

Abstract
Human skin uses millions of hairs and glands distributed across the body surface to function as an external barrier, thermoregulator, and stimuli sensor. The large-scale generation of human skin with these appendages would be beneficial, but is challenging. Here, we describe a detailed protocol for generating hair-bearing skin tissue entirely from a homogeneous population of human pluripotent stem cells in a three-dimensional in vitro culture system. Defined culture conditions are used over a 2-week period to induce differentiation of pluripotent stem cells to surface ectoderm and cranial neural crest cells, which give rise to the epidermis and dermis, respectively, in each organoid unit. After 60 days of incubation, the skin organoids produce hair follicles. By day ~130, the skin organoids reach full complexity and contain stratified skin layers, pigmented hair follicles, sebaceous glands, Merkel cells, and sensory neurons, recapitulating the cell composition and architecture of fetal skin tissue at week-18 of gestation. Skin organoids can be maintained in culture using this protocol for up to 150 days, enabling the organoids to be used to investigate basic skin biology, model disease, and further, the reconstruction or regeneration of skin tissue.
Introduction
The skin is our barrier to the outside world and protects the body against any thermal, chemical, or other noxious stimuli or substances1,2. The skin also helps regulate fluids and temperature within our bodies2,3. In addition, sensory nerve endings in the skin mediate information on touch, pain, heat, and cold2,4,5. Perhaps due to its critical role in various bodily functions, diseases of the skin are common, with >5.4 million cases of skin cancer every year and ~500,000 patients who receive treatment for burns or other skin trauma6–11. The mouse is a standard model for investigating skin development and disease in vivo12–17. Yet, there are critical differences between mouse and human skin, such as the mechanisms of wound closure, which have confounded efforts to translate therapies to the clinic18. Therefore, decades of research have been devoted to developing in vitro human skin equivalent models with layers composed of primary human skin cells and extracellular matrix components6,19–22. Skin equivalents are used to evaluate drug toxicity and therapeutic efficacy and to minimize the use of animals for research23,24. However, human skin equivalents have notable drawbacks, such as a lack of crucial skin tissue structures (e.g., hair follicles and sebaceous glands) and limited culture durations (e.g., 2–3 weeks)19–22. Therefore, new approaches to generate complete human skin models, as described here, have been developed.
This protocol provides step-by-step guidance to form human pluripotent stem cell (hPSC)-derived skin organoids, which we have previously shown can recapitulate many features of full-thickness human fetal skin and be xenografted on nude mice25 (Fig. 1). Below, we discuss the primary benefits of using the skin organoid system and provide detailed instructions for quality control during the cell differentiation process. This protocol can be performed in any laboratory setup for aseptic cell culture and by anyone trained in pluripotent stem cell (PSC) culture and cell differentiation techniques.
Fig. 1. Schematics comparing in vivo and in vitro skin organogenesis.
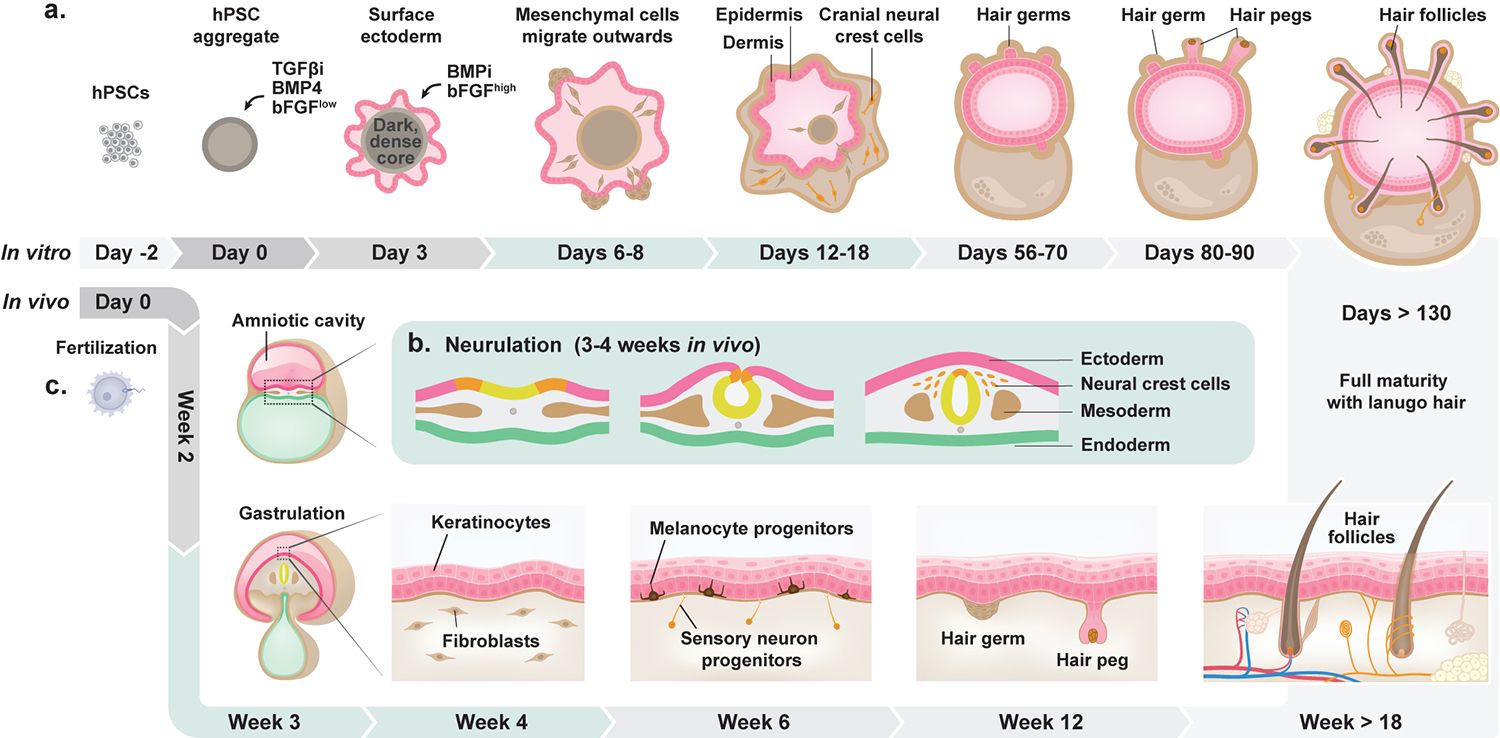
a, A timeline of in vitro skin organogenesis in the skin organoid model. hPSCs form aggregates on day 0 of differentiation. These aggregates are treated with TGF-β inhibitor (SB), BMP4, and a low concentration of bFGF, giving rise to surface ectoderm by day 3. The aggregates are then treated with BMP inhibitor (LDN) and a high concentration of bFGF. This day 3 treatment induces the development of non-epithelial cell populations consisting, in part, of cranial neural crest (CNC) cells, which further differentiate into diverse mesenchymal and neuro-glial cell populations contributing to the dermis layer of the organoid. Although rare, hair germs can be seen as early as day 56 of differentiation, with more mature hair pegs and hair follicles arising around day 70 through day 130 of differentiation. The fully mature skin organoid includes appendages, such as hair follicles, adipocytes, melanocytes, sebaceous glands, and sensory neurons. b, A schematic of neurulation in vivo. Neurulation occurs around 3–4 weeks of development, corresponding to days 6–18 of organoid culture. The ectoderm folds and pinches inward, resulting in the formation of the neural tube and, in the cranial region, delaminating CNC cells. CNC cells give rise to diverse cell lineages, such as chondrocytes, myocytes, fibroblasts, neurons, and Schwann cells. c, A timeline of in vivo skin development. Following fertilization, the zygote undergoes many rounds of rapid division. By day 12, the epiblast (amniotic cavity) and hypoblast (yolk sac) compose the bilaminar disk. The epiblast gives rise to the definitive germ layers, the ectoderm, mesoderm, and endoderm. In relation to the skin, the ectoderm gives rise to keratinocyte precursors (epidermis layer of skin), while the dermal fibroblasts (dermis layer) are derived primarily from the mesoderm in the body and CNCs in the face. By 6 weeks, melanocyte precursors, sensory neuron progenitors, and other diverse cell progenitors appear. Hair germs, which develop into hair pegs and finally into hair follicles, start to form around 12 weeks of gestation. Fully stratified skin with erupted lanugo hair is reached around week 18 of gestation. The skin further matures and develops diverse appendages, such as blood vessels, sweat glands, sebaceous glands, and a network of sensory neurons (thermoreceptors, mechanoreceptors, and nociceptors).
Development of the method and comparison with other methods
Skin development in the embryo involves coordinated interactions between epidermal and dermal cells, forming stratified skin layers and producing appendages, such as hair follicles and eccrine (sweat) glands. These higher-order skin structures arise by mechanical forces and chemical signaling cues, which are poorly understood and challenging to reproduce in cell cultures26–28. In an attempt to produce skin outside of the body, methods have been developed to isolate the two major skin cell types, keratinocytes (epidermal cells) and fibroblasts (dermal cells), from patients or by directly differentiating them from PSCs. In air-liquid interface cultures, keratinocytes and fibroblasts can be configured in a bilayer—with or without collagen matrices—to form stratified layers that mimic the general architecture of native skin, albeit without appendages19. To encourage appendage growth, undefined in vivo models, such as the mouse, have been used12,15,17,19,20. In these studies, the isolated or derived cells were mixed and transplanted into the hypodermis of the skin or subrenal capsules of the mice, either with12 or without17,20 extracellular matrix scaffolds (e.g., collagen and poly(ethylene terephthalate) membrane). Despite some advances, a clear demonstration that appendage-bearing skin could be recreated in culture remained elusive, until our publication where we demonstrated how to generate hair-bearing skin from mouse PSCs29.
While formulating our skin organoid method, we determined a chemically defined environment for co-induction of epidermal and dermal cell layers that properly modulated signaling cues according to the temporal progression of normal skin development. The skin organoid protocol arose from our work on a mouse inner ear organoid (IEO) model30, which we adapted initially from existing forebrain31 and retinal32 organoid protocols. These prior methods direct a 3D aggregate of PSCs into a neuroepithelium. We altered these existing methods for our IEO method by using a combination of a transforming growth factor-beta (TGF-β) inhibitor and recombinant bone morphogenetic protein-4 (BMP4) to induce surface ectoderm development in cell aggregates. Subsequently, we could induce placode development by activating the fibroblast growth factor (FGF) signaling pathway while simultaneously inhibiting the BMP signaling pathway, approximately 24–48 hours after surface ectoderm specification. We found that WNT signaling pathway activation was critical to stimulate otic development following these initial treatments. Using mouse PSCs, these signaling manipulations led to multi-lineage organoids containing both ectoderm (on-target; characterized by Tfap2a and E-cadherin expression) and mesoderm (off-target; characterized by Tbxt expression) cell types. Not surprisingly, we found that the surface ectoderm layers, induced early in the culture, formed both IEOs and epidermal organoids, mimicking the formation of the skin that covers the outer ear. However, we were surprised to find that the epidermal organoids formed an epidermis and a dermis, which generated hair follicles resembling bona fide mouse hair follicles. We suspected that the dermis arose from Tbxt+ paraxial mesodermal-like precursor cells. With a slight modification of the mouse IEO culture regimen (i.e., removing the WNT stimulus that is critical for otic induction), we could steer the cell aggregate toward primarily forming hairy skin tissue29. These findings marked the first demonstration that de novo hair could be generated entirely from mouse PSCs in culture.
To create human skin organoids, we strategically biased our human IEO differentiation culture toward the skin lineage (Figs. 1 and 2)25,33. A critical distinction between our human and mouse IEO inductions is that TBXT+ mesodermal lineage cells appear to be missing in the human IEO system, perhaps due to the higher dosage of a TGF-β pathway inhibitor used in the human protocol33. Thus, one of our initial concerns was whether a proper dermis could form in these cultures. However, our human IEO data suggested that a mesenchymal population with dermal fibroblast potential arises from cranial neural crest (CNC) cells that co-develop with human IEOs. It is also possible that a TBXT-independent mesoderm, such as lateral plate mesoderm34 or cranial paraxial mesoderm35, emerges during human skin organoid induction; however, this possibility is yet to be confirmed. While the body’s dermis arises from the mesoderm germ layer, the dermis of the face arises from CNC cells. To co-induce surface ectoderm and mesenchymal cells in a reproducible cystic structure, we meticulously modulated and optimized the major signaling manipulations (TGF-β, BMP4, and basic fibroblast growth factor (bFGF)). We screened for conditions that consistently produced cysts covered by a layer of mesenchyme as determined by an opaque appearance under phase-contrast imaging and follow-up immunohistochemistry (IHC) analysis for platelet-derived growth factor receptor α (PDGFRα) expression19,29,36. For long-term maturation, we transitioned statically cultured organoids (days 0–12 of culture) to a floating culture condition with constant medium agitation on an orbital shaker. Qualitatively, we found that hair-forming skin organoids arose more consistently and reproducibly with constant agitation; yet, instances of organoids forming hair follicles were also observed in a static culture. Over a month in culture, the epidermal layer of the organoid stratified inward, with the apical layers (suprabasal and peridermal layers) oriented toward the organoid core. Fibroblasts, representing a papillary dermis, entirely enveloped the basal layer of the epidermis, forming an outer “crust” of the skin organoid. After days 55–75 of differentiation, we observed the initial formation of hair placodes and germs, with further differentiation into hair pegs occurring during days 75–90 of differentiation (Fig. 3). By days 110–130, the hair pegs formed more elaborate hair follicles containing sebaceous glands (sebum-producing glands) and a bulge region with Merkel cells (touch-sensing cells) and nuclear factor of activated T cells 1 (NFATC1)+ hair follicle stem cell-like cells (Fig. 3)25. After follicle initiation, we have observed continuous initiation of new hair germs throughout the later stages of the culture; thus, by days 120–140 of culture, there is typically a mix of follicles at various developmental stages. The in vitro hair follicle developmental timeline corresponds well to that of human fetal hair follicle development in vivo: the hair placode, germ, peg, and follicle differentiation stages occur, mostly, between 80 and 170 days (12–25 gestational weeks), where the full anagen stage hair follicles develop by day 126 (18 gestational weeks; Fig. 1)37. We also observed adipocytes (fat cells), sensory neurons, Schwann cells, and melanocytes (pigment cells) developing in a proper spatial organization relative to native human skin. The neuronal and glial components of skin organoids appeared to be CNC-derived. Notably, the timing of the emergence of these various cell types mimics normal human development. We provide more details of the procedure to generate human skin organoids in ‘Experimental design’.
Fig. 2. Illustration of day-by-day differentiation protocol and representative checkpoint images.
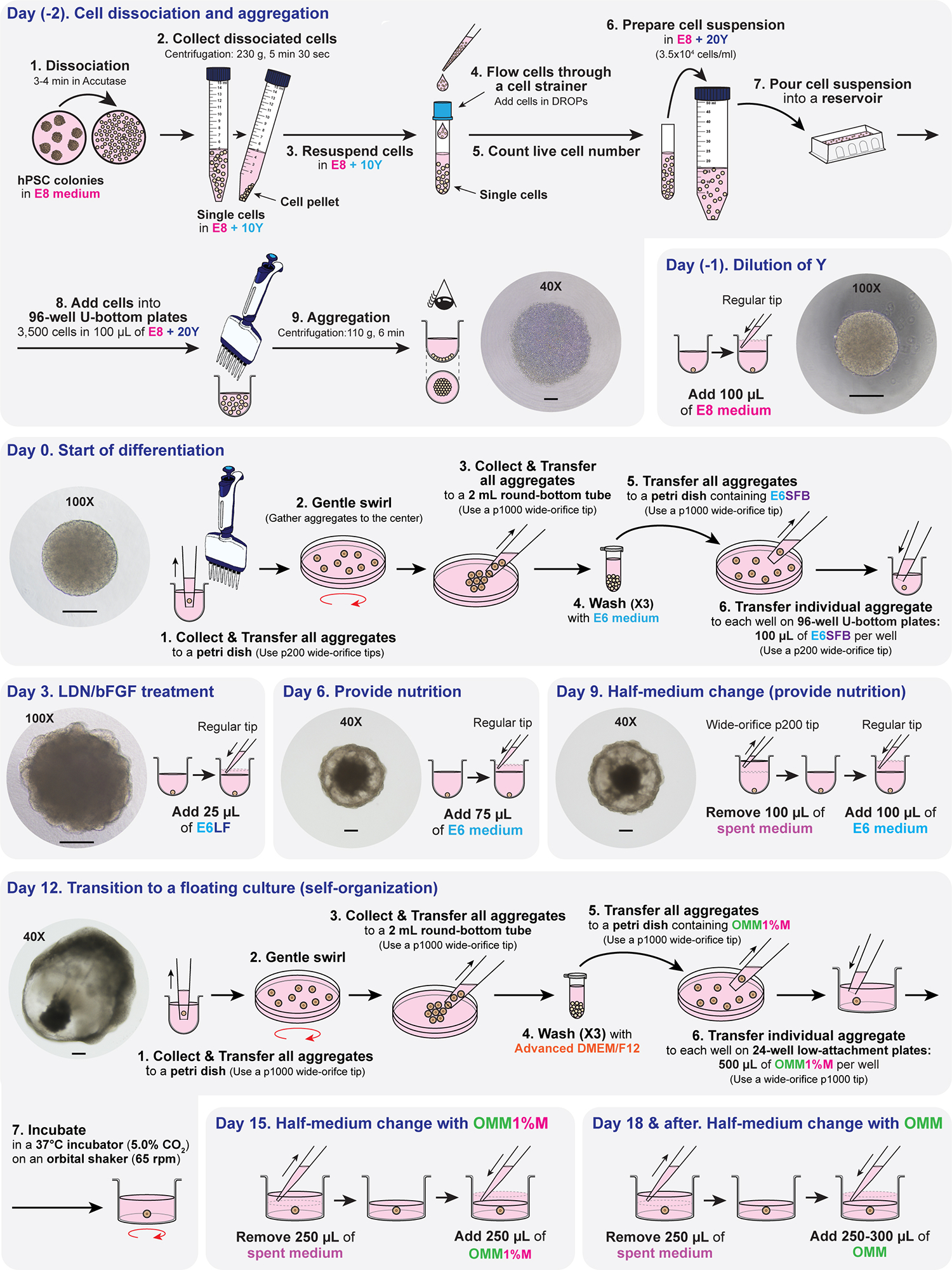
Briefly, on day (−2) of differentiation, the hPSCs cultured as colonies are dissociated into single cells and aggregated at a concentration of 3,500 cells in 100 μL of E8 + Y per well in a 96-well U-bottom plate. The E8 contains Y to inhibit apoptosis of cells when they become single-celled. After centrifugation, the single cells concentrate at the bottom of the 96-well U-bottom plate, forming a circular cell cluster layer. The single cells migrate and tightly bind each other, becoming a small sphere aggregate in the center of each well by day (−1). On day (−1), additional E8 is added to dilute Y and provide a better proliferative environment. During the process of introducing additional E8, any dead cells adhered to the aggregates get released so that the surface of the aggregates becomes clean. Starting on day 0, the differentiation initiates. All aggregates from the 96-well U-bottom plates are collected and washed thoroughly to remove any residues of E8 that may interfere with differentiation. The aggregates are individually transferred to each well of 96-well U-bottom plates in E6 medium containing SB, bFGF, and BMP4. SB is a TGF-β inhibitor that promotes ectoderm induction. BMP4 induces surface ectoderm formation. The combination of SB, BMP4, and bFGF (low concentration) at an optimal concentration and timing guides the outer layer of cells on the aggregate to differentiate purely into the surface ectoderm. By day 3 of differentiation, the aggregate will form a thin, bright, transparent-like epithelium surrounding the aggregate. Depending on the cell lines, the epithelium may appear wavy or linear and will be visible between days 3 and 5. Inhibition of the BMP signaling pathways with LDN and activation of FGF signaling with a high concentration of bFGF on day 3 induces the formation of NC cells. By day 6 of differentiation, the aggregate becomes more cystic, containing dark core and radial traces of migrating mesenchymal cells from the core to the epithelium. Fresh E6 is added to provide nutrition. On day 9 of differentiation, half the volume of spent medium is replenished with fresh E6, providing nutrition. The aggregate continues to grow larger, and the mesenchymal cells populate more on the surface of the aggregate. Around day 12 of differentiation, the mesenchymal cells typically start to concentrate on one pole of the aggregate, leaving the other pole more cystic. All aggregates are collected and washed thoroughly on day 12 of differentiation to remove any residual E6 differentiation medium. The individual aggregates are transferred to each well of the 24-well plate in OMM1%M and placed on an orbital shaker to provide a floating environment, where the aggregates self-organize further. The OMM is regularly replenished to provide sufficient nutrition during development. See Extended Data Fig. 1 and Supplementary information in ref. 25 for additional optimization details. Representative images are taken at 40X (4X microscope objective × 10X eyepiece), or 100X (10X microscope objective × 10X eyepiece) magnifications as noted in each image. Scale bars, 200 μm.
Fig. 3. Images from optimal time points to check differentiation and characterization of the resulting skin organoid structure.
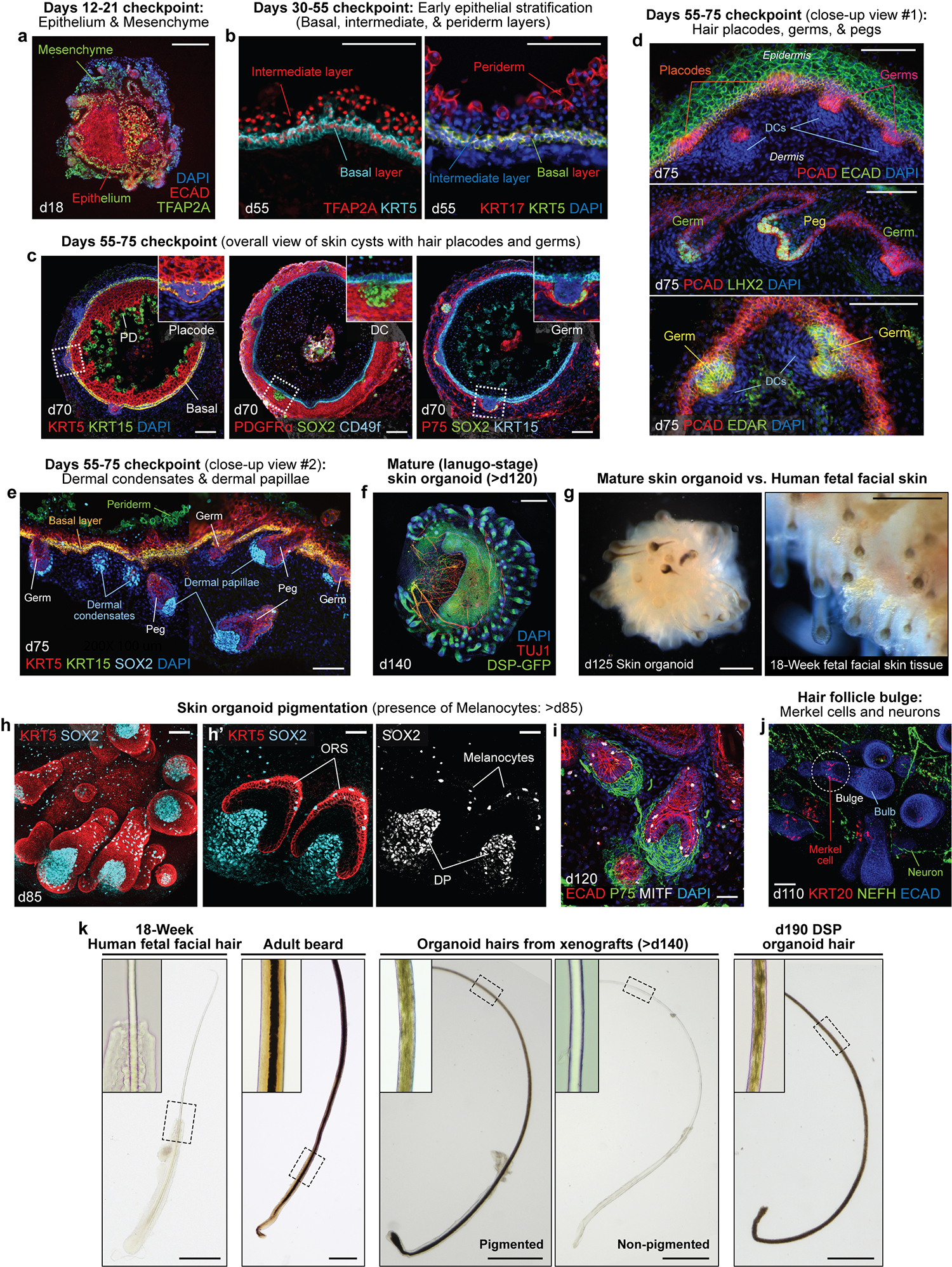
a-e, See Table 1 for further descriptions. Representative IHC images showing key checkpoints throughout the differentiation procedure of WA25 hESC-derived skin organoids. a, Day 18 image representing the ECAD+TFAP2A+ epithelium and TFAP2A+ CNC or mesenchymal cells surrounding the aggregate; the presence of these cell populations should be checked between days 6–20 of differentiation. b, Day 55 images showing TFAP2A+KRT5+ and KRT17+KRT5+ basal layer, TFAP2A+ intermediate layer, and KRT17+ periderm layer; the periderm layer is detectable only at early stages of differentiation, prior to formation of the granular and cornified layers; the periderm layer is visible in organoids around days 40–75 of differentiation. c, Overall-view image of day 70 skin cysts; the images show the major layers of skin that are required to form the skin, the epidermal and the dermal layers, and the initiating hair germs; the basal layer of skin is highlighted by KRT5+KRT15+ and CD49f+ fluorescence signals; the periderm layer is visualized by KRT15; the dermal layer (fibroblasts) is visualized by PDGFRα, and NC cell-derived mesenchymal cells within the population express P75; the SOX2+ cells represent dermal condensates at the tip of the hair germs; the initial hair placode and germ formations can be observed starting around day 55 of differentiation. PD, periderm; DC, dermal condensate. d,e, Day 75 high-magnification images representing PCAD+ hair placodes, PCAD+EDAR+LHX2+ hair germs, and PCAD+LHX2+ hair pegs; SOX2+ cells represent dermal condensates of hair germs and dermal papillae of hair pegs. f, A representative IHC image of a day 140 skin organoid. The endogenous green fluorescence from the DSP-GFP cell line visualizes epithelium of the skin cyst in the center and the hair follicles protruding from the surface of the cyst. TUJ1+ neurons are wrapping around and innervating the epithelium and the hair follicles. The skin organoids reach the lanugo-like mature stage around day 120 of differentiation. g, Representative darkfield images of a day 125 WA25 hESC-derived skin organoid (left) and dermal view of 18-week human fetal facial skin tissue (right). Skin organoids at days 120–140 resemble the mid-second trimester fetal skin tissue with (pigmented) hair follicles and adipocytes. h,i, Representative whole-mount immunostaining images of hair follicles with dermal papillae and melanocytes in a day 85 (h, left and middle) and a day 120 (i) WA25 hESC-derived skin organoids. KRT5 visualizes epithelium, outer root sheath (ORS) of hair follicles and newly forming hair germs (h, middle). SOX2 marks for melanocytes or Merkel cells present in the ORS of hair follicles and on the epithelium (h, right). MITF also specifies melanocytes in the ORS and on the epithelium. Dermal papillae of the hair follicles are also visualized by P75 (i). j, A representative whole-mount immunostaining image of a day 110 skin organoid hair follicles. The hair follicles contain a bulge region where KRT20+ touch-sensing Merkel cells are present. NEFH+ sensory neurons innervate the upper bulge region near Merkel cells. k, Representative brightfield images of plucked hairs from human fetal facial tissue at 18 weeks of gestation, adult male’s cheek (beard), skin organoid xenograft, and DSP skin organoid at day 190 of differentiation. Insets present a magnified area indicated with dash boxes. The medulla is only present in the adult beard. The medulla layer is not visible in xenograft hairs, either pigmented or non-pigmented. Darker hairs from a xenograft and a DSP skin organoid appear to contain pigmented cells that are scattered throughout the cortex, but no sign of medulla is detectable in the center of the hair shaft. See ref. 25 for additional images. The images are taken at the magnifications as follows: 200X (20X microscope objective × 10X eyepiece; b, d, e, h’); 100X (10X microscope objective × 10X eyepiece; a, c, h (left), i, and j); 50X (5X microscope objective × 10X eyepiece; g); 40X (4X microscope objective × 10X eyepiece; f and k (xenografts and organoid hairs)); 20X (2X microscope objective × 10X eyepiece; k (fetal hair and adult beard)). Scale bars, 500 μm (f, g, and k); 200 μm (a); 100 μm (b-e, and j); 50 μm (h (left) and i); 30 μm (h’).
Applications of the skin organoids
Modeling early development of human skin and hair follicles
The skin organoid system could be a tool for a wide range of skin-focused research, from basic developmental biology to translational projects. Skin organoids could be used to study the early development of human skin and hair follicles. Due to ethical, regulatory, and logistical challenges, human fetal tissue is not readily available for research use38. When available, the quality of tissue and gestational age of fetal specimens is highly variable39. Therefore, most skin studies have used murine models, resulting in a lack of mechanistic insight into the specification of cell types during human development. International consortium efforts to map developing cell lineages in organoids and human fetal tissue should shed light on these mechanisms40,41. Single-cell sequencing approaches, such as RNA and assay for transposase-accessible chromatin (ATAC) sequencing, can be performed on skin organoids. We provide a method for dissociating skin organoids to facilitate such studies. The skin organoid single-cell RNA sequencing (scRNA-seq) data can be compared with skin atlases available through the Human Cell Atlas data portal41,42. Based on our scRNA-seq and immunostaining data, the end-stage skin organoids (~150 days of culture) represent the human skin tissues at the second-trimester stage of the developing fetus25.
By the end-stage of culture, the basal keratinocyte layer of the skin that forms in the organoid differentiates and stratifies into all skin layers: spinous, granular, and cornified. The full-thickness skin produces lanugo-like hairs as in the human fetus at the second-trimester stage. Therefore, thoroughly tracing and analyzing the changes of gene expression levels and signaling pathways during the early developmental days of the skin organoid would provide immense insight into human skin development. At the end-stages of organoid culture, hair follicles remain in an anagen-like stage of development. Consequently, while the model may be suitable for studying hair follicle growth cycle initiation and perhaps the transition from anagen to catagen, it may be limiting for studies of telogen-stage (resting phase) follicles. Future studies could explore signaling cues to initiate the transition from anagen to catagen to telogen more rapidly in organoid culture. The model could potentially be used for studying the impact of various pharmacological or genetic factors on the rate of follicle growth during anagen stage. We have not performed extensive measurements of hair follicle lengths in the human skin organoids as it is difficult to make accurate measurements without terminating experiments. This is because the hair shafts grow inward and many hairs tend to wrap around the skin cyst as they grow, while few follicles protrude outward. However, if the experimenters plan to terminate experiments or collect samples at different timepoints, it is possible to pluck out individual hairs as shown in Fig. 3k and measure the lengths of the hairs for an additional period of time in culture. We also previously reported a method for approximating hair lengths in ref. 29. Briefly, we captured phase-contrast images of protruding mouse skin organoid follicles at different timepoints of differentiation. Using the captured images, the lengths of protruding pilosebaceous units could be measured and quantified for the approximate growth rate. Hair growth experiments can be facilitated by embedding the skin organoids in Matrigel during the initial stages of follicle induction to provide a matrix for the follicles to grow into.
For developmental and clinical studies, the skin organoid model will be excellent for studying skin innervation mechanisms. Nerves can be incorporated into existing skin equivalent cultures, but it remains uncertain how well these models replicate the innervation patterns and nerve endings of native skin43. Our characterization of the nerve endings in skin organoids has been limited. We have identified that sensory neurons in skin organoids form ganglia and fasciculated axons mimicking the architecture of a dorsal root or trigeminal ganglion/nerve in the body44. The model could be used to better understand how sensory neuronal diversity emerges during development. Regarding nerve endings, we have characterized rudimentary mechanosensory complex-like structures forming at the hair follicle bulge-nerve interface and, on rare occasions, we observe free nerve endings in the organoid epidermis. Future studies could use skin organoids to delve deeper into the mechanisms of skin innervation, brainstem or spinal cord innervation (i.e., using an assembloid approach)45, or disease/drug effects on peripheral neurons46.
Disease modeling
Skin organoids could be a useful disease model. Skin disease severity can range from relatively minor, like eczema and acne, to extreme, like skin cancer47–55. The three major types of skin cancer are basal cell carcinoma, squamous cell carcinoma, and melanoma50–55. The precursor cells of basal keratinocytes, squamous cells, and melanocytes are present within the skin organoid, setting the stage for researchers to investigate mechanisms of cancer initiation56. For example, researchers could expose organoids to external stress, such as ultraviolet radiation or toxic factors, to induce cancer formation in skin organoids. Alternatively, genetic alteration techniques (e.g., CRISPR/Cas9) can be applied to specifically mutate, knockdown, or overexpress target genes known to be involved as tumor suppressors or inducers of skin-associated cancers57,58. Furthermore, skin organoids can be readily generated from patient-derived cells and used to model genetic disorders, such as epidermolysis bullosa59–62. Finally, skin organoids could be treated with infectious agents (e.g., human papillomavirus or Ebola virus), inflammatory factors (e.g., cytokines or chemokines), or various types of drugs. The treatments can be microinjected into the core of the skin organoid cyst or applied within the medium, depending on how they would typically be introduced to the skin in the human body (topically or via circulation). This would lead to a better understanding of the mechanisms of infection, immune reactions, and drug responses. Thus, skin organoids would be beneficial for tracing the initiation and progression of diseases, screening novel targets for drug development, and testing responses to drugs.
Skin reconstruction and wound healing
Lastly, skin organoids could be a cell source for skin reconstruction and help facilitate wound healing processes. Using a xenograft mouse model, we engrafted intact hPSC-derived skin organoids into 1–2 mm incision sites on the backs of nude mice25. After 1 month of transplantation, we found that the skin organoid cysts had unfurled and integrated into the host skin, forming about 1–2 mm sized planar skin with outward-growing hairs. These observations indicate that organoids could be used for patients who need skin grafts because of skin or supportive tissue loss. In addition to minor incisional wound repair, we are currently testing whether multiple organoids or dissociated organoids could be used for more extensive excisional wound repair. Future work could investigate the mechanisms by which skin organoid cells rearrange from a cystic to a planar structure in the wound site or study whether organoids engage known wound repair mechanisms when wounded in vitro. This work could provide insights into critical developmental timing, cell populations, and signaling cues for skin regeneration.
For many of the applications above, we have demonstrated that the protocol works robustly with Allen Institute for Cell Science’s catalog of fluorescent reporter cell lines (e.g., the Desmoplakin-GFP cell line we used in ref. 25 and the SOX2-GFP cell line presented in this protocol). This growing catalog of reporter cell lines should be an excellent resource for developmental and translational skin studies. In addition, it should facilitate low- to mid-throughput image-based screens of organoids using confocal-equipped screening platforms (e.g., the Molecular Devices ImageXpress Micro Confocal).
Limitations
The major limitation of the hPSC-derived skin organoid model is its structure. Skin organoids form a sphere-like cyst where hair shafts sprout inward toward the fluid-filled core, with hair bulbs protruding outward from the surface of the cysts (Fig. 1)25,29. In normal human skin, the outermost cornified layer of skin functions as a physical barrier and sheds off when the cells age and die63. In skin organoids, however, the cystic structure limits shedding and accumulates cornified tissue in the core. Thus, at the late stages of culture (> day 150), we observe floating dead keratinocytes that have erupted to the surface of the organoid from the core of the cysts (Figs. 4a, b). In the same context, hair follicle shafts appear to have difficulty rupturing the core epidermis and become abnormal with kinked or curled morphologies (Fig. 4b). These limitations may be addressed by growing skin organoids in a planar format using microfluidic or air-liquid interface platforms. However, our unpublished attempts to form planar skin from organoid cysts suggest that this will be a significant challenge to accomplish.
Fig. 4. qCe3D whole-mount immunostaining.
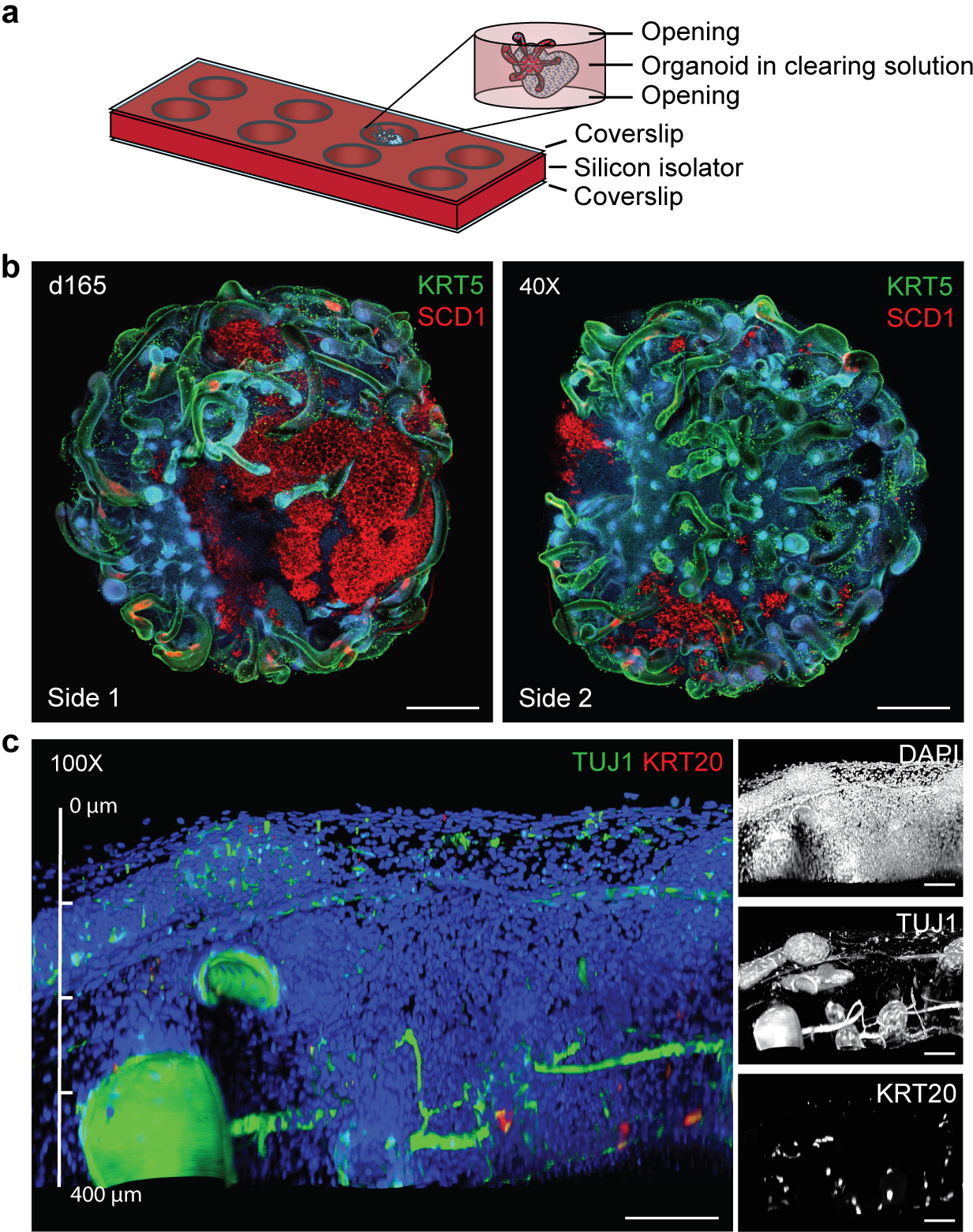
a, Schematic of mounting an immunostained organoid in a silicon isolator cassette after qCe3D clearing. A day 165 skin organoid is mounted with the qCe3D clearing solution in a well of a silicon isolator cassette. The silicone isolator is sealed with coverslips on both the top and the bottom, allowing the organoid to be imaged from both sides. b, Example of organoid imaging showing one side (left) and the other side (right) of the same organoid. KRT5 visualizes the epithelial layer of skin and ORS of hair follicles. SCD1 staining represents lipid-rich adipocytes and sebaceous glands. c, The application of the silicon isolator cassette allows for imaging up to 400 μm in depth of an organoid without loss of fluorescence signals. The panels on the right show extended depth of focus (EDF) images of individual channels of the image on the left. TUJ1 stains for neurons and KRT20 labels Merkel cells. Nuclei are visualized by DAPI. The images are taken at the magnification of 40X (4X microscope objective × 10X eyepiece; b) and 100X (10X microscope objective × 10X eyepiece; c). Scale bars, 500 μm (b); 100 μm (c).
Another limitation is that critical cell populations of normal human skin are missing from the skin organoid model. For example, the organoids currently lack sweat glands, blood vessels, arrector pili muscle (rarely observed), and immune cells, which may prevent them from fully maturing and preclude specific applications requiring these cells and tissues. Moreover, the hair follicles produced within the skin organoids do not contain a medulla layer. The medulla, the innermost layer of the hair, is a unique characteristic of adult terminal hair that is influenced by hormones64. Our comparative analysis with fetal hair suggests that the skin organoids in vitro are reminiscent of lanugo hair, which sheds just before or after birth (Fig. 3)25. Additional optimization of the protocol to co-induce or add in missing cellular compartments and accelerate the maturation of skin organoids to have adult-like features would expand the potential applications of skin organoids.
Lastly, off-target cell lineage induction is a limitation of the system. Although the neural crest (NC) lineage in skin organoids produces important skin lineage cells (i.e., melanocytes for pigmentation and neurons for touch/pain sensing), it also produces highly variable chondrocyte and myocyte populations. Therefore, meticulous analysis of the signaling cues during the early induction stages will be necessary to optimize the differentiation regimen, eliminating or minimizing undesired cell types (e.g., chondrocytes, Fig. 5) within the skin organoid.
Fig. 5. Morphologies of developing skin organoids during differentiation.
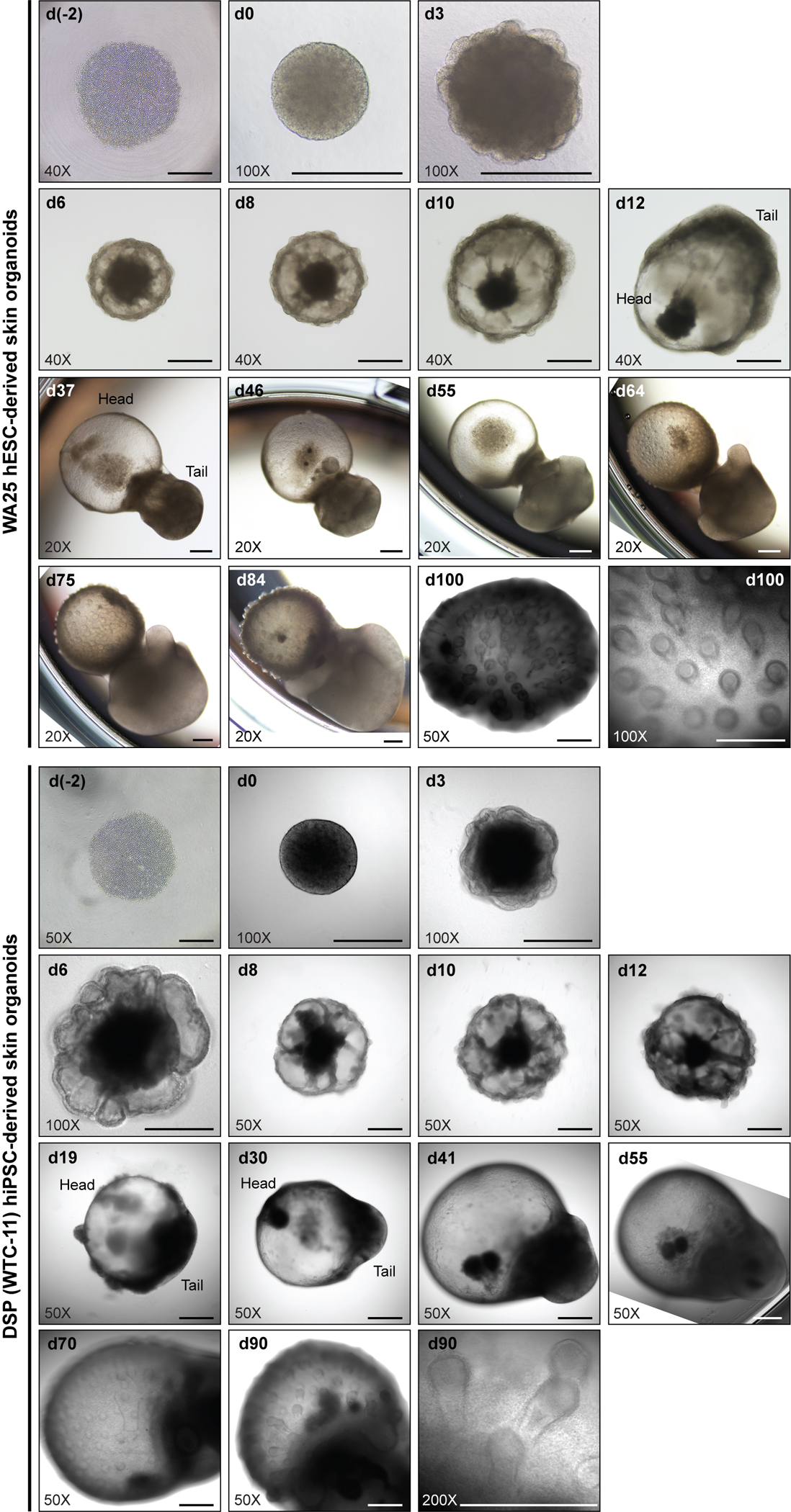
Representative phase-contrast (WA25; day (−2)-day 3), brightfield (WA25; day 6-day 84), and differential interference contrast (DIC) (WA25; day 100, DSP; day (−2)-day 90) images of skin organoids derived from WA25 hESCs and DSP-GFP (WTC-11) hiPSCs. By day 35 of differentiation, the skin organoids typically form two poles where one end is a skin cyst (referred to as ‘head’) and the other end is composed of mesenchymal cells (referred to as ‘tail’). The initiation of hair follicle formation (hair germs) is visible between days 55 and 75 of differentiation, depending on cell lines. The cartilage formation on the tail is apparent under a microscope as the skin organoids mature. See ref. 25 for additional images. Representative images are taken at 40X (4X microscope objective × 10X eyepiece), 50X (5X microscope objective × 10X eyepiece), 100X (10X microscope objective × 10X eyepiece), or 200X (20X microscope objective × 10X eyepiece) magnifications as noted in each image. Scale bars, 500 μm.
In summary, the current skin organoid culture system is under continuous development and needs to be further refined by our laboratory and others. Most desirably, creating skin organoids in a planar format and providing a sufficiently nutritious environment for extended in vitro culture will improve the skin organoid model to allow shedding of the outermost skin layers and eruption of hair shafts without the need for xenografting. Additionally, new exogenous signaling programs that stimulate missing skin features, such as sweat glands and blood vessels (e.g., BMP5 and vascular endothelial growth factor, respectively)65–69 or immune cells70, while minimizing off-target cells, would further increase the complexity and accuracy of the model.
Experimental design
Human skin organoids can be established from either embryonic stem cells (ESCs) or induced PSCs (iPSCs)59,71. To successfully generate skin, co-development of epidermis and dermis is required, and precise alterations of major signaling pathways (TGF-β, BMP4, and bFGF) are critical20,29,30,33,36,72. The most up-to-date schematic of the protocol applied in the Koehler laboratory is shown in Fig. 2. However, there are several factors that the experimenters should be aware of when preparing to execute the skin organoid method, such as characteristics of individual cell lines, reagents, and equipment.
Our laboratory uses E8 Flex medium and vitronectin-coated plates for PSC maintenance. Before initiating a skin organoid differentiation, we highly recommend that any cell lines that were cultured using different systems, including feeder-dependent cell lines, be adapted to the E8 Flex medium and vitronectin culture system (see ‘Troubleshooting’ for details). We suspect that the induction protocol is compatible with PSCs maintained using different platforms; however, skin organoid and hair generation may be suboptimal if other platforms are used.
The experimenters must be aware that outcomes are cell line dependent, as has been noted for other organoid models73,74. The inconsistency is potentially due to the different genetic backgrounds of the individual cell line, which leads to differentially expressed endogenous levels of TGF-β, BMP4, FGF, or other signaling factors. For example, we have found that the WA25 human ESC (hESC) line has a higher basal level of endogenous BMP4 compared with wild type C (WTC)-background cell lines and requires less exogenous recombinant BMP4 to induce surface ectoderm (Fig. 6). Therefore, some cell lines may require a slight modification to the differentiation regimen. Most critically, even though treatment of 5 ng/mL BMP4 on day 0 is recommended as the standardized regimen in this protocol, initial optimization of BMP4 treatment concentration and timing may be required to efficiently induce cell differentiation to surface ectoderm (Fig. 6, Table 1, and ‘Troubleshooting’). The optimization process must be performed sequentially; for example, when optimizing BMP4 concentrations on day 0, the experimenters must keep TGF-β inhibitor and bFGF concentrations the same but compare the different concentrations of BMP4. Differentiation day 3 is the best time to confirm whether surface ectoderm induction has been successful (Table 1 and ‘Troubleshooting’). By comparing the morphologies under a microscope (and with additional immunostaining for TFAP2A+ ECAD+ epithelium), it is possible to determine the optimal conditions in which the aggregates form a transparent epithelium surrounding the sphere, as shown in Figs. 2, 5, and 6. If the surface ectoderm fails to develop, the sphere will contain a comparatively thick epithelium, as shown in Fig. 6c, representing neuroepithelium formation. In addition, if the sphere forms a relatively large cyst with a very thin epithelium (without proper mesenchymal induction), as shown in Fig. 6d, the epithelium will fail to differentiate into higher-order skin layers. Once the day 0 regimen is complete, LDN-193189 (LDN) and bFGF treatment on day 3 can be optimized. Although LDN/bFGF treatment on day 3 appears to work with most PSC lines, various treatment timings between days 3 and 5 of differentiation might need to be tested to accommodate cell line-to-line idiosyncrasies. Additional morphological checks using microscope images should be undertaken on days 8 and 12 (Figs. 2, 5, and 6). By day 8, the epithelium forms as a surface lining of the sphere-like aggregate, and radial traces of migrating cells appear from the dark core to the epithelium. Mesenchymal cells covering the outermost layer of the aggregate should be visible. Between days 12 and 21, mesenchymal cells typically concentrate on one side of the aggregate, forming a mesenchymal cell pole (tail) on one end and a cystic pole on the other (head; Figs. 2, 3a, and 5). If the experiment is not optimal, the cell aggregates will be overpopulated with CNC cells and become opaque so that no sign of the epithelial cyst is visible. Additional timepoints (checkpoints) at which we recommend monitoring hair follicle formation are described in ‘Anticipated results’, Fig. 3, and Table 1.
Fig. 6. Morphological differences of skin organoids incubated with differing BMP4 concentrations.

a,b, Representative brightfield images of skin organoids derived from WA25 hESCs (a) and SOX2-GFP (WTC-11) hiPSCs (b) on (upper) day 3 and (lower) day 8 of differentiation. The aggregates are treated with BMP4 in a dose-dependent manner on day 0 of differentiation. This figure shows the importance of BMP4 concentration optimization depending on cell lines before initiating an experiment. The columns of optimal concentrations for each cell line are highlighted in blue. 2.5 ng/mL of BMP4 is sufficient for the WA25 cell line to produce a thin epithelium (surface ectoderm) by day 3 and further differentiate and form a transparent cystic morphology by day 8 of differentiation. On the other hand, 2.5 ng/mL of BMP4 has led the SOX2 cell line to form a thicker layer (reminiscent of neuroepithelium), which would develop into a cerebral organoid. By day 8 of differentiation, the 2.5 ng/mL BMP4 treated SOX2 cell line becomes an opaque, denser aggregate. For the SOX2 cell line and most of the WTC-11 background cell lines being used in the Koehler laboratory, 5 ng/mL of BMP4 is required to induce thin epithelium by day 3 and cystic aggregate by day 8 of differentiation. The day 8 aggregates also contain cells migrating from the core to the epithelium of the cystic aggregate. When evaluating BMP4 concentration, users should select the minimal concentration that produces this ‘spoked wheel’ morphology (highlighted in blue). The aggregates treated with slightly higher BMP4 concentrations (up to 10 ng/mL) than the minimally required concentrations ultimately mature into multi-layered skin generating hair follicles. However, be aware that the sizes of the cysts decrease, and morphological variabilities increase as higher BMP4 concentrations are introduced compared with the optimal BMP4 concentration. c, Representative day 3 DIC images of neuroepithelial formation (failed to differentiate into surface ectoderm) when BMP4 treatment is not applied on day 0 of differentiation. Thick neuroepithelium is visible surrounding the aggregates derived from SOX2 (left) and HIST 1H2BJ (right) (WTC-11 background) hiPSC lines. d, Representative brightfield images of aggregates that would fail to develop skin organoids. The images show SOX2 hiPSC-derived aggregate on day 12 (left) and day 28 (right) when treated with 5 ng/ml BMP4 (with SB and bFGF) on day 0 and only FGF (without LDN) on day 3. The aggregate develops into a thin-layered large cyst as shown in the day 12 image (left), which does not produce either skin layers or mesenchymal cells. Then, the cyst continues to shrink as shown in the day 28 image (right) and ultimately dies off. See Extended Data Fig. 1 and Supplementary information in ref. 25 for additional optimization details. The representative images are taken at 40X (4X microscope objective × 10X eyepiece) and 100X (10X microscope objective × 10X eyepiece) magnifications as noted in the figure. Scale bars, 500 μm (a and b (day 8), d); 200 μm (c); 100 μm (a and b (day 3)).
Table 1.
Major checkpoints
| Checkpoint | Questions | Continue | Terminate | IHC Markers |
|---|---|---|---|---|
| Day 3 | How does the layer surrounding the aggregate look? | Thin and clear as in Figs. 6a, b | Thick and opaque as Fig. 6c (neuroepithelium). Huge cyst with a thin layer as in Fig. 6d (will not differentiate to higher-order skin layers). |
Epithelial layer: ECAD+, NCAD− Neural tube: NCAD+ |
| Days 6–9 | Is the aggregate forming a cyst containing a dark core and radial traces of migrating mesenchymal cells from the core to the epithelium? (e.g., as in Figs. 2 and 5) | Yes | No The cyst forms a thin layer (representing an epithelium only) without any sign of other cell types. The aggregate is very opaque and dark, covered and overpopulated by mesenchymal cells. The cystic structure is not visible. |
Epithelial layer: ECAD+ |
| Days 12–21 | Is the epithelial cyst lightly covered by the mesenchymal cells? Is there a mesenchymal cell mass forming at one pole of the epithelial cyst? (e.g., as in Figs. 2, 3a, and 5) | Yes | No If there is no sign of mesenchymal cells but only a thin layered cyst exists, the aggregate will not develop multi-layered skin or hair follicles. |
Epithelial cyst: ECAD+, TFAP2A+ NC cells: ECAD−, TFAP2A+, PDGFRα+, P75+, SOX10+ |
| Days 35 | Are the basal and the intermediate layers of the epidermis present? (e.g., as in Fig. 3b) | Yes | No |
Basal layer: KRT5+, KRT17+, TFAP2A+, CD49f+ Intermediate layer: KRT10+, TFAP2A+ |
| Days 45 | Is the periderm layer present above the intermediate layer? (e.g., as in Figs. 3b, c) | Yes (a patchy layer of pearl-like cells should be visible on the skin organoid epithelium using phase-contrast imaging) |
No |
Periderm layer: KRT15+, KRT17+ |
| Days 55–75 | Are hair germs forming? (e.g., as in Figs. 3c-e) | Yes | No |
Hair placodes and germs: PCAD+, LHX2+, EDAR+ Dermal condensates/papillae: SOX2+ |
Lastly, regarding the reagents and equipment for skin organoid culture, the activity of each small molecule and protein may vary depending on vendor, lot, and in-lab handling. Diverse coating materials used on differentiation culture plates also lead to a higher variability and cell death in cultures. Therefore, when testing the protocol for the first time, we highly recommend using reagents and plasticware purchased from the vendors listed in this protocol and following our specific handling guidelines. Moreover, it should be confirmed that the small molecules and proteins are within the expiration date and reconstituted as suggested by the vendor before starting a differentiation culture. When beginning a differentiation culture with newly purchased or reconstituted reagents, we recommend that control groups be treated with individual culture reagents alongside experimental treatment groups to assess reagent activity.
Materials
Biological materials
We have used the WA25 (NIHhESC-12-0196, RRID:CVCL_E080, https://scicrunch.org/resolver/RRID:CVCL_E080, female) and WA01 (NIHhESC-10-0043, RRID:CVCL_9771, https://scicrunch.org/resolver/RRID:CVCL_9771, male) cells from WiCell Research Institute for hESC lines. For human iPSC (hiPSC) lines, we have used the DSP-GFP (AICS-0017 cl.65, RRID:CVCL_IR31, https://scicrunch.org/resolver/RRID:CVCL_IR31, male), SOX2-GFP (AICS-0074 cl.26, RRID:CVCL_WM15, https://scicrunch.org/resolver/RRID:CVCL_WM15, male), HIST1H2BJ-GFP (AICS-0061 cl.36, RRID:CVCL_UD17, https://scicrunch.org/resolver/RRID:CVCL_UD17, male), LMNB1-GFP (AICS-0013 cl.210, RRID:CVCL_IR32, https://scicrunch.org/resolver/RRID:CVCL_IR32, male), and TUBA1B-GFP (AICS-0012 cl.105, RRID:CVCL_IR34, https://scicrunch.org/resolver/RRID:CVCL_IR34, male) from Allen Institute for Cell Science and Coriell Institute (parental line WTC-11) and mND2-0 (MIRJT7i-mND2-0, RRID:CVCL_U173, https://scicrunch.org/resolver/RRID:CVCL_U173, male) cells from WiCell Research Institute. Detailed information on cell line validation and testing is available at https://www.wicell.org/home/stem-cells/catalog-of-stem-cell-lines/wa25.cmsx?closable=true and https://www.allencell.org/cell-catalog.html. We present representative images from mostly WA25, DSP-GFP, and SOX2-GFP cell lines, but all cell lines listed above have successfully produced hair follicles. ! CAUTION Cell lines should be routinely checked for any contamination (mycoplasma detection) or genetic abnormalities (karyotyping).
Reagents
Cell culture media and supplements
Essential 8 Flex medium kit (Gibco, cat. no. A2858501). Note that E8 Flex medium which is used for hPSC culture and differentiation can be substituted with widely used mTeSR Plus basal medium (STEMCELL Technologies, cat. no. 100-0276) with the same quality and outcome.
Essential 6 medium (Gibco, cat. no. A1516401)
Advanced Dulbecco’s modified Eagle medium (DMEM)/F12 (Gibco, cat. no. 12634010)
Neurobasal medium (Gibco, cat. no. 21103049)
GlutaMAX supplement (Gibco, cat. no. 35050061)
B-27 supplement, minus vitamin A (Gibco, cat. no. 12587010)
N2 supplement (Gibco, cat. no. 17502048)
2-Mercaptoethanol (Gibco, cat. no. 21985023)
Normocin (InvivoGen, cat. no. Ant-nr-1)
Cell dissociation and counting reagents
UltraPure 0.5 M EDTA (pH 8.0; Invitrogen, cat. no. 15575-038)
StemPro Accutase cell dissociation reagent (Gibco, cat. no. A1110501)
1X TrypLE express enzyme, phenol red (Gibco, cat. no. 12605010) ▲CRITICAL Light sensitive. Store in the dark at room temperature (RT, 20–25 °C), covered by aluminum foil, as recommended by the manufacturer.
Trypan blue solution (Gibco, cat. no. 15-250-061)
Small molecules, proteins, and matrices
Vitronectin recombinant human protein, truncated (Gibco, cat. no. A14700)
SB-431542 in solution (SB; Stemgent, cat. no. 04-0010-05) ▲CRITICAL Light sensitive. Store in the dark at −20 °C as recommended by the manufacturer.
Recombinant human bFGF (PeproTech, cat. no. 100-18B)
Recombinant human BMP4 (PeproTech, cat. no. 120-05 and STEMCELL Technologies, cat. no. 78211) ▲CRITICAL The activity of BMP4 may vary depending on vendor, lot, and in-lab handling. BMP4 treatments should be titrated when assessing new BMP4 from different vendors or lots and when starting experiments for the first time. ▲CRITICAL Using the same BMP4 lot for all experiments will generate the most consistent results.
LDN in solution (Stemgent, cat. no. 04-0074-02) ▲CRITICAL Light sensitive. Store in the dark at −20 °C as recommended by the manufacturer.
Y-27632 in solution (Stemgent, cat. no. 04-0012-02)
Matrigel, growth factor reduced (Corning, cat. no. 354230)
Sample fixation and cryo-embedding reagents
16% paraformaldehyde (PFA) solution (Electron Microscopy Sciences, cat. no. 15710)
Sucrose (MP Biomedicals, cat. no. 0219401891)
Tissue-Tek OCT compound (Sakura Finetek, cat. no. 4583)
Immunostaining and whole-mount staining reagents
Normal goat serum (Vector Laboratories, cat. no. s-1000-20)
Normal horse serum (Vector Laboratories, cat. no. s-2000-20)
Bovine serum albumin (BSA; Sigma-Aldrich, cat. no. A1470)
ProLong Gold antifade mountant (Invitrogen, cat. no. P10144)
ProLong Gold antifade mountant with DAPI (Invitrogen, cat. no. P36931)
DAPI (Invitrogen, cat. no. D3571)
Hoechst (Invitrogen, cat. no. H3570)
Quick Ce3D (qCe3D) organoid clearing solution reagents
N-methylacetamide (Sigma-Aldrich, cat. no. M26305-500G) ! CAUTION N-methylacetamide is hazardous. Manipulate in a fume hood. Exposure may damage the unborn child.
Histodenz (Sigma-Aldrich, cat. no. D2158)
1-Thioglycerol (Sigma-Aldrich, cat. no. M1753) ! CAUTION 1-Thioglycerol is hazardous. Avoid direct contact with skin and eyes. Wear rubber gloves and eye goggles. May cause respiratory irritation. Manipulate in a fume hood.
Buffers and other reagents
1X phosphate-buffered saline (PBS), pH 7.4 (Gibco, cat. no. 10010023)
10X PBS, pH 7.4 (Gibco, cat. no. 70011069)
1X Dulbecco’s PBS, no calcium, no magnesium (DPBS; Gibco, cat. no. 14190250)
1M Tris-HCl, pH 7.5 (Fisher BioReagents, cat. no. BP1757-100)
Citric acid monohydrate (Fisher Chemical, cat. no. A104-500)
Human serum albumin (HSA; Sigma-Aldrich, cat. no. A9731)
10% Triton X-100 (Sigma-Aldrich, cat. no. 93443)
Dimethyl sulfoxide (DMSO; Fisher BioReagents, cat. no. BP231-100)
Antibodies for immunostaining
▲CRITICAL See Tables 2 and 3. Antibodies list for details.
Table 2.
Primary antibodies
| Antibody | Vendor | Cat. no. | RRID | Host | Isotope | Dilution | Marker for: |
|---|---|---|---|---|---|---|---|
| CD49f | Thermo Fisher Scientific | 12-0495-81 | AB_891478 https://scicrunch.org/resolver/RRID:AB_891478 | Rat | IgG2a | 1:50 | Epidermal layer |
| Cytokeratin 5 (KRT5) | Thermo Fisher Scientific | MA5-12596 | AB_11008032 https://scicrunch.org/resolver/RRID:AB_11008032 | Mouse | IgG1 | 1:50 | Basal layer of skin |
| Cytokeratin 5 (KRT5) | Thermo Fisher Scientific | MA5-14473 | AB_10979451 https://scicrunch.org/resolver/RRID:AB_10979451 | Rabbit | IgG | 1:50 | Basal layer of skin |
| Cytokeratin 15 (KRT15) | Santa Cruz Biotechnology | sc-47697 | AB_627847 https://scicrunch.org/resolver/RRID:AB_627847 | Mouse | IgG2a | 1:50 | Periderm and basal layers of skin |
| Cytokeratin 17 (KRT17) | Santa Cruz Biotechnology | sc-393091 | AB_2893343 https://scicrunch.org/resolver/RRID:AB_2893343 | Mouse | IgG1 | 1:50 | Periderm and basal layers of skin |
| Cytokeratin 20 (KRT20) | Cell Signaling Technology | 13063 | AB_2798106 https://scicrunch.org/resolver/RRID:AB_2798106 | Rabbit | IgG | 1:100 | Merkel cells |
| E-cadherin (ECAD) | BD Bioscience | 610181 | AB_397580 https://scicrunch.org/resolver/RRID:AB_397580 | Mouse | IgG2a | 1:50 | Epithelia |
| EDAR | R&D Systems | AF745 | AB_355565 https://scicrunch.org/resolver/RRID:AB_355565 | Goat | IgG | 1:50 | Hair germs |
| LHX2 | Millipore | ABE1402 | AB_2722523 https://scicrunch.org/resolver/RRID:AB_2722523 | Rabbit | IgG | 1:750 | Hair germs and pegs |
| MITF | Abcam | ab122982 | AB_10902226 https://scicrunch.org/resolver/RRID:AB_10902226 | Rabbit | IgG | 1:20 | Melanocytes |
| Neurofilament-heavy chain (NEFH) | Cell Signaling Technology | 2836 | AB_10694081 https://scicrunch.org/resolver/RRID:AB_10694081 | Mouse | IgG1 | 1:100 | Sensory neurons |
| P75NTR (P75) | Cell Signaling Technology | 8238 | AB_10839265 https://scicrunch.org/resolver/RRID:AB_10839265 | Rabbit | IgG | 1:50 | Neural crest cells & dermal condensates |
| P-cadherin (PCAD) | Thermo Fisher Scientific | 32-4000 | AB_2533077 https://scicrunch.org/resolver/RRID:AB_2533077 | Mouse | IgG1 | 1:50 | Hair placodes, germs, and pegs |
| PDGFRα (D13C6) | Cell Signaling Technology | 5241 | AB_10692773 https://scicrunch.org/resolver/RRID:AB_10692773 | Rabbit | IgG | 1:50 | Dermal fibroblasts |
| SCD1 | Sigma-Aldrich | HPA012107 | AB_1856610 https://scicrunch.org/resolver/RRID:AB_1856610 | Rabbit | IgG | 1:50 | Sebaceous glands & adipocytes |
| SOX2 | BD Biosciences | 561469 | AB_10694256 https://scicrunch.org/resolver/RRID:AB_10694256 | Mouse | IgG1 | 1:100 | Dermal condensates and papillae, Merkel cells, Melanocytes |
| TFAP2A | DSHB | 3B5 | AB_528084 https://scicrunch.org/resolver/RRID:AB_528084 | Mouse | IgG2b | 1:5 | Epithelial & neural crest cells |
| Tubulin β class III (TUJ1) | BioLegend | 801202 | AB_10063408 https://scicrunch.org/resolver/RRID:AB_10063408 | Mouse | IgG2a | 1:100 | Neurons |
Table 3.
Secondary antibodies
| Host | Antibody | Isotope | Conjugate | Vendor | Cat. no. | RRID | Dilution |
|---|---|---|---|---|---|---|---|
| Goat | anti-Mouse | IgG1 | Alexa Fluor 488 | Thermo Fisher Scientific | A-21121 | AB_2535764 https://scicrunch.org/resolver/RRID:AB_2535764 | 1:2000 |
| Goat | anti-Mouse | IgG1 | Alexa Fluor 568 | Thermo Fisher Scientific | A-21124 | AB_2535766 https://scicrunch.org/resolver/RRID:AB_2535766 | 1:2000 |
| Goat | anti-Mouse | IgG1 | Alexa Fluor 647 | Thermo Fisher Scientific | A-21240 | AB_2535809 https://scicrunch.org/resolver/RRID:AB_2535809 | 1:2000 |
| Goat | anti-Mouse | IgG2a | Alexa Fluor 488 | Thermo Fisher Scientific | A-21131 | AB_2535771 https://scicrunch.org/resolver/RRID:AB_2535771 | 1:2000 |
| Goat | anti-Mouse | IgG2a | Alexa Fluor 568 | Thermo Fisher Scientific | A-21134 | AB_2535773 https://scicrunch.org/resolver/RRID:AB_2535773 | 1:2000 |
| Goat | anti-Mouse | IgG2a | Alexa Fluor 647 | Thermo Fisher Scientific | A-21241 | AB_2535810 https://scicrunch.org/resolver/RRID:AB_2535810 | 1:2000 |
| Goat | anti-Mouse | IgG2b | Alexa Fluor 488 | Thermo Fisher Scientific | A-21141 | AB_2535778 https://scicrunch.org/resolver/RRID:AB_2535778 | 1:2000 |
| Goat | anti-Mouse | IgG2b | Alexa Fluor 568 | Thermo Fisher Scientific | A-21144 | AB_2535780 https://scicrunch.org/resolver/RRID:AB_2535780 | 1:2000 |
| Goat | anti-Mouse | IgG2b | Alexa Fluor 647 | Thermo Fisher Scientific | A-21242 | AB_2535811 https://scicrunch.org/resolver/RRID:AB_2535811 | 1:2000 |
| Goat | anti-Rabbit | IgG | Alexa Fluor 488 | Thermo Fisher Scientific | A-11034 | AB_2576217 https://scicrunch.org/resolver/RRID:AB_2576217 | 1:2000 |
| Goat | anti-Rabbit | IgG | Alexa Fluor 568 | Thermo Fisher Scientific | A-11036 | AB_10563566 https://scicrunch.org/resolver/RRID:AB_10563566 | 1:2000 |
| Goat | anti-Rabbit | IgG | Alexa Fluor 647 | Thermo Fisher Scientific | A-21245 | AB_2535813 https://scicrunch.org/resolver/RRID:AB_2535813 | 1:2000 |
| Goat | anti-Rat | IgG | Alexa Fluor 488 | Thermo Fisher Scientific | A-11006 | AB_2534074 https://scicrunch.org/resolver/RRID:AB_2534074 | 1:2000 |
| Goat | anti-Rat | IgG | Alexa Fluor 568 | Thermo Fisher Scientific | A-11077 | AB_2534121 https://scicrunch.org/resolver/RRID:AB_2534121 | 1:2000 |
| Goat | anti-Rat | IgG | Alexa Fluor 647 | Thermo Fisher Scientific | A21247 | AB_141778 https://scicrunch.org/resolver/RRID:AB_141778 | 1:2000 |
| Donkey | anti-Goat | IgG | Alexa Fluor 488 | Thermo Fisher Scientific | A-11055 | AB_2534102 https://scicrunch.org/resolver/RRID:AB_2534102 | 1:2000 |
| Donkey | anti-Goat | IgG | Alexa Fluor 568 | Thermo Fisher Scientific | A-11057 | AB_2534104 https://scicrunch.org/resolver/RRID:AB_2534104 | 1:2000 |
| Donkey | anti-Goat | IgG | Alexa Fluor 647 | Thermo Fisher Scientific | A-21447 | AB_2535864 https://scicrunch.org/resolver/RRID:AB_2535864 | 1:2000 |
| Donkey | anti-Mouse | IgG | Alexa Fluor 488 | Thermo Fisher Scientific | A-21202 | AB_141607 https://scicrunch.org/resolver/RRID:AB_141607 | 1:2000 |
| Donkey | anti-Mouse | IgG | Alexa Fluor 647 | Thermo Fisher Scientific | A-31571 | AB_162542 https://scicrunch.org/resolver/RRID:AB_162542 | 1:2000 |
| Donkey | anti-Rabbit | IgG | Alexa Fluor 488 | Thermo Fisher Scientific | A-21206 | AB_2535792 https://scicrunch.org/resolver/RRID:AB_2535792 | 1:2000 |
| Donkey | anti-Rabbit | IgG | Alexa Fluor 568 | Thermo Fisher Scientific | A10042 | AB_2534017 https://scicrunch.org/resolver/RRID:AB_2534017 | 1:2000 |
| Donkey | anti-Rabbit | IgG | Alexa Fluor 647 | Thermo Fisher Scientific | A-31573 | AB_2536183 https://scicrunch.org/resolver/RRID:AB_2536183 | 1:2000 |
| Donkey | anti-Rat | IgG | Alexa Fluor Plus 488 | Thermo Fisher Scientific | A48269 | AB_2893137 https://scicrunch.org/resolver/RRID:AB_2893137 | 1:2000 |
| Donkey | anti-Rat | IgG | Alexa Fluor Plus 647 | Thermo Fisher Scientific | A48272 | AB_2893138 https://scicrunch.org/resolver/RRID:AB_2893138 | 1:2000 |
Primary antibodies:
CD49f (Thermo Fisher Scientific, cat. no. 12-0495-81, RRID: AB_891478, https://scicrunch.org/resolver/RRID:AB_891478)
Cytokeratin 5 (KRT5; Thermo Fisher Scientific, cat. no. MA5-12596, RRID: AB_11008032, https://scicrunch.org/resolver/RRID:AB_11008032)
Cytokeratin 5 (KRT5; Thermo Fisher Scientific, cat. no. MA5-14473, RRID: AB_10979451, https://scicrunch.org/resolver/RRID:AB_10979451)
Cytokeratin 15 (KRT15; Santa Cruz Biotechnology, cat. no. sc-47697, RRID: AB_627847, https://scicrunch.org/resolver/RRID:AB_627847)
Cytokeratin 17 (KRT17; Santa Cruz Biotechnology, cat. no. sc-393091, RRID: AB_2893343, https://scicrunch.org/resolver/RRID:AB_2893343)
Cytokeratin 20 (KRT20; Cell Signaling Technology, cat. no. 13063, RRID: AB_2798106, https://scicrunch.org/resolver/RRID:AB_2798106)
E-cadherin (ECAD; BD Bioscience, cat. no. 610181, RRID: AB_397580, https://scicrunch.org/resolver/RRID:AB_397580)
EDAR (R&D Systems, cat. no. AF745, RRID: AB_355565, https://scicrunch.org/resolver/RRID:AB_355565)
LHX2 (Millipore, cat. no. ABE1402, RRID: AB_2722523, https://scicrunch.org/resolver/RRID:AB_2722523)
MITF (Abcam, cat. no. ab122982, RRID: AB_10902226, https://scicrunch.org/resolver/RRID:AB_10902226)
Neurofilament-heavy chain (NEFH; Cell Signaling Technology, cat. no. 2836, RRID: AB_10694081, https://scicrunch.org/resolver/RRID:AB_10694081)
P75NTR (P75; Cell Signaling Technology, cat. no. 8238, RRID: AB_10839265, https://scicrunch.org/resolver/RRID:AB_10839265)
P-cadherin (PCAD; Thermo Fisher Scientific, cat. no. 32-4000, RRID: AB_2533077, https://scicrunch.org/resolver/RRID:AB_2533077)
PDGFRα (D13C6; Cell Signaling Technology, cat. no. 5241, RRID: AB_10692773, https://scicrunch.org/resolver/RRID:AB_10692773)
SCD1 (Sigma-Aldrich, cat. no. HPA012107, RRID: AB_1856610, https://scicrunch.org/resolver/RRID:AB_1856610)
SOX2 (BD Biosciences, cat. no. 561469, RRID: AB_10694256, https://scicrunch.org/resolver/RRID:AB_10694256)
TFAP2A (DSHB, cat. no. 3B5, RRID: AB_528084, https://scicrunch.org/resolver/RRID:AB_528084)
Tubulin β class III (TUJ1; BioLegend, cat. no. 801202, RRID: AB_10063408, https://scicrunch.org/resolver/RRID:AB_10063408)
Secondary antibodies:
Goat anti-Mouse IgG1, Alexa Fluor 488 (Thermo Fisher Scientific, cat. no. A-21121, RRID: AB_2535764, https://scicrunch.org/resolver/RRID:AB_2535764)
Goat anti-Mouse IgG1, Alexa Fluor 568 (Thermo Fisher Scientific, cat. no. A-21124, RRID: AB_2535766, https://scicrunch.org/resolver/RRID:AB_2535766)
Goat anti-Mouse IgG1, Alexa Fluor 647 (Thermo Fisher Scientific, cat. no. A-21240, RRID: AB_2535809, https://scicrunch.org/resolver/RRID:AB_2535809)
Goat anti-Mouse IgG2a, Alexa Fluor 488 (Thermo Fisher Scientific, cat. no. A-21131, RRID: AB_2535771, https://scicrunch.org/resolver/RRID:AB_2535771)
Goat anti-Mouse IgG2a, Alexa Fluor 568 (Thermo Fisher Scientific, cat. no. A-21134, RRID: AB_2535773, https://scicrunch.org/resolver/RRID:AB_2535773)
Goat anti-Mouse IgG2a, Alexa Fluor 647 (Thermo Fisher Scientific, cat. no. A-21241, RRID: AB_2535810, https://scicrunch.org/resolver/RRID:AB_2535810)
Goat anti-Mouse IgG2b, Alexa Fluor 488 (Thermo Fisher Scientific, cat. no. A-21141, RRID: AB_2535778, https://scicrunch.org/resolver/RRID:AB_2535778)
Goat anti-Mouse IgG2b, Alexa Fluor 568 (Thermo Fisher Scientific, cat. no. A-21144, RRID: AB_2535780, https://scicrunch.org/resolver/RRID:AB_2535780)
Goat anti-Mouse IgG2b, Alexa Fluor 647 (Thermo Fisher Scientific, cat. no. A21242, RRID: AB_2535811, https://scicrunch.org/resolver/RRID:AB_2535811)
Goat anti-Rabbit IgG, Alexa Fluor 488 (Thermo Fisher Scientific, cat. no. A-11034, RRID: AB_2576217, https://scicrunch.org/resolver/RRID:AB_2576217)
Goat anti-Rabbit IgG, Alexa Fluor 568 (Thermo Fisher Scientific, cat. no. A-11036, RRID: AB_10563566, https://scicrunch.org/resolver/RRID:AB_10563566)
Goat anti-Rabbit IgG, Alexa Fluor 647 (Thermo Fisher Scientific, cat. no. A-21245, RRID: AB_2535813, https://scicrunch.org/resolver/RRID:AB_2535813)
Goat anti-Rat IgG, Alexa Fluor 488 (Thermo Fisher Scientific, cat. no. A-11006, RRID: AB_2534074, https://scicrunch.org/resolver/RRID:AB_2534074)
Goat anti-Rat IgG, Alexa Fluor 568 (Thermo Fisher Scientific, cat. no. A-11077, RRID: AB_2534121, https://scicrunch.org/resolver/RRID:AB_2534121)
Goat anti-Rat IgG, Alexa Fluor 647 (Thermo Fisher Scientific, cat. no. A-21247, RRID: AB_141778, https://scicrunch.org/resolver/RRID:AB_141778)
Donkey anti-Goat IgG, Alexa Fluor 488 (Thermo Fisher Scientific, cat. no. A-11055, RRID: AB_2534102, https://scicrunch.org/resolver/RRID:AB_2534102)
Donkey anti-Goat IgG, Alexa Fluor 568 (Thermo Fisher Scientific, cat. no. A-11057, RRID: AB_2534104, https://scicrunch.org/resolver/RRID:AB_2534104)
Donkey anti-Goat IgG, Alexa Fluor 647 (Thermo Fisher Scientific, cat. no. A-21447, RRID: AB_2535864; https://scicrunch.org/resolver/RRID:AB_2535864)
Donkey anti-Mouse IgG, Alexa Fluor 488 (Thermo Fisher Scientific, cat. no. A-21202, RRID: AB_141607, https://scicrunch.org/resolver/RRID:AB_141607)
Donkey anti-Mouse IgG, Alexa Fluor 647 (Thermo Fisher Scientific, cat. no. A-31571, RRID: AB_162542, https://scicrunch.org/resolver/RRID:AB_162542)
Donkey anti-Rabbit IgG, Alexa Fluor 488 (Thermo Fisher Scientific, cat. no. A-21206, RRID: AB_2535792, https://scicrunch.org/resolver/RRID:AB_2535792)
Donkey anti-Rabbit IgG, Alexa Fluor 568 (Thermo Fisher Scientific, cat. no. A10042, RRID: AB_2534017, https://scicrunch.org/resolver/RRID:AB_2534017)
Donkey anti-Rabbit IgG, Alexa Fluor 647 (Thermo Fisher Scientific, cat. no. A-31573, RRID: AB_2536183, https://scicrunch.org/resolver/RRID:AB_2536183)
Donkey anti-Rat IgG, Alexa Fluor Plus 488 (Thermo Fisher Scientific, cat. no. A48269, RRID: AB_2893137, https://scicrunch.org/resolver/RRID:AB_2893137)
Donkey anti-Rat IgG, Alexa Fluor Plus 647 (Thermo Fisher Scientific, cat. no. A48272, RRID: AB_2893138, https://scicrunch.org/resolver/RRID:AB_2893138)
Equipment
For cell culture
STERILGARD 404 E3 (biosafety cabinet; Baker, cat no. SG404)
MCO-170AICUVL-PA CellIQ™ (incubator; PHC Corporation of North America, cat no. MCO-170AICUVL-PA)
CO2-resistant shaker (Thermo Scientific, cat. no. 88881101)
Universal aluminum platform for CO2-resistant shaker (Thermo Scientific, cat. no. 88-881-122)
Six-well culture plates (Eppendorf, cat. no. 0030720113)
U-bottom low-attachment 96-well plates (Thermo Scientific, cat. no. 174925 or S-Bio, cat. no. MS-9096UZ) ▲CRITICAL Different coating materials used from different vendors affect efficiency and quality of differentiation.
Low-attachment 24-well plates (Thermo Scientific, cat. no. 174930) ▲CRITICAL Different coating materials used from different vendors affect efficiency and quality of differentiation process.
Invitrogen Countess II automated cell counter (Invitrogen, cat. no. AMQAX1000)
10 mL reagent reservoirs (VistaLab Technologies, cat. no. 30542013)
25 mL reagent reservoirs (VistaLab Technologies, cat. no. 30541003)
5 mL round-bottom polystyrene test tubes with cell strainer snap caps; mesh size 35 μm (Falcon, cat. no. 352235)
250 μL wide-orifice LTS pipette tips, low retention, sterile (Rainin, cat. no. 30389250)
1000 μL wide-orifice LTS pipette tips, low retention, sterile (Rainin, cat. no. 30389221)
Bacteriological Petri dishes with lid (Falcon, cat. no. 351029)
2 mL Corning externally threaded cryogenic vials (Corning, cat. no. 430659)
Corning CoolCell FTS30 cell freezing vial containers (Corning, cat. no. 432008)
Corning CoolCell LX cell freezing vial containers (Corning, cat. no. 432002)
For reagent preparation
pH meter (METTLER TOLEDO, cat. nos. 30019029 and 51344055)
Stirrer with ceramic plate, Isotemp 30 °C to 540 °C (Fisher Scientific, cat. no. 11-300-49SHP)
Parafilm (Bemis, cat. no. PM996)
Hamilton concept fume hood
Steriflip sterile disposable vacuum filter (Millipore Sigma, cat. no. SE1M179M6)
0.22 μm sterile syringe filter (Millipore Sigma, cat. no. SLGPR33RS)
For cryosectioning and immunostaining
Cryostat (Leica, cat. no. 149491860us)
Tissue-Tek biopsy cryomolds 10 mm × 10 mm × 5 mm (Sakura Finetek, cat. no. 4565)
Tissue-Tek intermediate cryomolds 15 mm × 15 mm × 5 mm (Sakura Finetek, cat. no. 4566)
26 G sterile blunt needles (Strategic Applications Inc, cat. no. B26150)
Desiccator (Thermo Scientific, cat. no. 5311-0250)
Superfrost slides (Fisher Scientific, cat. no. 12-550-15)
Coverslip (Thermo Scientific, cat. no. 12460S)
PAP pen (Electron Microscopy Sciences, cat. no. 71310)
Coplin jar (IHC World LLC, cat. no. IW2501C)
Humidified slide staining chamber (Newcomer Supply, cat. no. 68432A)
Heated incubator / oven (PHC Corporation of North America, cat. no. MIR-H163)
Tube revolver / rotator (Thermo Scientific, cat. no. 88881001)
3D platform rotator (Fisher Scientific, cat. no. 88-861-045)
Eight-well silicone isolators 2.0 mm (Electron Microscopy Sciences, cat. no. 70339-44)
Eight-well silicone isolators 2.5 mm (Electron Microscopy Sciences, cat. no. 70339-46)
Cell imaging coverglasses with four chambers (Eppendorf, cat. no. 0030742028)
Cell imaging dishes with coverglass bottom, 145 μm (Eppendorf, cat. no. 0030740009)
Cell imaging dishes with coverglass bottom, 170 μm (Eppendorf, cat. no. 0030740017)
For imaging and image analysis
Nikon Ti2 (Nikon Instruments)
Nikon A1R (Nikon Instruments)
NIS-Elements Imaging Software (Nikon Instruments)
ImageJ
Common equipment
BD Slip Tip 1 mL sterile syringes (BD, cat. no. 309659)
Moria MC 17 BIS perforated spoon, diameter 15 mm (Fine Science Tools, cat. no. 1037018)
0.6 mL snap cap low-retention microcentrifuge tubes (Thermo Scientific, cat. no. 3446)
1.5 mL snap cap low-retention graduated microcentrifuge tubes (Thermo Scientific, cat. no. 3448)
2 mL round-bottom microcentrifuge tubes with locking snap cap (Fisher Scientific, cat. no. 14-666-315)
15 mL sterile conical tubes (Thermo Scientific, cat. no. 339650)
50 mL sterile conical tubes (Thermo Scientific, cat. no. 339652)
5 mL serological pipettes (Fisherbrand, cat. no. 13-678-11D)
10 mL serological pipettes (Fisherbrand, cat. no. 13-678-11E)
25 mL serological pipettes (Fisherbrand, cat. no. 13-678-11)
50 mL serological pipettes (Fisherbrand, cat. no. 13-678-11F)
Starter Kit including 4 LTS Pipet-Lite XLS+ manual single channel pipettes (Rainin, cat. no. 30386597)
20 μL LTS pipette tips, low retention, sterile (Rainin, cat. no. 30389229)
250 μL LTS pipette tips, low retention, sterile (Rainin, cat. no. 30389246)
1,000 μL LTS pipette tips, low retention, sterile (Rainin cat. no. 30389216)
Reagents setup
Human recombinant bFGF stock solution (200 μg/ml)
First, prepare 2 mL of 5 mM Tris-HCl (pH 7.5) by adding 10 μL of 1 M Tris-HCl in 1.99 mL of sterile water; vortex the solution to mix well. Using 1 mL of prepared 5 mM Tris-HCl, prepare 5 mM Tris-HCl containing 0.2% (wt/vol) HSA (carrier protein) by dissolving 2 mg of HSA in 1 mL of 5 mM Tris-HCl (hereafter, Tris-HCl + 0.2% HSA); vortex the solution to dissolve the HSA and flow through a 0.22 μm filter. Keep prepared 5 mM Tris-HCl and 5 mM Tris-HCl + 0.2% HSA solutions on ice. Then, start the reconstitution of the bFGF lyophilized powder (50 μg). Spin down the vial containing the powder before opening the cap and place the vial on ice. First, add 125 μL of 5 mM Tris-HCl (half of the final volume; 250 μL) to the lyophilized bFGF powder and leave the vial on ice for 5 min. After 5 min, mix the solution well by gently pipetting up and down. Do not vortex, and make sure to dissolve any remaining lyophilized powder on the inner wall of the vial. Then, add 125 μL (the remaining volume) of 5 mM Tris-HCl + 0.2% HSA, making the final volume of 250 μL containing 0.1% (final concentration) HSA; mix the solution well by gentle pipetting. Store bFGF solution in 2 μL and 7 μL aliquots at −80 °C and use within 6 months from the reconstituted date.
Human recombinant BMP4 stock solution (100 μg/mL)
PEPROTECH BMP4 (5 μg).
Lyophilized BMP4 from PEPROTECH needs to be reconstituted in 5 mM citric acid. First, prepare 50 mL of 1M citric acid stock solution (pH 3.0) by dissolving 9.606 g of citric acid (molecular weight 192.12 g/mol) in 50 mL of sterile water in a beaker on a stirrer, and adjust its pH to 3.0 using 10 N NaOH. Filter the prepared 1M citric acid solution through a Steriflip disposable vacuum filter and store the stock solution at 4 °C with the cap sealed with parafilm. Using this 1M citric acid stock solution, prepare 2 mL of 5 mM citric acid by adding 10 μL of 1 M citric acid in 1.99 mL of sterile water; vortex the solution to mix well. Using 1 mL of prepared 5 mM citric acid, prepare 5 mM citric acid containing 0.2% (wt/vol) of HSA by dissolving 2 mg of HSA in 1 mL of 5 mM citric acid (hereafter, citric acid + 0.2% HSA); vortex the solution to dissolve the HSA well, and flow through a 0.22 μm filter. Keep the prepared 5 mM citric acid and 5 mM citric acid + 0.2% HSA solutions on ice. Then, reconstitute the 5 μg of BMP4 lyophilized powder. Spin down the vial containing the powder before opening the cap and place the vial on ice. Add 25 μL of 5 mM citric acid (half of the final volume; 50 μL) to the powder, and leave it on ice for 5 min. After 5 min, mix gently by pipetting up and down. Do not vortex, and make sure to dissolve all lyophilized powder on the inner wall of the vial if there are any. Then, add 25 μL (the remaining volume) of 5 mM citric acid + 0.2% HSA, making the final volume of 50 μL containing 0.1% (final concentration) HSA; mix the solution well by gentle pipetting. Store BMP4 solution in 2 μL aliquots at −80 °C and use it within 6 months from the reconstituted date.
STEMCELL Technologies BMP4 (20 μg).
Lyophilized BMP4 from STEMCELL Technologies needs to be reconstituted in sterile water. First, prepare 1 mL of sterile water containing 0.2% (wt/vol) HSA by dissolving 2 mg of HSA in 1 mL of sterile water (hereafter, water + 0.2% HSA); vortex to dissolve the HSA and flow through a 0.22 μm filter. Keep the prepared water + 0.2% HSA and another 1 mL of sterile water on ice. Spin down the vial containing the lyophilized BMP4 before reconstitution to have all powder collected at the bottom of the vial. Add 100 μL (half of the final volume; 200 μL) of the chilled sterile water to the lyophilized powder, and keep the vial on ice for 5 min. After 5 min, mix the solution well by gentle pipetting. Do not vortex, and make sure that no residues of the power are remaining on the inner wall of the vial. Then, add 100 μL (the remaining volume) of the water + 0.2% HSA, making the final volume of 200 μL containing 0.1% (final concentration) HSA; mix the solution well by gentle pipetting. Store BMP4 solution in 2 μL aliquots at −80 °C and use it within 3 months after reconstitution.
SB in solution and LDN in solution
SB and LDN come in as ready-to-use in solution. Both small molecules are predissolved in DMSO. To avoid a freeze-thaw cycle, first, thaw both solutions at RT. Note that, as DMSO freezes at 4 °C or on ice, keep the solution at RT when thawing. Invert the vials or give gentle pipetting to mix the thawed solutions. Using a benchtop mini-centrifuge, give a brief spin-down (2–3 sec) to bring all solutions down to the bottom of each vial. Distribute SB solution in 32 μL and LDN solution in 2 μL aliquots, and store at −20 °C. Use the solutions within 6 months from the receiving date. ▲CRITICAL Both solutions are light sensitive. Store in dark storage boxes.
Essential 8 Flex medium containing normocin (E8)
Essential 8 Flex medium comes with a supplement as a kit. Thaw frozen supplement at 4 °C, and once it is thawed, transfer the whole volume to the Essential 8 Flex medium (~ 500 mL) and invert to mix well. Do not thaw the supplement at 37 °C. As the Essential 8 Flex medium containing supplement is used frequently, aliquot into 50 mL tubes (50 mL per tube) to decrease the chances of contamination and exposure to the air, which would change the pH of the medium. Label the name and date of the day that supplement has been added on the aliquoted 50 mL tubes. Store the medium at 4 °C and use it within 2 weeks from the day the supplement has been added. When using a fresh tube of 50 mL medium, add 100 μL of normocin (100 μg/mL) and invert to mix well. In this protocol, Essential 8 Flex medium containing supplement and normocin is stated as ‘E8’. ▲CRITICAL Do not warm the E8 stock bottles or aliquots at 37 °C. Always pre-warm the medium at RT.
Essential 6 medium containing normocin (E6)
As the Essential 6 medium is used only during the early differentiation period, calculate the volume that is needed for the experiment and aliquot out the volume directly from the medium bottle. When 30 mL of Essential 6 medium is needed, transfer 30 mL of the medium to a 50 mL tube, add 60 μL of normocin and invert to mix well. In this protocol, Essential 6 medium containing normocin is stated as ‘E6’.
E8 containing Y-27632 (Tables 4 and 5)
Table 4.
Day (−2): E8 + 10Y medium composition
| Components | Vendor | Cat. no. | Stock concentration | Final concentration | Volume (10 mL) |
|---|---|---|---|---|---|
| Essential 8 Flex medium | Gibco | A2858501 | - | 100% (v/v) | 10 mL |
| Y-27632 in solution | Stemgent | 04-0012-02 | 10 mM | 10 μM | 10 μL |
| Normocin | InvivoGen | Ant-nr-1 | 50 mg/mL | 100 μg/mL | 20 μL |
Table 5.
Day (−2): E8 + 20Y medium composition
| Components | Vendor | Cat. no. | Stock concentration | Final concentration | Volume (22 mL) |
|---|---|---|---|---|---|
| Essential 8 Flex medium | Gibco | A2858501 | - | 100% (v/v) | 22 mL |
| Y-27632 in solution | Stemgent | 04-0012-02 | 10 mM | 20 μM | 44 μL |
| Normocin | InvivoGen | Ant-nr-1 | 50 mg/mL | 100 μg/mL | 44 μL |
The E8 containing 10 μM Y-27632 is stated as ‘E8 + 10Y’ in this protocol and used during thawing, passaging, and freezing cells and preparing single cells for differentiation initiation and scRNA-seq experiments. The E8 containing 20 μM Y-27632 is stated as ‘E8 + 20Y’ in this protocol and used for cell suspension preparation on differentiation day −2. ▲CRITICAL Replenish or dilute out the E8 containing Y-27632 after 24 hrs of incubation.
Differentiation day 0 medium (E6SFB, Table 6)
Table 6.
Day 0: E6SFB differentiation medium composition
| Components | Vendor | Cat. no. | Stock concentration | Final concentration | Volume (30 mL) |
|---|---|---|---|---|---|
| Essential 6 medium | Gibco | A1516401 | - | 98% (v/v) | 29.4 mL |
| Matrigel, growth factor reduced | Corning | 354230 | - | 2% (v/v) | 600 μL |
| SB (TGF-β inhibitor) | Stemgent | 04-0010-05 | 10 mM | 10 μM | 30 μL |
| Recombinant human bFGF | PeproTech | 100-18B | 200 μg/mL | 4 ng/mL | 0.6 μL |
| Recombinant human BMP4 | PeproTech | 120-05 | 100 μg/mL | 5 ng/mL | 1.5 μL |
| Normocin | InvivoGen | Ant-nr-1 | 50 mg/mL | 100 μg/mL | 60 μL |
On day 0 of differentiation, E6 containing 2% Matrigel (vol/vol), 10 μM SB, 4 ng/ml bFGF, and 5 ng/ml BMP4 is needed. This day 0 medium is stated as ‘E6SFB’ in this protocol. When preparing E6SFB, make 2% Matrigel containing E6 first, and then add the small molecules and proteins. To make 30 mL of E6SFB, prepare 30 mL of E6 in a 50 mL tube and keep it on ice. From the 30 mL E6, remove 600 μL of the E6 (the volume of Matrigel that is going to be added), and add 600 μL of pre-thawed Matrigel. Mix well by inverting. Then, add 30 μL of SB, 0.6 μL of bFGF, and 1.5 μL of BMP4. Invert several times to mix well and keep on ice. ▲CRITICAL It takes about 2 hrs and 30 min to thaw aliquoted Matrigel on ice. It is recommended to thaw the Matrigel overnight on ice at 4 °C. Mix the thawed Matrigel by brief vortexing before use. ▲CRITICAL Always make the medium fresh. The concentration of BMP4 may vary depending on cell lines and vendors. See ‘Experimental design’ and ‘Troubleshooting’ for details. Reconstituted bFGF and BMP4 are temperature sensitive. Keep on ice once thawed.
Differentiation day 3 medium (E6LF, Table 7)
Table 7.
Day 3: E6LF differentiation medium composition
| Components | Vendor | Cat. no. | Stock concentration | Working concentration | Volume (5 mL) |
|---|---|---|---|---|---|
| Essential 6 medium | Gibco | A1516401 | - | 100% (v/v) | 5 mL |
| Recombinant human bFGF | PeproTech | 100-18B | 200 μg/mL | 250 ng/mL (50 ng/mL X5) | 6.25 μL |
| LDN (BMP inhibitor) | Stemgent | 04-0074-02 | 10 mM | 1 μM (200 nM X5) | 0.5 μL |
| Normocin | InvivoGen | Ant-nr-1 | 50 mg/mL | 100 μg/mL | 10 μL |
On day 3 of differentiation, LDN (BMP inhibitor) and bFGF will be treated at final concentrations of 200 nM and 50 ng/mL, respectively. Before the treatment of LDN and bFGF, there is 100 μL of medium per well in a 96-well round-bottom plate. The LDN and bFGF combination will be treated in a volume of 25 μL per well, making a final volume of 125 μL per well. As 25 μL is one-fifth the volume of the final volume, fivefold (5X) concentrated LDN (1 μM) and bFGF (250 ng/ml) need to be prepared in the medium to have 1X concentrations of LDN and bFGF in the final volume of 125 μL after treatment. Therefore, to make E6 containing 1 μM LDN and 250 ng/mL bFGF (hereafter, ‘E6LF’), prepare 5 mL of E6 in a 15 mL tube and add 0.5 μL of LDN and 6.25 μL of bFGF. Invert to mix well.
Organoid maturation medium (OMM, Table 8)
Table 8.
OMM composition
| Components | Vendor | Cat. no. | Stock concentration | Final concentration | Volume (50 mL) |
|---|---|---|---|---|---|
| Advanced DMEM/F12 | Gibco | 12634010 | - | 49% (v/v) | 24.5 mL |
| Neurobasal medium | Gibco | 21103049 | - | 49% (v/v) | 24.5 mL |
| GlutaMAX supplement | Gibco | 35050061 | 100X | 1X | 500 μL |
| B-27 supplement, minus vitamin A | Gibco | 12587010 | 50X | 0.5X | 500 μL |
| N2 supplement | Gibco | 17502048 | 100X | 0.5X | 250 μL |
| 2-Mercaptoethanol | Gibco | 21985023 | 55 mM | 0.1 mM | 91 μL |
| Normocin | InvivoGen | Ant-nr-1 | 50 mg/mL | 100 μg/mL | 100 μL |
To prepare 50 mL of OMM, combine 24.5 mL of advanced DMEM/F12, 24.5 mL of Neurobasal medium, 500 μL of GlutaMax supplement, 500 μL of B-27 supplement minus vitamin A, 250 μL of N2 supplement, 91 μL of 2-mercaptoethanol, and 100 μL of normocin. Make fresh OMM when needed, and the remaining medium can be stored at 4 °C and used within 1 week.
OMM containing 1% Matrigel (OMM1%M)
The OMM containing 1% Matrigel is referred to as ‘OMM1%M’ in this protocol. When making a medium containing Matrigel, always remove the same volume as the Matrigel that will be added first, and then add the Matrigel. For example, to make 30 mL of OMM1%M, prepare 30 mL of OMM in a 50 mL tube, remove 300 μL of OMM and add 300 μL of Matrigel. Invert several times to mix evenly and keep on ice until use. Make fresh OMM1%M when needed.
Freezing medium
For long-term storage of cells in a liquid nitrogen tank, cells need to be frozen in a freezing medium. The freezing medium is E8-based and contains 10% (vol/vol) DMSO. We recommend freezing down cells to three cryovials from a well of six-well plate when the cells have reached 75–80% confluency. Cells will be frozen in a 200 μL volume per cryovial. Therefore, when freezing down cells from three wells, nine cryovials and 1.8 mL of freezing medium will be needed. Considering any pipetting error, prepare a 2 mL freezing medium by adding 200 μL of DMSO to 1.8 mL of E8, and keep on ice. ▲CRITICAL Freezing medium does not contain Y-27632.
0.5 mM EDTA solution
To prepare 50 mL of 0.5 mM EDTA solution, add 50 μL of commercially available 0.5 M EDTA to 50 mL of 1X DPBS; mix well by vortexing or inverting the solution. Store the prepared solution at 4 °C and equilibrate to RT before use for dissociation.
4% PFA (vol/vol)
First, prepare 50 mL of 2X PBS by diluting 10 mL of 10X PBS in 40 mL of MilliQ water; vortex or invert to mix the solution. To prepare 36 mL of 4% PFA, first, transfer 9 mL of commercially available 16% PFA into a tube, followed by adding in 18 mL of 2X PBS; vortex or invert to mix the solution. Then, add 9 mL of MilliQ water and mix well by vortexing or inverting, making a final volume of 36 mL and final concentration of 4% PFA in 1X PBS. Diluted 4% PFA can be stored at 4 °C with the cap sealed with parafilm; make fresh and use it within 2 weeks. Any remaining undiluted 16% PFA can be stored at 4 °C with the cap sealed with parafilm; use within 1 month. ! CAUTION Procedures involving the usage of 16% PFA must be performed in a fume hood; inhalation of PFA is hazardous.
Sucrose solutions
30% sucrose.
To make 200 mL of 30% (wt/vol) sucrose stock solution, pour 200 mL of 1X PBS into a beaker containing a magnetic bar and mark the surface level of the buffer on the beaker with a marker. Pour out about a half volume of the 1X PBS to another beaker and keep it aside. Prepare 60 g of sucrose and pour into the beaker containing a magnetic bar. Place the sucrose-containing beaker on a magnetic stirrer and dissolve the sucrose by starting the stirrer. When sucrose fully dissolves, stop the stirrer and wait until the swirling movement settles. Then, pour in the 1X PBS that is kept aside until the volume reaches the level that is marked on the beaker. Give a last brief stirring on the magnetic stirrer to fully mix the volume. Transfer the full volume of 30% sucrose to a sterile bottle, label the bottle, and store it at 4 °C.
15% sucrose.
Using the 30% sucrose stock solution, dilute to 15% (vol/vol) in 1X PBS. Make fresh when needed.
Whole-mount immunostaining (qCe3D) washing buffer
Make 0.3% (vol/vol) Triton X-100 and 0.5% (vol/vol) 1-thioglycerol in 1X PBS. For example, when making 30 mL of washing buffer, remove 1.05 mL (total volume of other reagents that compose the washing buffer) from 30 mL of 1X PBS (or start with 28.95 mL of 1X PBS) and add 900 μL of 10% Triton X-100 and 150 μL of 1-thioglycerol. Mix well by placing the tube on a platform-rotator for ~20 min at RT. ! CAUTION Work with 1-thioglycerol in a fume hood to eliminate inhalation, which may cause respiratory irritation. It is malodorous.
Whole-mount immunostaining (qCe3D) blocking buffer
Make 0.3% (vol/vol) Triton X-100, 1% (vol/vol) normal goat or horse serum based on the antibodies, and 1% BSA (wt/vol) dissolved in 1X PBS. First, prepare 1% BSA solution in 1X PBS and place it on a platform-rotator for ~20 min or more at RT until the BSA powder is fully dissolved. After confirming that the BSA powder is completely dissolved, add the adequate volume of Triton X-100 and mix well. Then, add the adequate volume of serum. For example, when making 15 mL of blocking solution, first prepare 1% BSA solution by dissolving 150 mg of BSA in 15 mL of 1X PBS and placing it on a platform-rotator for 20 min at RT. From the 15 mL of 1% BSA solution, remove 600 μL (the total volume of other reagents that will be added) and then add 450 μL of 10% Triton X-100 and mix well by placing it on a platform-rotator for 5 min at RT. Then, add 150 μL of serum and mix well by inverting the tube a couple of times.
40% N-methylacetamide
N-methylacetamide is solid at RT. Place the entire bottle of N-methylacetamide at 37 °C overnight, until it fully liquefies. Pre-warm a 25 mL serological pipette at 37 °C together with the reagent. When the N-methylacetamide fully liquefies, using the pre-warmed 25 mL serological pipette, add 20 mL of N-methylacetamide to 30 mL of 1X PBS, making 50 mL of 40% (vol/vol) N-methylacetamide stock solution. Be quick when transferring N-methylacetamide to 1X PBS to inhibit the solidification of N-methylacetamide. Mix well by inverting the tube. Store at RT and use within 2 months. ! CAUTION Inhalation of N-methylacetamide may damage fertility or the unborn child. Perform all steps involving N-methylacetamide in a fume hood.
qCe3D clearing solution
Final solution comprises 22% N-methylacetamide, 80% Histodenz (wt/vol), 0.1% (vol/vol) Triton X-100, and 0.5% 1-thioglycerol. To make 10 mL of clearing solution, add the reagents in the following order: to a 15 mL tube, add 4 mL of 40% N-methylacetamide, 8 g of Histodenz, 1.5 mL of 40% N-methylacetamide, and then 100 μL of 10% Triton X-100. Seal the gaps between the tube and the caps using parafilm and incubate at 37 °C on a tube-rotator overnight (>1 hr) until all reagents are evenly mixed. Clumps of reagents in the solution will be observed if the solution is not well mixed. Give the last thorough mix using a 10 mL serological pipette. Lastly, add 50 μL of 1-thioglycerol and mix well by placing it on a platform-rotator for ~20 min. Store at RT and use within 1 month. ! CAUTION Inhalation of N-methylacetamide may damage fertility or the unborn child. Perform all steps involving N-methylacetamide in a fume hood. ! CAUTION 1-Thioglycerol is hazardous. Avoid direct contact with skin and eyes. Wear rubber gloves and eye goggles. May cause respiratory irritation. Manipulate in a fume hood.
Equipment setup
CO2-resistant shaker in an incubator
Sterilize the CO2-resistant shaker using 70% ethanol and place it on the lowest shelf of an incubator that is set to 37 °C with 5.0% CO2. Run the shaker at a speed of 65 rpm to provide consistent agitation to the medium for long-term cultures in 24-well low-attachment plates.
Vitronectin-coated plate preparation (1 mL per well in six-well culture plate)
Vitronectin is used at a final concentration of 5 μg/mL (0.5 μg/cm2, 1:100 dilution). Dilute 60 μL of vitronectin stock solution (0.5 mg/mL) in 6 mL of 1X DPBS and mix well by pipetting with a 5 mL serological pipette. Using the same serological pipette, distribute 1 mL of diluted vitronectin into each well of the six-well plate and gently swirl the plate to cover the whole area of each well. Incubate for at least 1 hr at RT before use. Vitronectin-coated plates can be stored at 4 °C for up to a week, sealed with parafilm; the solution should not dry up. When using the pre-coated plates stored at 4 °C, pre-warm to RT for at least 20 min before use. The volume of vitronectin solution should be adjusted according to the number of wells that will be coated. ▲CRITICAL Do not turn on UV of the biosafety cabinet while vitronectin coating/coated plates are placed in it. UV crosslinks the protein and changes its structure and character.
Procedure
▲CRITICAL Every step related to cell culture should be performed in a biosafety cabinet.
hPSC culture: thawing, maintenance, passaging, and freezing
▲CRITICAL Always pre-warm media at RT for 15–20 min. Do not warm the media in a water bath or 37 °C.
Thawing frozen hPSCs ●Timing 30 min
-
1
Prepare a vitronectin-coated six-well plate and a water bath at 37 °C.
▲CRITICAL STEP Different numbers (1–3) of vitronectin-coated wells may be needed depending on the cell density in a frozen vial. Typically, having three coated wells is enough for thawing a vial of cells.
-
2
Prepare E8 + 10Y by adding 13 μL of Y-27632 (Y) to 13 mL of E8 in a 15 mL tube.
-
3
Invert the tube to mix well, and pre-warm the E8 + 10Y at RT for 20 min.
-
4
Meanwhile, label on the lid of the six-well plate with cell line information, including the passage number written on the frozen vial.
-
5
From the pre-coated plate in Step 1, aspirate out the vitronectin solution and immediately add 1.5 mL of the E8 + 10Y into each well.
▲CRITICAL Perform this step quickly to avoid drying the surface of the wells.
■PAUSE POINT Store the plate containing E8 + 10Y in a 37 °C incubator with 5.0% CO2 while the cells are being prepared for seeding (<20 min).
-
6
Take out the frozen cell vial from dry ice, add 500 μL of pre-warmed E8 + 10Y and then thaw in a 37 °C water bath by giving a gentle swirl until a small piece of floating ice is observed.
▲CRITICAL STEP Perform this step quickly. The less time it takes, the higher the cell viability.
-
7
Bring the vial to the biosafety cabinet and spray with 70% ethanol. Using a p1000 W-O tip, give a gentle pipetting and collect the whole volume into a new 15 mL conical tube.
-
8
Rinse the vial that formerly contained the cells with 1 mL of E8 + 10Y using a p1000 W-O tip to collect any residual cells and transfer the volume to the 15 mL tube where cells are collected in Step 7.
-
9
Make a final volume to ~5.5 mL by adding 4 mL of E8 + 10Y to the 15 mL tube in Step 8.
-
10
Pellet the cells by centrifugation at a speed of 230g for 5 min 30 sec at RT (20 – 25 °C).
-
11
Carefully aspirate out the medium and add 750 μL of E8 + 10Y.
-
12
Using a p1000 W-O tip, give gentle pipetting to resuspend the cell pellet and add an additional 750 μL of E8 + 10Y to make a total volume of 1.5 mL, considering that the cells will be distributed in 500 μL per well to three wells. Give gentle pipetting to mix well.
-
13
Add 500 μL of the cell suspension in a drop-wise manner to the first well of the six-well plate prepared in Step 5, making a final volume of 2 mL in a well. Gently swirl and shake the plate to evenly distribute cells.
-
14
Check the cell seeding density under the microscope. See Fig. 7a as a reference. The recommended cell seeding density per well is ~7 × 105 cells per well of a six-well plate. Visually, under the microscope, floating cells appear to occupy ~50–55% of the area of a well when they adhere on the plate.
If the 500 μL of cell suspension reaches the recommended cell seeding density, distribute remaining 1 mL cell suspension in 500 μL (the equal cell suspension volume as in Step 13) per well to the following two vitronectin-coated wells. This brings to a final volume of 2 mL per well with cells in three wells of a six-well plate.
If the 500 μL of cell suspension is lower than the recommended cell seeding density, add an extra volume from the remaining cell suspension to the same well (the first well) to reach the desired confluency (e.g., add an extra 250 μL of cell suspension). If there is remaining cell suspension after adjusting the first well cell confluency (e.g., remaining 750 μL of cell suspension), add it to the next vitronectin-coated well. This brings to a final volume of 2.25 mL per well with cells in two wells of a six-well plate.
▲CRITICAL STEP As some cells fail to survive after the freezing-thawing process, we recommend seeding at a higher rather than a lower cell seeding density relative to the recommended density when the viability of the cells in the frozen cryovial is unclear.
-
15
Incubate cells in a 37 °C incubator with 5.0% CO2.
-
16
The next day (after 24 hrs), replenish medium with fresh E8 without Y (only E8 Flex medium containing normocin) and maintain the cells as mentioned in ‘Maintenance’. ▲CRITICAL STEP Y must be removed for normal cell proliferation.
? TROUBLESHOOTING
Fig. 7. Representative phase-contrast images of recommended cell seeding density and cell confluency.

a, A representative image showing ~50% of cell seeding density per well in a six-well plate, recommended for thawing and passaging procedures. Note that the cells are in tiny clusters; average six cells per cluster. Note that, when thawing a new vial of cells, seeding at a slightly higher cell density than what is shown in a is recommended. This is because the viability of cells in a vial could have decreased during the freezing-thawing cycle. b, A representative image of cell confluency and morphology, 1 day after thawing or passaging procedures. c, A representative image of the cell colonies that have reached 75–80% cell confluency. The cells shown in c are ready to be passaged or used for differentiation. Typically, the cells reach the 75–80% confluency after 4–5 days of passaging or thawing. All images are taken at 40X (4X microscope objective × 10X eyepiece) magnification as noted in each image. Scale bars, 500 μm.
hPSC maintenance ●Timing 10 min
▲CRITICAL STEP We recommend that newly thawed cells undergo the passaging process at least twice (i.e., this and next section) before being used for differentiation experiments. This ensures cells are healthier and better adapted to their growing environment.
-
17
Pre-warm E8 at RT for 15–20 min.
-
18
Replenish spent medium with E8 every other day by carefully aspirating spent medium.
-
19
Gently add 2 mL of E8 per well, toward the wall of each well using a 5 mL serological pipette. Increasing the E8 volume to 3 mL per well or replenishing E8 every day may be required as cells become confluent. When the cell confluency reaches ~75–80% in a well (See Fig. 7c for a reference image), generally every 4–5 days, the cells are ready to undergo the passaging process.
▲CRITICAL STEP Perform the steps gently but quickly enough to avoid drying out the cells.
? TROUBLESHOOTING
hPSC passaging ●Timing 30 min
▲CRITICAL This procedure is standardized to passaging cells into three wells of a six-well plate using accutase. Different dissociation enzymes (e.g., EDTA) are recommended for some cell lines. Check each cell line’s datasheet from the manufacturers for advice on optimal enzymes. See Fig. 7 for reference images of cell seeding density and confluency.
-
20
Prepare a vitronectin-coated six-well plate.
-
21
Prepare 17 mL of E8 + 10Y and pre-warm at RT for 20 min.
-
22
Equilibrate the accutase to RT (or pre-warm at 37 °C) for ~15 min.
-
23
When Steps 20–22 are ready, aspirate out the vitronectin solution from the six-well plate and immediately add 1.5 mL of E8 + 10Y into each well.
▲CRITICAL STEP Perform this step quickly to avoid drying out the surfaces of the wells.
■PAUSE POINT Store the plate containing E8 + 10Y in a 37 °C incubator with 5.0% CO2 while the detached cells are being readied for plating (<20 min).
-
24
From the well where cells have reached 75–80% confluency, carefully aspirate out the spent medium.
-
25
Wash two times with 2.5 mL of 1X DPBS using a serological pipette.
▲CRITICAL STEP Be gentle when adding 1X DPBS by adding solution toward the wall of the well.
-
26
After aspirating out the last washing solution, add 500 μL of pre-warmed accutase solution using a p1000 regular tip to the center of the well.
▲CRITICAL STEP Perform Steps 24–26 gently but quickly to avoid drying out the cells.
-
27
Gently swirl the plate to make sure that the solution fully covers the surface of the well.
-
28
Incubate the plate in a 37 °C incubator for a total of 3–5 min depending on cell lines. After 2–3 min, check under a microscope to see if the cells are rounding up and the cell colonies are detaching from the plate; gently rock the plate back and forth to make the loosen up cell colonies detach from the plate. If the cells are still attached on the surface of the plate, give an extra incubation with an increment of 20 sec.
▲CRITICAL STEP The detached cell clumps will be passaged in tiny clusters of cells (average six cells per cell cluster). See Fig. 7a for a reference image. Therefore, do not over-incubate the cells with the enzyme at 37 °C to avoid cell clumps dissociating to single cells.
-
29
Once the cell colonies are almost all detached, tilt the plate at ~45° angle and give a gentle window-wiper-like motion pipetting using a p1000 W-O tip to detach any cell colonies that are still slightly adhering on the surface.
-
30
Keeping the plate angled at 45°, collect all detached cells and transfer to a 15 mL tube.
-
31
Using a p1000 W-O tip, gently pipette to break down cell clumps into evenly sized tiny clusters (average six cells per cluster) within the cell suspension.
▲CRITICAL STEP Do not pipette aggressively at this step as the enzyme is still active.
-
32
Using a 5 mL serological pipette, slowly add 5 mL of E8 + 10Y to the 15 mL tube containing cell clusters in Step 31, making a final volume of 5.5 mL.
▲CRITICAL STEP Addition of medium (dilution of enzyme at least to a 1:4 ratio) inactivates the enzymatic activity of accutase. Practice tests are recommended to help experimenters get familiarized with the cell cluster sizes and optimal preparation.
-
33
Centrifuge at 230g for 5 min 30 sec at RT.
-
34
Carefully aspirate out the medium containing the accutase solution; remove as much as possible by tilting the tube to ~45° angle as the remaining volume in the tube decreases during aspiration.
-
35
Using a p1000 W-O tip, add 1 mL of E8 + 10Y and gently pipette up and down to resuspend the cell cluster pellet.
-
36
Using a 5 mL serological pipette, slowly add an extra 5 mL of E8 + 10Y (making a total volume of 6 mL) and gently pipette the entire cell cluster suspension twice to mix well. Note that, depending on the cell density in the tube, the final volume of cell suspension E8 + 10Y varies. The cell density in the tube can be predicted by the initial cell confluency in a well where the passaging process has initiated from and by the pellet size after the centrifugation in Step 33. If the cell confluency in a well was ~80% as shown in Fig. 7c, resuspend the cell pellet at a final volume of 6 mL. If the cell confluency was lower than the Fig. 7c representative image (e.g., 75% or less), resuspend the cells at a final volume of 5 mL or less.
▲CRITICAL STEP Several practice tests enable familiarization with adjusting the final cell suspension volume.
-
37
Using a 5 mL serological pipette, add five drops of cell cluster suspension into the first well of the six-well plate prepared in Step 23.
-
38
Gently swirl and shake the plate to evenly distribute the cell clusters within the well.
-
39
Check under the microscope and see if the cell seeding density is ~50% in the first well and the cell clusters are on average six-cell-sized. See Fig. 7a for a reference image. ▲CRITICAL STEP If the cell cluster sizes are still bigger than the desired size (e.g., containing more than 10 cells per cluster) or vary within the cell cluster suspension, gently pipette the whole cell suspension volume using a 5 mL serological pipette for two to three times. After the cell cluster sizes become even, add an extra one or two drops of the cell cluster suspension into the first well to reach ~50% cell seeding density (if the cell seeding density did not reach the recommended density in Step 39) or distribute to the following wells as described in Step 40.
-
40
According to the number of drops of cell cluster suspension added to the first well, add a sequentially decreasing number of cell cluster suspension drops to the following two wells. For example, if five drops of the cell cluster suspension were added in Step 37 (e.g., 50% cell density), add four drops (45%) to the second well and three drops (40%) to the third well.
▲CRITICAL STEP Sequentially different cell concentrations in one plate enables the well with the best cell confluency to be used at the time differentiation experiments or passaging are initiated.
-
41
Gently swirl and shake the plate to evenly distribute the cell clusters in each well.
-
42
Incubate cells in a 37 °C incubator with 5.0% CO2.
-
43
The next day (after 24 hrs), replenish medium to fresh E8 without Y and maintain the cells as mentioned in ‘Maintenance’ until they are ready to be passaged or used for differentiation experiments.
? TROUBLESHOOTING
Freezing hPSC ●Timing 40 min
▲CRITICAL This protocol is standardized to cryo-preserve cells from three wells of a six-well plate. Cells that have reached the confluency of 75–80% per well should be cryo-preserved in three cryovials per well in a 200 μL volume per cryovial. See Fig. 7c for a reference image of cell confluency.
-
44
Pre-chill the freezing containers in a 4 °C fridge, according to manufacturers.
-
45
Label cryovials with cell line information, passage number, freezing date, and the initial of the person’s name who is freezing the cells. Pre-chill the labeled cryovials in −20 °C for at least 1 hr.
-
46Prepare 2 mL of freezing medium by adding 200 μL of DMSO (10%, vol/vol) to 1.8 mL of E8 in a 2 mL tube and keep on ice to keep cold.
- Calculate the required volume of freezing medium: 3 wells × 3 cryovials × 200 μL per cryovial = 1.8 mL Adjust to make 2mL total to allow for pipetting error.
-
47
Pre-warm accutase and 6 mL of E8 + 10Y at RT for 15–20 min.
▲CRITICAL STEP As accutase needs to be diluted at least to a 1:4 ratio to become enzymatically inactive, higher volume of E8 + 10Y are required when collecting cells from more than three wells. For example, when collecting cells from five wells, a total of 2.5 mL accutase containing cell suspension is collected, and 10 mL of E8 + 10Y is needed for enzyme inactivation.
-
48
When ready, proceed with the same procedure as ‘Passaging’ Steps 24–34. Keep cell cluster sizes bigger (average ten cells per cluster) than during the normal passaging process to increase the viability of cells after freezing and thawing.
-
49
To the final cell cluster pellet, add 1 mL of the pre-chilled freezing medium, and resuspend the pellet by gentle pipetting using a p1000 W-O tip.
-
50
Using a p1000 W-O tip, add an additional 800 μL (the rest of the volume needed for freezing total nine vials, 1.8 mL) of pre-chilled freezing medium and gently pipette up and down to mix well. Keep the cell suspension on ice while uncapping the pre-chilled cryovials.
-
51
Uncap the pre-chilled cryovials in the biosafety cabinet.
▲CRITICAL STEP Keep the cryovials sterile. Do not touch the inner lining of the caps and the tubes.
-
52
Once all the cryovials are uncapped, using a p1000 W-O tip, give a final gentle pipetting to evenly mix the whole volume of the cell cluster suspension prepared in Step 50 and distribute in 200 μL volume per pre-chilled cryovial.
-
53
Transfer all cryovials to the pre-chilled freezing containers and store them in a −80 °C freezer overnight. For long-term storage, transfer the frozen cryovials to a liquid nitrogen tank within 72 hrs post-freezing process.
▲CRITICAL STEP Perform Steps 49–53 quickly. Reduced time results in higher cell viability.
Differentiation day (−2): cell aggregation ●Timing 50 min
▲CRITICAL This protocol is standardized to the preparation of culture in 2 plates of 96-well plate.
-
54
Media and enzyme preparation. Prepare 10 mL of E8 + 10Y in a 15 mL conical tube and 22 mL of E8 + 20Y in a 50 mL conical tube. Then, equilibrate prepared media to RT.
-
55
Pre-warm accutase at RT.
-
56
Cell dissociation. Aspirate out the spent medium of the cells in one well of a six-well culture plate.
-
57
Carefully wash with 2.5 mL of 1X DPBS twice and aspirate out the 1X DPBS from the last wash.
-
58
Add 500 μL of pre-warmed accutase and incubate for 3–4 min in a 37 °C incubator with 5.0% CO2. After 2–3 mins of the first incubation, gently rock and shake the plate to detach any loosened-up cells that are still adhering on the surface of the plate. Check under a microscope to confirm the detachment. If cell colonies are still attached tightly, incubate for further 20 sec increments.
-
59
When the cell colonies are all detached, gently and briefly pipette to break down cell clumps into single cells using a p1000 regular tip.
-
60
Transfer the cell suspension into a 15 mL conical tube.
-
61
Using a 5 mL serological pipette, add 5 mL of E8 + 10Y to the 15 mL tube that contains cell suspension from Step 60 and gently pipette to mix and break up any remaining cell clusters into single cells.
-
62
Centrifuge at 230g for 5 min 30 sec at RT.
-
63
Carefully aspirate almost all of the supernatant by tilting the tube to an angle and aspirating with the medium flowing away from the cell pellet.
▲CRITICAL Do not disturb the cell pellet.
-
64
Using a p1000 regular tip, resuspend the cells in 1 mL of E8 + 10Y by gentle pipetting.
-
65
Cell aggregation. To equilibrate the 35 μm mesh cell strainer, forcefully pipet 1 mL of E8 + 10Y through the strainer mesh.
▲CRITICAL Keep the strainer mesh intact. Do not scratch or poke the strainer mesh with the pipet tip.
-
66
Transfer resuspended cells in E8 + 10Y in Step 64 to the cell strainer prepared in Step 65 in a drop-wise manner.
-
67
To thoroughly collect cells, rinse the tube that formerly contained cells with 1 mL of E8 + 10Y and transfer the volume to the cell strainer in a drop-wise manner.
-
68
Carefully remove and discard the snap cap part of the cell strainer.
-
69
Determine the number of live cells that are collected in the Step 67 tube as follows:
Prepare 50 μL of trypan blue in a 500 μL tube.
Using a p1000 regular tip, gently pipette to mix the cells evenly in the tube.
Immediately, using a p200 regular tip, collect 50 μL of cell suspension from the center of the volume in the tube, and transfer it into the 50 μL of prepared trypan blue. This makes a 1:1 dilution of the cells in trypan blue.
Gently pipette to mix the cells evenly with trypan blue.
Add 11 μL of the cell-trypan blue mixture into both sides of a cell counting chamber slide for Invitrogen Countess II automated cell counter or on a hemocytometer.
Record the number of live cells per 1 mL shown on the automated counter. The Invitrogen Countess II automated cell counter gives automatically calculated cell numbers considering the trypan blue dilution. When manually counting the live cell number, be sure to incorporate trypan blue dilution in the calculation by multiplying by 2 (diluted factor).
Calculate the number of cells needed for a differentiation experiment. The final cell concentration of 35,000 cells/mL (3,500 cells in 100 μL per well) is needed for a differentiation culture. Therefore, 7.7 × 105 cells are needed in 22 mL.
Then, calculate the volume of single-cell suspension needed for the experiment (7.7 × 105 cells in 22 mL). For example, if the readout of the live cell number from the automated cell counter was 5 × 105 cells per mL, 1.54 mL of the cell suspension is needed in 22 mL. Equation: Volume of cell suspension needed for differentiation (mL) = total number of cells needed for differentiation (7.7 × 105 cells) / number of cells in 1 mL of cell suspension (5 × 105 cells) = 7.7 × 105 cells / 5 × 105 cells = 1. 54 mL
-
70
Based on the cell number calculation, remove the same volume of cell suspension that is needed for the experiment from 22 mL of E8 + 20Y prepared in Step 54. Then add the calculated volume of the cell suspension to the E8 + 20Y; i.e., remove 1.54 mL of E8 + 20Y from the 22 mL of E8 + 20Y prepared in Step 54, and add 1.54 mL of cell suspension prepared in Step 67 to the 20.46 mL of E8 + 20Y, which brings the final cell concentration to 7.7 × 105 cells in a total volume of 22 mL.
-
71
Gently invert a couple of times to mix the cell suspension evenly.
-
72
Pour the 22 mL cell suspension into a 25 mL reservoir.
-
73
Using a multichannel pipette and p200 regular tips, distribute 100 μL of cell suspension into each well of 96-well U-bottom plates.
▲CRITICAL When pipetting, be careful not to poke or scratch the interior of the wells with the tip ends; keep the coating of each well intact.
-
74
Centrifuge the plates at 110g for 6 min at RT.
-
75
Incubate the plates in a 37 °C incubator with 5.0% CO2 for 24 hrs.
Differentiation day (−1): dilution of Y solution ●Timing 15 min
-
76
Prepare and pre-warm 22 mL of fresh E8 (without Y).
-
77
Pour 22 mL of fresh E8 into a 25 mL reservoir.
-
78
Using a multichannel pipette and p200 regular tips, add 100 μL of E8 into each well, which brings the total volume to 200 μL per well.
-
79
Incubate the plates in a 37 °C incubator with 5.0% CO2 for 24 hrs.
-
80
Thaw 600 μL of Matrigel on ice overnight at 4 °C to be ready for the next step.
Differentiation day 0: transition to differentiation in E6SFB ●Timing 1 hr 30 min
▲CRITICAL Perform all procedures on ice.
-
81
Prepare 3 mL of E6 in a 15 mL conical tube for the washing step.
-
82
Prepare 30 mL of E6SFB and keep on ice.
▲CRITICAL STEP The E6SFB is composed of 2% Matrigel (vol/vol), 10 μM SB, 4 ng/ml bFGF, and 5 ng/ml BMP4 in E6.
? TROUBLESHOOTING
-
83
Collect all aggregates from 96-well U-bottom plates to a 2 mL round-bottom tube as follows.
Using p200 W-O tips and a multi-channel pipette - set the pipette to 180 μL (i.e., ~20 μL less than the total volume in each well) - collect all aggregates in a 100 mm Petri dish.
By gently swirling the Petri dish on a flat surface, concentrate all aggregates to the center of the dish.
Using a p1000 W-O tip, collect all aggregates into a 2 mL round-bottom tube.
-
84
When all aggregates settle at the bottom of the tube (typically takes <20 sec), carefully remove excessive E8 from the tube.
-
85
Wash with 1 mL of E6 three times to completely remove traces of E8.
-
86
Add 1 mL of E6SFB to the tube containing aggregates.
-
87
Place a new 100 mm Petri dish on ice and add ~15 mL of E6SFB.
-
88
Using a p1000 W-O tip, transfer all aggregates that are prepared in Step 86 to the Petri dish containing ~15 mL of E6SFB on ice.
-
89
Use an extra 1 mL of E6SFB to collect and transfer any remaining aggregates in the tube to the Petri dish.
-
90
Using a p200 W-O tip, transfer individual aggregate in 100 μL of E6SFB into each well in a new 96-well U-bottom plate (pour in more E6SFB to the Petri dish as it goes).
-
91
Incubate in a 37 °C incubator with 5.0% CO2.
-
92
Monitor morphological changes every day.
? TROUBLESHOOTING
Differentiation day 3: LDN and bFGF treatment ●Timing 20 min
-
93
Prepare 5 mL of E6LF and pre-warm at RT.
▲CRITICAL STEP The E6LF is composed of 1 μM LDN and 250 ng/mL bFGF in E6. The final concentrations of LDN and bFGF after the treatment will be 200 nM and 50 ng/ml, respectively.
-
94
Pour 5 mL of the E6LF into a 10 mL reservoir.
-
95
Using a multichannel pipette and p200 regular tips, add 25 μL of the E6LF per well into each well of 96-well U-bottom plates, making a final volume of 125 μL per well.
▲CRITICAL STEP Exclude wells in the first and last rows and columns of the 96-well U-bottom plate for experiments. Media in the edge wells evaporate during the differentiation process, altering the final volume, leading to inconsistent and variable outcomes.
-
96
Gently tap the plates to mix the medium.
-
97
Incubate in a 37 °C incubator with 5.0% CO2.
-
98
Monitor morphological changes every day.
? TROUBLESHOOTING
Differentiation day 6: providing nutrition ●Timing 15 min
-
99
Prepare and pre-warm 11 mL of E6 at RT.
-
100
Pour 11 mL of the E6 into a 10 mL reservoir.
-
101
Using a multichannel pipette and p200 regular tips, add 75 μL per well into each well of 96-well U-bottom plates, making a final volume of 200 μL per well.
-
102
Gently tap the plates to mix the medium.
-
103
Incubate in a 37 °C incubator with 5.0% CO2.
-
104
Monitor morphological changes every day.
? TROUBLESHOOTING
Differentiation day 9: providing fresh medium by half medium change ●Timing 30 min
-
105
Prepare and pre-warm 14 mL of E6 at RT.
-
106
Using p200 W-O tips and a multi-channel pipette, very carefully at ~60° angle, remove 100 μL of spent medium from each well of the 96-well U-bottom plate, leaving the remaining 100 μL of the medium in each well.
-
107
Pour 14 mL of fresh E6 into a 25 mL reservoir.
-
108
Using a multichannel pipette and p200 regular tips, add 100 μL of fresh E6 into each well, making a final volume of 200 μL per well.
-
109
Gently tap the plates to mix the medium.
-
110
Incubate in a 37 °C incubator with 5.0% CO2.
-
111
Monitor morphological changes every day.
? TROUBLESHOOTING
-
112
On day 11, thaw 630 μL of Matrigel on ice overnight at 4 °C.
Differentiation day 12: transition to floating culture in OMM1%M ●Timing 1 hr 30 min
-
113
Place and pre-chill 100 mm Petri dishes and 24-well low-attachment plates on ice.
-
114
Prepare 63 mL of OMM1%M and keep on ice. Note that the OMM1%M is composed of 1% Matrigel (vol/vol) in OMM.
-
115
Prepare 3 mL of Advanced DMEM/F12 medium in a 15 mL conical tube for washing steps.
-
116
Collect all aggregates except the ones in the first and last rows and columns of 96-well U-bottom plates in a 2 mL round-bottom tube as described below:
Using a p1000 W-O tip, transfer all aggregates individually to a 100 mm Petri dish at RT.
By gently swirling the dish on a flat surface, concentrate all aggregates to the center of the dish.
Using a p1000 W-O tip, collect all aggregates into a 2 mL round-bottom tube.
-
117
Carefully remove the excessive medium.
-
118
Wash aggregates with 1 mL of Advanced DMEM/F12 medium three times.
-
119
After removing all excessive washing medium residues, place the tube with aggregates on ice.
-
120
Add 1 mL of OMM1%M to the tube containing aggregates.
-
121
Pour ~15 mL of OMM1%M to the pre-chilled Petri dish placed on ice.
-
122
Using a p1000 W-O tip, transfer all aggregates that are prepared in Step 120 to the Petri dish containing ~15 mL of OMM1%M on ice in Step 121.
-
123
Use an extra 1 mL of OMM1%M to collect and transfer any remaining aggregates in the tube to the Petri dish.
-
124
Using a p1000 W-O tip, transfer individual aggregate in 500 μL of OMM1%M to each well in 24-well low-attachment plates (i.e., one aggregate per well in a total volume of 500 μL per well).
-
125
Gently swirl to make sure each well is completely covered by medium, and aggregates are not floating on the surface of the medium.
-
126
Place the aggregates containing 24-well plates on an orbital shaker inside the incubator and start constant agitation at a speed of 65 rpm.
▲CRITICAL STEP Hair-bearing organoids can also be formed in a static culture. Anecdotal accounts from other laboratories also suggested that the static culture can produce hair-bearing organoids. Based on the qualitative observations in the Koehler laboratory, however, the static condition, without constant medium agitation on an orbital shaker, leads to increased variability and decreased reproducibility of organoid formation within an experiment.
-
127
Incubate in a 37 °C incubator with 5.0% CO2.
-
128
Monitor morphological changes every three days.
? TROUBLESHOOTING
-
129
On day 14, thaw 320 μL of Matrigel on ice overnight at 4 °C.
Differentiation day 15: half medium change with OMM1%M ●Timing 30 min
-
130
Prepare and pre-chill 32 mL of OMM1%M on ice.
-
131
Remove 250 μL of spent medium from each well of the 24-well low-attachment plates, leaving ~250 μL per well.
-
132
Add 250 μL of freshly prepared OMM1%M to each well, making a final volume of ~500 μL per well.
-
133
Gently swirl the plates to evenly mix the medium.
▲CRITICAL STEP Make sure the aggregates are not floating on the surface of the medium.
-
134
Incubate in a 37 °C incubator with 5.0% CO2.
Differentiation day 18: half medium change for Matrigel dilution ●Timing 30 min
-
135
Prepare 32 mL of fresh OMM (without Matrigel).
-
136
Remove 250 μL of spent medium from each well of the 24-well low-attachment plates, leaving ~250 μL per well.
-
137
Add 250 μL of freshly prepared OMM to each well, making a final volume of ~500 μL per well.
-
138
Gently swirl the plates to evenly mix the medium.
▲CRITICAL STEP Make sure the aggregates are not floating on the surface of the medium.
-
139
Incubate in a 37 °C incubator with 5.0% CO2.
-
140
From differentiation day 18, perform a full medium change once every week by completely removing medium (500 μL) from each well and adding back fresh medium (500 μL). Within that one week, for experiments up to differentiation day 45, perform half medium changes every three days (i.e., full medium change on Monday, half medium changes on Thursday and Sunday, then full medium change on Tuesday). For longer experiments after differentiation day 45, perform half medium changes every other day. Also, based on the organoid sizes (i.e., larger) and medium consumption rate (i.e., color of medium changing to yellow), increase total volume to 1 mL per well to provide proper nutrition. Culture up to 150 days (see ‘Anticipated results’ for indicators of culture termination).
Downstream assays
-
141
At appropriate time points, cells can be dissociated (option A) or organoids can be prepared for cryo-embedding, sectioning, and immunostaining (option B) or whole-mount immunostaining with tissue clearing using qCe3D (option C).
A. Dissociation preparation for single-cell capture ●Timing 3 hrs 30 min
▲CRITICAL This procedure is standardized to dissociate six skin organoids randomly pooled at around day 30 of differentiation (one sample) that meet the recommended final cell concentration of 700–1,200 cells per μL for scRNA-seq. The average size of a day 30 skin organoid is 2 mm. Adaptation and optimization of dissociating samples at different time points is highly recommended (e.g., increasing or decreasing the length of enzyme treatment and introducing additional enzymes for samples that are older and hard to dissociate, such as dispase and collagenase). Anticipated approximate dissociated cell numbers per skin organoid would be: ~day 6 (1.6 × 105 cells); ~day 30 (2 × 105 cells); ~day 50 (2 × 105 cells without tail portion); ~day 130 (3 × 105 cells).
Aliquot 1 mL of TrypLE to a 1.5 mL tube, add 1 μL of Y (10 μM; TrypLE + 10Y), and pre-warm the TrypLE + 10Y to 37 °C.
Prepare 5 mL of 3% BSA solution containing 10 μM Y (3% BSA + 10Y) and 2 mL of 0.4% BSA solution containing 10 μM Y (0.4% BSA + 10Y) and pre-chill on ice. Note that this volume is for 1 sample (six of day 30 aggregates). Volume may vary based on sample numbers.
Prepare and pre-warm two wet Kimwipes on a dish and a lid of 60 mm culture or Petri dish (keep as sterile as possible) in a 37 °C incubator.
Collect six aggregates (may vary depending on aggregate sizes at different timepoints) into a 2 mL round-bottom tube, and carefully remove the residual medium.
Wash with 1 mL of 1X DPBS three times, and carefully remove residual 1X DPBS.
Add 300 μL of pre-warmed TrypLE + 10Y (50 μL per aggregates that are at around day 30). Note that the TrypLE + 10Y volume may change based on the size and the number of aggregates, which are different depending on the timepoints (e.g., a 100 μL of TrypLE + 10Y per day 85 skin organoid).
-
Incubate in a 37 °C incubator for a total of 30 min on an orbital shaker for a gentle swirl (65 rpm). Use a 60 mm dish containing wet and warm Kimwipes inside the incubator; the rim of the dish can be used to keep the tubes tilted. After placing the tubes tilted on the wet and warm Kimwipes in the dish, cover the bottom portion of tubes, where dissociation solution is at, with another wet and warm Kimwipes kept on the lid to provide and keep the warmth.
▲CRITICAL Later-stage organoids (after day 75) may require extra incubation time with the enzyme or treatment with different enzymes (e.g., pre-treatment with dispase or additional treatment with collagenase) for better dissociation.
Every 10 mins during 30 min incubation, give gentle pipetting to agitate and dissociate aggregates using a p1000 W-O tip. Small pieces of aggregates may still be visible after 10 min of incubation.
Check under a microscope and re-incubate in the incubator.
For the last pipetting after 30 min of incubation, give gentle pipetting using a p1000 W-O tip, followed by extra gentle pipetting using a p1000 regular tip.
-
Add 600 μL of pre-chilled 3% BSA + 10Y solution.
▲CRITICAL Dilution alone inactivates TrypLE.
-
Keep on ice for 10 min to inhibit any protein aggregation.
▲CRITICAL STEP From this step on, always place the cell suspension on ice.
Gently pipette using a p1000 regular tip.
Flow the cell suspension through a 70 μm Flowmi filter and collect in a new 2 mL round-bottom tube to remove any debris, including Matrigel, or any remaining cell chunks. ▲CRITICAL Cell chunks should not be present. If no debris is visible, a 40 μm Flowmi filter may be used instead. Be aware that there will be some cell loss during filtering steps. Therefore, minimize filtering steps.
Centrifuge at 230g for 5 min at 4 °C.
Carefully remove supernatant.
-
Perform the following wash three times:
Gently resuspend the cell pellet in 1 mL of pre-chilled 3% BSA + 10Y solution.
Centrifuge at 230g for 5 min at 4 °C.
Carefully remove supernatant.
After the last washing step, resuspend the cell pellet in 1 mL of pre-chilled 0.4% BSA + 10Y solution. Note that the final cell resuspension solution may vary depending on the purpose of the study and the requirement from genomics cores for scRNA-seq.
-
Carefully filter through 40 μm Flowmi filter. This is the final filtering step to remove any residual debris.
▲CRITICAL STEP This step may be skipped if no debris is present.
-
Count live-cell numbers using an automated cell counter as described below. Minimize the volume of cells for counting, to avoid losing too many cells.
Mix 15 μL of cell suspension and 15 μL of trypan blue solution (1:1 dilution).
Add 11 μL of the cell-trypan blue mixture to each side of the slide chamber.
Record the readouts of total cell number, live-cell number, and viability percentage from the automated counter.
Calculate and adjust the cell concentration as required for experimental purposes by re-pelleting the cells at 230g for 5 min at 4 °C and resuspending in an adequate volume of pre-chilled 0.4% BSA + 10Y solution. For scRNA-seq, a final cell concentration of 700–1,200 cells per μL with >90% viability is recommended.
B. Sample preparation, cryo-embedding, cryosectioning, and immunostaining ●Timing 2–3 days
Organoid fixation. Collect organoids in a 2 mL round-bottom tube using a p1000 W-O tip or a perforated spoon. Cutting the end of the p1000 tip to an appropriate diameter that can collect organoids intact is recommended. Do not poke the organoids. When collecting aggregates earlier than day 12, pre-equilibrate the pipette tip with 1X PBS before collecting the samples to minimize the chances of aggregates sticking inside and outside the tips.
Carefully remove any residual medium and rinse twice with 1 mL of 1X PBS at RT.
-
Add 1–1.5 mL of pre-chilled 4% PFA.
▲CRITICAL Generally, 10X volume of fixative is needed for fixing samples.
-
Adjust the incubation time and temperature based on the following suggestions:
Earlier than day 12: incubate at RT on a platform-rotator for 10–15 min.
Day 12 (standard): incubate at RT on a platform-rotator for 20 min.
Later or equal to day 12 of differentiation: incubate at 4 °C on a platform-rotator overnight or incubate at RT on a platform-rotator, increasing by 1 min over the standard time (20 min) as differentiation day increases (e.g., day 35 organoids: 43 min).
▲CRITICAL STEP Increase or decrease fixation times for RT incubation on the basis of the approximate timing listed above. Be careful not to over- or under-fix samples.
-
Wash fixed organoids three times with 1–1.5 mL of 1X PBS at RT on a platform-rotator for 10 min each.
■PAUSE POINT After replacing with fresh and sterile 1X PBS following the last wash, the samples can be stored at 4 °C until needed for further analysis.
Cryo-embedding. Remove 1X PBS and add 0.5–1 mL of 15% sucrose.
Incubate on a platform-rotator for about 30 min at RT until all organoids sink to the bottom of the tube.
Carefully remove 15% sucrose and add 0.5–1 mL of 30% sucrose.
Incubate on a platform-rotator for ~1 hr at RT until all organoids sink to the bottom of the tube.
Using a p1000 W-O tip (cut to an adequate diameter) or a perforated spoon, carefully transfer organoids to a cryo-mold.
Under a stereomicroscope, carefully remove all liquid in the cryo-mold using a syringe with a blunt-end needle and orient and place organoids in desired orientation and location.
-
Gently and slowly add OCT freezing medium to the center of the cryo-mold.
▲CRITICAL Minimize bubble formation.
-
Confirm that all organoids are sitting at the bottom of the cryo-mold.
▲CRITICAL STEP Some organoids may need to be reoriented, relocated, or gently pushed down to the bottom after introducing the OCT freezing medium.
-
(Optional) If lots of bubbles are visible by the organoids, place the cryo-mold in a desiccator for 15 minutes.
▲CRITICAL STEP Confirm that all organoids are settled at the bottom of the mold after desiccation as tiny organoids may float to the surface of the medium together with bubbles during the process.
-
Snap freeze the cryo-mold on dry ice until the OCT freezing medium turns fully opaque.
■PAUSE POINT Store the frozen cryo-mold in a sealed box at −80 °C until performing the cryosectioning.
-
Cryosectioning. Before initiating cryosectioning, place the cryo-molds stored at −80 °C in the cryostat for at least 20 min.
▲CRITICAL STEP Pre-equilibrating the temperature of the cryo-mold to the same temperature in the cryostat (typically −23 °C) enhances the ease and quality of the sectioning process.
Perform cryosectioning with a 12 μm (standard) setting for slice thickness. Note that thickness may be adjusted based on the experimenters’ needs.
-
Collect sliced sections on Superfrost slides in the order described for the below example of 15 slides:
Collect only one section on one end of the 1st slide.
Perform the same process from the 2nd slide to the 15th slide.
Then, do another round, collecting the 2nd slice per slide from the 1st to the 15th slide, right next to the first section on each slide.
Repeat the same process until no space remains for the next slice on the slide.
▲CRITICAL STEP By collecting sample slices as described, sections from different planes of individual organoids will be collected on one slide. This will provide more information on protein expression throughout the three-dimensional organoid in one slide when utilized for further analysis, such as immunostaining.
-
Label the slides, collect them in slide boxes, and desiccate them with box lids off for 1 hr at RT.
■PAUSE POINT Store the slide boxes at −80 °C with lids on until needed for analysis.
Immunostaining. Thaw the slides that were stored at −80 °C and dry slide at RT. ▲CRITICAL STEP Only thaw slides that are going to be used. Do not go through the thaw-freeze cycle.
Use a PAP pen to outline the sample area, and dry the outline for 1 min.
-
Transfer slides to a Coplin jar and hydrate in 1X PBS for 10 min. Meanwhile, prepare:
A slide staining chamber containing MilliQ water.
Blocking solution (10% (vol/vol) normal goat or horse serum diluted in 1X PBS containing 0.1% Triton X-100 (PBS-T)) and keep it on ice. Note that the type of serum required is determined by the host of the secondary antibodies.
-
Take out the slides from the Coplin jar and, using Kimwipes, carefully remove traces of 1X PBS outside of the PAP pen outlines on the slides.
▲CRITICAL STEP Do not allow the sections to dry out. Perform the following steps quickly.
Place slides in a humidified staining chamber.
Gently add 150–200 μL of blocking solution per slide in a drop-wise manner.
Close the lid of the humidified staining chamber and incubate for 1 hr at RT.
-
During the incubation period, prepare the primary antibodies mixture and keep it on ice:
Prepare 3% (vol/vol) normal goat or horse serum diluted in 1X PBS-T.
Calculate the total volume of 3% serum that is needed given that 100 μL of antibodies mixture will be applied per side. Prepare dilutions of primary antibodies based on the volume. Ensure that the isotypes (IgGs) of primary antibodies are not overlapping within the antibody mixture. Use the manufacturers’ recommended dilutions or concentration of the antibodies.
Remove the blocking solution by tilting the corner of the slides.
Gently add 100 μL of primary antibodies mixture per slide in a drop-wise manner.
Close the lid of the humidified staining chamber and incubate for 1 hr at RT.
-
During the incubation period, prepare the secondary antibodies mixture with Hoechst solution and keep it on ice in the dark:
Prepare 3% (vol/vol) normal goat or horse serum diluted in 1X PBS-T.
Consider that 150–200 μL of secondary antibodies mixture will be applied per side. Typically, 1 mL of 3% normal goat or horse serum is prepared per secondary antibodies mixture. The recommended dilution of the secondary antibodies and Hoechst solution is 1:2000. Add 0.5 μL of secondary antibodies and Hoechst solution to 1 mL of 3% serum in 1X PBS-T. Ensure the accurate IgGs of the primary antibodies are matched with non-overlapping fluorophores. The addition of Hoechst or DAPI solution to the secondary antibodies mixture is optional when using ProLong Gold antifade mountant with DAPI.
▲CRITICAL Secondary antibodies are light sensitive.
Remove the primary antibodies mixture by tilting the corner of the slides.
Wash three times with 1X PBS in a Coplin jar for 10 min per wash at RT.
-
Take out the slides from the Coplin jar and, using Kimwipes, carefully remove traces of 1X PBS outside of the PAP pen outlines on the slides.
▲CRITICAL STEP Do not allow the sections to dry out. Perform the following steps quickly.
Place slides in a humidified staining chamber.
Gently add 150–200 μL of secondary antibodies mixture in a drop-wise manner per slide.
Close the lid of the humidified staining chamber and incubate for 1 hr at RT.
Remove secondary antibodies mixture by tilting the corner of the slides.
-
Wash three times with 1X PBS in a Coplin jar for 10 min per wash at RT.
▲CRITICAL Light sensitive. Keep the lid of the Coplin jar on.
-
Take out the slides from the Coplin jar and carefully remove traces of 1X PBS using Kimwipes.
▲CRITICAL Do not allow the sections to dry out. Perform the following steps quickly.
-
Using a p1000 tip, gently add 3–4 drops of ProLong Gold antifade mountant per slide. Use ProLong Gold with DAPI if the samples were not stained with DAPI or Hoechst for visualizing nuclei. Use ProLong Gold without DAPI if nuclei are stained with DAPI or Hoechst.
▲CRITICAL STEP Pipette slowly as the mounting reagent is viscous. Using a W-O tip may help to ease the pipetting.
-
Cover the slide with a coverslip.
▲CRITICAL STEP Do not produce bubbles. Placing one end of the coverslip at one end of the slide at an angle and slowly releasing the other end of the coverslip to settle it on the slide will minimize bubble formation.
Empty the MilliQ water from the humidified staining chamber and place the mounted slides into the chamber.
Leave the chamber with the lid open overnight in dark to dry and stabilize the mounting solution.
-
(optional) The next day, use nail polish to seal any gaps along the edge of the slide and coverslip.
■PAUSE POINT Store the stained slides at 4 °C in a slide storage box in dark until imaging.
C. Whole-mount immunostaining with tissue clearing: quick Ce3D (qCe3D) ●Timing 9 days
▲CRITICAL This protocol is adapted from ref. 75 and optimized for staining skin organoids. Volumes are adjusted for samples placed in 2 mL U-bottom tubes.
Preparation. Prepare the washing and blocking buffers as described in ‘Reagents setup’.
Prepare fixed organoids as described in ‘Organoid fixation’.
Day 1 (early morning). Wash the prepared organoids three times at RT for 30 min per wash with a washing buffer on a platform-rotator. Use 1 mL of washing buffer per one to two organoids.
Carefully remove residual washing buffer.
-
Gently add a blocking buffer and incubate at 37 °C for 8 hr on a tube-rotator. Use 500 μL per one to two organoids.
▲CRITICAL STEP This step is to block and permeabilize the cells.
During the incubation period, prepare the primary antibodies mixture diluted in a blocking buffer and keep it on ice. Note that 500 μL of the primary antibodies mixture per one to two organoids will be needed.
Carefully remove residual blocking buffer.
-
Gently add primary antibodies mixture prepared in Step vi to the samples and incubate at 37 °C for ~67 hrs on a tube-rotator.
▲CRITICAL STEP Seal the gaps between the tube and its lid with Parafilm to prevent buffer evaporation and leakage. If samples are fragile, the sample tubes can be placed on a platform-rotator with gentle rocking.
Day 4 (afternoon). Carefully remove the primary antibodies mixture.
Wash the organoids with washing buffer at 37 °C for 4 hrs on a tube-rotator. Use 1 mL of washing buffer per organoid.
Carefully replace the washing buffer with fresh washing buffer and incubate at RT for overnight on a platform-rotator.
Day 5 (early morning). Carefully replace the washing buffer with fresh washing buffer and incubate at RT for 4 hrs on a platform-rotator.
-
During the incubation period, prepare the secondary antibodies and Hoechst (or DAPI) solution mixture diluted in a blocking buffer and keep it on ice in dark. 500 μL of the secondary antibody mixture with Hoechst solution per one to two organoids will be needed.
▲CRITICAL Light sensitive. Keep it in dark.
Carefully remove the washing buffer.
Gently add the secondary antibodies and Hoechst (or DAPI) solution mixture prepared in Step xiii and incubate at 37 °C for ~68 hrs on a tube-rotator. Seal the gaps between the tube and its lid with Parafilm to prevent evaporation and leakage of the solution. ▲CRITICAL Light sensitive. Keep it in the dark.
-
Day 6. Place the N-methylacetamide and a 25 mL serological pipette at 37 °C for overnight to melt and pre-warm, respectively.
▲CRITICAL STEP N-methylacetamide is solid at RT. Pre-warm the entire bottle in 37 °C for overnight (>1 hr) until it liquifies.
! CAUTION N-methylacetamide is hazardous. Manipulate in a fume hood. Exposure may damage the unborn child.
-
Day 7. Prepare a 40% N-methylacetamide as described in ‘Reagents setup’, and store at RT until its use on day 8.
! CAUTION N-methylacetamide is hazardous. Exposure may damage fertility of the unborn child. Manipulate in a fume hood.
-
Prepare qCe3D clearing solution as described in ‘Reagents setup’ and incubate at 37 °C for overnight on a tube-rotator.
! CAUTION N-methylacetamide and 1-thioglycerol are hazardous. Inhalation of N-methylacetamide may damage fertility or the unborn child. 1-Thioglycerol may cause respiratory irritation. Avoid direct contact with skin and eyes. Wear rubber gloves and eye goggles. Perform all steps involving N- methylacetamide and 1-thioglycerol in a fume hood.
Day 8 (early morning). Carefully remove the secondary antibodies with the Hoechst solution mixture.
Wash the organoids with washing buffer at 37 °C for 4 hrs on a tube-rotator. Use 1 mL of washing buffer per organoid.
-
Carefully replace the washing buffer with fresh washing buffer and incubate at RT for 2 hrs on a platform-rotator.
▲CRITICAL Light sensitive. Cover the tubes with aluminum foil.
Repeat Step xxi.
Carefully transfer organoids to a new 2 mL round-bottom tube (if they are in a larger-volume tube) and remove residual washing buffer.
-
Gently add qCe3D clearing solution and incubate at RT on a platform-rotator until the organoids become clear (>8 hrs, overnight). Use 1 mL of qCe3D clearing solution per organoid. Work quickly to prevent tissue dehydration.
▲CRITICAL Light sensitive. Cover the tubes with aluminum foil.
-
Day 9 (morning). Mount the immunostained and cleared organoid in a silicon isolator cassette as follows (see Fig. 4a for a schematic image):
Stick a coverslip to one side of a silicon isolator. Press gently and flip over to the other side.
Using a p1000 W-O tip, carefully transfer one immunostained and cleared organoid per well of the silicon isolator. Ensure the end of the pipet tip is sufficient diameter that an organoid can pass through intact. Another option is the use of a perforated spoon. Handle the organoids very carefully as they are fragile.
Fill up each well with qCe3D clearing solution until the solution level rises to just above the well.
Gently stick another coverslip on top of the silicon isolator by placing the coverslip at an angle at one side of the silicon isolator and releasing it gently on the other side. This technique will diminish the chance of forming bubbles and make the procedure easier.
Press the coverslip very gently to adhere it properly to the isolator.
Use nail polish to seal any gaps along the edge of the silicon isolator and coverslips.
▲CRITICAL STEP If organoids are too large to fit into a silicon isolator, an imaging coverglass with chambers or an imaging dish with coverglass bottom can be used as alternatives.
■PAUSE POINT Store immunostained and cleared organoids mounted in silicon isolator cassettes or on imaging dishes at 4 °C in a storage box in dark until imaging.
Image organoid using a confocal microscope. Both sides of the organoid can be imaged as presented in Fig. 4, by flipping the silicon isolator cassette to the other side without the burden of re-orienting samples.
Troubleshooting
Non-adherence of hPSCs on vitronectin-coated plate during thawing and passaging procedures (Steps 16 and 43)
This is potentially due to a failed plate coating procedure. Check the expiration date of vitronectin. Make sure that the vitronectin solution is diluted to the right concentration (a final concentration of 5 μg/mL; 0.5 μg/cm2, 1:100 dilution), the solution is covering the well of the plate evenly, and the solution is incubated for the recommended amount of time (at least 1 hr at RT). Then, re-seed the cells on the newly coated plate. When using any cell lines that have been grown on a feeder layer or any matrices other than vitronectin, adaptation to the feeder-layer-free, vitronectin-based environment is required. Likewise, any cell lines cultured in media other than E8 medium need to be adapted to E8 medium-based culture conditions. During the adaptation process, cell colonies may detach relatively easily. To overcome this issue, seed cells at a higher cell seeding density than recommended in the ‘Thawing’ and ‘Passaging’ sections and Fig. 7a. Take extra care when replenishing the medium for cell maintenance (e.g., adding medium extra gently and slowly toward the wall of a well). Together, cell attachment should improve over the course of several passages.
Differentiating cells appearing in the maintenance culture (Step 19)
This is potentially due to insufficient medium or overgrown cells in the well of a plate. Check the expiration dates on E8 Flex medium and E8 Flex supplement bottles. To minimize degradation of growth factors, do not warm the E8 Flex supplement or medium to 37 °C. We recommend thawing the supplement at 4 °C overnight before adding to the medium bottle. For each use, the complete E8 Flex medium should be warmed to RT. Check whether the E8 medium is in the 2 weeks window of usage after supplement addition. Perform the passaging procedure before the cells become over-confluent in the maintenance culture as shown in Fig. 7c. Note that the overgrown colonies form brownish cores, which represent the cells that are undergoing apoptosis or spontaneous differentiation. In addition, the maintenance cultures may benefit from eliminating any differentiated cells from time to time. This can be achieved by marking the area of differentiation with a marker on the bottom of the well and aspirating or scraping off the marked area. Perform this cleaning procedure at least a day before passaging or using the cells for differentiation to allow time for recovery.
The first thing to check whenever seeing any differentiation failures (Steps 82, 92, 98, 104, 111, and 128)
Any inactive or not accurately reconstituted reagents and media lead to failures of differentiation. Check individual reagent (small molecules and proteins; very critical) expiration dates and their reconstitution procedures and prepare fresh media.
Failed formation of surface ectoderm by days 3–5 (Step 92)
This is potentially due to different endogenous BMP4 expression levels and/or cell proliferation rates between cell lines. For efficient surface ectoderm induction, optimization of the treatment regime may be required depending on different cell lines by increasing or decreasing BMP4 concentration on day 0 and/or delaying the treatment timing (e.g., day 0 → day 1) may be required. If the issues persist, test multiple lots of recombinant BMP4 and bulk purchase the lots that produce consistent results.
Failed formation of CNC and mesenchyme by days 12–21 (Steps 111 and 128)
This is potentially due to different endogenous BMP4 and FGF expression levels and/or proliferation rates between cell lines. For efficient CNC and mesenchyme induction, optimizing the timing of BMP inhibition with activation of FGF signaling may be required depending on different cell lines, by advancing or delaying inhibition of BMP signaling and activation of FGF signaling (e.g., day 3 → day 4) may be required.
Condensation on the 96-well or 24-well plate lid (Steps 95, 101, 106,131, and 136)
This is mostly due to temperature differences surrounding the culture plates, typically observed when the plates are taken out from the 37 °C incubator to RT (or vice versa) for treatments, changing media, and imaging under a microscope. The solution to this issue is to wipe the condensation off using sterile Kimwipes and leave the inner side of the lid facing upward (place the lid deeper in the biosafety cabinet not to block the airflow and keep it sterile) while changing medium. Note that the condensation may slightly alter the medium volume per well and concentrations of medium components. However, in our experience, this has not shown any negative effects on the efficiency or quality of differentiation and long-term cultures.
Timing
Steps 1–16, thawing frozen hPSCs: 30 min
Steps 17–19, hPSC maintenance: 10 min
Steps 20–43, hPSC passaging: 30 min
Steps 44–53, freezing hPSCs: 40 min
-
Steps 54–140, differentiation:
Total time to observe hair germ formation: ~60 days
Total time to generate mature skin equipped with appendages: ~120 days onward
Steps 54–75 (day −2), cell aggregation: 50 min
Steps 76–80 (day −1), dilution of Y solution: 15 min
Steps 81–92 (day 0), transition to differentiation in E6SFB: 1 hr 30 min
Steps 93–98 (day 3), LDN and bFGF treatment: 30 min
Steps 99–104 (day 6), providing nutrition: 15 min
Steps 105–112 (day 9), providing fresh medium by half medium change: 30 min
Steps 113–129 (day 12), transition to floating culture in OMM1%M: 1 hr 30 min
Steps 130–134 (day 15), half medium change with OMM1%M: 30 min
Steps 135–140 (day 18 onward), half/full medium change with OMM: 30 min
-
Step 141, downstream assays:
Step 141A (i-xviii), dissociation preparation for single-cell capture: 3 hrs 30 min
Step 141B (i-xix), sample preparation, cryo-embedding, and cryosectioning: 1–3 days; varies depending on organoid sizes.
Step 141B (xx-xlv), immunostaining: 5 hrs
Step 141C (i-xxvi), whole-mount immunostaining with tissue clearing using qCe3D: 9 days
Anticipated results
If the differentiation experiment is performed correctly, a translucent epithelial cyst should form by day 8 of differentiation. The epithelial cyst will become covered by a layer of mesenchymal and neuroglial cells over the next week. After ~30 days of differentiation, the epithelial cyst should be composed of the stratified epidermis with basal, intermediate, and periderm layers (Fig. 3b, c). Between days 55 and 75 of differentiation depending on the cell lines, development of hair placodes, germs, and pegs will be visible under an inverted microscope at magnifications as low as 40X (4X microscope objective × 10X eyepiece) or 100X (10X microscope objective × 10X eyepiece; Fig. 5 and see Fig. 3c-e for immunostained images). During days 70–130 of differentiation, hair pegs will continue to elongate and differentiate to become mature hair follicles (Fig. 3f). Some hair follicles will have pigmentation such as that seen in 18-week fetal skin (Fig. 3g-i). As previously shown25, we anticipate that at least an average of 87% of skin organoids will generate hair follicles when incubated with 2.5 ng/mL BMP4 on day 0 of differentiation. Using the current protocol, which is standardized to 5 ng/mL BMP4, we anticipate that >90% of skin organoids will more consistently produce hairs. Even though 5 ng/mL BMP4 is used as a standardized concentration in this protocol, the optimal concentration must be determined cell line by cell line as we emphasized in the Fig. 6, ‘Experimental design’, and ‘Troubleshooting’. Beyond 150 days, the skin organoids display abnormal features, such as a compacted core of dead squamous cells and fused hair follicles (see abnormal follicles in a day 165 specimen in Fig. 4b). Before day 150, skin organoids can be extracted from culture and used for downstream assays (e.g., single-cell sequencing) or xenografting experiments (as demonstrated in ref. 25). The skin organoid system should be a suitable model for studying the development of human skin and its accessory tissues, as well as skin-related diseases. More details of differentiation checkpoint information are presented in Table 1, and additional representative images are available in ref. 25.
ACKNOWLEDGEMENTS
This work was supported by the Ralph W. and Grace M. Showalter Trust (K.R.K.), the Indiana CTSI (core pilot grant UL1 TR001108 to K.R.K. and Anantha Shekhar), the Indiana Center for Biomedical Innovation (Technology Enhancement Grant to K.R.K.), and the NIH (grants R01AR075018 and R01DC017461 to K.R.K.). Cell lines associated with this study were stored in a facility constructed with support from the NIH (grant C06 RR020128-01). We thank Matthew Steinhart, Stefan Heller, Benjamin Woodruff, Michael Rendl, Pantelis Rompolas, Leah Biggs, Muzlifah Haniffa, and Sara Wickström, for constructive feedback and technical assistance during the development of this method.
Footnotes
COMPETING INTERESTS
J.L. and K.R.K, with the Indiana University Research and Technology Corporation, have a patent covering the entire skin organoid induction method (US11021688B2), which is licensed to STEMCELL Technologies, Inc. for research use. The other authors declare no competing interests.
DATA AVAILABILITY
Examples of results obtained are included in the figures.
REFERENCES
- 1.Dąbrowska AK et al. The relationship between skin function, barrier properties, and body-dependent factors. Ski. Res. Technol. 24, 165–174 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Kolarsick PAJ, Kolarsick MA & Goodwin C Anatomy and physiology of the skin. J. Dermatol. Nurs. Assoc. 3, 203–213 (2011). [Google Scholar]
- 3.Nose H, Kamijo Y & Masuki S Chapter 25—interactions between body fluid homeostasis and thermoregulation in humans. Handb. Clin. Neurol. 156, 417–429 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Woo S-H, Lumpkin EA & Patapoutian A Merkel cells and neurons keep in touch. Trends Cell Biol. 25, 74–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zimmerman A, Bai L & Ginty DD The gentle touch receptors of mammalian skin. Science 346, 950–954 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun BK, Siprashvili Z & Khavari PA Advances in skin grafting and treatment of cutaneous wounds. Science 346, 941–945 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Karimkhani C et al. Global skin disease morbidity and mortality: an update from the Global Burden of Disease Study 2013. JAMA Dermatol. 153, 406–412 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He Z et al. Factors affecting health-related quality of life in patients with skin disease: cross-sectional results from 8,789 patients with 16 skin diseases. Health Qual. Life Outcomes 18, 298 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laughter MR et al. The burden of skin and subcutaneous diseases in the United States from 1990 to 2017. JAMA Dermatol. 156, 874–881 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laughter MR et al. The global burden of atopic dermatitis: lessons from the Global Burden of Disease Study 1990–2017*. Brit. J. Dermatol. 184, 304–309 (2021). [DOI] [PubMed] [Google Scholar]
- 11.Mehrmal S, Uppal P, Nedley N, Giesey RL & Delost GR The global, regional, and national burden of psoriasis in 195 countries and territories, 1990 to 2017: a systematic analysis from the Global Burden of Disease Study 2017. J. Am. Acad. Dermatol. 84, 46–52 (2021). [DOI] [PubMed] [Google Scholar]
- 12.Wu X, Scott L, Washenik K & Stenn K Full-thickness skin with mature hair follicles generated from tissue culture expanded human cells. Tissue Eng. 20, 3314–3321 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Klicznik MM et al. A novel humanized mouse model to study the function of human cutaneous memory T cells in vivo in human skin. Sci. Rep. 10, 11164 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agarwal Y et al. Development of humanized mouse and rat models with full-thickness human skin and autologous immune cells. Sci. Rep. 10, 14598 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salgado G, Ng YZ, Koh LF, Goh CSM & Common JE Human reconstructed skin xenografts on mice to model skin physiology. Differentiation 98, 14–24 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Takagi R et al. Bioengineering a 3D integumentary organ system from iPS cells using an in vivo transplantation model. Sci. Adv. 2, e1500887 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng Y et al. Organogenesis from dissociated cells: generation of mature cycling hair follicles from skinderived cells. J. Invest. Dermatol. 124, 867–876 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Zomer HD & Trentin AG Skin wound healing in humans and mice: challenges in translational research. J. Dermatol. Sci. 90, 3–12 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Itoh M et al. Generation of 3D skin equivalents fully reconstituted from human induced pluripotent stem cells (iPSCs). PLoS ONE 8, e77673 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang R et al. Generation of folliculogenic human epithelial stem cells from induced pluripotent stem cells. Nat. Commun. 5, 3071–3071 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gledhill K et al. Melanin transfer in human 3D skin equivalents generated exclusively from induced pluripotent stem cells. PLoS ONE 10, e0136713 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abaci HE et al. Tissue engineering of human hair follicles using a biomimetic developmental approach. Nat. Commun. 9, 5301 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Letsiou S Tracing skin aging process: a mini-review of in vitro approaches. Biogerontology 22, 261–272 (2021). [DOI] [PubMed] [Google Scholar]
- 24.Christian H, Hans & Yves P. in Skin Disease Models In Vitro and Inflammatory Mechanisms: Predictability for Drug Development 187– 218 (Springer International Publishing, 2021); 10.1007/164_2020_428 [DOI] [PubMed] [Google Scholar]
- 25.Lee J et al. Hair-bearing human skin generated entirely from pluripotent stem cells. Nature 582, 399–404 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee J & Koehler KR Skin organoids: a new human model for developmental and translational research. Exp. Dermatol. 30, 613–620 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biggs LC, Kim CS, Miroshnikova YA & Wickström SA Mechanical forces in the skin: roles in tissue architecture, stability, and function. J. Invest. Dermatol. 140, 284–290 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Wong R, Geyer S, Weninger W, Guimberteau J & Wong JK The dynamic anatomy and patterning of skin. Exp. Dermatol. 25, 92–98 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Lee J et al. Hair follicle development in mouse pluripotent stem cell-derived skin organoids. Cell Rep. 22, 242–254 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koehler KR, Mikosz AM, Molosh AI, Patel D & Hashino E Generation of inner ear sensory epithelia from pluripotent stem cells in 3D culture. Nature 500, 217–221 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eiraku M et al. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell 3, 519–532 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Eiraku M et al. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 472, 51–56 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Koehler KR et al. Generation of inner ear organoids containing functional hair cells from human pluripotent stem cells. Nat. Biotechnol. 35, 583–589 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prummel KD, Nieuwenhuize S & Mosimann C The lateral plate mesoderm. Development 147, dev175059 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guibentif C et al. Diverse routes toward early somites in the mouse embryo. Dev. Cell 56, 141–153.e6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilson PA & Hemmati-Brivanlou A Induction of epidermis and inhibition of neural fate by Bmp-4. Nature 376, 331–333 (1995). [DOI] [PubMed] [Google Scholar]
- 37.Duverger O & Morasso MI To grow or not to grow: hair morphogenesis and human genetic hair disorders. Semin. Cell Dev. Biol. 25, 22–33 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCune JM & Weissman IL The ban on US government funding research using human fetal tissues: how does this fit with the NIH mission to advance medical science for the benefit of the citizenry? Stem Cell Rep. 13, 777–786 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haniffa M et al. A roadmap for the Human Developmental Cell Atlas. Nature 597, 196–205 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu Q et al. Charting human development using a multi-endodermal organ atlas and organoid models. Cell 184, 3281–3298 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haniffa M et al. Human Developmental Cell Atlas: milestones achieved and the roadmap ahead. 10.21203/rs.3.rs-73986/v1 (2020). [DOI] [Google Scholar]
- 42.Regev A et al. The Human Cell Atlas. eLife 6, e27041 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yucha SEV, Tamamoto KA, Nguyen H, Cairns DM & Kaplan DL Human skin equivalents demonstrate need for neuro-immuno-cutaneous system. Adv. Biosyst. 3, 1800283 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Meltzer S, Santiago C, Sharma N & Ginty DD The cellular and molecular basis of somatosensory neuron development. Neuron 109, 3736–3757 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Andersen J et al. Generation of functional human 3D cortico-motor assembloids. Cell 183, 1913–1929.e26 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wainger BJ et al. Modeling pain in vitro using nociceptor neurons reprogrammed from fibroblasts. Nat. Neurosci. 18, 17–24 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oulès B et al. Contribution of GATA6 to homeostasis of the human upper pilosebaceous unit and acne pathogenesis. Nat. Commun. 11, 5067 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kanwar IL et al. Models for acne: a comprehensive study. Drug Discov. Ther. 12, 329–340 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Langan SM, Irvine AD & Weidinger S Atopic dermatitis. Lancet 396, 345–360 (2020). [DOI] [PubMed] [Google Scholar]
- 50.Condorelli AG, Dellambra E, Logli E, Zambruno G & Castiglia D Epidermolysis bullosa-associated squamous cell carcinoma: from pathogenesis to therapeutic perspectives. Int J. Mol. Sci. 20, 5707 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim DP, Kus KJB & Ruiz E Basal cell carcinoma review. Hematol. Oncol. Clin. North Am. 33, 13–24 (2019). [DOI] [PubMed] [Google Scholar]
- 52.Davis LE, Shalin SC & Tackett AJ Current state of melanoma diagnosis and treatment. Cancer Biol. Ther. 20, 1–14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Que SKT, Zwald FO & Schmults CD Cutaneous squamous cell carcinoma Incidence, risk factors, diagnosis, and staging. J. Am. Acad. Dermatol. 78, 237–247 (2018). [DOI] [PubMed] [Google Scholar]
- 54.Waldman A & Schmults C Cutaneous squamous cell carcinoma. Hematol. Oncol. Clin. North Am. 33, 1–12 (2019). [DOI] [PubMed] [Google Scholar]
- 55.Hogue L & Harvey VM Basal cell carcinoma, squamous cell carcinoma, and cutaneous melanoma in skin of color patients. Dermatol. Clin. 37, 519–526 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Yum MK et al. Tracing oncogene-driven remodelling of the intestinal stem cell niche. Nature 594, 442–447 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang L, Mali P, Kim-Kiselak C & Church G In Gene Correction. Methods in Molecular Biology (Methods and Protocols) (ed. Storici F.) Vol 1114, 245–267 (Humana Press, 2014); 10.1007/978-162703-761-7_16 [DOI] [PubMed] [Google Scholar]
- 58.Hendriks D, Clevers H & Artegiani B CRISPR-Cas tools and their application in genetic engineering of human stem cells and organoids. Cell Stem Cell 27, 705–731 (2020). [DOI] [PubMed] [Google Scholar]
- 59.Wenzel D et al. Genetically corrected iPSCs as cell therapy for recessive dystrophic epidermolysis bullosa. Sci. Transl. Med. 6, 264ra165–264ra165 (2014). [DOI] [PubMed] [Google Scholar]
- 60.Sebastiano V et al. Human COL7A1-corrected induced pluripotent stem cells for the treatment of recessive dystrophic epidermolysis bullosa. Sci. Transl. Med. 6, 264ra163–264ra163 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tolar J et al. Induced pluripotent stem cells from individuals with recessive dystrophic epidermolysis bullosa. J. Invest. Dermatol. 131, 848–856 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Itoh M, Kiuru M, Cairo MS & Christiano AM Generation of keratinocytes from normal and recessive dystrophic epidermolysis bullosa-induced pluripotent stem cells. Proc. Natl Acad. Sci. USA. 108, 8797–8802 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Candi E, Schmidt R & Melino G The cornified envelope: a model of cell death in the skin. Nat. Rev. Mol. Cell Biol. 6, 328–340 (2005). [DOI] [PubMed] [Google Scholar]
- 64.Langbein L, Yoshida H, Praetzel-Wunder S, Parry DA & Schweizer J The keratins of the human beard hair medulla: the riddle in the middle. J. Invest. Dermatol. 130, 55–73 (2010). [DOI] [PubMed] [Google Scholar]
- 65.Lu C & Fuchs E Sweat gland progenitors in development, homeostasis, and wound repair. Cold Spring Harb. Perspect. Med. 4, a015222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lu CP, Polak L, Keyes BE & Fuchs E Spatiotemporal antagonism in mesenchymal–epithelial signaling in sweat versus hair fate decision. Science 354, aah6102 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao X et al. Review on the vascularization of organoids and organoids-on-a-chip. Front. Bioeng. Biotechnol. 9, 637048 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Augustin HG, Koh GY, Thurston G & Alitalo K Control of vascular morphogenesis and homeostasis through the angiopoietin–Tie system. Nat. Rev. Mol. Cell Biol. 10, 165–177 (2009). [DOI] [PubMed] [Google Scholar]
- 69.Holloway EM et al. Differentiation of human intestinal organoids with endogenous vascular endothelial cells. Dev. Cell 54, 516–528.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bar-Ephraim YE, Kretzschmar K & Clevers H Organoids in immunological research. Nat. Rev. Immunol. 20, 279–293 (2020). [DOI] [PubMed] [Google Scholar]
- 71.Takahashi K & Yamanaka S Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006). [DOI] [PubMed] [Google Scholar]
- 72.Tchieu J et al. A modular platform for differentiation of human PSCs into all major ectodermal lineages. Cell Stem Cell 21, 399–410.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Volpato V & Webber C Addressing variability in iPSC-derived models of human disease: guidelines to promote reproducibility. Dis. Model Mech. 13, dmm042317 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vigilante A et al. Identifying extrinsic versus intrinsic drivers of variation in cell behavior in human iPSC lines from healthy donors. Cell Rep. 26, 2078–2087.e3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li W, Germain RN & Gerner MY High-dimensional cell-level analysis of tissues with Ce3D multiplex volume imaging. Nat. Protoc. 14, 1708–1733 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Examples of results obtained are included in the figures.