

Abstract
An intrinsic goal when designing synthetic methodology is to identify approaches whereby readily accessible precursors are converted into an array of products, which efficiently tap into new 3D-chemical space. In these studies, readily available bicyclic lactam-bromolactones have been interrogated in several fragment growth protocols by utilizing the halogen and lactone motifs as versatile linchpins for strategic construction of C–C, C–N, C–O, and C–S bonds. Diastereospecific C(sp3)–C(sp2) Kumada coupling of sterically imposing [5,5]-bicyclic lactam-bromolactones with several aryl Grignard reagents, under palladium catalysis, furnishes diarylmethane-tethered lactam-lactones in synthetically attractive yields, stereoinvertive fashion, and with a tolerance for many functional groups. When [5,6]-bicyclic lactam-bromolactones, which are prone to β-hydride elimination are employed, efficient arylation is observed only under Co(acac)3-catalyzed conditions. Importantly, these [5,6]-bicyclic lactam-bromolactones undergo retentive arylation, independent of the transition metal catalyst. A base-mediated cascade deconstructive amidation of the [5,6]-bicyclic lactam-bromolactones with primary aliphatic amines proceeds efficiently to afford epoxide-tethered lactam carboxamides, which bear four contiguous stereocenters. Furthermore, an unusual route to homoallylic thioesters has been uncovered through deconstructive contra-thermodynamic thioesterification of the lactam-fused bromolactone precursors.
Readily available lactam-bromolactones have been interrogated in several fragment growth protocols including diastereospecific Kumada cross-coupling with Grignard reagents, cascade deconstructive amidation, and contra-thermodynamic thioesterification.
Introduction
Functionalized γ-lactams bearing contiguous stereocenters (Fig. 1) constitute the core of several alkaloid natural products and pharmaceuticals, including pramanicin (antifungi), omuralide (proteasome inhibitor), and clausenamide (antidementia).1 The γ-lactam topology also presents an ideal platform for systematic scaffolding owing to its latent reactivity and the endless number of transformations it can undergo.2
Fig. 1. Examples of bioactive 2-pyrrolidinones bearing contiguous stereocenters.

Notable strategies for the construction of functionalized 2-pyrrolidinones include the ring-opening of aziridines,3 the aza-Heck reaction,4 and the use of cascade/multicomponent reactions.5 These methodological advances notwithstanding, approaches to the stereocontrolled synthesis of polysubstituted γ-lactams bearing contiguous stereocenters are still underdeveloped.6 As the drug development process continues to seek out more sophisticated nitrogen-containing heterocycles with higher degrees of saturation,7 the need for divergent methods for the stereocontrolled synthesis and post-diversification of sp3-rich 2-pyrrolidinones increases.
One of the key steps in drug discovery is to ‘grow’ fragment hits from potentially any position. Structural diversity is highly desirable given that molecular shape is among the most important factors that dictate the biological effects of molecules. Fragment libraries consisting of a variety of 3D scaffolds are expected to display a wider range of biological activities compared to single scaffold libraries. Within this context, and as part of a program aimed at leveraging the synthetic versatility of the 1,3-azadiene-anhydride reaction,8 our group has identified the catalytic halolactonization8g,h of lactam-bearing alkenoic acids of type 1 (Fig. 2A/B) and subsequent interrogation of the lactam-halolactones (see 2/3) in fragment growth protocols as an important research objective (Fig. 2C). Specifically, we sought to utilize the halogen and lactone motifs resident in 2/3 as versatile linchpins for strategic late-stage construction of C–C, C–N, C–O, and C–S bonds (see 4–7). Herein, we describe the efforts toward the realization of our objectives.
Fig. 2. Proposed plan for the diastereospecific arylation and deconstructive amidation/thioesterification of lactam-halolactones.

Results and discussion
Diastereospecific arylation of lactam-bromolactones
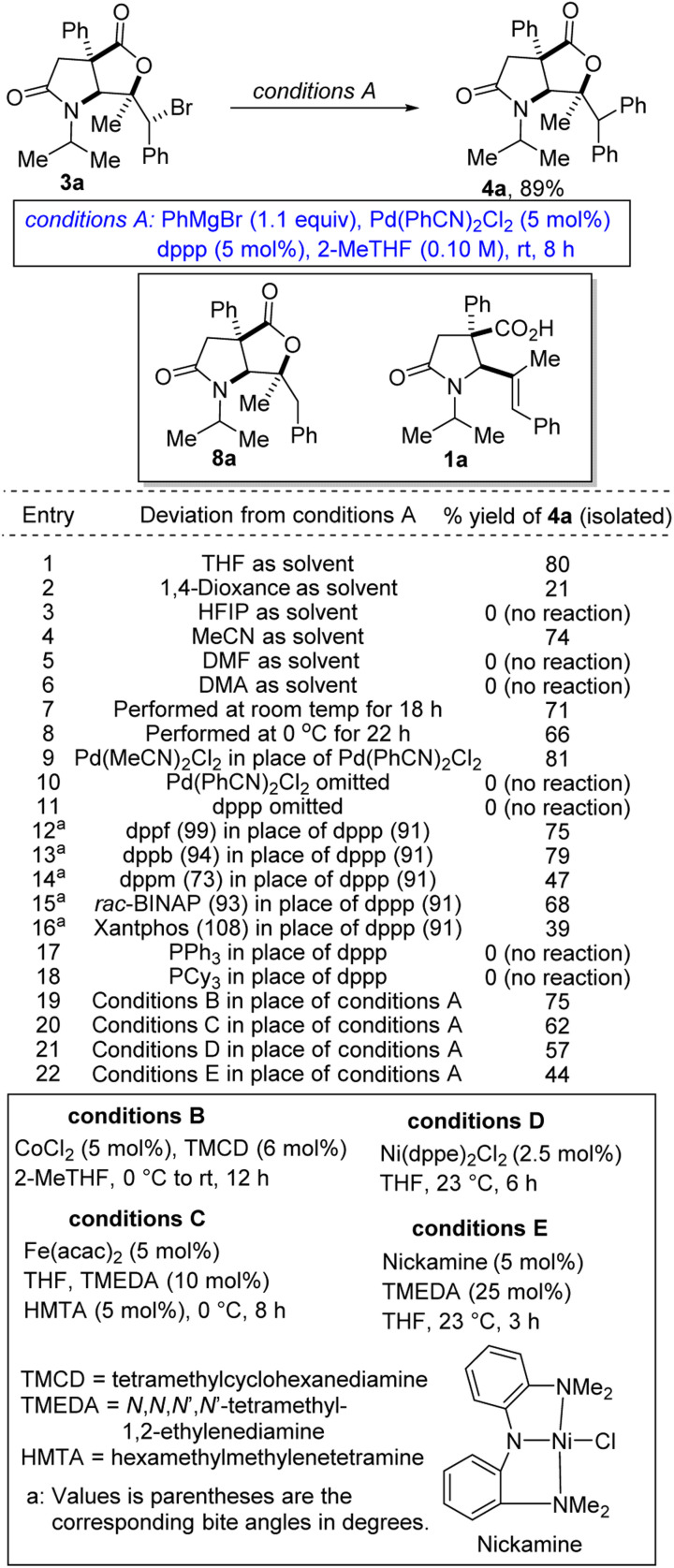
We first sought to explore the amenability of hindered alkyl bromides of type 3 to functional group-tolerant Kumada-Corriu cross-coupling with Grignard reagents.9 Such alkyl halides are typically challenging substrates owing to their reluctance to undergo oxidative addition. Furthermore, metal alkyl intermediates tend to induce unproductive β-hydride elimination.10 Recent methodological advances have however facilitated the formation of carbon–carbon bonds by cross-coupling reactions of less activated alkyl halides and Grignard reagents under palladium catalysis.11 In general, palladium complexes have an exceptional catalytic activity in Kumada cross-coupling, which is mainly attributed to their tendency to undergo a two-electron transfer process as well as to their tolerance for a wide range of functional groups.12 We initiated studies toward the arylation of lactam-bromolactones with aryl Grignard reagents by benchmarking our optimization efforts for the phenylation of bromolactone 3a with the reaction conditions described in Table 1.
Optimization of the arylation of lactam-bromolactone 3a with phenylmagnesium bromide.
|
At the outset, we were concerned about the possibility of reduction of the bromide to bicycle 8a. We also recognized that unwanted β-carboxylate elimination could instead lead to the generation of alkenoic acid 1a. Furthermore, we were concerned about any competing and undesirable nucleophilic addition of the Grignard reagent to the lactone/lactam motifs resident in 3a. Accordingly, we sought functional group-tolerant conditions that would not only minimize β-carboxylate elimination, but also facilitate oxidative addition, transmetalation, and reductive elimination. Ultimately, 2-methyltetrahydrofuran (2-MeTHF) emerged as the preferred reaction medium (entries 1–6). The reaction proceeds slowly at 0 °C (entry 8). We have found that Pd(MeCN)2Cl2 is slightly less efficient than the bulky Pd(PhCN)2Cl2 precatalyst (entry 9). No background cyclization reaction is observed in the absence of the palladium catalyst (entry 10). Knowing that the bite angle of the ligand can have a dramatic effect on the efficiency of cross-couplings, several ligands were evaluated (entries 12–18). Resounding success was mostly achieved when bis-1,2-diphenylphosphinopropane (dppp) was utilized. The results indicate that smaller or larger bite angles adversely affect the coupling. The respective bite angles are provided in parentheses. Other reaction conditions known to promote Kumada cross-couplings with alkyl halides were surveyed (entries 19–22). The reaction worked well under cobalt-catalyzed conditions (entry 19). Presumably, the TMCD ligand helped to suppress β-carboxylate elimination. In these cases, the mass balance was mostly accounted for by recovered starting material, reduction product 8a, and β-carboxylate elimination product 1a. The spectroscopic data for byproducts 8a and 1a are available in the ESI.† Under the optimized conditions (i.e., conditions A), diarylmethane-tethered lactam-lactone 4a was obtained in good yield.
The scope of the transformation with respect to the lactam-bromolactone has been surveyed (Scheme 1, see 4a–v). Knowing that the nature of the nitrogen substituent present on a nitrogen heterocycle can have a dramatic effect on its biological activity13 and reactivity, the effect of the N-substituent on the arylation was first explored. Encouragingly, N-alkyl-substituted lactam-bromolactones are competent substrates for the coupling (see 4a–d). The successful construction of arylated lactam-lactones harboring the N-phenethyl group (see 4e–g and 4i/j) is noteworthy given that the phenethyl group is often employed as a precursor to the indolizidine/quinolizidine scaffold. A readily removable benzyl group is well-tolerated (see 4h).
Scheme 1. Diastereospecific Kumada cross-coupling of various lactam-bromolactones with phenylmagnesium bromide.

When the phenyl group on the reactive center is replaced by an electron-deficient p-chlorophenyl or p-fluorophenyl group, the efficacy of the transformation is not compromised (see 4k–t). Similarly, the deployment of an electron-rich p-methoxyphenyl or p-tolyl group leads to efficient phenylation (see 4u/v). It is commendable that aryl halide-bearing substrates couple exclusively at the benzylic site (see 4g/i/k–v), without complications arising from aryl–aryl coupling. N-Arylated γ-lactam-bromolactones underwent productive cross-coupling with phenylmagnesium bromide (see 4m/u/v), which is noteworthy since N-aryl γ-lactams are embedded in several pharmacologically pertinent targets.14 The incorporation of a fluorinated moiety into organic molecules generally increases the solubility, lipophilicity and metabolic stability of the parent molecules, thus, explaining why ∼25% of existing preclinical drugs and 40% of agrochemicals contain at least one fluorine atom.15 It is therefore noteworthy that fluorinated products 4l–t are obtainable in satisfactory yields.
The scope of the arylation with respect to the Grignard reagent has been explored, albeit briefly (Scheme 2). Electron-deficient and electron-rich aryl Grignard reagents are competent coupling partners (4wvs.4x), suggesting that the coupling is less sensitive to its electronic environment. An ortho-substituted aryl Grignard reagent is marginally tolerated as exemplified through the synthesis of diarylmethane 4z2. A sterically imposing 2-naphthyl group can be installed using the corresponding Grignard reagent, but the efficiency is unsurprisingly modest (see 4z3). Indeed, our luck runs out when even more imposing 1-naphthylmagnesium bromide is employed as no coupling takes place (see 4z4). Additionally, no coupling is observed when highly π-deficient 2-pyridylmagnesium bromide is employed as the coupling partner (see 4z5). This indicates that transmetallation step is critical and probably rate-limiting. Our studies have revealed that the arylation reaction takes place stereoinvertively (as judged by NOESY) as the diarylmethane-tethered lactam-lactones are obtained in impeccable diastereoselectivities (see 4k–4z3). NOE correlations are shown in the ESI for compounds 4w–z. For example, clear NOEs are observed between protons Ha and Hb as well as between protons Hb and Hc (see 4w for the numbering).
Scheme 2. Kumada cross-coupling of lactam-bromolactones with electronically diverse Grignard reagents.

The palladium-catalyzed Kumada coupling discussed so far is widely understood to proceed through insertion of the Pd(0) catalyst into the C–Br bond of the benzylic bromide.10c Subsequent transmetalation with the Grignard reagent forms a hetero-organometallic complex, which undergoes isomerization and concomitant reductive elimination to furnish the diarylmethane-tethered lactam-lactone, with regeneration of the Pd(0) catalyst.
The amenability of [5,6]-bicyclic lactam-bromolactones of type 2 to cross-coupling with PhMgBr has been briefly investigated. To our delight, satisfactory cross-coupling was observed under the Co-catalyzed conditions described in Scheme 3. NOESY data revealed that diastereoretentive coupling too place (NOEs were observed between the α-amino and α-alkoxy protons resident in 5a and 5c). The PN ligand proved to be critical in suppressing undesirable β-hydride elimination. The strategic deployment of Co(acac)3 as the precatalyst is noteworthy given its relative stability and ease of storage. Although Kumada-type cross-couplings with cobalt have been extensively studied,16 sterically imposing halides of type 2 tend to react slowly. It is worth mentioning that using the reaction conditions developed for coupling of 3, the reaction of 2b (R = tert-Bu) with PhMgBr afforded mainly the β-hydride elimination product (i.e., 9) in 77% yield. The small amount of 5b that was formed in this case also displayed a preference for stereoinvertive arylation. This result indicates that the mode of attack of the aryl Grignard reagent is substrate-dependent rather than catalyst-dependent. Thus, whereas [5,6]-bicyclic lactam-bromolactones of type 2 undergo invertive arylation, the corresponding [5,5]-bicyclic lactam-bromolactones of type 3 display a preference for retentive arylation.
Scheme 3. Cobalt-catalyzed cross-coupling of lactam-bromolactones of type 2 with phenylmagnesium bromide.

Regarding the mechanistic underpinnings of this Co-catalyzed cross-coupling with PhMgBr, congruent with literature reports,16a we tentatively postulate that reduction of the Co(acac)3 pre-catalyst by PhMgBr furnishes the ‘active’ catalytic species (see 10, Fig. 3). The low-valent cobalt species (i.e., 10) undergoes single-electron transfer (SET) with lactam-bromolactone 2a to furnish secondary radical 12, (via the dissociation of radical-anion 11). Subsequent transmetalation between PhMgBr and cationic cobalt species 13 delivers intermediate 14, which unites with radical 12 to furnish complex 15. Concomitant reductive elimination of 15 gives rise to coupling product 5a with regeneration of active catalyst 10. The diastereoslective formation of 5a is presumably governed by steric effects. Indeed, when the two diastereomeric secondary bromides anti-2a and syn-2a (with respect to the bromine-bearing stereocenter) were reacted separately with phenylmagnesium bromide using the reaction conditions described in Scheme 3, the same stereoisomer of product 5a was obtained, indicating a diastereo-convergence of the cross-coupling. Such diastereo-convergent cross-couplings on 6-membered rings organic halides have previously been observed.16c It is worth reiterating that in the proposed mechanism, the true active catalyst as well as the order of the elementary steps are yet to be fully established.
Fig. 3. Tentative mechanism for Co-catalyzed diastereospecific arylation of lactam-bromolactones of type 2.

Diastereoselective synthesis of epoxide-tethered lactam carboxamides
The carboxamide motif is prevalent in natural products (e.g., penicillin), (bio)polymers (e.g., proteins and nylon), ligands, fragrances, pharmaceuticals, and agrochemicals.17 We reasoned that a strategy, which merges a functionalized γ-lactam, a highly substituted epoxide, and a carboxamide motif, would likely enhance the potential for the discovery of new small molecules with medicinal value. The preparation of carboxamides from carboxylic acids and amines is a high priority reaction, particularly from the standpoint of atom economy.18 However, it is marred by the high energy barrier for dehydration of a stable ammonium carboxylate.19 As such, the synthesis of carboxamides is traditionally achieved through the N-acylation of amines using moisture-sensitive acid chlorides19 or anhydrides.20 However, these protocols suffer from limited reagent stability and shelf life, hazardous reagent preparation, and their corrosive nature. Meanwhile, the use of coupling agents such as DCC,21 EDC,22 CDI,23 PyBOP,24 BOP,25 HBTU,26 and HATU (ref. 27) to achieve amidation results in acute safety hazards due to their explosive and allergenic natures. Sustainable approaches to epoxide-tethered γ-lactam carboxamides that do not rely on the aforementioned problematic reagents are therefore desirable. Intrinsic to our design was the prospect of employing cascade reactions to access epoxide-tethered γ-lactam carboxamides, given that the former are inherently step and atom-economical. Cascade reactions often lead to a reduction in the amount of waste and in the number of purification steps.28 Pleasingly, after surveying several bases and solvents, we found that fused bicyclic lactam-bromolactones of type 2 undergo efficient cascade deconstructive epoxy-amidation to afford the epoxylactam carboxamides depicted in Scheme 4. Nucleophilic addition of the amine to the lactone is accompanied by ring-opening and intramolecular backside attack of the displaced alkoxide on the organic bromide. Concomitant elimination of the bromide leaving group furnishes the epoxide.
Scheme 4. Construction of γ-lactam carboxamides from lactam-bromolactones of type 2.

Deconstructive thioesterification of lactam-bromolactones
Functionalized thioesters are useful building blocks in organic synthesis and biochemistry.29,30 Conventional routes for the preparation of thioesters involve the reaction of acyl chlorides with metal thiolates,31 the condensation of carboxylic acids with thiols, the displacement of halides with thiocarboxylates,32,33 the Mitsunobu reaction of alcohols with thioacetic acids,34 and carbonylation reactions in the presence of thiols.35 The conversion of more reactive thioesters to less reactive esters is readily achievable.36 In contrast, due to the high leaving group ability of thiolates, there is little to no driving force for the conversion from esters/lactones to thioesters/thiolactones under typical conditions. Specifically, to the best of our knowledge, there are no known methods for the synthesis of S-aryl thioesters from lactones. Efforts to overcome these methodological limitations have led to the discovery that lactam-bromolactones of type 2 are amenable to contra-thermodynamic cascade deconstructive aryl thioesterification under the conditions described in Scheme 5. The detailed mechanistic underpinnings of the transformation are currently under investigation.
Scheme 5. Construction of γ-lactam thioesters from lactam-bromolactones of type 2.

Conclusions
In summary, readily available lactam-bromolactones have been interrogated in three different fragment growth protocols. Diastereoretentive Pd-catalyzed Kumada cross-coupling of hindered benzylic bromides of type 3 with several aryl Grignard reagents has led to the synthesis of diarylmethane-tethered lactam-lactones such as 4, in synthetically attractive yields. Conversely, secondary nonbenzylic organic bromides of type 2, which are highly susceptible to β-hydride elimination undergo arylation under Co-catalyzed conditions to afford the corresponding products (i.e., 5) with complete inversion of configuration at the leaving group-bearing carbon. The protocol tolerates a variety of functional groups, which bodes well for late-stage modification. Furthermore, we have developed mild conditions for cascade deconstructive amidation and contra-thermodynamic thioesterification of lactam bromolactones such as 2. The epoxy-amidation reaction proceeds efficiently to afford pharmaceutically pertinent lactam carboxamides, which bear four contiguous stereocenters. Meanwhile, the thioesterification reaction furnishes lactam-tethered homoallylic thioesters in an unusual manner. It is anticipated that the structural diversity accomplished in these studies would endow it with some practical advantages over some existing diversity-oriented synthesis methodologies given that fragment libraries consisting of a variety of 3D scaffolds continue to display a wider range of biological activities compared to single scaffold libraries.
Author contributions
M. D. –investigation, data curation, validation; S. I. A. – investigation, methodology; T. K. B. – conceptualization, project administration, data curation, methodology, supervision, writing – original draft, internal funding acquisition.
Conflicts of interest
There are no conflicts of interest to declare.
Supplementary Material
Acknowledgments
We are grateful to Central Washington University for financial support through startup funds. The school of graduate studies and Provost DenBeste are thanked for research fellowships to S. I. A. and T. K. B.
Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data. See DOI: https://doi.org/10.1039/d3ra04690g
Notes and references
- (a) Feling R. H. Buchanan G. O. Mincer T. J. Kauffman C. A. Jensen P. R. Fenical W. Angew. Chem., Int. Ed. 2003;42:355–357. doi: 10.1002/anie.200390115. [DOI] [PubMed] [Google Scholar]; (b) Reddy L. R. Saravanan P. Corey E. J. J. Am. Chem. Soc. 2004;126:6230–6231. doi: 10.1021/ja048613p. [DOI] [PubMed] [Google Scholar]; (c) De Vreese R. D'Hooghe M. Beilstein J. Org. Chem. 2012;8:398–402. doi: 10.3762/bjoc.8.43. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zheng X. Dai X.-J. Yuan H.-Q. Ye C.-X. Ma J. Huang P.-Q. Angew. Chem., Int. Ed. 2013;52:3494. doi: 10.1002/anie.201210088. [DOI] [PubMed] [Google Scholar]; (e) Hu K.-Z. Ma J. Qiu S. Zheng X. Huang P.-Q. J. Org. Chem. 2013;78:1790. doi: 10.1021/jo301277n. [DOI] [PubMed] [Google Scholar]; (f) Lathrop S. P. Rovis T. Chem. Sci. 2013;4:1668. doi: 10.1039/C3SC22292F. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Armanino N. Carreira E. M. J. Am. Chem. Soc. 2013;135:6814. doi: 10.1021/ja4026787. [DOI] [PubMed] [Google Scholar]; (h) Kimmel K. L. Weaver J. D. Lee M. Ellman J. A. J. Am. Chem. Soc. 2012;134:9058. doi: 10.1021/ja3026196. [DOI] [PubMed] [Google Scholar]; (i) Satoh N. Yokoshima S. Fukuyama T. Org. Lett. 2011;13:3028. doi: 10.1021/ol200886j. [DOI] [PubMed] [Google Scholar]; (j) Shenvi R. A. Corey E. J. J. Am. Chem. Soc. 2009;131:5746. doi: 10.1021/ja901400q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Poulsen T. B. Dickmeiss G. Overgaard J. Jørgensen K. A. Angew. Chem., Int. Ed. 2008;47:4687. doi: 10.1002/anie.200800329. [DOI] [PubMed] [Google Scholar]; (l) Ma G. Nguyen H. Romo D. Org. Lett. 2007;9:2143. doi: 10.1021/ol070616u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Chauhan D. Catley L. Li G. Podar K. Hideshima T. Velankar M. Mitsiades C. Mitsiades N. Yasui H. Letai A. Ovaa H. Berkers C. Nicholson B. Chao T. H. Neuteboom S. T. Richardson P. Palladino M. A. Anderson K. C. Cancer Cell. 2005;8:407. doi: 10.1016/j.ccr.2005.10.013. [DOI] [PubMed] [Google Scholar]; (n) Fustero S. García de la Torre M. Sanz Cervera J. F. Ramírez de Arellano C. Piera J. Simón A. Org. Lett. 2002;4:3651. doi: 10.1021/ol026599k. [DOI] [PubMed] [Google Scholar]; (o) Barrett A. G. M. Head J. Smith M. L. Stock N. S. White A. J. P. Williams D. J. J. Org. Chem. 1999;64:6005. [Google Scholar]; (p) Corey E. J. Li W.-D. Z. Chem. Pharm. Bull. 1999;47:1. doi: 10.1248/cpb.47.1. [DOI] [PubMed] [Google Scholar]; (q) Fishwick C. W. G. Foster R. J. Carr R. E. Tetrahedron Lett. 1996;37:3915. [Google Scholar]
- (a) Gomez C. Gicquel M. Carry J.-C. Schio L. Retailleau P. Voituriez A. Marinetti A. J. Org. Chem. 2013;78:1488. doi: 10.1021/jo302460d. [DOI] [PubMed] [Google Scholar]; (b) Xiao K.-J. Wang A.-E. Huang P.-Q. Angew. Chem., Int. Ed. 2012;51:8314. doi: 10.1002/anie.201204098. [DOI] [PubMed] [Google Scholar]; (c) Shao C. Yu H.-J. Wu N.-Y. Tian P. Wang R. Feng C.-G. Lin G.-Q. Org. Lett. 2011;13:788. doi: 10.1021/ol103054a. [DOI] [PubMed] [Google Scholar]; (d) Xiao K.-J. Luo J.-M. Ye K.-Y. Wang Y. Huang P.-Q. Angew. Chem., Int. Ed. 2010;49:3037. doi: 10.1002/anie.201000652. [DOI] [PubMed] [Google Scholar]; (e) Chouhan G. Alper H. Org. Lett. 2008;10:4987. doi: 10.1021/ol802067y. [DOI] [PubMed] [Google Scholar]; (f) Agosti A. Britto S. Renaud P. Org. Lett. 2008;10:1417. doi: 10.1021/ol800145h. [DOI] [PubMed] [Google Scholar]; (g) Gheorghe A. Schulte M. Reiser O. J. Org. Chem. 2006;71:2173. doi: 10.1021/jo0524472. [DOI] [PubMed] [Google Scholar]; (h) Okino T. Hoashi Y. Furukawa T. Xu X. Takemoto Y. J. Am. Chem. Soc. 2005;127:119. doi: 10.1021/ja044370p. [DOI] [PubMed] [Google Scholar]; (i) Madan S. Milano P. Eddings D. B. Gawley R. E. J. Org. Chem. 2005;70:3066. doi: 10.1021/jo048484v. [DOI] [PubMed] [Google Scholar]
- Kas'yan L. I. Pal'chikov V. A. Bondarenko Y. S. Russ. J. Org. Chem. 2011;47:1609–1652. [Google Scholar]
- Shuler S. A. Yin G. Krause S. B. Vesper C. M. Watson D. A. J. Am. Chem. Soc. 2016;138:13830–13833. doi: 10.1021/jacs.6b08932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Burdzhiev N. T. Stanoeva E. R. Z. Naturforsch., B: J. Chem. Sci. 2008;63:313–320. [Google Scholar]; (b) Castagnoli, Jr N. J. Org. Chem. 1969;34:3187–3189. doi: 10.1021/jo01262a081. [DOI] [PubMed] [Google Scholar]
- (a) Kanbayashi N. Takenaka K. Okamura T. Onitsuka K. Angew. Chem., Int. Ed. 2013;52:4897. doi: 10.1002/anie.201300485. [DOI] [PubMed] [Google Scholar]; (b) Hatano M. Nishimura T. Angew. Chem., Int. Ed. 2015;54:10949. doi: 10.1002/anie.201505382. [DOI] [PubMed] [Google Scholar]; (c) Panger J. L. Denmark S. E. Org. Lett. 2020;22:2501. doi: 10.1021/acs.orglett.9b04347. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ashida K. Hoshimoto Y. Tohnai N. Scott D. E. Ohashi M. Imaizumi H. Tsuchiya Y. Ogoshi S. J. Am. Chem. Soc. 2020;142:1594. doi: 10.1021/jacs.9b12493. [DOI] [PubMed] [Google Scholar]; (e) Qian H. Sun S. Zhao W. Sun J. Chem. Sci. 2020;56:11295. doi: 10.1039/d0cc05188h. [DOI] [PubMed] [Google Scholar]; (f) Ramachandran P. V. Burghardt T. E. Chem.–Eur. J. 2005;11:4387. doi: 10.1002/chem.200401295. [DOI] [PubMed] [Google Scholar]
- Vitaku E. Smith D. T. Njardarson J. T. J. Med. Chem. 2014;57:10257–10274. doi: 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- (a) Braunstein H. Langevin S. Khim M. Adamson J. Hovenkotter K. Kotlarz L. Mansker B. Beng T. K. Org. Biomol. Chem. 2016;14:8864–8872. doi: 10.1039/c6ob01526c. [DOI] [PubMed] [Google Scholar]; (b) Beng T. K. Moreno A. New J. Chem. 2020;44:4257–4261. [Google Scholar]; (c) Beng T. K. Bauder M. Rodriguez M. J. Moreno A. New J. Chem. 2018;42:16451–16455. [Google Scholar]; (d) Hovenkotter K. Braunstein H. Langevin S. Beng T. K. Org. Biomol. Chem. 2017;15:1217–1221. doi: 10.1039/c6ob02652d. [DOI] [PubMed] [Google Scholar]; (e) Beng T. K. Moreno A. RSC Adv. 2020;10:8805–8809. doi: 10.1039/c9ra10888b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Garcia J. Eichwald J. Zesiger J. Beng T. K. RSC Adv. 2022;12:309–318. doi: 10.1039/d1ra07390g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Beng T. K. Rodriguez M. Borg C. RSC Adv. 2022;12:17617–17620. doi: 10.1039/d2ra02167f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Beng T. K. Borg C. Rodriguez M. RSC Adv. 2022;12:28685–28691. doi: 10.1039/d2ra04177d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- In 1986, the (dppf)Pd(0)-catalyzed cross-coupling reaction of a secondary alkyl iodide with Grignard reagents was reported: ; (a1) Castle P. L. Widdowson D. A. Tetrahedron Lett. 1986;27:6013. [Google Scholar]; , It has since been revealed that the reaction instead afforded reduction or β-elimination products rather than the coupling products: ; (b) Yuan K. Scott W. J. Tetrahedron Lett. 1989;30:4779. [Google Scholar]
- (a) Hu X. Chimia, International Journal for Chemistry. 2010;64:231–234. [Google Scholar]; (b) Yonova I. Johnson A. Osborne C. Moore C. Angew. Chem. 2014;53:2422–2427. doi: 10.1002/anie.201308666. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Knappke C. E. I. Jacobi von Wangelin A. Chem. Soc. Rev. 2011;40:4948–4962. doi: 10.1039/c1cs15137a. [DOI] [PubMed] [Google Scholar]
- (a) Galardon E. Ramdeehul S. Brown J. M. Cowley A. Hii K. K. Jutand A. Angew. Chem. 2002;41:1760–1763. doi: 10.1002/1521-3773(20020517)41:10<1760::aid-anie1760>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]; (b) Molander G. Canturk B. Angew. Chem. 2009;48:9240–9261. doi: 10.1002/anie.200904306. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Denmark S. E. Regens C. S. Acc. Chem. Res. 2008;41:1486–1499. doi: 10.1021/ar800037p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lopez-Perez A. Adrio J. Carretero J. C. Org. Lett. 2009;11:5514–5517. doi: 10.1021/ol902335c. [DOI] [PubMed] [Google Scholar]; (e) Cherney A. H. Kadunce N. T. Reisman S. E. Chem. Rev. 2015;115:9587–9652. doi: 10.1021/acs.chemrev.5b00162. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Xi Z. Liu B. Chen W. J. Org. Chem. 2008;73:3954–3957. doi: 10.1021/jo800197u. [DOI] [PubMed] [Google Scholar]; (g) Joshi-Pangu A. Wang C. Y. Biscoe M. R. J. Am. Chem. Soc. 2011;133:8478–8481. doi: 10.1021/ja202769t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Zhou Q. Cobb K. M. Tan T. Watson M. P. J. Am. Chem. Soc. 2016;138:12057–12060. doi: 10.1021/jacs.6b08075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Fernández-Rodríguez M. Hartwig J. J. Org. Chem. 2009;74:1663–1672. doi: 10.1021/jo802594d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kumar A. Kumar Rao G. Kumar S. Singh A. Organometallics. 2014;33:2921–2943. [Google Scholar]
- Beng T. K. Gawley R. E. J. Am. Chem. Soc. 2010;132:12216. doi: 10.1021/ja105772z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Qiao X. Cheney D. L. Alexander R. S. Smallwood A. M. King S. R. He K. Rendina A. R. Luettgen J. M. Knabb R. M. Wexler R. R. Lam P. Y. S. Bioorg. Med. Chem. Lett. 2008;18:4118–4123. doi: 10.1016/j.bmcl.2008.05.095. [DOI] [PubMed] [Google Scholar]; (b) Larsen M. A. Hennessy E. T. Deem M. C. Lam Y. Sauri J. Sather A. C. J. Am. Chem. Soc. 2020;142:726–732. doi: 10.1021/jacs.9b13114. [DOI] [PubMed] [Google Scholar]
- Liang T. Neumann C. N. Ritter T. Angew. Chem., Int. Ed. 2013;52:8214–8264. doi: 10.1002/anie.201206566. [DOI] [PubMed] [Google Scholar]
- (a) Guérinot A. Cossy J. Acc. Chem. Res. 2020;53:1351–1363. doi: 10.1021/acs.accounts.0c00238. [DOI] [PubMed] [Google Scholar]; (b) Cahiez G. Moyeux A. Chem. Rev. 2010;110:1435–1462. doi: 10.1021/cr9000786. [DOI] [PubMed] [Google Scholar]; (c) killion J. A. Darrow W. T. Brennan M. R. Leahy C. A. Fout A. R. Organometallics. 2022;41:1769–1776. [Google Scholar]; (d) Anderson C. Ferey V. Daumas M. Bernardelli P. Cossy J. Org. Lett. 2019;21:6241. doi: 10.1021/acs.orglett.9b00579. [DOI] [PubMed] [Google Scholar]; (e) Iwasaki T. Takagawa H. Singh S. P. Kuniyasu H. Kambe N. J. Am. Chem. Soc. 2013;135:9604. doi: 10.1021/ja404285b. [DOI] [PubMed] [Google Scholar]; (f) Affo W. Ohmiya H. Fujioka T. Ikeda Y. Nakamura T. Yorimitsu H. Oshima K. Imamura Y. Mizuta T. Miyoshi K. J. Am. Chem. Soc. 2006;128:8068. doi: 10.1021/ja061417t. [DOI] [PubMed] [Google Scholar]; (g) Kiefer G. Vrubel H. Scopelliti R. Severin K. Eur. J. Inorg. Chem. 2013;28:4916–4921. [Google Scholar]; (h) Kreyenschmidt F. Koszinowski K. Chem.–Eur. J. 2018;24:1168. doi: 10.1002/chem.201704547. [DOI] [PubMed] [Google Scholar]; (i) Kreyenschmidt F. Meurer S. E. Koszinowski K. Chem.–Eur. J. 2019;25:5912. doi: 10.1002/chem.201805964. [DOI] [PubMed] [Google Scholar]
- (a) Carey J. S. Laffan D. Thomson C. Williams M. T. Org. Biomol. Chem. 2006;4:2337–2347. doi: 10.1039/b602413k. [DOI] [PubMed] [Google Scholar]; (b) Pitzer J. Steiner K. J. Biotechnol. 2016;235:32–46. doi: 10.1016/j.jbiotec.2016.03.023. [DOI] [PubMed] [Google Scholar]
- (a) Constable D. J. C. Dunn P. J. Hayler J. D. Humphrey G. R. Leazer Jr J. L. Linderman R. J. Lorenz K. Manley J. Pearlman B. A. Wells A. Zaks A. Zhang T. Y. Green Chem. 2007;9:411–420. [Google Scholar]; (b) Bryan M. C. Dunn P. J. Entwistle D. Gallou F. Koenig S. G. Hayler J. D. Hickey M. R. Hughes S. Kopach M. E. Moine G. Richardson P. Roschangar F. Steven A. Weiberth F. J. Green Chem. 2018;20:5082–5103. [Google Scholar]
- (a) Charville H. Jackson D. Hodges G. Whiting A. Chem. Commun. 2010;46:1813–1823. doi: 10.1039/b923093a. [DOI] [PubMed] [Google Scholar]; (b) Goossen L. J. Ohlmann D. M. Lange P. P. Synthesis. 2009;209:160–164. [Google Scholar]; (c) Jursic B. S. Zdravkovski Z. Synth. Commun. 1993;23:2761–2770. [Google Scholar]
- Zhang L. Wang X. J. Wang J. Grinberg N. Krishnamurthy D. Senanayake C. H. Tetrahedron Lett. 2009;50:2964–2966. [Google Scholar]
- Coleman L. E. Bork J. F. Dunn H. J. Org. Chem. 1959;24:135–136. [Google Scholar]; Sheehan J. C. Hess G. P. J. Am. Chem. Soc. 1955;77:1067–1068. [Google Scholar]
- Goyal N. Synlett. 2010;20:335–336. [Google Scholar]
- Paul R. Anderson G. W. J. Am. Chem. Soc. 1960;82:4596–4600. [Google Scholar]
- Dourtoglou V. Gross B. Lambropoulou V. Zioudrou C. Synthesis. 1984;19:572–574. [Google Scholar]
- Carpino L. A. J. Am. Chem. Soc. 1993;115:4397–4398. [Google Scholar]
- (a) Bode J. W. Goossen L. J. Top. Organomet. Chem. 2012;44:13–33. [Google Scholar]; (b) Lundberg H. Tinnis F. Selander N. Adolfsson H. Chem. Soc. Rev. 2014;43:2714–2742. doi: 10.1039/c3cs60345h. [DOI] [PubMed] [Google Scholar]
- Ye F. Ge Y. Spannenberg A. Neumann H. Beller M. Nat. Commun. 2020;11:5383. doi: 10.1038/s41467-020-19110-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D. S. Knox C. Guo A. C. Shrivastava S. Hassanali M. Stothard P. Chang Z. Woolsey J. Nucleic Acids Res. 2006;34:D668–D672. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Lehninger A. L., Nelson D. L. and Cox M. M., Principles of Biochemistry, Worth Publishing, New York, 2002 [Google Scholar]; (b) de Duve C. Am. Sci. 1995;83:428–437. [Google Scholar]
- For reviews of sulfur-containing drugs, see: ; (a) Ilardi E. A. Vitaku E. Njardarson J. T. J. Med. Chem. 2014;57:2832–2842. doi: 10.1021/jm401375q. [DOI] [PubMed] [Google Scholar]; (b) Beno B. R. Yeung K. S. Bartberger M. D. Pennington L. D. Meanwell N. A. J. Med. Chem. 2015;58:4383–4438. doi: 10.1021/jm501853m. [DOI] [PubMed] [Google Scholar]; (c) Scott K. A. Njardarson J. T. Top. Curr. Chem. 2018;376:1–34. doi: 10.1007/s41061-018-0184-5. [DOI] [PubMed] [Google Scholar]; (d) Feng M. Tang B. Liang S. H. Jiang X. Curr. Top. Med. Chem. 2016;16:1200–1216. doi: 10.2174/1568026615666150915111741. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Wang N. Saidhareddy P. Jiang X. Nat. Prod. Rep. 2020;37:246–275. doi: 10.1039/c8np00093j. [DOI] [PubMed] [Google Scholar]
- Patai S., Chemistry of Functional Groups: Carboxylic Acids and Esters, Wiley, New York, 1969 [Google Scholar]
- Fujiwara S. Kambe N. Top. Curr. Chem. 2005;251:87–140. [Google Scholar]
- Mori Y. Seki M. Org. Synth. 2007;84:285. [Google Scholar]
- Volante R. Tetrahedron Lett. 1981;22:3119–3122. [Google Scholar]
- Hirschbeck V. Gehrtz P. H. Fleischer I. Chem.–Eur. J. 2018;24:7092–7107. doi: 10.1002/chem.201705025. [DOI] [PubMed] [Google Scholar]
- (a) Chan W. K. Masamune S. Spessard G. O. Org. Synth. 1983;61:48. [Google Scholar]; (b) McGrath N. A. Raines R. T. Acc. Chem. Res. 2011;44:752–761. doi: 10.1021/ar200081s. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.