

Abstract
Developing scalable electrical stimulating platforms for cell and tissue engineering applications is limited by external power source dependency, wetting resistance, microscale size requirements, and suitable flexibility. Here, a versatile and scalable platform is developed to enable tunable electrical stimulation for biological applications by harnessing the giant magnetoelastic effect in soft systems, converting gentle air pressure (100–400 kPa) to yield a current of up to 10.5 mA and a voltage of 9.5 mV. The platform can be easily manufactured and scaled up for integration in multiwell magnetoelastic plates via 3D printing. The authors demonstrate that the electrical stimulation generated by this platform enhances the conversion of fibroblasts into neurons up to 2-fold (104%) and subsequent neuronal maturation up to 3-fold (251%). This easily configurable electrical stimulation device has broad applications in high throughput organ-on-a-chip systems, and paves the way for future development of neural engineering, including cellular therapy via implantable self-powered electrical stimulation devices.
Keywords: cell reprogramming, electric stimulation, magnetoelastic effect, magnetoelastic generators, neural engineering
1. Introduction
The role of electrical stimulation (ES) in biological systems has been extensively exploited in preclinical research[1–6] as well as an innumerable number of therapeutic solutions.[7–10] With the importance of physical stimuli (including ES) becoming ever more apparent in processes including morphogenesis,[11,12] tissue regeneration,[2,13–16] and cellular differentiation,[17–21] developing an easily implementable and adaptable ES platform remains highly desired, especially for future adoption within in vivo studies aimed at therapeutic neuroregeneration.
Several technologies have been reported as substitutes for commercially available ES solutions, especially in the form of as-built ES devices designed specifically for laboratory use, but these are expensive, difficult to adapt, and lack in variety. Other options include the use of function generators and power amplifiers, which can be inconsistent in their output, tricky to implement, and which can potentially lead to dangerous live current exposures. Of relevance, utilizing passive energy sources associated with human biomechanical activities can help build self-sufficient, low-cost, and convenient ES platforms for in vivo use. Such platforms are of interest as they could be adopted and exploited for self-powered in vivo therapeutic ES in the treatment of neurodegenerative diseases, including for major clinical unmet needs such as spinal cord injury.[22] Although currently available platforms do provide self-powered solutions that are easy to fabricate and use inexpensive materials, they often lack biocompatibility, biocomformability, and cannot be operated in wetted environments – factors that have hindered their broader adoption into biomedical and in vivo research settings.[23–25] Furthermore, bioelectronics with these biomechanical-to-electrical energy conversion capabilities are often limited by low inherent current density and high internal impedance, stemming from their characteristic capacitive power generation mechanisms which are reliant on the electric dipole’s manipulation of the interfacing constitutive materials.[26–28]
The recent discovery of the giant magnetoelastic effect in soft systems[29] – exploiting magnetoelastic effect and electromagnetic induction coupling – offers a promising solution to produce electricity by harnessing passive and minimal forces including human biomechanical motions, and has led to the invention of the magnetoelastic generator (MEG).[29] MEGs are made with inexpensive materials, which are readily available, and can be fabricated and applied directly onto existing tissue culture platforms.[30] Considering the layered and adaptable nature of the MEG platform, materials such as polydimethylsiloxane (PDMS) can be utilized during their fabrication to render these stand-alone ES platforms tissue compliant and biocompatible, thereby allowing MEG platforms to be applied in in vivo studies, an essential validation step of ES devices during their transition to the clinic.
Herein, we developed a novel integrative approach that combines the assessment of cellular reprogramming ability while applying a stand-alone and one-body waterproof ES platform, which could potentially be used transcuteanously. This approach was carried out via air-induced pneumatic actuation and activation of the soft MEG, to help address the unmet medical of current state-of-the-art ES studies. To our knowledge, this is the first study for which in vitro ES will be directly integrated and built within an existing tissue culture and used to provide a novel soft system for in vitro ES with demonstrated biocompatibility. Through our study, we showed that our programmable ES platform, activated via means of magnetoelastic effect promoted the reprogramming of fibroblasts into neurons and enhance subsequent neuronal maturation.
From an operational perspective, MEGs are inherently self-powered and can be activated using gentle biomechanical stimuli, a concept we leveraged to enable their activation through air pressure pneumatic actuation. Considering the ubiquity of air pressure appliances in labs across the world, we devised a platform exploiting this simple modus operandi, and demonstrated the application of MEG for enhancing fibroblast reprogramming into neurons. This landmark study sets the foundations to develop magnetoelastic generators as new self-powered ES devices for neuronal regenerative medicine and tissue engineering. Indeed, specification to neuronal cell types was targeted based on their clinical relevance to the study and treatment of disorders of the nervous system and considering ES is a known modulator of their lineage commitment and maturation.[31–33]
2. ES Platform Design
Finding effective ways to readily generate electricity within the cellular microenvironment is paramount to the development of next generation tools for cell, organoid stimulation, and in vivo therapeutic solutions. Although the magnetoelastic effect has been recognized since the late 19th century, its application in miniaturized and soft systems has been limited by a number of factors including: the rigid nature of the ferromagnetic materials traditionally employed (e.g., Fe1−xCox, TbxDy1−xFe2 (Terfenol-D),[34] and GaxFe1−x (Galfenol)),[35,36] the need for an applied external magnetic field to the system, and the very large applied pressure, i.e., megapascal range[37] – required to generate the effect. The giant magnetoelastic effect has been recently discovered in soft systems.[29] Thorough testing has demonstrated the ability of this set up to bypass the need for external fields, as well as reducing pressure requirements by orders of magnitude that are within the range suitable for biological use. In order to form such a device, neodymium boron (NdFeB) nanoparticles can be homogeneously dispersed in a liquid silicon rubber solution and subsequently magnetized for intrastructural dipole alignment post curing, yielding a soft composite system (Figure 1a) that can be easily deformed using minimal and biologically tolerable force. Physically perturbing the system from a relaxed to a deformed state (whether compressed or stretched) (Figure 1b) leads to the atomic scale magnetic dipole–dipole interaction (MDDI) and microscale magnetic particle–particle interaction (MPPI), dynamically modifying the magnetic flux density of the system[38] and thus, creating the giant magnetoelastic effect (Figure 1c). Magnetic flux density changes can be exploited to yield an electromotive force (i.e., electricity, produced by a nonelectrical source) when coupled with an electrical conductor. Advantageously, the intensity of the magnetic fields generated from these systems is not adversely affected by wetted environments,[29] making them suitable for use within and in the study of biological systems and environments. The MEG holds a magnetomechanical coupling (MC) layer and a magnetic induction (MI) layer, which is thus able to act as an electricity generator via a coupling of giant magnetoelastic effect and magnetic induction. The MEGs can thus be used for ES in miniaturized formats (Figure 1d). Furthermore, beyond their inherent energy transducing (self-powering) and bioconformable nature, MEGs can be operated with physiologically tolerable pressure values (i.e., Young’s modulus of ≈446 kPa and ultimate strain of 140% (Figure 1e), can withstand prolonged exposure in wetted environments without compromising their functionality (Figure 1f), and can be made using biocompatible materials (Figure 1g) for easy integration into biological systems and research laboratory tools. Leveraging these capabilities, MEGs have demonstrated utility for biomonitoring[38,39] and energy generation[40,41] applications. Additionally, MEG manufacturing protocols are straightforward and easily integrated into a variety of device architectures, including electronic textiles[30] and films.[42] In our study, we developed a methodology for incorporating MEG substrates into commonly used tissue cultures plates to serve as platforms for applying electrical stimuli to cells. Mechanical deformation induced by applying air pressure to the base of the MEG substrate alters the inner MPPI and MDDI, leading to a change of its magnetic field that can be exploited to generate electricity via electromagnetic induction, a novel way to exploit and harness these systems for ES.
Figure 1.
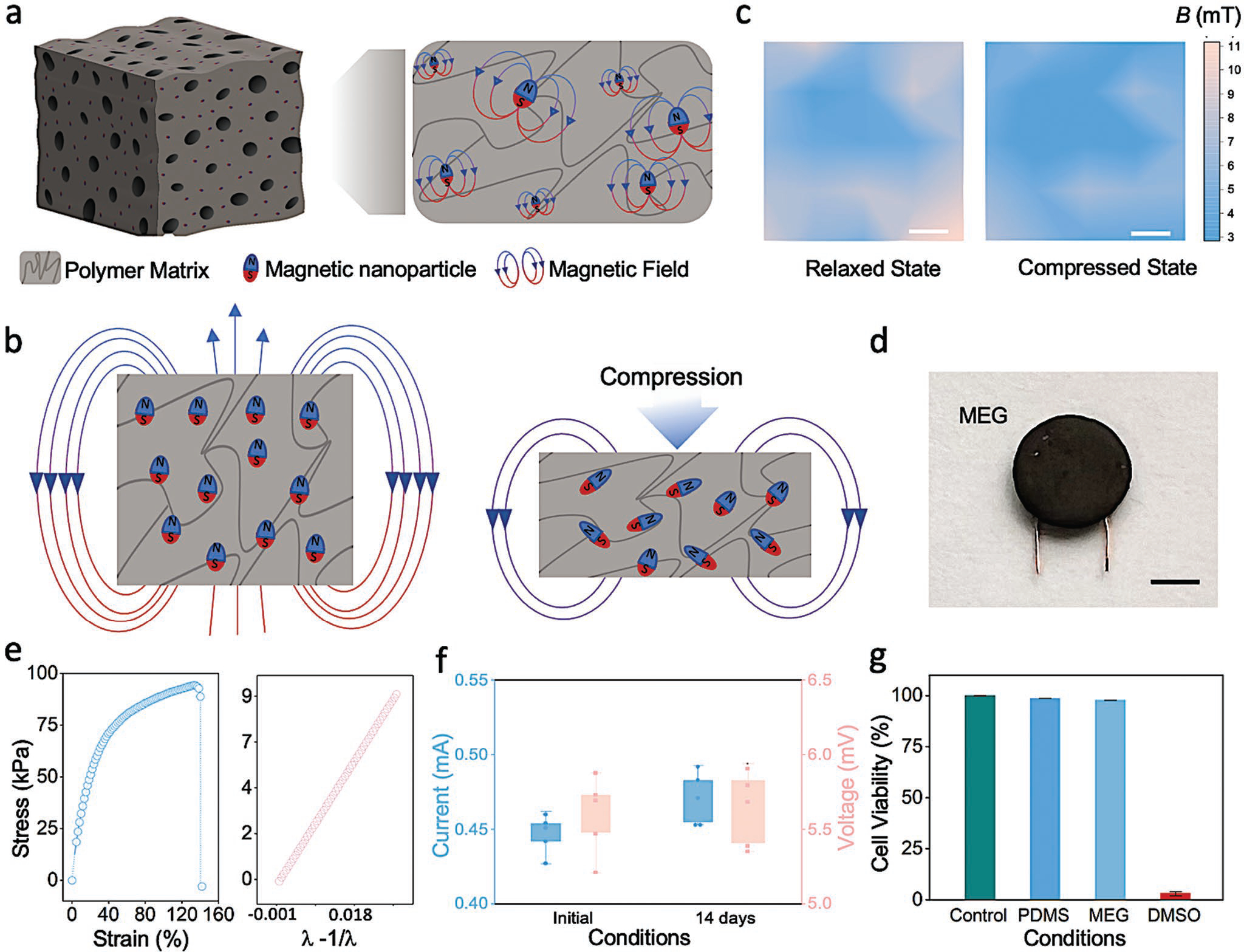
Magnetoelastic generators display suitability for bioengineering. a) Schematic representation of magnetic nanoparticles dispersed in a polymer matrix. b) Illustration of the magnetic dipole alignment changing the magnetic flux density of the soft magnetoelastic layer in the initial state and the compressed state. c) Vertical magnetic flux density (DB⊥) of the soft system at the South Pole surface; variation is mapped from a relaxed (0 N) state to a compressive (20 N) force state. Scale bar, 0.5 cm. d) Photograph of a miniaturized MEG. Scale bar: 0.5 cm. e) Stress and strain curve of the soft MEG composite assembly (left) and Young’s modulus calculated using a neo-Hookean model (right). f) MEG current and voltage outputs prior to and subsequent to water submersion for 14 days. Box plots represent mean ± standard deviation values, n = 5. g) Cell viability of mouse fibroblasts cultured on PDMS and MEG substrates compared to positive (cells maintained in untreated tissue culture plates) and negative (DMSO) controls, respectively, as determined using a PrestoBlue assay. Bar graphs show mean ± standard deviation of the results (n = 5). DMSO, dimethyl sulfoxide.
3. 3D Fabrication of the MEG ES Platform
Important factors that help determine the broader adoption of any new cell culture technology are its reproducibility and the ease and scalability of the manufacturing processes used to fabricate said cell culture devices. This also applies to the development of implantable self-powered electrical simulation devices, which the MEG can be expected to represent. To automate and improve the consistency and throughput of producing MEG devices, we established a 3D printing protocol for the direct in situ fabrication of MEGs within individual wells of 6-, 12-, 24-, 48-, and 96-well tissue culture plates (Movie S2, Supporting Information). Fabrication of MEG ES devices involved the systematic deposition of the MI and MC layers in configurations where the MC encapsulates the MI. The integrated MC-MI layers were then covered with a protective, biocompatible PDMS layer that was subsequently coupled to an indium–titanium oxide (ITO)-coated glass substrate (Figure 2a). The MEG devices were fabricated directly in commercially available tissue culture well plates (Figure 2b). Briefly, the initial MI layer was assembled by positioning copper (Cu) wire coils within the wells of the tissue culture plate. The MC layer was then prepared by pouring an uncured silicone rubber matrix that had been previously mixed with nonmagnetized NdFeB nanoparticles into the MI-containing wells. The MC-MI layer assemblies were then allowed to cure in an oven at 60 °C. Once the MC-MI layered MEG was fully cured, the MC layers were magnetized. Next, uncured PDMS was poured into the well plates to provide a protective biocompatible covering and hermetically seal the MEG platform, locking it in place and preventing the devices from dislodging from the tissue culture plate during operation (air pressure actuation). While the PDMS was curing, an ITO glass substrate cut to conform to the size of the tissue culture well was placed above and partially into the PDMS layer to secure it to the MEG platform. Space was allowed for the MI electrode wires to exit and connect (at the ITO glass’ antipodes). The fully-assembled platform was placed in an oven at 60 °C for 3 h to complete the curing process (Figure 2b). Prior to use, the ITO glass was functionalized using oxygen plasma to reduce its hydrophobicity (Figure S1, Supporting Information) to optimize wettability and cell adhesion. This processing step could also be carried out once the MEG ES platform was fully cured and integrated with the ITO glass substrate without affecting the functionality and structural integrity of the final MEG device assembly. For subsequent cell culturing experiments, the plasma treated ITO glass was coated with a thin layer of gelatin to ensure maximal cell adherence. Finally, a series of small 3 mm diameter holes (one per well) was carefully drilled at the bottom of each tissue culture well plate to permit subsequent air pressure activation during in vitro ES studies.
Figure 2.
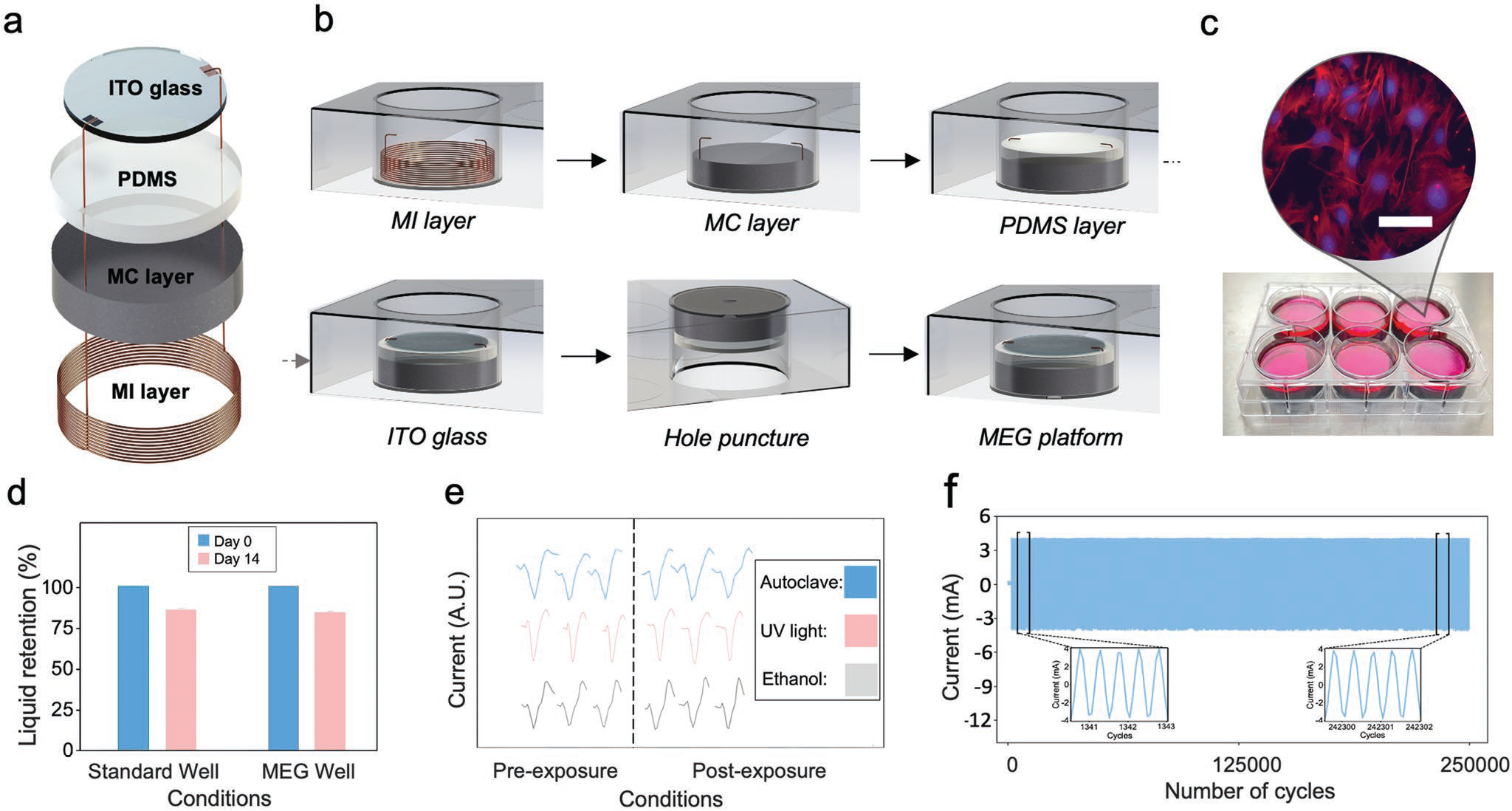
The MEG ES platform is simple to fabricate and provides a resilient in vitro tool. a) Exploded schematic view of a MEG ES device. b) Schematic illustrating the in situ fabrication of MEG devices within a commercially available tissue culture well plate. c) Photograph of a representative MEG-modified 6-well plate where fibroblasts were seeded on to the ITO glass surface and cultured in DMEM. The inset shows an immunofluorescent image of the seeded fibroblasts with nuclear (DAPI – blue) and cytoskeletal (Phalloidin –red) staining. Scale bar, 50 μm. d) Liquid retention in an MEG ES well as compared to a control standard tissue culture well, confirms the absence of absorption or leakage in the MEG ES platform. e) Exposing MEG assemblies to standard laboratory sterilization methods does not affect the electrical output and device performance. f) Durability testing to assess long-term performance and effects on output current following cyclic loading of the MEG ES platform.
Upon fabrication, tissue culture plates incorporating MEG devices were immediately ready for use in ES studies of cells in culture (Figure 2c). To assess the resilience and resistance of the MEG assemblies to exposure to various experimental conditions, we tested the integrity of the assembled platform against leakage (Figure 2d). Performance of MEG ES devices were also evaluated after exposure to a series of standard sterilization methods, including autoclaving, UV irradiation, and submerging the devices in ethanol. No tangible changes in MEG performance were observed in devices operated following any of these physical exposures (Figure 2e). These data are encouraging as the Curie temperature for conservation of magnetic properties of NdFeB is 300 °C, which is well above temperatures reached during autoclaving for steam generation (≈150 °C). Finally, as this platform is meant to be utilized for long-term cellular ES experiments, it is important to maintain consistent MEG performance when devices are operated repeatedly. No degradation of the electrical output was observed after 250,000 cycles (Figure 2f).
The PDMS layer provides a biocompatible and hermetically sealed environment where tissue culture medium can be safely deposited without leaks disrupting the activation of the MEG circuit. To complete the assembly of the MEG, an ITO glass substrate was placed on top of the PDMS layer and connected to the MI layer’s electrode wires. Cells seeded directly on the gelatin-coated ITO glass receive electrical stimuli from the MEG. ITO glass was chosen as the cell culture substrate as it possesses suitable optical properties and has been previously reported[43] to promote appropriate cell adhesion with minimal cytotoxicity (Figure S2, Supporting Information). Platform assembly required less than a day of preparation with the only time limiting steps being the curing of MC and PDMS layers, which can be carried out by leveraging heated plates built into the base of a 3D printer stand (Figure S3, Supporting Information).
Although other, more flexible ITO-coated polymers are available for use, their ITO coating process is laborious and requires specialized machinery.[44,45] In addition, electrically stimulated samples often need to be visualized under fluorescence microscopy, and the use of glass helps as ITO glass can be commercially sourced, easily cut into desired shapes, immunochemically stained, and mounted for post-experimental analysis. An alternative setup of the MEG ES platform was devised to enable parallel ES of various samples using a single external MEG, which is particularly useful for samples that need to be observed for extended periods using standard bifocal microscopy (Movie S1, Supporting Information). Additional details and operating parameters used for 3D printing are provided in the Experimental Section.
4. ES Platform Characterization
To evaluate the effectiveness and adaptability of the MEG ES platform, we tested the effect of several key parameters to determine how the architecture of the various components of the MEG devices influences the current and voltage of the output stimulus. This is an important factor to be considered not only for in vitro electrical platforms, but also for in vivo ES used in therapy. When the MC layer becomes deformed via air pressure, biomechanical-to-magnetoelastic energy conversion occurs, concurrent with magnetic induction in the MI layer, facilitating magnetic-to-electrical conversion and generation of an electric output signal (Figure 3a). We began by assessing how varying the number of coils in the MI layer affected output performance, where we observed a notable increase in both current and voltage output proportional to the increase in MI coil number (Figure 3b). We next tested the effects of modifying the MC layer. First, we altered the thickness of the MC layer, by pouring varying amounts of the uncured MC mixture into the well to produce MC layers of different thicknesses (ranging from 2 to 8 mm) (Figure 3c). Here, MEGs exhibit an initial increase in output signal with MC thickness with voltage and current peaking at 0.7 mA and 6.9 mV (6 mm), which we attribute to the increased rigidity of the thicker MC layers which coincides with a proportional decrease in magnetoelastic performance of the layer. Subsequently, we analyzed the effect on electric output caused by varying the magnitude of the air pressure input used to activate the MEG (Figure 3d). As stimulation frequency is an important parameter used to in cellular ES studies, we assessed whether there was a drop-off in the voltage and current output when exposing the MEG to various ES frequencies. Our data indicate that electric output peaked at 6 mV and 0.9 mA at 100 Hz. (Figures 3e). Finally, we observed increases in both current and voltage output when nanomagnet concentration (43%, 53%, 63%, 73%, and 83%) of the MEG’s MC layer was systematically increased (Figure 3f).
Figure 3.
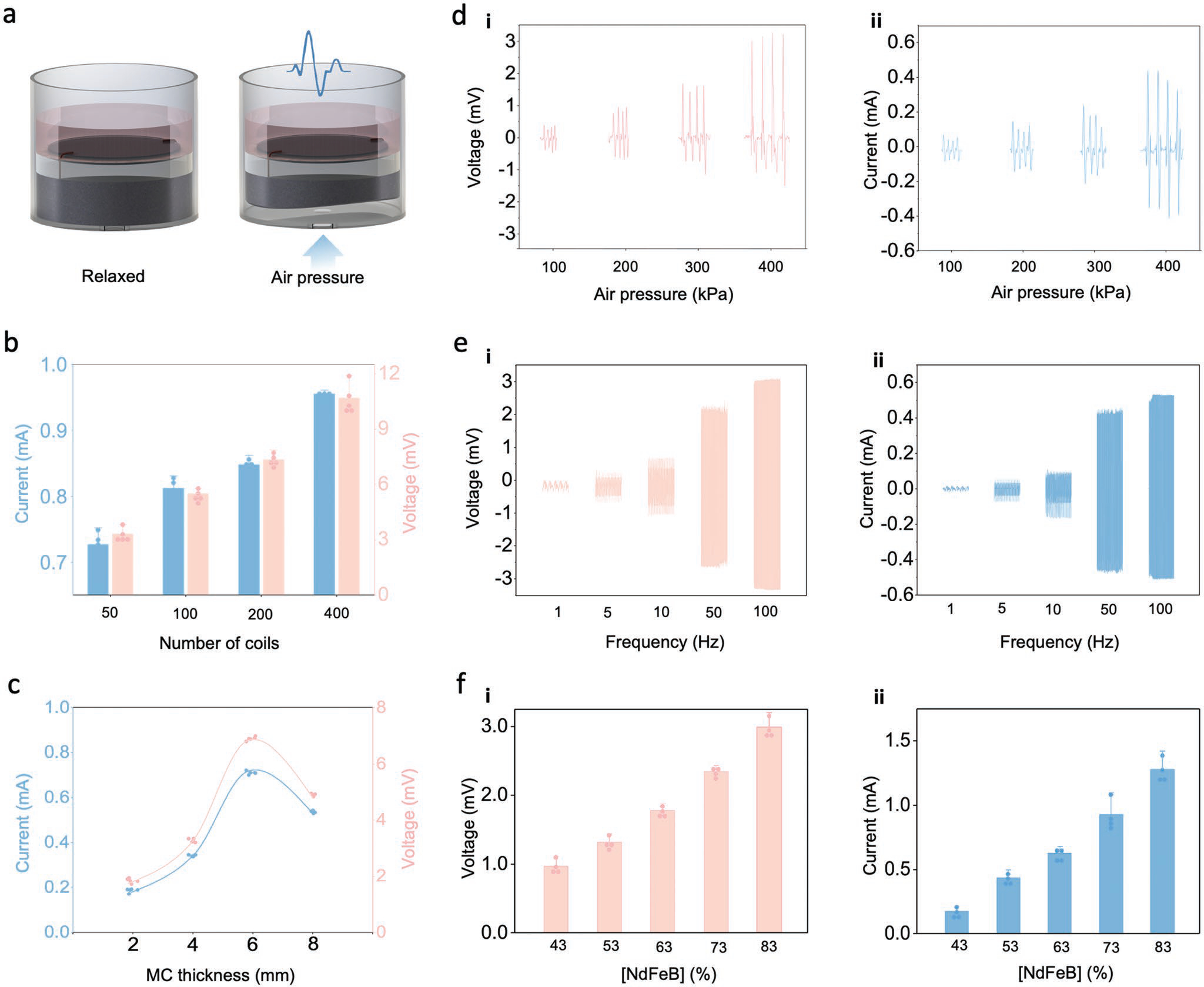
The MEG ES platform’s output can be tailored to required experimental conditions. a) Schematic illustration of the mechanism for MEG-mediated ES where air pressure-induced deformation of the MEG assembly activates the MC-MI layer to yield an electric output. b) Output current and voltage of the MEG ES platform can be adjusted by varying the number of MI layer coils. Bar graphs show mean ± standard deviation of the results (n = 5). c) MC-layer thickness is positively correlated to current and voltage output until a critical thickness is reached that hinders compression of the MC-MI layer. Curves indicate the general trend and elastic limit. d) Effect of air pressure intensity on i) voltage and ii) current output of the MEG ES platform. e) The MEG ES platform enables the use of different frequency parameters to yield specific i) voltage and ii) current values. f-i) Voltage and ii) current dependance on [NdFeB] concentration in the MC layer.
Modifying the number of coils used in the MI layer is a straightforward modification that can be used to fine tune the MEG ES platform’s desired output for both in vitro and future in vivo applications. The optimal weight concentration of NdFeB in the MC layer was 83%, which yielded the most effective balance of elastomer plasticity and electricity generation when the mechanical, magnetic, and magnetomechanical properties of the MEG were optimized for air pressure pneumatic stimulation. The most effective thickness for the MC layer in a standard 6-well tissue culture format was found to be 6 mm. Finally, much like for the MI layer, simply varying the input air pressure stimulus enables the MEG ES platform’s desired output to be tailored quickly and easily (Figures S4–S6, Supporting Information). Of note, the maximum input air pressure that can be applied to a MEG fitted to a 6-well tissue culture plate is ≈800 kPa before the device fails and/or dislodges from the well plate (Table S1, Supporting Information). Utilizing lower input air pressures to operate the MEG devices will improve the lifetime of the platform, particularly when used to study cell populations requiring long term ES.
5. ES via MEG Platform
ES platforms have been widely used for a variety of biotechnology and clinical experimental applications, and their effective development is paramount to developing new generation, effective in vivo ES therapeutic platforms. To facilitate effective translation from the bench to the bedside, these technologies need to be rigorously assessed and characterized in laboratory settings. To begin to bridge this gap, we tested whether the MEG-mediated ES could be applied to enhance the direct conversion of fibroblasts into neurons via cellular reprogramming.
The direct conversion of fibroblasts into neurons represents an effective way to obtain neuronal cells for basic research and the development of cell-based therapeutics. However, to date, the conversion efficiency and maturation rate of the neurons generated during reprogramming remains low,[46,47] which presents a barrier for applications such as high throughput drug screening and cell transplantation.[48] Traditionally, fibroblasts have been the preferred cell type to obtain neurons for a number of reasons, but especially as they can be easily sourced and demonstrate viability robustness during in vitro culturing.[48] If this can be achieved in vivo, the additional benefit of fibrosis reduction by conversion of fibroblasts into functional cells such as neurons, can provide a significant therapeutic indication. In addition, direct cell reprogramming does not carry the risk of tumorigenesis associated with other strategies, such as induced pluripotent stem cell-mediated differentiation.[48]
ES has been shown to facilitate cell reprogramming, including promoting neuronal induction. One disadvantage to these prior studies is their reliance on externally powered devices.[43] In the few instances where self-powered devices were used, the experimental setup needed to be decoupled from the stimulation environment. Establishing a stand-alone, easily fabricated, and self-powered MEG ES platform was therefore a primary objective of our study to provide a scalable and versatile solution for applying electrical stimuli to cellular populations. This represents a landmark study and application of MEG ES for future implantable therapeutic use, such as in the treatment of neurodegenerative diseases and spinal cord injuries. The contribution of this platform for self-powered programming of fibroblasts into neurons is thus two-fold.
5.1. MEG ES Increases the Efficiency of Converting Fibroblasts into Induced Neurons
In view of the clinical and research pertinence of ES, we investigated the biological effect of MEG ES on virally transduced fibroblasts[49] (Figure 4). Given the heterogeneity of the ES parameters used in prior studies where fibroblasts were converted into induced neurons, we first determined the ES tolerance range for adult mouse ear fibroblasts (Figure 4a). By exposing the cells to nA, μA, and mA range currents for varying intervals, we identified suitable, nontoxic values in the nA range that could be safely applied over a 30-min exposure period and 1-Hz impulse frequency as described in the Experimental Section.
Figure 4.
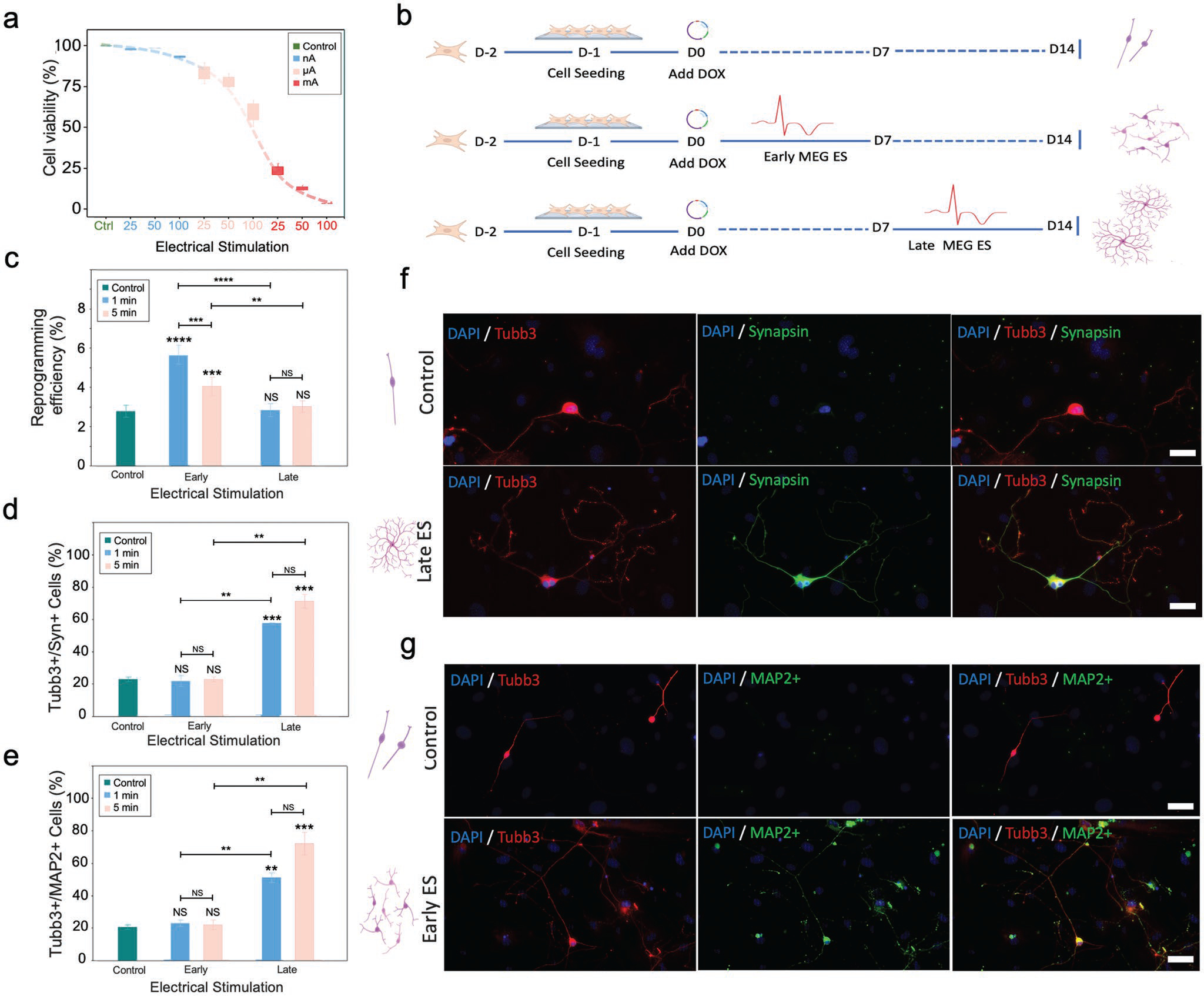
The MEG ES platform enhances efficiency and maturation of fibroblast reprogramming to induced neurons. a) Cell viability as a function of current used for MEG-mediated ES. Current values are plotted in the nano, micro, and milli Ampere ranges. Bar graph shows mean ± SD (n = 5). b) Schematic summarizing the experimental protocols used to assess the effect of MEG ES on cellular reprogramming. D = day, ES = ES, Dox = doxycycline. The expected morphology and relative number of neurons derived using each protocol are shown schematically. c) Reprogramming efficiency of fibroblasts directed to neuronal phenotypes obtained using each ES protocol (based on the number of Tubulin Beta 3 Class III+ (Tubb3+) cells identified on day 14 relative to the number of fibroblast cells initially seeded), Bar graph shows mean ± SD (n = 5), **p ≤ 0.01, ***p ≤0.001 **** p ≤ 0.0001, NS = not significant. Significance was determined using a one-way ANOVA and Tukey’s multiple comparison test. d) Quantification of mature neurons (based on the number of Tubb3+ and Synapsin+ expressing cells on day 14 relative to the number of Tubb3+ cells). Bar graph shows mean ± SD (n = 2), **p ≤ 0.01, ***p ≤0.001, NS = not significant. Significance was determined by a one-way ANOVA and Tukey’s multiple comparison test. (e) Quantification of mature neurons (based on Tubb3+ and microtubule associated protein 2 (MAP2+) cells on day 14 relative to the number of Tubb3+ cells). Bar graph shows mean ± SD (n = 2), **p ≤ 0.01, ***p ≤ 0.001, NS = not significant. Significance was determined by a one-way ANOVA and Tukey’s multiple comparison test. f) Immunofluorescent images of induced neurons generated without and with late MEG ES on day 14 post reprogramming. Cells were stained to indicate expression of the mature neuronal marker Synapsin. Images show neurite abundance and neuritic ramification. The relative morphology and number of the neurons is shown schematically for each condition. Scale bar, 50 μm. g) Immunofluorescent images of neurons generated without and with early MEG ES, on day 14, and stained to highlight expression of the mature neuronal marker MAP2. The images highlight increased efficiency with promoted induced neuron density and interconnection. Relative morphology and number of neurons yielded for each condition is shown schematically. Scale bar, 75 μm.
Subsequently, we tested various MEG-mediated ES protocols during the reprogramming process (Figure 4b). Using an MEG ES platform configured for a 6-well plate, we first coated plasma-treated ITO glass with 0.1% gelatin for 30 minutes, prior to seeding adult mouse ear fibroblasts that had been transduced with doxycycline (Dox)-inducible lentiviral vectors packaged with three reprogramming factors Brn2, Ascl1, and Mytl1 (BAM). The following day, Dox was added (marked as day 0) to induce BAM expression and initiate the reprogramming process. Cells were cultured in serum-free N2B27 medium for the remainder of the experiment as described in the Experimental Section. We first investigated whether ITO glass without electrical stimulation would have an effect on fibroblast reprogramming into induced neurons compared to traditional methods (i.e., standard tissue culture well plate), and found no significant difference in the reprogramming efficiency (Figure S7, Supporting Information), suggesting that ITO glass without electrical stimulation could serve as an appropriate control in these studies. Next, we configured MEG ES platforms to output 50 nA – confirmed using a low-noise current preamplifier – and examined the effect of MEG ES on reprogramming efficiency, which we defined as the percentage of the induced neurons expressing neuronal β-tubulin III (Tubb3+) at day 14. To do so, we pulsed the transduced fibroblasts using the following stimulation parameters: 1 Hz frequency, 50 nA current ES for 1 min and 5 min, respectively, at 48-h intervals for a duration of 7 days (i.e., the ES was applied during the first week of the reprogramming process). These specific parameters were chosen as preliminary testing showed them to be the most effective conditions for optimized reprogramming with the chosen fibroblast cells. The cultures were then left to grow until day 14 (Figure 4b). The experiment revealed a two-fold (104%) increase in the reprogramming efficiency (percentage of Tubb3+ cells) with 1 min stimulation (2.77% to 5.64%) and a 46% increase in reprogramming efficiency (number of Tubb3+ cells) with 5-min stimulation (2.77% to 4.05%) (Figure 4c), reinforcing the idea that applying MEG ES in the early-stage (i.e., first 7 days) of the reprogramming process helps to increase neuron yield. From a quantitative perspective, statistically significant differences were noted in the reprogramming efficiency of early-stage electrically stimulated samples versus control. Furthermore, the results seem to indicate that the duration of the electrical stimulus (i.e., 1 min vs 5 min) was important in enhancing direct conversion to neuronal phenotypes in early-stage stimulations, but not in late-stage ES (Figure 4c), a factor likely ascribed to the precise timing necessary to coordinate the activation of transcription factors involved in the reprogramming process. Considering the multitude of materials and physical forces at play, and in order to isolate the effect of the MEG ES as being the sole causative effect of reprogramming enhancement, we carried out a number of experiments with sham MEG ES devices which confirmed the role of MEG ES in induced neuron conversion (Figure S8, Supporting Information).
5.2. MEG ES Enhances Neuronal Maturation and Neuritic Ramification
We examined the effect of MEG ES on neuronal maturation, characterized using immunocytochemical staining and analysis of the reprogrammed cells as described in the Experimental Section. To understand whether MEG ES had any effect on neuronal maturation when applied during later stage reprogramming (i.e., during the second week of the reprogramming process when already formed induced neurons are present), we applied 1 and 5 min MEG electrical stimuli at 50 nA and 1 Hz every 48 h intervals on samples from day 8 to day 14 of the protocol (Figure 4b). While the samples did not show an increase in the reprogramming efficiency into induced neurons, we observed a marked increase in Tubb3+ cells expressing mature neuronal markers Synapsin and microtubule associated protein 2 (MAP2). Specifically, samples that had undergone late-stage MEG ES in the form of receiving 5 min stimulations, showed a more than three-fold (218%) increase (from 22.5% to 71.5%) in Tubb3+/Synapsin+ neurons (Figure 4d) and a more than three-fold (251%) increase (from 20.5% to 72%) in Tubb3+/MAP2+ neurons when compared to control samples (Figure 4e). Lowering the duration of MEG ES to 1 min, showed a modest, yet significant above two-fold increase (156%) (from 22.5% to 57.5%) and 173% (from 20.5% to 56%) in Tubb3+/Synapsin+ and Tubb3+/MAP2+ neurons, respectively, when compared to the controls. Conversely, samples that had been electrically stimulated only in the early stage of the reprogramming process (days 1–7) did not express a higher percentage of mature neuronal markers when compared to the control, aligning with evidence from previous studies that demonstrated that the influence of ES on reprogramming and maturity is distinct and depends on when the stimulus is applied.[50–52] Interestingly, although late-stage ES enhances maturation of neurons, its duration seemed to be less important in determining the degree of neuronal maturation (Figure 4d,e).
Another important difference that became apparent when visualizing induced neurons that were exposed to late-stage MEG ES under fluorescent microscopy was the stark difference in interconnectivity between samples that had undergone ES and those that did not. Induced neurons observed in control samples showed less articulate morphologies with far fewer and far less intrinsic neuritic ramifications, unlike for those encountered in electrically stimulated samples (Figure 4f). Clustering of induced neurons was also much more pronounced in electrically stimulated samples (Figure 4g), with the formation of highly interconnected neuronal networks comprised of as many as ten Tubb3+/Synapsin+ and/or Tubb3+/MAP2+ neurons.
6. Discussion
With this research we explored the first application and use of the soft magnetoelastic effect for tissue engineering purposes, devising a biocompatible electrical stimulating platform that is inexpensive, easy to fabricate, scalable using 3D printing, and adaptable for various culture conditions for in vitro use. We demonstrated a self-powered platform that could be adapted for both air pressure as well as mechanical activation. With this study we have furthermore set out the foundations for the development of a biocompatible implantable in vivo MEG for therapeutic ES in neural applications. To our knowledge, this is the first time a self-powered ES platform has been directly integrated and built within existing tissue culture research tools, providing a biocompatible, bioconformable soft system for in vitro electric stimulation of cultured cells and cellular architectures. This versatile platform may be configured straightforwardly to yield specific current and voltage outputs, as required per and during experimental protocols, and future implantable therapeutic ES use, and is compatible with common sterilization and sanitization processes used in biomedical research, with no loss of operational functionality following exposure to UV irradiation, ethanol soaking, and even autoclaving.
We also demonstrated the capability to 3D print of the MEG platform. Since the thickness of MC layer is proportionate to electricity generation, controlling the consistency in thickness within each well of a tissue culture plate is critical. Automating the MEG fabrication process with a 3D printer enabled precise control of layer height, allowing the input air pressure pneumatic stimulus to induce equal current output simultaneously within each well, increasing the consistency and repeatability of in vitro cell stimulation experiments. This 3D printing strategy drastically improved both the consistency and throughput of device fabrication. Manufacturing the device in a 3D printer also helps to improve sterility, reduce cross contamination from manual pipetting, and allows for direct cell seeding within the printing chamber, which is UV-sterilized and equipped with HEPA filtered laminar air flow. Furthermore, the use of conductive carbohydrate glass inks, which display thermoplastic properties during printing, could be applied to print conductive ITO glass-like coverslips in situ, potentially offering a one-step fabrication approach for producing MEG devices. This level of automation furthermore, bodes well for the development of implantable MEG ES devices for therapeutic applications in neurodegenerative diseases as well as spinal cord injuries.
Overall, assembling the MEGs onto transparent ITO glass enables treated cells to be analyzed using immunofluorescent staining while minimizing background autofluorescence. We applied the MEG ES platform in a model tissue engineering protocol for neuronal transdifferentiation, considering the clinical relevance of this tissue type and that they can be targeted in neuroregenerative therapeutic processes. When fibroblasts were stimulated in conjunction with viral transduction of genes encoding neuronal BAM transcription factors, reprogramming efficiency was increased up to two-fold (104%) when compared with gene delivery alone. The presence of mature neurons in the electrically stimulated populations was also improved by up to three-fold (251%) when compared to gene delivery alone. Furthermore, cells treated via MEG ES formed well-developed neurite networks and demonstrated intraneuronal connectivity. These results agree with previous studies that highlighted a role for ES in promoting both reprogramming efficiency and neuronal maturation. Although mechanistic studies were beyond the scope of the study, there are several potential mechanisms by which ES could modulate cellular reprogramming, as summarized in this comprehensive review.[53] Mechanistically, although not fully understood, it is thought that ES activates N-type Ca2+ channels during the early stages of reprogramming, inducing neuronal gene expression, and thus improving conversion efficiency. Similarly, in neuronal cultures, ES is thought to lead to an increase in Ca2+ mobility and indirect protein kinase C activation, which in turn promotes the phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2). This latter cascade is fundamental in cellular differentiation and its increased activation has been linked to neurite growth.[50]
Interestingly, in preliminary studies, we observed that when MEG ES was applied continuously to samples (from day 1 to day 18), there was an overall detrimental effect on cell survival, indicating that excessive frequency of ES was counterproductive. Similarly, excessively high current MEG ES leads to cellular detachment from the ITO glass surface and subsequent metabolic death (Movie S3, Supporting Information). Altogether, our studies suggest that similar MEG-mediated ES approaches could serve as important tools for probing cellular behaviors such as reprogramming, and investigate factors that inform normal development. Further development and validation of similar MEG-based platforms that can be applied in vivo would extend the reach of these technologies significantly toward the future development of MEG-based implantable devices. We anticipate that cell manipulation tools that leverage this technology will contribute to advancing studies across the spectrum of regenerative medicine and biophysics. For example, we envision and are eager to explore the design of MEG devices capable of mechanoelectrical co-stimulation that could inform improved methods and potential organ-on-a-chip systems for testing the functionality and the maintenance of reprogrammed cell types such as induced cardiomyocytes. Finally, this study represents a landmark foundational study for the future application of implantable MEG ES platforms for therapeutic use, including the treatment of neurodegenerative diseases and spinal cord injuries.
7. Conclusion
In summary, this is the first approach to investigate the magnetoelastic effect enabled ES in the reprogramming and maturation of fibroblasts into neuronal cells. Since magnetic fields can pass through water with negligible intensity loss, this MEG-based soft system can be encapsulated and is intrinsically waterproof and biocompatible. The mechanical actuation by air pressure can elicit evenly distributed current in multiple parallel units, and enable the scale-up of this MEG platform for in vitro studies. Furthermore, this high-throughput MEG ES system could promote the fundamental understanding of ES effects on cell reprogramming, neuronal cell behavior and other cell types (e.g., cardiomyocyte and muscle), providing a rational basis for ES therapeutics, and aiding in the optimization of current neurological diseases intervention with ES. Moreover, this MEG-driven ES can harness the biomechanical activities in human bodies and convert them into electrical signals for self-powered therapeutics. Beyond the reprogramming of fibroblasts into neurons, this technology can also be applied to investigate the conversion of other cell types, thereby potentially providing a general therapeutic approach for tissue engineering and regenerative medicine.
8. Experimental Section
Biological Suitability of MEG:
Magnetic flux density measurements were recorded as follows: Uniaxial stress (20 N) was applied onto a 25 mm by 25 mm magnetoelastic film. The vertical component of the magnetic field was measured using the axial probe of a digital Gauss meter (TD8620, Tunkia). Stress–strain curves were determined by stretching a magnetoelastic film of 15 mm (w) × 3 mm (h) × 23 mm (l) using a dynamic mechanical analyzer (Instron 5564, stretching rate of 5 mm s−1). The Young’s modulus was calculated by fitting the stress–strain data to a neo-Hookean model. A magnetized magnetoelastic layer (83 wt%) showed a Young’s modulus of 446 kPa and an ultimate strain of 140%. For water performance testing, a MC-MI layer of non PDMS-encapsulated MEG was soaked in water for 14 days with its current and voltage output tested pre- and postsoaking. For biocompatibility testing, mouse fibroblasts expanded in fibroblast medium consisting of Dulbecco’s modified Eagle medium (DMEM) basal medium (Gibco, 11965), 10% fetal bovine serum (Gibco, 26140079) and 1% penicillin/streptomycin (Gibco, 15140122) were seeded and grown on PDMS, a silicone (Cu/NdFeB-Ecoflex)-based MC-MI (“MEG”) layer, and in empty tissue culture well controls, and cultured in an incubator at 37 °C and 5% CO2. After 24 h, cell viability was assayed using the PrestoBlue Cell Viability Reagent (Invitrogen, A13261) according to the manufacturer’s protocol. Cells were incubated with 10% PrestoBlue reagent for 2 h and results were normalized to the control samples (for which fibroblasts seeded in the tissue culture plate were used). Dimethyl sulfoxide (DMSO, 99% ThermoFisher) was used to promote cell death and served as a negative control. Absorbance reading were recorded using an Enspire nanoplate reader (PerkinElmer, Massachusetts, USA).
Fabrication of MEG ES Platform:
This process was repeated for each of the wells in 6-well tissue culture plates (Falcon Polystyrene Nanoplates, ThermoFisher), in parallel. MI layer preparation: Cu wire (Enameled Cu Wire, 28 AWG, 28SNSP, Remington Industries) was spun in various coil turns (50, 100, 200, and 400) using a 25 mm diameter acrylic rod (TAP Plastics) and placed in the middle of a 6-well tissue culture plate with the Cu wire extremities positioned vertically at opposite ends, 15 mm in height. MC layer preparation: Standard MC layers were prepared by thoroughly mixing an uncured silicone rubber matrix with nonmagnetized NdFeB nanoparticles poured directly onto the MI layer, with the Cu wire extremities arranged vertically at opposite ends and then cured in an oven (60 °C for 2 h). Specifically, Ecoflex 00–30-part A, part B and NdFeB nanoparticles (MQFP-B-20076–088, Neo Magnequench) were blended thoroughly using a stirring rod. The weight ratio of Ecoflex 00–30-part A and part B was kept at 1:1 for all MC layers. NdFeB Nanomagnets with weight concentrations of 43%, 53%, 63%, 73%, and 83% were used to fabricate different MC layers for optimizing the mechanical, magnetic, and magnetomechanical properties of the soft system. For the characterization tests, varying amounts of MC layers were poured to yield MC layers with different thicknesses (2, 4, 6, and 8 mm). All the mixtures were cured at 60 °C in an oven (ThermoFisher) for 3 h, with the MI layer Cu wire extremities made to stand out of the MC mixture. The nonmagnetized soft system was magnetized by applying a magnetic pulse (≈2.655 T) using an impulse magnetizer (IM-10–30, ASC Scientific) to establish stable remnant magnetization. It is possible to perform this process either on cured MC-MI layers removed from the tissue culture well plate or on the entire the MEG tissue culture well plate following final assembly using a larger impulse magnetizer (Magnet-Physik, M-Series) and applying the same magnetic pulse (≈2.655 T). PDMS layer preparation: A thin PDMS layer (base:curing agent mixing ratio of 20:1, ≈1 mm, Dow SYLGARD 184, Ellsworth Adhesives) was placed over the MC-MI layer to avoid possible leakage, seal the platform, and maintain biocompatibility. A base to curing agent ratio of 20:1 was chosen as it provided a biocompatible, viscous yet elastic layer atop the MC-MI layer which would enable both a seal on the MEG ES platform, as well as a stable grip on the ITO glass. This would in turn prevent ITO glass free movement in the well whilst requiring minimal manual pressure for later dislodgment for sample retrieval. The Cu wire extremities were arranged so that they were positioned away from the curing solution. ITO layer preparation: A 1.1 mm thick ITO glass (10.1 Ohm sq−1, Geomatec), previously plasma treated for 5 min (Plasma Prep II, 2SPI), was cut to a 25 mm diameter circle using a circular glass cutter compass and placed atop the curing the PDMS layer after 1 h into the 2 h curing process in a 60 °C oven (ThermoFisher). Finally, the Cu wire extremities were cut to ≈4 mm above the ITO glass with the insulation removed by carefully cleaning the tips using a metal blade. The Cu wires were each bent atop the ITO glass and fixed in place using 3 mm long pieces of 5 mm strong adhesive tape (Kaisiking). The finalized device was placed at 60 °C to finish curing. A 3 mm diameter hole was drilled carefully at the bottom of each tissue culture well plate by first turning it upside down, and subsequently using an electric drill (DeWalt) mounted on a drill press to drill a hole that was the depth of the acrylic plastic.
Printing of MEG ES Devices:
The fabrication of the MC and PDMS layers of MEG substrates was automated using a pneumatic extrusion-based 3D bioprinter (BioX, Cellink, Gothenburg, Sweden). Fully assembled MEG substrates were printed layer-by-layer into individual wells of 12, 24, 48, and 96-well tissue culture plates (Falcon Polystyrene Nanoplates, ThermoFisher). Models and layouts for 3D printing were generated using computer-aided design (CAD) software (Fusion360, Autodesk). Print models were comprised of cylinders with diameters of 40, 20, 5, and 3 mm that correspond to the diameters of the 12, 24, 48, and 96-well plates. The height of each cylinder model was set at 6 mm. To produce the final desired print, a polymer-based ink is distributed in a concentric lattice structure (Movie S2, Supporting Information). MC layer: After mixing an uncured EcoFlex silicone-based ink with nonmagnetized NdFeB nanoparticles. The mixture was degassed under vacuum for 20 min and then carefully loaded into a 3D printer cartridge (3 ml cartridge, Cellink). After preheating the 3D printer’s print bed to 65 °C, the MC layer was printed at a rate of 10 mm/s with 15 kPa of air pressure through a 20-gauge tapered plastic nozzle. PDMS layer: After the printing of MC layer, well plates were left on the heated print bed at 65 °C for 30 min to allow the MC layer to cure briefly. The PDMS layer (base: curing agent mixing ratio of 20:1, Dow SYLGARD 184, Ellsworth Adhesives) was then 3D printed at an extrusion speed and pressure of 10 mm/s and 10 kPa, respectively, to a thickness of 1.5 mm on top of the MC-MI layer. With regards to both manual and 3D fabrication of the MEG ES devices, considering the final user of the MEG ES platform may require different experimental set ups (such as for instance, shallower or deeper wells for cell culturing), the thickness and matrices’ composition (i.e., Cu wire coil turn number, weight ratios) of these various layers can be modified and trialed to help yield the desired ES characteristics.
Performance of the MEG ES Platform:
For liquid retention testing, a MEG ES well plate, and a nontissue culture well plate (Standard), were each filled with 5 mL DMEM and left to rest at room temperature until day 14, with the liquid then collected from each sample and measured again. Sterilization testing of the MEG electrical platform was carried out with pre- and post-testing protocols, as follows: Durability of device performance was tested following exposure to UV irradiation for 30 min using the UV-C germicidal lamp of a biosafety cabinet (LabGard ES Nu-540, Class II, NuAire). Ethanol resistance was tested by filling each of the as-fabricated MEG wells with 5 mL of ethyl alcohol (HistoPrep 70% denatured, Fisherbrand) and leaving it to soak for 1 week. Resistance to autoclaving was tested by exposing a complete MEG ES device to the autoclaving process (2840EL-D, Tuttnauer, glass setting, steam at 134 °C, 340 kPa). Although those three approaches were tested to ensure they did not affect the platform’s performance (as they could be pertinent for future studies), in our current experimental protocols sterilization was performed by filling each of the as-fabricated MEG wells with 5 mL of ethyl alcohol (HistoPrep 70% denatured, Fisherbrand) and concomitantly exposing to 30 min of UV-C irradiation using the germicidal lamp of a biosafety cabinet (LabGard ES Nu-540, Class II, NuAire). For performance cycling and frequency testing of the MEG, a pulse generator comprised of a coupled arbitrary function generator (Tektronix afg1062), linear power amplifier (Labworks, PA-138) and electrodynamic transducer (Labworks, ET-127)) was used at 25 Hz (for 10 000 s – 250000 cycles) and varying (1, 5, 10, 50, 100 Hz) values, respectively. All the ES tests were performed in lab conditions under room temperature. The currents mentioned are short-circuit current (ISC) and the voltages mentioned are open-circuit voltage (VOC). More specifically, during testing, the voltage was measured with a Stanford low-noise voltage preamplifier (model SR560) across the output terminals of the proposed MEG, without connecting any load. On the other hand, the current was measured when the positive and negative terminals of the proposed MEG were connected through a Stanford low-noise current preamplifier (model SR570) in series. For air pressure testing, an air pressure gauge (PSL15–160, PneumaticPlus) was connected to a compressed air outlet (UCLA Bioengineering laboratory house line) and openings for 100, 200, 300, and 400 kPa outputs were devised for pulsed release. For the experiments in this study, the pneumatic actuation was applied by removing cell culture plates from the incubator and exposing them to an air pressure gauge (PSL15–160, PneumaticPlus), which was connected to a compressed air outlet (UCLA Bioengineering laboratory house line) with openings for 100, 200, 300, and 400 kPa outputs were devised for pulsed release (Movie S4, Figure S4 and Figure S5, Supporting Information). In our experiments the MEG ES air pressure actuation was only carried out on single MEG ES platforms/wells in order to obtain uniform and precise results in view of the initial stages of research in the field. Consequently, this current setup still requires a human interface to manually apply the pneumatic actuation.
MEG ES Viability Assays:
Fibroblasts were plated onto MEG devices and allowed to attach overnight. The following day, cells were stimulated at nA (25, 50, 100 nA), μA (25, 50, 100 μA), and mA (25, 50, 100 mA) values for 30 min, at 1 Hz frequency, using an alternative MEG ES platform (Movie S1, Supporting Information). The modified MEG platform yielded 2 mA current, with its output adjusted to the testing regime using an Ohm resistance box (Resistance Decade Box 380400, Extech Instruments). Cells were left to grow for 24 h, after which cell viability was assayed using the PrestoBlue Cell Viability Reagent (Invitrogen, A13261) according to the manufacturer’s protocol. Cells were incubated with the PrestoBlue Reagent for 2 h. Results were normalized to control (i.e., no stimulation) samples. The 1 mA value test was subsequently repeated whilst being visualized in time lapse using a Zeiss Axio Observer Z1 inverted fluorescence microscope with images recorded every minute, for 30 min, to show cell detachment and subsequent death, corroborating the PrestoBlue reagent results (Movie S3, Supporting Information).
Lentiviral Production and Transduction:
Doxycycline-inducible lentiviral vectors for Tet-O-FUW-Brn2, Tet-O-FUW-Ascl1, Tet-O-FUWMyt1l, and FUW-rtTA plasmids were used to transduce fibroblasts for ectopic expression of Brn2, Ascl1, Myt1L, and rtTA (Doxcycline-binding inducible promoter). Lentivirus was produced by using established calcium phosphate transfection methods, and Lenti-X Concentrator (Clontech, 631232) was utilized to concentrate viral particles according to the manufacturer’s protocol. Stable virus was aliquoted and stored at −80 °C. Fibroblasts were incubated with the virus for 24 h before being seeded onto gelatin-coated ITO glass.
Fibroblast Isolation, Culture, and Reprogramming:
Fibroblasts were isolated from ear tissue of one-month old adult C57BL/6 mice and expanded using fibroblast medium: DMEM (Gibco, 11965), 10% fetal bovine serum (FBS; Gibco 26140079), and 1% penicillin/streptomycin (GIBCO, 15140122). For all experiments, passage-2 cells were used. Fibroblasts were transduced with lentivirus-containing BAM constructs and seeded the following day onto ITO glass (10.1 Ohm sq−1, Geometec) coated with 0.1 mg mL−1 gelatin (30 min) at a density of 7000 cells per glass slide, with the ITO glass having previously been plasma treated for 5 min (Plasma Prep II, 2SPI). The following day (i.e., day 0), the medium was replaced with MEF medium containing doxycycline (2 ng mL−1, Sigma) to initiate the expression of the transgenes and thus, reprogramming. Twenty-four hours later (i.e., day 1), cells were cultured in N2B27 medium: DMEM/F12 (Gibco, 11 320 033), N-2 supplement (Gibco, 17 502 048), B-27 supplement (Gibco, 17 504 044), 1% penicillin/streptomycin, and Dox (2 ng mL−1), and half medium changes were performed every two days. Please note that the parameters described represent the protocol which yields the highest possible number of induced neurons within our experimental setup. MEG ES: For the early-stage MEG ES study using an ad hoc MEG ES platform yielding 50 nA, the transduced fibroblasts were pulsed with 1 Hz frequency, 50 nA current ES for 1 min and 5 min, at 48 h intervals for a duration of seven days starting at day 1 and cultures were left to grow until day 14. For the late-stage MEG ES study, the transduced fibroblasts were pulsed with 1 Hz frequency, 50 nA current ES for 1 and 5 min, at 48 h intervals, for a duration of seven days, starting on day 8 until day 14. On day 14, cells were fixed and stained for Tubb3 to determine the reprogramming efficiency, whilst other samples were also stained for Synapsin and MAP2 to determine maturation stage. Induced neuronal cells were identified based on positive Tubb3 staining and neuronal morphology (defined as Tubb3+/DAPI+, with the presence of at least two or more neurites [i.e., filopodia-like protrusions which exceeded the diameter of the cell body]). The reprogramming efficiency was determined as the percentage of Tubb3+ cells on day 14 relative to the number of the fibroblast cells initially seeded at 24 h. Reviewing previous literature that had utilized ES to enhance fibroblast reprogramming into induced neurons failed to reveal a framework which could be used to deduce the optimal ES conditions for our cell line and reprogramming protocol. Considering no nexus could be established, extensive preliminary testing underwent to identify the optimal ES parameters including current size (as shown in Figure 4), ES duration and ES frequency. It is anticipated this laborious trial-and-error method will be required for any novel cell line and reprogramming protocol. As 1 min and 5 min ES conditions maintained cell viability during preliminary testing, these values were chosen for subsequent reprogramming experiments. Sham MEG ES devices: sham devices were fabricated in the following fashion; a sham device with plain glass (no ITO) was prepared substituting ITO glass with a plain coverslip glass to show that the current flowing through the ITO was responsible for the observed effects; a sham device with a copper coil, MC-MI layer, PDMS layer and ITO glass, with the copper wires cut short and left insulated in the PDMS layer was prepared to exclude the effect of mechanic actuation; a sham device without a copper coil, but with the MC-MI layer, PDMS layer and ITO glass, was prepared to show that the effect could not be observed without the electric current flowing; and a sham device with a copper coil, with the MC-MI layer replaced with PDMS, a PDMS layer, and with ITO glass to show the need for a coupling of the MEG effect to create MEG ES.
Immunofluorescent Staining and Characterization of Neurons and Mature Neurons:
All samples collected for immunofluorescence staining at the indicated time points were washed once with 1× phosphate buffered saline (PBS) (Thermo Scientific PBS, 10× Solution) and fixed in 4% paraformaldehyde for 10 min. Samples were then washed three times with PBS for 5 min each and permeabilized using 0.5% Triton X-100 for 10 min. The ITO glass with fixed cells were dismounted from the MEG ES platform and, after three subsequent PBS washes, blocked with 5% normal donkey serum (NDS; Jackson Immunoresearch, 017000121) in PBS for 1 h. Samples were incubated with primary antibodies (Tubb3; 1:1000, Biolegend #802001, MAP2; 1:300, Biolegend #801801, Synapsin; 1:400, Abcam #ab254349) in antibody dilution buffer (5% NDS in PBS) overnight at 4 °C followed by three PBS washes and a 1 h incubation with Alexa Fluor 488- and/or Alexa Fluor 546-conjugated secondary antibodies (Molecular Probes). Nuclei were stained with DAPI (1:1000, Molecular Probes, Eugene, OR) in PBS for 10 min. For F-actin staining, cytoskeletons were stained using Phalloidin (1:200, Phalloidin-iFluor 514 Conjugate, AAT Bioquest). Epifluorescence images were collected using a Zeiss Axio Observer Z1 inverted fluorescence microscope and analyzed using ImageJ. Induced neurons were confirmed as such by nucleic DAPI+ and cellular Tubb3+ staining association as well as the presence of at least two or more neurites (filopodia-like protrusions which exceeded the diameter of the cell body), while mature neurons were confirmed by identifying cells with a DAPI+/Tubb3+/Synapsin+ and DAPI+/Tubb3+/MAP2+ staining association.
Statistical Analysis:
All data were presented as mean ± one standard deviation (SD), where n indicates the sample size from at least two independent experiments. All experiments were carried out with multiple (triplicate to quintuplicate) replicates. Comparisons among values for groups were performed using a one-way analysis of variance (ANOVA) followed by a Tukey’s post-hoc test. For comparison between two groups, a two-tailed, unpaired t-test was used. For all cases, significance level 𝛼 = 0.05 was set with a 95% confidence to detect a significant difference, and p-values less than 0.05 were considered statistically significant. GraphPad Prism 8.0.2 software was used for all statistical evaluations.
Supplementary Material
Acknowledgements
A.L., J.S., and J.X. contributed equally to this work. J.C. acknowledges the Henry Samueli School of Engineering & Applied Science and the Department of Bioengineering at the University of California, Los Angeles for the startup support. J.C. also acknowledges the Hellman Fellows Research Grant, the UCLA Pandemic Resources Program Research Award, the Research Recovery Grant by the UCLA Academic Senate, and the Brain & Behavior Research Foundation Young Investigator Grant, and the NIH NS126918. S.L. acknowledges the support of NIH (GM143485 and NS126918). S.J. acknowledges support from the NIH Common Fund through a NIH Director’s Early Independence Award (DP5OD028181).
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
The ORCID identification number(s) for the author(s) of this article can be found under https://doi.org/10.1002/adma.202206933.
Contributor Information
Alberto Libanori, Department of Bioengineering, University of California, Los Angeles, Los Angeles, CA 90095, USA.
Jennifer Soto, Department of Bioengineering, University of California, Los Angeles, Los Angeles, CA 90095, USA.
Jing Xu, Department of Bioengineering, University of California, Los Angeles, Los Angeles, CA 90095, USA.
Yang Song, Department of Bioengineering, University of California, Los Angeles, Los Angeles, CA 90095, USA.
Jana Zarubova, Department of Bioengineering, University of California, Los Angeles, Los Angeles, CA 90095, USA.
Trinny Tat, Department of Bioengineering, University of California, Los Angeles, Los Angeles, CA 90095, USA.
Xiao Xiao, Department of Bioengineering, University of California, Los Angeles, Los Angeles, CA 90095, USA.
Shouzheng Yue, California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA 90095, USA; Department of Pediatrics, David Geffen School of Medicine at the University of California, Los Angeles, Los Angeles, CA 90095, USA.
Steven J. Jonas, California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA 90095, USA Department of Pediatrics, David Geffen School of Medicine at the University of California, Los Angeles, Los Angeles, CA 90095, USA; Children’s Discovery and Innovation Institute, University of California, Los Angeles, Los Angeles, CA 90095, USA; Eli & Edythe Broad Center of Regenerative Medicine and Stem Cell Research, University of California, Los Angeles, Los Angeles, CA 90095, USA.
Song Li, Department of Bioengineering, University of California, Los Angeles, Los Angeles, CA 90095, USA.
Jun Chen, Department of Bioengineering, University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA 90095, USA.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- [1].Zhang J, Chen JDZ, Aliment. Pharmacol. Ther. 2006, 24, 991. [DOI] [PubMed] [Google Scholar]
- [2].Choi YS, Hsueh YY, Koo J, Yang Q, Avila R, Hu B, Xie Z, Lee G, Ning Z, Liu C, Xu Y, Lee YJ, Zhao W, Fang J, Deng Y, Lee SM, Vázquez-Guardado A, Stepien I, Yan Y, Song JW, Haney C, Oh YS, Liu W, Yun HJ, Banks A, MacEwan MR, Ameer GA, Ray WZ, Huang Y, Xie T, et al. , Nat. Commun. 2020, 11, 5990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hernández D, Millard R, Kong AM, Burns O, Sivakumaran P, Shepherd RK, Dusting GJ, Lim SY, Bioelectricity 2020, 2, 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chen C, Bai X, Ding Y, Lee I-S, Biomater. Res. 2019, 23, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lee GS, Kim MG, Kwon HJ, Biochem. Biophys. Res. Commun. 2019, 513, 990. [DOI] [PubMed] [Google Scholar]
- [6].Leppik L, Zhihua H, Mobini S, Thottakkattumana Parameswaran V, Eischen-Loges M, Slavici A, Helbing J, Pindur L, Oliveira KMC, Bhavsar MB, Hudak L, Henrich D, Barker JH, Sci. Rep. 2018, 8, 6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bestmann S, Walsh V, Curr. Biol. 2017, 27, R1258. [DOI] [PubMed] [Google Scholar]
- [8].Carmel JB, Martin JH, Front. Integr. Neurosci. 2014, 8, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Marquez-Chin C, Popovic MR, Biomed. Eng. Online 2020, 19, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Javeed S, Faraji AH, Dy C, Ray WZ, MacEwan MR, Interdiscip. Neurosurg. 2021, 24, 101117. [Google Scholar]
- [11].McCaig CD, Rajnicek AM, Song B, Zhao M, Physiol. Rev. 2005, 85, 943. [DOI] [PubMed] [Google Scholar]
- [12].Whited JL, Levin M, Curr. Opin. Genet. Dev. 2019, 57, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sullivan KG, Emmons-Bell M, Levin M, Commun. Integr. Biol. 2016, 9, e1192733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Du J, Zhen G, Chen H, Zhang S, Qing L, Yang X, Lee G, Mao H-Q, Jia X, Biomaterials 2018, 181, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Leppik L, Oliveira KMC, Bhavsar MB, Barker JH, Eur. J. Trauma Emerg. Surg. 2020, 46, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ferrigno B, Bordett R, Duraisamy N, Moskow J, Arul MR, Rudraiah S, Nukavarapu SP, Vella AT, Kumbar SG, Bioact. Mater. 2020, 5, 468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sun L-Y, Hsieh D-K, Lin P-C, Chiu H-T, Chiou T-W, Bioelectromagnetics 2009, 31, 209. [DOI] [PubMed] [Google Scholar]
- [18].Sun L-Y, Hsieh D-K, Yu T-C, Chiu H-T, Lu S-F, Luo G-H, Kuo TK, Lee OK, Chiou T-W, Bioelectromagnetics 2009, 30, 251. [DOI] [PubMed] [Google Scholar]
- [19].Tomaskovic-Crook E, Gu Q, Rahim SNA, Wallace GG, Crook JM, Cells 2020, 9, 658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Oh B, Wu YW, Swaminathan V, Lam V, Ding J, George PM, Adv. Sci. 2021, 8, 2002112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cheng H, Huang Y, Yue H, Fan Y, Stem Cells Int. 2021, 2021, 6697574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Libanori A, Chen G, Zhao X, Zhou Y, Chen J, Nat. Electron. 2022, 5, 142. [Google Scholar]
- [23].Conta G, Libanori A, Tat T, Chen G, Chen J, Adv. Mater. 2021, 33, 2007502. [DOI] [PubMed] [Google Scholar]
- [24].Deng W, Zhou Y, Libanori A, Chen G, Yang W, Chen J, Chem. Soc. Rev. 2022, 51, 3380. [DOI] [PubMed] [Google Scholar]
- [25].Chen G, Xiao X, Zhao X, Tat T, Bick M, Chen J, Chem. Rev. 2022, 122, 3259. [DOI] [PubMed] [Google Scholar]
- [26].Meng K, Xiao X, Wei W, Chen G, Nashalian A, Shen S, Xiao X, Chen J, Adv. Mater. 2022, 34, 2109357. [DOI] [PubMed] [Google Scholar]
- [27].Zi Y, Basset P, Chen J, EcoMat 2021, 3, e12129. [Google Scholar]
- [28].Gu L, Liu J, Cui N, Xu Q, Du T, Zhang L, Wang Z, Long C, Qin Y, Nat. Commun. 2020, 11, 1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhou Y, Zhao X, Xu J, Fang Y, Chen G, Song Y, Li S, Chen J, Nat. Mater. 2021, 20, 1670. [DOI] [PubMed] [Google Scholar]
- [30].Chen G, Zhao X, Andalib S, Xu J, Zhou Y, Tat T, Lin K, Chen J, Matter 2021, 4, 3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Rattay F, Neuroscience 1999, 89, 335. [DOI] [PubMed] [Google Scholar]
- [32].Michelson NJ, Eles JR, Vazquez AL, Ludwig KA, Kozai TDY, J. Neurosci. Res. 2019, 97, 620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Brocker DT, Grill WM, Handb. Clin. Neurol. 2013, 116, 3. [DOI] [PubMed] [Google Scholar]
- [34].Su Q, Morillo J, Wen Y, Wuttig M, J. Appl. Phys. 1996, 80, 3604. [Google Scholar]
- [35].Datta S, Atulasimha J, Mudivarthi C, Flatau AB, J. Magn. Magn. Mater. 2010, 322, 2135. [Google Scholar]
- [36].Jen SU, Lo Y, Pai L, J. Phys. D: Appl. Phys. 2016, 49, 145004. [Google Scholar]
- [37].Davino D, Giustiniani A, Visone C, Phys. B: Condens. Matter 2012, 407, 1427. [Google Scholar]
- [38].Zhao X, Chen G, Zhou Y, Nashalian A, Xu J, Tat T, Song Y, Libanori A, Xu S, Li S, Chen J, ACS Nano 2022, 16, 6013. [DOI] [PubMed] [Google Scholar]
- [39].Zhao X, Zhou Y, Xu J, Chen G, Fang Y, Tat T, Xiao X, Song Y, Li S, Chen J, Nat. Commun. 2021, 12, 6755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chen G, Zhou Y, Fang Y, Zhao X, Shen S, Tat T, Nashalian A, Chen J, ACS Nano 2021, 15, 20582. [DOI] [PubMed] [Google Scholar]
- [41].Zhao X, Nashalian A, Ock IW, Popoli S, Xu J, Yin J, Tat T, Libanori A, Chen G, Zhou Y, Chen J, Adv. Mater. 2022, 34, 2204238. [DOI] [PubMed] [Google Scholar]
- [42].Xu J, Tat T, Zhao X, Zhou Y, Ngo D, Xiao X, Chen J, Appl. Phys. Rev. 2022, 9, 031404. [Google Scholar]
- [43].Jin Y, Seo J, Lee JS, Shin S, Park H-J, Min S, Cheong E, Lee T, Cho S-W, Adv. Mater. 2016, 28, 7365. [DOI] [PubMed] [Google Scholar]
- [44].Boehme M, Charton C, Surf. Coatings Technol. 2005, 200, 932. [Google Scholar]
- [45].Løveng Sunde TO, Garskaite E, Otter B, Fossheim HE, Sæterli R, Holmestad R, Einarsrud M-A, Grande T, J. Mater. Chem. 2012, 22, 15740. [Google Scholar]
- [46].Gascón S, Masserdotti G, Russo GL, Götz M, Cell Stem Cell 2017, 21, 18. [DOI] [PubMed] [Google Scholar]
- [47].Wang H, Yang Y, Liu J, Qian L, Nat. Rev. Mol. Cell Biol. 2021, 22, 410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Xu Z, Su S, Zhou S, Yang W, Deng X, Sun Y, Li L, Li Y, Cell Biosci. 2020, 10, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Song Y, Soto J, Wang P, An Q, Zhang X, Hong SG, Lee LP, Fan G, Yang L, Li S, Adv. Sci. 2021, 8, 2003516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Chang Y-J, Hsu C-M, Lin C-H, Lu MS-C, Chen L, Biochim. Biophys. Acta, Gen. Subj. 2013, 1830, 4130. [DOI] [PubMed] [Google Scholar]
- [51].Brosenitsch TA, Katz DM, J. Neurosci. 2001, 21, 2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ghosh A, Greenberg ME, Science 1995, 268, 239. [DOI] [PubMed] [Google Scholar]
- [53].Heng W, Bhavsar M, Han Z, Barker JH, Curr. Stem Cell Res. Ther. 2020, 15, 441. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.