

Abstract
The European Commission asked EFSA to update their 2006 opinion on ochratoxin A (OTA) in food. OTA is produced by fungi of the genus Aspergillus and Penicillium and found as a contaminant in various foods. OTA causes kidney toxicity in different animal species and kidney tumours in rodents. OTA is genotoxic both in vitro and in vivo; however, the mechanisms of genotoxicity are unclear. Direct and indirect genotoxic and non‐genotoxic modes of action might each contribute to tumour formation. Since recent studies have raised uncertainty regarding the mode of action for kidney carcinogenicity, it is inappropriate to establish a health‐based guidance value (HBGV) and a margin of exposure (MOE) approach was applied. For the characterisation of non‐neoplastic effects, a BMDL 10 of 4.73 μg/kg body weight (bw) per day was calculated from kidney lesions observed in pigs. For characterisation of neoplastic effects, a BMDL 10 of 14.5 μg/kg bw per day was calculated from kidney tumours seen in rats. The estimation of chronic dietary exposure resulted in mean and 95th percentile levels ranging from 0.6 to 17.8 and from 2.4 to 51.7 ng/kg bw per day, respectively. Median OTA exposures in breastfed infants ranged from 1.7 to 2.6 ng/kg bw per day, 95th percentile exposures from 5.6 to 8.5 ng/kg bw per day in average/high breast milk consuming infants, respectively. Comparison of exposures with the BMDL 10 based on the non‐neoplastic endpoint resulted in MOEs of more than 200 in most consumer groups, indicating a low health concern with the exception of MOEs for high consumers in the younger age groups, indicating a possible health concern. When compared with the BMDL 10 based on the neoplastic endpoint, MOEs were lower than 10,000 for almost all exposure scenarios, including breastfed infants. This would indicate a possible health concern if genotoxicity is direct. Uncertainty in this assessment is high and risk may be overestimated.
Keywords: Ochratoxin, hazard characterisation, dietary exposure assessment, margin of exposure approach, risk characterisation
Short abstract
This publication is linked to the following EFSA Supporting Publications article: http://onlinelibrary.wiley.com/doi/10.2903/sp.efsa.2020.EN-1845/full
Summary
Following a request from the European Commission, the European Food Safety Authority (EFSA) Panel on Contaminants in the Food Chain (CONTAM Panel) evaluated the risks to human health related to the presence of ochratoxin A (OTA) in food. The previous risk assessment of OTA from EFSA of 2006 was used as a starting point for the evaluation, together with publications identified in a targeted literature search. EFSA guidance documents and general principles for risk assessment were applied for hazard and exposure assessment and risk characterisation in this opinion.
OTA is produced by various fungi of the genus Aspergillus and Penicillium, e.g. A. ochraceus, A. carbonarius and P. verrucosum. OTA is stable to moderate heating, but losses ranging up to 90% are observed at temperatures above 180°C.
OTA is rapidly absorbed and distributed but slowly eliminated and excreted leading to potential accumulation in the body, which is due mainly to binding to plasma proteins and a low rate of metabolism. Plasma half‐lifes range from several days in rodents and pigs to several weeks in non‐human primates and humans. The major metabolic pathway of OTA is hydrolysis to OTalpha, followed by conjugation with glucuronic acid. Formation of DNA‐reactive metabolites is either minor or absent under physiological conditions. Based on efficient degradation in the rumen, OTA levels in cow milk are low. In several studies, relatively high OTA concentrations have been found in human milk in comparison with those found in cow milk.
A series of studies on biomarkers of exposure of humans to OTA, published after the latest EFSA evaluation, have been evaluated. It was found that dietary exposure to OTA was reflected in OTA levels in plasma, serum, urine and breast milk. Reliable biomarkers of OTA‐specific effects, in particular kidney function, have not been identified.
The CONTAM Panel did not identify evidence suggesting that OTA is acutely toxic. OTA exerts adverse effects in repeated dose studies with mice, rats, rabbits and pigs. At high doses, various general signs of toxicity (e.g. reduced body and organ weights, changes in clinical chemistry) are observed. Also histopathological lesions, in particular in the kidney, immunotoxicity, neurotoxicity and developmental effects (associated with maternal toxicity) are observed. The critical effects occur in the kidney with the pig being the most susceptible species. In rats, kidney tumours have been observed in both sexes, with males being more sensitive. In mice, both liver and kidney tumours have been observed in both sexes albeit at higher doses as compared to the renal tumours in rats.
In vitro, OTA exposure induces gene mutations, single and double strand breaks (SSBs and DSBs) and chromosome damage in mammalian cells. This OTA‐dependent genetic damage is apparently independent of metabolic activation. Exposure to OTA increases the levels of reactive oxygen species (ROS) in vivo, and in vitro exposure increases 8‐hydroxydeoxyguanosine (8‐OHdG) in DNA, both of which indicate that some genotoxic effects may be secondary to oxidative stress. In vivo, OTA‐induced genetic damage (DSBs, aberrant mitoses and karyomegaly) is often associated with pathological findings in rat kidney. Gene mutations occurring in rats and mice are restricted to the cancer target site (the outer medulla of the kidney) with large deletions and base substitutions being the main mutagenic events. The molecular mechanisms underlying OTA genotoxicity in vivo remains unclear. OTA is a weak mutagen in vivo. Formation of covalent OTA‐DNA adducts remains controversial and the extremely low levels of DNA‐adducts reported do not explain the genotoxic effects of OTA. Nevertheless, OTA‐induced mutations in rat and mouse kidneys are clearly not a simple consequence of oxidative DNA damage. The mechanisms underlying OTA‐induced chromosomal damage are also unclear. Aberrant chromosome segregation and cell division may reflect damage to the mitotic spindle. Alternatively, abnormal anaphase figures might derive from un‐resolved ‘replication stress’ with break‐induced recombination of DSBs in the early stage of mitosis.
Possible associations between OTA exposure and kidney disease, bladder or hepatocellular cancer have been investigated in epidemiological studies using cross‐sectional designs, but it is not possible to establish a causal link between exposure to OTA and adverse effects in humans. The findings of raised protein concentrations in urine in a limited number of infants from Egypt exposed to high levels of OTA via their mothers (during pregnancy and lactation) raise health concern and would need confirmation in larger studies.
Multiple molecular events of OTA have been reported, but the MoAs of OTA toxicity have not been clarified. The susceptibility of the kidney appears to be largely due to OTA uptake via the organic anion transporters in the kidney. With regard to the MoAs for kidney carcinogenicity in rats, induction of apoptosis and autophagy, cell cycle arrest, alterations in the cellular proliferation response and cell signalling, oxidative stress and changes in gene expression have each been proposed as contributing factors. A role of genotoxicity in carcinogenesis cannot be excluded; however, a distinction between direct and indirect genotoxic modes of action which might each contribute to tumour formation could not be established.
Increased incidences of microscopic kidney lesions in a repeated dose study with female pigs have been identified as the critical non‐neoplastic effect for risk characterisation and a BMDL10 of 4.73 μg OTA/kg body weight (bw) per day was calculated from these effects and used as a non‐neoplastic reference point (RP).
Increased incidences of kidney tumours seen in male rats in a 2‐year study were identified as the critical neoplastic effect of OTA and a BMDL10 of 14.5 μg OTA/kg bw per day was calculated from these effects and used as a neoplastic RP.
Since the previous EFSA assessment (2006), more recent studies have raised uncertainty regarding the mode of action for kidney carcinogenicity. Following respective EFSA guidance, the CONTAM Panel considered that it was not appropriate to establish an HBGV and concluded that for both non‐neoplastic and neoplastic effects of OTA, an MOE approach needs to be applied for risk characterisation. As a consequence, the TWI of 120 ng/kg bw as established by the CONTAM Panel in 2006 is no longer valid.
For characterisation of chronic non‐neoplastic effects, an MOE of ≥ 200 between the selected non‐neoplastic RP and calculated exposures was considered as being of low health concern. This MOE was derived by applying a default uncertainty factor (UF) of 100 for intra‐ and interspecies toxicokinetic and toxicodynamic differences combined with an additional UF of 2 to account for extrapolation of a 3‐month study in pigs to a chronic situation in that species.
For characterisation of chronic neoplastic effects, an MOE of ≥ 10,000 between the selected neoplastic RP and calculated exposures would be of low health concern. This MOE was derived following EFSA guidance for substances that are both genotoxic and carcinogenic. In the interpretation of the MOE for the neoplastic risks, the Panel considered that the MOE of 10,000 for substances that are genotoxic and carcinogenic could be particularly conservative in this case because the evidence for a direct interaction of OTA with the DNA is inconclusive.
A total of 71,769 measurements of concentrations of OTA in food submitted within the last 10 years by 29 European countries and one Industry association were used for assessing dietary OTA exposures. The majority (about 55%) of the data came from Germany and the Netherlands. The proportion of left‐censored data (results below the limit of detection (LOD) or limit of quantification (LOQ)) was 75%. The highest mean concentrations of OTA were recorded in the categories ‘Plant extract formula’, ‘Flavourings or essences’ (both containing liquorice extracts) and ‘Chili pepper’.
For calculating chronic dietary exposure to OTA, food consumption and body weight data at the individual level were accessed in the EFSA Comprehensive Database. Mean occurrence levels and individual consumption data from different countries and age groups were linked at the relevant FoodEx level to estimate the chronic exposure.
The mean chronic exposure estimates ranged from 0.64 (minimum LB)/2.53 (minimum UB) to 9.13 (maximum LB)/17.79 (maximum UB) ng/kg bw per day across dietary surveys and age groups. The high (95th percentile) chronic dietary exposure estimates ranged from 2.40 (minimum LB)/5.13 (minimum UB) to 30.36 (maximum LB)/51.69 ng/kg (maximum UB) bw per day.
The most important contributors to the chronic dietary exposure to OTA were ‘Preserved meat’, ‘Cheese’ and ‘Grains and grain‐based products’. Dried and fresh fruits such as grapes, figs and dates as well as fruit juices and nectars were also contributing to the exposure of in some of the ‘Toddlers’ and ‘Other children’ groups, albeit, to a lesser extent than the three major categories. Non‐chocolate confectionary was a significant source of exposure in countries where liquorice‐based sweets are commonly consumed.
In a specific exposure scenario, occurrence data on OTA in breast milk in the public literature were used to assess the exposure of infants from breastfeeding (0–6 months of age). Based on the reported median concentration of OTA of 13 ng/L and a 95th percentile (P95) concentration of OTA of 43 ng/L in human milk (transitional and mature milk combined), the chronic dietary exposure to OTA ranged from 1.7 to 5.6 ng/kg bw per day for infants with an average milk consumption and from 2.6 to 8.5 ng/kg bw per day for infants with a high milk consumption.
The calculated MOEs for non‐neoplastic effects ranged from 7,391 (LB Min in ‘Infants’) to 266 (UB Max in ‘Toddlers’) at mean exposures and from 1971 (LB Min in ‘Very elderly’) to 92 (UB Max in ‘Toddlers’). Overall, they were above 200 in most of the dietary surveys for average and high consumers and therefore of low health concern and were below 200 at maximum LB in the age groups of ‘Infants’ and at maximum UB in the age group of ‘Infants’, ‘Toddlers’ and ‘Other children’ indicating a possible health concern for these age groups. In breastfed infants, the MOEs were above 200 in all scenarios indicating a low health concern.
The calculated MOEs for neoplastic effects ranged from 22,615 (LB Min in ‘Infants’) to 815 (UB Max in ‘Toddlers’) at mean exposures and from 6,042 (LB Min in ‘Very elderly’) to 281 (UB Max in ‘Infants’). With the exception of the minimum LB estimate for ‘Infants’ and ‘Very elderly’, the calculated MOEs for neoplastic effects were below 10,000 in most of the surveys, in particular for high consumers and breastfed infants indicating a possible health concern for these consumer groups.
For neoplastic effects of OTA, the CONTAM Panel emphasises, that it was not possible to make a clear distinction between a direct and an indirect mechanism of genotoxicity. As a result, the risk characterisation will be either not sufficiently cautious or overcautious depending on which MoAs actually take place. The Panel points out that the MOE of 10,000 is likely to be most conservative in this case as the evidence for a direct interaction of OTA with the DNA is inconclusive and alternative thresholded mechanisms may play a role in the formation of kidney tumours.
Pending elucidation of the MoAs for the genotoxicity/carcinogenicity of OTA, the Panel concluded that an MOE of 10,000 to the neoplastic reference point is warranted for the risk characterisation of OTA, albeit being the most conservative approach until the MoAs are clarified.
The overall uncertainty associated with the present assessment is considered as high. The assessment is more likely to overestimate than to underestimate the risk.
The CONTAM Panel recommended that more studies elucidating the sequence of critical events at the carcinogenic target site in the kidney are needed as well as more data on the levels of OTA in human milk and the differential toxicokinetics of OTA in different species including transfer to the fetus.
More information on the specific signature of OTA gene mutations in appropriate animal models is also needed. Reliable and representative investigations of the levels of OTA in human breast milk are needed. More occurrence data on OTA in cheese paste in comparison to cheese rinds are needed. More data on occurrence and toxicity of modified OTA are needed.
1. Introduction
1.1. Background and Terms of Reference as provided by the requestor
1.1.1.
Background
Maximum levels for ochratoxin A are established at EU level for a wide range of foodstuffs.1 Recently, findings of high levels of ochratoxin A are observed in food for which no maximum levels were set at EU level. Therefore, the possible setting of maximum levels for ochratoxin A in these foods was considered. During these discussions, certain delegations indicated that new toxicity studies have become available since the EFSA opinion of 2006.2 It is therefore appropriate to request EFSA to assess these toxicity studies and to consider, if an update of the scientific opinion is necessary.
Terms of Reference
In accordance with Art. 29 (1) of Regulation (EC) No 178/2002, the European Commission asks the European Food Safety Authority to Authority to
-
–
To assess the toxicity studies on ochratoxin A which have become available since the EFSA opinion on ochratoxin A in 2006 and to update the scientific opinion if necessary as regards hazard characterisation.
-
–
To provide an updated exposure assessment taking into account recent occurrence data and the Comprehensive European Food Consumption Database.
-
–
To update the section on risk characterisation, as necessary.
2. Additional information
2.1. Chemistry and formation of ochratoxin A
Ochratoxins are mycotoxins produced by various fungi of the genus Aspergillus and Penicillium, e.g. A. ochraceus, A. carbonarius and P. verrucosum. Their chemical structures contain the amino acid L‐phenylalanine linked via an amide bond to a substituted dihydroisocoumaric acid (Figure 1). The most prevalent and toxic ochratoxin is ochratoxin A (OTA), which is the subject of this opinion. A notable feature of the OTA structure is the chlorine substituent (R2 in Figure 1), which appears to be important for the toxicity of OTA. Ochratoxin B (OTB) is the non‐chlorinated form of OTA, whereas ochratoxin C represents the ethyl ester of OTA (Figure 1). Other related mycotoxins formed at low levels by OTA‐producing fungi are methyl esters of OTA and OTB, free dihydroisocoumaric acid (ochratoxin alpha, OTalpha see also Section 4.1.1 on Toxicokinetics) and amides of OTalpha with serine, hydroxyproline and lysine (Malir et al., 2016).
Figure 1.
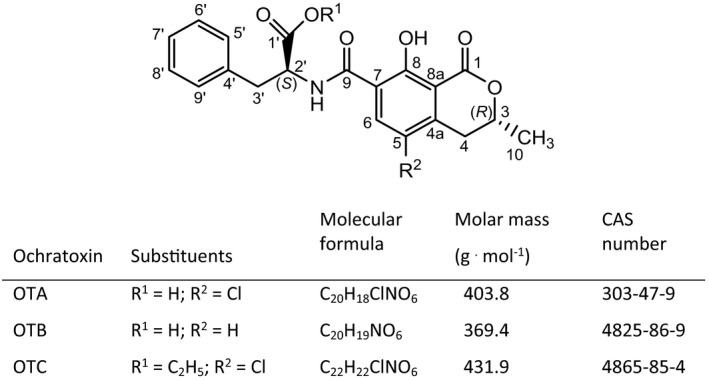
Chemical structure of ochratoxin A, B and C
Although the biosynthetic pathways of ochratoxins have not been established completely, it is known that the phenylalanine moiety originates from the shikimate pathway and the dihydroisocoumarin moiety from a polyketide (pentaketide) pathway (Heussner and Bingle, 2015).
OTA is a white, odourless, crystalline solid compound with melting point 168–173°C (Köszegi and Poor, 2016). It has a poor solubility in water (about 0.42 mg/L at 25°C) and a moderate solubility in polar organic solvents such as chloroform, ethanol and methanol. Due to its carboxylic group and phenolic hydroxyl group, OTA exists in non‐ionic, monoanionic (OTA‐) and dianionic (OTA2‐) forms in aqueous solvents, depending on the pH. Its pKa values are 4.3 and 7.2. Due to its isocoumarin moiety, OTA exhibits a strong fluorescence after absorption of ultraviolet light, which depends strongly on the pH (Steinbrück et al., 2008). For details on the physico‐chemical properties of OTA, including spectral data, see Pohland et al. (1982) and El Khoury and Atoui (2010).
2.1.1. Modified forms of OTA
Parent mycotoxins as produced by the fungi may undergo alterations of their chemical structure by chemical or biological reactions, e.g. during food processing or metabolism. Due to their altered structures, such modified mycotoxins are usually not detected by the analytical methods aimed at the parent toxins (Berthiller et al., 2013; Rychlik et al., 2014). However, these modified forms may contribute to the overall exposure and toxicity, if they are toxic in their own right or if they can release the parent toxin after absorption. Although OTA is quite stable to moderate heating (exhibiting for example only 30% reduction after dry heating to 150°C for 1 h), losses ranging from 70% to > 90% have been observed at temperatures above 180°C, such as used for coffee roasting (Cramer et al., 2008). Two OTA degradation products were identified by Cramer et al. (2008) in samples of roasted coffee: The major one was 2'R‐OTA (Figure 2), which was originally reported as 14R‐OTA and is the diastereomer of OTA. The second product is decarboxy‐OTA (Figure 2) and constitutes only trace amounts compared to 2'R‐OTA. More recently, OTalpha amide and OTA alpha (Figure 2) were identified as minor thermal degradation products of OTA (Bittner et al., 2013). The formation of OTA alpha amide had before been observed when OTA was exposed to light at 470 nm wavelength (Schmidt‐Heydt et al., 2012), and OTalpha arises also from the hydrolysis of the peptide bond of OTA catalysed by numerous peptidases and lipases (Abrunhosa et al., 2010). Finally, Bittner et al. (2013) provided evidence for the covalent binding of OTA to coffee polysaccharides via esterification of its carboxyl group as a further thermal reaction. It is likely that some of the esters are hydrolysed during coffee brewing and the liberated OTA is released into the coffee brew, because higher amounts of OTA have been observed in brewed coffee than in roasted coffee beans (Studer‐Rohr et al., 1995).
Figure 2.
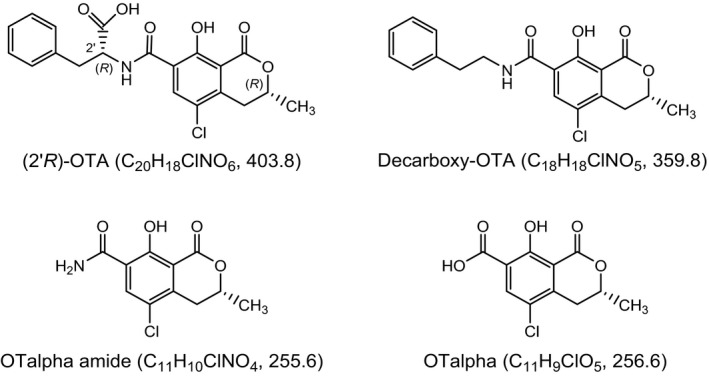
Modified forms of OTA arising from food processing
Although roasted coffee appears to be the major source of 2'R‐OTA due to the high temperatures of > 120°C needed for its formation, small amounts of this isomerisation product of OTA have recently also been detected in malt coffee and in traditionally baked rye bread (Sueck et al., 2019).
In addition to OTA modifications occurring during food processing, OTA has also been shown to be metabolised by plants. Modified mycotoxins formed in plants are also termed ‘masked’ mycotoxins (Rychlik et al., 2014). Ruhland et al. (1994, 1996a) demonstrated that (4S)‐ and (4R)‐4‐hydroxy‐OTA and their monoglucosides are formed in wheat and maize cell suspension cultures (Figure 3). Moreover, OTalpha (see Figure 2) and the methyl esters of OTA and of both 4‐hydroxy‐OTA isomers (Figure 3) were detected, together with several minor unidentified metabolites. A nearly complete conversion of OTA to some of these metabolites was observed in cultures of several other crop plants (Ruhland et al., 1996b). OTalpha and the 4‐hydroxy‐OTA isomers were also identified in several vegetables and cereals after exposure of the plants to OTA (Ruhland et al., 1997). Therefore, modified forms of OTA may contribute to the health risk posed by this mycotoxin.
Figure 3.
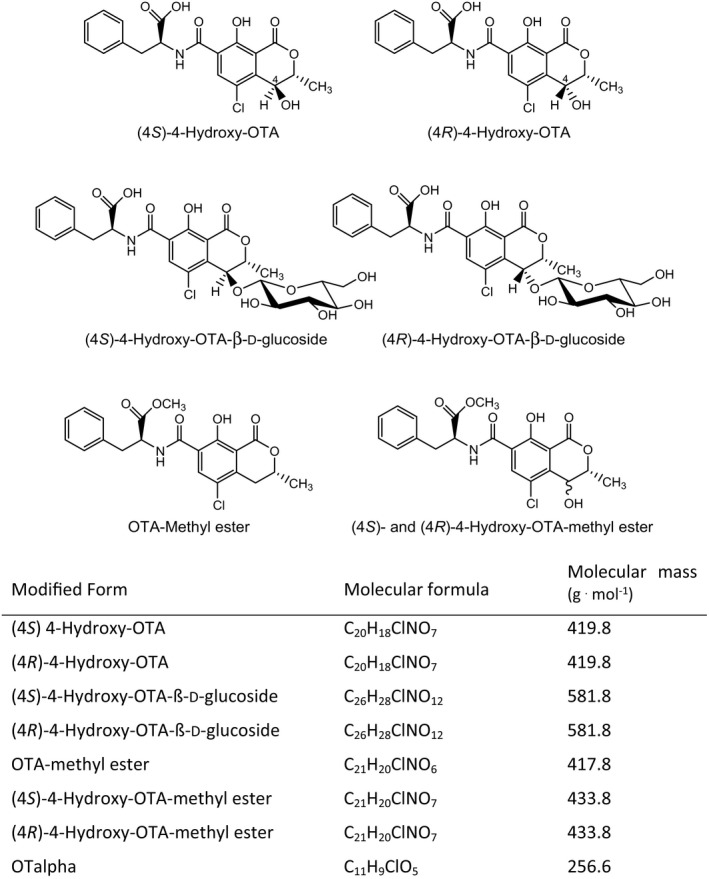
Modified forms of OTA arising from plant metabolism
Another modified form of OTA is represented by OTC, the ethyl ester of OTA (see Figure 1). Fuchs et al. (1984) demonstrated that a single oral dose of OTC administered to rats gives rise to the same blood levels of OTA over 48 h as an equivalent dose of OTA, including the same maximum concentration of OTA 60 min after dosing. This implies that OTC is rapidly converted to OTA after oral administration.
Modified forms of OTA may also be formed as mammalian metabolites. For example, OTA glucuronides and sulfates have been reported (see Section 3.1.1 on Toxicokinetics), but in all these reports, OTA conjugates were determined indirectly by using enzymatic hydrolysis. No definite evidence for the formation of OTA conjugates and no complete structural characterisation are available. Likewise, conjugates of OTA with a hexose and pentose were described (see Section 3.1.1 on Toxicokinetics), but the structure elucidation of such OTA glycosides is still lacking.
2.1.2. Analytical methods
The analysis of OTA in food relies on well‐established methods, and reliable results generally are obtained as evidenced by the results from proficiency testing.
Due to the chemical nature of the molecule, acid or alkali extraction from the matrix is commonly used. In general, alkaline extraction provides better recoveries; however, in the case of complex matrices (i.e. meat or sausages), an alkali–acid liquid–liquid partition can be used to increase selectivity.
Fluorescence‐based methods, when positive results are obtained, are often followed by a confirmatory analysis based on OTA‐methylation or OTA cleavage by carboxypeptidase treatment (Hult et al., 1979; Stander et al., 2001).
LC‐MS methods are widely used for OTA, and usually cover OTB and minor metabolites such as OTalpha, as well (Hautbergue et al., 2017; Limay‐Rios et al., 2017). Mass spectrometry is also extensively used for studying possible transformation occurring to the compound upon processing (Duarte et al., 2010; Bittner et al., 2013). The same approach based on LC‐MS/MS is in use for modified forms of OTA originating upon processing (Bittner et al., 2013).
Classic analysis involves a C18 solid phase or immunoaffinity clean up prior to HPLC determination with fluorescence detection. Available validated methods cover almost all the possible foodstuffs, among them cereals (Spanjer et al., 2008; Desmarchelier et al., 2014), coffee (Drunday and Pacin, 2013; Nielsen et al., 2015; Chen et al., 2016; Kokina et al., 2016) and cocoa (Amézqueta et al., 2004), pork meat and sausages (Duarte et al., 2013; Brera et al., 2014), milk (Sørensen and Elbæk, 2005; Beltrán et al., 2011), dried fruits (Spanjer et al., 2008; Desmarchelier et al., 2014), wine (Visconti et al., 1999; Sáez et al., 2004; Rodríguez‐Cabo et al., 2016), beer (Sáez et al., 2004; Ventura et al., 2006; Limay‐Rios et al., 2017) and liquorice (Ariño et al., 2007; Lerda et al., 2013).
Over the past decade, OTA determination was included in the so‐called ‘multi‐toxin’ methods, based on LC‐MS and covering hundreds of mycotoxins in the same analysis. The MS‐based analysis of OTA is simple, with a good sensitivity and accuracy.
OTA and metabolites thereof are commonly analysed as biomarkers in biological fluids, such as blood, plasma, urine and tissues. Methods are based on LC‐MS/MS using either direct detection (González‐Arias et al., 2017; Sueck et al., 2019) or enzymatic cleavage (Ali et al., 2017; Muñoz et al., 2017).
When LC‐MS is used, sample clean‐up is often avoided, and a ‘dilute‐and‐shoot’ approach is adopted. In addition, confirmatory analysis is not required. Multi‐toxin methods often suffer from lower accuracy and recoveries compared to single‐analyte methods, due to the analytical compromise adopted to cover a wide spectrum of different compounds. However, parameters for OTA are usually of good analytical quality, as confirmed in proficiency testing (De Girolamo et al., 2013; Solfrizzo et al., 2013).
Besides chromatographic methods, immune‐based analysis is often used for OTA determination (Barthelmebs et al., 2011; Cichna‐Markl, 2011; Meneely and Elliott, 2014; Sun et al., 2017). A wide range of ELISA kits, dipsticks and lateral flow devices are commercially available, with established procedures for screening purposes. In the last decade, several biosensors have been proposed and implemented for rapid detection, often involving specific antibodies, aptamers or molecular imprinting polymers involved in the recognition process.
2.2. Previous risk assessments
In 2006, the EFSA CONTAM Panel published an opinion related to OTA in food (EFSA, 2006). The CONTAM Panel concluded that there is a lack of convincing evidence that Balkan Endemic Nephropathy (BEN) is associated with OTA exposure. OTA is nephrotoxic in all animal species tested and there is evidence that these effects are associated with oxidative stress. It also causes immuno‐ and neurotoxicity and teratogenicity at higher doses. OTA accumulates in the kidneys and upon chronic exposure to doses toxic to the kidney it also induces kidney and liver tumours in rodents. The results from genotoxicity tests are inconsistent. The overall evidence supports the hypothesis that DNA damage is induced by oxidative stress rather than by direct interaction. The Panel identified the effects on the kidneys in rats and pigs as the critical endpoint for OTA and established a tolerable weekly intake (TWI) of 120 ng OTA/kg bw based on an LOAEL of 8 μg/kg bw per day identified for early markers for renal toxicity in pigs and by applying a composite uncertainty factor (UF) of 450. This UF is composed of a default UF of 10 for intraspecies variability and a default UF of 2.5 for toxicodynamic and an OTA‐specific UF of 6 for toxicokinetic interspecies variability (instead of the default 4.0) because the body burden resulting from the same external dose of OTA will be six times higher in humans than in pigs. An additional UF of 3 was used accounting for an LOAEL instead of an NOAEL. Main contributors to OTA dietary exposure were cereals and cereal products, wine, beer, grape juice, brewed coffee, cocoa and cocoa products and pork meat. Estimated exposure levels for average consumers varied between 15 and 20 ng/kg bw per week and for high consumers between 40 and 60 ng/kg bw per week and thus were below the TWI of 120 ng/kg bw. It was noted that these estimates are conservative for the adult population but that it cannot be excluded that infants and children and certain high consumers could have higher exposures.
The JECFA (Joint FAO/WHO Expert Committee on Food Additives) has issued several evaluations of OTA (FAO/WHO, 1991, 1996, 2001, 2008). In 1991, a provisional tolerable weekly intake (PTWI) of 112 ng/kg bw was established, based on an LOEL of 8 μg/kg bw per day for deterioration of renal function in pigs, and by application of an UF of 500. This UF was applied by the Committee because NOELs were frequently not demonstrated for OTA and the effects were observed already after a small proportion of a pig's lifetime and that consequently a 500‐fold margin needs to be applied to the LOELs. In 1995, the PTWI was confirmed but rounded off to 100 ng/kg bw and was then retained in the JECFA evaluation that followed in 2002. In their most recent evaluation on OTA (FAO/WHO, 2008) in which new evidence and data becoming available since their last evaluation were considered, the JECFA again confirmed the PTWI of 100 ng OTA/kg bw. The estimates of dietary exposure to OTA using occurrence data (mainly from Europe) ranged from 8 to 17 ng/kg bw per week, and thus were well below the PTWI.
Health Canada have re‐evaluated the appropriateness of the EFSA TWI to see if it would be applicable if OTA were to be regulated by a threshold approach and carried out a dietary risk assessment of OTA applying a non‐threshold approach assuming that the compound is a genotoxic carcinogen (Kuiper‐Goodman et al., 2010). Using the data on decreases in TM (transport maximum) renal clearance expressed relative to body weight or to inulin clearance seen in a 90‐day pig study (Krogh et al., 1974, 1979), they have derived a BMD10 (a benchmark dose defined as a 10% increase over the background) of 1.56 μg OTA/kg bw per day). Applying a default UF of 10 for intra‐species variability and a default UF of 2.5 for toxicodynamic and a specific UF of 10 for toxicokinetic inter‐species variability (instead of the UF of 6 used by EFSA) and a factor of 2 for extrapolating from a subchronic study to a chronic health‐based guidance value (HBGV), a tolerable daily intake (TDI) of 3 ng/kg bw per day (which would correspond to a TWI of 21 ng/kg bw) was considered appropriate instead of the 17 ng/kg bw per day (corresponding to 120 ng/kg bw per week) as previously established by EFSA. For their risk assessment of OTA, Health Canada used a negligible cancer risk intake (NCRI) that was defined as the exposure associated with a cancer risk of 1:100,000. This is equivalent in units to a TDI derived using a tumourigenic dose 5% (TD05, a 5% increase in incidence over background in the observable dose response curve) of 27.4 μg/kg bw per day from kidney tumours seen in a 2‐year rat study. This value was corrected to 19.6 μg/kg bw per day considering that animals were dosed only 5 days per week (NTP, 1989). Applying a safety factor of 5,000 that results from dividing the TD05 by 5,000, which is equivalent to a 10−5 risk on a linear extrapolation to zero exposure, an NCRI of 4 ng OTA/kg bw per day resulted. Dietary exposures to OTA in a Canadian population were below the NCRI except for 1‐ to 4‐year‐old children.
In 2017, the UK Committee on Toxicity of Chemicals in Food Consumer Products and the Environment (COT) published a review of potential risks from OTA in the diet of infants aged 0–12 months and children aged 1–5 years (COT, 2018). The Committee evaluated all relevant in vivo studies published since the 2006 EFSA opinion and concluded that there is no new data available suggesting that the TWI of 120 ng/kg bw was established by EFSA in 2006, needs to be changed. Estimated exposure for breastfed infants ranged from 0.68 to 55 ng/kg bw per day for average consumers and from 1.02 to 82 ng/kg bw per day for high consumers. At low or average concentrations of OTA in breast milk exposures are below the TDI but at high concentrations infants estimated exposures exceeded the TWI by up to fivefold. The COT noted that such high exposures are unlikely to occur.
Mitchell et al. (2017) carried out a risk assessment of dietary OTA in the United States. For the risk characterisation, calculated exposures for different age groups in the ‘total population’ and ‘consumers’ (individuals reporting consumption of specific foodstuffs prone to contain OTA) were compared either with the JECFA PTWI of 100 ng/kg bw or the Health Canada NCRI of 4 ng/kg bw per day (for both HBGV values see text above in this section) and a margin of safety (MOS) approach was applied. Overall, MOS was > 1 in all age groups (both total population and consumers) applying either reference value. The only MOS ≤ 1 was seen when 95th percentile exposures of ‘consumers’ aged between 1 and 5 years were compared with the NCRI as established by Health Canada.
2.3. Legislation
Council Regulation (EEC) No 315/933 stipulates that food containing a contaminant in an amount unacceptable for public health shall not be placed on the market, that contaminant levels should be kept as low as can reasonably be achieved and that, if necessary, the European Commission may establish maximum levels for specific contaminants. These maximum levels are laid down in the Annex of Commission Regulation (EC) No 1881/20064 and may include limits for the same contaminants in different foods, analytical detection limits and reference to the sampling and analysis methods to be used. Maximum levels for OTA have been established, for instance, for unprocessed cereals, dried vine fruits (currants, raisins and sultanas), green coffee, roasted coffee beans, ground roasted coffee, soluble coffee, wine and grape juice and range from 0.5 to 10 μg/kg. Commission Recommendation (EC) 2006/576/EC5 on the presence of deoxynivalenol, zearalenone, OTA, T‐2 and HT‐2 toxin and fumonisins in products intended for animal feeding provides guidance levels for OTA for feed materials and complementary and complete feedingstuffs ranging from 0.05 to 0.25 mg/kg.
3. Data and methodologies
3.1. Collection and appraisal of data collected from public literature
Following the terms of reference, the EFSA opinion of 2006 has been used as a starting point for the present assessment. On 26 July 2018, a literature search has been carried out for studies published in the period from 2006 to 2018. The databases used were Web of Science6 and Pubmed7 and references retrieved were managed using Endnote.8 The precise search terms for the different areas used are listed in detail in Appendix A.
Upon screening of the abstracts and by applying expert judgement, for the field of ‘Mode of action’, 156 publications were considered as potentially relevant for the assessment. The corresponding figures for the field of ‘Toxicokinetics, were 37, for the field of ‘Toxicity’, they were 254 and for the field of ‘Human evidence and biomarkers’, they were 27. Studies dealing with compounds ameliorating the toxic effects of OTA have been included as potentially relevant. Studies on combined effects of OTA and other compounds have only been considered insofar they were potentially relevant for the assessment of OTA alone. Overall, the total number of publications considered as relevant in these fields was 451 and full‐text originals were retrieved. In addition to these original studies also the previous assessments on OTA of JECFA (FAO/WHO, 2008), Kuiper‐Goodman et al. (2010) and the UK COT (COT, 2018) and the studies referenced therein were considered for the present assessment. During the development of the opinion, additional publications were collected by applying a ‘snowballing approach’9 and considered for the assessment where relevant.
3.2. Occurrence data submitted to EFSA
3.2.1. Data collection and validation
Following an European Commission mandate to EFSA, a call for annual collection of chemical contaminant occurrence data in food and feed, including OTA, was issued by EFSA's DATA unit (the former EFSA Dietary and Chemical Monitoring Unit) in December 2010 with a closing date of 1 October of each year. European national authorities and similar bodies, research institutions, academia, food business operators and other stakeholders were invited to submit analytical data on OTA in food and feed.
The data submission to EFSA followed the requirements of the EFSA Guidance on Standard Sample Description (SSD) for Food and Feed (EFSA, 2010a). Occurrence data were managed following the EFSA standard operational procedures (SOPs) on ‘Data collection and validation’ and on ‘Data analysis of food consumption and occurrence data’.
Data on OTA in food available in the EFSA database from 2009 until the end of December 2018 were used for the present assessment. Data received after this date were not included in the present assessment.
3.2.2. Data analysis
The data received were evaluated by EFSA and special attention was paid to the identification of duplicates and to the accuracy of different parameters, such as ‘Analytical methods’, ‘Reporting unit’ and the coding of the different samples under FoodEx classification. Upon identification of potential inconsistencies, data providers were contacted to provide further clarification.
The left‐censored data (analytical data below the limit of detection (LOD) or limit of quantification (LOQ)) were treated by the substitution method as recommended in the ‘Principles and Methods for the Risk Assessment of Chemicals in Food’ (WHO/IPCS, 2009). The same method is described in the EFSA scientific report ‘Management of left‐censored data in dietary exposure assessment of chemical substances’ (EFSA, 2010b) as an option for the treatment of left‐censored data. The guidance suggests that the lower bound (LB) and upper bound (UB) approach should be used for chemicals likely to be present in the food. At the LB, results below the LOQ or LOD were replaced by zero; at the UB, the results below the LOD were replaced by the numerical values of the LOD and those below the LOQ were replaced by the value reported as LOQ.
The use of different cut‐off values on the reported LOQs was also evaluated in order to reduce the uncertainty associated with the exposure estimations (Annex, Tables 2 and 5).
For certain chemical contaminants such as mycotoxins, it is common to have a high percentage of left‐censored data in several food groups. Existing maximum levels (MLs) in Commission Regulation (EC) No 1881/20064 have been used in the corresponding food categories as cut‐off values for the LOQs so that results obtained with analytical methods with an LOQ higher than the MLs were not included. In addition, in those food groups which are important sources of dietary exposure, the distribution of the reported LOQ was assessed, and a cut‐off value was selected based on selected percentiles (see Table 12) to eliminate extreme high LOQs.
Table 12.
LOD/LOQ cut‐off values (μg/kg) applied for selected food categories
| Food category | Cut‐off (μg/kg) | Justification |
|---|---|---|
| Ad hoc cut‐off values | ||
| ‘Grains and grain‐based products' (covers the ML of 5 μg/kg for ‘Grains for human consumption’, ‘Grains as crops’, ‘Unprocessed cereals’ and the ML of 3 for ‘Bread and rolls’, ‘Breakfast cereals’, ‘Fine bakery wares’, ‘Pasta (Raw)’, ‘Grain milling products’, All products derived from unprocessed cereals, including processed cereal products and cereals intended for direct human consumption) | 1.5 | P75 of corresponding LOQs |
| ‘Fruit nectar’ | 2 | P95 of corresponding LOQs |
| ‘Mixed fruit juice’ | 2 | P95 of corresponding LOQs |
| ‘Ready‐to‐eat meal for infants and young children’ | 2 | P95 of corresponding LOQs |
| ‘Cereal‐based dishes’ | 2 | P75 of corresponding LOQs |
| ‘Juice, apple’ | 2 | P95 of corresponding LOQs |
| ‘Fruit compote’ | 2 | P75 of corresponding LOQs |
| ML‐based cut‐off values | ||
| Dried vine fruits (currants, raisins and sultanas) | 10 | n.a. |
| Coffee beans and coffee products (solid) | 5 | n.a. |
| Instant coffee, powder | 10 | n.a. |
| Wine (including sparkling wine, excluding liqueur wine and wine with an alcoholic strength of not less than 15% vol) and fruit wine | 2 | n.a. |
| Aromatised wine, aromatised wine‐based drinks and aromatised wine‐product cocktails | 2 | n.a. |
| Grape juice, concentrated grape juice as reconstituted, grape nectar, grape must and concentrated grape must as reconstituted | 2 | n.a. |
| Processed cereal‐based food for infants and young children | 0.5 | n.a. |
| Pepper, black and white (Piper nigrum), Nutmeg (Myristica fragans), Ginger (Zingiber officinale), Turmeric (Curcuma), Piper spp. | 15 | n.a. |
| Chilli pepper (Capsicum frutescens), Cayenne pepper (Capsicum frutescens), Paprika powder (Capsicum spp.) (dried fruits thereof, whole or ground, including chillies, chilli powder, cayenne and paprika) | 20 | n.a. |
| Mixtures of spices containing one of the above mentioned | 15 | n.a. |
| ‘Liquorice extract’ (flavouring) | 80 | n.a. |
| ‘Liquorice’ as root candies and liquorice for herbal infusion | 20 | n.a. |
ML; maximum level; LOQ: limit of quantification; n.a.: not applicable; P75: 75th percentile; P95: 95th percentile;
3.3. Food consumption data
The EFSA Comprehensive European Food Consumption Database (Comprehensive Database) provides a compilation of existing national information on food consumption at individual level and was first built in 2010 (EFSA, 2011a; Huybrechts et al., 2011; Merten et al., 2011). Details on how the Comprehensive Database is used are published in the Guidance of EFSA (EFSA, 2011a). The latest version of the Comprehensive Database, updated in 2018, contains results from a total of 60 different dietary surveys carried out in 25 different Member States covering 119,458 individuals.
Within the dietary studies, subjects are classified in different age classes as follows:
Infants: < 12 months old
Toddlers: ≥ 12 months to < 36 months old
Other children: ≥ 36 months to < 10 years old
Adolescents: ≥ 10 years to < 18 years old
Adults: ≥ 18 years to < 65 years old
Elderly: ≥ 65 years to < 75 years old
Very elderly: ≥ 75 years old
Four additional surveys provided information on specific population groups: ‘Pregnant women’ (≥ 15 years to ≤ 45 years old, Latvia; 17 years old to 46 years old, Portugal) and ‘Lactating women’ (≥ 28 years to ≤ 39 years old, Greece; 18 years old to 45 years old, Estonia).
For chronic exposure assessment, food consumption data were available from 53 different dietary surveys carried out in 22 different European countries. When for one particular country and age class two different dietary surveys were available, only the most recent one was used. This resulted in a total of 38 dietary surveys selected to estimate chronic dietary exposure.
In Table 4 of the Annex, these dietary surveys and the number of subjects available for the acute and chronic exposure assessment are described.
The food consumption data gathered by EFSA in the Comprehensive Database are the most complete and detailed data currently available in the EU. Consumption data were collected using single or repeated 24‐ or 48‐h dietary recalls or dietary records covering from 3 to 7 days per subject. Because of the differences in the methods used for data collection, direct country‐to‐country comparisons can be misleading.
3.4. Food classification
Consumption and occurrence data were classified according to the FoodEx classification system (EFSA, 2011b). FoodEx is a food classification system developed by EFSA in 2009 with the objective of simplifying the linkage between occurrence and food consumption data when assessing the exposure to hazardous substances. The system consists of a large number of individual food items aggregated into food groups and broader food categories in a hierarchical parent–child relationship. It contains 20 main food categories (first level), which are further divided into subgroups having 140 items at the second level, 1,261 items at the third level and reaching about 1,800 endpoints (food names or generic food names) at the fourth level.
3.5. Methodology for exposure assessment
The CONTAM Panel considered that only chronic dietary exposure had to be assessed. As suggested by the EFSA Working Group on Food Consumption and Exposure (EFSA, 2011c), dietary surveys with only one day per subject were not considered for chronic exposure as they are not adequate to assess repeated exposure. Similarly, subjects who participated only one day in the dietary studies, when the protocol prescribed more reporting days per individual, were also excluded for the chronic exposure assessment. Not all countries provided consumption information for all age groups, and in some cases the same country provided more than one consumption survey.
For calculating chronic dietary exposure to OTA, food consumption and body weight data at the individual level were accessed in the Comprehensive Database. Occurrence data and consumption data were linked at the relevant FoodEx level.
The mean and the high (95th percentile) chronic dietary exposures were calculated by combining mean occurrence values for each food collected in different countries (pooled European occurrence data) with the average daily consumption of each food at individual level in each dietary survey and age class. Consequently, individual average exposures per day and body weight were obtained for all individuals. Based on the distributions of individual exposures, the mean and 95th percentile exposures were calculated per survey and per age class. Dietary exposure was assessed using overall European LB and UB mean occurrence of OTA. All analyses were run using the SAS Statistical Software (SAS enterprise guide 7.15).
In the present assessment, occurrence levels above the MLs as laid down in Commission Regulation (EC) No 1881/2006 have been considered as the occurrence data sampled from products which are actually on the market. This is in accordance with the general principles of EFSA exposure assessments. To evaluate their impact on the overall assessment, an ad hoc scenario with the exclusion of non‐compliant samples was carried out which showed only a very minor impact on the exposure results (see Section 4.1.1.2).
3.6. Methodology for risk characterisation
The CONTAM Panel applied the general principles of the risk assessment process for chemicals in food as described by the WHO/IPCS (2009), which include hazard identification and characterisation, exposure assessment and risk characterisation. Additional to the principles described by the WHO/IPCS (2009), EFSA guidance pertaining to risk assessment has been applied for the present assessment. The EFSA guidance covers the procedures currently used within EFSA for the assessment of dietary exposure to different chemical substances and the uncertainties arising from such assessments. EFSA guidance documents applied in the present opinion are the guidances on approaches for the assessment for substances that are both genotoxic and carcinogenic (EFSA, 2005; EFSA Scientific Committee, 2012b), uncertainties in dietary exposure assessment (EFSA, 2007), transparency in scientific aspects of risk assessments (EFSA, 2009), standard sample description for food and feed (EFSA, 2010a), management of left‐censored data in dietary exposure assessments (EFSA, 2010b), use of the EFSA comprehensive food consumption database in intakes assessment (EFSA, 2011a), procedures used for dietary exposure assessment (EFSA, 2011b), evaluation of FoodEx (EFSA, 2011c), genotoxicity testing (EFSA Scientific Committee, 2011), selected default values to be used in the absence of data (EFSA Scientific Committee, 2012a), risk assessment terminology (EFSA Scientific Committee, 2012b) and on the use of the benchmark dose approach (EFSA Scientific Committee, 2017).
4. Assessment
4.1. Hazard identification and characterisation
4.1.1. Toxicokinetics
In 2006, EFSA characterised the toxicokinetics of OTA as follows: OTA is rapidly absorbed after oral ingestion with a bioavailability ranging from 40% to 66% depending on the species and the dose. In the systemic circulation, OTA is extensively (up to 99.98% in humans) bound to albumin and other serum proteins, which gives rise to long elimination half‐lifes in blood (3–10 days in various rat strains, 5–6 days in pigs, 19–21 days in non‐human primates and up to 35 days in one human individual). In rodents, the major route of excretion is via bile and faeces, whereas renal excretion prevails in other species including monkeys and humans. Although a few hydroxylated and conjugated metabolites of OTA have been described in vitro and in vivo, biotransformation of OTA in general appears to be low and restricted mostly to hydrolysis of the amide bond leading to OTalpha in vivo. A striking feature observed in in vivo animal studies is the accumulation of OTA in kidneys, associated with marked differences between species and sexes. Both organic anion transporters (OAT10) and members of the ATP‐dependent transporter family appear to be involved in the uptake of OTA into renal cells and its efflux (EFSA, 2006).
Toxicokinetic studies on OTA conducted more recently, i.e. after 2006, have confirmed the earlier observations. The following chapters will discuss the toxicokinetics of OTA in more detail with an emphasis on recently published data. For recent reviews, see Ringot et al. (2006), Vettorazzi et al. (2014) and Köszegi and Poor (2016).
4.1.1.1. Absorption
After oral ingestion, OTA is rapidly absorbed in several animal species, reaching peak blood levels within a few hours. For example, in a recent study in male SD rats using LC‐MS technology and 13C‐labelled OTA as internal standard, a peak concentration of 1.9 μg OTA/mL blood plasma was reached 4.8 h after oral gavage of a single dose of 0.2 mg OTA/kg bw, followed by a decrease with a half‐life of 76 h (Han et al., 2013a). Systemic availability via passive diffusion from the stomach and particularly from the proximal jejunum is believed to be greatly facilitated by the high binding affinity of OTA to plasma proteins (see Section 4.1.1.2 on Distribution). Absorption from the jejunum can take place against a concentration gradient and appears to involve a transport by OAT in addition to passive diffusion. The bioavailability of OTA is in the range of 50%, e.g. 66% in pigs, 56% in rats and rabbits and 40% in chicken in the study by Galtier et al. (1981) and depends mainly on the species, dose, vehicle and presence of food in the stomach at the time of OTA administration. In a male adult volunteer with an empty stomach ingesting tritium‐labelled OTA at a dose of 0.02 nmol/kg bw, 93% of the administered radioactivity was found in the blood within 8 h of ingestion, with 4–10% of the radioactivity in the blood located in the erythrocytes and over 90% in the plasma (Studer‐Rohr et al., 2000).
4.1.1.2. Distribution
A major characteristic of OTA in humans and many animal species is its very high non‐covalent binding to serum proteins, particularly to albumin. After an oral dose of 50 ng OTA/g bw, only 0.02% of the OTA in blood remained unbound in humans, 0.08% in monkeys, 0.1% in mice and pigs, but 22% in fish (Hagelberg et al., 1989). The high binding to serum proteins facilitates absorption of OTA but delays its elimination from blood and its excretion and explains the long half‐life of OTA in the body. After a single oral dose of tritium‐labelled OTA, a fast elimination from plasma with a half‐life of about 20 h was observed in humans during the first 6 days, followed by a much slower elimination phase with a half‐life of 35 days and a renal clearance of about 0.1 mL/min (Studer‐Rohr et al., 2000). Mantle (2008) reported considerable strain differences in the plasma half‐life of OTA, ranging from 8 to 10 days in Fischer rats to 2–3 days in Dark Agouti rats. The influence of the plasma protein binding of OTA is illustrated by the observation that the half‐life of OTA in albumin‐deficient rats was much shorter than in normal rats and that the concentration of OTA in bile and urine was 20‐ to 70‐fold higher in the deficient animals than in control rats (Kumagai, 1985). OTA interacts with albumin as a dianion, and two binding sites for OTA were identified on human serum albumin (Perry et al., 2003).
The differences between species in the binding of OTA to plasma albumin have a profound impact on the proportion of free (unbound) OTA in plasma. For instance, using the different binding constants for Wistar rats (4.0 × 104 M−1), pigs (7.1 × 104 M−1) and humans (5.2 × 106 M−1) as reported by Perry et al. (2003), and assuming the same total plasma concentration of 10 nM OTA (bound plus unbound OTA), the concentration of free OTA in plasma is 400 pM in rats, 230 pM in pigs and 3.2 pM in humans. For the calculation of these values, see Appendix D, and for their consideration in deriving uncertainty factors, see Section 4.6 on Establishment of a HBGV vs. application of an MOE approach.
OTA is distributed into every organ, although the concentrations of the toxin appear to depend on the animal species, the dose administered and the design of the kinetic study. Many studies report the highest concentrations in the kidney, followed by liver and muscle. After removal of residual blood from organs, Zepnik et al. (2003) found 40‐, 100‐, and 270‐fold higher concentrations of OTA in rat kidney than in rat liver at 24 h, 48 h and 72 h after a single oral dose of 0.5 mg OTA/kg bw. In the study by Han et al. (2013a), the order of tissues with the highest OTA concentrations were kidney > lung > liver = heart > spleen > brain after a single oral dose of 0.2 mg OTA/kg bw; all tissue levels peaked at 4 h after dosing. After repeated doses, levels in kidney and liver tend to be more similar. In addition, redistribution and deposition of OTA in lipid‐rich organs may occur. For example, when OTA was orally administered to male Sprague‐Dawley rats at a daily dose of 0.1 mg/kg bw for 20 days, the concentration of OTA was somewhat higher in the lung (96 ± 14 ng/g wet weight) than in the liver (76 ± 10), followed by heart (62 ± 4), kidney 56 ± 5), spleen (25 ± 2) and brain (4 ± 1) (Han et al., 2013a). The ratio of OTA levels in kidney to liver was about 2 after 4 h of a single dose and 0.7 after repeated dosing for 20 days (Han et al., 2013a). When male and female Dark Agouti rats were fed a diet containing four different concentrations of OTA (ranging from 2.5 to 100 μg per kg diet) for 28 days, the lung was found to contain the highest concentration of OTA, with slightly lower levels in the liver and about half the lung concentration in the kidney (Tozlovanu et al., 2012).
In order to determine placental transfer of OTA, the kinetics of tritium‐labelled OTA was studied in human perfused term placenta (Woo et al., 2012). Only minimal passage of OTA through the placenta was observed in this in vitro study and the fetal/maternal ratio of OTA at the end of the perfusions (up to 20 h) was only about 0.02. Further studies showed that inhibition of the placental efflux transporters ABCG2 and ABCC2 did not increase the transfer of OTA to the fetal circulation (Woo et al., 2012). However, several studies showed in utero transfer in mammalian species such as rat, mouse and swine (Ringot et al., 2006 and literature cited therein). Differences in fetal uptake of OTA were observed in mice after different durations of gestation, suggesting that the transfer is influenced by the developmental stage of the placenta. In individuals from Switzerland and Poland eating a normal diet, OTA concentrations in placenta and fetal serum were found to be about twice as high as in maternal serum, and the involvement of placental OATs have been proposed to explain this active placental transfer (Zimmerli and Dick, 1995; Postupolski et al., 2006).
OTA is also transferred to breast milk in animals and humans. The results of some animal studies have been reviewed by Ringot et al. (2006). A low milk/plasma ratio of 0.015 was observed with lactating rabbits fed a naturally contaminated diet, and the concentration of OTA in milk did not change throughout the lactation period. No OTA was detected in the milk of lactating sows on a naturally contaminated diet. In contrast, studies in lactating rats receiving a single oral dose of OTA or repeated doses over 8 weeks showed a milk to blood concentrations of OTA ranging between 0.4 and 0.7. The transfer of OTA into human milk is lower (0.2) and is discussed in detail in Section 4.1.1.4.1. Only trace amounts of OTA are commonly detected in the milk of cows, which is the major provider of dairy products in human nutrition; this is attributed to the efficient presystemic degradation of OTA by the ruminal microflora, as described in Section 4.1.1.6.
4.1.1.3. Metabolism
The recent report by Wu et al. (2011) reviews extensively the metabolism of OTA in various animal species, humans, plants and microorganisms. The established biotransformation pathways in animals and humans are presented in Figure 4. For references, see Wu et al. (2011). For details of studies on the biotransformation of OTA in in vitro systems, in animals in vivo and in humans see Appendix E, Tables E.1, E.2 and E.3, respectively.
Figure 4.
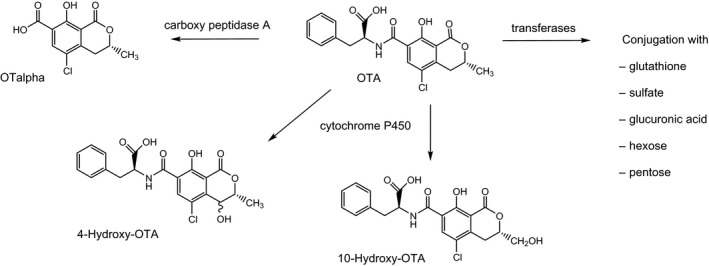
Metabolic pathways of OTA in animals and humans
Table E.1.
In vitro studies on biotransformation of OTA
| Experimental system | Species | Purity of OTA | Metabolites | Analytical method | Structure confirmation | Comment | Reference |
|---|---|---|---|---|---|---|---|
| Liver microsomes | Rabbit, human, pig | 4(R)‐HO‐OTA;4(S)‐HO‐OTA | TLC | MS, NMR | Enzyme kinetics (Vmax + KM) | Størmer et al. (1981) | |
| Liver microsomes +/− phenobarbital | Rabbit | Not specified | 4(R)‐HO‐OTA; 4(S)‐HO‐OTA; 10‐HO‐OTA |
TLC HPLC‐UV |
MS, NMR | Enzyme kinetics (Vmax + KM) | Størmer et al. (1983) |
| Kidney cells (Vero cells) | Monkey | Not specified | OTalpha; 4(R)‐HO‐OTA; 4(S)‐HO‐OTA; OTB | HPLC‐FLD | – | No kinetics | Grosse et al. (1995) |
| Kidney microsomes +/− phenobarbital | Rabbit | Not specified |
4(R)‐HO‐OTA; 4(S)‐HO‐OTA 10‐HO‐OTA; OTB; 3 unidentified metabolites |
HPCL‐FLD | No kinetics | El Adlouni et al. (2000) | |
| Bronchial epithelial cells +/− phenobarbital | Human | Not specified | OTalpha; 4(R)‐HO‐OTA; 4(S)‐HO‐OTA; 10‐HO‐OTA; OTB; 3 unidentified metabolites | ||||
| Liver microsomes +/− GSH and cytosol | Rat ♂♀ | [U‐3H]OTA (18 Ci/mmol) radiochemical purity > 98% | 4(R)‐HO‐OTA | HPLC‐radioactivity detector | MS, NMR | Enzyme kinetics (Vmax) | Gautier et al. (2001) |
| Liver microsomes +/− GSH and cytosol | Human | 4(R)‐HO‐OTA | No evidence of GSH conjugates | ||||
| Liver S‐9 +/− GSH | Rat | 4(R)‐HO‐OTA | No evidence of GSH conjugates | ||||
| Kidney microsomes +/− GSH and cytosol | Rat, Human | – | No evidence of GSH conjugates | ||||
| CYP1A1, 1A2, 2E1, 3A4 | Human | 4(R)‐HO‐OTA | |||||
| CYP1A2, 2C11 | Rat | – | |||||
| Seminal vesicle microsomes | Ram | Unidentified metabolite | |||||
| Seminal vesicle microsomes enriched in prostaglandin H‐synthase Horseradish peroxidase | – | ||||||
| Liver microsomes +/− inducers of CYPs | Rat, mice ♂♀ | > 99% (HPLC) | 4(R)‐HO‐OTA; 4(S)‐HO‐OTA | HPLC‐FLD | MS/MS | Enzyme kinetics (Vmax + KM) | Zepnik et al. (2001) |
| CYP1A2, 2C9‐1, 3A4 supersomes® a | Human | > 99% (HPLC) | 4(R)‐HO‐OTA; 4(S)‐HO‐OTA | MS/MS | Enzyme kinetics (Vmax) | ||
| Semipurified GSTs +/− liver and kidney cytosol | Rat | > 99% (HPLC) | 4(R)‐HO‐OTA; 4(S)‐HO‐OTA | MS/MS | Enzyme kinetics (Vmax) | ||
| Kidney microsomes or cytosol fortified with NADPH and GSH; Liver cytosol | Rat | > 99% (HPLC) | – | – | – | ||
|
Horseradish peroxidase/H2O2 Soybean lipoxygenase |
– | > 99% (HPLC) | – | – | – | ||
| Primary hepatocytes +/− induction by 3‐methylcholanthrene | Rat, human | 3H‐OTA (0.6 Ci/mmol) |
4(R)‐HO‐OTA; OTA‐hexose OTA‐pentose |
HPLC‐radioactivity detection/quantification system | MS/MS | No effect of β‐Gluc, sulfatase or GGT on metabolite retention times, suggesting that phase‐II‐conjugates were not formed | Gross‐Steinmeyer et al. (2002) |
| Liver microsomes | Rat | Not specified | 4‐HO‐OTA; OTHQ‐GSH | LC‐MS/MS | OTHQ‐GSH generated in less than 1% yield | Dai et al. (2002) | |
| Horseradish peroxidase/H2O2 | – | ||||||
| Fe2(NH4)2(SO4)2/H2O2 | OTHQ‐GSH | ||||||
| Kidney cells | Opossum | Not specified |
OTalpha; 4(R)‐HO‐OTA 4(S)‐HO‐OTA; OTB; OTHQ; OP‐OTA; Several unidentified metabolites |
HPLC‐FLD nano‐ESI‐IT‐MS |
No kinetics | Faucet‐Marquis et al. (2006) | |
| Liver microsomes | Rat, human, pig, cow, chicken, goat | 99% |
4(R)‐HO‐OTA; 4(S)‐HO‐OTA Hydroxylated metabolites tentatively identified as 9′‐HO‐OTA: 7′‐HO‐OTA, 5′‐HO‐OTA; OTB |
UPLC‐Q/TOF‐MS |
No enzyme kinetics No OTA‐derived glucuronides detected |
Yang et al. (2015) |
GGT:γ‐glutamyltranspeptidase; β‐Gluc:β‐glucuronidase; GSH: glutathione; HO‐OTA: hydroxy ochratoxin; HPLC‐FLD: high‐performance liquid chromatography with fluorescence detection; KM:Michaelis constant, concentration of substrate which permits the enzyme to achieve half Vmax; NADPH: nicotinamide adeninedinucleotide phosphate; OTA: ochratoxin A; OTalpha: ochratoxin alpha; OTB: ochratoxin B; OTHQ: ochratoxin hydroquinone; LC‐MS/MS: liquid chromatography‐mass spectrometry and tandem mass spectrometry; MS: mass spectroscopy; nano‐ESI‐IT‐MS: TLC: thin layer chromatography; TLC HPLC‐UV: thin layer chromatography high‐performance liquid chromatography with ultraviolet detection; UPLC‐Q/TOF‐MS: ultra‐high‐performance liquid chromatography‐quadrupole time‐of‐flight mass spectrometry; Vmax: maximum velocity of an enzymatic reaction.
Corning Supersomes® enzymes are recombinantly expressed drug metabolising enzyme reagents, consisting of microsomes prepared from insect cells infected with a virus engineered to express a CYP isoform.
Table E.2.
In vivo studies on biotransformation of OTA in rats
| Strain/sex | Treatment regimen | Purity of OTA | Specimen analysed | Metabolites | Analytical method | Comment | Reference |
|---|---|---|---|---|---|---|---|
| Wistar rats ♂ | 15 mg/kg bw; single oral dose; 14C‐OTA labelled inisocoumarin moiety | Not specified | Urine, faeces | OTalpha | TLC‐ Liquid scintillation counting | Suzuki et al. (1977) | |
| Sprague Dawley rats ♂ | 2.7 mg/kg bw; single i.v. dose; 14C‐OTA uniformly labelled | 96% radio‐chemical purity (TLC) | Urine, faeces | OTalpha; 5 unidentified metabolites | TLC‐Autoradiography | Galtier et al. (1979) | |
| Albino rats ♂ | 6.6 mg/kg bw; single oral or i.p. dose | Not specified | Urine, faeces | OTalpha; 4(R)‐HO‐OTA | TLC‐UV | Confirmation of OTalpha by MS | Støren et al. (1982) |
| Sprague Dawley rats ♂ | 4 mg/kg bw; single i.p. dose | Not specified | Urine | OTalpha; 4(R)‐HO‐OTA; 2 unidentified metabolites | HPLC‐FLD | OTB and OTB‐derived metabolites not detected | Xiao et al. (1996) |
| Dark agouti rats ♀; Lewis rats ♀ | 0.5, 2.5, 5 mg/kg bw single dose; 1.5 mg/kg per day repeated oral dose for 8 weeks (5 days/week) | Not specified | Urine | 4(R)‐HO‐OTA | HPLC‐FLD | Castegnaro et al. (1989) | |
| Sprague Dawley rats ♀ | ~ 333 μg/kg bw single i.v. dose | Not specified | Urine, bile | OP‐OTA; 2 unidentified metabolites | HPLC‐FLD | Li et al. (2000) | |
| F344 rats ♂ | 1 mg/kg bw; single oral dose 40‐55 μCi of [3H]OTA uniformly labelled | > 98% radiochemically pure | Urine, faeces | OTalpha; 2 unidentified metabolites | HPLC‐radioactive detector | Unknown metabolites did not correspond to OP‐OTA, 4(R)‐OH‐OTA or OTB | Gautier et al. (2001) |
| F344 rats ♀♂ | 0.5 mg/kg bw; single oral dose | 99,99% (HPLC‐FLD) | Liver, kidney plasma, urine, faeces | OTalpha; OTA‐hexose OTA‐pentose | HPLC‐FLD; LC‐MS/MS | 4(R,S)‐OH‐OTA, 10‐OH‐OTA, OTB, OTHQ and OTA derived GSH conjugates and glucuronides not detected | Zepnik et al. (2003) |
| F344 rats ♂ | 2 mg/kg bw per day; repeated oral dose for 2 weeks (5 days/week) | 99% (HPLC‐FLD); Impurities: OTB (LC‐MS/MS) | Liver, kidney plasma, urine, faeces | OTalpha; OTA‐hexose; OTA‐pentose | LC‐MS/MS | Traces of OTB and a hydroxylated metabolite (presumably 4‐OH‐OTB (Mally et al., 2005c)) initially suggested to present OTHQ detected in urine. Authors consider this due to OTB present as impurity in OTA used to treat animals | Mally et al. (2004), personal communication, Angela Mally, University of Würzburg |
| Dark agouti rats ♀♂ | 100 μg/kg diet | Not specified | Lung, liver, kidney | OTalpha; OTB‐GSH OTHQ‐GSH | HPLC‐FLD | Tozlovanu et al. (2012) | |
| Sprague‐Dawley rats ♂ | 0.2 mg/kg bw; single oral dose | Not specified | Plasma, liver, heart, spleen, lung, kidney, brain | OTbeta, phenylalanine, OTB‐methyl ester | LC‐MS/MS; LC‐TOF‐MS | OTalpha not detected; OTB not detected; OTA used to treat animals did not contain OTbeta or phenylalanine, presence of OTB not specified | Han et al. (2013a) |
| Wistar rats ♀♂ | 5 mg/kg bw; single oral dose | 99% |
4(R)‐HO‐OTA,4(S)‐HO‐OTA; Hydroxylated metabolites tentatively identified as 9′‐HO‐OTA, 7′‐HO‐OTA, 5′‐HO‐OTA OTB |
UPLC‐Q/TOF‐MS |
No OTA‐derived glucuronides detected No synthetic reference compounds to support the chemical structure of metabolites |
(Yang et al., 2015) |
GSH: glutathione; HO‐OTA: hydroxy ochratoxin; HPLC‐FLD: high‐performance liquid chromatography with fluorescence detection; OTA: ochratoxin A; OTalpha: ochratoxin alpha; OTB: ochratoxin B; OTbeta: ochratoxin beta; OTHQ: ochratoxin hydroquinone; LC‐MS/MS: liquid chromatography‐mass spectrometry and tandem mass spectrometry; MS: mass spectroscopy; nano‐ESI‐IT‐MS: TLC: thin layer chromatography; TLC HPLC‐UV: thin layer chromatography high‐performance liquid chromatography with ultraviolet detection; UPLC‐Q/TOF‐MS: ultra‐high‐performance liquid chromatography‐quadrupole time‐of‐flight mass spectrometry; Vmax: maximum velocity of an enzymatic reaction.
Table E.3.
Studies on biotransformation products of OTA in humans
| Human | OTA exposure | Specimen analysed | Metabolites | Analytical method | Comment | Reference |
|---|---|---|---|---|---|---|
| Families affected by Balkan endemic nephropathy ♀♂ | Unknown | Urine | HPLC‐FLD | OTA present; 4‐OH‐OTA not detected | Castegnaro et al. (1991) | |
| Human volunteer ♂ | 395 ng 3H‐OTA, uniformly labelled (3.8 μCi); radiochemical purity > 98% | Urine, plasma | 42–54% of radioactivity in urine coeluted with OTA standard 14–20% of radioactivity in urine suggested to present metabolite (s) of smaller size or higher polarity that OTA | HPLC‐FLD; Liquid scintillation counting of 2‐min HPLC fractions | Studer‐Rohr et al. (2000) | |
| Volunteers from Portugal ♀♂ | Unknown | Urine | Indirect evidence for OTA‐derived glucuronide | HPLC‐FLD +/− enzymatic cleavage of conjugates by β‐Gluc | Structure of OTA‐glucuronide not identified | Pena et al. (2006) |
| Healthy volunteers from Germany ♀♂ | Unknown | Urine, plasma | OTalpha; Indirect evidence for OTalpha‐conjugates | HPLC‐FLD +/− enzymatic cleavage of conjugates by β‐Gluc/ArylS |
OTA glucuronide not detected Structure of OTalpha‐conjugates not identified (e.g. OTalpha‐glucuronide or OTalpha‐sulfate) |
Muñoz et al. (2010) |
| Volunteers from Spain ♀♂ | Unknown | Urine | OTalpha | HPLC‐FLD | Coronel et al. (2011) | |
| Pregnant women from Croatia | Unknown | Urine | OTalpha; indirect evidence for OTalpha‐conjugates | HPLC‐FLD +/− enzymatic cleavage of conjugates by β‐Gluc/ArylS | Structure of OTalpha‐conjugates not identified (e.g. OTalpha‐glucuronide or OTalpha‐sulfate) | Klapec et al. (2012) |
| Breastfed infants from Germany and Turkey; German adults ♀♂ | Unknown | Urine | Indirect evidence for OTA‐conjugates in Turkish infants and German adults | HPLC‐LC‐MS/MS +/− enzymatic cleavage of conjugates by β‐Gluc/ArylS | Proposed to represent OTA‐8‐β‐glucuronide but structure of OTA‐derived conjugates in urine needs yet to be confirmed | Muñoz et al. (2017) |
| German adults ♀♂ | Unknown | Urine | OTB‐NAC | LC‐MS/MS | Sueck et al. (2019) |
ArylS; Aryl synthetase; β‐Gluc: β‐glucuronidase; HO‐OTA: hydroxy ochratoxin; HPLC‐FLD: high‐performance liquid chromatography with fluorescence detection; HPLC‐LC‐MS/MS: high‐performance: liquid chromatography liquid chromatography tandem mass spectroscopy; OTA: ochratoxin A; OTalpha: ochratoxin alpha; OTB: ochratoxin B; OTB‐NAC:OTB N‐acetyl‐L‐cysteine; OTHQ: ochratoxin hydroquinone; LC‐MS/MS: liquid chromatography‐mass spectrometry and tandem mass spectrometry; MS: mass spectroscopy; TLC HPLC‐UV: thin layer chromatography high‐performance liquid chromatography with ultraviolet detection; UPLC‐Q/TOF‐MS: ultra‐high‐performance liquid chromatography‐quadrupole time‐of‐flight mass spectrometry.
The major OTA metabolite is OTalpha, which is formed when the amide bond between phenylalanine and dihydroisocoumaric acid is hydrolysed. OTalpha has first been found in the caecum and large intestine of rats and in the excreted faeces after oral administration of OTA. It is believed to be mostly generated by the intestinal microbiome in non‐ruminants including humans (Ali et al., 2017) and by the rumen microbiome in cows, sheep and other ruminants. The degradation of OTA to OTalpha is catalysed by numerous hydrolases, among which carboxypeptidase A appears to be particularly active. The formation of OTalpha is considered as an important detoxification pathway of OTA. Although partly absorbed from the intestine, OTalpha does not accumulate in the kidney but is rapidly excreted with the urine, also as a glucuronide. In humans, OTalpha‐glucuronide is a major urinary metabolite.
Cytochrome P450‐mediated hydroxylation, a common phase I metabolic reaction of many lipophilic compounds, has also been demonstrated for OTA. The respective metabolites are 4‐hydroxy‐OTA and 10‐hydroxy‐OTA. Because of the chirality of the C‐4 atom of 4‐hydroxy‐OTA, two stereoisomers (4R and 4S) are formed. These metabolites are consistently formed in microsomal incubations and in cell cultures in vitro (see Appendix E, Table E.1), and have also been detected in some studies at very low levels in urine of rats treated with OTA (see Appendix E, Table E.1). Overall, in vitro and in vivo studies on OTA biotransformation show that hydroxylation of the isocoumarin moiety plays only a minor role in OTA metabolism.
In addition to these well‐established hydrolytic and oxidative pathways of OTA, several other phase I and phase II biotransformation pathways have been proposed. These include i) hydroxylation of OTA at the phenylalanine moiety, ii) opening of the lactone ring, iii) dechlorination of OTA leading to OTB, a hydroquinone (OTHQ)/quinone (OTQ) redox couple and corresponding glutathione‐S (GSH)‐conjugates, and iv) conjugation with a hexose and pentose, glucuronic acid and sulfate. These pathways are discussed in more detail below.
4.1.1.3.1. Hydroxylation at the phenylalanine moiety
Besides hydroxylation of the isocoumarin moiety of OTA, a recent study suggests that OTA may also be hydroxylated at the phenylalanine moiety. Using LC‐MS/MS analysis of liver microsomal incubations and urine and faeces samples obtained from rat and chickens exposed to OTA, Yang et al. (2015) detected three further hydroxylated metabolites in addition to 4(R)‐ and 4(S)‐hydroxy‐OTA and tentatively identified these as 9′‐hydroxy‐OTA, 7′‐hydroxy‐OTA, and 5′‐hydroxy‐OTA based on mass spectral data. Synthetic reference compounds to support the chemical structure of the proposed metabolites and to allow quantitative analysis were not available.
4.1.1.3.2. Lactone ring opening
A metabolite with an open lactone ring (OP‐OTA) has been detected in urine and bile of rats dosed with OTA (Xiao et al., 1996; Li et al., 2000). The high toxicity of this metabolite may be due to its ability to undergo reversion of lactone ring opening, which would lead to OTA.
4.1.1.3.3. Dechlorination of OTA and conjugation with GSH
Electrochemical and photochemical reaction of OTA has been shown to generate an OTHQ/OTQ redox couple (Figure 5) that may react with tissue nucleophiles (Calcutt et al., 2001; Dai et al., 2002). This reaction involves loss of the chlorine atom and presumably proceeds via an aryl radical, which may also form OTB (Manderville, 2005; Pfohl‐Leszkowicz and Manderville, 2012). Whether dechlorination of OTA leading to formation of OTB, OTHQ and OTQ and associated GSH‐conjugates can also occur under physiological conditions is of key interest as this may indicate formation of reactive metabolic intermediates and may thus have a bearing on the question of OTA genotoxicity. OTB has been repeatedly reported as a minor metabolite of OTA (usually in the range of 1% of OTA) in vitro (El Adlouni et al., 2000; Faucet‐Marquis et al., 2006; Yang et al., 2015) (see Appendix E, Table E.1) and in vivo (Mally et al., 2004; Yang et al., 2015) (see Appendix E, Table E.2). However, because the OTA used in metabolic studies is invariably of fungal origin and only very rarely analysed for OTB, it is difficult to discriminate the OTB introduced as contaminant of OTA from OTB potentially arising from the metabolism of OTA. Han et al. (2013b), when studying the microsomal glucuronidation of OTA in vitro, detected OTB both in active and in control incubations in similar amounts as found in the standard solution of OTA and concluded that the OTB was the impurity from the standard and not formed as a metabolite. LC‐MS/MS analyses of OTA from a commercial source and another laboratory showed the presence of about 1% OTB (personal communication, Angela Mally, University of Würzburg). A metabolite suggested by the authors to be OTHQ was detected in kidney cells in vitro (Faucet‐Marquis et al., 2006) and in rat urine (Mally et al., 2004). Both studies also reported the presence of OTB. Since 4‐hydroxy‐OTB, which is a metabolite of OTB, differs from OTHQ only in the position of the hydroxyl group and has the same mass, mass fragmentation pattern and similar chromatographic conditions as OTHQ (Mally et al., 2005c), it is possible that 4‐hydroxy‐OTB may be mistaken for OTHQ. In contrast to 4‐hydroxy‐OTB, OTHQ is unstable in the presence of oxygen and is easily oxidised to OTQ, which itself decomposes in aqueous solution to several products (Gillman et al., 1999) and must be expected to react with nucleophilic components of biological materials, e.g. GSH. OTQ itself has not been found to date in metabolic studies in vivo or in vitro.
Figure 5.
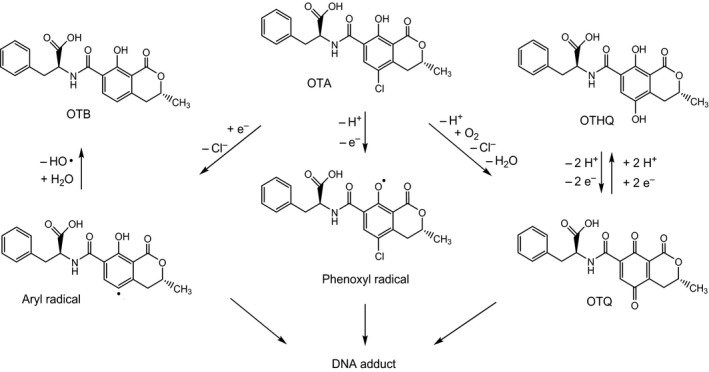
Hypothetical reactive metabolites of OTA and their products
The proposed reactive metabolites include (1) an aryl radical formed from OTA by reductive dechlorination, (2) a phenoxyl radical formed from OTA by one‐electron oxidation and (3) OTQ formed from OTA by oxidation mediated by peroxidases or cytochrome P450.
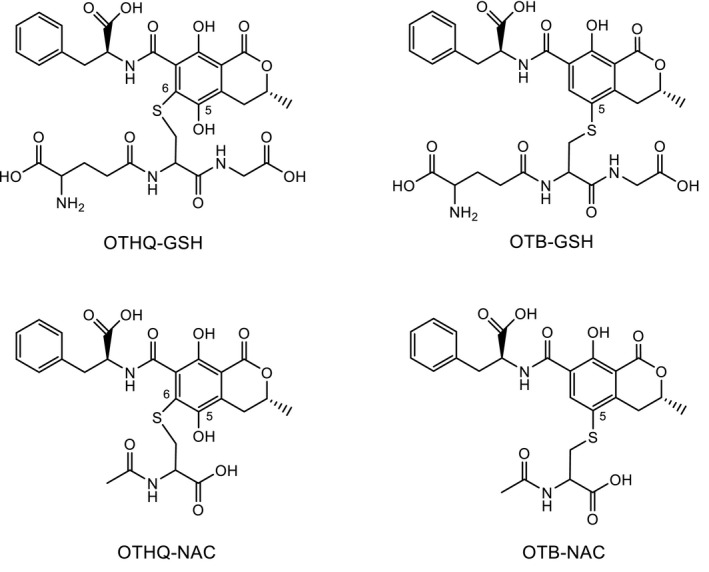
Dai et al. (2002) first reported that OTHQ photoreacted with GSH to a product identified by mass spectrometry (MS) and nuclear magnetic resonance (NMR) spectroscopy as OTHQ with a covalently bonded GSH at C‐6 (Figure 6); this OTHQ‐GSH conjugate was formed in about 1% yield in incubations of OTA with rat liver microsomes/NADPH or horseradish peroxidase/H2O2 or free Fe2+. While other reports found no evidence for formation of GSH‐conjugate in vitro (Gautier et al., 2001; Gross‐Steinmeyer et al., 2002) or in vivo (Gautier et al., 2001; Zepnik et al., 2003). Tozlovanu et al. (2012) reported the detection of OTHQ‐GSH in livers and kidneys of male and female Dark Agouti rats dosed with 100 μg/kg bw per day for 28 days. In the same studies, another GSH conjugate was detected which carries the GSH at C‐5 of OTA (Figure 6), thus replacing the chlorine atom and therefore named OTB‐GSH; the amount of OTB‐GSH exceeded that of OTHQ, particularly in the kidney of male rats. GSH conjugates are degraded to their corresponding N‐acetyl‐L‐cysteine (NAC) conjugates (also termed mercapturic acids) in the kidney prior to renal excretion. In a very recent study using LC‐MS/MS analysis and authentic reference compounds, OTB‐NAC (Figure 6) has been reported to occur in the urine of 11 out of 18 male and female volunteers (Sueck et al., 2020). No correlation between the urinary concentration of OTB‐NAC and OTA was observed.
Figure 6.
Conjugates of OTA metabolites with GSH and their corresponding mercapturic acids
4.1.1.3.4. Further OTA‐derived conjugates
The formation of glucuronic acid and sulfate conjugates in OTA metabolism has so far been concluded from the fact that more OTA could be detected after treatment with ß‐glucuronidase and aryl sulfatase. In a more recent in vitro study with rat liver microsomes, small amounts of all three possible glucuronides of OTA, i.e. with the glucuronic acid located at the phenolic hydroxyl group (C‐8), the carboxyl group (C‐1’) and the amide group, were tentatively identified by mass spectrometry (Han et al., 2013b). Moreover, OTA‐methyl ester, OTalpha and OTalpha glucuronide were detected in this study.
OTA conjugates with a hexose and a pentose have been identified in incubations of OTA with rat and human hepatocytes (Gross‐Steinmeyer et al., 2002) and in the urine of OTA‐treated rats (Zepnik et al., 2003) based on evidence from LC‐MS/MS, but the exact chemical structures are yet unknown.
4.1.1.3.5. Summary of OTA metabolism
With the exception of hydrolysis of the peptidic bond of OTA leading to OTalpha (which occurs mainly in the intestine), all the described biotransformation reactions appear to represent minor pathways in the mammalian metabolism of OTA. For example, when rats were administered a single low dose of OTA and urine, faeces, blood, liver and kidney were analysed by a sensitive LC‐MS/MS method, OTalpha mainly as a glucuronic acid conjugate was detected as the major OTA metabolite in urine and faeces, together with low concentrations of OTA‐hexose and OTA‐pentose in urine (Zepnik et al., 2003). OTalpha was also detected in blood but not in liver and kidney. 4‐ and 10‐Hydroxy‐OTA, lactone ring open OTA, glutathione and glucuronide conjugates were not present in urine, blood, liver or kidney in detectable concentrations (Zepnik et al., 2003). These data are in good agreement with detection of OTalpha, 4(R)‐OH‐OTA and two unidentified metabolites in urine of male rats administered uniformly labelled [3H]OTA (Gautier et al., 2001). The unidentified metabolites did not correspond to OP‐OTA, 4(R)‐hydroxy‐OTA or OTB and may be identical to the OTA‐hexose‐ and pentose derivatives as judged by their chromatographic behaviour.
In another study with tritium‐labelled OTA in a male human, > 80% of the radioactivity in blood was unchanged OTA and no metabolites were observed. In urine, about 50% was OTA and the rest was probably metabolites, which however were not further characterised (Studer‐Rohr et al., 2000).
Overall, the rate of in vivo biotransformation of OTA in non‐ruminant animals and in humans appears to be low.
A critical and much disputed question is whether OTA can be activated to electrophilic metabolites capable of covalent binding to DNA and other cellular macromolecules. The potential formation of such genotoxic metabolites is highly relevant for risk assessment of this carcinogenic mycotoxin. Several pathways have been proposed for the metabolic activation of OTA (Manderville, 2005; Manderville and Wetmore, 2017) as depicted in Figure 5. However, formation of reactive metabolites capable of covalent binding to DNA, such as those demonstrated in photochemical and iron‐containing systems, appears to be very minor or absent under physiological conditions. (Appendix E, Tables E.1 and E.2). For example, in vitro studies by Gautier et al. (2001) showed the formation of 4R‐hydroxy‐OTA upon incubation of OTA with rat or human liver microsomes, but no evidence for oxidative metabolites was found with rat, mouse or human kidney microsomes or postmitochondrial supernatants, several recombinant human cytochrome P450s, prostaglandin H synthase or horseradish peroxidase.
4.1.1.4. Excretion
OTA and its metabolites are excreted with the urine and the faeces. The urinary route appears to predominate in humans and non‐human primates, while biliary and faecal excretion appears to be preferred in rats (Dietrich et al., 2005). Both routes are slow, owing to the high binding of OTA to plasma proteins and the low rate of metabolism. Studer‐Rohr et al. (2000) reported that about 3% of a single oral dose of tritium‐labelled OTA was excreted with the daily urine by a male human during the first 6 days, with a total urinary excretion of 62% of the dose after 75 days. Studies in rats and mice have demonstrated that OTA and some of its metabolites are prone to enterohepatic circulation, and Fuchs and Hult (1992) have proposed that differences in the extent of enterohepatic circulation of OTA might, in part, be responsible for the differences in plasma half‐life between the species.
Renal excretion of OTA is of particular interest because the kidney is the major target organ of OTA toxicity. Because of its high plasma protein binding, OTA undergoes tubular secretion rather than glomerular filtration, followed by reabsorption at all segments of the nephron (Ringot et al., 2006). According to Jutabha et al. (2011), the transport of OTA into the proximal tubule cells of human kidneys is mediated by OAT1 and 3 from the basolateral (blood) side and by OAT4 from the apical (proximal tubule) side, whereas multidrug‐resistant protein (MRP) 2 and efflux transporter NPT4 mediate the secretion from the proximal tubule cell into the tubule. Because OAT1, 3 and 4 are more efficient than MRP2 and NPT4, high intracellular concentrations of OTA are reached (Jutabha et al., 2011). The reabsorption of secreted and filtered OTA not only leads to its accumulation in the renal tissue but also delays its excretion.
4.1.1.4.1. Transfer/excretion to human milk
Several studies have reported the detection of OTA in human milk, and a correlation between the OTA concentration in milk and the dietary intake has been shown (Skaug et al., 2001). Studies from European countries report levels ranging between 10 and 400 ng OTA/L (Degen et al., 2013). According to the literature, OTA levels are significantly higher in milk of habitual consumers of bread, bakery products, cured pork meat and sweets. In addition to OTA, the occurrence of OTalpha was also monitored in milk and blood from nine lactating women (Muñoz et al., 2010). The concentration levels of unconjugated OTalpha in breast milk were significantly lower than those observed for OTA (40 ± 30 ng/L vs. 106 ± 45 ng/L). However, OTalpha concentrations were almost eight times higher (840 ± 256 ng/L) after an enzymatic treatment of breast milk with ß‐glucuronidase/sulfatase, while the OTA concentration remained the same. This was an indication of the occurrence of OTalpha conjugates in milk. The lactational transfer of OTA was later reported by the same group by comparing 21 mother–child pairs with parallel collection of maternal blood, milk and of infant urine samples over a period of up to 6 months (Muñoz et al., 2014). OTA was detected in almost all maternal blood plasma samples, at concentrations ranging between 72 and 639 ng/L. An average milk/plasma (M/P) ratio of 0.25 was reported. The authors observed that a higher fraction of circulating OTA was excreted in colostrum (M/P 0.4) than with mature milk (M/P ≤ 0.2). However, the M/P ratios varied substantially between individuals. OTA concentrations in infant urine correlated with OTA levels in concurrently collected milk samples.
4.1.1.5. Summary of toxicokinetics
OTA is a cumulative mycotoxin with relatively rapid absorption and distribution but slow elimination and excretion, which is mostly due to its high extent of binding to plasma proteins in particular albumin and its low rate of metabolism. Both faecal and urinary excretion are important for the plasma clearance of OTA. Plasma half‐life range from several days in rodents and pigs to several weeks in humans and non‐human primates. The major metabolic pathway of OTA is its hydrolysis to OTalpha, which is particularly pronounced in ruminants, and subsequent glucuronic acid conjugation while oxidative metabolism appears to be of minor importance. Likewise, formation of reactive metabolites capable of covalent binding to DNA, although demonstrated in photochemical and iron‐containing systems, appears to be very minor or absent under in vivo conditions.
4.1.1.6. Transfer to food of animal origin
In addition to food derived from OTA‐contaminated plant products, e.g. coffee, beer and wine, some animal‐derived food items may contain significant levels of OTA. Because OTA is frequently present in the feed of food‐producing animals and has a high bioavailability and long half‐life in some monogastric farm animals such as pigs, preruminant calves and rabbits, OTA may accumulate in the meat and organs of such animals (Battacone et al., 2010; Duarte et al., 2011).
Poultry species appear to eliminate OTA faster than monogastric mammalian species, resulting in a low OTA accumulation in the blood and tissues (Galtier et al., 1981; Schiavone et al., 2008). Likewise, transfer of OTA into eggs appears to occur only when OTA intake is very high. For example, no OTA could be detected in eggs when hens were fed diets containing 0.3 or 1 mg OTA/kg of feed, but OTA was found in eggs of laying hens fed OTA at 10 mg/kg bw (cited in Battacone et al., 2010). Bozzo et al. (2011) did not detect OTA in the eggs of laying hens fed a diet containing 2 mg OTA/kg of feed.
In contrast to monogastric animals, ruminants such as cows, sheep and goats are capable of degrading OTA to the virtually non‐toxic OTalpha in their rumen by the microflora (Fink‐Gremmels, 2008; Battacone et al., 2010). Hydrolysis of OTA to OTalpha (Figure 2) has also been observed in vitro with cow rumen fluid and is attributed to its protozoan fraction (Kiessling et al., 1984). Consequently, only a small amount of intact OTA is absorbed, which explains the comparably high tolerance of cattle to OTA toxicity and the low levels of OTA in bovine tissues (Zhang et al., 2019 and literature cited therein). However, the ability to detoxify OTA is strictly related to a functional rumen and may change when the feed composition is altered, e.g. a high proportion of protein‐rich concentrates in feed which may modify the hydrolytic capacity of rumen microorganisms (Fink‐Gremmels, 2008; Mobashar et al., 2012). Similar to cattle, small ruminant species such as sheep and goats are protected against the toxicity of OTA by their rumen microorganisms (Battacone et al., 2010).
In accordance with the low level of intact OTA reaching the circulation of ruminants, transfer into cow milk is expected to be very low (Fink‐Gremmels, 2008). Indeed, analysis of commercially available milk samples in Italy showed that only 3 out of 83 samples contained OTA at levels between 70 and 110 ng/L, with an LOQ of 5 ng/L (Pattono et al., 2011). Out of 132 French milk samples, only three were found to contain OTA levels above the LOQ of 5 ng/L (Boudra et al., 2007). Galtier (1991) estimated that 0.01% of the OTA in cattle feed would be transferred into cow milk. The very low transfer of OTA from feed into the milk and tissues of cows was confirmed in a recent experiment by Hashimoto et al. (2016). When lactating Holstein cows were fed a diet containing 100 μg OTA per kg of dry matter for 28 days, no OTA was detected in the milk or in liver, kidney, muscle or fat with an LOQ of 0.1 μg/kg. For dairy ewes, Boudra et al. (2013) also determined a very low transfer rate (< 0.02%) for OTA from feed into milk.
Although levels of OTA in the milk of ruminants appear to be very low, OTA occurrence may be higher in dairy products such as cheese. Dall'Asta et al. (2008) first reported on the occurrence of OTA in commercial samples of blue‐mould ripened cheeses at levels ranging from 0.25 to 3.0 μg/kg, and provided circumstantial evidence that the OTA does not originate from a contamination of the milk but is formed during ripening and storage of the cheese. More recently, higher levels of OTA were detected in the rind (1–262 μg/kg) and interior (18–146 μg/kg) of traditional handmade semi‐hard cheeses (Pattono et al., 2013). More information on the occurrence of OTA in cheese is reported in Section 4.7.2.
The toxicokinetics of OTA was previously studied in rainbow trout (Fuchs et al., 1986), carp (Hagelberg et al., 1989) and more recently in Atlantic salmon (Bernhoft et al., 2017). The oral bioavailability of OTA appears to be low and it distributes mainly to liver and kidney. Binding to proteins in blood is low and elimination is rapid. Only trace amounts are transferred to edible tissue, i.e. muscle, making fish a negligible source of exposure.
4.1.1.6.1. Summary of transfer to animal‐derived food
Because of its long half‐life OTA may accumulate in tissues of monogastric farm animals such as pigs and thus be present in meat and meat products. Due to efficient degradation in the rumen OTA levels in the milk and edible tissues of cows and other ruminants are low. In fish, OTA has a short half‐life and very low tissue levels.
4.1.2. Biomarkers of exposure
The issue of possible adverse effects observations in humans, including Balkan endemic nephropathy and other endemic nephropathies, is discussed in Section 4.2.6 Observations in humans.
Concerning OTA biomarkers of exposure, MS‐based methods have been developed for biological fluids (i.e. blood, urine, breast milk) and used extensively for biomonitoring studies. Besides OTA, its major metabolites are detected and (semi)quantified as well.
In the previous EFSA opinion (EFSA, 2006), several, then considered recent, studies on OTA concentrations in human blood, urine and milk were presented and compared with previous findings. In the seven studies presented, mean plasma concentrations of OTA in blood, serum or plasma ranged from 0.17 to 0.56 ng/mL while individual blood concentrations ranged between 0.1 and 10 ng/mL, overall confirming results obtained in previous studies. The CONTAM Panel then concluded that there was a declining trend in OTA plasma levels in recent studies compared to earlier results. It was noted that relating dietary intake to OTA in blood is hampered by its long half‐life in plasma due to protein binding and that plasma concentration represents the integrated dietary OTA intake over a longer period of time. Determination of OTA in urine appears to be a more suitable marker for recent exposure as detection methods are more sensitive (about 0.01 ng/mL). Since OTA is excreted in breast milk, analysis of human milk samples could serve as an additional exposure marker (OTA levels detected in breast milk range from 1.2 to 182 ng/L).
Since 2006, a series of new studies on biomarkers of exposure became available.
4.1.2.1. OTA and metabolites in plasma/serum/blood
Coronel et al. (2011) reported OTA levels of 0.50 (median) and 0.06–10.92 ng/mL (range) in 325 inhabitants from Spain and also reviewed earlier studies reporting levels of OTA in serum/plasma and found no new studies since 2006.
Duarte et al. (2010, 2011) reported levels of OTA from Portugal. Values varied slightly between geographical areas and mean values of 0.46–1.01 ng/mL were found in men whereas women had somewhat lower mean values, 0.38–0.60 ng/mL. In Italy, OTA values in serum/plasma in 324 individuals were 0.029–2.9 ng/mL with a mean value of 0.229 ng/mL. Less than 5% of the samples were above 0.5 ng/mL (di Giuseppe et al., 2012). Soto et al. (2016) provided a review of more recent studies covering the period from 2005 to 2015. Median OTA values in serum/plasma varied from 0.15 to 18.0 ng/mL. The higher value being from Tunisian patients with renal failure. In European samples, the median values ranged from 0.15 to 1.098 ng/mL. The LOD of the methods employed varied considerably and the fraction of positive samples varied accordingly.
In more recent studies from Bangladesh and Germany (Ali et al., 2016, 2017, 2018), OTalpha was determined. This metabolite was also to a large extent conjugated and the concentration varied more than OTA. The OTalpha/OTA ratio varied from 0.09 to 2.29 in samples from Bangladesh. There was limited success in correlating serum/plasma values with dietary intake of OTA (Coronel et al., 2010).
Cramer et al. (2015), using dried blood spots, reported the presence of OTA and the modified OTA isomer, 2R’‐OTA, that may form upon roasting coffee (see Section 2.1.1). 2'R‐OTA was only detected in samples from coffee drinkers, whereas OTA was found at a similar mean concentration (0.21 ng/mL) in both non‐coffee drinkers and in coffee drinkers. In the group of coffee drinkers (n = 34), the mean and SD concentration was 0.11 ± 0.093 ng/mL with a range of 0.021–0.414 ng/mL. On average, the 2'R‐OTA concentrations were about half of that of OTA. In a Swedish study on adolescents (about 3,000 students) from the 5th, 8th and 11th grade (Warensjö Lemming et al., 2015), 2'R‐OTA was found in 8.3% of the serum samples, all from the 8th and 11th grade. Average concentrations varied between 0.028 and 0.037 ng/mL with a maximum of 0.136 ng/mL, and the 2'R‐OTA concentrations varied with coffee consumption. OTA was detected in 49–69% of the serum samples from all groups and mean concentration were 0.050–0.059 ng/mL with a maximum of 0.658 ng/mL.
Attempts have been made to estimate OTA intakes from plasma concentrations taking into account the plasma clearance and bioavailability of OTA assuming only glomerular filtration (Coronel et al., 2010). Different values for the plasma clearance have been used resulting in different daily intake estimates of OTA as described by Coronel et al. (2010). Using a plasma clearance of 0.99 mL/kg bw per day and 50% bioavailability, median plasma concentrations between 0.15 and 1.10 ng/mL reported from Europe would correspond to daily intakes of 0.30–2.17 ng/kg bw. With the lower clearance value (0.67 mL/kg bw per day), intake values would be 30% lower. Although being highly uncertain, the estimated intake values were within the same order of magnitude as those obtained in the exposure assessment (see Section 4.8.1).
In summary, although no formal trend analyses have been conducted there seems to be little change in OTA concentrations in serum/plasma/blood reported in studies published since the last EFSA CONTAM opinion on OTA.
4.1.2.2. OTA and metabolites in urine
Sensitive analytical techniques are necessary for determination of the low urinary OTA concentrations. In addition, OTA metabolites which include phase II metabolites of OTA, OTalpha and also OTalpha glucuronides also make up a considerable fraction of OTA and OTA metabolites excreted into urine (Ali et al., 2017; Muñoz et al., 2017). Application of enzymatic hydrolysis of the urine sample prior to MS‐analysis is recommended because phase II metabolites may escape direct detection because of lower ionisation. Reported urinary concentrations of OTA and OTalpha with and without prior enzymatic hydrolysis were reviewed by Ali and co‐workers (Ali et al., 2017). European populations had a range of mean OTA concentrations without hydrolysis between 0.009 and 0.04 ng/mL. OTalpha was not determined in these studies. Using hydrolysis prior to analysis, the mean values reported varied for OTA from 0.07 to 0.46 ng/mL, while OTalpha had a mean concentration range of 0.44–2.88 ng/mL. In a Swedish study (Mitropoulou et al., 2018), OTA was determined after enzymatic hydrolysis in urine samples from 250 adults and 50 children. There were 51% positive adult and 96% positive child samples. Positive adult samples had a mean and SD concentration of 0.89 ± 0.50 ng/mL, range 0.12–2.76 ng/mL. Adjusting for density (1.015) gave the following figures for adults: 1.20 ± 1.68 mg/mL, range 0.09–16.5 mg/mL. Corresponding values for children were, unadjusted: 0.18 ± 0.15 ng/mL, range 0.05–0.93 ng/mL. Density (1.019) adjusted figures were 0.13 ± 0.08 mg/mL, range 0.06–0.52 ng/mL. The rate of positive samples was within a large range of 12.5–100% positive samples in other studies, but the mean crude total OTA concentration (0.89 ± 0.50 ng/mL, range 0.12–2.76 ng/mL) was about three times higher than other European studies indicating a higher exposure.
Recently, Sueck et al. (2019) identified a new metabolite of OTA in urine i.e. ochratoxin‐N‐acetyl‐L‐cystein (OTB‐NAC) derived from a glutathione conjugation reaction of OTA. The metabolite was detected in 11 of 18 urine samples and could be quantified in five ranging from 0.023 to 0.176 ng/mg creatinine.
Attempts have been made to estimate daily intake of OTA based on urinary concentrations in spot samples, but the results are considered questionable because of long half‐life and strong plasma protein binding and uncertain excretion rates (values from 2.6% to 50% have been used) (Solfrizzo et al., 2014; Degen, 2016; Mitropoulou et al., 2018).
4.1.2.3. OTA in breast milk
There are only few studies on the transfer of OTA from plasma to breast milk in humans (see Warth et al., 2016). Muñoz et al. (2014) reported a study from Chile in 21 mother–children pairs that were followed for up to 6 months. The authors reported, overall, an average level in breast milk from this cohort of 52 ± 46 ng/L. The milk to plasma ratio varied considerably changing from the first days with colostrum (0.40 ± 0.26) to later stages (0.15 ± 0.26 after 15–30 days, 0.09 ± 0.06 after 2 months and 0.26 ± 0.19 after 4 months). Breast milk mean concentrations were 86 ± 59 ng/L in the first week, 33 ± 27 after 15–30 days, 27 ± 19 ng/L after 2 months, 30 ± 14 ng/L after 4 months. No sample was collected at 6 months. Corresponding OTA mean concentrations in infant urine were 156 ± 36 ng/L in the first week, 36 ± 37 ng/L after 15–30 days, 51 ± 38 ng/L after 2 months, 36 ± 23 ng/L after 4 months and 47 ± 7 ng/L after 6 months.
Dietary exposures to OTA are reflected in OTA levels in plasma/serum, blood, urine and breast milk. Overall, in Europe, these levels have not changed since the last EFSA evaluation.
4.2. Toxicity
4.2.1. Acute toxicity
In the EFSA opinion of 2006, acute toxicity of OTA was not discussed and an acute reference dose (ARfD) was not established.
JECFA (FAO/WHO, 1991) reported LD50 values for OTA in various species based on literature compilations by Harwig et al. (1983). Oral LD50 values ranged from 46 to 58.3 mg/kg bw in mouse and 20–30.3 mg/kg bw in rat. LD50 values in rat neonate, dog, pig and chicken were 3.9, 0.2, 1 and 3.3 mg/kg bw, respectively. In a study with male rats given a single doses (gavage) of 0, 17 or 22 mg OTA/kg bw, respectively, earliest changes identified were multifocal haemorrhages and fibrin thrombi in different organs. Other changes were hepatic and lymphoid necrosis, villous atrophy in particular in the jejunum and nephrosis (Albassam et al., 1987). In subsequent JECFA evaluations (FAO/WHO, 1996, 2001, 2008), no further data on acute toxicity on OTA were presented (and no ARfD was derived). An ARfD was not established in any of their evaluations.
The CONTAM Panel did not identify new studies (published after the EFSA opinion of 2006) relevant for the assessment of acute toxicity of OTA.
4.2.2. Repeated dose studies
4.2.2.1. Introduction
The repeated dose toxicity of OTA has been previously reviewed by EFSA (EFSA, 2006). Experimental data identified the kidney as the main target organ for the adverse effects of OTA. Immunotoxic, neurotoxic and teratogenic effects were observed at high dose levels. OTA has been shown to induce dose‐ and time‐dependent nephrotoxicity in all mammalian species tested, including mice, rats, dogs and pigs, with marked differences in sensitivity to OTA toxicity between sex and species. Long‐term administration of OTA causes renal tumours in rats and mice. Rats are more sensitive to renal tumours formation by OTA than mice, and higher incidences of renal tumours are observed in male animals as compared to females. Renal tumours produced by OTA in rats are preceded by evidence of renal toxicity as shown in short‐term toxicity studies. The effects of OTA on the kidneys in rats and pigs were identified as the most sensitive and pivotal endpoints of OTA. In a 2‐year gavage study with Fischer 344/N rats (NTP, 1989), the NOAEL and LOAEL for OTA‐induced nephrotoxicity were 21 and 70 μg OTA/kg bw per day when dosed 5 days per week, respectively, equivalent to 15 and 50 μg/kg bw per day. In a 90‐day study in Wistar rats, mild and reversible renal changes were reported at a dose of 200 μg OTA/kg diet (Munro et al., 1974), equivalent to 15 μg OTA/kg bw per day (FAO/WHO, 2001). On the basis of a series of studies in female pigs, an LO(A)EL for minimal effects on the kidney (minimal morphological changes, renal enzymes and renal function tests) was identified by EFSA (and previously by JECFA) at 8 μg/kg bw per day from a 90‐day feeding study (FAO/WHO, 1991, 1996, 2001, 2008; EFSA, 2006). EFSA concluded that ‘8 μg OTA/kg bw. per day is a LOAEL that represents an early marker of renal toxicity in experimental animals (i.e. female pigs) and likely to be close to a NOAEL, as the observed changes in biochemical parameters indicate transient changes in the kidneys’ (EFSA, 2006). The CONTAM Panel re‐evaluated the pivotal studies in rats and pigs used as a basis to establish a HBGV in the previous EFSA opinion (EFSA, 2006). In addition, new in vivo toxicity studies with mice, rats, rabbits and pigs published since the 2006 opinion of EFSA was reviewed to identify possible new evidence relevant for the hazard identification and characterisation of OTA.
4.2.2.2. Repeated dose studies in rats considered in the previous EFSA assessment
Details of the pivotal repeated dose toxicity studies in rats published before 2006 are summarised in Appendix C. On the basis of these studies, an LOAEL for minimal renal changes at 15 μg OTA/kg bw was derived from a 90‐day feeding study in Wistar rats (Munro et al., 1974).
4.2.2.3. Repeated dose studies in rats published since 2006
Most of the studies in rats conducted since 2006 were designed to elucidate the mechanism of OTA‐induced nephrotoxicity and thus employed dose levels known to be toxic. In some studies, hepatotoxic or immunotoxic effects of OTA were investigated. Table 1 provides a description of new subacute and subchronic toxicity studies in rats. None of the studies described in Table 1 (and briefly summarised in the text below) provided NOAELs/LOAELs lower than the LOAEL of 15 μg/kg bw derived from the 90‐day feeding study in Wistar rats from Munro et al. (1974).
Table 1.
Repeated dose studies in rats published after 2006
| Strain | Dose and animals per group | Duration/time of observation | Main effects observed | Effect level | Reference |
|---|---|---|---|---|---|
| F344/N (♂) | 0, 21, 70, 210 μg/kg bw per day (gavage, corn oil, 5 days/week)( a ), n = 5 | 14, 28, 90 days |
|
NOAEL 21 μg/kg bw per day (5 days/week)( b ) (based on renal histopathology, gene expression, urinary KIM‐1 and urine biochemistry) | Rached et al. (2007) |
|
Rached et al. (2008) | ||||
|
Hoffmann et al. (2010) | ||||
|
Sieber et al. (2009) | ||||
|
Adler et al. (2009) | ||||
| F344 gpt delta rats (♂, ♀) | 0, 5 mg/kg diet corresponding to 0, 360 μg/kg bw (♂); 380 μg/kg bw (♀), n = 4–5 | 4, 13 weeks |
|
5 mg/kg diet (corresponding to 360 μg/kg bw) (based on renal histopathology) | Hibi et al. (2011) |
| 4 weeks |
|
Hibi et al. (2013a) | |||
| F344 gpt delta rats (♂) | 0, 70, 210, 630 μg/kg bw (gavage, sodium bicarbonate, 7 days/weeks), n = 5 | 4 weeks |
|
LOAEL* 70 μg/kg bw per day (7 days/weeks) (based on kidney weight, DNA strand breaks) | Kuroda et al. (2014) |
| F344 (♂) | 0, 70, 210 μg/kg bw (gavage, corn oil, 5 days/weeks)( c ), n = 6 | 4, 13 weeks |
|
LOAEL* 70 μg/kg bw per day (5 days/weeks)( d ) (based on renal histopathology) | Qi et al. (2014a) |
| F344 (♂) | 0, 70, 210 μg/kg bw (gavage, corn oil, 5 days/weeks)( c ), n = 6 | 2, 4, 13, 26 weeks |
|
LOAEL* 70 μg/kg bw per day (5 days/week)( d ) (based on renal histopathology, renal/hepatic mRNA and miRNA expression and changes in gut microflora) | Dai et al. (2014) |
|
Qi et al. (2014b) | ||||
|
Guo et al. (2014) | ||||
| Wistar (♂) | 0, 1, 4 mg/kg bw (gavage, corn oil, daily), n = 6 | 7 days |
|
LOAEL* 1 mg/kg bw per day (7 days/week) (based on organ and body weight, renal histopathology, blood biochemistry) | Zhu et al. (2016a) |
| Wistar (♂) | 0, 289 μg/kg bw (gavage, 0.1 M NaHCO3; every 2nd day)( e ), n = 8 | 90 days |
|
289 μg/kg bw every 2nd day( e ) | Gagliano et al. (2006) |
| Wistar (♂) | 0, 50, 125, 250, 500 μg/kg bw (gavage, 51 mM NaHCO3, every 2nd day)( f ), n = 4 | 10 days |
|
LOAEL * 50 μg/kg bw every 2nd day( g ) (based on renal histopathology) | Zlender et al. (2009) |
| F344/NSIc (♂) | 0( h ), 210 μg/kg bw (gavage, 0.1 M NaHCO3, daily), n = 10 | 28 days |
|
210 μg/kg bw per day (based on renal histopathology) | Taniai et al. (2012a) |
|
Taniai et al. (2012b) | ||||
| F344/NSlc (♂) | 0( h ), 210 μg/kg bw (gavage, 0.1 M NaHCO3; 7 days/week), n = 15–16 | 28 days |
|
210 μg/kg bw per day (7 days/weekweek) (based on renal histopathology) | Taniai et al. (2014) |
| F344/IcoCrl (♂,♀) | 0, 210, 500 μg/kg bw (gavage, 0.1 M NaHCO3, 7 days/week), n = 6 | 7, 21 days |
|
LOAEL* 210 μg/kg bw per day (based on renal histopathology) | Pastor et al. (2018) |
|
Enciso et al. (2018) |
ALT: alanine aminotransferase; AST: aspartate aminotransferase; BrdU: bromo‐desoxy‐uridine; BUN: blood urea nitrogen; clusterin: protein associated with clearance of cellular debris and apoptosis; DNA: deoxyribonucleic acid; γ‐H2AX: H2A histone family member X phosphorylated on Ser 139, marker for DNA damage; G2/M arrest: gap2/mitotic arrest; Gpt: Glutamic‐pyruvic transaminase gene; GSH; glutathione; GSH/GSSG ratio: ratio of reduced GSH to oxidised GSH (GSSG); GST: glutathione S‐transferase; 8‐OH‐dG: 8‐hydroxy‐deoxyguanine; Ki‐67: marker protein for proliferation; KIM‐1; Kidney injury molecule‐1; LDH: lactate dehydrogenase; LOAEL: lowest observed adverse effect level; LOEL: lowest observed effect level; MDA: malondialdehyde; miRNA: micro RNA; mRNA: messenger RNA; n: number; n.d.: not determined; NOAEL; no observed adverse effect level; OAT; organic anion transporter; OSOM: outer stripe of the outer medulla; OTA: ochratoxin; PCNA: proliferating cell nuclear antigen ROS: reactive oxygen species; S3 segment: Straight segments of the proximal tubule that descend into the outer medulla; SOD: superoxide dismutase; spi− – protein spitz precursor (a proto oncogene); TIMP‐1: tissue inhibitor of metalloproteinase‐1 (a protein); wk: week.
*: NOAEL/LOAEL not explicitly reported but derived from data presented in study.
Equivalent to 0, 15, 50, 150 μg/kg bw per day.
Equivalent to 15 μg/kg bw per day.
Equivalent to 0, 50, 150 μg/kg bw per day.
Equivalent to 50 μg/kg bw per day.
Equivalent to 145 μg/kg bw per day.
Equivalent to 25, 62.5, 125, 250 μg/kg bw per day.
Equivalent to 25 μg/kg bw per day.
Untreated control animals were maintained on the basal diet and tap water without any treatment during the experimental period.
Subacute and subchronic studies were conducted in male F344/N rats and F344 gpt delta rats employing dosing regimens similar to the 2‐year gavage study in F344/N rats (NTP, 1989). Essentially, these confirmed renal changes at a dose of 50 μg/kg bw per day observed previously in this rat strain (Rached et al., 2007, 2008; Adler et al., 2009; Sieber et al., 2009; Hoffmann et al., 2010; Dai et al., 2014; Guo et al., 2014; Kuroda et al., 2014; Qi et al., 2014a,b; Zhu et al., 2016a). Consistent with the NOAEL of 15 μg/kg bw per day derived from the NTP study (NTP, 1989), no effects on renal histology, urinary biomarkers indicative of kidney injury, and expression of kidney injury marker genes or genes mechanistically linked to OTA toxicity were observed after exposure of male F344/N rats to 15 μg/kg bw per day for up to 90 days (Rached et al., 2007, 2008; Adler et al., 2009; Sieber et al., 2009; Hoffmann et al., 2010).
4.2.2.4. Repeated dose studies in mice published since 2006
New repeated dose toxicity studies in mice published since the 2006 EFSA opinion is described in Table 2. Consistent with the lower sensitivity of mice to OTA toxicity as compared with rats and pigs, none of the new studies provided NOAELs/LOAELs that were lower than the LOAELs of 15 and 8 μg/kg bw previously established in rats and pigs, respectively (FAO/WHO, 2001; EFSA, 2006).
Table 2.
Repeated dose studies in mice published after 2006
| Strain | Dose and animals per group | Duration/time of observation | Main effects observed | Effect level | Reference |
|---|---|---|---|---|---|
| Inbred Swiss albino (♂) | 0( a ), 1.5, 3 mg/kg bw per day (gavage, olive oil, 7 days/week) n = 10 | 45 days |
|
*LOAEL 1.5 mg/kg bw per day (based on renal non‐enzymatic and enzymatic antioxidants) | Chakraborty and Verma (2010b) |
| C57BL/6J – p53 (p53 gene) proficient homozygous Gpt delta C57BL/6J – p53‐deficient homozygous Gpt delta (♂) | 1 and 5 mg/kg bw per day (gavage, 0.1 M NaHCO3) n = not provided | 4 weeks |
|
LOAEL* 1 mg/kg bw per day (based on renal histopathology) | Hibi et al. (2013b) |
|
C57BL/6 × FVB ‐ HO‐1+/+ C57BL/6 × FVB ‐ HO‐1−/− (♂, ♀) |
0, 2.5 mg/kg bw per day (i.p. 0.1 M NaHCO3 every 2nd day), n = not provided | 20 days |
|
2.5 mg/kg bw every 2nd day, i.p. | Loboda et al. (2017a) |
| C57BL/6 ‐ Nrf2+/+, C57BL/6 ‐ Nrf2−/− (♂, ♀) | 0, 2.5 mg/kg bw per day (i.p., 0.1 M NaHCO3, every 2nd day), n=3‐5 | 20 days |
|
2.5 mg/kg bw every 2nd day, i.p. | Loboda et al. (2017b) |
DNA; deoxyribonucleic acid; GPx: Glutathione peroxidase; GSH; glutathione; GSR: Glutathione reductase, i.p.: intraperitoneal; HO‐1: heme oxygenase 1, LOAEL; lowest observed adverse effect level; Nrf2: nuclear factor erythroid 2–related factor 2 (a protein that regulates expression of antioxidant proteins that protect against oxidative damage; n= number; RNA: ribonucleic acid; TBARS: thiobarbituric acid reactive substances; SOD: superoxide dismutase; spi− ‐ protein spitz precursor (a proto oncogene); wk: week.
*: NOAEL/LOAEL not explicitly reported but derived from data presented in study.
Untreated controls were maintained without any treatment.
4.2.2.5. Repeated dose studies in rabbits published since 2006
Table 3 provides a description of subacute and subchronic studies in rabbits published since 2006. Clinical signs of toxicity, renal histopathological changes and biochemical changes indicative of liver and kidney toxicity were reported in rabbits given OTA in diet corresponding to approximately 30 μg/kg bw per day for up to 2 months. Thus, studies in rabbits did not provide more sensitive endpoints of OTA toxicity compared to the LOAELs of 15 and 8 μg/kg bw previously established in rats and pigs, respectively (FAO/WHO, 2001; EFSA, 2006).
Table 3.
Repeated dose studies in rabbits published after 2006
| Strain | Dose and animals per group | Duration/time of observation | Main effects observed | Effect level | Reference |
|---|---|---|---|---|---|
| New Zealand White (♂) | 0, 1, 2 mg/kg diet (corresponding to 30 and 60 μg/kg bw per day b); n = 6–12 | 8 weeks |
|
LOAELa 30 μg/kg bw per day (based on blood biochemistry) | Mir and Dwivedi (2010) |
| New Zealand White (♂) | 0, 1 mg/kg diet (corresponding to approx. 33 μg/kg bw per dayb), n = 4 | 30, 60 days |
|
33 μg/kg bw per day (based on renal histopathology & clinical signs) | Prabu et al. (2013) |
ALP: alanine phosphatase; ALT: alanine aminotransferase; AST: aspartate aminotransferase; LOAEL: lowest observed adverse effect level; n: number; PCT: proximal convoluted tubular.
NOAEL/LOAEL not explicitly reported but derived from data presented in study.
4.2.2.6. Repeated dose studies in pigs considered in the previous EFSA assessment
In previous assessments, nephrotoxicity of OTA in pigs was identified as the most sensitive endpoint of OTA toxicity, and an LOAEL of 8 μg/kg bw derived from a series of studies in pigs was used to establish a HBGV (FAO/WHO, 1991, 1996, 2001, 2008; EFSA, 2006). From these studies (for details see Appendix C, Table C.2), the CONTAM Panel identified the study of Krogh et al. (1974) selected as the pivotal 90‐day study in the previous EFSA opinion and is therefore described in further detail below.
Table C.2.
Studies in pigs used in previous assessments to establish HBGVs
| Strain | Dose and animals per group | Duration/time of observation | Main effects observed | Effect level | Reference |
|---|---|---|---|---|---|
| Danish Landrace‐Duroc (♀) | Gelatin capsules containing OTA equivalent to 0, 0.2 and 1 mg/kg feed, n = 6 | 1–5 weeks |
|
0.2 mg/kg feed (based on kidney lesions) | Krogh et al. (1988), Meisner and Krogh (1986) |
| Danish Landrace (♀) | 0, 0.2, 1, 4 mg/kg feed (corresponding to ~ 8, 40, and 160 μg/kg bw), n = 4–9 | 3–4 months (i.e. from 20 to 90 kg bw) |
|
0.2 mg/kg feed (corresponding to 8 μg/kg bw) (based on kidney lesions) | Krogh et al. (1974) |
| Danish Landrace (♀) | 0, 30–50 μg/kg bw (gelatin capsule) (corresponding to 0 and 1 mg/kg feed), n = 3–6 | 3 months |
|
0, 30‐50 μg/kg bw (corresponding to 0 and 1 mg/kg feed) (based on kidney lesions) | Krogh et al. (1976, 1979), Elling (1979a, 1979b) |
| 2 years |
|
0, 30–50 μg/kg bw (corresponding to 0 and 1 mg/kg feed) (based on kidney lesions) | Krogh et al. (1979), Elling (1979b) |
ACP: acid phosphatase; ALP: alkaline phosphatase; GLDH: glutamate dehydrogenase; ɣ‐GT: ɣ‐glutamyl transpeptidase; LAP: leucine aminopeptidase; NADH‐TR: NADH‐tetrazolium reductase; OTA: ochratoxin A; PEPCK: cytosolic phosphoenolpyruvate carboxykinase; SDH: succinate dehydrogenase; TmPAH: average maximal tubular excretion of PAH; TmPAH/CInulin: ratio between TmPAH and average renal clearance of inulin.
In this study (Krogh et al., 1974), female pigs (Danish Landrace) were given doses (via feed) of 0 (control group), 8, 40 and 160 μg/kg bw per day for 3–4 months (i.e. from 20 to 90 kg bw). Increased water consumption and increased concentrations of creatinine and urea were observed in animals administered OTA at 160 μg/kg bw per day. Increased glucose was detected in urine of pigs given the two highest dose levels (40 and 160 μg/kg bw per day). The average renal clearance of inulin CInulin and p‐aminohippuric acid (PAH) measured after 3–3.5 months was significantly reduced at the highest dose. The average maximal tubular excretion of PAH (TmPAH) and TmPAH/CInulin was reduced in a dose‐related manner, with significant changes at all doses. In contrast, reabsorption of glucose and the ability to concentrate urine (as assessed by urine specific gravity and osmolality) were only affected at the two highest dose levels (40 and 160 μg/kg bw per day). Alterations in the enzymatic activities of glutamate dehydrogenase (GLDH), leucine aminopeptidase (LAP) in renal cortex were also recorded at the two highest dose levels. However, microscopic kidney lesions were observed even at the lowest dose. At this dose, these changes were confined to the proximal tubules and consisted of tubule dilation with reduced brush boarder, pycnotic nuclei, mitotic figures, desquamation of tubule cells and a slight increase of interstitial fibrous tissue. The microscopic kidney lesions in the higher dose groups included degenerative changes of the proximal tubule epithelium, dilation of cortical tubules, tubular atrophy, thickened basement membrane, glomerulosclerosis and interstitial fibrosis. The frequency of animals with kidney lesions at the end of the exposure period in the control, 8, 40 and 160 μg/kg bw per day group was 0/9, 4/9, 9/9 and 4/4, respectively.
While the previous evaluation in 2006, used dose‐related reduction in TmPAH and TmPAH/CInulin which occurred in parallel with microscopic lesions, as the critical effect and identified the same LOAEL of 8 μg/kg bw, it is now well established that renal transport of OTA is mediated by OATs. Therefore, it was concluded that reduced tubular excretion of PAH (a prototype OAT substrate), which was previously interpreted as evidence of impaired renal function by OTA, is likely also to be due to interference by OAT‐mediated transport and cannot be considered as adverse.
However, based on evidence of microscopic kidney lesions, the CONTAM Panel confirmed 8 μg/kg bw as the LOAEL for nephrotoxicity in this study.
4.2.2.7. Repeated dose studies in pigs published since 2006
The repeated dose studies in pigs published since 2006 are described in Table 4 below. A series of studies published after 2006 investigated the effects of OTA in female cross‐bred weaned piglets (TOPIGS‐40) exposed to OTA via feed at the recommended EU guidance value of 50 μg OTA/kg diet11 (corresponding to approximately 2.7 μg/kg bw based on reported body weights and feed intakes) for 33 days (Marin et al., 2016, 2017a,b, 2018). Exposure to OTA had no effect on growth performance or organ weights. Biochemical analysis of plasma revealed a small but statistically significant decrease in total protein and albumin, accompanied by a significant increase in alanine aminotransferase and triglycerides. The activity of superoxide dismutase (SOD) was reported to be significantly increased in animals given OTA as compared to controls. The biochemical changes were interpreted by the authors of the study as an indication of hepatocellular injury (Marin et al., 2016, 2017a,b). Histopathological evaluation of liver tissue to confirm hepatotoxic effects was not performed. There was no evidence of kidney injury. However, genome‐wide expression profiling revealed significant alterations in 105 different renal transcripts related to immune response, oxidative stress response, cell growth and cell death (Marin et al., 2017a). Changes in parameters indicative of altered immune and oxidative stress response were also reported to occur in the gut (Marin et al., 2017b). The authors of these studies emphasised the need to perform confirmatory in vivo studies on larger number of animals to provide a reliable database for regulatory purposes (Marin et al., 2016). In a subsequent study from the same group, 15 cross‐bred TOPIG hybrid [(Landrace × Large white) × (Duroc × Pietrain)] pigs (n = 5) were exposed to a diet contaminated with 250 μg OTA/kg feed (corresponding to a dose of 12.5 μg OTA/kg bw per day) for 28 days (Marin et al., 2018). In contrast to the decrease in total protein and albumin reported in Marin et al. (2016), exposure to OTA resulted in a significant increase in the serum concentration of total protein, albumin, triglycerides and creatinine accompanied by a decreased total antioxidant capacity in liver and kidney (Marin et al., 2018).
Table 4.
Repeated dose studies in pigs published after 2006
| Strain | Dose and animals per group | Duration/time of observation | Main effects observed | Effect level | Reference |
|---|---|---|---|---|---|
| TOPIGS‐40, cross‐bred weaned piglets (♀) | 0 and 50 μg/kg diet corresponding to approx. 2.7 μg OTA/kg bw per day** ( 1 ) n = 6 | 30–33 days |
|
50 μg/kg diet corresponding to approx. 2.7 μg OTA/kg bw per day** (based on biochemical changes) | Marin et al. (2016) |
|
Marin et al. (2017a) | ||||
|
Marin et al. (2017b) | ||||
| TOPIG hybrid [(Landrace × Large white) × (Duroc × Pietrain)] piglets (sex not specified) | 0 and 250 μg/kg diet corresponding to approx. 12.5 μg/kg bw per day**; n = 5 | 28 days |
|
250 μg/kg diet corresponding to 12.5 μg/kg bw per day** (based on biochemical changes) | Marin et al. (2018) |
| cross‐bred ([Landrace × Yorkshire] × Duroc) piglets (sex not specified) | 0, 400, 800 μg/kg diet corresponding to approx. 20 and 36 μg/kg bw per day** n = 12 | 42 days |
|
LOAEL* 400 μg/kg diet corresponding to approx. 20 μg/kg bw per day (based on reduced growth performance, renal histopathology and blood biochemistry) | Zhang et al. (2016) |
| 0, 400, 800 μg/kg diet corresponding to approx. 19 and 36 μg/kg bw per day** n=9 | 42 days |
|
LOAEL* 400 μg OTA/kg diet corresponding to approx. 19 μg/kg bw per day (based on reduced growth performance, renal histopathology and immunological parameters) | Gan et al. (2017b) | |
| Large‐White weaned pigs (castrated ♂) | ~ 0.45 (control), 180 μg/kg diet corresponding to approx. 9 μg/kg bw per day **; n = 30 | 43 days |
|
180 μg/kg diet corresponding to approx. 9 μg/kg bw per day** | Bernardini et al. (2014) |
| Hungarian Large White × Hungarian Landrace F1 weaned piglets (♂: ♀ = 1:1) | 0, 380 – 338 μg/kg diet corresponding to approx. 14 μg/kg bw per day**; n = 12 | 49 days |
|
379.6–338.1 μg/kg diet corresponding to approx. 14 μg/kg bw per day** | Balogh et al. (2007) |
| Zegers hybrid (♂) | 0, 300 μg/kg diet( 2 ) corresponding to approx. 9 μg/kg bw per day***; n = 6 | 30 days |
|
300 μg/kg diet( 2 ) corresponding to approx 9 μg/kg bw per day*** | Pleadin et al. (2012) |
| Landrace × Bulgarian white (♂: ♀ = 1:1) | 0, 500 μg/kg diet corresponding to approx. 15 μg/kg bw per day***; n = 6 | 3 months |
|
500 μg/kg OTA corresponding to approx. 15 μg/kg bw per day*** | Stoev et al. (2012) |
ALT: alanine aminotransferase; AST: aspartate aminotransferase; LOAEL: lowest observed adverse effect level; NOAEL; no observed adverse effect level; n: number; PCT: proximal convoluted tubular; GPx: Glutathione peroxidase; TCR: T‐cell receptor.
NOAEL/LOAEL not explicitly reported but derived from data presented in study.
Calculated using parameters (bw, feed intake) provided in the paper.
Calculated using default values provided in EFSA Scientific Committee (2012a), EFSA FEEDAP Panel (2012), WHO/IPCS (2009).
The experimental diet also contained low levels of fumonisin B1 (88 μg/kg corresponding to approx. 5 μg/kg bw per day), which is below the NOAEL for fumonisins in pigs.
Unclear reporting of route of administration: ‘The experimental group was treated with feed contaminated with 300 μg OTA/kg (300 ppb). OTA as a pure chemical (standard) was mixed with lactose and distributed in gelatin capsules that were applied directly into pig mouth.’
Indications for cellular stress and inflammatory response (increased TNF‐a and IL‐10 in plasma, decreased HO‐1 in kidney) were reported in 43‐day study in weaned pigs (Large‐White, castrated male) fed on a diet containing 181 ± 34 μg OTA /kg feed (corresponding to 9 μg/kg bw per day, calculated based on body weight and food consumption data provided in the paper) (Bernardini et al., 2014).
Treatment of weaned piglets (Hungarian Large White × Hungarian Landrace F1, sex ratio: 1:1) with experimental diets contaminated with OTA at levels corresponding to an intake of approximately 12 μg/kg bw per day for 42 days resulted in reduced feed consumption and daily weight gain and indications of oxidative stress (increased malondialdehyde in liver, decreased glutathione peroxidase activity in liver and kidney) (Balogh et al., 2007).
In male pigs (Zegers hybrid), 30‐day treatment with OTA was reported to cause an increase in serum creatinine and urea and a decrease in serum glucose and total protein on days 10, 20 and 30 (Pleadin et al., 2012). Histopathological evaluation of tissues was not conducted. The CONTAM Panel noted the unclear reporting of the route of administration (OTA in feed vs. OTA administered via gelatine capsule into mouth of pigs) and the apparent lack of time‐matched controls. Based on default values (WHO/IPCS, 2009; EFSA FEEDAP Panel, 2012; EFSA Scientific Committee, 2012a), the CONTAM Panel calculated an approximate daily intake of OTA of 9 μg/kg bw.
In a series of experiments in cross‐bred ([Landrace × Yorkshire] × Duroc) piglets administered OTA at 0, 400, 800 μg/kg diet (corresponding to approx. 20 and 36 μg/kg bw per day) for 42 days, decreased growth performance and renal histopathological changes were observed. These consisted of hyperchromatic nuclei and cytoplasm, nuclear atrophy, necrosis and exfoliation of proximal tubule cells and were consistently reported at doses equal or greater than 400 μg OTA/kg diet (Zhang et al., 2016; Gan et al., 2017b). Treatment with OTA at these dose levels also lead to a dose‐related decrease in TCR‐induced T lymphocyte viability in peripheral blood lymphocytes and splenocytes, accompanied by decreased concentrations of IL‐2 and increased concentrations of TNF‐α in serum. Increased GRP78 expression, increased p38 and ERK1/2 phosphorylation and increased expression of the autophagy‐related gene Atg5 and LC3II genes were observed in kidney and spleen of OTA‐treated pigs (Gan et al., 2017b), suggesting to the authors that nephrotoxicity and immunotoxicity of OTA may involve ER stress, activation of MAPK signalling and autophagy (Gan et al., 2017b,c).
Proximal tubule injury accompanied by increased serum creatinine and urea was reported in male and female pigs (Landrace×Bulgarian White) fed a mouldy diet containing 500 μg OTA/kg feed (corresponding to approximately 15 μg/kg bw per day) for 3 months (Stoev et al., 2012). Slight degenerative changes were also recorded in the duodenal or jejunal mucosa. At the same dose, a strong decrease in antibody titre was observed after immunisation against Morbus Aujesky (Pseudorabies), suggesting that OTA affects humoral immune response (Stoev et al., 2012).
4.2.2.7.1. Summary remarks on studies in pigs published since 2006
Overall, the CONTAM Panel concluded that, the new studies in pigs, even though most of them are of shorter duration, support OTA‐mediated effects at dose levels close to the previously established LOAEL of 8 μg/kg bw per day but did not provide LOAELs that were below this dose. The CONTAM Panel noted that the subtle changes in biochemical parameters reported in the 33‐day feeding study using only one dose by Marin et al. (2016) could not be considered as being clearly adverse and are inconsistent with a subsequent report from the same group (Marin et al., 2018). Therefore, the CONTAM Panel considered that the study by Marin et al. (2016) was not suitable for hazard characterisation.
4.2.3. Chronic toxicity and carcinogenicity studies
4.2.3.1. Studies with rodents published before 2006
OTA has been shown to cause increased incidences of renal tumours in rats and mice, with male rats being most sensitive, and liver tumours in mice (both sexes). Since it has been considered as a pivotal study for characterisation of carcinogenic hazards by OTA in several previous assessments (FAO/WHO, 1991, 1996, 2001, 2008; EFSA, 2006), the 2‐year gavage study with from NTP (1989) is described here again in detail. Groups of male and female F344/N rats (n = 80/group) were administered doses of 0, 21, 70 and 210 μg/kg bw OTA (98% pure) in corn oil by gavage 5 days per week for up to 2 years (equivalent to 0, 15, 50 and 150 μg OTA/kg bw per day), with interim sacrifices at 9 and 15 months (n = 15/group per sex) (NTP, 1989). After 9 and 15 months, increased incidences of renal tubular cell neoplasms were observed in males along with non‐neoplastic kidney lesions characterised by hyperplasia, degeneration and karyomegaly of renal tubular epithelial cells in both males and females. At both time points, renal tubular epithelial karyomegalic changes were evident in 100% of male and female rats in the 50 and 150 μg/kg bw dose group, whereas no such changes were recorded in rats given the lower dose of 15 μg/kg bw. After 2 years, increased incidences of kidney tubule adenoma and carcinoma accompanied by proliferative and degenerative kidney lesions (degeneration, hyperplasia, karyomegaly, proliferation) were observed in the 50 and 150 μg/kg dose group of both sexes, with highest incidences in males (Table 5).
Table 5.
Incidence of renal tumours and karyomegaly in male F344/N rats (NTP, 1989)
| Dose in μg/kg bw per day | Adenomas | Carcinomas | Adenomas and carcinomas | Karyomegaly |
|---|---|---|---|---|
| 0 | 1/50 (2%) | 0/50 (0%) | 1/50 (2%) | 0/50 (0%) |
| 15 | 1/50 (2%) | 0/50 (0%) | 1/50 (2%) | 1/50 (2%) |
| 50 | 6/51 (12%) | 16/50 (31%) | 20/51 (39%) | 51/51 (100%) |
| 150 | 10/50 (20%) | 30/50 (60%) | 36/50 (78%) | 50/50 (100%) |
Compared with rats, mice appear to be less susceptible to renal carcinogenicity of OTA. In a 24‐month feeding study in C57BL/6J X C3H)F1 (B6C3F1) mice, renal adenomas (26/49) and carcinomas (14/49) were observed in male mice fed diet containing 40 mg/kg OTA (the crude preparation contained 84% OTA, 7% OTB and 9% benzene) (Bendele et al., 1985), corresponding to approximately 4.5 mg/kg bw per day (Huff, 1991). No renal neoplasms were found in female B6C3F1 mice and in males fed OTA at 1 mg/kg in diet, whereas the incidence of hepatocellular adenomas and carcinomas was slightly increased in male and female B6C3F1 mice administered OTA‐contaminated diet (Bendele et al., 1985). Renal cystadenomas and renal cell tumours as well as liver tumours were also recorded in male ddY and DDD mice exposed to OTA via feed (Kanisawa and Suzuki, 1978; Huff, 1991), although the number of animals per group was considered insufficient to derive adequate information on tumour incidence (Bendele et al., 1985).
4.2.3.2. Studies with rodents published after 2006
New studies investigating the carcinogenic effects of OTA in rodents (Table 6) are briefly summarised below.
Table 6.
Summary of chronic toxicity studies, including carcinogenicity studies published since 2006
| Species | Dose and animals per group | Duration/time of observation | Main effects observed | Effect level | Reference |
|---|---|---|---|---|---|
| Chronic toxicity studies in rats | |||||
| F344 (♂) | 0, 70, 210 μg/kg bw (gavage, corn oil, 5 days/week) equivalent to 0, 50, 150 μg/kg bw per day n = 6 | 2, 4, 13, 26 weeks |
|
LOAEL* 50 μg/kg bw per day (based on renal histopathology) | Dai et al. (2014) |
| F344 (♀) | 0, 3 mg/kg diet (corresponding to approx. 300 μg/kg bw per day**), n = 7 | 6, 9 and 24 weeks |
|
300 μg/kg bw per day**) | Mor et al. (2017) |
| Carcinogenicity studies in rats | |||||
| F344 (♂) | 0, ~ 1 mg/kg feed corresponding to ~ 50 μg/kg/bw per day’ n = 34 (treatment), n = 15 (controls) | Up to 2 years (105‐week exposure) |
|
~ 50 μg/kg bw per day | Mantle and Kulinskaya (2010) |
| F344 (♂) |
~ 250 μg/kg bw per day( a ) No information on controls and total number of animals provided |
Up to 2 years (interim sacrifices at 7, 21 days 4, 7, 12 months; n = 4/dose/time point) |
|
~ 250 μg/kg bw per day( a ) | Mantle et al. (2005) |
|
Marin‐Kuan (2006) | ||||
|
Marin‐Kuan et al. (2007) | ||||
| Sprague‐Dawley ♀ × F344 ♂ | 0, 5 mg/kg diet (5 μg/kg) for 36 weeks (corresponding to ~ 150 μg/kg bw per day (♂) and ~200 μg/kg bw per day (♀)) followed by sodium barbitate (500 μg/kg in drinking water) for life, n = 5 ♂ (OTA); n = 5 ♂ + 3 ♀ (OTA + barbitate); n = 3 ♀ (controls) | 36‐week exposure to OTA followed by sodium barbitate (500 μg/kg in drinking water) for life |
|
~ 150 μg/kg bw per day (based on renal tumours) | Mantle et al. (2010a) |
| Dark Agouti (♂) | 5 mg/kg diet (5 μg/kg) (640–250 μg/kg bw per day) the dose falling as animals gained weight, n = 20 (treatment) No controls | 3, 6, 9 months exposure (observation throughout natural life) |
|
~ 640–250 μg/kg bw per day (based on renal tumours) | Mantle (2009) |
| 400 μg/kg diet (corresponding to ~20‐30 μg/kg bw per day) n = 20 (treatment) No controls | up to 2 years |
|
NOAEL* ~ 20–30 μg/kg bw per day (based on renal tumours) | Mantle (2009) | |
| F344 (♂) | 0, 5 mg/kg diet (300 μg/kg bw per day) n = 5 (treatment, 1st year of life) n = 12 (treatment, lifetime exposure n = 13 (controls) | 10‐month exposure in 1st year of life vs. lifetime exposure |
|
5 mg/kg diet (~ 300 μg/kg bw per day) (based on renal tumours) | Mantle (2016) |
| Chronic toxicity and carcinogenicity studies in mice | |||||
| Swiss albino mice (females) | A single topical application of DMBA (30 μg/mice) or OTA (80 μg/mice) followed by twice weekly application of TPA (2.5 mg) after a week of initiation | 24 weeks |
|
LOAEL as initiator: OTA (20 μg/mice) | Kumar et al. (2012) |
| Swiss albino mice (females) | Single topical application of DMBA (24 μg/mouse) followed by twice weekly application of TPA (2.4 μg/mouse) or OTA (20 μg/mouse) after a week of initiation; n = 10 | 24 weeks |
|
LOAEL as promoter: OTA (20 μg/mouse) NOAEL: OTA (10 μg/mice) | Kumar et al. (2013) |
| P53N5‐T (p53+/−) and P53N5‐W (p53+/+) mice (♂) | 0, 0.5, 2 or 10 mg/kg diet( b ) (corresponding to ~ 0, 30, 200, 1,500 μg/kg bw per day) n = 10 | 26 weeks |
|
LOAEL( c ) 200 μg OTA/kg bw per day (based on non‐neoplastic kidney lesions) | Bondy et al. (2015) |
DMBA: 7,12‐dimethyl benz[α]anthracene; ELK1/2 : nuclear substrate ETS protein 1/2; ERK1/2: extracellular signal‐regulated kinase 1/2; HNF4α: hepatocyte nuclear factor 4 alpha; LOAEL: lowest observed adverse effect level; MAPK: mitogen‐activated protein kinase; NOAEL: no observed adverse effect level; Nrf2: nuclear factor‐erythroid2‐related factor 2; OSOM: outer stripe of the outer medulla; OTA: ochratoxin A; p90RSK; ribosomal‐S6 kinase; PKC: protein kinase C; TPA: 12‐O‐ tetradecanoylphorbol‐13‐acetate.
*: NOAEL/LOAEL not explicitly reported but derived from data presented in study.
‘As from their initial weight of ~ 175 g, daily dietary intake was 300 μg OTA/kg bw, but was held at 100 μg/rat after animals reached 333 g’.
Due to rapid weight loss of animals during initial 2 weeks of treatment, the concentration of OTA in diet was adjusted from 0, 1, 15, 40 mg/kg diet (weeks 1–2) to 0, 0.5, 2, or 10 mg/kg diet for the remaining exposure period (weeks 3–26). Mean daily OTA intake during week 1 was 0, 0.12, 1.44, 4.42 mg/kg bw (p53+/+) and 0, 0.09, 1.30, 3.94 mg/kg bw (p53+/−). Mean daily OTA intake in weeks 3–26 was 0, 0.03, 0.20, 1.46 mg/kg bw (p53+/+) and 0, 0.03, 0.19, 1.55 mg/kg bw (p53+/−).
Note that the study authors designated an LOEL, but the CONTAM Panel considers this a LOAEL.
In a group of 34 male F344 rats administered OTA in diet corresponding to 50 μg OTA/kg bw per day for up to 105 weeks, two renal adenomas and four renal carcinomas corresponding to a renal carcinoma incidence of 12% were observed (Mantle and Kulinskaya, 2010). The same group reported a renal carcinoma incidence of 20% following chronic dietary administration of OTA to male F344 rats at approximately 250 μg OTA/kg bw per day. In this study, male F344 rats were given OTA via feed at a dose corresponding to 300 μg OTA/kg bw until rats reached a body weight of 333 g, and were then maintained at a dose of 100 μg/rat (Mantle et al., 2005). No details such as initial number of animals per dose group or number of controls were presented. Leukaemia occurred in 50% of animals. A total number of 64 OTA‐treated rats were used as a basis to establish renal tumour incidence. In these animals, a total of 16 renal tumours were discovered after ≥ 75 weeks of OTA treatment (i.e. total renal tumour incidence 25%). Most tumours were unilateral carcinomas, while renal tumours in three animals were classified as adenomas (i.e. renal carcinoma incidence 20%). The lower tumour incidences in these feeding studies as compared to the 2‐year oral gavage study (NTP, 1989) were considered by the authors to be due to the different routes of administration, i.e. feed vs. gavage, with the lower potency of OTA administered via feed being more relevant to humans (Mantle et al., 2005; Mantle and Kulinskaya, 2010). Considering the high incidence of mononuclear leukaemia in both controls (69%) and rats dosed with OTA (47% and 50% in animals given OTA at 50 and 250 μg/kg bw per day) that resulted in premature death of animals these studies (Mantle and Kulinskaya, 2010), a pilot study was conducted in hybrid rats (offspring of female Sprague Dawley crossed with male F344) to minimise the confounding factor of spontaneous leukaemia during ageing. Rats (10 males, 3 females) were administered feed contaminated with OTA (5 mg/kg) for 36 weeks, which was estimated to correspond to a dose of ~ 150 μg OTA/kg bw in males and ~200 μg OTA/kg bw in females (Mantle et al., 2010a). A subgroup of animals (5 males, 3 females) was then given sodium barbitate in drinking water for life to test if renal tumour formation could be accelerated by the tumour promoter. Three female rats were used as controls. Renal tumours were found in 2/5 (40%) male rats given OTA, and in 3/5 (60%) male rats given OTA followed by sodium barbitate in drinking water. A small renal tumour (adenoma) was found in a female rat given OTA followed by sodium barbitate. All three females in this group developed mammary tumours. In the control group, a mammary tumour was observed. Barbitate treatment was considered to only slightly accelerate the development of OTA‐induced renal tumours (Mantle et al., 2010a). Renal tumours were also observed in male Dark Agouti rats given OTA in diet at 5 μg/kg for 6 and 9 months and monitored throughout natural life, whereas no renal tumours were found in a group of rats administered OTA at 400 μg/kg in the diet for up to 2 years (Mantle, 2009). Based on these findings, the authors suggested OTA at 400 μg/kg in diet (corresponding to ~ 20–30 μg OTA/kg bw) as a threshold dose for OTA‐induced renal carcinogenicity in this rat strain (Mantle, 2009).
In a further study in male F344 rats (n = 5; 10 weeks old at beginning of OTA exposure) by the same group, 10‐month exposure to OTA at 5 mg/kg diet corresponding to an intake of 300 μg OTA/kg bw per day within the first year of life followed by an exposure‐free period for life was reported to cause a 100% incidence of renal carcinomas. In the same study, seven renal carcinomas were found in 12 animals continuously exposed to OTA at the same dose for life, corresponding to a renal carcinoma incidence of 58% (Mantle, 2016). The lower tumour incidence in the continuous exposure may be related to the high incidence of mononuclear leukaemia (41%) and reduced longevity as compared to the 10‐month exposure group (75% of rats in the continuous exposure group had already deceased before tumours were discovered in the 10‐month exposure group). High incidences of mononuclear leukaemia (69%), but no renal tumours were found in controls.
No renal tumours were observed in p53 heterozygous (p53+/−) and p53 homozygous (p53+/+) mice after chronic exposure to OTA in diet for 26 weeks. In this study, the levels of OTA in diet were adjusted from 1, 15 and 40 mg OTA/kg diet to 0.5, 2 and 10 mg OTA/kg diet (equivalent to ~ 0.03, 0.2 and 1.5 mg OTA/kg bw) due to a rapid weight loss during the first 2 weeks of exposure. Minimal nuclear variability in size (anisokaryosis) was recorded at the low dose of 0.5 mg OTA/kg diet, whereas apoptosis, karyomegaly and tubular degeneration were only at 2 and 10 mg OTA/kg diet. Based on these findings, the authors derived an LOEL (note that the CONTAM Panel considers this to be a LOAEL) for renal changes in p53+/− and p53+/+ mice of 2 mg/kg diet (corresponding to 200 μg/kg bw per day) (Bondy et al., 2015).
In a two‐stage mouse skin tumorigenesis protocol, a single topical application of OTA (80 μg/mouse) followed by twice weekly application of 12‐O‐ tetradecanoylphorbol‐13‐acetate (TPA) for 24 weeks lead to the development of skin tumours in 40% of the animals. These data indicate that OTA has skin tumour initiating properties. Early events induced by single OTA skin application included increased levels of DNA damage (SSBs and γH2AX), induction of the DNA damage response (p53 and p21) and of several markers of oxidative stress (see also section 4.3 on Mode of action) (Kumar et al., 2012). In a follow‐up study by the same group, a single topical application of 7,12‐dimethyl benz[α]anthracene (DMBA) as tumour initiator followed by twice weekly application for 24 weeks of OTA (20 μg/mouse) as tumour promoter resulted in the development of skin papillomas. Tumours developed in 100% and 40% of mice in DMBA/TPA‐ and in DMBA/OTA‐treated groups, respectively, indicating that OTA is a weak tumour promoter in comparison to TPA (Kumar et al., 2013).
4.2.3.2.1. Summary on toxicity
The CONTAM Panel noted the inadequate study design and unclear reporting of the new studies (published after 2006) investigating the carcinogenic effects of OTA and considered that these provided only limited additional information on OTA carcinogenicity that could serve as a basis for quantitative risk assessment (Table 6). Long‐term OTA administration of OTA to female Wistar rats was reported to cause toxic effects on the endocrine pancreas, suggesting diabetogenic potential of OTA (Mor et al., 2017). These effects were evident at a dose higher than those required for nephrotoxicity and renal tumour formation (Table 6), and therefore did not provide a more sensitive endpoint of toxicity.
4.2.4. Genotoxicity
4.2.4.1. Introduction
In the previous EFSA opinion on OTA (EFSA, 2006), the CONTAM Panel concluded that ‘DNA damage and genotoxic effects of OTA were most likely attributable to cellular oxidative damage and within the limit of sensitivity of the analytical procedures, there was no clear evidence for the formation of specific OTA‐containing DNA adducts’. The evidence available at the time included:
a) negative mutation assays in Salmonella Typhimurium tester strains (with exception of a single study reporting positive results in TA1535, TA98 and TA1538 strains incubated with murine kidney microsomes in the presence of either NADP or arachidonic acid as cofactors (Obrecht‐Pflumio et al., 1999),
b) in vitro evidence of increased mutations in V79 cells, and
c) increased micronuclei and single strand breaks (SSBs) seen in comet assays in vitro.
Few in vivo studies were available at the time. Here we review data published from 2006 to 2018 addressing the potential genotoxicity of OTA both in cultured mammalian cells and animal models. The findings are summarised below, and further details are presented in Tables 7 (in vitro studies) and 8 (in vivo studies).
Table 7.
In vitro studies on OTA genotoxicity
| Test system | Cells | Concentration/Treatment time | Results | Comments | Reference |
|---|---|---|---|---|---|
| SSBs by comet assays | NIH/3T3 cells expressing human CYP450s, 1A1, 1A2, 2C9, 3A4 and human oxidoreductase (hOR) | 10, 25, 50, 100, 150, 200 μM (8 h) | Positive: Only in CYP2C9‐hOR. No toxicity (MTT and neutral red, no data presented) | Increased ROS. Poor presentation of the results | SimarroDoorten et al. (2006) |
| SSBs by comet assays +/− Fpg/EndoIII | Human renal proximal tubular epithelial cells (HK‐2) | 50, 100, 200, 400, 600 μM +/− rat liver S9 (3 and 6 h) | Positive: Slight increase at 6 h (400 and 600 μM, 50–60% survival); Survival and SSB unaffected by S9; SSB increased by Fpg and EndoIII (unconvincing data) | Increased ROS levels; slight protection by NAC | Arbillaga et al. (2007) |
| SSBs by comet assays, polyploidy by flow cytometry | CHO cells | 0.2, 0.8, 1 mM (3 h) |
Positive: Comet assay. Extremely toxic doses (cytotoxic at > 10 μM) Positive: polyploidy |
Dose‐dependent inhibition of TopoII (in vitro assay) | Cosimi et al. (2009) |
|
SSBs in comet assays; DSBs in γH2AX assay 8‐OHdG with HPLC‐ECD |
Human peripheral blood mononuclear cells | 5, 10, 20 μM (24 h) | Positive: SSB, DSBs, 8‐OHdG (dose‐dependent increases). No information on the toxicity of the treatment | Increased ROS at all doses. SSB partially inhibited by NAC‐pre‐treatment. Accumulation in G1 phase | Liu et al. (2012) |
| SSB by comet assays, DSBs with γH2AX (IF detection and western blotting), 8‐OHdG detection with HPLC‐ECD | Human gastric cell line (GES‐1) | 5, 10, 20 μM (24 h) | Positive: SSB, DSBs, 8‐OHdG (dose‐dependent increases). No information on the toxicity of the treatment | Increased ROS; G2 phase arrest; Phosphorylation of ATM, CHK2, p53 | Cui et al. (2013) |
| SSBs in comet assays +/− pg; MN | CHO and human TK6 cells | MN: 5, 7.5, 10, 12.5, 15, 17.5, 20, 22.5, 25 μM (24‐27 h). Comet assays: 5, 10, 20, 30, 40, 50 μM (4 h) |
Positive: SSB +/− Fpg in TK6 Positive: SSB only + Fpg in CHO cells Positive: MN in TK6 and possibly in CHO cells (borderline levels of toxicity) |
Hypodiploid cells in both cell lines (scored in the MN, assays). Evidence of both direct and indirect DNA damage | Ali et al. (2011) |
| SSBs by comet assays; MN | Green monkey kidney Vero cell line | 10 μM (24 h) |
Positive: SSB Positive: MN. Experiments at 80% survival |
Limitation: Single dose observations | Ramyaa and Padma (2013) |
| SSBs in comet assays; MN | Human lymphocytes | Comet assays: 0.075, 0.15, 1.5, 5, 15 μM; MN: 0.075, 0.15, 1.5, 5 μM (3 h) |
Positive: Small increases in SSB at single points, no dose response Positive: Small increases in micronuclei (1.5 and 5 μM). Experiments at > 80% survival |
González‐Arias et al. (2014) | |
| SSBs in comet assays; DSBs by γH2AX | Human embryonic kidney cells (HEK 293) | 12.5, 25, 50 μM (24 h) | Positive: Dose‐dependent increase in SSB and DSBs. Limited toxicity (MTT assay) | Increased ROS; cell cycle arrest at S phase; downregulation of S‐phase proteins (Cyclin A2 and E1 and CDK2); decreased mitochondrial membrane potential | Yang et al. (2014) |
| SSBs in comet assays; CA | Human oesophageal epithelium immortalised cells (Het‐1A) | 2.5, 5, 10, 20 μM (24 h) |
Positive: dose response increase in SSB (IC50: 65.9 μM in MTT assay) Positive: dose response increase in CA |
G2 phase arrest (10–20 μM). Decreased expression of Cyclin B1, Cdc2 and Phospho‐Cdc2 | Liu et al. (2015) |
| SSBs in comet assays | Porcine epithelial kidney PK15 cells; human leucocytes | 1 and 5 μM (1 and 24 h) | Positive: dose‐ and time‐dependent increases (both cell lines). Non‐toxic doses by MTT assay | Combined effects with BEA | Klarić et al. (2010) |
| SSBs in comet assays | Primary rat hepatocytes | 2, 4, 6 nM (12 h) | Positive: at 4 and 6 nM (viability with MTT: 78% and 62%, respectively) | Limited study | Goyary et al. (2014) |
| SSBs in comet assays | Human peripheral blood lymphocytes | 1, 2, 4 μM (1 h) | Positive: SSB only at 4 μm/L (no toxicity with ethidium bromide/acridine orange) | Prevention by sodium copper chlorophyllin. Limited study | Domijan et al. (2015) |
|
SSBs in comet assays Plasmid DNA damage assay |
Mouse neuroblastoma Neuro‐2a cell line | 100, 250, 500 nM (24 h) | Positive: All doses (toxicity > 50%). Plasmid DNA damage assay (non‐informative) | Increased ROS; Small protection by NAC | Bhat et al. (2016) |
| SSBs by comet assays +/− Fpg; MN | Green monkey kidney Vero cell line | 7.5, 15, 25, 50 μM (24 h) |
Positive: SSB (only at 15μM in the presence of Fpg) Positive: MN (only at 25 μM) |
No dose response in MN and comet assays. Limited information | Costa et al. (2016) |
| DSBs by γH2AX (western blotting) | Human gastric cells (GES‐1) | 5, 10, 20 μM (24 h) | Positive: DSBs (small increase at 20 μM). No information on toxicity | Decreased Rad51 levels. Overexpression of Rad51 decreases OTA‐induced DSBs. Limited study | Lian et al. (2014) |
| MN | V79 cells and primary porcine urothelial bladder epithelial cells (PUBEC) | 0.01, 0.03, 0.1, 0.3, 1, 3, 10 μM (18 and 24 h for V79, 24 h for PUBEC) | Positive: V79 (0.3–10 μM but no dose response); PUBEC (3–10 μM) | Unclear levels of toxicity | Föllmann et al. (2007) |
| MN, SCE, CA endoreduplication, polyploidy | Human lymphocytes; V79 cells | 23.6‐1149 μM (+/− rat kidney and liver S9) |
Negative: SCEs, MN, CA Positive: Endoreduplications and polyploidy (V79 + kidney S9); (human lymphocytes +/− liver S9) |
Metaphases with abnormally separated chromatids | Mosesso et al. (2008) |
| Aberrant mitoses | Immortalised human kidney epithelial cells (IHKE) | 1, 10, 50 μM (12 and 24 h) | Positive: Dose‐response increase in giant cells with enlarged/multiple nuclei. Asymmetric/multipolar mitotic spindles | Inhibition of microtubule assembly in a cell‐free in vitro assay (200 & 400 μM) | Rached et al. (2006) |
| Chromosome condensation and loss of sister chromatid separation | Immortalised human kidney epithelial cells (IHKE) | 0, 1, 5, 10, 25, 50 μM (up to 15 h) | Positive: Chromosome over‐condensation and aberrant separation of sister chromatids | Decreased phosphorylation of histone H3Th3 and decreased acetylation of histones H3 and H4 | Czakai et al. (2011) |
| MN | Porcine epithelial kidney cells (PK15) | 0.05, 0.5, 5 μg/mL (corresponding to 0.12, 1.25, 12.5 μM (24 and 48 h) | Positive: All concentrations: No information on toxicity | Combined effects with BEA and FB1 | Klarić et al. (2008) |
| SCEs | Human hepatocellular carcinoma cells (Hep3B) | 10−12−2 × 10−4 M (72 h) | Positive: range 10−12–×10−6 M. No dose response; no information on toxicity (by MTT only at 24 and 48 h) | Combined effects with sterigmatocystin and citrinin | Anninou et al. (2014) |
| Mutations at Hprt and Tk genes | Chinese hamster V79 cells and mouse lymphoma cells (L5178Y) |
V79 (7, 11, 20, 35, 46, 108, 251 μM; 3h and 24 h); +/− rat kidney and liver S9 L5178Y (3, 81, 188, 438μM, 3 h) +/− rat kidney S9 |
Positive: Increased mutation frequency in both cell lines. No effects of S9. Mutational spectrum similar to the endogenous one (V79); point mutations and large deletions (L5178Y) | G2‐phase/M‐phase cell cycle arrest (V79 cells) | Palma et al. (2007) |
| Mutations in the SupF gene in plasmid Ps189 | Human embryonic kidney cells (Ad293) | 0.5, 1, 5 μM +/− rat liver microsomes or Fe(III)/Cu(II) metals | Positive: All doses, only + S9 or metal activation. (OTHQ: positive without metabolic activation) | No dose response. No proper controls for metal activation | Akman et al. (2012) |
| Mutations Tk+/− SSB by comet assays +/− Fpg | Mouse lymphoma cells (L5178Y) | 0, 5, 10, 25, 50, 100 μM +/− S9 rat liver (4 h) |
Positive: Dose‐dependent increase in mutations (from 25 μM) Positive: Dose‐dependent increase in SSB (increased by Fpg). No effect of S9 for both endpoints |
Large colonies (gene mutations). All assays at survival levels > 70% | Ali et al. (2014) |
| 8‐OHdG by oxyblot | Canine umbilical cord matrix‐mesenchymal stem cells (UCM‐MSC) | 0.025, 2.5 nM (4 and 7 days) | Positive: Similar increases in DNA 8‐OHdG levels at 0.025 and 2.5 nM. Less than 50% cell growth and cells showing fragmented chromatin | Rutigliano et al. (2015) | |
| 8‐OHdG by oxyblot | Bovine mammary epithelial (BME‐UV1) cells; Madin‐Darby canine kidney cells (MDCK) | 0.3, 0.6, 1.25 μg/mL (corresponding to 0.75, 1.5, 3.1 μM) |
Positive: BME‐UV1 (only 1.25 μg/mL, survival 40%) Negative: MDCK (range survival 100%–20%) |
No changes in the global content of 5‐meC | Giromini et al. (2016) |
| AP sites | Rat kidney cells (NRK) and rat hepatocytes | 3, 6 μM (24 h) | Positive: similar increases at 3 and 6 μM (unclear toxicity) | Non‐standard test | Cavin et al. (2007, 2009) |
| UDS | Swine lymphocytes, swine kidney cells (SK6) | 0.01–50 μM (72 h) |
Positive: swine lymphocytes (1 and 2 μM, cytotoxic concentration > 5 μM) ositive: SK6 (0.5–4 μM) |
Stec et al. (2006) |
8‐OHdG: 8‐hydroxydeoxyguanosine; AP: Apurinic/apyrimidinic; ATM; ataxia telangiectasia mutated gene; BEA: Beauvericin; CA: chromosome aberrations; cdc2; cell division control protein; CDK2: cyclin‐dependent kinase 2; CHK2; checkpoint kinase 2; CHO; Chinese hamster ovary; CTN: citrinin; CYP450: cytochrome P‐450); DSBs: Double strand breaks; EndoIII: endonuclease III; FB1: Fumonisin B1; fpg; formamido‐pyrimidine‐DNA glycosylase; γH2AX: Histone variant H2AX in which the Ser‐139 residue variant is phosphorylated (an early cellular response to induction of DSBs); G2: gap phase 2; HPLC‐ECD: High‐Performance Liquid Chromatography with Electrochemical Detection; IF: immunofluorescence; MN: micronuclei; MTT; 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide; NAC: N‐acetyl‐ L‐cysteine; OTHQ: ochratoxin hydroquinone metabolite; p53; tumour protein p53; ROS: reactive oxygen species; SSBs: single strand breaks; SCE: sister chromatid exchange; SupF: E. coli tyrosine amber suppressor tRNA gene; Tk: Thymidin kinase; TopoII; topoisomerase II; UDS: unscheduled DNA synthesis; V79 cells: Chinese hamster lung cells clone V79.
4.2.4.2. In vitro genotoxicity studies
No mutagenicity assays in S. Typhimurium published after 2006 were identified. In contrast, the genotoxic potential of OTA has been extensively investigated in comet assays with induction of SSBs as endpoint. Positive results were reported in established rodent, porcine, canine, bovine, green monkey and human cell lines as well as in primary cultures. Several of these cell lines are of kidney origin (see Table 8). With a single exception (SimarroDoorten et al., 2006), SSB induction was observed in the absence of rat liver S9. It is possible that some of these OTA‐associated SSBs are secondary to oxidative stress. Inclusion of formamido‐pyrimidine‐DNA glycosylase (Fpg)/endonuclease III (EndoIII) digestion in the Comet assays to reveal the presence of oxidised purines and pyrimidines, increased the yield of SSBs. This observation is consistent with oxidative DNA damage (Arbillaga et al., 2007; Ali et al., 2011, 2014; Costa et al., 2016). The induction of DNA 8‐hydroxydeoxyguanosine (8‐OHdG) by OTA‐treatment of cells was confirmed by direct measurements (Liu et al., 2012; Cui et al., 2013; Rutigliano et al., 2015; Giromini et al., 2016). Consistent with these findings, OTA exposure increased the levels of cellular reactive oxygen species (ROS) (SimarroDoorten et al., 2006; Arbillaga et al., 2007; Liu et al., 2012; Cui et al., 2013; Yang et al., 2014; Bhat et al., 2016).
Table 8.
In vivo studies on OTA genotoxicity
| Test system | Animals | Concentration/Treatment | Result | Comments | Reference |
|---|---|---|---|---|---|
| SSBs by comet assays +/−Fpg (kidney) | Wistar rats (males) | 0.005, 0.05 and 0.5 mg/kg bw/15 days (oral) | Positive: all doses. SSBs increased by Fpg | Combined effects with FB1: synergistic increase in SSB | Domijan et al. (2006) |
| SSBs by comet assays in liver, kidney, lymphocytes | Sprague–Dawley rat (males) | 0.5 mg/kg bw/14 days (gavage) |
Positive: liver & kidney Negative: lymphocytes |
Protection by lycopene | Aydin et al. (2013) |
| SSBs by comet assays +/−Fpg in kidney | Fisher rats (males & females) |
0.5 mg/kg bw/7 days 0.21 and 0.5 mg/kg bw/21 days (gavage) |
Negative: in both males and females | No changes in total GST and SOD activity, total GSH and GSSG levels | Enciso et al. (2018) |
| SSBs by comet assays +/− hOGG1 in kidney and liver | Wistar rats (males) | 0.125 and 0.250 mg/kg bw/21 days (gavage) |
Negative: kidney Positive: liver (only at highest does) but no control data for comet assays are provided |
Combined effects with CTN Limited value (no controls) |
Rašić et al. (2018) |
| SSBs by comet assays in kidney | Wistar rats (female) | 0.5 mg/kg bw/7, 14, and 21 days (i.p.) | Positive | Unreliable study (0 values in controls) | Zeljezić et al. (2006) |
|
SSBs by comet assays assays +/− Fpg (kidney, liver, spleen, blood) CA in splenocytes |
Wistar rats (males) |
OTA: 0.25, 0.5, 1 and 2 mg/kg bw/2 weeks (oral) OTB: 2 mg/kg bw/2 weeks (oral) |
Positive (OTA): SSBs in kidney (all doses), liver and spleen (0.5–2 mg/kg). Increased by Fpg in liver and blood (all doses), kidney (only highest dose) Negative (OTA): CA in splenocytes (non‐significant increase) |
Increased polyploidy in renal medulla (but not in cortical regions of the kidney) OTB‐induced SSBs only in liver and spleen |
Mally et al. (2005b) |
| SSBs by comet assays in blood, micronuclei in bone marrow | BALB/c mice (males) | 0.85 mg/kg bw i.p. Analyses at 24 h |
Positive: SSBs in blood Positive: micronuclei in bone marrow |
Combined effects with Polyphenols. Limited study (single dose) | Cariddi et al. (2015) |
|
SSBs by comet assays +/− Fpg in liver and kidney Micronuclei in bone marrow |
F344 rats (males) | 0.5 mg/kg bw/3h and 24 h (oral). No clinical signs of toxicity |
Positive: SSBs in kidney (only +Fpg) Negative: SSBs in liver Negative: micronuclei |
Combined treatment with AFB1: antagonistic effects | Corcuera et al. (2015) |
| Micronuclei in bone marrow | Sprague–Dawley rats (males) | 3 mg/kg bw/15 days (oral) | Positive: single and very high dose (30% of death in OTA group) | Combined treatment with plant extracts (Inulacrithmoides). Limited study | Abdel‐Wahhab et al. (2008) |
| DSBs by γH2AX | F344 rats (males) | 0.21 mg/kg bw/28 days (gavage) | Positive: DSBs in outer stripe of outer medulla | Increased cdc‐2‐positive and P‐Chk2 cells. Karyomegaly in outer medulla | Taniai et al. (2012a) |
| CA in bone marrow | BALB/c (males) | 0.6, 1.2, and 2.4 mg/kg bw (corresponding to 2, 4, and 8% of the mouse LD50, 31 mg/kg bw) (sacrifice 24 h after single ip injection) | Positive: dose‐response increase in CA at 1.2 and 2.4 mg/kg bw (no statistical significance provided) | Bouslimi et al. (2008) | |
| Mutations in Gpt and Red/Gam genes (Spi − ) in kidney | Gpt‐delta F344 rats (males & females) | 5 ppm (gavage)/4 & 13 wks) (corresponding to 0.36–0.38 mg/kg bw/4 wks and 32.7–34 mg/kg bw/13 wks for males & females, respectively). Decreased absolute/relative kidney weights (both times) |
Negative: gpt and Spi − mutations in the whole kidney (4 and 13 wks). No changes in mutational spectra of gptmutants Positive: Spi − mutations in outer medulla, but not in inner medulla or renal cortex (4 wks) |
Karyomegaly in outer medulla (both times) No increase in DNA 8‐OHdG levels in the cortex or outer medulla (13 wks) |
Hibi et al. (2011) |
| Mutations in Gpt and Red/Gam (Spi − ) in kidney | Gpt‐delta p53+/+ and p53−/− C57BL/6J mouse | 1 and 5 mg/kg bw/4 weeks (gavage)(males). Decreased body weights & absolute/relative kidney weights (highest dose) |
Negative: no increase in mutation frequency at both loci in p53+/+ mice. No difference in mutational spectra at Gpt Positive: increased Spi ‐ mutations in p53−/− mice in outer medulla (only at 5 mg/kg bw) |
Increased p53 in p53+/+ (5 mg/kg bw). Higher levels of apoptosis & karyomegaly in p53−/− kidneys (evident also in the cortex) | Hibi et al. (2013b) |
| Mutations in Red/Gam (Spi − ) SSBs by comet assays, DSBs by γ‐H2AX (IF and western blotting) in kidney outer medulla | Gpt‐delta F344 rats (males) |
Spi‐ mutations: 5 ppm/4 weeks (0.36 mg/kg bw) SSBs & DSBs: 0.07, 0.21, 0.63 mg/kg bw/4 weeks (gavage) |
Positive: Increased Spi− mutation frequency in outer medulla (large deletions, range 1–6 kb) Positive: SSBs (all doses) Positive: DSBs (2 highest doses) |
Dose‐dependent increased expression of homologous recombination genes and genes involved in G2/M arrest and S/G2 phase | Kuroda et al. (2014) |
| Mutations in Red/Gam (Spi − ), SSBs by comet assays, DSBs by γ‐H2AX in kidney | Gpt‐delta p53+/+ and p53−/− C57BL/ 6J mouse (males) |
Spi‐ mutations &DSBs: 5 mg/kg bw/4 wks SSBs: 5 mg/kg bw/3 days (gavage) |
Positive: increased Spi‐ mutations only in p53−/− mice (single bp deletions in repetitive G/C sequences, insertions and base substitutions) Positive: SSBs (no difference p53+/+/ p53−/− mice) Positive: DSBs (preferentially in outer medulla of p53+/+ mice). Higher DSBs in p53−/− mice (also in the cortex) |
Increased apoptotic & karyomegalic cells (higher in p53−/− mice) Several differentially expressed genes affected by p53 status identified by global expression analysis by microarrays |
Kuroda et al. (2015) |
| DNA 8‐OHdG by LC/MS/MS in liver and kidney | F344 rats (males) | 0.25, 0.5, 1, 2 mg/kg bw/ 2 weeks (oral) | Negative | Increased apoptosis and polyploidy in cells of outer stripe of outer medulla | Mally et al. (2005a) |
|
Micronuclei in bone marrow 8‐OHdG by HPLC/ECD in kidney and liver |
Wistar rats (males) | 1 and 4 mg/kg bw/7 days | Negative: both micronuclei & 8‐OHdG | No consistent evidence of oxidative stress in the liver or kidney (ROS, SOD, GSH, MDA) | Zhu et al. (2016a) |
|
8‐OHdG by HPLC/ECD in kidney and liver SSB by comet assays |
Fisher 344 Rats (males) | 70 and 210 μg/kg bw/4 weeks and 13 weeks |
Positive: 8‐OHdG in liver (only at 4 weeks, both doses, no dose response) Positive: 8‐OHdG in kidney (only at 13 weeks, both doses, no dose response) |
Negative: SSB (but data not shown) | Qi et al. (2014a) |
| AP sites in kidney | Fisher 344 Rats (males) | Diet containing 300 μg//kg bw decreased to 100 μg/rat after animals reached 333 g (21 days and 12 months) |
Negative: 21 days Positive: 12 months |
Non‐standard test | Cavin et al. (2007) |
AFB1: Aflatoxin B1; AP: purinic/apyrimidinic; CA: Chromosome aberrations; CTN: citrinin; γ‐H2AX: phosphorylated form of histone protein of the H2A family formed when double‐strand breaks appear. Gpt: Glutamic‐pyruvic transaminase gene; GST: Glutathione‐S‐transferase; SOD: Superoxide dismutase; Spi: protein spitz precursor (a proto oncogene); GST: Glutathione synthetase; GSSG: Glutathione disulfide; 8‐OHdG: 8‐hydroxydeoxyguanosine; DSBs: Double strand breaks; hOR: human oxidoreductase; FB1: Fumonisin B1 ; Fpg: Fapy glycosylase gene; ROS: reactive oxygen species; MDA: Malondialdehyde; SSBs: Single strand breaks; STER: Sterigmatocystin.
Several studies reported evidence of OTA‐induced micronuclei, chromosomal aberrations and double strand breaks (DSBs) (Ali et al., 2011; Liu et al., 2012, 2015; Ramyaa and Padma, 2013; González‐Arias et al., 2014; Lian et al., 2014; Yang et al., 2014). DSBs were associated with a pronounced gap phase 2 (G2) arrest, ATM (ataxia telangiectasia mutated), CHK2 (checkpoint kinase 2) and p53 phosphorylation and decreased expression of several cyclins (see also Section 3.3 on Mode of Action). OTA also induced aberrant mitoses, chromosome hypercondensation with abnormally separated chromatids, asymmetric/multipolar mitotic spindles as well as endoreduplications and polyploidy (Rached et al., 2006; Mosesso et al., 2008; Cosimi et al., 2009; Czakai et al., 2011). Sister Chromatid Exchange (SCE) formation was not consistently associated with OTA exposure (Mosesso et al., 2008; Anninou et al., 2014).
OTA induced mutations in the Hprt gene of V79 cells (Palma et al., 2007) and at the L5178Y tk locus (Palma et al., 2007; Ali et al., 2014). Sequencing of Hprt mutants indicated that the OTA‐induced and spontaneous mutational spectra were similar. Analysis of OTA‐induced Tk mutant colonies revealed both point mutations and large chromosomal rearrangements. Increased mutation frequencies were also observed in the SupF gene in a plasmid treated in vitro with OTA and transfected into the human kidney Ad293 cell line (Akman et al., 2012).
4.2.4.3. In vivo genotoxicity studies
OTA exposure increased SSBs levels in kidneys of both rats and mice (Mally et al., 2005b; Domijan et al., 2006; Kuroda et al., 2014, 2015), with the single exception of a negative report in rats (Enciso et al., 2018). These positive results were confirmed in a series of more limited studies in which the synergistic and/or antagonistic effects of other chemicals (including mycotoxins) on single‐dose exposures to OTA were investigated (Aydin et al., 2013; Cariddi et al., 2015; Corcuera et al., 2015). In one report, Comet assays detection of OTA‐induced SSBs was possible only following DNA digestion with Fpg/EndoIII (Corcuera et al., 2015), although a direct increase in SSBs levels was reported in other studies (Mally et al., 2005b; Domijan et al., 2006). Overall these findings are consistent with the induction of oxidative DNA damage by OTA exposure in vivo. However, the few reports that addressed this possibility by direct measurements of rat kidney DNA 8‐OHdG were generally negative (Mally et al., 2005a; Hibi et al., 2011; Zhu et al., 2016a). With regard to non‐kidney tissues, there are both negative and positive results of OTA‐induced SSBs formation in rat liver (Mally et al., 2005b; Aydin et al., 2013; Corcuera et al., 2015), rat lymphocytes and spleen and mouse blood (Mally et al., 2005b; Aydin et al., 2013; Cariddi et al., 2015).
There are several reports of OTA‐induced DSBs (as measured by immunofluorescence or western blotting) in the tubular epithelial cells located in the outer stripe of the renal outer medulla of mouse and rat kidneys (Taniai et al., 2012a; Kuroda et al., 2014, 2015).
The impact of p53 status on OTA‐induced DNA breakage was investigated in mouse kidney. The levels of OTA‐induced SSBs were similar in p53+/+ and p53−/− mice. The frequencies of DSBs were, however, significantly higher in p53−/− mice. The authors suggest that p53 protein plays a major role in preventing the progression of OTA‐induced DNA damage into DSBs (Kuroda et al., 2015).
Whether OTA can induce chromosomal damage in the bone marrow remains unclear. Conflicting data on OTA induction of micronuclei have been obtained in mice (Cariddi et al., 2015) and rats (Abdel‐Wahhab et al., 2008; Corcuera et al., 2015; Zhu et al., 2016a). A single study reported increased chromosomal aberrations in the bone marrow of treated mice (Bouslimi et al., 2008). No increase of chromosome aberrations was observed in splenocytes of OTA‐treated rats (Mally et al., 2005b). In the same study, a dose‐dependent increase in hyperdiploid cells was observed in the renal medulla, but not in cortical regions, of kidneys from OTA‐treated animals.
Mutation induction by OTA has been thoroughly investigated in rat (Hibi et al., 2011; Kuroda et al., 2014) and mouse (Hibi et al., 2013b; Kuroda et al., 2015) kidney. In rats, mutations were analysed at the Gpt and Red/Gam (Spi‐ mutants) loci. The latter assay selectively detects deletion and frameshift mutations. After a 4‐ or 13‐week OTA treatment, no mutations were identified in the cortex. After a 4‐week OTA treatment, an increased frequency of Spi‐ (but not Gpt) mutant was specifically observed at the carcinogenic target site, i.e. in the outer medulla. OTA treatment was associated with deletions (1‐6kb), insertions as well as base substitutions (Hibi et al., 2013b). No cortical changes or increased levels of DNA 8‐oxodG were observed in the outer medulla (Hibi et al., 2011).
The role of p53 in OTA‐induced mutagenesis was also investigated in p53+/+ and p53−/− mice (Hibi et al., 2013b; Kuroda et al., 2015). OTA‐induced mutagenesis was only observed in p53‐deficient mouse kidneys. Mutations were confined to the Red/Gam locus and were not observed at the Gpt locus. The spectrum of OTA‐induced Spi‐mutants comprised single base deletions at G/C repetitive sequences, single base substitutions and insertions. The authors noted that the mutational spectra in rats and p53−/− mice were only partially overlapping.
4.2.4.4. DNA adduct formation
The possible formation of specific OTA‐induced DNA adducts remains highly controversial. An oxidative pathway leading to covalent OTA‐DNA adducts via biotransformation of OTA to the quinone OTQ (see Figure 5) has been proposed (Pfohl‐Leszkowicz and Manderville, 2012). In vitro reaction of the synthetic OTA hydroquinone OTHQ (see Figure 5) with DNA was reported to give rise to adduct spots detected by 32P‐post‐labelling (Tozlovanu et al., 2006). The chemical structure of the adducts was not identified. The DNA adduct spots ascribed to covalent attachment of the OTQ electrophile generated by OTHQ autoxidation were also detected by 32P post‐labelling after in vitro OTA treatment of salmon sperm DNA (in the presence of microsomes/NADH) as well as in WI26 and HK2 human cell lines (Tozlovanu et al., 2006). A further adduct spot observed by 32P‐post‐labelling in human kidney cells and in vivo in the kidneys of OTA‐treated rats was reported to co‐migrate with a post‐labelled carbon‐linked C8 OTA‐3′dGMP adduct standard generated by photoreaction of OTA with DNA (Tozlovanu et al., 2006; Mantle et al., 2010b). This adduct was proposed to be formed via reductive dehalogenation of OTA to generate an aryl radical (see Figure 5) that reacts with the C8 site of dG (Pfohl‐Leszkowicz and Manderville, 2012). The levels of this principle adduct in rat kidney were reported to be in the range of 20–70 × 109 nucleotides (Mantle et al., 2010b). However, these results in rat kidneys were not reproduced by other laboratories using 32P post‐labelling and stable isotope dilution LC‐MS/MS with an LOD of 3.5 OTA‐dG × 109 nucleotides (Mally et al., 2005a,b; Delatour et al., 2008).
An approach based on structure–activity relationships proposed a dissociation between the cytotoxic and mutagenic properties of OTA with the first one being due to ROS production and the second one to DNA adduction (Hadjeba‐Medjdoub et al., 2012). More recently, molecular modelling studies (molecular dynamics simulations) were used to predict the impact of the sequence context of OTA‐dG adducts on their mutagenic potential (Kathuria et al., 2017, 2018). Finally, oligonucleotides containing C‐linked C8 aryl‐dG model adducts, inserted in the NarI sequence, were used in primer extension assays with model DNA polymerases for investigating the levels of base misincorporation and DNA polymerases blockage and slippage (Manderville and Wetmore, 2017). Evidence of stalled DNA replication possibly leading to deletions and base substitution mutations at repetitive sequences were obtained (Sproviero et al., 2014). It remains unclear whether and to what extent these DNA adducts are formed in vivo and which metabolic pathway(s) are responsible for the formation. In any event, it seems unlikely that these very low numbers of DNA adducts could contribute significantly to either the observed DNA breakage or mutation.
4.2.4.5. Summary of genotoxicity
OTA exposure induces SSBs, DSBs and chromosome damage in numerous mammalian cell lines and in primary cultures from various tissues. DNA break induction does not require metabolic activation. Since OTA increases in vitro levels of ROS and DNA 8‐OHdG, some OTA‐associated SSBs may be secondary to oxidative stress. SSBs induced in rat kidney may be associated with pathological findings, although these are not accompanied by measurable increases in DNA 8‐OHdG.
OTA induces a narrow spectrum of chromosomal damage: chromosome hypercondensation, abnormally separated chromatids, multipolar mitotic spindles, endoreduplications, polyploidy and aneuploidy. Importantly, aberrant mitoses and karyomegaly were concentrated in the target area for OTA carcinogenesis (the kidney outer medulla).
OTA is a weak mutagen in vitro and in vivo. Mutations restricted to the cancer target site are detected in rats after a short (4 weeks) exposure to a carcinogenic OTA dose. Large deletions and base substitutions are the main mutagenic events. DNA 8‐OHdG at the carcinogenic target site was not detectable. The mutational spectrum in the kidney of OTA‐treated p53−/− mice is similar.
The molecular mechanism underlying OTA genotoxicity remains unclear. The formation of covalent OTA‐DNA adducts remains controversial. The extremely low reported levels of DNA adducts (10/109 nucleotides) are difficult to reconcile with the genotoxic effects of OTA and the proposed OTA‐DNA adducts are likely to be, at best, minor contributors. Nevertheless, the OTA‐induced mutations in rat and mouse kidneys are clearly not a simple consequence of oxidative DNA damage.
The mechanism underlying OTA‐induced chromosomal damage is also unclear. Abnormal anaphase figures might derive from unresolved ‘replication stress’ with break‐induced recombination of DSBs in the early stage of mitosis. In addition, aberrant chromosome segregation and cell division may reflect damage to the mitotic spindle.
4.2.5. Reproductive toxicity
In their opinion of 2006, EFSA noted that no adequate studies on reproductive toxicity with OTA were available but that several studies described in previous assessments (FAO/WHO, 1991, 1996, 2001; SCF, 1998) together with recently published studies suggest that OTA is teratogenic in rodents (EFSA, 2006). It was also noted that the lowest teratogenic dose in rodents was about 12 times higher than the LOAEL identified for OTA (8 μg/kg bw per day for renal effects in a 90‐day study with pigs). Table 9 presents new in vivo studies on reproductive toxicity published after the last EFSA evaluation.
Table 9.
In vivo reproductive and developmental toxicity studies published since 2006
| Animals | Study design | Observations | Effect level | Reference |
|---|---|---|---|---|
| Pregnant Wistar rats |
Preliminary study: Gavage; single doses of 0.0, 2.0, 2.5, 2.75, 3.0, 3.5 and 4.0 mg OTA/kg bw on one day in the period of GD 6–15. Sacrifice on GD 20 Main study: Gavage, single doses of 0.0 and 2.75 mg OTA/kg bw per day in the period of GD 6–15. Sacrifice on GD 20. N = 10 per group |
Preliminary study: Doses of ≥ 3.0 mg/kg bw: pronounced maternal and developmental toxicity Doses of ≤ 2.0 mg/kg bw: no effects Main study: Dams: anorexia, polydipsia, polyuria, lacrimation, in particular when treated on GD6–8. Resorptions and post implantation loss when treated on GD6–7 Fetuses: Hydrocephaly, incomplete closure of skull and omphalocele, microphthalmia, enlarged renal pelvis and renal hypoplasia; malformed skulls, sternebrae, vertebrae, ribs, in particular in dams treated on GD6–7 |
2.75 mg OTA/kg bw per day (only one dose level tested) | Patil et al. (2006) |
| Male and female 45–49 days old Fischer rats | Diet, 0.0, 0.16, 0.4, 1.0 and 2.5 mg OTA/kg feed, corresponding to 0.0, 8.9, 21.7, 55.2 and 141.8 μg/kg bw per day(males) and 0.0, 11.9, 33.9, 73.3 and 167.0 μg/kg bw per day (females); From day 1–70 (F0 males and females pre‐mating, mating), from day 28–70 (F0 males, pregnant and non‐pregnant F0 females); From day 49–70 or PND1‐21 (F1 pups). N = 10 per group and sex |
F0 males and females: mild increases in severity scores for tubular epithelial cell degeneration at 0.16 and 2.5 mg/kg in females and from 0.4 mg/kg onwards in males F0 males: ↓relative kidney weights at 1.0 and 2.5 mg/kg feed. At 2.5 mg/kg feed ↑ in albumin, sodium, total protein and bilirubin at 2.5 mg/kg feed F0 females: ↓relative kidney weights, bw, absolute ovary weight, cholesterol, RBC, haemoglobin, haematocrit, RDW, monocytes, neutrophils ↓ implantations at 2.5 mg/kg feed In F1 males from dams given 1.0 mg/kg feed ↓bw in on PND4, 7, 14, 21; In F1 females from dams given 1.0 mg/kg feed ↓ bw on PND4, 7; ↑ tubular degeneration in F1 males of dams given 0.16 mg/kg onward sand in F1 females from dams given 0.4 mg/kg onwards; In F1 males from dams given 1.0 mg/kg ↓relative kidney/liver weights, ↑ plasma cholesterol and in F1 males from dams given 0.4 mg/kg ↑ plasma phosphorous In F1 females from dams given 1.0 mg/kg ↓ relative liver weight, ↑ cholesterol and in F1 females from dams given 0.4 mg/kg onwards ↓ relative kidney weight |
The authors set an LOAEL for ↓ body weight for female F0 rats at 167 μg/kg bw and a LOAEL for ↓ kidney weight in F0 males at 55.2 μg/kg bw per day and for F0 females at 167 μg/kg bw per day Authors set an LOAEL for ↓ bw for F1 males and females at 55 and 73.3μg/kg bw per day, respectively, was set and an LOAEL for ↓ relative kidney weight in F1 males at 55 μg/kg bw per day and for F1 females at 33.9 μg/kg bw per day |
Bondy et al. (2018) |
| Pregnant rabbits | Diet, 0.0, 0.03, 0.06 mg OTA/kg bw for 15 days (GD 6–20); N = 5 per group |
At 0.03 mg/kg bw: ↑ ALT, ↓ RBC, ↑MCV in dams, ↓fetus length and weight At 0.06 mg/kg bw: ↑ urea, AST, ALT; ↓ RBC, Hb, PCV; ↑MCV in dams, ↓ fetus length and weight At both doses, dams had mild to moderate degenerative changes (pyknosis, necrosis of glomeruli, congestion of tubular epithelial cells) in kidney, at high dose also accumulation of eosinophilic material. At high dose moderate degenerative changes in the liver (cellular shrinkage, widening of sinusoidal spaces, vacuolation). No effects on the intestine observed |
0.03 mg/kg bw per day | Jan et al. (2017) |
| Young male inbred Swiss albino mice | Gavage, 0.0, 1.5, 3.0 mg/kg bw for 45 days. N = 10 per group | At 1.5 and 3.0 mg OTA/kg bw ↓ sperm count (−22% and ‐69%), sperm motility (−45% and −72%), sperm viability (−34% and‐59%) fertility rate (−50 and −77%) | 1500 μg/kg bw per day | Chakraborty and Verma (2010a) |
| Groups of 12 mated female Crl:CD (SD) rats | Diet, 0.0, 012, 0.6, 3.0 mg/kg OTA from GD6‐21 (corresponding to 0.0, 8.0, 39.3, 203.6 μg/kg bw per day during gestation and 0.0, 16.1, 76.0 and 378.6 μg/kg bw per day during lactation period). Sacrifice of dams on PND 22. Offspring maintained until PND 77 (27 males and 10 females per group) without OTA. Interim sacrifice of offspring on PND 21 (10 males per group) |
No effect on maternal parameters (behaviour, brain and kidney weight) At 3.0 mg/kg bw male and female offspring (F1) showed transient body weight decrease after weaning. Changes in hippocampal neurogenesis‐related parameters were seen in males at PND 21. Female and male offspring showed neuroprotective actions against OTA‐induced neurogenesis. All changes were reversed by PND77 |
Authors set the NOAEL for offspring neurogenesis at 0.6 mg OTA/kg feed, corresponding to 39.3–76.0 μg/kg bw per day | Tanaka et al. (2016) |
ALT: alanine transaminase; AST: aspartate transaminase; F0: parent generation; F1: first filial generation; F2; second filial generation; GD: gestation day; Hb: haemoglobin; LOAEL: lowest observed adverse effect level; MCV: mean corpuscular volume; NOAEL: no observed adverse effect level; OTA: ochratoxin; PCV; packed cell volume; PND: post‐natal day; RBC: red blood cells; RDW: red cell distribution.
Summarising briefly the in vivo studies presented in Table 9. A developmental LOAEL of 2.75 mg OTA/kg bw per day can be derived from the study of Patil et al. (2006) and is based on occurrence of malformations paralleled by pronounced maternal toxicity at the single dose for which a thorough toxicological evaluation has been carried out. From the study of Bondy et al. (2018), lower LOAELs for OTA have been identified. The authors set a maternal LOAEL of 167 μg OTA/kg bw per day based on reduced body and kidney weights, while the paternal LOAEL for reduced kidney weights was 55.2 μg OTA/kg bw per day. The corresponding LOAELs for reduced body and kidney weight in male offspring are 141.8 μg OTA/kg bw per day and in females 167 μg and 33.9 μg OTA/kg bw per day, respectively. Mild increases in severity scores for tubular degeneration in males were significant starting from 8.9 μg/kg bw per day and increased with dose. In females, these were significant at 33.9 μg/kg bw per day and at the highest dose. It is not clear from this publication to which extent the offspring was exposed to OTA during lactation (i.e. the relative exposure via milk or feed). A maternal and developmental LOAEL of 0.03 mg/kg bw per day (lowest dose tested) can be derived from Jan et al. (2017) based on reduced fetus lengths and weights in the presence of altered blood chemistry in the dams. Tanaka et al. (2016) identified a developmental NOAEL for neurogenesis of 33.9–76.0 μg OTA/kg bw per day. In this study, no effects on behaviour, brain or kidney weight were observed in dams up to the highest dose of 203.6–378.6 μg OTA/kg bw per day. It must be noted that the adverse nature of the parameters used for derivation of a developmental NOAEL is questionable and that these parameters were also not assessed in the dams. Finally, an LOAEL of 1,500 μg/kg bw per day (lowest dose tested) for male fertility can be derived from the study of Chakraborty and Verma (2010a) based on alterations of sperm parameters.
Several in vitro studies indicate a teratogenic potential of OTA. Based on a mean half maximal inhibitory concentration (IC50) for cell viability and differentiation of brain cells exposed to 2.52 ± 0.062 μg/mL OTA, it was suggested by the authors that OTA is a strong potential teratogen acting via its general toxicity (Wilk‐Zasadna and Minta, 2009). OTA‐induced morphological defects (neural tube defects (NTD), stunted limb bud development, curling of tail and hypoplasia of mandibular arches) in rat embryo explants (Balasaheb Wangikar et al., 2007). When mouse oocytes were incubated with OTA before in vitro maturation (IVM) or OTA was applied to female mice before collection of the oocytes for IVM, a series of adverse effects was observed in these cells (Huang and Chan, 2016).
4.2.5.1. Summary on reproductive toxicity
The new reproduction studies show that OTA induces developmental toxicity. However, the doses at which effects are observed differ by orders of magnitude in the different investigations and thus LOAELs derived in these studies (albeit for different endpoints and from studies with deviating designs) vary from 33.9 to 2,750 μg OTA/kg bw per day. Notably, adverse effects on offspring are usually paralleled by pronounced maternal toxicity. In vitro data suggest that OTA developmental effects might be mediated via its general toxicity which would support the in vivo findings. Overall, the new studies corroborate the conclusion of EFSA (2006) that OTA is a developmental toxicant but that developmental effects are generally observed at doses higher than those causing adverse effects in the kidney of pigs (EFSA, 2006).
4.2.6. Observations in humans
4.2.6.1. Balkan Endemic Nephropathy
In previous EFSA (EFSA, 2006) and JECFA (FAO/WHO, 2001, 2008) assessments, it was concluded that the available data from old epidemiological studies did not allow establishing an aetiological link between OTA, BEN and associated urinary tract tumours. It is now generally agreed that these conditions occurring in the Balkan and are rather associated with exposure to toxins other than OTA, i.e. aristolochic acid (Gifford et al., 2017; Jadot et al., 2017).
4.2.6.2. Chronic interstitial nephropathy of unknown aetiology
In 1995, an endemic chronic interstitial nephropathy (CIN) of unknown aetiology with striking similarities with that of BEN was described in Tunisia (Maaroufi et al., 1995). Because of high food contamination levels and high serum or plasma OTA concentrations, the authors suggested that OTA might be related to this group of renal disease. In summary, they reported that in healthy controls, concentrations of OTA ranged from 0.1 to 16.6 μg/kg in food and from 0.1 to 2.3 ng/mL in serum/plasma concentrations, and in nephropathy patients OTA values were 0.3–46,830 μg/kg in food and 0.7–1,136 ng/mL in serum/plasma. In surveys conducted during the 1990s and published later, they found significantly higher mean serum concentrations of OTA in patient groups with CIN of unknown cause (total n = 954, 44.4 ± −19 μg/L to 55.6 ± 19 μg/L) than in healthy controls recruited from the general population (total n = 205, 1.22 ± −1.2 μg/L to 3.35 ± −2.32 μg/L) (note that OTA values in this study was given per L not per mL as in the other studies below) (Abid et al., 2003). This was followed up in a later study (Hassen et al., 2004) on patients and healthy individuals from the centre of Tunisia previously reported to be a hot spot of chronic renal failure with high OTA exposure. In a cross‐sectional study, they included 40 patients with CIN of unknown cause, 60 patients with CIN of known aetiology (not specified) and 40 healthy individuals. The percentage of positive OTA plasma samples in the respective groups were 100, 78 and 62%, with OTA concentrations of (mean and SD): 50.4 ± 8.2 ng/mL (range: 18.4–171.25 ng/mL), 12.36 ± 4.4 ng/mL (range: 1.68–29 ng/mL) and 1.22 ± 1.2 ng/mL (range: 0–3.2 ng/mL). The OTA concentrations in the group with unknown aetiology were significantly different from that of both the healthy subjects and the group with known aetiology. The mean levels of ß2‐microglobinuria in the groups were 1,960 ± 990 μg/L, 1,450 ± 90 μg/L and 120 ± 70 μg/L, respectively, and the value of the group with unknown aetiology was significantly different from that of both the healthy subjects and the group with known aetiology (Hassen et al., 2004).
Two studies from a second research group investigated plasma or serum concentrations of OTA in patients with renal diseases and in healthy subjects (Hmaissia Khlifa et al., 2008, 2012). In the first study, blood samples were collected from healthy individuals (healthy group, HG) (n = 105) and five different groups of nephropathic patients suffering from CIN of known aetiology (n = 22), CIN of unknown aetiology (CINI) (n = 30), chronic vascular nephropathy (CVN) (n = 26), chronic glomerular nephropathy (CGN) (n = 26) and renal transplanted subjects (RTS) (n = 27). The samples were from various regions of Tunisia. The following fraction of OTA‐positive subjects and mean and SD of plasma of the different groups were: HG, 28%, 0.49 ± 0.67 ng/mL; CIN, 45%, 0.92 ± 0.83 ng/mL; CINI, 80%, 1.25 ± 1.22 ng/mL; CVN, 26%, 0.75 ± 0.88 ng/mL; CGN, 26%, 0.21 ± 0.14 ng/mL; RTS, 85%, 0.64 ± 1.38 ng/mL. The CINI group had the highest fraction of OTA‐positive individuals and highest OTA plasma value, both being significantly higher than the controls (Hmaissia Khlifa et al., 2008). In the second study, they collected samples from the Military Hospital of Tunisia and determined serum OTA concentrations in healthy subjects (n = 44) and patients with CIN of unknown aetiology (n = 22). For each group, 44 samples of food consumed every day or several times a week were collected. Analyses of the food samples showed that OTA was detectable in 52.3% of the samples and none of the samples from the healthy controls were above 6.1 ng/g with a mean value of 0.77 ng/g. The corresponding values for the nephropathic patients were 88.6%, 33.8 ng/g and 9.28 ng/g. Of the healthy subjects, the 34% with detectable OTA concentration in serum had a mean value of 0.22 ± 1.39 ng/mL (range 0.12–1.5 ng/mL). Eighty per cent of the patients were OTA positive and these had a mean OTA serum value of 1.25 ± 1.22 ng/mL (range: 0.12–3.8 ng/mL). Both the fraction of OTA positive and mean serum concentration of OTA were significantly higher than in the controls (Hmaissia Khlifa et al., 2012).
Finally, in an additional cross‐sectional study from the first research group, Zaied et al. (2011) measured serum concentration of OTA in blood samples collected in Tunisian hospitals from patient with different chronic renal diseases (n = 270, 115 males and 115 females). The renal disease groups included patients with chronic vascular nephropathy (n = 49), chronic glomerular nephropathy (n = 61) and CIN with known (n = 77) and unknown (n = 83) aetiology, respectively. Healthy volunteers were included as controls (total, n = 138, 91 males and 47 females). The group with CIN of unknown aetiology had the highest rate of OTA positive serum samples, and the highest mean concentration of OTA (18 ng/mL, range: 1.8–65 ng/mL). In comparison, the other groups with renal disease had the following OTA detection rates and mean and range of serum concentrations of OTA: chronic vascular nephropathy, 37%, 5.5, 1.5–16 ng/mL; chronic glomerular nephropathy, 49%, 5.8, 1.1–16.3 ng/mL; CIN with known aetiology, 62%, 5.5, 1.0–21.6 ng/mL. The healthy controls had 49% OTA‐positive samples and a mean of 3.3 ng/mL (1.7–8.5 ng/mL). Mean intakes of OTA calculated from serum OTA values were 26 ng/kg bw in the group with CIN of unknown aetiology as compared with around 8 ng/kg bw in the other groups of renal disease and 4.4 ng/kg bw in the control group.
The CONTAM Panel notes that these studies on CIN all conducted in Tunisia, which all had cross‐sectional designs, did not allow concluding on a possible causal association between OTA exposure and chronic interstitial nephropathy or other renal diseases.
4.2.6.3. Renal function in children
Hassan et al. (2016) investigated the relationship between OTA in breast milk and maternal and infant serum, and kidney function of infants as assessed by urinary ß2‐microglobulin and microalbuminuria in mother infant pairs from Egypt. The mean and standard deviation (SD) of the age of the breastfeeding mothers was 27 ± 5.77 years. Their infants were exclusively breast‐fed for 4 months. OTA levels in serum and milk were determined using LC with microfluorimetric detection. The limit of detection was not given. Of the 50 mothers included, 36 had detectable OTA concentrations in serum (mean, SD; 4.28 ± 3.97 ng/mL) and milk (mean, SD; 1.89 ± 0.98 ng/mL). All their breastfed infants had detectable OTA in serum (mean, SD; 1.26 ± 1.1 ng/mL). Univariate comparison of infants of women that had non‐detectable and detectable level of OTA in serum revealed significant higher urine ß2‐microglobulin in the OTA‐positive vs. OTA‐negative mothers (mean, SD; 260 ± 162.5 μg/mL and 30.3 ± 7.9 μg/mL, respectively) and for microalbumin in urine of infants of OTA‐positive and OTA‐negative mothers (mean, SD; 29.7 ± 13.9 mg/L and 11.6 ± 7.7 mg/L, respectively). However, upon multivariate logistic regression analysis, these differences did not remain statistically significant. Grouping the infants of OTA‐positive mothers into those who had OTA in serum above and below 2 ng/mL, showed that the former group had higher urinary levels of ß2‐microglobulin (mean, SD; 322 ± 188.9 μg/mL and 196 ± 107.9 μg/mL, respectively) and of microalbumin (mean, SD; 37.7 ± 12.9 mg/L and 22.5 ± 10.7 mg/L). Using multivariate logistic regression analyses, the authors found a significant positive association between infant serum level of OTA and the degree of microalbuminuria, but not ß2‐microglobulinuria.
The CONTAM Panel notes that in this study, albumin and ß2‐microglobulin were determined in ‘freshly collected’ urine samples of the infants indicating that these were spot samples. Microalbuminuria, indicative of leaky kidney filtration, is defined as an albumin excretion between 30 and 300 mg/24h, or in spot samples between 30 and 300 mg/L. The reference values for ß2‐microglobulin, indicative of tubular kidney dysfunction, are commonly given as < 200–300 μg/L in urine, which is in the range of 10‐3 of the values given in this study, the μg/mL range, possibly being a misprint.
4.2.6.4. Cancer
In a study on bladder cancer patients from Pakistan, OTA concentrations in plasma were compared with levels in healthy controls (Aslam et al., 2012). Ninety‐six bladder cancer cases and 31 controls were recruited. There were no differences between the groups. The mean and SD OTA concentrations in plasma of the cases and the controls were 0.33 ± 0.42ng/mL (range: 0.03–3.41 ng/mL) and 0.31 ± 0.29 ng/mL plasma (range: 0.04–1.24 ng/mL), respectively.
The association between OTA and bladder cancer was also investigated in a cross‐sectional study including 120 cases and 120 healthy controls from the Cote d'Ivoire (Thé et al., 2015). OTA was determined in blood samples and the mean and SD concentrations were 1.16 ± 1.85 μg/L in the cases and 0.79 ± 1.2 μg/L in the controls. These figures were not significantly different.
The association between exposure to OTA and hepatocellular cancer (HCC) was investigated in a cross‐sectional study in Egypt (Ibrahim et al., 2013). Thirty‐nine HCC cases were identified and matched by age, sex, residence and date of recruitment with 22 healthy controls. OTA was determined in sera from the participants. In the HCC group, the concentration of OTA ranged between 0.129 and 10.93 ng/mL with a mean value ± SE of 1.11 ± 0.3 ng/mL, which was significantly higher than that of the healthy controls ranging between 0.005 and 0.50 ng/mL with a mean value ± SE 0.201 ± 0.02 ng/mL. Odd ratio (OR) was calculated using a cut‐off value for OTA of 0.207 ng/mL based on receiver operating curve12 analysis. An increased risk for HCC upon OTA exposure, OR = 9.8 (95 pct. confidence interval: 2.91–32.98), was observed.
4.2.6.5. Other studies
In a study (Ates et al., 2011) from Turkey, 180 healthy individuals from different cities delivered morning urine samples and filled in a questionnaire about sex, age, smoking habits and alcohol and coffee consumption. OTA, malondialdehyde (MDA), 8‐OH‐dG and creatinine were determined in urine. OTA, MDA and 8‐OH‐dG were expressed relative to the amount of creatinine. Means and SD were: OTA 9.68 ± 4.21 ng/g creatinine, 8‐OH‐dG 2.91 ± 1.05 ng/mg creatinine, MDA 5.80 ± 3.67 nmol/mg creatinine. MDA was significantly lower in men than in women and in those below 35 years of age. There were no differences in urinary OTA and 8OH‐dG between the sexes, non‐smokers and smokers and coffee non‐consumers and consumers. There was a significant correlation between OTA and 8‐OH‐dG (r = 0.34) and MDA (r = 0.5), and between MDA and 8‐OH‐dG (r = 0.35) in urine.
4.2.6.6. Summary of human studies
With regard to the studies on CIN of unknown cause in Tunisia and OTA exposure, the CONTAM Panel considered not possible to causally link high levels of OTA exposure to this condition because of the cross‐sectional nature of these studies. The findings of raised protein concentrations in urine of limited number of infants from Egypt exposed to high levels of OTA via their mothers (during pregnancy and lactation) need confirmation in larger controlled studies. In one cross‐sectional study, OTA was associated with an increased risk of HCC; however, due to the small sample size and the study design limitations, no inferences on causality could be drawn. In two studies, OTA appeared not to be associated with bladder cancer. In one study, OTA in urine correlated with urinary 8OH‐dG and MDA.
The CONTAM Panel did not identify studies in humans providing reliable biomarkers of OTA‐specific effects, in particular on kidney function.
4.3. Evaluation of the mode of action of OTA
4.3.1. MoA on cellular toxicity in liver and kidney
4.3.1.1. Mechanism of induction of apoptosis and autophagy
OTA is well established as being toxic to a range of cell types in vitro in which apoptosis is implicated and in vivo the kidney proximal tubular cells are particularly sensitive due, at least in part, to the facilitated uptake. OTA is transported into the kidney by OAT1 (Tsuda et al., 1999) coded by the SLC22A6 gene which is expressed predominantly in the kidney13 explaining the high specificity of OTA toxicity towards this organ. It is noted, however, that additional protein(s) in the kidney appear to have a high affinity for binding OTA (based on substrate competition data, Heussner et al. (2002)) and which may also contribute to concentration of OTA in the kidney and thus to this target organ specificity.
The association between oxidative stress and the loss of mitochondrial membrane potential associated with apoptosis produced by OTA was noted in a range of cell types in vitro (Bouaziz et al., 2008; Hsuuw et al., 2013; Liang et al., 2015; Wang et al., 2017). Caspase 3 activation associated with upregulation of the levels of p53 and p21 appeared responsible for apoptosis in oocytes and blastocysts treated with OTA (Huang and Chan, 2016). In contrast, p53 activation supplied a cell survival action against apoptosis produced by OTA in several kidney cell lines by suppressing pro‐apoptotic JunN‐terminal kinase (JNK, Li et al., 2011). It appears that OTA may also induce apoptosis independent of mitochondria since in human H9 T cells an increase of tumour necrosis factor‐α (TNFα) and activation of caspase 8 also occurred (Darif et al., 2016).
Autophagy has also been implicated in human embryonic kidney (HEK293) cells treated with OTA and Nix (a selective autophagic receptor) was shown to play a critical role in this protective autophagic response (Shen et al., 2013). Endoplasmic reticulum (ER) stress was evident in the pig kidney along with decreased antioxidant capacity, increased p38 and phosphorylation of extracellular signal‐regulated kinase 1/2 (ERK 1/2) and increased expression of genes involved in autophagy following treatment of pigs with OTA in the diet (Gan et al., 2017a; see Table 11). As a possible explanation for glomerulopathy, Sheu et al. (2017) found that OTA‐activated nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and calpain and caused an endoplasmic reticulum (ER) stress response and oxidative stress associated with apoptosis in mouse and rat glomerular mesangial cells.
Table 11.
Effect of OTA on the expression of genes and microRNAs and associated proteomic analyses relevant to the kidney and liver
| Test system | Methodology employed | Modulation observed | Associated changes or observations | Reference |
|---|---|---|---|---|
| Mouse, 10 mg OTA/kg bw, single dose; after 24 h kidney was analysed | Proteomic analyses | Modulation of proteins in peritoneal macrophages involved in inflammation | Associated with kidney and liver toxicity | Ferrante et al. (2006) |
| Rat; 300 then reduced to 100 μg OTA/kg bw per day; Kidney and liver analysed after 7 days and 12 months | Transcriptomic analyses | In kidney greater degree of differentially expressed genes than in liver. Modulation of expression of genes as markers of kidney injury, cell and pathways regulated by HNF4a and Nrf2. Downregulation of regucalcin | Authors noted little evidence of a DNA‐damage response or of apoptosis and concluded that the effects suggested impairment of defence mechanisms. The authors noted the role of regucalcin in the regulation of calcium homoeostasis. Downregulation of Nrf2 was associated with oxidative DNA damage | Marin‐Kuan et al. (2006) |
| Rat, 300 then reduced to 100 μg OTA/kg bw per day; Kidney analysed after 21 days and 12 months | Assessment of proteins regulated by Nrf2 | Decreased protein markers regulated by Nrf2 | Cavin et al. (2007) | |
| Rat, gavaged daily with 500μg OTA/kg bw; Kidney and liver analysed at 7 and 21 days | Transcriptomic analyses | Kidney greater degree of differentially expressed genes than in liver. Downregulation of genes was the predominant effect notably for genes involved in oxidative stress, calcium homoeostasis, metabolism and transport. Upregulation of genes involved in cell survival and proliferation | Kidney toxicity | Arbillaga et al. (2008) |
| Rat, 21, 70 and 210 μg OTA/kg bw per day for up to 90 days (kidneys analysed) | qPCR | Modulation of the expression of genes relating to chromosomal instability and control of cell cycle | Changes were associated with a proliferative response | Adler et al. (2009) |
| Rat, 3 mg OTA /kg per day for 1, 3 and 7 days via gavage, Analysis of kidneys and various kidney cells in vitro | Transcriptomic analyses | Modulation of expression of genes involved in cytoskeleton, nucleosome regulation, translation, transcription, ubiquitination and cell cycle pathways | Jennings et al. (2012) | |
| Rat; 70 and 210 μg OTA/kg bw up to 26 weeks; Kidney analysed | MicroRNA profiling |
Alteration of microRNAs involved in MAPK signalling Suppression of microRNAs involved in the production of microRNAs themselves |
Dai et al. (2014) | |
| Rat, (70 or 210 μg OTA/kg bw per day up to 26 weeks; Liver analysed |
MicroRNA profiling; Transcriptomic analyses; Proteomic analyses |
Modulation of thiol metabolism, PPAR signalling, bile acid synthesis, arginine and proline metabolism and xenobiotic metabolism | Authors concluded the changes were indicative of liver toxicity and metabolic disease | Qi et al. (2014b) |
| Rat, 210 μg OTA/kg bw per day for 28 days | qPCR | Modulation of expression of genes involved in oxidative stress | Changes were associated with apoptosis, cell proliferation and disruption of cell cycle control | Taniai et al. (2014) |
| Human primary proximal tubule cells | qPCR | Alteration of expression of genes involved in inflammation. Malignant transformation and epithelial‐to‐mesenchymal transition. Notable upregulation of Wnt‐1 | Authors noted the role of Wnt‐1 as a growth factor involved in oncogenesis and fibrosis | Hennemeier et al. (2012) |
| HepG2 cells | Transcriptomic analyses | Enhanced expression of genes involved in inflammation, oxidative stress, cell pro‐survival, energy and xenobiotic metabolism | Hundhausen et al. (2008) | |
| Pig, 0.05 mg OTA /kg feed; Kidney analysed after 30 days | Transcriptomic analyses | Modulation of expression of genes related to immune response, oxidative stress and the growth, proliferation, death and survival of cells | Marin et al. (2017a,b) | |
| Human and rat renal proximal tubule cells | Transcriptomic analyses | Enhanced expression of genes involved in cytoskeleton, nucleosome regulation, translation, transcription, ubiquitination and cell cycle. Notable no effect of Nrf2 | Jennings et al. (2012) | |
| Human embryonic kidney cells (HEK293) | Proteomic analyses | Increased expression of proteins in mitochondria involved in electron transport, protein synthesis, stress response and cell death | Shen et al. (2013) | |
| Porcine proximal tubule cells | Specific product analyses | Attenuation of Nrf2 and Haem oxygenase via induction of microRNAs (miR‐132 and miR‐200c) and pro‐fibrotic TGFβ | Stachurska et al. (2013) | |
| HEK293 cells | Specific product analyses | Post‐transcriptional reduction of miR‐29B | Collagen formation (marker of fibrosis) | Hennemeier et al. (2014) |
| Mouse spermatid cells (GC‐2) | MicroRNA analyses | Expression of MiR‐122 | Caspase‐3 mediated apoptosis | Chen et al. (2015) |
| Rat hepatocytes in vitro and in vivo and in HEPG2 cells | MicroRNA analyses | Expression of MiR‐122 | Apoptosis | Zhu et al. (2016b) |
| HepG2 and HEK293 cells also in rat; 70 and 210 μg OTA /kg bw for 13 weeks; kidney analysed | MicroRNA profiling | Deregulation of microRNAs involved in signal transduction and human cancer | Zhao et al. (2017) |
HNF4a: hepatocyte nuclear factor 4 α; MiR; microRNA; Nrf2: Nuclear factor erythroid 2‐related factor 2; OTA: ochratoxin A; qPCR: quantitative polymerase chain reaction; TGFβ: tumour growth factor β; PPAR: peroxisome proliferator‐activated receptor; RNA: ribonucleic acid; MMTV: mouse mammary tumour virus; Wnt‐1: wingless‐type MMTV integration site family, member 1, a proto‐oncogene protein.
4.3.1.2. The cellular proliferation response
Stimulation of cell proliferation by OTA is also evident in rodent kidney. In rats and mice, renal toxicity was associated with a dose‐dependent increase in positive proliferating cell nuclear antigen (PCNA) signals in the kidneys (Rached et al., 2007; Qi et al., 2014a; Bondy et al., 2015; Zhu et al., 2016a). Taniai et al. (2014) and Mally et al. (2005a) also found evidence of stimulation of cell proliferation in the rat kidney along with increased incidence of apoptosis. The proliferative response was associated with changes in the expression of genes involved in response to DNA damage and cell cycle (Rached et al., 2007; Adler et al., 2009; Taniai et al., 2014). The sections below on cell signalling and gene expression help to understand the basis of the cell proliferative and apoptotic processes.
4.3.1.3. Effects mediated by altered cell signalling
Altered cell signalling by OTA has been demonstrated in various cell types in vitro and in vivo (see Table 10). Prominent in these effects is the modulation of signalling involving nuclear factor kappa light chain enhancer of activated B cells (NF‐kB) (associated with inflammation and fibrosis) and mitogen‐activated protein kinases (MAPK) specifically ERK 1/2, JNK and extracellular‐regulated protein kinase 38 (p38) involved in inflammatory, apoptotic and fibrotic processes (Sauvant et al., 2005; Wang et al., 2012). Increase of Ikβ kinase (IKK) and phosphorylation of NFkB was evident in HEK293 cells (Raghubeer et al., 2017), but this anti‐apoptotic, pro‐inflammatory change was reversed as the concentration of OTA increased such that pro‐apoptotic signals became evident. In gastric human epithelial (GES‐1) cells, apoptosis and G2 phase arrest caused by OTA appears to involve Cdc25C, Cdc2 and cyclin B1 (Cui et al., 2010).
Table 10.
Modulation of cell signalling by OTA
| Test system | Cellular signalling system activated | Associated changes | Reference |
|---|---|---|---|
| Fischer 344 rats; 300 μg OTA/kg bw per day; Kidney analyses | Phosphorylation of atypical PKC; ERK 1/2; ELK12 (nuclear substrate); ribosomal‐S kinase (cytosolic substrate) | Authors proposed a role in stress response and cell proliferation | Mantle et al. (2005) |
| Rat, 300 then reduced to 100 μg/ kg bw/day; up to 12 months | MAPK‐ERK via phosphorylation of atypical PKC | Authors highlighted the roles of the altered signalling in the processes of cell proliferation, apoptosis, cell survival and potentially carcinogenesis | Marin‐Kuan et al. (2007) |
| Mouse skin; 80 mg/mouse; 12–72h | ERK1/2; p38 and JNK MAPK; nuclear translocation of Nrf2 | Oxidative stress, Cell cycle arrest and apoptosis | Kumar et al. (2011) |
| Pig; Kidney analysed | p38; ERK 1/2 | ER stress, decreased antioxidant capacity | Gan et al. (2017c) |
| Rat kidney proximal tubule cells (NRK‐52E) | NFkB; ERK 1/2; JNK and p38 (MAPK) | Apoptosis | Sauvant et al. (2005) |
| Human proximal tubule cells | NFκB; collagen III; fibronectin | Hypertrophy | Schwerdt et al. (2007) |
| Gastric human epithelial cells (GES‐1) | Cdc25C; cdc2; cyclinB1 | Apoptosis and G2 phase arrest | Cui et al. (2010) |
| Different kidney cell lines | p53 (and associated suppression of pro‐apoptosis JNK | Decreased apoptosis | Li et al. (2011) |
| Gastric human epithelial cells (GES‐1) | ERK; p38 (MAPK) | G2 arrest | Wang et al. (2012) |
| Primary mouse keratinocytes | EGFR and downstream MAPKs and Akt pathways | Cell proliferation | Kumar et al. (2013) |
| Human proximal tubule cells (HK) | Deliberate experimental chemical inhibition of ERK 1/2 | Decreased apoptosis | Özcan et al. (2015) |
| Oocyte and blastocyst cells | Caspase 3; p53; p21 | Apoptosis | Huang and Chan (2016) |
| Human H9T cells | TNFα; Caspase 8 | Apoptosis | Darif et al. (2016) |
| PK15 cells | p38 | Apoptosis | Gan et al. (2016) |
| Porcine primary splenocytes | ERK | Apoptosis | Gan et al. (2017b) |
| Rat and mouse glomerular mesangial cells | NADPH oxidase; calpain | ER stress, oxidative stress, Apoptosis | Sheu et al. (2017) |
| HEK293 cells | Anti‐apoptotic signalling (IKK and phosphorylation of NFkB) (Pro‐apoptotic as the OTA concentration increased) | Raghubeer et al. (2017) | |
| Rat kidney proximal tubule cells (HKC) | p53, p21, p16‐pRB pathways | Senescence phenotype associated with cell cycle arrest | Yang et al. (2017) |
Akt: protein kinase B; Cdc: cell division cycle; ERK: extracellular signal regulated kinase; EGFR: epidermal growth factor receptor; ELK: ETS domain containing protein; ETS: E26 transformation specific; ER: Endoplasmatic reticulum; G2: gap 2 phase; IGF: insulin‐like growth factor; IKK: Ikβ kinase; JNK: c‐Jun N‐terminal kinase; MAPK: mitogen‐activated protein kinase; NADPH: nicotinamide adenine dinucleotide phosphate; NfkB: Nuclear factor kB; Nrf2: nuclear factor erythroid 2‐related factor 2; p16, p21, p38, p53; extracellular‐regulated protein kinase p16, p21, p38, p53; PDK: Phosphoinositide‐dependent kinase; PKC: protein kinase C.
Further studies in vivo focused on interference of signal transduction in the kidneys of Fischer 344 rats given OTA in the diet at 300 μg/kg bw per day (Mantle et al., 2005). There was an increase in the phosphorylation of atypical protein kinase C (PKC) and a downstream activation of ERK1/2 and of their nuclear substrate ETS‐domain protein 1 (ELK 1/2) and cytosolic substrate, ribosomal‐S6 kinase (p90RSK). PKC activation appeared to be due to mobilisation of the insulin‐like growth factor‐1 receptor (IGF1R) and phosphoinositide‐dependent kinase‐1 system (PDK1). The authors considered that signal transduction modulation may be involved in stimulation of a stress response and cell proliferation. MAPK‐ERK activation, via phosphorylation of atypical protein kinase C, was also evident in the kidneys of rats fed OTA (Marin‐Kuan et al., 2007). The roles of PKC and MAPK pathways in cell proliferation, apoptosis, cell survival and potentially carcinogenesis were highlighted.
4.3.1.4. Alteration of gene expression
Various studies have investigated altered gene expression induced by OTA. These findings relating to kidney or liver both in vitro and in vivo are summarised in Table 11 along with changes in microRNA and proteomic profiles. Prominent in these findings are changes in the expression of genes involved in inflammation, fibrosis, malignant transformation and epithelial to mesenchymal transition. In HepG2 cells, enhanced expression of genes related to inflammation, oxidative stress, cell pro‐survival, energy and xenobiotic metabolism was observed (Hundhausen et al., 2008). A set of OTA‐inducible genes was detected using transcriptomic analyses in human and rat renal proximal tubular cells. Major biological processes implicated were related to cytoskeleton, nucleosome regulation, translation, transcription, ubiquitination and cell cycle. Surprisingly, the oxidative stress nuclear factor‐erythroid 2 p45‐related factor (Nrf2) pathway was not affected in this study (Jennings et al., 2012).
Important in vivo studies showed that, in rats treated with OTA, the degree of differentially expressed genes was greater in kidney than in liver. Genes involved in oxidative stress, calcium homoeostasis, metabolism and transport were prominent in downregulation whereas genes involved in cell survival and proliferation were elevated in expression (Arbillaga et al., 2008). In further transcriptomic analyses, rats were given OTA in the feed (Marin‐Kuan et al., 2006; see Section 3.2.2). Gene expression was assessed in kidney and liver at 7 days and at 12 months. There was little evidence of a DNA damage response with the most prominent changes in gene expression being kidney transcription factors hepatocyte nuclear factor 4α (HNF4α) and Nrf2 and genes regulated by these factors suggesting impairment of defence mechanisms. A strong downregulation of regucalcin was seen. Regucalcin is involved in the intracellular calcium homoeostasis in kidney tubule cells. Downregulation of Nrf2 gene expression paralleled by induction of DNA damage measured as formation of abasic DNA sites was observed both in kidney NRK cells and in vivo (Cavin et al., 2007). Based on these results, the authors proposed that the loss of Nrf2 protection against oxidative stress may play an important role in kidney toxicity and carcinogenesis. Indeed, pretreatment of primary isolated rat hepatocytes with activators of Nrf2 reduced oxidative stress, protein nitration and cytotoxicity induced by OTA (Cavin et al., 2009). When Nrf2 was lacking in porcine or mouse kidney tubular epithelial cells and in mice in vivo, this exacerbated OTA‐induced toxicity (Loboda et al., 2017a). Haem oxygenase (HO‐1) induction by cobalt protoporphyrin attenuated the nephrotoxic effect of OTA in mice and HO‐1‐deficient mice were more sensitive (Loboda et al., 2017b). Moreover, Stachurska et al. (2013) showed that OTA attenuates Nrf2 and HO‐1 via induction of microRNAs (miR‐132 and miR‐200c) that contributes to elevation of ROS and pro‐fibrotic TGFβ expression in porcine renal proximal tubule cells.
Microarray analyses were also carried out in kidney samples taken from pigs given OTA in the feed (Marin et al., 2017a). Assessment of biological function of altered gene expressions indicated changes relating to immune response, oxidative stress and the growth, proliferation, death and survival of cells. Alterations in gene expression in the kidney in which there was OTA‐induced toxicity have also been associated with transcriptional changes in genes involved in DNA damage response and apoptosis, response to oxidative stress and inflammatory reactions (Arbillaga et al., 2008; Jennings et al., 2012).
OTA has also been found to modulate the expression of microRNAs, particularly in kidney cells in vivo and in vitro (see Table 11). Many of the altered miRNAs are involved in the MAPK signalling pathways, in support of the signalling changes summarised above, but of particular interest was the suppression of miRNAs involved in the production of microRNAs themselves which could have implications for toxicity. The microRNA MiR‐122 was involved in the induction of apoptosis (Chen et al., 2015; Zhu et al., 2016b).
Proteomic approaches have also been used to assess factors involved in OTA toxicity (see Table 11). Notable are the enhanced expression of mitochondrial proteins involved in electron transport, protein synthesis, stress response and cell death and modulation of proteins involved in inflammation (Ferrante et al., 2006; Shen et al., 2013). Qi et al. (2014b) provided a comprehensive analysis of molecular changes in liver based on profiling of microRNAs, mRNAs and proteins following treatment of rats with OTA by gavage. The combined analyses provided evidence of five predominant pathways that were modulated (thiol metabolism, PPAR signalling, bile acid synthesis, arginine and proline metabolism and xenobiotic metabolism) indicative of liver toxicity and metabolic disease.
4.3.1.5. Role of oxidative stress in OTA‐toxicity
The downregulation of Nrf2 (see above) is a major contributor to the induction of cellular oxidative stress induced by OTA. There is also evidence that an Fe3+ complex with OTA can produce hydroxyl radicals in the presence of NADPH and NADPH‐cytochrome‐P‐450 reductase (Hasinoff et al., 1990). Evidence for oxidative DNA damage in rat kidney and in liver has been noted (Kamp et al., 2005; Mally et al., 2005a) although oxidative stress in the rat kidney was only minimal in the study by Zhu et al. (2016a). In cultured porcine kidney tubule cells (LLC‐PK1), OTA decreased superoxide dismutase, elevated reactive oxygen species and downregulated expression of glutathione S‐transferase (GST) associated with a lowered activation of Nrf‐2 (Bösch‐Saadatmandi et al., 2008, 2009). The ratio of glutathione (GSH)/glutathione disulfide (GSSG) was reduced as were total thiols and heat shock proteins were downregulated in PK15 porcine kidney cells treated with OTA (Klarić et al., 2014). Oxidative stress is also evidenced in pigs and in rats by altered activity of superoxide dismutase (SOD), increased protein carbonyls and modulation of the expression of genes involved in response to oxidative stress (Domijan et al., 2006; Zhang et al., 2016) along with downregulation of gene expression of markers of inflammation (interleukin (IL)‐6, IL‐8, IL‐12, IL‐17A, IL‐18 –colon; IL‐17A and IL‐10‐kidney). These effects correlated with renal toxicity and the modulation of the expression of genes functionally associated with renal necrosis, kidney failure, renal proliferation, renal dysplasia and hypoplasia (Marin et al., 2017a,b). In support of the evidence for oxidative stress in playing a role in OTA adverse effects, protection is afforded by antioxidants and scavengers of free radicals. See Tables B.1 and B.2 of Appendix B.
Table B.1.
In vivo evidence for protective effect of antioxidants/free radical scavengers against OTA
| Test system | Protective agent | Results | Reference |
|---|---|---|---|
| Pre‐ or Co‐treatment of rats after 4 weeks of OTA treatment | Melatonin | Inhibition of liver and kidney toxicity. Reduction of indicators of oxidative stress | Meki and Hussein (2001), Ozçelik et al. (2004), Abdel‐Wahhab et al. (2005), Sutken et al. (2007) |
| Rats | Red wine | Inhibition of kidney toxicity. Reduction of oxidative damage | Bertelli et al. (2005) |
| Rats | 2‐mercaptoethane sulfonate (thiol) | Reduced karyomegaly but did not reduce the incidence of renal tumours | Pfohl‐Leszkowicz and Manderville (2007) |
| Mice | Emblica officinalis | Prevented loss of DNA, RNA and protein from liver and kidney | Verma and Chakraborty (2008) |
| Mice | Emblica officinalis | Reduced lipid peroxidation in liver and kidney | Chakraborty and Verma (2010b) |
| Mice | Red wine | Reduced oxidative stress and DNA strand breaks in kidney | Cipriani et al. (2011) |
| Rats | Lycopene | Reduced liver and kidney DNA strand breaks | Aydin et al. (2013) |
| Rats | Myricetin | Reduced oxidative stress and kidney toxicity based on histopathology | El‐Haleem et al. (2016) |
| Rats | Recombinant mitochondrial manganese containing superoxide dismutase | Prevention of hypertension and reduced glomerular filtration rate; prevention of renal and tubular degeneration | Ciarcia et al. (2016) |
| Rats | Inulacrithmoides extract | Reduced toxicity, reduced liver and kidney oxidative stress; reduced bone marrow micronuclei | Abdel‐Wahhab et al. (2008) |
| Rats | Mitochondrial manganese containing superoxide dismutase | Prevented nephrotoxicity and reduced the loss of sodium reabsorption in kidney proximal tubules | Damiano et al. (2018a) |
| Rats | Delta‐tocotrienol | Prevention of hypertension and reduced glomerular filtration rate and lowering of kidney antioxidant enzymes | Damiano et al. (2018b) |
DNA: deoxynucleic acid; OTA: ochratoxin; RNA: ribonucleic acid.
Table B.2.
In vitro evidence for protective effect of antioxidants/free radical scavengers against OTA
| Test system | Protective agent | Results | Reference |
|---|---|---|---|
| Human hepatoma cells (HepG2) | Alpha‐tocopherol, alpha‐tocopherolphosphate, epigallocatechin gallate, quercetin, catechin and rosmarinic acid | No protection against cytotoxicity | Bösch‐Saadatmandi et al. (2006) |
| Human proximal kidney tubule cells (HK‐2) | N‐Acetyl‐L‐cysteine | Reduction of reactive oxygen species and DNA strand breaks | Arbillaga et al. (2007) |
| Pretreatment of pig kidney cell line (LLC‐PK1) | Epigallocatechin gallate and epicatechin gallate (antioxidant catechins) | Reduction of cell death, reactive oxygen and DNA fragmentation | Costa et al. (2007) |
| Gut epithelial cell line (Caco‐2) | N‐acetylcysteine | Protected against loss of claudins from membranes associated with tight junctions | Lambert et al. (2007) |
| Intestinal cells (Caco‐2\TC7) | De‐alcoholated Red wine |
Increased tight junction permeability Increased relocation of claudin‐4; Increased apoptosis |
Ranaldi et al. (2007) |
| Bovine mammary epithelial cells; Porcine primary fibroblasts; Madin‐Darby canine kidney cells (MDCK) | Alpha‐tocopherol | Decreased apoptotic cell death | Fusi et al. (2008, 2010, 2018) |
| Human hepatoma cells (HepG2) | Ebselen or vitamin E | No protection against cytotoxicity | El Golli Bennour et al. (2009) |
| Human Caco‐2\TC7 cells | Zinc chelation | Increased apoptosis and permeability of tight junctions (reversed by zinc supplementation) | Ranaldi et al. (2009) |
| Pig kidney cells (LLC‐PK1) | Leontopodic acid pretreatment | Reduced production of reactive oxygen species but did not inhibit cytotoxicity | Costa et al. (2010) |
| Primary rat hepatocytes | Silibinin | Decrease in apoptotic cell death; decreased lipid peroxidation | Essid and Petzinger (2011), Essid et al. (2012) |
| Human peripheral blood mononuclear cells | N‐acetylcysteine | Reduction of reactive oxygen, DNA damage, cell cycle G1 arrest and downregulation of CDK4 and cyclinD1 | Liu et al. (2012) |
| Monkey kidney cells (Vero cell line) | Quercetin |
Reduced apoptosis and oxidative stress Reduced DNA damage |
Ramyaa and Padma (2013) |
| Madin‐Darby canine kidney cells (MDCK) | Diosmetin flavonoid | Prevents ATP depletion | Poór et al. (2014) |
| Human embryonic kidney (HEK293) cells | N‐acetylcysteine | Reduced reactive oxygen production; reduced loss of mitochondrial membrane potential; reduced loss of superoxide dismutase activity; reduced DNA strand breaks and cell cycle arrest | Yang et al. (2014) |
| Intestinal cells (Caco‐2) | Resveratrol | Increased cytotoxicity without an increase in reactive oxygen species | Cano‐Sancho et al. (2015) |
| Vero cells, and lymphocytes in vitro and mice in vivo | Polyphenols (caffeic acid, luteolin and chlorogenic acid | Reduction of cytotoxicity (chlorogenic acid and caffeic acid); inhibition of DNA strand breaks (caffeic acid and chlorogenic acid) and inhibition of micronucleation (chlorogenic acid) | Cariddi et al. (2015) |
| Human peripheral blood lymphocytes | Sodium copper chlorophyllin | Reduced cytotoxicity and reduced DNA damage | Domijan et al. (2015) |
| Porcine kidney cells (PK15) | Selenomethionine; overexpressed selenoprotein S | Reduced cytotoxicity (by selenomethionine, evidently via induction of glutathione peroxidase); Inhibition of promotion of viral replication (selenoprotein S) | Gan et al. (2015, 2016) |
| Human hepatoma cells (HepG2) | Vitamin E | Reduced cytotoxicity and reactive oxygen production | Gayathri et al. (2015) |
| Human embryonic kidney cells (HEK293) | Resveratol | Reduced DNA strand breaks | Raghubeer et al. (2015) |
| Neuro‐2a cells | N‐acetylcysteine pretreatment | Protected against apoptotic cell death | Bhat et al. (2016) |
| Monkey kidney cells (Vero cell line) | Superoxide dismutase mimic (MnTnHex‐2‐PyP) |
Decreased reactive oxygen concentration Decreased cytotoxicity |
Costa et al. (2016) |
| Porcine kidney cells (PK‐15) |
N‐acetylcysteine Selenomethionine |
Reduced autophagy | Qian et al. (2017, 2018) |
ATP: adenosin triphosphate; CDK4: Cyclin‐dependent kinase 4; DNA: deoxyribonucleic acid; OTA; ochratoxin A; G1: Gap 1 phase.
4.3.1.6. Summary of OTA – induced cell toxicity via modulation of gene expression and cell signalling
In summary, OTA causes cell death in a wide range of cell types both in vitro and in vivo. Both mitochondrial/caspase 3 and caspase 8–mediated apoptotic pathways have been implicated. The kidney proximal tubule cells are particularly sensitive due to the presence of anion transport systems for uptake. OTA alters the expression of proteins via transcriptional, microRNA and protein post‐translational modifications to modulate cell signalling systems responsible for apoptosis and also pathways related to stress responses, inflammation, fibrosis and the balance between cell survival, cell proliferation, cell division and cell death. Activation of PKC and MAPK‐ERK signalling pathways play an important role in the stress response and cell proliferation. Particularly important is the downregulation of Nrf2 which contributes to the oxidative stress induced by OTA (see below).
4.3.1.7. Potential for toxicity in cell types other than those in the kidney and liver
4.3.1.7.1. Immune system
There is evidence mainly from in vitro studies, for effects of OTA on neutrophils and macrophages including oxidative stress, apoptosis, phosphorylation of the ERK1/2 and release of TNFα via NF‐kB pathways by OTA (Al‐Anati et al., 2010; Liu et al., 2012; Giromini et al., 2016; Kupski et al., 2016; Brennan et al., 2017; Xu et al., 2017). Further observations in human blood mononuclear cells (Rossiello et al., 2008) indicated that OTA reduced the ability of lipopolysaccharide, phorbol myristate, IL‐1β or TNFα to produce tissue factor and plasminogen activator inhibitor 2 which led the authors to propose a MoA in immunosuppression. Although Ferrante et al. (2006) found infiltration of polymorphonuclear infiltration into the duodenum following a single dose of OTA in mice, the role of the immune response towards oxidative stress in the kidney in vivo has not been demonstrated.
4.3.1.7.2. Intestine
The ability of OTA to affect intestinal function has also been assessed in vitro using Caco‐2 cells, porcine intestinal epithelial (IPEC‐J2) cells or mouse rectal CMT93‐II cells. OTA disrupted barrier dysfunction through reduction of claudins (Lambert et al., 2007; Maresca et al., 2008; Romero et al., 2016; Nakayama et al., 2018; Wang et al., 2018). The significance of these finding in vivo is yet to be established.
4.3.1.7.3. Nervous system
Neurological toxicity of OTA during embryonic development has been noted above (see Section 4.2.5 on Reproductive toxicity). Following treatment of mice with OTA behavioural studies showed adversity resembling Parkinsonian features which were ameliorated with L‐Dopa (Bhat et al., 2018). Oxidative stress and inflammation produced by OTA are associated with neuronal toxicity in vitro and in vivo (Sava et al., 2006; Bang et al., 2009; Yoon et al., 2009; Zhang et al., 2009; Razafimanjato et al., 2010; Bhat et al., 2016) also found that OTA could inhibit glutamate uptake by rat astrocytes but the relevance in vivo is uncertain.
4.3.1.7.4. Endocrine systems
OTA was found not to interact with steroid receptors, but in H295R cells, it increased production of oestradiol and increased the aromatase protein at 1 μmol/mL (Frizzell et al., 2013). Furthermore, Woo et al. (2013) reported that OTA upregulated 3β‐hydroxysteroid dehydrogenase/isomerase expression and there was an associated increase in progesterone secretion in human placental JEG‐3 cells. Kumar et al. (2011) found alteration of the blood levels of a range of hormones (triiodothyronine, thyroxin, testosterone, insulin, prolactin and cortisol) following dosage of OTA in the feed of rats for 30 days, but the mechanisms of these changes were not clear.
4.3.2. Mode of action in carcinogenesis
The results of genotoxicity tests with OTA are provided in Section 4.2.4 on Genotoxicity. A key question concerns the ability of OTA, or a metabolite, to form DNA adducts which could be responsible for the DNA damage and mutational spectra observed in rodent kidney. The weight of evidence suggests that metabolic activation of OTA to a reactive metabolite does not occur (see Section 4.1.1 on Toxicokinetics). Evidence for the formation of OTA‐derived DNA adducts in the kidney outer medulla site of carcinogenicity is controversial (see Section 4.2.2 on Repeated dose studies for details of studies). Whereas P32‐post‐labelling studies have suggested the presence of DNA adducts (albeit uncharacterised), other studies using accelerator mass spectrometry or isotope dilution have failed to detect such adducts. Thus, DNA adducts are either not produced from OTA at all or (at most) the level of adducts is extremely low (no more than 10 adducts/cell).
OTA induces gene mutations in rats and mice only in the outer medulla of the kidney and only at the Red/Gam locus (Spi‐mutants), which has been specifically devised to detect deletion mutations (Hibi et al., 2011; Kuroda et al., 2014). Hibi et al. (2013b) found altered expression of genes involved in the DNA damage response and cell cycle regulation in the outer medulla of rats following dietary administration of OTA for 4 weeks. The authors considered the pattern of altered gene expression as evidence of DNA DSBs. Indeed, large deletion mutations and DSBs were found in rat kidney associated with increased γ‐H2AX protein expression and increased expression of homologous recombination repair‐related genes and genes involved in cell cycle arrest following administration of OTA by gavage for 4 weeks to rats (Kuroda et al., 2014, 2015).
In an attempt to understand the occurrence of Spi − mutations in the kidneys of p53‐deficient Gpt mice (but not in p53‐proficient mice), Kuroda et al. (2015) considered that homologous recombination repair was responsible for the single base mutations seen in the absence of p53 since only in the presence of p53 did DNA DSBs arise. Importantly, Bondy et al. (2015) found no increased sensitivity to OTA tumourigenicity as a result of p53 heterozygosity in mice following administration in the diet for 26 weeks. The authors considered this as evidence of a non‐genotoxic MoA. Bloch et al. (2012) attempted to distinguish between genotoxic and non‐genotoxic renal carcinogens based on their profile of alteration of gene expression in NRK‐52E cells, using OTA to represent the non‐genotoxic class of carcinogen. However, several pathways were common to both genotoxic and non‐genotoxic categories making distinction not possible. The production of a senescence phenotype associated with cell cycle arrest was detected in rat kidney proximal tubular cells (HKC) exposed to OTA and this was associated with activation of the p53‐p21 and p16‐pRB pathways (Yang et al., 2017). Disturbance of mitosis and chromosomal segregation appear to be important (Mosesso et al., 2008; Czakai et al., 2011). The processes involved in DNA damage following disturbance of the cell cycle and cytokinesis are discussed in detail by Hayashi and Karlseder (2013) and by Bizard and Hickson (2018). It is possible that the mutations arise as a result of the evident replication stress as discussed by Gelot et al. (2015). The mechanism leading to replicative stress is, however, unclear.
In immortalised human kidney epithelial cells exposed to OTA, increases in apoptosis and decreases in the rate of mitosis were associated with an increase in aberrant mitotic figures with misaligned chromosomes and formation of giant cells with abnormally large or multiple nuclei. The authors concluded that OTA might promote tumour formation via interference with microtubule dynamics and mitotic spindle formation, resulting in apoptosis or interruption of mitosis to explain the observed cytogenetic abnormalities. A cell‐free in vitro tubulin polymerisation assay showed that OTA inhibited microtubule assembly (Rached et al., 2006). However, the mechanism is unclear and the inhibitory concentration of OTA was relatively high. Similar effects have been observed with citrinin, another mycotoxin (Pfeiffer et al., 1998).
Nuclear enlargement was also seen in kidney proximal tubule cells in OTA‐treated F344 rats (Rached et al., 2007). Moreover, in rats, mRNA levels for various regulators of mitosis and genes linked to chromosomal instability were over‐expressed in kidney of OTA‐treated rats, whereas Cx32 and regucalcin genes were downregulated (Adler et al., 2009). The authors suggest that aberrant mitosis may play a role in kidney carcinogenesis. However, the Panel notes that the gene expression changes could potentially be causative or a consequence of carcinogenesis. OTA can interfere with DNA topoisomerase II and chromosome segregation during cell division (Cosimi et al., 2009) and disruption of chromosome separation and progression of mitosis as a non‐DNA reactive mechanism was also proposed by Mosesso et al. (2008) as evidenced by detection of endoreduplication and chromatid separation in V79 cells.
In conclusion, post‐translational modification of proteins and the alteration of gene expression detailed above may contribute to modulation of the cell cycle and spindle function leading to aberrant alignment of chromosomes, increased ploidy and karyomegaly. In association with the cell proliferative responses and the effect on DNA repair enzymes described above, these changes appear to lead to genomic instability. The processes involved in DNA damage may follow disturbance of the cell cycle and cytokinesis.
4.3.2.1. The contribution of additional non‐DNA reactive mechanisms
4.3.2.1.1. Secondary oxidative genotoxicity
The question arises as to whether DNA damage may result from secondary production of ROS (see above). In several cellular systems in vitro the ability of DNA repair enzymes (formamido‐pyrimidine‐DNA glycosylase (Fpg) or endonuclease III) used in the Comet assay to convert oxidative DNA damage into SSBs was shown. This provided evidence of DNA oxidation (Kamp et al., 2005; Arbillaga et al., 2007; Ali et al., 2014). However, despite oxidative stress under these conditions, there was no change in the expression of genes involved in a DNA damage response (DNA repair, apoptosis and cell cycle control; Arbillaga et al., 2007). Similar evidence for oxidative DNA damage comes from some studies on SSBs (Comet assay) observed in cells derived from liver and kidney of Fischer rats treated with OTA (Mally et al., 2005b; Domijan et al., 2006). However, the major oxidised base 8‐OHdG was not detected in vivo in rat kidney (Mally et al., 2005a, 2005b) and GC:TA transversions (typically associated with DNA oxidation) were not seen in rats treated with OTA (Hibi et al., 2011).
Kumar et al. (2012) assessed tumour formation in mouse skin. A single topical application of OTA (80 mg/mouse) followed by twice weekly application of the tumour promoting agent 12‐Otetradecanoylphorbol‐13‐acetate for 24 weeks lead to tumour formation. The authors associated this effect to the evident oxidative stress, MAPKs signalling and DNA damage.
4.3.2.1.2. Altered cell signalling and cell proliferation
Cell proliferation in the kidney of rats given OTA was considered by Qi et al. (2014a) to be a major factor in kidney carcinogenesis. They found that, in association with kidney toxicity, there was a dose‐related increase seen in proliferating cell nuclear antigen (PCNA). p53 gene expression was also decreased in both kidney and liver. Abassi et al. (2016) found that OTA‐induced clonogenicity in human colon carcinoma cells and fetal lung fibroblast‐like cells transformed with MYC. In an attempt to understand the apparent tumour promoting action of OTA on mouse skin, primary murine keratinocytes were treated with OTA which was found to cause cell proliferation and activation of EGFR and downstream MAPKs and Akt pathways (Kumar et al., 2013). Further evidence of the importance of proliferative changes in rat kidney carcinogenesis come from the observation that OTA activated Topo IIα, Ubd and apoptosis that cooperate in inducing a marked proliferative response (Taniai et al., 2012a). In this study, proximal tubule cell proliferation was accompanied by increased levels of topoisomerase IIα protein and evidence of DNA damage and cell cycle disruption (G2M transition) (Taniai et al., 2012b). Using the same treatment regime, karyomegaly and aberrations of the cell cycle were seen in the kidney tubular cells without evidence of oxidative stress (Taniai et al., 2014).
4.3.2.1.3. Effect on intercellular communication
Many non‐genotoxic carcinogens interfere with connexin‐mediated intercellular communication (e.g. Chipman et al., 2003). The ability of OTA to inhibit gap junctional intercellular communication was studied in Madin Darby canine kidney (MDCK) cells (Mally et al., 2006). In the absence of cytotoxicity, there was a significant reduction of intercellular communication which was associated with a small, but dose‐dependent, decrease in the concentration of the gap junction protein connexin 43. Adler et al. (2009) found that there was a downregulation of Cx32 in rat kidney following 90‐day treatment with OTA. In the liver of rats given OTA (289 μg/ kg bw alternate days for 90 days), there was a reduction of E‐cadherin and the connexins 26, 32 and 43 implying interference with intercellular gap junctional communication (Gagliano et al., 2006).
4.3.2.1.4. Epigenetic mechanisms
Various studies in cell cultures have demonstrated modulation of histone acetylation (Czakai et al., 2011; Giromini et al., 2016; Limbeck et al., 2018). In Fischer 344 rats given OTA in the diet at 300 μg/kg bw per day (Mantle et al., 2005) an increased histone deacetylase (HDAC) enzymatic activity associated with enhanced HDAC3 protein levels was measured which may give rise to epigenetic modulation (Oh et al., 2013). Genome wide analysis of DNA methylation in rat kidney following treatment of rats with OTA (210 μg/ kg by gavage 5 days/week for 90 days) revealed that various categories of genes were either hypermethylated or hypomethylated including those involved in protein kinase activity and mTOR cell signalling (Ozden et al., 2015). Li et al. (2015) gave rats 70 or 210 μg/kg body weight OTA by gavage for up to 26 weeks and measured parameters relating to DNA methylation in the kidney. Dynamic and dose‐dependent alteration of global DNA methylation was seen in this study with hypermethylation of the promoters of E‐cadherin and N‐cadherin (along with reduced expression) and increased expression of Wnt and P13K/AKT pathways.
4.3.2.2. Overall conclusion on the MoA in carcinogenesis
Non‐genotoxic mechanisms appear to contribute to carcinogenesis. This is based on a stress response (including an oxidative stress) involving alteration of gene expression (in part mediated by epigenetic modifications) contributing to modulation of DNA repair enzymes, disruption of cell cycle and an imbalance of apoptosis and cell proliferation. Spindle function is impaired leading to aberrant alignment of chromosomes. There is increased ploidy, aneuploidy and karyomegaly. In combination, these effects appear to lead to genomic instability and large deletions of DNA in surviving proliferating cells. The precise mechanism whereby OTA leads to the observed low level of mutations at the target site in rat kidney remains unclear. The mutational spectrum is not consistent with reactive oxygen damage. One possibility is through interaction of OTA with DNA, although the evidence for DNA adducts is weak. A further possibility is that the mutations arise as a result of the evident replication stress. In that event, there remains uncertainty also as to the cause of the replication stress which may result both from initial DNA damage (direct or secondary to oxidation) or from modulation of proteins that regulate the cell cycle including those coded by oncogenes. In conclusion, there is insufficient evidence to support either direct DNA damage or indirect DNA damage in OTA carcinogenesis.
4.4. Identification of critical effects
4.4.1. Identification of critical acute effects
The evidence of acute effects of OTA in laboratory animals is very limited and no relevant studies published since the last EFSA assessment in 2006 has been identified (see Section 4.2.1 on Acute toxicity). No acute effects upon exposure to OTA in humans have been reported (see Section 4.2.6 on Observations in humans). Thus, the CONTAM Panel concluded that acute toxicity is not a critical effect for the hazard assessment of OTA.
4.4.2. Identification of critical chronic effects
In the previous OTA opinion (EFSA, 2006), the CONTAM Panel concluded that the kidney is the main target organ for the adverse effects of OTA and that immunotoxic, neurotoxic and teratogenic effects were observed at high dose levels. OTA has been shown to induce nephrotoxicity in mice, rats, dogs and pigs. Long‐term administration of OTA caused renal tumours in rats and mice and a NOAEL/LOAEL of 15/50 μg/kg bw per day (extrapolated from the doses from 5 days treatment per week) for tumour induction was derived from a 2‐year gavage study with Fischer 344/N rats (NTP, 1989). The mechanisms underlying the genotoxicity associated with OTA exposure were unclear and the Panel was unable to conclude whether OTA causes DNA damage by indirect or direct effects. Epidemiological studies were insufficient to classify OTA as renal carcinogen or link it to BEN or other endemic nephropathies of unknown cause. Considering a series of repeated dose studies in female pigs, the Panel identified a LOAEL of 8 μg/kg bw per day from a 90‐day feeding study with pigs for minimal morphological changes in kidney, changes in renal enzymes and impaired renal function (Krogh et al., 1974) as critical for the setting of a chronic HBGV (i.e. a TWI).
For the present opinion, the pivotal studies in pigs used as a basis to establish a HBGV previously have been re‐evaluated. In addition, new in vivo toxicity studies with mice, rats, rabbits and pigs published since the 2006 opinion of EFSA were reviewed to identify if any studies would provide observations of adverse effects lower than the LOAEL of 8 μg/kg bw per day previously identified in pigs (EFSA, 2006). Additionally, human evidence published since 2006 was also considered.
The new repeated doses studies are described in detail in Section 4.2 on Toxicity. In brief, the new subacute/subchronic studies with rats were mainly designed to elucidate the mechanism of OTA induced nephrotoxicity and thus employed dose levels known to be toxic. None of these studies provided observations of adverse effects lower than the LOAEL of 15 μg/kg bw derived from the 90‐day feeding study in Wistar rats from Munro et al. (1974). In new repeated dose studies with rabbits, liver and kidney toxicity was reported in rabbits, albeit not at doses as low as the LOAELs of 15 and 8 μg/kg bw previously identified in subchronic/subacute studies with rats and pigs, respectively (FAO/WHO, 2001; EFSA, 2006). The results of the new subacute/subchronic dose toxicity studies in mice are consistent with the lower sensitivity of mice to OTA toxicity as compared to rats and pigs. Likewise, none of the new studies provided observations to suggest NOAELs/LOAELs lower than the LOAELs of 15 and 8 μg/kg bw previously identified in rats and pigs, respectively (FAO/WHO, 2001; EFSA, 2006). Overall, the CONTAM Panel concluded that the new studies in pigs, even though most of them are of shorter duration, support OTA‐mediated adverse effects at dose levels close to the previously established LOAEL of 8 μg/kg bw per day.
The CONTAM Panel noted that the new chronic studies with rats were inadequate for risk assessment based on their study design. The new evidence on possible toxicity in humans is described in detail in Section 4.2.6 Observations in humans. The CONTAM Panel concluded that it is not possible to establish a causal link of exposure to OTA and adverse effects observed in epidemiological studies in humans.
Based on the above, the CONTAM Panel concluded that the results obtained in the 3‐month study in female pigs (Krogh et al., 1974) and in which an LOAEL of 8 μg/kg bw per day had been previously established were the most relevant for assessment of non‐neoplastic chronic health hazards (for details on the study see Appendix C.2). The CONTAM Panel considered the increase in total incidence of microscopic kidney lesions and the microscopic tubular lesions in the kidney occurring in 45% of the pigs at 8 μg/kg bw per day as the most appropriate endpoint for characterisation of non‐neoplastic hazards.
The CONTAM Panel also considered an increased percentage of glucose excretion as a potential critical effect for the characterisation of non‐neoplastic hazards. However, the CONTAM Panel noted the big standard deviations in all treated groups as well as in the control presented a large uncertainty and limited the use of this endpoint for BMD analysis. Furthermore, it was not possible to identify a biologically relevant BMR for this endpoint. Nevertheless, the observed effects on glucose, indicative of impaired tubular reabsorption of glucose, give supportive evidence for the use of microscopic kidney lesion for assessment of non‐neoplastic hazards. In a previous evaluation reduced p‐aminohippuric acid renal transport maximum (TmPAH) (reduced maximum tubular excretion of PAH) observed in the same experiment on pigs (Krogh et al., 1974) was used as a reference point for hazard characterisation (FAO/WHO, 2008). However, as OTA and PAH use the same transporters, OATs, in the renal tubular cells, this effect might have been the result of competitive inhibition rather than a toxic response.
The mode of action for OTA‐induced carcinogenesis is described in detail in Section 4.3.2 on Mode of action. OTA causes kidney tumours in mice and rats, the male rat being most sensitive (NTP, 1989). There is a large, but inconsistent database on possible direct and indirect interactions of OTA with DNA including also many recent experiments.
Following respective EFSA guidance documents (EFSA, 2005; EFSA Scientific Committee, 2012a), the CONTAM Panel concluded that the kidney tumours occurring in male rats (at much higher dose than the non‐neoplastic effects seen in kidney in pigs or rats) are of relevance for the risk characterisation of the compound. There is insufficient evidence to rule out that direct DNA damage contributes to their formation. The kidney tumours observed in male rats in a 2‐year feeding study (NTP, 1989) were considered as critical and thus to be used for characterisation of neoplastic hazard characterisation of OTA.
4.5. Calculation of benchmark doses
The benchmark dose (BMD) analyses were performed following the guidance of the Scientific Committee on BMD modelling (EFSA Scientific Committee, 2017).
4.5.1. Non‐neoplastic endpoints
For microscopic kidney lesions seen in the study of Krogh et al. (1974) using a standard benchmark response (BMR) of 10% for quantal data, a BMD confidence interval (BMDL10–BMDU10) of 4.73–7.17 μg OTA/kg bw per day was calculated. A detailed description of the BMD analysis can be found in Appendix F.1.
4.5.2. Neoplastic endpoints
Using the standard BMR of 10% for quantal data a BMD (BMDL10–BMDU10) of 14.5–36 μg OTA/kg bw per day for the combined incidences of adenoma and carcinoma in male rats seen in a 2‐year oral study (NTP, 1989) was calculated and used for the neoplastic risk characterisation. A detailed description of the BMD analysis can be found in Appendix F.2.
4.5.3. Consideration of toxicokinetic differences between species
As described in Section 2.2 on Previous assessments, in the EFSA assessment of 2006, an UF of 6 for toxicokinetic interspecies variability instead of the default value of 4 was applied for derivation of a TWI because of the longer half‐life of humans as compared to pigs. However, as discussed in the present opinion in Section 3.1 on Toxicokinetics, OTA exhibits very high non‐covalent binding to plasma proteins, in particular albumin, in laboratory animals and humans. In general, only the free (unbound) proportion of a substance present in blood plasma undergoes glomerular filtration in the kidneys. Therefore, high plasma protein binding (PPB) decreases the elimination of compounds from blood plasma and leads to increases in plasma half‐lifes and, after repeated exposure, to accumulation. Due to differences in the extent of PPB between species, the proportion of free OTA in the blood and the accumulation of OTA differ markedly between humans and experimental animals.
According to the different affinity constants for PPB in humans, pigs and rats, and assuming the same total OTA concentration (bound plus unbound) in blood, it has been calculated (see Appendix D) that the plasma concentration of free OTA is about 70‐fold lower in humans than in pigs, and about 120‐fold lower in humans than in rats. On the other hand, if humans (with a plasma half‐life of 35 days), pigs (5–6 days) and rats (5–6 days) are exposed to the same level of OTA in their diet and the total plasma concentration of OTA at steady state will be about six times higher in humans than in pigs and about six times higher in humans than in rats. This assumes the same bioavailability and rate of metabolism in each species. Taking into account both the accumulation and the PPB at equal exposure (same external dose), the plasma levels of free OTA in humans should still be about 10 times lower in humans than in pigs and 18 times lower in humans than in rats. Therefore, the CONTAM Panel considered that an extra UF factor for accumulation in humans is not necessary and the default factor of 4 is sufficient to account for toxicokinetic interspecies differences.
4.6. Establishment of a health‐based guidance value (HBGV) vs. application of a margin of exposure (MOE) approach
As described in Section 4.4 on Identification of critical effects, the CONTAM Panel concluded that there is clear evidence for carcinogenic effects of OTA. The mechanisms underlying the genotoxicity associated with OTA exposure are unclear and the Panel was unable to conclude whether OTA causes DNA damage by indirect or direct effects. Following respective EFSA guidance on substances that are both genotoxic and carcinogenic (EFSA, 2005; EFSA Scientific Committee, 2012a), the CONTAM Panel considered it was not appropriate to establish a tolerable daily intake (TDI) or a TWI and concluded that for both non‐neoplastic and neoplastic effects of OTA an MOE approach needs to be applied for risk characterisation. As a consequence, the TWI of 120 ng OTA/kg bw as established by the CONTAM Panel in 2006 is no longer valid.
The increased incidence of microscopic kidney lesions seen in a 3‐month feeding study with pigs (Krogh et al., 1974) was considered as the most appropriate endpoint of non‐neoplastic effects of OTA and the resulting BMDL of 4.73 μg/kg bw per day was used for comparison with chronic exposures. The CONTAM Panel concluded, that an MOE of 200 needs to be applied to this BMDL consisting of the default UFs of 100 for toxicodynamic and toxicokinetic inter‐ and intraspecies differences and an additional UF of 2 considering the short study duration of Krogh et al. (1974) (3 months) that does not cover the entire lifespan of the animals (pigs) tested. In this context, it needs to be noted that in the previous EFSA opinion (EFSA, 2006) for derivation of a TWI, a specific UF of 6 for toxicokinetic differences between pigs and humans (instead of the default 4) was introduced because of the longer half‐life of OTA in humans (see Section 2.2 on Previous risk assessments). Upon a comprehensive re‐evaluation of the toxicokinetic data of OTA in different species including OTA plasma protein binding, the CONTAM Panel concluded that such additional UF is not needed (for details see Section 4.1.1 on Toxicokinetics).
The EFSA guidance on default values (EFSA Scientific Committee, 2012a) issued after publication of the previous EFSA opinion on OTA (EFSA, 2006) proposes the use of an UF of 2 for extrapolation from subchronic to chronic duration in rodents. The CONTAM Panel noted that no differences were observed between NOAELs derived from subacute, subchronic and chronic toxicity studies on OTA in rats (i.e. the ratios between NOAEL from both subacute and subchronic to chronic toxicity studies in rats were 1, see Sections 4.2.2 and 4.2.3) and considered that this is also likely to be the case in pigs.
However, as 3 months represent a much smaller fraction of the total lifespan of pigs as compared to rats, the CONTAM Panel considered it appropriate to apply an UF of 2 for extrapolating the 3‐month study in pigs to chronic duration. The CONTAM Panel recognised the uncertainty associated with applying the UF of 2.
For neoplastic effects of OTA, the CONTAM Panel emphasise that a clear distinction between direct and indirect mechanism of genotoxicity was not possible. As a result, the risk characterisation will be either not sufficiently cautious or overcautious depending on which MoAs actually occur.
In the absence of elucidated MoAs for the genotoxicity/carcinogenicity of OTA, the Panel concluded that an MOE of 10,000 needs to be applied to the BMDL10 of 14.5 μg/kg bw per day for neoplastic effects (kidney tumours) in the rat. The Panel points out that this MOE is likely to be particularly conservative in this case as the evidence for a direct interaction of OTA with the DNA is inconclusive and other threshold mechanisms may play a role in the formation of kidney tumours. As it was not possible to quantify these variables, the default MOE of 10,000 was applied.
4.7. Occurrence data
4.7.1. Occurrence data used in the present assessment
For the present assessment, it was decided to use data submitted to EFSA between 2009 and 2018 in order to have a data set representing recent/current occurrence levels while retaining a sufficiently high number of samples. The resulting data set included a total of 90,147 analytical results on OTA in food.
Data providers were contacted to clarify inconsistencies identified during the data check. The following modifications were made to the initial data set based on the feedback received:
2,008 occurrence values identified as suspect samples were excluded. In the final data set, 53% of the data were taken by selective sampling, 44% by objective sampling while the remaining 3% were either convenient or import sampling, or with no information provided. It was decided to retain the samples with no specific sampling strategy as the % of left‐censored samples were in accordance with the ones taken by other sampling strategies.
The product description of a number of records allowed a more accurate FoodEx classification. In these cases, the samples were reclassified to a more specific, lower level.
Special attention was given for the ‘Product treatment’ information. Samples classified as fresh fruit or vegetable with the PRODTREAT field ‘dehydration’, ‘processed’, etc. were reclassified accordingly as dried or processed. In the case of figs, samples reported as unprocessed or without any information on the treatment were all assumed to be dried based on the comparison with the levels found in dried figs. As the original occurrence data referred to fresh figs and it cannot be excluded that figs can be contaminated in their fresh form, a factor of 2.86 (taken from EFSA's Raw Primary Commodity Model Database, EFSA, 2019) was applied to adjust the contamination levels for fresh figs taking into account their water content. Without this assumption, the exposure to OTA from consumption of figs would most likely be more overestimated.
Analytical results with an LOQ above the ML as defined by Commission Regulation (EC) No 1881/2006 were not used for the assessment. In addition, exclusion criteria were also set for those samples where upon carefully checking the occurrence levels and distribution of the LOQ/LOD values, high percentiles (P95 or P75) of LODs/LOQs were chosen to eliminate extremely high ones reported. The method was applied only for categories considered as an important source of exposure in order to decrease overestimation with the UB exposure scenario (see Table 12). More information about the distribution of LOQ values is provided in Annex, Table 7.
Results obtained without information on the analytical method were retained for the assessment if their LOD/LOQ values were sufficiently low comparing to the majority of other samples reported with accepted analytical method.
Analytical results of samples reported to be measured with PCR‐ELISA, Enzyme Immunoassay Tests (EIA), IgG ELISA, electrochemical tests based on potentiometry, AAS, Competitive ELISA) were not considered for the present assessment for reasons of lack of sensitivity, specificity, possible false positive/negatives and/or frequency of use of these methods.
In total 16,256 samples were discarded from the assessment for several reasons including suspect sampling, taken within a total diet survey or subject of a duplicate submission (see Annex, Table 1 for more details).
4.7.1.1. Expression of results
Most of the analytical results were expressed as whole weight. In cases where the results were reported as dry weight, the values were adjusted with the reported moisture content to whole weight. Where information on ‘whole weight’ or ‘dry weight’ was not reported (e.g. some samples of coffee beans, spices, paprika powder), these were, upon re‐checking, assumed to be whole weights when they were in the range of the other reported occurrence levels within the corresponding category.
4.7.1.2. Occurrence summary statistics
Food samples were submitted by 29 European countries and FEDIOL, the European Vegetable Oil and Protein meal Industry (FEDIOL) (Figure 7) between the years 2009 and 2018 (Figure 8). Mean and P95 levels for OTA in a number of food categories are shown in Table 13. More data on summary statistics of occurrence values are presented in the Annex, Table 2. The proportion of left‐censored data (results below the LOD or LOQ) was 75%.
Figure 7.
Number of food samples in the final data set encoded by European countries: n = 73,891 food samples with results for OTA
Figure 8.
Number of food samples in the final data set reported by year for the n = 73,891 food samples with results for OTA
Table 13.
Highest 15 mean LB and UB occurrence levels in decreasing order for OTA among food groups with at least six samples (see Annex for additional food items)
| FoodEx level2 | FoodEx Level3 | No of samples | % LC | Mean LB (μg/kg) | Mean UB (μg/kg) |
|---|---|---|---|---|---|
| Dietary supplements | Plant extract formula | 19 | 42 | 35.62 | 35.79 |
| Fruiting vegetables | Chilli pepper (Capsicum frutescens) | 49 | 27 | 10.57 | 10.88 |
| Confectionery (non‐chocolate) | Liquorice candies | 112 | 66 | 9.47 | 10.30 |
| Spices | Paprika powder | 2,670 | 15 | 8.92 | 9.07 |
| Flavourings or essences | Liquorice (Glycyrrhiza glabra) | 146 | 62 | 6.27 | 7.13 |
| Spices | Chilli powder | 450 | 24 | 5.65 | 5.89 |
| Spices | Allspice (Pimenta dioica) | 25 | 76 | 5.49 | 6.08 |
| Spices | Cayenne pepper (Capsicum frutescens) | 226 | 24 | 4.34 | 4.72 |
| Bulb vegetables | Garlic, bulb (Allium sativum) | 19 | 74 | 3.70 | 5.28 |
| Dried fruits | Dried figs (Ficus carica) | 1,936 | 75 | 3.63 | 4.12 |
| Preserved meat | Ham, pork | 54 | 46 | 3.03 | 3.09 |
| Spices | Nutmeg (Myristica fragans) | 1,069 | 54 | 2.95 | 3.71 |
| Herb and spice mixtures | Curry powder | 299 | 42 | 2.6 | 3.1 |
| Herb and spice mixtures | Mixed herbs | 56 | 73 | 2.5 | 3.7 |
| Cheese | Cheese (type not specified)(a) | 15 | 73 | 2.2 | 2.9 |
LC: left censored; LB: lower bound; UB: upper bound;
Occurrence levels of five samples of Grana Padano cheese are not included here, but considered in the exposure assessment (Annex, Tables 2 and 3).
The occurrence data for OTA on 73,891 food samples is available at: http://doi.org/10.5281/zenodo.3739292.
4.7.1.3. Grouping and selection of food categories for the exposure assessment
In view of the exposure assessment, food data were grouped at different FoodEx levels, and other merged categories used as supplements to FoodEx for the present assessment, taking into consideration several factors including the similarities between food categories, the number of samples and the concentrations observed.
At the most detailed level (FoodEx level 3), the food was retained if more than six samples were available in the category. If less than six samples were available, the levels were compared with similar foods belonging to other categories:
If the levels were similar, the samples were either grouped together with the similar food, resulting in a new category, or the food was taken into account at a higher (parent) level (FoodEx level 2) if this broader category was well‐represented by the available categories at FoodEx level 3.
If the levels reported were very different, the category was excluded as it was considered insufficiently covered. In order to avoid excluding a specific, but potentially relevant category, a single exception was made for Chocolate and chocolate products for diabetics (n = 4).
Samples in the categories of the least detailed FoodEx classification (FoodEx level 1) were excluded in cases where no further information was available for a more specific reclassification.
Categories where the analytical results were 100% left censored and OTA contamination was not expected were not taken into account in the assessment. Where in similar food categories on the same level quantified values were found, the samples were grouped together with the similar food according to the same principle as in the first bullet point. For instance in the category ‘Black tea, infusion’, there were only three left‐censored samples, but as in unspecified tea and other tea categories we had quantified values, all tea samples were pooled together (in case of solid teas, after application of a dilution factor) and the mean was calculated based on all of them. For the detailed list of the categories considered, see Annex, Table 2.
Only one honey sample was quantified out of a total 77. Because in the literature no evidence can be found for the presence of OTA in honey, the Panel decided not to consider this category as it would very likely lead to an overestimation of the exposure.
Considering that it is unlikely that fresh cheese gets contaminated with OTA, the contamination levels analysed in cheese (not defined) were not applied on these types.
In case of powdered or concentrated foods (such as coffee, beverage concentrate), dilution factors (see Annex, Table 3) were applied in order to get the ready‐to‐consume form and match with the consumption data.
Overall, 2,122 samples were excluded from the assessment from the 73,891 samples as they did not fulfil the above selection criteria. In total, 209 categories were created from the 71,769 analytical results for the linking between food occurrence and food consumption data (see Annex, Table 3).
4.7.2. Occurrence of OTA as reported in the public literature
OTA has been detected in foods of plant and animal origin. In foods of plant origin, OTA is frequently found in cereal products, beer, coffee, cacao, chocolate, vegetables, green tea, raisins, grape juice, pistachios, figs, wine, liquorice, chestnuts and spices (e.g. dried red pepper, chilli powder, black pepper, coriander, ginger, cayenne pepper, curcuma and nutmeg).
According to the Rapid Alert System for Food and Feed (RASFF) portal,14 in the period between January 2016 and June 2019, 25 of in total 87 notifications of OTA were on cereal‐based products.
OTA frequently occurs in baby food and breakfast cereals. Rice, barley, oats and wheat are the most contaminated ingredients (Piacentini et al., 2019).
Coffee is frequently infected with ochratoxigenic fungi and temperature affects significantly growth, germination of fungi, as well as the toxin production. Contamination of roasted coffee with OTA is directly related to the processing quality throughout the coffee production chain, from the farming to the roasting processes (Khaneghah et al., 2019).
OTA occurs also frequently in capsicum powder. In the EU, in 2017 and 2018 alone, 41 cases of pepper contamination were reported by the RASFF. Among them, eight notifications were on OTA level exceeding the maximum level (ML). Production of the pepper crop, its transportation, processing and storage are crucial for food safety. OTA has previously been reported in red pepper samples (Santos et al., 2010; Ham et al., 2016), dried chilli pod (Thirumala‐Devi et al., 2000; Jalili and Jinap, 2012; Yogendrarajah et al., 2014), chilli powder (Ozbey and Kabak, 2012; Iqbal et al., 2013), red pepper flakes (Tosun and Ozden, 2015), chilli sauce (Iqbal et al., 2011), sweet pepper (Gambacorta et al., 2018) and paprika (Fazekas et al., 2005; Hierro et al., 2008; Ahn et al., 2010). One of the main issues affecting spices is the possible co‐occurrence with other mycotoxins such as aflatoxins, zearalenone and deoxynivalenol, due to multiple fungal infection (Santos et al., 2010, 2011).
Several studies reported the occurrence of OTA in tea and herbal infusions. It has been shown that toxigenic fungi, mainly Aspergillus niger and Penicillium spp., grow on tea and herbs leaves, thus inducing OTA accumulation (Haas et al., 2013). OTA was observed in only few samples, but there often at high concentrations, suggesting that under certain conditions, toxin production can be high. The transfer rate during the infusion process of tea was calculated by Malir et al. (2014), who found a mean value of 34.8% ± 1.3% for green tea. Transfer rates for green and black teas were reported by Carraturo et al. (2018), with values in the range from 33% to 55%.
A number of studies deal with the occurrence of OTA in botanicals, dietary supplements and traditional herbal remedies on the European market (Ariño et al., 2007; Diana Di Mavungu et al., 2009; Santos et al., 2010; Vaclavic et al., 2013; Veprikova et al., 2015; Gottschalk et al., 2016). Various contaminants may be present in the raw plant material and OTA may be found in dietary supplements obtained from green coffee, liquorice and spices because in commonly used extraction procedures of plant material co‐isolation is not avoided.
Contamination of foods of animal origin such as pork meat, pork blood products, poultry kidney or liver with OTA results from consumption of OTA‐contaminated feed. Meat products, such as raw ham muscle, cured meats, salami or dry‐cured ham can also be directly contaminated during processing or storage. OTA can be produced by Penicillium nordicum, which can grow on pork meat during ripening (Rodríguez et al., 2012; Merla et al., 2018).
The Italian Ministry of Health has recommended a guideline value of 1 μg/kg in pork meat products.15 Therefore, over the last years, a number of studies were performed in Italy on dry cured meat, to better understand the transfer of OTA from feed to pork meat. Several trials have been reported to establish the OTA transfer rate from feed to tissues (Bertuzzi et al., 2013; Altafini et al., 2017). Studies focused on the OTA distribution in tissues, consistently reported the order blood plasma > lung > kidney, heart > bile > liver > fat > muscle, following a dose resposnse. Besides direct contamination because of transfer from the feed the authors identified very high OTA levels in several outer dry‐cured ham samples, due to environmental contamination and subsequent mycelia growth on ham surfaces (Dall'Asta et al., 2010; Bertuzzi et al., 2013). OTA occurrence was monitored also in traditional Croatian pork cured meat products (Pleadin et al., 2014) and in French pig liver samples (Hort et al., 2018) with consistent results.
Over the last decade, several studies reported the possible occurrence of OTA in ripened cheese (Dall'Asta et al., 2010; Biancardi et al., 2013; Pattono et al., 2013; Decontardi et al., 2017; Sakin et al., 2018; Anelli et al., 2019; Ramos‐Pereira et al., 2019). Considering that the transfer of OTA from feed to ruminants milk is negligible, due to the hydrolytic activity of rumen bacteria, the OTA occurrence in cheese is very likely due to environmental contamination leading to fungal growth on the cheese surface (Kure and Skaar, 2019). Although the common fungi growing on cheese surface have been reported as non‐OTA producers (Lund et al., 1995), the growth of uncontrolled moulds during ripening and ageing may occur causing spoilage and possibly mycotoxin production (Ropars et al., 2012; Camardo Leggieri et al., 2020). Very recently, the possible migration of OTA from the crust as far as 1.6 cm in depth was demonstrated in French semi‐hard Comté cheese following artificial inoculation with OTA‐producing strains (Coton et al., 2019).
Overall, it can be concluded that the occurrence data reported in the public literature are consistent with those available in the EFSA database and used for the exposure assessment in the present opinion.
4.7.3. Effect of processing on OTA occurrence
Many studies have been performed over years to understand the effect of food processing on the concentration of OTA in the final product. Some treatments have been recognised as an effective mitigation strategy, while the efficiency of others is still less known.
In particular, sorting and other cleaning procedures have been demonstrated to be effective in decreasing the OTA content also in food commodities such as nuts, cocoa and coffee beans, through removal of ‘hot spots’ of contamination. Controlled environmental conditions, mainly humidity and temperature, may avoid fungal growth during storage. High temperatures, which are achieved e.g. during the roasting process, were shown to considerably decrease OTA by thermal degradation.
It is known that cleaning procedures such as gravity separation, dehulling, optical sorting, grading and sieving may represent an effective mitigation step in grain, on account of the distribution of fungal biomass, and the subsequent mycotoxin accumulation, on the kernel external layers (Scudamore and Patel, 2000; Cheli et al., 2013). In general, milling can cause a redistribution of mycotoxins among fractions, with a decrease in toxins in flour and an increase in milling by‐products, among them bran (Mousavi Khaneghah et al., 2018; Schaarschmidt and Fauhl‐Hassek, 2018). Thermal treatments such as kilning, baking and toasting, are commonly recognised to decrease the overall content of mycotoxins, among them OTA. Besides the effect of the thermal processing, mycotoxins might be affected by other accompanying factors such as additives, ingredients or fermentation (Schaarschmidt and Fauhl‐Hassek, 2018).
Although sorting and milling may decrease OTA content in the final fractions, no significant mitigation has been described during the bakery process (Vidal et al., 2014; Schaarschmidt and Fauhl‐Hassek, 2018). Extrusion processing, widely used in breakfast cereals manufacture, may reduce mycotoxin levels to varying degrees (e.g. up to 80% in rice). The use of baking soda increases the degradation rate of OTA (Ryu et al., 2019).
Besides cereals, beer can represent another source of OTA exposure (Peters et al., 2017). In brewery, steeping, kilning, malting and fermentation may lead to a significant decrease in OTA. Bertuzzi et al. (2011) reported an average decrease of OTA in the range of 70–90% along the beer production process.
In dried fruits such as figs, removal of damaged fruits also following an automated approach (i.e. IR or UV spectroscopy) may reduce final OTA content, especially in combination with proper drying techniques such as controlled ventilation (Mutlu‐Ingok and Karbancioglu‐Guler, 2014; Benalia et al., 2015; Petrić et al., 2018). Novel technologies such as ozonation and treatment with ionising radiation were shown to reduce OTA in different types of nuts and dried fruit (Pankaj et al., 2018). However, there is a lack of knowledge on the chemistry of the possible degradation products, and on the effect on the overall quality of food material. Therefore, these techniques are not routinely applied in food production.
Herbs and spices are subject to minimal processing before use, hand or optical sorting being the main steps for mycotoxin mitigation. However, in some cases, fumigation and steam treatment are used which may also contribute to the reduction of mycotoxin load. Although seeds, rhizomes and fruits are at risk of field contamination, it has been demonstrated that a principal concern is downstream contamination due to the use of traditional preparation practices particularly the dehydration of fruits through prolonged sun drying of bell and red peppers (Sahar et al., 2017; Weil et al., 2020). Another example is insufficient control of water activity during storage of bulk ground spices, particularly in tropical and subtropical regions which can be mitigated by controlling drying processes and storage conditions.
Fungal growth may affect cocoa beans during the fermentation and drying processes, if not adequately controlled. Cocoa beans surrounded by their pulp are traditionally fermented in heaps. Available data indicate the importance of fermentation length to avoid mycotoxin production, which should not exceed 7 days to minimise OTA contamination (FAO, 2013). At that stage, also lowering the pH and/or the addition of mild organic acid has been shown to reduce to accumulation of OTA to some extent (Copetti et al., 2012). Roasting (generally conducted at 190–210°C for 20–30 min) may significantly reduce the content of OTA via thermal degradation (Manda et al., 2009).
In coffee beans, wet‐drying methods are considered more effective in OTA mitigation than the traditional drying method. Proper drying to 11% moisture content is essential to avoid fungal growth during storage and transport.
Several studies have shown that roasting efficiently reduces OTA concentrations (Blanc et al., 1998; Pérez de Obanos et al., 2005; Ferraz et al., 2010; Castellanos‐Onorio et al., 2011). Roasting levels (stronger roasting leads to lower OTA levels) and particle size (coarse particles lead to lesser OTA contents than fine ones) seem to be the most relevant factors affecting the final OTA concentration, as reported by Oliveira et al. (2013). Roasting degradation products have been studied and formation of 2'R‐OTA and 2‐decarboxy‐OTA, explain about 40% of the total loss over processing. The remaining degradation is probably due to the binding of OTA to coffee polysaccharides via esterification as a further thermal reaction (Bittner et al., 2013). More recently, OTalpha amide and OTalpha (see Figure 2 of Section 4.1.1 on Toxicokinetics) were identified as minor thermal degradation products of OTA (Bittner et al., 2015). The final occurrence of OTA in the coffee beverage is affected by the preparation method (e.g. brewing, espresso, filtration). Malir et al. (2014) reported that median values for OTA transfer rate from ground roasted coffee to the drinks were the following: ‘lungo’ (54.9%) > ‘americano’ (51.1%) > ‘espresso’ (32.9%) > ‘doppio’ (29.8%) > ‘ristretto’ (22.7%). The authors also demonstrated a positive correlation between the OTA transfer rate and the water volume used for the preparation. Slightly acidic conditions were also found positively correlated with a higher OTA extraction.
4.8. Exposure assessment
4.8.1. Current exposure assessment
4.8.1.1. Mean and high dietary exposure
Chronic dietary exposure was estimated across Europe following the methodology described in Section 3.5. A total of 38 dietary surveys, carried out in 22 different Member States, were selected for this assessment. These dietary surveys and the number of subjects available per age class are described in the Annex, Table 4).
Table 14 summarises the chronic dietary exposure estimates for OTA across the 38 dietary surveys. Detailed summary statistics on the exposure estimates calculated for each dietary survey are presented in the Annex, Table 5.
Table 14.
Mean and 95th percentile of chronic exposure estimates at the LB and UB for OTA
| Age classa | Number of surveys | Mean dietary exposure (ng/kg bw per day) | |||||
|---|---|---|---|---|---|---|---|
| Minimum | Median | Maximum | |||||
| LB | UB | LB | UB | LB | UB | ||
| Infants | 11 | 0.64 | 2.53 | 1.93 | 5.66 | 8.64 | 15.71 |
| Toddlers | 14 | 3.32 | 7.34 | 5.72 | 12.33 | 9.13 | 17.79 |
| Other children | 19 | 2.91 | 7.40 | 5.03 | 10.78 | 8.05 | 14.36 |
| Adolescents | 18 | 2.20 | 5.15 | 3.26 | 6.02 | 4.54 | 8.09 |
| Adults | 19 | 1.66 | 3.47 | 2.41 | 5.09 | 3.84 | 6.03 |
| Elderly | 18 | 1.30 | 3.09 | 2.07 | 4.54 | 3.57 | 6.18 |
| Very elderly | 14 | 1.10 | 2.62 | 2.08 | 4.52 | 3.59 | 5.32 |
| Pregnant women | 2 | 1.49 | 3.28 | 1.88 | 3.81 | 2.26 | 4.33 |
| Lactating women | 2 | 1.66 | 4.07 | 2.11 | 4.48 | 2.57 | 4.88 |
| Age classa | Number of surveys | 95th percentile dietary exposure (ng/kg bw per day)b | |||||
|---|---|---|---|---|---|---|---|
| Minimum | Median | Maximum | |||||
| LB | UB | LB | UB | LB | UB | ||
| Infants | 10 | 2.58 | 7.34 | 6.86 | 14.22 | 30.36 | 51.69 |
| Toddlers | 12 | 9.97 | 16.59 | 12.20 | 22.01 | 22.23 | 37.13 |
| Other children | 19 | 6.26 | 14.40 | 10.09 | 19.44 | 17.92 | 28.06 |
| Adolescents | 17 | 4.82 | 9.93 | 6.55 | 11.38 | 10.46 | 15.70 |
| Adults | 19 | 3.86 | 7.56 | 5.44 | 10.36 | 8.88 | 12.65 |
| Elderly | 18 | 3.16 | 6.68 | 4.30 | 8.61 | 8.60 | 13.71 |
| Very elderly | 10 | 2.40 | 5.13 | 3.97 | 8.45 | 5.22 | 9.19 |
| Pregnant women | 2 | 3.07 | 6.21 | 4.06 | 7.23 | 5.04 | 8.26 |
| Lactating women | 2 | 3.11 | 6.40 | 4.51 | 8.23 | 5.92 | 10.06 |
bw: body weight; LB: lower bound; UB: upper bound.
Section 2.3 describes the age range within each age class.
The 95th percentile estimates obtained on dietary surveys/age classes with less than 60 observations may not be statistically robust (EFSA, 2011a). Those estimates were not included in this table.
As can be seen in Table 14, the mean exposure estimates ranged from 0.64 (minimum LB)/2.53 (minimum UB) to 9.13 (maximum LB)/17.79 (maximum UB) ng/kg bw per day across dietary surveys and age groups. At the 95th percentile, exposure estimates ranged from 2.40 (minimum LB)/5.13 (minimum UB) to 30.36 (maximum LB)/51.69 ng/kg (maximum UB) bw per day.
Dietary exposure in specific groups of the population, namely pregnant and lactating women (each represented with two survey), was within the range of exposure estimates for the adult population.
4.8.1.2. Contribution of different food groups to the total dietary exposure
Figures 9, 10 and 11 summarise the proportions of contributors of exposure (LB) in 11 broad food groups together with the magnitude of exposure for each the surveys in age groups ‘Toddlers’, ‘Other Children’, ‘Adolescents’, ‘Adults’, ‘Elderly’ and ‘Very elderly’. As shown in the figures, preserved meat, cheese, grains and grain‐based products are main contributors across all surveys. However, contribution of the first two varies to a larger extent, while contribution of grain‐based products remains more constant. Dried fruits, and fresh fruits such as grapes, figs and dates (see occurrence values and categories considered in the Annex, Table 3) as well as fruit juices and nectars were also contributing to the exposure in some of the ‘Toddlers’ and ‘Other children’ surveys, albeit, to a lesser extent than the three major contributor groups. Non‐chocolate confectionary is a significant source of exposure in countries where liquorice‐based sweets are frequently consumed (in particular Finland, the Netherlands, Sweden and Denmark). All subcategories as designated and depicted in Figures 9, 10 and 11 are listed in the Annex, Tables 3 and 8 where information about the percentages is provided. Occurrence levels of selected subcategories of the main contributing foods are shown in Table 15, except those from ‘Grain and grain‐based products’ where there are 55 subcategories and thus, they could not be included in the table.
Figure 9.
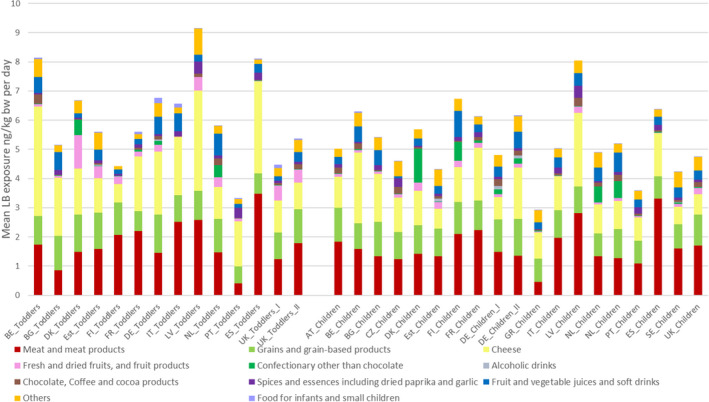
Contributors of exposure of OTA among different countries/surveys in ‘Toddlers’ and ‘Other Children’
Figure 10.
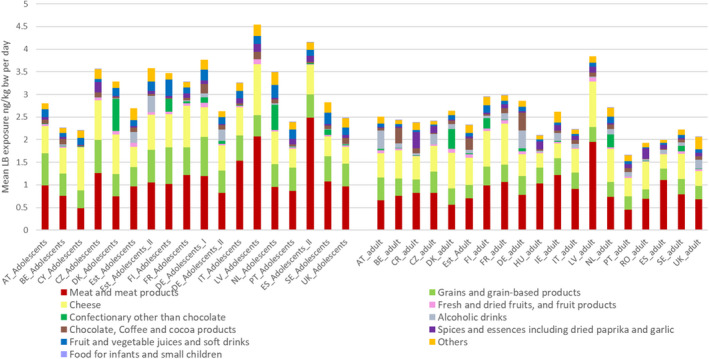
Contributors of exposure of OTA among different countries/surveys in ‘Adolescents’ and ‘Adults’
Figure 11.
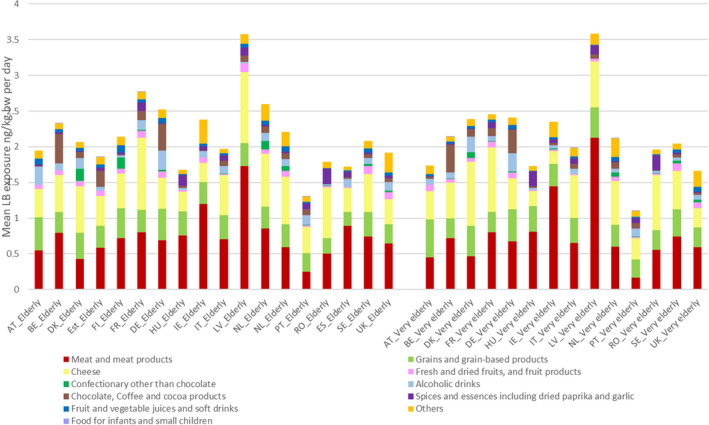
Contributors of exposure of OTA among different countries/surveys in ‘Elderly’ and ‘Very Elderly’
Table 15.
Occurrence levels and subcategories of the main contributing foods considered in the exposure assessment and shown in Figures 9, 10 and 11
| Category as listed in Figures 9, 10 and 11 | Linking categoryc | LB MEAN μg/kg | UB MEAN μg/kg |
|---|---|---|---|
| Cheese | Cheese (on Foodex2 level)a | 1.68 | 2.43 |
| Meat and meat products | Boar meat (Sus scrofa) | 0.05 | 0.13 |
| Ham, pork | 3.03 | 3.09 | |
| Liver and kidney, farmed animals | 0.04 | 4.67 | |
| Pastes, pâtés and terrines | 0.07 | 0.12 | |
| Preserved meat | 2.20 | 2.27 | |
| Sausages | 0.94 | 1.11 | |
| Fresh and dried fruits, and fruit products | Dried apricots (Prunus armeniaca) | 0.28 | 1.12 |
| Dried dates (Phoenix dactylifera) | 1.83 | 2.27 | |
| Dried figs (Ficuscarica) | 3.63 | 4.12 | |
| Dried fruits (not defined) | 2.16 | 2.74 | |
| Dried vine fruits (currants, raisins and sultanas) | 1.86 | 2.44 | |
| Berries and small fruits (not defined) | 0.13 | 1.61 | |
| Dates (Phoenix dactylifera) | 0.01 | 1.72 | |
| Figsb | 1.27 | 1.44 | |
| Fruit, canned | 0.02 | 0.25 | |
| Jam, marmalade and other fruit spreads | 0.13 | 0.65 | |
| Grains and grain‐based productsd | Bread and rolls | 0.16 | 0.34 |
| Breakfast cereals | 0.15 | 0.42 | |
| Fine bakery wares | 0.05 | 0.26 | |
| Muesli | 0.23 | 0.43 | |
| Pasta (Raw) | 0.07 | 0.36 | |
| Pastries and cakes | 0.15 | 0.48 |
Fresh cheeses, such as mozzarella, robiola, etc. were not taken into consideration.
Derived from occurrence levels of ‘Dried figs’.
Category used in the exposure assessment for matching the occurrence and consumption data.
Includes ‘Grains for human consumption’, ‘Grain milling products’, ‘Bread and rolls’, ‘Pasta (Raw)’, ‘Breakfast cereals’, ‘Fine bakery wares’ in 55 subcategories (Annex, Table 3).
In the case of infants, cheese and meat products are the most important contributors to OTA exposures together with various grains and grain‐based products (such as bread and rolls, various cereal flakes, rice, popped cereals). Food groups contributing more than 5% to the LB exposure estimates in at least one dietary survey are summarised in the Annex, Table 6.
The Panel acknowledged that data from the younger age groups (i.e. ‘Toddlers’, ‘Children’ and ‘Adolescents’) showed some levels of intake from consumption of alcoholic beverages. These exposure estimates (which are very low) are most likely a result of the indirect consumption of alcoholic beverages as ingredients of composite foods.
As already noted above, Figures 9, 10 and 11 below summarise the contribution of different food categories to OTA exposures in different surveys. Notably the categories used in these figures do not correspond to FoodEx categories.
4.8.1.3. Comparison of occurrence levels and exposure with the previous EFSA opinion on OTA
In 2006, data from a SCOOP (Socioeconomic sciences: communicating outcomes oriented to policy) report from 2002 (European Community, 2002) were used as a basis for the assessment of the exposure. It included seven food categories representing the supposed main contributors to OTA exposure. These categories were cereals and cereal products, wine, beer, grape juice, brewed coffee, cocoa and cocoa products as well as pork meat (see Table 16).
Table 16.
Comparison of occurrence levels used in the 2006 EFSA opinion with levels detected in the current database in the corresponding food categories
| 2006 occurrence data – taken from European Community (2002) | 2019 occurrence data – taken from EFSA data warehouse | ||||
|---|---|---|---|---|---|
| Food category | N | Mean (μg/kg)(a) | Food category | N | LB/UB Mean (μg/kg) |
| Cereals and cereal products | 5,180 | 0.29 | Breakfast cereals | 1,882 | 0.18/0.44 |
| Grain milling products | 4,409 | 0.29/0.55 | |||
| Grains for human consumption | 6,852 | 0.22/0.50 | |||
| Beer | 496 | 0.03 | Beer and beer‐like beverages | 1,755 | 0.04/0.26 |
| Wine | 1,470 | 0.36 | Wine | 5,692 | 0.06/0.25 |
| Grape juice | 146 | 0.55 | Grape juice | 1,809 | 0.18/0.27 |
| Cocoa and cocoa products | 547 | 0.24 | Cocoa and cocoa products | 1,204 | 0.57/0.83 |
| Pork meat/edible offal | 1,860 | 0.20 | Ham, pork | 54 | 3.03/3.09 |
| Pork meat | 112 | 0.0/0.2 (100% left censored) | |||
| Pork kidney | 2,327 | 0.04/5.1 | |||
| Roasted ground coffee (dry weight) | 1,184 | 0.72 | Roasted ground coffee (whole weight) | 1,651 | 0.56/0.94 |
LB: lower bound; N: number of samples; UB: upper bound;
Mean was calculated according to the following criteria: 1) If LOD and LOQ were available, mean level was calculated using LOD/2 for results lower than the LOD. For results between LOD and LOQ, numerical values, if available, were used. 2) If only LOQ was available, or if numerical values between LOD and LOQ were not available, LOQ/2 for values below the LOQ were used (European Community, 2002).
As can be seen in Table 16, the occurrence levels in the database used for the present assessment are similar to those in the seven main food categories used in the EFSA assessment of 2006. However, the current exposure estimate considered 210 subcategories in a wide range of food products including also data on other grain‐based products, cheese, various meat products, tree nuts, liquorice‐containing products and dried fruits that were previously not considered. This resulted in slightly higher exposure estimates. Moreover, because in 2006, the EFSA Comprehensive Food Consumption Database was not yet available the Concise Database was used, allowing only less refined estimates. Thus, in 2006, mean exposure of an average adult consumer varied between 2 and 3 ng/kg bw per day while in the current assessment, mean LB/UB exposures range between 1.68/3.51 and 3.84/6.05 ng/kg bw per day in ‘Adults’. In addition, in 2006, the use of the Concise Database did not allow an accurate assessment of the dietary exposures of age classes other than ‘Adults’.
4.9. Exposure assessment of infants fed on human milk
4.9.1. Occurrence of OTA in human milk in Europe
The occurrence of OTA in human milk in European populations was investigated intensely over the last two decades. In terms of analytical performance, methods are significantly improved over the last three decades in particular in terms of sensitivity and selectivity. Therefore, the CONTAM Panel decided to list in Table 17 only those papers reporting sufficient information on the analytical performance and the data collected.
Table 17.
Studies on OTA levels in breast milk in Europe
| Study population | Analytical method | No of samples (No of positive samples) | Median of positive samples (ng/L) | Mean ± SD positive samples (ng/L) | Mean ± SD all samples (ng/L) | Concentration range of positive samples (ng/L) | LOD (ng/L) | LOQ (ng/L) | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Lactating women from mid north Sweden. Milk sampled 6 weeks after delivery (mature milk) and 2–3 h after the previous feed. No selection/exclusion criteria reported | LC‐FD | 40 (23) | n.r. | n.r. | n.r. | 10.0–40.0 | 10 | 40 | Breitholtz‐Emanuelsson et al. (1993) |
| Milk samples from Swiss mothers living north of the Alps. No further selection criteria reported | LC‐FD | 40 (4) | n.r. | n.r. | n.r. | 5–14 | n.r. | 5–10 | Zimmerli and Dick (1995) |
| Samples obtained May–August in different regions of Norway. Elverum (south east); Trondheim (middle coast); Bodo (north). No selection/exclusion criteria reported | LC‐FD |
Total: 115 (38) Elverum: 48 (20) Trondheim: 19 (11) Bodo: 48 (7) |
Total: 30 |
Total: n.r. Elverum: 43.2 Trondheim: 49.0 Bodo: 27.4 |
n.r. | 10–130 | n.r. | 10 | Skaug et al. (1998) |
| Samples from healthy women (19–35 years old) from one (not the first) morning feed. Mothers were asked for but not selected because of their dietary habits | LC‐FD | 80 (17) | 16 | 30 | n.r. | 10–182 | n.r. | 10 | Skaug et al. (2001) |
| Samples of mature milk from Polish mothers. No information on selection criteria | ELISA | 78 (40) | n.r. | 6.8 ± 4.9 | n.r. | 2.4–16.0 | n.r. | 5 | Karwowska et al. (2004) |
| Italian mothers from different parts of Lombardy. Milk sampled 3–4 days after birth. No preselection of mothers based on dietary habits | LC‐FD | 231 (198) | 4 | 6.01 ± 8.31 | n.r. | 0.5–17 | 0.5 | n.r. | Turconi et al. (2004) |
| Milk from Polish mothers obtained 3–‐4 days after labour | LC‐FD | 13 (5) | n.r. | 5.6 ± 4 | n.r. | 5.3–17 | 5 | 15 | Postupolski et al. (2006) |
| Milk obtained from Slovak mothers from 0 to 6 months after delivery | LC‐FD | 76 (23) | n.r. | n.r. | n.r. | 4.8–60.3 | 4.8 | 14.4 | Dostal et al. (2008) |
| Mature milk samples collected from Italian mothers 30 days after delivery. Only healthy non‐smoking, non‐ drinking mothers with healthy babies were enrolled in the study | LC‐FD | 82 (61) | n.r. | 30.43 ±.88 | n.r. | 5–405 | 2 | 5 | Galvano et al. (2008) |
| Samples from 57 Italian and non‐Italian mothers obtained 3–4 days after delivery in a hospital in Northern Italy | LC‐FD | 57 (41) | n.r. | 10.0 ± 15.6 | n.r. | 1.0–75.1 | 0.5 | 1.0 | Biasucci et al. (2011) |
| Milk collected from 90 German mothers, 30 in Nordrhein Westfalen (NRW) and 60 in Niedersachsen (NS) | LC‐MS |
90 in total NRW 30 (7) NS NS 60 (6) |
n.r. | n.r. |
24.41 ± 21.1 (NRW) 14.4 ± 15.1 (NS) |
n.r. | 10 | 30 | Muñoz et al. (2013) |
| Healthy (30) and ceoliac (35, on a gluten‐free diet) breast feeding mothers. Samples collected three times daily for three consecutive days between 2 weeks and 4 months after birth. No further selection/exclusion criteria reported | LC‐FD | 453 (7) | n.r. | n.r. | n.r. | 17–123 | n.r. | 34 | Valitutti et al. (2018) |
ELISA: enzyme‐linked immunosorbent assay; LC‐FD: liquid chromatography‐fluorescence detection; LC‐MS: liquid chromatography‐mass spectroscopy; LOD: limit of quantification; LOQ: limit of quantification; n.r.: not reported; SD: standard deviation.
Table 17 presents an overview of recent studies on occurrence of OTA in human milk potentially useful for derivation of occurrence levels for OTA in breast milk. In all these studies consistently lower OTA values (all in the same order of magnitude) are reported compared to the older studies. In Table 17 study design, concentration median/mean/range, LOQ and LODs are indicated.
Overall, it can be seen that occurrence values are affected by a large variability within studies, probably owed to study design and specificity and sensitivity of methods. For most of the studies, individual data were not provided. In several studies only concentration ranges but no mean values have been reported (Breitholtz‐Emanuelsson et al., 1993; Zimmerli and Dick, 1995; Dostal et al., 2008; Valitutti et al., 2018). The studies from Skaug et al. (1998, 2001) showed a high variability, probably due to the lower specificity of the analytical methods compared to those currently used. The study from Postupolski et al. (2006) had a rather small study population while the values derived by Galvano et al. (2008) had a rather high variability.
After evaluation of the data as reported, the CONTAM Panel decided to base the exposure assessment on the most recent studies, i.e. Muñoz et al. (2013), Biasucci et al. (2011) and Valitutti et al. (2018). In order to provide a transparent exposure assessment, the study authors were contacted and the raw data kindly provided by Gisela Degen (Muñoz et al., 2013), Fillipo Rossi (Biasucci et al., 2011) and Carlo Brera and Barbara de Santis (Valitutti et al., 2018) and are presented in Appendix G.
Breast milk has a different nutritional composition in different stages and is designated as colostrum (1–5 days post‐partum), transitional milk (6–14 days post‐partum) and mature milk (15 days to 15 months post‐partum). In particular colostrum has a higher protein content as compared to transitional or mature milk (EFSA NDA Panel, 2013). Muñoz et al. (2014) found in a comparative study that milk‐to‐plasma ratio, measured over a period of 4 months, is significantly higher (p = 0.001) during the first week of lactation (0.40 ± 0.26) compared to the following period (i.e. 0.26 ± 0.19 at 4 months), probably in consideration of the high OTA affinity with proteins. In the same study, the authors also found that individual OTA levels in mature milk varied considerably individually during the study period but overall mean concentration levels did not vary substantially over a period of 6 months (mature milk was measured at 15–30 days, 2, 4 and 6 months). Notably several subjects dropped out early during the study and only in four mothers was it possible to receive milk samples until the very end of the study. Considering the particular composition of colostrum in comparison to mature milk and the possibly different excretion rate of OTA to colostrum and the short duration of feeding of colostrum, the CONTAM Panel concluded that such samples are likely not representative of exposure via breast feeding during the first 6 months of the life period of an infant. Therefore, the data from Biasucci et al. (2011), which were exclusively colostrum samples (see Appendix G, Table G.2), were not considered in the analysis.
Table G.2.
OTA levels (ng/L) in milk samples (colostrum) from Biasucci et al. (2011)
| Mothers ID | Nationality | OTA concentration (ng/L) |
|---|---|---|
| 115741 | Italian | 20.7 |
| 115519 | Italian | 6.4 |
| 115543 | Italian | 16.7 |
| 115501 | Italian | 3.5 |
| 115733 | Italian | 7.6 |
| 115782 | Non‐Italian | 6.0 |
| 115493 | Non‐Italian | 1.9 |
| 115634 | Italian | 2.5 |
| 115824 | Non‐Italian | 10.9 |
| 115725 | Italian | 1.5 |
| 114835 | Italian | 4.2 |
| 115550 | Italian | 20.9 |
| 115626 | Italian | 11.8 |
| 115758 | Non‐Italian | 2.6 |
| 115972 | Non‐Italian | 2.1 |
| 115766 | Italian | 28.1 |
| 115642 | Italian | n.d. |
| 114660 | Non‐Italian | 3.1 |
| 116004 | Italian | 3.7 |
| 115881 | Non‐Italian | 6.2 |
| 115840 | Italian | n.d. |
| 115774 | Italian | 3.8 |
| 115584 | Non‐Italian | 3.9 |
| 115196 | Italian | 68.8 |
| 115600 | Italian | n.d. |
| 115899 | Non‐Italian | 4.5 |
| 115873 | Italian | n.d. |
| 115691 | Italian | n.d. |
| 115857 | Non‐Italian | 13.9 |
| 115816 | Italian | 3.4 |
| 115683 | Italian | 5.4 |
| 115900 | Italian | 6.4 |
| 115618 | Italian | 3.9 |
| 115576 | Italian | n.d. |
| 115832 | Italian | n.d. |
| 115527 | Non‐Italian | n.d. |
| 115709 | Italian | 75.1 |
| 114702 | Italian | n.d. |
| 115865 | Non‐Italian | 2.6 |
| 115592 | Non‐Italian | 3.8 |
| 115956 | Italian | 10.3 |
| 115659 | Non‐Italian | n.d. |
| 114611 | Non‐Italian | 2.7 |
| 115949 | Non‐Italian | 21.8 |
| 114637 | Non‐Italian | 3.9 |
| 115915 | Non‐Italian | 4.3 |
| 115980 | Non‐Italian | 3.6 |
| 115931 | Italian | 3.0 |
| 115964 | Italian | 1.3 |
| 115808 | Non‐Italian | n.d. |
| 115923 | Italian | 1.9 |
| 115998 | Non‐Italian | 3.5 |
n.d.: not detected; OTA: ochratoxin.
In the study from Valitutti et al. (2018), mature milk from groups of healthy and coeliac mothers was collected three times daily over a period of 3 days (see Appendix G, Tables G.3 and G.4). However, out of a total of 453 milk samples, OTA was detected only in seven samples and these came mainly from the same individual. It was concluded that these results could not be used for a representative exposure assessment because this would be driven mainly by non‐detects and by high OTA levels detected in a single individual.
Table G.4.
OTA levels (ng/L) in milk samples from ceoliac mothers from Valitutti et al. (2018)
| 1st day | 2nd day | 3rd day | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Sample ID | 4 h after lunch | 2 h after dinner | Morning | 4 h after lunch | 2 h after dinner | Morning | 4 h after lunch | 2 h after dinner | Morning |
| COD01 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD02 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.t. | n.d. |
| COD03 | n.d. | n.d. | n.d. | n.t. | n.d. | n.t. | n.d. | n.d. | n.d. |
| COD04 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD05 | n.t. | n.d. | n.d. | n.t. | n.d. | n.t. | n.t. | n.d. | n.t. |
| COD06 | n.d. | n.d. | n.d. | n.d. | n.t. | n.d. | n.t. | n.d. | n.d. |
| COD07 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD08 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.t. | n.d. | n.d. |
| COD09 | n.d. | n.d. | n.d. | n.d. | n.d. | n.t. | n.d. | n.d. | n.d. |
| COD11 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD13 | n.d. | n.d. | n.d. | n.d. | n.d. | n.t. | n.d. | n.t. | n.d. |
| COD14 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD16 | 123 | n.d. | n.d. | n.d. | n.d. | n.t. | n.d. | n.d. | n.t. |
| COD17 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD18 | n.t. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD20 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD25 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD36 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD42 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD43 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD44 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD50 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD55 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD56 | n.d. | n.t. | n.t. | n.t. | n.t. | n.d. | n.d. | n.d. | n.d. |
| COD58 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD72 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD73 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD74 | n.d. | n.t. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD75 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD76 | n.d. | 64.6 | n.d. | 62.1 | n.d. | 80.2 | n.d. | 83.9 | 56.5 |
| COD83 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD89 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD108 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
n.d.: not detected; n.t.: not tested; OTA: ochratoxin.
The CONTAM Panel concluded that the study from Muñoz et al. (2013), obtained using a confirmatory LC‐MS method, presenting a good sensitivity and enrolling a sufficient amount of individuals/breast milk samples is the most appropriate investigation for deriving a reliable and representative mean OTA occurrence level for the exposure assessment from human milk. In this study (see also Table 17), a total of 90 breast milk samples from two different cities in Germany (Dortmund and Hannover) were analysed (for individual data, see Appendix G Table G.1). For reasons already described above the data on colostrum were omitted from use in the exposure assessment and only data obtained for transitory and mature milk were used for deriving OTA exposure for breastfed infants for the period from birth to 6 months of age. For the remaining 71 milk samples, median and P95 LB levels were 13 and 43 ng/mL, respectively.
Table G.1.
OTA levels (ng/L) in milk samples from Muñoz et al. (2013)
| Mothers ID | Sample taken (Breastfeeding day) | OTA concentration (ng/L) | Milk type |
|---|---|---|---|
| BFD_1 | 3 | 26 | Colostrum |
| BFD_2 | 4 | 17 | Colostrum |
| BFD_3 | 4 | 100 | Colostrum |
| BFD_4 | 4 | n.d. | Colostrum |
| BFD_5 | 5 | n.d. | Colostrum |
| BFD_6 | 5 | 53 | Colostrum |
| BFD_7 | 6 | n.d. | Colostrum |
| BFD_8 | 6 | 30 | Colostrum |
| BFD_9 | 6 | n.d. | Colostrum |
| BFD_10 | 6 | 36 | Colostrum |
| BFH_1 | 4 | 29 | Colostrum |
| BFH_2 | 4 | 47 | Colostrum |
| BFH_3 | 3 | 14 | Colostrum |
| BFH_4 | 5 | n.d. | Colostrum |
| BFH_5 | 1 | 22 | Colostrum |
| BFH_6 | 6 | n.d. | Colostrum |
| BFH_7 | 4 | n.d. | Colostrum |
| BFH_8 | 5 | n.d. | Colostrum |
| BFH_9 | 5 | n.d. | Colostrum |
| BFD_11 | 7 | 16 | Transitory milk |
| BFD_12 | 7 | 24 | Transitory milk |
| BFD_13 | 7 | 29 | Transitory milk |
| BFD_14 | 8 | 28 | Transitory milk |
| BFD_15 | 8 | 18 | Transitory milk |
| BFD_16 | 10 | n.d. | Transitory milk |
| BFD_17 | 11 | 26 | Transitory milk |
| BFD_18 | 12 | 17 | Transitory milk |
| BFD_19 | 14 | n.d. | Transitory milk |
| BFD_20 | 14 | 21 | Transitory milk |
| BFH_10 | 8 | 14 | Transitory milk |
| BFH_11 | 9 | n.d. | Transitory milk |
| BFH_12 | 11 | n.d. | Transitory milk |
| BFH_13 | 13 | 21 | Transitory milk |
| BFH_14 | 8 | 40 | Transitory milk |
| BFH_15 | 13 | 40 | Transitory milk |
| BFH_16 | 16 | n.d. | Transitory milk |
| BFH_17 | 14 | n.d. | Transitory milk |
| BFH_18 | 15 | n.d. | Transitory milk |
| BFH_19 | 17 | 26 | Transitory milk |
| BFD_21 | 17 | 22 | Mature milk |
| BFD_22 | 17 | n.d. | Mature milk |
| BFD_23 | 18 | n.d. | Mature milk |
| BFD_24 | 19 | 43 | Mature milk |
| BFD_25 | 30 | 10 | Mature milk |
| BFD_26 | 35 | 26 | Mature milk |
| BFD_27 | 35 | 26 | Mature milk |
| BFD_28 | 36 | 19 | Mature milk |
| BFD_29 | 56 | 68 | Mature milk |
| BFD_30 | 60 | 42 | Mature milk |
| BFH_20 | 32 | 16 | Mature milk |
| BFH_21 | 25 | n.d. | Mature milk |
| BFH_22 | 30 | 13 | Mature milk |
| BFH_23 | 32 | 19 | Mature milk |
| BFH_24 | 34 | n.d. | Mature milk |
| BFH_25 | 33 | 29 | Mature milk |
| BFH_26 | 57 | n.d. | Mature milk |
| BFH_27 | 51 | 26 | Mature milk |
| BFH_28 | 33 | 21 | Mature milk |
| BFH_29 | 55 | 30 | Mature milk |
| BFH_30 | 24 | n.d. | Mature milk |
| BFH_31 | 61 | 27 | Mature milk |
| BFH_32 | 58 | 18 | Mature milk |
| BFH_33 | 30 | n.d. | Mature milk |
| BFH_34 | 32 | 23 | Mature milk |
| BFH_35 | 33 | n.d. | Mature milk |
| BFH_36 | 52 | n.d. | Mature milk |
| BFH_37 | 55 | 78 | Mature milk |
| BFH_38 | 56 | n.d. | Mature milk |
| BFH_39 | 54 | n.d. | Mature milk |
| BFH_40 | 50 | n.d. | Mature milk |
| BFH_41 | 58 | n.d. | Mature milk |
| BFH_42 | 32 | n.d. | Mature milk |
| BFH_43 | 33 | n.d. | Mature milk |
| BFH_44 | 34 | n.d. | Mature milk |
| BFH_45 | 30 | n.d. | Mature milk |
| BFH_46 | 31 | n.d. | Mature milk |
| BFH_47 | 52 | n.d. | Mature milk |
| BFH_48 | 34 | n.d. | Mature milk |
| BFH_49 | 58 | n.d. | Mature milk |
| BFH_50 | 30 | n.d. | Mature milk |
| BFH_51 | 34 | 66 | Mature milk |
| BFH_52 | 34 | 15 | Mature milk |
| BFH_53 | 25 | 18 | Mature milk |
| BFH_54 | 25 | 20 | Mature milk |
| BFH_55 | 25 | n.d. | Mature milk |
| BFH_56 | 54 | 15 | Mature milk |
| BFH_57 | 35 | n.d. | Mature milk |
| BFH_58 | 26 | n.d. | Mature milk |
| BFH_59 | 50 | n.d. | Mature milk |
| BFH_60 | 25 | n.d. | Mature milk |
n.d.: not detected; OTA: ochratoxin.
The CONTAM Panel concluded that the median and the 95th percentile of the concentrations are the most appropriate parameters to be used for the exposure assessment. These provide information on the median exposure of infants and also on the infants consuming milk with high OTA concentrations. The approach is further supported by the consideration that infants are a sensitive population, and they are consuming only breast milk from one mother with a daily exposure to a toxin that has the potential to accumulate in the body.
4.9.2. Breast milk consumption and exposure assessment of infants fed on human milk
Thus, for the exposure assessment of infants below 6 months of age to OTA via breast milk, a value of 3 months was selected, assuming an average body weight of 6.1 kg, with an estimated average daily consumption of 800 mL per day and a high consumption of 1,200 mL per day of human milk (EFSA NDA Panel, 2013) was combined with the median and P95 occurrence levels mentioned above. Based on the reported median concentration of OTA of 13 ng/L and a 95th percentile (P95) concentration of OTA of 43 ng/L in human milk (transitional and mature milk combined) the dietary exposure to OTA ranged from 1.7 to 5.6 ng/kg bw per day for infants with an average milk consumption and from to 2.6 to 8.5 ng/kg bw per day for infants with a high milk consumption.
These intakes are actually in the lower part of those based on consumption of other food (Table 14), being for the mean LB exposure 0.65 to 8.64 ng/kg bw per day (lowest LB‐highest LB) and for high exposure (P95) 2.57 to 30.36 ng/kg bw per day (lowest LB‐highest LB). It should be noted that in these calculations on exposure from food, the class of infants actually covers the period up to 12 months. Breastfeeding may also cover a period of 12 months, but after a number of months, the infants will receive additional food.
4.10. Risk characterisation
4.10.1. Non‐neoplastic effects
The CONTAM Panel selected the BMDL10 of 4.73 μg OTA/kg bw per day for increased incidence of microscopic kidney lesions seen in a 3‐month study with female pigs (Krogh et al., 1974) as a reference point for the risk characterisation of non‐neoplastic effects. Comparison of the chronic dietary exposure to OTA across dietary surveys and age groups as reported in Table 14 of Section 4.7 Exposure assessment to this BMDL10 results in MOE values that range from 7391 (lowest minimum LB exposure across national consumption surveys) to 266 (highest maximum UB exposure across national consumption surveys) for the mean exposure estimates, and from 1971 (lowest minimum LB) to 92 (highest maximum UB exposure) for the 95th percentile exposure estimates across dietary surveys and age groups (see Table 18 below).
Table 18.
Margins of exposure (MOE) values of the different age groups to the BMDL10 of 4.73 μg OTA/kg bw per day for increased incidence of microscopic kidney lesions upon OTA exposure of pigs
| Age group | MOE calculated from mean dietary exposure | MOE calculated from P95 dietary exposure | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Minimum | Median | Maximum | Minimum | Median | Maximum | |||||||
| LB | UB | LB | UB | LB | UB | LB | UB | LB | UB | LB | UB | |
| Infants | 7,391 | 1,870 | 2,451 | 836 | 547 | 301 | 1,833 | 644 | 690 | 333 | 156 | 92 |
| Toddlers | 1,425 | 644 | 827 | 384 | 518 | 266 | 474 | 285 | 388 | 215 | 213 | 127 |
| Other children | 1,625 | 639 | 940 | 439 | 588 | 329 | 756 | 328 | 469 | 243 | 264 | 169 |
| Adolescents | 2,150 | 918 | 1,451 | 786 | 1,042 | 585 | 981 | 476 | 722 | 416 | 452 | 301 |
| Adults | 2,849 | 1,363 | 1,963 | 929 | 1,232 | 784 | 1,225 | 626 | 869 | 457 | 533 | 374 |
| Elderly | 3,638 | 1,531 | 2,285 | 1,042 | 1,325 | 765 | 1,497 | 708 | 1,100 | 549 | 550 | 345 |
| Very elderly | 4,300 | 1,805 | 2,274 | 1,046 | 1,318 | 889 | 1,971 | 922 | 1,191 | 560 | 906 | 515 |
| Pregnant women | 3,174 | 1,442 | 2,516 | 1,241 | 2,093 | 1,092 | 1,541 | 762 | 1,165 | 654 | 938 | 573 |
| Lactating women | 2,849 | 1,162 | 2,242 | 1,056 | 1,840 | 969 | 1,521 | 739 | 1,049 | 575 | 799 | 470 |
LB: lower bound; UB: upper bound; P95: 95th percentile.
Comparison of dietary exposure of infants from breastfeeding, with this BMDL10 results in MOE values of 2783 and 1819 at median OTA concentrations in milk and of 845 and 556 at 95th percentile OTA concentrations in milk in average and high breast milk consumers, respectively.
The CONTAM Panel considered that based on the available toxicity data and taking inter‐ and intraspecies variations and the relatively short duration of the pig study (3 months) used for deriving a non‐neoplastic chronic BMDL10, an MOE of 200 would be sufficient to conclude on a low health concern for non‐neoplastic effects of OTA. As can be seen in Table 19, comparison of exposures with the BMDL10 based on the non‐neoplastic endpoint resulted in MOEs of more than 200 in most consumer groups. Exceptions are 95th percentile exposures at maximum LB in the age group of ‘Infants’ and at maximum UB in the age group of ‘Infants’, ‘Toddlers’ and ‘Other children’ indicating a possible health concern for these age groups.
Table 19.
Margins of exposure (MOE) values of the different age groups to the BMDL10 of 14.5 μg OTA/kg bw per day calculated from increased combined incidences of kidney tumours in male rats
| Age group | MOE calculated from mean dietary exposure | MOE calculated from P95 dietary exposure | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Minimum | Median | Maximum | Minimum | Median | Maximum | |||||||
| LB | UB | LB | UB | LB | UB | LB | UB | LB | UB | LB | UB | |
| Infants | 22,656 | 5,731 | 7,513 | 2,562 | 1,678 | 923 | 5,620 | 1,975 | 2,114 | 1,020 | 478 | 281 |
| Toddlers | 4,367 | 1,975 | 2,535 | 1,176 | 1,588 | 815 | 1,454 | 874 | 1,189 | 659 | 652 | 391 |
| Other children | 4,983 | 1,959 | 2,883 | 1,345 | 1,801 | 1,010 | 2,316 | 1,007 | 1,437 | 746 | 809 | 517 |
| Adolescents | 6,591 | 2,816 | 4,448 | 2,409 | 3,194 | 1,792 | 3,008 | 1,460 | 2,214 | 1,274 | 1,386 | 924 |
| Adults | 8,735 | 4,179 | 6,017 | 2,849 | 3,776 | 2,405 | 3,756 | 1,918 | 2,665 | 1,400 | 1,633 | 1,146 |
| Elderly | 11,154 | 4,693 | 7,005 | 3,194 | 4,062 | 2,346 | 4,589 | 2,171 | 3,372 | 1,684 | 1,686 | 1,058 |
| Very elderly | 13,182 | 5,534 | 6,971 | 3,208 | 4,039 | 2,726 | 6,042 | 2,827 | 3,652 | 1,716 | 2,778 | 1,578 |
| Pregnant women | 9,732 | 4,421 | 7,713 | 3,806 | 6,416 | 3,349 | 4,723 | 2,335 | 3,571 | 2,006 | 2,877 | 1,755 |
| Lactating women | 8,735 | 3,563 | 6,872 | 3,237 | 5,642 | 2,971 | 4,662 | 2,266 | 3,215 | 1,762 | 2,449 | 1,441 |
LB: lower bound; UB: upper bound; P95: 95th percentile.
MOEs in breastfed infants were all above 200 indicating a low health concern.
The CONTAM Panel concluded that these MOEs indicate a possible health concern for non‐neoplastic effects of OTA for high consumers in the young age groups.
4.10.2. Neoplastic effects
The CONTAM Panel selected the BMDL10 of 14.5 μg OTA/kg bw per day for increased combined incidences of adenomas and carcinomas in male rats in a chronic study (NTP, 1989) as a reference point for the risk characterisation of neoplastic effects. Comparison of the chronic dietary exposure to OTA across dietary surveys and age groups reported in Table 14 of Section 4.7 Exposure assessment to this BMDL10 results in MOE values (see Table 20 below) that range from 22,656 (lowest minimum LB exposure) to 815 (highest maximum UB exposure) for the mean exposure estimates, and from 6,042 (lowest minimum LB exposure) to 281 (highest maximum UB exposure) for the 95th percentile exposure estimates across dietary surveys and age groups.
Table 20.
Summary of major uncertainties in the risk assessment of OTA in food
| Sources of uncertainty | Directiona |
|---|---|
| Assumption that OTA‐induced mutations result from direct genotoxicity | + |
| Assumption that OTA‐induced mutations result from indirect genotoxicity | – |
| Use of kidney lesion incidences for non‐neoplastic effect characterisation | + |
| Use of an uncertainty factor of 2 for extrapolation from a 3‐month study in pigs to chronic effects | – |
| Small sample size in human study where raised protein concentrations were seen | +/– |
| Extrapolation of the occurrence data submitted mainly by two countries to the whole of Europe | +/– |
| Methodology used to estimate 95th percentile chronic exposure based on consumption surveys only covering a few days | + |
| Consideration of all cheese with equal contamination level | + |
| Extrapolation of occurrence data from one study used for exposure assessment for breastfed infants to the whole of Europe | +/– |
+ = uncertainty with potential to cause overestimation of exposure/risk; – = uncertainty with potential to cause underestimation of exposure/risk. Extent of potential over/underestimation might differ in direction.
Comparison of dietary exposure of infants from breastfeeding, with this BMDL10 results in MOE values of 8,529 and 5,577 at median OTA concentrations in milk and 2,589 and 1,706 at 95th percentile OTA concentrations in milk in average and high breast milk consumers, respectively.
For substances that are both genotoxic and carcinogenic, EFSA (2005) and EFSA Scientific Committee (2012a) stated that an MOE of 10,000 or higher if based on a BMDL10 from an animal carcinogenicity study would be of low health concern.
However, as can be seen in Table 20, the calculated MOEs are below 10,000 in most of the dietary surveys, in particular for 95th percentile exposures which are all far below 10,000. In breastfed infants, the MOEs are below 10,000 in all scenarios. The CONTAM Panel noted that these MOEs would indicate a possible health concern but the CONTAM Panel also notes the uncertainties associated with the interpretation of the mode of action for carcinogenicity (see below Section 4.11 on Uncertainties).
For neoplastic effects of OTA, the CONTAM Panel emphasises, that it was not possible to make a clear distinction between a direct and an indirect mechanism of genotoxicity. As a result, the risk characterisation will be either not sufficiently cautious or overcautious depending on which MoAs actually take place. The Panel points out that the MOE of 10,000 is likely to be most conservative in this case as the evidence for a direct interaction of OTA with the DNA is inconclusive and alternative thresholded mechanisms may play a role in the formation of kidney tumours.
Pending the elucidation of the MoAs for the genotoxicity/carcinogenicity of OTA, the Panel concluded that an MOE of 10,000 to the neoplastic reference point is warranted for the risk characterisation of OTA, albeit being the most conservative approach until the MoAs are clarified.
4.11. Uncertainty analysis
The evaluation of the inherent uncertainties in the present assessment was performed following the guidance of the Scientific Committee related to uncertainties in dietary exposure assessment (EFSA, 2007). The CONTAM Panel took note of the new guidance on uncertainties of the Scientific Committee (EFSA Scientific Committee, 2018), but it was not implemented in this opinion.
4.11.1. Assessment objectives
The objectives of the assessment are clearly laid down in the terms of reference.
4.11.2. Occurrence data/consumption data/exposure assessment
4.11.2.1. Occurrence data
The number of occurrence data submitted varied considerably between the different food categories. Most samples belonged to category ‘Grains’ and ‘Grain‐based products’. The lower the number of measured samples, the higher the uncertainty related to the levels of OTA in food commodities which was an issue for example for cheese and certain preserved meat products where relatively few data were available.
It was noted that the uncertainties regarding the UB mean occurrence levels can be higher for non‐homogenous food samples where contamination levels below the LOQ/LOD are less likely to occur.
About 55% of the occurrence data used for the exposure assessment were collected from two EU countries (Germany and The Netherlands) and thus the assessment might not fully represent the contamination of food in other countries or the EU as a whole.
To reduce the uncertainty in the levels and inherent exposure assessment, LOQ/LOD cut‐off values were applied based on the existing MLs in Commission Regulation (EC) No 1881/2006 and also to certain important contributing food categories where particularly high LOQs/LODs were found and where the differences between the mean UB/LB values were large. In the final database used for the assessment, 75% of the samples were left‐censored. An ad hoc scenario was carried out without the application of LOQ cut‐off values in order to check the impact of their application (see Annex, Table 5). The results show slightly higher UB exposures i.e. slightly greater differences between LB and UB estimates. Comparing the ranges, mean and 95th percentile levels in the current assessment are ranging from 0.64 to 17.79 and from 2.4 to 51.7 ng/kg bw per day, while in the ad hoc scenario, the same parameters were 0.61 to 17.36 and 2.23 to 50.53 ng/kg bw per day.
Likewise, the lack of information on the analytical method used to analyse some food samples (relevant for about 16% of the data) adds uncertainty regarding reported vs. actual concentrations of OTA.
Re‐classifications have been carried out regarding the ‘product treatment’ field of the occurrence data records (e.g. where processed/dehydrated fruits were classified as fresh have been reclassified to the corresponding category). However, not all the data providers indicated the product treatment information, thus in some cases the reclassification was made based on assumption (e.g. in the case of figs). This may result in over‐ or underestimation of the exposure.
In the final occurrence database 678 analytical results were found to be above the MLs of Commission Regulation (EC) No 1881/2006. To evaluate their impact on the overall assessment, an ad hoc scenario with the exclusion of non‐compliant samples was carried out which showed only a very minor impact on the exposure results.
4.11.2.2. Consumption data
Uncertainties and limitations related to the use of the EFSA Comprehensive Food Consumption Database were described by EFSA (EFSA, 2011a) and are therefore not further detailed in this opinion. Generally, these limitations and uncertainties relate to the use of different dietary survey methodologies, standard portion sizes, representativeness of samples included in surveys or to the inclusion of consumption surveys covering only few days to estimate high percentiles of chronic exposure.
4.11.2.3. Dietary exposure assessment
Further sources of uncertainty are related to the different assumptions done for the present assessment, mainly for the linkage between the occurrence and consumption data. In particular:
A number of foodstuffs were grouped at the second level of the FoodEx, assuming homogeneity of the contamination levels, which could lead to both over‐ and underestimation of the exposure.
According to the literature (see Section 4.7.2 Occurrence of OTA as reported in public literature), certain types of cheese, particularly ripened ones might have higher OTA contamination levels and predominantly in the crust as they get contaminated with OTA during manufacturing or storing. Consequently, cheeses with edible rind as well as the grated ones where the outer part is also included in the final product are assumed to be the main contributors of exposure originated from cheese. However, most of the occurrence data were available on unspecified cheese, and also in the majority of the food consumption records in the Comprehensive Database the type of cheese was not reported either. Thus, the only possible refinement was to exclude the fresh types of cheeses from the assessment, where it was reported in the consumption surveys. As the impact of this refinement was minor, especially on those surveys where only unspecified cheese was reported, OTA exposure from cheese consumption is most likely overestimated.
In case of food supplements, based on the text description of the samples, highest contamination levels were found in the ones containing liquorice and green coffee extractions. However, this level of specification was not possible to be distinguished from the consumption side, which most likely results in overestimation of the exposure as the levels were applied to all ‘Plant based formula’ consumed.
4.11.2.4. Exposure assessment for breast fed infants
The exposure assessment was based on relatively few measured occurrence data from a single study in Germany with a limited number of samples (Muñoz et al., 2013) together with default values for consumption. These occurrence data are probably not representative of the situation in the entire EU.
4.11.3. Hazard characterisation
For the microscopic kidney lesions used for calculation of a non‐neoplastic BMD only incidence but no severity data were available. The EFSA guidance on default values (EFSA Scientific Committee, 2012a) proposes the use of an UF of 2 from subchronic to chronic duration in rodents. The use of an UF of 2 for extrapolating from a 3‐month study in pigs to a chronic effect is an assumption associated with uncertainty as 3 months represent a relatively shorter fraction of the natural lifespan of a pig as compared to the lifespan of rats a larger UF could be applied. Indirect mechanism of genotoxicity and non‐ genotoxic MoAs are likely to play major roles in the carcinogenic activity of OTA, albeit to which extent cannot be quantified. The application of an MOE of 10,000, following EFSA guidance is particularly conservative in this case, because such an MOE applies for compounds that are directly genotoxic and carcinogenic and the evidence for a direct interaction of OTA with the DNA is inconclusive. The interpretation of the relevance of the findings of the raised protein concentrations in urine in infants exposed to high levels of OTA via their mothers is hampered by the small sample size in this study.
4.11.4. Summary of uncertainties
In Table 20, a summary of the uncertainty evaluation is presented, highlighting the main sources of uncertainty and indicating an estimate of whether the source of uncertainty leads to over/underestimation of the resulting risk.
The overall uncertainty incurred with the present assessment is high. The assessment is more likely to overestimate than to underestimate the risk.
5. Conclusions
5.1. General conclusions
OTA is produced by various fungi of the genus Aspergillus and Penicillium, e.g. A. ochraceus, A. carbonarius and P. verrucosum. OTA is stable to moderate heating but losses ranging up to 90% are observed at temperatures above 180°C. OTA in food is analysed by LC‐MS, LC‐MS/MS and HPLC methods.
5.2. Toxicokinetics
OTA is rapidly absorbed and distributed following ingestion.
OTA is slowly eliminated from blood, with plasma half‐lifes ranging from several days in rodents and pigs to several weeks in non‐human primates and humans. Due to the slow elimination and excretion, and low rate of metabolism, OTA has the potential to bioaccumulate in the organism.
The major metabolic pathway of OTA is hydrolysis to OTalpha, followed by conjugation with glucuronic acid.
5.3. Transfer to food
Because of its long half‐life OTA may accumulate in animal tissues and thus is found in meat and meat products in particular of non‐ruminants such as pigs. In fish, half‐life of OTA is shorter and OTA does not accumulate in edible parts, i.e. muscle tissue.
Based on efficient degradation in the rumen, OTA levels in cow milk are low.
Biomarkers for dietary exposure are reflected in OTA levels in plasma, serum, urine and breast milk and overall levels in Europe have not changed since the last EFSA evaluation.
Reliable biomarkers of OTA‐specific effects, in particular on kidney function, have not been identified.
5.4. Toxicity in experimental animals and in vitro
OTA exerts a variety of adverse effects in repeated dose studies in mice, rats, rabbits and pigs including immunotoxicity, neurotoxicity, developmental effects at maternally toxic doses. The critical effects occur in the kidney, the pig being the most sensitive animal species.
Upon chronic oral exposure to OTA, increased incidences of kidney tumours have been observed in mice and rats, the rat being the most sensitive species.
OTA induces gene mutations, DNA breaks and chromosomal damage of a specific type both in vitro and in vivo.
5.5. Observations in humans
Possible associations between OTA exposure and kidney disease, bladder or hepatocellular cancer have been investigated in epidemiological studies, but it is not possible to establish a causal link between exposure to OTA and adverse effects in humans.
The findings of raised protein concentrations in urine, indicative of impaired renal function, in a limited number of infants from Egypt exposed to high levels of OTA via their mothers (during pregnancy and lactation) raise concern and would need confirmation in larger studies.
5.6. Mode of action
Multiple molecular events of OTA have been observed but the MoAs of OTA toxicity and renal carcinogenicity have not been clarified.
Non‐DNA reactive mechanisms play a role in OTA‐induced kidney carcinogenesis. There is evidence that cycle arrest, induction of apoptosis and autophagy, alterations in cell proliferation, cell signalling, gene expression, redox homoeostasis and epigenetic modifications as well as DNA and chromosomal damage may each contribute to tumour formation.
The molecular mechanism underlying OTA genotoxicity remains unclear and it is not possible to conclude whether OTA causes DNA damage by indirect or direct effects.
5.7. Critical effects and derivation of reference points
Increased incidence of microscopic kidney lesions seen in a repeated dose study with female pigs has been identified as the critical effect to be used for characterising non‐neoplastic effects of OTA. A BMDL10 of 4.73 μg OTA/kg bw per day derived from these effects was established.
The increased incidences of kidney tumours seen in male rats in a 2‐year study were identified as the critical effect for characterising neoplastic effects of OTA. A BMDL10 of 14.5 μg OTA/kg bw per day derived from these effects was established.
5.8. Occurrence
A total of 73,891 analytical results for OTA in food, respectively, fulfilled the quality criteria applied and from these 71,769 were selected and used in the assessment after grouping the categories and matching with the food consumption data.
Current occurrence levels were similar to the ones in the previous assessment (EFSA, 2006) in case of foodstuffs where data were available that time. However, several new food categories were considered in the present assessment (e.g. other grain‐based products, cheese, various meat products, tree nuts and dried fruits).
The proportion of left‐censored data was 75%, which resulted in a relatively big difference between the LB and UB occurrence values and exposure results.
The highest mean concentrations of OTA were recorded in categories of ‘Plant extract formula’, ‘Flavourings or essences’ (both containing liquorice extracts) and ‘Chili pepper’.
5.9. Exposure
The mean exposure estimates ranged from 0.64 (minimum LB)/2.53 (minimum UB) to 9.13 (maximum LB)/17.79 (maximum UB) ng/kg bw per day across dietary surveys and age groups. At the 95th percentile, exposure estimates ranged from 2.40 (minimum LB)/5.13 (minimum UB) to 30.36 (maximum LB)/51.69 ng/kg (maximum UB) bw per day.
The most important contributors to the chronic dietary exposure to OTA were ‘Preserved meat’, ‘Cheese’ and ‘Grains and grain‐based products’. Dried and fresh fruit such as grapes, figs and dates as well as fruit juices and nectars were also contributing to the exposure in some of the ‘Toddlers’ and ‘Other children’ groups, albeit, to a lesser extent than the three major categories. ‘Non‐chocolate confectionary’ was a significant source of exposure in countries where liquorice‐based sweets are commonly consumed.
In a specific exposure scenario, occurrence data on OTA in breast milk from one study in the public literature were used, to assess the exposure of infants from breastfeeding.
Based on a median concentration of OTA of 13 ng/L and a 95th percentile (P95) concentration of OTA of 43 ng/L in human milk, the chronic dietary exposure to OTA ranged from 1.7 to 5.6 ng/kg bw per day for infants with an average milk consumption and from to 2.6 to 8.5 ng/kg bw per day for infants with a high milk consumption.
5.10. Risk characterisation
Since the previous EFSA assessment (2006), more recent studies have raised uncertainty regarding the mode of action for kidney carcinogenicity. The CONTAM Panel considered it was not appropriate to establish a HBGV and concluded that for both non‐neoplastic and neoplastic effects of OTA, an MOE approach needs to be applied.
The TWI of 120 ng/kg bw as established by the CONTAM Panel in 2006 is no longer valid.
For non‐neoplastic effects, an MOE of 200 or higher between the selected RP and the estimated dietary exposures would be of low health concern.
The calculated MOEs for non‐neoplastic effects were above 200 in most of the dietary surveys for average and high consumers and therefore of low health concern.
However, in high consumers of the age group of ‘Infants’, the MOEs were below 200 at maximum LB in ‘Infants’ and at maximum in ‘Infants’, ‘Toddlers’ and ‘Other children’, indicating a possible health concern for these age groups.
The calculated MOEs for non‐neoplastic effects were above 200 in all exposure scenarios for breastfed infants and thus of low health concern.
Following EFSA guidance for substances that are both genotoxic and carcinogenic, an MOE of 10,000 or higher between the RP and the estimated dietary exposure would be of low health concern.
The calculated MOEs for neoplastic effects in most of the surveys, in particular those for high consumers and for breastfed infants in all scenarios were below 10,000 and thus indicate a possible health concern for these consumer groups.
In the interpretation of the MOE for the neoplastic risks, the Panel considered that the MOE of 10,000 for substances that are directly genotoxic and carcinogenic may not be appropriate in this case because the evidence for a direct interaction of OTA with the DNA is inconclusive.
However, in the absence of elucidated MoAs for the genotoxicity/carcinogenicity of OTA, the Panel concluded that an MOE of 10,000 needs to be applied to the BMDL10 of 14.5 μg/kg bw per day for neoplastic effects (kidney tumours) in the rat.
6. Recommendations
More studies elucidating the sequence of critical events at the carcinogenic target site in the kidney are needed.
More information on the specific signature of OTA gene mutations in appropriate animal models is needed
More studies on the differential toxicokinetics of OTA in laboratory animals and humans are needed including transfer of OTA to the fetus.
More studies relating biomarkers of exposure, i.e. OTA in plasma/serum, to external daily intakes are needed for more reliable estimates of OTA exposure.
Reliable and representative investigations of the levels of OTA in human breast milk are needed.
More occurrence data on OTA in cheese paste vs. cheese rind are needed.
More data on occurrence and toxicity of modified OTA are needed.
Documentation provided to EFSA
Data on individual OTA concentrations in breast milk samples were kindly provided by Gisela Degen (Leibniz‐Research Centre for Working Environment and Human Factors (IfADo), Dortmund, Germany) on 3 October 2019 (data from Muñoz et al., 2013); Filippo Rossi (Institute of Food Science and Nutrition, Faculty of Agriculture, Universita` Cattolica del Sacro Cuore, Piacenza, Italy) on 10 October 2019 (data from Biasucci et al., 2011); Carlo Brera and Barbara de Santis (Laboratory for Mycotoxins, Istituto Superiore di Sanità, Rome, Italy) with the consent of the data owner AIC (Associazione Italiana Celiaca) on 17 October 2019 (data from Valitutti et al., 2018).
Abbreviations
- 8‐OHdG
8‐hydroxydeoxyguanosine
- Ad293
Human embryonic kidney (cells)
- ADME
absorption, distribution, metabolism and excretion
- AFB1
Aflatoxin B1
- AhR
Aryl hydrocarbon receptor
- Akt
Protein kinase B
- ALP
Alkaline phosphatase
- ALT
Alanine aminotransferase
- AP
Apurinic/apyrimidinic
- ArylS
Aryl synthetase
- AST
aspartate transaminase
- Atk
protein kinase B
- ATM
ataxia telangiectasia mutated gene
- BD10
Benchmark dose defined as a 10% increase over the background
- BEA
Beauvericin
- BEN
Balkan endemic nephropathy
- BMD
Benchmark dose
- BMDL10
Benchmark dose lower confidence limit for an extra cancer risk of 10%
- BMDL05
Benchmark dose lower confidence limit for an extra cancer risk of 5%
- BMDL1SD
Benchmark dose lower confidence limit for an extra cancer risk of 1 standard deviation
- BMDU10
Benchmark dose upper confidence limit for an extra cancer risk of 10%
- BMDU05
Benchmark dose upper confidence limit for an extra cancer risk of 5%
- BMDU1SD
Benchmark dose upper confidence limit for an extra cancer risk of 1 standard deviation
- BMR
Benchmark response
- BrdU
Bromodeoxyuridine
- BUN
blood urea nitrogen
- bw
Body weight
- CA
Chromosome aberrations
- CAR
constitutive activated/androstane receptor
- cdc2
Cell division control protein
- CDK2
Cyclin dependent kinase 2
- CGN
chronic glomerular nephropathy
- CHK2
Checkpoint kinase 2
- Cinulin
Inulin concentration
- CHO
Chinese hamster ovary
- CIN
Chronic interstitial nephropathy
- CINI
CIN of unknown aetiology
- CONTAM
Panel on Contaminants in the Food Chain
- COT
Committee on Toxicology
- CTN
citrinin
- CYP
Cytochrome P450
- DATA Unit
EFSA former EFSA Dietary and Chemical Monitoring Unit
- DMBA
7,12‐dimethyl benz[α]anthracene
- DMSO
Dimethyl sulfoxide
- DNA
Deoxyribonucleic acid
- DSBs
Double strand breaks
- EEC
European Economic Community
- EGFR
Epidermal growth factor receptor
- ELISA
Enzyme‐linked immunosorbent assay
- ELK
ETS domain containing protein
- EndoIII
endonuclease III
- ER
Endoplasmatic reticulum
- ERK 1/2
extracellular signal‐regulated kinase 1/2
- ETS
E26 transformation specific
- EU
European Union
- f
female
- FAO
Food and Agriculture Organization
- FB1
Fumonisin B1
- FD
fluorescence detection
- FEEDAP
EFSA Panel on additives and products or substances used in animal feed
- FEDIOL
Vegetable oil and protein meal industry association
- Fpg
Formamido‐pyrimidine‐DNA glycosylase
- GC
Gas chromatography
- GD
Gestation day
- GI
Gastrointestinal
- GGT
γ‐glutamyltranspeptidase
- GLDH
Glutamate dehydrogenase
- GPx
Glutathione peroxidase
- GSH
Glutathione
- GSSG
Glutathione disulfide
- GSR
Glutathione reductase
- GST
glutathione S‐transferase
- Hb
Haemoglobin
- HBGV
Health‐based guidance value
- HCC
Hepatocellular cancer
- HDAC
Histone deacetylase
- HEK293
human embryonic kidney
- HKC
Rat kidney proximal tubule cells
- HNF4α
Hepatocyte nuclear factor 4 alpha
- HO‐1
Heme oxygenase 1
- hOR
human oxidoreductase
- HO‐OTA
Hydroxy ochratoxin
- HPLC
High‐performance liquid chromatography
- HPLC‐ECD
High‐performance liquid chromatography with electrochemical detection
- HPLC‐FLD
High‐performance liquid chromatography with fluorescence detection
- IARC
International Agency for Research on Cancer
- IC50
mean half maximum inhibitory concentration
- IF
Immunofluorescence
- IFN
Interferon
- IGF
Insulin‐like growth factor
- IHKE
Immortalised human kidney epithelial (cells)
- IKK
Ikβ kinase
- IL
interleukin
- i.p.
intraperitoneal
- IPCS
International Programme on Chemical Safety
- i.v.
Intravenous
- IVM
in vitro maturation
- JECFA
Joint FAO/WHO Expert Committee on Food Additives
- JNK
JunN‐terminal kinase
- KIM‐1
Kidney injury molecule‐1
- KM:
Michaelis constant
- LAP
leucine aminopeptidase
- LB
Lower bound
- LC‐MS
Liquid chromatography coupled to mass spectrometry
- LC‐MS/MS
Liquid chromatography coupled to quadrupole tandem mass spectrometry
- LC‐FD
Liquid chromatography coupled to fluorescence detector
- LD50
Lethal dose killing 50% of the animals
- LDH
lactate dehydrogenase
- LOD
Limit of detection
- LOAEL
Lowest‐observed‐adverse‐effect‐level
- LOEL
Lowest‐observed‐ effect‐level
- LOQ
Limit of quantification
- m
Male
- MAPK
Mitogen activated protein kinases
- MCV
mean corpuscular volume
- MDA
malondialdehyde
- MDCK
Madin Darby canine kidney (cell)
- miRNA
micro RNA
- ML
Maximum level
- MMTV
mouse mammary tumour virus
- MN
Micronuclei
- mRNA
messenger RNA
- MoA
Mode of action
- MOE
Margin of exposure
- MRP
multidrug resistant protein
- MS
Mass spectrometry
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide
- NAC
N‐acetyl‐L‐cysteine
- NADPH
Nicotinamide adenine dinucleotide phosphate
- NCRI
Negligible cancer risk intake
- NfkB
Nuclear factor kB
- NMR
Nuclear magnatic resonance
- NOEL
no‐observed‐effect‐level
- NOAEL
no‐observed‐adverse‐effect‐level
- Nrf2
Nuclear factor erythroid 2‐related factor 2
- NTD
neural tube defects
- NTP
National Toxicology Programme
- OAT
Organic anion transporters
- OR
Odds ratio
- OSOM
Outer stripe of the outer medulla
- OTA
Ochratoxin A
- OTalpha
Ochratoxin alpha
- OTB
Ochratoxin B
- OTC
Ochratoxin C
- OTHQ
Hydroquinone
- OTQ
Quinona
- p53
tumour protein p53
- P95
95th percentile
- PAH
p‐aminohippuric acid
- PCNA
Proliferating cell nuclear antigen
- PCV
packed cell volume
- PCT
proximal convoluted tubular
- PDK
Phosphoinositide dependent kinase
- PEPCK
phosphoenolpyruvate carboxykinase
- PKC
protein kinase C
- PND
Postnatal day
- PPAR
peroxisome proliferator‐activated receptor
- PPB
Plasma protein binding
- PTWI
Provisional tolerable weekly intake
- PXR
Pregnane X receptor
- RBC
Red blood cells
- qPCR
quantitative polymerase chain reaction
- RDW
red cell distribution
- RNA
Ribonucleic acid
- ROS
Reactive oxygen species
- RTS
renal transplanted subjects
- SCE
Sister chromatid exchange
- SD
Standard deviation
- SOD
Superoxide dismutase
- SOP
Standard operating procedure
- SSBs
Single strand breaks
- SSD
Standard Sample Description
- STER
Sterigmatocystin
- TBARS
Thiobarbituric acid reactive substances
- TCR
T‐cell receptor
- TD05
Tumorigenic dose 5%
- TDI
Tolerable daily intake
- Tk
Thymidin kinase
- TK
Toxicokinetics
- TIMP‐1
Tissue inhibitor of metalloproteinase‐1
- TLC
Thin layer chromatography
- TLC HPLC‐UV
thin layer chromatography high‐performance liquid chromatography with ultraviolet detection
- TmPAH
Average maximal tubular excretion of PAH
- Tm
Time to maximum concentration
- TNF
Tumour necrosis factor
- TopoII
Topoisomerase II
- TPA
12‐O‐ tetradecanoylphorbol‐13‐acetate
- TWI
Tolerable weekly intake
- UB
Upper bound
- UCM‐MSC
Canine umbelical cord matrix‐mesenchymal stem cells
- UDS
Unscheduled DNA synthesis
- UF
Uncertainty factor
- UGT
Uridine 5’‐diphospho‐glucuronosyltransferase (UGT)
- UPLC
Ultra performance liquid chromatography
- UPLC‐Q/TOF‐MS
ultra‐high‐performance liquid chromatography‐quadrupole time‐of‐flight mass spectrometry
- V79 cells
Chinese hamster lung cells clone V79
- Vmax
maximum velocity of an enzymatic reaction
- WG
working group
- Wnt‐1
Wingless ‐type MMTV integration site family, member 1
- WHO
World Health Organization
- wk
week
Appendix A – Identification and selection of relevant scientific literature and reports
1.
| Chemistry and Analysis | |
|---|---|
| Search terms | TOPIC: ochratoxin OR ochratoxin A OR OTA AND TOPIC: analys* OR determ* OR method* OR identif* OR character* |
| Numbers of papers found | 4,882 |
| Papers selected as potentially relevant | Not applicable |
| Formation Occurrence Exposure | |
| Search terms | TOPIC: ochratoxin OR ochratoxin A OR OTA AND TOPIC: occurrence OR expos* OR level* OR concentrat* OR formation OR food OR breast milk OR consump* OR foodstuff* OR diet* |
| Numbers of papers found | 5,050 |
| Papers selected as potentially relevant | Not applicable |
| Mode of Action | |
| Search terms | TOPIC: ochratoxin OR ochratoxin A OR OTA AND TOPIC: mode OR mechanis* OR mode of action OR mechanism of action |
| Numbers of papers found | 1,052 |
| Papers selected as potentially relevant | 156 |
| Toxicokinetics | |
| Search terms | TOPIC: ochratoxin OR ochratoxin A OR OTA AND TOPIC: toxicokinetic* OR metabolis* OR distribut* OR excret* OR absor* OR distribut* OR biotransform* OR eliminat* tissue OR kinetic* |
| Numbers of papers found | 871 |
| Papers selected as potentially relevant | 37 |
| Toxicity | |
| Search terms | TOPIC: ochratoxin OR ochratoxin A OR OTA AND TOPIC: toxic* OR tox* OR mutagen* OR carcino* OR genotox* OR aneugen* OR RNA OR reprotox* OR nephrotox* OR neurotox* OR hepatotox* OR immunotox* OR haemotox* OR hematotox* OR cytotox* OR develop* OR DNA* OR clastogen* OR cancer OR tumour OR tumor OR teratogen* OR rat OR mouse OR mice OR rabbit* OR in vitro OR in vivo OR muta* OR damage OR repair OR inhibit* OR chromosom* OR genetic* OR oxidative OR proliferation |
| Numbers of papers found | 4,274 |
| Papers selected as potentially relevant | 254 |
| Human evidence and biomarkers | |
| Search terms | TOPIC: ochratoxin OR ochratoxin A OR OTA AND TOPIC: biomarker OR biological marker OR case stud* OR poison* OR epidem* human* OR cohort OR case control OR cross‐section* |
| Numbers of papers retrieved | 157 |
| Papers selected as relevant | 27 |
| Database used | Web of Science and PubMed |
| Time limit | 2006–2018 |
| Date of search | 15/7/2018 |
| Total number considered potentially relevant in sections Mode of action, toxicokinetics, toxicity and human evidence a | 451 |
Note that there was a considerable overlap, i.e. many publications were found in more than one of the individual searches.
Appendix B – Evidence for protective effects of antioxidants and free radical scavengers against OTA‐induced toxicity
1.
Appendix C – Description of repeated dose studies already evaluated in previous assessments
C.1. Studies with rats
In Fischer 344/N rats administered OTA at 0, 21, 70, or 210 μg OTA /kg bw in corn oil by gavage (5 days per week) equivalent to 0, 15, 50 and 150 μg/kg bw per day for up to 2 years, increased incidences of neoplastic (kidney tubule adenoma and carcinoma) and non‐neoplastic renal lesions involving degeneration of the renal tubular epithelium, proliferation of the tubular epithelium and karyomegaly were observed in kidney of male and female rats at 70 and 210 μg OTA/kg bw (NTP, 1989). No renal lesions were recorded at 21 μg OTA/kg bw (NTP, 1989). Thus, the NOAEL and LOAEL for OTA nephrotoxicity in Fischer 344/N rats were 21 and 70 μg OTA /kg bw per day, 5 days per week, respectively, equivalent to 15 and 50 μg/kg bw per day. In contrast, minimal renal changes consisting of eosinophilic granular or hyaline changes in proximal convoluted tubules were reported in a 90‐day feeding study in Wistar rats (Munro et al., 1974). In this study, groups of male and female weanling Wistar rats were fed semi‐purified diets containing 0, 0.2, 1 and 5 mg OTA/kg (Munro et al., 1974), equivalent to 0, 15, 75 and 37 μg OTA/kg bw(FAO/WHO, 2001), for 90 days. The authors of the study considered that these changes may present a phenomenon of ageing accelerated by OTA exposure (Munro et al., 1974). Based on this study, an (sensitive) LOAEL for OTA nephrotoxicity in Wistar rats of 15 μg OTA/kg bw was derived (FAO/WHO, 2001; EFSA, 2006).
Table C.1.
Key studies with rats published before 2006
| Strain | Dose and animals per group | Duration/time of observation | Main effects observed | Effect level | Reference |
|---|---|---|---|---|---|
| Wistar (♂, ♀) | 0, 0.2, 1, 5 mg/kg feed (corresponding to 0, 15, 75, 37 μg/kg bw( a )) | 90 days |
|
LOAEL* 15 μg/kg bw per day( a ) | Munro et al. (1974) |
| F344/N (♂,♀) | 0, 21, 70, 210 μg/kg bw (gavage, 5 days/week)( b ) n = 50 | 2 years |
|
NOAEL* 21 μg/kg bw (5 days/week)( c ) LOAEL* 70 μg/kg bw (5 days/week)( d ) |
NTP (1989) |
LOAEL: lowest observed adverse effect level; NOAEL: no observed adverse effect level.
As estimated by FAO/WHO (2001).
Equivalent to 0, 15, 50, 150 μg/kg bw per day.
Equivalent to 15 μg/kg bw per day.
Equivalent to 50 μg/kg bw per day.
*: NOAEL/LOAEL not explicitly reported by the authors of the study but derived from data presented in study.
C.2. Studies with pigs
In previous assessments, a series of studies in pigs was used to establish a HBGV (FAO/WHO, 1991, 1996, 2001, 2008; EFSA, 2006). These studies are briefly summarised below and presented in more detail in Table C.2.
Porcine nephropathy induced by administration of crystalline OTA for periods of 5 days, 3 months and 2 years was reported in a series of papers (Elling, 1979a,b; Krogh et al., 1979). Groups of female pigs (Danish Landrace) were daily administered gelatin capsules containing a crystalline OTA preparation (90% OTA and 5% OTB as determined by thin layer chromatography) at an average dose of 30–50 μg OTA/kg bw corresponding to a feed level of 1 mg/kg bw for 3 months (n = 3) or 2 years (n = 6) (Krogh et al., 1976, 1979). Control animals were given capsules containing lactose. Consistent with the previous study (Krogh et al., 1974), increased serum creatinine and urinary glucose accompanied by a decrease in TmPAH and TmPAH/CInulin and a decreased ability to concentrate urine were reported in animals administered OTA for 3 months. Microscopic lesions in kidneys consistent primarily of degenerative changes within the proximal tubule, tubular atrophy and interstitial fibrosis (Krogh et al., 1976). Using histochemical staining of kidney sections, changes in the enzymatic activity of NADH‐tetrazolium reductase, succinate dehydrogenase, acid phosphatase and alkaline phosphatase within the pars convolute and/or pars recta of the proximal tubules were observed (Elling, 1979b). After 2 years exposure to OTA, essentially the same changes were evident as after 3 months, except that renal histopathological lesions were more widely distributed throughout the renal cortex (Elling, 1979b; Krogh et al., 1979). Terminal renal failure was not observed (Krogh et al., 1979).
In a subsequent study by the same group, renal enzyme activities were measured in needle biopsies obtained from kidneys of female pigs (Danish Landrace – Duroc) given gelatine capsules containing OTA equivalent to 0.2 and 1 mg OTA/kg feed for up to 5 weeks (Krogh et al., 1988). A dose‐related decrease in the enzymatic activities of cytosolic phosphoenolpyruvate carboxykinase (PEPCK) and the brush boarder enzyme gamma‐glutamyl transpeptidase accompanied by decrease in TmPAH and TmPAH/CInulin were observed (Meisner and Krogh, 1986; Krogh et al., 1988).
Appendix D – Species differences regarding free and bound OTA
1.
Calculations for the differences in the concentrations of free OTA in the blood plasma of humans, pigs and rats because of the different plasma protein binding (PPB) of OT (in particular to serum albumin, Alb), and the different accumulation.
Blood plasma levels of free (unbound) OTA in humans, pigs and rats assuming the same total concentration (free plus albumin‐bound)
In general terms, the binding constant (or association constant, Ka) is the equilibrium constant for the binding of a ligand L to a receptor R according to the reaction R + L ↔ RL and defined as Ka = [RL]/[R] × [L] (indicating the concentrations of R, L and RL at equilibrium).
In this case, Ka refers to the equilibrium Alb + OTA ↔ Alb × OTA and therefore
For the concentration of free (unbound) OTA: [OTA] = [Alb × OTA]/[Alb] × Ka
The following assumptions were made:
Total plasma levels of OTA (bound and unbound): 10 nanomolar (= 10–8 M, corresponding to the upper limit of normally exposed humans)
Concentration of albumin in the plasma of humans, pigs and rats: 600 micromolar (= 6 × 10–4 M)
Because of the large excess of Alb over OTA: [Alb] = 6 × 10–4 M at equilibrium
Because of the high binding: [Alb × OTA] = 10–8 M (virtually all the OTA is bound)
Binding of OTA to human albumin (Ka = 5.2 × 106 M−1):
[OTA] = 10−8 M/6 × 10−4 M × 5.2 × 106 M−1 = 10−8 M/31.2 × 102 = 3.2 × 10−12 M = 3.2 picomolar free OTA
9,997 picomolar bound OTA
Binding of OTA to pig albumin (Ka = 7.1 × 104 M−1):
[OTA] = 10−8 M/6 × 10−4 M × 7.1 × 104 M−1 = 10−8 M/42.6 × 100 = 2.3 × 10−10 M = 230 picomolar free OTA
9,770 picomolar bound OTA
Binding of OTA to rat albumin (Ka = 4.0 × 104 M−1):
[OTA] = 10−8 M/6 × 10−4 M × 4.0 × 104 M−1 = 10−8 M/24 × 100 = 4 × 10−10 M = 400 picomolar free OTA
9,600 picomolar bound OTA
Conclusion: At the same total plasma concentration of OTA in humans, pigs and rats, the plasma concentration of free OTA is more than 70‐fold lower in humans than in pigs and 120‐fold lower in humans than in rats.
Blood plasma levels of free (unbound) OTA in humans, pigs and rats taking into account the different accumulation at steady state (assuming the same bioavailability and rate of metabolism)
The free plasma concentration will be dependent on both the elimination half‐life t1/2 and free fraction
The following free fractions and plasma elimination half‐lifes were used:
Human: 3.2 × 10−4 M, 35 days
Pig: 230 × 10‐4 M, 5–6 days
Rat: 400 × 10‐4 M, 5–6 days
Conclusion:
At the repeated intake of the same amount of OTA, given the same bioavailability and at steady state, the free fraction of OTA in human plasma is about 10 times lower than in the pig and 18 times lower than in the rat.
Appendix E – Studies on biotransformation of OTA
1.
Appendix F – BMD analyses
1.
1.1.
1.1.1.
F.1. BMD analysis of microscopic kidney lesions in female pigs (Krogh et al., 1974)
A) Data Description
The endpoint to be analysed is: animals with microscopic kidney lesions.
B) Data used for analysis
| μg OTA/kg bw per day | Number of animals with microscopic kidney lesions | Total number of animals |
|---|---|---|
| 0 | 0 | 9 |
| 8 | 4 | 9 |
| 40 | 9 | 9 |
| 160 | 4 | 4 |
C) Selection of the BMR
The benchmark response (BMR) used is an extra risk of 10%.
The benchmark dose (BMD) is the dose corresponding to the BMR of interest.
A 90% confidence interval for the BMD will be estimated, with lower and upper bound denoted BMDL and BMDU, respectively.
D) Software Used
Results are obtained using the EFSA web‐tool for BMD analysis, which uses the R‐package PROAST, version 66.40, for the underlying calculations.
E) Results
Response variable: animals with microscopic kidney lesions
Fitted Models
| Model | No.par | loglik | AIC | Accepted | BMDL | BMDU | BMD | Conv |
|---|---|---|---|---|---|---|---|---|
| null | 1 | −21.34 | 44.68 | NA | NA | NA | NA | |
| full | 4 | −6.18 | 20.36 | NA | NA | NA | NA | |
| two.stage | 3 | −6.18 | 18.36 | Yes | 0.6880 | 5.440 | 3.34 | No |
| log.logist | 3 | −6.18 | 18.36 | Yes | 0.9080 | 7.920 | 6.78 | Yes |
| Weibull | 3 | −6.18 | 18.36 | Yes | 0.1820 | 7.920 | 3.62 | Yes |
| log.prob | 3 | −6.18 | 18.36 | Yes | 0.7940 | 7.950 | 5.93 | Yes |
| gamma | 3 | −6.18 | 18.36 | Yes | 0.0488 | 7.640 | 5.14 | Yes |
| logistic | 2 | −6.18 | 16.36 | Yes | 2.3200 | 0.856 | 7.18 | Yes |
| probit | 2 | −6.18 | 16.36 | Yes | 2.1200 | 6.890 | 6.27 | Yes |
| LVM: Expon. m3‐ | 3 | −6.18 | 18.36 | Yes | 0.1620 | 7.820 | 6.38 | Yes |
| LVM: Hill m3‐ | 3 | −6.18 | 18.36 | Yes | 0.2920 | 7.690 | 5.50 | Yes |
F) Estimated Model Parameters
two.stage
estimate for a‐ : 1e‐06
estimate for BMD‐ : 3.337
estimate for c : 3505
log.logist
estimate for a‐ : 1e‐06
estimate for BMD‐ : 6.782
estimate for c : 11.95
Weibull
estimate for a‐ : 1e‐06
estimate for BMD‐ : 3.621
estimate for c : 2.168
log.prob
estimate for a‐ : 1e‐06
estimate for BMD‐ : 5.926
estimate for c : 3.805
gamma
estimate for a‐ : 1e‐06
estimate for BMD‐ : 5.135
estimate for cc : 8.164
logistic
estimate for a‐ : ‐19.48
estimate for BMD‐ : 7.18
probit
estimate for a‐ : ‐5.409
estimate for BMD‐ : 6.267
EXP
estimate for a‐ : 4.249
estimate for CED‐ : 6.382
estimate for d‐ : 1
estimate for th(fixed) : 0
estimate for sigma(fixed) : 0.25
HILL
estimate for a‐ : 4.357
estimate for CED‐ : 5.503
estimate for d‐ : 1.053
estimate for th(fixed) : 0
estimate for sigma(fixed) : 0.25
G) Weights for Model Averaging
| two.stage | log.logist | Weibull | log.prob | gamma | logistic | probit | EXP | HILL |
|---|---|---|---|---|---|---|---|---|
| 0.08 | 0.08 | 0.08 | 0.08 | 0.08 | 0.22 | 0.22 | 0.08 | 0.08 |
H) Final BMD Values
| subgroup | BMDL | BMDU |
|---|---|---|
| 4.73 | 7.17 |
Confidence intervals for the BMD are based on 500 bootstrap data.
J) Visualisation
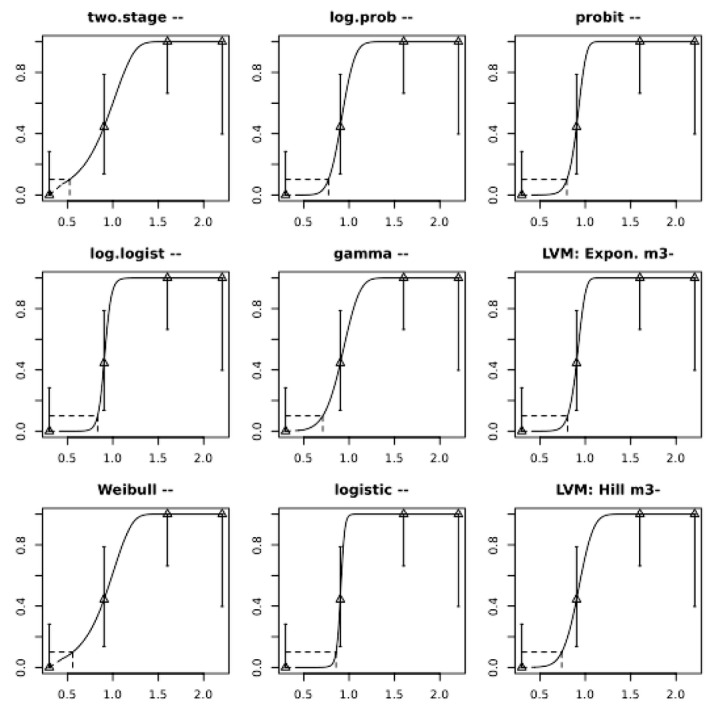
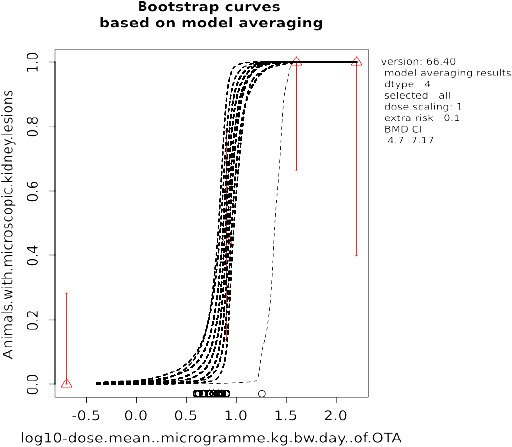
F.2. BMD analysis of combined incidence of adenoma and carcinoma in male rats (NTP, 1989)
A) Data Description
The endpoint to be analysed is incidence kidney adenoma and carcinoma.
B) Data used for analysis:
| μg OTA/kg bw per day | Incidence of kidney adenoma and carcinoma | N |
|---|---|---|
| 0 | 1 | 50 |
| 15 | 1 | 50 |
| 50 | 20 | 51 |
| 150 | 36 | 50 |
C) Selection of the BMR
The BMR used is an extra risk of 10% compared to the controls.
The BMD is the dose corresponding with the BMR of interest.
A 90% confidence interval for the BMD will be estimated, with the lower and upper bound denoted BMDL and BMDU, respectively.
D) Software Used
Results are obtained using the EFSA web‐tool for BMD analysis, which uses the R‐package PROAST, version 66.40, for the underlying calculations.
E) Results
Response variable: incidence of kidney adenoma and carcinoma
Fitted Models
| Model | No.par | loglik | AIC | Accepted | BMDL | BMDU | BMD | conv |
|---|---|---|---|---|---|---|---|---|
| null | 1 | −120.77 | 243.54 | NA | NA | NA | NA | |
| full | 4 | −73.61 | 155.22 | NA | NA | NA | NA | |
| two.stage | 3 | −77.29 | 160.58 | No | NA | NA | 17.2 | Yes |
| log.logist | 3 | −75.57 | 157.14 | Yes | 14.7 | 31.6 | 22.7 | Yes |
| Weibull | 3 | −76.68 | 159.36 | No | NA | NA | 20.3 | Yes |
| log.prob | 3 | −75.05 | 156.10 | Yes | 15.9 | 32.4 | 23.6 | Yes |
| gamma | 3 | −76.36 | 158.72 | No | NA | NA | 22.0 | Yes |
| logistic | 2 | −82.11 | 168.22 | No | NA | NA | 37.3 | Yes |
| probit | 2 | −81.23 | 166.46 | No | NA | NA | 35.0 | Yes |
| LVM: Expon. m5‐ | 4 | −73.61 | 155.22 | Yes | 20.9 | 42.4 | 39.3 | Yes |
| LVM: Hill m3‐ | 3 | −76.86 | 159.72 | No | NA | NA | 19.4 | Yes |
Estimated Model Parameters
two.stage
estimate for a‐ : 0.01331
estimate for BMD‐ : 17.24
estimate for c : 0.6374
log.logist
estimate for a‐ : 0.01334
estimate for BMD‐ : 22.66
estimate for c : 1.759
Weibull
estimate for a‐ : 0.01368
estimate for BMD‐ : 20.32
estimate for c : 1.287
log.prob
estimate for a‐ : 0.01385
estimate for BMD‐ : 23.64
estimate for c : 1.061
gamma
estimate for a‐ : 0.01394
estimate for BMD‐ : 21.97
estimate for cc : 1.551
logistic
estimate for a‐ : ‐2.58
estimate for BMD‐ : 37.31
probit
estimate for a‐ : ‐1.563
estimate for BMD‐ : 35.04
EXP
estimate for a‐ : 1.676
estimate for CED‐ : 39.3
estimate for c‐ : 0.5159
estimate for d‐ : 4
estimate for th(fixed) : 0
estimate for sigma(fixed) : 0.25
HILL
estimate for a‐ : 1.791
estimate for CED‐ : 19.38
estimate for d‐ : 0.6204
estimate for th(fixed) : 0
estimate for sigma(fixed) : 0.25
Weights for Model Averaging
| two.stage | log.logist | Weibull | log.prob | gamma | logistic | probit | EXP | HILL |
|---|---|---|---|---|---|---|---|---|
| 0.03 | 0.15 | 0.05 | 0.26 | 0.07 | 0 | 0 | 0.4 | 0.04 |
F) Final BMD Values
| subgroup | BMDL | BMDU |
|---|---|---|
| 14.5 | 36 |
Confidence intervals for the BMD are based on 500 bootstrap data sets.
G) Visualisation
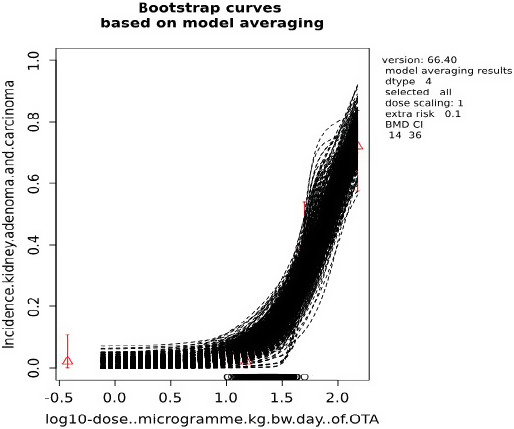
Appendix G – Original data used for the exposures assessment of infants fed with breast milk
1.
Table G.3.
OTA levels (ng/L) in milk samples from healthy mothers from Valitutti et al. (2018)
| 1st day | 2nd day | 3rd day | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Sample ID | 4 h after lunch | 2 h after dinner | Morning | 4 h after lunch | 2 h after dinner | Morning | 4 h after lunch | 2 h after dinner | Morning |
| COD15 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD21 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD22 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.t. | n.d. |
| COD28 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD30 | n.d. | n.d. | n.d. | n.d. | n.d. | n.t. | n.d. | n.d. | n.t. |
| COD31 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.t. | n.t. |
| COD37 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD38 | n.t. | n.t. | n.d. | n.d. | n.t. | n.d. | n.t. | n.t. | n.t. |
| COD40 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD45 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD46 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD54 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD60 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD66 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 56 |
| COD68 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD70 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD77 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD78 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD82 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD93 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| COD97 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
n.d.: not detected; n.t.: not tested; OTA: ochratoxin.
Annex – Summary statistics on occurrence and consumption data and exposure assessment results
1.
The Annex is provided as separate Excel files containing summary statistics on occurrence and consumption data and exposure assessment results on ochratoxin A and is available on the EFSA Knowledge Junction community on Zenodo at: https://doi.org/10.5281/zenodo.3739292
At the zenodo link above, we have also included the occurrence data of OTA ‐ please see Section 4.7 Occurrence data.
Suggested citation: EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain) , Schrenk D, Bodin L, Chipman JK, del Mazo J, Grasl‐Kraupp B, Hogstrand C, Hoogenboom L, Leblanc J‐C, Nebbia CS, Nielsen E, Ntzani E, Petersen A, Sand S, Schwerdtle T, Vleminckx C, Wallace H, Alexander J, Dall'Asta C, Mally A, Metzler M, Binaglia M, Horváth Z, Steinkellner H and Bignami M, 2020. Scientific Opinion on the risk assessment of ochratoxin A in food. EFSA Journal 2020;18(5):6113, 150 pp. 10.2903/j.efsa.2020.6113
Requestor: European Commission
Question number: EFSA‐Q‐2018‐00699
Panel members: Margherita Bignami, Laurent Bodin, James Kevin Chipman, Jesús del Mazo, Bettina Grasl‐Kraupp, Christer Hogstrand, Laurentius (Ron) Hoogenboom, Jean‐Charles Leblanc, Carlo Stefano Nebbia, Elsa Nielsen, Evangelia Ntzani, Annette Petersen, Salomon Sand, Dieter Schrenk, Tanja Schwerdtle, Christiane Vleminckx and Heather Wallace.
Adopted: 31 March 2020
This publication is linked to the following EFSA Supporting Publications article: http://onlinelibrary.wiley.com/doi/10.2903/sp.efsa.2020.EN-1845/full
Amendment: On page 75, before Figure 7, and on page 150, we have added text clarifying that the occurrence data for OTA on 73,891 food samples is available at the Zenodo link mentioned under the Annex section. On page 22 a chemical structure in Figure 6 has been revised to show the correct structure of the OTB‐GSH conjugate. To avoid confusion, the original version of the document has been removed from the EFSA Journal, but is available on request, as is a version showing all the changes made.
Amended: 15 Jul 2020
Notes
Commission Regulation (EC) No 1881/2006 of 19 December 2006 setting maximum levels for certain contaminants in foodstuffs. OJ L 364, 20.12.2006, p. 5–24.
Council Regulation (EEC) No 315/93 of February 1993 laying down Community procedures for contaminants in food. OJ L 37, 13.2.1993, p. 1–5.
Regulation (EC) No 1881/2006 of the European Parliament and the Council of 19 December 2006 setting maximum levels for certain contaminants in foodstuffs. OJ L 364, 20.12.2006, p. 5–24.
Commission Recommendation of 17 August 2006 on the presence of deoxynivalenol, zearalenone, ochratoxin A, T‐2 and HT‐2 and fumonisins in products intended for animal feeding. OJ L 118 M, 8.5.2007, p. 1111–1113.
Web of Science (WoS), formally ISI Web of Knowledge, Thomson Reuters. http://thomsonreuters.com/thomson-reuters-web-of-science/
EndNote X5, Thomson Reuters. http://endnote.com/
Identifying articles that have been cited in articles found in a search (see Jalali and Wohlin, 2012).
OATs are a subgroup of solute carriers (SLCs).
Commission Recommendation of 17 August 2006 on the presence of deoxynivalenol, zearalenone, ochratoxin A, T‐2 and HT‐2 and fumonisins in products intended for animal feeding. OJ L 229, 23.8.2006, p. 7–9.
A receiver operating characteristic curve, or ROC curve, is a graphical plot that illustrates the diagnostic ability of a binary classifier system as its discrimination threshold is varied.
Ministero della Sanità, 1999. Circolare 09.06.1999. In Sanità MD (ed.). Circolare 09.06.1999 (vol. 135) 11.06.1999. Gazzetta Ufficiale Repubblica Italiana. Available online: https://www.gazzettaufficiale.it/eli/id/1999/06/26/099A5104/sg
References
- Abassi H, Ayed‐Boussema I, Shirley S, Abid S and Bacha H, 2016. Ochratoxin A and T‐ toxin induce clonogenicity and cell migration in human colon carcinoma and fetal lung fibroblast cell lines. Journal of Biochemical and Molecular Toxicology, 30, 128–135. 10.1002/jbt.21771 [DOI] [PubMed] [Google Scholar]
- Abdel‐Wahhab MA, Abdel‐Galil MM and El‐Lithey M, 2005. Melatonin counteracts oxidative stress in ratsfed an ochratoxin A contaminated diet. Journal of Pineal Research, 38, 130–135. [DOI] [PubMed] [Google Scholar]
- Abdel‐Wahhab MA, Abdel‐Azim SH and El‐Nekeety AA, 2008. Inulacrithmoides extract protects against ochratoxin A‐induced oxidative stress, clastogenic and mutagenic alterations in male rats. Toxicon, 52, 566–573. [DOI] [PubMed] [Google Scholar]
- Abid S, Hassen W, Achour A, Skhiri H, Maaroufi K, Ellouz F, Creppy E and Bacha H, 2003. Ochratoxin A and human chronic nephropathy in Tunisia: is the situation endemic? Human and Experimental Toxicology, 22, 77–84. [DOI] [PubMed] [Google Scholar]
- Abrunhosa L, Paterson RRM and Venancio A, 2010. Biodegradation of ochratoxin A for food and feed decontamination. Toxins, 2, 1078–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler M, Muller K, Rached E, Dekant W and Mally A, 2009. Modulation of key regulators of mitosis linked to chromosomal instability is an early event in ochratoxin A carcinogenicity. Carcinogenesis, 30, 711–719. [DOI] [PubMed] [Google Scholar]
- Ahn J, Kim D, Jang HS, Kim Y, Shim WB and Chung DH, 2010. Occurrence of ochratoxin A in Korean red paprika and factors to be considered in prevention strategy. Mycotoxin Research, 4, 279–286. [DOI] [PubMed] [Google Scholar]
- Akman SA, Adams M, Case D, Park G and Manderville RA, 2012. Mutagenicity of ochratoxin A and its hydroquinone metabolite in the SupF gene of the mutation reporter plasmid Ps189. Toxins (Basel), 4, 267–280. 10.3390/toxins4040267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Anati L, Essid E, Stenius U, Beuerlein K, Schuh K and Petzinger E, 2010. Differential cell sensitivity between OTA and LPS upon releasing TNF‐α. Toxins (Basel), 2, 1279–1299. 10.3390/toxins2061279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albassam MA, Yong SI, Bhatnagar R, Sharma AK and Prior MG, 1987. Histopathologic and electron microscopic studies on the acute toxicity of ochratoxin A in rats. Veterinary Pathology, 424, 427–435. [DOI] [PubMed] [Google Scholar]
- Ali R, Mittelstaedt RA, Shaddock JG, Ding W, Bhalli JA, Khan QM and Heflich RH, 2011. Comparative analysis of micronuclei and DNA damage induced by Ochratoxin A in two mammalian cell lines. Mutation Research, 723, 58–64. [DOI] [PubMed] [Google Scholar]
- Ali R, Guo X, Lin H, Khan QM, Ismail M, Waheed U, Ali T and Bhalli JA, 2014. Mutant frequency in comparison to oxidative DNA damage induced by ochratoxin A in L5178Y tk+/‐ (3.7.2C) mouse lymphoma cells. Drug and Chemical Toxicology, 37, 227–232. [DOI] [PubMed] [Google Scholar]
- Ali N, Blaszkewicz M, Alim A, Hossain K and Degen GH, 2016. Urinary biomarkers of ochratoxin A and citrinin exposure in two Bangladeshi cohorts: follow‐up study on regional and seasonal influences. Archives of Toxicology, 90, 2683–2697. [DOI] [PubMed] [Google Scholar]
- Ali N, Muñoz K and Degen GH, 2017. Ochratoxin A and its metabolites in urines of German adults ‐an assessment of variables in biomarker analysis. Toxicology Letters, 5, 19–26. 10.1016/j.toxlet.2017.04.013 [DOI] [PubMed] [Google Scholar]
- Ali N, Hossain K and Degen GH, 2018. Blood plasma biomarkers of citrinin and ochratoxin A exposure in young adults in Bangladesh. Mycotoxin Research, 34, 59–67. 10.1007/s12550-017-0299-5 [DOI] [PubMed] [Google Scholar]
- Altafini A, Armorini S, Zaghini A, Sardi L and Roncada P, 2017. Tissue distribution of ochratoxin A in pigs after administration of two‐levels contaminated diets. World Mycotoxin Journal, 10, 263–272. 10.3920/WMJ2016.2152 [DOI] [Google Scholar]
- Amézqueta S, González‐Peñas E, Murillo M and López de Cerain A, 2004. Validation of a high‐performance liquid chromatography analytical method for ochratoxin A quantification in cocoa beans. Food Additives and Contaminants, 21, 1096–1106. [DOI] [PubMed] [Google Scholar]
- Anelli P, Haidukowski M, Epifani F, Cimmarusti MT, Moretti A, Logrieco A and Susca A, 2019. Fungal mycobiota and mycotoxin risk for traditional artisan Italian cave cheese. Food Microbiology, 78, 62–72. [DOI] [PubMed] [Google Scholar]
- Anninou N, Chatzaki E, Papachristou F, Pitiakoudis M and Simopoulos C, 2014. Mycotoxins activity at toxic and sub‐toxic concentrations: differential cytotoxic and genotoxic effects of single and combined administration of sterigmatocystin, ochratoxin A and citrinin on the hepatocellular cancer cell line Hep3B. International Journal of Environmental Research and Public Health, 11, 1855–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbillaga L, Azqueta A, Ezpeleta O and Lopez de Cerain A, 2007. Oxidative DNA damage induced by Ochratoxin A in the HK‐2 human kidney cell line: evidence of the relationship with cytotoxicity. Mutagenesis, 22, 35–42. [DOI] [PubMed] [Google Scholar]
- Arbillaga L, Vettorazzi A, Gil AG, van Delft JH, García‐Jalón JA and López de Cerain A, 2008. Gene expression changes induced by ochratoxin A in renal and hepatic tissues of male F344 rat after oral repeated administration. Toxicology and Applied Pharmacology, 15;230, 197–207. 10.1016/j.taap.2008.02.018 [DOI] [PubMed] [Google Scholar]
- Ariño A, Herrera M, Estopañan G and Juan T, 2007. High levels of ochratoxin A in licorice and derived products. International Journal of Food Microbiology, 114, 366–369. [DOI] [PubMed] [Google Scholar]
- Aslam M, Rivzi SA, Beg AE, Blaszkewicz M, Golka K and Degen GH, 2012. Analysis of ochratoxin A blood levels in bladder cancer cases and healthy persons from Pakistan. Journal of Toxicology and Environmental Health A, 75, 1176–1184. [DOI] [PubMed] [Google Scholar]
- Ates I, Ulker OC, Akdemir C and Karakaya A, 2011. Correlation of Ochratoxin A exposure to urinary levels of 8‐hydroxydeoxyguanosine and malondialdehyde in a Turkish population. Bulletin of Environmental Contamination and Toxicology, 86, 258–262. 10.1007/s00128-011-0225-z [DOI] [PubMed] [Google Scholar]
- Aydin S, Palabiyik SS, Erkekoglu P, Sahin G, Başaran N and Giray BK, 2013. The carotenoid lycopene protects rats against DNA damage induced by Ochratoxin A. Toxicon, 73, 96–103. [DOI] [PubMed] [Google Scholar]
- Balasaheb Wangikar P, Sinha N, Dwivedi P and Sharma AK, 2007. Teratogenic effects of ochratoxin A and aflatoxin B1 alone and in combination on post‐implantation rat embryos in culture. Journal of the Turkish German Gynecological Association, 8, 357–364. [Google Scholar]
- Balogh K, Hausenblasz J, Weber M, Erdelyi M, Fodor J and Mezes M, 2007. Effects of ochratoxin a on some production traits, lipid peroxide and glutathione redox status of weaned piglets. Acta Veterinaria Hungarica, 55, 463–470. [DOI] [PubMed] [Google Scholar]
- Bang YJ, Yoon SM, Cong WT, Lee SN, Kim CS, Lee KY, Choi JK and Choi HJ, 2009. Proteomics approach to ochratoxin A‐induced apoptosis and cytotoxicity in HT22 cell. Journal of Neurochemistry, 110(Suppl. 2), 227–269. [Google Scholar]
- Barthelmebs L, Jonca J, Hayat A, Prieto‐Simon B and Marty J‐L, 2011. Enzyme‐Linked Aptamer Assays (ELAAs), based on a competition format for a rapid and sensitive detection of Ochratoxin A in wine. Food Control, 22, 737–743. [Google Scholar]
- Battacone G, Nudda A and Pulina G, 2010. Effects of ochratoxin A on livestock production. Toxins, 2, 1796–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrán E, Ibáñez M, Sancho JV, Cortés MA, Yus V and Hernández F, 2011. UHPLC‐MS/MS highly sensitive determination of aflatoxins, the aflatoxin metabolite M1 and ochratoxin A in baby food and milk. Food Chemistry, 126, 737–744. [Google Scholar]
- Benalia S, Bernardi B, Cubero S, Leuzzi A, Larizza M and Blasco J, 2015. Preliminary trials on Hyperspectral imaging implementation to detect Mycotoxins in dried figs. Chemical Engineering Transactions, 44, 157–162. [Google Scholar]
- Bendele AM, Carlton WW, Krogh P and Lillehoj E, 1985. Ochratoxin A carcinogenesis in the (C57BL/6J× C3H) F1 mouse. Journal of the National Cancer Institute, 75, 733–742. [PubMed] [Google Scholar]
- Bernardini C, Grilli E, Duvigneau JC, Zannoni A, Tugnoli B, Gentilini F, Bertuzzi T, Spinozzi S, Camborata C, Bacci ML, Piva A and Forni M, 2014. Cellular stress marker alteration and inflammatory response in pigs fed with an ochratoxin contaminated diet. Research in Veterinary Science, 97, 244–250. [DOI] [PubMed] [Google Scholar]
- Bernhoft A, Høgåsen HR, Rosenlund G, Ivanova L, Berntssen MHG, Alexander J, Eriksen GS and Kruse Fæste C, 2017. Tissue distribution and elimination of deoxynivalenol and ochratoxin A in dietary‐exposed Atlantic salmon (Salmo salar). Food Additives and Contaminants Part A, 34, 1211–1224. 10.1080/19440049.2017.1321149 [DOI] [PubMed] [Google Scholar]
- Bertelli AA, Migliori M, Filippi C, Gagliano N, Donetti E, Panichi V, Scalori V, Colombo R, Mannari C, Tillement JP and Giovannini L, 2005. Effect of ethanol and red wine on ochratoxin a‐induced experimental acute nephrotoxicity. Journal of Agricultural and Food Chemistry, 53, 6924–6929. [DOI] [PubMed] [Google Scholar]
- Berthiller F, Crews C, Dall'Asta C, Saeger SD, Haesaert G, Karlovsky P, Oswald IP, Seefelder W, Speijers G and Stroka J, 2013. Masked mycotoxins: a review. Molecular Nutrition and Food Research, 57, 165–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertuzzi T, Rastelli S, Mulazzi A, Donadini G and Pietri A, 2011. Mycotoxin occurrence in beer produced in several European countries. Food Control, 22, 2059–2064. [Google Scholar]
- Bertuzzi T, Gualla A, Morlacchini M and Pietri A, 2013. Direct and indirect contamination with ochratoxin A of ripened pork products. Food Control, 34, 79–83. [Google Scholar]
- Bhat PV, Pandareesh MD, Khanum F and Tamatam A, 2016. Cytotoxic effects of ochratoxin A in Neuro‐2a cells: role of oxidative stress evidenced by N‐acetylcysteine. Frontiers in Microbiology, 7, 1142. 10.3389/fmicb.2016.01142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat PV, Anand T, Mohan Manu T and Khanum F, 2018. Restorative effect of l‐Dopa treatment against Ochratoxin A induced neurotoxicity. Neurochemistry International, 118, 252–263. 10.1016/j.neuint.2018.04.003 [DOI] [PubMed] [Google Scholar]
- Biancardi A, Piro R, Galaverna G and Dall'Asta C, 2013. A simple and reliable liquid chromatography‐tandem mass spectrometry method for determination of ochratoxin A in hard cheese. International Journal of Food Sciences and Nutrition, 64, 632–640. [DOI] [PubMed] [Google Scholar]
- Biasucci G, Calabrese G, Di Giuseppe R, Carrara G, Colombo F, Mandelli B, Maj M, Bertuzzi T, Pietri A and Rossi F, 2011. The presence of ochratoxin A in cord serum and in human milk and its correspondence with maternal dietary habits. European Journal of Nutrition, 50, 211–218. 10.1007/s00394-010-0130-y [DOI] [PubMed] [Google Scholar]
- Bittner A, Cramer B and Humpf HU, 2013. Matrix binding of ochratoxin A during roasting. Journal of Agricultural and Food Chemistry, 61, 12737–12743. [DOI] [PubMed] [Google Scholar]
- Bittner A, Cramer B, Harrer H and Humpf HU, 2015. Structure elucidation and in vitro cytotoxicity of ochratoxin α amide, a new degradation product of ochratoxin A. Mycotoxin Research, 31, 83–90. [DOI] [PubMed] [Google Scholar]
- Bizard AH and Hickson ID, 2018. Anaphase: a fortune‐teller of genomic instability. Current Opinion in Cell Biology, 52, 112–119. 10.1016/j.ceb.2018.02.012 [DOI] [PubMed] [Google Scholar]
- Blanc M, Pittet A, Muñoz R and Viani R, 1998. Behavior of ochratoxin a during green coffee roasting and soluble coffee manufacture. Journal of Agricultural and Food Chemistry, 46, 673–675. [DOI] [PubMed] [Google Scholar]
- Bloch KM, Yaqoob N, Evans AR, Radford R, Jennings P, Boei JWA, McMorrow T, Slattery C, Ryan MP, Gmuender H, van Delft JHM and Lock EA, 2012. Detection of genotoxic and non‐genotoxic renal carcinogens in vitro in NRK‐52E cells using a transcriptomics approach. Toxicology Research, 1, 211–219. ISSN 2045–452X. [Google Scholar]
- Bondy GS, Caldwell DS, Aziz SA, Coady LC, Armstrong CL, Curran IH, Koffman RL, Kapal K, Lefebvre DE and Mehta R, 2015. Effects of chronic ochratoxin A exposure on p53 heterozygous and p53 homozygous mice. Toxicologic Pathology, 43, 715–729. [DOI] [PubMed] [Google Scholar]
- Bondy GS, Coady L, Ross N, Caldwell D, Gannon AM, Kwong K, Hayward S, Lefebvre DE, Liston V, Raju J, Pantazopoulos P and Curran I, 2018. A reproductive and developmental screening study of the fungal toxin ochratoxin A in Fischer rats. Mycotoxin Research, 34, 241–255. 10.1007/s12550-018-0319-0 [DOI] [PubMed] [Google Scholar]
- Bösch‐Saadatmandi C, Hundhausen C, Jofre‐Monseny L, Blank R, Wolffram S and Rimbach G, 2006. Ochratoxin A‐induced cytotoxicity in liver (HepG2) cells: impact of serum concentration, dietary antioxidants and glutathione‐modulating compounds. Journal of Applied Botany and Food Quality, 80, 179–186. [Google Scholar]
- Bösch‐Saadatmandi C, Loboda A, Jozkowicz A, Huebbe P, Blank R, Wolffram S, Dulak J and Rimbach G, 2008. Effect of ochratoxin A on redox‐regulated transcription factors, antioxidant enzymes and glutathione‐S‐transferase in cultured kidney tubulus cells. Food and Chemical Toxicology, 46, 2665–2671. 10.1016/j.fct.2008.04.023 [DOI] [PubMed] [Google Scholar]
- Bösch‐Saadatmandi C, Wagner AE, Graeser AC, Hundhausen C, Wolffram S and Rimbach G, 2009. Ochratoxin A impairs Nrf2‐dependent gene expression in porcine kidney tubulus cells. Journal of Animal Physiology and Animal Nutrition, 93, 547–554. 10.1111/j.1439-0396.2008.00838.x [DOI] [PubMed] [Google Scholar]
- Bouaziz C, Sharaf El Dein O, El Golli E, Abid‐Essefi S, Brenner C, Lemaire C and Bacha H, 2008. Different apoptotic pathways induced by zearalenone, T‐2 toxin and ochratoxin A in human hepatoma cells. Toxicology, 254, 19–28. 10.1016/j.tox.2008.08.020 [DOI] [PubMed] [Google Scholar]
- Boudra H, Barnouin J, Dragacci S and Morgavi DP, 2007. Aflatoxin M1 and ochratoxin A in raw bulk milk from French dairy herds. Journal of Dairy Science, 90, 3197–3201. [DOI] [PubMed] [Google Scholar]
- Boudra H, Saivin S, Buffiere C and Morgavi DP, 2013. Toxicokinetics of ochratoxin A in dairy ewes and carryover to milk following a single or long‐term ingestion of contaminated feed. Journal of Dairy Science, 96, 6690–6696. [DOI] [PubMed] [Google Scholar]
- Bouslimi A, Bouaziz C, Ayed‐Boussema I, Hassen W and Bacha H, 2008. Individual and combined effects of ochratoxin A and citrinin on viability and DNA fragmentation in cultured Vero cells and on chromosome aberrations in mice bone marrow cells. Toxicology, 251, 1–7. [DOI] [PubMed] [Google Scholar]
- Bozzo G, Bonerba E, Ceci E, Colao V and Tantillo G, 2011. Determination of ochratoxin A in eggs and target tissues of experimentally drugged hens using HPLC‐FLD. Food Chemistry, 126, 1278–1282. [Google Scholar]
- Breitholtz‐Emanuelsson A, Olsen M, Oskarsson A, Palminger I and Hult K, 1993. Ochratoxin A in cow's milk and in human milk with corresponding human blood samples. Journal of AOAC International, 76, 842–846. [PubMed] [Google Scholar]
- Brennan KM, Oh SY, Yiannikouris A, Graugnard DE and Karrow NA, 2017. Differential gene expression analysis of bovine macrophages after exposure to the penicillium mycotoxins citrinin and/or ochratoxin A. Toxins (Basel), 9, 366. 10.3390/toxins9110366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brera C, Pannunzi E, Guarino C, Debegnach F, Gregori E and De Santis B, 2014. Ochratoxin a determination in cured ham by high performance liquid chromatography fluorescence detection and ultra‐performance liquid chromatography tandem mass spectrometry: a comparative study. Journal of Liquid Chromatography and Related Technologies, 37, 2036–2045. [Google Scholar]
- Calcutt MW, Gillman IG, Noftle RE and Manderville RA, 2001. Electrochemical oxidation of ochratoxin A: correlation with 4‐chlorophenol. Chemical Research in Toxicology, 14, 1266–1272. [DOI] [PubMed] [Google Scholar]
- Camardo Leggieri M, Pietri A and Battilani P, 2020. Modelling fungal growth, mycotoxin production and release in grana cheese. Microorganisms, 8 , art. no. 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano‐Sancho G, González‐Arias CA, Ramos AJ, Sanchis V and Fernández‐Cruz ML, 2015. Cytotoxicity of the mycotoxins deoxynivalenol and ochratoxin A on Caco‐2 cell line in presence of resveratrol. Toxicology in Vitro, 29, 1639–1646. 10.1016/j.tiv.2015.06.020 [DOI] [PubMed] [Google Scholar]
- Cariddi LN, Sabini MC, Escobar FM, Montironi I, Mañas F, Iglesias D, Comini LR, Sabini LI and Dalcero AM, 2015. Polyphenols as possible bioprotectors against cytotoxicity and DNA damage induced by ochratoxin A. Environmental Toxicology and Pharmacology, 39, 1008–1018. [DOI] [PubMed] [Google Scholar]
- Carraturo F, De Castro O, Troisi J, De Luca A, Masucci A, Cennamo P, Trifuoggi M, Aliberti F and Guida M, 2018. Comparative assessment of the quality of commercial black and green tea using microbiology analyses. BMC Microbiology, 18, 4. 10.1186/s12866-017-1142-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castegnaro M, Bartsch H, Bereziat JC, Arvela P, Michelon J and Broussolle L, 1989. Polymorphic ochratoxin A hydroxylation in rat strains phenotyped as poor and extensive metabolizers of debrisoquine. Xenobiotica, 19, 230. [DOI] [PubMed] [Google Scholar]
- Castegnaro M, Maru V, Petkova‐Bocharova T, Nikolov I and Bartsch H, 1991. Concentrations of ochratoxin A in the urine of endemic nephropathy patients and controls in Bulgaria: lack of detection of 4‐hydroxyochratoxin A. IARC Scientific Publications, 115, 165–169. [PubMed] [Google Scholar]
- Castellanos‐Onorio O, Gonzalez‐Rios O, Guyot B, Fontana Tachon A, Guiraud JP and Schorr‐Galindo S, 2011. Effect of two different roasting techniques on the ochratoxin A (OTA) reduction in coffee beans (Coffee arabica). Food Control, 22, 1184–1188. [Google Scholar]
- Cavin C, Delatour T, Marin‐Kuan M, Holzhäuser D, Higgins L, Bezençon C, Guignard G, Junod S, Richoz‐Payot J, Gremaud E, Hayes JD, Nestler S, Mantle P and Schilter B, 2007. Reduction in antioxidant defenses may contribute to ochratoxin A toxicity and carcinogenicity. Toxicological Sciences, 96, 30–39. [DOI] [PubMed] [Google Scholar]
- Cavin C, Delatour T, Marin‐Kuan M, Fenaille F, Holzhäuser D, Guignard G, Bezençon C, Piguet D, Parisod V, Richoz‐Payot J and Schilter B, 2009. Ochratoxin A‐mediated DNA and protein damage: roles of nitrosative and oxidative stresses. Toxicological Sciences, 110, 84–94. [DOI] [PubMed] [Google Scholar]
- Chakraborty D and Verma R, 2010a. Spermatotoxic effect of ochratoxin and its amelioration by emblica officinalis aqueous extract. Acta Poloniae Pharmaceutica ‐ Drug Research, 66, 689–695. [PubMed] [Google Scholar]
- Chakraborty D and Verma R, 2010b. Ameliorative effect of Emblica officinalis aqueous extract on ochratoxin‐induced lipid peroxidation in the kidney and liver of mice. International Journal of Occupational Medicine and Environmental Health, 23, 63–73. [DOI] [PubMed] [Google Scholar]
- Cheli F, Pinotti L, Rossi L and Dell'Orto V, 2013. Effect of milling procedures on mycotoxin distribution in wheat fractions: a review. Lwt‐Food Science and Technology, 54, 307–314. [Google Scholar]
- Chen R, Deng L, Yu X, Wang X, Zhu L, Yu T, Zhang Y, Zhou B, Xu W, Chen L and Luo H, 2015. MiR‐122 partly mediates the ochratoxinA‐induced GC‐2cell apoptosis. Toxicology In Vitro, 30(1 Pt B): 264–273. 10.1016/j.tiv.2015.10.011 [DOI] [PubMed] [Google Scholar]
- Chen W‐L, Chang C‐W and Chen C‐Y, 2016. Measuring ochratoxin A concentrations in coffee beverages with immunoaffinity columns and ultra‐performance liquid chromatography/tandem mass spectrometry. Journal of AOAC International, 99, 469–474. [DOI] [PubMed] [Google Scholar]
- Chipman JK, Mally A and Edwards GO, 2003. Disruption of gap junctions in toxicity and carcinogenicity. Toxicological Sciences, 71, 146–153. [DOI] [PubMed] [Google Scholar]
- Ciarcia R, Damiano S, Squillacioti C, Mirabella N, Pagnini U, Florio A, Severino L, Capasso G, Borrelli A, Mancini A, Boffo S, Romano G, Giordano A and Florio S, 2016. Recombinant mitochondrial manganese containing superoxide dismutase protects against ochratoxin A‐induced nephrotoxicity. Journal of Cellular Biochemistry, 117, 1352–1358. 10.1002/jcb.25425 [DOI] [PubMed] [Google Scholar]
- Cichna‐Markl M, 2011. New strategies in sample clean‐up for mycotoxin analysis. World Mycotoxin Journal, 4, 203–215. [Google Scholar]
- Cipriani V, Spertino S, Marsano F, Patrone M and Cavaletto M, 2011. Red wine prevents ochratoxin a damage in mouse kidney and liver. FEBS Journal, 278, 74–445. [Google Scholar]
- Copetti MV, Iamanaka BT, Mororo RC, Pereira JL, Frisvad JC and Taniwaki MH, 2012. The effect of cocoa fermentation and weak organic acids on growth and ochratoxin A production by Aspergillus species. International Journal of Food Microbiology, 155, 158–164. [DOI] [PubMed] [Google Scholar]
- Corcuera LA, Vettorazzi A, Arbillaga L, Pérez N, Gil AG, Azqueta A, González‐Peñas E, García‐Jalón JA and López de Cerain A, 2015. Genotoxicity of Aflatoxin B1 and Ochratoxin A after simultaneous application of the in vivo micronucleus and comet assay. Food and Chemical Toxicology, 76, 116–124. [DOI] [PubMed] [Google Scholar]
- Coronel MB, Sanchis V, Ramos AJ and Marin S, 2010. Review. Ochratoxin A: presence in human plasma and intake estimation. Food Science and Technology International, 16, 5–18. 10.1177/1082013209353359 [DOI] [PubMed] [Google Scholar]
- Coronel MB, Marin S, Tarragó M, Cano‐Sancho G, Ramos AJ and Sanchis V, 2011. Ochratoxin A and its metabolite ochratoxin alpha in urine and assessment of the exposure of inhabitants of Lleida, Spain. Food and Chemical Toxicology, 49, 1436–1442. 10.1016/j.fct.2011.03.03 [DOI] [PubMed] [Google Scholar]
- Cosimi S, Orta L, Mateos S and Cortés F, 2009. The mycotoxin ochratoxin A inhibits DNA topoisomerase II and induces polyploidy in cultured CHO cells. Toxicology In Vitro, 23, 1110–1115. [DOI] [PubMed] [Google Scholar]
- Costa S, Utan A, Cervellati R, Speroni E and Guerra MC, 2007. Catechins: natural free‐radicals scavengers against ochratoxin A‐induced cell damage in a pig kidney cell line (LLC‐PK1). Food and Chemical Toxicology, 45, 1910–1917. [DOI] [PubMed] [Google Scholar]
- Costa S, Cervellati R, Speroni E, Guerra M and Greco E, 2010. Free radicals and antioxidants in two oxidative‐stress cell models exposed to ochratoxin A and amyloid β: unexpected results. World Mycotoxin Journal, 3, 257–261. 10.3920/WMJ2010.1221 [DOI] [Google Scholar]
- Costa JG, Saraiva N, Guerreiro PS, Louro H, Silva MJ, Miranda JP, Castro M, Batinic‐Haberle I, Fernandes AS and Oliveira NG, 2016. Ochratoxin A‐induced cytotoxicity, genotoxicity and reactive oxygen species in kidney cells: an integrative approach of complementary endpoints. Food and Chemical Toxicology, 87, 65–76. [DOI] [PubMed] [Google Scholar]
- COT (Committee on Toxicity of Chemicals in Food Consumer Products and the Environment), 2018. Review of potential risks from ochratoxin A (OTA) in the diet of infants aged 0 to 12 months and children aged 1 to 5 years. Available online: https://cot.food.gov.uk/committee/committee-on-toxicity/cotstatements/cotstatementsyrs/cot-statements-2018/cot-statement-on-potential-risks-from-ochratoxin-a-ota-in-the-diet-of-infants-aged-0-to-12-months-and-children-aged-1-to-5-year
- Coton M, Auffret A, Poirier E, Debaets S, Coton E and Dantigny P, 2019. Production and migration of ochratoxin A and citrinin in Comté cheese by an isolate of Penicillium verrucosum selected among Penicillium spp. mycotoxin producers in YES medium. Food Microbiology, 82, 551–559. [DOI] [PubMed] [Google Scholar]
- Cramer B, Königs M and Humpf HU, 2008. Identification and in vitro cytogenicity of ochratoxin A degradation products formed during coffee roasting. Journal of Agricultural and Food Chemistry, 56, 5673–5681. [DOI] [PubMed] [Google Scholar]
- Cramer B, Osteresch B, Muñoz KA, Hillmann H, Sibrowski W and Humpf HU, 2015. Biomonitoring using dried blood spots: detection of ochratoxin A and its degradation product 2'R‐ochratoxin A in blood from coffee drinkers. Molecular Nutrition and Food Research, 59, 1837–1843. 10.1002/mnfr.201500220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Xing L, Li Z, Wu S, Wang J, Liu J, Wang J, Yan X and Zhang X, 2010. Ochratoxin A induces G(2) phase arrest in human gastric epithelium GES‐1 cells in vitro . Toxicology Letters, 193, 152–158. 10.1016/j.toxlet.2009.12.019 [DOI] [PubMed] [Google Scholar]
- Cui J, Liu J, Wu S, Wang Y, Shen H, Xing L, Wang J, Yan X and Zhang X, 2013. Oxidative DNA damage is involved in ochratoxin A‐induced G2 arrest through ataxia telangiectasia‐mutated (ATM) pathways in human gastric epithelium GES‐1 cells in vitro . Archives of Toxicology, 87, 1829–1840. [DOI] [PubMed] [Google Scholar]
- Czakai K, Muller K, Mosesso P, Pepe G, Schulze M, Gohla A, Patnaik D, Dekant W, Higgins J and Mally A, 2011. Perturbation of mitosis through inhibition of histone acetyltransferases: the key to ochratoxin A toxicity and carcinogenicity? Toxicological Sciences, 122, 317–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Park G, Wright MW, Adams M, Akman SA and Manderville RA, 2002. Detection and characterization of a glutathione conjugate of ochratoxin A. Chemical Research in Toxicology, 15, 1581–1588. [DOI] [PubMed] [Google Scholar]
- Dai Q, Zhao J, Qi X, Xu W, He X, Guo M, Dweep H, Cheng WH, Luo Y, Xia K, Gretz N and Huang K, 2014. MicroRNA profiling of rats with ochratoxin A nephrotoxicity. BMC Genomics, 15, 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dall'Asta C, De Dea Lindner J, Galaverna G, Dossena A, Neviani E and Marchelli R, 2008. The occurrence of ochratoxin A in blue cheese. Food Chemistry, 106, 729–734. [Google Scholar]
- Dall'Asta C, Galaverna G, Bertuzzi T, Moseriti A, Pietri A, Dossena A and Marchelli R, 2010. Occurrence of ochratoxin A in raw ham muscle, salami and dry‐cured ham from pigs fed with contaminated diet. Food Chemistry, 120, 978–983. [Google Scholar]
- Damiano S, Navas L, Lombari P, Montagnaro S, Forte IM, Giordano A, Florio S and Ciarcia R, 2018a. Effects of δ‐tocotrienol on ochratoxinA‐induced nephrotoxicity in rats. Journal of Cell Physiology, 233, 8731–8739. 10.1002/jcp.26753 [DOI] [PubMed] [Google Scholar]
- Damiano S, Puzio MV, Squillacioti C, Mirabella N, Zona E, Mancini A, Borrelli A, Astarita C, Boffo S, Giordano A, Avallone L, Florio S and Ciarcia R, 2018b. Effect of rMnSOD on sodium reabsorption in renal proximal tubule in ochratoxin A‐treated rats. Journal of Cell Biochemistry, 119, 424–430. 10.1002/jcb.26197 [DOI] [PubMed] [Google Scholar]
- Darif Y, Mountassif D, Belkebir A, Zaid Y, Basu K, Mourad W and Oudghiri M, 2016. Ochratoxin A mediates MAPK activation, modulates IL‐2 and TNF‐α mRNA expression and induces apoptosis by mitochondria‐dependent and mitochondria‐independent pathways in human H9 T cells. Journal of Toxicological Sciences, 41, 403–416. 10.2131/jts.41.403 [DOI] [PubMed] [Google Scholar]
- De Girolamo A, Solfrizzo M, Lattanzio VMT, Stroka J, Alldrick A, Van Egmond HP and Visconti A, 2013. Critical evaluation of LC‐MS‐based methods for simultaneous determination of deoxynivalenol, ochratoxin A, zearalenone, aflatoxins, fumonisins and T‐2/HT‐2 toxins in maize. World Mycotoxin Journal, 6, 317–334. [Google Scholar]
- Decontardi S, Mauro A, Lima N and Battilani P, 2017. Survey of Penicillia associated with Italian grana cheese. International Journal of Food Microbiology, 246, 25–31. [DOI] [PubMed] [Google Scholar]
- Degen GH, Muñoz K and Hengstler JG, 2013. Occurrence of mycotoxins in breast milk. In: Zibadi S, Watson RR and Preedy VR (eds). Handbook of Dietary and Nutritional Aspects of Human Breast Milk. Wageningen Academic Publishers, the Netherlands, 813–831. [Google Scholar]
- Degen GH, 2016. Are we ready to estimate daily ochratoxin A intake based on urinary concentrations? Environment International, 97, 254–255. 10.1016/j.envint.2015.10.010 [DOI] [PubMed] [Google Scholar]
- Delatour T, Mally A, Richoz J, Özden S, Dekant W, Ihmels H, Otto D, Gasparutto D, Marin‐Kuan M, Schilter B and Cavin C, 2008. Absence of 2’‐deoxyguanosine‐carbon 8‐bound ochratoxin A adduct in rat kidney DNA monitored by isotope dilution LC‐MS/MS. Molecular Nutrition and Food Research, 52, 472–482. [DOI] [PubMed] [Google Scholar]
- Desmarchelier A, Tessiot S, Bessaire T, Racault L, Fiorese E, Urbani A, Chan W‐C, Cheng P and Mottier P, 2014. Combining the quick, easy, cheap, effective, rugged and safe approach and clean‐up by immunoaffinity column for the analysis of 15 mycotoxins by isotope dilution liquid chromatography tandem mass spectrometry. Journal of Chromatography A, 1337, 75–84. [DOI] [PubMed] [Google Scholar]
- di Giuseppe R, Bertuzzi T, Rossi F, Rastelli S, Mulazzi A, Capraro J, de Curtis A, Iacoviello L and Pietri A, 2012. Plasma ochratoxin A levels, food consumption, and risk biomarkers of a representative sample of men and women from the Molise region in Italy. European Journal of Nutrition, 51, 851–860. [DOI] [PubMed] [Google Scholar]
- Diana Di Mavungu J, Monbaliu S, Scippo ML, Maghuin‐Rogister G, Schneider YJ, Larondelle Y, Callebaut A, Robbens J, Van Peteghem C and De Saeger S, 2009. LC‐MS/MS multi‐analyte method for mycotoxin determination in food supplements. Food Additives and Contaminants Part A, 26, 885–895. 10.1080/02652030902774649 [DOI] [PubMed] [Google Scholar]
- Dietrich DR, Heussner AH and O'Brien E, 2005. Ochratoxin A: comparative pharmacokinetics and toxicological implications (experimental and domestic animals and humans). Food Additives and Contaminants, Supplement, 1, 45–52. [DOI] [PubMed] [Google Scholar]
- Domijan AM, Zeljezić D, Kopjar N and Peraica M, 2006. Standard and Fpg‐modified comet assay in kidney cells of ochratoxin A‐ and fumonisinB (1)‐treated rats. Toxicology, 222, 53–59. [DOI] [PubMed] [Google Scholar]
- Domijan AM, Gajski G, Novak Jovanovic I, Geric M and Garaj‐Vrhova V, 2015. In vitro genotoxicity of mycotoxins ochratoxin A and fumonisin B1 could be prevented by sodium copper chlorophyllin – Implication to their genotoxic mechanism. Food Chemistry, 170, 455–462. [DOI] [PubMed] [Google Scholar]
- Dostal A, Jakusova L, Cajdova J and Hudeckova H, 2008. Results of the first studies of occurrence of ochratoxin A in human milk in Slovakia. Bratislavske lekarske listy, 109, 276–278. [PubMed] [Google Scholar]
- Drunday V and Pacin A, 2013. Occurrence of ochratoxin A in coffee beans, ground roasted coffee and soluble coffee and method validation. Food Control, 30, 675–678. [Google Scholar]
- Duarte SC, Pena A and Lino CM, 2010. Review on ochratoxin occurrence and effects of processing of cereal and cereal derived food products. Food Microbiology, 27, 187–198. 10.1016/j.fm.2009.11.016 [DOI] [PubMed] [Google Scholar]
- Duarte SC, Lino CM and Pena A, 2011. Ochratoxin A in feed of food‐producing animals: an undesirable mycotoxin with health and performance effects. Veterinary Microbiology, 154, 1–13. [DOI] [PubMed] [Google Scholar]
- Duarte SC, Lino CM and Pena A, 2013. Novel IAC‐LC‐ESI‐MS2 analytical set‐up for ochratoxin A determination in pork. Food Chemistry, 138, 1055–1061. [DOI] [PubMed] [Google Scholar]
- EFSA (European Food Safety Authority), 2005. Opinion of the Scientific Committee on a request from EFSA related to a harmonised approach for risk assessment of substances which are both genotoxic and carcinogenic. EFSA Journal 2005;3(10):282, 33 pp. 10.2903/j.efsa.2005.282 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2006. Opinion of the Scientific Panel on Contaminants in the Food Chain on a request from the Commission related to ochratoxin A in food. EFSA Journal 2006;4(6):365, 56 pp. 10.2903/j.efsa.2006.365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA (European Food Safety Authority), 2007. Guidance of the Scientific Committee on a request from EFSA related to Uncertainties in Dietary Exposure Assessment. EFSA Journal 2007;4(12):438, 54 pp. 10.2903/j.efsa.2007.438 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2009. Guidance of the Scientific Committee on transparency in the scientific aspects of risk assessments carried out by EFSA. Part 2: general principles. EFSA Journal 2009;7(5):1051, 22 pp. 10.2903/j.efsa.2009.1051 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2010a. Standard sample description for food and feed. EFSA Journal 2010;8(1):1457, 54 pp. 10.2903/j.efsa.2010.1457 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2010b. Management of left‐censored data in dietary exposure assessment of chemical substances. EFSA Journal 2010;8(3):1557, 96 pp. 10.2903/j.efsa.2010.1557 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2011a. Use of the EFSA Comprehensive European Food Consumption Database in Intakes Assessment. EFSA Journal 2011;9(3):2097, 34 pp. 10.2903/j.efsa.2011.2097 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2011b. Overview of the procedures currently used at EFSA for the assessment of dietary exposure to different chemical substances. EFSA Journal 2011;9(12):2490. 33 pp. 10.2903/j.efsa.2011.2490 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2011c. Evaluation of the FoodEx, the food classification system applied to the development of the EFSA Comprehensive European Food Consumption Database. EFSA Journal 2011;9(3):1970, 27 pp. 10.2903/j.efsa.2011.1970 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), Dujardin B and Kirwan L, 2019. Technical report on the raw primary commodity (RPC) model: strengthening EFSA's capacity to assess dietary exposure at different levels of the food chain, from raw primary commodities to foods as consumed. EFSA supporting publication 2019;EN‐1532, 30 pp. 10.2903/sp.efsa.2019 .EN‐ 1532 [DOI]
- EFSA (European Food Safety Authority), 2020. Outcome of a public consultation on the risk assessment of ochratoxin A in food. EFSA supporting publication 2020‐EN.1845. 10.2903/sp.efsa.2020.1845 [DOI] [Google Scholar]
- EFSA FEEDAP Panel (EFSA Panel on Additives and Products or Substances used in Animal Feed), 2012. Guidancefor the preparation of dossiers for sensory additives. EFSA Journal 2012;10(1):2534, 26 pp. 10.2903/j.efsa.2012.2534 [DOI] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies), 2013. Scientific Opinion on nutrient requirements and dietary intakes of infants and young children in the European Union. EFSA Journal 2013;11(10):3408, 103 pp. 10.2903/j.efsa.2013.3408 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2011. Scientific Opinion on genotoxicity testing strategies applicable to food and feed safety assessment. EFSA Journal 2011;9(9):2379, 69 pp. 10.2903/j.efsa.2011.2379 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2012a. Guidance on selected default values to be used by the EFSA Scientific Committee, Scientific Panels and Units in the absence of actual measured data. EFSA Journal 2012;10(3):2579, 32 pp. 10.2903/j.efsa.2012.2579 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2012b. Scientific Opinion on Risk Assessment Terminology. EFSA Journal 2012;10(5):2664, 43 pp. 10.2903/j.efsa.2012.2664 [DOI] [Google Scholar]
- EFSA Scientific Committee , Hardy A, Benford D, Halldorsson T, Jeger MJ, Knutsen KH, More S, Mortensen A, Naegeli H, Noteborn H, Ockleford C, Ricci A, Rychen G, Silano V, Solecki R, Turck D, Aerts M, Bodin L, Davis A, Edler L, Gundert‐Remy U, Sand S, Slob W, Bottex B, Abrahantes JC, Marques DC, Kass G and Schlatter JR, 2017. Update: use of the benchmark dose approach in risk assessment. EFSA Journal 2017;15(1):4658, 41 pp. 10.2903/j.efsa.2017.4658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , Benford D, Halldorsson T, Jeger MJ, Knutsen HK, More S, Naegeli H, Noteborn H, Ockleford C, Ricci A, Rychen G, Schlatter JR, Silano V, Solecki R, Turck D, Younes M, Craig P, Hart A, Von Goetz N, Koutsoumanis K, Mortensen A, Ossendorp B, Martino L, Merten C, Mosbach‐Schulz O and Hardy A, 2018. Guidance on uncertainty analysis in scientific assessments. EFSA Journal 2018;16(1):5123, 39 pp. 10.2903/j.efsa.2018.5123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Adlouni C, Pinelli E, Azemar B, Zaoui D, Beaune P and Pfohl‐Leszkowicz A, 2000. Phenobarbital increases DNA adduct and metabolites formed by ochratoxin A: Role of CYP 2C9 and microsomal glutathione‐S‐transferase. Environmental and Molecular Mutagenesis, 35, 123–131. [DOI] [PubMed] [Google Scholar]
- El Golli Bennour E, Bouaziz C, Ladjimi M, Renaud F and Bacha H, 2009. Comparative mechanisms of zearalenone and ochratoxin A toxicities on cultured HepG2 cells: is oxidative stress a common process? Environmental Toxicology, 24, 538–548. 10.1002/tox.20449 [DOI] [PubMed] [Google Scholar]
- El Khoury A and Atoui A, 2010. Ochratoxin A: general overview and actual molecular status. Toxins, 2, 461–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Haleem MR, Kattaia AA, El‐Baset SA and Mostafa Hel S, 2016. Alleviative effect of myricetin on ochratoxin A‐induced oxidative stress in rat renal cortex: histological and biochemical study. Histology and Histopathology, 31, 441–451. 10.14670/hh-11-689 [DOI] [PubMed] [Google Scholar]
- Elling F, 1979a. Enzyme histochemical studies of ochratoxin A‐induced mycotoxic porcine nephropathy. 17 pp. [DOI] [PubMed] [Google Scholar]
- Elling F, 1979b. Ochratoxin A‐induced mycotoxic porcine nephropathy: alterations in enzyme activity in tubular cells. Acta Pathologica, Microbiologica, et Immunologica Scandinavica, 87, 237–243. [DOI] [PubMed] [Google Scholar]
- Enciso JM, López de Cerain A, Pastor L, Azqueta A and Vettorazzi A, 2018. Is oxidative stress involved in the sex‐dependent response to ochratoxin A renal toxicity? Food and Chemical Toxicology, 116, 379–387. [DOI] [PubMed] [Google Scholar]
- Essid E and Petzinger E, 2011. Silibinin pretreatment protects against ochratoxin A‐mediated apoptosis in primary rat hepatocytes. Mycotoxin Research, 27, 167–176. 10.1007/s12550-011-0092-9 [DOI] [PubMed] [Google Scholar]
- Essid E, Dernawi Y and Petzinger E, 2012. Apoptosis induction by OTA and TNF‐α in cultured primary rat hepatocytes and prevention by silibinin. Toxins, 4, 1139–1156. 10.3390/toxins4111139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- European Community , 2002. Assessment of dietary intake of ochratoxin A by the population of EU Member States. Report of the Scientific Cooperation, Task 3.2.7. Directorate‐General Health and Consumer Protection, European Commission. Available online: europa.eu.int/comm/food/fs/scoop/3.2.7_en.pdf
- FAO (Food and Agricultural Organization of the United Nations), 2013. Code of practice for the prevention and reduction of ochratoxin A contamination in cocoa. Available online: http://www.fao.org/fao-who-codexalimentarius/sh-proxy/en/?lnk=1&url=https%253A%252F%252Fworkspace.fao.org%252Fsites%252Fcodex%252FStandards%252FCXC%2B72-2013%252FCXP_072e.pdf [Google Scholar]
- FAO/WHO (Food an Agricultural Organization/World Health Organization), 1991. Evaluation of certain food additives and contaminants (Thirty‐seventh report of the Joint FAO/WHO Expert Committee on Food Additives). WHO Technical Report Series, No. 806 and corrigenda. [PubMed]
- FAO/WHO (Food and Agriculture Organisation/World Health Organisation), 1996. Toxicological evaluation of certain food additives and contaminants. Joint FAO/WHO Expert Committee on Food Additives (JECFA). WHO Food Additive Series 35. World Health Organisation, Geneva, Switzerland.
- FAO/WHO (Food and Agriculture Organisation/World Health Organisation), 2001. Ochratoxin A. In: Safety evaluation of certain mycotoxins in food, Prepared by the 56th Meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA). WHO Food Additives Series 47, 281–387. World Health Organisation, Geneva, Switzerland.
- FAO/WHO (Food an Agricultural Organization/World Health Organization), 2008. Safety evaluation of certain food additives and contaminants. Prepared by the sixty eighth Meeting of the Joint FAO/WHO Expert Committee on Food Additives). WHO Food Additives Series 59. Ochratoxin A (addendum), 357‐ 426.
- Faucet‐Marquis V, Pont F, Stormer FC, Rizk T, Castegnaro M and Pfohl‐Leszkowicz A, 2006. Evidence of a new dechlorinated ochratoxin A derivative formed in opossum kidney cell cultures after pretreatment by modulators of glutathione pathways: correlation with DNA‐adduct formation. Molecular Nutrition and Food Research, 50, 530–542. [DOI] [PubMed] [Google Scholar]
- Fazekas B, Tar A and Kovács M, 2005. Aflatoxin and ochratoxin A content of spices in Hungary. Food Additives and Contaminants, 22, 856–863. [DOI] [PubMed] [Google Scholar]
- Ferrante MC, Bilancione M, Raso GM, Esposito E, Iacono A, Zaccaroni A and Meli R, 2006. Expression of COX‐2 and hsp72 in peritoneal macrophages after an acute ochratoxin A treatment in mice. Life Sciences, 79, 1242–1247. [DOI] [PubMed] [Google Scholar]
- Ferraz MBM, Farah A, Iamanaka BT, Perrone D, Copetti MV, Marques VX, Alfredo A, Vitali M and Taniwaki H, 2010. Kinetics of ochratoxin A destruction during coffee roasting. Food Control, 21, 872–877. [Google Scholar]
- Fink‐Gremmels J, 2008. Mycotoxins in cattle feed and carry‐over to dairy milk: a review. Food Additives and Contaminants, 25, 172–180. [DOI] [PubMed] [Google Scholar]
- Föllmann W, Behm C and Degen GH, 2007. Induction of micronuclei by ochratoxin A is a sensitive parameter of its genotoxicity in cultured cells. Mycotoxin Research, 23, 101–109. 10.1007/BF02946034 [DOI] [PubMed] [Google Scholar]
- Frizzell C, Verhaegen S, Ropstad E, Elliott CT and Connolly L, 2013. Endocrine disrupting effects of ochratoxin A at the level of nuclear receptor activation and steroidogenesis. Toxicology Letters, 217, 243–250. 10.1016/j.toxlet.2012.12.018 [DOI] [PubMed] [Google Scholar]
- Fuchs R, Hult K, Peraica M, Radic B and Plestina R, 1984. Conversion of ochratoxin C into ochratoxin A in vivo . Applied and Environmental Microbiology, 48, 41–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs R, Appelgren L‐E and Hult K, 1986. Distribution of 14Cochratoxin A in the rainbow trout (Salmo gairdneri). Acta Pharmacologica et Toxicologica, 59, 220–227. [DOI] [PubMed] [Google Scholar]
- Fuchs R and Hult K, 1992. Ochratoxin A in blood and its pharmacokinetic properties. Food Chemistry and Toxicology, 30, 201–204. [DOI] [PubMed] [Google Scholar]
- Fusi E, Rebucci R, Pecorini C, Rossi L, D'Ambrosio F and Baldi A, 2008. Evaluation of the damage induced by ochratoxin A and the protective role of alpha‐tocopherol in cultured bovine mammary epithelial cells. Veterinary Research Communications, 32(Suppl 1), S343–S345. 10.1007/s11259-008-9144-9 [DOI] [PubMed] [Google Scholar]
- Fusi E, Rebucci R, Pecorini C, Campagnoli A, Pinotti L, Saccone F, Cheli F, Purup S, Sejrsen K and Baldi A, 2010. Alpha‐tocopherol counteracts the cytotoxicity induced by ochratoxin A in primary porcine fibroblasts. Toxins (Basel), 2, 1265–1278. 10.3390/toxins2061265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusi E, Giromini C, Rebucci R, Pinotti L, Caprarulo V, Cheli F, Vitari F, Domeneghini C and Baldi A, 2018. Ochratoxin A cytotoxicity on Madin‐Darby canine kidney cells in the presence of alpha‐tocopherol: effects on cell viability and tight junctions. Journal of Animal Physiology and Animal Nutrition (Berl), 102, 350–355. 10.1111/jpn.12682 [DOI] [PubMed] [Google Scholar]
- Gagliano N, Donne ID, Torri C, Migliori M, Grizzi F, Milzani A, Filippi C, Annoni G, Colombo P, Costa F, Ceva‐Grimaldi G, Bertelli AA, Giovannini L and Gioia M, 2006. Early cytotoxic effects of ochratoxin A in rat liver: a morphological, biochemical and molecular study. Toxicology, 225, 214–224. [DOI] [PubMed] [Google Scholar]
- Galtier P, Charpenteau JL, Alvinerie M and Labouche C, 1979. The pharmacokinetic profile of ochratoxin A in the rat after oral and intravenous administration. Drug Metablism and Disposition, 7, 429–434. [PubMed] [Google Scholar]
- Galtier P, Alvinerie M and Charpenteau JL, 1981. The pharmacokinetic profiles of ochratoxin A in pigs, rabbits and chickens. Food and Cosmetics Toxicology, 19, 735–738. [DOI] [PubMed] [Google Scholar]
- Galtier P, 1991. Pharmacokinetics of ochratoxin A in animals. IARC Scientific Publications, 115, 187–200. [PubMed] [Google Scholar]
- Galvano F, Pietri A, Bertuzzi T, Gagliardi L, Ciotti S, Luisi S, Bognanno M, La Fauci L, Iacopino AM, Nigro F, Li Volti G, Vanella L, Giammanco G, Tina GL and Gazzolo D, 2008. Maternal dietary habits and mycotoxin occurrence in human mature milk. Molecular Nutrition and Food Research, 52, 496–501. 10.1002/mnfr.200700266 [DOI] [PubMed] [Google Scholar]
- Gambacorta L, Magistà D, Perrone G, Murgolo S, Logrieco AF and Solfrizzo M, 2018. Co‐occurrence of toxigenic moulds, aflatoxins, ochratoxin A, Fusarium and Alternaria mycotoxins in fresh sweet peppers (Capsicum annuum) and their processed products. World Mycotoxin Journal., 2018, 159–174. [Google Scholar]
- Gan F, Xue H, Huang Y, Pan C and Huang K, 2015. Selenium alleviates porcine nephrotoxicity of ochratoxin A by improving selenoenzyme expression in vitro . PLoS ONE, 10, e0119808. 10.1371/journal.pone.0119808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan F, Hu Z, Huang Y, Xue H, Huang D, Qian G, Hu J, Chen X, Wang T and Huang K, 2016. Overexpression of pig selenoprotein S blocks OTA‐induced promotion of PCV2 replication by inhibiting oxidative stress and p38 phosphorylation in PK15 cells. Oncotarget, 7, 20469–20485. 10.18632/oncotarget.7814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan F, Hu Z, Zhou Y and Huang K, 2017a. Overexpression and low expression of selenoprotein S impact Ochratoxin A‐induced porcine cytotoxicity and apoptosis in vitro . Journal of Agricultural and Food Chemistry, 65, 6972–6981. 10.1021/acs.jafc.7b02115 [DOI] [PubMed] [Google Scholar]
- Gan F, Hou LL, Zhou YJ, Liu YH, Huang D, Chen XX and Huang KH, 2017b. Effects of ochratoxin A on ER stress, MAPK signaling pathway and autophagy of kidney and spleen in pigs. Environmental Toxicology, 32, 2277–2286. [DOI] [PubMed] [Google Scholar]
- Gan F, Zhou Y, Hou L, Qian G, Chen X and Huang K, 2017c. Ochratoxin A induces nephrotoxicity and immunotoxicity through different MAPK signaling pathways in PK15 cells and porcine primary splenocytes. Chemosphere, 182, 630–637. 10.1016/j.chemosphere.2017.05.030. [DOI] [PubMed] [Google Scholar]
- Gautier JC, Richoz J, Welti DH, Markovic J, Gremaud E, Guengerich FP and Turesky RJ, 2001. Metabolism of ochratoxin A: absence of formation of genotoxic derivatives by human and rat enzymes. Chemical Research in Toxicology, 14, 34–45. [DOI] [PubMed] [Google Scholar]
- Gayathri L, Dhivya R, Dhanasekaran D, Periasamy VS, Alshatwi AA and Akbarsha MA, 2015. Hepatotoxic effect of ochratoxin A and citrinin, alone and in combination, and protective effect of vitamin E: in vitro study in HepG2 cell. Food and Chemical Toxicology, 83, 151–163. 10.1016/j.fct.2015.06.009 [DOI] [PubMed] [Google Scholar]
- Gelot C, Indiana M and Lopez BS, 2015. Replication stress in mammalian cells and its consequences for mitosis. Genes, 6, 267–298. 10.3390/genes6020267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifford FJ, Gifford RM, Eddleston M and Neeraj D, 2017. Endemic nephropathy around the world. Kidney International Reports, 2, 282–292. 10.1016/j.ekir.2016.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillman IG, Clark TN and Manderville RA, 1999. Oxidation of ochratoxin A by an Fe‐porphyrin system: model for enzymatic activation and DNA cleavage. Chemical Research in Toxicology, 12, 1066–1076. [DOI] [PubMed] [Google Scholar]
- Giromini C, Rebucci R, Fusi E, Rossi L, Saccone F and Baldi A, 2016. Cytotoxicity, apoptosis, DNA damage and methylation in mammary and kidney epithelial cell lines exposed to ochratoxin A. Cell Biology and Toxicology, 32, 249–258. [DOI] [PubMed] [Google Scholar]
- González‐Arias CA, Benitez‐Trinidad AB, Sordo M, Robledo‐Marenco L, Medina‐Díaz IM, Barrón‐Vivanco BS, Marín S, Sanchis V, Ramos AJ and Rojas‐García AE, 2014. Low doses of ochratoxin A induce micronucleus formation and delay DNA repair in human lymphocytes. Food and Chemical Toxicology, 74, 249–254. [DOI] [PubMed] [Google Scholar]
- González‐Arias CA, Marín S, Rojas‐García AE, Sanchis V and Ramos AJ, 2017. UPLC‐MS/MS analysis of ochratoxin A metabolites produced by Caco‐ and HepG2 cells in a co‐culture system. Food and Chemical Toxicology, 109(Pt 1), 333–340. 10.1016/j.fct.2017.09.011 [DOI] [PubMed] [Google Scholar]
- Gottschalk C, Biermaier B, Gross M, Schwaiger K and Gareis M, 2016. Ochratoxin A in brewer's yeast used as food supplement. Mycotoxin Research, 32, 1–5. 10.1007/s12550-015-0230-x [DOI] [PubMed] [Google Scholar]
- Goyary D, Chattopadhyay P, Giri S, Aher V, Upadhyay A and Veer V, 2014. Ochratoxin A induces cytotoxicity, DNA damage and apoptosis in rat hepatocyte primary cell culture at nanomolar concentration. World Mycotoxin Journal, 7, 379–386. [Google Scholar]
- Grosse Y, Baudrimont I, Castegnaro M, Betbeder A‐M, Ekué Creppy E, Dirheimer G and Pfohl‐Leszkowicz A, 1995. Formation of ochratoxin A metabolites and DNA‐adducts in monkey kidney cells. Chemico‐Biological Interactions, 95, 175–187. [DOI] [PubMed] [Google Scholar]
- Gross‐Steinmeyer K, Weymann J, Hege HG and Metzler M, 2002. Metabolism and lack of DNA reactivity of the mycotoxin ochratoxin A in cultured rat and human hepatocytes. Journal of Agricultural and Food Chemistry, 50, 938–945. [DOI] [PubMed] [Google Scholar]
- Guo M, Huang K, Chen S, Qi X, He X, Cheng WH, Luo Y, Xia K and Xu W, 2014. Combination of metagenomics and culture‐based methods to study the interaction between ochratoxin A and gut microbiota. Toxicological Sciences, 141, 314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas D, Pfeifer B, Reiterich C, Partenheimer R, Reck B and Buzina W, 2013. Identification and quantification of fungi and mycotoxins from Pu‐erh tea. International Journal of Food Microbiology, 166, 316–322. [DOI] [PubMed] [Google Scholar]
- Hadjeba‐Medjdoub K, Tozlovanu M, Pfohl‐Leszkowicz A, Frenette C, Paugh RJ and Manderville RA, 2012. Structure‐activity relationships imply different mechanisms of action for ochratoxin A‐mediated cytotoxicity and genotoxicity. Chemical Research in Toxicology, 25, 181–190. [DOI] [PubMed] [Google Scholar]
- Hagelberg S, Hult K and Fuchs R, 1989. Toxicokinetics of ochratoxin A in several species and its plasma‐binding properties. Journal of Applied Toxicology, 9, 91–96. [DOI] [PubMed] [Google Scholar]
- Ham H, Kim S, Kim MH, Lee S, Hong SK, Ryu JG and Lee T, 2016. Mycobiota of ground red pepper and their aflatoxigenic potential. Journal of Microbiology, 54, 832–837. [DOI] [PubMed] [Google Scholar]
- Han Z, Zhao Z, Shi J, Liao Y, Zhao Z, Zhang D Wu Y, De Saeger S and Wu A, 2013a. Combinatorial approach of LC‐MS/MS and LC‐TOF‐MS for uncovering in vivo kinetics and biotransformation of ochratoxin A in rat. Journal of Chromatography B, 925, 46–53. [DOI] [PubMed] [Google Scholar]
- Han Z, Tangni EK, Di Mavungu JD, Vanhaecke L, De Saeger S, Wu A and Callebaut A, 2013b. In vitro glucuronidation of ochratoxin A by rat liver microsomes. Toxins, 5, 2671–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harwig J, Kuiper‐Goodman T and Scott PM, 1983. Microbial food toxicants: ochratoxins. In: Rechcigl M (ed). Handbook of Foodborne Diseases of Biological Origin. CRC Press, Boca Raton, FL. pp. 193–238. [Google Scholar]
- Hashimoto Y, Katsunuma Y, Nunokawa M, Minato H and Yonemochi C, 2016. Influence of repeated ochratoxin A ingestion on milk production and its carry‐over into the milk, blood and tissues of lactating cows. Animal Science Journal, 87, 541–546. [DOI] [PubMed] [Google Scholar]
- Hasinoff BB, Rahimtula AD and Omar RF, 1990. NADPH‐cytochrome‐P‐450 reductase promoted hydroxyl radical production by the iron(III)‐ochratoxin A complex. Biochimica et Biophysica Acta, 1036, 78–81. [DOI] [PubMed] [Google Scholar]
- Hassan AM, Sheashaa HA, Fattah MF, Ibrahim AZ, Gaber OA and Sobh MA, 2016. Study of ochratoxin A as an environmental risk that causes renal injury in breast‐fed Egyptian infants. Pediatric Nephrology, 21, 102–105. [DOI] [PubMed] [Google Scholar]
- Hassen W, Abid S, Achour A, Creppy EE and Bacha H, 2004. Ochratoxin A and ‐2 microglobulinuria in healthy individuals and in chronic interstitial nephropathy patients in the Centre of Tunisia: a hot spot of Ochratoxin A exposure. Toxicology, 199, 185–193. [DOI] [PubMed] [Google Scholar]
- Hautbergue T, Puel O, Tadrist S, Meneghetti L, Péan M, Delaforge M, Debrauwer L, Oswald IP and Jamin EL, 2017. Evidencing 98 secondarymetabolites of Penicillium verrucosum using substrate isotopic labeling and high‐resolution mass spectrometry. Journal of Chromatography B Analytical Technologies in the Biomedical and Life Sciences , 1071, 29–43. 10.1016/j.jchromb.2017.03.011 [DOI] [PubMed] [Google Scholar]
- Hayashi MT and Karlseder J, 2013. DNA damage associated with mitosis and cytokinesis failure. Oncogene, 32, 4593–4601. 10.1038/onc.2012.615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennemeier I, Humpf HU, Gekle M and Schwerdt G, 2012. The food contaminant and nephrotoxin ochratoxin A enhances Wnt1 inducible signaling protein 1 and tumor necrosis factor‐α expression in human primary proximal tubule cells. Molecular Nutrition and Food Research, 56, 1375–1384. 10.1002/mnfr.201200164 [DOI] [PubMed] [Google Scholar]
- Hennemeier I, Humpf HU, Gekle M and Schwerdt G, 2014. Role of microRNA‐29b in the ochratoxin A‐induced enhanced collagen formation in human kidney cells. Toxicology, 3, 116–122. 10.1016/j.tox.2014.07.012 [DOI] [PubMed] [Google Scholar]
- Heussner AH and Bingle LEH, 2015. Comparative ochratoxin toxicity: a review of the available data. Toxins, 7, 4253–4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heussner AH, O'Brien E and Dietrich DR, 2002. Species‐ and sex‐specific variations in binding of ochratoxin A by renal proteins in vitro . Experimental and Toxicologic Pathology, 54, 151–159. [DOI] [PubMed] [Google Scholar]
- Hibi D, Suzuki Y, Ishii Y, Jin M, Watanabe M, Sugita‐Konishi Y, Yanai T, Nohmi T, Nishikawa A and Umemura T, 2011. Site‐specific in vivo mutagenicity in the kidney of gpt delta rats given a carcinogenic dose of ochratoxin A. Toxicological Sciences, 122, 406–414. [DOI] [PubMed] [Google Scholar]
- Hibi D, Kijima A, Kuroda K, Suzuki Y, Ishii Y, Jin M, Nakajima M, Sugita‐Konishi Y, Yanai T, Nohmi T, Nishikawa A and Umemura T, 2013a. Molecular mechanisms underlying ochratoxin A‐induced genotoxicity: global gene expression analysis suggests induction of DNA double‐strand breaks and cell cycle progression. Journal of Toxicological Sciences, 38, 57–69. [DOI] [PubMed] [Google Scholar]
- Hibi D, Kijima A, Suzuki Y, Ishii Y, Jin M, Sugita‐Konishi Y, Yanai T, Nishikawa A and Umemura T, 2013b. Effects of p53 knockout on ochratoxin A‐induced genotoxicity in p53‐deficient gpt delta mice. Toxicology, 304, 92–99. [DOI] [PubMed] [Google Scholar]
- Hierro JMH, Garcia‐Villanova J, Torreno P and Fonseca IMT, 2008. Aflatoxins and ochratoxin A in red paprika for retail sale in Spain: occurrence and evaluation of a simultaneous analytical method. Journal of Agricultural and Food Chemistry, 56, 751–756. [DOI] [PubMed] [Google Scholar]
- Hmaissia Khlifa K, Ghali R, Mezigh C, Aouni Z, Ghorbel H, Harrzallah K, Machgoul S and Hedhili A, 2008. Serum levels of ochratoxin A in healthy subjects and in nephropathic patients in Tunisia. Annales de biologie clinique (Paris), 66, 631–636. 10.1684/abc.2008.0278 [DOI] [PubMed] [Google Scholar]
- Hmaissia Khlifa K, Ghali R, Mazigh C, Aouni Z, Machgoul S and Hedhili A, 2012. Ochratoxin A levels in human serum and foods from nephropathy patients in Tunisia: where are you now? Experimental and Toxicologic Pathology, 64, 509–512. 10.1016/j.etp.2010.11.006 [DOI] [PubMed] [Google Scholar]
- Hoffmann D, Adler M, Vaidya V, Rached E, Mulrane L, Gallagher WM, Callanan JJ, Gautier JC, Matheis K, Staedtler F, Dieterle F, Brandenburg A, Sposny A, Hewitt P, Ellinger‐Ziegelbauer H, Bonventre JV, Dekant W and Mally A, 2010. Performance of novel kidney biomarkers in preclinical toxicity studies. Toxicological Sciences, 116, 8–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hort V, Nicolas M, Minvielle B, Maleix C, Desbourdes C, Hommet F, Dragacci S, Dervilly‐Pinel G, Engel E and Guérin T, 2018. Ochratoxin A determination in swine muscle and liver from French conventional or organic farming production systems. Journal of Chromatography B: Analytical Technologies in the Biomedical and Life Sciences, 1092, 131–137. [DOI] [PubMed] [Google Scholar]
- Hsuuw YD, Chan WH and Yu JS, 2013. Ochratoxin A inhibits mouse embryonic development by activating a mitochondrion‐dependent apoptotic signaling pathway. International Journal of Molecular Sciences, 14, 935–953. 10.3390/ijms14010935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang FJ and Chan WH, 2016. Effects of ochratoxin A on mouse oocyte maturation and fertilization, and apoptosis during foetal development. Environmental Toxicology, 31, 724–735. 10.1002/tox.22085 [DOI] [PubMed] [Google Scholar]
- Huff JE, 1991. Carcinogenicity of ochratoxin A in experimental animals. IARC Scientific Publications, 115, 229–244. [PubMed] [Google Scholar]
- Hult K, Hokby E, Hagglund U, Gatenbeck S, Rutqvist L and Sellyey G, 1979. Ochratoxin A in pig blood: method of analysis and use as a tool for feed studies. Applied and Environmental Microbiology, 38, 772–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hundhausen C, Boesch‐Saadatmandi C, Matzner N, Lang F, Blank R, Wolffram S, Blaschek W and Rimbach G, 2008. Ochratoxin a lowers mRNA levels of genes encoding for key proteins of liver cell metabolism. Cancer Genomics & Proteomics, 5, 319–332. [PubMed] [Google Scholar]
- Huybrechts I, Sioen I, Boon PE, Ruprich J, Lafay L, Turrini A, Amiano P, Hirvonen T, De Neve M, Arcella D, Moschandreas J, Westerlund A, Ribas‐Barba L, Hilbig A, Papoutsou S, Christensen T, Oltarzewski M, Virtanen S, Rehurkova I, Azpiri M, Sette S, Kersting M, Walkiewicz A, Serra‐Majem L, Volatier J‐L, Trolle E, Tornaritis M, Busk L, Kafatos A, Fabiansson S, De Henauw S and Van Klaveren JD, 2011. Dietary exposure assessments for children in Europe (the EXPOCHI project): rationale, methods and design. Archives of Public Health, 69, 4. 10.1186/0778-7367-69-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim AS, Zaghloul H and Badria FA, 2013. Case report evidence of relationships between hepatocellular carcinoma and ochratoxicosis, 2013. PLoS ONE, 8, e71423. 10.1371/journal.pone.0071423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal Q, Amjad M, Asi MR and Arin A, 2011. Assessment of hot peppers for aflatoxin and mold proliferate during storage. Journal of Food Prottection, 74, 830–835. [DOI] [PubMed] [Google Scholar]
- Iqbal SZ, Asi MR, Zuber M, Akhtar J and Saif MJ, 2013. Natural occurrence of aflatoxins and ochratoxin A in commercial chilli and chilli sauce samples. Food Control, 2, 621–625. [Google Scholar]
- Jadot I, Declèves A‐E, Nortier J and Caron N, 2017. An Integrated view of aristolochic acid nephropathy: update of the literature. International Journal of Molecular Sciences, 18, 297. 10.3390/ijms18020297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalali S and Wohlin C, 2012. Systematic literature studies: database searches vs. backward snowballing. 6th ACM‐IEEE International Symposium on Empirical Software Engineering and Measurement Lund. Available online: http://www.diva-portal.org/smash/get/diva2:834640/FULLTEXT01.pdf
- Jalili M and Jinap S, 2012. Natural occurrence of aflatoxins and ochratoxin A in commercial dried chili. Food Control, 24, 160–164. [Google Scholar]
- Jan R, Sadique U, Hassan ZU, Farid K, Ahmad S, Khan S and Khan H, 2017. Toxico‐pathological and reproductive effects of concurrent oral administration of ochratoxin A and endosulfan in pregnant rabbits (Oryctolagus cuniculus). Pakistan Veterinary Journal, 37, 19–24. [Google Scholar]
- Jennings P, Weiland C, Limonciel A, Bloch KM, Radford R, Aschauer L, McMorrow T, Wilmes A, Pfaller W, Ahr HJ, Slattery C, Lock EA, Ryan MP and Ellinger‐Ziegelbauer H, 2012. Transcriptomic alterations induced by Ochratoxin A in rat and human renal proximal tubular in vitro models and comparison to a rat in vivo model. Archives of Toxicology, 86, 571–589. 10.1007/s00204-011-0780-4 [DOI] [PubMed] [Google Scholar]
- Jutabha P, Anzai N, Hayashi K, Domae M, Uchida K, Endou H and Sakurai H, 2011. A novel human organic anion transporter NPT4 mediates the transport of ochratoxin A. Journal of Pharmacological Sciences, 116, 392–396. [DOI] [PubMed] [Google Scholar]
- Kamp HG, Eisenbrand G, Janzowski C, Kiossev J, Latendresse JR, Schlatter J and Turesky RJ, 2005. Ochratoxin A induces oxidative DNA damage in liver and kidney after oral dosing to rats. Molecular Nutrition and Food Research, 49, 1160–1167. [DOI] [PubMed] [Google Scholar]
- Kanisawa M and Suzuki S, 1978. Induction of renal and hepatic tumors in mice by ochratoxin A, a mycotoxin. Gan to Kagaku Ryoho, 69, 599–600. [PubMed] [Google Scholar]
- Karwowska W, Pierzynowska J, Janicki A, WaszkiewiczRobak B and Przybylska A, 2004. Qualitative and quantitative analysis of filamentous fungi in air, food and ochratoxin A in human milk. Polish journal of food and nutrition sciences, 13(Suppl. 2), 41–44. [Google Scholar]
- Kathuria P, Sharma P, Manderville RA and Wetmore SD, 2017. Molecular modeling of the major DNA adduct formed from food mutagen ochratoxin a in nari two‐base deletion duplexes: Impact of sequence context and adduct ionization on conformational preference and mutagenicity. Chemical Research in Toxicology, 30, 1582–1591. [DOI] [PubMed] [Google Scholar]
- Kathuria P, Sharma P, Manderville RA and Wetmore SD, 2018. Molecular dynamics simulations of mismatched DNA duplexes associated with the major c8‐linked 2’‐deoxyguanosine adduct of the food mutagen ochratoxin A: influence of opposing base, adduct ionization state, and sequence on the structure of damaged DNA. Chemical Research in Toxicology, 31, 712–720. [DOI] [PubMed] [Google Scholar]
- Khaneghah AM, Fakhri Y, Abdi L, Coppa CFSC, Franco LT and de Oliveira CAF, 2019. The concentration and prevalence of ochratoxin A in coffee and coffee‐based products: a global systematic review, meta‐analysis and meta‐regression. Fungal Biology, 123, 611–617. [DOI] [PubMed] [Google Scholar]
- Kiessling KH, Pettersson H, Sandholm K and Olsen M, 1984. Metabolism of aflatoxin, ochratoxin, zearalenone, and three trichothecenes by intact rumen fluid, rumen protozoa, and rumen bacteria. Applied Environmental Microbiology, 47, 1070–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapec T, Šarkanj B, Banjari I and Strelec I, 2012. Urinary ochratoxin A and ochratoxin alpha in pregnant women. Food and Chemical Toxicology, 50, 4487–4492. [DOI] [PubMed] [Google Scholar]
- Klarić MS, Pepeljnjak S and Rozgaj R, 2008. Genotoxicity of fumonisin B1, Beauvericin and Ochratoxin A in Porcine Kidney PK15 cells: effects of individual and combined treatment. Croatica Chemica Acta, 81, 139–146. [DOI] [PubMed] [Google Scholar]
- Klarić MS, Darabos D, Rozgaj R, Kasuba V and Pepeljnjak S, 2010. Beauvericin and ochratoxin A genotoxicity evaluated using the alkaline comet assay: single and combined genotoxic action. Archives of Toxicology, 84, 641–650. [DOI] [PubMed] [Google Scholar]
- Klarić MS, Medić N, Hulina A, Grubišić TZ and Rumora L, 2014. Disturbed Hsp70 and Hsp27 expression and thiol redox status in porcine kidney PK15 cells provoked by individual and combined ochratoxin A and citrinin treatments. Food and Chemical Toxicology, 71, 97–105. 10.1016/j.fct.2014.06.002 [DOI] [PubMed] [Google Scholar]
- Kokina A, Pugajeva I and Bartkevics V, 2016. Improved sensitivity of ochratoxin A analysis in coffee using high‐performance liquid chromatography with hybrid triple quadrupole‐linear ion trap mass spectrometry (LC‐QqQLIT‐MS/MS). Food Additives and Contaminants ‐ Part A Chemistry, Analysis, Control, Exposure and Risk Assessment, 33, 693–702. [DOI] [PubMed] [Google Scholar]
- Köszegi T and Poor M, 2016. Ochratoxin A: Molecular interactions, mechanisms of toxicity and prevention at the molecular level. Toxins, 8, 111. 10.3390/toxins8040111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh P, Axelsen NH, Elling F, Gyrd‐Hansen N, Hald B, Hyldgaard‐Jensen J, Larsen AE, Madsen A, Mortensen HP, Moller T, Petersen OK, Ravnskov U, Rostgaard M and Aalund O, 1974. Experimental porcine nephropathy. Changes of renal function and structure induced by ochratoxin A‐ contaminated feed. Acta Pathologica, Microbiologica, et Immunologica Scandinavica [A], 1–21. [PubMed] [Google Scholar]
- Krogh P, Elling F, Gyrd‐Hansen N, Hald B, Larsen AE, Lillehoj EB, Madsen A, Mortensen HP and Ravnskov U, 1976. Experimental porcine nephropathy: changes of renal function and structure perorally induced by crystalline ochratoxin. A. Acta Pathologica, Microbiologica, et Immunologica Scandinavica [A], 84, 429–434. [PubMed] [Google Scholar]
- Krogh P, Elling F, Friis C, Hald B, Larsen AE, Lillehoj EB, Madsen A, Mortensen HP, Rasmussen F and Ravnskov U, 1979. Porcine nephropathy induced by long‐term ingestion of ochratoxin A. Veterinary Pathology, 16, 466–475. [DOI] [PubMed] [Google Scholar]
- Krogh P, Gyrd‐Hansen N, Hald B, Larsen S, Neilsen JP, Smith M, Ivanoff C and Meisner H, 1988. Renal enzyme activities in experimental ochratoxin A‐induced porcine nephropathy: diagnostic potential of phosphoenolpyruvate carboxykinase and gamma‐glutamyl transpeptidase activity. Journal of Toxicology and Environmental Health, 23, 1–14. [DOI] [PubMed] [Google Scholar]
- Kuiper‐Goodman T, Hilts C, Billiard SM, Kiparissis Y, Richard ID and Hayward S, 2010. Health risk assessment of ochratoxin A for all age‐sex strata in a market economy. Food Additives and Contaminants Part A‐Chemistry Analysis Control Exposure and Risk Assessment, 27, 212–240. 10.1080/02652030903013278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai S, 1985. Ochratoxin A: plasma concentration and excretion into bile and urine in albumin‐deficient rats. Food and Chemical Toxicology, 23, 941–943. [DOI] [PubMed] [Google Scholar]
- Kumar SN, Telang AG, Singh KP, Jain AK, Afroz M and Patil RD, 2011. Experimentally induced toxicity of ochratoxin A and endosulfan in male wistar rats: a hormonal disorder. Journal of Animal and Veterinary Advances, 10, 1750–1755. 10.3923/javaa.2011.1750.1755 [DOI] [Google Scholar]
- Kumar R, Ansari KM, Chaudhari BP, Dhawan A, Dwivedi PD, Jain SK and Das M, 2012. Topical Application of Ochratoxin A Causes DNA Damage and Tumor Initiation in Mouse Skin. PLoS ONE, 7, e47280. 10.1371/journal.pone.0047280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Alam S, Chaudhari BP, Dwivedi PD, Jain SK, Ansari KM and Das M, 2013. Ochratoxin A‐induced cell proliferation and tumor promotion in mouse skin by activating the expression of cyclin‐D1 and cyclooxygenase‐2 through nuclear factor‐kappa B and activator protein‐1. Carcinogenesis, 34, 647–657. 10.1093/carcin/bgs368 [DOI] [PubMed] [Google Scholar]
- Kupski L, Freitas M, Ribeiro D, Furlong EB and Fernandes E, 2016. Ochratoxin A activates neutrophils and kills these cells through necrosis, an effect eliminated through its conversion into ochratoxin α. Toxicology, 10, 91–102. 10.1016/j.tox.2016.09.001 [DOI] [PubMed] [Google Scholar]
- Kure CF and Skaar I, 2019. The fungal problem in cheese industry. Current Opinion in Food Science, 29, 14–19. [Google Scholar]
- Kuroda K, Hibi D, Ishii Y, Takasu S, Kijima A, Matsushita K, Masumura K, Watanabe M, Sugita‐Konishi Y, Sakai H, Yanai T, Nohmi T, Ogawa K and Umemura T, 2014. Ochratoxin A induces DNA double‐strand breaks and large deletion mutations in the carcinogenic target site of gpt delta rats. Mutagenesis, 29, 27–36. [DOI] [PubMed] [Google Scholar]
- Kuroda K, Hibi D, Ishii Y, Yokoo Y, Takasu S, Kijima A, Matsushita K, Masumura K, Kodama Y, Yanai T, Sakai H, Nohmi T, Ogawa K and Umemura T, 2015. Role of p53 in the progression from ochratoxin A‐induced DNA damage to gene mutations in the kidneys of mice. Toxicological Sciences, 144, 65–76. [DOI] [PubMed] [Google Scholar]
- Lambert D, Padfield PJ, McLaughlin J, Cannell S and O'Neill CA, 2007. Ochratoxin A displaces claudins from detergent resistant membrane microdomains. Biochemical and Biophysical Research Communications, 358, 632–636. [DOI] [PubMed] [Google Scholar]
- Lerda D, Ambrosio M, Kunsagi Z and Stroka J, 2013. Determination of ochratoxin a in licorice and licorice extracts by high‐performance liquid chromatography coupled with fluorescence detection: Collaborative study. Journal of AOAC International, 96, 331–340. [DOI] [PubMed] [Google Scholar]
- Li S, Marquardt RR and Frohlich AA, 2000. Identification of ochratoxins and some of their metabolites in bile and urine of rats. Food and Chemical Toxicology, 38, 141–152. [DOI] [PubMed] [Google Scholar]
- Li J, Yin S, Dong Y, Fan L and Hu H, 2011. p53 activation inhibits ochratoxin A‐induced apoptosis in monkey and human kidney epithelial cells via suppression of JNK activation. Biochemical and Biophysical Research Communications, 411, 458–463. 10.1016/j.bbrc.2011.06.190 [DOI] [PubMed] [Google Scholar]
- Li X, Gao J, Huang K, Qi X, Dai Q, Mei X and Xu W, 2015. Dynamic changes of global DNA methylation and hypermethylation of cell adhesion‐related genes in rat kidneys in response to ochratoxin A. World Mycotoxin Journal, 8, 465–476. [Google Scholar]
- Lian H, Cui J, Wang Y, Liu J, Wang J, Shen H, Xing L, Wang J, Yan X and Zhang X, 2014. Downregulation of Rad51 participates in OTA‐induced DNA double‐strand breaks in GES‐1 cells in vitro . Toxicology Letters, 226, 214–221. [DOI] [PubMed] [Google Scholar]
- Liang R, Shen XL, Zhang B, Li Y, Xu W, Zhao C, Luo Y and Huang K, 2015. Apoptosis signal‐regulating kinase 1promotes OchratoxinA‐induced renal cytotoxicity. Science Reports, 28, 8078. 10.1038/srep08078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limay‐Rios V, Miller JD and Schaafsma AW, 2017. Occurrence of Penicillium verrucosum, ochratoxin A, ochratoxin B and citrinin in on‐farm stored winter wheat from the Canadian Great Lakes Region. PLoS ONE, 12, e0181239. 10.1371/journal.pone.0181239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limbeck E, Vanselow JT, Hofmann J, Schlosser A and Mally A, 2018. Linking site‐specific loss of histone acetylation to repression of gene expression by the mycotoxin ochratoxin A. Archives of Toxicology, 92, 995–1014. 10.1007/s00204-017-2107-6 [DOI] [PubMed] [Google Scholar]
- Liu J, Wang Y, Cui J, Xing L, Shen H, Wu S, Lian H, Wang J, Yan X and Zhang X, 2012. Ochratoxin A induces oxidative DNA damage and G1 phase arrest in human peripheral blood mononuclear cells in vitro . Toxicology Letters, 211, 164–171. [DOI] [PubMed] [Google Scholar]
- Liu J, Wu S, Shen H, Cui J, Wang Y, Xing L, Wang J, Yan X and Zhang X, 2015. Ochratoxin A induces DNA damage and G2 phase arrest in human esophageal epithelium Het‐1A cells in vitro . Journal of Toxicological Sciences, 40, 657–665. [DOI] [PubMed] [Google Scholar]
- Loboda A, Stachurska A, Podkalicka P, Sobczak M, Mucha O, Witalisz‐Siepracka A, Jozkowicz A and Dulak J, 2017a. Effect of heme oxygenase‐1 on ochratoxin A‐induced nephrotoxicity in mice. International Journal of Biochemistry and Cell Biology, 84, 46–57. [DOI] [PubMed] [Google Scholar]
- Loboda A, Stachurska A, Sobczak M, Podkalicka P, Mucha O, Jozkowicz A and Dulak J, 2017b. Nrf2 deficiency exacerbates ochratoxin A‐induced toxicity in vitro and in vivo . Toxicology, 389, 42–52. [DOI] [PubMed] [Google Scholar]
- Lund F, Filtenborg O and Frisvad JC, 1995. Associated mycoflora of cheese. Food Microbiology, 12, 173–180. [Google Scholar]
- Maaroufi K, Achour A, Betbeder AM, Hammami M, Ellouz F, Creppy EE and Bacha H, 1995. Foodstuffs and human blood contamination by the mycotoxin ochratoxin A: correlation with chronic interstitial nephropathy in Tunisia. Archives of Toxicology, 69, 552–558. [DOI] [PubMed] [Google Scholar]
- Malir F, Ostry V, Pfohl‐Leszkowicz A, Toman J, Bazin I and Roubal T, 2014. Transfer of ochratoxin A into tea and coffee beverages. Toxins (Basel), 6, 3438–3453. 10.3390/toxins6123438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malir F, Osrty V, Pfohl‐Leszkowicz A, Malir J and Toman J, 2016. Ochratoxin A: 50 years of research. Toxins, 8, 191. 10.3390/toxins8070191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mally A, Zepnik H, Wanek P, Eder E, Dingley K, Ihmels H, Volkel W and Dekant D, 2004. Ochratoxin A: Lack of formation of covalent DNA adducts. Chemical Research in Toxicology, 17, 234–242. [DOI] [PubMed] [Google Scholar]
- Mally A, Völkel W, Amberg A, Kurz M, Wanek P, Eder E, Hard G and Dekant W, 2005a. Functional, biochemical, and pathological effects of repeated oral administration of ochratoxin A to rats. Chemical Research in Toxicology, 18, 1242–1252. [DOI] [PubMed] [Google Scholar]
- Mally A, Pepe G, Ravoori S, Fiore M, Gupta RC, Dekant W and Mosesso P, 2005b. Ochratoxin A causes DNA damage and cytogenetic effects but no DNA adducts in rats. Chemical Research in Toxicology, 18, 1253–1261. [DOI] [PubMed] [Google Scholar]
- Mally A, Keim‐Heusler H, Amberg A, Kurz M, Zepnik H, Mantle P, Völkel W, Hard GC and Dekant W, 2005c. Biotransformation and nephrotoxicity of ochratoxin B in rats. Toxicology and Applied Pharmacology, 206, 43–53. [DOI] [PubMed] [Google Scholar]
- Mally A, Decker M and Dekant W, 2006. Ochratoxin A alters cell adhesion and gap junction intercellular communication in MDCK cells. Toxicology, 223, 15–25. [DOI] [PubMed] [Google Scholar]
- Manda P, Dano DS, Kouadio JH, Diakite A, Sangare‐Tigori B, Ezoulin MJM, Soumahoro A, Dembele A and Fourny G, 2009. Impact of industrial treatments on ochratoxin A content in artificially contaminated cocoa beans. Food Additives and Contaminants Part A‐Chemistry Analysis Control Exposure and Risk Assessment, 26, 1081–1088. [DOI] [PubMed] [Google Scholar]
- Manderville RA, 2005. A case for the genotoxicity of ochratoxin A by bioactivation and covalent DNA adduction. Chemical Research in Toxicology, 18, 1091–1097. [DOI] [PubMed] [Google Scholar]
- Manderville RA and Wetmore SD, 2017. Mutagenicity of ochratoxin A: Role for a carbon‐linked C8‐deoxyguanosine adduct? Journal of Agricultural and Food Chemistry, 65, 7097–7105. [DOI] [PubMed] [Google Scholar]
- Mantle P, Kulinskaya E and Nestler S, 2005. Renal tumourigenesis in male rats in response to chronic dietary ochratoxin A. Food Additives and Contaminants, 22(Suppl 1), 58–64. [DOI] [PubMed] [Google Scholar]
- Mantle PG, 2008. Interpretation of the pharmacokinetics of ochratoxin A in blood plasma of rats, during and after acute or chronic ingestion. Food and Chemical Toxicology, 46, 1808–1816. [DOI] [PubMed] [Google Scholar]
- Mantle PG, 2009. Minimum tolerable exposure period and maximum threshold dietary intake of ochratoxin A for causing renal cancer in male Dark Agouti rats. Food and Chemical Toxicology, 47, 2419–2424. [DOI] [PubMed] [Google Scholar]
- Mantle P and Kulinskaya E, 2010. Lifetime, low‐dose ochratoxin A dietary study on renal carcinogenesis in male Fischer rats. Food Additives and Contaminants Part A ‐ Chemistry Analysis Control Exposure and Risk Assessment, 27, 1566–1573. [DOI] [PubMed] [Google Scholar]
- Mantle PG, Dobrota M, Gillett CE, Odell EW and Pinder SE, 2010a. Oncological outcomes in rats given nephrocarcinogenic exposure to dietary ochratoxin A, followed by the tumour promoter sodium barbital for life: a pilot study. Toxins (Basel), 2, 552–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantle PG, Faucet‐Marquis V, Manderville RA, Squillaci B and Pfohl‐Leszkowicz A, 2010b. Structures of covalent adducts between DNA and ochratoxin A: A new factor in debate about genotoxicity and human risk assessment. Chemical Research in Toxicology, 23, 89–98. [DOI] [PubMed] [Google Scholar]
- Mantle P, 2016. Rat Kidney Cancers Determined by Dietary Ochratoxin A in the First Year of Life. Journal of Kidney Cancer and VHL, 3, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maresca M, Yahi N, Younès‐Sakr L, Boyron M, Caporiccio B and Fantini J, 2008. Both direct and indirect effects account for the pro‐inflammatory activity of enteropathogenic mycotoxins on the human intestinal epithelium: stimulation of interleukin‐8 secretion, potentiation of interleukin‐1beta effect and increase in the transepithelial passage of commensal bacteria. Toxicology and Applied Pharmacology, 228, 84–92. 10.1016/j.taap.2007.11.013 [DOI] [PubMed] [Google Scholar]
- Marin DE, Motiu M, Pistol GC, Gras MA, Israel‐Roming F, Calin L, Stancu M and Taranu I, 2016. Diet contaminated with ochratoxin A at the highest level allowed by EU recommendation disturbs liver metabolism in weaned piglets. World Mycotoxin Journal, 9, 587–596. [Google Scholar]
- Marin DE, Braicu C, Gras MA, Pistol GC, Petric RC, Berindan Neagoe I, Palade M and Taranu I, 2017a. Low level of ochratoxin A affects genome‐wide expression in kidney of pig. Toxicon, 136, 67–77. [DOI] [PubMed] [Google Scholar]
- Marin DE, Pistol GC, Gras MA, Palade ML and Taranu I, 2017b. Comparative effect of ochratoxin A on inflammation and oxidative stress parameters in gut and kidney of piglets. Regulatory Toxicology and Pharmacology, 89, 224–231. [DOI] [PubMed] [Google Scholar]
- Marin DE, Pisto GC, Gras M, Palade M and Taranu I, 2018. A comparison between the effects of ochratoxin A and aristolochic acid on the inflammation and oxidative stress in the liver and kidney of weanling piglets. Naunyn‐Schmiedeberg's Archives of Pharmacology, 391, 1147–1156. [DOI] [PubMed] [Google Scholar]
- Marin‐Kuan M, Nestler S, Verguet C, Bezençon C, Piguet D, Mansourian R, Holzwarth J, Grigorov M, Delatour T, Mantle P, Cavin C and Schilter B, 2006. A toxicogenomics approach to identify new plausible epigenetic mechanisms of ochratoxin a carcinogenicity in rat. Toxicological Sciences, 89, 120–134. [DOI] [PubMed] [Google Scholar]
- Marin‐Kuan M, Nestler S, Verguet C, Bezencon C, Piguet D, Delatour T, Mantle P, Cavin C and Schilter B, 2007. MAPK‐ERK activation in kidney of male rats chronically fed ochratoxin A at a dose causing a significant incidence of renal carcinoma. Toxicology and Applied Pharmacology, 224, 174–181. [DOI] [PubMed] [Google Scholar]
- Meisner H and Krogh P, 1986. Phosphoenolpyruvate carboxykinase as a selective indicator of ochratoxin A induced nephropathy. Developments in Toxicology and Environmental Science, 14, 199–206. [PubMed] [Google Scholar]
- Meki AR and Hussein AA, 2001. Melatonin reduces oxidative stress induced by ochratoxin A in rat liver and kidney. Comparative Biochemistry and Physiology Part C: Pharmacology, Toxicology and Endocrinology, 130, 305–313. [DOI] [PubMed] [Google Scholar]
- Meneely JP and Elliott CT, 2014. Rapid surface plasmon resonance immunoassays for the determination of mycotoxins in cereals and cereal‐based food products. World Mycotoxin Journal, 7, 491–505. [Google Scholar]
- Merla C, Andreoli G, Garino C, Vicari N, Tosi G, Guglielminetti ML, Moretti A, Biancardi A, Arlorio M and Fabbi M, 2018. Monitoring of ochratoxin A and ochratoxin‐producing fungi in traditional salami manufactured in Northern Italy. Mycotoxin Research, 34, 107–116. [DOI] [PubMed] [Google Scholar]
- Merten C, Ferrari P, Bakker M, Boss A, Hearty A, Leclercq C, Lindtner O, Tlustos C, Verger P, Volatier JL and Arcella D, 2011. Methodological characteristics of the national dietary surveys carried out in the European Union as included in the European Food Safety Authority (EFSA) Comprehensive European Food Consumption Database. Food Additives and Contaminants. Part A, 28, 975–995. 10.1080/19440049.2011.576440 [DOI] [PubMed] [Google Scholar]
- Mir MS and Dwivedi P, 2010. Ochratoxin A‐induced serum biochemical alterations in New Zealand white rabbits (Oryctolagus cuniculus). Turkish Journal of Veterinary and Animal Sciences, 34, 525–531. © TUBİTAK, 10.3906/vet-0901-23 [DOI] [Google Scholar]
- Mitchell NJ, Chen C, Palumbo JD, Bianchini A, Cappozzo J, Stratton J, Ryu D and Wu F, 2017. A risk assessment of dietary ochratoxin A in the United States. Food and Chemical Toxicology, 100, 265–273. 10.1016/j.fct.2016.12.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitropoulou A, Gambacorta L, Warensjö Lemming E, Solfrizzo M and Olsen M, 2018. Extended evaluation of urinary multi‐biomarker analyses of mycotoxins in Swedish adults and children. World Mycotoxin Journal, 11, 647–659. [Google Scholar]
- Mobashar M, Blank R, Hummel J, Westphal A, Tholen E and Südekum KH, 2012. Ruminal ochratoxin A degradation – Contribution of the different microbial populations and influence of diet. Animal Feed Science and Technology, 17, 85–97. [Google Scholar]
- Mor F, Sengul O, Topsakal S, Kilic MA and Ozmen O, 2017. Diabetogenic Effects of Ochratoxin A in Female Rats. Toxins (Basel), 9, piiE144. 10.3390/toxins9040144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosesso P, Cinelli S, Piñero J, Bellacima R and Pepe G, 2008. In vitro cytogenetic results supporting a DNA nonreactive mechanism for ochratoxin A, potentially relevant for its carcinogenicity. Chemical Research in Toxicology, 21, 1235–1243. [DOI] [PubMed] [Google Scholar]
- Mousavi Khaneghah A, Fakhri Y and Sant'Ana AS, 2018. Impact of unit operations during processing of cereal‐based products on the levels of deoxynivalenol, total aflatoxin, ochratoxin A, and zearalenone: A systematic review and meta‐analysis. Food Chemistry, 268, 611–624. 10.1016/j.foodchem.2018.06.072. [DOI] [PubMed] [Google Scholar]
- Muñoz K, Campos V, Blaszkewicz M, Vega M, Alvarez A, Neira J and Degen GH, 2010. Exposure of neonates to ochratoxin A: first biomonitoring results in human milk (colostrum) from Chile. Mycotoxin Reearch, 26, 59–67. 10.1007/s12550-009-0040-0 [DOI] [PubMed] [Google Scholar]
- Muñoz K, Wollin KM, Kalhoff H and Degen GH, 2013. Occurrence of the mycotoxin ochratoxin a in breast milk samples from Germany. Gesundheitswesen, 75, 194–197. 10.1055/s-0033-1341442 [DOI] [PubMed] [Google Scholar]
- Muñoz K, Blaszkewicz M, Campos V, Vega M and Degen GH, 2014. Exposure of infants to ochratoxin A with breast milk. Archives of Toxicology, 88, 837–846. [DOI] [PubMed] [Google Scholar]
- Muñoz K, Cramer B, Dopstadt J, Humpf HU and Degen GH, 2017. Evidence of ochratoxin A conjugates in urine samples from infants and adults. Mycotoxin Research, 33, 39–47. 10.1007/s12550-016-0261-y [DOI] [PubMed] [Google Scholar]
- Munro IC, Moodie CA, Kuiper‐Goodman T, Scott PM and Grice HC, 1974. Toxicologic changes in rats fed graded dietary levels of ochratoxin A. Toxicology and Applied Pharmacology, 28, 180–188. [DOI] [PubMed] [Google Scholar]
- Mutlu‐Ingok A and Karbancioglu‐Guler F, 2014. Effect of temperature on the growth and ochratoxin a production of the Aspergillus section Nigri members isolated from dried figs. Journal of Food Safety, 34, 333–339. [Google Scholar]
- Nakayama H, Kitagawa N, Otani T, Iida H, Anan H and Inai T, 2018. Ochratoxin A, citrinin and deoxynivalenol decrease claudin‐2 expression in mouse rectum CMT93‐II cells. Microscopy, 67, 99–111. 10.1093/jmicro/dfy005 [DOI] [PubMed] [Google Scholar]
- Nielsen KF, Ngemela AF, Jensen LB, De Medeiros LS and Rasmussen PH, 2015. UHPLC‐MS/MS determination of ochratoxin a and fumonisins in coffee using QuEChERS extraction combined with mixed‐mode SPE purification. Journal of Agricultural and Food Chemistry, 63, 1029–1034. 10.1021/jf504254q [DOI] [PubMed] [Google Scholar]
- NTP (National Toxicology Program), 1989. Toxicology and Carcinogenesis Studies of Ochratoxin A (CAS No. 303‐47‐9) in F344/N Rats (Gavage Studies). National Toxicology Programme, Technical Reports Series, 358, 1–142. [PubMed]
- Obrecht‐Pflumio S, Chassat T, Dirheimer G and Marzin D, 1999. Genotoxicity of ochratoxin A by Salmonella mutagenicity test after bioactivation by mouse kidney microsomes. Mutation Research, 446, 95–102. [DOI] [PubMed] [Google Scholar]
- Oh SY, Balch CG, Cliff RL, Sharma BS, Boermans HJ, Swamy HV, Quinton VM and Karrow NA, 2013. Exposure to Penicillium mycotoxins alters gene expression of enzymes involved in the epigenetic regulation of bovine macrophages (BoMacs). Mycotoxin Research, 29, 235–243. 10.1007/s12550-013-0174-y [DOI] [PubMed] [Google Scholar]
- Oliveira G, Daiani M, Silva R, Gualberto Fonseca A, Pereira L, Paiva C, Prado G and Batista LR, 2013. Effect of different roasting levels and particle sizes on ochratoxin A concentration in coffee beansss. Food Control, 34, 651–656. [Google Scholar]
- Ozbey F and Kabak B, 2012. Natural co‐occurrence of aflatoxins and ochratoxin A in spices. Food Control, 2012, 354–361. [Google Scholar]
- Özcan Z, Gül G and Yaman I, 2015. Ochratoxin A activates opposing c‐MET/PI3K/Akt and MAPK/ERK 1‐2 pathways in human proximal tubule HK‐2 cells. Archives of Toxicology, 89, 1313–1327. 10.1007/s00204-014-1311-x [DOI] [PubMed] [Google Scholar]
- Ozçelik N, Soyöz M and Kilinç I, 2004. Effects of ochratoxin a on oxidative damage in rat kidney: protective role of melatonin. Journal of Applied Toxicology, 24, 211–215. [DOI] [PubMed] [Google Scholar]
- Ozden S, Turgut Kara N, Sezerman OU, Durasi İM, Chen T, Demirel G, Alpertunga B, Chipman JK and Mally A, 2015. Assessment of global and gene‐specific DNA methylation in rat liver and kidney in response to non‐genotoxic carcinogen exposure. Toxicology and Applied Pharmacology, 289, 203–212. 10.1016/j.taap.2015.09.023 [DOI] [PubMed] [Google Scholar]
- Palma N, Cinelli S, Sapora O, Wilson SH and Dogliotti E, 2007. Ochratoxin A‐induced mutagenesis in mammalian cells is consistent with the production of oxidative stress. Chemical Research in Toxicology, 20, 1031–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankaj SK, Wan Z and Keener KM, 2018. Effects of Cold Plasma on Food Quality. A Review. Foods (Basel, Switzerland), 7, piiE4. 10.3390/foods7010004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor L, Vettorazzi A, Enciso JM, Gonzalez‐Penas E, Garcia‐Jalon JA, Monreal JI and de Certain AL, 2018. Sex differences in ochratoxin a toxicity in F344 rats after 7 and 21 days of daily oral administration. Food and Chemical Toxicology, 111, 363–373. [DOI] [PubMed] [Google Scholar]
- Patil RD, Dwivedi P and Sharma AK, 2006. Critical period and minimum single oral dose of ochratoxin A for inducing developmental toxicity in pregnant Wistar rats. Reproductive Toxicology, 22, 679–687. [DOI] [PubMed] [Google Scholar]
- Pattono D, Gallo PF and Civera T, 2011. Detection and quantification of ochratoxin A in milk produced in organic farms. Food Chemistry, 127, 374–377. [Google Scholar]
- Pattono D, Grosso A, Stocco PP, Pazzi M and Zeppa G, 2013. Survey of the presence of patulin and ochratoxin A in traditional semi‐hard cheeses. Food Control, 33, 54–57. [Google Scholar]
- Pena A, Seifrtová M, Lino C, Silveira I and Solich P, 2006. Estimation of ochratoxin A in Portuguese population: new data on the occurrence in human urine by high performance liquid chromatography with fluorescence detection. Food and Chemical Toxicology, 44, 1449–1454. [DOI] [PubMed] [Google Scholar]
- Pérez de Obanos A, González‐Peñas E and López de Certain A, 2005. Influence of roasting and brew preparation on the ochratoxin A content in coffee infusion. Food Additives and Contaminants, 22, 463–471. [DOI] [PubMed] [Google Scholar]
- Perry JL, Christensen T, Goldsmith MR, Toone EJ, Beratan DN and Simon JD, 2003. Binding of ochratoxin A to human serum albumin stabilized by a protein – ligand ion pair. Journal of Physical Chemistry, 107, 7884–7888. [Google Scholar]
- Peters J, Van Dam R, Van Doorn R, Katerere D, Berthiller F, Haasnoot W and Nielen MWF, 2017. Mycotoxin profiling of 1000 beer samples with a special focus on craft beer. PLoS ONE, 12, art. no. e0185887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrić J, Šarkanj B, Mujić I, Mujić A, Sulyok M, Krska R, Šubarić D and Jokić S, 2018. Effect of pretreatments on mycotoxin profiles and levels in dried figs. Arhiv za Higijenu Rada i Toksikologiju, 69, 328–333. [DOI] [PubMed] [Google Scholar]
- Pfeiffer E, Gross K and Metzler M, 1998. Aneuploidogenic and clastogenic potential of the mycotoxins citrinin and patulin. Carcinogenesis, 19, 1313–1318. [DOI] [PubMed] [Google Scholar]
- Pfohl‐Leszkowicz A and Manderville RA, 2007. Ochratoxin A: an overview on toxicity and carcinogenicity in animals and humans. Molecular Nutrition and Food Research, 51, 61–99. [DOI] [PubMed] [Google Scholar]
- Pfohl‐Leszkowicz A and Manderville RA, 2012. An update on direct genotoxicity as a molecular mechanism of ochratoxin A carcinogenicity. Chemical Research in Toxicology, 25, 252–262. [DOI] [PubMed] [Google Scholar]
- Piacentini KC, Ferranti LS, Pinheiro M, Bertozzi BG and Rocha LO, 2019. Mycotoxin contamination in cereal‐based baby foods. Current Opinion in Food Science, 30, 73–78. [Google Scholar]
- Pleadin J, Persi N, Mitak M, Terzic S, Milic D, Vulic A and Brstilo M, 2012. Biochemical Changes in Pig Serum After Ochratoxin A Exposure. Bulletin of Environmental Contamination and Toxicology, 88, 1043–1047. [DOI] [PubMed] [Google Scholar]
- Pleadin J, Perši N, Kovačević D, Vulić A, Frecec J and Markovc K, 2014. Ochratoxin A reduction in meat sausages using processing methods practiced in households. Food Additives and Contaminants: Part B, 7, 239–246. 10.1080/19393210.2014.900119 [DOI] [PubMed] [Google Scholar]
- Pohland AE, Schuller PL, Steyn PS and Van Egmond HP, 1982. Physicochemical data for some selected mycotoxins. Pure and Applied Chemistry, 54, 2219–2284. [Google Scholar]
- Poór M, Veres B, Jakus PB, Antus C, Montskó G, Zrínyi Z, Vladimir‐Knežević S, Petrik J and Kőszegi T, 2014. Flavonoid diosmetin increases ATP levels in kidney cells and relieves ATP depleting effect of ochratoxin A. Journal of Photochemistry and Photobiology B, 132, 1–9. 10.1016/j.jphotobiol.2014.01.016 [DOI] [PubMed] [Google Scholar]
- Postupolski J, Karlowski K and Kubik P, 2006. Ochratoxin A in maternal and foetal blood and in maternal milk. Roczniki Panstwowego Zakladu Higieny (Annals of the National Institute of Hygiene), 75, 23–30. [PubMed] [Google Scholar]
- Prabu PC, Dwivedi P and Sharma AK, 2013. Toxicopathological studies on the effects of aflatoxin B(1), ochratoxin A and their interaction in New Zealand White rabbits. Experimental and Toxicologic Pathology, 65, 277–286. [DOI] [PubMed] [Google Scholar]
- Qi X, Yu T, Zhu L, Gao J, He X, Huang K, Luo Y and Xu W, 2014a. Ochratoxin A induces rat renal carcinogenicity with limited induction of oxidative stress responses. Toxicology and Applied Pharmacology, 280, 543–549. [DOI] [PubMed] [Google Scholar]
- Qi XZ, Yang X, Chen SY, He XY, Dweep H, Guo MZ, Cheng WH, Xu WT, Luo YB, Gretz N, Dai Q and Huang KL, 2014b. Ochratoxin A induced early hepatotoxicity: new mechanistic insights from microRNA, mRNA and proteomic profiling studies. Science Reports, 4. [Google Scholar]
- Qian G, Liu D, Hu J, Gan F, Hou L, Chen X and Huang K, 2017. Ochratoxin A‐induced autophagy in vitro and in vivo promotes porcine circovirus type 2 replication. Cell Death and Disease, 8, e2909. 10.1038/cddis.2017.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian G, Liu D, Hu J, Gan F, Hou L, Zhai N, Chen X and Huang K, 2018. SeMet attenuates OTA‐induced PCV2 replication promotion by inhibiting autophagy by activating the AKT/mTOR signaling pathway. Veterinary Research, 49, 15. 10.1186/s13567-018-0508-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rached E, Pfeiffer E, Dekant W and Mally A, 2006. Ochratoxin A: apoptosis and aberrant exit from mitosis due to perturbation of microtubule dynamics? Toxicological Sciences, 92, 78–86. [DOI] [PubMed] [Google Scholar]
- Rached E, Hard GC, Blumbach K, Weber K, Draheim R, Lutz WK, Ozden S, Steger U, Dekant W and Mally A, 2007. Ochratoxin A: 13‐week oral toxicity and cell proliferation in male F344/N rats. Toxicological Sciences, 97, 288–298. [DOI] [PubMed] [Google Scholar]
- Rached E, Hoffmann D, Blumbach K, Weber K, Dekant W and Mally A, 2008. Evaluation of putative biomarkers of nephrotoxicity after exposure to ochratoxin A in vivo and in vitro . Toxicological Sciences, 103, 371–381. [DOI] [PubMed] [Google Scholar]
- Raghubeer S, Nagiah S, Phulukdaree A and Chuturgoon A, 2015. The phytoalexin resveratrol ameliorates ochratoxin A toxicity in human embryonic kidney (HEK293) cells. Journal of Cellular Biochemistry, 116, 2947–2955. 10.1002/jcb.25242 [DOI] [PubMed] [Google Scholar]
- Raghubeer S, Nagiah S and Chuturgoon AA, 2017. Acute Ochratoxin A exposure induces inflammation and apoptosis in human embryonic kidney (HEK293) cells. Toxicon, 137, 48–53. 10.1016/j.toxicon.2017.07.013 [DOI] [PubMed] [Google Scholar]
- Ramos‐Pereira J, Mareze J, Patrinou E, Santos JA and López‐Díaz T‐M, 2019. Polyphasic identification of Penicillium spp. isolated from Spanish semi‐hard ripened cheeses. Food Microbiology, 84, art. no. 103253, [DOI] [PubMed] [Google Scholar]
- Ramyaa P and Padma VV, 2013. Ochratoxin‐induced toxicity, oxidative stress and apoptosis ameliorated by quercetin – Modulation by Nrf2. Food and Chemical Toxicology, 62, 205–216. [DOI] [PubMed] [Google Scholar]
- Ranaldi G, Mancini E, Ferruzza S, Sambuy Y and Perozzi G, 2007. Effects of red wine on ochratoxin A toxicity in intestinal Caco‐2/TC7 cells. Toxicology In Vitro, 21, 204–210. [DOI] [PubMed] [Google Scholar]
- Ranaldi G, Caprini V, Sambuy Y, Perozzi G and Murgia C, 2009. Intracellular zinc stores protect the intestinal epithelium from Ochratoxin A toxicity. Toxicology In Vitro, 23, 1516–1521. 10.1016/j.tiv.2009.08.012 [DOI] [PubMed] [Google Scholar]
- Rašić D, MLadinić M, Želježić D, Pizent A, Stefanović S, Milićević D, Konjevoda P and Peraica M, 2018. Effects of combined treatment with ochratoxin A and citrinin on oxidative damage in kidneys and liver of rats. Toxicon, 146, 99–105. [DOI] [PubMed] [Google Scholar]
- Razafimanjato H, Garmy N, Guo XJ, Varini K, Di Scala C, Di Pasquale E, Taïeb N and Maresca M, 2010. The food‐associated fungal neurotoxin ochratoxin A inhibits the absorption of glutamate by astrocytes through a decrease in cell surface expression of the excitatory amino‐acid transporters GLAST and GLT‐1. Neurotoxicology, 31, 475–484. 10.1016/j.neuro.2010.06.003 [DOI] [PubMed] [Google Scholar]
- Ringot D, Chango A, Schneider YJ and Larondelle Y, 2006. Toxicokinetics and toxicodynamics of ochratoxin A, an update. Chemico‐Biological Interactions, 159, 18–46. [DOI] [PubMed] [Google Scholar]
- Rodríguez A, Rodríguez M, Martín A, Delgado J and Córdoba JJ, 2012. Presence of ochratoxin A on the surface of dry‐cured Iberian ham after initial fungal growth in the drying stage. Meat Science, 92, 728–734. [DOI] [PubMed] [Google Scholar]
- Rodríguez‐Cabo T, Rodríguez‐Cabo I, Ramil M and Cela R, 2016. Liquid chromatography quadrupole time‐of‐flight mass spectrometry selective determination of ochratoxin A in wine. Food Chemistry, 199, 401–408. [DOI] [PubMed] [Google Scholar]
- Romero A, Ares I, Ramos E, Castellano V, Martínez M, Martínez‐Larrañaga MR, Anadón A and Martínez MA, 2016. Mycotoxins modify the barrier function of Caco‐2cells through differential gene expression of specific claudin isoforms: Protective effect of illite mineral clay. Toxicology, 15, 21–33. 10.1016/j.tox.2016.05.003 [DOI] [PubMed] [Google Scholar]
- Ropars J, Cruaud C, Lacoste S and Dupont J, 2012. A taxonomic and ecological overview of cheese fungi. International Journal of Food Microbiology, 155, 199–210. [DOI] [PubMed] [Google Scholar]
- Rossiello MR, Rotunno C, Coluccia A, Carratù MR, Di Santo A, Evangelista V, Semeraro N and Colucci M, 2008. Ochratoxin A inhibits the production of tissue factor and plasminogen activator inhibitor‐2 by human blood mononuclear cells: another potential mechanism of immune‐suppression. Toxicology and Applied Pharmacology, 229, 227–231. 10.1016/j.taap.2008.01.004 [DOI] [PubMed] [Google Scholar]
- Ruhland M, Engelhardt G, Wallnoefer PR and Schaefer W, 1994. Transformation of the mycotoxin ochratoxin A in wheat and maize cell suspension cultures. Naturwissenschaften, 81, 453–454. [DOI] [PubMed] [Google Scholar]
- Ruhland M, Engelhardt G, Schaefer W and Wallnoefer PR, 1996a. Transformation of the mycotoxin ochratoxin A in plants: 1. Isolation and identification of metabolites formed in cell suspension cultures of wheat and maize. Natural Toxins, 4, 254–260. [DOI] [PubMed] [Google Scholar]
- Ruhland M, Engelhardt G and Wallnoefer PR, 1996b. Transformation of the mycotoxin ochratoxin A in plants: 2. Time course and rates of degradation and metabolite production in cell‐suspension cultures of different crop plants. Mycopathologia, 134, 97–102. [DOI] [PubMed] [Google Scholar]
- Ruhland M, Engelhardt G and Wallnoefer PR, 1997. Transformation of the mycotoxin ochratoxin A in artificially contaminated vegetables and cereals. Mycotoxin Research, 13, 54–60. [DOI] [PubMed] [Google Scholar]
- Rutigliano L, Valentini L, Martino NA, Pizzi F, Zanghì A, Dell'Aquila ME and Minervini F, 2015. Ochratoxin A at low concentrations inhibits in vitro growth of canine umbilical cord matrix mesenchymal stem cells through oxidative chromatin and DNA damage. Reproductive Toxicology, 57, 121–129. [DOI] [PubMed] [Google Scholar]
- Rychlik M, Humpf HU, Marko D, Dänicke S, Mally A, Berthiller F, Klaffke H and Lorenz N, 2014. Proposal of a comprehensive definition of modified and other forms of mycotoxins including ‘masked’ mycotoxins. Mycotoxin Research, 30, 197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu D, Kowalski RJ, Ganjyal G and Lee HJ, 2019. Reduction of ochratoxin A in oats and rice by twin‐screw extrusion processing with baking soda. Food Control, 105, 21–25. 10.1016/j.foodcont.2019.05.014 [DOI] [PubMed] [Google Scholar]
- Sáez JM, Medina A, Gimeno‐Adelantado JV, Mateo R and Jiménez M, 2004. Comparison of different sample treatments for the analysis of ochratoxin A in must, wine and beer by liquid chromatography. Journal of Chromatography A, 1029, 125–133. [DOI] [PubMed] [Google Scholar]
- Sahar N, Yousaf S, Shibli S, Khurshid S, Mehwish Iqbal H, Akbar Q‐U‐A and Arif S, 2017. The Impact of Sun Drying on the Occurrence of Aflatoxin in Red Chilies. Journal of Basic and Applied Sciences, 13, 632–637. [Google Scholar]
- Sakin FT, Ozan İ, Yipel M and Kürekci C, 2018. Occurrence and health risk assessment of aflatoxins and ochratoxin a in Sürk, a Turkish dairy food, as studied by HPLC. Food Control, 90, 317–323. 10.1016/j.foodcont.2018.03.012 [DOI] [Google Scholar]
- Santos L, Marín S, Sanchis V and Ramos AJ, 2010. Co‐occurrence of aflatoxins, ochratoxin A and zearalenone in Capsicum powder samples available on the Spanish market. Food Chemistry, 122, 826–830. [Google Scholar]
- Santos L, Marín S, Mateo EM, Gil‐Serna J, Valle‐Algarra FM, Patiño B and Ramos AJ, 2011. Mycobiota and co‐occurrence of mycotoxins in Capsicum powder. International Journal of Food Microbiology, 151, 270–276. 10.1016/j.ijfoodmicro.2011.09.011 [DOI] [PubMed] [Google Scholar]
- Sauvant C, Holzinger H and Gekle M, 2005. The nephrotoxin ochratoxin A induces key parameters of chronic interstitial nephropathy in renal proximal tubular cells. Cellular Physiology and Biochemistry, 15, 125–134. [DOI] [PubMed] [Google Scholar]
- Sava V, Reunova O, Velasquez A, Harbison R and Sánchez‐Ramos J, 2006. Acute neurotoxic effects of the fungal metabolite ochratoxin‐A. Neurotoxicology, 27, 82–92. [DOI] [PubMed] [Google Scholar]
- SCF (Scientific Committee on Food), 1998. Opinion on of the Scientific Committee on Food (SCF) on ochratoxin A. Expressed on 17 September 1998. Available online: http://europa.eu.int/comm/food/fs/sc/scf/out14_en.htmL.
- Schaarschmidt S and Fauhl‐Hassek C, 2018. The fate of mycotoxins during the processing of wheat for human consumption. Comprehensive Reviews in Food Science and Food Safety, 17, 556–593. 10.1111/1541-4337.12338 [DOI] [PubMed] [Google Scholar]
- Schiavone A, Cavallero C, Girotto L, Pozzo L, Antoniazzi S and Cavallin L, 2008. A survey on the occurrence of ochratoxin A in feeds and sera collected in conventional and organic poultry farms in Northern Italy. Italian Journal of Animal Science, 7, 495–503. [Google Scholar]
- Schmidt‐Heydt M, Cramer B, Graf I, Lech S, Humpf HU and Geisen R, 2012. Wavelength‐dependent degradation of ochratoxin and citrinin by light in vitro and in vivo and its implications on Penicillium . Toxins, 4, 1535–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwerdt G, Holzinger H, Sauvant C, Königs M, Humpf HU and Gekle M, 2007. Long‐term effects of ochratoxin A on fibrosis and cell death in human proximal tubule or fibroblast cells in primary culture. Toxicology, 22;232(1–2), 57–67. [DOI] [PubMed] [Google Scholar]
- Scudamore KA and Patel S, 2000. Survey for aflatoxins, ochratoxin A, zearalenone and fumonisins in maize imported into the United Kingdom. Food Additives and Contaminants, 17, 407–416. [DOI] [PubMed] [Google Scholar]
- Shen XL, Zhang Y, Xu W, Liang R, Zheng J, Luo Y, Wang Y and Huang K, 2013. An iTRAQ‐based mitoproteomics approach for profiling the nephrotoxicity mechanisms of ochratoxin A in HEK 293 cells. Journal of Proteomics, 78, 398–415. 10.1016/j.jprot.2012.10.010 [DOI] [PubMed] [Google Scholar]
- Sheu ML, Shen CC, Chen YS and Chiang CK, 2017. Ochratoxin A induces ER stress and apoptosis in mesangial cells via a NADPH oxidase‐derived reactive oxygen species‐mediated calpain activation pathway. Oncotarget, 21;8(12), 19376–19388. 10.18632/oncotarget.14270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber M, Wagner S, Rached E, Amberg A, Mally A and Dekant W, 2009. Metabonomic study of ochratoxin A toxicity after repeated administration: Phenotypic anchoring enhances ability for biomarker discovery. Chemical Research in Toxicology, 22, 1221–1231. [DOI] [PubMed] [Google Scholar]
- SimarroDoorten Y, Nijmeijer S, de Nijs‐Tjon L and Fink‐Gremmels J, 2006. Metabolism‐mediated Ochratoxin A genotoxicity in the single‐cell gel electrophoresis (Comet) assay. Food and Chemical Toxicology, 44, 261–270. [DOI] [PubMed] [Google Scholar]
- Skaug MA, Størmer FC and Saugstad OD, 1998. Ochratoxin A: a naturally occurring mycotoxin found in human milk samples from Norway. Acta Pædiatrica, 87, 1275–1278. [DOI] [PubMed] [Google Scholar]
- Skaug MA, Helland I, Solvoll K and Saugstad OD, 2001. Presence of ochratoxin A in human milk in relation to dietary intake. Food Additives and Contaminants, 18, 321–327. [DOI] [PubMed] [Google Scholar]
- Solfrizzo M, De Girolamo A, Lattanzio VMT, Visconti A, Stroka J, Alldrick A and van Egmond HP, 2013. Results of a proficiency test for multi‐mycotoxin determination in maize by using methods based on LC‐MS/(MS). Quality Assurance and Safety of Crops and Foods, 5, 15–48. [Google Scholar]
- Solfrizzo M, Gambacorta L and Visconti A, 2014. Assessment of multi‐mycotoxin exposure in southern Italy by urinary multi‐biomarker determination. Toxins (Basel)., 6, 523–538. 10.3390/toxins6020523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen LK and Elbæk TH, 2005. Determination of mycotoxins in bovine milk by liquid chromatography tandem mass spectrometry. Journal of Chromatography B: Analytical Technologies in the Biomedical and Life Sciences, 820, 183–196. [DOI] [PubMed] [Google Scholar]
- Soto JB, Ruiz MJ, Manyes L and Juan‐García A, 2016. Blood, breast milk and urine: potential biomarkers of exposure and estimated daily intake of ochratoxin A: a review. Food Additives and Contaminants Part A Chemistry, Analysis, Control, Exposure and Risk Assessment, 33, 313–328. 10.1080/19440049.2015.1118160 [DOI] [PubMed] [Google Scholar]
- Spanjer MC, Rensen PM and Scholten JM, 2008. LC–MS/MS multi‐method for mycotoxins after single extraction, with validation data for peanut, pistachio, wheat, maize, cornflakes, raisins and figs. Food Additives and Contaminants ‐ Part A Chemistry, Analysis, Control, Exposure and Risk Assessment, 25, 472–489. [DOI] [PubMed] [Google Scholar]
- Sproviero M, Verwey AM, Rankin KM, Witham AA, Soldatov DV, Manderville RA, Fekry MI, Sturla SJ, Sharma P and Wetmore SD, 2014. Structural and biochemical impact of C8‐aryl‐guanine adducts within the NarI recognition DNA sequence: influence of aryl ring size on targeted and semi‐targeted mutagenicity. Nucleic Acids Research, 42, 13405–13421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stachurska A, Ciesla M, Kozakowska M, Wolffram S, Boesch‐Saadatmandi C, Rimbach G, Jozkowicz A, Dulak J and Loboda A, 2013. Cross‐talk between microRNAs, nuclear factor E2‐related factor 2, and heme oxygenase‐1 in ochratoxin A‐induced toxic effects in renal proximal tubular epithelial cells. Molecular Nutrition and Food Research, 57, 504–515. 10.1002/mnfr.201200456 [DOI] [PubMed] [Google Scholar]
- Stander MA, Steyn PS, van der Westhuizen FH and Payne BE, 2001. A kinetic study into the hydrolysis of the ochratoxins and analogues by carboxypeptidase A. Chemical Research in Toxicology, 14, 302–304. [DOI] [PubMed] [Google Scholar]
- Stec J, Rachubik J and Kuźmak J, 2006. Effects of ochratoxin A, for DNA repair in cultures of swine lymphocytes and kidney cell. Bulletin of the Veterinary Institute in Pulawy, 50, 521–526. [Google Scholar]
- Steinbrück D, Rasch C and Kumke MU, 2008. Photophysics of ochratoxin A in aqueous solution. Zeitschrift für Naturforschung, 63b, 1321–1326. [Google Scholar]
- Stoev SD, Gundasheva D, Zarkov I, Mircheva T, Zapryanova D, Denev S, Mitev Y, Daskalov H, Dutton M, Mwanza M and Schneider YJ, 2012. Experimental mycotoxic nephropathy in pigs provoked by a mouldy diet containing ochratoxin A and fumonisin B1. Experimental and Toxicologic Pathology, 64, 733–741. [DOI] [PubMed] [Google Scholar]
- Støren O, Holm H and Størmer FC, 1982. Metabolism of ochratoxin A by rats. Applied and Environmental Microbiology, 44, 785–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Størmer FC, Hansen CE, Pedersen JI, Hvistendahl G and Aasen AJ, 1981. Formation of (4R)‐ and (4S)‐4‐hydroxyochratoxin A from ochratoxin A by liver microsomes from various species. Applied and Environmental Microbiology, 42, 1051–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Størmer FC, Støren O, Hansen CE, Pedersen JI and Aasen AJ, 1983. Formation of (4R)‐ and (4S)‐4‐hydroxyochratoxin A and 10‐hydroxyochratoxin A from Ochratoxin A by rabbit liver microsomes. Applied and Environmental Microbiology, 45, 1183–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer‐Rohr L, Dietrich DR, Schlatter J and Schlatter C, 1995. The occurrence of ochratoxin A in coffee. Food and Chemical Toxicology, 33, 341–355. [DOI] [PubMed] [Google Scholar]
- Studer‐Rohr I, Schlatter J and Dietrich DR, 2000. Kinetic parameters and intraindividual fluctuations of ochratoxin A plasma levels in humans. Archives of Toxicology, 74, 499–510. [DOI] [PubMed] [Google Scholar]
- Sueck F, Hemp V, Specht J, Torres O, Cramer B and Humpf HU, 2019. Occurrence of the ochratoxin A degradation product 2'R‐ochratoxin A in coffee and other food: An update. Toxins, 11, 329–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sueck F, Specht J, Cramer B and Humpf HU, 2020. Identification of ochratoxin‐N‐acetyl‐L‐cysteine as a new ochratoxin A metabolite and potential biomarker in human urine. Mycotoxin Research, 36, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun AL, Zhang YF, Sun G‐P, Wang X‐N and Tang D, 2017. Homogeneous electrochemical detection of ochratoxin A in foodstuff using aptamer–graphene oxide nanosheets and DNase I‐based target recycling reaction. Biosensors and Bioelectronics, 89, 659–665. [DOI] [PubMed] [Google Scholar]
- Sutken E, Aral E, Ozdemir F, Uslu S, Alatas O and Colak O, 2007. Protective role of melatonin and coenzyme Q10 in ochratoxin A toxicity in rat liver and kidney. International Journal of Toxicology, 26, 81–87. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Satoh T and Yamazaki M, 1977. The pharmacokinetics of ochratoxin A in rats. Japanese Journal of Pharmacology, 27, 735–744. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Hasegawa‐Baba Y, Watanabe Y, Mizukami S, Kangawa Y, Yoshida T and Shibutani M, 2016. Maternal exposure to ochratoxin A targets intermediate progenitor cells of hippocampal neurogenesis in rat offspring via cholinergic signal downregulation and oxidative stress responses. Reproductive Toxicology, 65, 113–122. 10.1016/j.reprotox.2016.06.018 [DOI] [PubMed] [Google Scholar]
- Taniai E, Hayashi H, Yafune A, Watanabe M, Akane H, Suzuki K, Mitsumori K and Shibutani M, 2012a. Cellular distribution of cell cycle‐related molecules in the renal tubules of rats treated with renal carcinogens for 28 days: relationship between cell cycle aberration and carcinogenesis. Archives of Toxicology, 86, 1453–1464. [DOI] [PubMed] [Google Scholar]
- Taniai E, Yafune A, Hayashi H, Itahashi M, Hara‐Kudo Y, Suzuki K, Mitsumori K and Shibutani M, 2012b. Aberrant activation of ubiquitin D at G2 phase and apoptosis by carcinogens that evoke cell proliferation after 28‐day administration in rats. Journal of Toxicological Sciences, 37, 1093–1111. [DOI] [PubMed] [Google Scholar]
- Taniai E, Yafune A, Nakajima M, Hayashi SM, Nakane F, Itahashi M and Shibutani M, 2014. Ochratoxin A induces karyomegaly and cell cycle aberrations in renal tubular cells without relation to induction od oxidative stress responses in rats. Toxicology Letters., 224, 64–72. [DOI] [PubMed] [Google Scholar]
- Thé Y, Manda P, Elleingand E, Yapi FH, Yapo FA, N'Guessan JD, Dano SD and Djaman JA, 2015. Rôle de l'ochratoxine A dans le développement des tumeurs de la vessie chez les patients ivoiriens. Toxicologie Analytique & Clinique, 27, 66–71. [Google Scholar]
- Thirumala‐Devi K, Mayo MA, Reddy G, Reddy SV, Delfosse P and Reddy DVR, 2000. Production of polyclonal antibodies against ochratoxin A and its detection in chilies by ELISA. Journal of Agricultural and Food Chemistry, 48, 5079–5082. [DOI] [PubMed] [Google Scholar]
- Tosun A and Ozden S, 2015. Ochratoxin A in red pepper flakes commercialised in Turkey. Food Additives and Contaminants., 1, 46–50. [DOI] [PubMed] [Google Scholar]
- Tozlovanu M, Faucet‐Marquis V, Pfohl‐Leszkowicz A and Manderville RA, 2006. Genotoxicity of the hydroquinone metabolite of ochratoxin A: structure‐activity relationships for covalent DNA adduction. Chemical Research in Toxicology, 19, 1241–1247. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Sekine T, Takeda M, Cha SH, Kanai Y, Kimura M and Endou H, 1999. Transport of ochratoxin A by renal multispecific organic anion transporter 1. Journal of Pharmacology and Experimental Therapeutics, 289, 1301–1305. [PubMed] [Google Scholar]
- Turconi G, Guarcello M, Livieri C, Comizzoli S, Maccarini L, Castellazzi AM, Pietri A, Piva G and Roggi C, 2004. Evaluation of xenobiotics in human milk and ingestion by the newborn–an epidemiological survey in Lombardy (Northern Italy). European Journal of Nutrition, 43, 191–197. [DOI] [PubMed] [Google Scholar]
- Vaclavic L, Vaclavikova M, Begley TH, Krynitsky AJ and Rader JI, 2013. Determination of multiple mycotoxins in dietary supplements containing green coffee bean extracts using ultra high performance liquid chromatography−tandem mass spectrometry (UHPLC‐MS/MS). Journal of Agricultural and Food Chemistry, 61, 4822–4830. [DOI] [PubMed] [Google Scholar]
- Valitutti F, De Santis B, Trovato CM, Montuori M, Gatti S, Oliva S, Brera C and Catassi C, 2018. Assessment of Mycotoxin Exposure in Breastfeeding Mothers with Celiac Disease. Nutrients, 10, pii: E336. 10.3390/nu10030336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura M, Guillén D, Anaya I, Broto‐Puig F, Lliberia JL, Agut M and Comellas L, 2006. Ultra‐performance liquid chromatography/tandem mass spectrometry for the simultaneous analysis of aflatoxins B1, G1, B2, G2 and ochratoxin A in beer. Rapid Communications in Mass Spectrometry, 20, 3199–3204. [DOI] [PubMed] [Google Scholar]
- Veprikova Z, Zachariasova M, Dzuman Z, Zachariasova A, Fenclova M, Slavikova P, Vaclavikova M, Mastovska K, Hengst D and Hajslova J, 2015. Mycotoxins inplant‐based dietary supplements: hidden health risk for consumers. Journal of Agricultural and Food Chemistry, 29, 6633–6643. [DOI] [PubMed] [Google Scholar]
- Verma R and Chakraborty D, 2008. Alterations in DNA, RNA and protein contents in liver and kidney of mice treated with ochratoxin and their amelioration by Emblica officinalis aqueous extract. Acta Poloniae Pharmaceutica, 65, 3–9. [PubMed] [Google Scholar]
- Vettorazzi A, Gonzales‐Penas E and Lopez de Cerain A, 2014. Ochratoxin A kinetics: A review of analytical methods and studies in rat model. Food and Chemical Toxicology, 72, 273–288. [DOI] [PubMed] [Google Scholar]
- Vidal A, Morales H, Sanchis V, Ramos AJ and Marín S, 2014. Stability of DON and OTA during the breadmaking process and determination of process and performance criteria. Food Control, 40, 234–242. [Google Scholar]
- Visconti A, Pascale M and Centonze G, 1999. Determination of ochratoxin A in wine by means of immunoaffinity column clean‐up and high‐performance liquid chromatography. Journal of Chromatography A, 864, 89–101. [DOI] [PubMed] [Google Scholar]
- von Tobel JS, Antinori P, Zurich MG, Rosset R, Aschner M, Glück F, Scherl A and Monnet‐Tschudi F, 2014. Repeated exposure to Ochratoxin A generates a neuroinflammatory response, characterized by neurodegenerative M1 microglial phenotype. Neurotoxicology., 44, 61–70. 10.1016/j.neuro.2014.04.005 [DOI] [PubMed] [Google Scholar]
- Wang Y, Liu J, Cui J, Xing L, Wang J, Yan X and Zhang X, 2012. ERK and p38MAPK signaling pathways are involved in ochratoxinA‐induced G2 phase arrest in human gastric epithelium cells. Toxicology Letters, 209, 186–192. 10.1016/j.toxlet.2011.12.011 [DOI] [PubMed] [Google Scholar]
- Wang H, Chen Y, Zhai N, Chen X, Gan F, Li H and Huang K, 2017. Ochratoxin A‐induced apoptosis of IPEC‐J2cells through ROS‐mediated mitochondrial permeability transition pore opening pathway. Journal of Agricultural and Food Chemistry, 6;65(48), 10630–10637. 10.1021/acs.jafc.7b04434 [DOI] [PubMed] [Google Scholar]
- Wang H, Zhai N, Chen Y, Fu C and Huang K, 2018. OTA induces intestinal epithelial barrier dysfunction and tight junction disruption in IPEC‐J2 cells through ROS/Ca2+‐mediated MLCK activation. Environmental Pollution, 242(Pt A), 106–112. 10.1016/j.envpol.2018.06.062 [DOI] [PubMed] [Google Scholar]
- Warensjö Lemming E, Montano Montes A, Schmidt J, Cramer B, Humpf HU, Moraeus L and Olsen M, 2015. Mycotoxins in blood and urine of Swedish adolescents‐possible associations to food intake and other background characteristics. Mycotoxin Research, 10.1007/s12550-019-00381-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warth B, Braun D, Ezekiel CN, Turner PC, Degen GH and Marko D, 2016. Biomonitoring of Mycotoxins in Human Breast Milk: Current State and Future Perspectives. Chemical Research in Toxicology, 29, 1087–1097. 10.1021/acs.chemrestox.6b00125 [DOI] [PubMed] [Google Scholar]
- Weil M, Remize F, Durand N, Alter P, Hoarau M and Meile JC, 2020. Effect of processing on microbial safety of wild pepper (Piper borbonense) from Reunion Island. Food Control, 111, art. no. 107061. [Google Scholar]
- WHO/IPCS (World Health Organization/International Programme on Chemical Safety), 2009. Principles and Methods for the Risk Assessment of Chemicals in Food. WHO Library Cataloguing‐in‐Publication Data. Environmental Health Criteria 240. ISBN 978 92 4 157240 8.
- Wilk‐Zasadna I and Minta M, 2009. Developmental toxicity of Ochratoxin A in rat embryo midbrain micromass cultures. International Journal of Molecular Sciences, 10, 37–49. 10.3390/ijms10010037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo CS, Partanen H, Myllynen P, Vähäkangas K and El‐Nezami H, 2012. Fate of the teratogenic and carcinogenic ochratoxin A in human perfused placenta. Toxicology Letters, 208, 92–99. [DOI] [PubMed] [Google Scholar]
- Woo CS, Wan ML, Ahokas J and El‐Nezami H, 2013. Potential endocrine disrupting effect of ochratoxin A on human placental 3β‐hydroxysteroid dehydrogenase/isomerase in JEG‐3cells at levels relevant to human exposure. Reproductive Toxicology, 38, 47–52. 10.1016/j.reprotox.2013.02.034 [DOI] [PubMed] [Google Scholar]
- Wu QH, Dohnal V, Huang LL, Kuca K, Wang X, Chen GY and Yuan ZH, 2011. Metabolic pathways of ochratoxin A. Current Drug Metabolism, 12, 1–10. [DOI] [PubMed] [Google Scholar]
- Xiao H, Madhyastha S, Marquardt RR, Li S, Vodela JK, Frohlich AA and Kemppainen BW, 1996. Toxicity of ochratoxin A, its opened lactone form and several of its analogs: Structure‐activity relationships. Toxicology and Applied Pharmacology, 137, 182–192. [DOI] [PubMed] [Google Scholar]
- Xu H, Hao S, Gan F, Wang H, Xu J, Liu D and Huang K, 2017. In vitro immune toxicity of ochratoxin A in porcine alveolar macrophages: A role for the ROS‐relative TLR4/MyD88 signaling pathway. Chemico‐Biological Interactions, 25, 107–116. 10.1016/j.cbi.2017.05.016 [DOI] [PubMed] [Google Scholar]
- Yang Q, He X, Li X, Xu W, Luo Y, Yang X, Wang Y, Li Y and Huang K, 2014. DNA damage and S phase arrest induced by Ochratoxin A in human embryonic kidney cells (HEK 293). Mutation Research, 765, 22–31. [DOI] [PubMed] [Google Scholar]
- Yang S, Zhang H, De Saeger S, De Boevre M, Sun F, Zhang S, Cao X and Wang Z, 2015. In vitro and in vivo metabolism of ochratoxin A: a comparative study using ultra‐performance liquid chromatography‐quadrupole/time‐of‐flight hybrid mass spectrometry. Analytical and Bioanalytical Chemistry, 407, 3579–3589. [DOI] [PubMed] [Google Scholar]
- Yang X, Liu S, Huang C, Wang H, Luo Y, Xu W and Huang K, 2017. Ochratoxin A induced premature senescence in human renal proximal tubular cells. Toxicology, 382, 75–83. 10.1016/j.tox.2017.03.009 [DOI] [PubMed] [Google Scholar]
- Yogendrarajah P, Jacxsens L, Saeger S and Meulenaer B, 2014. Co‐occurrence of multiple mycotoxins in dry chilli (Capsicum annum L.) samples from the markets of Sri Lanka and Belgium. Food Control, 46, 26–34. [Google Scholar]
- Yoon S, Cong WT, Bang Y, Lee SN, Yoon CS, Kwack SJ, Kang TS, Lee KY, Choi JK and Choi HJ, 2009. Proteome response to ochratoxin A‐induced apoptotic cell death in mouse hippocampal HT22 cells. Neurotoxicology, 30, 666–676. 10.1016/j.neuro.2009.04.013 [DOI] [PubMed] [Google Scholar]
- Zaied C, Bouaziz C, Azizi I, Bensassi F, Chour A, Bacha H and Abid S, 2011. Presence of ochratoxin A in Tunisian blood nephropathy patients. Exposure level to OTA. Experimental and Toxicologic Pathology, 63, 613–618. 10.1016/j.etp.2010.05.001 [DOI] [PubMed] [Google Scholar]
- Zeljezić D, Domijan AM and Peraica M, 2006. DNA damage by ochratoxin A in rat kidney assessed by the alkaline comet assay. Brazilian Journal of Medical and Biological Research, 39, 1563–1568. [DOI] [PubMed] [Google Scholar]
- Zepnik H, Pähler A, Schauer U and Dekant W, 2001. Ochratoxin A‐induced tumor formation: is there a role of reactive ochratoxin A metabolites? Toxicological Sciences, 59, 59–67. [DOI] [PubMed] [Google Scholar]
- Zepnik H, Voelkel W and Dekant W, 2003. Toxicokinetics of the mycotoxin ochratoxin A in F344 rats after oral administration. Toxicology and Applied Pharmacology, 192, 36–44. [DOI] [PubMed] [Google Scholar]
- Zhang X, Boesch‐Saadatmandi C, Lou Y, Wolffram S, Huebbe P and Rimbach G, 2009. Ochratoxin A induces apoptosis in neuronal cells. Genes and Nutrition, 4, 41–48. 10.1007/s12263-008-0109-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Gan F, Xue H, Liu Y, Huang D, Khan AZ, Chen X and Huang K, 2016. Nephropathy and hepatopathy in weaned piglets provoked by natural ochratoxin A and involved mechanisms. Experimental and Toxicologic Pathology, 68, 205–213. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Fan Z, Nie D, Zhao Z and Han Z, 2019. Analysis of the carry‐over of ochratoxin A from feed to milk, blood, urine, and different tissues of dairy cows based on the establishment of a reliable LC‐MS/MS method. Molecules, 24, 2823–2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T, Xiao Shen XL, Chen W, Liao X, Yang J, Wang Y, Zou Y and Fang C, 2017. Advances in research of nephrotoxicity and toxic antagonism of ochratoxin A. Toxin Reviews, 36, 39–44. 10.1080/15569543.2016.1243560 [DOI] [Google Scholar]
- Zhu LY, Yu T, Qi XZ, Gao J, Huang KL, He XY, Luo HS and Xu WT, 2016a. Limited Link between Oxidative Stress and Ochratoxin A‐Induced Renal Injury in an Acute Toxicity Rat Model. Toxins, 8, pii: E373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Yu T, Qi X, Yang B, Shi L, Luo H, He X, Huang K and Xu W, 2016b. miR‐122 plays an important role in ochratoxin A‐induced hepatocyte apoptosis in vitro and in vivo. Toxicological Research, 5, 160–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerli B and Dick R, 1995. Determination of ochratoxin A at the ppt level in human blood, serum, milk and some foodstuffs by high‐performance liquid chromatography with enhanced fluorescence detection and immunoaffinity column cleanup: methodology and Swiss data. Journal of Chromatography B, 666, 85–99. [DOI] [PubMed] [Google Scholar]
- Zlender V, Breljak D, Ljubojevic M, Flajs D, Balen D, Brzica H, Domijan AM, Peraica M, Fuchs R, Anzai N and Sabolic I, 2009. Low doses of ochratoxin A upregulate the protein expression of organic anion transporters Oat1, Oat2, Oat3 and Oat5 in rat kidney cortex. Toxicology and Applied Pharmacology, 239, 284–296. [DOI] [PubMed] [Google Scholar]