

SUMMARY:
Arthropod-borne viruses, including the alphavirus chikungunya virus (CHIKV), cause acute disease in millions of people and utilize potent mechanisms to antagonize and circumvent innate immune pathways including the Type I interferon (IFN) pathway. In response, hosts have evolved antiviral counterdefense strategies which remain incompletely understood. Recent studies have found that long noncoding RNAs (lncRNAs) regulate classical innate immune pathways; how lncRNAs contribute to additional antiviral counterdefenses remains unclear. Using high-throughput genetic screening, we identified a cytoplasmic antiviral lncRNA that we named Antiviral LncRNA Prohibiting Human Alphaviruses (ALPHA), which is transcriptionally induced by alphaviruses and functions independently of IFN to inhibit the replication of CHIKV and its closest relative, O’nyong’nyong virus (ONNV), but not other viruses. Furthermore, we showed that ALPHA interacts with CHIKV genomic RNA and restrains viral RNA replication. Together, our findings reveal that ALPHA and potentially other lncRNAs, can mediate non-canonical antiviral immune responses against specific viruses.
In Brief:
Basavappa, et al. identify the alphavirus infection-induced, cytoplasmic, antiviral lncRNA ALPHA using high-throughput, loss-of-function screening. ALPHA specifically inhibits the replication of the related alphaviruses, chikungunya and O’nyong’nyong virus, but not other alphaviruses. ALPHA functions independently of Type I IFN signaling, directly binding viral genomic RNA to block viral RNA replication.
Graphical Abstract
INTRODUCTION:
Innate immunity is the first line of defense against all infectious microbes. During viral infection, unusual nucleic acid structures encoded by the virus are “sensed” by germline encoded pattern recognition receptor (PRR) families including the retinoic acid inducible gene I (RIG-I)-like receptors (RLRs) and the toll-like receptors (TLRs) (Chow et al., 2018; Rehwinkel and Gack, 2020a). Engagement of these PRRs leads to the activation of signaling adaptor proteins including mitochondrial antiviral signaling (MAVS) and TIR-domain-containing adapter inducing interferon β (TRIF) (Hou et al., 2011; Seth et al., 2005; Ullah et al., 2016). These adaptors stimulate downstream signaling pathways and de novo transcription of diverse innate immune effectors such as the canonical antiviral cytokine family, Type I interferons (IFN). IFN functions in an autocrine and paracrine manner to upregulate hundreds of IFN-stimulated genes (ISGs) that restrict viral infections (Schneider et al., 2014; Schoggins, 2019). In response, many viruses have developed methods to antagonize and evade IFN signaling. These pathways thus lie at the center of an ever-present evolutionary arms race wherein viruses continually evolve mechanisms to circumvent antiviral immunity while hosts attempt to overcome virus-mediated innate immune antagonism. Since most viruses are ultimately cleared, additional layers of immune regulation and function must exist to counter these viral evasion strategies.
Mosquito-borne viruses represent a diverse subset of medically relevant pathogens with few therapeutic options. Chikungunya virus (CHIKV) reemerged in the early 2000s and has since infected millions of individuals across the globe. CHIKV belongs to the alphavirus family, possesses an ~11.8 kb positive single stranded (ss)RNA genome, and replicates within the cytoplasm of infected host cells (Solignat et al., 2009). CHIKV displays broad tropism infecting epithelia, endothelia, and a subset of myeloid cells causing disease characterized by debilitating and often chronic arthralgia (Matusali et al., 2019; Schwartz and Albert, 2010). Like many RNA viruses, CHIKV can be sensed by RLR and TLR family members resulting in the induction of Type I IFN and ISGs (Fox and Diamond, 2016). However, CHIKV has evolved mechanisms to evade and attenuate these pathways. For example, CHIKV non-structural protein (nsp)2 both shuts down global transcription by inhibiting the polymerase (Pol)II cofactor, retinol binding protein (RBP)1, and blocks the phosphorylation of signal transducer and activator (STAT)1 limiting both IFN and ISG production (Akhrymuk et al., 2012; Akhrymuk et al., 2019; Fros et al., 2010; Fros et al., 2015; Fros and Pijlman, 2016; Goertz et al., 2018). The full spectrum of host antiviral factors that inhibit CHIKV replication is unknown.
While many of the protein coding pathways involved in antiviral defense have been characterized, how long noncoding RNAs (lncRNAs) contribute to antiviral immunity is unclear. LncRNAs are defined as any noncoding RNA that is greater than 200 nucleotides in length. These transcripts are Pol II transcribed, 5’ capped and can be polyadenylated and spliced thus closely mirroring messenger RNAs in terms of their biogenesis except for the definitive lack of an open reading frame (ORF) producing a polypeptide of >100 amino acids (aa). LncRNAs have been shown to regulate various aspects of innate immunity including innate immune cell development, RLR and TLR signaling, and immune gene expression (Agarwal et al., 2020; Agliano et al., 2019; Atianand et al., 2017; Basavappa et al., 2019; Chen et al., 2017; Hadjicharalambous and Lindsay, 2019; Jiang et al., 2018; Kotzin et al., 2016; Lin et al., 2019; Mowel et al., 2018; Mowel et al., 2017; Vierbuchen and Fitzgerald, 2021; Wang et al., 2014; Yi et al., 2019). These studies have largely focused on lncRNAs in the context of classical innate immune pathways. Whether lncRNAs play additional roles in the control of RNA viruses and/or counteract immune evasion strategies is not known.
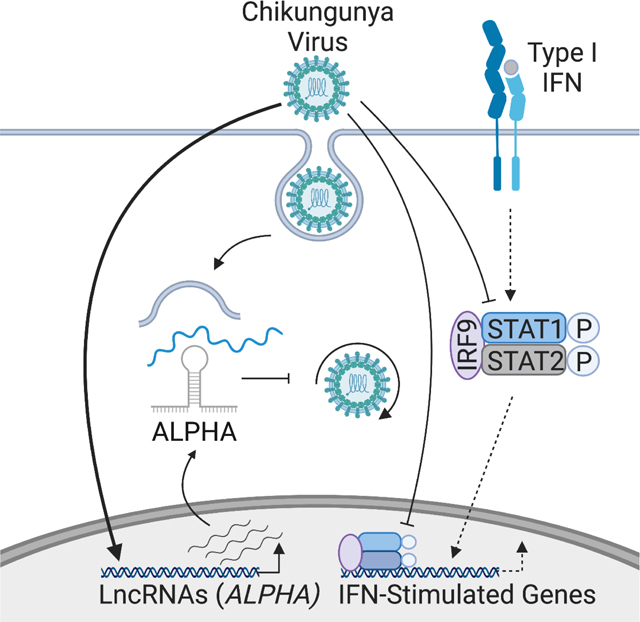
Using RNA-sequencing, we found that lncRNAs are induced by infection in a virus-specific manner. To assess the impact of lncRNAs in anti-CHIKV responses, we performed an unbiased, loss-of-function screen targeting 2200 human lncRNAs in CHIKV-infected endothelial cells. Using this approach, we identified a subset of lncRNAs with putative antiviral activity against CHIKV including the previously uncharacterized, Antiviral LncRNA Prohibiting Human Alphaviruses (ALPHA). Strikingly, ALPHA is induced in response to diverse alphavirus infections but only inhibits a subset of alphaviruses specifically CHIKV and its closest relative, O’nyong’nyong virus (ONNV). Mechanistically, we show that ALPHA functions independently of canonical, IFN-dependent responses and instead binds directly to CHIKV genomic RNA to inhibit viral RNA replication. Together, our findings provide evidence that lncRNAs can serve as potent and specific antiviral effectors, adding a new layer to innate antiviral immunity.
RESULTS
Chikungunya virus (CHIKV) is a mosquito-borne alphavirus which infects humans and primates and causes symptomatic disease characterized by arthralgia (Burt et al., 2017; Silva and Dermody, 2017). To promote its replication, CHIKV has evolved potent mechanisms to antagonize canonical, Type I interferon (IFN)-dependent signaling (Akhrymuk et al., 2012; Akhrymuk et al., 2019; Fros et al., 2010; Fros et al., 2015; Fros and Pijlman, 2016; Fros et al., 2013; Goertz et al., 2018; Meshram et al., 2019). We set out to identify additional mechanisms used by host cells to counteract these CHIKV evasion strategies. We began by exploring the transcriptional response to CHIKV infection. Since endothelial cells are an important target for many viruses including CHIKV, we performed RNA-sequencing (RNA-seq) in human brain microvascular endothelial cells (HBMEC) in the presence and absence of CHIKV or the phylogenetically disparate arbovirus, Zika virus (ZIKV). Inclusion of ZIKV allowed us to define pathways commonly induced by viral infection (e.g., IFN) from those that are virus-specific. As expected, analysis of differentially induced coding RNAs (mRNAs) revealed significant changes in canonical transcriptional programs including IFNs, IFN-stimulated genes (ISGs), and NFκB signaling in both CHIKV and ZIKV-infected cells (Figure S1A–B). Notably, both the total number of significantly upregulated transcripts and the relative levels of many induced canonical innate immune genes were reduced in CHIKV-infected cells compared to ZIKV-infected cells (Figure S1A, C). This suggests that CHIKV evades canonical signaling to a greater extent than ZIKV as has been previously suggested (Nelemans and Kikkert, 2019). Strikingly, we also found that long noncoding RNAs (lncRNAs) comprise a substantial portion of the total transcripts induced upon infection and that, unlike classical immune coding genes, the majority of these RNAs are either CHIKV- or ZIKV-specific (Figure 1A, S1A–C). These data suggest that lncRNAs may serve as key mediators of virus-specific antiviral responses.
Figure 1: High-throughput, loss-of-function, genome-wide screens reveal antiviral lncRNAs against CHIKV.

(A) Heat-map depicting differentially upregulated long noncoding RNAs (lncRNAs) with a log2 fold change greater than 1, read cutoff of 10, and adjusted p value less than 0.05 in uninfected, CHIKV, or ZIKV-infected HBMEC at 24h post-infection in three independent experiments. The y-axis represents individual annotated lncRNAs. (B) Results from high-throughput screens targeting 2200 lncRNAs across the human genome in CHIKV-infected HBMEC. The screen was performed in duplicate, Z scores were calculated from percent infection values and replicates were plotted against each other. The 9 anti-CHIKV lncRNAs identified in the screen (Z>2 in both replicates) are highlighted in light blue. ALPHA is demarcated by the enlarged, dark blue data point. The 21 proviral lncRNAs are highlighted in purple. (C) Secondary screens were performed for 6 of the 9 anti-CHIKV candidate lncRNAs using three pooled siRNAs per target followed by infection with CHIKV-mKate for 24h at the indicated MOIs and the percentage of infected cells was quantified. ALPHA is highlighted by dark blue symbols and a blue asterisk. (D) The relative intracellular localization of ALPHA in uninfected HBMEC measured by qPCR and displayed as the percentage of total transcript. GAPDH was used a positive control for cytoplasmic enrichment; MALAT1 was used as a positive control for nuclear enrichment. (E-F) Control and ALPHA-depleted HBMEC were infected with CHIKV for 30h at the indicated MOIs. Infection levels were quantified by (E) quantitative reverse transcription PCR (qPCR) for viral RNA or (F) TCID50s for viral titers. (G) ALPHA was depleted using three independent siRNAs followed by infection with CHIKV at MOI 0.05 for 24h. Viral RNA was quantified by qPCR. (H) ALPHA−/− HBMEC were generated using CRISPR/Cas9 and infected with CHIKV for 24h at the indicated MOIs. CHIKV RNA was measured by qPCR. (I) HBMEC clones stably expressing ALPHA cDNA were infected with MOI 0.2 for 24h. The percentage of cells infected was quantified by immunofluorescence and automated microscopy. Data are presented as fold change vs. control cells in (E) and (G-I). GAPDH was used as a loading control gene for all qPCR experiments unless otherwise noted. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; error bars represent S.E.M; Screens shown in (B) were performed in duplicate, n=3–4 for all other experiments as indicated; Statistical analyses were performed using Student’s (unpaired, two-tailed) t-test with Holm-Sidak correction for multiple comparisons (C, E, F, H), one-way ANOVA with Tukey correction for multiple comparisons (G); one-way ANOVA with Dunnet correction for multiple comparisons (I). See also Figures S1 and S2.
To identify lncRNAs with anti-CHIKV activity, we performed a high-throughput RNAi screen targeting 2200 lncRNAs (designed by Ambion) in HBMEC followed by infection with CHIKV engineered to express an mKate fluorescent reporter during viral replication (CHIKV-mKate, Figure S1D–E) (Long et al., 2016; Moser et al., 2016). The screen contained a subset of the lncRNAs identified by RNA-seq. We used automated imaging and imaging analysis to quantify the total number of cells (nuclei counts) and percentage of cells infected (mKate+) (Figure S1D). This screen was performed in duplicate and Z scores were calculated for cell number and percent infection (Figure 1B, S1F). As a negative control, we used a scrambled siRNA (siCON); as positive controls for RNAi efficiency, we used siRNAs targeting both pro-mitotic and anti-apoptotic RNAs (siKIF11 and siDEATH respectively) (Figure S1G). As a positive control for an antiviral effect, we targeted the canonical immune factor zinc finger antiviral protein (ZAP) (Figure S1H) (Bick et al., 2003; MacDonald et al., 2007). We removed cytotoxic genes (Z<−2 for cell number) and plotted Z-scores calculated from percent infection replicate data (Figure 1B, S1F). Using a cutoff of Z>2, we identified 9 previously uncharacterized, potentially antiviral lncRNAs where RNA depletion resulted in an increase in the percentage of CHIKV-infected cells (Figure 1B). We also identified 21 putative “proviral” lncRNAs using a cutoff of Z<−2. It is possible that a subset of these “proviral” lncRNAs represent negative regulators of innate immunity which may be critical to a complete understanding of anti-CHIKV responses. However, we initially focused on the narrower set of 9 antiviral lncRNAs, validating 6 in secondary screens (Figure 1C). In all, these findings identify new lncRNAs with putative activity against CHIKV.
LncRNAs can have nuclear or cytoplasmic activities; nuclear lncRNAs often regulate proximal genes in cis while cytoplasmic lncRNAs can have more divergent functions (Basavappa et al., 2019; Mowel et al., 2018). Since CHIKV RNA replication occurs within the cytoplasm of infected cells, we were particularly interested in lncRNAs localized in this compartment. To this end, we isolated nuclear and cytoplasmic fractions from uninfected HBMEC and measured the relative enrichment of lncRNA transcripts in each compartment. We used GAPDH (a cytoplasmic mRNA) and MALAT1 (a nuclear lncRNA) as controls. Using this approach, we found that the ENST00000452500 transcript is localized in the cytoplasm; we have named this lncRNA Antiviral LncRNA Prohibiting Human Alphaviruses (ALPHA, Figure 1B and 1D). The ALPHA locus encodes a human-specific (non-conserved), long intergenic noncoding RNA (lincRNA) located on chromosome 21 (Figure S2A). ALPHA exists as a single isoform of 530 bp containing 3 exons (Figure S2A). To verify that ALPHA is indeed noncoding, we demonstrated that ALPHA is not enriched in polysomes (Figure S2B–C). In order to explore ALPHA antiviral function more extensively, we assessed how ALPHA depletion affects viral protein, RNA, and newly produced virions. We observed significant increases in all three parameters upon ALPHA knockdown in HBMEC when compared to control cells, recapitulating the screening results (Figure 1E–F and S2D–E). Strikingly, loss of ALPHA led to >10-fold increases in both viral RNA and titers, closely mirroring the effects observed by depletion of the anti-alphaviral protein ZAP, indicating that ALPHA is a potent inhibitor of CHIKV infection (Figure 1E–F). To eliminate the possibility that ALPHA’s antiviral effects were a result of RNAi-associated off-target effects, we used three independent siRNAs targeting non-overlapping regions of exons 2 and 3 and measured CHIKV replication. Again, we observed significant increases in viral RNA upon ALPHA depletion using each of these siRNAs (Figure 1G). In addition, we generated an HBMEC clone with genetic deletion of the ALPHA locus using CRISPR/Cas9. Similar to our RNAi results, we observed significant increases in viral RNA, viral titers, and the percentage of infected cells across different MOIs in ALPHA−/− cells (Figure 1H and S2F–H). Finally, we generated HBMEC clones which stably overexpress the single ALPHA isoform containing 3 exons and found that CHIKV is significantly attenuated in these cells compared to control infected cells (Figure 1I and S2I). Together, these data indicate that ALPHA is a cytoplasmic lncRNA with potent antiviral activity against CHIKV.
To understand ALPHA transcriptional regulation during CHIKV infection, we infected HBMEC with CHIKV for 24h and measured ALPHA expression (Figure 2A and S3A). We found that ALPHA RNA is potently induced by CHIKV infection in a dose-dependent manner (Figure 2A and S3A). We next assessed whether ALPHA is specifically induced by CHIKV or is more broadly upregulated by viral infection. To this end, we infected HBMEC with a set of related alphaviruses including CHIKV, O’nyong’nyong (ONNV), Mayaro (MAYV), and Sindbis virus (SINV), or the unrelated arbovirus, ZIKV at MOI 5 for 24h and quantified ALPHA levels (Figure 2B). We found that infection with all alphaviruses tested resulted in a ≥10-fold increase in ALPHA levels; however, ZIKV infection did not induce ALPHA expression (Figure 2B). Thus, ALPHA induction may be specific to alphaviral infection. We next addressed whether viral replication is required for this upregulation or whether sensing of incoming particles alone is sufficient. To test this, we infected HBMEC with either wildtype (WT) or UV-inactivated CHIKV (Figure 2C). WT CHIKV infection led to a dose-dependent increase in ALPHA expression, while UV-inactivated CHIKV did not (Figure 2C). Altogether, these data demonstrate that ALPHA is induced in an alphavirus-specific and replication-dependent manner.
Figure 2: ALPHA induction and function is independent of canonical interferon pathways.

(A) HBMEC were infected with CHIKV for 24h at the indicated MOIs. Viral RNA and ALPHA levels were quantified by qPCR. (B) HBMEC were infected with the related alphaviruses CHIKV, O’nyong’nyong (ONNV), Mayaro (MAYV), and Sindbis virus (SINV) or the phylogenetically disparate, ZIKV at MOI 5 for 24h. ALPHA was measured by qPCR. (C) ALPHA transcript levels were measured by qPCR following infection with either wildtype or UV-inactivated CHIKV at the indicated MOIs for 24h. (D) HBMEC were stimulated with either Sendai virus (SeV, 100 HAU/mL) or recombinant IFNβ (10 ng/mL) for the indicated timepoints and ALPHA was quantified by qPCR. (E) Control and ALPHA-depleted HBMEC were infected with CHIKV for 24h at the indicated MOIs. Viral RNA, IFNB1, and ISG56 transcript levels were measured by qPCR. (F) HBMEC were treated with either control or pooled ALPHA siRNAs and infected with CHIKV at MOI 1 for 24h. IFIT1 protein expression was quantified by immunofluorescence and automated microscopy. (G) Control and ALPHA-depleted HBMEC were pre-treated with Ruxolitinib at the indicated concentrations for 2h. The cells were then spin infected with CHIKV at MOI 1 for 24h. The percentage of CHIKV-infected cells was measured by immunofluorescence and automated microscopy. Scale bars represent 200 μm. GAPDH was used as a loading control in all qPCR experiments. Data are presented as fold change relative to uninfected or unstimulated controls (A-D, F) or siCON (E, G). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; error bars are S.E.M.; n=3–6 as indicated; Statistical analyses were performed using one-way ANOVA with Dunnett correction for multiple comparisons (A), one-way ANOVA with Tukey correction for multiple comparisons (B), two-way ANOVA with Tukey correction for multiple comparisons (C, D), Student’s (unpaired, two-tailed) t test with Holm-Sidak correction for multiple corrections (E, F, G). See also Figures S3 and S4.
Classical cytokines, IFNs, and ISGs are robustly upregulated upon viral infection. As these response mechanisms are not unique to CHIKV (and are induced by ZIKV), it seemed unlikely that these pathways regulated ALPHA induction. However, to directly test whether IFN-dependent pathways induce ALPHA, we stimulated HBMEC with Sendai virus (SeV), a potent stimulator of RLRs which leads to robust induction of both type I IFN and ISGs, for 24h and measured ALPHA expression (Figure 2D and S3B) (Seth et al., 2005). Notably, although the cells were effectively stimulated as measured by IFNB1 and ISG56 transcript levels, ALPHA remained unchanged (Figure 2D and S3B). Similarly, direct treatment with recombinant IFNβ for 24h induced ISGs but did not affect ALPHA expression (Figure 2D and S3B). In addition to Type I IFN, IL-1β and TNFα are also important cytokines induced during CHIKV infection (Kelvin et al., 2011; Ng et al., 2009; Tanabe et al., 2018; Venugopalan et al., 2014). However, we found that stimulation of HBMEC with either IL-1β or TNFα for 24h did not lead to ALPHA induction (Figure S3C–D). Additionally, we evaluated the contribution of MAPK and JNK signaling to ALPHA regulation as these cascades have previously been implicated in CHIKV infection (Nayak et al., 2019; Varghese et al., 2016). Although CHIKV infection was modestly decreased upon treatment with the JNK inhibitor SP600125 and the MAPK inhibitor PD98059 as previously described, ALPHA induction was not significantly impacted (Figure S3E–F) (Nayak et al., 2019; Varghese et al., 2016).
We further mined our own transcriptomic data for CHIKV-induced pathways in HBMEC. One of the most highly induced genes was the prostaglandin F2α (PGF2α) receptor (PTGFR, 100-fold). To test whether PGF2α signaling contributes to ALPHA induction, we treated HBMEC with a PGF2α antagonist (AL8810), infected cells with CHIKV and measured ALPHA levels (Figure S3G). We observed that PTGFR activity was not required for CHIKV-induced ALPHA upregulation and did not impact infection (Figure S3G). We also tested the contribution of Ca2+ signaling to ALPHA regulation as this is a classical signaling pathway used throughout the immune system. To assess whether Ca2+ flux impacts ALPHA induction, we stimulated HBMEC with the calcium ionophore ionomycin for 24h and observed no significant difference in ALPHA levels (Figure S3H). Together, these data indicate that ALPHA induction is specifically induced during alphavirus infection and is independent of classical cytokines, canonical innate immune signaling pathways, and other characterized CHIKV-induced pathways.
We further assessed ALPHA induction across other cell types including primary human monocytes, A549 cells (lung epithelial carcinoma cells), and U2OS cells (osteosarcoma cells). CHIKV replicates in these cells as indicated by both CHIKV RNA levels and IFN induction (Figure S3I–K). We found that ALPHA is neither expressed at baseline nor induced by infection in either monocytes or A549s (Figure S3I–J). Interestingly, ALPHA is expressed in U2OS at baseline to similar levels as HBMEC but is not induced by CHIKV infection (Figure S3K). These data suggest that ALPHA expression is regulated in both a virus- and cell type-specific manner. This supports previous studies demonstrating that lncRNAs typically have more restricted cell expression patterns relative to messenger RNAs (Cabili et al., 2011; Hon et al., 2017; Werner et al., 2017).
We next set out to define the mechanism by which ALPHA attenuates CHIKV infection. While we found that ALPHA induction is independent of IFN, we wanted to assess whether ALPHA inhibits infection by regulating IFN-dependent signaling as has been shown for other lncRNAs. (Agarwal et al., 2020; Atianand et al., 2017; Basavappa et al., 2019; Chen et al., 2017; Mowel et al., 2018; Vierbuchen and Fitzgerald, 2021). To this end, we compared IFNB1 and ISG56 transcript levels in control and ALPHA-depleted cells infected with CHIKV (Figure 2E). If ALPHA promotes this pathway, we would have expected decreased levels of IFNB1 and ISG56 upon loss of ALPHA. In contrast, we observed elevated levels of these transcripts compared to control cells likely as a consequence of the increased viral infection that occurs upon ALPHA depletion (Figure 2E). We further explored the impact of ALPHA on IFN by generating a HBMEC line which stably expresses an IFN-mCherry reporter. Using this tool, we assessed the contribution of ALPHA to IFN regulation outside of CHIKV infection. As expected, at baseline there was no detectable IFN-mCherry expression; however, upon infection with SeV, reporter expression was robustly induced in a dose-dependent manner (Figure S4A–B). We depleted ALPHA in these cells followed by infection with SeV for 24h and quantified reporter signal by automated microscopy. As a positive control, we included siRNAs targeting MAVS, a canonical adaptor protein required for IFN induction downstream of SeV infection (Seth et al., 2005). As expected, MAVS depletion caused a significant loss in both the percentage of IFN-mCherry+ cells as well as total mCherry protein as measured by mean fluorescent intensity (MFI, Figure S4A–B). In contrast, we observed no difference in IFN-mCherry expression in ALPHA-depleted cells compared to control (Figure S4A–B). Given that cytoplasmic lncRNAs can also modulate protein translation in diverse biological contexts, we also tested whether ALPHA regulates ISG translation (Basavappa et al., 2019; Mowel et al., 2018). To test this, we depleted ALPHA in HBMEC, infected with CHIKV for 24h, and quantified IFIT1 protein levels by immunofluorescence and automated microscopy (Figure 2F). Again, we found that ALPHA is not required for ISG protein production. Instead, we observed that IFIT1 protein levels were increased upon ALPHA knockdown (Figure 2F). Together, these data suggest that ALPHA antiviral function is independent of IFN and ISG transcription or translation.
To address whether ALPHA antiviral activity is dependent on IFN signaling, we infected control or ALPHA-depleted HBMEC with CHIKV in the presence of the janus kinase (JAK)1/2 inhibitor, Ruxolitinib, which potently inhibits ISG induction (Figure 2G and S4C) (Chiappinelli et al., 2015; Stewart et al., 2014). As expected, Ruxolitinib treatment led to a marked attenuation in CHIKV-induced IFIT1 production (Figure S4C). Moreover, we observed that in ALPHA-depleted cells, the percentage of infected cells was significantly elevated compared to control cells in both untreated and Ruxolitinib-treated cells (Figure 2G). These findings together indicate that ALPHA anti-CHIKV activity is independent of IFN signaling.
A hallmark of Type I IFN is its broad potency against diverse viral families. However, we found that ALPHA is specifically induced by alphaviruses and has anti-CHIKV activity independent of classical IFN signaling. This led us to hypothesize that ALPHA may instead have restricted antiviral activity against alphaviruses. To test this possibility, we depleted ALPHA in HBMEC and infected with diverse alphaviruses including CHIKV, ONNV, SINV, MAYV, and Ross River virus (RRV, phylogeny shown in Figure 3A). In addition to increasing CHIKV infection, ALPHA depletion resulted in a significant increase in ONNV infected cells, viral RNA, and titers (Figure 3B–C). Strikingly however, there was no effect on more distant alphaviruses including MAYV, SINV, and RRV (Figure 3D). In addition, we also tested ALPHA’s antiviral activity against other unrelated viruses including ZIKV, Influenza A (IAV), Rift Valley Fever (RVFV), La Crosse (LACV), and Herpes Simplex I (HSV-1) and found that loss of ALPHA had no effect on the replication of any these viruses (Figure 3E, S4D–G). In contrast, knockdown of the broadly acting anti-alphaviral factor ZAP, resulted in an increase in infection across multiple alphaviruses (Figure S4H). Furthermore, we infected both ALPHA−/− and ALPHA-overexpressing HBMEC with ZIKV and observed no change in infection in either cell population compared to controls (Figure 3F–G). Altogether, these results demonstrate that the antiviral activity of ALPHA is highly restricted to the closely related alphaviruses CHIKV and ONNV.
Figure 3: ALPHA antiviral activity is restricted to CHIKV and ONNV.

(A) Phylogenetic tree of the indicated alphaviruses generated using whole genome nucleotide sequences (in VIPR). (B, D) Control and ALPHA-depleted HBMEC were infected for 24h with CHIKV-mKate (MOI 0.03), ONNV (MOI 0.1), MAYV (MOI 0.1), SINV-mKate (MOI 0.1), and Ross River-GFP (RRV-GFP, MOI 0.3). The percentage of cells infected was measured by immunofluorescence and automated microscopy. (C) Viral RNA measured by qPCR and viral titers measured by TCID50 in control and ALPHA-depleted HBMEC infected with ONNV at the indicated MOIs for 30h. Viral RNA data are displayed as fold change vs. control cells. (E) Control and ALPHA-depleted HBMEC were infected with ZIKV (MOI 0.03) for 24h. Percent infection was measured using immunofluorescence and automated microscopy. (F) ALPHA+/+ and ALPHA−/− HBMEC were infected with ZIKV at the indicated MOIs for 24h. Viral RNA was quantified by qPCR. (G) ALPHA-overexpressing cells were infected with ZIKV MOI 0.1 for 24h. The percentage of cells infected was quantified by immunofluorescence and automated microscopy. Data are displayed as fold change relative to control. **p<0.01, ***p<0.001, ****p<0.0001; error bars are S.E.M.; n=3; Statistical analyses were performed using Student’s (unpaired, two-tailed) t test (B, D, E), Student’s (unpaired, two-tailed) t test with Holm-Sidak correction for multiple comparisons (C, F), one-way ANOVA with Dunnett correction for multiple comparisons (G). See also Figure S4.
We next explored how ALPHA interferes with the CHIKV replication cycle. Briefly, after binding to its cognate receptor on the plasma membrane, CHIKV is internalized into the endocytic compartment and fuses to the endosome upon acidification, releasing its genome into the cytoplasm. The 5’ ORF is translated by host ribosomes to produce the non-structural (nsp)1–4 polyprotein which is cleaved to form the replicase complex. The replicase then synthesizes anti-genome templates, new genome copies and subgenomic RNAs (encoded in the 3’ ORF and required to produce structural proteins). New virions are assembled in the cytoplasm, exit the cell at the plasma membrane and infect additional cells leading to secondary infection and viral spread. To first test viral entry, we infected HBMEC in the presence of cycloheximide (CHX) which inhibits viral translation, the first step in intracellular viral replication. Measuring internalized viral RNA in this context thus allows for the specific quantification of only viral genomes which have entered into the cells. Using this approach, we observed equivalent levels of viral genomic RNA in control and ALPHA-depleted cells indicating that ALPHA does not regulate early entry (Figure S5A). We also assessed viral spread by treating cells with ammonium chloride (NH4Cl) after virus entry. This inhibits endocytic acidification required for secondary infection. As expected, blocking spread reduced overall viral levels in both control and ALPHA-depleted HBMEC (Figure S5B). However, viral RNA levels remained significantly increased upon ALPHA depletion even in the presence of NH4Cl, demonstrating that ALPHA does not affect viral spread (Figure S5B). Together, these data indicate that ALPHA activity modulates an intracellular step of the CHIKV life cycle.
A crucial step in the cytoplasmic CHIKV life cycle is the generation of genome and anti-genome copies. These transcripts form double stranded (ds)RNA intermediates which can be detected using a specific antibody (J2). We depleted ALPHA in HBMEC followed by infection with CHIKV at MOI 20 for 8h, immunostained with J2, and quantified dsRNA puncta by confocal microscopy. Using this approach, we observed a significant increase in dsRNA puncta in ALPHA knockdown conditions compared to control cells (Figure 4A and S5C). Interestingly, a bifurcation in the dsRNA+ population appears specifically in ALPHA-depleted cells. This may reflect the nonsynchronized manner of infection wherein different cells become infected at different times and those that express higher levels of ALPHA are more restrictive in control cells. Overall, these data suggest that ALPHA attenuates early viral RNA replication.
Figure 4: ALPHA directly binds CHIKV genomes in the cytoplasm to inhibit viral RNA replication.

(A) Control and ALPHA-depleted HBMEC were infected with CHIKV at MOI 20 for 8h and dsRNA/J2+ puncta were detected by immunofluorescence and confocal microscopy. The number of puncta per infected cell was quantified by ImageJ in >50 cells per experiment in three independent experiments. Scale bars represent 10 μm. (B) Anti-genome copies were quantified using strand-specific RT-qPCR in control and ALPHA-depleted HBMEC infected with CHIKV at MOI 0.5 for 8h. (C) Schematic detailing RNA-PLA. Fixed, permeabilized CHIKV-infected HBMEC were incubated with paired, DNA probes containing a 35–45 nt antisense sequence complementary to target RNAs of interest (i.e., GAPDH, CHIKV, and ALPHA), a polyA linker, and a unique PLA oligo sequence. Two PLA connector oligos which can bind the PLA probes are introduced into the cells along with T4 ligase. If the PLA probes are near each other (indicating that the target RNAs are in close proximity), the PLA connectors will be ligated into a circle which can then serve as a template for rolling circle amplification (RCA). The resulting can then be detected using a Cy5-conjugated antisense DNA probe and confocal microscopy. (D-E) HBMEC were infected with CHIKV at MOI 5 for 24h and hybridized with PLA probe pairs GAPDH + CHIKV (G + C), GAPDH + ALPHA (G+A), CHIKV + CHIKV (C + C), or ALPHA + CHIKV (A + C). PLA amplicons were visualized as described above. Scale bars represent 10 μm. (E) PLA puncta per cell were quantified for 3 independent experiments in 40–50 cells per experiment using ImageJ. (F) ALPHA-overexpressing HBMEC were either uninfected or infected with CHIKV or SINV at MOI 2 for 20h and subjected to glutaraldehyde crosslinking, overnight anti-sense probe hybridization, streptavidin pulldown, and quantification of both target RNAs (GAPDH, CHIKV genomic RNA, or SINV genomic RNA) as well as co-precipitated RNAs (ALPHA) by qPCR. Displayed is the relative enrichment of ALPHA in each conditions. All data were first normalized to input levels of each transcript and displayed as fold change relative to GAPDH pulldown levels (GAPDH RP). *p<0.05, **p<0.01, ****p<0.0001; error bars represent S.E.M.; n=3 (A, C, D), n=4 (B); Statistical analyses were performed using Student’s (unpaired, two-tailed) t test (A), two-way ANOVA with Sidak correction for multiple comparisons (B), one-way ANOVA with Tukey correction for multiple comparisons (D), one-way ANOVA with Sidak correction for multiple comparisons. See also Figures S5 and S6.
We next assessed which specific step of viral RNA replication is impacted by ALPHA. The first step in the production of new viral genomes is the generation of anti-genome RNA. An impact on this initial step would consequently alter the downstream levels of both genomic and sub-genomic RNAs as well as viral titers. We quantified the number of CHIKV anti-genome copies present in control and ALPHA-depleted HBMEC using strand-specific RT-qPCR. We found a significant increase in anti-genome copies upon ALPHA knockdown compared to control (Figure 4B). These data show that ALPHA affects early viral replication by inhibiting anti-genome production.
Given the observed decrease in CHIKV RNA replication as well as the specificity of ALPHA antiviral function, we hypothesized that ALPHA may directly bind to CHIKV genomic RNA. Indeed, analysis of the alphaviruses used in this study revealed that the nucleotide sequences between alphaviral genomes are more divergent than the amino acid sequences they encode (Figure S5D). ONNV is closest to CHIKV at both the nucleotide and protein level (Figure S5D). We thus hypothesized that ALPHA may bind directly to CHIKV genomic RNA to mediate its antiviral effect. To test this hypothesis, we adapted a previously described proximity ligation assay (PLA) to visualize ALPHA-CHIKV RNA interactions in situ (Figure 4C) (Fredriksson et al., 2002; Soderberg et al., 2006; Zhang et al., 2016). We designed tripartite, antisense, DNA probes containing i.) a 35–45mer complementary to either GAPDH, ALPHA, or CHIKV RNA, ii.) a poly(A) linker, and iii.) a non-specific PLA oligonucleotide overhang. We paired the CHIKV probe with either the GAPDH probe as a negative control (CHIKV + GAPDH), a second CHIKV probe 50 bp downstream of the original probe as a positive control (CHIKV + CHIKV) or the ALPHA probe (CHIKV + ALPHA). As an additional negative control, we also paired the GAPDH probe with an ALPHA probe (GAPDH + ALPHA). We allowed these partnered probes to hybridize with RNAs within fixed, permeabilized, CHIKV-infected HBMEC. This was followed by incubation with T4 ligase and PLA “connector” oligos complementary to the probe PLA overhangs. If the two partner probes are in close proximity, the connector oligos will be ligated to form a circle which serves as a template for rolling circle amplification. The PCR product can then be detected using a fluorescently conjugated antisense DNA probe specific to the amplicon. As expected, we observed very few PLA puncta in GAPDH + CHIKV and GAPDH + ALPHA samples and many puncta in CHIKV + CHIKV samples (Figure 4D–E and S5E). Interestingly, we also found a high number of PLA puncta in ALPHA + CHIKV samples that was significantly greater than that observed in either negative control (Figure 4D–E and S5E). These results suggest that ALPHA interacts with CHIKV genomic RNA. These data also reveal that this potential interaction occurs in the cytoplasm as is expected given that both ALPHA RNA and CHIKV RNA replication are localized in this compartment.
To assess RNA-RNA interactions more directly, we used a modified Chromatin Isolation by RNA Pulldown (ChIRP) approach (Figure S6A) (Chu et al., 2012). Briefly, we used glutaraldehyde to crosslink uninfected, CHIKV-, or SINV-infected ALPHA-overexpressing HBMEC. In addition to serving as a control for ALPHA’s binding specificity, inclusion of SINV-infected cells also provides an important control for ALPHA transcript levels as SINV induces ALPHA to similar levels as CHIKV but is not sensitive to ALPHA antiviral activity (Figure 2B and 3D). Lysates were generated and incubated with 500mer biotinylated, antisense oligonucleotide (ASO) probes designed against target RNAs of interest specifically GAPDH (as a negative control), CHIKV genomic RNA, and SINV genomic RNA. Following hybridization, the biotinylated probes were enriched using streptavidin and relative RNA levels of both the target RNAs and any co-precipitated RNAs were quantified. Using this assay, we effectively enriched for viral genomes using viral probes compared to the control GAPDH probe (Figure S6BC). Conversely, the control GAPDH probe successfully enriched for GAPDH compared to either viral probe (Figure S6D). Importantly, the relative levels of GAPDH enrichment were similar across conditions indicating a lack of bias between samples (Figure S6D). We further quantified ALPHA levels in both control GAPDH and viral RNA pulldowns. As ALPHA was not directly targeted by a probe, any positive ALPHA signal detected in this assay is a result of co-precipitation and reveals interactions with our target RNAs. Strikingly, we observed a significant and specific enrichment in ALPHA upon CHIKV RNA pulldown but not SINV RNA pulldown (Figure 4F). Importantly, ALPHA enrichment was absent in uninfected cells incubated with the CHIKV genomic RNA probe, demonstrating a requirement for CHIKV RNA (Figure 4F). We repeated these experiments in wildtype HBMEC and observed a similar, specific enrichment of endogenous ALPHA only upon CHIKV RNA pulldown (Figure S6E–H).
We next explored whether the ALPHA-CHIKV RNA interaction can form independently of other cellular factors. We used purified RNAs to quantify the interaction between ALPHA and CHIKV RNA in vitro. We in vitro transcribed and biotinylated full-length ALPHA as well as a truncated GAPDH RNA of equivalent length, as a negative control (500 nt, Figure 5A, S6I–J). As a positive control we also generated a biotinylated, antisense probe complementary to the nsp3 region of the CHIKV genome and of similar length to ALPHA (500 nt, Figure 5A, S6I–J). We incubated each of these RNAs with unbiotinylated CHIKV replicon RNA which contains only the 5’ end of the CHIKV genome encoding nsp1–4 (7.5 kb) (Jones et al., 2017). We then enriched for each biotinylated RNA (GAPDH, nsp3, or ALPHA) using streptavidin-conjugated beads and quantified the amount of co-precipitated CHIKV replicon RNA (Figure 5A). We observed significant enrichment of the CHIKV replicon by our positive control compared to GAPDH confirming specificity (Figure 5B). Interestingly, we also found that replicon RNA is highly enriched upon pulldown of full-length ALPHA (Figure 5B). Importantly, these results further demonstrate that the ALPHA-CHIKV RNA interaction does not require protein and pinpoints the interaction within the 5’ end of the CHIKV genome.
Figure 5: ALPHA exon 1 and CHIKV nsp1 interact.

(A-B) In vitro transcribed, biotinylated GAPDH, antisense CHIKV probe (Anti-CHIKV), or full-length ALPHA, were incubated with CHIKV replicon RNA and enriched using streptavidin-conjugated beads. The relative levels of co-precipitated replicon RNA were quantified by qPCR. (C) In vitro transcribed RNAs spanning ALPHA exon 1/2 (Ex1/2), exon 3 (Ex3), and exon 2/part of exon 3 (Ex2/3) were biotinylated and used as in (A). (D) In vitro transcribed RNAs spanning ALPHA exon 1 alone (Ex1), exon 2 alone (Ex2), and exon 1 with a deletion of the last 12 nt (Ex1Truncated) were biotinylated and used as in (A). (E-H) Individual nsps1–4 were in vitro transcribed, biotinylated, and mixed with either truncated GAPDH or ALPHA. The relative levels of unbiotinylated GAPDH or ALPHA was measured by qPCR. Data are presented as fold enrichment versus GAPDH (B-D) or ALPHA + full-length CHIKV replicon RNA (E-H). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; error bars represent S.E.M.; n=3–7 as indicated; Statistical analyses were performed using one-way ANOVA with Dunnett correction for multiple comparisons (B,C, E-H) or multiple, unpaired, two-tailed Student’s t tests (D). See also Figure S6 and S7.
We utilized this in vitro RNA interaction system to begin mapping the sequence requirements within ALPHA to bind CHIKV RNA. We first generated ~250 nt truncations in ALPHA spanning Exons 1 and 2 (Ex1/2), Exon 3 alone (Ex3), and Exon 2 and half of Exon 3 (Ex2/3, encoding the middle 250 nt of the ALPHA transcript). These RNAs were biotinylated and subsequently used as probes to assess in vitro interaction with unbiotinylated CHIKV replicon RNA. We observed that Exons 1 and 2 are essential for binding to CHIKV RNA while Exon 3 is dispensable (Figure 5C, S6I–J). Next, we made additional smaller truncations in Exons 1 and 2 which were again assessed for binding to CHIKV RNA. These probes include Exon 1 only (Ex1), Exon 2 only (Ex2) and a truncated Ex1 missing the last 12 nt (Ex1Truncated). We found that Exon 1 is required for interaction with CHIKV RNA (Figure 5D, S6I–J). Interestingly, deletion of the last 12 nt of Exon 1 results in loss of CHIKV RNA enrichment closely mirroring levels seen for the GAPDH negative control (Figure 5D, S6I–J). These results indicate that the 3’ end of ALPHA Exon 1 is minimally required for binding to the CHIKV genome.
We also used this in vitro RNA interaction system to identify regions within the CHIKV replicon RNA that are bound by ALPHA. The CHIKV genome is 11.8 kb in length; the replicon RNA we used in our initial interaction mapping experiments contains the first 7.5 kb, encoding the nonstructural proteins (nsp)1–4. We generated biotinylated RNAs encoding each nsp and tested their interactions with unbiotinylated, full-length ALPHA. As negative controls, we paired unbiotinylated GAPDH with full-length biotinylated CHIKV replicon RNA or we paired unbiotinylated ALPHA with biotinylated GAPDH (Figure 5E–H, S6K–L). As a positive control, we paired unbiotinylated ALPHA with biotinylated full-length CHIKV replicon RNA, paralleling the control conditions from our previous in vitro experiments but with the opposite RNA biotinylated (Figure 5E–H, S6K–L). As above, we incubated partnered RNAs together to form interactions, enriched for biotinylated RNAs using streptavidin-conjugated beads, and quantified the relative levels of the unbiotinylated RNA partners, GAPDH or ALPHA. As anticipated based on our previous results, ALPHA was significantly enriched by full-length biotinylated replicon RNA relative to GAPDH in all experiments (Figure 5E–H, S6K–L). Similarly, ALPHA was not bound to biotinylated GAPDH, paralleling our findings by RNA-PLA (Figure 4C–D, 5E–H, S6K–L). When we assessed the individual nsps, we found a striking enrichment in ALPHA upon pulldown with nsp1, similar to the levels observed for full-length replicon (Figure 5E, S6K–L). Notably, ALPHA did not interact with nsp2, nsp3 or nsp4, closely mirroring our negative controls (Figure 5F–H, S6K–L). These results thus indicate that ALPHA interacts within nsp1 of the CHIKV genome.
Together, these data identify ALPHA as a new antiviral lncRNA required for cytoplasmic recognition and control of CHIKV infection and suggests a larger role for lncRNAs as antiviral effectors capable of directly inhibiting viral infection independently of canonical innate immune responses (Figure S7A).
DISCUSSION:
Classical innate antiviral immune pathways lie at the center of a constant evolutionary arms race between host and virus. To survive infection, hosts use diverse mechanisms to overcome virus-mediated immune evasion. (Ma and Suthar, 2015; Nelemans and Kikkert, 2019). LncRNAs have recently been shown to be critical regulators of canonical innate immune responses including Type I IFN signaling (Agliano et al., 2019; Atianand et al., 2017; Basavappa et al., 2019; Carpenter and Fitzgerald, 2018; Chen et al., 2017; Hadjicharalambous and Lindsay, 2019; Mowel et al., 2018; Rehwinkel and Gack, 2020b; Yi et al., 2019). Here, we identified a set of both putative antiviral and proviral lncRNAs including ALPHA which functions as an anti-CHIKV effector independently of canonical IFN signaling. Indeed, we found that ALPHA specifically inhibits CHIKV and its closest relative ONNV but not other viruses tested. Thus, our findings demonstrate that lncRNAs can selectively target specific subsets of viruses outside of IFN-mediated pathways. Whether the other 5 validated antiviral lncRNAs identified in this study function in a similar way to ALPHA will be essential in understanding whether antiviral specificity and IFN-independence are a broader hallmark of antiviral lncRNAs. Further investigation of the proviral lncRNAs identified in this study may also reveal new mechanisms by which lncRNAs can facilitate viral infection and/or serve as negative regulators of innate immunity, potentially adding another layer to our understanding of innate immune signaling.
Our transcriptomics revealed that hundreds of lncRNAs are induced upon infection in a virus-specific manner. Furthermore, we found that ALPHA is highly induced upon infection with alphaviruses but not the unrelated arbovirus ZIKV. This suggests that viral-specificity may be a shared characteristic of non-canonical, antiviral lncRNA transcriptional regulation. Further exploration into the signal that drives ALPHA induction showed that unlike other innate lncRNAs, broadly acting immune pathways including IFN are dispensable for ALPHA transcriptional induction (Atianand et al., 2017; Basavappa et al., 2019; Elling et al., 2016; Mowel et al., 2018; Vierbuchen and Fitzgerald, 2021). These data therefore suggest that ALPHA expression is controlled by an unknown alphavirus-specific pathway. This is particularly provocative as alphavirus-encoded nsp2 localizes to the nucleus of infected cells to inhibit the PolII cofactor RBP1 and attenuate global transcription as a potent immune evasion strategy (Akhrymuk et al., 2012; Akhrymuk et al., 2019). Disruption of canonical PolII complexes has been shown to result in a redistribution of PolII to lncRNA loci (Nojima et al., 2018). Thus, it is possible that alphaviral nsp2-mediated transcriptional shutoff results in an indirect induction of ALPHA. Future work will focus on defining the molecular signal which drives ALPHA upregulation during infection.
Much of our current understanding of innate immune-associated lncRNAs has focused on nuclear-resident lncRNAs that regulate immune gene expression. We found that ALPHA, which is localized in the cytoplasm, can bind directly to CHIKV genomic RNA establishing a new paradigm. How the ALPHA-CHIKV RNA interaction attenuates viral replication is still unclear. Alignment studies between full-length ALPHA and CHIKV genomic RNA did not reveal obvious regions of extensive complementarity (data not shown). However, previous work has shown that functional RNA-RNA interactions can be formed between short, tiled sequences (Lee et al., 2015). More restricted analysis of CHIKV nsp1 and ALPHA ex1 reveal two, consecutive, tiled, 7 nt regions of complementarity within the first 100 nt of ALPHA and nucleotides 380–490 of CHIKV nsp1 (Figure S7B–C). While the second of these sites (starting at nucleotide 484) is similarly conserved across alphaviruses, the first site spanning nucleotides 380–386 is highly conserved in ONNV (~86%), but poorly conserved in MAYV (~29%) and absent in SINV (Figure S7C). Since ALPHA also controls ONNV, our data suggest that regions of similarity between these viruses are targeted. In addition to nucleotides 380–386, comparison of the full nsp1 sequence between related alphaviruses uncovered a region at the 3’ end which displays relatively high conservation with ONNV (78.9%), reduced conservation with MAYV (60.9%), and no conservation with SINV (Figure S7B). Notably, this region contains a mapped structural element in CHIKV spanning nucleotides 1377–1506 whose function remains unclear (Madden et al., 2020). Further analysis of the sequences underlying this structure reveals gaps in the MAYV genome at the 3’ end of this region but not in ONNV; this could consequently result in mismatched pairing and altered structure that may potentially affect ALPHA binding (Figure S7D). Indeed, given the highly structured nature of the CHIKV genome, the ALPHA-CHIKV RNA interaction may be mediated by secondary structure-to-structure contacts (Kendall et al., 2019; Madden et al., 2020). While we found that the 3’ end of ALPHA ex1 is required for binding to CHIKV RNA, it remains unclear whether this is sequence-dependent. Predicted secondary structures of both full-length and truncated ALPHA ex1 generated using RNAfold show that truncated ex1 remains highly structured but lacks a stem loop present in full-length ex1 (Figure S7E–F). It is possible that this stem loop is required for engagement with CHIKV RNA.
Together, our results provide evidence that cytoplasmic lncRNAs can have direct antiviral activity against closely related viruses. Furthermore, we demonstrate that a host lncRNA can physically interact with viral genomes to disrupt viral replication. More broadly, these findings may indicate an important role for lncRNAs as direct, antiviral effectors which complement canonical innate immune signaling pathways to control infection. Indeed, lncRNAs may be uniquely suited to this function as they are subject to reduced selective pressure relative to coding genes allowing for more rapid acquisition of immune activity in the context of the ever-present evolutionary arms race between host and virus. Our observation that infection-dependent lncRNA induction is also virus-specific further supports the hypothesis that lncRNAs may represent a new avenue to discover antiviral RNAs. Future studies will define the full spectrum of lncRNAs that like ALPHA, serve as important antiviral effectors which can function independently of canonical IFN-mediated innate immune responses to directly inhibit infection.
LIMITATIONS OF THE STUDY:
While our results demonstrate that ALPHA binds CHIKV genomic RNA, we have not definitively linked this interaction to the inhibition of CHIKV replication. Furthermore, the exact molecular signal which drives ALPHA induction following alphavirus infection has yet to be defined.
METHODS:
RESOURCE AVAILABILITY:
Lead Contact
Questions and requests for reagents should be directed to and will be fulfilled by the Lead Contact: Sara Cherry (cherrys@pennmedicine.upenn.edu).
Materials Availability
Reagents generated within this study are available upon reasonable request to the Lead Contact.
Data and Code Availability
All sequencing data generated in this manuscript are available through the NCBI Gene Expression Omnibus using accession number GSE184306.
All unprocessed images can be found at the following Mendeley link: https://data.mendeley.com/datasets/yxxg7×5c6y/draft?a=28296dfe-76bf-448c-8a94-662582b96df6
This paper does not report original code.
Any additional information required to reanalyze data reported in this paper can be requested from the Lead Contact.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cells and Viruses:
Human brain microvascular endothelial cells (HBMEC) (Bayer et al., 2016) were maintained in Roswell Park Memorial Institute 1640 medium (RPMI 1640 with L-glutamine, Corning) containing 10% fetal bovine serum (FBS, Hyclone), 10% Nu-Serum (Corning), 1% penicillin/streptomycin (Sigma), 1% sodium pyruvate (Sigma), 1% non-essential amino acids (Sigma), 1% MEM vitamins (Sigma), and 10 μg/mL endothelial cell growth supplement (Corning). BHK-21, Vero, A549, U2OS, and HEK293T cells were acquired from American Type Culture Collections (ATCC) and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% FBS, 1% P/S, and 0.5% L-glutamine. Primary human monocytes were isolated from patient blood by the University of Pennsylvania Human Immunology Core and cultured in RPMI 1640 (w/ L-glutamine) containing 10% FBS, and 1% P/S for a maximum length of 48h. All cell lines were confirmed mycoplasma-negative.
Chikungunya (CHIKV)-mKate (strain 181/25) and Sindbis (SINV)-mKate (strain Girdwood) were provided by Dr. Mark Heise (University of North Carolina). CHIKV (strain Ross) was provided by Dr. David Weiner (University of Pennsylvania). O’nyong’nyong (ONNV, strain SG650) was a gift from Dr. Thomas E. Morrison (University of Colorado). Mayaro (MAYV, strain BeH407), Zika (ZIKV, strain MR766), Rift Valley Fever (RVFV, strain MP12), and La Crosse (LACV) were gifts from Dr. Michael Diamond (Washington University in St. Louis). Ross River (RRV)-GFP was a gift from Dr. Richard Kuhn (Purdue University). Influenza A (IAV, strain A/Puerto Rico/8/34) was a gift from Dr. Scott Hensley (University of Pennsylvania). Sendai (SeV, strain Cantelli) was acquired from Charles River Laboratories (#10100774). Herpes Simplex (HSV)-1-GFP was a gift from Dr. Carolyn Coyne (University of Pittsburgh). CHIKV, SINV, MAYV, and ZIKV were all propagated in C636 mosquito cells. ONNV, RRV and HSV-1 were propagated in Vero-E6 cells. RVFV and LACV were propagated in BHK-21 cells. Supernatants from infected C636, Vero-E6, or BHK-21 cells were collected, aliquoted and subjected to only a single freeze-thaw. Titers (represented as pfu/mL) were calculated by plaque assays or TCID50 assays performed on BHK-21 or Vero-E6 cells.
METHOD DETAILS
RNA-sequencing and analysis:
HBMEC were either left uninfected (mock) or infected with CHIKV or ZIKV at MOI 5 for 24h. Cells were then directly lysed in TRIzol and RNA was extracted using the Zymogen RNA Clean and Concentrator Kit with on-column DNase I treatment. RNA quality was measured using the Agilent BioAnalyzer and quantified using the Qubit Fluorescent Quantification System (ThermoFisher Scientific). Libraries were prepared using the Illumina TruSeq Stranded Total RNA Kit with ribosomal RNA depletion. Paired-end sequencing with 150bp read-length was performed with the NextSeq 500.
Raw fastq files were trimmed to remove adapters and low quality reads with bbduk 38.56 using the parameters “ref=/bbmap/resources/adapters.fa ktrim=r k=23 mink=11 hdist=1 minlength=35 tpe tbo qtrim=r trimq=10” (Bushnell). Next transcripts were counted using salmon 0.13.1 in pseudoalignment mode by mapping to Homo sapiens (human) genome assembly GRCh38 (build 94) with the parameters “--validateMappings -rangeFactorizationBins 4 --seqBias –gcBias” (Patro et al., 2017). Transcript counts were collapsed to the gene level using the R package tximport v1.16.1 and differential abundance was analyzed using DESeq2 v1.28.1(Love et al., 2014; Soneson et al., 2015).
Genes that were potentially lncRNAs were identified based on ENSEMBL annotations (Howe et al., 2021). Reactome’s “Interferon Signaling” pathway (R-HSA-913531) was used for the list of interferon stimulated genes (Fabregat et al., 2018). Heatmaps were generated with the ComplexHeatmap v2.4.3 R package implementing Ward’s method for clustering (Gu et al., 2016; Ward, 1963). Data management was performed using base R and dplyr v1.0.2 (RCoreTeam, 2018; Wickham, 2017).
All sequencing data in this manuscript has been deposited with NCBI GEO and is available under accession number GSE184306.
High-throughput RNAi screening and analysis:
Three, pooled, LNA-modified siRNAs per target were spotted into each well of a 384-well black tissue culture treated plate. siRNAs were acquired from a pre-designed library targeting 2200 lncRNAs across the human genome (Ambion). Briefly, 0.5 μL of HiPerfect diluted in 9.5 μL of Opti-MEM was added to each well using a ThermoFisher Scientific Matrix WellMate automated dispenser. Each plate containing the siRNA/HiPerfect mixture was then incubated at room temperature for 5–10 minutes. HBMEC (2500 cells/40 μL) were then added to each well using the WellMate and incubated for 3 days at 37°C, 5% CO2. The final concentration of siRNA in each well was 30 nM. Following knockdown, CHIKV-mKate virus (MOI 0.05) was dispensed into each well followed by spinoculation at 2500 RPM for 1h at 4°C. Cells were then incubated at 37°C, 5% CO2 for an additional 24h to allow infection to proceed. The cells were fixed with 4% formaldehyde/PBS for 10 min at room temperature. Finally, the cells were washed 3X with PBS, stained with Hoechst 333432, and subjected to automated microscopy (ImageXpress, 10X objective). Four sites per well were acquired in two fluorescent channels.
For each parameter measured using the Cell Scoring module in MetaXpress (percent infection and total cell number) the data were first log transformed and the plate median and interquartile range (IQR) were calculated. Z scores were further calculated based on these values ((log10(%infection) − log10(median)/(IQR × 0.74)). Any condition that resulted in Z<−2 for total cell number was removed from the infection datasets to avoid the potentially confounding effects of cytotoxicity on viral infection levels.
Cytokines and inhibitors:
Human interferon β (IFNβ), tumor necrosis factor α (TNFα), and interleukin-1 β (IL-1β) were purchased from Peprotech, resuspended in phosphate buffered saline (PBS) and used at 10 ng/mL unless otherwise specified. AL8810 (PGF2α inhibitor) was purchased from Santa Cruz Biotechnologies. SP600125 (JNK inhibitor), PD98059 (MEK1/2 inhibitor), and Ruxolitinib (JAK/STAT inhibitor) were purchased from Invivogen. AL8810, SP600125, and PD98095 were reconstituted per the manufacturer’s instructions, used at the indicated concentrations, and were added to cells at the same time as viral inoculation. Ruxolitinib was also reconstituted per the manufacturer’s instructions and used at the specified concentrations, but cells were instead pre-treated for 2h prior to infection. Ionomycin was purchased from Sigma, reconstituted in PBS, and used at the specified concentrations and time points. Mock and untreated controls were “stimulated” with the appropriate vehicle only at the highest volume used for the given experiment and reagent concentration.
RNA Interference:
All knockdowns were performed using the Qiagen HiPerfect Fast-Forward protocol. For viral RNA and titer quantification, 10 μL of HiPerfect and 1.5 μL of 20 μM stock siRNA were diluted in 120 μL of Opti-MEM. These contents were vortexed for 5s and incubated at room temperature for 5–10 minutes. During incubation, 5×104 cells were plated per well in a 12-well tissue culture-treated plate and complete HBMEC media was added to a volume of 875 μL. 125 μL of siRNA transfection mix was then added dropwise into each appropriate well. The final concentration of siRNA equated to 30 nM. Cells were then incubated at 37°C, 5% CO2 for 72h followed by subsequent downstream treatments and infections. For viral breadth studies measured by immunofluorescence, a 384-well format was used. Briefly, 1.5 μL of 1 μM siRNA stock was spotted into each well in triplicate for each condition. 0.5 μL of HiPerfect was diluted in 9.5 μL of Opti-MEM per well, added to each well, and incubated at room temperature for 5–10 minutes. 2.5×103 cells suspended in 40 μL of complete HBMEC media were then added to each well and incubated at 37°C, 5% CO2 for 72h at which point downstream treatment and infections were performed.
Transfection and transduction:
HBMEC were transfected using X-treme Gene 9 following the manufacturer’s instructions. Cells were plated at a density of 1×105 cells per well of a 6-well plate the day prior to transfection. 1 μg of plasmid was introduced at a 1:2 ratio with X-treme Gene 9 and incubated at 37°C, 5% CO2 for 48h before further processing.
To produce lentivirus, HEK293T cells were simultaneously transfected with 1 μg lentiviral plasmid containing the cDNA of interest, 0.7 μg CMV-VSV-g (Addgene) and 1 μg of psPAX2 (Addgene) in a 12-well format using Lipofectamine 2000 per the manufacturer’s protocol. Following a 24h incubation at 37°C, 5% CO2, the media was replaced with 750 μL of fresh media followed by an additional 24h incubation. The supernatants were then collected and filtered through a 0.45 μm syringe filter and stored at −80°C. To transduce HBMEC, 5×104 cells were plated in a 6-well plate in 2 mL complete media containing 100 μL viral supernatant and 10 μg/mL polybrene. Transduction was allowed to proceed for 48h followed by antibiotic selection and other downstream applications.
Immunofluorescence and microscopy:
Cells were fixed in 4% formaldehyde/PBS for 10 minutes at room temperature (covered if fluorescently tagged viruses were used). Cells were then washed 3X in PBS and incubated in 0.5% Triton-X100/PBS (PBST) for 10 min at room temperature to permeabilize cell membranes. Cells were then blocked in 2% bovine serum albumin (BSA)/PBST for 30 min at room temperature followed by overnight incubation at 4°C in primary antibody diluted in 2% BSA/PBST. Cells were washed 3X with PBST followed by a 1h room temperature incubation in fluorescently conjugated secondary antibody diluted in 2% BSA/PBST. Cells were washed 3X in PBST and stored in PBS. Viral protein quantification (for lncRNA screens, viral breadth, and viral phenotyping in ALPHA knockdown, knockout, and overexpression studies) were performed using a Molecular Devices automated imager and analyzed with MetaXpress (Mulitwavelength Cell Scoring Module). Negative-strand quantification (measured by J2 staining) was imaged using a Leica DM5500 Q confocal microscope and quantified using ImageJ.
Nuclear/Cytoplasmic Fractionation:
HBMEC (5×105) were trypsinized, collected via centrifugation, and resuspended in 200 μL of cytoplasmic lysis buffer (CLB; 30 mM HEPES pH 7.4, 2mM MgOAC, 0.1% NP-40). Lysates were then incubated on ice for 10 minutes and pipetted up and down gently every 2–3 minutes to promote lysis. The lysate was then centrifuged at 2300 RPM for 20 minutes at 4°C. The supernatant was removed, placed in a fresh 1.5 mL tube and kept on ice (cytoplasmic fraction). The nuclear pellet was then washed 3X with CLB (nuclei were centrifuged at 2300 RPM for 5 min between each wash). Nuclei were subsequently lysed in 200 μL nuclear lysis buffer (NLB; CLB + 150 mM KOAc) and incubated on ice for 10 minutes, vortexing every 2–3 minutes to promote lysis. Both the cytoplasmic and nuclear lysates were sonicated 2X, 30s on and 30 off for 3 min (equating to roughly 1000 J of exposure). Lysates were then cleared at 10,000 RPM for 10 minutes at 4°C. Supernatants were removed, placed in 1.5 mL tubes, and 1 mL TRIzol was added for downstream RNA isolation. Primers for GAPDH and MALAT1 were used as controls for the quality of the cytoplasmic and nuclear fractionations respectively. The relative enrichment of the RNA targets was quantified as a percentage of the total amplification of the combined nuclear and cytoplasmic values.
Viral entry, dsRNA quantification, viral spread, and anti-genome quantification:
Both control and ALPHA-depleted cells were infected with CHIKV, MOI 20 in the presence of 10 μg/mL cycloheximide (CHX) for 4h. Cells were then incubated in 0.25% trypsin-EDTA for 7 min at 37°C, 5% CO2 to remove bound virions that had not yet entered. Trypsin was neutralized 1:1 with complete media, cells were collected by centrifugation, and lysed in 1 mL TRIzol for downstream RNA isolation. To measure dsRNA production, control and ALPHA-depleted cells were plated on glass coverslips in a 24-well plate and infected with CHIKV, MOI 20 for 8h. Following infection, the cells were fixed with 4% formaldehyde for downstream immunofluorescence staining using J2 antibody. The coverslips were mounted onto glass slides using Vectashield (Vector Laboratories) prior to imaging. Viral spread was assayed by infecting control and ALPHA-depleted cells for 4h with CHIKV, MOI 0.05 followed by the addition of 20 mM NH4Cl. Cells were lysed in TRIzol 20 hpi for downstream RNA isolation. Anti-genome quantification was performed as previously described (Meertens et al., 2019). Briefly, control and ALPHA-depleted HBMEC were infected for 8h at MOI 0.5. The cells were lysed in TRIzol and RNA was extracted as described below. Strand-specific reverse transcription was performed using a primer complementary to anti-genome at a final concentration of 100 nM following the manufacturer’s protocol. The cDNA was diluted 1:10 prior to qPCR with anti-genome-specific primers. A gBlock encoding the 133 bp amplicon produced from these primers was synthesized by IDT and used to generate a standard curve ranging from 1×108 to 10 DNA copies. Anti-genome copies were calculated based off of this standard curve.
CRISPR knockout generation:
CRISPR reagents were generated as previously described (Sanjana et al., 2014; Shalem et al., 2014). Briefly, guide RNAs (gRNAs) flanking the 5’ and 3’ ends of the ALPHA locus (ENSG00000227075) were designed using the CRISPOR design algorithm. These gRNAs were cloned into lentiCRISPRv2 and lentivirus was generated as described above. HBMEC were transduced and selected using 1 μg/mL puromycin. Single cell clonal populations were genotyped and ALPHA levels quantified. Control cells were generated in an identical manner using two predefined, non-targeting gRNAs (Kearns et al., 2014; Mimee et al., 2015).
Stable ectopic expression cell line generation:
pcDNA3.1-ALPHA (empty pcDNA3.1 was used to generate control cells) or pmCherry-N1-IFNb were transfected into HBMEC as described above and selected for 7 days with 1.5 mg/mL of G418. pcDNA3.1-ALPHA was synthesized by Genscript and pmCherry-N1-IFNb was a gift from Drs. Yueh-Ming Loo and Michael Gale (University of Washington). For ALPHA overexpressing clones, ALPHA levels were quantified by qPCR to confirm overexpression. For IFN-mCherry reporter HBMEC, cells were stimulated with SeV (100 HAU/mL) for 24h and imaged by immunofluorescence to confirm positive clones.
RNA isolation and quantitative reverse transcription PCR
Samples were lysed in TRIzol and RNA was extracted using the RNA Clean and Concentrator Kit (Zymogen) with on-column DNase I treatment per the manufacturer’s instructions. Complementary DNA (cDNA) was generated using moloney murine leukemia virus (M-MLV) reverse transcriptase (Ambion) per the manufacturer’s protocol. Quantitative PCR (qPCR) was performed in technical triplicate using 2X Power SYBR green/Rox qPCR Master Mix (ThermoScientific) and analyzed using relative Ct values. GAPDH was used as a normalization control for all experiments unless otherwise specified.
Polysome fractionation
HBMEC were grown to ~90% confluence in two 15 cm2 tissue-culture treated plates. The media was removed, cells were washed 1X with PBS and fresh media containing 100 μg/mL cycloheximide (CHX) was added to the cells and allowed to incubate for 7 min at 37°C. The media was then removed and cells were collected and pelleted at 1200 RPM for 5 min. Cell pellets were washed 2X with PBS containing 100 μg/mL CHX and lysed in 250 μL Polysome Lysis Buffer (PLB; 100 mM KCl, 5 mM MgCl2, 10 mM Hepes pH 7.4, and 0.5% NP-40) containing 100 μg/mL CHX, SUPERase-In (1 μL/1 mL PLB), and 1X protease inhibitor. Lysates were incubated on ice for 20 min (without vortexing). During the lysate incubation, a 10–50% sucrose gradient was prepared using a Seton Gradient Maker in a 10 mL ultracentrifuge tube. Following incubation, the lysates were then cleared at 13,200 RPM for 3 min at 4°C. Cleared lysate was then loaded on top of the prepared sucrose gradient and balanced to the hundredth decimal place. The sample was then spun using a SW-40i rotor at 35,000 RPM for 2.5 hours at 4°C. Fractions were then collected into 1.5 mL tubes using a Biocomp Piston Gradient Fractionator per the manufacturer’s protocol. Each fraction was additionally split into 250 μL volumes, lysed in TRIzol, and stored at −80°C for further RNA extraction.
UV-inactivation of virus:
Virus (300 μL) was placed in a 60 mm2 tissue culture dish and rotated to spread and maximize the surface area for exposure. With the lid removed, virus was exposed to 1200 μJ x 100 of 254 nm UV light using a Stratalinker 2400. This exposure was repeated 5X, rotating the plate between each UV treatment. The UV-inactivated virus was placed in a fresh 1.5 mL tube and kept on ice until further use. Equivalent volumes of either WT or UV-inactivated virus were used based on the calculated WT MOI.
Viral Titer Quantification:
To measure viral titers produced in control or ALPHA-depleted HBMEC, cells were infected for 6h at the indicated MOIs. The media was then removed and the cells were washed 3X with PBS. Fresh, complete HBMEC media was added to each well and the infection was allowed to continue for 24h from that point. Supernatants were then used for TCID50 assays on either BHK-21 or Vero cells. The day before inoculation, 1.5×104 cells were plated in quadruplicate in a 96-well black tissue-culture treated plate. Ten-fold serial dilutions of viral supernatants were prepared in Opti-MEM in 10 μL volumes and added to the appropriate wells. The cells were then incubated for 24h at 37°C, 5% CO2 followed by fixation with 4% formaldehyde. Infection was quantified by immunofluorescence and automated imaging as described. TCID50s were calculated using the Reed-Muench Method (Reed, 1938).
RNA-RNA Interactions Probe Generation:
Templates for in vitro transcription were generated by PCR amplification from either uninfected HBMEC cDNA (GAPDH) or cDNA generated from purified viral RNA (CHIKV and SINV genomic RNA) with the addition of T7 (5’) and/or SP6 (3’) promoter sequences to the amplicon ends. PCR was performed using Phusion DNA Polymerase per the manufacturer’s protocol and products were purified using the Qiagen PCR Purification Kit. Probe RNA was synthesized using either a T7 (sense RNA) or SP6 (antisense RNA) MEGAscript Kit (ThermoFisher Scientific) following the manufacturer’s instructions and purified by lithium chloride extraction. The probes were then biotinylated using the Pierce™ RNA 3’ End Biotinylation Kit (ThermoFisher Scientific) following the manufacturer’s instructions (incorporating 50 pmol of RNA per reaction).
In cellulo RNA-RNA Interactions:
HBMEC (1×107 cells, either HBMEC or ALPHA-overexpressing HBMEC) were either uninfected or infected with CHIKV or SINV at a MOI 2 for 20h. Following infection, the media was removed and cells were trypsinized and centrifuged at 1200 RPM for 5 min to pellet. The cell pellets were washed 1X with PBS and centrifuged again at 1200 RPM for 5 min. Each pellet was then resuspended in 1% glutaraldehyde/PBS and incubated with orbital rotation for 10 min at room temperature. The glutaraldehyde crosslinking solution was subsequently quenched with 1/10th (1 mL) volume of 1 M glycine and incubated with orbital rotation for 5 min. The cells were then pelleted at 2000 RCF for 5 min at room temperature and washed 1X with PBS. Cells were then pelleted at 2000 RCF for 5 min at 4°C and resuspended in 1 mL of PBS. The cell suspension was transferred to a 1.5 mL tube and centrifuged at 2000 RCF for 5 min at room temperature. The supernatants were removed, the pellets were flash frozen on dry ice, and stored at −80°C until further processing.
Frozen cell pellets were thawed quickly at room temperature followed by a pulse spin to collect and remove any remaining supernatant. The pellets were then lysed in 300 μL Lysis Buffer (LB; 50 mM Tris-Cl pH 7.0, 10 mM EDTA, 1% SDS) containing protease inhibitor (Roche Applied Science), PMSF, and SUPERase-In (Ambion, stock treated as 200X) and immediately subjected to sonication. The lysates were then cleared at 10,000 RPM for 10 min, placed in fresh 1.5 mL tubes, and kept on ice. 20 μL of lysate (6.7%) was removed as input control and stored at −80°C. The remaining lysate was split into 1.5 mL tubes containing 250 μL Hybridization Buffer (HB; 750 mM NaCl, 50 mM Tris-Cl, 1 mM EDTA, 1% SDS, 15% formamide) with added protease inhibitor, PMSF, and SUPERase-In, and 12.5 pmol of the biotinylated, 500mer, antisense oligonucleotide probe designed to target either a control transcript (GAPDH), CHIKV genomic RNA, or SINV genomic RNA. The probes were hybridized at 37°C with rotation for 16–20h. Streptavidin-conjugated magnetic beads (50 μL/sample) were washed 3X in unsupplemented LB and resuspended in the original volume of LB containing protease inhibitor, PMSF, and SUPERase-In. Washed beads were then added to the lysate/hybridization solution and incubated for 1h at 37°C with rotation. Samples were placed on a DynaMag-2 magnet to collect beads. Supernatants were removed and discarded. Beads were resuspended in Wash Buffer (WB; 2X SSC, 0.5% SDS) containing PMSF and placed in fresh 1.5 mL tubes. The samples were then incubated at 37°C for 4 min with rotation. Samples were placed on a DynaMag-2 magnet to collect beads and then resuspended in WB containing PMSF. These wash steps were performed 5X in total. Following the final wash, the beads were pulse spun and placed back on the magnet to remove residual WB. The beads were then resuspended in 95 μL Proteinase K Buffer (PKB; 100 mM NaCl, 10 mM Tris-Cl, 1 mM EDTA, 0.5% SDS). Input samples were concordantly thawed at room temperature and 75 μL of PKB was added to each. 5 μL of Proteinase K (final concentration: 5 μg/mL) was added to each sample and incubated at 50°C with 250 RPM orbital rotation for 45 min. The samples were then heated to 95°C for 10 min and immediately placed on ice. Each sample was then resuspended in TRIzol and stored at −80°C until further processing.
In vitro RNA-RNA Interactions:
For Figures 5B–D, 200 ng of each biotinylated probe was mixed with 200 ng unbiotinylated CHIKV replicon RNA in 300 μL Binding Buffer (20 mM Tris-HCL ph 7.4, 1 M NaCl, 2 mM EDTA, 0.1% SDS, (Gorbea et al., 2017)) and incubated for 2h at room temperature with rotation. For Figures 5E–H, 300 ng of biotinylated, full-length CHIKV replicon RNA was combined with 30 ng of unbiotinylated ALPHA, representing a ~1:1 molar ratio. The quantities of biotinylated nsp1–4 used were calculated to equate to 300 ng of the full-length replicon by molar ratios. Streptavidin-conjugated beads (15 μL per sample) were washed 3X with Binding Buffer prior to being added to each sample. The beads were then incubated with the samples at room temperature for 30 min with rotation. Samples were then placed on a Dynmag-2 magnet to collect beads and the supernatants were discarded. The beads were washed 3X with Binding Buffer, resuspended in 500 μL of TRIzol and stored at −80 until ready for RNA extraction, cDNA synthesis, and qPCR.
RNA Proximity Ligation Assay (RNA-PLA):
HBMEC (2 × 104 cells) were plated on 12 mm2 round coverslips in a 24-well plate the day prior to infection. Cells were infected with CHIKV, MOI 5 for 24h and fixed using 4% formaldehyde/PBS for 10 min at room temperature. Cells were then permeabilized using 0.2% Triton-X 100/PBS for 10 min at room temperature and blocked for 1h in PLA Blocking Buffer (10 mM Tris-Acetate, 10 mM magnesium acetate, 50 mM potassium acetate, 250 mM NaCl, 0.25 μg/μL BSA, 0.05% Tween-20) at 37°C. Coverslips were then incubated overnight with 200 nM PLA probe solution (100 nM each of priming and non-priming probe diluted in PLA Blocking Buffer) at 37°C in a sealed humidity chamber. The specific probe pairings are as follows: GAPDH-Priming + CHIKV-Non-Priming, CHIKV(50 nt downstream)-Priming + CHIKV-Non-Priming, ALPHA-Priming + CHIKV-Non-Priming, and GAPDH-Priming + ALPHA-Non-Priming. The same CHIKV-Non-Priming probe was used in each condition where CHIKV RNA was targeted. Cells were washed 3X with PBS, 5 min each at room temperature. Ligation mix was prepared immediately before placement on coverslips containing 125 nM PLA connector, 125 nM PLA linker, and 1 μL T4 ligase in 1X T4 ligation buffer. Ligation was carried out at 37°C for 30 min followed by 3X washes with PBS, 5 min each at room temperature. Rolling circle amplification and amplicon detection was performed using 10 nM PLA amplicon probe (Cy5-conjugated), 100 μM dNTPS, 10 μg/mL BSA, and 1 μL phi29 polymerase in 1X phi29 reaction buffer at 37°C for 1h, 40 min. Cells were then washed 1X in PBS containing Hoechst solution for 10 min at room temperature followed by 2X washes in PBS. Coverslips were mounted using Vectashield and imaged using a Leica DM5500 Q confocal microscope. Puncta from 40–60 cells per condition were counted in each replicate using ImageJ.
All PLA oligonucleotide sequences were designed as previously described containing a three-part structure: i.) a 40–50 nucleotide sequence complementary to a target RNA of interest ii.) a 17–20 nucleotide polyA linker and iii.) an assay-specific, non-targeting PLA sequence (Fredriksson et al., 2002; Soderberg et al., 2006; Zhang et al., 2016). The complementary region was designed using the Stellaris Probe Designer tool (https://www.biosearchtech.com/support/tools/design-software/stellaris-probe-designer ). The polyA linker and PLA sequences were then appended to the 3’ end as is shown in the Key Resources Table
Key Resources Table:
| RESOURCES/REAGENTS | SOURCE | IDENTIFIER |
|---|---|---|
| ANTIBODIES | ||
| Anti-IFIT1 (D2X9Z) | Cell Signaling | Cat#14769S |
| Anti-dsRNA (J2) | Cherry Laboratory | N/A |
| Anti-CHIKV E2 | Dr. Michael Diamond, Washington University in St. Louis | N/A |
| Anti-IAV NP | Dr. Scott Hensley, University of Pennsylvania |
N/A |
| Streptavidin-HRP | Cell Signaling | Cat#3999S |
| BACTERIAL AND VIRUS STRAINS | ||
| CHIKV | Dr. David Weiner, University of Pennsylvania | N/A |
| CHIKV-mKate (181/25) | Dr. Mark Heise, University of North Carolina | N/A |
| ONNV (SG650) | Dr. Thomas E. Morrison, University of Colorado | N/A |
| MAYV (BeH407) | Dr. Michael Diamond, Washington University in St. Louis | N/A |
| RRV-GFP | Dr. R. Kuhn, Purdue University of Pennsylvania | N/A |
| SINV-mKate (Girdwood) | Dr. Mark Heise, University of North Carolina | N/A |
| ZIKV (MR766) | Dr. Michael Diamond, Washington University in St. Louis | N/A |
| RVFV (MP12) | Dr. Michael Diamond, Washington University in St. Louis | N/A |
| LACV | Dr. Michael Diamond, Washington University in St. Louis | N/A |
| IAV (A/Puerto Rico/8/34) | Dr. Scott Hensley, University of Pennsylvania | N/A |
| SeV (Cantelli) | Charles River Laboratories | Cat#10100774 |
| HSV-1-GFP | Dr. Carolyn Coyne, University of Pittsburgh | N/A |
| One Shot Stbl3 Chemically Competent Cells | Thermo Fisher | Cat#C737303 |
| CHEMICALS, PEPTIDES, AND RECOMBINANT PROTEINS | ||
| Cycloheximide | Sigma Aldrich | Cat#01810 |
| Recombinant human IFNβ | Peprotech | Cat#300–02BC |
| Recombinant human IL-1β | Peprotech | Cat#200–01B |
| Recombinant human TNFα | Peprotech | Cat#300–01A |
| Ruxolitinib | Invivogen | Cat#tlrl-rux |
| AL8810 | Santa Cruz Biotechnologies | Cat#sc-205204 |
| SP600125 | Invivogen | Cat#tlrl-sp60 |
| PD98095 | Invivogen | Cat#tlrl-pd98 |
| Ionomycin | Sigma-Aldrich | Cat#I0634 |
| TRIzol | Life Technologies | Cat#15596018 |
| HiPerfect | Qiagen | Cat#301705 |
| Lipofectamine 2000 | Invitrogen | Cat#11668–019 |
| Polybrene | Fisher Scientific | Cat#TR1003G |
| Puromycin | Takara Bio | Cat#63130 |
| G418 | Cell Center, University of Pennsylvania | N/A |
| DYNAL MyOne Dynabeads Streptavidin C1 | Thermo Fisher | Cat#65–001 |
| Hybond-N+ Membrane | GE Healthcare | Cat#RPN303B |
| ECL | Amersham | Cat#GERPN2106 |
| Metaphor Agarose | Fisher Scientific | Cat#BMA50181 |
| Ethidium Bromide | Sigma-Aldrich | Cat#E1510 |
| Quick-Load Purple 2-Log Ladder | New England Biotechnologies | Cat#N0550 |
| T4 DNA Ligase | New England Biotechnologies | Cat#M0202 |
| Phi29 Polymerase | New England Biotechnologies | Cat#M0269 |
| Vectashield | Fisher Scientific | Cat#NC9265087 |
| CRITICAL COMMERCIAL ASSAYS | ||
| RNA Clean and Concentrator Kit | Zymogen Research | Cat#R1018 |
| M-MLV Reverse Transcriptase | Life Technologies | Cat#28025021 |
| Power SYBR Green PCR MasterMix | Life Technologies | Cat#4368708 |
| TruSeq Stranded Total RNA Kit | Illumina | Cat#20020596 |
| Phusion High Fidelity DNA Polymerase Kit | New England Biotechnologies | Cat#M0530 |
| RNA 3’ End Biotinylation Kit | Thermo Fisher | Cat#20160 |
| T7 MEGAscript Kit | Life Technologies | Cat#AMB13345 |
| SP6 MEGAscript Kit | Thermo Fisher | Cat#AM1330 |
| DEPOSITED DATA | ||
| RNA-seq datasets | This paper | GSE184306 |
| Microscope images | Mendeley | https://data.mendeley.com/datasets/yxxg7×5c6y/draft?a=28296dfe-76bf-448c-8a94-662582b96df6 |
| EXPERIMENTAL MODELS: CELL LINES | ||
| HBMEC | Dr. Carolyn Coyne, University of Pittsburgh | N/A |
| BHK-21 | ATCC | CCL-10 |
| Vero | ATCC | CCL-81 |
| U2OS | ATCC | HTB-96 |
| HEK293T | ATCC | CRL-3216 |
| Primary Human Peripheral Blood Monocytes | Human Immunology Core, University of Pennsylvania | N/A |
| RECOMBINANT DNA | ||
| lentiCRISPRv2-Puro | Addgene | 98290 |
| pLenti-Puro | Addgene | 39481 |
| psPAX2 | Addgene | 12260 |
| pCMV-VSV-G | Addgene | 8454 |
| pcDNA3(+)-ALPHA | This paper (generated by Genscript) | N/A |
| pJMH40-CHIKV Replicon | Dr. Mark Heise, University of North Carolina | N/A |
| OLIGONUCLEOTIDES | ||
| qPCR: CHIKVnsp2-F: GGCAGTGGTCCCAGATAATTCAAG |
This paper | N/A |
| qPCR: CHIKVnsp2-R: ACTGTCTAGATCCACCCCATACATG |
This paper | N/A |
| qPCR: ALPHA-F: CCTTGCTGCCCTCATGATAATTC |
This paper | N/A |
| qPCR: ALPHA-R: TCACAGCAGGACACACTATG |
This paper | N/A |
| qPCR: GAPDH-F: ACCAAATCCGTTGACTCCGACCTT |
This paper | N/A |
| qPCR: GAPDH-R: TCGACAGTCAGCCGCATCTTCTTT |
This paper | N/A |
| qPCR: IFNB1-F: GCTTCTCCACTACAGCTCTTTC |
This paper | N/A |
| qPCR: IFNB1-R: CAGTATTCAAGCCTCCCATTCA |
This paper | N/A |
| qPCR: ISG56-F: CAACCAAGCAAATGTGAGGA |
This paper | N/A |
| qPCR: ISG56-R: AGGGGAAGCAAAGAAAATGG |
This paper | N/A |
| qPCR: NEAT1-F: GATCTTTTCCACCCCAAGAGTACATAA |
This paper | N/A |
| qPCR: NEAT1-R: CTCACACAAACACAGATTCCACAAC |
This paper | N/A |
| qPCR: MALAT1-F: TTCCGGGTGTTGTAGGTTTC |
This paper | N/A |
| qPCR: MALAT1-R: AAACCCACAAACTTGCCATC |
This paper | N/A |
| qPCR: CHIKV Anti-Genome cDNA Primer: GGCAGTATCGTGAATTCGATGCCG CTGTACCGTCCCCATTCC |
Meertens, et al. | N/A |
| qPCR: CHIKV Anti-Genome-F: GGCAGTATCGTGAATTCGATGC |
Meertens, et al. | N/A |
| qPCR: CHIKV Anti-Genome-R: ACTGCTGAGTCCAAAGTGGG |
Meertens, et al. | N/A |
| qPCR: gBlock CHIKV Anti-Genome Amplicon Standard: GGCAGTATCGTGAATTCGATGCCGCT GTACCGTCCCCATTCCAGAACACACTAC AGAATGTACTGGCAGCAGCCACGAAAA GAAACTGCAACGTCACACAGATGAG GGAATTCACCACTTTGGACTCAGCAGT |
Meertens, et al. | N/A |
| RNA-PLA: CHIKV-NonPriming: AGAGACATAGCTGTGTCAC GCGTCTCCGCTGTTTCTTGT AAAAAAAAAAAAAAAAAGACG CTAATAGTTAAGACGCTT [UUU] |
This paper | N/A |
| RNA-PLA: CHIKV50bpdown-Priming: TTGGTGCACCGAAGGAGAT CGGCGGGTGACTCAGTTC CGTAAAAAAAAAAAAAAAAA AAATATGACAGAACTAGACACTCTT |
This paper | N/A |
| RNA-PLA: ALPHA-Priming: TCACAGCAGGACACACTAT GTAATTCATATCAACATTTGG GAAAAAAAAAAAAAAAAAA AATATGACAGAACTAGACACTCTT |
This paper | N/A |
| RNA-PLA: GAPDH-Priming: GCTGGCGACGCAAAAGAA GATGCGGCTGACTGTCGA ACAGAAAAAAAAAAAAAAAAAA AATATGACAGAACTAGACACTCTT |
This paper | N/A |
| RNA-PLA: Connector: CTATTAGCGTCCAGTGAATG CGAGTCCGTCTAAGAGAGT AGTACAGCAGCCGTCAAGAGTGTCTA |
Soderberg, et al. | N/A |
| RNA-PLA: Linker: GTTCTGTCATATTTAAGCGTCTTAA |
Soderberg, et al. | N/A |
| RNA-PLA: Amplicon Probe: /5Cy5/CAGTGAATGCGAGTCCGTCT |
Soderberg, et al. | N/A |
| ChIRP/IVP: CHIKVnsp3probe-F: TAATACGACTCACTATAGGGAG ACTGCGATATTGTTCGCGTGC |
This paper | N/A |
| ChIRP/IVP: CHIKVnsp3oprobe-R: ATTTAGGTGACACTATAGAAGG GACGTGATTGTACTCGCCTCC |
This paper | N/A |
| ChIRP: SINVnsp3probe-F: TAATACGACTCACTATAGGGAG AGAAAGTGATCCACGCGGTTG |
This paper | N/A |
| ChIRP: SINVnsp3probe-R: ATTTAGGTGACACTATAGAAGG GGCTCGTTGCTTTCCTGGTCA |
This paper | N/A |
| ChIRP: GAPDHprobe-F: TAATACGACTCACTATAGGGA GAATCTTCTTTTGCGTCGCCAG |
This paper | N/A |
| ChIRP:GAPDHprobe-R: ATTTAGGTGACACTATAGAAGG GTGCAGGAGGCATTGCTGATG |
This paper | N/A |
| IVP: GAPDHprobe-F: TAATACGACTCACTATAGGGAG AAAAATCAAGTGGGGCGATGC |
This paper | N/A |
| IVP: GAPDHprobe-R: ATTTAGGTGACACTATAGAAGG GTCTAGACGGCAGGTCAGGTC |
This paper | N/A |
| IVP: ALPHAprobe-F: TAATACGACTCACTATAGGG AGAATTTGGCTCTGTGTCCCTA |
This paper | N/A |
| IVP: ALPHAprobe-R: ATTTAGGTGACACTATAGAAG GGTCATAATCCCCGGTGTTGG |
This paper | N/A |
| IVP: ALPHA5Pprobe-F: TAATACGACTCACTATAGGGAG AATTTGGCTCTGTGTCCCTAC |
This paper | N/A |
| IVP: ALPHA5Pprobe-R: TATATAGCCTTCTGATGCTC |
This paper | N/A |
| IVP: ALPHA3Pprobe-F: TAATACGACTCACTATAGGGAG AACCTGCTCAGATTCTGGGAAG |
This paper | N/A |
| IVP: ALPHA3Pprobe-R: CGGTGTTGGGGGTGGAACC |
This paper | N/A |
| IVP: ALPHAMidprobe-F: TAATACGACTCACTATAGGGAG ACCTTGCTGCCCTCATGATAA |
This paper | N/A |
| IVP: ALPHAMidprobe-R: TTTAAAAATCTGTAGTACC |
This paper | N/A |
| IVP: ALPHAEx1probe-R: TATCATGAGGGCAGCAAG |
This paper | N/A |
| IVP: ALPHAEx2probe-F: TAATACGACTCACTATAGGGAG AATTCTGTGTTCAGTCAAAATAG |
This paper | N/A |
| IVP: ALPHAEx2probe-R: CTTCTGATGCTCACAGCAG |
This paper | N/A |
| IVP: ALPHAEx1truncprobe-R: GAAAATCTGCCCCCGTGATC |
This paper | N/A |
| IVP: CHIKVnsp1(SL15649)-F: TAATACGACTCACTATAGGGAG AATGGATCCTGTGTACGTGGAC |
This paper | N/A |
| IVP: CHIKVnsp1(SL15649)-R: TGCGCCCGCTCTGTCCTCAAGC |
This paper | N/A |
| IVP: CHIKVnsp2(SL15649)-F: TAATACGACTCACTATAGGGAGAG GAATAATAGAGACTCCGAGAGG |
This paper | N/A |
| IVP: CHIKVnsp2(SL15649)-R: ACATCCTGCTCGGGTGACCTG |
This paper | N/A |
| IVP: CHIKVnsp3(SL15649)-F: TAATACGACTCACTATAGGGAG AGCACCGTCGTACCGGGTAAAAC |
This paper | N/A |
| IVP: CHIKVnsp3(SL15649)-R: CCACCTGCCCTGTCTAGTC |
This paper | N/A |
| IVP: CHIKVnsp4(SL15649)-F: TAATACGACTCACTATAGGGAG ATATATATTCTCGTCGGACACCG |
This paper | N/A |
| IVP: CHIKVnsp4(SL15649)-R: TTTAGGACCGCCGTACAAAG |
This paper | N/A |
| CRISPR: sgControl1-S: CACCGTTCCGCGTTACATAACTTA |
Kearns, et al. | N/A |
| CRISPR: sgControl1-AS: AAACTAAGTTATGTAACGCGGAAC |
Kearns, et al. | N/A |
| CRISPR: sgControl2-S: CACCCTGGAATGAATTGGCCTATG |
Mimee, et al. | N/A |
| CRISPR: sgControl2-AS: AAACCATAGGCCAATTCATTCCAG |
Mimee, et al. | N/A |
| CRISPR: sgALPHA-5Prime-S: CACCGCAAAACACCCCTTCTCTAT | This paper | N/A |
| CRISPR: sgALPHA-5Prime-AS: AAACATAGAGAAGGGGTGTTTTGC | This paper | N/A |
| CRISPR: sgALPHA-3Prime-S: CACCGATGTTAGTGGTGTAGCTCTA | This paper | N/A |
| CRISPR: sgALPHA-3Primer-AS: AAACTAGAGCTACACCACTAACATC | This paper | N/A |
| SOFTWARE AND ALGORITHMS | ||
| ImageJ | Wayne Rasband (NIH) | https://imagej.nih.gov/ij/ |
| MetaXpress | Molecular Devices | N/A |
Quantification and Statistical Analysis:
All statistics performed on fold change data were log2 transformed prior to analysis. Each dot represents an individual experiment. All analyses were performed as indicated using Prism or R.
Supplementary Material
Highlights:
High-throughput screening identifies the anti-CHIKV lncRNA ALPHA.
ALPHA inhibits CHIKV and its closest relative, O’nyong’nyong virus.
ALPHA impacts viral RNA replication independently of Type I interferon signaling.
ALPHA directly binds to CHIKV genomic RNA.
ACKNOWLEDGEMENTS:
We thank the Cherry, Henao-Mejia, Liu, Lynch and Beiting labs for scientific discussion and assistance with data analysis, assays, and reagents. We thank the High Throughput Screening Core at the University of Pennsylvania for technical support with the lncRNA screens. We thank C. Coyne for HBMEC and HSV-1-GFP, M. Heise for CHIKV-mKate and SINV-mKate, D. Weiner for CHIKV, T.E. Morrison for ONNV, M. Diamond for MAYV, ZIKV, RVFV, and LACV, R. Kuhn for RRV-GFP, and S. Hensley for IAV. We thank Y. Loo and M. Gale for the IFNβ-mCherry plasmid and Addgene for lentivirus and CRISPR plasmids. Funding: This work was supported by grants from the National Institutes of Health to S.C. (R01AI074951, R01AI122749, 1R21AI151882, and R01AI140539) and J.H-M. (R21AI128060, R21DK111755, and RO1HL136572) as well as funding to S.C. from the Penn Center for Precision Medicine, Mercatus, and the Gates Foundation. J.H-M. is a recipient of the P.E.W. Biomedical Scholars Award. S.C. and J.H-M. are recipients of the Burroughs Wellcome Investigators in the Pathogenesis of Infectious Disease Award. Schematics in Figures 4, 5, S6, and S7 were generated using Biorender.com.
Footnotes
DECLARATION OF INTERESTS:
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES:
- Agarwal S, Vierbuchen T, Ghosh S, Chan J, Jiang Z, Kandasamy RK, Ricci E, and Fitzgerald KA (2020). The long non-coding RNA LUCAT1 is a negative feedback regulator of interferon responses in humans. Nat Commun 11, 6348. 10.1038/s41467-020-20165-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agliano F, Rathinam VA, Medvedev AE, Vanaja SK, and Vella AT (2019). Long Noncoding RNAs in Host-Pathogen Interactions. Trends Immunol 40, 492–510. 10.1016/j.it.2019.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhrymuk I, Kulemzin SV, and Frolova EI (2012). Evasion of the innate immune response: the Old World alphavirus nsP2 protein induces rapid degradation of Rpb1, a catalytic subunit of RNA polymerase II. J Virol 86, 7180–7191. 10.1128/JVI.00541-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhrymuk I, Lukash T, Frolov I, and Frolova EI (2019). Novel Mutations in nsP2 Abolish Chikungunya Virus-Induced Transcriptional Shutoff and Make the Virus Less Cytopathic without Affecting Its Replication Rates. J Virol 93. 10.1128/JVI.02062-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atianand MK, Caffrey DR, and Fitzgerald KA (2017). Immunobiology of Long Noncoding RNAs. Annu Rev Immunol 35, 177–198. 10.1146/annurev-immunol-041015-055459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basavappa M, Cherry S, and Henao-Mejia J. (2019). Long noncoding RNAs and the regulation of innate immunity and host-virus interactions. J Leukoc Biol 106, 83–93. 10.1002/JLB.3MIR0918-354R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer A, Lennemann NJ, Ouyang Y, Bramley JC, Morosky S, Marques ET Jr., Cherry S, Sadovsky Y, and Coyne CB (2016). Type III Interferons Produced by Human Placental Trophoblasts Confer Protection against Zika Virus Infection. Cell Host Microbe 19, 705–712. 10.1016/j.chom.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bick MJ, Carroll JW, Gao G, Goff SP, Rice CM, and MacDonald MR (2003). Expression of the zinc-finger antiviral protein inhibits alphavirus replication. J Virol 77, 11555–11562. 10.1128/jvi.77.21.11555-11562.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt FJ, Chen W, Miner JJ, Lenschow DJ, Merits A, Schnettler E, Kohl A, Rudd PA, Taylor A, Herrero LJ, et al. (2017). Chikungunya virus: an update on the biology and pathogenesis of this emerging pathogen. Lancet Infect Dis 17, e107–e117. 10.1016/S1473-3099(16)30385-1. [DOI] [PubMed] [Google Scholar]
- Bushnell B. BBMap. https://sourceforge.net/projects/bbmap/. [Google Scholar]
- Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, and Rinn JL (2011). Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25, 1915–1927. 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter S, and Fitzgerald KA (2018). Cytokines and Long Noncoding RNAs. Cold Spring Harb Perspect Biol 10. 10.1101/cshperspect.a028589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YG, Satpathy AT, and Chang HY (2017). Gene regulation in the immune system by long noncoding RNAs. Nat Immunol 18, 962–972. 10.1038/ni.3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, et al. (2015). Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 162, 974–986. 10.1016/j.cell.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow KT, Gale M Jr., and Loo YM (2018). RIG-I and Other RNA Sensors in Antiviral Immunity. Annu Rev Immunol 36, 667–694. 10.1146/annurev-immunol-042617-053309. [DOI] [PubMed] [Google Scholar]
- Chu C, Quinn J, and Chang HY (2012). Chromatin isolation by RNA purification (ChIRP). J Vis Exp. 10.3791/3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elling R, Chan J, and Fitzgerald KA (2016). Emerging role of long noncoding RNAs as regulators of innate immune cell development and inflammatory gene expression. Eur J Immunol 46, 504–512. 10.1002/eji.201444558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B, et al. (2018). The Reactome Pathway Knowledgebase. Nucleic Acids Res 46, D649–D655. 10.1093/nar/gkx1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox JM, and Diamond MS (2016). Immune-Mediated Protection and Pathogenesis of Chikungunya Virus. J Immunol 197, 4210–4218. 10.4049/jimmunol.1601426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson S, Gullberg M, Jarvius J, Olsson C, Pietras K, Gustafsdottir SM, Ostman A, and Landegren U. (2002). Protein detection using proximity-dependent DNA ligation assays. Nat Biotechnol 20, 473–477. 10.1038/nbt0502-473. [DOI] [PubMed] [Google Scholar]
- Fros JJ, Liu WJ, Prow NA, Geertsema C, Ligtenberg M, Vanlandingham DL, Schnettler E, Vlak JM, Suhrbier A, Khromykh AA, and Pijlman GP (2010). Chikungunya virus nonstructural protein 2 inhibits type I/II interferon-stimulated JAK-STAT signaling. J Virol 84, 10877–10887. 10.1128/JVI.00949-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fros JJ, Major LD, Scholte FEM, Gardner J, van Hemert MJ, Suhrbier A, and Pijlman GP (2015). Chikungunya virus non-structural protein 2-mediated host shut-off disables the unfolded protein response. J Gen Virol 96, 580–589. 10.1099/vir.0.071845-0. [DOI] [PubMed] [Google Scholar]
- Fros JJ, and Pijlman GP (2016). Alphavirus Infection: Host Cell Shut-Off and Inhibition of Antiviral Responses. Viruses 8. 10.3390/v8060166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fros JJ, van der Maten E, Vlak JM, and Pijlman GP (2013). The C-terminal domain of chikungunya virus nsP2 independently governs viral RNA replication, cytopathicity, and inhibition of interferon signaling. J Virol 87, 10394–10400. 10.1128/JVI.00884-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goertz GP, McNally KL, Robertson SJ, Best SM, Pijlman GP, and Fros JJ (2018). The Methyltransferase-Like Domain of Chikungunya Virus nsP2 Inhibits the Interferon Response by Promoting the Nuclear Export of STAT1. J Virol 92. 10.1128/JVI.01008-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbea C, Mosbruger T, and Cazalla D. (2017). A viral Sm-class RNA base-pairs with mRNAs and recruits microRNAs to inhibit apoptosis. Nature 550, 275–279. 10.1038/nature24034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Eils R, and Schlesner M. (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- Hadjicharalambous MR, and Lindsay MA (2019). Long Non-Coding RNAs and the Innate Immune Response. Noncoding RNA 5. 10.3390/ncrna5020034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon CC, Ramilowski JA, Harshbarger J, Bertin N, Rackham OJ, Gough J, Denisenko E, Schmeier S, Poulsen TM, Severin J, et al. (2017). An atlas of human long non-coding RNAs with accurate 5’ ends. Nature 543, 199–204. 10.1038/nature21374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou F, Sun L, Zheng H, Skaug B, Jiang QX, and Chen ZJ (2011). MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 146, 448–461. 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe KL, Achuthan P, Allen J, Allen J, Alvarez-Jarreta J, Amode MR, Armean IM, Azov AG, Bennett R, Bhai J, et al. (2021). Ensembl 2021. Nucleic Acids Res 49, D884–D891. 10.1093/nar/gkaa942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Zhang S, Yang Z, Lin H, Zhu J, Liu L, Wang W, Liu S, Liu W, Ma Y, et al. (2018). Self-Recognition of an Inducible Host lncRNA by RIG-I Feedback Restricts Innate Immune Response. Cell 173, 906–919 e913. 10.1016/j.cell.2018.03.064. [DOI] [PubMed] [Google Scholar]
- Jones JE, Long KM, Whitmore AC, Sanders W, Thurlow LR, Brown JA, Morrison CR, Vincent H, Peck KM, Browning C, et al. (2017). Disruption of the Opal Stop Codon Attenuates Chikungunya Virus-Induced Arthritis and Pathology. mBio 8. 10.1128/mBio.01456-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearns NA, Genga RM, Enuameh MS, Garber M, Wolfe SA, and Maehr R. (2014). Cas9 effector-mediated regulation of transcription and differentiation in human pluripotent stem cells. Development 141, 219–223. 10.1242/dev.103341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelvin AA, Banner D, Silvi G, Moro ML, Spataro N, Gaibani P, Cavrini F, Pierro A, Rossini G, Cameron MJ, et al. (2011). Inflammatory cytokine expression is associated with chikungunya virus resolution and symptom severity. PLoS Negl Trop Dis 5, e1279. 10.1371/journal.pntd.0001279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall C, Khalid H, Muller M, Banda DH, Kohl A, Merits A, Stonehouse NJ, and Tuplin A. (2019). Structural and phenotypic analysis of Chikungunya virus RNA replication elements. Nucleic Acids Res 47, 9296–9312. 10.1093/nar/gkz640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotzin JJ, Spencer SP, McCright SJ, Kumar DBU, Collet MA, Mowel WK, Elliott EN, Uyar A, Makiya MA, Dunagin MC, et al. (2016). The long non-coding RNA Morrbid regulates Bim and short-lived myeloid cell lifespan. Nature 537, 239–243. 10.1038/nature19346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee N, Moss WN, Yario TA, and Steitz JA (2015). EBV noncoding RNA binds nascent RNA to drive host PAX5 to viral DNA. Cell 160, 607–618. 10.1016/j.cell.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Jiang M, Liu L, Yang Z, Ma Z, Liu S, Ma Y, Zhang L, and Cao X. (2019). The long noncoding RNA Lnczc3h7a promotes a TRIM25-mediated RIG-I antiviral innate immune response. Nat Immunol 20, 812–823. 10.1038/s41590-019-0379-0. [DOI] [PubMed] [Google Scholar]
- Long KM, Ferris MT, Whitmore AC, Montgomery SA, Thurlow LR, McGee CE, Rodriguez CA, Lim JK, and Heise MT (2016). γδ T Cells Play a Protective Role in Chikungunya Virus-Induced Disease. J Virol 90, 433–443. 10.1128/JVI.02159-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma DY, and Suthar MS (2015). Mechanisms of innate immune evasion in re-emerging RNA viruses. Curr Opin Virol 12, 26–37. 10.1016/j.coviro.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald MR, Machlin ES, Albin OR, and Levy DE (2007). The zinc finger antiviral protein acts synergistically with an interferon-induced factor for maximal activity against alphaviruses. J Virol 81, 13509–13518. 10.1128/JVI.00402-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden EA, Plante KS, Morrison CR, Kutchko KM, Sanders W, Long KM, Taft-Benz S, Cruz Cisneros MC, White AM, Sarkar S, et al. (2020). Using SHAPE-MaP To Model RNA Secondary Structure and Identify 3’UTR Variation in Chikungunya Virus. J Virol 94. 10.1128/JVI.00701-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matusali G, Colavita F, Bordi L, Lalle E, Ippolito G, Capobianchi MR, and Castilletti C. (2019). Tropism of the Chikungunya Virus. Viruses 11. 10.3390/v11020175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meertens L, Hafirassou ML, Couderc T, Bonnet-Madin L, Kril V, Kummerer BM, Labeau A, Brugier A, Simon-Loriere E, Burlaud-Gaillard J, et al. (2019). FHL1 is a major host factor for chikungunya virus infection. Nature 574, 259–263. 10.1038/s41586-019-1578-4. [DOI] [PubMed] [Google Scholar]
- Meshram CD, Lukash T, Phillips AT, Akhrymuk I, Frolova EI, and Frolov I. (2019). Lack of nsP2-specific nuclear functions attenuates chikungunya virus replication both in vitro and in vivo. Virology 534, 14–24. 10.1016/j.virol.2019.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimee M, Tucker AC, Voigt CA, and Lu TK (2015). Programming a Human Commensal Bacterium, Bacteroides thetaiotaomicron, to Sense and Respond to Stimuli in the Murine Gut Microbiota. Cell Syst 1, 62–71. 10.1016/j.cels.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser LA, Ramirez-Carvajal L, Puri V, Pauszek SJ, Matthews K, Dilley KA, Mullan C, McGraw J, Khayat M, Beeri K, et al. (2016). A Universal Next-Generation Sequencing Protocol To Generate Noninfectious Barcoded cDNA Libraries from High-Containment RNA Viruses. mSystems 1. 10.1128/mSystems.00039-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowel WK, Kotzin JJ, McCright SJ, Neal VD, and Henao-Mejia J. (2018). Control of Immune Cell Homeostasis and Function by lncRNAs. Trends Immunol 39, 55–69. 10.1016/j.it.2017.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowel WK, McCright SJ, Kotzin JJ, Collet MA, Uyar A, Chen X, DeLaney A, Spencer SP, Virtue AT, Yang E, et al. (2017). Group 1 Innate Lymphoid Cell Lineage Identity Is Determined by a cis-Regulatory Element Marked by a Long Non-coding RNA. Immunity 47, 435–449 e438. 10.1016/j.immuni.2017.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak TK, Mamidi P, Sahoo SS, Kumar PS, Mahish C, Chatterjee S, Subudhi BB, Chattopadhyay S, and Chattopadhyay S. (2019). P38 and JNK Mitogen-Activated Protein Kinases Interact With Chikungunya Virus Non-structural Protein-2 and Regulate INF Induction During Viral Infection in Macrophages. Front Immunol 10, 786. 10.3389/fimmu.2019.00786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelemans T, and Kikkert M. (2019). Viral Innate Immune Evasion and the Pathogenesis of Emerging RNA Virus Infections. Viruses 11. 10.3390/v11100961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng LF, Chow A, Sun YJ, Kwek DJ, Lim PL, Dimatatac F, Ng LC, Ooi EE, Choo KH, Her Z, et al. (2009). IL-1beta, IL-6, and RANTES as biomarkers of Chikungunya severity. PLoS One 4, e4261. 10.1371/journal.pone.0004261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nojima T, Tellier M, Foxwell J, Ribeiro de Almeida C, Tan-Wong SM, Dhir S, Dujardin G, Dhir A, Murphy S, and Proudfoot NJ (2018). Deregulated Expression of Mammalian lncRNA through Loss of SPT6 Induces R-Loop Formation, Replication Stress, and Cellular Senescence. Mol Cell 72, 970–984 e977. 10.1016/j.molcel.2018.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14, 417–419. 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RCoreTeam (2018). R: A language and environment for statistical computing. R Found. Stat. Comput. Vienna, Austria: https://www.r-project.org/). [Google Scholar]
- Reed LJa.M., H. (1938). A simple method of estimating fifty per cent endpoints. American Journal of Epidemiology 27, 493–497. [Google Scholar]
- Rehwinkel J, and Gack MU (2020a). RIG-I-like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol 20, 537–551. 10.1038/s41577-020-0288-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehwinkel J, and Gack MU (2020b). RIG-I-like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol. 10.1038/s41577-020-0288-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F. (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784. 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, and Rice CM (2014). Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 32, 513–545. 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW (2019). Interferon-Stimulated Genes: What Do They All Do? Annu Rev Virol 6, 567–584. 10.1146/annurev-virology-092818-015756. [DOI] [PubMed] [Google Scholar]
- Schwartz O, and Albert ML (2010). Biology and pathogenesis of chikungunya virus. Nat Rev Microbiol 8, 491–500. 10.1038/nrmicro2368. [DOI] [PubMed] [Google Scholar]
- Seth RB, Sun L, Ea CK, and Chen ZJ (2005). Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122, 669–682. 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, and Zhang F. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva LA, and Dermody TS (2017). Chikungunya virus: epidemiology, replication, disease mechanisms, and prospective intervention strategies. J Clin Invest 127, 737–749. 10.1172/JCI84417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, and Landegren U. (2006). Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 3, 995–1000. 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- Solignat M, Gay B, Higgs S, Briant L, and Devaux C. (2009). Replication cycle of chikungunya: a re-emerging arbovirus. Virology 393, 183–197. 10.1016/j.virol.2009.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C, Love MI, and Robinson MD (2015). Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 4, 1521. 10.12688/f1000research.7563.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart CE, Randall RE, and Adamson CS (2014). Inhibitors of the interferon response enhance virus replication in vitro. PLoS One 9, e112014. 10.1371/journal.pone.0112014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe ISB, Tanabe ELL, Santos EC, Martins WV, Araujo I, Cavalcante MCA, Lima ARV, Camara NOS, Anderson L, Yunusov D, and Bassi EJ (2018). Cellular and Molecular Immune Response to Chikungunya Virus Infection. Front Cell Infect Microbiol 8, 345. 10.3389/fcimb.2018.00345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullah MO, Sweet MJ, Mansell A, Kellie S, and Kobe B. (2016). iRIh-dependent ilk signaling, its functions in host defense and inflammation, and its potential as a therapeutic target. J Leukoc Biol 100, 27–45. 10.1189/jlb.2RI1115-531R. [DOI] [PubMed] [Google Scholar]
- Varghese FS, Thaa B, Amrun SN, Simarmata D, Rausalu K, Nyman TA, Merits A, McInerney GM, Ng LFP, and Ahola T. (2016). The Antiviral Alkaloid Berberine Reduces Chikungunya Virus-Induced Mitogen-Activated Protein Kinase Signaling. J Virol 90, 9743–9757. 10.1128/JVI.01382-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venugopalan A, Ghorpade RP, and Chopra A. (2014). Cytokines in acute chikungunya. PLoS One 9, e111305. 10.1371/journal.pone.0111305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T, and Fitzgerald KA (2021). Long non-coding RNAs in antiviral immunity. Semin Cell Dev Biol 111, 126–134. 10.1016/j.semcdb.2020.06.009. [DOI] [PubMed] [Google Scholar]
- Wang P, Xue Y, Han Y, Lin L, Wu C, Xu S, Jiang Z, Xu J, Liu Q, and Cao X. (2014). The STAT3-binding long noncoding RNA lnc-DC controls human dendritic cell differentiation. Science 344, 310–313. 10.1126/science.1251456. [DOI] [PubMed] [Google Scholar]
- Ward JH (1963). Hierarchical Grouping to Optimize an Objective Function. J Am Stat Assoc 58, 236-&. Doi 10.2307/2282967. [DOI] [Google Scholar]
- Werner MS, Sullivan MA, Shah RN, Nadadur RD, Grzybowski AT, Galat V, Moskowitz IP, and Ruthenburg AJ (2017). Chromatin-enriched lncRNAs can act as cell-type specific activators of proximal gene transcription. Nat Struct Mol Biol 24, 596–603. 10.1038/nsmb.3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H, Francois R, Henry L, Müller K. (2017). dplyr: A Grammar of Data Manipulation. R Packag. version 0.7.4 https://cran.r-project.org/package=dply. [Google Scholar]
- Yi K, Zhang Y, Wang Y, Wang Z, Xie M, Jin Z, and Zhao T. (2019). Long noncoding RNA and its role in virus infection and pathogenesis. Front Biosci (Landmark Ed) 24, 777–789. [DOI] [PubMed] [Google Scholar]
- Zhang W, Xie M, Shu MD, Steitz JA, and DiMaio D. (2016). A proximity-dependent assay for specific RNA-protein interactions in intact cells. RNA 22, 1785–1792. 10.1261/rna.058248.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data generated in this manuscript are available through the NCBI Gene Expression Omnibus using accession number GSE184306.
All unprocessed images can be found at the following Mendeley link: https://data.mendeley.com/datasets/yxxg7×5c6y/draft?a=28296dfe-76bf-448c-8a94-662582b96df6
This paper does not report original code.
Any additional information required to reanalyze data reported in this paper can be requested from the Lead Contact.