the neuromuscular junction: Paradigm lost. Curr Opin Neurobiol 16: 74–82, 2006. [DOI] [PubMed] [Google Scholar]
- 242.Kurihara T Seronegative myasthenia gravis and muscle atrophy of the tongue. Intern Med 44: 536–537, 2005. [DOI] [PubMed] [Google Scholar]
- 243.Labovitz SS, Robbins N, Fahim MA. Endplate topgraphy of denervated and disused rat neuromuscular junctions: Comparison by scanning and light microscopy. Neuroscience 11 (4): 963–971, 1984. [DOI] [PubMed] [Google Scholar]
- 244.Lambert EH, Lindstrom JM, Lennon VA. End-plate potentials in experimental autoimmune myasthenia gravis in rats. Ann N Y Acad Sci 274: 300–318, 1976. [DOI] [PubMed] [Google Scholar]
- 245.Land BR, Harris WV, Salpeter EE, Salpeter MM. Diffusion and binding constants for acetylcholine derived from the falling phase of miniature endplate currents. Proc Natl Acad Sci U S A 81: 1594–1598, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Land BR, Salpeter EE, Salpeter MM. Kinetic parameters for acetylcholine interaction in intact neuromuscular junction. Proc Natl Acad Sci U S A 78: 7200–7204, 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Landmesser L The relationship of intramuscular nerve branching and synaptogenesis to motoneuron survival. J Neurobiol 23: 1131–1139, 1992. [DOI] [PubMed] [Google Scholar]
- 248.Langley JN. On the reaction of cells and of nerve-endings to certain poisons, chiefly as regards the reaction of striated muscle to nicotine and to curari. J Physiol 33: 374–413, 1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Laskowski MB, Sanes JR. Topographic mapping of motor pools onto skeletal muscles. J Neurosci 7: 252–260, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Laughlin SB, de Ruyter van Steveninck RR, Anderson JC. The metabolic cost of neural information. Nat Neurosci 1: 36–41, 1998. [DOI] [PubMed] [Google Scholar]
- 251.Lebrasseur NK, Cote GM, Miller TA, Fielding RA, Sawyer DB. Regulation of neuregulin/ErbB signaling by contractile activity in skeletal muscle. Am J Physiol Cell Physiol 284: C1149–C1155, 2003. [DOI] [PubMed] [Google Scholar]
- 252.Lee CW, Zhang H, Geng L, Peng HB. Crosslinking-induced endocytosis of acetylcholine receptors by quantum dots. PLoS One 9: e90187, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Lee KM, Chand KK, Hammond LA, Lavidis NA, Noakes PG. Functional decline at the aging neuromuscular junction is associated with altered laminin-alpha4 expression. Aging (Albany NY) 9: 880–899, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Lee YI, Li Y, Mikesh M, Smith I, Nave KA, Schwab MH, Thompson WJ.Neuregulin1 displayed on motor axons regulates terminal Schwann cell-mediated synapse elimination at developing neuromuscular junctions. Proc Natl Acad Sci U S A 113: E479–E487, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Lennon VA, Lambert EH. Myasthenia gravis induced by monoclonal antibodies to acetylcholine receptors. Nature 285: 238–240, 1980. [DOI] [PubMed] [Google Scholar]
- 256.Lewis WH. The development of the muscular system. In: Manual of Human Embryology. Philadelphia, PA: Lippincott, 1910, p. 454–522. [Google Scholar]
- 257.Li P, Steinbach JH. The neuronal nicotinic alpha4beta2 receptor has a high maximal probability of being open. Br J Pharmacol 160: 1906–1915, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Li XM,Dong XP, Luo SW, Zhang B, Lee DH, Ting AK, Neiswender H, Kim CH, Carpenter-Hyland E, Gao TM, Xiong WC, Mei L. Retrograde regulation of motoneuron differentiation by muscle beta-catenin. Nat Neurosci 11: 262–268, 2008. [DOI] [PubMed] [Google Scholar]
- 259.Liddell EGT, Sherrington CS. Recruitment and some other factors of reflex inhibition. Proc Roy Soc Lond (Biol) 97: 488–518, 1925. [Google Scholar]
- 260.Lin S, Landmann L, Ruegg MA, Brenner HR. The role of nerve- versus muscle-derived factors in mammalian neuromuscular junction formation. J Neurosci 28: 3333–3340, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Ling G, Gerard RW. The normal membrane potential of frog sartorius fibers. J Cell Comp Physiol 34: 383–396, 1949. [DOI] [PubMed] [Google Scholar]
- 262.Liu C, Bickford LS, Held RG, Nyitrai H, Sudhof TC, Kaeser PS. The active zone protein family ELKS supports Ca2+ influx at nerve terminals of inhibitory hippocampal neurons. J Neurosci 34: 12289–12303, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Lubischer JL, Bebinger DM. Regulation of terminal Schwann cell number at the adult neuromuscular junction. J Neurosci 19: RC46, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Lupa MT, Gordon H, Hall ZW. A specific effect of muscle cells on the distribution of presynaptic proteins in neurites and its absence in a C2 muscle cell variant. Dev Biol 142: 31–43, 1990. [DOI] [PubMed] [Google Scholar]
- 265.Lytton J, Westlin M, Burk SE, Shull GE, MacLennan DH. Functional comparisons between isoforms of the sarcoplasmic or endoplasmic reticulum family of calcium pumps. J Biol Chem 267: 14483–14489, 1992. [PubMed] [Google Scholar]
- 266.Ma KH, Duong P, Moran JJ, Junaidi N, Svaren J. Polycomb repression regulates Schwann cell proliferation and axon regeneration after nerve injury. Glia 66: 2487–2502, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Madhavan R, Zhao XT, Ruegg MA, Peng HB. Tyrosine phosphatase regulation of MuSK-dependent acetylcholine receptor clustering. Mol Cell Neurosci 28: 403–416, 2005. [DOI] [PubMed] [Google Scholar]
- 268.Maggio S, Ceccaroli P, Polidori E, Cioccoloni A, Stocchi V, Guescini M. Signal exchange through extracellular vesicles in neuromuscular junction establishment and maintenance: From physiology to pathology. Int J Mol Sci 20: 2804, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Mallart A, Dreyer F, Peper K. Current-voltage relation and reversal potential at junctional and extrajunctional ACh-receptors of the frog neuromuscular junction. Pflugers Arch 362: 43–47, 1976. [DOI] [PubMed] [Google Scholar]
- 270.Mantilla CB, Fahim MA, Sieck GC. Functional development of respiratory muscles. In: Polin RA, Fox WW, Abman SH, editors. Fetal and Neonatal Physiology (4th ed). W.B. Saunders, 2011, p. 937–952. DOI: 10.1016/B978-1-4160-3479-7.10085-0. [DOI] [Google Scholar]
- 271.Mantilla CB, Rowley KL, Fahim MA, Zhan WZ, Sieck GC. Synaptic vesicle cycling at type-identified diaphragm neuromuscular junctions. Muscle Nerve 30: 774–783, 2004. [DOI] [PubMed] [Google Scholar]
- 272.Mantilla CB, Rowley KL, Zhan WZ, Fahim MA, Sieck GC. Synaptic vesicle pools at diaphragm neuromuscular junctions vary with motoneuron soma, not axon terminal, inactivity. Neuroscience 146: 178–189, 2007. [DOI] [PubMed] [Google Scholar]
- 273.Mantilla CB, Seven YB, Zhan WZ, Sieck GC. Diaphragm motor unit recruitment in rats. Respir Physiol Neurobiol 173: 101–106, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Mantilla CB, Sieck GC. Invited Review:Mechanisms underlying motor unit plasticity in the respiratory system. J Appl Physiol 94: 1230–1241, 2003. [DOI] [PubMed] [Google Scholar]
- 275.Mantilla CB, Sieck GC. Key aspects of phrenic motoneuron and diaphragm muscle development during the perinatal period. J Appl Physiol 104: 1818–1827, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Mantilla CB, Sieck GC. Key aspects of phrenic motoneuron and diaphragm muscle development during the perinatal period. J Appl Physiol, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Mantilla CB, Sill RV, Aravamudan B, Zhan WZ, Sieck GC. Developmental effects on myonuclear domain size of rat diaphragm fibers. J Appl Physiol 104: 787–794, 2008. [DOI] [PubMed] [Google Scholar]
- 278.Mantilla CB, Zhan WZ, Sieck GC. Neurotrophins improve neuromuscular transmission in the adult rat diaphragm. Muscle Nerve 29: 381–386, 2004. [DOI] [PubMed] [Google Scholar]
- 279.Mantilla CB, Zhan WZ, Sieck GC. Retrograde labeling of phrenic motoneurons by intrapleural injection. J Neurosci Methods 182: 244–249, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Manuel M, Zytnicki D. Molecular and electrophysiological properties of mouse motoneuron and motor unit subtypes. Curr Opin Physio 8: 23–29, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Marques MJ, Conchello JA, Lichtman JW. From plaque to pretzel: Fold formation and acetylcholine receptor loss at the developing neuromuscular junction. J Neurosci 20: 3663–3675, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Martignago S, Fanin M, Albertini E, Pegoraro E, Angelini C. Muscle histopathology in myasthenia gravis with antibodies against MuSK and AChR. Neuropathol Appl Neurobiol 35: 103–110, 2009. [DOI] [PubMed] [Google Scholar]
- 283.Martin A Junctional transmission II. Presynaptic mechanisms. Compr Physiol: 329–355, 2011. [Google Scholar]
- 284.Martin AR. Amplification of neuromuscular transmission by postjunctional folds. Proc Biol Sci 258: 321–326, 1994. [DOI] [PubMed] [Google Scholar]
- 285.Martineau E, Di Polo A, Vande Velde C, Robitaille R. Dynamic neuromuscular remodeling precedes motor-unit loss in a mouse model of ALS. elife 7: e41973, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Martineau E, Di Polo A, Vande Velde C, Robitaille R. Sex-specific differences in motor-unit remodeling in a mouse model of ALS. eNeuro 7, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Martinez-Valencia A, Ramirez-Santiago G, De-Miguel FF. Dynamics of neuromuscular transmission reproduced by calcium-dependent and reversible serial transitions in the vesicle fusion complex. Front Synaptic Neurosci 13: 785361, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Matthews-Bellinger JA, Salpeter MM. Fine structural distribution of acetylcholine receptors at developing mouse neuromuscular junctions. J Neurosci 3: 644–657, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Mazala DA, Pratt SJP, Chen D, Molkentin JD, Lovering RM, Chin ER. SERCA1 overexpression minimizes skeletal muscle damage in dystrophic mouse models. Am J Physiol Cell Physiol 308: C699–C709, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.McMahan UJ. The agrin hypothesis. Cold Spring Harb Symp Quant Biol 55: 407–418, 1990. [DOI] [PubMed] [Google Scholar]
- 291.McMorrow C, Fredsted A, Carberry J, O’Connell RA, Bradford A, Jones JF, O’Halloran KD. Chronic hypoxia increases rat diaphragm muscle endurance and sodium-potassium ATPase pump content. Eur Respir J 37: 1474–1481, 2011. [DOI] [PubMed] [Google Scholar]
- 292.Megighian A, Zordan M, Pantano S, Scorzeto M, Rigoni M, Zanini D, Rossetto O, Montecucco C. Evidence for a radial SNARE supercomplex mediating neurotransmitter release at the Drosophila neuromuscular junction. J Cell Sci 126: 3134–3140, 2013. [DOI] [PubMed] [Google Scholar]
- 293.Merlie JP, Heinemann S, Einarson B, Lindstrom JM. Degradation of acetylcholine receptor in diaphragms of rats with experimental autoimmune myasthenia gravis. J Biol Chem 254: 6328–6332, 1979. [PubMed] [Google Scholar]
- 294.Mier-Jedrzejowicz AK, Brophy C, Green M. Respiratory muscle function in myasthenia gravis. Am Rev Respir Dis 138: 867–873, 1988. [DOI] [PubMed] [Google Scholar]
- 295.Milton RL, Lupa MT, Caldwell JH. Fast and slow twitch skeletal muscle fibres differ in their distribution of Na channels near the endplate. Neurosci Lett 135: 41–44, 1992. [DOI] [PubMed] [Google Scholar]
- 296.Mirsky R, Jessen KR, Brennan A, Parkinson D, Dong Z, Meier C, Parmantier E, Lawson D. Schwann cells as regulators of nerve development. J Physiol Paris 96: 17–24, 2002. [DOI] [PubMed] [Google Scholar]
- 297.Miyazawa A, Fujiyoshi Y, Stowell M, Unwin N. Nicotinic acetylcholine receptor at 4.6 A resolution: Transverse tunnels in the channel wall. J Mol Biol 288: 765–786, 1999. [DOI] [PubMed] [Google Scholar]
- 298.Miyoshi S, Tezuka T, Arimura S, Tomono T, Okada T, Yamanashi Y. DOK7 gene therapy enhances motor activity and life span in ALS model mice. EMBO Mol Med 9: 880–889, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Molenaar PC, Polak RL,Miledi R,Alema S, Vincent A, Newsom-Davis J. Acetylcholine in intercostal muscle from myasthenia gravis patients and in rat diaphragm after blockade of acetylcholine receptors. Prog Brain Res 49: 449–458, 1979. [DOI] [PubMed] [Google Scholar]
- 300.Montecucco C, Schiavo G, Pantano S. SNARE complexes and neuroexocytosis: How many, how close? Trends Biochem Sci 30: 367–372, 2005. [DOI] [PubMed] [Google Scholar]
- 301.Mora M, Lambert EH, Engel AG. Synaptic vesicle abnormality in familial infantile myasthenia. Neurology 37: 206–214, 1987. [DOI] [PubMed] [Google Scholar]
- 302.Mori S, Kubo S, Akiyoshi T, Yamada S, Miyazaki T, Hotta H, Desaki J, Kishi M, Konishi T, Nishino Y, Miyazawa A, Maruyama N, Shigemoto K. Antibodies against muscle-specific kinase impair both presynaptic and postsynaptic functions in a murine model of myasthenia gravis. Am J Pathol 180: 798–810, 2012. [DOI] [PubMed] [Google Scholar]
- 303.Morsch M, Reddel SW, Ghazanfari N, Toyka KV, Phillips WD. Pyridostigmine but not 3, 4-diaminopyridine exacerbates ACh receptor loss and myasthenia induced in mice by muscle-specific kinase autoantibody. J Physiol 591: 2747–2762, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Muniak CG, Kriebel ME, Carlson CG. Changes in MEPP and EPP amplitude distributions in the mouse diaphragm during synapse formation and degeneration. Dev Brain Res 5: 123–138, 1982. [DOI] [PubMed] [Google Scholar]
- 305.Muppidi S, Guptill JT, Jacob S, Li Y, Farrugia ME, Guidon AC, Tavee JO, Kaminski H, Howard JF Jr, Cutter G, Wiendl H, Maas MB, Illa I, Mantegazza R, Murai H, Utsugisawa K, Nowak RJ, Group C-MS. COVID-19-associated risks and effects in myasthenia gravis (CAREMG). Lancet Neurol 19: 970–971, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Murphy RM, Larkins NT, Mollica JP, Beard NA, Lamb GD. Calsequestrin content and SERCA determine normal and maximal Ca2+ storage levels in sarcoplasmic reticulum of fast- and slow-twitch fibres of rat. J Physiol 587: 443–460, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Nagwaney S, Harlow ML, Jung JH, Szule JA, Ress D, Xu J, Marshall RM, McMahan UJ. Macromolecular connections of active zone material to docked synaptic vesicles and presynaptic membrane at neuromuscular junctions of mouse. J Comp Neurol 513: 457–468, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Nakashima H, Ohkawara B, Ishigaki S, Fukudome T, Ito K, Tsushima M, Konishi H, Okuno T, Yoshimura T, Ito M, Masuda A, Sobue G, Kiyama H, Ishiguro N, Ohno K. R-spondin 2 promotes acetylcholine receptor clustering at the neuromuscular junction via Lgr5. Sci Rep 6: 28512, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Narayanan CH, Fox MW, Hamburger V. Prenatal development of spontaneous and evoked activity in the rat (Rattus norvegicus albinus). Behaviour 40: 100–134, 1971. [DOI] [PubMed] [Google Scholar]
- 310.Nascimento F, Pousinha PA, Correia AM, Gomes R, Sebastiao AM, Ribeiro JA. Adenosine A2A receptors activation facilitates neuromuscular transmission in the pre-symptomatic phase of the SOD1(G93A) ALS mice, but not in the symptomatic phase. PLoS One 9: e104081, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Nastuk WLH, A. L The electrical activity of singlemuscle fibers. J Cell Comp Physiol 35: 39–73, 1950. [Google Scholar]
- 312.Nemoto Y, Kuwabara S, Misawa S, Kawaguchi N, Hattori T, Takamori M, Vincent A. Patterns and severity of neuromuscular transmission failure in seronegative myasthenia gravis. J Neurol Neurosurg Psychiatry 76: 714–718, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Ngo ST, Baumann F, Ridall PG, Pettitt AN, Henderson RD, Bellingham MC, McCombe PA. The relationship between Bayesian motor unit number estimation and histological measurements of motor neurons in wild-type and SOD1(G93A) mice. Clin Neurophysiol 123: 2080–2091, 2012. [DOI] [PubMed] [Google Scholar]
- 314.Ngo ST, Cole RN, Sunn N, Phillips WD, Noakes PG. Neuregulin-1 potentiates agrin-induced acetylcholine receptor clustering through muscle-specific kinase phosphorylation. J Cell Sci 125: 1531–1543, 2012. [DOI] [PubMed] [Google Scholar]
- 315.Nguyen QT, Prasadanian AS, Snider WD, Lichtman JW. Hyperinnervation of neuromuscular junctions caused by GDNF overexpression in muscle. Science 279: 1725–1729, 1998. [DOI] [PubMed] [Google Scholar]
- 316.Niks EH, Kuks JB, Wokke JH, Veldman H, Bakker E, Verschuuren JJ, Plomp JJ. Pre- and postsynaptic neuromuscular junction abnormalities in musk myasthenia. Muscle Nerve 42: 283–288, 2010. [DOI] [PubMed] [Google Scholar]
- 317.Nishimaru H, Iizuka M, Ozaki S, Kudo N. Spontaneous motoneuronal activity mediated by glycine and GABA in the spinal cord of rat fetuses in vitro. J Physiol 497 (Pt 1): 131–143, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Norenberg MD, Rao KV. The mitochondrial permeability transition in neurologic disease. Neurochem Int 50: 983–997, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Nykjaer A, Willnow TE, Petersen CM. p75NTR--live or let die. Curr Opin Neurobiol 15: 49–57, 2005. [DOI] [PubMed] [Google Scholar]
- 320.O’Brien RA, Ostberg AJ, Vrbova G. Observations on the elimination of polyneuronal innervation in developing mammalian skeletal muscle. J Physiol 282: 571–582, 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.O’Brien RAD, Ostberg AJC, Vrbova G. Observations on the elimination of polyneuronal innervation in developing mammalian skeletal muscle. J Physiol 282: 571–582, 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Oda K. Age changes of motor innervation and acetylcholine receptor distribution on human skeletal muscle fibres. J Neurol Sci 66: 327–338, 1984. [DOI] [PubMed] [Google Scholar]
- 323.Ogata T. A histochemical study on the structural differences of motor endplate in the red, white and intermediate muscle fibers of mouse limb muscle. Acta Med Okayama 19: 149–153, 1965. [PubMed] [Google Scholar]
- 324.Ohnishi T,Yanazawa M, Sasahara T, Kitamura Y, Hiroaki H, Fukazawa Y, Kii I, Nishiyama T, Kakita A, Takeda H, Takeuchi A, Arai Y, Ito A, Komura H, Hirao H, Satomura K, Inoue M, Muramatsu S, Matsui K, Tada M, Sato M, Saijo E, Shigemitsu Y, Sakai S, Umetsu Y, Goda N, Takino N, Takahashi H, Hagiwara M, Sawasaki T, Iwasaki G, Nakamura Y, Nabeshima Y, Teplow DB, Hoshi M. Na, K-ATPase alpha3 is a death target of Alzheimer patient amyloid-beta assembly. Proc Natl Acad Sci U S A 112: E4465–E4474, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Okada K, Inoue A, Okada M, Murata Y, Kakuta S, Jigami T, Kubo S, Shiraishi H, Eguchi K, Motomura M, Akiyama T, Iwakura Y, Higuchi O, Yamanashi Y. The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science 312: 1802–1805, 2006. [DOI] [PubMed] [Google Scholar]
- 326.Oppenheim RW. The neurotrophic theory and naturally occurring motoneuron death. Trends Neurosci 12: 252–255, 1989. [DOI] [PubMed] [Google Scholar]
- 327.Oppenheim RW.Neurotrophic survivalmolecules formotoneurons: An embarrassment of riches. Neuron 17: 195–197, 1996. [DOI] [PubMed] [Google Scholar]
- 328.Oppenheim RW, Caldero J, Cuitat D, Esquerda J, Ayala V, Prevette D, Wang S. Rescue of developing spinal motoneurons from programmed cell death by the GABA(A) agonist muscimol acts by blockade of neuromuscular activity and increased intramuscular nerve branching. Mol Cell Neurosci 22: 331–343, 2003. [DOI] [PubMed] [Google Scholar]
- 329.Oppenheim RW, Caldero J, Cuitat D, Esquerda J, McArdle JJ, Olivera BM, Prevette D, Teichert RW. The rescue of developing avian motoneurons from programmed cell death by a selective inhibitor of the fetal muscle-specific nicotinic acetylcholine receptor. Dev Neurobiol 68: 972–980, 2008. [DOI] [PubMed] [Google Scholar]
- 330.Oppenheim RW, Houenou LJ, Parsadanian AS, Prevette D, Snider WD, Shen L. Glial cell line-derived neurotrophic factor and developing mammalian motoneurons: Regulation of programmed cell death among motoneuron subtypes. J Neurosci 20: 5001–5011, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Oppenheim RW, Nunez R. Electrical stimulation of hindlimb increases neuronal cell death in chick embryo. Nature 295: 57–59, 1982. [DOI] [PubMed] [Google Scholar]
- 332.Oppenheim RW, Prevette D, D’Costa A, Wang S, Houenou LJ, McIntosh JM. Reduction of neuromuscular activity is required for the rescue of motoneurons from naturally occurring cell death by nicotinic-blocking agents. J Neurosci 20: 6117–6124, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Oppenheim RW, Prevette D, Houenou LJ, Pincon-Raymond M, Dimitriadou V, Donevan A, O’Donovan M, Wenner P, McKemy DD, Allen PD. Neuromuscular development in the avian paralytic mutant crooked neck dwarf (cn/cn): Further evidence for the role of neuromuscular activity in motoneuron survival. J Comp Neurol 381: 353–372, 1997. [DOI] [PubMed] [Google Scholar]
- 334.Oppenheim RW, Prevette D, Qin-Wei Y, Collins F,MacDonald J. Control of embryonic motoneuron survival in vivo by ciliary neurotrophic factor. Science 251: 1616–1618, 1991. [DOI] [PubMed] [Google Scholar]
- 335.Ottenheijm CA, Heunks LM, Dekhuijzen RP. Diaphragm adaptations in patients with COPD. Respir Res 9: 12, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Owe JF, Daltveit AK, Gilhus NE. Causes of death among patients with myasthenia gravis in Norway between 1951 and 2001. J Neurol Neurosurg Psychiatry 77: 203–207, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Padua L, Tonali P, Aprile I, Caliandro P, Bartoccioni E, Evoli A. Seronegative myasthenia gravis: Comparison of neurophysiological picture in MuSK+ and MuSK− patients. Eur J Neurol 13: 273–276, 2006. [DOI] [PubMed] [Google Scholar]
- 338.Palay SL. Synapses in the central nervous system. J Biophys Biochem Cytol 2: 193–202, 1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Pannuzzo M, Grassi A, Raudino A. Hydrodynamic enhancement of the diffusion rate in the region between two fluctuating membranes in close opposition: A theoretical and computational study. J Phys Chem B 118: 8662–8672, 2014. [DOI] [PubMed] [Google Scholar]
- 340.Pearse BM. Clathrin: A unique protein associated with intracellular transfer of membrane by coated vesicles. Proc Natl Acad Sci U S A 73: 1255–1259, 1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Pehar M, Cassina P, Vargas MR, Castellanos R, Viera L, Beckman JS, Estevez AG, Barbeito L. Astrocytic production of nerve growth factor in motor neuron apoptosis: Implications for amyotrophic lateral sclerosis. J Neurochem 89: 464–473, 2004. [DOI] [PubMed] [Google Scholar]
- 342.Perez V, Bermedo-Garcia F, Zelada D, Court FA, Perez MA, Fuenzalida M, Abrigo J, Cabello-Verrugio C, Moya-Alvarado G, Tapia JC, Valenzuela V, Hetz C, Bronfman FC, Henriquez JP. The p75(NTR) neurotrophin receptor is required to organize themature neuromuscular synapse by regulating synaptic vesicle availability. Acta Neuropathol Commun 7: 147, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Perez-Garcia MJ, Burden SJ. Increasing MuSK activity delays denervation and improves motor function in ALS mice. Cell Rep 2: 497–502, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Personius KE, Balice-Gordon RJ. Activity-dependent editing of neuromuscular synaptic connections. Brain Res Bull 53: 513–522, 2000. [DOI] [PubMed] [Google Scholar]
- 345.Pittman R, Oppenheim RW. Cell death of motoneurons in the chick embryo spinal cord. IV. Evidence that a functional neuromuscular interaction is involved in the regulation of naturally occurring cell death and the stabilization of synapses. J Comp Neurol 187: 425–446, 1979. [DOI] [PubMed] [Google Scholar]
- 346.Plomp JJ, Morsch M, Phillips WD, Verschuuren JJ. Electrophysiological analysis of neuromuscular synaptic function in myasthenia gravis patients and animal models. Exp Neurol 270: 41–54, 2015. [DOI] [PubMed] [Google Scholar]
- 347.Plomp JJ, Van Kempen GT, De Baets MB, Graus YM, Kuks JB, Molenaar PC. Acetylcholine release in myasthenia gravis: Regulation at single end-plate level. Ann Neurol 37: 627–636, 1995. [DOI] [PubMed] [Google Scholar]
- 348.Plomp JJ, van Kempen GT, Molenaar PC. Adaptation of quantal content to decreased postsynaptic sensitivity at single endplates in alpha-bungarotoxin-treated rats. J Physiol 458: 487–499, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Prakash YS, Fournier M, Sieck GC. Effects of prenatal undernutrition on developing rat diaphragm. J Appl Physiol 75: 1044–1052, 1993. [DOI] [PubMed] [Google Scholar]
- 350.Prakash YS, Gosselin LE, Zhan WZ, Sieck GC. Alterations of diaphragm neuromuscular junctions with hypothyroidism. J Appl Physiol 81: 1240–1248, 1996. [DOI] [PubMed] [Google Scholar]
- 351.Prakash YS, Mantilla CB, Zhan WZ, Smithson KG, Sieck GC. Phrenic motoneuron morphology during rapid diaphragm muscle growth. J Appl Physiol 89: 563–572, 2000. [DOI] [PubMed] [Google Scholar]
- 352.Prakash YS, Miller SM, Huang M, Sieck GC. Morphology of diaphragm neuromuscular junctions on different fibre types. J Neurocytol 25: 88–100, 1996. [DOI] [PubMed] [Google Scholar]
- 353.Prakash YS, Miyata H, Zhan WZ, Sieck GC. Inactivity-induced remodeling of neuromuscular junctions in rat diaphragmatic muscle. Muscle Nerve 22: 307–319, 1999. [DOI] [PubMed] [Google Scholar]
- 354.Prakash YS, Sieck GC. Age-related remodeling of neuromuscular junctions on type-identified diaphragm fibers. Muscle Nerve 21: 887–895, 1998. [DOI] [PubMed] [Google Scholar]
- 355.Prakash YS, Smithson KG, Sieck GC. Measurements of phrenic motoneuron somal volumes using laser scanning confocal microscopy: Comparisons with estimates using the Cavalieri principle the nucleator. Neurosci Abstr 19: 1112, 1993. [Google Scholar]
- 356.Prakash YS, Smithson KG, Sieck GC. Growth-related alterations in motor endplates of type-identified diaphragm muscle fibres. J Neurocytol 24: 225–235, 1995. [DOI] [PubMed] [Google Scholar]
- 357.Prakash YS, Zhan WZ,Miyata H, Sieck GC. Adaptations of diaphragm neuromuscular junction following inactivity. Acta Anat 154: 147–161, 1995. [DOI] [PubMed] [Google Scholar]
- 358.Pun S, Sigrist M, Santos AF, Ruegg MA, Sanes JR, Jessell TM, Arber S, Caroni P. An intrinsic distinction in neuromuscular junction assembly and maintenance in different skeletal muscles. Neuron 34: 357–370, 2002. [DOI] [PubMed] [Google Scholar]
- 359.Punga AR, Lin S, Oliveri F, Meinen S, Ruegg MA. Muscle-selective synaptic disassembly and reorganization in MuSK antibody positive MG mice. Exp Neurol 230: 207–217, 2011. [DOI] [PubMed] [Google Scholar]
- 360.Qaisar R, Bhaskaran S, Ranjit R, Sataranatarajan K, Premkumar P, Huseman K, Van Remmen H. Restoration of SERCA ATPase prevents oxidative stress-related muscle atrophy and weakness. Redox Biol 20: 68–74, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Qaisar R, Pharaoh G, Bhaskaran S, Xu H, Ranjit R, Bian J, Ahn B, Georgescu C, Wren JD, Van Remmen H. Restoration of sarcoplasmic reticulum Ca(2+) ATPase (SERCA) activity prevents age-related muscle atrophy and weakness in mice. Int J Mol Sci 22: 37, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Raja MK, Preobraschenski J, Del Olmo-Cabrera S, Martinez-Turrillas R, Jahn R, Perez-Otano I, Wesseling JF. Elevated synaptic vesicle release probability in synaptophysin/gyrin family quadruple knockouts. elife 8: e40744, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Rathish D, Karalliyadda M. Takotsubo syndrome in patients with myasthenia gravis: A systematic review of previously reported cases. BMC Neurol 19: 281, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Reddy LV, Koirala S, Sugiura Y, Herrera AA, Ko CP. Glial cells maintain synaptic structure and function and promote development of the neuromuscular junction in vivo. Neuron 40: 563–580, 2003. [DOI] [PubMed] [Google Scholar]
- 365.Redfern P. Neuromuscular transmission in new-born rats. J Physiol 209: 701–709, 1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Reid B, Slater CR, Bewick GS. Synaptic vesicle dynamics in rat fast and slow motor nerve terminals. J Neurosci 19: 2511–2521, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Reim K, Mansour M, Varoqueaux F,McMahon HT, Sudhof TC, Brose N, Rosenmund C. Complexins regulate a late step in Ca2+-dependent neurotransmitter release. Cell 104: 71–81, 2001. [DOI] [PubMed] [Google Scholar]
- 368.Reist NE, Smith SJ. Neurally evoked calcium transients in terminal Schwann cells at the neuromuscular junction. Proc Natl Acad Sci U S A 89: 7625–7629, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Rhodes SJ, Konieczny SF. Identification of MRF4: A new member of the muscle regulatory factor gene family. Genes Dev 3: 2050–2061, 1989. [DOI] [PubMed] [Google Scholar]
- 370.Richards DA, Guatimosim C, Rizzoli SO, Betz WJ. Synaptic vesicle pools at the frog neuromuscular junction. Neuron 39: 529–541, 2003. [DOI] [PubMed] [Google Scholar]
- 371.Richman DP, Gomez CM, Berman PW, Burres SA, Fitch FW, Arnason BG. Monoclonal anti-acetylcholine receptor antibodies can cause experimental myasthenia. Nature 286: 738–739, 1980. [DOI] [PubMed] [Google Scholar]
- 372.Richman DP, Nishi K, Morell SW, Chang JM, Ferns MJ, Wollmann RL, Maselli RA, Schnier J, Agius MA. Acute severe animal model of anti-muscle-specific kinase myasthenia: Combined postsynaptic and presynaptic changes. Arch Neurol 69: 453–460, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Riethmacher D, Sonnenberg-Riethmacher E, Brinkmann V, Yamaai T, Lewin GR, Birchmeier C. Severe neuropathies in mice with targeted mutations in the ErbB3 receptor. Nature 389: 725–730, 1997. [DOI] [PubMed] [Google Scholar]
- 374.Rizzoli SO, Betz WJ. The structural organization of the readily releasable pool of synaptic vesicles. Science 303: 2037–2039, 2004. [DOI] [PubMed] [Google Scholar]
- 375.Rizzoli SO, Betz WJ. Synaptic vesicle pools. Nat Rev Neurosci 6: 57–69, 2005. [DOI] [PubMed] [Google Scholar]
- 376.Rizzoli SO, Jahn R. Kiss-and-run, collapse and ‘readily retrievable’ vesicles. Traffic 8: 1137–1144, 2007. [DOI] [PubMed] [Google Scholar]
- 377.Rizzoli SO, Richards DA, Betz WJ. Monitoring synaptic vesicle recycling in frog motor nerve terminals with FM dyes. J Neurocytol 32: 539–549, 2003. [DOI] [PubMed] [Google Scholar]
- 378.Robbins N. Compensatory plasticity of aging at the neuromuscular junction. Exp Gerontol 27: 75–81, 1992. [DOI] [PubMed] [Google Scholar]
- 379.Robbins N, Fahim MA. Progression of age changes in mature mouse motor nerve terminals and its relation to locomotor activity. J Neurocytol 14: 1019–1036, 1985. [DOI] [PubMed] [Google Scholar]
- 380.Robitaille R. Purinergic receptors and their activation by endogenous purines at perisynaptic glial cells of the frog neuromuscular junction. J Neurosci 15: 7121–7131, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Robitaille R. Modulation of synaptic efficacy and synaptic depression by glial cells at the frog neuromuscular junction. Neuron 21: 847–855, 1998. [DOI] [PubMed] [Google Scholar]
- 382.Robitaille R, Adler EM, Charlton MP. Strategic location of calcium channels at transmitter release sites of frog neuromuscular synapses. Neuron 5: 773–779, 1990. [DOI] [PubMed] [Google Scholar]
- 383.Robitaille R, Garcia ML, Kaczorowski GJ, Charlton MP. Functional colocalization of calcium and calcium-gated potassium channels in control of transmitter release. Neuron 11: 645–655, 1993. [DOI] [PubMed] [Google Scholar]
- 384.Rocha MC, Pousinha PA, Correia AM, Sebastiao AM, Ribeiro JA. Early changes of neuromuscular transmission in the SOD1(G93A) mice model of ALS start long before motor symptoms onset. PLoS One 8: e73846, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Roche SL, Sherman DL, Dissanayake K, Soucy G, Desmazieres A, Lamont DJ, Peles E, Julien JP, Wishart TM, Ribchester RR, Brophy PJ, Gillingwater TH. Loss of glial neurofascin155 delays developmental synapse elimination at the neuromuscular junction. J Neurosci 34: 12904–12918, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Rochon D, Rousse I, Robitaille R. Synapse-glia interactions at the mammalian neuromuscular junction. J Neurosci 21: 3819–3829, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Rome S, Forterre A, Mizgier ML, Bouzakri K. Skeletal muscle-released extracellular vesicles: State of the art. Front Physiol 10: 929, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Rosenheimer JL, Smith DO. Differential changes in the endplate architecture of functionally diverse muscles during aging. J Neurophysiol 53: 1567–1581, 1985. [DOI] [PubMed] [Google Scholar]
- 389.Rowley KL, Mantilla CB, Ermilov LG, Sieck GC. Synaptic vesicle distribution and release at rat diaphragm neuromuscular junctions. J Neurophysiol 98: 478–487, 2007. [DOI] [PubMed] [Google Scholar]
- 390.Ruegsegger C, Maharjan N, Goswami A, Filezac de L’Etang A, Weis J, Troost D, Heller M, Gut H, Saxena S. Aberrant association of misfolded SOD1 with Na(+)/K(+)ATPase-alpha3 impairs its activity and contributes to motor neuron vulnerability in ALS. Acta Neuropathol 131: 427–451, 2016. [DOI] [PubMed] [Google Scholar]
- 391.Ruff RL. Sodium channel slow inactivation and the distribution of sodium channels on skeletal muscle fibres enable the performance properties of different skeletal muscle fibre types. Acta Physiol Scand 156: 159–168, 1996. [DOI] [PubMed] [Google Scholar]
- 392.Ruff RL. Endplate contributions to the safety factor for neuromuscular transmission. Muscle Nerve 44: 854–861, 2011. [DOI] [PubMed] [Google Scholar]
- 393.Ruff RL, Lennon VA. How myasthenia gravis alters the safety factor for neuromuscular transmission. J Neuroimmunol 201-202: 13–20, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Ruiz R, Cano R, Casanas JJ, Gaffield MA, Betz WJ, Tabares L. Active zones and the readily releasable pool of synaptic vesicles at the neuromuscular junction of the mouse. J Neurosci 31: 2000–2008, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Ryu JK, Min D, Rah SH, Kim SJ, Park Y, Kim H, Hyeon C, Kim HM, Jahn R, Yoon TY. Spring-loaded unraveling of a single SNARE complex by NSF in one round of ATP turnover. Science 347: 1485–1489, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Sabatini BL, Regehr WG. Timing of neurotransmission at fast synapses in the mammalian brain. Nature 384: 170–172, 1996. [DOI] [PubMed] [Google Scholar]
- 397.Saied Z, Rachdi A, Thamlaoui S, Nabli F, Jeridi C, Baffoun N, Kaddour C, Belal S, Ben SS. Myasthenia gravis and COVID-19: A case series and comparison with literature. Acta Neurol Scand 144 (3): 334–340, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Salanova M, Schiffl G, Blottner D. Atypical fast SERCA1a protein expression in slow myofibers and differential S-nitrosylation prevented by exercise during long term bed rest. Histochem Cell Biol 132: 383–394, 2009. [DOI] [PubMed] [Google Scholar]
- 399.Salpeter MM, Elderfrawi ME. Sizes of end plate compartments, densities of acetylcholine receptor and other quantitative aspects of neuromuscular transmission. J Histochem Cytochem 21: 769–778, 1973. [DOI] [PubMed] [Google Scholar]
- 400.Sancho S, Young P, Suter U. Regulation of Schwann cell proliferation and apoptosis in PMP22-deficient mice and mouse models of Charcot-Marie-Tooth disease type 1A. Brain 124: 2177–2187, 2001. [DOI] [PubMed] [Google Scholar]
- 401.Sandow A. Fundamental mechanics of skeletal muscle contraction. Am J Phys Med 31: 103–125, 1952. [PubMed] [Google Scholar]
- 402.Sandrock AW Jr, Dryer SE, Rosen KM, Gozani SN, Kramer R, Theill LE, Fischbach GD. Maintenance of acetylcholine receptor number by neuregulins at the neuromuscular junction in vivo. Science 276: 599–603, 1997. [DOI] [PubMed] [Google Scholar]
- 403.Sanes JR, Lichtman JW. Development of the vertebrate neuromuscular junction. Annu Rev Neurosci 22: 389–442, 1999. [DOI] [PubMed] [Google Scholar]
- 404.Sanes JR, Lichtman JW. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci 2: 791–805, 2001. [DOI] [PubMed] [Google Scholar]
- 405.Sanes JR, Marshall LM, McMahan UJ. Reinnervation of muscle fiber basal lamina after removal ofmyofibers. Differentiation of regenerating axons at original synaptic sites. J Cell Biol 78: 176–198, 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Saroussi S, Nelson N. The little we know on the structure and machinery of V-ATPase. J Exp Biol 212: 1604–1610, 2009. [DOI] [PubMed] [Google Scholar]
- 407.Schiaffino S, Ausoni S, Gorza L, Saggin I, Gundersen K, Lomo T. Myosin heavy chain isoforms and velocity of shortening of type 2 skeletal muscle fibres. Acta Physiol Scand 134: 575–576, 1988. [DOI] [PubMed] [Google Scholar]
- 408.Schiaffino S, Gorza L, Ausoni S. Muscle fiber types expressing different myosin heavy chain isoforms. Their functional properties and adaptive capacity. In: Pette D, editor. The Dynamic State of Muscle Fibers. Berlin: De Gruyter, 1990, p. 329–341. [Google Scholar]
- 409.Schikorski T, Stevens CF. Quantitative ultrastructural analysis of hippocampal excitatory synapses. J Neurosci 17: 5858–5867, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Schleiden MJ. Beitrage zur Phytogenesis. Arch Anat Physiol Wiss Med: 137–176, 1838. [Google Scholar]
- 411.Schwann T, Hünseler F. Mikroskopische Untersuchungen über die Ubereinstimmung in der Struktur und dem Wachstume der Tiere und Pflanzen. W Engelmann 176, 1910. [Google Scholar]
- 412.Scurry AN, Heredia DJ, Feng CY, Gephart GB, Hennig GW, Gould TW. Structural and functional abnormalities of the neuromuscular junction in the Trembler-J homozygote mouse model of congenital hypomyelinating neuropathy. J Neuropathol Exp Neurol 75: 334–346, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Seeburger JL, Tarras S, Natter H, Springer JE. Spinal cord motoneurons express p75NGFR and p145trkB mRNA in amyotrophic lateral sclerosis. Brain Res 621: 111–115, 1993. [DOI] [PubMed] [Google Scholar]
- 414.Segal JR, Ceccarelli B, Fesce R, Hurlbut WP. Miniature endplate potential frequency and amplitude determined by an extension of Campbell’s theorem. Biophys J 47: 183–202, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Seven YB, Mantilla CB, Sieck GC. Recruitment of rat diaphragmmotor units across motor behaviors with different levels of diaphragm activation. J Appl Physiol 117: 1308–1316, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Seven YB, Mantilla CB, Zhan WZ, Sieck GC. Frequency-domain analysis of diaphragm muscle EMG activity across ventilatory and non-ventilatory motor behaviors. FASEB J 25: 1111.1124, 2011. [Google Scholar]
- 417.Seyfarth EA. Julius Bernstein (1839-1917): Pioneer neurobiologist and biophysicist. Biol Cybern 94: 2–8, 2006. [DOI] [PubMed] [Google Scholar]
- 418.Shaw PJ, Chinnery RM, Thagesen H, Borthwick GM, Ince PG. Immunocytochemical study of the distribution of the free radical scavenging enzymes Cu/Zn superoxide dismutase (SOD1); MN superoxide dismutase (MN SOD) and catalase in the normal human spinal cord and in motor neuron disease. J Neurol Sci 147: 115–125, 1997. [DOI] [PubMed] [Google Scholar]
- 419.Shi L, Shen QT, Kiel A, Wang J, Wang HW, Melia TJ, Rothman JE, Pincet F. SNARE proteins: One to fuse and three to keep the nascent fusion pore open. Science 335: 1355–1359, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Shortt CM, Fredsted A, Bradford A, O’Halloran KD. Diaphragm muscle remodeling in a rat model of chronic intermittent hypoxia. J Histochem Cytochem 61: 487–499, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Shrivastava AN, Redeker V, Fritz N, Pieri L, Almeida LG, Spolidoro M, Liebmann T, Bousset L, Renner M, Lena C, Aperia A, Melki R, Triller A. alpha-synuclein assemblies sequester neuronal alpha3-Na+/K+-ATPase and impair Na+ gradient. EMBO J 34: 2408–2423, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Sieb JP. Myasthenia gravis: An update for the clinician. Clin Exp Immunol 175: 408–418, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Sieck GC. Neural control of the inspiratory pump. NIPS 6: 260–264, 1991. [Google Scholar]
- 424.Sieck GC. Physiological effects of diaphragm muscle denervation and disuse. Clin Chest Med 15: 641–659, 1994. [PubMed] [Google Scholar]
- 425.Sieck GC, Ferreira LF, Reid MB, Mantilla CB. Mechanical properties of respiratory muscles. Compr Physiol 3: 1553–1567, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Sieck GC, Fournier M. Diaphragm motor unit recruitment during ventilatory and nonventilatory behaviors. J Appl Physiol 66: 2539–2545, 1989. [DOI] [PubMed] [Google Scholar]
- 427.Sieck GC, Fournier M, Enad JG. Fiber type composition ofmuscle units in the cat diaphragm. Neurosci Lett 97: 29–34, 1989. [DOI] [PubMed] [Google Scholar]
- 428.Sieck GC, Han YS, Prakash YS, Jones KA. Cross-bridge cycling kinetics, actomyosin ATPase activity and myosin heavy chain isoforms in skeletal and smooth respiratory muscle. Comp Biochem Physiol 119: 435–450, 1997. [DOI] [PubMed] [Google Scholar]
- 429.Sieck GC, Prakash YS. Fatigue at the neuromuscular junction: Branch point vs. presynaptic vs. postsynaptic mechanisms. In: Stuart DG, Gandevia S, Enoka RM, McComas AJ, Thomas CK, editors. Neural and Neuromuscular Aspects of Muscle Fatigue. New York, NY: Plenum Press, 1995, p. 83–100. [PubMed] [Google Scholar]
- 430.Sieck GC, Van Balkom RH, Prakash YS, Zhan WZ, Dekhuijzen PN. Corticosteroid effects on diaphragm neuromuscular junctions. J Appl Physiol 86: 114–122, 1999. [DOI] [PubMed] [Google Scholar]
- 431.Sieck GC, Zhan WZ, Prakash YS, Daood MJ, Watchko JF. SDH and actomyosin ATPase activities of different fiber types in rat diaphragm muscle. J Appl Physiol 79: 1629–1639, 1995. [DOI] [PubMed] [Google Scholar]
- 432.Slater CR. Postnatal maturation of nerve-muscle junctions in hindlimb muscles of the mouse. Dev Biol 94: 11–22, 1982. [DOI] [PubMed] [Google Scholar]
- 433.Slater CR. The functional organization of motor nerve terminals. Prog Neurobiol 134: 55–103, 2015. [DOI] [PubMed] [Google Scholar]
- 434.Slater CR. The structure of human neuromuscular junctions: Some unanswered molecular questions. Int J Mol Sci 18: 2183, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Slater CR, Lyons PR, Walls TJ, Fawcett PR, Young C. Structure and function of neuromuscular junctions in the vastus lateralis of man. A motor point biopsy study of two groups of patients. Brain 115 (Pt 2): 451–478, 1992. [PubMed] [Google Scholar]
- 436.Smith DO, Chapman MR. Acetylcholine receptor binding properties at the rat neuromuscular junction during aging. J Neurochem 48: 1834–1841, 1987. [DOI] [PubMed] [Google Scholar]
- 437.Smith DO, Williams KD, Emmerling M. Changes in acetylcholine receptor distribution and binding properties at the neuromuscular junction during aging. Int J Dev Neurosci 8: 629–642, 1990. [DOI] [PubMed] [Google Scholar]
- 438.Smith IW, Mikesh M, Lee Y, Thompson WJ. Terminal Schwann cells participate in the competition underlying neuromuscular synapse elimination. J Neurosci 33: 17724–17736, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Soendenbroe C, Heisterberg MF, Schjerling P, Karlsen A, Kjaer M, Andersen JL,Mackey AL.Molecular indicators of denervation in aging human skeletal muscle. Muscle Nerve 60: 453–463, 2019. [DOI] [PubMed] [Google Scholar]
- 440.Sole G, Mathis S, Friedman D, Salort-Campana E, Tard C, Bouhour F, Magot A, Annane D, Clair B, Le Masson G, Soulages A, Duval F, Carla L, Violleau MH, Saulnier T, Segovia-Kueny S, Kern L, Antoine JC, Beaudonnet G, Audic F, Kremer L, Chanson JB, Nadaj-Pakleza A, Stojkovic T, Cintas P, Spinazzi M, Foubert-Samier A, Attarian S. Impact of coronavirus disease 2019 in a French cohort of myasthenia gravis. Neurology 96 (16): e2109–e2120, 2021. [DOI] [PubMed] [Google Scholar]
- 441.Sotelo C. Camillo Golgi and Santigo Ramon y Cajal: The anatomical organization of the cortex of the cerebellum. Can the neuron doctrine still support our actual knowledge on the cerebellar structural arrangement? Brain Res Rev 66 (1-2): 16–34, 2011. [DOI] [PubMed] [Google Scholar]
- 442.Soykan T, Maritzen T, Haucke V. Modes and mechanisms of synaptic vesicle recycling. Curr Opin Neurobiol 39: 17–23, 2016. [DOI] [PubMed] [Google Scholar]
- 443.Sterz R, Pagala M, Peper K. Postjunctional characteristics of the endplates in mammalian fast and slow muscles. Pflugers Arch 398: 48–54, 1983. [DOI] [PubMed] [Google Scholar]
- 444.Steyn FJ, Lee K, Fogarty MJ, Veldhuis JD, McCombe PA, Bellingham MC, Ngo ST, Chen C. Growth hormone secretion is correlated with neuromuscular innervation rather than motor neuron number in earlysymptomatic male amyotrophic lateral sclerosis mice. Endocrinology 154: 4695–4706, 2013. [DOI] [PubMed] [Google Scholar]
- 445.Stockbridge N. Differential conduction at axonal bifurcations. II. Theoretical basis. J Neurophysiol 59: 1286–1295, 1988. [DOI] [PubMed] [Google Scholar]
- 446.Stockbridge N, Stockbridge LL. Differential conduction at axonal bifurcations. I. Effect of electrotonic length. J Neurophysiol 59: 1277–1285, 1988. [DOI] [PubMed] [Google Scholar]
- 447.Stryker E, Johnson KG. LAR, liprin alpha and the regulation of active zone morphogenesis. J Cell Sci 120: 3723–3728, 2007. [DOI] [PubMed] [Google Scholar]
- 448.Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci 27: 509–547, 2004. [DOI] [PubMed] [Google Scholar]
- 449.Sudhof TC. Neurotransmitter release: The last millisecond in the life of a synaptic vesicle. Neuron 80: 675–690, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Szule JA, Jung JH, McMahan UJ. The structure and function of ’active zone material’ at synapses. Philos Trans R Soc Lond Ser B Biol Sci 370: 20140189, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Taetzsch T, Tenga MJ, Valdez G. Muscle fibers secrete FGFBP1 to slow degeneration of neuromuscular synapses during aging and progression of ALS. J Neurosci 37: 70–82, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Takamori M. Myasthenia gravis: From the viewpoint of pathogenicity focusing on acetylcholine receptor clustering, trans-synaptic homeostasis and synaptic stability. Front Mol Neurosci 13: 86, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Takamori S, Holt M, Stenius K, Lemke EA, Gronborg M, Riedel D, Urlaub H, Schenck S, Brugger B, Ringler P, Muller SA, Rammner B, Grater F, Hub JS, De Groot BL, Mieskes G, Moriyama Y, Klingauf J, Grubmuller H, Heuser J, Wieland F, Jahn R. Molecular anatomy of a trafficking organelle. Cell 127: 831–846, 2006. [DOI] [PubMed] [Google Scholar]
- 454.Takeuchi A. Junctional transmission I. Postsynaptic mechanisms. Compr Physiol: 295–327, 2011.23737174 [Google Scholar]
- 455.Talmadge RJ, Roy RR, Bodine-Fowler SC, Pierotti DJ, Edgerton VR. Adaptations in myosin heavy chain profile in chronically unloaded muscles. Basic Appl Myol 5: 117–137, 1995. [PubMed] [Google Scholar]
- 456.Tanaka H, Furuya T, Kameda N, Kobayashi T, Mizusawa H. Triad proteins and intracellular Ca2+ transients during development of human skeletal muscle cells in aneural and innervated cultures. J Muscle Res Cell Motil 21: 507–526, 2000. [DOI] [PubMed] [Google Scholar]
- 457.Tang J, Landmesser L. Reduction of intramuscular nerve branching and synaptogenesis is correlated with decreased motoneuron survival. J Neurosci 13: 3095–3103, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Tansey EM. Henry Dale and the discovery of acetylcholine. C R Biol 329: 419–425, 2006. [DOI] [PubMed] [Google Scholar]
- 459.Tapia JC, Wylie JD, Kasthuri N, Hayworth KJ, Schalek R, Berger DR, Guatimosim C, Seung HS, Lichtman JW. Pervasive synaptic branch removal in the mammalian neuromuscular system at birth. Neuron 74: 816–829, 2012. [DOI] [PubMed] [Google Scholar]
- 460.Taxt T. Local and systemic effects of tetrodotoxin on the formation and elimination of synapses in reinnervated adult rat muscle. J Physiol 340: 175–194, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Thomas MM, Khan W, Betik AC, Wright KJ, Hepple RT. Initiating exercise training in late middle age minimally protects muscle contractile function and increases myocyte oxidative damage in senescent rats. Exp Gerontol 45: 856–867, 2010. [DOI] [PubMed] [Google Scholar]
- 462.Thompson CB, McDonough AA. Skeletal muscle Na, K-ATPase alpha and beta subunit protein levels respond to hypokalemic challenge with isoform and muscle type specificity. J Biol Chem 271: 32653–32658, 1996. [DOI] [PubMed] [Google Scholar]
- 463.Thompson DM, Buettner HM. Neurite outgrowth is directed by schwann cell alignment in the absence of other guidance cues. Ann Biomed Eng 34: 161–168, 2006. [DOI] [PubMed] [Google Scholar]
- 464.Thompson W. Synapse elimination in neonatal rat muscle is sensitive to pattern of muscle use. Nature 302: 614–616, 1983. [DOI] [PubMed] [Google Scholar]
- 465.Todd KJ, Darabid H, Robitaille R. Perisynaptic glia discriminate patterns of motor nerve activity and influence plasticity at the neuromuscular junction. J Neurosci 30: 11870–11882, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Tokumaru H, Umayahara K, Pellegrini LL, Ishizuka T, Saisu H, Betz H, Augustine GJ, Abe T. SNARE complex oligomerization by synaphin/complexin is essential for synaptic vesicle exocytosis. Cell 104: 421–432, 2001. [DOI] [PubMed] [Google Scholar]
- 467.Tomlinson BE, Irving D. The numbers of limb motor neurons in the human lumbosacral cord throughout life. J Neurol Sci 34: 213–219, 1977. [DOI] [PubMed] [Google Scholar]
- 468.Tong JJ. Mitochondrial delivery is essential for synaptic potentiation. Biol Bull 212: 169–175, 2007. [DOI] [PubMed] [Google Scholar]
- 469.Trachtenberg JT, Thompson WJ. Nerve terminal withdrawal from rat neuromuscular junctions induced by neuregulin and Schwann cells. J Neurosci 17: 6243–6255, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Trelease RB, Sieck GC, Harper RM. A new technique for acute and chronic recording of crural diaphragm EMG in cats. Electroencephalogr Clin Neurophysiol 53: 459–462, 1982. [DOI] [PubMed] [Google Scholar]
- 471.Tremblay E, Martineau E, Robitaille R. Opposite synaptic alterations at the neuromuscular junction in an ALS mouse model: When motor units matter. J Neurosci 37: 8901–8918, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Trinidad JC, Fischbach GD, Cohen JB. The Agrin/MuSK signaling pathway is spatially segregated from the neuregulin/ErbB receptor signaling pathway at the neuromuscular junction. J Neurosci 20: 8762–8770, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Turner MR, Hardiman O, Benatar M, Brooks BR, Chio A, de Carvalho M, Ince PG, Lin C, Miller RG, Mitsumoto H, Nicholson G, Ravits J, Shaw PJ, Swash M, Talbot K, Traynor BJ, Van den Berg LH, Veldink JH, Vucic S, Kiernan MC. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol 12: 310–322, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Van Hoecke A, Schoonaert L, Lemmens R, Timmers M, Staats KA, Laird AS, Peeters E, Philips T, Goris A, Dubois B, Andersen PM, Al-Chalabi A, Thijs V, Turnley AM, vanVught PW, Veldink JH, Hardiman O, Van Den Bosch L, Gonzalez-Perez P, Van Damme P, Brown RH Jr, van den Berg LH, Robberecht W. EPHA4 is a disease modifier of amyotrophic lateral sclerosis in animal models and in humans. NatMed 18: 1418–1422, 2012. [DOI] [PubMed] [Google Scholar]
- 475.van Lunteren E, Moyer M, Kaminski HJ. Adverse effects of myasthenia gravis on rat phrenic diaphragm contractile performance. J Appl Physiol (1985) 97: 895–901, 2004. [DOI] [PubMed] [Google Scholar]
- 476.Vanlandingham PA, Barmchi MP, Royer S, Green R, Bao H, Reist N, Zhang B. AP180 couples protein retrieval to clathrin-mediated endocytosis of synaptic vesicles. Traffic 15: 433–450, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Vernon EM, Oppenheim RW, Johnson JE. Distinct muscle targets do not vary in the developmental regulation of brain-derived neurotrophic factor. J Comp Neurol 470: 330–337, 2004. [DOI] [PubMed] [Google Scholar]
- 478.Viana Di Prisco G. Hebb synaptic plasticity. Prog Neurobiol 22: 89–102, 1984. [DOI] [PubMed] [Google Scholar]
- 479.Vila L, Barrett EF, Barrett JN. Stimulation-induced mitochondrial [Ca2+] elevations in mouse motor terminals: Comparison of wild-type with SOD1-G93A. J Physiol 549: 719–728, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Vilmont V, Cadot B, Vezin E, Le Grand F, Gomes ER. Dynein disruption perturbs post-synaptic components and contributes to impaired MuSK clustering at the NMJ: Implication in ALS. Sci Rep 6: 27804, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Vock VM, Ponomareva ON, Rimer M. Evidence for muscle-dependent neuromuscular synaptic site determination in mammals. J Neurosci 28: 3123–3130, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Vos M, Lauwers E, Verstreken P. Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front Synaptic Neurosci 2: 139, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Wainger BJ, Macklin EA, Vucic S, McIlduff CE, Paganoni S, Maragakis NJ, Bedlack R, Goyal NA, Rutkove SB, Lange DJ, Rivner MH, Goutman SA, Ladha SS, Mauricio EA, Baloh RH, Simmons Z, Pothier L, Kassis SB, La T, Hall M, Evora A, Klements D, Hurtado A, Pereira JD, Koh J, Celnik PA, Chaudhry V, Gable K, Juel VC, Phielipp N, Marei A, Rosenquist P, Meehan S, Oskarsson B, Lewis RA, Kaur D, Kiskinis E, Woolf CJ, Eggan K, Weiss MD, Berry JD, David WS, Davila-Perez P, Camprodon JA, Pascual-Leone A, Kiernan MC, Shefner JM, Atassi N, Cudkowicz ME. Effect of ezogabine on cortical and spinal motor neuron excitability in amyotrophic lateral sclerosis: A randomized clinical trial. JAMA Neurol 78: 186–196, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Walrond JP, Reese TS. Structure of axon terminals and active zones at synapses on lizard twitch and tonic muscle fibers. J Neurosci 5: 1118–1131, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Wernig A, Herrera AA. Sprouting and remodelling at the nerve-muscle junction. Prog Neurobiol 27: 251–291, 1986. [DOI] [PubMed] [Google Scholar]
- 486.Westerberg E, Punga AR. Mortality rates and causes of death in Swedish myasthenia gravis patients. Neuromuscul Disord 30: 815–824, 2020. [DOI] [PubMed] [Google Scholar]
- 487.Whelchel DD, Brehmer TM, Brooks PM, Darragh N, Coffield JA. Molecular targets of botulinum toxin at the mammalian neuromuscular junction. Mov Disord 19 (Suppl 8): S7–S16, 2004. [DOI] [PubMed] [Google Scholar]
- 488.Willadt S, Nash M, Slater CR. Age-related fragmentation of the motor endplate is not associated with impaired neuromuscular transmission in the mouse diaphragm. Sci Rep 6: 24849, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Williams AH, Liu N, van Rooij E, Olson EN. MicroRNA control of muscle development and disease. Curr Opin Cell Biol 21: 461–469, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Williams AH, Valdez G, Moresi V, Qi X, McAnally J, Elliott JL, Bassel-Duby R, Sanes JR, Olson EN. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science 326: 1549–1554, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Willshaw DJ. The establishment and the subsequent elimination of polyneuronal innervation of developing muscle: Theoretical considerations. Proc R Soc Lond B Biol Sci 212: 233–252, 1981. [DOI] [PubMed] [Google Scholar]
- 492.Wong-Riley MT. Cytochrome oxidase: An endogenous metabolic marker for neuronal activity. Trends Neurosci 12: 94–101, 1989. [DOI] [PubMed] [Google Scholar]
- 493.Wood SJ, Slater CR. Safety factor at the neuromuscular junction. Prog Neurobiol 64: 393–429, 2001. [DOI] [PubMed] [Google Scholar]
- 494.Wood SJS, C.R Quantal content at neuromuscular junctions that lack postsynaptic folds. J Physiol Lond 511: 142P–142P, 1998. [Google Scholar]
- 495.Wray SH. Innervation ratios for large and small limb muscles in the baboon. J Comp Neurol 137: 227–250, 1969. [DOI] [PubMed] [Google Scholar]
- 496.Wu J, Yan Z, Li Z, Qian X, Lu S, Dong M, Zhou Q, Yan N. Structure of the voltage-gated calcium channel Ca(v)1.1 at 3.6 A resolution. Nature 537: 191–196, 2016. [DOI] [PubMed] [Google Scholar]
- 497.Wu KD, Lytton J. Molecular cloning and quantification of sarcoplasmic reticulum Ca(2+)-ATPase isoforms in rat muscles. Am J Phys 264: C333–C341, 1993. [DOI] [PubMed] [Google Scholar]
- 498.Wu W, Li L, Yick LW, Chai H, Xie Y, Yang Y, Prevette DM, Oppenheim RW. GDNF and BDNF alter the expression of neuronal NOS, c-Jun, and p75 and prevent motoneuron death following spinal root avulsion in adult rats. J Neurotrauma 20: 603–612, 2003. [DOI] [PubMed] [Google Scholar]
- 499.Wuerker RB, McPhedran M, Henneman E. Properties of motor units in a heterogeneous pale muscle (M. gastrocnemius) of the cat. J Neurophysiol 28: 85–99, 1965. [DOI] [PubMed] [Google Scholar]
- 500.Xu K, Jha S, Hoch W, Dryer SE. Delayed synapsing muscles are more severely affected in an experimental model of MuSK-induced myasthenia gravis. Neuroscience 143: 655–659, 2006. [DOI] [PubMed] [Google Scholar]
- 501.Yang DP, Zhang DP, Mak KS, Bonder DE, Pomeroy SL, Kim HA. Schwann cell proliferation during Wallerian degeneration is not necessary for regeneration and remyelination of the peripheral nerves: Axon-dependent removal of newly generated Schwann cells by apoptosis. Mol Cell Neurosci 38: 80–88, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Yang X, Arber S,William C, Li L, Tanabe Y, Jessell TM, Birchmeier C, Burden SJ. Patterning of muscle acetylcholine receptor gene expression in the absence of motor innervation. Neuron 30: 399–410, 2001. [DOI] [PubMed] [Google Scholar]
- 503.Yao C, Wang Q, Wang Y, Wu J, Cao X, Lu Y, Chen Y, Feng W, Gu X, Dun XP, Yu B. Loc680254 regulates Schwann cell proliferation through Psrc1 and Ska1 as a microRNA sponge following sciatic nerve injury. Glia 69: 2391–2403, 2021. [DOI] [PubMed] [Google Scholar]
- 504.York AL, Zheng JQ. Super-resolution microscopy reveals a nanoscale organization of acetylcholine receptors for trans-synaptic alignment at neuromuscular synapses. eNeuro 4, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Yoshihara T, Ishii T, Iwata M, Nomoto M. Ultrastructural and histochemical study of the motor end plates of the intrinsic laryngeal muscles in amyotrophic lateral sclerosis. Ultrastruct Pathol 22: 121–126, 1998. [DOI] [PubMed] [Google Scholar]
- 506.Zahler R, Sun W, Ardito T, Zhang ZT, Kocsis JD, Kashgarian M. The alpha3 isoform protein of the Na+, K(+)-ATPase is associated with the sites of cardiac and neuromuscular impulse transmission. Circ Res 78: 870–879, 1996. [DOI] [PubMed] [Google Scholar]
- 507.Zajac FE, Faden JS. Relationship among recruitment order, axonal conduction velocity, and muscle-unit properties of type-identified motor units in cat plantaris muscle. J Neurophysiol 53 (5): 1303–1322, 1985. [DOI] [PubMed] [Google Scholar]
- 508.Zengel JE, Reid SA, Sypert GW, Munson JB. Membrane electrical properties and prediction of motor-unit type of medial gastrocnemius motoneurons in the cat. J Neurophysiol 53 (5): 1323–1344, 1985. [DOI] [PubMed] [Google Scholar]
- 509.Zhan WZ,Mantilla CB, Sieck GC. Regulation of neuromuscular transmission by neurotrophins. Sheng Li Xue Bao 55: 617–624, 2003. [PubMed] [Google Scholar]
- 510.Zhang B, Koh YH, Beckstead RB, Budnik V, Ganetzky B, Bellen HJ. Synaptic vesicle size and number are regulated by a clathrin adaptor protein required for endocytosis. Neuron 21: 1465–1475, 1998. [DOI] [PubMed] [Google Scholar]
- 511.Zhang L,Morris KJ, Ng YC. Fiber type-specific immunostaining of the Na+, K+-ATPase subunit isoforms in skeletal muscle: Age-associated differential changes. Biochim Biophys Acta 1762: 783–793, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512.Zhu X, Lai C, Thomas S, Burden SJ. Neuregulin receptors, erbB3 and erbB4, are localized at neuromuscular synapses. EMBO J 14: 5842–5848, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]