

Abstract
Purpose of review:
To provide an update on state-of-the-art evidence on the role of immunometabolism reprogramming in the pathogenesis of systemic lupus erythematosus (SLE).
Recent findings:
Mitochondrial dysfunction and enhanced oxidative stress, along with specific defects in other metabolic pathways, can promote dysregulation of innate and adaptive immune responses in SLE. These abnormalities appear to be driven by genetic and epigenetic factors, modulated by stochastic events. In addition to extensive descriptions of abnormalities in immunometabolism of lupus lymphocytes, recent studies support the critical role of dysregulation of metabolic pathways in innate immune cells including neutrophils, macrophages and dendritic cells, in SLE pathogenesis. Recent abnormalities described in lipid metabolism have been associated with SLE disease activity and related damage. Promising therapeutic strategies that target these metabolic abnormalities have recently been described in SLE.
Summary:
Fundamental new insights regarding the role of mitochondrial dysfunction in innate immune dysregulation in SLE pathogenesis have recently emerged. Defects in specific molecular pathways pertinent to immunometabolism in SLE have been described. New insights in translational medicine and promising therapeutic targets have been proposed based on these recent findings.
Keywords: Systemic lupus erythematosus, immunometabolism, mitochondrial dysfunction, oxidative stress, glycolysis
Introduction: Relevance of immunometabolism in the context of SLE.
Immunometabolism comprises the different metabolic pathways involved in immune cells’ effector functions in response to energy-demanding environmental challenges.(1) In many immune cells, energetic demands of resting cells rely on fatty acid oxidation (FAO) and oxidative phosphorylation (OXPHOS). Upon activation, increasing energetic requirements promote a metabolic switch to promptly generate ATP from other metabolic pathways including basic nutrients such as glucose and glutamine, in order to fulfill rapid cell proliferation and differentiation rates.(2, 3) SLE is a highly heterogenous autoimmune syndrome characterized by immune dysregulation that promotes the generation of autoantibodies directed to nuclear antigens, as well as an enhanced activation, differentiation and effector function of immune cells, which culminates in tissue damage. Metabolic reprogramming of adaptive and innate immune elements might contribute to the disturbed immune homeostasis in SLE.(3) Accordingly, the aforementioned pathways have been described to be dysregulated in SLE. In addition, mitochondrial dysfunction, oxidative stress and enhanced activity of the mechanistic target of rapamycin (mTOR) pathway have been reported to play pathogenic roles in SLE.(4)(Figure 1) The translational implications for these findings are substantial and may lead to novel therapeutic targets.(5)
Figure 1. Mitochondrial dysfunction and oxidative stress in SLE.
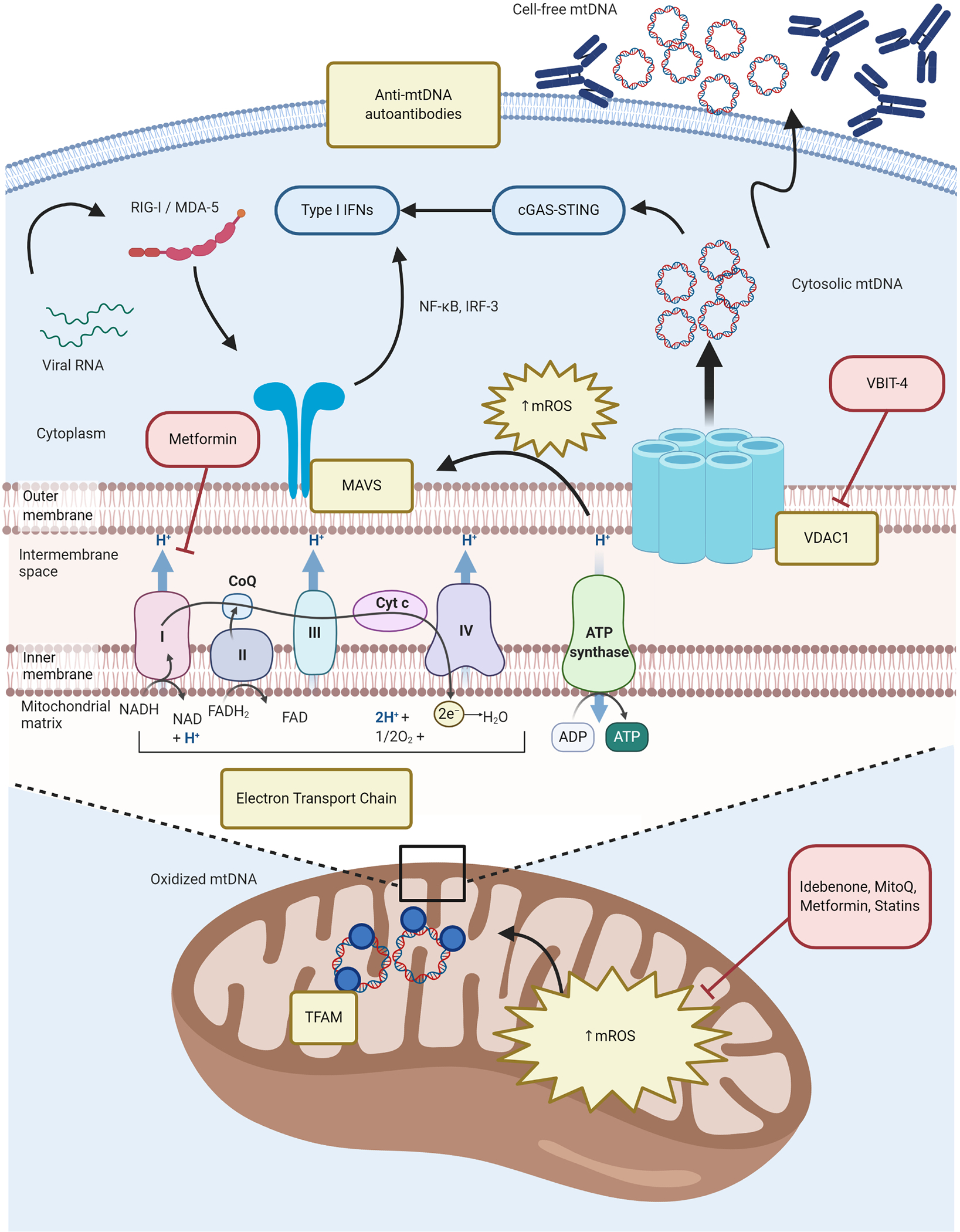
Chronically increased OXPHOS in immune cells promotes enhanced generation of mROS and enhanced type I IFN responses. Excessive mROS synthesis promotes MAVS oligomerization in the MOM that is essential for antiviral immune responses but dysregulated in SLE. Enhanced oxidative stress along with a putative defective disassociation of mtDNA from TFAM in the mitochondrial matrix, facilitates the oxidation of mtDNA. Increased VDAC oligomers in the MOM interact with oxidized mtDNA fragments and facilitating their release into the cytoplasm, where they activate the cGAS-STING pathway and upregulate ISG expression. Additionally, autoantibodies to free-cell mtDNA may contribute to lupus immune dysregulation. Perturbations in immunometabolism pathways described in SLE are depicted in yellow boxes; novel therapeutic strategies targeting some of these specific defects are depicted in red boxes. cGAS-STING = cyclic guanosine monophosphate-–adenosine monophosphate synthase (cGAS)-Stimulator of Interferon Genes (STING); CoQ = coenzyme Q10; Cyt c = cytochrome c; IFNs = interferons; IRF-3 = Interferon regulatory factor 3; MAVS = mitochondrial antiviral stimulator; MDA-5 = melanoma differentiation-associated protein 5; mROS = mitochondrial reactive oxygen species (ROS); mtDNA = mitochondrial DNA; NF-κB = nuclear factor kappa-light-chain-enhancer of activated B cells; OXPHOS = oxidative phosphorylation; RIG-I = retinoic acid-inducible gene I; SLE = systemic lupus erythematosus; VDAC1 = voltage-dependent anion channel 1.
Text of review:
Mitochondrial dysfunction and oxidative stress
❖. Mitochondrial DNA (mtDNA) oxidation and release and formation of anti-mtDNA autoantibodies.
The role of mitochondrial dysfunction in the pathogenesis of SLE has been extensively studied recently. Among the several abnormalities described in immune cells are mitochondrial membrane hyperpolarization, enhanced reactive oxygen species (ROS) production and defective ATP generation and, particularly in T cells, decreased intracellular glutathione (GSH) levels, which promotes their inappropriate activation and aberrant immune responses along with enhanced apoptosis and defective clearance of apoptotic bodies.(6, 7) In neutrophils, enhanced synthesis of mitochondrial ROS (mROS), driven by immune complexes and observed in lupus low density granulocytes (LDGs), promotes mtDNA oxidation and neutrophil extracellular trap (NET) formation.(8) The externalization of oxidized mtDNA in NETs promotes interferogenic responses in target cells through the cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS)-Stimulator of Interferon Genes (STING) pathway and the development of lupus-like disease.(8) In a recent study, Kim et al. described that aberrant oligomerization of the voltage dependent anion channel (VDAC) in the mitochondrial outer membrane (MOM) during oxidative stress, results in its interaction with short mtDNA fragments and the release of this DNA into the cytoplasm.(9) There, mtDNA cytoplasmic release also activates the cGAS-STING pathway, promoting intracellular upregulation of type I interferon (IFN)-stimulated genes (ISGs).(8, 9) Increased levels of VDAC1 oligomers in lupus murine splenocytes and human SLE mononuclear cells have been documented. Furthermore, pharmacological inhibition of VDAC oligomerization using a small molecule promoted the attenuation of clinical and immunologic features of murine lupus, decreased levels of circulating mtDNA and NET formation.(9) These recent studies suggest that the generation of oxidized genomic and mitochondrial DNA fragments can promote proinflammatory responses with deleterious effects on immune regulation.
Oxidized mtDNA has been reported to be retained within the mitochondria in SLE, due to a defective disassociation from transcription factor A mitochondria (TFAM), which in normal conditions is a necessary step prior to the lysosomal degradation of oxidized mtDNA.(10) In addition, Becker et al. recently reported increased levels of autoantibodies to MOM and mtDNA in both murine and human SLE, with anti-mtDNA antibodies being associated with lupus nephritis (LN) and elevated anti-dsDNA antibodies.(11) Pisetsky et al. found that anti-MOM antibodies were associated with lupus disease activity and specific clinical and laboratory features such as arthritis, renal disease, anti-dsDNA levels and complement consumption.(12) While these findings need to be confirmed in larger prospective cohorts, these studies support a putative pathogenic role for autoantibodies and other responses to mitochondrial components in SLE.
❖. Oxidative stress in SLE.
Supporting the role of mROS in promoting the type I IFN signature in SLE, Fortner et al. documented increased oxidative and glycolytic metabolism in CD4−CD8− TCR-αβ+ (B220+) T cells from lupus-prone MRL-lpr mice, along with enlarged and rounded-shape mitochondria, which favored spontaneous oligomerization of the mitochondrial antiviral stimulator (MAVS) protein and in parallel with the upregulation of ISGs expression in that T cell subset.(13) In addition, enhanced type I IFN signature in SLE CD4+ T cells has been recently described to depend on mitochondrial respiration, since in vitro treatment with metformin inhibited electron transport chain (ETC) complex I and thus reduced ISGs expression in healthy control and SLE CD4+ T cells upon exposure to IFN-α.(14)
mTOR signaling pathway abnormalities
❖. Aberrant activation of the mTOR pathway in SLE.
Multifactorial enhancement of the phosphoinositide-3 kinase (PI3K)/AKT/mTOR pathway in SLE has been extensively studied in murine models and humans and described to play pathogenic roles in immune dysregulation in this disease. Katsuyama et al. recently studied the role of serine/arginine-rich splicing factor 1 (SRSF1), an RNA-binding protein, whose expression is decreased in lupus T cells and that in normal conditions suppresses mTOR complex 1 (mTORC1) activity by promoting the expression of its negative regulator phosphatase and tensin homolog (PTEN).(15) Low SRSF1 levels associated with disease activity in SLE subjects, and correlated with lymphopenia and levels of the anti-apoptotic protein Bcl-xL in their T cells.(16) Additionally, some genetic and epigenetic factors have been found to contribute to the increased mTORC1 activity in SLE.(17–20)
❖. Functional consequences of enhanced mTOR pathway.
Enhanced mTOR signaling pathway in SLE has been described to play pathogenic roles in SLE through various mechanisms including the promotion of B and T cell differentiation, the latter associated with shifts toward Th1, Th17 and Th22 differentiation, while hindering Treg differentiation and autophagy..(21, 22) Zheng et al. recently reported that mTOR pathway activation favors enhancer of zeste homolog 2 (EZH2) upregulation in lupus T cells, which promotes proinflammatory epigenetic changes, by increasing glycolysis and then suppressing the microRNAs miR-26a and miR-101.(23) The role of the mTOR pathway in SLE pathogenesis goes beyond T cells, as recently reported by Murayama et al., who found that the mTOR inhibitor rapamycin suppressed 2’3’-cGAMP-induced IFN-α production in lupus monocytes, conventional dendritic cells (cDCs) and plasmacytoid DCs (pDCs), by downregulating the cGAS-STING pathway.(24) Moreover, the mTOR pathway has been described to be important in promoting the expansion and function of myeloid cells induced by TLR7 agonists and type I IFNs in two murine lupus models.(25) However, the precise mechanisms involved in this induction remain to be elucidated.
Glucose metabolism abnormalities
Various glucose pathways defects have been implicated in SLE.(3) Aerobic glycolysis is characteristically increased in activated SLE B and T cells, including CD4−CD8− TCR-αβ+ T cells, which parallels rapid proliferation followed by enhancement of spontaneous cell death. In this regard, Secinaro et al. recently described the role of methylation-controlled J protein (MCJ), another negative regulator of ETC complex I, which favors caspase-3 activity in coordination with glycolysis and, therefore, prevents characteristic OXPHOS-driven T cell survival. MCJ expression was found to be enhanced in the highly proliferative and glycolytic CD4−CD8− TCR-αβ+ T cells in vivo from MRL-lpr mice, promoting upregulated caspase-3 activation and increased rates of cell death that might contribute to inflammatory responses.(26) Enhanced glycolysis seemed to be required for the ability of naive CD4+ T cells to differentiate into Th17 cells in the same mouse model, and this was partially dependent of glutamine metabolism, as will be further discussed below.(27) Furthermore, a 10-fold increased surface expression of the glucose receptor GLUT1 and increased transcription of high glycolysis-related genes were documented in CD4+ T cells from active SLE patients during Th17 polarizing conditions.(28) This phenomenon was facilitated by calcium/calmodulin-dependent protein kinase 4 (CaMK4), which binds to pyruvate kinase M2 that is the final rate-limiting enzyme in glycolysis, to promote its activity.(29, 30) Additionally, pyruvate dehydrogenase (PDH) enzymatic activity is inhibited in lupus Th17 cells to promote conversion of pyruvate to lactate rather than pyruvate to acetyl coenzyme A (acetyl-CoA) that enters the tricarboxylic acid (TCA) cycle. This was in turn modulated by overexpressed cAMP response element modulator/inducible cAMP early repressor (CREM/ICER) through the downregulation of PDH phosphatase expression.(31) Of note, CaMK4 expression in CD4+ T cells was involved in mTOR pathway upregulation and correlated with SLE disease activity.(22, 30) A recent report on GLUT1-deficient macrophages supports the crucial role of this receptor in the metabolism of macrophages and in the development of proinflammatory subsets and oxidative stress.(32) Thus, GLUT1-mediated glycolysis may be an important player in the adaptive and innate immune abnormalities seen in the context of autoimmunity.
Abnormalities in the metabolism of amino acids
Amino acids provide another source of energy for immune cell effector function, and dysregulation of their metabolic pathways has been associated with T cell activation disturbances. Kono et al. recently highlighted the critical role of glutaminolysis, a stepwise process in which the most abundant amino acid in circulation, glutamine, is converted to α-ketoglutarate, in modulation of Th17 differentiation in a lupus model. Specifically, pharmacologic and genetic silencing of glutaminolysis first enzyme glutaminase 1 (Gls1), which converts glutamine to glutamate, in naive MRL-lpr CD4+ T cells cultured under Th17-polarization conditions, showed decreased Th17 differentiation rates and glycolysis. This effect was mediated, at least in part, by reducing Hypoxia-inducible factor 1α (HIF-1α) protein levels.(27)
Lipids metabolism abnormalities
While abnormalities in lipid metabolism have been less explored in the pathogenesis of SLE, some recent findings suggest a relevant role due to their high association with oxidative stress, given that they are main components of cellular membranes. An increased generation of oxidized lipids and their products in SLE patients has been well described and suggested as putative metabolic biomarkers in this disease and its associated vascular damage.(33) The proatherogenic “lupus lipid profile” is present in many SLE subjects and consists of increased low-density-lipoproteins (LDL), very-LDL (VLDL) and triglycerides, along with reduced levels of high-density-lipoproteins (HDL).(34, 35) Furthermore, lipidomic analyses have unveiled increased serum levels of oxidized LDL (ox-LDL) in SLE along with higher levels of autoantibodies and immune complexes to ox-LDL in those active subjects.(36) Additionally, several proteomic and lipidomic changes in SLE HDL promotes the loss of their cholesterol efflux capacity and anti-inflammatory and anti-oxidative properties.(34) Abnormalities in lipid metabolism could also contribute to T cell dysfunction in SLE, including abnormal glycosphingolipid metabolism that can promote T cell signaling defects and has been suggested as a potential therapeutic target in a lupus prone model.(33, 37) Current evidence suggests that intake of fiber-derived short-chain and omega-3 polyunsaturated fatty acids (FA) could attenuate lupus disease activity by dampening B cell differentiation and class-switched autoantibodies, ISGs and chemokine-related gene expression.(38, 39)
Immunometabolism: From the bench to the bedside.
There have been several recent reports that translate the metabolic abnormalities observed in preclinical models. For instance, Carlucci et al. reported that increased levels of lupus LDGs are associated with impaired HDL cholesterol efflux.(40, 41) This is considered to be driven, at least in part, by the ability of LDGs to synthesize higher amounts of NETs which in turn oxidize HDL through myeloperoxidase and other enzymes contained in these lattices.(42) Purmalek et al. recently reported that non-calcified coronary plaque burden (NCB) in SLE subjects was negatively associated with HDL and positively associated with VLDL, as measured by nuclear MR (NMR) spectroscopy. Circulating glycoprotein acetylation (GlycA), a novel inflammatory marker that has been related with cardiovascular events in several conditions, was indeed positively associated with NCB and insulin resistance. Moreover, dysregulation of lipoprotein fractions in these patients was significantly associated with SLE disease activity and specific autoantibody profiles.(35) These findings reinforce that pro-inflammatory and proatherogenic HDL particles in SLE promote vascular damage, at least in part, by dampening cholesterol efflux capacity.
Novel therapeutic strategies regarding immunometabolism in SLE
Since mROS and oxidative stress are determinant in the formation of oxidized mtDNA-enriched NETs and in the enhanced type I IFN signature in SLE patients, some therapeutic strategies targeting their pathogenic role have been recently emerged.(Table 1) For instance, Blanco et al. demonstrated that idebenone, a synthetic coenzyme Q10 analog with potent antioxidant effects and previously reported to be effective in other conditions associated with mitochondrial dysfunction, ameliorated lupus disease activity, organ damage and immune dysregulation in two mouse models of lupus. This included significant improvements in overall survival, kidney function, vasculopathy and inflammatory gene expression, at least in part, due to the modulation of mitochondrial metabolism and mROS-driven NET formation.(43) Similarly, administration of MitoQ, another mitochondria-targeted antioxidant containing ubiquinol attached to a lipophilic cation that facilitates membrane permeabilization and mitochondrial matrix access, led to improvements in renal disease, neutrophil mitochondrial metabolism and NET formation in MRL-lpr mice. Moreover, CD4−CD8− TCR-αβ+ T cells from MitoQ-treated mice revealed decreased levels of MAVS oligomerization, along with reduced serum IFN-α.(13) In contrast with the model that used idebenone, the role of MitoQ in other organ manifestations and modulation of immune cell subsets is less clear and requires further study.(13, 43) The therapeutic potential of metformin should be further studied but data reported so far supports that targeting mitochondrial respiration could be beneficial in SLE as another add-on therapy, along with its known positive effects on lipid and glucose metabolism, oxidative stress and immune regulation.(14, 44, 45) Cornaby et al. recently reported the therapeutic efficacy of the combination of metformin with Cytotoxic T-Lymphocyte Antigen 4 (CTLA-4)-Ig (abatacept) on preventing the development of lupus nephritis in a murine model.(46) Promising treatments targeting mitochondrial dysfunction in SLE, such as the above mentioned VDAC oligomerization small molecule inhibitor, have recently emerged.(9, 47)
Table 1.
Novel therapeutic strategies that target immunometabolism in SLE.
| Biological process targeted | Targeted molecule | Drug | Model | Proposed mechanism in SLE | Reference |
|---|---|---|---|---|---|
| Mitochondrial dysfunction and oxidative stress | mROS | Idebenone | MRL-lpr, NZM2328 | • Mitochondrial metabolism and mROS-NETosis modulation. | Blanco et al.(43) |
| MitoQ | MRL-lpr | • Mitochondrial metabolism and mROS-NETosis modulation. • Reduced MAVS oligomerization and IFN-α. |
Fortner et al.(13) | ||
| HRES-1/Rab4 and Rab5 | 3-PEHPC | MRL-lpr | • Downregulation of IL-10. • Reversal of Drp1 protein depletion; reduction of mitochondrial mass in T and B cells and macrophages. |
Caza et al.(47) | |
| VDAC1 | VBIT-4 | MpJ-Faslpr | • Decreased cell-free mtDNA and ISG expression. • mROS-NETosis modulation. |
Kim et al.(34) | |
| mTOR pathway | mTORC1 | Rapamycin (sirolimus) | MRL-lpr, NZB/W F1, Srsf1-cKO mice, human SLE | • Downregulation of proinflammatory cytokines. • Decreased Tfh cell infiltration. • Restoring Th17/Treg balance and increasing CD8+ memory T cells. • Targeting cGAS-STING pathway and ISG expression in myeloid cells. |
Katsuyama et al.(15) Zhang et al.(51) Eriksson et al.(50) Zhao et al.(52) Yap et al.(53) Murayama et al.(24) |
| GSH | NAC | NZB × NZW F1, human SLE | • Increased GSH levels in PBLs and reversed mTOR activity. • Enhanced mitochondrial mass and hyperpolarization, and apoptosis of CD4−CD8−TCR-αβ+ T cells. • Increased Treg cells count. |
Lai et al.(54) | |
| mTORC1/2 | INK-128 | MRL/lpr, Pristane-induced lupus mice | • Impairing CD11b+Gr1+ cell differentiation. | Shi et al.(25) | |
| AMPK, ETC | Metformin | B6.Sle1.Sle2.Sle3, NZB/W mice, Roquinsan/san mice, human SLE | • Synergistic effect with 2-DG to reduce glycolysis and OXPHOS, and IFN-γ production. • Restored IL-2 production and decreased ISGs expression in CD4+ T cells. • Reduced GC B cells and autoantibodies production. • Restoring Th17/Treg cells balance. |
Yin et al.(44) Titov et al.(14) Lee et al.(45) | |
| Glycolysis | CAMK4 | KN-93 | MRL-lpr, human SLE | • Restoring Th17/Treg cells balance. • Decreased glycolysis-related proteins expression in T cells under Th17-polarizing conditions. • mTOR pathway downregulation. |
Koga et al.(30, 55) |
| Glucose transporters | CG-5 | B6.NZM2410.Sle1.Sle2.Sle3, human SLE | • Decreased glycolysis-related proteins expression and increased FAO and PPP in activated CD4+ T cells. • Restoring Th17/Treg cells balance. • Reduced GC B cells and autoantibodies production. |
Li et al.(56) | |
| Glutaminolysis | Gs1 | BPTES | MRL-lpr, human SLE | • Decreased CD4−CD8−TCR-αβ+ T cells counts and Th17 differentiation. • Decreased glycolysis favored by reduced HIF-α levels. |
Kono et al.(27) |
| Lipids metabolism | HMG-CoA reductase | Statins | gld.apoE−/− mice, NZB/W F1, human SLE | • Downregulation of proinflammatory cytokines, TLR2 and MHC-II expression. • Cholesterol and lipid metabolism, oxidative stress and mitochondrial activity modulation in monocytes. |
Tan et al.(57) Ruiz-Limón et al.(58) |
| GlcCer synthase | Genz-667161 | MRL-lpr | • Reduction of GSL hexosylceramide and ganglioside GM3 levels. | Nowling et al.(37) |
2-DG = 2-deoxyglucose; AMPK = AMP-activated protein kinase; CaMK4 = calcium/calmodulin-dependent protein kinase 4; cGAS-STING = cyclic guanosine monophosphate-–adenosine monophosphate synthase (cGAS)-Stimulator of Interferon Genes (STING); Drp1 = dynamin-related protein 1; ETC = electron transport chain; FAO = fatty acid oxidation; GC = germinal center; GlcCer synthase = UDP-glucose:ceramide glucosyltransferase; Gls1 = glutaminase 1; GSH = glutathione; GSL = glycosphingolipids; HIF-1α = hypoxia-inducible factor 1α; HMG-CoA = 3-hydroxy-3-methyl-glutaryl-coenzyme A; ISGs = type I interferon (IFN)-stimulated genes; MAVS = mitochondrial antiviral stimulator; MHC-II = major histocompatibility complex class-II; mROS = mitochondrial reactive oxygen species (ROS); mtDNA = mitochondrial DNA; mTOR = mammalian target of rapamycin; mTORC1 = mTOR complex 1; mTORC1/2 = mTOR complex 1 and 2; NAC = N-acetylcysteine; OXPHOS = oxidative phosphorylation; PBLs = peripheral blood lymphocytes; PPP = pentose phosphate pathway; SLE = systemic lupus erythematosus; SRSF1 = serine/arginine-rich splicing factor 1; VDAC1 = voltage-dependent anion channel 1.
Novel therapeutic strategies for SLE targeting mTOR and glycolysis pathways have been proposed.(30, 48, 49) For instance, recent reports have reinforced beneficial effects of rapamycin or sirolimus for renal and non-renal manifestations of SLE.(50–53) Similarly, the beneficial effect of the GSH precursor N-acetylcysteine (NAC) on lupus disease activity by inhibiting mTOR in T cells was demonstrated in a clinical trial.(54) Additionally, the mTOR inhibitor INK128 has reported promising effects on lupus development by targeting inflammation-induced myeloid cells and by impairing their differentiation into macrophages in glomerulonephritis, even more effectively than rapamycin.(30) CaMK4 inhibition favored decreased expression of glycolysis-related proteins during T cell activation as well as Th17 differentiation in MRL-lpr mice and active SLE subjects.(30, 55) In the same way and similar to other autoimmune conditions(28), direct inhibition of glucose transporters has also demonstrated to attenuate lupus features in two murine models by reducing glycolysis in activated T cells, which is compensated by an increased FAO and pentose phosphate pathway (PPP). Furthermore, in vitro experiments have revealed decreases in T cell expansion and Th1Th17 polarization and enhanced Treg differentiation.(56) Nevertheless, the precise molecular mechanisms involved in this phenomenon and the putative clinical impact of this T cell modulation remains to be determined, as there are some discrepancies between the in vivo and in vitro findings. As previously mentioned, glutaminolysis is another potential therapeutic target in SLE, given that Gls1 inhibition showed beneficial effects in vitro and in vivo.(27) Finally, along with their favorable impact on primary and secondary prevention of cardiovascular events, the lipid-lowering drugs statins can attenuate SLE disease activity through different immunomodulatory effects, such as downregulation of proinflammatory cytokines, TLR2 signaling and major histocompatibility complex class-II (MHC-II) expression.(57, 58) Taking this together, targeting metabolic reprogramming in SLE might complement immunosuppressive standard therapies in the future, although more evidence is needed to clarify the efficacy and safety of these new therapeutic strategies in the context of an extraordinarily complex syndrome such as SLE.
Conclusions
Increasing evidence supports an important role for disturbances in immunometabolic pathways in the pathogenesis of SLE. These disturbances involve numerous immune cell subsets of the innate and adaptive arms. As more is learned about how various molecular metabolic pathways impact immune function, more promising therapeutic targeting strategies may emerge that could positively impact SLE by complementing current therapies and limiting the adverse effects of immunosuppressive drugs.
Figure 2. Glycolysis, glutaminolysis and mTOR pathway defects in SLE.
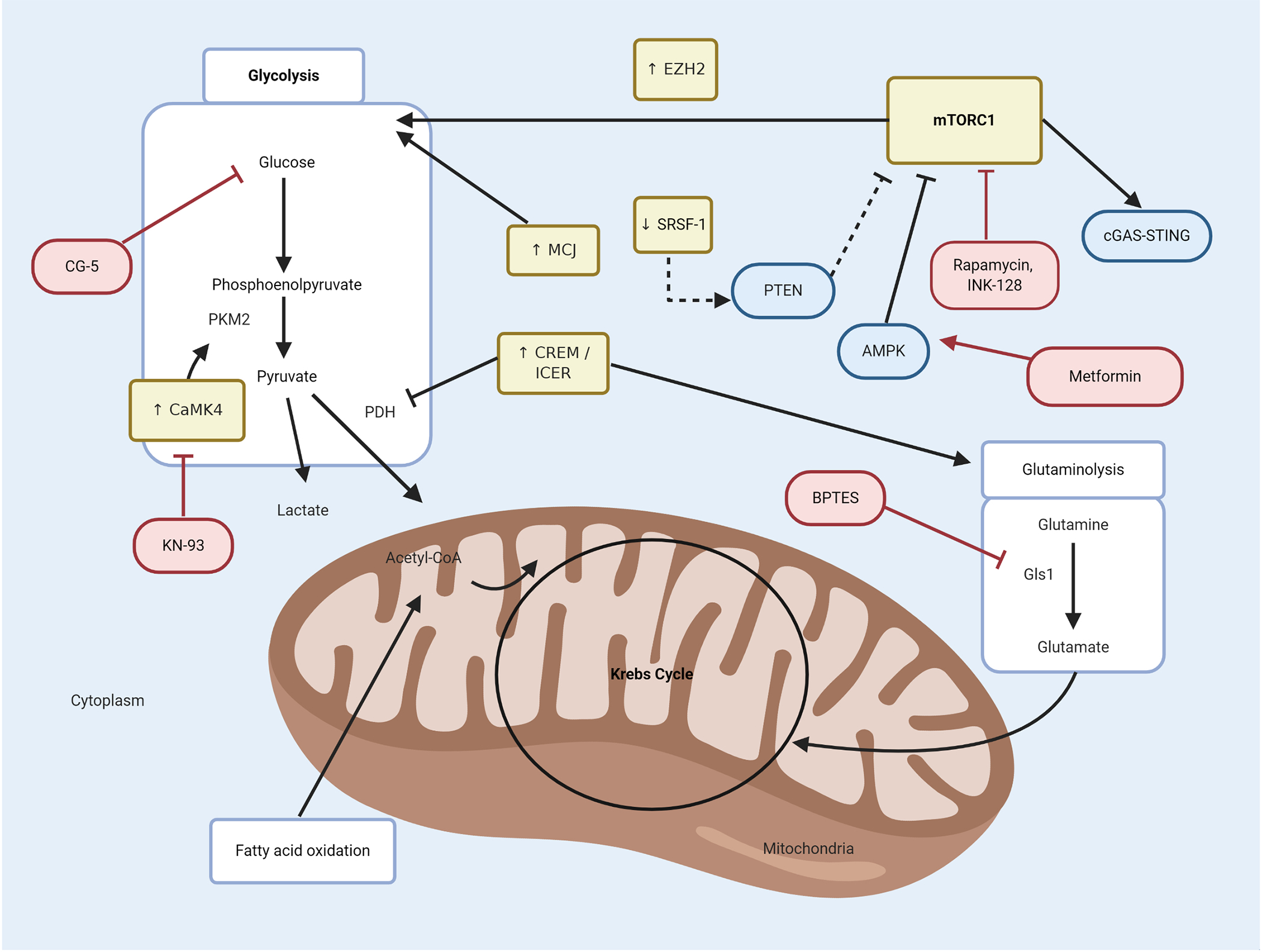
Specific defects in immunometabolism pathways in SLE, such as mTOR, glycolysis and glutaminolysis, interact in an orchestrated manner to promote dysregulation of immune cells. GLUT1 and high glycolysis-related genes are enhanced in lupus. In addition, upregulation of mTOR pathway, increased levels of MCJ protein and enhanced CaMK4 (which binds to PKM2) perpetuate a highly glycolytic state in lymphocytes. The mTOR pathway is triggered by low levels of SRSF-1 protein which, in normal conditions promotes the expression of the negative regulator of mTORC1, PTEN. Treatment with metformin inhibits mTOR pathway and attenuates lupus-like disease by enhancing AMPK. In lupus Th17 cells, overexpressed CREM/ICER inhibits PDH enzymatic activity, which favors the shift from pyruvate to lactate instead of pyruvate to acetyl-CoA. Furthermore, CREM/ICER activates the glutaminolysis pathway, with a putative role in SLE, as the inhibition of the Gs1 enzyme attenuates lupus-like disease. Perturbations in immunometabolism pathways described in SLE are depicted in yellow boxes; novel therapeutic strategies targeting some of these specific defects are depicted in red boxes. Acetyl-CoA = acetyl coenzyme A; AMPK = AMP-activated protein kinase; CaMK4 = calcium/calmodulin-dependent protein kinase 4; cGAS-STING = cyclic guanosine monophosphate-–adenosine monophosphate synthase (cGAS)-Stimulator of Interferon Genes (STING); CREM/ICER = cAMP response element modulator/inducible cAMP early repressor; EZH2 = enhancer of zeste homolog 2; Gls1 = glutaminase 1; MCJ = methylation-controlled J protein; mTORC1 = mammalian target of rapamycin complex 1; PDH = pyruvate dehydrogenase; PKM2 = pyruvate kinase M2; PTEN = phosphatase and tensin homolog; SRSF1 = serine/arginine-rich splicing factor 1.
Key points.
Immunometabolism reprogramming is critical in the pathogenesis of SLE through a complex crosstalk between innate and adaptive immune responses.
Mitochondrial dysfunction and oxidative stress contribute to the distinctive increased type I IFN signature in SLE.
Abnormalities in various immunometabolism pathways such as mTOR, glycolysis and glutaminolysis favor the differentiation and skewing towards proinflammatory immune cell subsets in SLE.
Abnormalities in lipid metabolism may contribute to drive to immune dysregulation and premature cardiovascular disease in SLE.
Targeting immunometabolism defects in SLE might complement immunosuppressive standard therapies.
Financial support and sponsorship
Supported by the Intramural Research Program at NIAMS/NIH; ZIA AR041199 and by the Fulbright Scholar Program.
Footnotes
Conflicts of interest
There are no conflicts of interest.
Disclosure: Supported by the Intramural Research Program at NIAMS/NIH; ZIA AR041199 and by the Fulbright Scholar Program.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
* of special interest
** of outstanding interest
- 1.Teng X, Brown J, Choi SC, Li W, Morel L. Metabolic determinants of lupus pathogenesis. Immunol Rev. 2020;295(1):167–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stathopoulou C, Nikoleri D, Bertsias G. Immunometabolism: an overview and therapeutic prospects in autoimmune diseases. Immunotherapy. 2019;11(9):813–29. [DOI] [PubMed] [Google Scholar]
- 3.Morel L Immunometabolism in systemic lupus erythematosus. Nat Rev Rheumatol. 2017;13(5):280–90. [DOI] [PubMed] [Google Scholar]
- 4.Xiao YB, Guo MY, Zuo XX. [Immunometabolism and systemic lupus erythematosus]. Beijing Da Xue Xue Bao Yi Xue Ban. 2018;50(6):1120–4. [PubMed] [Google Scholar]
- 5.Piranavan P, Bhamra M, Perl A. Metabolic Targets for Treatment of Autoimmune Diseases. Immunometabolism. 2020;2(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang SK, Zhang HR, Shi SP, Zhu YQ, Song N, Dai Q, et al. The role of mitochondria in systemic lupus erythematosus: A glimpse of various pathogenetic mechanisms. Curr Med Chem. 2018. [DOI] [PubMed] [Google Scholar]
- 7.Vukelic M, Kono M, Tsokos GC. T cell Metabolism in Lupus. Immunometabolism. 2020;2(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. 2016;22(2):146–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim J, Gupta R, Blanco LP, Yang S, Shteinfer-Kuzmine A, Wang K, et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science. 2019;366(6472):1531–6. * This report supports the role of VDAC oligomers on forming mitochondrial pores to release mtDNA fragments to the cytoplasm, where they promote ISG expression ultimately by activating cGAS-STING pathway. Importantly, lupus-like disease was blocked by an inhibitor of VDAC oligomerization in vivo.
- 10.Takeshima Y, Iwasaki Y, Fujio K, Yamamoto K. Metabolism as a key regulator in the pathogenesis of systemic lupus erythematosus. Semin Arthritis Rheum. 2019;48(6):1142–5. [DOI] [PubMed] [Google Scholar]
- 11.Becker Y, Loignon RC, Julien AS, Marcoux G, Allaeys I, Levesque T, et al. Anti-mitochondrial autoantibodies in systemic lupus erythematosus and their association with disease manifestations. Sci Rep. 2019;9(1):4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pisetsky DS, Spencer DM, Mobarrez F, Fuzzi E, Gunnarsson I, Svenungsson E. The binding of SLE autoantibodies to mitochondria. Clin Immunol. 2020;212:108349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fortner KA, Blanco LP, Buskiewicz I, Huang N, Gibson PC, Cook DL, et al. Targeting mitochondrial oxidative stress with MitoQ reduces NET formation and kidney disease in lupus-prone MRL-lpr mice. Lupus Sci Med. 2020;7(1). * This study demonstrates for the first time the therapeutic potential of the antioxidant MitoQ in a lupus prone model, by dampening mROS generation, NET formation, MAVS oligomerization and type I IFN production in immune cells.
- 14. Titov AA, Baker HV, Brusko TM, Sobel ES, Morel L. Metformin Inhibits the Type 1 IFN Response in Human CD4(+) T Cells. J Immunol. 2019;203(2):338–48. ** This study underscores the beneficial impact of metformin in SLE cells through reduction of oxidative stress and the mTOR pathway. Furthermore, the authors demonstrate that mitochondrial respiration is required for ISG expression in SLE immune cells.
- 15. Katsuyama T, Li H, Comte D, Tsokos GC, Moulton VR. Splicing factor SRSF1 controls T cell hyperactivity and systemic autoimmunity. J Clin Invest. 2019;129(12):5411–23. ** This study reveals the critical role of the protein SRSF1 in modulating T cell activation, at least in part, by targeting the mTOR pathway. In addition, along with previous reports of low levels of SRSF1 in active SLE, the study suggests a relevant role for this molecule in the pathogenesis of this disease.
- 16.Katsuyama T, Martin-Delgado IJ, Krishfield SM, Kyttaris VC, Moulton VR. Splicing factor SRSF1 controls T cell homeostasis and its decreased levels are linked to lymphopenia in systemic lupus erythematosus. Rheumatology (Oxford). 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saravani M, Shahraki-Ghadimi H, Maruei-Milan R, Mehrabani M, Mirzamohammadi S, Nematollahi MH. Effects of the mTOR and AKT genes polymorphisms on systemic lupus erythematosus risk. Mol Biol Rep. 2020. [DOI] [PubMed] [Google Scholar]
- 18.Li X, Luo F, Li J, Luo C. MiR-183 delivery attenuates murine lupus nephritis-related injuries via targeting mTOR. Scand J Immunol. 2019;90(5):e12810. [DOI] [PubMed] [Google Scholar]
- 19.Chen S, Wang Y, Qin H, Lin J, Xie L, Chen S, et al. Downregulation of miR-633 activated AKT/mTOR pathway by targeting AKT1 in lupus CD4(+) T cells. Lupus. 2019;28(4):510–9. [DOI] [PubMed] [Google Scholar]
- 20.Godavarthy A, Kelly R, Jimah J, Beckford M, Caza T, Fernandez D, et al. Lupus-associated endogenous retroviral LTR polymorphism and epigenetic imprinting promote HRES-1/RAB4 expression and mTOR activation. JCI Insight. 2020;5(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang J, Yang X, Wang L, Li M. B cells control lupus autoimmunity by inhibiting Th17 and promoting Th22 cells. Cell Death Dis. 2020;11(3):164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He J, Ma J, Ren B, Liu A. Advances in systemic lupus erythematosus pathogenesis via mTOR signaling pathway. Semin Arthritis Rheum. 2020;50(2):314–20. [DOI] [PubMed] [Google Scholar]
- 23.Zheng X, Tsou PS, Sawalha AH. Increased Expression of EZH2 Is Mediated by Higher Glycolysis and mTORC1 Activation in Lupus CD4(+) T Cells. Immunometabolism. 2020;2(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Murayama G, Chiba A, Kuga T, Makiyama A, Yamaji K, Tamura N, et al. Inhibition of mTOR suppresses IFNalpha production and the STING pathway in monocytes from systemic lupus erythematosus patients. Rheumatology (Oxford). 2020. * This report highlights the role of mTOR signaling pathway in different innate immune cells in SLE, by promoting IFN-α production through the cGAS-STING pathway.
- 25. Shi G, Li D, Li X, Ren J, Xu J, Ding L, et al. mTOR inhibitor INK128 attenuates systemic lupus erythematosus by regulating inflammation-induced CD11b(+)Gr1(+) cells. Biochim Biophys Acta Mol Basis Dis. 2019;1865(1):1–13. * This study adds additional evidence that targeting mTOR pathway in lupus may be beneficial and modulates myeloid-derived CD11b+Gr1+ cells in two murine models.
- 26.Secinaro MA, Fortner KA, Collins C, Rincon M, Budd RC. Glycolysis Induces MCJ Expression That Links T Cell Proliferation With Caspase-3 Activity and Death. Front Cell Dev Biol. 2019;7:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kono M, Yoshida N, Maeda K, Suarez-Fueyo A, Kyttaris VC, Tsokos GC. Glutaminase 1 Inhibition Reduces Glycolysis and Ameliorates Lupus-like Disease in MRL/lpr Mice and Experimental Autoimmune Encephalomyelitis. Arthritis Rheumatol. 2019;71(11):1869–78. ** This is the first report to demonstrate the therapeutic potential of a glutaminase 1 selective inhibitor in a lupus prone model by dampening Th17 differentiation. Likewise, it highlights the critical role of glucose and glutamine metabolism pathways in promoting aberrant immune responses in SLE.
- 28.Zezina E, Sercan-Alp O, Herrmann M, Biesemann N. Glucose transporter 1 in rheumatoid arthritis and autoimmunity. Wiley Interdiscip Rev Syst Biol Med. 2020:e1483. [DOI] [PubMed] [Google Scholar]
- 29. Kono M, Maeda K, Stocton-Gavanescu I, Pan W, Umeda M, Katsuyama E, et al. Pyruvate kinase M2 is requisite for Th1 and Th17 differentiation. JCI Insight. 2019;4(12). * This report provides new evidence of a critical checkpoint in glycolysis pathway for T cell differentiation into proinflammatory subsets, relevant in the context of autoimmune diseases.
- 30. Koga T, Sato T, Furukawa K, Morimoto S, Endo Y, Umeda M, et al. Promotion of Calcium/Calmodulin-Dependent Protein Kinase 4 by GLUT1-Dependent Glycolysis in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2019;71(5):766–72. ** This study underscores the critical role of CaMK4 in Th17 differentiation by enhancing different metabolic pathways in T cells.
- 31.Kono M, Yoshida N, Maeda K, Skinner NE, Pan W, Kyttaris VC, et al. Pyruvate dehydrogenase phosphatase catalytic subunit 2 limits Th17 differentiation. Proc Natl Acad Sci U S A. 2018;115(37):9288–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Freemerman AJ, Zhao L, Pingili AK, Teng B, Cozzo AJ, Fuller AM, et al. Myeloid Slc2a1-Deficient Murine Model Revealed Macrophage Activation and Metabolic Phenotype Are Fueled by GLUT1. J Immunol. 2019;202(4):1265–86. * This report highlights the importance of GLUT1-mediated glycolysis in macrophage differentiation into proinflammatory subsets and oxidative stress, relevant in the context of autoimmune diseases.
- 33.Ferreira HB, Pereira AM, Melo T, Paiva A, Domingues MR. Lipidomics in autoimmune diseases with main focus on systemic lupus erythematosus. J Pharm Biomed Anal. 2019;174:386–95. [DOI] [PubMed] [Google Scholar]
- 34.Kim SY, Yu M, Morin EE, Kang J, Kaplan MJ, Schwendeman A. High-Density Lipoprotein in Lupus: Disease Biomarkers and Potential Therapeutic Strategy. Arthritis Rheumatol. 2020;72(1):20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Purmalek MM, Carlucci PM, Dey AK, Sampson M, Temesgen-Oyelakin Y, Sakhardande S, et al. Association of lipoprotein subfractions and glycoprotein acetylation with coronary plaque burden in SLE. Lupus Sci Med. 2019;6(1):e000332. * This study reports significant associations between lipoprotein fraction dysregulation and coronary plaque burden in SLE patients.
- 36. Ye Y, Wu T, Zhang T, Han J, Habazi D, Saxena R, et al. Elevated oxidized lipids, anti-lipid autoantibodies and oxidized lipid immune complexes in active SLE. Clin Immunol. 2019;205:43–8. * This study provides new evidence on the link between lipidomic analysis findings in SLE patients and aberrant humoral immune responses that might contribute to disease activity and tissue damage.
- 37.Nowling TK, Rodgers J, Thiyagarajan T, Wolf B, Bruner E, Sundararaj K, et al. Targeting glycosphingolipid metabolism as a potential therapeutic approach for treating disease in female MRL/lpr lupus mice. PLoS One. 2020;15(3):e0230499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sanchez HN, Moroney JB, Gan H, Shen T, Im JL, Li T, et al. B cell-intrinsic epigenetic modulation of antibody responses by dietary fiber-derived short-chain fatty acids. Nat Commun. 2020;11(1):60. ** This report provides evidence on how an environmental factor, such as dietary intake of fiber-derived short-chain FA, could modulate B cells intrinsic function and attenuate autoantibody responses and autoimmunity in a lupus model.
- 39.Benninghoff AD, Bates MA, Chauhan PS, Wierenga KA, Gilley KN, Holian A, et al. Docosahexaenoic Acid Consumption Impedes Early Interferon- and Chemokine-Related Gene Expression While Suppressing Silica-Triggered Flaring of Murine Lupus. Front Immunol. 2019;10:2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carlucci PM, Purmalek MM, Dey AK, Temesgen-Oyelakin Y, Sakhardande S, Joshi AA, et al. Neutrophil subsets and their gene signature associate with vascular inflammation and coronary atherosclerosis in lupus. JCI Insight. 2018;3(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mistry P, Nakabo S, O’Neil L, Goel RR, Jiang K, Carmona-Rivera C, et al. Transcriptomic, epigenetic, and functional analyses implicate neutrophil diversity in the pathogenesis of systemic lupus erythematosus. Proc Natl Acad Sci U S A. 2019;116(50):25222–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith CK, Vivekanandan-Giri A, Tang C, Knight JS, Mathew A, Padilla RL, et al. Neutrophil extracellular trap-derived enzymes oxidize high-density lipoprotein: an additional proatherogenic mechanism in systemic lupus erythematosus. Arthritis Rheumatol. 2014;66(9):2532–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Blanco LP, Pedersen HL, Wang X, Lightfoot YL, Seto N, Carmona-Rivera C, et al. Improved Mitochondrial Metabolism and Reduced Inflammation Following Attenuation of Murine Lupus With Coenzyme Q10 Analog Idebenone. Arthritis Rheumatol. 2020;72(3):454–64. * This report provides new evidence of the positive impact of coenzyme Q10 analog idebenone on two lupus models, by counteracting mitochondrial dysfunction and mROS-driven NET formation.
- 44.Yin Y, Choi SC, Xu Z, Perry DJ, Seay H, Croker BP, et al. Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med. 2015;7(274):274ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee SY, Moon SJ, Kim EK, Seo HB, Yang EJ, Son HJ, et al. Metformin Suppresses Systemic Autoimmunity in Roquin(san/san) Mice through Inhibiting B Cell Differentiation into Plasma Cells via Regulation of AMPK/mTOR/STAT3. J Immunol. 2017;198(7):2661–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cornaby C, Elshikha AS, Teng X, Choi SC, Scindia Y, Davidson A, et al. Efficacy of the Combination of Metformin and CTLA4Ig in the (NZB x NZW)F1 Mouse Model of Lupus Nephritis. Immunohorizons. 2020;4(6):319–31. [DOI] [PubMed] [Google Scholar]
- 47.Caza TN, Fernandez DR, Talaber G, Oaks Z, Haas M, Madaio MP, et al. HRES-1/Rab4-mediated depletion of Drp1 impairs mitochondrial homeostasis and represents a target for treatment in SLE. Ann Rheum Dis. 2014;73(10):1888–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wei S, Xie S, Yang Z, Peng X, Gong L, Zhao K, et al. Allogeneic adipose-derived stem cells suppress mTORC1 pathway in a murine model of systemic lupus erythematosus. Lupus. 2019;28(2):199–209. [DOI] [PubMed] [Google Scholar]
- 49.Yang J, Yang X, Yang J, Li M. Baicalin ameliorates lupus autoimmunity by inhibiting differentiation of Tfh cells and inducing expansion of Tfr cells. Cell Death Dis. 2019;10(2):140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eriksson P, Wallin P, Sjowall C. Clinical Experience of Sirolimus Regarding Efficacy and Safety in Systemic Lupus Erythematosus. Front Pharmacol. 2019;10:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang C, Chan CCY, Cheung KF, Chau MKM, Yap DYH, Ma MKM, et al. Effect of mycophenolate and rapamycin on renal fibrosis in lupus nephritis. Clin Sci (Lond). 2019;133(15):1721–44. [DOI] [PubMed] [Google Scholar]
- 52.Zhao C, Chu Y, Liang Z, Zhang B, Wang X, Jing X, et al. Low dose of IL-2 combined with rapamycin restores and maintains the long-term balance of Th17/Treg cells in refractory SLE patients. BMC Immunol. 2019;20(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yap DYH, Tang C, Chan GCW, Kwan LPY, Ma MKM, Mok MMY, et al. Longterm Data on Sirolimus Treatment in Patients with Lupus Nephritis. J Rheumatol. 2018;45(12):1663–70. [DOI] [PubMed] [Google Scholar]
- 54.Lai ZW, Hanczko R, Bonilla E, Caza TN, Clair B, Bartos A, et al. N-acetylcysteine reduces disease activity by blocking mammalian target of rapamycin in T cells from systemic lupus erythematosus patients: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2012;64(9):2937–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koga T, Hedrich CM, Mizui M, Yoshida N, Otomo K, Lieberman LA, et al. CaMK4-dependent activation of AKT/mTOR and CREM-alpha underlies autoimmunity-associated Th17 imbalance. J Clin Invest. 2014;124(5):2234–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li W, Qu G, Choi SC, Cornaby C, Titov A, Kanda N, et al. Targeting T Cell Activation and Lupus Autoimmune Phenotypes by Inhibiting Glucose Transporters. Front Immunol. 2019;10:833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tan MKX, Heng TYJ, Mak A. The Potential Use of Metformin, Dipyridamole, N-Acetylcysteine and Statins as Adjunctive Therapy for Systemic Lupus Erythematosus. Cells. 2019;8(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ruiz-Limon P, Barbarroja N, Perez-Sanchez C, Aguirre MA, Bertolaccini ML, Khamashta MA, et al. Atherosclerosis and cardiovascular disease in systemic lupus erythematosus: effects of in vivo statin treatment. Ann Rheum Dis. 2015;74(7):1450–8. [DOI] [PubMed] [Google Scholar]