

Abstract
Ferrocene-based phosphines equipped with additional functional groups are versatile ligands for coordination chemistry and catalysis. This contribution describes a new compound of this type, combining phosphine and stibine groups at the ferrocene backbone, viz. 1-(diphenylphosphino)-1′-(diphenylstibino)ferrocene (1). Phosphinostibine 1 and the corresponding P-chalcogenide derivatives Ph2P(E)fcSbPh2 (1E, fc = ferrocene-1,1′-diyl, E = O, S, Se) were synthesized and further converted to the corresponding stiboranes Ph2P(E)fcSb(O2C6Cl4)Ph2 (6 and 6E) by oxidation with o-chloranil. All compounds were characterized by spectroscopic methods, X-ray diffraction analysis, cyclic voltammetry, and theoretical methods. Both NMR spectroscopy and DFT calculations confirmed the presence of P → Sb and P=O → Sb donor–acceptor interactions in 6 and 6O, triggered by the oxidation of the stibine moiety into Lewis acidic stiborane. The corresponding interactions in 6S and 6Se were of the same type but significantly weaker. A coordination study with AuCl as the model metal fragment revealed that the phosphine group acts as the “primary” coordination site, in line with its higher basicity. The obtained Au(I) complexes were applied as catalysts in the Au-catalyzed cyclization of N-propargylbenzamide and in the oxidative [2 + 2 + 1] cyclization of ethynylbenzene with acetonitrile and pyridine N-oxides. The catalytic results showed that the stibine complexes had worse catalytic performance than their phosphine counterparts, most likely due to the formation of weaker coordination bonds and hence poorer stabilization of the active metal species. Nevertheless, the stibine moiety could be used to fine-tune the properties of the ligated metal center by changing the oxidation state or substituents at the “remote” Sb atom.
Short abstract
Manipulation of the functional substituents in 1-(diphenyphosphino)-1′-(diphenylstibino)ferrocene can be used to trigger intramolecular P···Sb and P=E···Sb (E = O, S, Sb) interactions, which have been described using a combination of experimental and theoretical approaches. Oxidation of the stibine moiety into a stiborane unit and the presence of the polarized P=O bond result in the strongest donor-acceptor interaction of this type. As a ligand, the title compound uses the phosphine moiety as a primary donor site.
Introduction
Hybrid ligands1 possessing distinct donor groups often exhibit coordination behavior and catalytic properties different from those of the corresponding monofunctional derivatives. In particular, phosphinoamine (P,N) ligands, combining the homologous donor moieties, stand out due to their structural versatility, the specific chemical properties and reactivity of the two donor groups, and the possibility of tuning their properties through substituents, resulting in wide catalytic applications.2,3 A generally similar situation is encountered in the case of phosphinostibine (P,Sb) ligands possessing heavier pnictogen atoms, which have been studied much less thus far. Even in this case, the donor groups significantly differ. Due to an inefficient mixing of the valence s and p orbitals and their more diffuse nature,4 stibines are worse σ-donors and π-acceptors than phosphines5 and can even behave as electron density acceptors.6 The Lewis acidity of stibines can be enhanced by introducing electron-withdrawing substituents and, alternatively, by their oxidation to Sb(V) compounds (stiboranes), which differentiates them from their phosphorus analogues. Compared to the corresponding phosphines, stibines are less sterically demanding due to longer C–Sb bonds and smaller angles between the substituents, which can result in different coordination preferences.5,7 When combined in one molecule, the phosphine moiety often behaves as the “primary” coordination site, while the stibine group remains uncoordinated or forms additional interactions with Lewis acids or bases.8,9 Prominent examples of phosphinostibine ligands (Scheme 1) include compounds whose functional groups are connected by methylene or phenylene spacers (A10 and B(11)) and the multidonor ligands C and D.12,13
Scheme 1. Examples of Phosphinostibine Donors (Top, for A–D: R = Various Alkyl and Aryl Groups; X = Cl or R) and Ferrocene Stibines (Bottom, for E: Z = NMe2, NHR, NMe3+I–, OH, OR, SR, etc.; for F: n = 2, 3) Studied to Date and the Structure of 1.
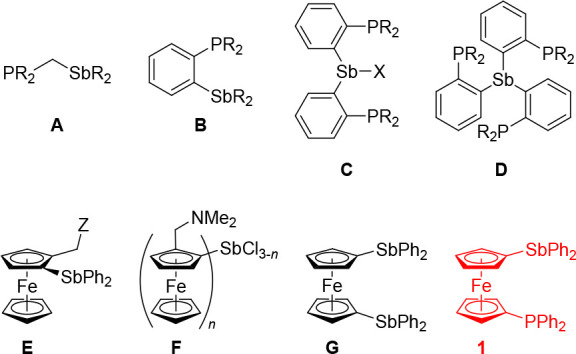
In the chemistry of ferrocene ligands,14 stibine functional groups have only rarely been used. Until recently, ferrocene stibines were limited mainly to compounds E and F comprising a 1,2-disubstituted ferrocene backbone, which have been studied with a focus on the possible D → Sb interactions (D = adjacent donor moiety; for examples, see Scheme 1).15 Earlier this year, we reported the synthesis of ferrocene distibine G (Scheme 1),16 a congener of the iconic ferrocene ligand 1,1′-bis(diphenylphosphino)ferrocene (dppf).17 The facile conversion of this compound into isolable stiboranes and the differences in the reaction behavior of G and dppf led us to focus now on the mixed-donor analogue 1, which represents the missing link between the two symmetrical ligands (Scheme 1). In this contribution, we describe the preparation of this compound and various oxidized derivatives, viz. phosphine-stiborane and phosphine chalcogenide-stiboranes. The resulting compounds are analyzed in view of the difference between the pnictogen donor groups and their possible interactions, which are studied through a combination of experimental and theoretical approaches. Also reported are the results of our preliminary coordination study employing Au(I) as a probe metal ion and the applications of the prepared complexes in gold-catalyzed reactions.
Results and Discussion
Synthesis of Phosphinostibine 1 and the Corresponding P-Chalcogenides and Stiboranes
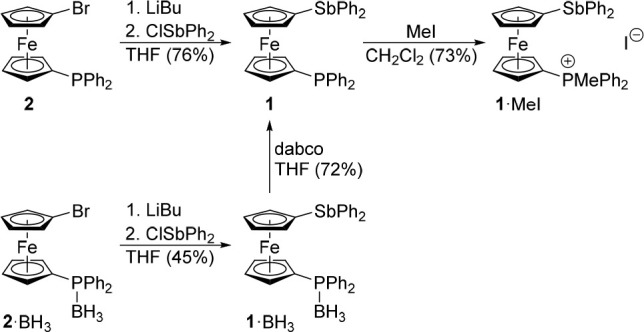
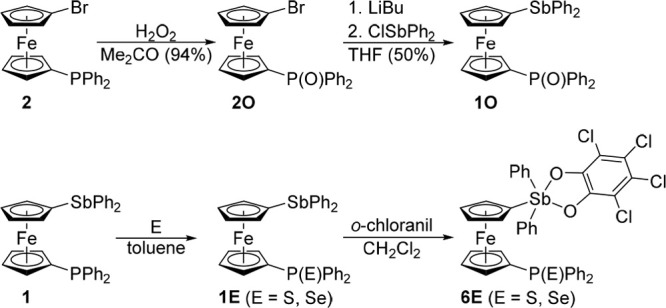
Phosphinostibine 1 was prepared by lithiation of 1-bromo-1′-(diphenylphosphino)ferrocene (2) with n-butyllithium followed by the reaction of the in situ generated lithio intermediate with chlorodiphenylstibine (Scheme 2) and was isolated as an air-stable, orange crystalline solid in 76% yield after column chromatography and crystallization. A similar reaction employing 1-bromo-1′-(diphenylphosphino)ferrocene–borane (1:1) (1·BH3) produced the P-protected phosphinostibine 1·BH3, which was smoothly deprotected18 with 1,4-diazabicyclo[2.2.2]octane (dabco)19 to give 1.
Scheme 2. Synthesis of 1 and Its Reaction with MeI.
Compounds 1 and 1·BH3 were characterized by NMR spectroscopy, ESI MS, and elemental analysis. The 1H and 13C{1H} NMR spectra displayed characteristic signals due to the asymmetrically 1,1′-disubstituted ferrocene units and the phenyl rings, whereas the 31P{1H} NMR spectra showed a sharp singlet for 1 (δP −16.4; cf. −16.2 for (diphenylphosphino)ferrocene20) and a broad doublet-like signal for 1·BH3 (δP 16.4). Although compound 1 crystallized readily, its structure could not be determined with sufficient precision due to disorder. In the crystal, the P and Sb atoms alternated in their positions with only a minor effect on the overall arrangement, which controlled the crystal packing. Notably, this property was also observed for P-chalcogenides Ph2P(E)fcSbPh2 (fc = ferrocene-1,1′-diyl) with lighter chalcogen atoms (O and S), which formed shorter P=E bonds (vide infra). In contrast, the BH3 moiety in 1·BH3 sufficiently “differentiated” the substituents and, thus, allowed the crystal structure to be determined (Figure 1). The structure of 1·BH3 comprises a regular ferrocene unit with parallel cyclopentadienyl rings (dihedral angle 1.5(2)°) and substituents in approximately anti positions (the torsion angle τ = C1–Cg1–Cg2–C6, where Cg1 and Cg2 denote the centroids of the cyclopentadienyl rings C(1–5) and C(6–10), respectively, is 160.3(2)°; see Figure S17). The arrangement of the stibine substituent was similar to that in G(16) or 1-(diphenylstibino)-2-vinylferrocene,15b i.e., with Sb–C(Ph) distances slightly longer than the Sb–C(ferrocenyl) bond and with C(Ph)–Sb–C(Ph) angles wider than the C(ferrocenyl)–Sb–C(Ph) angles. In turn, the geometry of the phosphine part compared well with that in dppf·2BH321 or 1-(diphenylphosphino)-1-methylferrocene–borane (1:1).22
Figure 1.
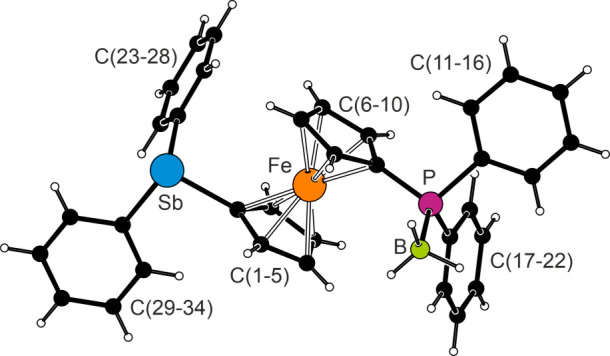
Molecular structure of 1·BH3. Selected distances and angles (in Å and deg): Fe–C(1–10) (range) 2.031(3)–2.058(3), Sb–C1 2.131(3), Sb–C23 2.156(3), Sb–C29 2.165(3), C1–Sb–C23 95.6(1), C1–Sb–C29 94.2(1), C23–Sb–C29 96.4(1), P1–B 1.909(4), P–C6 1.789(2), P–C11 1.816(3), P–C17 1.807(3), C6–P–C11 104.5(1), C6–P–C17 107.4(1), C11–P–C17 104.8(1), B–P–C (range) 111.4(1)-114.6(1). A Displacement ellipsoid plot is available in the Supporting Information.
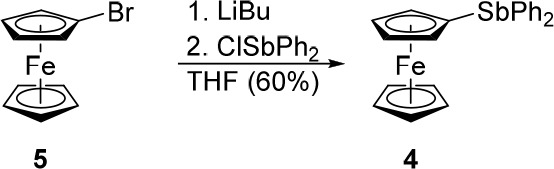
To compare the donor groups present in 1 and to follow their possible interactions, we also prepared the corresponding monofunctional compounds, viz. (diphenylphosphino)ferrocene (3) and (diphenylstibino)ferrocene (4). The previously unreported stibine 4 was synthesized analogously to 1 (Scheme 3), i.e., by the lithiation of bromoferrocene (5) with n-butyllithium and subsequent reaction of chlorodiphenylstibine. The compound was isolated as an air-stable, orange crystalline solid in 60% yield by crystallization and was fully characterized, including structure determination (Supporting Information; Figure S13).
Scheme 3. Synthesis of (Diphenylstibino)ferrocene (4).
To quantify the differences between the phosphine and stibine donor groups, we calculated the methyl cation affinities (MCA)23 of 1, 3, and 4 (Table 1). Defined as the enthalpy of [LB–CH3]+ dissociation (LB = Lewis base), larger MCA values are obtained for stronger Lewis bases (LB). In the present case, the MCA values clearly differentiated the pnictogen donor groups in the model compounds, suggesting that the stibine derivative had lower basicity. The presence of the other substituent in the molecule of 1 had only a minor effect on the MCA values estimated for the individual pnictogen substituents (cf. the respective values for 1 and 3/4). The inclusion of solvation phenomena significantly influenced the MCA values, but the general trend remained the same.
Table 1. Methyl Cation Affinities (MCAs in kJ mol–1 at 298.15 K) for 1, 3, and 4a.
| compound | vacuum | chloroform |
|---|---|---|
| 1 (P) | 672 | 532 |
| 1 (Sb) | 549 | 413 |
| 3 | 666 | 533 |
| 4 | 546 | 430 |
Calculated at the PBE0(d3)/def2-TZVP:sdd(Fe,Sb) level of theory. Solvent effects have been approximated using the PCM model. For 1, the site at which methylation occurred is specified.
These theoretical results corresponded with the outcome of a simple reaction test showing that methylation of 1 with methyl iodide proceeded selectively at the phosphorus atom to afford 1·MeI in 73% isolated yield (Scheme 2), whereas the stibine group remained intact even when 2 equiv of MeI was used.24 Selective formation of 1·MeI was manifested in the NMR spectra, which displayed only one signal attributable to the methyl group, split into a doublet because of interaction with 31P (δH 3.05, 2JPH = 13.2 Hz; δC 11.64, 1JPC = 60 Hz), while the 31P{1H} NMR signal was observed downfield relative to that of the parent compound (δP 24.2; cf. 22.6 for 3·MeI in CD3CN25). An ultimate structure confirmation was provided by X-ray diffraction analysis (see the Supporting Information, Figures S9 and S10). Correspondingly, no borane scrambling between the phosphine and stibine moieties was observed for borane adduct 1·BH3 in solution, consistent with the higher basicity of the phosphine group that renders the P–B adduct more stable.26
No evidence of a P → Sb donor interaction in 1 was observed. To increase the Lewis acidity of the Sb atom and thus make it amenable for the formation of P → Sb dative interactions, we converted phosphinostibine into phosphinostiborane 6 by oxidation with 3,4,5,6-tetrachloro-1,2-benzoquinone (o-chloranil)27 (Scheme 4). According to the results of the NMR analysis, the reaction of o-chloranil (1 equiv) with 1 in dichloromethane (1 h/room temperature) produced a mixture of the expected compound 6, the corresponding phosphine oxide 6O, and unreacted 1 (ratio of 6:6O:1 = 28%:35%:37%). The oxidation reaction lacked the selectivity observed in the similar oxidation of 1,2-Ph2PC6H4SbPh2, during which only the stibine moiety was oxidized.28 Nevertheless, our experiments suggested that the stibine moiety in 1 was oxidized preferentially because the compound Ph2P(O2C6Cl4)fcSbPh2 with an intact stibine moiety was not detected in the crude reaction mixture. The formation of 6O can be explained by the 2-fold oxidation of 1 producing Ph2P(O2C6Cl4)fcSbPh2(O2C6Cl4) and subsequent (partial) hydrolysis by traces of water.29 Indeed, performing the reaction under dry conditions but with commercial o-chloranil improved the yield of 6 and decreased the amount of hydrolysis product 6O (6:6O:1 = 37%:29%:34%). The components of the reaction mixture were separated by chromatography. Despite the changes in the crude product composition, the isolated yield of 6 remained at approximately 20% due to reactions of this compound with the stationary phase used (silica gel) that resulted in irreversible binding, presumably after hydrolysis of the stiborane moiety. The facile hydrolysis of the presumed doubly oxidized intermediate Ph2P(O2C6Cl4)fcSbPh2(O2C6Cl4) was advantageously used to prepare 6O, which was obtained as the main product upon adding 2 equiv of o-chloranil to a dichloromethane solution of 1 containing a few drops of water. Subsequent workup and crystallization afforded 6O in 65% isolated yield (Scheme 4).
Scheme 4. Oxidation of 1 with o-Chloranil.
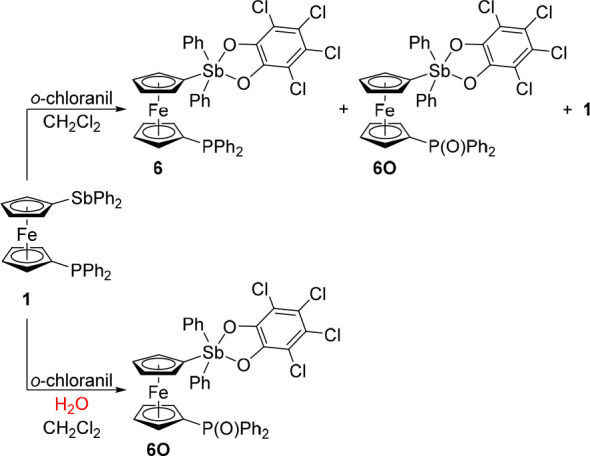
Next, the family of stibine and stiborane derivatives was expanded by compounds featuring heavier P-chalcogenides30 to establish the possible influence of the chalcogenide donor atom on the E → Sb interaction (Scheme 5). The oxidation changes not only the possible donor atom and the donor···Sb distance but also the electron density distribution in the system. Thus, phosphine oxide 1O was obtained in two steps from phosphine-bromide 2, which was oxidized by hydrogen peroxide to 2O and subsequently lithiated and reacted with ClSbPh2 to produce 1O (47% yield over the two steps after crystallization; N.B. the direct oxidation of 1 with hydrogen peroxide was not used due to side reactions at the stibine moiety). Phosphine sulfide 1S and selenide 1Se were obtained directly by reacting 1 with the corresponding chalcogens in refluxing toluene. The yields were 92% and 83% after crystallization, respectively. Compounds 1S and 1Se underwent clean oxidations with o-chloranil (1 equiv) to produce 6S and 6Se, respectively (∼95%; ∼5% of 1E remained unreacted). Despite practically complete conversion, the sulfide was purified by column chromatography and isolated in only 67% yield because it remained partly adsorbed on the silica gel column (most likely after hydrolysis, vide supra). Selenide 1Se could not be purified similarly due to decomposition and adsorption on the column. Alternatively, it was crystallized from hot heptane (66% yield). Increasing the amount of oxidant to 1.1 equiv resulted in complete conversion but also led to decomposition during isolation.
Scheme 5. Synthesis of Phosphine Chalcogenides 1E and the Corresponding Stiboranes.
The oxidized phosphine moieties in stibines 1E (E = O, S, and Se) showed characteristic, downfield-shifted 31P NMR signals (Table 2) and increased JPC coupling constants31 compared to 1. Notably, the chemical shifts were similar to the values reported for chalcogenides derived from 3 (FcP(E)Ph2; E = O, δP 30.3,32 E = S, 41.2 in C6D6,33 and E = Se, 32.7;20b Fc = ferrocenyl), which indicated the absence of significant P=E···Sb interactions. The1JSeP coupling constant determined for 1Se (735 Hz) was higher than that for FcP(Se)Ph2 (731 Hz),20b suggesting a lower basicity of the phosphine group34 in 1, which can be ascribed to the presence of an electron-withdrawing stibine substituent that decreased electron density at the ferrocene unit and thus rendered the phosphine less basic.
Table 2. 31P NMR Shifts (δP in ppm) of Compounds 1 and 6a.
| compound | δP | compound | δP | ΔδPc |
|---|---|---|---|---|
| 1 | –16.4 | 6 | –9.7 | +6.7 |
| 1O | 29.3 | 6O | 39.1 | +9.8 |
| 1S | 41.9 | 6S | 41.4 | –0.5 |
| 1Se | 32.1 [735]b | 6Se | 31.8 [726]b | –0.3 |
The spectra were recorded in CDCl3 at 25 °C.
1JSeP coupling constant in Hz.
Chemical shift difference between 6 and 1.
The structure determination of 1S and 1Se ruled out the presence of Sb···E interactions even in the solid state (the structure of 1O was severely disordered and could not be satisfactorily refined). The sulfide 1S crystallized as a racemic twin (monoclinic space group Cc) with positional disorder of the SbPh2 and P(S)Ph2 moieties, similar to 1 (vide supra). No such problems were encountered in the structure of 1Se. The structures of 1S and 1Se were generally similar (Figure 2 and Table 3) with parallel cyclopentadienyl rings and substituents in approximately anti positions (see the τ angles in Table 3). A difference was observed in the mutual positioning of the substituents as the Ph2P(S) group was directed with its S atom away from the lone pair at the Sb atom, while the Se atom in 1Se pointed in the same direction (Figure S17). The parameters of the stibine group were similar to those in G and 1·BH3, while the geometry of the phosphorus substituents compared well with those observed in FcP(S)Ph2,33 dppfE2,35 and related compounds.36
Figure 2.
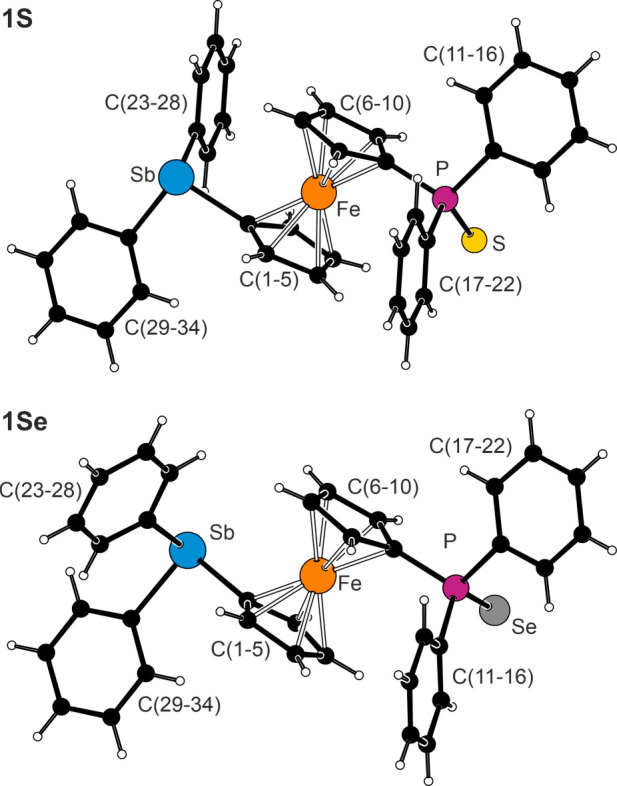
Molecular structures of 1S and 1Se (for the sake of clarity, only one position of the disordered substituents in 1S is shown).
Table 3. Selected Distances and Angles for 1S and 1Se (in Å and deg).
| parametera | 1S (E = S)c | 1Se (E = Se) |
|---|---|---|
| Sb–C1 | 2.125(3) | 2.130(2) |
| Sb–C23/C29 | 2.160(4)/2.146(4) | 2.156(2)/2.165(2) |
| C–Sb–Cb | 94.9(2)–97.2(2) | 94.06(7)–96.74(7) |
| P=E | 1.954(2) | 2.1034(7) |
| P–C6 | 1.796(4) | 1.784(2) |
| P–C11/C17 | 1.816(4)/1.806(4) | 1.810(2)/1.811(2) |
| C–P–Cb | 104.9(2)–106.8(2) | 104.72(8)–107.25(8) |
| Fe–C | 2.036(4)–2.060(4) | 2.034(2)–2.059(2) |
| tilt | 2.0(2) | 1.2(1) |
| τ | 169.0(3) | –161.4(1) |
Fe–C is the range of the Fe–C(1–10) bond lengths, tilt stands for the dihedral angle of the least-squares cyclopentadienyl planes C(1–5) and C(6–10), and τ denotes the torsion angle C1–Cg1–Cg2–C6, where Cg1 and Cg2 are the centroids of the respective cyclopentadienyl rings.
The range of the C1–Sb–C23/29 and C23–Sb–C29 angles.
Data for the major orientation.
In contrast, the NMR spectra of stiboranes 6 and 6O suggested possible P → Sb and P=O → Sb interactions, as the 31P NMR signals shifted downfield relative to those of respective stibines 1 and 1O (Table 2). For phosphinostiborane 6, the interaction was further indicated by the splitting of the 13C{1H} NMR signals due to CH and Cipso carbons in the Sb-bound C5H4 ring and Cipso of SbPh2 with 31P, while no such coupling was observed for 1. Conversely, the 1H NMR spectra remained virtually unaffected, displaying only signals attributable to a conformationally unconstrained, PIII-substituted ferrocene-1,1′-diyl unit, namely, three apparent triplets and one apparent quartet due to the C5H4 rings, albeit at a lower field compared to 1 because of the increased electron-withdrawing character of the stiborane substituent (this is consistent with the trend in the redox potentials of ferrocene oxidation, vide infra).
In addition, the 1H and 13C{1H} NMR spectra of 6O were broadened, suggesting a dynamic structure on the NMR time scale. This was confirmed by a VT 1H NMR study (Figure 3 and Figure S1): the spectrum recorded at −50 °C displayed eight separate signals for the ferrocene CH groups, which became diastereotopic due to a fixed conformation at a low temperature (the ferrocene moiety became axially chiral). Upon increasing the temperature, the signals broadened, and at 25 °C, only three signals were observed for the C5H4 protons due to time averaging.
Figure 3.
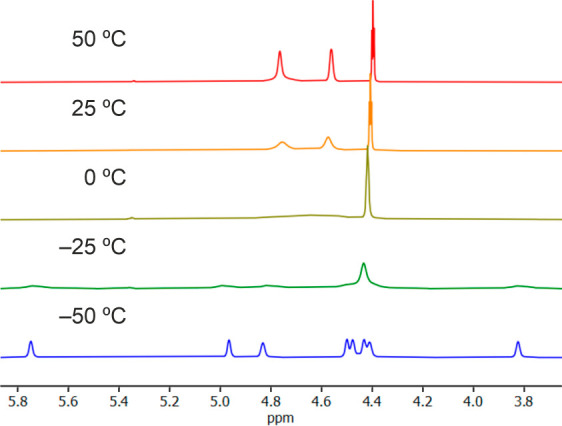
VT 1H NMR spectra (400 MHz, CDCl3) of 6O showing the region of ferrocene protons (complete spectra are available in the Supporting Information).
Attempts to disrupt the intramolecular P=O → Sb interaction through the addition of competing Lewis bases failed. The NMR spectrum of 6O remained unchanged upon addition of 4-(dimethylamino)pyridine (1.0 equiv), triethylphosphine oxide (1.3 or 10 equiv), or triphenylphosphine chalcogenides (1.5 equiv; Figure 4 and Figures S2–S4) in CDCl3, and even the spectrum recorded in DMSO-d6 as a strongly donating solvent suggested that an intramolecular interaction was present. Conversely, the addition of BF3·OEt2 (1 or 5 equiv) as a competing Lewis acid to 6O cleaved the P=O → Sb dative bond, presumably with concomitant formation of the phosphine oxide–borane adduct Ph2P(O)fcSbPh2(O2C6Cl4)·BF3 (Figure S5). Analogous reaction with B(C6F5)3 resulted in decomposition. Similar competing experiments with 6 showed that the intramolecular P → Sb interaction was efficiently canceled by adding 1.5 equiv of Et3PO, very likely with concomitant formation of 6·Et3PO (Figure 4).
Figure 4.
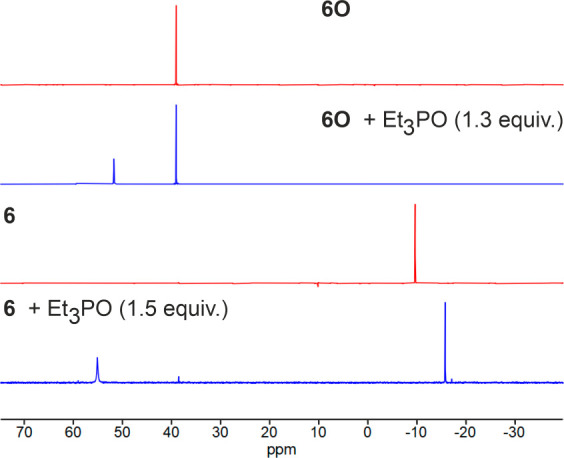
31P{1H} NMR spectra (162 MHz, CDCl3, 25 °C) of 6, 6O, and their mixtures with Et3PO.
The intramolecular interactions were clearly detected in the crystal structures of 6·C6H14 and 6O·CHCl3 (Figure 5 and Table 4); compounds 6S and 6Se did not provide suitable crystals despite numerous attempts. The P → Sb interaction in the molecule of 6 was suggested by the short P···Sb distance (3.0987(6) Å), which is approximately halfway between the sum of the van der Waals radii (3.86 Å)37 and the sum of the covalent radii (2.46 Å) of these atoms.38 Compared to 1·BH3 containing an intact SbPh2 group, the substituents at the Sb atom were moved apart to provide space for the phosphorus lone pair. For 6·C6H14, this can be illustrated by wider C–Sb–C angles (∼99–102°) and, mainly, by the τ5 index of 0.03, which was close to the value expected for an ideal square pyramid (τ5 = 0; an ideal trigonal bipyramid yields τ5 = 1).39 The Sb atom was located 0.28 Å above the {O1, O2, C1, and C29} basal plane, whose minor distortion resulted from the narrower O1–Sb–O2 angle (78.93(5)°) associated with the chelating catecholate ligand (the remaining angles between the basal donor atoms were 86.84(6)–101.95(6)°).
Figure 5.
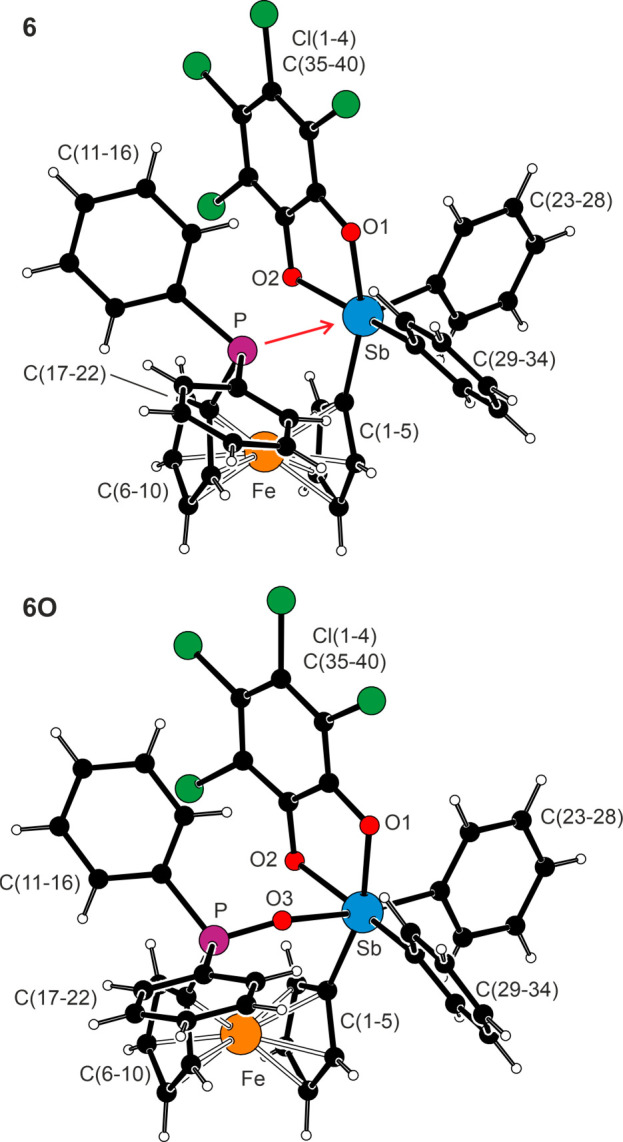
Molecular structures of 6·C6H14 and 6O·CHCl3 (the solvent molecules and the less populated orientation of phenyl ring C(17–22) in the molecule of 6 have been omitted for clarity). The P → Sb interaction in molecule 6 is indicated by a red arrow.
Table 4. Selected Distances and Angles for 6 and 6O·CHCl3 (in Å and deg).
| parametera | 6·C6H14 (X = P) | 6O·CHCl3 (X = O3)d |
|---|---|---|
| Sb···X | 3.0987(6) | 2.256(1) |
| Sb–O1/2 | 2.068(1)/2.082(1) | 2.064(1)/2.076(1) |
| Sb–C1 | 2.127(2) | 2.116(2) |
| Sb–C23/C29 | 2.124(2)/2.138(2) | 2.147(2)/2.126(2) |
| C–Sb–Ob | 161.65(5)/159.94(5) | 160.93(6)/162.59(6) |
| P–C6 | 1.811(2) | 1.779(2) |
| P–C11/C17 | 1.839(2) | 1.803(2)/1.796(2) |
| C6–P–C11/C17 | 101.90(8)/–c | 107.07(7)/107.07(8) |
| C11–P–C17 | 101.3(1) | 106.04(8) |
| Fe–C | 2.022(2)–2.057(2) | 2.034(2)–2.049(2) |
| tilt | 4.0(1) | 2.9(1) |
| τ | –14.1(1) | 5.1(1) |
Despite the vicinity of the phosphoryl oxygen atom O3, the geometry around the Sb atom in 6O·CHCl3 remained square pyramidal (τ5 = 0.03) and was similarly distorted. The Sb atom was displaced by 0.23 Å from the basal plane, and the O3–Sb–C23 angle was nearly linear (172.05(5)°). Even in this case, the Sb···O3 separation (2.256(1) Å) was well below the sum of the van der Waals radii (3.58 Å) but longer than the sum of the covalent radii (2.05 Å). Notably, the O···Sb distance in 6O·CHCl3 was significantly shorter than that in (2-Ph2P(O)C6H4SbPh3)[BF4] (2.432(2) Å), where the P–O···Sb fragment is bent (∼116°) due to the geometric constraints imposed by the o-phenylene backbone.40 Compared to 6, compound 6O showed shorter P–C bonds and wider C–P–C angles, which is a trend detectable also in the FcPPh2 (3)/FcP(O)Ph2 pair.41
Analysis of the Bonding Situation
The nature of the P/P=E → Sb interactions was studied by DFT calculations. Initially, we analyzed the calculated electron densities using the quantum theory of atoms in molecules (QTAIM) approach.42 The key parameters are summarized in Table 5 and in the Supporting Information (Figure S24).
Table 5. Electron Densities (ρbcp), Laplacians of the Electron Density (∇2ρbcp), Total Electron Densities (H), Ratios of Potential and Kinetic Energy Density (|V|/G), and Ratios of Kinetic (G/ρbcp) and Total Energy Density (H/ρbcp) to Electron Density at the Bond Critical Point (bcp) Located between the Antimony Atom and a Donor Atom and the Experimental and Calculated Bond Distances.
| bond length
(Å) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| compd | bond | exp | calca | ρbcp (e Å–3) | ∇2ρ(r) (e Å–5) | H (au) | |V|/G (au) | G/ρbcp (au) | H/ρbcp (au) |
| 6 | P···Sb | 3.0987(6) | 2.975 | 0.037 | 0.028 | –0.75 × 10–2 | 1.77 | 0.26 | –0.20 |
| 6O | O···Sb | 2.256(1) | 2.293 | 0.055 | 0.173 | –1.22 × 10–2 | 1.44 | 0.51 | –0.22 |
| 6S | S···Sb | nab | 2.915 | 0.034 | 0.044 | –0.56 × 10–2 | 1.48 | 0.35 | –0.17 |
| 6Se | Se···Sb | nab | 2.996 | 0.034 | 0.034 | –0.62 × 10–2 | 1.60 | 0.30 | –0.18 |
Calculated at the PBE0(d3)/def2-TZVP:sdd(Sb,Fe) level of theory.
Not available.
The Laplacian of the electron density at the bond critical points (∇2ρbcp) between antimony and the donor atom was positive for all stiboranes, indicating some type of closed-shell interaction (ionic, dative, or van der Waals). These weak noncovalent interactions can be distinguished from dative bonds by comparing the relative amounts of potential and kinetic energy at the bond critical point (bcp).43,44 Specifically, a covalent bonding interaction is indicated by a potential energy density (Vbcp, always negative) greater than the kinetic energy density (Gbcp, always positive), which yields a negative total energy density (H = V + G < 0) or, alternatively, |V|/G > 1. The pertinent values indicated that the bonding interactions in stiboranes 6 and 6E were dative bonds (P → Sb or P=E → Sb). Further inspection of the ratios of the kinetic and total energy density to the electron density (ρbcp), viz. G/ρbcp and H/ρbcp, revealed a slightly higher electrostatic contribution for the P=E → Sb interactions (reflected by more positive G/ρbcp values) than for the P → Sb bond and a comparable degree of covalency (reflected by similarly negative H/ρbcp values). Both indices were the highest for stiborane 6O, suggesting the strongest interaction in this compound, which corresponds with the experimental results.
This was consistent with the calculated energy difference between the “open” and “closed” forms of stiboranes 6 and 6O (Table 6), which clearly favored the closed form, where the phosphorus groups and stiborane moieties are oriented toward each other and interact (for the structure diagrams, see the Supporting Information, Figure S25). The energy difference, which can be taken as a measure of the strength of the interaction (N.B. the closed form is expected to be destabilized sterically, which hampers the interaction), in absolute values, decreased from 6O to 6 to 6S/6Se for isolated species under a vacuum, and the same trend was noted even when considering the solvent effects. However, while the inclusion of solvation phenomena resulted in almost no change in the energy difference for 6, the values for compounds featuring polar P=E bonds were more affected. In particular, the energy difference for the phosphine oxide 6O decreased by 12 kJ mol–1 upon accounting for the solvation effects, which was attributed to the strong polarization of the P=O bond toward P+–O–.45 Notably, the energy differences determined for 6 and 6O were significantly higher than the energy barrier for the rotation of the cyclopentadienyl rings in ferrocene itself, which was estimated to be approximately 4 kJ mol–1 in the gas phase;46 those in 6S and 6Se were of the same order. The P → Sb interaction in 6 was also manifested by a decreased fluoride ion affinity47 (305 kJ mol–1) compared to the model compound FcSbPh2(O2C6Cl4) (341 kJ mol–1), indicating a decreased Lewis acidity of the stiborane moiety in 6 as the result of P → Sb donation.48
Table 6. Free Energy Differences (ΔG in kJ mol–1 at 298.15 K) between the Open and Closed Isomers of Stiboranes 6, 6O, 6S, and 6Sea.
| compound | vacuum | chloroform |
|---|---|---|
| 6 | –26 | –25 |
| 6O | –49 | –37 |
| 6S | –10 | –5 |
| 6Se | –10 | –5 |
Defined as ΔG = G(closed) – G(open) and calculated at the PBE0(d3)/def2-TZVP:sdd(Fe,Sb) level of theory. Solvent effects have been approximated using the PCM model.
The presence of dative interactions was further indicated by the calculated Mayer bond orders (MBOs) and Wiberg bond indices (WBIs) (Table 7), which represent the number of electrons shared between two interacting atoms. The lowest values found for stiborane 6O seemingly did not correspond to the experimental and theoretical results but indicated a more pronounced role for the electrostatic contribution to bonding. The dative interactions between the stiborane and phosphine/phosphine-chalcogenide moieties were visualized using intrinsic bond orbital (IBO) analysis.49 The identified IBOs corroborated the donation of electron density from lone electron pairs located on either the phosphorus or chalcogenide atom to antimony (Figure 6).
Table 7. Selected Mayer Bond Orders (MBOs) and Wiberg Bond Indices (WBIs) for 6 and 6Ea.
| MBO |
WBI |
|||
|---|---|---|---|---|
| compound | P/E···Sb | P=E | P/E···Sb | P=E |
| 6 | 0.49 | na | 0.22 | na |
| 6O | 0.13 | 1.44 | 0.19 | 1.92 |
| 6S | 0.55 | 1.44 | 0.21 | 1.65 |
| 6Se | 0.57 | 1.35 | 0.25 | 1.49 |
Calculated at the PBE0(d3)/def2-TZVP:sdd(Fe,Sb) level of theory. na = not applicable.
Figure 6.

Selected intrinsic bond orbitals (IBOs) of stiboranes 6, 6O, 6S, and 6Se. Values in parentheses indicate the fraction of bonding electrons assigned to the individual atoms (lp = lone electron pair).
The IBO analysis further revealed bonding differences between the phosphine chalcogenide moieties (see the Supporting Information, Figures S26–S28), although the overall description complied well with the generally accepted bonding scheme.50 The IBO corresponding to the P–O σ-bond in 6O was largely located at the oxygen atom, confirming the highly polarized nature of this bond. In contrast, the corresponding IBOs in 6S and 6Se showed an equal distribution of the bonding electron pairs between the two atoms (phosphorus and chalcogen). Similarly, the π-component of the P–O bond in 6O differed from those of its heavier congeners, as all three oxygen lone electron pairs were involved in π-interactions with the phosphorus atom, although two of them were involved only to a limited extent. For 6S and 6Se, only two electron pairs were involved in the π-bonding interaction, leaving one electron pair essentially nonbonding at the chalcogen atom.51
Electrochemistry
The electrochemical behavior of compounds 1 and 6 was studied by cyclic voltammetry on a Pt-disk electrode in dichloromethane by using Bu4N[PF6] as the supporting electrolyte. In the accessible range (approximately −1.5 to 2 V vs the ferrocene/ferrocenium reference52), compound 1 showed a single redox transition (Figure 7), which was essentially reversible and diffusion-controlled, as indicated by ipa ∝ ν1/2 (ipa is the anodic peak current and ν the scan rate; Figures S18 and S19). Such behavior contrasted with the redox response of dppf53 and analogous compounds,54 whose electrochemical oxidation was typically associated with follow-up reactions that decreased the reversibility of the electrochemical oxidation. In line with the electron-withdrawing nature of the substituents in 1, the oxidation occurred 0.14 V more positive than that of ferrocene itself (Table 8) but at a slightly lower potential than the oxidation of dppf under similar conditions (E°′ = 0.17 V vs ferrocene/ferrocenium),53b reflecting the lower electronegativity of Sb. The redox responses of 1O and 1S were similar, except that the redox waves were shifted toward more positive potentials as a result of the increased electron-withdrawing character of the P(E)Ph2 substituents (Figure 7). The voltammogram of 1S displayed an additional reductive wave when the scan range was extended toward more positive potentials (Figure S20). Conversely, the oxidation of 1Se was irreversible and multielectron in nature.55 At more positive potentials, the selenide underwent another ill-defined oxidation (Epa ≈ 0.89 V) and showed several weak reductive waves upon a reverse scan (Figure S20).
Figure 7.
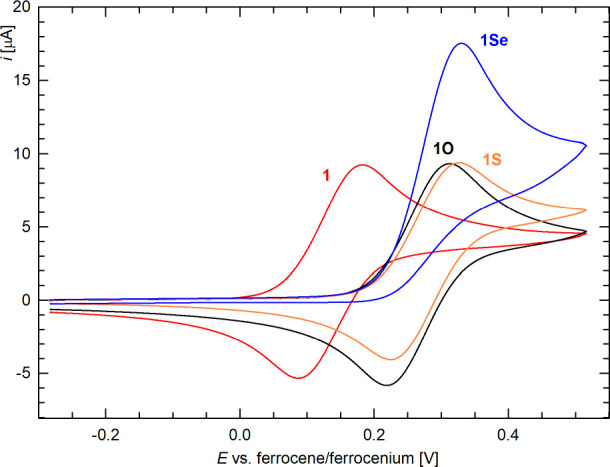
Cyclic voltammograms of 1 and 1E (Pt-disk electrode, CH2Cl2, 0.1 M Bu4N[PF6], scan rate 0.1 V s–1).
Table 8. Summary of the Electrochemical Dataa.
The measurements were conducted at the Pt-disk electrode in dichloromethane containing 0.1 M Bu4N[PF6]. The potentials are given in volts relative to ferrocene/ferrocenium references (for details, see the Supporting Information). The potentials for reversible processes were determined as the average of anodic (Epa) and cathodic (Epc) peak potentials, E°′ = 1/2 (Epa + Epc). The separation of the peaks in the cyclic voltammograms was approximately 90 mV due to a large resistance. The decamethylferrocene standard showed similar values.
Irreversible wave. The anodic peak potential (Epa) at a scan rate of 100 mV s–1 is given.
Based on the results of DFT calculations, the primary oxidation of 1 was assigned to the ferrocene/ferrocenium transition. Although inspection of the frontier orbitals (Figure 8) using natural atomic orbitals (NAOs) showed that the HOMO of 1 corresponds mainly to the lone electron pair of the PPh2 group, being composed of the phosphorus 3s (∼11%) and 3p (∼47%) orbitals with a contribution from the 2p orbitals of the proximal carbon atoms (∼29%) and the iron 3d orbitals (∼5%), a change in the electron density from 1 to 1+, ρ(1+) – ρ(1), mapped at the equilibrium geometry of 1,56 occurred exclusively at the ferrocene iron, which supported the assignment of the electrochemical oxidation as ferrocene-based. Conversely, the LUMO of 1 was the antibonding combination of carbon 2p orbitals in the π-system of one phenyl ring at the PPh2 group.
Figure 8.
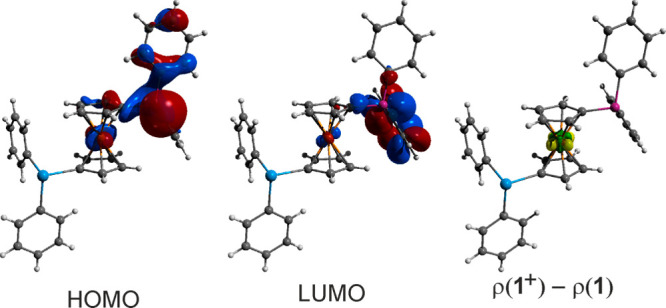
Frontier orbitals (isosurface at ±0.04 au) and the electron difference map ρ(1) – ρ(1+ mapped at the geometry of 1) (isosurface at ±0.02 au) at the PBE0(d3)/def2TZVP level of theory.
The first oxidation of catecholatostiboranes 6, 6O, and 6S (Figure 9 and Figures S21–S23) was also reversible when they were scanned separately (i.e., when the switching potential was set just after the first redox wave). At higher potentials, however, the compounds displayed several irreversible oxidations, which affected the overall response (e.g., by decreasing the intensity of the first reductive wave and through additional broad reduction features) and resulted in weak reductive waves during the reverse scans. In the case of 6Se, even the first oxidation was irreversible, and several weak reductive waves were observed, suggesting the instability of the electrochemically generated species. Compared to that of the corresponding stibines, the oxidation of 6 and 6E was shifted by 0.13–0.16 V toward more positive potentials (Table 8), which indicated a decrease in the electron density at the ferrocene unit upon converting the stibine group into the strongly electron-withdrawing stiboranyl moiety.57
Figure 9.
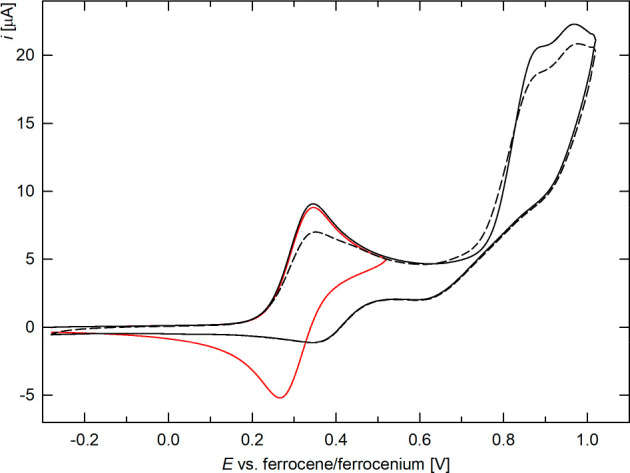
Cyclic voltammograms of 6 (Pt-disk electrode, CH2Cl2, 0.1 M Bu4N[PF6], scan rate of 0.1 V s–1). The second scan is shown by a dashed line. The peak potentials of the irreversible oxidations are approximately 0.87 and 0.97 V.
Gold Complexes
In view of the intended catalytic testing, the coordination properties of 1 were investigated through reactions with Au(I) precursors. Thus, the reaction of 1 with [AuCl(SMe2)] at a 1:1 molar ratio in dichloromethane produced the phosphine complex [AuCl(1-κP)] (7) as the sole product (Scheme 6). When the amount of the gold(I) precursors was increased to 2 equiv, a similar reaction afforded the digold(I) complex [(μ(P,Sb)-1)(AuCl)2] (8). Compared to 7, however, digold complex 8 was much less stable, decomposing rapidly in solution and even when stored as a solid at low temperatures in the dark. Attempts to prepare P,Sb-bridged digold complexes via removal of chloride from 7 with Ag[SbF6] or by the reaction of 1 with [Au(tht)2][SbF6] (Au:1 = 1:1) containing the easily dissociating tetrahydrothiophene ligands (tht) were unsuccessful. Reactions of model compounds 3 and 4 with [AuCl(SMe2)] (1 equiv) produced the respective chlorogold(I) complexes, [AuCl(FcPPh2-κP)] (9)58 and [AuCl(FcSbPh2-κP)] (10) (Scheme 6). Even in this pair, the stibine complex was considerably less stable than its phosphine analogue, decomposing in both solution and the solid state.
Scheme 6. Synthesis of AuCl Complexes.
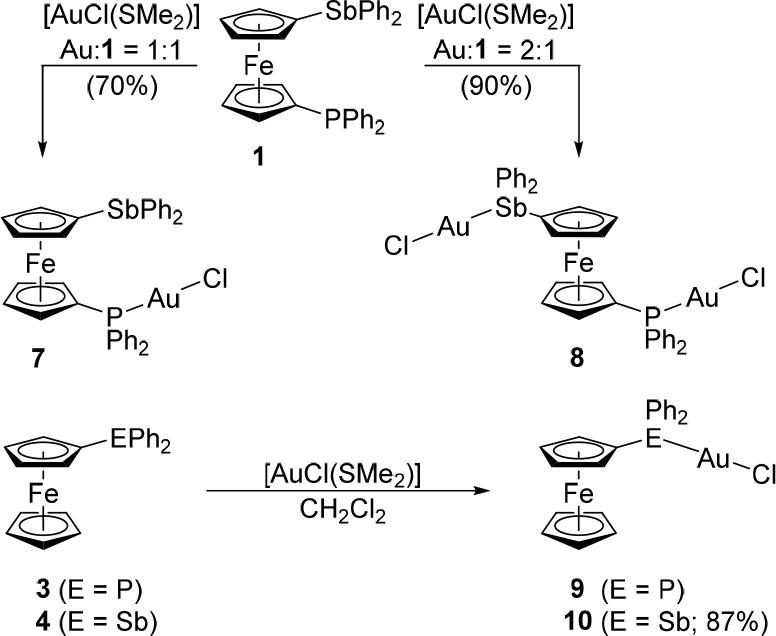
The coordination of the phosphine moiety was indicated by a downfield shift of the 31P{1H} NMR signal (δP 28.9 and 27.4 for 7 and 8, respectively) and changes in the 1H and 13C{1H} NMR spectra (e.g., the 13C{1H} NMR signal due to Cipso-P in 7 was shifted to a higher field, and the 1JPC coupling constant increased to 73 Hz from 7 Hz in the free ligand). The coordination of the stibine moiety had no distinct marker (such as the 31P chemical shift) but was indicated by changes in the 1H and 13C{1H} NMR spectra.59 For 9, coordination increased the 1H NMR chemical shifts due to ferrocene protons, and the signal due to ferrocene Cipso shifted upfield (signals due to CH groups experienced smaller changes). The ESI MS of 7 and 8 showed ions due to [M – Cl]+; the spectrum of 10 displayed a major peak due to [Au(4)2]+ resulting by ligand redistribution and a minor peak of [M – Cl + Me2CO]+.
Complex 7 reacted cleanly with o-chloranil and thionyl chloride to produce stable complexes 11 and 12, respectively, which were isolated in 78% and 96% yields (Scheme 7). The NMR spectra of these compounds showed the expected signals, including those due to the tetrachlorocatecholate ligand for 11. The 31P{1H} NMR resonances were only marginally affected (δP ≈ 28), and ESI MS revealed ions attributable to the sodiated species [M + Na]+.
Scheme 7. Oxidation of Complex 7.
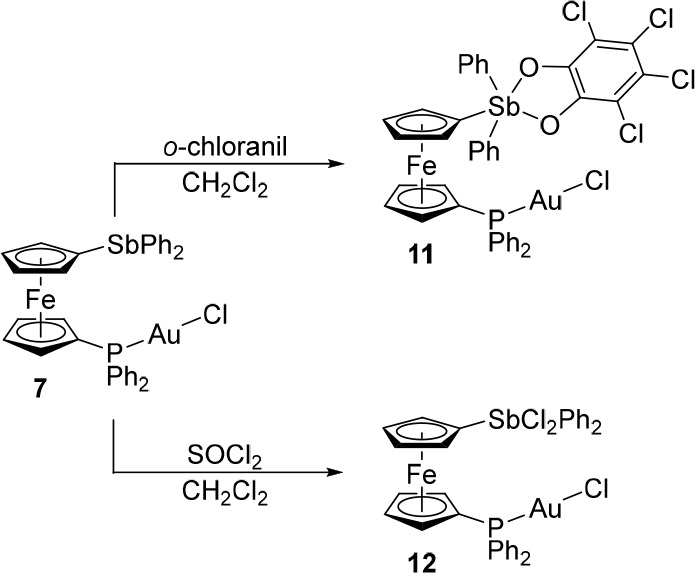
The structures of 7 and 12·0.4CHCl3 (Figure 10 and Table 9) comprised linear P–Au–Cl moieties (∼175°) with Au–P and Au–Cl distances similar to those in [AuCl(PPh3)]60 and 9.58 The oxidation changed the geometry at the antimony atom from ψ-tetrahedral to trigonal bipyramidal and shortened the Sb–C bonds (cf. the structures of G and the corresponding bis(stiborane), [Fe(η5-C5H4SbPh2Cl2)2]).16 The τ5 parameter for the stiborane moiety in 12 was 0.86, reflecting that although the Cl–Sb–Cl (axial) angle was close to the ideal 180°, the C–Sb–C angles varied (∼115–128°) for steric reasons. The ferrocene units assumed their regular geometry and were negligibly tilted, but the increased steric bulk of the Sb substituent in 12 was reflected by a more open conformation of the ferrocene unit.
Figure 10.
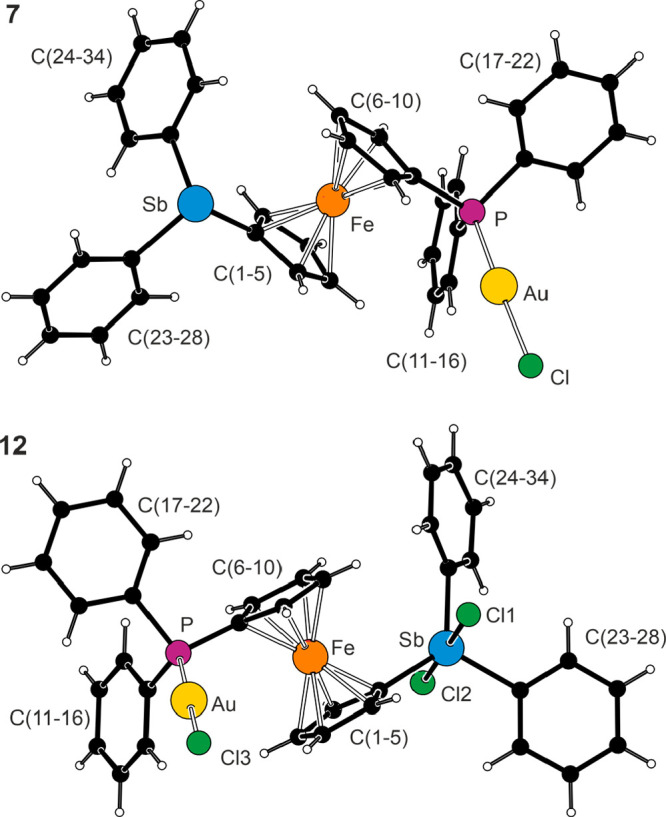
Views of the complex molecules in the structures of 7 and 12·0.4CHCl3.
Table 9. Selected Distances and Angles for 7 and 12·0.4CHCl3 (in Å and deg)a.
| Parameter | 7 (X = void) | 12·0.4CHCl3 (X = Cl)b |
|---|---|---|
| Au–Cl | 2.2922(7) | 2.2864(7) |
| Au–P | 2.2268(7) | 2.2286(6) |
| P–Au–Cl | 174.47(3) | 175.34(3) |
| Sb–X1/2 | na | 2.4356(6)/2.4875(6) |
| Sb–C1 | 2.130(2) | 2.096(2) |
| Sb–C23/C29 | 2.157(3)/2.152(2) | 2.107(2)/2.121(3) |
| C1–Sb–C23/C29 | 95.03(9)/95.16(9) | 117.08(8)/128.10(9) |
| C23–Sb–C29 | 95.95(9) | 114.71(9) |
| P–C6 | 1.787(2) | 1.786(2) |
| P–C11/C17 | 1.841(2)/1.818(3) | 1.811(2)/1.819(2) |
| C6–P–C11/17 | 106.2(1)/103.9(1) | 108.1(1)/103.7(1) |
| C11–P–C17 | 105.4(1) | 103.7(1) |
| Fe–C | 2.028(3)–2.064(3) | 2.034(2)–2.062(2) |
| tilt | 2.1(2) | 1.1(2) |
| τ | 160.8(2) | 174.0(2) |
The molecular structure of stibine complex 10 (Figure 11) was unexceptional in view of the data determined for 7, 12·0.4CHCl3, and [(μ(Sb,Sb)-G)(AuCl)2]16 (vide supra). The prominent feature that differentiated 10 from the reference compounds was the presence of intermolecular aurophilic interactions61 between two independent molecules present in the structure (Z′ = 2). The Au1···Au2 distance was 2.9992(5) Å, and the interacting P–Au–Cl units were approximately perpendicular to each other (torsion angle Cl1–Au1···Au2–Cl2 was 102.89(4)°). In turn, this suggested that aurophilic interactions can also be responsible for the multiplication of the molecules in the asymmetric unit62 by linking them into supramolecular molecular arrays that behave as the real repeating unit. This hypothesis was verified through a search in the Cambridge Structural Database63 for structures with intramolecular Au···Au distances in the arbitrary 2.7–3.2 Å range and with Z′ > 1 (i.e., with two or more formula units per asymmetric unit), which resulted in approximately 120 hits (duplicate structures were excluded).64
Figure 11.
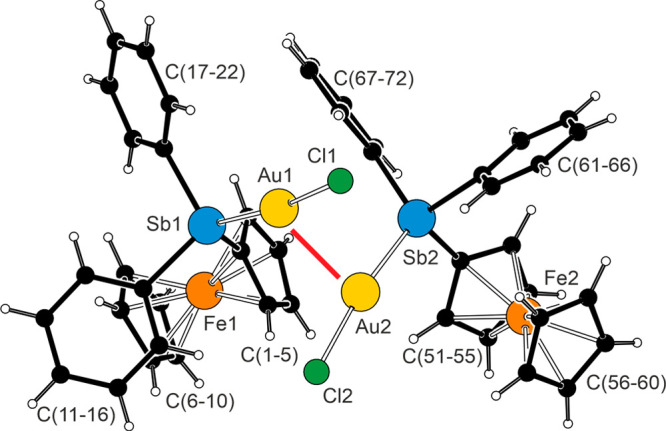
View of the two crystallographically independent molecules of 10. The Au···Au interaction (Au1···Au2 = 2.9992(5) Å) is indicated by a red line. Selected distances and angles (in Å and deg) for molecule 1 [molecule 2]: Au–Sb 2.4939(4) [2.4945(5)], Au–Cl 2.288(1) [2.3034(9)], Sb–Au–Cl 172.76(3) [169.36(3)].
Catalytic Experiments
Gold(I) complexes 7, 9, 10, and 12,65 activated in situ with AgNTf2, were applied in Au-mediated cyclization of N-propargylbenzamide (13) into 4,5-dihydro-5-methylene-2-phenyloxazole (14) (Scheme 8).66 The reactions were performed with 1 mol % of the gold catalyst in CD2Cl2 at 25 °C and followed by 1H NMR spectroscopy.67
Scheme 8. Gold-Catalyzed Cyclization of N-Propargylbenzamide (13) to Oxazole 14.
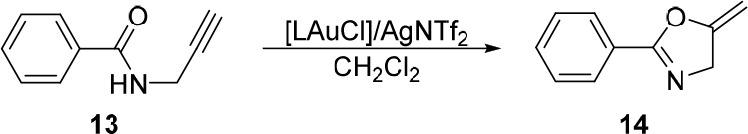
The cyclization reactions proceeded selectively; no other products were detected in the spectra. The kinetic profiles shown in Figure 12 indicate a superior catalytic performance of complex 9 containing phosphine 3, which maintained a relatively high activity, comparable to the prototypical catalyst [Au(MeCN)(PPh3)][SbF6],67,68 and resulted in a 97% NMR yield after 3 h (complete conversion was achieved after 6 h). The yield of 14 obtained with phosphine complex 7 was similar but was reached at a slower reaction rate. Notably, oxidation of the stibine moiety, such as in 12, accelerated the reaction at the initial stages (indicated by a visual comparison of the reaction rates during the first 10–20 min of the reaction), but then, the reaction rate decreased, very likely due to catalyst decomposition. The catalyst with the slowest reaction rate was obtained from complex 10, which presumably reflected the lower stability of this compound and, consequently, easier catalyst decomposition. Nevertheless, the yield of 14 was 82% after a 3 h reaction time. AgNTf2 itself did not catalyze the reaction (N.B.: a further analysis, e.g., an estimation of the initial reaction rates, was not performed, as it could be misleading due to catalyst activation and decomposition).
Figure 12.
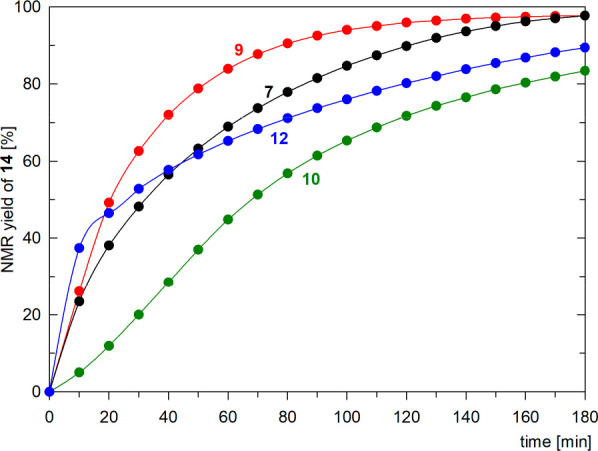
Kinetic profiles for Au-catalyzed cyclization of 13 into oxazole 14 using complexes 7, 9, 10, and 12 as precatalysts. The solid lines are shown only as a guide for an eye.
Next, we investigated the more challenging Au-catalyzed oxidative [2 + 2 + 1] cyclization of ethynylbenzene (15) with acetonitrile, used as a solvent, and pyridine N-oxides as the oxidants to afford 2-methyl-5-phenyloxazole (16; Scheme 9).69 The initial screening (Table 10, entries 1–8) using catalysts generated in situ from the defined Au(I) complexes (5 mol %), AgNTf2 (1 equiv), and pyridine N-oxide (17), performed at 60 °C for 24 h, showed that only phosphine complexes efficiently mediated this reaction. The yields of 16 achieved with complexes 7 and 9 were 37% and 51%, respectively. A lower yield, 27%, was obtained with compound 12, whereas no appreciable reaction was observed when the stibine complex 10 was used as the precatalyst. Subsequent experiments focused on complex 7 showed that this compound alone (i.e., without the silver(I) salt) was also active but resulted in a significantly lower yield (4%). Adding 2 or 3 equiv of AgNTf2 to complex 12 improved the yield of the cyclization product to approximately 40%.
Scheme 9. Gold-Catalyzed Oxidative [2 + 2 + 1] Annulation of Ethynylbenzene, Acetonitrile, and Pyridine N-Oxides.
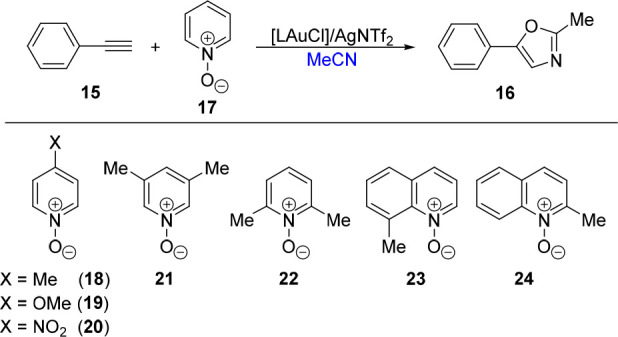
Table 10. Summary of the Catalytic Results for the Au-Catalyzed Formation of Oxazole 16a.
| entry | Au complex | oxidant | yield of 16 (%) |
|---|---|---|---|
| 1 | 7 | 17 | 37 |
| 2 | 9 | 17 | 51 |
| 3 | 10 | 17 | 0 |
| 4 | 12 | 17 | 27 |
| 5b | 12 | 17 | 4 |
| 6 | none | 17 | 0 |
| 7c | 12 | 17 | 40 |
| 8d | 12 | 17 | 38 |
| 9 | 7 | 18 | 23 |
| 10 | 7 | 19 | 3 |
| 11 | 7 | 20 | 19 |
| 12 | 7 | 21 | 28 |
| 13 | 7 | 22 | 15 |
| 14 | 7 | 23 | 73 (61)f |
| 15 | 7 | 24 | 11 |
| 16e | 7 | 23 | 69 (56)f |
Conditions unless specified otherwise: alkyne 17 (c = 0.10 M) was added to a mixture of gold complex (5 mol %), AgNTf2 (5 mol %), and oxidant (1.3 equiv) in MeCN at 60 °C for 24 h. The yields were determined by 1H NMR spectroscopy using anisole (1 equiv) as an internal standard and are an average of two independent runs.
No silver salt was used.
2 equiv of AgNTf2 was added.
3 equiv of AgNTf2 was added.
Ag[SbF6] was used instead of AgNTf2.
Isolated yield in parentheses.
Since the outcome of this catalytic reaction69 is known to depend on the N-oxide component, we screened several N-oxides (Table 10, entries 9–15). The yields achieved with substituted pyridine N-oxides 18-22 were substantially lower than those achieved with the parent compound 17, irrespective of the electronic properties of the substituents and steric bulk. An improvement to a 73% yield of 16 was observed when using the sterically encumbered 8-methylquinoline N-oxide (23), while the reaction in the presence of the isomeric 2-methylquinoline N-oxide (24) gave only an 11% yield. When AgNTf2 was replaced with Ag[SbF6] in the reaction with 7 and the best-performing oxidant 23, the yield of the cyclization product decreased slightly (to 68%), very likely for solubility reasons (AgNTf2 is more soluble in organic solvents).
Conclusion
Reported here are the synthesis, detailed structural characterization, and reactivity studies of 1-(diphenyphosphino)-1′-(diphenylstibino)ferrocene (1), which is the first ferrocene-based phosphinostibine ligand, falling halfway between the widely studied 1,1′-bis(diphenylphosphino)ferrocene (dppf)17 and 1,1′-bis(diphenylstibino)ferrocene (G) reported only recently.16 Although compounds combining phosphine and stibine donor groups are not unprecedented, phosphinostibine 1 is unique thanks to the particular combination of steric and electronic properties of its central ferrocene backbone.14f,70 As a strong electron donor, the ferrocene moiety increases the electron density at the Sb atom and, thus, decreases its acceptor properties. In addition, the ferrocene scaffold allows mutual reorientation of the functional pnictogen groups attached in positions 1 and 1′ by acting as molecular ball bearing with only low energy barrier. This markedly differentiates compounds 1, 1E, 6, and 6E (E = O, S, Se) from the previously reported compounds, wherein the P and Sb substituents were typically brought into close proximity by a rigid backbone. Importantly, the functional substituents at the ferrocene unit can be manipulated independently, which allowed the selective synthesis of P(III)/Sb(V), P(V)/Sb(III), and P(V)/Sb(V) derivatives, which were subsequently investigated for possible interactions between the pnictogen groups. While stibines 1 and 1E virtually lacked donor–acceptor interactions, the more Lewis acidic stiboranes 6 and 6O, obtained by oxidation of 1 with o-chloranil, formed distinct intramolecular interactions of the P → Sb(V) and P=O → Sb(V) type, which were manifested in both the crystal structures and the spectroscopic properties. In contrast, the analogous P=S → Sb(V) and P=Se → Sb(V) interactions in phosphine chalcogenide-stiboranes 6S and 6Se were weaker and did not result in the significant stabilization of a particular conformation. DFT calculations were used to investigate the nature of these interactions, and the results showed lone pair donation from phosphine phosphorus (in 6) or phosphoryl oxygen (in 6O) to the stiborane Sb atom at the other cyclopentadienyl ring. Although similar in nature, these interactions differed in strength and electrostatic contribution, with that in 6O being stronger and more ionic due to the strong polarization of the P=O bond.
From another viewpoint, compound 1 can be considered a typical example of a hybrid and potentially hemilabile ligand that forms coordination bonds of different strengths. This is reflected in the stability of the coordination compounds obtained from this ligand and affects the catalytic properties of the obtained complexes. The data collected for complexes containing the soft Au(I) metal ion suggest that stibine coordination does not sufficiently stabilize the active metal species, which is thus prone to decomposition. In turn, this leads to a shorter catalyst lifetime and poorer catalytic performance. A similar observation was previously made for the analogous Pd complexes featuring dppf and G as the ligands, among which the phosphine complexes showed better catalytic properties in Suzuki–Miyaura cross-coupling.16 From this perspective, compound 1 should be considered a functional phosphine whose P atom acts as the primary coordination site for soft metal ions, while the Sb moiety can be used as a secondary donor moiety or a reactive group whose chemical transformations can be used to control the overall electronic properties and an ability to form structure-directing intra- and intermolecular secondary interactions in the complexes.
Acknowledgments
This work was supported by the Czech Science Foundation (project no. 21-02316S). Computational resources were provided by the e-INFRA CZ project (ID: 90254), supported by the Ministry of Education, Youth and Sports of the Czech Republic.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.3c02075.
Author Contributions
The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Jeffrey J. C.; Rauchfuss T. B. Metal Complexes of Hemilabile Ligands. Reactivity and Structure of Dichlorobis(o-(diphenylphosphino)anisole)ruthenium(II). Inorg. Chem. 1979, 18, 2658–2666. 10.1021/ic50200a004. [DOI] [Google Scholar]
- b Braunstein P.; Naud F. Hemilability of Hybrid Ligands and the Coordination Chemistry of Oxazoline-Based Systems. Angew. Chem., Int. Ed. 2001, 40, 680–699. . [DOI] [PubMed] [Google Scholar]; a Slone C. S.; Weinberger D. A.; Mirkin C. A.; Karlin K. D. The Transition Metal Coordination Chemistry of Hemilabile Ligands. Prog. Inorg. Chem. 2007, 233–350. 10.1002/9780470166499.ch3. [DOI] [Google Scholar]
- For an overview of the chemistry of ferrocene-based P,N-ligands, see:; Dwadnia N.; Roger J.; Pirio N.; Cattey H.; Hierso J.-C. Input of P, N-(phosphanyl, amino)-ferrocene hybrid derivatives in late transition metals catalysis. Coord. Chem. Rev. 2018, 355, 74–100. 10.1016/j.ccr.2017.07.015. [DOI] [Google Scholar]; See also ref (12).
- Kutzelnigg W. Chemical Bonding in Higher Main Group Elements. Angew. Chem., Int. Ed. 1984, 23, 272–295. 10.1002/anie.198402721. [DOI] [Google Scholar]
- a Champness N. R.; Levason W. Coordination chemistry of stibine and bismuthine ligands. Coord. Chem. Rev. 1994, 133, 115–217. 10.1016/0010-8545(94)80058-8. [DOI] [Google Scholar]; b Werner H. The Way into the Bridge: A New Bonding Mode of Tertiary Phosphanes, Arsanes and Stibanes. Angew. Chem., Int. Ed. 2004, 43, 938–954. 10.1002/anie.200300627. [DOI] [PubMed] [Google Scholar]; c Levason W.; Reid G. Developments in the coordination chemistry of stibine ligands. Coord. Chem. Rev. 2006, 250, 2565–2594. 10.1016/j.ccr.2006.03.024. [DOI] [Google Scholar]; d Greenacre V. K.; Levason W.; Reid G. Developments in the chemistry of stibine and bismuthine complexes. Coord. Chem. Rev. 2021, 432, 213698. 10.1016/j.ccr.2020.213698. [DOI] [Google Scholar]; e Lipshultz J. M.; Li G.; Radosevich A. T. Main Group Redox Catalysis of Organopnictogens: Vertical Periodic Table Trends and Emerging Opportunities in Group 15. J. Am. Chem. Soc. 2021, 143, 1699–1721. 10.1021/jacs.0c12816. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Hollingsworth W. M.; Hill E. A. Exploring the potential role of heavy pnictogen elements in ligand design for new metal-ligand cooperative chemistry. J. Coord. Chem. 2022, 75, 1436–1466. 10.1080/00958972.2022.2124863. [DOI] [Google Scholar]
- Benjamin S. L.; Reid G. Neutral organoantimony(III) and organobismuth(III) ligands as acceptors in transition metal complexes – Role of substituents and co-ligands. Coord. Chem. Rev. 2015, 297–298, 168–180. 10.1016/j.ccr.2015.02.003. [DOI] [Google Scholar]
- For recent examples, see:; a Benjamin S. L.; Krämer T.; Levason W.; Light M. E.; Macgregor S. A.; Reid G. [Pd4(μ3-SbMe3)4(SbMe3)4]: A Pd(0) Tetrahedron with μ3-Bridging Trimethylantimony Ligands. J. Am. Chem. Soc. 2016, 138, 6964–6967. 10.1021/jacs.6b04060. [DOI] [PubMed] [Google Scholar]; b Jolleys A.; Lake B. R. M.; Krämer T.; Benjamin S. L. A Five-Membered PdSbn Coordination Series. Organometallics 2018, 37, 3854–3862. 10.1021/acs.organomet.8b00556. [DOI] [Google Scholar]; c Benjamin S. L.; Levason W.; Reid G.; Warr R. P. Halostibines SbMeX2 and SbMe2X: Lewis Acids or Lewis Bases?. Organometallics 2012, 31, 1025–1034. 10.1021/om2010996. [DOI] [Google Scholar]
- The affinity of Lewis acidic stibine and stiborane groups toward Lewis bases was exploited to design ion sensors. For representative examples, see:; a Wade C. R.; Ke I.-S.; Gabbaï F. P. Sensing of Aqueous Fluoride Anions by Cationic Stibine-Palladium Complexes. Angew. Chem., Int. Ed. 2012, 51, 478–481. 10.1002/anie.201106242. [DOI] [PubMed] [Google Scholar]; b Hirai M.; Gabbaï F. P. Lewis acidic stiborafluorenes for the fluorescence turn-on sensing of fluoride in drinking water at ppm concentrations. Chem. Sci. 2014, 5, 1886–1893. 10.1039/C4SC00343H. [DOI] [Google Scholar]; c Christianson A. M.; Gabbaï F. P. A Lewis Acidic, π-Conjugated Stibaindole with a Colorimetric Response to Anion Binding at Sb(III). Organometallics 2017, 36, 3013–3015. 10.1021/acs.organomet.7b00419. [DOI] [Google Scholar]
- For other examples, see:; a Rat C. I.; Silvestru C.; Breunig H. J. Hypervalent organoantimony and -bismuth compounds with pendant arm ligands. Coord. Chem. Rev. 2013, 257, 818–879. 10.1016/j.ccr.2012.07.026. [DOI] [Google Scholar]; b Benjamin S. L.; Levason W.; Reid G.; Rogers M. C. Hybrid dibismuthines and distibines as ligands towards transition metal carbonyls. Dalton Trans. 2011, 40, 6565–6574. 10.1039/c1dt10447k. [DOI] [PubMed] [Google Scholar]; c Benjamin S. L.; Karagiannidis L.; Levason W.; Reid G.; Rogers M. C. Hybrid Dibismuthines and Distibines: Preparation and Properties of Antimony and Bismuth Oxygen, Sulfur, and Nitrogen Donor Ligands. Organometallics 2011, 30, 895–904. 10.1021/om1010148. [DOI] [Google Scholar]
- a Kauffmann T.; Joußen R.; Klas N.; Vahrenhorst A. Neue Reagenzien, XXV. [(Diphenylstibino)methyl]lithium und -kupfer(I); Synthese und präparative Anwendungen. Chem. Ber. 1983, 116, 473–478. 10.1002/cber.19831160207. [DOI] [Google Scholar]; b Manger M.; Wolf J.; Laubender M.; Teichert M.; Stalke D.; Werner H. The First Peralkylated Phosphino(stibino)methanes and Their Organometallic Rhodium Complexes. Chem. Eur. J. 1997, 3, 1442–1450. 10.1002/chem.19970030910. [DOI] [Google Scholar]; c Manger M.; Gevert O.; Werner H. Unusual Dinuclear Hydridorhodium(III) Complexes Containing Bulky Phosphinyl(stibanyl)methanes as Chelating Ligands. Chem. Ber. 1997, 130, 1529–1531. 10.1002/cber.19971301028. [DOI] [Google Scholar]; For the synthesis of Sb(CH2PPh2)3, see:; d Karsch H. H.; Witt E. Phosphinomethanides and Group 15 element halides: Redox reactions, rearrangements and novel heterocycles. J. Organomet. Chem. 1997, 529, 151–169. 10.1016/S0022-328X(96)06578-3. [DOI] [Google Scholar]
- Selected examples:; a Levason W.; McAuliffe C. A. Bidentate ligands containing very soft donor atoms. Nickel(II) complexes of arylarsines and arylstibines. Inorg. Chim. Acta 1974, 11, 33–40. 10.1016/S0020-1693(00)93689-2. [DOI] [Google Scholar]; b Levason W.; McAuliffe C. A. Bidentate Group VB ligands. Part XVII. Palladium(II), platinum(II), and rhodium(III) complexes of o-phenylenebis(diphenylphosphine), (o-diphenylphosphinophenyl)diphenylstibine, and (o-diphenylarsinophenyl)diphenylstibine. Inorg. Chim. Acta 1976, 16, 167–172. 10.1016/S0020-1693(00)91707-9. [DOI] [Google Scholar]; c Levason W.; Smith K. G.; McAuliffe C. A.; McCullough F. P.; Sedgwick R. D.; Murray S. G. Synthesis and properties of group 5B ligand analogues of o-phenylenebis(dimethylarsine), o-C6H4(EMe2)(E′Me2) where E, E′ = P, N, As, or Sb. J. Chem. Soc., Dalton Trans. 1979, 1718–1724. 10.1039/DT9790001718. [DOI] [Google Scholar]; d Talay R.; Rehder D. Carbonylvanadium, -manganese and -molybdenum complexes of the ligands o-C6H4EPh2(E′Ph2) (E,E′ = P, As, Sb, Bi) and cis-Ph2PCH = CHPPh2. Z. Naturforsch. 1981, 36b, 451–462. 10.1515/znb-1981-0411. [DOI] [Google Scholar]; e Gray L. R.; Hale A. L.; Levason W.; McCullough F. P.; Webster M. Diphosphine and diarsine complexes of chromium(III). Crystal and molecular structure of tetra-n-propylammonium [cis-1,2-bis(diphenylphosphino)ethene]tetrachlorochromate(III). J. Chem. Soc., Dalton Trans. 1983, 2573–2580. 10.1039/dt9830002573. [DOI] [Google Scholar]; f Black J. R.; Levason W.; Spicer M. D.; Webster M. Synthesis and solution multinuclear magnetic resonance studies of homoleptic copper(I) complexes of Group 15 donor ligands. J. Chem. Soc.; Dalton Trans. 1993, 3129–3136. 10.1039/dt9930003129. [DOI] [Google Scholar]; g Jewiss H. C.; Levason W.; Spicer M. D.; Webster M. Coordination chemistry of higher oxidation states. 25. Synthesis and properties (including 59Co NMR Spectra) of cobalt(III) complexes of ligands containing two tertiary stibine groups. Crystal structure of trans-[Co{o-C6H4(SbMe2)2}2Cl2]2[CoCl4]. Inorg. Chem. 1987, 26, 2102–2016. 10.1021/ic00260a018. [DOI] [Google Scholar]; h Chalmers B. A.; Bühl M.; Arachige K. S. A.; Slawin A. M. Z.; Kilian P. Structural, Spectroscopic and Computational Examination of the Dative Interaction in Constrained Phosphine-Stibines and Phosphine-Stiboranes. Chem. Eur. J. 2015, 21, 7520–7531. 10.1002/chem.201500281. [DOI] [PubMed] [Google Scholar]; i Chalmers B. A.; Meigh C. B. E.; Nejman P. S.; Bühl M.; Lébl T.; Woollins J. D.; Slawin A. M. Z.; Kilian P. Geminally Substituted Tris(acenaphthyl) and Bis(acenaphthyl) Arsines, Stibines, and Bismuthine: A Structural and Nuclear Magnetic Resonance Investigation. Inorg. Chem. 2016, 55, 7117–7125. 10.1021/acs.inorgchem.6b01079. [DOI] [PubMed] [Google Scholar]; j Jones J. S.; Gabbaï F. P. Activation of an Au–Cl Bond by a Pendent SbIII Lewis Acid: Impact on Structure and Catalytic Activity. Chem. Eur. J. 2017, 23, 1136–1144. 10.1002/chem.201604521. [DOI] [PubMed] [Google Scholar]; For a binaphthyl derivative, see:; k Yasuike S.; Kawara S.; Okajima S.; Seki H.; Yamaguchi K.; Kurita J. Non-C2-symmetrical antimony–phosphorus ligand, (R/S)-2-diphenylphosphano-2′-di(p-tolyl)stibano-1,1′-binaphthyl (BINAPSb): preparation and its use for asymmetric reactions as a chiral auxiliary. Tetrahedron Lett. 2004, 45, 9135–9138. 10.1016/j.tetlet.2004.10.020. [DOI] [Google Scholar]
- a Dawson J. W.; Venanzi L. M. Phosphorus-31 nuclear magnetic resonance studies of coordination compounds. I. Stereochemistry of some complexes with multidentate ligands. J. Am. Chem. Soc. 1968, 90, 7229–7233. 10.1021/ja01028a010. [DOI] [Google Scholar]; b Higginson B. R.; McAuliffe C. A.; Venanzi L. M. Anomalous ligand field effects in complexes of quadridentate ligands containing Group V donors. Inorg. Chim. Acta 1971, 5, 37–40. 10.1016/S0020-1693(00)95876-6. [DOI] [Google Scholar]; c Wade C. R.; Gabbaï F. P. Two-Electron Redox Chemistry and Reversible Umpolung of a Gold–Antimony Bond,. Angew. Chem., Int. Ed. 2011, 50, 7369–7372. 10.1002/anie.201103109. [DOI] [PubMed] [Google Scholar]; d Ke I.-S.; Gabbaï F. P. σ-Donor/Acceptor-Confused Ligands: The Case of a Chlorostibine. Inorg. Chem. 2013, 52, 7145–7151. 10.1021/ic400736b. [DOI] [PubMed] [Google Scholar]; e Ke I.-S.; Gabbaï F. P. Cu3(μ2-Cl)3 and Ag3(μ2-Cl)3 Complexes Supported by Tetradentate Trisphosphino-stibine and -bismuthine Ligands: Structural Evidence for Triply Bridging Heavy Pnictines. Aust. J. Chem. 2013, 66, 1281–1287. 10.1071/CH13260. [DOI] [Google Scholar]; f Jones J. S.; Wade C. R.; Gabbaï F. P. Redox and Anion Exchange Chemistry of a Stibine-Nickel Complex: Writing L, X, Z Ligand Alphabet with a Single Element. Angew. Chem., Int. Ed. 2014, 53, 8876–8879. 10.1002/anie.201404156. [DOI] [PubMed] [Google Scholar]; g Ke I.-S.; Jones J. S.; Gabbaï F. P. Anion-Controlled Switching of an X Ligand into a Z Ligand: Coordination Non-innocence of a Stiboranyl Ligand. Angew. Chem., Int. Ed. 2014, 53, 2633–2637. 10.1002/anie.201309132. [DOI] [PubMed] [Google Scholar]; h Yang H.; Gabbaï F. P. Activation of an Hydroamination Gold Catalyst by Oxidation of a Redox-Noninnocent Chlorostibine Z-Ligand. J. Am. Chem. Soc. 2015, 137, 13425–13432. 10.1021/jacs.5b07998. [DOI] [PubMed] [Google Scholar]; i You D.; Gabbaï F. P. Unmasking the Catalytic Activity of a Platinum Complex with a Lewis Acidic, Non-innocent Antimony Ligand. J. Am. Chem. Soc. 2017, 139, 6843–6846. 10.1021/jacs.7b03287. [DOI] [PubMed] [Google Scholar]; j Jones J. S.; Wade C. R.; Yang M.; Gabbaï F. P. On the coordination non-innocence of antimony in nickel(II) complexes of the tetradentate (o-(Ph2PC6H4)3Sb ligand. Dalton Trans. 2017, 46, 5598–5604. 10.1039/C6DT04817J. [DOI] [PubMed] [Google Scholar]; k Sen S.; Ke I.-S.; Gabbaï F. P. T-Shaped Gold→Stiborane Complexes as Carbophilic Catalysts: Influence of the Peripheral Substituents. Organometallics 2017, 36, 4224–4230. 10.1021/acs.organomet.7b00654. [DOI] [Google Scholar]; l Piesch M.; Gabbaï F. P.; Scheer M. Phosphino-Stibine Ligands for the Synthesis of Heterometallic Comlexes. Z. Anorg. Allg. Chem. 2021, 647, 266–278. 10.1002/zaac.202000249. [DOI] [Google Scholar]; For similar ligands with acenaphthene scaffold, see:; m Furan S.; Hupf E.; Boidol J.; Brünig J.; Lork E.; Mebs S.; Beckmann J. Transition metal complexes of antimony centered ligands based upon acenaphthyl scaffolds. Coordination non-innocent or not?. Dalton Trans. 2019, 48, 4504–4513. 10.1039/C9DT00088G. [DOI] [PubMed] [Google Scholar]
- For a recent example of Sb,N- and Bi,N-ligands, see:; Garcia-Romero A.; Waters J. E.; Jethwa R. B.; Bond A. D.; Colebatch A. L.; Garcia-Rodriguez R.; Wright D. S. Highly Adaptive Nature of Group 15 Tris(quinolinyl) Ligands–Studies with Coinage Metals. Inorg. Chem. 2023, 62, 4625–4636. 10.1021/acs.inorgchem.3c00057. [DOI] [PubMed] [Google Scholar]
- a Ferrocenes. Homogeneous Catalysis. Organic Synthesis. Materials Science; Togni A., Hayashi T.; Eds.; VCH: 1995. [Google Scholar]; b Atkinson R. C. J.; Gibson V. C.; Long N. J. The syntheses and catalytic applications of unsymmetrical ferrocene ligands. Chem. Soc. Rev. 2004, 33, 313–328. 10.1039/b316819k. [DOI] [PubMed] [Google Scholar]; c Gomez Arrayas R.; Adrio J.; Carretero J. C. Recent Applications of Chiral Ferrocene Ligands in Asymmetric Catalysis. Angew. Chem., Int. Ed. 2006, 45, 7674–7715. 10.1002/anie.200602482. [DOI] [PubMed] [Google Scholar]; d Ferrocenes: Ligands, Materials and Biomolecules, Štěpnička P.; Ed.; Wiley: 2008. [Google Scholar]; e Cunningham L.; Benson A.; Guiry P. J. Recent developments in the synthesis and applications of chiral ferrocene ligands and organocatalysts in asymmetric catalysis. Org. Biomol. Chem. 2020, 18, 9329–9370. 10.1039/D0OB01933J. [DOI] [PubMed] [Google Scholar]; f Štěpnička P. Forever young: the first seventy years of ferrocene. Dalton Trans. 2022, 51, 8085–8102. 10.1039/D2DT00903J. [DOI] [PubMed] [Google Scholar]
- a Sharma P.; Lopez J. G.; Ortega C.; Rosas N.; Cabrera A.; Alvarez C.; Toscano A.; Reyes E. First ferrocenylstibines and their molecular structures. Inorg. Chem. Commun. 2006, 9, 82–85. 10.1016/j.inoche.2005.09.029. [DOI] [Google Scholar]; b Vázquez J.; Sharma P.; Cabrera A.; Toscano A.; Hernández S.; Pérez J.; Gutiérrez R. Formation of (vinyl-ferrocenyl)stibines involving β-elimination: Hypervalent Sb–N bonding. J. Organomet. Chem. 2007, 692, 3486–3491. 10.1016/j.jorganchem.2007.04.020. [DOI] [Google Scholar]; c Ortiz A. M.; Sharma P.; Pérez D.; Rosas N.; Velasco L.; Toscano A.; Hernández S. New 1,2-disubstituted ferrocenyl stibines containing N-heterocyclic pendant arm: Sb–N hypervalent compounds. J. Organomet. Chem. 2009, 694, 2037–2042. 10.1016/j.jorganchem.2009.01.051. [DOI] [Google Scholar]; d Pérez D.; Sharma P.; Cabrera A.; Rosas N.; Arellano I.; Toscano A.; Hernández S. Preparation of new 1,2-disubstituted ferrocenyl stibines containing ether/thioether arm from a quaternarny ferrocenyl ammonium salt. Polyhedron 2009, 28, 3115–3119. 10.1016/j.poly.2009.06.067. [DOI] [Google Scholar]; For 1,1′-ferrocene derivatives, see:; e Perez D.; Herrera C.; Sharma M.; Gutierrez R.; Hernández S.; Toscano A.; Sharma P. Synthesis of C3-symmetric tris(1,1′-formylferrocenyl)stibine and bismuthine: Rare examplex of tris 1,1′-asymmetrically ferrocenyl substituted group V compounds. J. Organomet. Chem. 2013, 743, 97–101. 10.1016/j.jorganchem.2013.06.015. [DOI] [Google Scholar]
- Schulz J.; Antala J.; Císařová I.; Štěpnička P. Beyond phosphorus: synthesis, reactivity, coordination behaviour and catalytic properties of 1,1′-bis(diphenylstibino)ferrocene. Dalton Trans. 2023, 52, 1198–1211. 10.1039/D2DT03770J. [DOI] [PubMed] [Google Scholar]
- a Gan K.-S.; Hor T. S. A.. 1,1′-Bis(diphenylphosphino)ferrocene. Coordination Chemistry, Organic Syntheses, and Catalysis. In Ferrocenes: Homogeneous Catalysis, Organic Synthesis, Materials Science; Togni A., Hayashi T., Eds.; Wiley-VCH: 1995; Chapter 1, pp 3–104. [Google Scholar]; b Chien S. W.; Hor T. S. A.. The Coordination and Homogeneous Catalytic Chemistry of 1,1′-Bis(diphenylphosphino)ferrocene and its Chalcogenide Derivatives. In Ferrocenes: Ligands, Materials and Biomolecules; Štěpnička P., Ed.; Wiley: 2008; Chapter 2, pp 33–116. [Google Scholar]; c Colacot T. J.; Parisel S.. Synthesis, Coordination Chemistry and Catalytic Use of dppf Analogs. In Ferrocenes: Ligands, Materials and Biomolecules; Štěpnička P., Ed.; Wiley: 2008; Chapter 3, pp 117–140. [Google Scholar]; d Bandoli G.; Dolmella A. Ligating ability of 1,1′-bis(diphenylphosphino)ferrocene: a structural survey (1994–1998). Coord. Chem. Rev. 2000, 209, 161–196. 10.1016/S0010-8545(00)00229-0. [DOI] [Google Scholar]; e Dey S.; Pietschnig R. Chemistry of sterically demanding dppf-analogs. Coord. Chem. Rev. 2021, 437, 213850. 10.1016/j.ccr.2021.213850. [DOI] [Google Scholar]
- Brunel J. M.; Faure B.; Maffei M. Phosphane–boranes: synthesis, characterization and synthetic applications,. Coord. Chem. Rev. 1998, 178–180, 665–698. 10.1016/S0010-8545(98)00072-1. [DOI] [Google Scholar]
- Brisset H.; Gourdel Y.; Pellon P.; Le Corre M. Phosphine-borane complexes; direct use in asymmetric catalysis. Tetrahedron Lett. 1993, 34, 4523–4526. 10.1016/0040-4039(93)88075-T. [DOI] [Google Scholar]
- a Estevan F.; Lahuerta P.; Latorre J.; Peris E.; García-Granda S.; Gómez-Beltrán F.; Aguirre A.; Salvadó M. A. Synthesis and electrochemical studies of new ferrocene-labelled dinuclear rhodium(II) complexes. Crystal structures of [Rh2(O2CMe)2{[(C6H4)PhP(C5H4)]Fe(C5H5)}2(HO2CMe)2] and [Rh2(O2CMe)2{[(C6H4)PhP(C5H4)]2Fe}(HO2CMe)]·CH2Cl2. J. Chem. Soc., Dalton Trans. 1993, 1681–1688. 10.1039/DT9930001681. [DOI] [Google Scholar]; b Muller A.; Otto S.; Roodt A. Rapid phosphorus(III) ligand evaluation utilising potassium selenocyanate. Dalton Trans. 2008, 650–657. 10.1039/B712782K. [DOI] [PubMed] [Google Scholar]
- Donaghy K. J.; Carroll P. J.; Sneddon L. G. Reactions of 1,1′-Bis(diphenylphosphino)ferrocene with Boranes, Thiaboranes, and Carboranes. Inorg. Chem. 1997, 36, 547–553. 10.1021/ic9611913. [DOI] [Google Scholar]
- Štěpnička P.; Císařová I. Selective borane reduction of phosphinoferrocene carbaldehydes to phosphinoalcohol–borane adducts. The coordination behaviour of 1-(diphenylphosphino)-1′-(methoxymethyl)ferrocene, a new ferrocene O,P-hybrid donor prepared from such an adduct. Dalton Trans. 2013, 42, 3373–3389. 10.1039/C2DT32511J. [DOI] [PubMed] [Google Scholar]
- a Lindner C.; Maryasin B.; Richter F.; Zipse H. Methyl cation affinity (MCA) for phosphanes. J. Phys. Org. Chem. 2010, 23, 1036–1042. 10.1002/poc.1726. [DOI] [Google Scholar]; b Lindner C.; Tandon R.; Maryasin B.; Larionov E.; Zipse H. Cation affinity numbers of Lewis bases. Beilstein J. Org. Chem. 2012, 8, 1406–1442. 10.3762/bjoc.8.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkylation of triphenylstibine requires a stronger alkylating agent, e.g., the Meerwein salt:; Henry M. C.; Wittig G. The Organometallic Alkylidene Reaction. J. Am. Chem. Soc. 1960, 82, 563–564. 10.1021/ja01488a017. [DOI] [Google Scholar]
- Kübler P.; Sundermeyer J. Ferrocenyl-phosphonium ionic liquids–synthesis, characterisation and electrochemistry. Dalton Trans. 2014, 43, 3750–3766. 10.1039/c3dt53402b. [DOI] [PubMed] [Google Scholar]
- BH3 scrambling was observed in ferrocene acylphosphines of the type R2PfcC(O)PR′2:; Vosáhlo P.; Císařová I.; Štěpnička P. Synthesis, coordination behavior, and catalytic properties of dppf congeners with an inserted carbonyl moiety. New J. Chem. 2022, 46, 21536–21552. 10.1039/D2NJ04270C. [DOI] [Google Scholar]
- Holmes R. R.; Day R. O.; Chandrasekhar V.; Holmes J. M. Pentacoordinated molecules. 67. Formation and structure of cyclic five-coordinated antimony derivatives. The first square-pyramidal geometry for a bicyclic stiborane. Inorg. Chem. 1987, 26, 157–163. 10.1021/ic00248a031. [DOI] [Google Scholar]
- Tofan D.; Gabbaï F. P. Fluorinated antimony(V) derivatives: strong Lewis acidic properties and application to the complexation of formaldehyde in aqueous solutions. Chem. Sci. 2016, 7, 6768–6778. 10.1039/C6SC02558G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Arduengo A. J. III; Stewart C. A.; Davidson F.; Dixon D. A.; Becker J. Y.; Culley S. A.; Mizen M. B. The Synthesis, Structure, and Chemistry of 10-Pn-3 Systems: Tricoordinate Hypervalent Pnictogen Compounds. J. Am. Chem. Soc. 1987, 109, 627–647. 10.1021/ja00237a001. [DOI] [Google Scholar]; b Chishiro A.; Akioka I.; Sumida A.; Oka K.; Tohnai N.; Yumura T.; Imoto H.; Naka K. Tetrachlorocatecholates of triarylarsines as a novel class of Lewis acids. Dalton Trans. 2022, 51, 13716–13724. 10.1039/D2DT02145E. [DOI] [PubMed] [Google Scholar]
- Štěpnička P.; Horký F. The coordination and catalytic chemistry of phosphanylferrocene chalcogenides. Eur. J. Inorg. Chem. 2022, 2022, e202200276 10.1002/ejic.202200276. [DOI] [Google Scholar]
- Kühl O.Phosphorus-31 NMR Spectroscopy: A Concise Introduction for the Synthetic Organic and Organometallic Chemist; Springer: 2008. [Google Scholar]
- Baillie C.; Zhang L.; Xiao J. Ferrocenyl Monophosphine Ligands: Synthesis and Applications in the Suzuki–Miyaura Coupling of Aryl Chlorides. J. Org. Chem. 2004, 69, 7779–7782. 10.1021/jo048963u. [DOI] [PubMed] [Google Scholar]
- Verschoor-Kirss M. J.; Hendricks O.; Verschoor C. M.; Conry R.; Kirss R. U. Chemical oxidation of ferrocenyl(phenyl)phosphines and ferrocenyl(phenyl)phosphine chalcogenides. Inorg. Chim. Acta 2016, 450, 30–38. 10.1016/j.ica.2016.05.010. [DOI] [Google Scholar]
- Beckmann U.; Süslüyan D.; Kunz P. C. Is the 1JPSe Coupling Constant a Reliable Probe for the Basicity of Phosphines? A 31P NMR Study. Phosphorus, Sulfur, Silicon, Relat. Elem. 2011, 186, 2061–2070. 10.1080/10426507.2010.547892. [DOI] [Google Scholar]
- a Fang Z.-G.; Hor T. S. A.; Wen Y.-S.; Liu L.-K.; Mak T. C. W. Molecular structures of 1,1′-bis(diphenylphosphino) ferrocene oxide and sulphide and their thermal properties. Polyhedron 1995, 14, 2403–2409. 10.1016/0277-5387(95)00072-Z. [DOI] [Google Scholar]; b Pilloni G.; Longato B.; Bandoli G.; Corain B. Bonding ability of 1,1′-bis(diphenylthiophosphoryl)ferrocene (dptpf) and its selenium analogue towards copper(I). Crystal structure of [Cu(dptpf)]BF4. J. Chem. Soc., Dalton Trans. 1997, 819–826. 10.1039/a606877d. [DOI] [Google Scholar]
- Selected examples:; a Štěpnička P.; Císařová I. Synthesis of [1′-(diphenylthiophosphoryl)ferrocenyl]ethyne and alkyne-metal complexes thereof. J. Organomet. Chem. 2006, 691, 2863–2871. 10.1016/j.jorganchem.2006.02.027. [DOI] [Google Scholar]; b Kahn S. L.; Breheney M. K.; Martinak S. L.; Fosbenner S. M.; Seibert A. R.; Kassel W. S.; Dougherty W. G.; Nataro C. Synthesis, Characterization, and Electrochemistry of Compounds Containing 1-Diphenylphosphino-1′-(di-tert-butylphosphino)ferrocene (dppdtbpf). Organometallics 2009, 28, 2119–2126. 10.1021/om800850c. [DOI] [Google Scholar]; c Fernandes T. A.; Solařová H.; Císařová I.; Uhlík F.; Štícha M.; Štěpnička P. Synthesis of phosphinoferrocene amides and thioamides from carbamoyl chlorides and the structural chemistry of Group 11 metal complexes with these mixed-donor ligands. Dalton Trans. 2015, 44, 3092–3108. 10.1039/C4DT03279A. [DOI] [PubMed] [Google Scholar]; d Schulz J.; Vosáhlo P.; Uhlík F.; Císařová I.; Štěpnička P. Probing the Influence of Phosphine Substituents on the Donor and Catalytic Properties of Phosphinoferrocene Carboxamides: A Combined Experimental and Theoretical Study. Organometallics 2017, 36, 1828–1841. 10.1021/acs.organomet.7b00181. [DOI] [Google Scholar]; e Vosáhlo P.; Císařová I.; Štěpnička P. Comparing the asymmetric dppf-type ligands with their semi-homologous counterparts. J. Organomet. Chem. 2018, 860, 14–29. 10.1016/j.jorganchem.2018.01.009. [DOI] [Google Scholar]
- Mantina M.; Chamberlin A. C.; Valero R.; Cramer C. J.; Truhlar D. G. Consistent van der Waals Radii for the Whole Main Group. J. Phys. Chem. A 2009, 113, 5806–5812. 10.1021/jp8111556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero B.; Gómez V.; Platero-Prats A. E.; Revés M.; Echeverría J.; Cremades E.; Barragán F.; Alvarez S. Covalent radii revisited. Dalton Trans. 2008, 2832–2838. 10.1039/b801115j. [DOI] [PubMed] [Google Scholar]
- Addison A. W.; Rao T. N.; Reedijk J.; van Rijn J.; Verschoor G. C. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen–sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate. J. Chem. Soc., Dalton Trans. 1984, 1349–1356. 10.1039/DT9840001349. [DOI] [Google Scholar]
- Gonzalez V. M.; Park G.; Yang M.; Gabbai F. Fluoride anion complexation and transport using a stibonium cation stabilized by an intramolecular P = O → Sb pnictogen bond. Dalton Trans. 2021, 50, 17897–17900. 10.1039/D1DT03370K. [DOI] [PubMed] [Google Scholar]
- a Adeleke J. A.; Liu L.-K. Diphenylphosphinoferrocene. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1993, 49, 680–682. 10.1107/S0108270192010436. [DOI] [Google Scholar]; b Kim T.-J.; Lee J.-H.; Kwon S.-C.; Kwon K.-H.; Uhm J.-K.; Lee H.; Byum S.-I. Reaction and Coordination Chemistry of Ferrocenylphosphines with (η5-C5H5)Co(CO)2-Crystal structures of Two Ferrocenylphosphine Oxides. Bull. Korean Chem. Soc. 1991, 12, 116–118. [Google Scholar]
- Bader R. F. W. A Quantum Theory of Molecular Structure and its Applications. Chem. Rev. 1991, 91, 893–928. 10.1021/cr00005a013. [DOI] [Google Scholar]
- Cremer D.; Kraka E. Chemical-Bonds without Bonding Electron-Density – Does the Difference Electron-Density Analysis Suffice for a Description of the Chemical-Bond?. Angew. Chem., Int. Ed. Engl. 1984, 23, 627–628. 10.1002/anie.198406271. [DOI] [Google Scholar]
- a Hupf E.; Lork E.; Mebs S.; Beckmann J. Intramolecularly Coordinated (6-(Diphenylphosphino)acenaphth-5-yl)stannanes. Repulsion vs Attraction of P- and Sn-Containing Substituents in the peri Positions. Organometallics 2014, 33, 2409–2423. 10.1021/om500133a. [DOI] [Google Scholar]; b Hupf E.; Lork E.; Mebs S.; Checińska L.; Beckmann J. Probing Donor-Acceptor Interactions in peri-Substituted Diphenylphosphinoacenaphthyl-Element Dichlorides of Group 13 and 15 Elements. Organometallics 2014, 33, 7247–7259. 10.1021/om501036c. [DOI] [Google Scholar]; c Hupf E.; Do T. G.; Nordheider A.; Wehrhahn M.; Sanz Camacho P.; Ashbrook S. E.; Lork E.; Slawin A. M. Z.; Mebs S.; Woollins J. D.; Beckmann J. Selective Oxidation and Functionalization of 6-Diphenylphosphinoacenaphthyl-5-tellurenyl Species 6-Ph2P-Ace-5-TeX (X = Mes, Cl, O3SCF3). Various Types of P–E···Te(II,IV) Bonding Situations (E = O, S, Se). Organometallics 2017, 36, 1566–1579. 10.1021/acs.organomet.7b00133. [DOI] [Google Scholar]; d Mokrai R.; Barrett J.; Apperley D. C.; Benkö Z.; Heift D. Tweaking the Charge Transfer: Bonding Analysis of Bismuth(III) Complexes with a Flexidentate Phosphane Ligand. Inorg. Chem. 2020, 59, 8916–8924. 10.1021/acs.inorgchem.0c00734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Streitweiser A. Jr; Rajca A.; McDowell R. S.; Glaser R. Semipolar P–O and P–C Bonds: A Theoretical Study of Hypophosphite and Related Methylenephosphoranes. J. Am. Chem. Soc. 1987, 109, 4184–4188. 10.1021/ja00248a010. [DOI] [Google Scholar]; b Chesnut D. B. An Ab Initio Nuclear Magnetic Resonance and Atoms-in-Molecules Study of the PO Bond in Phosphine Oxides. J. Am. Chem. Soc. 1998, 120, 10504–10510. 10.1021/ja9822198. [DOI] [Google Scholar]; c Dobado J. A.; Martínez-García H.; Molina J. M.; Sundberg M. R. Chemical Bonding in Hypervalent Molecules Revised. Application of the Atoms in Molecules Theory to Y3X and Y3XZ (Y = H or CH3; X = N, P or As; Z = O or S) Compounds. J. Am. Chem. Soc. 1998, 120, 8461–8471. 10.1021/ja980141p. [DOI] [Google Scholar]; d Chesnut D. B.; Savin A. The Electron Localization Function (ELF) Description of the PO Bond in Phosphine Oxide. J. Am. Chem. Soc. 1999, 121, 2335–2336. 10.1021/ja984314m. [DOI] [Google Scholar]; e Yamada K.; Koga N. Variationally Determined Electronic States for the Theoretical Analysis of Intramolecular Interaction. II. Qualitative Nature of the P–O Bond in Phosphine Oxides. J. Comput. Chem. 2013, 34, 149–161. 10.1002/jcc.23118. [DOI] [PubMed] [Google Scholar]; f Yang T.; Andrada D. M.; Frenking G. Dative versus electron-sharing bonding in N-oxides and phosphane oxides R3EO and relative energies of the R2EOR isomers (E = N, P; R = H, F, Cl, Me, Ph). A theoretical study. Phys. Chem. Chem. Phys. 2018, 20, 11856–11866. 10.1039/C8CP00951A. [DOI] [PubMed] [Google Scholar]; g Lindquist-Kleissler B.; Wenger J. S.; Johnstone T. C. Analysis of Oxygen–Pnictogen Bonding with Full Bond Path Topological Analysis of the Electron Density. Inorg. Chem. 2021, 60, 1846–1856. 10.1021/acs.inorgchem.0c03308. [DOI] [PubMed] [Google Scholar]
- Haaland A.; Nilsson J. E.; et al. The Determination of Barriers to Internal Rotation by Means of Electron Diffraction. Ferrocene and Ruthenocene. Acta Chem. Scand. 1968, 22, 2653–2670. 10.3891/acta.chem.scand.22-2653. [DOI] [Google Scholar]
- Erdmann P.; Leitner J.; Schwarz J.; Greb L. An Extensive Set of Accurate Fluoride Ion Affinities for p-Block Element Lewis Acids and Basic Design Principles for Strong Fluoride Ion Acceptors. ChemPhysChem 2020, 21, 987–994. 10.1002/cphc.202000244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calculated at the PW6B95(d3bj)/def2-qzvpp//PBE0(d3)/def2-TZVP:sdd(Fe,Sb) level of theory in vacuum at 298.15 K.
- a Knizia G. Intrinsic Atomic Orbitals: An Unbiased Bridge between Quantum Theory and Chemical Concepts. J. Chem. Theory Comput. 2013, 9, 4834–4843. 10.1021/ct400687b. [DOI] [PubMed] [Google Scholar]; b Knizia G.; Klein J. E. M. N. Electron Flow in Reaction Mechanisms–Revealed from First Principles. Angew. Chem., Int. Ed. 2015, 54, 5518–5522. 10.1002/anie.201410637. [DOI] [PubMed] [Google Scholar]
- a Sandblom N.; Ziegler T.; Chivers T. A density functional study of the bonding in tertiary phosphine chalcogenides and related molecules. Can. J. Chem. 1996, 74, 2363–2371. 10.1139/v96-263. [DOI] [Google Scholar]; See also ref (45c).
- Yamada K.; Koga N. Variationally Determined Electronic States for the Theoretical Analysis of Intramolecular Interaction. II. Qualitative Nature of the P–O Bond in Phosphine Oxides. J. Comput. Chem. 2013, 34, 149–161. 10.1002/jcc.23118. [DOI] [PubMed] [Google Scholar]
- a Gritzner G.; Kůta J. Recommendations on reporting electrode potentials in nonaqueous solvents. Pure Appl. Chem. 1984, 56, 461–466. 10.1351/pac198456040461. [DOI] [Google Scholar]; b Gagné R. R.; Koval C. A.; Lisensky G. C. Ferrocene as an Internal Standard for Electrochemical Measurements. Inorg. Chem. 1980, 19, 2854–2855. 10.1021/ic50211a080. [DOI] [Google Scholar]
- a Pilloni G.; Longato B.; Corain B. Heteropolymetallic complexes of 1,1′-bis(diphenylphosphino)ferrocene (dppf) VII. Redox behaviour of dppf. J. Organomet. Chem. 1991, 420, 57–65. 10.1016/0022-328X(91)86445-V. [DOI] [Google Scholar]; b Zanello P.; Opromolla G.; Giorgi G.; Sasso G.; Togni A. Redox behaviour of ferrocene derivatives VIII. 1,1′-Bis(diphenylphosphino)ferrocenes. J. Organomet. Chem. 1996, 506, 61–65. 10.1016/0022-328X(95)05720-A. [DOI] [Google Scholar]; c Nataro C.; Campbell A. N.; Ferguson M. A.; Incarvito C. D.; Rheingold A. L. Group 10 metal complexes of 1,1′-bis(diphenylphosphino)ferrocene (dppf) and 1,1′-bis(diphenylphosphino)ruthenocene: A structural and electrochemical investigation. X-ray structures of [MCl2(dppr)] (M = Ni, Pd). J. Organomet. Chem. 2003, 673, 47–55. 10.1016/S0022-328X(03)00155-4. [DOI] [Google Scholar]
- Representative examples:; a Podlaha J.; Štěpnička P.; Ludvík J.; Císařová I. 1′-(Diphenylphosphino)ferrocenecarboxylic acid and Its P-Oxide and Methylester: Synthesis, Characterization, Crystal Structure, and Electrochemistry. Organometallics 1996, 15, 543–550. 10.1021/om950528q. [DOI] [Google Scholar]; b Ghent B. L.; Martinak S. L.; Sites L. A.; Golen J. A.; Rheingold A. L.; Nataro C. Electrochemistry and complexation of Josiphos ligands. J. Organomet. Chem. 2007, 692, 2365–2374. 10.1016/j.jorganchem.2007.02.039. [DOI] [Google Scholar]; c Bennett M. A.; Bhargava S. K.; Bond A. M.; Burgar I. M.; Guo S.-X.; Kar G.; Privér S. H.; Wagler J.; Willis A. C.; Torriero A. A. J. Synthesis, X-ray structure and electrochemical oxidation of palladium(II) complexes of ferrocenyldiphenylphosphine. Dalton Trans. 2010, 39, 9079–9090. 10.1039/c0dt00016g. [DOI] [PubMed] [Google Scholar]
- a Swartz B. D.; Nataro C. Anodic Electrochemistry of Ferrocenylphosphine and Ruthenocenylphosphine Chalcogenide Complexes and Lewis Acid Adducts. Organometallics 2005, 24, 2447–2451. See also 10.1021/om050074p. [DOI] [Google Scholar]; b Barriere F.; Kirss R. U.; Geiger W. E. Anodic Electrochemistry of Multiferrocenyl Phosphine and Phosphine Chalcogenide Complexes in Weakly Nucleophilic Electrolytes. Organometallics 2005, 24, 48–52. 10.1021/om040123i. [DOI] [Google Scholar]
- For a description and further examples of this approach, see:; a Schulz J.; Uhlík F.; Speck J. M.; Císařová I.; Lang H.; Štěpnička P. Synthesis, Crystal Structures, and Electrochemical Behavior of Fe–Ru Heterobimetallic Complexes with Bridged Metallocene Units. Organometallics 2014, 33, 5020–5032. 10.1021/om500505n. [DOI] [Google Scholar]; b Škoch K.; Císařová I.; Uhlík F.; Štěpnička P. Comparing the reactivity of isomeric phosphinoferrocene nitrile and isocyanide in Pd(ii) complexes: synthesis of simple coordination compounds vs. preparation of P-chelated insertion products and Fischer-type carbenes. Dalton Trans. 2018, 47, 16082–16101. 10.1039/C8DT03564D. [DOI] [PubMed] [Google Scholar]; c Škoch K.; Schulz J.; Císařová I.; Štěpnička P. Pd(II) Complexes with Chelating Phosphinoferrocene Diaminocarbene Ligands: Synthesis, Characterization, and Catalytic Use in Pd-Catalyzed Borylation of Aryl Bromides. Organometallics 2019, 38, 3060–3073. 10.1021/acs.organomet.9b00398. [DOI] [Google Scholar]; d Vosáhlo P.; Schulz J.; Císařová I.; Štěpnička P. Cyclopalladation of a ferrocene acylphosphine and the reactivity of the C–H activated products. Dalton Trans. 2021, 50, 6232–6244. 10.1039/D1DT00668A. [DOI] [PubMed] [Google Scholar]
- Oxidation of stibine to a stiborane moiety has been shown to affect physical properties. For a representative example, see:; Lo Y.-H.; Gabbaï F. P. Controlling the Properties of a 2,2′-bipy–Platinum Dichloride Complex via Oxidation of a Peripheral Stibine Moiety. Organometallics 2018, 37, 2500–2506. 10.1021/acs.organomet.8b00296. [DOI] [Google Scholar]
- Rössler K.; Rüffer T.; Walfort B.; Packheiser R.; Holze R.; Zharnikov M.; Lang H. Synthesis, characterization and electrochemical behavior of unsymmetric transition metal-terminated biphenyl ethynyl thiols. J. Organomet. Chem. 2007, 692, 1530–1545. 10.1016/j.jorganchem.2006.12.002. [DOI] [Google Scholar]
- The solution 13C{1H} NMR spectrum of 7 could not be obtained due to sample decomposition.
- Dunstan S. P. C.; Healy P. C.; Sobolev A. N.; Tiekink E. R. T.; White A. H.; Williams M. L. Isomorphism in the structural chemistry of two-coordinate adducts of diphenyl(2-formylphenyl)phosphine and triphenylphosphine with gold(I) halides. J. Mol. Struct. 2014, 1072, 253–259. 10.1016/j.molstruc.2014.05.020. [DOI] [Google Scholar]
- a Schmidbaur H. The Aurophilicity Phenomenon: A Decade of Experimental Findings, Theoretical Concepts and Emerging Applications. Gold Bull. 2000, 33, 3–10. 10.1007/BF03215477. [DOI] [Google Scholar]; b Schmidbaur H.; Schier A. A briefing on aurophilicity. Chem. Soc. Rev. 2008, 37, 1931–1951. 10.1039/b708845k. [DOI] [PubMed] [Google Scholar]; c Schmidbaur H.; Schier A. Aurophilic interactions as a subject of current research: an up-date. Chem. Soc. Rev. 2012, 41, 370–412. 10.1039/C1CS15182G. [DOI] [PubMed] [Google Scholar]
- a Steed K. M.; Steed J. W. Packing Problems: High Z′ Crystal Structures and Their Relationship to Cocrystals, Inclusion Compounds, and Polymorphism. Chem. Rev. 2015, 115, 2895–2933. 10.1021/cr500564z. [DOI] [PubMed] [Google Scholar]; b Taylor R.; Cole J. C.; Groom C. R. Molecular Interactions in Crystal Structures with Z′ > 1. Cryst. Growth Des. 2016, 16, 2988–3001. 10.1021/acs.cgd.6b00355. [DOI] [Google Scholar]
- Cambridge Structural Database, version 5.34 of November 2021, with updates from March, June and September 2022.
- For an example of an Au(I)-ferrocenebisphosphole complex, where aurophilic interactions give rise to a different polymorph, see:; Orthaber A.; Borucki S.; Shen W.; Réau R.; Lescop C.; Pietschnig R. Coordination Behaviour of a Hexadentate1,1′-Ferrocenylene-Bridged Bisphosphole towards CoinageMetal Centres. Eur. J. Inorg. Chem. 2014, 2014, 1751–1759. 10.1002/ejic.201301281. [DOI] [Google Scholar]
- Complex 11 was not included in catalytic testing because of facile hydrolysis of the catecholato-stiborane moiety that could influence the catalytic results.
- a Hashmi A. S. K.; Weyrauch J. P.; Frey W.; Bats J. W. Gold Catalysis: Mild Conditions for the Synthesis of Oxazoles from N-Propargylcarboxamides and Mechanistic Aspects. Org. Lett. 2004, 6, 4391–4394. 10.1021/ol0480067. [DOI] [PubMed] [Google Scholar]; b Weyrauch J. P.; Hashmi A. S. K.; Schuster A.; Hengst T.; Schetter S.; Littmann A.; Rudolph M.; Hamzic M.; Visus J.; Rominger F.; Frey W.; Bats J. W. Cyclization of Propargylic Amides: Mild Access to Oxazole Derivatives. Chem. Eur. J. 2010, 16, 956–963. 10.1002/chem.200902472. [DOI] [PubMed] [Google Scholar]
- Bárta O.; Císařová I.; Schulz J.; Štěpnička P. Assessing the influence of phosphine substituents on the catalytic properties of self-stabilised digold(I) complexes with supporting ferrocene phosphinonitrile ligands. New J. Chem. 2019, 43, 11258–11262. 10.1039/C9NJ02555C. [DOI] [Google Scholar]
- Vosáhlo P.; Štěpnička P. Assessing the role of substituents in ferrocene acylphosphines and their impact on gold-catalysed reactions. New J. Chem. 2023, 47, 4510–4520. 10.1039/D3NJ00201B. [DOI] [Google Scholar]
- He W.; Li C.; Zhang L. An Efficient [2 + 2 + 1] Synthesis of 2,5-Disubstituted Oxazoles via Gold-Catalyzed Intermolecular Alkyne Oxidation. J. Am. Chem. Soc. 2011, 133, 8482–8485. 10.1021/ja2029188. [DOI] [PubMed] [Google Scholar]
- Astruc D. Why is Ferrocene so Exceptional?. Eur. J. Inorg. Chem. 2017, 2017, 6–29. 10.1002/ejic.201600983. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.