

Key Points
Question
Can muvalaplin, an orally administered small molecule inhibitor of lipoprotein(a) (Lp[a]) formation, achieve safe and tolerable plasma concentrations adequate to reduce steady-state Lp(a) levels without modulating plasminogen activity in humans?
Findings
In this first-in-human phase 1 study involving healthy participants, muvalaplin administered orally as single ascending doses ranging from 1 mg to 800 mg and as multiple ascending doses ranging from 30 mg to 800 mg for 14 days caused dose-dependent plasma concentration increases. Muvalaplin administration was not associated with concerns about safety or tolerability, and it reduced Lp(a) levels but not plasminogen activity.
Meaning
The observed safety, tolerability, pharmacokinetics, and exploratory pharmacodynamics of muvalaplin in healthy participants support further clinical evaluation in patients with elevated Lp(a) levels.
Abstract
Importance
Lipoprotein(a) (Lp[a]) is associated with atherosclerotic disease and aortic stenosis. Lp(a) forms by bonding between apolipoprotein(a) (apo[a]) and apo B100. Muvalaplin is an orally administered small molecule that inhibits Lp(a) formation by blocking the apo(a)-apo B100 interaction while avoiding interaction with a homologous protein, plasminogen.
Objective
To determine the safety, tolerability, pharmacokinetics, and pharmacodynamic effects of muvalaplin.
Design, Setting, and Participants
This phase 1 randomized, double-blind, parallel-design study enrolled 114 participants (55 assigned to a single-ascending dose; 59 assigned to a multiple-ascending dose group) at 1 site in the Netherlands.
Interventions
The single ascending dose treatment evaluated the effect of a single dose of muvalaplin ranging from 1 mg to 800 mg or placebo taken by healthy participants with any Lp(a) level. The multiple ascending dose treatment evaluated the effect of taking daily doses of muvalaplin (30 mg to 800 mg) or placebo for 14 days in patients with Lp(a) levels of 30 mg/dL or higher.
Main Outcomes and Measures
Outcomes included safety, tolerability, pharmacokinetics, and exploratory pharmacodynamic biomarkers.
Results
Among 114 randomized (55 in the single ascending dose group: mean [SD] age, 29 [10] years, 35 females [64%], 2 American Indian or Alaska Native [4%], 50 White [91%], 3 multiracial [5%]; 59 in the multiple ascending dose group: mean [SD] age 32 [15] years; 34 females [58%]; 3 American Indian or Alaska Native [5%], 6 Black [10%], 47 White [80%], 3 multiracial [5%]), 105 completed the trial. Muvalaplin was not associated with tolerability concerns or clinically significant adverse effects. Oral doses of 30 mg to 800 mg for 14 days resulted in increasing muvalaplin plasma concentrations and half-life ranging from 70 to 414 hours. Muvalaplin lowered Lp(a) plasma levels within 24 hours after the first dose, with further Lp(a) reduction on repeated dosing. Maximum placebo-adjusted Lp(a) reduction was 63% to 65%, resulting in Lp(a) plasma levels less than 50 mg/dL in 93% of participants, with similar effects at daily doses of 100 mg or more. No clinically significant changes in plasminogen levels or activity were observed.
Conclusion
Muvalaplin, a selective small molecule inhibitor of Lp(a) formation, was not associated with tolerability concerns and lowered Lp(a) levels up to 65% following daily administration for 14 days. Longer and larger trials will be required to further evaluate safety, tolerability, and effect of muvalaplin on Lp(a) levels and cardiovascular outcomes.
Trial Registration
ClinicalTrials.gov Identifier: NCT04472676
This phase 1 randomized clinical trial evaluates the safety, tolerability, pharmacokinetics, and exploratory pharmacodynamic biomarker effects of muvalaplin in humans.
Introduction
The residual risk of cardiovascular events, despite optimal lowering of low-density lipoprotein (LDL) cholesterol concentration, supports the need to develop new therapies that modify additional risk factors involved in the pathogenesis of atherosclerotic cardiovascular disease. Increasing evidence has suggested a causal role of lipoprotein(a) (Lp[a]) in both atherosclerotic cardiovascular disease and aortic stenosis.1 Observations from cohort2 and genetic3,4,5 studies directly associate increasing Lp(a) levels with increased risk of cardiovascular disease. Although Lp(a) apheresis may be used to lower Lp(a) levels in some patients, there are no pharmacological agents currently approved for lowering Lp(a) levels.6 In recent years, several therapeutic approaches have been developed that selectively target Lp(a) by reducing hepatic synthesis of apolipoprotein(a) (apo[a]). Initial studies demonstrated that Lp(a) was lowered by approximately 80% with an antisense oligonucleotide7 and by up to 98% with RNA interference.8,9 These agents are now being investigated in large clinical trials to evaluate the effect Lp(a) lowering has on cardiovascular outcomes. However, each of these approaches involves injectable therapies. Development of an oral agent that specifically lowers Lp(a) could provide a therapeutic alternative that may enable broader use.
Lp(a) is formed via a covalent bond of apo(a) to an apo B100 protein on an LDL-like particle.10,11 This process involves initial noncovalent binding of apo(a) kringle IV domains 7 and 8 to lysine residues of apo B100 in the hepatocyte and the space of Disse, followed by the formation of a covalent disulfide bond. Muvalaplin is a small molecule Lp(a) inhibitor that disrupts the initial noncovalent interaction between apo(a) and apo B100, thus preventing the disulfide bond and the formation of Lp(a). This approach mimics naturally occurring variants in apo(a) that are unable to interact with apo B100, resulting in low Lp(a) levels (Figure 1).12,13 Preclinical studies demonstrated that muvalaplin-mediated reductions in Lp(a) formation in assembly reactions in vitro and in circulating Lp(a) levels in cynomolgus monkeys treated with muvalaplin (N. Diaz, et al, unpublished data, Eli Lilly and Co, 2023). Muvalaplin is the first oral agent specifically developed to lower Lp(a) levels. The objective of the current study was to evaluate the safety, tolerability, pharmacokinetics, and exploratory pharmacodynamic biomarker effects of muvalaplin in a phase 1 trial, representing the initial evaluation of its effects in humans.
Figure 1. Lipoprotein(a) Biology and Therapeutic Approaches to Lower Lipoprotein(a).
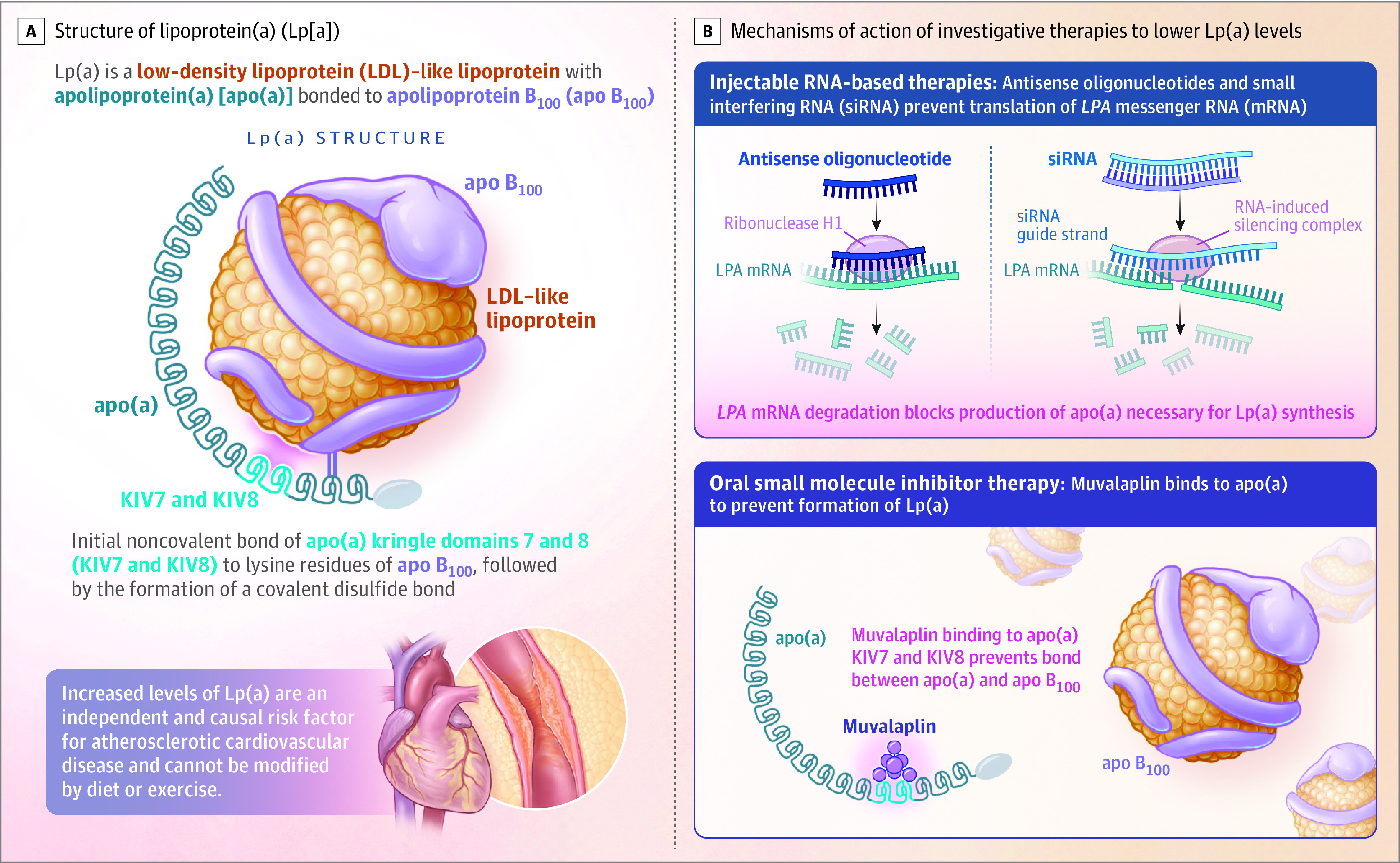
Methods
Study Design
This single-center, randomized, double-blind, placebo-controlled, phase 1 study of muvalaplin (LY3473329) was conducted in 2 parts at the ICON facility in Groningen, the Netherlands (Figure 2). The first part of study was to evaluate the effect of only 1 dose of muvalaplin in ascending increments (range, 1 mg to 800 mg) by healthy participants. The second part of the study was to evaluate the effect of muvalaplin taken as a single daily dose in ascending increments (range, 30 mg to 800 mg) taken over 14 days by healthy participants with elevated Lp(a) plasma levels (≥30 mg/dL).
Figure 2. Flow of Patients Through Both Parts of the Study of Muvalaplin for Lipoprotein(a) Inhibition.
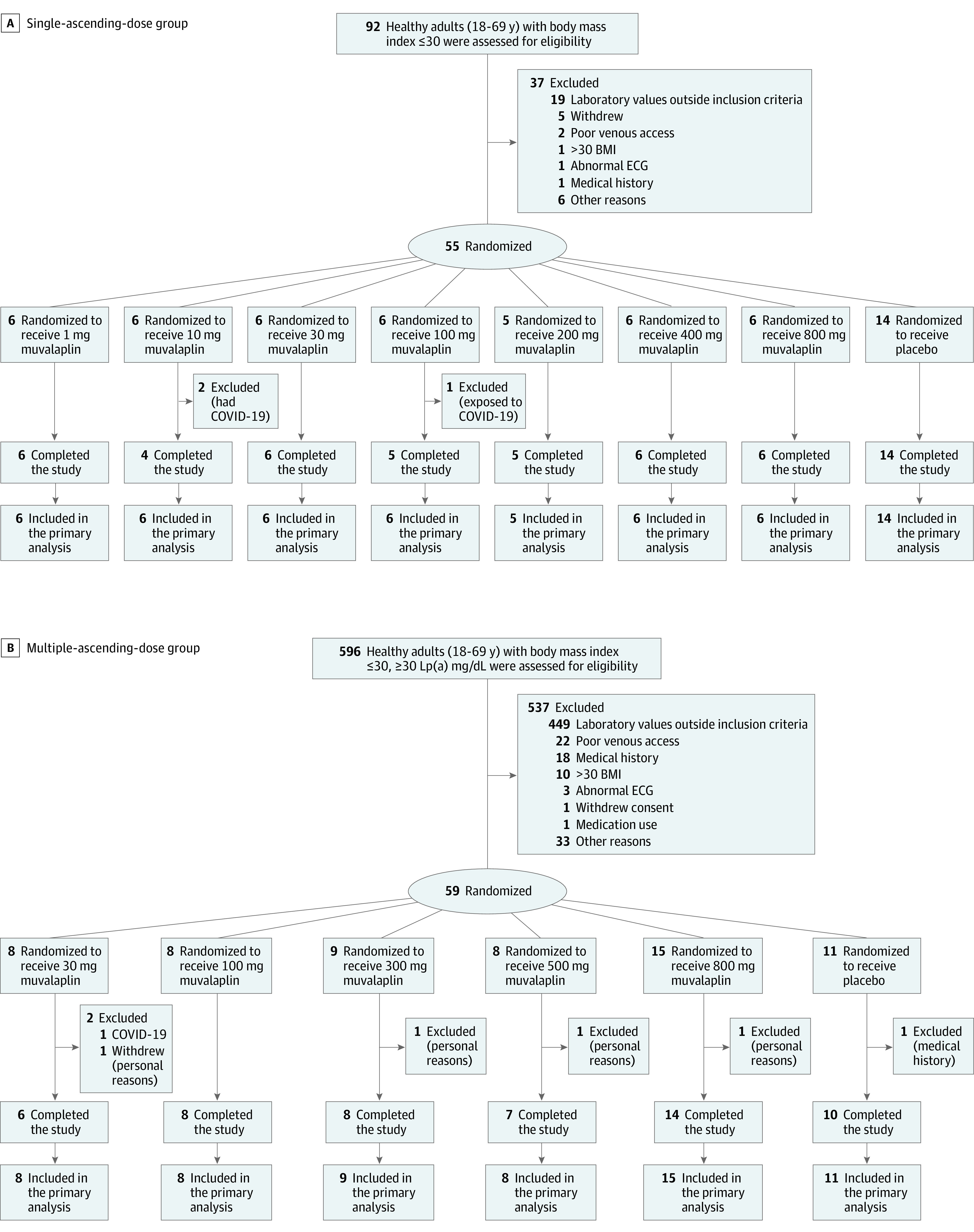
Within each study part, participants were randomly assigned to receive either muvalaplin or placebo in a 6:2 ratio for the single ascending dose part, and in a 8:2 ratio for the first 4 cohorts and 15:2 ratio in the last cohort of the multiple ascending dose study part. Two participants were given sentinel 1-mg doses in the single ascending dose part of the study prior to dosing the full cohort. The primary analysis assessed safety.
Abbreviations: BMI, body mass index, calculated as weight in kilograms divided by height in meters squared; ECG, electrocardiogram.
The study was sponsored and designed by Eli Lilly and Company. The study protocol was approved by an independent ethics committee and was carried out in accordance with the Declaration of Helsinki and in compliance with current regulations and standards of Good Clinical Practice. All participants provided written informed consent. The sponsor collected the data, and the statistical analyses were performed by ICON, a contract research organization. The data tables with all trial results were transferred to the sponsor and the Victorian Heart Institute, Monash University, for further analysis. The manuscript figures and tables were produced by academic and sponsor-employed authors. The first draft of the manuscript was written by the lead author and modified based on comments by other authors including those employed by the sponsor; however, the final decision on content was retained by the lead author (S.J.N.), who vouches for the accuracy and completeness of the data and for the fidelity of the trial to the protocol, available in Supplement 1. (The statistical analysis plan is available in Supplement 2.)
Study Population
Eligible participants were healthy adults aged 18 through 69 years, with a body mass index of 30 or less (calculated as weight in kilograms divided by height in meters squared). Participants randomized to the single ascending dose group were enrolled independently of their Lp(a) levels. Participants randomized to the multiple ascending dose group were required to have an Lp(a) concentration of 30 mg/dL or more. Female participants with childbearing potential and male participants agreed to use a reliable method of birth control throughout the study and for 105 days following the last dose of study drug. Race was determined by self-selection from closed categories, which we collect as standard practice.
Treatment Procedures
Participants in the single ascending dose group were randomly assigned to receive either muvalaplin or placebo in a 6:2 ratio. For the first cohort, 2 participants received sentinel dosing (1 each with muvalaplin and placebo), with subsequent administration to the remaining participants in each cohort following review by the sponsor of the safety data through the 24-hour period following dosing. The single ascending dose levels studied were 1 mg, 10 mg, 30 mg, 100 mg, 200 mg, 400 mg, and 800 mg (eFigure 1 in Supplement 3).
Participants in the multiple ascending dose group were randomly assigned to receive either muvalaplin or placebo in an 8:2 ratio in all but the last cohort, which used a 15:2 ratio. After an 8-hour fast, participants orally ingested either the study drug or placebo once a day for 14 days (eFigure 1 in Supplement 3). The planned dosage levels for the multiple ascending dose group were selected based on safety and pharmacokinetic data from the single ascending dose part of the study: 30 mg, 100 mg, 300 mg, 500 mg, and 800 mg daily. Participants, investigators, and clinical research unit staff who conducted trial-related activities or with the ability to influence study outcomes were blinded to muvalaplin and placebo treatment.
Outcome Measures
Safety assessments included monitoring adverse events, clinical laboratory evaluations, vital signs, 12-lead electrocardiogram, physical examination, and body weight. Muvalaplin plasma pharmacokinetics were estimated via noncompartmental analysis of plasma concentration-time data. Exploratory pharmacodynamic monitoring involved assessment of Lp(a), total cholesterol, LDL cholesterol, high-density lipoprotein cholesterol, triglyceride, and apo B100 levels. Quantitative measurement of Lp(a) in human serum was measured with the Atellica CH Analyzer (Siemens Healthcare Diagnostics), calibrated using Atellica CH Lp(a) CAL. The Lp(a) assay provides results from 10.00 mg/dL to 85.00 mg/dL. Because a high percentage of sequence identity is shared between apo(a) and plasminogen,11 additional pharmacodynamic biomarkers were monitored, including plasminogen concentration, plasminogen activity, high-sensitivity C-reactive protein, plasminogen activator inhibitor 1, tissue plasminogen activator antigen, and α2-antiplasmin.
Statistical Analysis
The sample size for the single ascending dose cohort was typical for first-in-human studies designed to address the safety, tolerability, and pharmacokinetic objectives with no formal power analyses. The sample size for the multiple ascending dose cohort was typical for a first-in-human multiple-dose study, except for the final cohort. The sample size for the final cohort was determined through simulations using FACTS version 6.4 software, to detect an Lp(a) decrease of at least 60% (placebo-adjusted) 90% of the time if the true decrease was 75% or more. Placebo-treated participants from each cohort were pooled into a single-treatment group within each part of the study. Pharmacokinetic and exploratory pharmacodynamic analyses were conducted using data from all participants who received at least 1 dose of muvalaplin drug and had evaluable data. In addition, exploratory pharmacodynamic analyses included participants who received at least 1 dose of placebo. Results are summarized by dosage level using descriptive statistics. Analyses for the primary objective of safety were conducted for all enrolled participants.
Results
Study Population
Screening of participants commenced in August 2020 and the final follow-up visit was conducted in November 2021. A total of 114 participants were enrolled in the study (55 in the single ascending dose group and 59 in the multiple ascending dose group). Of these participants, 89 were treated with muvalaplin and 25 were treated with placebo (Figure 2). A total of 105 participants completed the study (Figure 2). Baseline demographics and medical history were similar between participants treated with muvalaplin and placebo in each treatment group (Table 1). Participants in the single ascending dose group were a mean (SD) age of 29 (10) years, 35 females (64%), 2 American Indian or Alaska Native (4%), 50 White (91%), and 3 multiracial individuals (5%). Participants in the multiple ascending dose group were a mean (SD) age of 32 (15) years; 34 females (58%); 3 American Indian or Alaska Native (5%), 6 Black (10%), 47 White (80%), and 3 multiracial individuals (5%).
Table 1. Demographic Characteristics.
| Variable | Single ascending dose group | Multiple ascending dose group | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Muvalaplin | Placebo (n = 14) | Muvalaplin | Placebo (n = 11) | |||||||||||
| 1 mg (n = 6) | 10 mg (n = 6) | 30 mg (n = 6) | 100 mg (n = 6) | 200 mg (n = 5) | 400 mg (n = 6) | 800 mg (n = 6) | 30 mg (n = 8) | 100 mg (n = 8) | 300 mg (n = 9) | 500 mg (n = 8) | 800 mg (n = 15) | |||
| Characteristics | ||||||||||||||
| Age, median (IQR), y | 26.5 (25-35) | 35 (30-48) | 31.5 (29-44) | 27.5 (21-37) | 23 (23-24) | 21.5 (20-27) | 27 (24-34) | 23.5 (20-25) | 27.5 (25.5-32) | 28.5 (23-35) | 20 (20-26) | 22.5 (20.5-37.5) | 54 (23-64) | 27 (21-34) |
| Sex, No. (%) | ||||||||||||||
| Female | 2 (33) | 3 (50) | 2 (33) | 5 (83) | 4 (80) | 4 (67) | 6 (100) | 9 (64) | 4 (50) | 5 (63) | 8 (89) | 6 (75) | 4 (27) | 7 (64) |
| Male | 4 (67) | 3 (50) | 4 (67) | 1 (17) | 1 (20) | 2 (33) | 5 (36) | 4 (50) | 3 (38) | 1 (11) | 2 (25) | 11 (73) | 4 (36) | |
| BMI, median (IQR) | 22.3 (20.0-29.1) | 24.3 (22.6-28.2) | 25.6 (25.0-27.8) | 23.3 (21.5-23.9) | 23.3 (21.0-23.7) | 23.0 (21.3-23.2) | 24.2 (22.0-28.7) | 22.7 (21.3-24.4) | 25.9 (22.4-28.0) | 23.6 (21.6-28.1) | 24.5 (23.5-27.2) | 26.7 (25.1-27.2) | 24.7 (23.3-27.5) | 22.9 (20.0-26.7) |
| Race and ethnicity, No. (%) a | ||||||||||||||
| Ethnicity | ||||||||||||||
| Not Hispanic or Latino | 6 (100) | 6 (100) | 5 (83) | 6 (100) | 4 (80) | 6 (100) | 5 (83) | 14 (100) | 7 (88) | 7 (88) | 7 (78) | 7 (88) | 15 (100) | 11 (100) |
| Hispanic or Latino | 1 (17) | 1 (20) | 1 (17) | 1 (13) | 1 (13) | 2 (22) | 1 (13) | |||||||
| Race | ||||||||||||||
| American Indian or Alaska Native | 1 (20) | 1 (17) | 1 (13) | 1 (11) | 1 (13) | |||||||||
| Black | 1 (13) | 1 (13) | 2 (22) | 1 (7) | 1 (9) | |||||||||
| White | 6 (100) | 5 (83) | 6 (100) | 6 (100) | 4 (80) | 6 (100) | 4 (67) | 13 (93) | 6 (75) | 6 (75) | 5 (56) | 7 (88) | 13 (87) | 10 (91) |
| Multiracial | 1 (7) | 1 (17) | 1 (7) | 1 (13) | 1 (11) | 1 (7) | ||||||||
| Laboratory results, median (IQR), mg/dL b | ||||||||||||||
| Lp(a) | 15.9 (9.9-47.5) | 9.9 (9.9-9.9) | 36.5 (11.0-78.0) | 18.2 (15.1-24.5) | 9.9 (9.9-9.9) | 9.9 (9.9-47.5) | 10.5 (9.9-41.2) | 10.2 (9.9-19.6) | 56.7 (47.8-91.3) | 42.0 (36.6-48.8) | 64.4 (36.1-80.0) | 75.9 (53.0-81.1) | 52.1 (36.7-95.0) | 59.8 (38.4-70.5) |
| Total cholesterol | 164.4 (154.7-181.8) | 166.3 (143.1-170.2) | 177.9 (166.3-185.6) | 177.9 (139.2-185.6) | 139.2 (112.1-143.1) | 156.6 (135.4-181.8) | 166.3 (162.4-181.8) | 166.3 (143.1-212.7) | 162.4 (158.6-191.4) | 176.0 (160.5-203.0) | 170.2 (154.7-174.0) | 172.1 (154.7-197.2) | 189.5 (154.7-205.0) | 174.0 (166.3-189.5) |
| LDL cholesterol | 110.2 (104.4-116.0) | 98.6 (96.7-119.9) | 127.6 (108.3-135.4) | 104.4 (100.5-123.7) | 77.3 (54.1-81.2) | 87.0 (73.5-104.4) | 90.9 (85.1-108.3) | 114.1 (92.8-147.0) | 102.5 (96.7-125.7) | 119.9 (92.8-135.4) | 108.3 (92.8-112.1) | 119.9 (106.3-141.2) | 119.9 (92.8-131.5) | 119.9 (112.1-131.5) |
| HDL cholesterol | 56.1 (38.7-65.7) | 48.3 (42.5-61.9) | 54.1 (42.5-58.0) | 52.2 (50.3-58.0) | 50.3 (46.4-54.1) | 50.3 (50.3-73.5) | 58.0 (38.7-73.5) | 50.3 (50.3-58.0) | 52.2 (42.5-54.1) | 54.1 (46.4-61.9) | 58.0 (54.1-58.0) | 46.4 (40.6-52.2) | 54.1 (42.5-61.9) | 50.3 (46.4-58.0) |
| Triglycerides (IQR) | 95.7 (71.7-156.8) | 66.0 (56.7-117.8) | 105.9 (65.5-108.9) | 110.7 (71.7-132.0) | 62.9 (60.2-85.9) | 64.7 (53.1-124.9) | 77.5 (47.8-81.5) | 81.0 (71.7-96.5) | 62.0 (55.4-122.7) | 86.4 (66.4-115.1) | 60.2 (49.6-68.2) | 82.4 (74.9-94.8) | 83.3 (62.0-94.8) | 92.1 (70.0-130.2) |
| apo B100 | 70.3 (64.9-71.1) | 60.7 (60.3-73.0) | 81.3 (73.8-86.6) | 68.2 (66.6-80.8) | 51.8 (38.0-61.0) | 63.6 (51.7-70.0) | 62.9 (59.6-66.9) | 71.0 (59.7-92.9) | 70.4 (60.3-80.2) | 75.3 (63.4-86.6) | 68.6 (60.9-75.6) | 66.9 (63.4-80.1) | 71.8 (63.7-81.2) | 75.0 (69.7-81.1) |
Abbreviations: Apo B100, apolipoprotein B100; BMI, body mass index, calculated as weight in kilograms divided by height in meters squared; HDL, high-density lipoprotein; LDL, low-density lipoprotein; Lp(a), lipoprotein(a).
SI conversion factors: To convert HDL, LDL, and total cholesterol from mg/dL to mmol/L, multiply by 0.0259; triglyceride from mg/dL to mmol/L, multiply by 0.0113.
Race was determined by self-selection from closed categories.
Normal ranges for select analyses were Lp(a), 0 to 30 mg/dL; total cholesterol, 0 to 247 mg/dL; LDL cholesterol, 0 to 151 mg/dL; HDL cholesterol, 38.6 mg/dL; triglycerides, 0 to 238.1 mg/dL; apo B100 (male) 46 to 174 mg/dL and (female) 46 to 142 mg/dL.
Among participants in the single ascending dose group, the baseline median Lp(a) levels were 10.3 mg/dL (IQR, 9.9-41.2 mg/dL) and the LDL cholesterol levels, 104.4 mg/dL (IQR, 81.2-123.7 mg/dL), and among the multiple-ascending group, the median Lp(a) levels were 58.3 mg/dL (IQR, 38.4-79.8 mg/dL), and the LDL cholesterol levels, 116.0 mg/dL (IQR, 100.5-127.6 mg/dL; eFigures 4 and 5 in Supplement 3). (To convert LDL cholesterol from mg/dL to mmol/L, multiply by 0.0259.) Medication usage during the study included 3 participants who were administered ibuprofen (2 participants took it once, 1 took it as needed for 2 days) and 3 participants who were administered COVID-19 vaccines. No participant was administered statin therapy.
Pharmacokinetic Measurements
Pharmacokinetic measurements for both parts of the study are presented in Table 2. In the single ascending dose group, plasma muvalaplin levels were below the limit of quantitation (1.0 ng/mL) in the 1-mg dose cohort. Muvalaplin reached maximum observed plasma concentration between 2 and 5 hours after dosing, across the 10-mg to 800-mg dose range and exhibited a multiphasic decline thereafter (Figure 3, panel A). Elimination half-life ranged from 12.1 hours to 67.0 hours across the 30-mg to 800-mg dose range, increasing with dose. The maximum observed plasma concentration of muvalaplin and area under the concentration-time curve (AUC) from time 0 extrapolated to infinite time for muvalaplin increased with increasing single doses (Table 2) but was not dose proportional (eTable 1 in Supplement 3).
Table 2. Muvalaplin Pharmacokinetic Parameters by Dose Levela.
| Single ascending dose of muvalaplinb | Day | Multiple ascending dose of muvalaplin per dayb | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 mg (n = 2)c | 10 mg (n = 6)d | 30 mg (n = 6) | 100 mg (n = 6) | 200 mg (n = 5) | 400 mg (n = 6) | 800 mg (n = 6) | 30 mg (n = 7) | 100 mg (n = 8) | 300 mg (n = 8) | 500 mg (n = 8) | 800 mg (n = 14) | ||
| Maximum plasma concentration, ng/mL | 1.86 (6.9) | 3.72 (93.1) | 16.0 (49.9) | 45.5 (64.1) | 52.3 (57.3) | 64.0 (55.2) | 152 (36.9) | 1 | 23.0 (57.7) | 46.4 (23.5) | 117 (38.7) | 181 (47.7) | 212 (46.0) |
| 14 | 28.1 (53.3) | 51.0 (19.8) | 114 (22.8) | 169 (35.2) | 203 (47.2) | ||||||||
| Time to reach maximum plasma clearance, he | 5.0 (4.0-6.0) | 4.0 (2.0-6.0) | 4.0 (4.0-8.0) | 5.0 (2.0-6.0) | 4.0 (2.0-8.0) | 4.0 (4.0-6.0) | 2.0 (2.0-4.0) | 1 | 5.9 (4.0-6.0) | 6.0 (2.0-6.0) | 4.0 (2.0-6.0) | 4.0 (2.0-8.0) | 2.0 (1.0-6.0) |
| 14 | 4.0 (1.0-4.1) | 4.0 (2.0-8.0) | 4.0 (2.0-8.0) | 4.0 (1.0-12.0) | 2.0 (2.0-8.0) | ||||||||
| AUC0-last, ng × h/mLf | 15.9 (15.3) | 37.5 (219.5) | 270 (215.8) | 870 (71.4) | 869 (161.7) | 1615 (114.6) | 3661 (97.3) | ||||||
| AUC0-inf, ng × h/mLg | 311 (209.8) | 964 (63.8) | 997 (152.3) | 1696 (114.7) | 3802 (97.8) | ||||||||
| AUC0-24, ng × h/mLh | 1 | 350 (52.3) | 661 (29.2) | 1846 (24.8) | 2968 (29.5) | 3005 (49.1) | |||||||
| AUC0-tau, ng × h/mLi | 14 | 460 (57.4) | 698 (31.0) | 1848 (18.0) | 2819 (34.0) | 3194 (45.3) | |||||||
| Elimination half-life, h | 12.1 (154.5) | 31.7 (73.3) | 33.9 (127.3) | 26.9 (185.0) | 67.0 (167.9) | 14 | 70.9 (101.2) | 159 (74.0) | 315 (65.4) | 414 (33.5) | 364 (36.9) | ||
| Apparent oral clearance, L/h | 96.5 (209.8) | 104 (63.8) | 201 (152.3) | 236 (114.7) | 210 (97.8) | 14 | 65.2 (57.4) | 143 (31.0) | 162 (18.0) | 177 (34.0) | 250 (45.3) | ||
| Apparent volume of distribution after oral dosing, L | 1689 (29.6) | 4742 (60.0) | 9808 (19.2) | 9136 (68.3) | 20 347 (65.7) | 14 | 6673 (43.6) | 32 834 (48.8) | 73 888 (80.9) | 110 565 (48.7) | 131 533 (42.1) | ||
| Accumulation ratio AUCj | 14 | 1.41 (76.7) | 1.06 (28.1) | 0.96 (19.5) | 0.95 (39.5) | 1.06 (32.8) | |||||||
Data are shown as geometric mean (coefficient of variation %) unless otherwise indicated.
Pharmacokinetic end points were estimated based on the following pharmacokinetic sampling schedules: in the single ascending dose part of the study, plasma muvalaplin concentrations were measured at predose (0 hour) and at 1, 2, 4, 6, 8, 12, 24, 48, 72, 168, 336, 504, 1008, 1512, 2016, and 2520 hours postdose; in the multiple ascending dose part of the study, plasma muvalaplin concentrations were measured predose (day 1) and 1, 2, 4, 6, 8, 12, 24 hours postdose; on day 5, day 8, and day 11 only predose; and on day 14 predose; and 1, 2, 4, 6, 8, 12, 24, 96, 192, 360, 696, 1200, 1704, 2208, and 2952 hours postdose.
For the 1-mg dose level only maximum plasma concentration, time to reach maximum clearance, and area under the concentration-time curve (AUC0-last) are reported for 2 participants because there were insufficient values higher than the lower limit of quantitation to calculate all parameters.
For the 10-mg dose level, the maximum plasma concentration, time to reach maximum clearance, and AUC0-last were reported for all participants. AUC0-inf could only be calculated for 1 participant due to insufficient values higher than the lower limit of quantitation in the terminal phase to calculate a reliable elimination half-life. Percent extrapolated was 15% for this subject, but because n = 1, no summary statistics are reported.
Presented as median (minimum-maximum).
Time 0 to the time of the last measured concentration.
Time 0 extrapolated to infinity time.
Time 0 to 24 hours.
Time 0 to the end of the dosing interval.
Accumulation ratio calculated as AUC0-tau day 14 divided by AUC0-tau day 1.
Figure 3. Plasma Muvalaplin Concentration-Time Profile Following Single and Multiple Daily Doses of Muvalaplin.
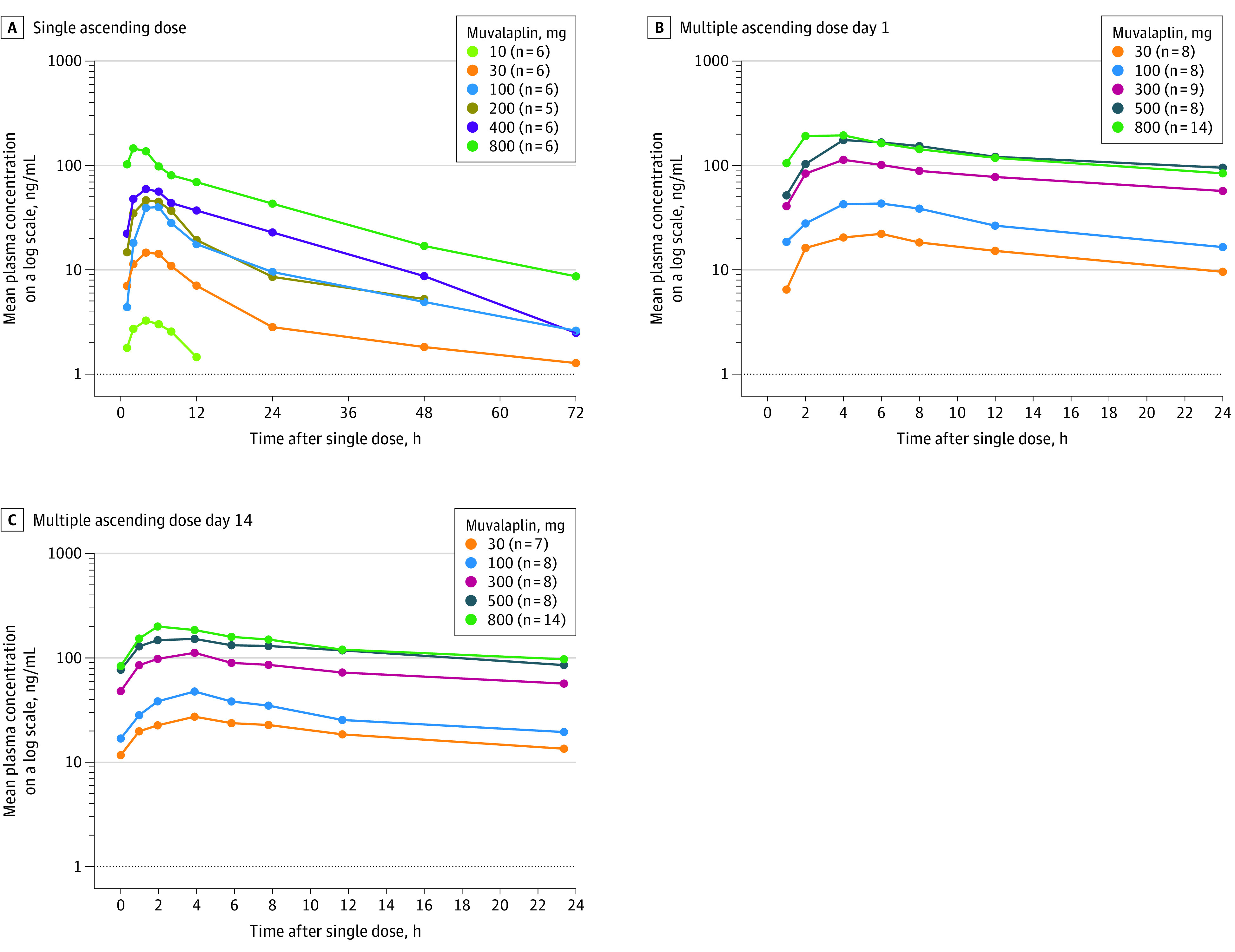
A, Plasma muvalaplin concentration-time profile after a single oral administration of muvalaplin in healthy participants.
B, Plasma muvalaplin concentration-time profiles 0 to 24 hours of participants with elevated lipoprotein(a) levels (≥30 mg/dL) after receiving the first dose on day 1.
C, Plasma muvalaplin concentration-time profiles 0 to 24 hours in participants with elevated lipoprotein(a) levels (≥30 mg/dL) after receiving the last dose on day 14.
In the multiple ascending dose group, the time of the maximum observed plasma concentration of muvalaplin occurred between 2 and 6 hours after dosing on day 1 and between 2 and 4 hours after dosing on day 14, across the 30-mg to 800-mg dose range, and exhibited a multiphasic decline thereafter (Table 2 and Figure 2, panels B and 2C). The elimination half-life of muvalaplin ranged from 70.9 to 414 hours and increased with doses from 30 mg to 500 mg. The muvalaplin maximum observed plasma concentration and AUC during a dosing interval also increased with dose after multiple dosing, and there was minimal accumulation between days 1 and 14. Analysis of increases in the maximum observed plasma concentration on days 1 and 14 and AUC during a dosing interval was not dose proportional (eTable 1 in Supplement 3).
Exploratory Pharmacodynamic Measurements
Effects of muvalaplin administration on Lp(a) levels and other biomarkers are illustrated in Figure 3 and eFigures 2 and 3 in Supplement 3. Reductions in Lp(a) levels from baseline were observed as early as day 2 with multiple dosing. The placebo-controlled reduction in Lp(a) was 63% to 65% at doses of 100 mg or more, occurring on days 14 and 15. Lp(a) levels returned to baseline by day 29 for the 30-mg dose, day 43 for the 100-mg dose, and day 64 for the 300-mg to 800-mg doses. Changes in levels of other parameters, including total cholesterol, LDL cholesterol, HDL cholesterol, triglyceride, and apo B100 levels, were not significant for any dose of muvalaplin compared with placebo (eFigure 2 in Supplement 3). Modest correlations were observed between changes in Lp(a) and both apo B100 and LDL cholesterol levels (eFigure 6 in Supplement 3).
Safety and Tolerability
Safety outcomes assessed during the studies are summarized in Table 3 and eTables 2 and 3 in Supplement 3. Most adverse events associated with treatment were mild in severity, transient, and resolved without sequelae. There was no dose dependency for the frequency of participants reporting adverse events. No deaths or serious adverse events were reported. Four participants discontinued the study due to COVID-19 infection. In the single ascending dose group, 34 participants (62%) reported a total of 71 adverse events, the most common being headache (33%), back pain (13%), and fatigue (11%). Seven adverse events (headache, fatigue, and vomiting) were considered by the investigator to be related to study drug for 5 participants, 4 of whom received the 800-mg dose. The other participant received placebo. In the multiple ascending dose group, 47 participants (80%) reported a total of 175 adverse events, the most common being headache (31%), diarrhea (20%), abdominal pain (15%), nausea (10%), and fatigue (10%). One adverse event (acne) was considered by the investigator to be related to study drug. No discernible prolongation of the corrected QT interval was noted with any dose of muvalaplin. No hematological or hepatic biochemical adverse events were observed.
Table 3. Treatment-Emergent Adverse Events.
| Single ascending dose part of the study (n = 55) | Multiple ascending dose part of the study(n = 59) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Muvalaplin, No. (%) | Placebo (n = 14) | Muvalaplin, No. (%) | Placebo (n = 11) | |||||||||||||
| 1 mg (n = 6) | 10 mg (n = 6) | 30 mg (n = 6) | 100 mg (n = 6) | 200 mg (n = 5) | 400 mg (n = 6) | 800 mg (n = 6) | 30 mg (n = 8) | 100 mg (n = 8) | 300 mg (n = 9) | 500 mg (n = 8) | 800 mg (n = 15) | |||||
| Adverse event | 3 (50) | 4 (67) | 2 (33) | 4 (67) | 4 (80) | 4 (67) | 6 (100) | 7 (50) | 7 (88) | 5 (63) | 7 (78) | 7 (88) | 12 (80) | 9 (82) | ||
| Mild | 3 (50) | 4 (67) | 2 (33) | 4 (67) | 4 (80) | 4 (67) | 6 (100) | 7 (50) | 7 (88) | 5 (63) | 7 (78) | 6 (75) | 11 (73) | 9 (82) | ||
| Moderate | 0 | 0 | 0 | 0 | 0 | 0 | 1 (17)a | 2 (14)b | 0 | 1 (13)c | 0 | 1 (13)d | 1 (7)e | 0 | ||
| Severe | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| Adverse event related to study drug | ||||||||||||||||
| Mild | 0 | 0 | 0 | 0 | 0 | 0 | 3 (50)f | 1 (7)g | 1 (13)h | 0 | 0 | 0 | 0 | 0 | ||
| Moderate | 0 | 0 | 0 | 0 | 0 | 0 | 1 (17)i | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| Severe | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| Adverse events reported in ≥3 (in the overall group), event/No. (%) of participants | ||||||||||||||||
| Diarrhea | 1/1 (13) | 3/2 (25) | 4/4 (44) | 1/1 (13) | 3/3 (20) | 1/1 (9) | ||||||||||
| Abdominal pain | 3/3 (38) | 0 | 4/4 (44) | 0 | 0 | 4/2 (18) | ||||||||||
| Nausea | 1/1 (13) | 1/1 (13) | 2/2 (22) | 0 | 0 | 2/2 (18) | ||||||||||
| Gastrointestinal sounds abnormal | 1/1 (11) | 1/1 (13) | 1/1 (7) | |||||||||||||
| Headache | 1/1 (17) | 2/2 (33) | 2/2 (33) | 2/2 (40) | 2/2 (33) | 4/4 (67) | 5/5 (36) | 5/4 (50) | 1/1 (13) | 7/4 (44) | 3/2 (25) | 4/3 (20) | 4/4 (36) | |||
| Dizziness | 1/1 (17) | 2/2 (14) | 1/1 (11) | 1/1 (13) | 1/1 (7) | |||||||||||
| Paraesthesia | 3/3 (38) | |||||||||||||||
| Fatigue | 1/1 (17) | 1/1 (20) | 1/1 (17) | 3/3 (21) | 4/3 (33) | 2/2 (25) | 1/1 (7) | |||||||||
| Catheter site pain | 3/2 (25) | 2/2 (25) | 1/1 (11) | |||||||||||||
| Vessel puncture site bruise | 1/1 (13) | 3/2 (22) | 2/2 (18) | |||||||||||||
| Back pain | 1/1 (17) | 2/2 (33) | 1/1 (17) | 3/3 (50) | 1/1 (3) | 2/2 (22) | 1/1 (9) | |||||||||
| Arthralgia | 1/1 (13) | 2/1 (11) | 1/1 (9) | |||||||||||||
| Acne | 1/1 (13) | 2/2 (25) | 1/1 (11) | 1/1 (9) | ||||||||||||
| Dry skin | 2/2 (25) | 1/1 (11) | ||||||||||||||
| Epistaxis | 1/1 (13) | 3/2 (22) | ||||||||||||||
| Dysmenorrhoea | 2/2 (22) | 1/1 (9) | ||||||||||||||
| Overall for any adverse event in each part, event/No. (%)j | ||||||||||||||||
| Total | 71/34 (62) | 175/47 (80) | ||||||||||||||
| Mild | 65/34 (62) | 172/45 (76) | ||||||||||||||
| Moderate | 6/3 (5) | 3/3 (5) | ||||||||||||||
| Severe | 0 | 0 | ||||||||||||||
Headache in 1 participant.
One episode of presyncope in a participant, and 1 shoulder dislocation in another participant.
Influenzalike illness in 1 participant.
External ear inflammation in 1 participant.
Wrist fracture in 1 participant.
Headache in 3 participants.
Headache and fatigue in 1 participant.
Acne in 1 participant.
Two episodes of vomiting in 1 participant.
Mild indicates asymptomatic or mild symptoms; moderate, minimal local or noninvasive intervention indicated; severe, medically significant but not life threatening.
Given the close homology between apo(a) and plasminogen,11 it is theoretically possible that lowering Lp(a) concentration may impact hemostatic parameters, and accordingly, additional analyses were performed to evaluate hemostasis-related biomarkers and plasminogen activity and levels in the multiple ascending dose study. Small reductions in plasminogen activity at the 2 highest doses (maximum reduction of approximately 14% with the 500-mg dose) were observed (Figure 4). These differences were most evident on day 14 and returned to baseline after dosing cessation. No dose or time-dependent changes were observed in plasminogen concentration, plasminogen activator inhibitor 1, tissue plasminogen activity antigen or α2-antiplasmin (eFigure 3 in Supplement 3). No significant changes were observed in high-sensitivity C-reactive protein levels at day 14 (eFigure 3 in Supplement 3).
Figure 4. Effect of Multiple Daily Doses of Muvalaplin on Lipoprotein(a) and Plasminogen Activity.
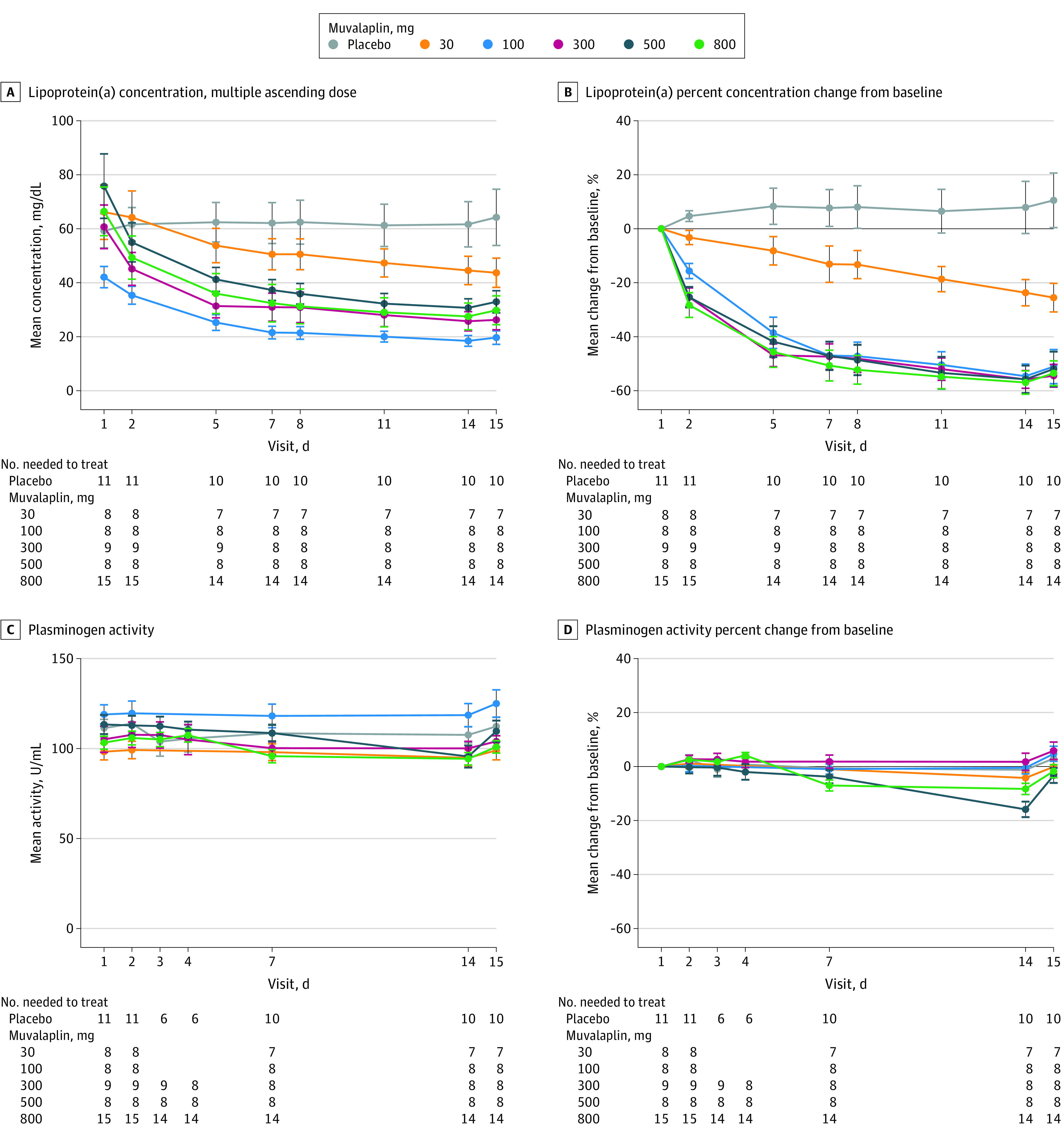
Dosing began on day 1, and the values shown from day 1 are from before dosing began.
A, The absolute change in lipoprotein(a) (Lp[a]) levels in participants with levels of 30 mg/dL or higher.
B, The mean percent change from baseline in Lp(a) levels over time.
C, The absolute change in plasminogen activity.
D, The mean percent change from baseline in plasminogen activity in the same participants and during the same time shown in panels A and B.
Data markers indicate the mean; error bars, SEM.
Discussion
Muvalaplin is a small molecule that inhibits formation of Lp(a) by interfering with the binding of apo(a) to apo B100. This first-in-human, placebo-controlled, phase 1 study demonstrated that administration of muvalaplin for up to 14 days was not associated with tolerability concerns or any serious adverse events. Daily administration of muvalaplin produced dose-dependent lowering of Lp(a) by up to 65% by day 14 of daily dosing. Reduced Lp(a) levels, compared with baseline, persisted up to 50 days after the last muvalaplin dose. In those participants with elevated Lp(a) levels, treatment with muvalaplin at the highest dose resulted in 93% of participants with Lp(a) trough levels of 50 mg/dL or less at the end of the 14-day treatment period. Muvalaplin had no significant effects on other lipids in healthy participants with elevated Lp(a) levels.
Increasing epidemiological and genetic evidence suggests that Lp(a) has a causal role in atherosclerotic cardiovascular disease and calcific aortic valve disease. Evaluation of 460 506 participants of the UK Biobank, with median follow-up of 11.2 years, demonstrated a linear relationship of Lp(a) concentrations with incident atherosclerotic cardiovascular disease, with a hazard ratio of 1.11 (95% CI, 1.10-1.12) per 50-nmol/L increment in Lp(a) concentration observed in the overall population.14 Estimation of Lp(a) lowering needed to reduce major adverse cardiovascular events from the Copenhagen General Population Study suggests that lowering Lp(a) by 50 mg/dL within a 5-year period would potentially reduce the risk of cardiovascular disease events by 20% for secondary prevention.15 This contrasts with estimates of potential need for greater Lp(a) lowering in analyses from the UK Biobank, which included a much broader range of individuals across the cardiovascular risk spectrum.5
Recent developments of therapeutics that inhibit hepatic production of apo(a) through nucleic acid therapeutics have demonstrated effective Lp(a) lowering and good tolerability.7,8,9 These agents are currently undergoing further evaluation in large clinical cardiovascular outcome trials, and they have the potential to transform the treatment of patients with high Lp(a) levels. However, the requirement for parenteral administration may present a potential barrier to widespread use. Muvalaplin is distinct, given both its once daily oral administration and mechanism of action that disrupts Lp(a) formation by blocking the initial noncovalent interaction between apo(a) and apo B100. Maximal dose-dependent reduction in Lp(a) concentration with muvalaplin was 65%, and the molecule was not associated with any serious adverse effects in this short-term study. The current study involved participants younger than those in the trials evaluating nucleic acid therapeutics, pelacarsen, and olpasiran.8,9 Although muvalaplin lowers Lp(a) levels to a lesser degree than parenteral therapies currently in development, the extent of reduction required for clinical benefit has not been established and remains controversial.16
Current treatment guidelines consider elevated Lp(a) levels as a risk-enhancing factor, supporting the need for intensification of lipid-lowering therapy.17 The only established treatment option, apheresis, is reserved for those patients with severe Lp(a) elevations and is cumbersome.18,19 Post hoc analyses of clinical trials of proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors demonstrated that moderate reduction of Lp(a) levels was associated independently with a reduction in cardiovascular events.20,21 The degree of Lp(a) lowering observed in these trials was modest (approximately 5 mg/dL, or 25%).
An additional area of importance involves the potential impact of muvalaplin on plasminogen activity. A high-percent sequence identity is shared between apo(a) and plasminogen, a factor involved in the fibrinolytic system.11 Preclinical studies in rats demonstrated that multivalent molecules, similar to muvalaplin, bind to rat plasminogen kringle IV domains in vitro and cause reductions in plasminogen activity in vivo. However, species comparison of plasminogen primary protein sequences revealed a greater number of targeted interaction motifs in rat plasminogen compared with human plasminogen, suggesting the possibility of a lesser impact of muvalaplin on human plasminogen. In this study, minimal effects on plasminogen were observed with muvalaplin at the 500-mg dose (maximal 14% reduction in activity), with no dose effect noted, suggesting that this agent will not adversely affect fibrinolysis. The clinical consequences of such lowering of plasminogen levels in patients with high Lp(a) levels or with cardiovascular disease are unknown.
In developing Lp(a)-lowering therapies, the absence of standardized assays of Lp(a) levels remains an ongoing challenge. Variable reporting of Lp(a) levels according to mass or molar quantity and no standardization of conversion factors between the 2 limits direct comparisons between research studies.22 The anti-apo(a) polyclonal antibody–based clinical immunoturbidometric assays to quantify Lp(a) levels have the potential to underestimate Lp(a) lowering during treatment with apo(a)-apo B100 disrupters because these methods also likely measure apo(a) bound to muvalaplin in the circulation. Accordingly, the degree of true Lp(a) lowering with muvalaplin may be greater than observed with currently used assays.
Limitations
Several limitations should be noted. First, this is a phase 1 study involving a small number of participants to establish an initial characterization of Lp(a) lowering and tolerability of muvalaplin during administration for 14 days. Establishing the safety profile of muvalaplin will require larger and longer clinical trials in more diverse populations, including patients with established cardiovascular disease. Second, the study included evaluation of the effect of muvalaplin in participants with both low and moderately elevated Lp(a) levels. However, this drug would likely be used in the clinical setting of participants with greater Lp(a) elevations. Third, the effect of muvalaplin on additional factors related to platelet activation in the setting of elevated Lp(a) levels has not been investigated. Fourth, it remains uncertain whether Lp(a) lowering with muvalaplin will reduce cardiovascular risk.
Conclusions
In summary, increasing evidence has implicated Lp(a) as a causal factor in both atherosclerosis and aortic stenosis. Accordingly, there is interest in developing effective approaches that will lower Lp(a) levels and reduce cardiovascular risk. Muvalaplin is the first oral agent specifically developed to lower levels of Lp(a) by disrupting its formation with once daily administration, introducing a different option for targeting Lp(a) in clinical development. These initial phase 1 clinical findings demonstrate that muvalaplin effectively lowers Lp(a) with no serious adverse effects.
Study Protocol
Statistical Analysis Plan
eTable 1. Summary Statistics of Muvalaplin Dose Proportionality Analysis
eTable 2. Treatment-Emergent Adverse Events by System Organ Class and Preferred Term (Single Ascending Dose Part)
eTable 3. Treatment-Emergent Adverse Events by System Organ Class and Preferred Term (Multiple Ascending Dose Part)
eFigure 1. Study Design
eFigure 2. Lipoprotein Panel Change vs. Time (Multiple Ascending Dose Part)
eFigure 3. Other Biomarkers Panel Change vs. Time (Multiple Ascending Dose Part)
eFigure 4. Frequency Distribution of Lp(a) Concentration at Baseline
eFigure 5. Lp(a) Concentration at Baseline on a Per-Patient Basis
eFigure 6. Scatterplot of Concentration Changes From Baseline
Data Sharing Statement
References
- 1.Kronenberg F, Mora S, Stroes ESG, et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: a European Atherosclerosis Society consensus statement. Eur Heart J. 2022;43(39):3925-3946. doi: 10.1093/eurheartj/ehac361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Erqou S, Kaptoge S, Perry PL, et al. ; Emerging Risk Factors Collaboration . Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302(4):412-423. doi: 10.1001/jama.2009.1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301(22):2331-2339. doi: 10.1001/jama.2009.801 [DOI] [PubMed] [Google Scholar]
- 4.Clarke R, Peden JF, Hopewell JC, et al. ; PROCARDIS Consortium . Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361(26):2518-2528. doi: 10.1056/NEJMoa0902604 [DOI] [PubMed] [Google Scholar]
- 5.Burgess S, Ference BA, Staley JR, et al. ; European Prospective Investigation Into Cancer and Nutrition–Cardiovascular Disease (EPIC-CVD) Consortium . Association of LPA variants with risk of coronary disease and the implications for lipoprotein(a)-lowering therapies: a MENDELIAN randomization analysis. JAMA Cardiol. 2018;3(7):619-627. doi: 10.1001/jamacardio.2018.1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanssen R, Gouni-Berthold I. Lipoprotein(a) management: pharmacological and apheretic treatment. Curr Med Chem. 2017;24(10):957-968. doi: 10.2174/0929867324666170112110928 [DOI] [PubMed] [Google Scholar]
- 7.Tsimikas S, Karwatowska-Prokopczuk E, Gouni-Berthold I, et al. ; AKCEA-APO(a)-LRx Study Investigators . Lipoprotein(a) reduction in persons with cardiovascular disease. N Engl J Med. 2020;382(3):244-255. doi: 10.1056/NEJMoa1905239 [DOI] [PubMed] [Google Scholar]
- 8.O’Donoghue ML, Rosenson RS, Gencer B, et al. ; OCEAN(a)-DOSE Trial Investigators . Small interfering RNA to reduce lipoprotein(a) in cardiovascular disease. N Engl J Med. 2022;387(20):1855-1864. doi: 10.1056/NEJMoa2211023 [DOI] [PubMed] [Google Scholar]
- 9.Nissen SE, Wolski K, Balog C, et al. Single ascending dose study of a short interfering RNA targeting lipoprotein(a) production in individuals with elevated plasma lipoprotein(a) levels. JAMA. 2022;327(17):1679-1687. doi: 10.1001/jama.2022.5050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berg K. A new serum type system in man—the Lp(a) system. Acta Pathol Microbiol Scand. 1963;59:369-382. doi: 10.1111/j.1699-0463.1963.tb01808.x [DOI] [PubMed] [Google Scholar]
- 11.McLean JW, Tomlinson JE, Kuang WJ, et al. cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature. 1987;330(6144):132-137. doi: 10.1038/330132a0 [DOI] [PubMed] [Google Scholar]
- 12.Ogorelkova M, Gruber A, Utermann G. Molecular basis of congenital Lp(a) deficiency: a frequent apo(a) ‘null’ mutation in Caucasians. Hum Mol Genet. 1999;8(11):2087-2096. doi: 10.1093/hmg/8.11.2087 [DOI] [PubMed] [Google Scholar]
- 13.Ogorelkova M, Kraft HG, Ehnholm C, Utermann G. Single nucleotide polymorphisms in exons of the apo(a) kringles IV types 6 to 10 domain affect Lp(a) plasma concentrations and have different patterns in Africans and Caucasians. Hum Mol Genet. 2001;10(8):815-824. doi: 10.1093/hmg/10.8.815 [DOI] [PubMed] [Google Scholar]
- 14.Patel AP, Wang M, Pirruccello JP, et al. Lp(a) (lipoprotein[a]) concentrations and incident atherosclerotic cardiovascular disease: new insights from a large national biobank. Arterioscler Thromb Vasc Biol. 2021;41(1):465-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Madsen CM, Kamstrup PR, Langsted A, Varbo A, Nordestgaard BG. Lipoprotein(a)-lowering by 50 mg/dL (105 nmol/L) may be needed to reduce cardiovascular disease 20% in secondary prevention: a population-based study. Arterioscler Thromb Vasc Biol. 2020;40(1):255-266. doi: 10.1161/ATVBAHA.119.312951 [DOI] [PubMed] [Google Scholar]
- 16.Kronenberg F. Therapeutic lowering of lipoprotein(a): how much is enough? Atherosclerosis. 2019;288:163-165. doi: 10.1016/j.atherosclerosis.2019.07.003 [DOI] [PubMed] [Google Scholar]
- 17.Mach F, Baigent C, Catapano AL, et al. ; ESC Scientific Document Group . 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41(1):111-188. doi: 10.1093/eurheartj/ehz455 [DOI] [PubMed] [Google Scholar]
- 18.Roeseler E, Julius U, Heigl F, et al. ; Pro(a)LiFe-Study Group . Lipoprotein apheresis for lipoprotein(a)-associated cardiovascular disease: prospective 5 years of follow-up and apolipoprotein(a) characterization. Arterioscler Thromb Vasc Biol. 2016;36(9):2019-2027. doi: 10.1161/ATVBAHA.116.307983 [DOI] [PubMed] [Google Scholar]
- 19.Kayikcioglu M. LDL Apheresis and Lp (a) apheresis: a clinician’s perspective. Curr Atheroscler Rep. 2021;23(4):15. doi: 10.1007/s11883-021-00911-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Donoghue ML, Fazio S, Giugliano RP, et al. Lipoprotein(a), PCSK9 inhibition, and cardiovascular risk. Circulation. 2019;139(12):1483-1492. doi: 10.1161/CIRCULATIONAHA.118.037184 [DOI] [PubMed] [Google Scholar]
- 21.Bittner VA, Szarek M, Aylward PE, et al. ; ODYSSEY OUTCOMES Committees and Investigators . Effect of alirocumab on lipoprotein(a) and cardiovascular risk after acute coronary syndrome. J Am Coll Cardiol. 2020;75(2):133-144. doi: 10.1016/j.jacc.2019.10.057 [DOI] [PubMed] [Google Scholar]
- 22.Reyes-Soffer G, Ginsberg HN, Berglund L, et al. ; American Heart Association Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular Radiology and Intervention; and Council on Peripheral Vascular Disease . Lipoprotein(a): a genetically determined, causal, and prevalent risk factor for atherosclerotic cardiovascular disease: a scientific statement from the American Heart Association. Arterioscler Thromb Vasc Biol. 2022;42(1):e48-e60. doi: 10.1161/ATV.0000000000000147 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Study Protocol
Statistical Analysis Plan
eTable 1. Summary Statistics of Muvalaplin Dose Proportionality Analysis
eTable 2. Treatment-Emergent Adverse Events by System Organ Class and Preferred Term (Single Ascending Dose Part)
eTable 3. Treatment-Emergent Adverse Events by System Organ Class and Preferred Term (Multiple Ascending Dose Part)
eFigure 1. Study Design
eFigure 2. Lipoprotein Panel Change vs. Time (Multiple Ascending Dose Part)
eFigure 3. Other Biomarkers Panel Change vs. Time (Multiple Ascending Dose Part)
eFigure 4. Frequency Distribution of Lp(a) Concentration at Baseline
eFigure 5. Lp(a) Concentration at Baseline on a Per-Patient Basis
eFigure 6. Scatterplot of Concentration Changes From Baseline
Data Sharing Statement