

Abstract
Small rings that contain allenes are unconventional transient compounds that have been known since the 1960s. Despite being discovered around the same time as benzyne and offering a number of synthetically advantageous features, strained cyclic allenes have seen relatively little use in chemical synthesis. We report a concise total synthesis of the manzamine alkaloid lissodendoric acid A, which hinges on the development of a regioselective, diastereoselective, and stereospecific trapping of a fleeting cyclic allene intermediate. This key step swiftly assembles the azadecalin framework of the natural product, allows for a succinct synthetic endgame, and enables a 12-step total synthesis (longest linear sequence; 0.8% overall yield). These studies demonstrate that strained cyclic allenes are versatile building blocks in chemical synthesis.
Geometrically constrained organic molecules have long fascinated chemists. One subclass of such compounds are small cyclic molecules that bear functional groups of typically linear geometry. Embedding typically linear functional groups within small ring systems forces bent geometries, leading to a high degree of strain. Small rings that contain triple bonds were speculated in the early 1900s (1), ultimately leading to the validation of benzyne (1) (2, 3) and cyclohexyne (2) (4) in the 1950s (Fig. 1A) (5–9). Although these in situ–generated intermediates exhibit substantial strain energies of ~40 to 50 kcal/mol (10, 11) and short lifetimes (12, 13), scientists have learned how to leverage these highly reactive intermediates in a range of useful chemical transformations and biological applications. Examples of aromatic and nonaromatic cyclic alkyne trapping experiments can be seen in the synthesis of important ligands, such as XPhos, medicines, agrochemicals, materials, naturally occurring compounds, and other areas (9). Moreover, bioorthogonal chemistry (14), recently recognized by the Nobel Prize in Chemistry, relies on cyclooctynes (3) and other strained intermediates.
Fig. 1. Strained intermediates and overview of current study.
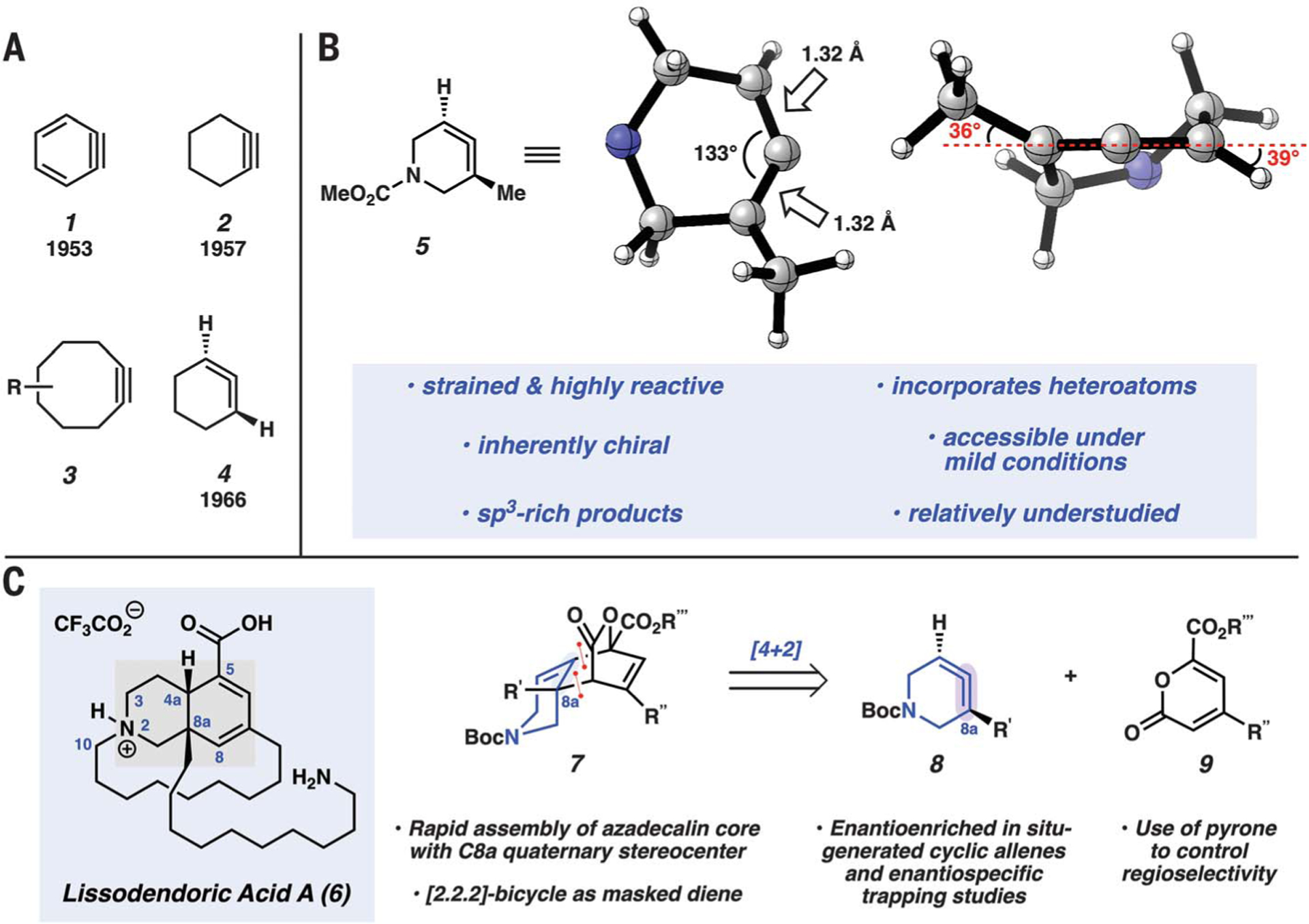
(A) Seminal strained cyclic intermediates 1, 2, and 4 and bioorthogonal cyclooctyne reagents 3. (B) Geometry-optimized structure of strained azacyclic allene 5 [ωB97XD/6–31G(d)] and key features. CO2Me is omitted from the three-dimensional representations for clarity. (C) Our synthetic target, lissodendoric acid A (6), and our synthetic approach by using a [4+2] cycloaddition to access 7 from cyclic allene 8 and pyrone 9 (R’, R”, R”’ = alkyl).
Cyclic allenes, such as 1,2-cyclohexadiene (4), are a related class of strained molecules that contain a typically linear functional group that is constrained in a small ring (Fig. 1A). Cyclic allene 4 was first validated in 1964 (15) and again in 1966 (16). This intermediate, along with other cyclic allenes, has received far less attention compared with that of strained cyclic alkynes, despite having a number of advantages that warrant their further investigation. Our laboratory has been interested in heterocyclic allenes, such as azacyclic allene 5, because of their properties and potential use in chemical synthesis (9, 17, 18). The geometry-optimized structure of this species is depicted in Fig. 1B. As shown, the allene C=C bonds are each 1.32 Å, and the central allene carbon bears an internal angle of 133°. Additionally, the allene C–H and C–CH3 bonds are twisted out of the allene C=C=C plane by 39° and 36°, respectively. Several features result from this distinctive structure, including the high reactivity of cyclic allenes due to strain; their capacity to undergo cycloadditions, nucleophilic additions, or metal-catalyzed reactions; and the capability to form two new bonds in a single transformation, with introduction of up to three C(sp3) stereocenters. As such, strained cyclic allenes have recently been used to prepare highly substituted, sp3-rich compounds (9, 17–24) and have even been used to access DNA-encoded libraries (25). Further underscoring their attractiveness is that cyclic allenes are axially chiral and could serve as unconventional yet valuable building blocks for stereoselective synthesis. Moreover, from a practical perspective, strained cyclic allenes can possess heteroatoms within the ring and can be accessed under mild conditions (such as fluoride-based reactions or ambient temperatures) (19). Last, because of the relatively scarce body of literature surrounding strained cyclic allenes, there exist numerous opportunities for discovery with regard to reactivity and selectivity patterns in synthetic applications. Cyclic allenes have rarely been used in the synthesis of complex natural products, which is notable given that their close relatives, arynes and strained cyclic alkynes, have been used in more than 100 total syntheses to date (6–8). One notable example of the use of a cyclic allene in total synthesis was reported in 1997 by Marshall and Sehon and involves a relatively unstrained allene embedded in a macrocyclic 14-membered ring (26).
With the aim of developing the chemistry of strained cyclic allenes and evaluating their utility in total synthesis, we considered lissodendoric acid A (6) (27), a structurally complex member of the manzamine family of alkaloids (28) (Fig. 1C). Manzamine alkaloids have been popular targets for total synthesis because of their complex structures and promising biological activities (29, 30). The cumulative synthetic efforts toward manzamine alkaloids, as recently highlighted by Dixon and Jakubec (29), have involved more than 25 research groups to date. With regard to lissodendoric acid A (6), this natural product was isolated in 2017 from the marine sponge Lissodendoryx florida and has been shown to reduce reactive oxygen species (ROS) in a Parkinson’s disease model consisting of Neuro 2a cells treated with 6-hydroxydopamine (~50% reduction in ROS levels at concentrations of 0.1 and 10 μM) (27). The natural product’s complex structure poses a daunting synthetic challenge that in turn would push the limits of strained cyclic allene chemistry. The core of lissodendoric acid A (6) is an azadecalin scaffold bearing a conjugated diene, a carboxylic acid substituent, and two stereogenic centers, one of which is quaternary at C8a. In addition, the natural product bears a 14-membered macrocycle tethered beneath the C8a aminodecane substituent. A total synthesis of lissodendoric acid A (6) has not been reported.
We questioned whether the azadecalin core and C8a quaternary stereocenter (for example, 7) could be introduced simultaneously by means of a [4+2] cycloaddition between cyclic allene 8 and 2-pyrone 9 (Fig. 1C) (31). The strain associated with cyclic allene 8 would be crucial to the success of the proposed reaction because such intermolecular cycloadditions of analogous cyclic alkene substrates are challenging (32, 33). The use of 2-pyrones in [4+2] cycloadditions with cyclic allenes was previously unknown but, if successful, could enable control of regioselectivity through an inverse electron-demand Diels-Alder process. In addition, the use of a pyrone would furnish a [2.2.2]-bicyclic product (7), which in turn would function as a masked 1,3-diene, enabling the strategic unveiling of this potentially sensitive motif at a later stage through thermal expulsion of CO2. Last, we were intrigued by the possibility of accessing cyclic allene 8 in enantioenriched form and developing enantiospecific trapping reactions.
Our successful total synthesis of lissodendoric acid A (6) was enabled by the development of a regioselective, diastereoselective, and stereospecific trapping reaction of a fleeting cyclic allene intermediate. The total synthesis is concise owing to generation of substantial structural complexity in the key cyclic allene trapping step.
Development of regio- and stereoselective cycloadditions
Because the success of our approach would hinge on the ability to control the regioselectivity and stereoselectivity in the key [4+2] cycloaddition step, we performed preliminary studies with furan 11 and pyrone 13 (Fig. 2). These dienes were selected so that we could compare and contrast the electronic effects of the cyclic allene trapping partners (electron-rich furan versus electron-poor pyrones). Pyrone 13 was chosen because it was easily accessible and possesses potentially useful synthetic handles, whereas furan (11) had previously been used in cyclic allene [4+2] cycloadditions (17). The syntheses of 12, 16, and 18, which function as precursors to cyclic allene 8 (Fig. 1C), are detailed in the supplementary materials and relied on chiral separation technology to access enantioenriched material. Treatment of silyltriflate 12 with furan (11) and CsF in acetonitrile at 23°C delivered the undesired regioisomer 10, which is consistent with prior experiments that used the N-Cbz derivative of 12 (17). In a crucial result, we found that treatment of 12 with readily available pyrone 13 (34) gave cycloadduct 14, which possesses the desired connectivity and the necessary C8a quaternary center, without substantial loss of stereochemical integrity. The reaction is endo-selective, which is consistent with theoretical studies of 1,2-cyclohexadienes (35), and occurs with the expected regioselectivity based on this being an inverse electron-demand process. These results demonstrate that regioselectivity in cyclic allene [4+2] cycloadditions can be modulated by judicious selection of the trapping agent.
Fig. 2. Regioselectivity and stereospecificity studies using variable cyclic allene precursors and [4+2] cycloaddition partners.
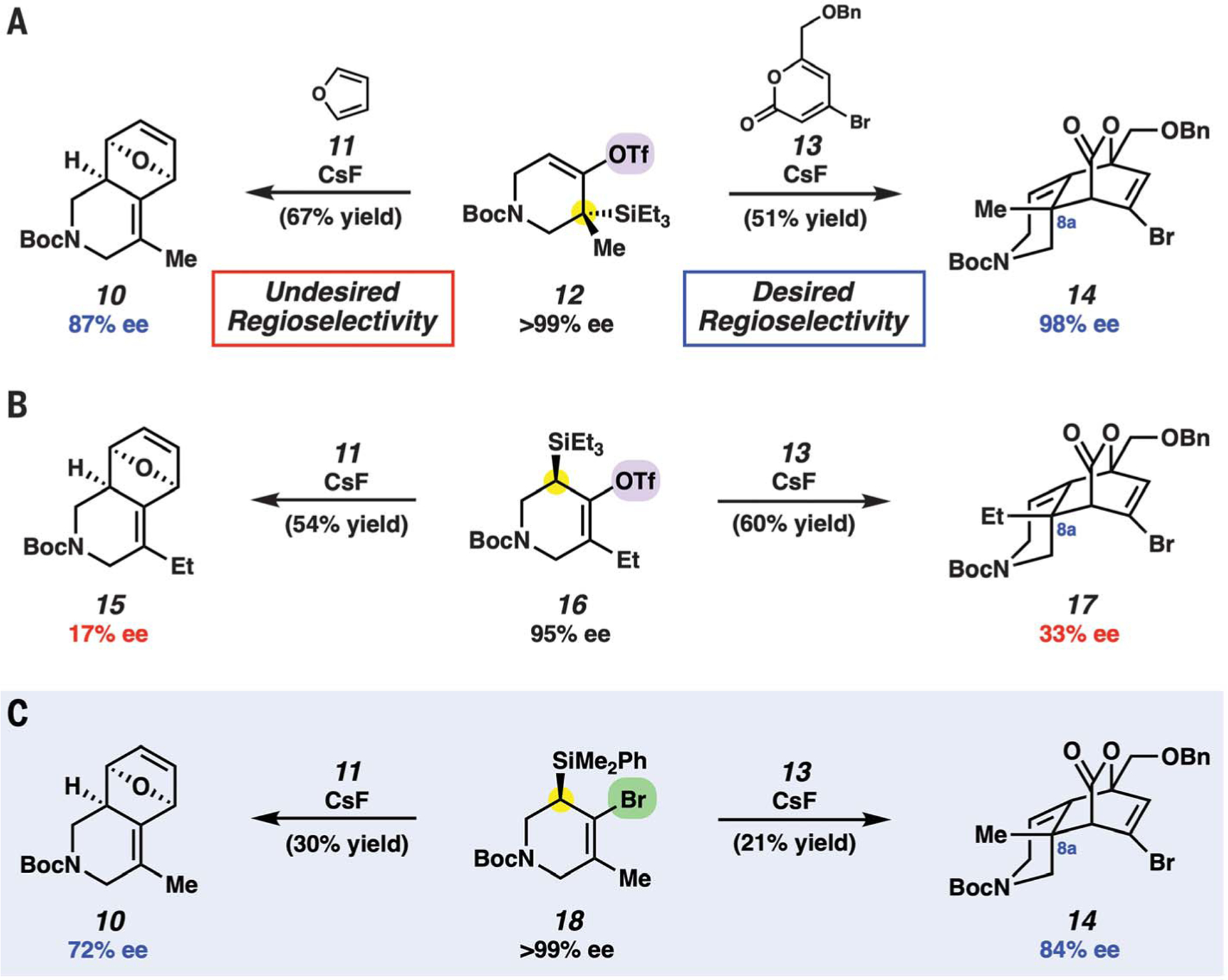
(A) Use of cyclic allene precursor 12 allows for efficient and stereospecific cycloadditions. (B) Stereospecificity is modest when using pseudo-isomeric silyltriflate 16. (C) Switching to silylbromide 18 as the cyclic allene precursor allows for regioselective and stereospecific cycloaddition reactions, while providing a plausible entryway to enantioenriched material. Highlights indicate the primary differences between cyclic allene precursors 12, 16, and 18. Reaction conditions are provided in the supplementary materials.
Although these results were encouraging, we were unable to access compounds such as 12 in high enantiomeric excess (ee) without the use of chiral separation technologies. As such, we evaluated alternative precursors 16 and 18, in which the silicon substituent would be placed on a less-substituted carbon, in the corresponding cycloaddition reactions (36). As shown in Fig. 2B, the use of pseudo-isomeric silyltriflate 16 (albeit with Et in place of Me owing to substrate synthesis logistics), furnished the expected cycloadducts 15 and 17. However, regardless of the trapping agent, poor stereoretention was observed for reasons that are not well understood presently. Last, we examined silylbromide 18, which is analogous to an approach to cyclic allene generation developed by West and coworkers (Fig. 2C) (22). Although yields were modest in these initial studies, notable stereoretention was observed in the formation of cycloadducts 10 and 14 (37). Silylbromide precursors to cyclic allenes had not been synthesized previously in enantioenriched fashion, but we were optimistic that derivatives of 18 could be prepared without the need for chiral separations. Before describing the translation of these studies to the total synthesis of lissodendoric acid A (6), we also highlight several key features of cyclic allene chemistry that are reflected in the results shown in Fig. 2. Specifically, all reactions proceed under mild conditions, lead to the formation of two C–C bonds, occur with high endo diastereoselectivity (35), and provide access to complex heterocyclic products containing three stereocenters, one of which is quaternary in the cases of the pyrone cycloadducts.
Fragment synthesis and assembly of azadecalin core
Having established the feasibility of the key cyclic allene trapping step, we sought to perform analogous studies on a more elaborate substrate. Pursuing this endeavor could allow us to push the limits of strained cyclic allene chemistry while enabling the synthesis of lissodendoric acid A (6). Pyrone 22 was prepared in two steps from commercially available carboxylic acid 19 (Fig. 3A). Double tosylation, followed by the addition of t-BuOH, furnished tosylate 20, bearing a t-butyl ester substituent. Subsequent Negishi coupling with organozinc reagent 21 provided the alkylated pyrone 22, which contains a terminal alkene necessary for the eventual installation of the macrocycle by using ring-closing metathesis. The desired cyclic allene precursor, silylbromide 27, was prepared from bromotriflate 23, which was obtained commercially (Fig. 3B). Treatment of 23 with alkylzirconium reagent 24 gave ketone 25 through 1,4-addition and ejection of the triflate leaving group (38). Corey-BakshiShibata (CBS) reduction (39), followed by treatment with ethylchloroformate, gave 26 in 90 to 92% ee. Displacement of the carbonate with a silyl cuprate nucleophile (22, 40, 41) delivered cyclic allene precursor 27 without loss in enantioenrichment. This step was thought to proceed with inversion of stereochemistry on the basis of prior studies by Oestreich (41). The conversion of bromoketone 25 to silylbromide 27 parallels a general racemic strategy pioneered by Lofstrand et al. (22), but the enantioselective reduction and stereospecific displacement of the carbonate had not been demonstrated previously.
Fig. 3. Assembly of the azadecalin core of lissodendoric acid A (6) by using a strained cyclic allene.
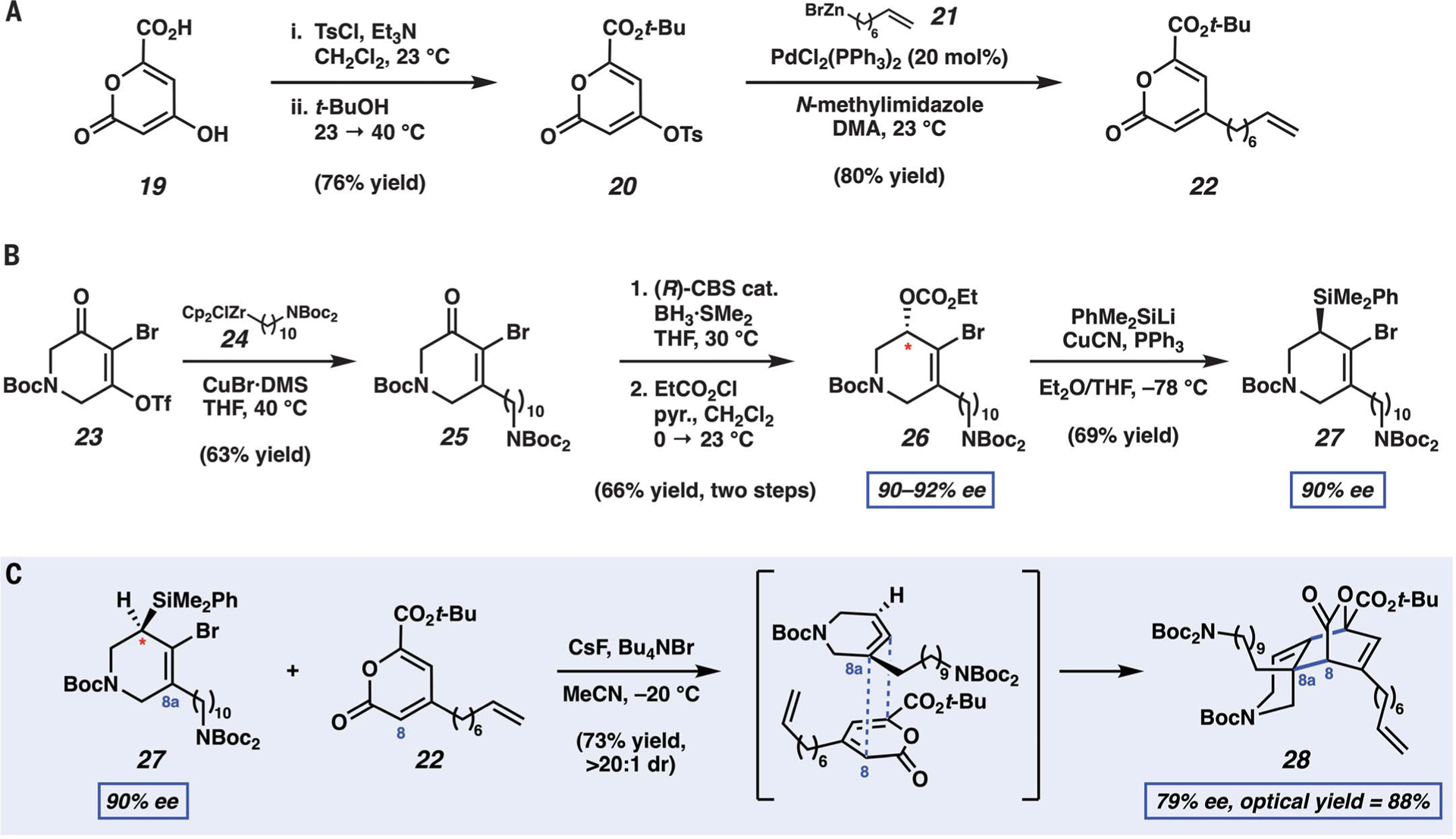
(A) Synthesis of pyrone 22. (B) Enantioselective route to silyltriflate 27. (C) Cycloaddition of the strained cyclic allene derived from 27 with pyrone 22 proceeds with regioselectivity, diastereoselectivity, and stereospecificity to deliver enantioenriched adduct 28 with the desired quaternary stereocenter at C8a.
With pyrone 22 and cyclic allene precursor 27 in hand, we evaluated the key [4+2] cycloaddition (Fig. 3C). We found that by simply treating these reactants with CsF, the desired cycloadduct, 28, could be obtained. Through optimization, we found that the optical yield (42, 43), or percent stereoretention, was greatly improved at lower reaction temperatures, which in turn required the use of Bu4NBr to improve the solubility of fluoride ion in solution. Ultimately, –20°C was found to be the optimal temperature, enabling the formation of 28 in 73% isolated yield, presumably by means of the depicted transition structure proposed on the basis of prior computational studies of cyclic allene Diels-Alder reactions (17, 35, 44).
Several aspects of this complexity-generating step should be noted, especially the regioselectivity and stereoselectivity. The regioselectivity is thought to depend on both the cyclic allene and pyrone components. First considering the cyclic allene component, either olefin of the allene could potentially undergo trapping; however, the more electron-rich substituted olefin of the cyclic allene engages with the electron-deficient pyrone, presumably because of the inverse electron-demand nature of this cycloaddition. With regard to the pyrone component, the diene in pyrone 22 aligns with the cyclic allene to promote bond formation between C8a and C8. The pyrone carbonyl, which is conjugated to the diene in 22, is thought to provide a dominant electronic effect that leads to this selectivity. The origins of stereoselectivity in this transformation are also notable. The reaction is thought to proceed in an endo fashion, with favorable orbital overlap between the diene and the nonreactive olefin of the cyclic allene, and with favorable approach of the pyrone opposite the C8a alkyl substituent. This gives rise to 28 in >20:1 diastereomeric ratio. The endo approach and facial selectivity are consistent with prior computational studies of cyclic allene Diels-Alder reactions (17, 35). Despite that four cycloadducts could be envisioned (not accounting for enantiomers discussed below), cycloadduct 28, which bears considerable structural complexity and functionality, is formed selectively as the sole observable product of the reaction.
The absolute stereochemistry and optical yield also warrant particular attention. Cyclic allene precursor 27 bears a single stereocenter (Fig. 3B, asterisk). Upon generation of the cyclic allene intermediate, point chirality in 27 is transmitted to axial chirality in the cyclic allene intermediate. Then, cycloaddition reintroduces point chirality, with the generation of two tertiary stereocenters and the C8a quaternary stereocenter. The sole stereocenter present in reactant 27 is ablated in this process. The optical yield (42, 43), or stereoretention, in forming 28 at –20°C is 88%, indicating that strain-driven cycloaddition between two highly substituted reactants is favorable over facile enantiomerization of the strained cyclic allene at low temperature. The barrier for enantiomerization for related cyclic allenes has been estimated to be ~14 kcal/mol (45).
Completion of total synthesis
To complete the total synthesis of lissodendoric acid A (6), we developed the concise late-stage sequence shown in Fig. 4 using (±)-28. The first challenge was to introduce the C4a hydrogen substituent chemoselectively, without reduction of the other olefins. The C4/C4a olefin proved recalcitrant toward a range of reduction conditions; however, we ultimately achieved a selective reduction by first oxidizing C3 with pyridinium dichromate (PDC)/t-BuOOH (46, 47) and then treating the resulting α,β-unsaturated lactam intermediate with a copper hydride source (48) to effect diastereoselective 1,4-reduction and furnish cis-azadecalin 29. The CO2 bridge embedded in the [2.2.2]-bicycle of 28 (installed in the aforementioned cyclic allene trapping) was important for the success of this sequence because it masked the 1,3-diene motif that would otherwise be in conjugation with the C4/C4a olefin. Next, we arrived at tetraene 30 through a sequence involving thermal extrusion of CO2 (49), copper triflate–promoted removal of two of the three Boc groups (50), and amidation of the piperidine nitrogen with acryloyl chloride. Access to 30 set the stage for macrocyclization, which we achieved using ring-closing metathesis promoted by the robust Grubbs’ 2nd Generation catalyst (Sigma-Aldrich) (51, 52). The desired macrocycle 31 was obtained in 83% yield and as an inconsequential mixture of E:Z olefin isomers (ratio ranging from 2:1 to 3.5:1). All that remained to complete the total synthesis was to selectively reduce the α,β-unsaturated imide and perform a global deprotection. We achieved the former using a reduction protocol reported by Das et al. (53), which led to saturation of the C3 amide and the C10 α,β-unsaturated amide selectively without reduction of the ester or diene functional groups. Although the overall yield of this transformation is modest (38%), given that three functional groups are being reduced (~72% yield per reduction), we deemed the yield acceptable. Last, treatment of the reduced intermediate with trifluoroacetic acid furnished lissodendoric acid A (6). The total synthesis is 12 steps from 23 (longest linear sequence; 0.8% overall yield).
Fig. 4.
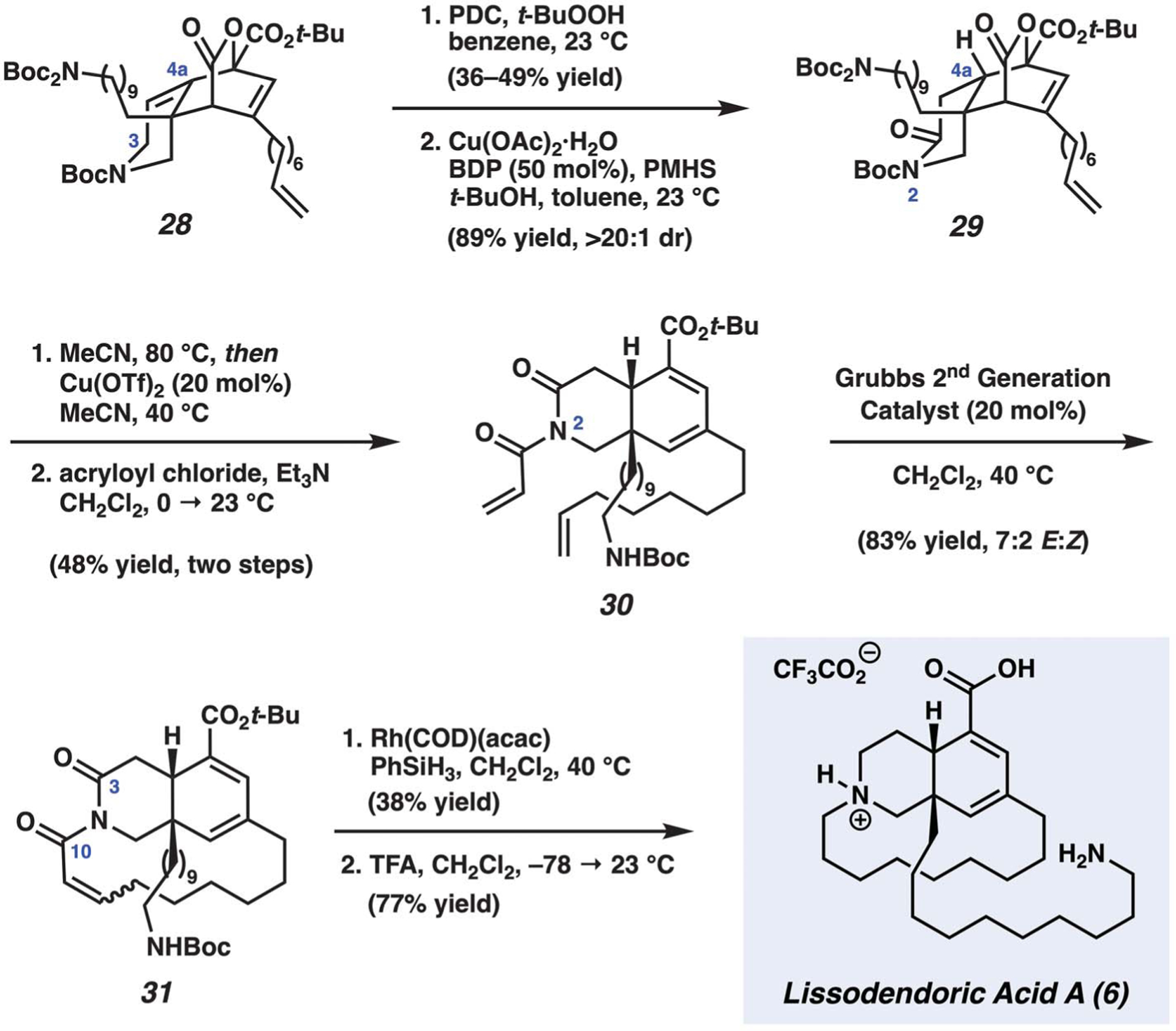
Completion of the total synthesis of lissodendoric acid A (6).
Another matter of interest is the absolute stereochemical configuration of lissodendoric acid A. On the basis of experimental and computational circular dichroism studies, the isolation chemists had postulated the absolute configuration of (–)-6 to be as we have depicted throughout this study (27). To validate this hypothesis, our enantioenriched material was elaborated to the natural product. The circular dichroism spectrum for our synthetic sample matched that of a natural sample. We conclude that the natural configuration of (–)-6 proposed by the isolation chemists was indeed correct.
These studies demonstrate that cyclic allenes are powerful tools for complex molecule synthesis and should prompt the further investigation and application of these unusual compounds.
Supplementary Material
ACKNOWLEDGMENTS
We thank E. Lyakhova (G. B. Elyakov Pacific Institute of Bioorganic Chemistry, Russia) for providing an authentic sample of (-)-6. We thank A. Doyle (UCLA) for use of her laboratory’s chiral HPLC.
Funding:
The authors are grateful to the NIH-NIGMS (R01 GM123299 and R35 GM139593 to N.K.G., F32 GM139289 to N.J.A., and F32-GM122245 to E.R.D.), the NSF (DGE-2034835 to F.M.I. and L.G.W.), Bristol-Myers Squibb (to J.S.D.), the UCLA Graduate Division (to J.S.D.), and the Trueblood family (to N.K.G.). These studies were supported by shared instrumentation grants from the NSF (CHE-1048804) and the NIH NCRR (S10RR025631).
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability:
Experimental procedures and characterization data are provided in the supplementary materials and (54). Correspondence and requests for materials should be addressed to N.K.G. (neilgarg@chem.ucla.edu).
REFERENCES AND NOTES
- 1.Stoermer R, Kahlert B, Ber. Dtsch. Chem. Ges 35, 1633–1640 (1902). [Google Scholar]
- 2.Roberts JD, Simmons HE Jr., L. A. Carlsmith, C. W. Vaughan, J. Am. Chem. Soc 75, 3290–3291 (1953). [Google Scholar]
- 3.Wittig G, Pohmer L, Angew. Chem 67, 348 (1955). [Google Scholar]
- 4.Scardiglia F, Roberts JD, Tetrahedron 1, 343–344 (1957). [Google Scholar]
- 5.Wenk HH, Winkler M, Sander W, Angew. Chem. Int. Ed 42, 502–528 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Tadross PM, Stoltz BM, Chem. Rev 112, 3550–3577 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Gampe CM, Carreira EM, Angew. Chem. Int. Ed 51, 3766–3778 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Takikawa H, Nishii A, Sakai T, Suzuki K, Chem. Soc. Rev 47, 8030–8056 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Anthony SM, Wonilowicz LG, McVeigh MS, Garg NK, JACS Au 1, 897–912 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liebman JF, Greenberg A, Chem. Rev 76, 311–365 (1976). [Google Scholar]
- 11.Angus RO Jr., Schmidt MW, Johnson RP, J. Am. Chem. Soc 107, 532–537 (1985). [Google Scholar]
- 12.Berry RS, Clardy J, Schafer ME, J. Am. Chem. Soc 86, 2738–2739 (1964). [Google Scholar]
- 13.Diau EW-G, Casanova J, Roberts JD, Zewail AH, Proc. Natl. Acad. Sci. U.S.A 97, 1376–1379 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sletten EM, Bertozzi CR, Angew. Chem. Int. Ed 48, 6974–6998 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moser MW, The reactions of gem-dihalocyclopropanes with organometallic reagents. thesis, Massachusetts Institute of Technology (1964).
- 16.Wittig G, Fritze P, Angew. Chem. Int. Ed 5, 846 (1966). [Google Scholar]
- 17.Barber JS et al. , Nat. Chem 10, 953–960 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamano MM et al. , Angew. Chem. Int. Ed 58, 5653–5657 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quintana I, Peña D, Pérez D, Guitián E, Eur. J. Org. Chem 2009, 5519–5524 (2009). [Google Scholar]
- 20.Lofstrand VA, West FG, Chem. Eur. J 22, 10763–10767 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Barber JS et al. , J. Am. Chem. Soc 138, 2512–2515 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lofstrand VA, McIntosh KC, Almehmadi YA, West FG, Org. Lett 21, 6231–6234 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Yamano MM et al. , Nature 586, 242–247 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kelleghan AV, Witkowski DC, McVeigh MS, Garg NK, J. Am. Chem. Soc 143, 9338–9342 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Westphal MV et al. , J. Am. Chem. Soc 142, 7776–7782 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marshall JA, Sehon CA, J. Org. Chem 62, 4313–4320 (1997). [DOI] [PubMed] [Google Scholar]
- 27.Lyakhova EG et al. , Org. Lett 19, 5320–5323 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Radwan M, Hanora A, Khalifa S, Abou-El-Ela SH, Cell Cycle 11, 1765–1772 (2012). [DOI] [PubMed] [Google Scholar]
- 29.Dixon DJ, Jakubec P, Total synthesis of manzamine alkaloids, in Marine Natural Products, Kiyota H, Ed. (Springer, 2021), pp. 23–59. [Google Scholar]
- 30.Ashok P, Ganguly S, Murugesan S, Drug Discov. Today 19, 1781–1791 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Hopf H, Sherburn MS, Synthesis 54, 864–886 (2002). [Google Scholar]
- 32.Control experiments are available in the supplementary materials.
- 33.Afarinkia K, Vinader V, Nelson TD, Posner GH, Tetrahedron 48, 9111–9171 (1992). [Google Scholar]
- 34.Luo T, Dai M, Zheng S-L, Schreiber SL, Org. Lett 13, 2834–2836 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramirez M, Svatunek D, Liu F, Garg NK, Houk KN, Angew. Chem. Int. Ed 60, 14989–14997 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Strategically, an enantioenriched cyclic allene precursor bearing a tri-substituted stereocenter was expected to be more easily accessible as compared with a precursor with a fully substituted stereocenter.
- 37.The relationship between stereoretention and the structure of the cyclic allene precursor (identity and location of the silyl group and the nature of the leaving group) is not well understood presently and will be the subject of further investigation.
- 38.Wipf P, Smitrovich JH, J. Org. Chem 56, 6494–6496 (1991). [Google Scholar]
- 39.Corey EJ, Helal CJ, Angew. Chem. Int. Ed 37, 1986–2012 (1998). [DOI] [PubMed] [Google Scholar]
- 40.Ito H, Horita Y, Sawamura M, Adv. Synth. Catal 354, 813–817 (2012). [Google Scholar]
- 41.Weickgenannt A, Oestreich M, Chem. Eur. J 16, 402–412 (2010). [DOI] [PubMed] [Google Scholar]
- 42.Although the term “stereoretention” is commonly used by synthetic chemists, “optical yield” is the preferred International Union of Pure and Applied Chemistry (IUPAC) term when comparing the ratio of the optical purity of the product with that of the reactant.
- 43.IUPAC, Compendium of Chemical Terminology, McNaught AD, Wilkinson A, Eds. (Blackwell Scientific Publications, ed. 2, 1997); 10.1351/goldbook. [DOI] [Google Scholar]
- 44.The use of five or more equivalents of pyrone 22 led to optimal results. However, using fewer equivalents of pyrone 22 was also synthetically useful. For example, when using two equivalents of pyrone 22, cycloadduct 28 was obtained in slightly lower isolated yield and optical yield of 62 and 85%, respectively.
- 45.Daoust KJ et al. , J. Org. Chem 71, 5708–5714 (2006). [DOI] [PubMed] [Google Scholar]
- 46.This oxidation step proceeds by means of a peroxide intermediate, which can also be isolated and recycled (supplementary materials).
- 47.Chidambaram N, Chandrasekaran S, J. Org. Chem 52, 5048–5051 (1987). [Google Scholar]
- 48.Baker BA, Bosković ZV, Lipshutz BH, Org. Lett 10, 289–292 (2008). [DOI] [PubMed] [Google Scholar]
- 49.Abdullahi MH, Thompson LM, Bearpark MJ, Vinader V, Afarinkia K, Tetrahedron 72, 6021–6024 (2016). [Google Scholar]
- 50.Evans V, Mahon MF, Webster RL, Tetrahedron 70, 7593–7597 (2014). [Google Scholar]
- 51.Chatterjee AK, Morgan JP, Scholl M, Grubbs RH, J. Am. Chem. Soc 122, 3783–3784 (2000). [Google Scholar]
- 52.Chatterjee AK, Choi T-L, Sanders DP, Grubbs RH, J. Am. Chem. Soc 125, 11360–11370 (2003). [DOI] [PubMed] [Google Scholar]
- 53.Das S, Li Y, Lu L-Q, Junge K, Beller M, Chem. Eur. J 22, 7050–7053 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Ippoliti FM et al. , Data from: Total synthesis of lissodendoric acid A via stereospecific trapping of a strained cyclic allene. Dryad (2023); doi: 10.5068/D1Q38N. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Experimental procedures and characterization data are provided in the supplementary materials and (54). Correspondence and requests for materials should be addressed to N.K.G. (neilgarg@chem.ucla.edu).