

Abstract
Sarcomas are a group of diverse and complex cancers of mesenchymal origin that remains poorly understood. Recent developments in cancer immunotherapy have demonstrated a potential for better outcomes with immune checkpoint inhibition in some sarcomas compared to conventional chemotherapy. Immune checkpoint inhibitors (ICIs) are key agents in cancer immunotherapy, demonstrating improved outcomes in many tumor types. However, most patients with sarcoma do not benefit from treatment, highlighting the need for identification and development of predictive biomarkers for response to ICIs. In this review, we first discuss United States (US) Food and Drug Administration (FDA)-approved and European Medicines Agency (EMA)-approved biomarkers, as well as the limitations of their use in sarcomas. We then review eight potential predictive biomarkers and rationalize their utility in sarcomas. These include gene expression signatures (GES), circulating neutrophil-to-lymphocyte ratio (NLR), indoleamine 2,3-dioxygenase (IDO), lymphocyte activation gene 3 (LAG-3), T cell immunoglobin and mucin domain-containing protein 3 (TIM-3), TP53 mutation status, B cells, and tertiary lymphoid structures (TLS). Finally, we discuss the potential for TLS as both a predictive and prognostic biomarker for ICI response in sarcomas to be implemented in the clinic.
Keywords: Immune checkpoint inhibitors, Predictive biomarkers, Sarcomas, Tertiary lymphoid structures
Background
Sarcomas are a diverse and complex group of cancers of mesenchymal origin that often have very poor prognosis, with median survival of about 18 months with metastatic disease [1]. In soft-tissue sarcomas (STS), the 5-year survival rates for localized, regional, and metastatic disease are 81%, 56% and 16% respectively [2]. Comparatively, in osteosarcoma, the 5-year survival rates are 74%, 66% and 27% respectively [3]. Lastly, the 5-year survival rates in Ewing sarcoma are 81%, 67% and 38% respectively [4]. The systemic treatment of sarcomas has relied on conventional chemotherapy that has remained widely unchanged over several decades. Doxorubicin and ifosfamide represent the current standard of care in most subtypes of advanced and metastatic sarcomas [5]. However, response to treatment remains poor and more efficacious treatment options are needed. In a phase III trial comparing doxorubicin monotherapy against intensified doxorubicin with ifosfamide in advanced or metastatic STS, treatment with doxorubicin alone yielded an overall response rate of 14%, compared to 26% in patients treated with doxorubicin and ifosfamide. Importantly, there was no significant difference in overall survival (OS) between the two groups, with a median OS of 12.8 months (95.5% confidence interval (CI), 10.5–14.3) in the doxorubicin-only group, compared to 14.3 months (95.5% CI, 12.5–16.5) in the combination group [6]. Alternative agents such as gemcitabine and docetaxel are reserved for patients who have failed or are unable to tolerate doxorubicin and ifosfamide. Gemcitabine is commonly used alone or in combination with docetaxel, with complete or partial response, or stable disease after at least 25 weeks being achieved by 27% in the gemcitabine-only group and 32% in the combination group [7]. These response rates are in stark contrast to other tumors such as lymphomas, leukemias, germ cell tumors and others with response rates of > 70% with chemotherapy [8]. While targeted therapies are available, only less than 5% of STS are amenable to these treatments [9–11]. Limited treatment options compounded by poor treatment response necessitates the exploration of more treatment options with better outcomes and side effect profiles.
Research in treatment for sarcomas has faced many challenges. Sarcomas are rare cancers representing only 1% of adult malignancies [12], making it difficult to recruit sufficient clinical trial participants to generate rapid and robust evidence for treatment efficacy. Furthermore, heterogeneity in their histology and genetic drivers of oncogenic pathways in sarcomas gives rise to a wide variation in their biology, as well as degree of immune infiltration. As such, each subtype exhibits different clinical characteristics, often requiring patient-specific treatment approaches [13] since different patients may not respond to the same therapy.
Amidst these challenges, immune checkpoint inhibitor (ICI) therapy has emerged as an attractive treatment option [14]. ICIs target immune checkpoints that under physiologic conditions restrict the strength and duration of immune responses to avoid immune-mediated tissue damage, but which can be exploited by tumors to evade immune-mediated elimination. Efficacy of treatment with ICIs has been established in several cancers [15], including advanced renal cell carcinoma (RCC) [16], cervical cancer [17], classical Hodgkin lymphoma [18], gastric carcinoma [19], hepatocellular carcinoma (HCC) [20], melanoma [21–23], Merkel cell carcinoma [24, 25], non-small cell lung cancer (NSCLC) [26], primary mediastinal large B-cell lymphoma [27], small cell lung cancer [28], head and neck squamous cell cancer (HNSCC) [29], triple negative breast cancer [30], and urothelial cancer [31]. In an exciting step forward in the treatment of sarcoma, the United States (US) Food and Drug Administration (FDA) recently approved the first ICI for use in the treatment of STS, with atezolizumab being approved for use in the treatment of unresectable or metastatic alveolar soft-part sarcomas (ASPS) [32]. Atezolizumab as the first agent of its class being indicated for ASPS could set the stage for more ICIs to be indicated for the treatment of more STS subtypes and offers exciting possibilities for further evaluation.
In fact, although STS have been traditionally thought to be immune “cold” [33], as a whole, the response of STS to immune checkpoint inhibition does not differ too much from that of all cancers considered together. In 2019, Haslam and Prasad estimated that the percentage of US patients with cancer that respond to ICIs was 12.46% (95% CI, 12.37–12.54%) [34], which is comparable to the results of the SARC028 trial (NCT02301039), where 18% of patients with STS had an objective response to pembrolizumab [35]. Additionally, ICI therapy has shown improved outcomes in the clinical management of selected populations in sarcomas [36–38]. Within STS subtypes, liposarcomas (LPS), undifferentiated pleomorphic sarcomas (UPS) and ASPS have demonstrated better responses than other subtypes, while leiomyosarcomas (LMS) and synovial sarcomas (SS) have been reported to be resistant to ICI monotherapy [39]. Table 1 outlines a comprehensive list of studies using ICIs, both as monotherapy and in combination, and the respective clinical outcomes in sarcomas. Aside from clinical efficacy, another concern that clinicians have to consider is the potential for immune-related adverse events (irAEs) that range from mild adverse conditions like diarrhea and rashes to life-threatening conditions like cardiomyopathy and toxic epidermal necrolysis [40]. Thus, there is an urgent need to identify biomarkers that can guide clinical use of ICIs in potential responders while sparing non-responders from potentially life-threatening irAEs.
Table 1.
Overview of studies using immune checkpoint inhibitors (ICIs) alone or in combination with other drugs in sarcomas
| ICI | Combination | NCT | Phase (Status) | Type of Tumor | Clinical Efficacy | ≥ G3 TRAE |
|---|---|---|---|---|---|---|
| Atezolizumab | Cabozantinib | NCT05019703 | Phase II (recruiting) | OGS | NA | NA |
| ± CMB305 | NCT02609984 | Phase II (terminated due to failure to meet efficacy objective) | NY-ESO-1 + sarcoma |
Atezolizumab only: 0 CR, 0 PR, 17 SD, 25 PD (n = 44) Atezolizumab + CMB305: 1.8% ORR (95%CI: 0.8–4.2%), 0 CR, 1 PR, 23 SD, 19 SD (n = 45) mPFS: 1.6 months in atezolizumab only arm (n = 43), 2.6 months in atezolizumab + CMB305 arm (n = 45) (HR: 0.9, 95% CI: 0.6–1.3) mOS: 18 months in both arms (atezolizumab only arm: 95% CI, 15.3 to 26.5 and atezolizumab + CMB305 arm: 95% CI, 10.1 to 22.1; HR, 1.2; p = 0.47) |
13 ≥ G3 TRAE reported in atezolizumab only arm 18 ≥ G3 TRAE reported in atezolizumab + CB305 arm |
|
| Bevacizumab | NCT03141684 | Phase II (recruiting) | ASPS | 1 CR, 14 PR, 1 unconfirmed PR, 25 SD (n = 43) | 10 ≥ G3 TRAE | |
| Bevacizumab + rucaparib | NCT03694262 | Phase II (active, not recruiting) | Endometrial cancer, uterine carcinosarcoma | 1 CR, 9 PR, 13 SD (n = 26) | ≥ G3 TRAE reported in 50% patients | |
| Cobimetinib | NCT04216953 | Phase I/II (recruiting) | STS | NA | NA | |
| Irinotecan + temozolomide + vincristine | NCT04796012 | Phase I/II (recruiting) | Rhabdomyosarcoma, solid tumor | NA | NA | |
| NA | NCT04273061 | Phase II (recruiting) | Cancers (breast, gastrointestinal, genitourinary, gynecologic, head and neck, lung, skin, unknown primary tumor), sarcoma | NA | NA | |
| NCT04458922 | Phase II (active, not recruiting) | Chondrosarcoma, clear cell sarcoma of soft tissue |
3 SD (n = 9 in grade 2/3 chondrosarcoma cohort) No RECIST objective responses observed (n = 9 in dedifferentiated chondrosarcoma cohort) |
Grade 3 TRAEs occurred in 2 patients in dedifferentiated chondrosarcoma cohort (22%), included infusion reaction, myonecrosis, and anemia | ||
| RT + surgical resection | NCT03474094 | Phase II (recruiting) | STS | NA | NA | |
| SABR | NCT02992912 | Phase II (unknown) | Metastatic tumors (colorectal cancer, NSCLC, RCC, sarcoma) | NA | NA | |
| Selinexor | NCT05333458 | Phase II (recruiting) | ASPS, STS | NA | NA | |
| Tiragolumab | NCT05286801 | Phase I/II (recruiting) | Epithelioid sarcoma, SMARCB1 or SMARCA4 deficient tumors | NA | NA | |
| Tivozanib | NCT05000294 | Phase I/II (recruiting) | Bile duct cancer, breast cancer, gall bladder cancer, neuroendocrine cancer, ovarian cancer, pancreatic adenocarcinoma, prostate cancer, STS, vulvar cancer | NA | NA | |
| ± Atezolizumab | SBRT | NCT03548428 | Phase II (recruiting) | Sarcoma | NA | NA |
| Avelumab | NA | NCT03006848 | Phase II (active, not recruiting) | OGS |
No objective responses occurred (17 PD) (n = 18) mPFS: 8 weeks (95% CI: 6.7–9.1 months) |
6 ≥ G3 TRAE |
| Trabectedin | NCT03074318 | Phase I/II (terminated due to investigator leaving institute) | LMS, LPS |
2 DLT reported (n = 6) 2 PR (1 confirmed), 11 SD (n = 23) mPFS: 23.4 months |
Most common G3 TRAE attributed to study drug were neutropenia and ALT increase No G4/5 TRAE at the Phase 2 dose |
|
| Camrelizumab | Apatinib | NCT04239443 | Phase II (unknown) | NSCLC, STS, uterine cancer | NA | NA |
| Cisplatin + doxorubicin + ifosfamide + methotrexate | NCT04294511 | Phase II (recruiting) | OGS | 31 showed good response (n = 65) | Most common grade 3–4 adverse events were decreased platelet count (44.0%), decreased white blood cell (37.3%), decreased neutrophil count (29.3%), oral mucositis (14.7%), increased alanine aminotransferase (12.0%), and increased aspartate aminotransferase (10.7%) | |
| Ifosfamide + liposome doxorubicin | NCT04606108 | Phase II (recruiting) | STS | NA | NA | |
| ± Camrelizumab | Famitinib ± ifosfamide | NCT04044378 | Phase I/II (withdrawn due to toxicity) | OGS | NA | NA |
| Durvalumab + ipilimumab + pembrolizumab | NA | NCT05187338 | Phase I/II (recruiting) | Sarcoma, solid tumors | NA | NA |
| Envafolimab ± ipilimumab | NA | NCT04480502 | Phase II (recruiting) | MFS, UPS | NA | NA |
| Envafolimab + YH001 (anti-CTLA4 antibody) | ± Doxorubicin | NCT05448820 | Phase I/II (recruiting) | Sarcoma | NA | NA |
| FAZ053 (anti-PD-L1 antibody) ± spartalizumab | NA | NCT02936102 | Phase I (active, not recruiting) | ASPS, chordoma, solid tumors, TNBC | NA | NA |
| Ipilimumab | CD4+ T cells + cyclophosphamide | NCT02210104 | Phase I (withdrawn due to issues with tetramer staining) | Melanoma, sarcoma | NA | NA |
| Dasitinib | NCT01643278 | Phase I (completed) | GIST, STS |
DLT included grade 3 gastric hemorrhage and anemia 0 CR, 0 PR (n = 28) mPFS: 2.8 months (95% CI: 2.7–3.0 months) (n = 18) mOS: 13.5 months (95% CI: 11.4 months – NR) |
19 ≥ G3 TRAE | |
| NA | NCT00140855 | Phase II (terminated due to poor accrual) | SS | 0 CR, 0 PR, 0 SD, 6 PD (n = 6) | 3 ≥ G3 TRAE | |
| NCT01445379 | Phase I (completed) | Lymphoma, neuroblastoma, sarcoma, Wilms’ tumor |
DLT observed at 10 mg/kg (n = 2) 6 SD for four to ten cycles (clear cell sarcoma, melanoma, OGS, SS) |
11 ≥ G3 TRAE | ||
| Ipilimumab + nivolumab | Cabozantinib | NCT04149275 | Phase II (withdrawn due to stoppage of funding by sponsor) | Gynecologic carcinosarcoma | NA | NA |
| NCT04551430 | Phase II (active, not recruiting) | STS | NA | NA | ||
| ± Cabozantinib | NCT05836571 | Phase II (not yet recruiting) | Extraskeletal myxoid chondrosarcoma, LMS, LPS, UPS | NA | NA | |
| Cryoablation | NCT04118166 | Phase II (active, not recruiting) | STS | 0 CR, 3 PR, 7 SD, 19 PD (n = 29) | 41 ≥ G3 TRAE | |
| NCT05302921 | Phase II (recruiting) | ES, hepatoblastoma, hepatocellular carcinoma, melanoma, neuroblastoma, OGS, rhabdomyosarcoma, Wilms’ tumor | NA | NA | ||
| Lurbinectedin | NCT05876715 | Phase II (recruiting) | STS | NA | NA | |
| NA | NCT02982486 | Phase II (unknown) | BS, STS | NA | NA | |
| NCT03219671 | Phase II (unknown) | Classic Kaposi sarcoma | 87% ORR (n = 15) | 2 ≥ G3 TRAE | ||
| NCT04416568 | Phase II (recruiting) | Epithelioid sarcoma, INI1-negative cancers | NA | NA | ||
| NCT04465643 | Phase I (recruiting) | MPNST | NA | NA | ||
| Pazopanib alone | NCT04741438 | Phase III (recruiting) | Sarcoma | NA | NA | |
| Tazemetostat | NCT05407441 | Phase I/II (recruiting) | INI1-negative/SMARCA4-deficient cancers | NA | NA | |
| Trabectedin | NCT03138161 | Phase I/II (recruiting) | STS |
8 CR, 11 PR, 58 SD and 11 PD with 21.6% BORR and 87.5% DCR (n = 88) mPFS: 7 months (1–44 months) mOS: 14 months (1–46 months) |
76 ≥ G3 TRAE | |
| ± Ipilimumab | XmAb23104 | NCT03752398 | Phase I (recruiting) | Solid tumors, UPS |
No DLT reported (n = 62) 3 PR in HNSCC, RCC, sarcoma |
≥ G3 TRAE reported in 6 patients 2 ≥ G3 irAEs |
| ± Ipilimumab or pembrolizumab | INT230-6 | NCT03058289 | Phase I/II (completed) | Cancer, sarcoma | No DLT reported |
Incidence of ≥ G3 TRAE was 11% and 14% in INT230-6 only and INT230-6 + pembrolizumab arm 1 G4 neutrophil count decrease reported in INT230-6 + pembrolizumab arm |
| ± Ipilimumab with nivolumab | Aldesleukin + autologous TIL LN-145 + autologous TIL LN-145-S1 | NCT03449108 | Phase II (recruiting) | Anaplastic thyroid cancer, BS, STS, relapsed/refractory ovarian cancer, TNBC, undifferentiated high grade pleomorphic sarcoma of bone | NA | NA |
| LAG525 + spartalizumab | NA | NCT03365791 | Phase II (completed) | Solid and hematologic malignancies, STS |
7.3% ORR (n = 75) mPFS: 2.8 months (95% CI: 2.6–3.1 months) |
Serious adverse events in 35 patients reported (n = 76) |
| Nivolumab | Anlotinib hydrochloride | NCT04165330 | Phase I/II (active, not recruiting) | NSCLC, SCLC, STS | NA | NA |
| ± Azacitidine | NCT03628209 | Phase I/II (recruiting) | OGS, sarcoma | NA | NA | |
| Bempegaldesleukin | NCT03282344 | Phase II (active, not recruiting) | Sarcoma |
9 PR (n = 77) mPFS: 1.8–7.3 months mOS: 5.9–21.7 months (NR in ASPS and angiosarcoma) |
32 ≥ G3 TRAE 1 possible treatment related death |
|
| NCT04730349 | Phase I/II (terminated due to changes in business objectives) | ES, recurrent/treatment-resistant cancers | NA | NA | ||
| BMS-986205 | NCT04106414 | Phase II (closed to accrual due to lack of observed clinical efficacy) | Endometrial adeno-, carcino-sarcoma |
No response in nivolumab only arm (n = 12) 1 PR in nivolumab + BMS-986205 arm (n = 12) mPFS: 7.3 weeks (80% CI: 6.4–15.1 weeks) (nivolumab only), 12.3 weeks (80% CI: 4.1–22.1 weeks) (nivolumab + BMS-986205) mOS: 27.5 weeks (80% CI: 17-NA) (nivolumab only), NR (nivolumab + BMS-986205) |
3 ≥ G3 TRAE in nivolumab only arm 2 ≥ G3 TRAE in nivolumab + BMS-986205 arm |
|
| BO-112 + RT + surgical resection | NCT04420975 | Phase I (active, not recruiting) | STS | NA | NA | |
| Cabozantinib | NCT04514484 | Phase I (recruiting) | Advanced cancer, HIV, Kaposi sarcoma | NA | NA | |
| ± Cabozantinib S-malate or paclitaxel or paclitaxel only | NCT04339738 | Phase II (active, not recruiting) | Angiosarcoma |
Taxane only: 13 PR (n = 21), 13 ORR (n = 18) mPFS: 9.6 months (5.3 months – NR) mOS: 20.5 months (14.4 months – NR) |
G3 hypertension reported in 10% patients only | |
| Cisplatin + dacarbazine + doxorubicin + epirubicin + ifosfamide + methotrexate + sunitinib | NCT03277924 | Phase I/II (recruiting) | BS, STS |
1 CR, 1 PR, 22 SD, 16 PD (n = 40) mPFS: 3.7 months (95% CI: 3.4–4 months) mOS: 14.2 months (95% CI: 7.1–21.3 months) |
21 ≥ G3 TRAE | |
| Docetaxel + doxorubicin + gemcitabine | NCT04535713 | Phase II (recruiting) | Sarcoma |
8 PR, 44SD, 7 PD (n = 59 in intention-to-treat cohort) mPFS: 5.1 months (2.837–7.363 months) mOS: 15.3 months (95%CI: 5.48–25.12 months) |
60 ≥ G3 TRAE | |
| NA | NCT03241745 | Phase II (active, not recruiting) | Carcinosarcoma, clear cell carcinoma, endometrial carcinoma, high grade endometrial stromal sarcoma, LMS, undifferentiated sarcoma, uterine cancer | NA | NA | |
| NCT03316274 | Phase I (completed) | HIV/AIDS, Kaposi sarcoma | NA | NA | ||
| NCT03465592 | Phase I/II (recruiting) | Sarcoma | NA | NA | ||
| NCT05224999 | Phase II (recruiting) | Carcinosarcoma | NA | NA | ||
| Nab-rapamycin | NCT03190174 | Phase I/II (completed) | Sarcoma and certain cancers | Two DLTs reported at 150 mg/m2 (grade 3 aspartate aminotransferase elevation and grade 4 thrombocytopenia) and 125 mg/m2 (grade 3 suicidal ideation and grade 3 hypophosphatemia) each (n = 26) | 12 ≥ G3 TRAE | |
| ± Pazopanib | NCT03149120 | Phase II (withdrawn) | STS | NA | NA | |
| Pomalidomide | NCT04902443 | Phase I (recruiting) | Kaposi sarcoma, viral Associated Malignancies | NA | NA | |
| Regorafenib | NCT04803877 | Phase II (active, not recruiting) | OGS | NA | NA | |
| Rucaparib | NCT04624178 | Phase II (active, not recruiting) | LMS | NA | NA | |
| Trabectedin | NCT03590210 | Phase II (completed) | STS |
mPFS: 5.5 months in LMS/LPS cohort (n = 43), 2.3 months in others (n = 49) mOS: 18.7 months in LMS/LPS cohort (n = 43), 5.6 months in others (n = 49) |
NA | |
| Trabectedin + T-VEC | NCT03886311 | Phase II (recruiting) | Sarcoma |
3 PR, 30 SD, 6 PD, 7.7% BORR (n = 39) mPFS: 7.8 months (95% CI: 4.1–13.1 months) mOS: 19.3 months (95% CI: 12.8 months-NR) |
3 ≥ G3 TRAE related to nivolumab 38 ≥ G3 TRAE related to trabectedin 1 ≥ G3 TRAE related to T-VEC |
|
| ± Nivolumab | Bempegaldesleukin ± NKTR-262 | NCT03435640 | Phase I/II (terminated due to poor overall results) | CRC, HNSCC, melanoma, Merkel cell carcinoma, RCC, sarcoma, TNBC |
1 DLT reported at 3.84 mg NKTR-262 2 PR (n = 17) |
Most frequent treatment-related adverse events were flu-like symptoms, fatigue, nausea, and pruritus |
| TPST-1120 | NCT03829436 | Phase I (active, not recruiting) | Advanced cancer, sarcoma |
G3 hypertension reported in TPST-1120 monotherapy 3 G3 TRAE reported in combination therapy arm 10 SD (n = 19 in monotherapy arm) |
3 ≥ G3 TRAE in combination therapy arm | |
| Nivolumab ± Ipilimumab | NA | NCT02304458 | Phase I/II (completed) | Lymphoma, recurrent/refractory solid tumors or sarcomas |
No DLT reported (n = 12) Hodgkin lymphoma (n = 10): 1 CR, 2 PR, 5 SD Neuroblastoma (n = 10): 5 SD Sarcoma (n = 33): 11 SD |
54 ≥ G3 TRAE |
| NCT02428192 | Phase II (active, not recruiting) | LMS |
mPFS: 1.8 months (95% CI: 0.8 months – unknown) (n = 12) mOS: NR |
14 ≥ G3 TRAE | ||
| NCT02500797 | Phase II (active, not recruiting) | Sarcoma |
Nivolumab only: 3 PR, 5% ORR (92% CI:1–15%) (n = 38) Nivolumab + Ipilimumab arm: 15% adjusted ORR (92% CI: 6–30%) (n = 41) mPFS: 1.7 months (95% CI: 1.4–4.3 months) (n = 42 in nivolumab only arm), 4.1 months (95% CI: 2.6–4.7 months) (n = 41 in nivolumab + ipilimumab arm) mOS: 10.7 months (95% CI: 5.5–15.4 months) (n = 42 in nivolumab only arm), 14.3 months (95% CI: 9.6 months – not estimable) (n = 41 in nivolumab + ipilimumab arm) |
44 ≥ G3 TRAE in nivolumab only arm 66 ≥ G3 TRAE in nivolumab + ipilimumab arm |
||
| RT | NCT03463408 | Phase I (active, not recruiting) | Sarcoma | NA | NA | |
| ± RT | NCT03307616 | Phase II (active, not recruiting) | DDLPS, UPS |
mPFS: 18 months (IQR:8 months – NR in DDLPS), NR (IQR:19 – NR in UPS) mOS: NR |
NA | |
| Nivolumab ± relatlimab | NA | NCT04095208 | Phase II (recruiting) | STS | NA | NA |
| ONC-392 (anti-CTLA4 IgG1 monoclonal antibody) ± pembrolizumab | NA | NCT04140526 | Phase I/II (recruiting) | Sarcoma, solid tumors | NA | NA |
| ± PD-1 inhibitor (not specified) | Anlotinib hydrochloride | NCT05193188 | Phase II (recruiting) | Chondrosarcoma | NA | NA |
| CAB-AXL-ADC | NCT03425279 | Phase I/II (recruiting) | BS, ES, LMS, LPS, melanoma, NSCLC, OGS, refractory sarcoma, solid tumor, SS, STS | NA | NA | |
| Pembrolizumab | Antiretroviral therapy | NCT02595866 | Phase I (active, not recruiting) | HIV/AIDS related cancer, Kaposi sarcoma | NA | ≥ G3 TRAE reported in 20% of patients |
| APG-115 | NCT03611868 | Phase I/II (recruiting) | Melanomas, MPNST, solid tumors |
Cutaneous/uveal melanoma:2 CR, 2 PR (n = 17) Melanoma: 2 CR, 3 PR (n = 38) MPNST: 4 SD (n = 10) LPS: 1 PR (n = 17) |
≥ G3 TRAE reported in ≥ 5% patients | |
| Axitinib | NCT02636725 | Phase II (completed) | STS |
0 CR, 8 PR, 9 SD (n = 32) mPFS: 4.7 months in intention-to-treat analysis (95% CI: 3.0–9.4 months) (n = 33), 6.9 months in per-protocol analysis (95% CI: 3.0–9.4 months) (n = 30) mOS: 18.7 months (95% CI: 12.0 months – NR) (n = 33) |
26 ≥ G3 TRAE | |
| Cabozantinib | NCT05182164 | Phase II (recruiting) | ES, OGS, STS | NA | NA | |
| Cyclophosphamide | NCT02406781 | Phase II (unknown) | Sarcoma |
9 PR, 10 SD (n = 30) mPFS: 4.1 months (95%CI: 1.4–12.5 months) mOS: 18.3 months (95%CI: 8.5 months – NR) |
9 ≥ G3 TRAE (n = 35) | |
| Cyclophosphamide + fludarabine | NCT03697824 | Phase II (withdrawn due to internal decision, study will be replaced with a larger monotherapy trial) | NY-ESO-1 and/or LAGE-1a + SS | NA | NA | |
| Dactinomycin + melphalan | NCT04332874 | Phase II (recruiting) | ASPS, myxofibrosarcoma, UPS | NA | NA | |
| Docetaxel + gemcitabine or + gemcitabine or gemcitabine + vinorelbine or irinotecan or liposomal doxorubicin | NCT02331251 | Phase I/II (terminated as investigator is no longer at site) | Advanced cancer, sarcoma | 2 DLT reported | ≥ G3 TRAE reported in 12 patients (n = 17) | |
| Doxorubicin | NCT03056001 | Phase II (completed) | STS |
1 CR, 8 PR, 12 SD, 33% ORR (n = 27) mPFS: 6.9 months mOS: 15 months |
26 ≥ G3 TRAE | |
| Doxorubicin hydrochloride | NCT02888665 | Phase I/II (completed) | Sarcoma |
No DLT reported Overall: 7 PR, 2 unconfirmed PR, 11 SD, 19% ORR (n = 37) Phase II: 4 PR (n = 31) mPFS: 8.1 months (95%CI: 7.6–10.8 months) mOS: 27.6 months (95%CI: 18.7%—NR) |
24 ≥ G3 TRAE Notable pembrolizumab-related toxic effects included grade 3 adrenal insufficiency (n = 1) and hypothyroidism (n = 7) |
|
| Epacadostat | NCT03414229 | Phase II (active, not recruiting) | Sarcoma |
1 PR, 47% DCR (CR + PR + SD) (n = 30) mPFS: 7.6 weeks (95% CI: 6.9–26.7 weeks) mOS: 16.9 weeks (95% CI: 9.4 weeks – not estimable) |
7 ≥ G3 TRAE | |
| Eribulin | NCT03899805 | Phase II (active, not recruiting) | LPS, LMS, UPS |
1 PR, 5SD, 5.3% ORR (n = 19 in LMS cohort) mPFS: 11.1 weeks in LMS cohort |
68% ≥ G3 TRAE in LMS cohort | |
| Gemcitabine | NCT03123276 | Phase I/II (unknown) | LMS, UPS |
DLT observed at gemcitabine 1000 mg/m2, but not confirmed in the expansion cohort LMS: 8 SD, 3 PD (n = 11) UPS: 2 PR (n = 2) mPFS: 5.1 months (95% CI: 2–9 months) |
NA | |
| IFN-γ-1β | NCT03063632 | Phase II (active, not recruiting) | Mycosis Fungoides and Sezary syndrome, myxoid LPS, round cell LPS, SS | NA | NA | |
| Lenvatinib | NCT04784247 | Phase II (recruiting) | Sarcoma | NA | NA | |
| NCT05147558 | Phase II (recruiting) | Uterine carcinosarcoma | NA | NA | ||
| NCT05846724 | Phase II (not yet recruiting) | Relapsed/refractory Kaposi sarcoma | NA | NA | ||
| Modified vaccinia virus Ankara vaccine expressing p53 | NCT02432963 | Phase I (not recruiting) | Solid tumors, STS |
1 DLT reported 3 SD (n = 11) |
1 fatal G5 myocarditis reported 10 ≥ G3 TRAE |
|
| NA | NCT02301039 | Phase II (completed) | BS, STS |
5.0% PR (95% CI: 71.0–16.9%) (n = 40 in BS), 17.5% PR (95% CI: 7.3–32.8%) (n = 40 in STS), 13.0% PR (95% CI: 5.5–25.3%) (n = 53 in expansion cohort) mPFS: 8 weeks (95% CI: 7–9 weeks) (n = 39 in BS), 18 weeks (95% CI: 8–22 weeks) (n = 37 in STS), 8 weeks (95% CI: 7–13 weeks) (n = 53 in expansion cohort) mOS: 52 weeks (95% CI: 40–72 weeks) (n = 42 in BS), 49 weeks (95% CI: 34–73 weeks) (n = 42 in STS), 57 weeks (95% CI: 33–86 weeks) (n = 60 in expansion cohort) |
15 ≥ G3 TRAE in BS 19 ≥ G3 TRAE in STS cohort 19 ≥ G3 TRAE in expansion cohort |
|
| NCT02691026 | Phase II (terminated due to slow enrollment as a result of low incidence of MPNST and the COVID-19 pandemic) | MPNST | NA | NA | ||
| NCT03012620 | Phase II (active, not recruiting) | CNS neoplasm, germ cell/embryonal neoplasms, neuroendocrine carcinoma, NK/T cell lymphoma, ovarian neoplasm, sarcoma, thyroid cancer |
1 CR, 14 PR, 33 SD (n = 98) mPFS: 2.75 months (n = 98 in overall), 7.5 months (ASPS), 6.6 months (chordoma), 2.1 months (DSRCT) mOS: 19/7 months (n = 98 in overall), 10 months (DSRCT) |
NA | ||
| NCT03013127 | Phase II (terminated due to poor clinical benefits) | OGS |
9 PD with no clinical benefit after 18 weeks of treatment (n = 12) mPFS: 1.7 months (95% CI: 1.2–2.2 months) mOS: 6.6 months (95% CI: 3.8–9.3 months) |
0 ≥ G3 TRAE | ||
| NCT03316573 | Phase II (suspended due to low accrual) | Follicular dendritic cell sarcoma, histiocytic sarcoma, interdigitating dendritic cell sarcoma, lymphoma | NA | NA | ||
| NCT03469804 | Phase II (active, not recruiting) | Classic and endemic Kaposi sarcoma | 2 CR, 10 PR, 5 SD, 71% BORR (95%CI: 44–90%) (n = 17) | 2 ≥ G3 TRAE | ||
| Olaparib | NCT05156268 | Phase II (recruiting) | Endometrial carcinosarcoma | NA | NA | |
| Olaratumab | NCT03126591 | Phase I (completed) | STS |
0 CR, 6 CR, 9 SD (n = 28) mPFS: 2.7 months (95% CI:1.3–4.07 months) mOS: 14.8 months (95% CI: 12.6 months – NR) |
≥ G3 TRAE in 2 patients reported | |
| ± Pazopanib | NCT05679921 | Phase II (not yet recruiting) | STS | NA | NA | |
| RT | NCT03338959 | Phase I/II (active, not recruiting) | STS | NA | NA | |
| + RT or SOC alone | NCT03092323 | Phase II (recruiting) | STS | NA | NA | |
| T-VEC | NCT03069378 | Phase II (active, not recruiting) | Cutaneous angiosarcoma, epithelioid sarcoma, MFS, UPS (expansion cohort) |
43% BORR (95%CI: 0.1–0.82) (n = 7 in cutaneous angiosarcoma cohort), 0% BORR (n = 3 in epithelioid sarcoma), 11% BORR (95% CI: 0.0–0.48) (n = 9 in MFS/UPS cohort) mPFS: 54 weeks (95% CI: 3 weeks – NR in cutaneous angiosarcoma cohort), NA in cutaneous angiosarcoma cohort, 14.9 weeks (95% CI: 7–110 weeks in MFS/UPS cohort) |
1 ≥ G3 TRAE in cutaneous angiosarcoma cohort | |
| Ziv-Aflibercept | NCT02298959 | Phase I (active, not recruiting) | Advanced cancer, sarcoma |
No DLT reported Melanoma: 1 CR, 1 PR Mesothelioma: 1 PR RCC: 1 PR mOS: 3.3 months (CRC), (90% CI: 0.6–3.4 months), NR (melanoma), 12.5 months (ovarian), (90% CI: 3.8–13.6 months), NR (others), 15.7 months (RCC) (90% CI: 2.5–15.7 months), |
G3 TRAE reported in 19 patients (n = 33) | |
| ± Pembrolizumab | Bevacizumab ± pegcetacoplan | NCT04919629 | Phase II (recruiting) | Fallopian tube carcinosarcoma, primary peritoneal cancer, recurrent ovarian, fallopian tube cancer | NA | NA |
| BT-001 | NCT04725331 | Phase I/II (recruiting) | Sarcoma, solid tumors | NA | NA | |
| Eribulin mesylate | NCT05619913 | Phase II (recruiting) | Ovarian carcinosarcoma, uterine carcinosarcoma | NA | NA | |
| GI-101 ± lenvatinib or RT | NCT04977453 | Phase I/II (recruiting) | Advanced solid tumors, sarcoma | 1 PR (n = 16 in GI-101 monotherapy), 2 PR (n = 9 in GI-101 + pembrolizumab arm) |
≥ G3 TRAE reported in 3 patients in GI-101 monotherapy arm No ≥ G3 TRAE reported in GI-101 + pembrolizumab arm |
|
| KVA12123 | NCT05708950 | Phase I/II (recruiting) | Sarcoma, solid tumors | NA | NA | |
| MQ719 | NCT05859074 | Phase I (recruiting) | Kaposi sarcoma, solid tumors | NA | NA | |
| Mupadolimab ± or ciforadenant | NCT03454451 | Phase I (active, not recruiting) | Advanced cancer, sarcoma | No objective responses by RECIST criteria were observed (n = 34) | 28 ≥ G3 TRAE | |
| Nanatinostat + valganciclovir | NCT05166577 | Phase I/II (recruiting) | EBV + LMS, EBV + sarcoma, EBV + solid tumors | NA | NA | |
| RT | NCT05488366 | Phase I (recruiting) | STS | NA | NA | |
| T3011 | NCT04370587 | Phase I/II (recruiting) | HNSCC, melanoma, NSCLC, sarcoma, solid tumor, squamous cell carcinoma | No DLT reported | No treatment related serious adverse events reported | |
| LY3435151 | NCT04099277 | Phase I (terminated due to strategic business decision) | LMS, solid tumors, UPS | NA | NA | |
| Pembrolizumab/nivolumab | Autologous HER2 CAR T cells | NCT04995003 | Phase I (recruiting) | HER2 + sarcoma | NA | NA |
| Retifanlimab | Docetaxel + gemcitabine | NCT04577014 | Phase I/II (recruiting) | STS | 17% ORR (95% CI: 1%-64%) and 50% (95%: 19%-81%) in the run in (n = 7) and de-escalation (n = 6) cohort, 100% DCR (95% CI: 52%-100%) | 11 ≥ G3 TRAE |
| ± Retifanlimab | Doxorubicin + ifosfamide | NCT04968106 | Phase II (recruiting) | Resectable sarcoma | NA | NA |
| Sintilimab | Doxorubicin hydrochloride + ifosfamide | NCT04356872 | Phase II (unknown) | DDLPS, myxoid liposarcoma, UPS, SS | 62.5% ORR (n = 24) | 1/6 DLT |
| Surufatinib + RT | NCT05839275 | Phase Ib/II (recruiting) | High risk localized STS | NA | NA | |
| Spartalizumab | NA | NCT04802876 | Phase II (active, not recruiting) | PD-1-high mRNA expressing tumors, sarcoma | NA | NA |
| Toripalimab | NA | NCT03474640 | Phase I (active, not recruiting) | Advanced malignancies, chondrosarcoma, STS | NA | NA |
AIDS Acquired immunodeficiency syndrome, ASPS Alveolar soft part sarcoma, BORR Best overall response rate, BS Bone sarcoma, CAB-AXL-ADC Conditionally active biologic AXL-targeted antibody drug conjugate, CAR Chimeric antigen receptor, CI Confidence interval, CNS Central nervous system, CR Complete response, CRC Colorectal cancer, CTLA4 Cytotoxic T-lymphocyte-associated protein 4, DCR Disease control rate, DDLPS Dedifferentiated liposarcoma, DLT Dose-limiting toxicity, DSRCT Desmoplastic small round cell tumor, EBV Epstein-Barr virus, ES Ewing sarcoma, GIST Gastrointestinal stromal tumor, HER2 Human epidermal growth factor receptor 2, HIV Human immunodeficiency virus, HNSCC Head and neck squamous cell carcinoma, HR Hazard ratio, IFN-γ-1β Interferon-γ-1β, IgG Immunoglobulin G, INI1 Integrase interactor 1, IQR Interquartile range, irAEs Immune-related adverse events, LMS Leiomyosarcoma, LPS Liposarcoma, MFS Myxofibrosarcoma, mOS Median overall survival, mPFS Median progression-free survival, MPNST Malignant peripheral nerve sheath tumor, mRNA Messenger ribonucleic acid, NA Not available, NCT National Clinical Trial, NK cells Natural killer cells, NR Not reached, NSCLC Non-small cell lung cancer, NY-ESO-1 New York Esophageal Squamous Cell Carcinoma 1 gene, OGS Osteosarcoma, ORR Objective response rate, PD-1 Programmed cell death 1, PD-L1 Programmed death-ligand 1, PD Progressive disease, PR Partial response, RCC Renal cell carcinoma, RT Radiotherapy, SABR Stereotactic ablative radiotherapy, SBRT Stereotactic body radiation therapy, SCLC Small cell lung cancer, SD Stable disease, SOC Standard of care, SS Synovial sarcoma, STS Soft-tissue sarcoma, TIL Tumor infiltrating lymphocyte, TNBC Triple-negative breast cancer, TRAE Treatment-related adverse event (G3 = grade 3), T-VEC Talimogene Laherparepvec, UPS Undifferentiated pleomorphic sarcoma
In this review, we will consider existing US FDA-approved and European Medicines Agency (EMA)-approved biomarkers for ICIs in clinical practice and evaluate their applicability in sarcomas. We then discuss exploratory biomarkers and evidence for their potential utility in sarcomas. Predictive biomarkers covered in this review are illustrated in Fig. 1.
Fig. 1.

Overview of approved and exploratory biomarkers for immune checkpoint inhibitors (ICIs) in cancer. Tumor and immune features can influence response to ICIs and serve as predictive biomarkers for response. FDA- and EMA-approved biomarkers for ICIs in cancer are indicated in blue, while exploratory biomarkers are indicated in red. MSI and a high TMB contribute to the expression of tumor neoantigens presented by MHC I molecules on tumor cells that can be recognized by the TCR on CD8+ T cells, leading to antitumor T cell activity. In gastrointestinal cancers, the expression of immunogenic neoantigens in tumors with high TMB is dependent on certain mutational signatures [41]. On the other hand, binding of PD-L1 on tumor cells to PD-1 on T cells leads to the suppression of T cell antitumor activity. Additionally, exhausted T cells may also express the exhaustion markers TIM-3 and LAG-3. In lung adenocarcinoma, TP53 mutations are correlated with higher TMB and neoantigen expression, while TP53 missense but not nonsense mutations are associated with increased PD-L1 expression [42]. Various GES have also been associated with response to ICIs. IDO contributes to T cell suppression and its expression was induced in resistant HCC after ICI therapy [43]. The presence of B cells and TLS have been associated with improved prognosis and response to ICIs in several cancers, including sarcomas. Within the blood, a higher baseline circulating NLR has also been found to correlate with poorer outcomes in patients receiving ICIs in lung cancer [44].
Biomarkers approved for immune checkpoint inhibition in cancer
ICI therapy is indicated without biomarker requirement in several cancer settings because of studies demonstrating improved clinical outcomes [45]. These indications include patients with advanced melanoma [46–48], relapsed or refractory Hodgkin lymphoma [49, 50], cisplatin-ineligible patients with urothelial carcinoma [49, 50], patients with relapsed or refractory primary mediastinal large B-cell lymphoma [51, 52], second-line treatment for patients with HCC [49, 53], patients with Merkel cell carcinoma [49, 53], patients with recurrent or metastatic HNSCC [24, 54] and Bacillus Calmette-Guérin-unresponsive high risk non-muscle invasive bladder cancer [55]. In contrast, there are cancer types such as sarcoma [35], breast, prostate and colon cancers [56] that demonstrate lower frequency of response to ICI therapy, and would therefore require biomarkers to distinguish between responders and non-responders.
Currently, only three predictive biomarkers have been approved by the FDA for ICI therapy in cancers, namely programmed death-ligand 1 (PD-L1), microsatellite instability (MSI) or defective mismatch repair (dMMR), and tumor mutational burden (TMB), while only two predictive biomarkers, namely PD-L1 and MSI/dMMR have been approved by the EMA [57]. Variability in the antibody clones, expression thresholds, scoring systems and the cell types expressing PD-L1 among FDA/EMA-approved PD-L1 assays across multiple cancer types can pose difficulty of interpretation for researchers and clinicians. PD-L1 assays were previously described by Wang et al. to have poor diagnostic accuracy, poor predictability, and low negative predictive value in cancers [58], also limiting its clinical use in sarcomas. For the detection of MSI-high (MSI-H) tumors, approved assay methods include immunohistochemistry (IHC), polymerase chain reaction (PCR) and whole exome sequencing (WES). Both IHC and PCR are established methods and are widely available in the pathology laboratory. However, IHC is limited by its low analytic sensitivity and accuracy, while PCR may be unable to capture full MSI profiles that results in missing 0.3% to 10% of MSI-H cases [58, 59]. Circumventing the limitations of PCR, WES provides better predictive power compared to PCR and can be used for all tumor types [58]. Additionally, TMB can be derived from WES and may provide a better prediction of response to ICIs [58]. On the other hand, WES is characterized by high cost, limited availability, potentially complicated pipelines and requires technical expertise that may hinder its clinical utility [60]. Table 2 summarizes FDA- and EMA-approved predictive biomarkers for ICIs in selected cancers.
Table 2.
Overview of Food and Drug Administration (FDA)- and European Medicines Agency (EMA)-approved predictive biomarkers for patient selection for immune checkpoint inhibition
| Predictive Biomarkers | Assay Methods | Antibody | Expression Threshold | Cancers | Regulatory Authority | NCT Number | Author, Year |
|---|---|---|---|---|---|---|---|
| PD-L1 | PD-L1 IHC 22C3 pharmDx assay | Monoclonal mouse anti PD-L1 clone 22C3 | PD-L1 CPS ≥ 20 and CPS ≥ 1 | TNBC | FDA | NCT02622074 | Schmid et al., 2020 [61] |
| CPS ≥ 1 | HNSCC | FDA/EMA | NCT02358031 | Burtness et al., 2019 [62] | |||
| TPS ≥ 50% | NSCLC | FDA/EMA | NCT02142738 | Reck et al., 2019 [63] | |||
| CPS ≥ 10 | UC | EMA | NCT02256436 | Bellmunt et al., 2017 [64] | |||
| PD-L1 IHC 28–8 pharmDx assay | Monoclonal rabbit anti PD-L1 clone 28–8 | TPS ≥ 1% | NSCLC | FDA/EMA | NCT02477826 | Hellmann et al., 2019 [65] | |
| VENTANA SP142 PD-L1 IHC assay | Monoclonal rabbit anti PD-L1 clone SP142 | IC ≥ 1% | TNBC | FDA/EMA | NCT02425891 | Schmid et al., 2018 [30] | |
| TC ≥ 50% or IC ≥ 10% | NSCLC | FDA | NCT02008227 | Rittmeyer et al., 2017 [66] | |||
| IC ≥ 5% | UC | FDA/EMA | NCT02108652 | Rosenberg et al., 2016 [67] | |||
| VENTANA SP263 assay | Monoclonal rabbit anti PD-L1 clone SP263 | TC ≥ 25% or IC ≥ 25% | UBC | FDA | NCT01693562 | Massard et al., 2016 [65, 68] | |
| MSI | PCR or IHC | - | MSI-H/dMMR | Colorectal cancer | FDA/EMA | NCT02460198 | Le et al., 2020 [69] |
| Fluorescent Multiplex PCR-based method | - | MMR-deficient or proficient | Progressive metastatic carcinomas | FDA | NCT01876511 | Le et al., 2015 [70] | |
| TMB | FoundationOne CDx assay | - | tTMB-high ≥ 10 mutations per Mb | Advanced solid tumors | FDA | NCT02628067 | Marabelle et al., 2020 [71] |
| WES | - | NA | Advanced solid tumors | FDA | NCT02054806 | Ott et al., 2019 [72] |
Year = year of publication
CPS Combined positive score, dMMR Deficient mismatch repair, HNSCC Head and neck squamous cell carcinoma, IC Percentage of tumor-infiltrating immune cells within the tumor area expressing PD-L1, IHC Immunohistochemistry, MMR Mismatch repair, MSI Microsatellite instability, MSI-H Microsatellite instability-high, NCT National Clinical Trial, NSCLC Non-small cell lung cancer, PCR Polymerase chain reaction, PD-1 Programmed cell death 1, PD-L1 Programmed death-ligand 1, TC Percentage of tumor cells within total tumor cells expressing PD-L1, TMB Tumor mutational burden, TNBC Triple-negative breast cancer, TPS Tumor proportion score, tTMB Tissue tumor mutational burden, UBC Urothelial bladder cancer, UC Urothelial carcinoma, WES Whole exome sequencing
Programmed death-ligand 1 (PD-L1)
PD-L1 is a ligand for the T cell immune checkpoint receptor programmed cell death 1 (PD-1) and is expressed by a variety of normal and immune cells. Interaction between PD-1 and PD-L1 serves to promote self-tolerance through the suppression of T cell activation. Cancer cells have been found to exploit the PD-1/PD-L1 axis for immune evasion through the overexpression of PD-L1 [73]. Thus, PD-1 and PD-L1 expression provide an attractive avenue to predict response to ICI therapy. At present, there are four FDA- and three EMA-approved PD-L1 assays (Table 2). For further reading, a detailed review on the key parameters for the FDA-approved PD-L1 assays has been conducted by Wang et al., describing different test methods and challenges [58].
The diverse and dynamic PD-L1 expression on specific cell types within the tumor microenvironment (TME) has made the correlation of global PD-L1 expression with response to ICI therapy challenging. Noguchi et al. demonstrated that PD-L1 expression in tumor-associated macrophages are partially dependent on interferon-γ (IFN-γ) [74]. Further studies by Lau et al. in PD-L1-depleted mouse models highlighted that although immune evasion occurs at a repressed rate, infiltrating myeloid cells may contribute to immune evasion through compensatory PD-L1 expression [75]. There is also contradicting evidence demonstrating that efficacy of PD-L1 blockade is independent of PD-1/PD-L1 expression on tumor cells [76]. Instead, PD-L1 expression on dendritic cells (DCs) and macrophages correlates to clinical response in melanoma and ovarian cancer patients [76]. Given that PD-L1 expression level in the TME is highly variable, global PD-L1 positivity alone may not be sufficient to predict response to ICIs [77]. Instead, understanding the effects of differential expression of PD-L1 in specific immune and tumor cells in the TME may reveal mechanisms of the PD-1/PD-L1 axis that could be exploited to better predict response to ICI therapy.
In sarcomas, PD-L1 expression levels have shown conflicting association with ICI response [78]. Indeed, levels of PD-L1 expression can vary widely between different histological subtypes [79] (Fig. 2) that is further complicated by the heterogenous TME present in primary and metastatic lesions [78, 80]. This high degree of heterogeneity in PD-L1 expression, coupled with limited studies clarifying the relationship between PD-L1 expression and response to ICI warrants further investigation of the use of PD-L1 testing in sarcomas. Additionally, Patel et al. demonstrated that pre-treatment with radiotherapy (RT) prior to surgical resection increased PD-L1 expression in 10.9% of patient STS tumors (p = 0.056) while post-operative radiation therapy did not elicit PD-L1 expression in any STS resection samples [81]. These findings suggest that PD-L1 expression can be influenced by other treatment modalities, though much work remains to be done due to the small study sample sizes and limited studies available in sarcomas.
Fig. 2.

Prevalence of PD-L1 expression in soft-tissue sarcomas across published studies. This figure shows the levels of PD-L1 expression in different sarcoma subtypes that has been reported across a number of studies [79, 81–90]. Inter- and intra-variability of PD-L1 expression among different sarcoma subtypes warrants extensive studies to establish the use of existing PD-L1 assays as a reliable predictive biomarker to immune checkpoint inhibition in soft tissue sarcomas (STS). ARMS: Alveolar rhabdomyosarcoma; ASPS: Alveolar soft part sarcoma; DDLPS: Dedifferentiated liposarcoma; ERMS: Embryonal rhabdomyosarcoma; ES: Ewing sarcoma; LMS: Leiomyosarcoma; LPS: Liposarcoma; MFS: Myxofibrosarcoma; MPNST: Malignant peripheral nerve sheath tumor; OGS: Osteosarcoma; PRMS: Pleomorphic rhabdomyosarcoma; SS: Synovial sarcoma; UPS: Undifferentiated pleomorphic sarcoma; WD-LPS: Well differentiated liposarcoma
Microsatellite Instability (MSI)/ Deficient Mismatch Repair (dMMR)
MSI occurs when dMMR results in hypermutation in short stretches of DNA (microsatellites). MSI-H have higher potential to code for tumor-associated neoantigens [91] that can be recognized by the immune system, eliciting an antitumor response. A phase II study by Le et al. demonstrated that high levels of somatic mutations in dMMR colorectal tumors was associated with increased expression of tumor-associated antigens compared to proficient mismatch repair (pMMR) colorectal tumors [70]. In the same study, 40% of patients with dMMR tumors responded to PD-1 inhibition, while none of the patients with pMMR tumors achieved an objective response, thus highlighting the role of dMMR as a predictive biomarker for ICI response.
Currently, IHC, PCR and next-generation sequencing (NGS) are used to assess MSI [92]. In the same review mentioned previously, Wang et al. has provided a comprehensive evaluation of the three assays in use [58].
A meta-analysis by Lorenzi et al. reported the prevalence of dMMR among six common tumor types, including colorectal, endometrial, esophageal, gastric, renal and ovarian cancers, which suggested that the prevalence of dMMR/MSI differs between tumor types and cancer stages [93] (Fig. 3). Notably, MSI/dMMR accounts for only approximately 1% of sarcomas, with the exception of pleomorphic rhabdomyosarcoma (PRMS), embryonal rhabdomyosarcomas (ERMS), LMS and malignant peripheral nerve sheath tumor (MPNST) that have higher rates of MSI/dMMR [94]. Given the low prevalence of MSI-H tumors in sarcomas and the lack of trials evaluating the role of MSI in predicting ICI treatment response in sarcomas, MSI/dMMR may be of limited use in guiding the clinical decision-making for ICIs in sarcomas.
Fig. 3.
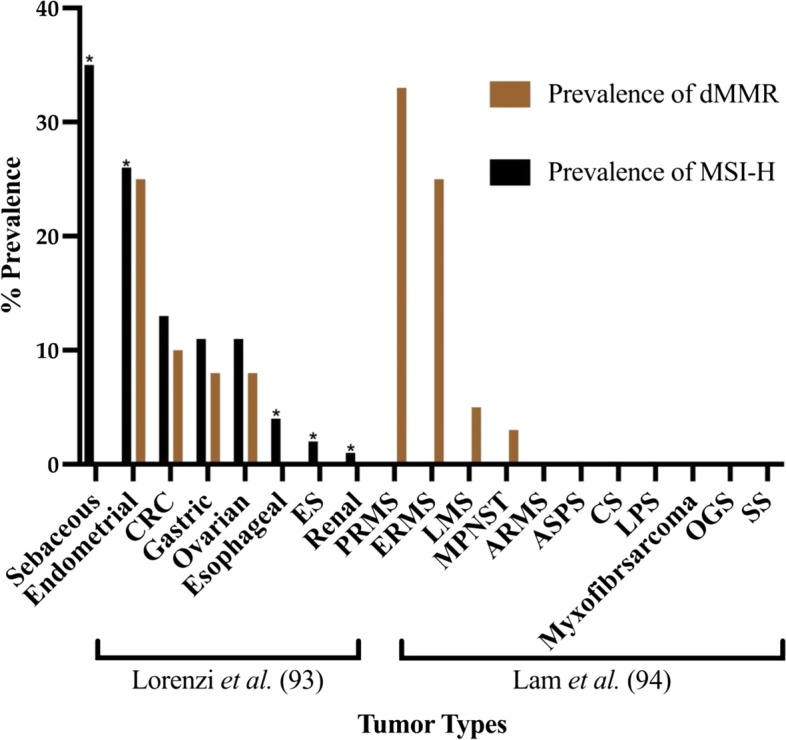
Pooled prevalence of MSI-H and dMMR among different tumor types. Bar graphs show the prevalence of MSI-H and dMMR in various cancers as summarized by Lorenzi et al. and Lam et al. [93, 94]. Low prevalence of MSI-H in Ewing sarcoma (ES) and wide variation of dMMR between sarcoma subtypes warrants further studies to explore the correlation between MSI-H / dMMR and clinical response to immune checkpoint inhibition. Results from Lorenzi et al. were pooled from various studies. Lam et al. did not evaluate for MSI-H. Asterisk indicates analysis for dMMR was not feasible. ARMS: Alveolar rhabdomyosarcoma; ASPS: Alveolar soft part sarcoma; CRC: Colorectal cancer; CS: Chondrosarcoma; ERMS: Embryonal rhabdomyosarcoma; ES: Ewing sarcoma; LMS: Leiomyosarcoma; MPNST: Malignant peripheral nerve sheath tumor; OGS: Osteosarcoma; PRMS: Pleomorphic rhabdomyosarcoma; SS: Synovial sarcoma. Asterisk indicates analysis for dMMR was not included
Tumor Mutation Burden (TMB)
Cancer neoantigens are tumor-specific antigens that arise from genetic mutations within tumor cells that can be recognized by the immune system. Hence, highly mutated tumors are more likely to express neoantigens and provide an opportunity for ICIs to reinvigorate the immune system and stimulate an antitumor response [95]. As predicted, improved survival after ICI treatment was indeed observed in patients with high TMB in multiple cancer types [96, 97].
However, the use of high TMB as a predictive biomarker for ICI response has demonstrated conflicting results in gastrointestinal cancers, with most studies reporting the lack of a significant association between high TMB and response to ICIs [71, 98–101]. A retrospective study by Wang et al. analyzed the mutational signatures of microsatellite-stable gastrointestinal tumors with high TMB and found that not all genes associated with high TMB correlated with an enhanced antitumor response, hence suggesting that the types of mutational signatures in tumors could play a role in the expression of immunogenic neoantigens [41].
TMB is defined as the number of somatic mutations in the tumor exome [96] and can be classified into low (1–5 mutations per Mb), intermediate (6–19 mutations per Mb) and high (≥ 20 mutations per Mb) [102]. TMB can be measured using WES, but clinical implementation has been limited due to the large amount of genomic deoxyribonucleic acid (DNA) required, long sequencing time, availability of matched samples and costs [103]. To circumvent the limitations of WES, targeted NGS panels have been developed to accurately recapitulate WES-derived genomic information while sequencing less DNA [60, 96, 104]. In assessing TMB, both WES and targeted NGS panels can be influenced by various factors from sample collection, processing, sequencing, data analysis to the lack of harmonization in reporting cut-offs, thus limiting the independent clinical utility of TMB [58].
Studies analyzing genomic profiles in sarcomas have suggested low somatic mutation burden across most sarcomas. A study of the molecular landscape of adult STS demonstrated an average of 1.06 mutations per Mb across 206 sarcomas of different histological subtypes [105], while genomic profiling of over 6,100 sarcoma cases showed a median of 1.7 mutations per Mb [106]. Additionally, even in dMMR sarcomas, TMB appears lower than that in other dMMR tumor types, with a median TMB of 16 mutations per Mb compared to 28 mutations per Mb [107]. The exception appears to be head and neck angiosarcomas, where 63.4% of cases have high TMB defined as ≥ 10 mutations per Mb [108]. Even so, in a phase II clinical trial of metastatic or unresectable angiosarcoma treated with combined ipilimumab and nivolumab (NCT02834013), the objective response rate (ORR) was only 25% and six-month progression-free survival (PFS) was 38% [109].
Overall, the lack of studies examining the use of TMB as a predictive biomarker of ICI response in sarcomas, poor stratification of TMB classification, as well as a low median TMB across most sarcomas may limit the clinical utility of TMB in directing ICI use in sarcomas.
Exploratory biomarkers for immune checkpoint inhibition in sarcomas
In this section, we discuss eight exploratory biomarkers that may predict response to ICI therapy in sarcomas, including gene expression signatures (GES), circulating neutrophil-to-lymphocyte ratio (NLR), indoleamine 2,3-dioxygenase (IDO), lymphocyte activation gene 3 (LAG-3), T cell immunoglobulin and mucin domain-containing protein 3 (TIM-3), TP53 mutation status, B cells, and tertiary lymphoid structures (TLS).
Gene Expression Signatures (GES)
GES are presented as a group of genes whose differential expression has been found to be associated with a particular outcome, and have been used in the determination of diagnosis, prognosis, and the prediction of therapeutic outcomes [110]. Methods used to measure gene expression levels include ribonucleic acid (RNA) microarray and RNA sequencing [111, 112], as well as newer methods including single-cell RNA sequencing, single-nucleus RNA sequencing [113] and spatial transcriptomics [114].
In several cancers, various GES have been found to be capable of predicting ICI response, including in melanoma [115–117], NSCLC [118–121], gastric cancer [122], lower-grade glioma [123] and some across multiple cancer types such as in both NSCLC and melanoma [124]. In addition, a pan-tumor signature predictive of ICI response was derived from 220 patients across HNSCC, gastric cancer, triple-negative breast cancer, bladder, anal canal, biliary, colorectal, esophageal, and ovarian cancers. This pan-tumor signature defined by Ayers et al. contains IFN-γ- and T cell-associated inflammatory genes, and high expression of this gene signature correlated well with objective response to pembrolizumab (1-sided p-value < 0.001) [125].
In STS, given the heterogeneity in genomic alterations across the various histological subtypes [126], identifying a robust GES that is able to be used in multiple subtypes may prove to be challenging. Nonetheless, Petitprez et al. identified a B lineage signature associated with improved response to ICI therapy in STS [127], and this will be discussed in further detail in the section on B cells below.
Presently, the implementation of routine gene sequencing is costly, and the complexity of its results require expertise to analyze and interpret before they can be used to guide clinical decision making [128, 129]. There is a thus a need to identify a GES with minimal number of genes to be sequenced in order to determine response to ICIs, with its accuracy subsequently being validated in a prospective trial.
Circulating Neutrophil-to-Lymphocyte Ratio (NLR)
Compared to other biomarkers that may require patients’ tumor samples, NLR can be easily derived from whole blood as a less invasive procedure with minimal risk of complications. The ease of sample acquisition and minimal patient risk has led to extensive studies of its use in cardiovascular diseases, infectious diseases, and cancers where it has been found to correlate with prognosis [130].
In the published literature, there is a lack of clearly defined cutoffs as well as contrasting evidence for the use of NLR across and within the different cancer types [131]. In a retrospective study of 509 patients with advanced cancer, a non-linear response trend during ICI treatment was observed and significant decreases or increases in NLR on-treatment correlated to poorer prognostic outcomes [132]. Conversely, in a meta-analysis by Jing et al., higher NLR at baseline across 23 studies correlated to lower OS in lung cancer patients receiving ICIs [44]. In STS, Strong et al. found that high baseline NLR, defined as ≥ 4.5, was not independently associated with worse survival outcomes in patients with extremity STS [133]. On the other hand, Chan et al. used receiver operating curve analysis to determine a cutoff of high NLR at > 2.5, and demonstrated high baseline NLR to be an independent marker for poor prognosis in STS patients [134].
Overall, while the use of NLR in the clinic is less invasive and more convenient, the lack of harmonization in key parameters such as a standardized baseline NLR may hinder the use of NLR as a predictor of response to ICIs in sarcomas. The establishment of clearly defined cutoffs would be essential to support its use.
Indoleamine 2,3-Dioxygenase (IDO)
IDO is a heme-containing enzyme that catalyzes the conversion of tryptophan into kynurenine. IDO contributes to an immunosuppressive effect involving both CD4+ and CD8+ T cells via the rapid depletion of tryptophan [135]. Subsequent downstream activation of stress response mediator general control nonderepressible 2 (GCN2) kinase results in cell cycle arrest [136], thus inhibiting T cell proliferation. Additionally, IDO has been demonstrated to upregulate regulatory T cell (Treg) activation and activity [137, 138]. Thus, IDO has been suggested for use as a prognostic marker.
In a meta-analysis by Wang et al., high expression of IDO in tumor tissues was associated with poor prognosis (pooled hazard ratio (HR) 1.92, 95% CI, 1.52–2.43, p < 0.001) and tumor progression (pooled HR = 2.25, 95% CI, 1.58–3.22, p < 0.001) in cancer patients [135]. An in vitro study has also shown that ICI therapy induces IDO in resistant HCC through upregulation of IFN-γ that consequently results in adaptive immune evasion [43]. These studies shed light on alternative immune evasion pathways conferred in the TME.
In sarcomas, Hiroshi et al. analyzed 47 patient specimens in which 96% of high-grade osteosarcoma of the extremities are IDO-positive [139]. Consequently, IDO positivity has been correlated to decreased progression free survival (PFS) (p = 0.016) and OS (p = 0.005) [139]. To circumvent IDO-induced resistance, IDO inhibitors have been proposed to be included in combination treatment with ICIs. Imatinib, a tyrosine kinase inhibitor used in the treatment of gastrointestinal stromal tumor (GIST), has demonstrated inhibition of IDO expression in GIST mouse models [140]. However, clinical trials testing for combination treatment with ipilimumab and imatinib demonstrated limited efficacy and antitumor immune response in GISTs [141].
In conclusion, IDO has been recognized as an immune target in the TME, and the combination of IDO inhibitors with ICIs has also shown efficacy in several phase I/II clinical trials [142]. However, the phase III trial of epacadostat with pembrolizumab in unresectable or metastatic melanoma (NCT02752074) failed to demonstrate better efficacy versus placebo and pembrolizumab [143]. Taken together, there is a need for deeper understanding of the role that IDO plays in the TME before establishing IDO as a biomarker.
Lymphocyte-activation gene 3 (LAG-3)
In March 2022, the FDA approved a LAG-3 ICI (relatlimab) given in combination with the PD-1 inhibitor nivolumab, expanding the list of immunotherapeutic options in advanced melanoma [144]. LAG-3 is an inhibitory molecule expressed by activated T cells and associates with the T cell receptor (TCR) and CD3 at the T cell surface [145]. The intracellular region of LAG-3 is responsible for transducing inhibitory signals to suppress T cell activation, but the molecular mechanisms governing this remain under investigation [146]. The known ligands of LAG-3 include major histocompatibility complex (MHC) class II [147, 148], galectin-3 [149] and fibrinogen-like protein 1 (FGL1) [150]. The utility of LAG-3 ICIs remains to be seen, but an early phase I/II study of combination treatment with LAG-3 and PD-1 inhibitor showed synergistic activity albeit with modest antitumor response [151]. For further reading, Huo et al. recently reviewed the clinical development of these novel agents [152], which will not be further elaborated on in this review.
In STS, analysis of blood samples from patients and healthy donors found that LAG-3 expression in peripheral T cells was correlated with the degree of intratumoral CD8+ T cell infiltration and poor prognosis [153]. Due to the novelty of anti-LAG-3 antibodies, there have been limited clinical trials regarding the use of LAG-3 as a potential immune biomarker for ICI response. As ongoing and future research uncovers more about the role of LAG-3 in suppressing T cell activation and the molecular mechanisms governing this, we would then be able to better understand its place in cancer immunotherapy and as a predictive biomarker for ICI response in sarcomas.
T-cell immunoglobulin and mucin domain-containing protein 3 (TIM-3)
TIM-3 is an immune checkpoint receptor that has been found to be expressed on many types of immune cells, including CD4+ and CD8+ T cells [154], Treg cells [155], myeloid cells [156], natural killer (NK) cells [157] and mast cells [158, 159]. In CD8+ T cells, co-expression of TIM-3 and PD-1 has been observed on the most exhausted subset of tumor-infiltrating lymphocytes [160, 161].
TIM-3 has several ligands that bind to different regions on the receptor, including galectin-9 (Gal-9), phosphatidylserine, high mobility group protein B1 (HMGB1) and carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) [159]. Gal-9 is expressed and secreted by many hematopoietic cells and some tumor cells, and its binding has been reported to result in T cell inhibition and cell death [159, 162]. HMGB1 binds to DNA from dying cells and is also secreted by tumor cells. HMGB1 binding to DNA facilitates their uptake and activation of toll-like receptors (TLRs), but it can also be bound by TIM-3, which sequesters it and prevents its activation of TLRs, thereby dampening antitumor immunity [159, 163]. CEACAM1 is expressed by T cells [164], DCs [165], monocytes [166] and macrophages [167], and its binding results in TCR signaling inhibition [164].
In mouse models of lung adenocarcinoma, Koyama et al. observed that in tumors which progressed following initial response to anti-PD-1 therapy, there was an upregulation of other immune checkpoint receptors, particularly TIM-3, on PD-1 antibody-bound T cells. Subsequent administration of combined anti-PD-1 and anti-TIM-3 therapy resulted in improved survival. The upregulation of TIM-3 was also seen in two patients who developed adaptive resistance to anti-PD-1 therapy, presenting TIM-3 upregulation as a possible biomarker of PD-1 therapy resistance [168].
Several anti-TIM-3 antibodies are being tested in phase I/II clinical trials, with some in combination with anti-PD-1/-PD-L1 antibodies, in the contexts of acute myelogenous leukemia, myelodysplastic syndrome, and various solid tumors. This combination has been demonstrated to be generally well-tolerated in early data and some anti-TIM-3 antibodies have displayed activity in lung cancer [169]. Nonetheless, the efficacy of these novel agents remains to be explored in sarcomas.
There have also been some studies evaluating the prognostic value of TIM-3 expression. Zang et al. demonstrated that TIM-3 was an independent prognostic indicator for poor OS in patients with malignant tumors (HR = 1.54; 95% CI, 1.19–1.98; p = 0.001) based on multivariate Cox regression analysis of 28 studies, and this was also observed in The Cancer Genome Atlas (TCGA) patient cohorts (HR = 1.2; p < 0.001). When stratified by tumor type, however, TIM-3 expression was not associated with OS in sarcoma (3 studies with 780 cases; p = 0.232) [170]. In contrast, Pu et al. reported that among 38 osteosarcoma tumor samples, 36 samples expressed TIM-3, and TIM-3 overexpression was associated with poorer OS (p < 0.001) [171].
Overall, anti-TIM-3 targeted therapy is still in its early stages of development, and more robust data on TIM-3 is needed to evaluate its role as a predictive biomarker for ICI therapy in sarcomas. Clinical trials evaluating the efficacy of anti-PD-1/PD-L1 antibodies combined with anti-TIM-3 antibodies could uncover more information on the relationship between immune checkpoint receptors within the TME.
TP53 mutation status
The tumor suppressor protein p53 is critical in the prevention of oncogenesis [172]. TP53 is the most frequently mutated gene among human cancers [172–174] and TP53 mutations commonly result in both loss of tumor suppressor function and gain of oncogenic function [175].
In sarcomas, TP53 is also one of the most frequently altered genes, albeit widely varying across histological subtypes [42, 127, 176–178]. Nassif et al. reported that TP53 mutation in sarcomas is associated with shorter disease-free survival (HR = 1.63; 95% CI, 1.04–2.54; Cox p = 0.032) and better treatment outcomes with anthracyclines (OR = 3.70; 95% CI, 1.20–11.97; p = 0.02) [42, 176, 177, 179, 180]. However, there has been a lack of studies evaluating the use of TP53 as an immune biomarker for ICI therapy in sarcomas.
Nevertheless, TP53 mutation status has been observed to be significantly correlated with PD-L1 expression [42] and response to ICI therapy in NSCLC [181–184]. In NSCLC and colorectal cancer (CRC), Agersborg et al. explored the relationship between mutation profile and PD-L1 expression and found that tumors with TP53 mutation in the NSCLC cohort had significantly higher PD-L1 expression (p = 0.01), though this was not observed in the CRC cohort (p = 0.5). In fact, the CRC cohort had significantly lower expression of PD-L1 (p = 0.0005) compared to the NSCLC cohort despite similar rates of TP53 mutation across both cancers, suggesting that varying mechanisms regulate PD-L1 expression across different tumor types [185].
In addition, Sun et al. compared lung adenocarcinoma TMB data of TP53-missense-mutant and TP53-nonsense-mutant groups to TP53-wild-type groups from Memorial Sloan Kettering Cancer Center (MSKCC) (p < 0.01 and p < 0.05 respectively), TCGA (p < 0.0001 for both) and GENE + (p < 0.0001 for both) databases using a Wilcoxon test and reported that both TP53-mutant groups demonstrated elevated TMB and neoantigen levels compared to the TP53-wild-type group [42].
Taken together, TP53 mutation status appears to be correlated with other biomarkers of ICI therapy in NSCLC. However, whether this is also true in sarcoma remains to be seen, as further investigation into the relationship between TP53 mutation status and response to ICIs is needed.
B Cells
B cells are responsible for the humoral arm of the adaptive immune system. Activation of naïve B cells by CD4+ T cells results in B cell proliferation, somatic hypermutation of immunoglobulin genes and class switching. Subsequently, activated B cells differentiate into plasmablasts and long-lived plasma cells which produce antigen-specific antibodies that are responsible for the clearance of antigens [186].
The role of B cells in the TME remains controversial, with conflicting evidence across different studies. A comprehensive review of publications investigating the prognostic value of tumor-infiltrating B cells in cancer found that 50% of studies reported a positive prognostic effect for B cells, while 9% and 40% reported a negative or neutral effect respectively [187]. An in vitro study showed that B cells suppress tumor immunity by downregulating the expression of IFN-γ in CD8+ T cells, a cytokine possessing antitumor activity [188], while increasing interleukin-10 (IL-10) production that further inhibits IFN-γ production by T cells [189]. Interestingly, co-culture of B cells with different cancer cell lines yielded different expression levels of IL-10, with sarcoma cells failing to stimulate IL-10 production in B cells, in contrast to Friend murine leukemia virus gag-expressing and melanoma cells which induced B cell IL-10 secretion [189]. In contrast, a separate study highlighted the antibody-mediated antitumor response of activated B cells in murine models of metastatic pulmonary tumors [190]. These conflicting reports of the role of B cells in antitumor immunity are likely due to heterogeneity of the B cell population within the TME, which could ultimately influence clinical outcomes.
Various subtypes of B cells are found in the TME. In tertiary lymphoid structures (TLS) within the TME, B cells are thought to be mainly involved in antigen presentation, where they help to activate both CD4+ and CD8+ T cells [191–194]. Subsequent antigen-driven maturation of B cells into plasma cells leads to the generation of in situ tumor antigen-specific antibodies [191]. Thus, B cells are instrumental in the generation of antitumor activity initiated within TLS. An immunosuppressive subset of B cells within the TME has also been described, commonly referred to as regulatory B cells. These cells act by secreting immunosuppressive cytokines [189] and have been identified in the TME of several cancers, including breast cancer [195], HCC [196], tongue squamous carcinoma [197], gastric cancer [198] and prostate cancer [199].
Increasing numbers of studies on immune subsets in the TME have led to the development of predictive biomarkers focused on the B cell compartment. In melanoma and RCC, B cell markers were enriched in tumors from responders versus non-responders to ICI therapy [178]. In another study involving the gene expression analysis of 3585 patients, a B cell-related gene signature comprising nine cytokine signaling genes was predictive of clinical response to ICI therapy in melanoma [200].
In STS, Petitprez et al. identified the overexpression of the B lineage signature as a distinctive feature of an immune class of sarcomas with high immune infiltration (p = 1.8 × 10–29) and found that it was also significantly associated with improved OS (p = 4.25 × 10–4). Patients in this immune class also demonstrated the best response to pembrolizumab defined by the percentage change in size of target lesions from baseline (n = 45, p = 0.026) in the SARC028 trial [127].
In conclusion, the role that B cells play in the TME is not clearly understood, given the numerous B cell subtypes present. Nonetheless, there is evidence for B cells playing a crucial role in response to ICI therapy in sarcomas and other cancers, as seen from the B cell-related gene signatures. Characterization of B cell subtypes in the TME as well as further validation of these gene signatures in larger cohorts and prospective trials could help identify the specific B cell populations and their cell states as a predictor for response to ICIs.
Tertiary Lymphoid Structures (TLS)
TLS are ectopic lymphoid structures that have been found to develop in response to chronic inflammation [201] and in various solid tumor types [202, 203]. Within the cancer literature, definitions of what constitutes a TLS as well as its maturation state vary significantly. Sautès-Fridman et al. and Vanhersecke et al. defined TLS as lymphoid aggregates consisting of B lymphocytes that are closely associated with plasma cells and T lymphocytes, making the distinction that mature TLS (mTLS) have at least one CD23+ follicular dendritic cell, while immature TLS (iTLS) are CD23− [201, 204]. In contrast, Lin et al. classified TLS into two categories based on their morphology – TLS aggregates, which are simply small clusters of lymphocytes; and TLS follicles, which are large clusters of lymphocytes that can be further distinguished based on the presence or absence of germinal centers [205].
TLS have been found to benefit prognosis [204–207] and are also associated with favorable ICI treatment outcomes [127, 204, 208–211] in several cancers. In a retrospective analysis of patient samples comprising 11 different tumor types from three independent cohorts by Vanhersecke et al., a higher proportion of patients with mTLS demonstrated objective response to ICIs compared to patients with iTLS or no TLS (36.9% versus 19.3% versus 19%, respectively, p = 0.015). Importantly, mTLS were predictive of response to ICIs regardless of PD-L1 expression [204]. Remarkably, in the phase II PEMBROSARC trial (NCT02406781) cohort, TLS-positive patients (n = 30) demonstrated a 6-month non-progression rate (NPR) and ORR of 40% (95% CI, 22.7–59.4) and 30% (95% CI, 14.7–49.4) respectively, compared to a 6-month NPR and ORR of 4.9% (95% CI, 0.6–16.5) and 2.4% (95% CI, 0.1–12.9) respectively, in the unselected all-comer cohorts [210]. Interestingly, in the study by Petitprez et al. mentioned in the previous section, at least one TLS was found in the TME of nine out of eleven tumors (82%) in the immune-high class of STS [127]. Taken together, this class of tumors is characterized by a high expression of the B lineage signature and the presence of TLS, further supporting the significance of the role that B cells and TLS play in the TME.
This significant improvement in clinical benefit highlights the potential for the presence of TLS to be utilized as a biomarker for the selection of patients with STS for ICI therapy.
Although TLS are emerging as key players in the TME, the exact mechanisms of their antitumor activity have not been fully elucidated. It has been proposed that TLS provide a favorable environment for antigen presentation and the differentiation and proliferation of lymphocytes in the TME as well as the generation of effector memory T cells, memory B cells and plasma cells [191, 201, 205]. In some TLS, spatial visualization through IHC has shown that B cells in TLS express markers of germinal center B cells, including activation-induced deaminase, the proliferation marker Ki67 and transcription factor B-cell lymphoma 6 (BCL6) [212]. The expression of these markers suggests an ongoing humoral immune response generated within TLS.
The growing evidence for TLS predicting response to ICI therapy thus gives rise to the important question of whether their use as predictive biomarkers can be implemented in clinical workflows. This will be discussed in the following section.
Clinical relevance of TLS as a predictive biomarker for ICI response in sarcomas
Of all the exploratory predictive biomarkers for response to ICI in sarcomas, the presence of TLS appears most promising thus far based on the results from the PEMBROSARC trial [210] and the study by Petitprez et al. [127]. However, the identification of TLS via multiplex IHC involves a complex laboratory workflow that requires substantial runtime and is not available in most pathology laboratories. As such, several automated methodologies have been suggested to simplify the workflow for TLS identification.
Panagiotis et al. described the use of a deep learning algorithm to quantitatively identify hematoxylin and eosin (H&E)-stained TLS [213]. The proposed computational methodology has accurately identified TLS comparable to a human counterpart and circumvents TLS that may not be identified by specific IHC staining in lung cancer [213]. However, the algorithm is not without limitations, as it does not discriminate between the various maturation states of TLS described in the literature [204, 213]. Nevertheless, preliminary identification of TLS through digital pathology provides a novel option to incorporate into the clinical workflow.
Subsequently, downstream processes to characterize TLS can include various immunostaining techniques such as multiplex IHC and immunohistofluorescence (IHF) [214]. Currently, there is a lack of standardized marker panels to robustly quantify TLS [201]. Vanhersecke et al. adopted a previously described method consisting of H&E, CD3 and CD20 staining to assess the preliminary TLS status of pathological samples [127], followed by a 5-marker multiplex IHF panel consisting of CD4, CD8, CD20, CD21 and CD23 to differentiate between CD23-positive mTLS and CD23-negative iTLS [204]. Similarly, the phase II PEMBROSARC trial cohort screened for TLS using H&E, CD3 and CD20 staining [127], followed by three different multiplex IHF panels to visualize the immune environment of TLS [210]. Other studies have suggested the use of genomic probes to identify the presence of TLS in melanoma through a 12-chemokine gene signature [215].
Although screening with a wide coverage of immune markers could improve sensitivity and specificity in TLS detection, using more markers for every patient sample would also inevitably translate to increased costs and turnaround time which would not be ideal in the clinical setting. Additionally, the lack of standardized immune markers in TLS detection could lead to inconsistencies in the identification of TLS in the clinic. Hence, there is an urgent need to streamline and define a standardized panel of markers that can be adopted in the clinical setting.
It is important to also take into consideration that the presence of TLS alone may not always be able to predict response to ICIs due to the complex interplay of factors within the TME. For example, tumors may have innate resistance to ICIs, or even acquire resistance after treatment. Jenkins et al. attributed ICI treatment failure to three broad causes – inadequate formation of antitumor T cells, impaired function of tumor-specific T cells, or impaired formation of memory T cells [216]. Hence, the use of biomarkers to infer the states of immune cells in the TME together with the presence or absence of TLS may be able to better predict response to ICIs.
Conclusion
Presently in sarcomas, there is still a lack of robust predictive biomarkers that can be implemented in the clinic. Putative biomarkers will need to be tested in clinical trials to establish their roles in the treatment of sarcomas using ICIs. As new mechanisms emerge, this list will also expand, but it is also critically important that tests are simple and cost-effective with a short turnaround time, so as to be applicable in centers worldwide. Patients matched to biomarkers that accurately predict response to ICI will change the paradigm for systemic treatment in sarcomas and likely supersede the current standard of care.
Acknowledgements
Not applicable.
Abbreviations
- AIDS
Acquired immunodeficiency syndrome
- ARMS
Alveolar rhabdomyosarcoma
- ASPS
Alveolar soft-part sarcoma
- BCL6
B-cell lymphoma 6
- BORR
Best overall response rate
- BS
Bone sarcoma
- CAB-AXL-ADC
Conditionally active biologic AXL-targeted antibody drug conjugate
- CAR
Chimeric antigen receptor
- CEACAM1
Carcinoembryonic antigen-related cell adhesion molecule 1
- CI
Confidence interval
- CNS
Central nervous system
- CPS
Combined positive score
- CR
Complete response
- CRC
Colorectal cancer
- CS
Chondrosarcoma
- CTLA4
Cytotoxic T-lymphocyte-associated protein 4
- DCR
Disease control rate
- DCs
Dendritic cells
- DDLPS
Dedifferentiated liposarcoma
- DLT
Dose-limiting toxicity
- dMMR
Defective mismatch repair
- DNA
Deoxyribonucleic acid
- DSRCT
Desmoplastic small round cell tumor
- EBV
Epstein-Barr virus
- EMA
European Medicines Agency
- ERMS
Embryonal rhabdomyosarcoma
- ES
Ewing sarcoma
- FDA
Food and Drug Administration
- FGL1
Fibrinogen-like protein 1
- Gal-9
Galectin-9
- GCN2
General control nonderepressible 2
- GES
Gene expression signatures
- GIST
Gastrointestinal stromal tumor
- H&E
Hematoxylin and eosin
- HCC
Hepatocellular carcinoma
- HER2
Human epidermal growth factor receptor 2
- HIV
Human immunodeficiency virus
- HMGB1
High mobility group protein B1
- HNSCC
Head and neck squamous cell carcinoma
- HR
Hazard ratio
- IC
Percentage of tumor-infiltrating immune cells within the tumor area expressing PD-L1
- ICI
Immune checkpoint inhibitor
- IDO
Indoleamine 2,3-dioxygenase
- IFN-γ
Interferon-γ
- IFN-γ-1β
Interferon-γ-1β
- IgG
Immunoglobulin G
- IHC
Immunohistochemistry
- IHF
Immunohistofluorescence
- IL-10
Interleukin-10
- INI1
Integrase interactor 1
- IQR
Interquartile range
- irAEs
Immune-related adverse events
- iTLS
Immature tertiary lymphoid structures
- LAG-3
Lymphocyte activation gene 3
- LMS
Leiomyosarcoma
- LPS
Liposarcoma
- MHC
Major histocompatibility complex
- MFS
Myxofibrosarcoma
- mOS
Median overall survival
- mPFS
Median progression free survival
- MPNST
Malignant peripheral nerve sheath tumor
- mRNA
Messenger ribonucleic acid
- MSI
Microsatellite instability
- MSI-H
Microsatellite instability-high
- MSKCC
Memorial Sloan Kettering Cancer Center
- mTLS
Mature tertiary lymphoid structures
- NA
Not available
- NCT
National Clinical Trial
- NGS
Next generation sequencing
- NK cells
Natural killer cells
- NLR
Neutrophil-to-lymphocyte ratio
- NPR
Non-progression rate
- NR
Not reached
- NSCLC
Non-small cell lung cancer
- NY-ESO-1
New York Esophageal Squamous Cell Carcinoma 1 gene
- OGS
Osteosarcoma
- ORR
Objective response rate
- OS
Overall survival
- PCR
Polymerase chain reaction
- PD-1
Programmed cell death 1
- PD-L1
Programmed death ligand 1
- PD
Progressive disease
- PFS
Progression free survival
- pMMR
Proficient mismatch repair
- PR
Partial response
- PRMS
Pleomorphic rhabdomyosarcoma
- RCC
Renal cell carcinoma
- RNA
Ribonucleic acid
- RT
Radiotherapy
- SABR
Stereotactic ablative radiotherapy
- SBRT
Stereotactic body radiation therapy
- SCLC
Small cell lung cancer
- SD
Stable disease
- SOC
Standard of care
- SS
Synovial sarcoma
- STS
Soft-tissue sarcoma
- TC
Percentage of tumor cells within total tumor cells expressing PD-L1
- TCGA
The Cancer Genome Atlas
- TCR
T cell receptor
- TIL
Tumor-infiltrating lymphocyte
- TIM-3
T cell immunoglobulin and mucin domain-containing protein 3
- TLR
Toll-like receptor
- TLS
Tertiary lymphoid structures
- TMB
Tumor mutational burden
- TME
Tumor microenvironment
- TNBC
Triple-negative breast cancer
- TPS
Tumor proportion score
- TRAE
Treatment-related adverse event
- Treg cells
Regulatory T cells
- tTMB
Tissue tumor mutational burden
- T-VEC
Talimogene Laherparepvec
- UBC
Urothelial bladder cancer
- UC
Urothelial carcinoma
- UPS
Undifferentiated pleomorphic sarcoma
- US
United States
- WD-LPS
Well-differentiated liposarcoma
- WES
Whole exome sequencing
Authors’ contributions
V.S.Y designed the work and guided the preparation of this manuscript. C.S.Y. and T.P.L. reviewed the literature and drafted the manuscript. C.S.Y., T.P.L, T.B.T., V.Y.L., and V.S.Y. reviewed and revised the manuscript. All authors have read and approved the final manuscript.
Funding
V.S.Y. is supported by the National Medical Research Council Transition Award (TA20nov-0020), SingHealth Duke-NUS Oncology Academic Clinical Programme (08/FY2021/EX/17-A47) and (08/FY2020/EX/67-A143), the Khoo Pilot Collaborative Award (Duke-NUS-KP(Coll)/2022/0020A), the National Medical Research Council Clinician Scientist-Individual Research Grant-New Investigator Grant (CNIGnov-0025) and the Terry Fox Foundation International Research Grant (I1056).
Availability of data and materials
Not applicable.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Chin Sern Yiong and Tzu Ping Lin contributed equally to this work.
References
- 1.Italiano A, Mathoulin-Pelissier S, Cesne AL, Terrier P, Bonvalot S, Collin F, et al. Trends in survival for patients with metastatic soft-tissue sarcoma. Cancer. 2011;117(5):1049–1054. doi: 10.1002/cncr.25538. [DOI] [PubMed] [Google Scholar]
- 2.Cancer.Net Editorial Board. Sarcomas, Soft Tissue: Statistics: American Society of Clinical Oncology; 2022. Available from: https://www.cancer.net/cancer-types/sarcomas-soft-tissue/statistics.
- 3.Cancer.Net Editorial Board. Osteosarcoma - Childhood and Adolescence: Statistics: American Society of Clinical Oncology; 2022. Available from: https://www.cancer.net/cancer-types/osteosarcoma-childhood-and-adolescence/statistics.
- 4.Cancer.Net Editorial Board. Ewing Sarcoma - Childhood and Adolescence: Statistics: American Society of Clinical Oncology; 2022. Available from: https://www.cancer.net/cancer-types/ewing-sarcoma-childhood-and-adolescence/statistics.
- 5.Spira AI, Ettinger DS. The use of chemotherapy in soft-tissue sarcomas. Oncologist. 2002;7(4):348–359. doi: 10.1634/theoncologist.7-4-348. [DOI] [PubMed] [Google Scholar]
- 6.Judson I, Verweij J, Gelderblom H, Hartmann JT, Schöffski P, Blay J-Y, et al. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: a randomised controlled phase 3 trial. Lancet Oncol. 2014;15(4):415–423. doi: 10.1016/S1470-2045(14)70063-4. [DOI] [PubMed] [Google Scholar]
- 7.Maki RG, Wathen JK, Patel SR, Priebat DA, Okuno SH, Samuels B, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002. J Clin Oncol. 2007;25(19):2755–2763. doi: 10.1200/JCO.2006.10.4117. [DOI] [PubMed] [Google Scholar]
- 8.Maldonado EB, Parsons S, Chen EY, Haslam A, Prasad V. Estimation of US patients with cancer who may respond to cytotoxic chemotherapy. Future Sci OA. 2020;6(8):FSO600. doi: 10.2144/fsoa-2020-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gounder MM, Mahoney MR, Van Tine BA, Ravi V, Attia S, Deshpande HA, et al. Sorafenib for advanced and refractory desmoid tumors. N Engl J Med. 2018;379(25):2417–2428. doi: 10.1056/NEJMoa1805052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kheder ES, Hong DS. Emerging targeted therapy for tumors with NTRK fusion proteins. Clin Cancer Res. 2018;24(23):5807–5814. doi: 10.1158/1078-0432.CCR-18-1156. [DOI] [PubMed] [Google Scholar]
- 11.Pollack SM, Ingham M, Spraker MB, Schwartz GK. Emerging targeted and immune-based therapies in sarcoma. J Clin Oncol. 2018;36(2):125–135. doi: 10.1200/JCO.2017.75.1610. [DOI] [PubMed] [Google Scholar]
- 12.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 13.Skubitz KM, Pambuccian S, Manivel JC, Skubitz AP. Identification of heterogeneity among soft tissue sarcomas by gene expression profiles from different tumors. J Transl Med. 2008;6:23. doi: 10.1186/1479-5876-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hargadon KM, Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: an overview of FDA-approved immune checkpoint inhibitors. Int Immunopharmacol. 2018;62:29–39. doi: 10.1016/j.intimp.2018.06.001. [DOI] [PubMed] [Google Scholar]
- 15.Vaddepally RK, Kharel P, Pandey R, Garje R, Chandra AB. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers. 2020;12(3):738. doi: 10.3390/cancers12030738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373(19):1803–1813. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chung HC, Ros W, Delord J-P, Perets R, Italiano A, Shapira-Frommer R, et al. Efficacy and safety of pembrolizumab in previously treated advanced cervical cancer: results from the phase II KEYNOTE-158 study. J Clin Oncol. 2019;37(17):1470–1478. doi: 10.1200/JCO.18.01265. [DOI] [PubMed] [Google Scholar]
- 18.Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372(4):311–319. doi: 10.1056/NEJMoa1411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fuchs CS, Doi T, Jang RW, Muro K, Satoh T, Machado M, et al. Safety and efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer: phase 2 clinical KEYNOTE-059 trial. JAMA Oncol. 2018;4(5):e180013. doi: 10.1001/jamaoncol.2018.0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088):2492–2502. doi: 10.1016/S0140-6736(17)31046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McDermott D, Haanen J, Chen TT, Lorigan P, O'Day S. Efficacy and safety of ipilimumab in metastatic melanoma patients surviving more than 2 years following treatment in a phase III trial (MDX010-20) Ann Oncol. 2013;24(10):2694–2698. doi: 10.1093/annonc/mdt291. [DOI] [PubMed] [Google Scholar]
- 22.Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372(21):2006–2017. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nghiem P, Bhatia S, Lipson EJ, Sharfman WH, Kudchadkar RR, Brohl AS, et al. Durable tumor regression and overall survival in patients with advanced merkel cell carcinoma receiving pembrolizumab as first-line therapy. J Clin Oncol. 2019;37(9):693–702. doi: 10.1200/JCO.18.01896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaufman HL, Russell J, Hamid O, Bhatia S, Terheyden P, D'Angelo SP, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016;17(10):1374–1385. doi: 10.1016/S1470-2045(16)30364-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non–small-cell lung cancer. N Engl J Med. 2015;373(2):123–135. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zinzani P, Thieblemont C, Melnichenko V, Osmanov D, Bouabdallah K, Walewski J, et al. Efficacy and safety of pembrolizumab in relapsed/refractory primary mediastinal large B-cell lymphoma (rrPMBCL): interim analysis of the KEYNOTE-170 phase 2 trial. Hematol Oncol. 2017;35(S2):62–63. [Google Scholar]
- 28.Antonia SJ, López-Martin JA, Bendell J, Ott PA, Taylor M, Eder JP, et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol. 2016;17(7):883–895. doi: 10.1016/S1470-2045(16)30098-5. [DOI] [PubMed] [Google Scholar]
- 29.Seiwert TY, Burtness B, Mehra R, Weiss J, Berger R, Eder JP, et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol. 2016;17(7):956–965. doi: 10.1016/S1470-2045(16)30066-3. [DOI] [PubMed] [Google Scholar]
- 30.Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018;379(22):2108–2121. doi: 10.1056/NEJMoa1809615. [DOI] [PubMed] [Google Scholar]
- 31.Sharma P, Retz M, Siefker-Radtke A, Baron A, Necchi A, Bedke J, et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol. 2017;18(3):312–322. doi: 10.1016/S1470-2045(17)30065-7. [DOI] [PubMed] [Google Scholar]
- 32.Naqash AR, O'Sullivan Coyne GH, Moore N, Sharon E, Takebe N, Fino KK, et al. Phase II study of atezolizumab in advanced alveolar soft part sarcoma (ASPS) J Clin Oncol. 2021;39(15_suppl):11519. [Google Scholar]
- 33.WHO Classification of Tumours Editorial Board . Soft tissue and bone tumours. 5. Lyon: IARC; 2020. [Google Scholar]
- 34.Haslam A, Prasad V. Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Network Open. 2019;2(5):e192535. doi: 10.1001/jamanetworkopen.2019.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tawbi HA, Burgess M, Bolejack V, Van Tine BA, Schuetze SM, Hu J, et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017;18(11):1493–1501. doi: 10.1016/S1470-2045(17)30624-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Conley AP, Trinh VA, Zobniw CM, Posey K, Martinez JD, Arrieta OG, et al. Positive tumor response to combined checkpoint inhibitors in a patient with refractory alveolar soft part sarcoma: a case report. J Glob Oncol. 2018;4:1–6. doi: 10.1200/JGO.2017.009993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marcrom S, De Los Santos JF, Conry RM. Complete response of mediastinal clear cell sarcoma to pembrolizumab with radiotherapy. Clin Sarcoma Res. 2017;7:14. doi: 10.1186/s13569-017-0079-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guram K, Nunez M, Einck J, Mell LK, Cohen E, Sanders PD, et al. Radiation therapy combined with checkpoint blockade immunotherapy for metastatic undifferentiated pleomorphic sarcoma of the maxillary sinus with a complete response. Front Oncol. 2018;8:435. doi: 10.3389/fonc.2018.00435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roulleaux Dugage M, Nassif EF, Italiano A, Bahleda R. Improving immunotherapy efficacy in soft-tissue sarcomas: a biomarker driven and histotype tailored review. Front Immunol. 2021;12:775761. doi: 10.3389/fimmu.2021.775761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Myers G. Immune-related adverse events of immune checkpoint inhibitors: a brief review. Curr Oncol. 2018;25(5):342–347. doi: 10.3747/co.25.4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J, Xiu J, Farrell A, Baca Y, Arai H, Battaglin F, et al. Mutational analysis of microsatellite-stable gastrointestinal cancer with high tumour mutational burden: a retrospective cohort study. Lancet Oncol. 2023;24(2):151–161. doi: 10.1016/S1470-2045(22)00783-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun H, Liu SY, Zhou JY, Xu JT, Zhang HK, Yan HH, et al. Specific TP53 subtype as biomarker for immune checkpoint inhibitors in lung adenocarcinoma. EBioMedicine. 2020;60:102990. doi: 10.1016/j.ebiom.2020.102990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brown ZJ, Yu SJ, Heinrich B, Ma C, Fu Q, Sandhu M, et al. Indoleamine 2,3-dioxygenase provides adaptive resistance to immune checkpoint inhibitors in hepatocellular carcinoma. Cancer Immunol Immunother. 2018;67(8):1305–1315. doi: 10.1007/s00262-018-2190-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jin J, Yang L, Liu D, Li W. Association of the neutrophil to lymphocyte ratio and clinical outcomes in patients with lung cancer receiving immunotherapy: a meta-analysis. BMJ Open. 2020;10(6):e035031. doi: 10.1136/bmjopen-2019-035031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Twomey JD, Zhang B. Cancer immunotherapy update: FDA-approved checkpoint inhibitors and companion diagnostics. AAPS J. 2021;23(2):39. doi: 10.1208/s12248-021-00574-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann Oncol. 2019;30(4):582–588. doi: 10.1093/annonc/mdz011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Michielin O, van Akkooi ACJ, Ascierto PA, Dummer R, Keilholz U. Cutaneous melanoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up††approved by the ESMO guidelines Committee: February 2002, last update September 2019. Ann Oncol. 2019;30(12):1884–1901. doi: 10.1093/annonc/mdz411. [DOI] [PubMed] [Google Scholar]
- 48.Garbe C, Amaral T, Peris K, Hauschild A, Arenberger P, Bastholt L, et al. European consensus-based interdisciplinary guideline for melanoma. Part 2: treatment – update 2019. Eur J Cancer. 2020;126:159–77. doi: 10.1016/j.ejca.2019.11.015. [DOI] [PubMed] [Google Scholar]
- 49.Armand P, Rodig S, Melnichenko V, Thieblemont C, Bouabdallah K, Tumyan G, et al. Pembrolizumab in Relapsed or Refractory Primary Mediastinal Large B-Cell Lymphoma. J Clin Oncol. 2019;37(34):3291–9. [DOI] [PMC free article] [PubMed]
- 50.Chen R, Zinzani PL, Lee HJ, Armand P, Johnson NA, Brice P, et al. Pembrolizumab in relapsed or refractory Hodgkin lymphoma: 2-year follow-up of KEYNOTE-087. Blood. 2019;134(14):1144–1153. doi: 10.1182/blood.2019000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Balar AV, Castellano D, O'Donnell PH, Grivas P, Vuky J, Powles T, et al. First-line pembrolizumab in cisplatin-ineligible patients with locally advanced and unresectable or metastatic urothelial cancer (KEYNOTE-052): a multicentre, single-arm, phase 2 study. Lancet Oncol. 2017;18(11):1483–1492. doi: 10.1016/S1470-2045(17)30616-2. [DOI] [PubMed] [Google Scholar]
- 52.Suzman DL, Agrawal S, Ning YM, Maher VE, Fernandes LL, Karuri S, et al. FDA approval summary: atezolizumab or pembrolizumab for the treatment of patients with advanced urothelial carcinoma ineligible for cisplatin-containing chemotherapy. Oncologist. 2019;24(4):563–569. doi: 10.1634/theoncologist.2018-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zinzani PL, Ribrag V, Moskowitz CH, Michot JM, Kuruvilla J, Balakumaran A, et al. Safety and tolerability of pembrolizumab in patients with relapsed/refractory primary mediastinal large B-cell lymphoma. Blood. 2017;130(3):267–270. doi: 10.1182/blood-2016-12-758383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bradford D, Demko S, Jin S, Mishra-Kalyani P, Beckles AR, Goldberg KB, et al. FDA accelerated approval of pembrolizumab for recurrent locally advanced or metastatic merkel cell carcinoma. Oncologist. 2020;25(7):e1077–e1082. doi: 10.1634/theoncologist.2020-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kamat AM, Shore N, Hahn N, Alanee S, Nishiyama H, Shariat S, et al. KEYNOTE-676: phase III study of BCG and pembrolizumab for persistent/recurrent high-risk NMIBC. Future Oncol. 2020;16(10):507–516. doi: 10.2217/fon-2019-0817. [DOI] [PubMed] [Google Scholar]
- 56.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168(4):707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Orellana García LP, Ehmann F, Hines PA, Ritzhaupt A, Brand A. Biomarker and companion diagnostics-a review of medicinal products approved by the European medicines agency. Front Med (Lausanne) 2021;8:753187. doi: 10.3389/fmed.2021.753187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Y, Tong Z, Zhang W, Zhang W, Buzdin A, Mu X, et al. FDA-approved and emerging next generation predictive biomarkers for immune checkpoint inhibitors in cancer patients. Front Oncol. 2021;11:683419. doi: 10.3389/fonc.2021.683419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berg KD, Glaser CL, Thompson RE, Hamilton SR, Griffin CA, Eshleman JR. Detection of microsatellite instability by fluorescence multiplex polymerase chain reaction. J Mol Diagn. 2000;2(1):20–28. doi: 10.1016/S1525-1578(10)60611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Klempner SJ, Fabrizio D, Bane S, Reinhart M, Peoples T, Ali SM, et al. Tumor mutational burden as a predictive biomarker for response to immune checkpoint inhibitors: a review of current evidence. Oncologist. 2020;25(1):e147–e159. doi: 10.1634/theoncologist.2019-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schmid P, Salgado R, Park YH, Muñoz-Couselo E, Kim SB, Sohn J, et al. Pembrolizumab plus chemotherapy as neoadjuvant treatment of high-risk, early-stage triple-negative breast cancer: results from the phase 1b open-label, multicohort KEYNOTE-173 study. Ann Oncol. 2020;31(5):569–581. doi: 10.1016/j.annonc.2020.01.072. [DOI] [PubMed] [Google Scholar]
- 62.Burtness B, Harrington KJ, Greil R, Soulières D, Tahara M, de Castro G, Jr, et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet. 2019;394(10212):1915–28. doi: 10.1016/S0140-6736(19)32591-7. [DOI] [PubMed] [Google Scholar]
- 63.Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Updated analysis of KEYNOTE-024: pembrolizumab versus platinum-based chemotherapy for advanced non–small-cell lung cancer with PD-L1 tumor proportion score of 50% or greater. J Clin Oncol. 2019;37(7):537–546. doi: 10.1200/JCO.18.00149. [DOI] [PubMed] [Google Scholar]
- 64.Bellmunt J, de Wit R, Vaughn DJ, Fradet Y, Lee JL, Fong L, et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med. 2017;376(11):1015–1026. doi: 10.1056/NEJMoa1613683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim SW, Carcereny Costa E, et al. Nivolumab plus ipilimumab in advanced non-small-cell lung cancer. N Engl J Med. 2019;381(21):2020–2031. doi: 10.1056/NEJMoa1910231. [DOI] [PubMed] [Google Scholar]
- 66.Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389(10066):255–265. doi: 10.1016/S0140-6736(16)32517-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387(10031):1909–1920. doi: 10.1016/S0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Massard C, Gordon MS, Sharma S, Rafii S, Wainberg ZA, Luke J, et al. Safety and Efficacy of Durvalumab (MEDI4736), an anti-programmed cell death ligand-1 immune checkpoint inhibitor, in patients with advanced urothelial bladder cancer. J Clin Oncol. 2016;34(26):3119–3125. doi: 10.1200/JCO.2016.67.9761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Le DT, Kim TW, Van Cutsem E, Geva R, Jäger D, Hara H, et al. Phase II open-label study of pembrolizumab in treatment-refractory, microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer: KEYNOTE-164. J Clin Oncol. 2020;38(1):11–19. doi: 10.1200/JCO.19.02107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020;21(10):1353–1365. doi: 10.1016/S1470-2045(20)30445-9. [DOI] [PubMed] [Google Scholar]
- 72.Ott PA, Bang Y-J, Piha-Paul SA, Razak ARA, Bennouna J, Soria J-C, et al. T-cell–inflamed gene-expression profile, programmed death ligand 1 expression, and tumor mutational burden predict efficacy in patients treated with pembrolizumab across 20 cancers: KEYNOTE-028. J Clin Oncol. 2018;37(4):318–327. doi: 10.1200/JCO.2018.78.2276. [DOI] [PubMed] [Google Scholar]
- 73.Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, et al. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol. 2017;8:561. doi: 10.3389/fphar.2017.00561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Noguchi T, Ward JP, Gubin MM, Arthur CD, Lee SH, Hundal J, et al. Temporally distinct PD-L1 expression by tumor and host cells contributes to immune escape. Cancer Immunol Res. 2017;5(2):106–117. doi: 10.1158/2326-6066.CIR-16-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lau J, Cheung J, Navarro A, Lianoglou S, Haley B, Totpal K, et al. Tumour and host cell PD-L1 is required to mediate suppression of anti-tumour immunity in mice. Nat Commun. 2017;8:14572. doi: 10.1038/ncomms14572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lin H, Wei S, Hurt EM, Green MD, Zhao L, Vatan L, et al. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J Clin Invest. 2018;128(2):805–815. doi: 10.1172/JCI96113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yi M, Niu M, Xu L, Luo S, Wu K. Regulation of PD-L1 expression in the tumor microenvironment. J Hematol Oncol. 2021;14(1):10. doi: 10.1186/s13045-020-01027-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Le Cesne A, Marec-Berard P, Blay JY, Gaspar N, Bertucci F, Penel N, et al. Programmed cell death 1 (PD-1) targeting in patients with advanced osteosarcomas: results from the PEMBROSARC study. Eur J Cancer. 2019;119:151–157. doi: 10.1016/j.ejca.2019.07.018. [DOI] [PubMed] [Google Scholar]
- 79.Kelany M, Barth TF, Salem D, Shakweer MM. Prevalence and prognostic implications of PD-L1 expression in soft tissue sarcomas. Pathol Oncol Res. 2021;27:1609804. doi: 10.3389/pore.2021.1609804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sundara YT, Kostine M, Cleven AHG, Bovée JVMG, Schilham MW, Cleton-Jansen A-M. Increased PD-L1 and T-cell infiltration in the presence of HLA class I expression in metastatic high-grade osteosarcoma: a rationale for T-cell-based immunotherapy. Cancer Immunol Immunother. 2017;66(1):119–128. doi: 10.1007/s00262-016-1925-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Patel KR, Martinez A, Stahl JM, Logan SJ, Perricone AJ, Ferris MJ, et al. Increase in PD-L1 expression after pre-operative radiotherapy for soft tissue sarcoma. Oncoimmunology. 2018;7(7):e1442168. doi: 10.1080/2162402X.2018.1442168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Boxberg M, Steiger K, Lenze U, Rechl H, von Eisenhart-Rothe R, Wörtler K, et al. PD-L1 and PD-1 and characterization of tumor-infiltrating lymphocytes in high grade sarcomas of soft tissue - prognostic implications and rationale for immunotherapy. Oncoimmunology. 2018;7(3):e1389366. doi: 10.1080/2162402X.2017.1389366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim JR, Moon YJ, Kwon KS, Bae JS, Wagle S, Kim KM, et al. Tumor infiltrating PD1-positive lymphocytes and the expression of PD-L1 predict poor prognosis of soft tissue sarcomas. PLoS ONE. 2013;8(12):e82870. doi: 10.1371/journal.pone.0082870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim C, Kim EK, Jung H, Chon HJ, Han JW, Shin KH, et al. Prognostic implications of PD-L1 expression in patients with soft tissue sarcoma. BMC Cancer. 2016;16:434. doi: 10.1186/s12885-016-2451-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.D'Angelo SP, Shoushtari AN, Agaram NP, Kuk D, Qin LX, Carvajal RD, et al. Prevalence of tumor-infiltrating lymphocytes and PD-L1 expression in the soft tissue sarcoma microenvironment. Hum Pathol. 2015;46(3):357–365. doi: 10.1016/j.humpath.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chowdhury F, Dunn S, Mitchell S, Mellows T, Ashton-Key M, Gray JC. PD-L1 and CD8+PD1+ lymphocytes exist as targets in the pediatric tumor microenvironment for immunomodulatory therapy. OncoImmunology. 2015;4(10):e1029701. [Google Scholar]
- 87.Que Y, Xiao W, Guan YX, Liang Y, Yan SM, Chen HY, et al. PD-L1 expression is associated with FOXP3+ regulatory T-Cell infiltration of soft tissue sarcoma and poor patient prognosis. J Cancer. 2017;8(11):2018–2025. doi: 10.7150/jca.18683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pollack SM, He Q, Yearley JH, Emerson R, Vignali M, Zhang Y, et al. T-cell infiltration and clonality correlate with programmed cell death protein 1 and programmed death-ligand 1 expression in patients with soft tissue sarcomas. Cancer. 2017;123(17):3291–3304. doi: 10.1002/cncr.30726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.van Erp AEM, Versleijen-Jonkers YMH, Hillebrandt-Roeffen MHS, van Houdt L, Gorris MAJ, van Dam LS, et al. Expression and clinical association of programmed cell death-1, programmed death-ligand-1 and CD8(+) lymphocytes in primary sarcomas is subtype dependent. Oncotarget. 2017;8(41):71371–71384. doi: 10.18632/oncotarget.19071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Paydas S, Bagir EK, Deveci MA, Gonlusen G. Clinical and prognostic significance of PD-1 and PD-L1 expression in sarcomas. Med Oncol. 2016;33(8):93. doi: 10.1007/s12032-016-0807-z. [DOI] [PubMed] [Google Scholar]
- 91.Kloor M, von Knebel DM. The immune biology of microsatellite-unstable cancer. Trends Cancer. 2016;2(3):121–133. doi: 10.1016/j.trecan.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 92.Luchini C, Bibeau F, Ligtenberg MJL, Singh N, Nottegar A, Bosse T, et al. ESMO recommendations on microsatellite instability testing for immunotherapy in cancer, and its relationship with PD-1/PD-L1 expression and tumour mutational burden: a systematic review-based approach. Ann Oncol. 2019;30(8):1232–1243. doi: 10.1093/annonc/mdz116. [DOI] [PubMed] [Google Scholar]
- 93.Lorenzi M, Amonkar M, Zhang J, Mehta S, Liaw K-L. Epidemiology of Microsatellite Instability High (MSI-H) and Deficient Mismatch Repair (dMMR) in solid tumors: a structured literature review. J Oncol. 2020;2020:1807929. [Google Scholar]
- 94.Lam SW, Kostine M, de Miranda NFCC, Schöffski P, Lee C-J, Morreau H, et al. Mismatch repair deficiency is rare in bone and soft tissue tumors. Histopathology. 2021;79(4):509–520. doi: 10.1111/his.14377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zamora AE, Crawford JC, Thomas PG. Hitting the target: how t cells detect and eliminate tumors. J Immunol. 2018;200(2):392. doi: 10.4049/jimmunol.1701413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kim JY, Kronbichler A, Eisenhut M, Hong SH, van der Vliet HJ, Kang J, et al. Tumor mutational burden and efficacy of immune checkpoint inhibitors: a systematic review and meta-analysis. Cancers (Basel) 2019;11(11):1798. doi: 10.3390/cancers11111798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med. 2017;377(25):2500–2501. doi: 10.1056/NEJMc1713444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang F, Wei XL, Wang FH, Xu N, Shen L, Dai GH, et al. Safety, efficacy and tumor mutational burden as a biomarker of overall survival benefit in chemo-refractory gastric cancer treated with toripalimab, a PD-1 antibody in phase Ib/II clinical trial NCT02915432. Ann Oncol. 2019;30(9):1479–1486. doi: 10.1093/annonc/mdz197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Valero C, Lee M, Hoen D, Zehir A, Berger MF, Seshan VE, et al. Response rates to Anti-PD-1 immunotherapy in microsatellite-stable solid tumors with 10 or more mutations per megabase. JAMA Oncol. 2021;7(5):739–743. doi: 10.1001/jamaoncol.2020.7684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kim ST, Cristescu R, Bass AJ, Kim K-M, Odegaard JI, Kim K, et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med. 2018;24(9):1449–1458. doi: 10.1038/s41591-018-0101-z. [DOI] [PubMed] [Google Scholar]
- 101.Hara H, Fukuoka S, Takahashi N, Kojima T, Kawazoe A, Asayama M, et al. Regorafenib plus nivolumab in patients with advanced colorectal or gastric cancer: an open-label, dose-finding, and dose-expansion phase 1b trial (REGONIVO, EPOC1603) Ann Oncol. 2019;30:124. doi: 10.1200/JCO.19.03296. [DOI] [PubMed] [Google Scholar]
- 102.Pillozzi S, Bernini A, Palchetti I, Crociani O, Antonuzzo L, Campanacci D, et al. Soft tissue sarcoma: an insight on biomarkers at molecular, metabolic and cellular level. Cancers (Basel) 2021;13(12):3044. doi: 10.3390/cancers13123044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Meléndez B, Van Campenhout C, Rorive S, Remmelink M, Salmon I, D'Haene N. Methods of measurement for tumor mutational burden in tumor tissue. Transl Lung Cancer Res. 2018;7(6):661–667. doi: 10.21037/tlcr.2018.08.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Allgäuer M, Budczies J, Christopoulos P, Endris V, Lier A, Rempel E, et al. Implementing tumor mutational burden (TMB) analysis in routine diagnostics-a primer for molecular pathologists and clinicians. Transl Lung Cancer Res. 2018;7(6):703–715. doi: 10.21037/tlcr.2018.08.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Abeshouse A, Adebamowo C, Adebamowo SN, Akbani R, Akeredolu T, Ally A, et al. Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell. 2017;171(4):950–65.e28. doi: 10.1016/j.cell.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Trabucco SE, Ali SM, Sokol E, Schrock AB, Albacker LA, Chung J, et al. Frequency of genomic biomarkers of response to immunotherapy in sarcoma. J Clin Oncol. 2018;36(15_suppl):11579. [Google Scholar]
- 107.Doyle LA, Nowak JA, Nathenson MJ, Thornton K, Wagner AJ, Johnson JM, et al. Characteristics of mismatch repair deficiency in sarcomas. Mod Pathol. 2019;32(7):977–987. doi: 10.1038/s41379-019-0202-3. [DOI] [PubMed] [Google Scholar]
- 108.Espejo-Freire AP, Elliott A, Rosenberg A, Costa PA, Barreto-Coelho P, Jonczak E, et al. Genomic landscape of angiosarcoma: a targeted and immunotherapy biomarker analysis. Cancers (Basel) 2021;13(19):4816. doi: 10.3390/cancers13194816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wagner MJ, Othus M, Patel SP, Ryan C, Sangal A, Powers B, et al. Multicenter phase II trial (SWOG S1609, cohort 51) of ipilimumab and nivolumab in metastatic or unresectable angiosarcoma: a substudy of dual anti-CTLA-4 and anti-PD-1 blockade in rare tumors (DART) J Immunother Cancer. 2021;9(8):e002990. doi: 10.1136/jitc-2021-002990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chibon F. Cancer gene expression signatures – the rise and fall? Eur J Cancer. 2013;49(8):2000–2009. doi: 10.1016/j.ejca.2013.02.021. [DOI] [PubMed] [Google Scholar]
- 111.Singh KP, Miaskowski C, Dhruva AA, Flowers E, Kober KM. Mechanisms and measurement of changes in gene expression. Biol Res Nurs. 2018;20(4):369–382. doi: 10.1177/1099800418772161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rahman R, Zatorski N, Hansen J, Xiong Y, van Hasselt JGC, Sobie EA, et al. Protein structure–based gene expression signatures. Proc Natl Acad Sci. 2021;118(19):e2014866118. doi: 10.1073/pnas.2014866118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Slyper M, Porter CBM, Ashenberg O, Waldman J, Drokhlyansky E, Wakiro I, et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat Med. 2020;26(5):792–802. doi: 10.1038/s41591-020-0844-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Marx V. Method of the year: spatially resolved transcriptomics. Nat Methods. 2021;18(1):9–14. doi: 10.1038/s41592-020-01033-y. [DOI] [PubMed] [Google Scholar]
- 115.Auslander N, Zhang G, Lee JS, Frederick DT, Miao B, Moll T, et al. Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma. Nat Med. 2018;24(10):1545–1549. doi: 10.1038/s41591-018-0157-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell. 2018;175(4):984–97.e24. doi: 10.1016/j.cell.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165(1):35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Budczies J, Kirchner M, Kluck K, Kazdal D, Glade J, Allgäuer M, et al. A gene expression signature associated with B cells predicts benefit from immune checkpoint blockade in lung adenocarcinoma. OncoImmunology. 2021;10(1):1860586. doi: 10.1080/2162402X.2020.1860586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.De Marchi P, Ferro Leal L, da Silva LS, de Oliveira Cavagna R, Ferreira da Silva FA, da Silva VD, et al. LungTS: a new gene expression signature for prediction of response to checkpoint inhibitors in non-small cell lung cancer. J Clin Oncol. 2022;40(16_suppl):e21143. [Google Scholar]
- 120.Chen H, Lin R, Lin W, Chen Q, Ye D, Li J, et al. An immune gene signature to predict prognosis and immunotherapeutic response in lung adenocarcinoma. Sci Rep. 2022;12(1):8230. doi: 10.1038/s41598-022-12301-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hwang S, Kwon A-Y, Jeong J-Y, Kim S, Kang H, Park J, et al. Immune gene signatures for predicting durable clinical benefit of anti-PD-1 immunotherapy in patients with non-small cell lung cancer. Sci Rep. 2020;10(1):643. doi: 10.1038/s41598-019-57218-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yuan B, Jiang C, Chen L, Wen L, Cui J, Chen M, et al. A novel DNA repair gene signature for immune checkpoint inhibitor-based therapy in gastric cancer. Front Cell Dev Biol. 2022;10:893546. doi: 10.3389/fcell.2022.893546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lai G, Li K, Deng J, Liu H, Xie B, Zhong X. Identification and validation of a gene signature for lower-grade gliomas based on pyroptosis-related genes to predict survival and response to immune checkpoint inhibitors. J Healthc Eng. 2022;2022:8704127. doi: 10.1155/2022/8704127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Thompson JC, Davis C, Deshpande C, Hwang W-T, Jeffries S, Huang A, et al. Gene signature of antigen processing and presentation machinery predicts response to checkpoint blockade in non-small cell lung cancer (NSCLC) and melanoma. J Immunother Cancer. 2020;8(2):e000974. doi: 10.1136/jitc-2020-000974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J Clin Investig. 2017;127(8):2930–2940. doi: 10.1172/JCI91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Du XH, Wei H, Zhang P, Yao WT, Cai QQ. Heterogeneity of soft tissue sarcomas and its implications in targeted therapy. Front Oncol. 2020;10:564852. doi: 10.3389/fonc.2020.564852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Petitprez F, de Reyniès A, Keung EZ, Chen TW-W, Sun C-M, Calderaro J, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature. 2020;577(7791):556–60. doi: 10.1038/s41586-019-1906-8. [DOI] [PubMed] [Google Scholar]
- 128.Bacher U, Shumilov E, Flach J, Porret N, Joncourt R, Wiedemann G, et al. Challenges in the introduction of next-generation sequencing (NGS) for diagnostics of myeloid malignancies into clinical routine use. Blood Cancer J. 2018;8(11):113. doi: 10.1038/s41408-018-0148-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhang L, Chen D, Song D, Liu X, Zhang Y, Xu X, et al. Clinical and translational values of spatial transcriptomics. Signal Transduct Target Ther. 2022;7(1):111. doi: 10.1038/s41392-022-00960-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Song M, Graubard BI, Rabkin CS, Engels EA. Neutrophil-to-lymphocyte ratio and mortality in the United States general population. Sci Rep. 2021;11(1):464. doi: 10.1038/s41598-020-79431-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Howard R, Kanetsky PA, Egan KM. Exploring the prognostic value of the neutrophil-to-lymphocyte ratio in cancer. Sci Rep. 2019;9(1):19673. doi: 10.1038/s41598-019-56218-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Li M, Spakowicz D, Burkart J, Patel S, Husain M, He K, et al. Change in neutrophil to lymphocyte ratio during immunotherapy treatment is a non-linear predictor of patient outcomes in advanced cancers. J Cancer Res Clin Oncol. 2019;145(10):2541–2546. doi: 10.1007/s00432-019-02982-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Strong EA, Park SH, Ethun CG, Chow B, King D, Bedi M, et al. High neutrophil-lymphocyte ratio is not independently associated with worse survival or recurrence in patients with extremity soft tissue sarcoma. Surgery. 2020;168(4):760–767. doi: 10.1016/j.surg.2020.06.017. [DOI] [PubMed] [Google Scholar]
- 134.Chan JY, Zhang Z, Chew W, Tan GF, Lim CL, Zhou L, et al. Biological significance and prognostic relevance of peripheral blood neutrophil-to-lymphocyte ratio in soft tissue sarcoma. Sci Rep. 2018;8(1):11959. doi: 10.1038/s41598-018-30442-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang S, Wu J, Shen H, Wang J. The prognostic value of IDO expression in solid tumors: a systematic review and meta-analysis. BMC Cancer. 2020;20(1):471. doi: 10.1186/s12885-020-06956-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013;34(3):137–143. doi: 10.1016/j.it.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22(5):633–642. doi: 10.1016/j.immuni.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 138.Sharma MD, Baban B, Chandler P, Hou DY, Singh N, Yagita H, et al. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest. 2007;117(9):2570–2582. doi: 10.1172/JCI31911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Urakawa H, Nishida Y, Nakashima H, Shimoyama Y, Nakamura S, Ishiguro N. Prognostic value of indoleamine 2,3-dioxygenase expression in high grade osteosarcoma. Clin Exp Metas. 2009;26(8):1005–1012. doi: 10.1007/s10585-009-9290-7. [DOI] [PubMed] [Google Scholar]
- 140.Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med. 2011;17(9):1094–1100. doi: 10.1038/nm.2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Reilley MJ, Bailey A, Subbiah V, Janku F, Naing A, Falchook G, et al. Phase I clinical trial of combination imatinib and ipilimumab in patients with advanced malignancies. J Immunother Cancer. 2017;5(1):35. doi: 10.1186/s40425-017-0238-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tang K, Wu Y-H, Song Y, Yu B. Indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors in clinical trials for cancer immunotherapy. J Hematol Oncol. 2021;14(1):68. doi: 10.1186/s13045-021-01080-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol. 2019;20(8):1083–1097. doi: 10.1016/S1470-2045(19)30274-8. [DOI] [PubMed] [Google Scholar]
- 144.United States Food & Drug Administration. FDA approves Opdualag for unresectable or metastatic melanoma, 2022. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-opdualag-unresectable-or-metastatic-melanoma.
- 145.Goldberg MV, Drake CG. LAG-3 in Cancer Immunotherapy. Curr Top Microbiol Immunol. 2011;344:269–278. doi: 10.1007/82_2010_114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Maruhashi T, Sugiura D, Okazaki IM, Okazaki T. LAG-3: from molecular functions to clinical applications. J Immunother Cancer. 2020;8(2):e001014. doi: 10.1136/jitc-2020-001014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Baixeras E, Huard B, Miossec C, Jitsukawa S, Martin M, Hercend T, et al. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J Exp Med. 1992;176(2):327–37. doi: 10.1084/jem.176.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Maruhashi T, Okazaki IM, Sugiura D, Takahashi S, Maeda TK, Shimizu K, et al. LAG-3 inhibits the activation of CD4(+) T cells that recognize stable pMHCII through its conformation-dependent recognition of pMHCII. Nat Immunol. 2018;19(12):1415–1426. doi: 10.1038/s41590-018-0217-9. [DOI] [PubMed] [Google Scholar]
- 149.Kouo T, Huang L, Pucsek AB, Cao M, Solt S, Armstrong T, et al. Galectin-3 shapes antitumor immune responses by suppressing CD8+ T cells via LAG-3 and inhibiting expansion of plasmacytoid dendritic cells. Cancer Immunol Res. 2015;3(4):412–423. doi: 10.1158/2326-6066.CIR-14-0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell. 2019;176(1–2):334–47.e12. doi: 10.1016/j.cell.2018.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Schöffski P, Tan DSW, Martín M, Ochoa-de-Olza M, Sarantopoulos J, Carvajal RD, et al. Phase I/II study of the LAG-3 inhibitor ieramilimab (LAG525) ± anti-PD-1 spartalizumab (PDR001) in patients with advanced malignancies. J Immunother Cancer. 2022;10(2):e003776. doi: 10.1136/jitc-2021-003776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Huo J-L, Wang Y-T, Fu W-J, Lu N, Liu Z-S. The promising immune checkpoint LAG-3 in cancer immunotherapy: from basic research to clinical application. Front Immunol. 2022;13:956090. doi: 10.3389/fimmu.2022.956090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Que Y, Fang Z, Guan Y, Xiao W, Xu B, Zhao J, et al. LAG-3 expression on tumor-infiltrating T cells in soft tissue sarcoma correlates with poor survival. Cancer Biol Med. 2019;16(2):331–340. doi: 10.20892/j.issn.2095-3941.2018.0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415(6871):536–541. doi: 10.1038/415536a. [DOI] [PubMed] [Google Scholar]
- 155.Gao X, Zhu Y, Li G, Huang H, Zhang G, Wang F, et al. TIM-3 expression characterizes regulatory T Cells in tumor tissues and is associated with lung cancer progression. PLoS One. 2012;7(2):e30676. doi: 10.1371/journal.pone.0030676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Anderson AC, Anderson DE, Bregoli L, Hastings WD, Kassam N, Lei C, et al. Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science. 2007;318(5853):1141–1143. doi: 10.1126/science.1148536. [DOI] [PubMed] [Google Scholar]
- 157.Ndhlovu LC, Lopez-Vergès S, Barbour JD, Jones RB, Jha AR, Long BR, et al. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood. 2012;119(16):3734–3743. doi: 10.1182/blood-2011-11-392951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Phong BL, Avery L, Sumpter TL, Gorman JV, Watkins SC, Colgan JD, et al. Tim-3 enhances FcεRI-proximal signaling to modulate mast cell activation. J Exp Med. 2015;212(13):2289–2304. doi: 10.1084/jem.20150388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wolf Y, Anderson AC, Kuchroo VK. TIM3 comes of age as an inhibitory receptor. Nat Rev Immunol. 2020;20(3):173–185. doi: 10.1038/s41577-019-0224-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207(10):2187–2194. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yang R, Sun L, Li C-F, Wang Y-H, Yao J, Li H, et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat Commun. 2021;12(1):832. doi: 10.1038/s41467-021-21099-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Rangachari M, Zhu C, Sakuishi K, Xiao S, Karman J, Chen A, et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3–mediated cell death and exhaustion. Nat Med. 2012;18(9):1394–1400. doi: 10.1038/nm.2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Curtin JF, Liu N, Candolfi M, Xiong W, Assi H, Yagiz K, et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 2009;6(1):e1000010. doi: 10.1371/journal.pmed.1000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Huang Y-H, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature. 2015;517(7534):386–390. doi: 10.1038/nature13848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kammerer R, Stober D, Singer BB, Öbrink B, Reimann J. Carcinoembryonic antigen-related cell adhesion molecule 1 on murine dendritic cells is a potent regulator of T Cell stimulation. J Immunol. 2001;166(11):6537. doi: 10.4049/jimmunol.166.11.6537. [DOI] [PubMed] [Google Scholar]
- 166.Horst AK, Bickert T, Brewig N, Ludewig P, van Rooijen N, Schumacher U, et al. CEACAM1+ myeloid cells control angiogenesis in inflammation. Blood. 2009;113(26):6726–6736. doi: 10.1182/blood-2008-10-184556. [DOI] [PubMed] [Google Scholar]
- 167.Coutelier J-P, Godfraind C, Dveksler GS, Wysocka M, Cardellichio CB, Noël H, et al. B lymphocyte and macrophage expression of carcinoembryonic antigen-related adhesion molecules that serve as receptors for murine coronavirus. Eur J Immunol. 1994;24(6):1383–1390. doi: 10.1002/eji.1830240622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501. doi: 10.1038/ncomms10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Acharya N, Sabatos-Peyton C, Anderson AC. Tim-3 finds its place in the cancer immunotherapy landscape. J Immunother Cancer. 2020;8(1):e000911. doi: 10.1136/jitc-2020-000911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zang K, Hui L, Wang M, Huang Y, Zhu X, Yao B. TIM-3 as a prognostic marker and a potential immunotherapy target in human malignant tumors: a meta-analysis and bioinformatics validation. Front Oncol. 2021;11:579. doi: 10.3389/fonc.2021.579351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Pu F, Chen F, Zhang Z, Qing X, Lin H, Zhao L, et al. TIM-3 expression and its association with overall survival in primary osteosarcoma. Oncol Lett. 2019;18(5):5294–5300. doi: 10.3892/ol.2019.10855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Levine AJ. p53: 800 million years of evolution and 40 years of discovery. Nat Rev Cancer. 2020;20(8):471–480. doi: 10.1038/s41568-020-0262-1. [DOI] [PubMed] [Google Scholar]
- 173.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Zhu G, Pan C, Bei J-X, Li B, Liang C, Xu Y, et al. Mutant p53 in cancer progression and targeted therapies. Front Oncol. 2020;10:595187. doi: 10.3389/fonc.2020.595187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Miller M, Shirole N, Tian R, Pal D, Sordella R. The evolution of TP53 mutations: from loss-of-function to separation-of-function mutants. J Cancer Biol Res. 2016;4(4):1091. [PMC free article] [PubMed] [Google Scholar]
- 176.Biton J, Mansuet-Lupo A, Pécuchet N, Alifano M, Ouakrim H, Arrondeau J, et al. TP53, STK11, and EGFR mutations predict tumor immune profile and the response to Anti–PD-1 in lung adenocarcinoma. Clin Cancer Res. 2018;24(22):5710–5723. doi: 10.1158/1078-0432.CCR-18-0163. [DOI] [PubMed] [Google Scholar]
- 177.Dong Z-Y, Zhong W-Z, Zhang X-C, Su J, Xie Z, Liu S-Y, et al. Potential predictive value of TP53 and KRAS mutation status for response to PD-1 blockade immunotherapy in lung adenocarcinoma. Clin Cancer Res. 2017;23(12):3012–3024. doi: 10.1158/1078-0432.CCR-16-2554. [DOI] [PubMed] [Google Scholar]
- 178.Yu X-Y, Zhang X-W, Wang F, Lin Y-B, Wang W-D, Chen Y-Q, et al. Correlation and prognostic significance of PD-L1 and P53 expression in resected primary pulmonary lymphoepithelioma-like carcinoma. J Thorac Dis. 2018;10(3):1891–1902. doi: 10.21037/jtd.2018.03.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Assoun S, Theou-Anton N, Nguenang M, Cazes A, Danel C, Abbar B, et al. Association of TP53 mutations with response and longer survival under immune checkpoint inhibitors in advanced non-small-cell lung cancer. Lung Cancer. 2019;132:65–71. doi: 10.1016/j.lungcan.2019.04.005. [DOI] [PubMed] [Google Scholar]
- 180.Lin X, Wang L, Xie X, Qin Y, Xie Z, Ouyang M, et al. Prognostic biomarker TP53 mutations for immune checkpoint blockade therapy and its association with tumor microenvironment of lung adenocarcinoma. Front Mol Biosci. 2020;7:602328. doi: 10.3389/fmolb.2020.602328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Nassif EF, Auclin E, Bahleda R, Honoré C, Mir O, Dumont S, et al. TP53 mutation as a prognostic and predictive marker in sarcoma: pooled analysis of MOSCATO and ProfiLER precision medicine trials. Cancers (Basel) 2021;13(13):3362. doi: 10.3390/cancers13133362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Pérot G, Chibon F, Montero A, Lagarde P, de Thé H, Terrier P, et al. Constant p53 pathway inactivation in a large series of soft tissue sarcomas with complex genetics. Am J Pathol. 2010;177(4):2080–2090. doi: 10.2353/ajpath.2010.100104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Thoenen E, Curl A, Iwakuma T. TP53 in bone and soft tissue sarcomas. Pharmacol Ther. 2019;202:149–164. doi: 10.1016/j.pharmthera.2019.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Taubert H, Meye A, Würl P. Soft tissue sarcomas and p53 mutations. Mol Med. 1998;4(6):365–372. [PMC free article] [PubMed] [Google Scholar]
- 185.Agersborg S, Jiang S, Chen W, Ma W, Albitar M. PD-L1 expression correlation with TP53 gene mutation status in lung cancer but not in colorectal cancer. J Clin Oncol. 2016;34(15):11557. [Google Scholar]
- 186.Cyster JG, Allen CDC. B Cell responses: cell interaction dynamics and decisions. Cell. 2019;177(3):524–540. doi: 10.1016/j.cell.2019.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wouters MCA, Nelson BH. Prognostic significance of tumor-infiltrating B cells and plasma cells in human cancer. Clin Cancer Res. 2018;24(24):6125–6135. doi: 10.1158/1078-0432.CCR-18-1481. [DOI] [PubMed] [Google Scholar]
- 188.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3(11):991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 189.Inoue S, Leitner WW, Golding B, Scott D. Inhibitory effects of B cells on antitumor immunity. Can Res. 2006;66(15):7741–7747. doi: 10.1158/0008-5472.CAN-05-3766. [DOI] [PubMed] [Google Scholar]
- 190.Li Q, Teitz-Tennenbaum S, Donald EJ, Li M, Chang AE. In vivo sensitized and in vitro activated B cells mediate tumor regression in cancer adoptive immunotherapy. J Immunol. 2009;183(5):3195–3203. doi: 10.4049/jimmunol.0803773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Fridman WH, Meylan M, Petitprez F, Sun C-M, Italiano A, Sautès-Fridman C. B cells and tertiary lymphoid structures as determinants of tumour immune contexture and clinical outcome. Nat Rev Clin Oncol. 2022;19(7):441–457. doi: 10.1038/s41571-022-00619-z. [DOI] [PubMed] [Google Scholar]
- 192.Bruno TC, Ebner PJ, Moore BL, Squalls OG, Waugh KA, Eruslanov EB, et al. Antigen-presenting intratumoral B cells affect CD4(+) TIL phenotypes in non-small cell lung cancer patients. Cancer Immunol Res. 2017;5(10):898–907. doi: 10.1158/2326-6066.CIR-17-0075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kinker GS, Vitiello GAF, Ferreira WAS, Chaves AS, Cordeiro de Lima VC, Medina TDS. B cell orchestration of anti-tumor immune responses: a matter of cell localization and communication. Front Cell Dev Biol. 2021;9:678127. doi: 10.3389/fcell.2021.678127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Nielsen JS, Nelson BH. Tumor-infiltrating B cells and T cells: working together to promote patient survival. Oncoimmunology. 2012;1(9):1623–1625. doi: 10.4161/onci.21650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Lee-Chang C, Bodogai M, Martin-Montalvo A, Wejksza K, Sanghvi M, Moaddel R, et al. Inhibition of breast cancer metastasis by resveratrol-mediated inactivation of tumor-evoked regulatory B cells. J Immunol. 2013;191(8):4141–4151. doi: 10.4049/jimmunol.1300606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Shao Y, Lo CM, Ling CC, Liu XB, Ng KT, Chu AC, et al. Regulatory B cells accelerate hepatocellular carcinoma progression via CD40/CD154 signaling pathway. Cancer Lett. 2014;355(2):264–272. doi: 10.1016/j.canlet.2014.09.026. [DOI] [PubMed] [Google Scholar]
- 197.Zhou X, Su YX, Lao XM, Liang YJ, Liao GQ. CD19(+)IL-10(+) regulatory B cells affect survival of tongue squamous cell carcinoma patients and induce resting CD4(+) T cells to CD4(+)Foxp3(+) regulatory T cells. Oral Oncol. 2016;53:27–35. doi: 10.1016/j.oraloncology.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 198.Wang WW, Yuan XL, Chen H, Xie GH, Ma YH, Zheng YX, et al. CD19+CD24hiCD38hiBregs involved in downregulate helper T cells and upregulate regulatory T cells in gastric cancer. Oncotarget. 2015;6(32):33486–33499. doi: 10.18632/oncotarget.5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Roya N, Fatemeh T, Faramarz MA, Milad SG, Mohammad-Javad S, Najmeh SV, et al. Frequency of IL-10+CD19+ B cells in patients with prostate cancer compared to patients with benign prostatic hyperplasia. Afr Health Sci. 2020;20(3):1264–1272. doi: 10.4314/ahs.v20i3.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Lundberg A, Li B, Li R. B cell-related gene signature and cancer immunotherapy response. Br J Cancer. 2022;126(6):899–906. doi: 10.1038/s41416-021-01674-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Sautès-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer. 2019;19(6):307–325. doi: 10.1038/s41568-019-0144-6. [DOI] [PubMed] [Google Scholar]
- 202.Sautès-Fridman C, Lawand M, Giraldo NA, Kaplon H, Germain C, Fridman WH, et al. Tertiary lymphoid structures in cancers: prognostic value, regulation, and manipulation for therapeutic intervention. Front Immunol. 2016;7:407. doi: 10.3389/fimmu.2016.00407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Dieu-Nosjean MC, Giraldo NA, Kaplon H, Germain C, Fridman WH, Sautès-Fridman C. Tertiary lymphoid structures, drivers of the anti-tumor responses in human cancers. Immunol Rev. 2016;271(1):260–275. doi: 10.1111/imr.12405. [DOI] [PubMed] [Google Scholar]
- 204.Vanhersecke L, Brunet M, Guégan J-P, Rey C, Bougouin A, Cousin S, et al. Mature tertiary lymphoid structures predict immune checkpoint inhibitor efficacy in solid tumors independently of PD-L1 expression. Nat Cancer. 2021;2(8):794–802. doi: 10.1038/s43018-021-00232-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Lin Q, Tao P, Wang J, Ma L, Jiang Q, Li J, et al. Tumor-associated tertiary lymphoid structure predicts postoperative outcomes in patients with primary gastrointestinal stromal tumors. OncoImmunology. 2020;9(1):1747339. doi: 10.1080/2162402X.2020.1747339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Ladányi A, Kiss J, Mohos A, Somlai B, Liszkay G, Gilde K, et al. Prognostic impact of B-cell density in cutaneous melanoma. Cancer Immunol Immunother. 2011;60(12):1729–1738. doi: 10.1007/s00262-011-1071-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Goc J, Germain C, Vo-Bourgais TK, Lupo A, Klein C, Knockaert S, et al. Dendritic cells in tumor-associated tertiary lymphoid structures signal a Th1 cytotoxic immune contexture and license the positive prognostic value of infiltrating CD8+ T cells. Cancer Res. 2014;74(3):705–715. doi: 10.1158/0008-5472.CAN-13-1342. [DOI] [PubMed] [Google Scholar]
- 208.Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577(7791):561–565. doi: 10.1038/s41586-019-1914-8. [DOI] [PubMed] [Google Scholar]
- 209.Groeneveld CS, Fontugne J, Cabel L, Bernard-Pierrot I, Radvanyi F, Allory Y, et al. Tertiary lymphoid structures marker CXCL13 is associated with better survival for patients with advanced-stage bladder cancer treated with immunotherapy. Eur J Cancer. 2021;148:181–189. doi: 10.1016/j.ejca.2021.01.036. [DOI] [PubMed] [Google Scholar]
- 210.Italiano A, Bessede A, Pulido M, Bompas E, Piperno-Neumann S, Chevreau C, et al. Pembrolizumab in soft-tissue sarcomas with tertiary lymphoid structures: a phase 2 PEMBROSARC trial cohort. Nat Med. 2022;28(6):1199–1206. doi: 10.1038/s41591-022-01821-3. [DOI] [PubMed] [Google Scholar]
- 211.Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577(7791):549–555. doi: 10.1038/s41586-019-1922-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Germain C, Gnjatic S, Tamzalit F, Knockaert S, Remark R, Goc J, et al. Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am J Respir Crit Care Med. 2014;189(7):832–844. doi: 10.1164/rccm.201309-1611OC. [DOI] [PubMed] [Google Scholar]
- 213.Barmpoutis P, Di Capite M, Kayhanian H, Waddingham W, Alexander DC, Jansen M, et al. Tertiary lymphoid structures (TLS) identification and density assessment on H&E-stained digital slides of lung cancer. PLoS One. 2021;16(9):e0256907. doi: 10.1371/journal.pone.0256907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Tan WCC, Nerurkar SN, Cai HY, Ng HHM, Wu D, Wee YTF, et al. Overview of multiplex immunohistochemistry/immunofluorescence techniques in the era of cancer immunotherapy. Cancer Commun (Lond) 2020;40(4):135–153. doi: 10.1002/cac2.12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Messina JL, Fenstermacher DA, Eschrich S, Qu X, Berglund AE, Lloyd MC, et al. 12-Chemokine gene signature identifies lymph node-like structures in melanoma: potential for patient selection for immunotherapy? Sci Rep. 2012;2(1):765. doi: 10.1038/srep00765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. 2018;118(1):9–16. doi: 10.1038/bjc.2017.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.