

Summary
Acute myeloid leukemia (AML) is a hematologic malignancy for which several epigenetic regulators have been identified as therapeutic targets. Here we report the development of cereblon-dependent degraders of IKZF2 and casein kinase 1 alpha (CK1α) termed DEG-35 and DEG-77. We utilized a structure-guided approach to develop DEG-35 as a nanomolar degrader of IKZF2, a hematopoietic-specific transcription factor that contributes to myeloid leukemogenesis. DEG-35 possesses additional substrate specificity for the therapeutically-relevant target CK1α, which was identified through unbiased proteomics and a PRISM screen assay. Degradation of IKZF2 and CK1α blocks cell growth and induces myeloid differentiation in AML cells through CK1α–p53- and IKZF2-dependent pathways. Target degradation by DEG-35 or a more soluble analog DEG-77 delays leukemia progression in murine and human AML mice models. Overall, we provide a strategy for multi-targeted degradation of IKZF2 and CK1α to enhance efficacy against AML that may be expanded to additional targets and indications.
Keywords: targeted protein degradation, acute myeloid leukemia, cereblon, casein kinase 1 alpha, IKZF2
Graphical Abstract

eTOC
Park et al. report the development of monofunctional degraders of AML targeting IKZF2 and CK1α. These degraders induce cell cycle arrest, differentiation and apoptosis in AML cells and significantly prolong survival in AML mouse models. Degraders targeting IKZF2 and CK1α can be therapeutic strategies for AML.
Introduction
Acute myeloid leukemia (AML) is an aggressive hematologic malignancy characterized by uncontrolled expansion of immature myeloid cells.1 This disease is caused by mutations and chromosomal translocations that target a wide variety of factors that include kinases and epigenetic regulators.2 Although several promising therapies are in development, there remains an unmet need for additional therapeutic strategies.3,4 Therapy resistance frequently occurs in AML patients, giving rise to different clones at diagnosis and relapse. Thus, utilizing a multi-targeted therapeutic strategy may be advantageous for AML treatment.
Previously we found that the IKZF2 zinc finger transcription factor is highly expressed in leukemia stem cells (LSCs) and is required for maintaining survival in mouse and human AML cells.5 In the MLL-AF9 leukemic mouse model, IKZF2 regulates the HOXA9 self-renewal gene expression program and inhibits the C/EBP-driven myeloid differentiation program. Although therapeutically targeting transcription factors remains a pharmacological challenge, recent studies with thalidomide analogs, lenalidomide and pomalidomide, have revealed a potential opportunity for degradation of C2H2 zinc fingers like IKZF2. Lenalidomide and pomalidomide bind to the E3 ubiquitin ligase adapter cereblon (CRBN) to recruit the related Ikaros family transcription factors IKZF1 and IKZF3, which promotes their ubiquitination and proteasomal degradation.6,7 Although lenalidomide and pomalidomide do not degrade IKZF2 or IKZF4, structural studies have yielded degraders of IKZF2 as a potential immunotherapeutic target.8,9,10 The rational design of degraders for IKZF2 and other synergistic targets may provide new therapeutic opportunities for AML.
Results
1. Development of an IKZF2 degrader
As we previously found that IKZF2 contributes to AML survival and cell growth, we sought to develop novel IKZF2 degraders. Selectivity of lenalidomide-induced degradation of IKZF1 and IKZF3, but not the homologs IKZF2 and IKZF4, has been traced to a single amino acid variant where substitution of Q147 in IKZF3 to histidine (IKZF3 Q147H) blocks lenalidomide-induced degradation (Figure 1A).6 Recent crystal structures show that the ternary complex between IKZF1, lenalidomide, and CRBN is dependent on a key hydrogen bond that is lost upon replacement of glutamine found in IKZF1 and IKZF3 with histidine found in IKZF2 and IKZF4.8
Figure 1. DEG-35 is a potent degrader of IKZF2 with leukemic cell killing activity.
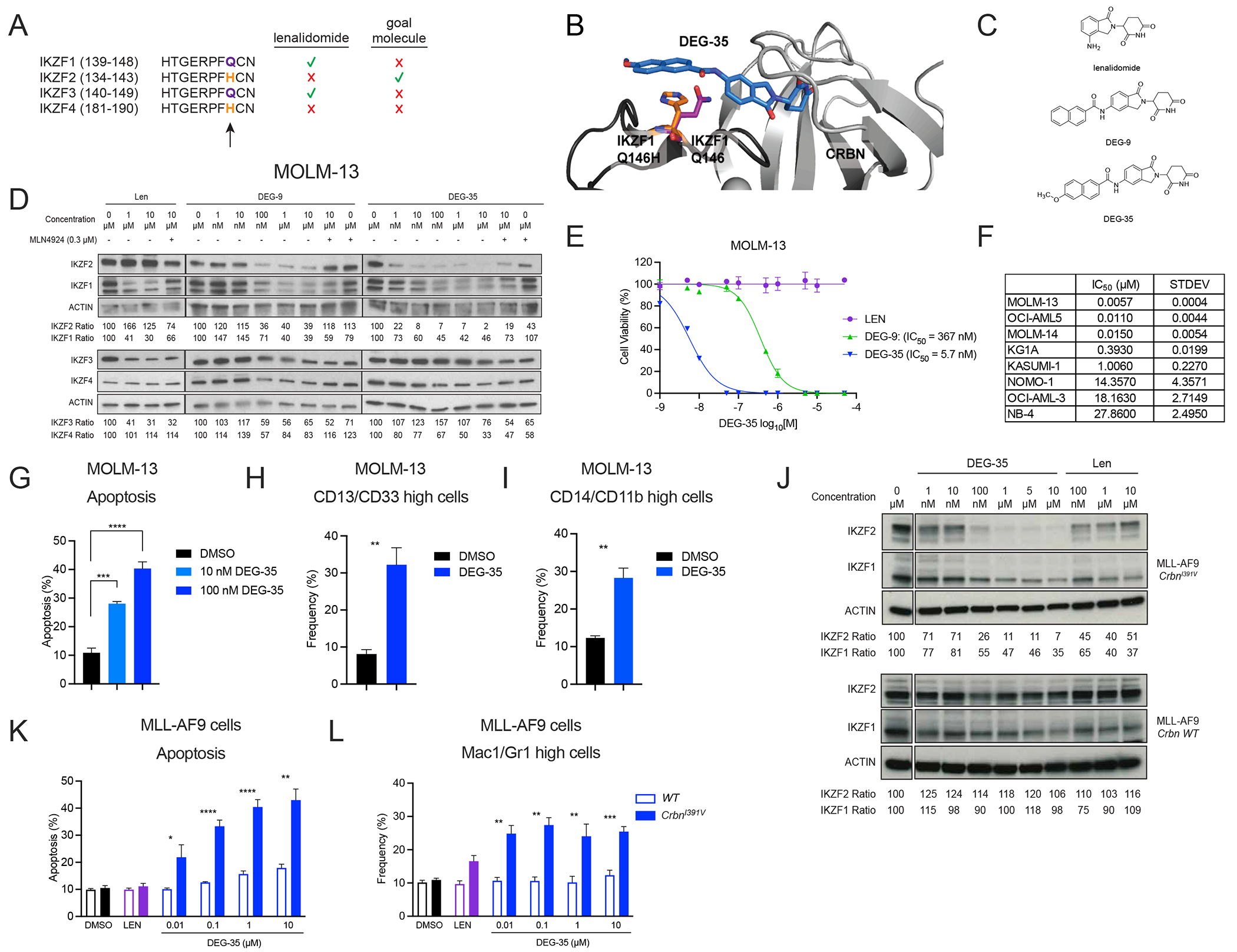
(A) Schematic diagram showing the sequence of critical amino acids required for binding of the IKAROS family members to IMiDs. (B) Docking model of DEG-35 binding to IKZF2 and CRBN. (C) Chemical structures of lenalidomide, DEG-9, and DEG-35. (D) Western blot analysis of IKAROS family members in MOLM-13 cells treated with the indicated drugs for 24 h with or without MLN4924 1 h pretreatment. (E) Viability assay performed in MOLM-13 cells treated with indicated drugs for 72 h to obtain IC50 values. Data shown is the representative graph of three independent experiments. Mean ± SEM of triplicates. (F) IC50 values of DEG-35 in different AML cell lines treated for 72 h. Mean ± SEM of three independent experiments are shown. (G) Apoptosis was measured by flow cytometry using Annexin V as a marker. (H) CD13/CD33 and (I) CD14/CD11b were stained on MOLM-13 cells and measured by flow cytometry for differentiation at day 2 post-treatment with 100 nM DEG-35. (J) Western blot analysis for indicated proteins in MLL-AF9 CrbnI391V or WT cells treated with DEG-35 or lenalidomide for 24 h. (K,L) (K) Apoptosis and (L) differentiation were measured by flow cytometry of cells stained with Annexin V and myeloid markers, Mac1/Gr1 respectively. Flow cytometry was performed 24 h post-treatment with DEG-35 or 10 μM lenalidomide. Mean ± SEM of three independent experiments for results in (G-I, K and L). Student’s t test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. See also Figure S1 and Table S1.
We hypothesized that we could selectively target IKZF2 by designing lenalidomide analogs that introduce pi-stacking interactions with the aromatic IKZF2 H141 to enhance ternary complex formation with CRBN and thus promote IKZF2 degradation. Based on models derived from a crystal structure of IKZF1, lenalidomide, and CRBN,8 we proposed that projection of aromatic groups from the 5-position of lenalidomide via an amide linkage would induce these desired interactions (Figure 1B). We synthesized a small compound library and screened for IKZF2 degradation. We found that the addition of a naphthamide to lenalidomide induced dose-dependent degradation of IKZF2 (DEG-9, Figures 1C–D). Structure-activity relationship studies revealed that adding a methoxy group to the 6-position of the naphthalene greatly increased IKZF2 degradation (DEG-35, Figures 1C–D). DEG-35 efficiently degraded IKZF2 with limited activity against IKZF1, IKZF3 and IKZF4 in a Cullin-dependent manner (Figure 1D). Unlike lenalidomide, DEG-9 and DEG-35 inhibited cellular growth that correlated with the extent of IKZF2 degradation in MOLM-13 cells (Figure 1E).
DEG-35 inhibited growth activity in a panel of AML cell lines representing various oncogenic mutations with IC50 values from single digit nM to low μM concentrations (Figure 1F, S1A and Table S1) with MOLM-13 cells being the most sensitive cell line with an IC50 value of 5.7nM. Similarly, DEG-35 treatment at 10 nM and 100 nM in MOLM-13 cells led to significant dose-dependent apoptosis induction and myeloid differentiation after 2 days (Figures 1G–I and S1B–D). These data indicate that DEG-35 degrades IKZF2, which corresponds with a more potent induction of cell death in MOLM-13 cells as compared to lenalidomide.
We performed a time course and measured apoptosis and differentiation from DEG-35 treatment for 5 days (Figures S1E–L). We found that DEG-35-treated MOLM-13 cells exist in a spectrum of differentiation states in which apoptosis is largely occurring at earlier time points (day 1–3) with the detection of two populations that include apoptosis in the absence of differentiation and cells that express increased positivity for both differentiation markers and the apoptotic marker Annexin V. At later time points (day 4) there is an increase in cells that are differentiating without yet being Annexin V positive and another subset of cells that are both differentiating and undergoing apoptosis (Figures S1H–K).
Unlike human CRBN, the mouse ortholog is resistant to lenalidomide-mediated degradation of substrates.11,12 Nevertheless, mutation of isoleucine 391 to valine in the mouse Crbn gene enables efficient substrate degradation by lenalidomide in mice.11 Thus, we generated MLL-AF9 leukemia using Lin−Sca-1+c-Kit+ (LSK) cells from CrbnI391V mice. Consistent with a CRBN-mediated mechanism, DEG-35 treatment in the MLL-AF9 CrbnI391V cells led to a more significant reduction of IKZF2 as compared to IKZF1 and resulted in apoptosis and myeloid differentiation at nanomolar concentrations (10 nM, 24 h post treatment, Figures 1J–L and S1M–N) in a manner that was dependent on humanized CRBN. These data suggest that the anti-proliferative effects of DEG-35 are through CRBN-mediated degradation of substrates including IKZF2.
2. DEG-35 recruits and degrades CK1α
To investigate the direct targets of DEG-35 in an unbiased manner, we performed global quantitative proteomics with MOLM-13 cells treated with 50 nM DEG-35 for 2 h. Interestingly, the most significantly downregulated protein upon DEG-35 treatment was CK1α (Figures 2A, Table S2). DEG-35 depleted CK1α at concentrations as low as 1–10 nM, which was more efficient than DEG-9 or lenalidomide (Figure 2B). We additionally confirmed that DEG-35 degrades both IKZF2 and CK1α in additional AML cell lines (Figure S2A). The degradation of CK1α and IKZF2 was quantified in HEK293T cells stably expressing either a CK1α-GFP or IKZF2(zinc finger 2)-GFP fusion protein following DEG-35 treatment for 5 h or 24 h, respectively (consistent with the timepoint of maximal degradation, Figures 2C–D). In line with the degradation efficiency of the endogenous proteins, DEG-35 was a potent degrader of CK1α (DC50 = 1.4 nM, DCmax = 84%) and IKZF2 (DC50 = 4.4 nM, DCmax = 92%). Notably, lenalidomide did not induce significant degradation of either substrate over the same concentration ranges and treatment times (DCmax CK1α = 24%, DCmax IKZF2 = 14%). By comparison, HEK293T cells stably expressing IKZF1(zinc finger 2)-GFP and GSPT1-GFP showed more moderate responses to DEG-35 treatment (DCmax IKZF1-GFP = 12%, DCmax GSPT1-GFP = 24%), whereas known degraders of these targets elicited strong responses (Figures S2B–C). Similarly, we observed endogenous GSPT1 degradation by Western blot at micromolar concentrations but limited degradation at nanomolar concentrations (Figure S2D).
Figure 2. DEG-35 degrades dual targets IKZF2 and CK1α via Cullin-CRBN dependent pathway.
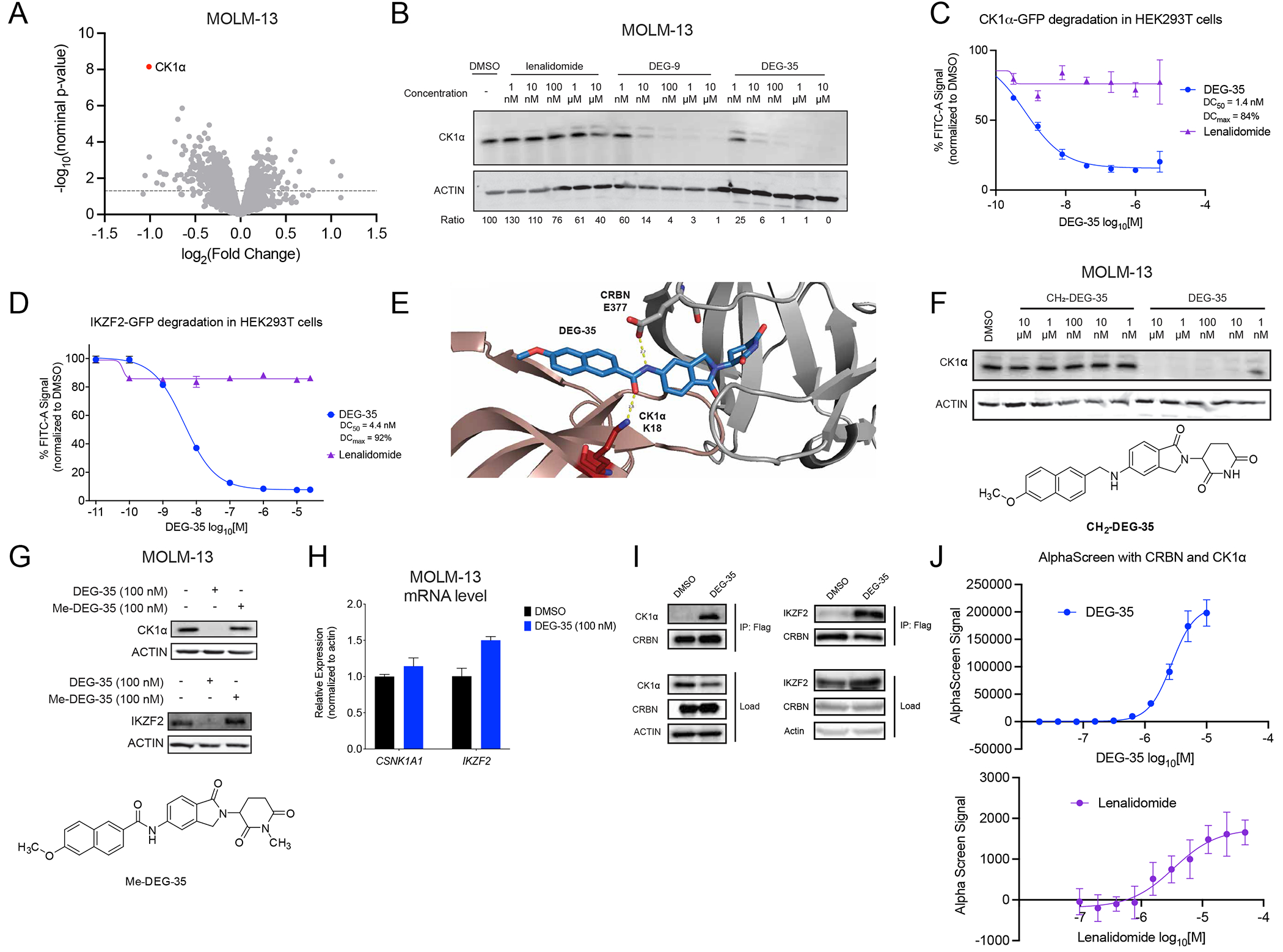
(A) Quantitative global proteomics analysis of MOLM-13 cells treated for 2 h with 50 nM DEG-35. (B) Western blot analysis of CK1α in MOLM-13 cells treated for 24 h with indicated drugs. (C,D) Flow cytometry analysis of HEK293T cells expressing (C) GFP-CK1α or (D) GFP-IKZF2 zinc finger 2 treated with DEG-35 for 5 h or 24 h respectively. Data shown is the mean ± SEM of three biological replicates and is representative of two independent experiments. (E) Docking model of DEG-35 with CK1α and CRBN. (F) Structure of CH2-DEG-35 and Western blot analysis of CK1α and IKZF2 in MOLM-13 cells treated with DEG-35 or CH2-DEG-35. (G) Structure of Me-DEG-35 and western blot analysis of CK1α and IKZF2 in MOLM-13 cells treated with DEG-35 or Me-DEG-35. (H) qRT-PCR analysis of CK1α and IKZF2 mRNA levels in MOLM-13 cells treated 24 h with 100 nM DEG-35. Data shown is the mean ± SEM of three biological replicates. (I) Western blot analysis of co-immunoprecipitation of endogenous CK1α and transiently expressed IKZF2 with Flag-CRBN in HEK293T. (J) In vitro analysis for lenalidomide or DEG-35 between His-CULT and streptactin-CK1α by AlphaScreen. Data shown is the mean ± SEM of three biological replicates and is representative of two independent experiments. See also Figure S2 and Table S2.
Based on our prior work with the 5-NH2-lenalidomide scaffold,13 we hypothesized that DEG-35 efficiently degrades CK1α by mediating a hydrogen bond network between CRBN E37714 and CK1α K18, to form a bridge between the proteins and strengthen the ternary complex formation (Figure 2E). To test this model, we synthesized an analog of DEG-35 that lacked the amide mediating these interactions (CH2-DEG-35) and tested its ability to degrade CK1α (Figure 2F). Consistent with our model, DEG-35 but not CH2-DEG-35 efficiently degraded CK1α in MOLM-13 cells.
Degradation of both substrates was inhibited by the neddylation inhibitor MLN4924 indicative of Cullin-dependent degradation (Figures 1D and S2E). Methylation of the glutarimide on DEG-35 (Me-DEG-35), which blocks engagement of CRBN, rescued IKZF2 and CK1α degradation (Figure 2G). Additionally, DEG-35 was competitively inhibited by pomalidomide, which binds to CRBN but does not degrade IKZF2 and CK1α (Figure S2F). RNA expression levels of CSNK1A1 (CK1α) remained unchanged at 24 h of treatment and IKZF2 RNA expression increased modestly, consistent with a post-translational degradation mechanism (Figure 2H).
The enhanced degradation of IKZF2 and CK1α by DEG-35 may be connected to the stronger engagement with CRBN in a ternary complex. CK1α was readily co-immunoprecipitated with Flag-CRBN in HEK293T cells treated with DEG-35. Similarly, IKZF2 was co-immunoprecipitated with Flag-CRBN in HEK293T cells transiently expressing IKZF2 (Figure 2I). DEG-35 has higher affinity (KD = 472.6 nM) than lenalidomide (KD = 804.7 nM) for CRBN, as measured by HTR-FRET (Figures S2G–H). DEG-35 also induced a significantly stronger ternary complex between CRBN and CK1α than lenalidomide, as measured by AlphaScreen (Figure 2J). Collectively, these data show that DEG-35 induces a strong ternary complex between substrates, like IKZF2 or CK1α, and CRBN to exert its anti-proliferative effects in AML cells.
3. DEG-35 activates the p53 apoptosis pathway
Given that DEG-35 is a dual degrader of IKZF2 and CK1α, we next evaluated whether DEG-35 possesses broad spectrum or selective anti-proliferative effects across cancer cell lines to further probe the mechanism of action. We leveraged the PRISM (Profiling Relative Inhibition Simultaneously in Mixtures) platform that uses barcoded cell lines from 20 lineages that are treated with DEG-35 at eight concentrations (4.6 nM–10 μM) for 5 days. The sensitivity of each cell line to DEG-35 is then measured based on detection of the barcode (Figure 3A).15,16 Evaluation of DEG-35 against these 770 cancer cell lines revealed that a subset of cell lineages exhibited enhanced sensitivity to DEG-35, including some cell lines in the AML lineage (Figures S3A–B). As expected, gene expression and copy number aberrations of CRBN correlated with DEG-35 sensitivity (Table S3). Correlation analysis against CRISPR knockout data for genetic dependency showed that DEG-35 sensitivity strongly correlated with cellular dependence on MDM4, a p53 tumor suppressor, mutations in TP53, and inhibitors of p53–MDM2 interaction, such as nutlin and AMG-232 (Figure 3B–D, Table S3). Interestingly, a corresponding profile of nutlin-3a revealed correlations with both MDM2 and MDM4 that drive resistance, which differed from the profile we observed for DEG-35 (Figure S3C, Table S3). These data point to the primary function of DEG-35 in cancer cell lines through its degradation of CK1α, as CK1α is known to regulate MDM2/MDM4 and TP53 stability. 16–18 Interestingly among the different hematologic malignancies, only AML cell lines with TP53 wildtype exhibited significantly increased sensitivity to DEG-35 compared to those with TP53 mutations (Figures 3E, Figure S3D–E).
Figure 3. DEG-35 leads to activation of the p53 pathway via CK1α degradation.
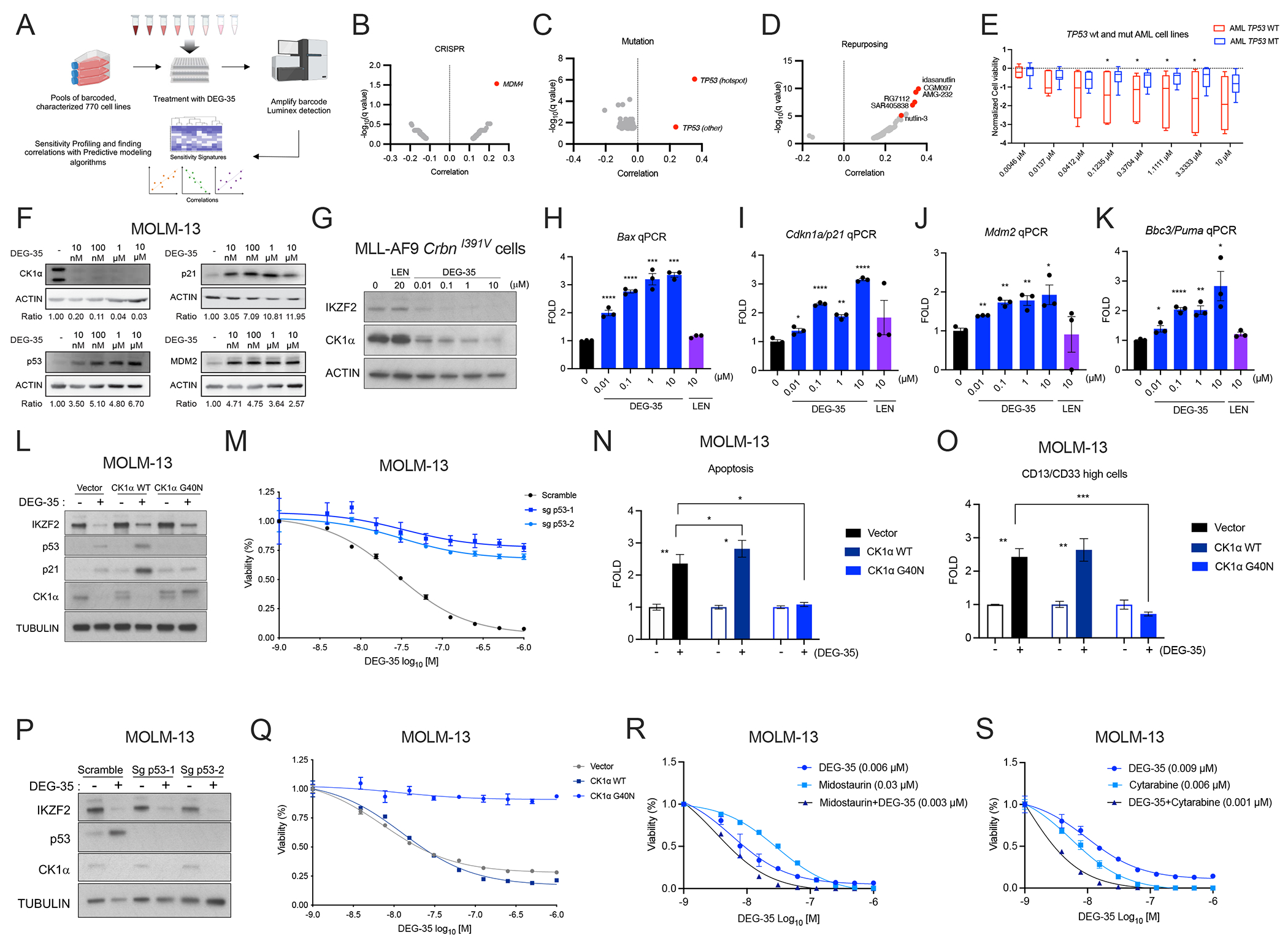
(A) Scheme showing the layout of the PRISM platform. (B-D) Correlation results showing the sensitivity signature of 770 human cell lines to DEG-35 to (B) genes knocked out by CRISPR screens (C) mutations harbored in cell lines and (D) repurposed drugs from PRISM data. (E) Normalized cell viability of AML cell lines with wildtype and p53 mutation shown as box and whiskers plot presenting maximum and minimum values with median as the midline. Student’s t test was used, *p < 0.05. (F) Western blot analysis of p53 targets p21 and MDM2 in MOLM-13 cells treated with different doses of DEG-35 for 24 h. (G) Western blot analysis of CK1α and IKZF2 in MLL-AF9 CrbnI391V cells treated with different doses of DEG-35 for 24 h. (H-K) qPCR analysis of p53 targets (H) Bax, (I) Cdkn1a/p21, (J) Mdm2 and (K) Bbc3/Puma in MLL-AF9 CrbnI391V cells treated with different doses of DEG-35 for 24 h. (L) Western blot analysis of CK1α, IKZF2, p53, and p21 in MOLM-13 cells overexpressing CK1α WT, CK1α G40N, or vector control. (M) MOLM-13 cells overexpressing CK1α WT, CK1α G40N, or vector control were treated with DEG-35. 3 days post treatment cell viability was measured by Cell titer Glo assay. Data shown is a representative result of three independent experiments. Mean ± SEM of triplicates. (N,O) (N) Apoptosis and (O) differentiation were examined in MOLM-13 cells treated with DEG-35 for 2 days. (P) Western blot analysis showing protein levels of IKZF2, p53, and CK1α in MOLM-13 cas9 cells expressing sgRNA for TP53 and treated with DMSO or DEG-35. (Q) Cell viability was measured in MOLM-13 cells knocked out for p53 which were treated with DEG-35 for 3 days. Data shown is a representative result of three independent experiments. Mean ± SEM of triplicates. (R,S) Combination treatment of DEG-35 with (R) Midostaurin or (S) Cytarabine in MOLM-13 cells for 72 h, measured by Cell-titer glo assay. Representative data is shown from 3 independent experiments mean ± SEM of triplicates for (Q–S). Statistics for (H–K, N–O) are mean ± SEM of three independent experiments: Student’s t test, *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001. See also Figure S3 and Table S3 and S4.
Consistent with this pathway activation, mRNA and protein levels of CDKN1A/p21 and MDM2 increased at 24 h in MOLM-13 cells and maintained for 5 days (Figures 3F, S3F–G). Cell cycle analysis also indicated that cells undergo G1 cell cycle arrest from day 1 until day 5 after a single DEG-35 treatment (Figure S3H). Activation of the p53 pathway was also observed in the MLL-AF9 CrbnI391V cells in which CK1α was degraded, and p53 targets were transcriptionally induced when treated with DEG-35 (Figures 3G–K).
To evaluate the contribution of the CK1α–TP53 axis in DEG-35-mediated cytotoxicity in myeloid leukemia cells, we utilized the degradation resistant mutant CK1α G40N, which is unable to engage lenalidomide-bound CRL4CRBN.19 As expected, DEG-35 could not degrade the CK1α G40N mutant, which blocked upregulation of p53 and p21 (Figure 3L). Furthermore, cytotoxicity, apoptosis, and differentiation were predominantly rescued by the CK1α G40N mutation (Figures 3M–O). Single cell clones deleted for TP53 were mostly resistant to effects from DEG-35 where we observed enhanced cell viability and resistance to differentiation and apoptosis (Figures 3P–Q and Figure S3I–K). Importantly, TP53 loss did not alter degradation of CK1α by DEG-35 (Figure 3P). These data indicate that degradation of CK1α activates p53-dependent apoptosis by reduction of the MDM4-p53 interaction.17,20
We next investigated whether DEG-35 could be used in combination with other AML drugs. We performed viability assays treating MOLM-13 and MOLM-14 cells with DEG-35 alone or in combination with midostaurin, cytarabine, venetoclax, or methothrexate (Figures 3R–S and S3L–M). Interestingly, we found that cells were more sensitive to DEG-35 when co-treated with midostaurin or cytarabine but not with venetoclax or methotrexate.
4. DEG-35 activates IKZF2-dependent differentiation
We hypothesized that IKZF2 also contributes to the cellular phenotypes of DEG-35 based on our finding that the IC50 values do not correlate solely with CK1α degradation in the various AML cell lines. We analyzed the list of shRNA genes and gene expression (positive correlation coeff >0.2, q-value <0.05) from the PRISM data with a gene set enrichment analysis tool, ENRICHR.21 DEG-35 sensitivity from the PRISM screen analysis was associated with c-MYC and FoxP3 transcription factors, which we previously found associated with IKZF2-dependent pathways (Table S4).
To further understand how DEG-35 alters myeloid leukemia cell growth, apoptosis, and differentiation, we performed RNA-sequencing in MOLM-13 cells treated with DMSO or DEG-35 at 50 nM for 24 h. We found 643 genes were upregulated and 646 genes were downregulated in DEG-35 treated cells compared to DMSO control with log2-fold change >1 and p-adjusted <0.05 (Figure 4A). In agreement with our PRISM data showing the p53 pathway is activated by DEG-35, we found up-regulation of p53 targets upon DEG-35 treatment (annotated in black Figures 4A and S4A–B). Interestingly, we also could detect the same genes that are differentially expressed in MLL-AF9 LSCs that were Ikzf2-deleted in the DEG-35 treated MOLM-13 cells, indicating that the IKZF2 pathway is inhibited (annotated in red, Figures 4A and S4C–D). Surprisingly three of the up-regulated IKZF2 knockout genes, C3, ICA1, and CSF1R, are C/EBP regulated genes22–24 suggesting that the C/EBP pathway, which has been previously shown to be up-regulated in Ikzf2-knockout LSCs, is also up-regulated in DEG-35 treated cells.
Figure 4. DEG-35 treated cells exhibit IKZF2 knockout leukemia signature and can be rescued by IKZF2 mutants.

(A) Heatmap of differentially expressed genes from RNA-seq result obtained from MOLM-13 cells treated with DMSO or DEG-35 at 50 nM for 24 h. Heatmap shows 637 up and 615 down regulated genes in DEG-35 treated cells compared to DMSO control, with log 2-fold change >1 with p adjusted <0.05. P53 targets are marked in black and genes overlapping with MLL-AF9 Ikzf2 knockout LSCs are marked in red. (B-D) GSEA analysis showing enrichment of upregulated and downregulated genes in MOLM-13 cells treated with DEG-35 with signatures of (B) genes altered in IKZF2 knockout leukemic stem cells, (C) myeloid differentiation, (D) genes regulated by HOXA9-MEIS1 and MYC. (E) Western blot analysis showing reduction of HOXA9 and MYC by DEG-35 treatment at 24 h post-treatment in MOLM-13 cells. (F) Western blot analysis showing MOLM-13 cells expressing WT or mutant IKZF2 treated with DEG-35 for 24h. (G-H) Differentiation myeloid markers (G) CD33/CD13 and (H) CD14/CD11b were measured in MOLM-13 cells expressing WT or mutant human IKZF2 treated with DEG-35 for 2 days. MOLM-13 cells were transduced with vectors expressing IKZF2 with IRES GFP. For measuring differentiation, cells were gated on GFP+ cells before gating on myeloid markers. For results in (G, H), mean ± SEM of three independent experiments; Student’s t test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. See also Figure S4 and Tables S4-S6.
We performed gene set enrichment analysis (GSEA) using the rank list of differentially expressed genes from the MOLM-13 cells treated with DMSO and DEG-35 on all curated gene sets in the Molecular Signatures Database25 combined with an additional set of relevant gene sets (92 gene sets from our experimentally derived or published hematopoietic self-renewal and differentiation signatures, CK1 knockout keratinocyte signature) (Tables S5 and S6). Consistent with the PRISM screen and our rescue experiments, we observed enrichment for genes upregulated in CK1α knockout keratinocytes and genes induced by a GSK3 inhibitor (Figures S4E–F) which could be due to stabilization of BETA-CATENIN upon CK1α loss.26 Additionally, genes related to the p53 pathway (DNA damage agents such as cisplatin and irradiation) were identified in the GSEA (Figure S4G) and p53 targets including CDKN1A/p21, BAX, and MDM2 were up-regulated in DEG-35 treated MOLM-13 cells (Figures 4A and S4A–B). We then examined if genes associated with IKZF2 loss are enriched in DEG-35 treated MOLM-13 cells. In agreement with DEG-35 targeting IKZF2, we found significant enrichment with gene sets from the Ikzf2-deleted leukemic stem cells from the mouse MLL-AF9 model (Figure 4B). We could also observe enrichment for myeloid differentiation (Figure 4C) and loss of MYC and HOXA9 self-renewal signatures (Figure 4D) as described with Ikzf2-knockout LSCs. This is consistent with the reduction of c-MYC and HOXA9 protein abundance by DEG-35 treatment (Figure 4E). Signatures involving the loss of targets for the transcription factor FOXP3, an associated marker of T regs which is also known to be regulated by IKZF2, were also found27 (Figure S4H). These data support that DEG-35 can target both CK1α and IKZF2 molecular gene expression programs through degradation of these targets.
To understand the contribution of IKZF2 degradation to DEG-35 activity, we designed mutations in the amino acids that have been suggested to interact with CRBN in mouse and human IKZF2 that could render IKZF2 resistant to CRBN mediated degradation. The human IKZF2 resistant mutant, IKZF2H141Q was overexpressed in MOLM-13 cells and was resistant to DEG-35 mediated degradation (Figure 4F). Overexpression of wildtype and IKZF2 mutant both rescued the myeloid differentiation effects (Figures 4G–H and S4I–K); however, DEG-35 induced apoptosis was not affected (Figure S4L). As the H141Q mutation used in hIKZF2 did not confer resistance to DEG-35 in mIKZF2, mIKZF2L155/L156R was used to rescue DEG-35 effect in MLL-AF9 CrbnI391V cells (Figure S4M). Overexpression of mIKZF2L155R/L156R resistant mutant also rescued myeloid differentiation induced by DEG-35 in MLL-AF9 CrbnI391V cells (Figure S4N). Consistently, DEG-35 induced apoptosis was not affected by overexpression of IKZF2 mutant in MLL-AF9 CrbnI391V cells similar to the human system, supporting the previously reported inhibitory role of IKZF2 in differentiation (Figures S4N–O).5 These results indicate that degradation of both targets, IKZF2 and CK1α, by DEG-35 contribute to its cellular effects.
5. DEG-35 has preferential cytotoxic activity in AML cells compared to normal cells
We then determined DEG-35’s differential sensitivity between mouse normal and leukemic cells. Murine hematopoietic stem and progenitor cells (HSPCs; lineage low Sca1+ c-Kit+ cells; LSK) and LSK-derived MLL-AF9 leukemic cells from CrbnI391V mice were sensitive to DEG-35 whereas the wildtype counterparts were not (Figure 5A). MLL-AF9 CrbnI391V leukemic cells exhibited reduction of colonies by half at 10 nM whereas LSK derived colonies were not affected at this dose. Moreover, proliferation studies identified the IC50 for normal bone marrow CrbnI391V cells as 100 nM whereas MLL-AF9 leukemic bone marrow CrbnI391V cells have an IC50 of 9.2 nM, demonstrating that the leukemic cells are more sensitive to DEG-35 than the normal cells (Figure 5B).
Figure 5. DEG-35 has preferential cytotoxic activity in AML cells compared to normal cells.

(A) Normalized colony assay of wt LSK cells, CrbnI391V LSK cells, MLL-AF9 wt cells and MLL-AF9 CrbnI391V cells treated with different concentrations of DEG-35 plated in M3434 methylcellulose for 5 days. Cells from n=3 mice for wt LSK cells, CrbnI391V LSK cells, MLL-AF9 CrbnI391V and n=2 mice for MLL-AF9 wt cells. Data shown is the mean ± SEM of the different biological replicates tested from two different experiments. (B) Viability assay of wt LSK, CrbnI391V LSK, MLL-AF9 wt and MLL-AF9 CrbnI391V cells treated with DEG-35 for 3 days. Data shown is the mean ± SEM of three different biological replicates tested. (C) Normalized colony assay of eight primary AML patient cells and four normal bone marrow or CD34+ HSPC cord blood cells treated with DEG-35 plated in H443 methylcellulose for 7 days. The mean ± SEM of indicated samples tested from more than three different experiments is shown. (D) Viability assay of AML PDX cells and normal BM cells treated with DEG-35 for 2 days. Data shown is the combined result of two experiments using normal BM (n=2) and PDX cells (n=2). Mean ± SEM of triplicates. (E) Western blot analysis of AML PDX sample 1 treated with different concentration of DEG-35 for 24h. For results in (A, C), Student’s t test, *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S5.
Similarly, in primary AML patient samples (n = 8) we found reduced colony formation (10 nM) compared to adult bone marrow (BM) cells from healthy human donors or cord blood cells (Figure 5C). Using hCD45 and mCD45 to differentiate PDX cells from the host cells (Figure S5A), we found that PDX cells underwent apoptosis (Figures S5B–D) and differentiation (Figures S5E–F). AML PDX cells exhibited lower IC50 concentrations of 2.8 nM and 5.2 nM compared to healthy bone marrow cells which exhibited an IC50 of 15.7 nM (Figure 5D). Furthermore, DEG-35 treatment in AML PDX2 cells led to increased apoptosis compared to normal bone marrow cells (Figures S5G–H). AML PDX cells also recapitulated the degradation of IKZF2 and CK1α after DEG-35 treatment and upregulation of p53 and p21 (Figure 5E). Thus, DEG-35 has differential activity in both mouse and human leukemic cells compared to that in normal cells.
6. In vivo efficacy of DEG-35 in leukemic mice
To determine if DEG-35 can effectively degrade its targets in vivo, we first performed pharmacodynamic experiments in the MLL-AF9 mouse model. A single dose of DEG-35 at 50 mg/kg was injected in BL6 mice transplanted with MLL-AF9 CrbnI391V cells, and leukemic cells were isolated and sorted (Figure 6A). After 24 h and 72 h, we found efficient degradation of CK1α and IKZF2 in the MLL-AF9 leukemic cells from bone marrow and spleen (Figures 6B and S6A). A single dose of DEG-35 led to increased apoptosis, myeloid differentiation, and reduced kit high frequency (representing the LSC population) in the leukemic cells (Figures S6B–D). Leukemic cells from DEG-35 treated mice had reduced colony forming activity (Figure 6C). We next performed pharmacokinetic studies and found DEG-35 to have a half-life of 2.5 h in the plasma of the mice when administered intraperitoneally (Figure S6E).
Figure 6. DEG-35 can degrade targets in AML cells in vivo and delay leukemia progression.
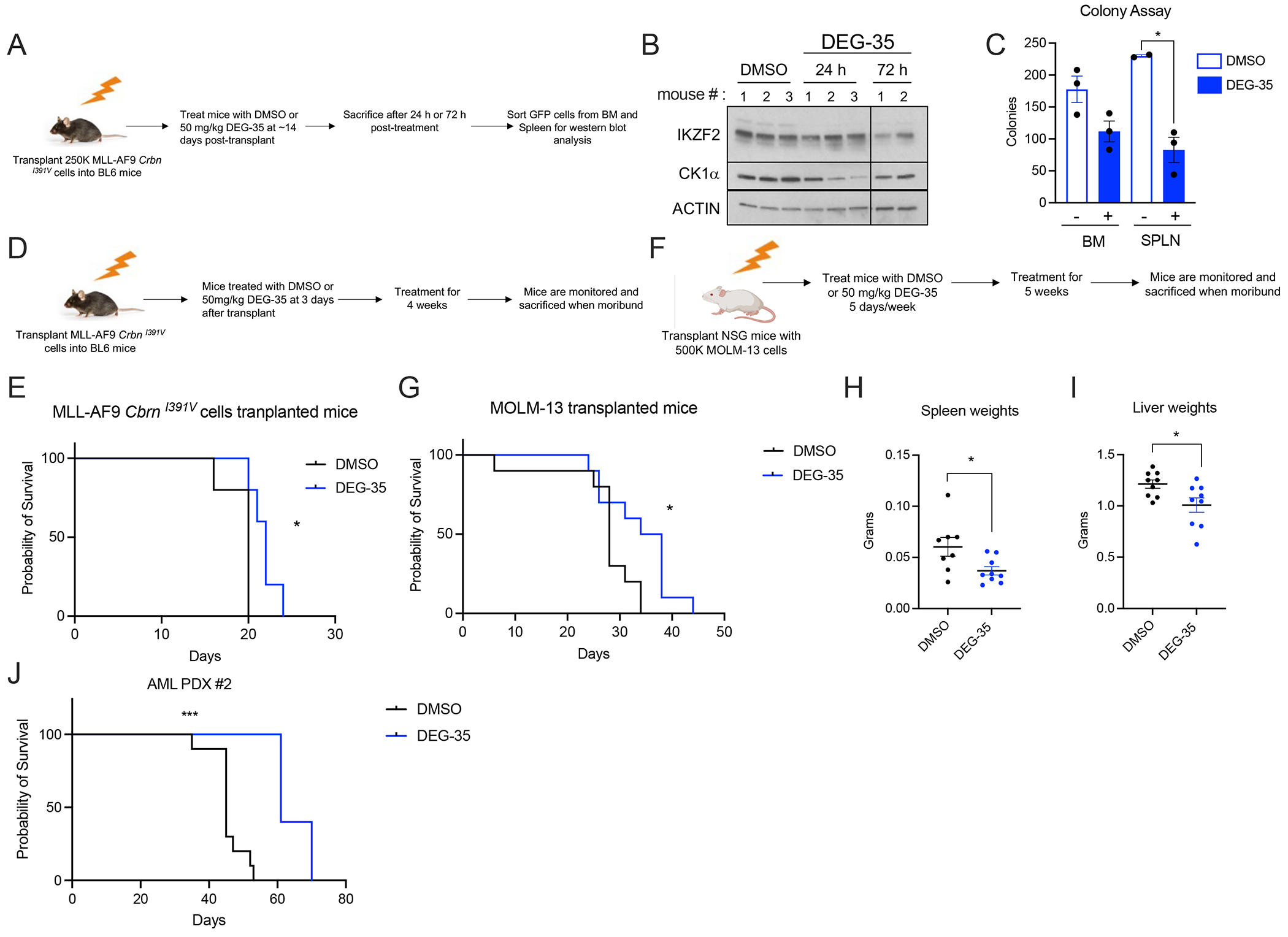
(A) Scheme strategy for analyzing target pharmacodynamics of DEG-35 in the MLL-AF9 CrbnI391V mice model. (B) Western blot analysis of targets IKZF2 and CK1α of leukemic bone marrow cells from mice treated with DEG-35 at 50 mg/kg, 24 h and 72 h post-treatment. (C) Colony assay of leukemic cells from bone marrow and spleen from mice treated with DMSO or DEG-35 24 h post treatment. Data shown is the mean ± SEM of three different biological replicates tested. Student’s t test was used, *p < 0.05. (D) Scheme showing treatment strategy for DMSO or DEG-35 in BL6 mice transplanted with MLL-AF9 CrbnI391V cells. (E) Kaplan-Meier survival curve for MLL-AF9 CrbnI391V transplanted BL6 mice treated with DMSO or 50 mg/kg DEG-35. n=5 mice for each group. (F) Scheme showing treatment strategy for DMSO or DEG-35 in MOLM-13 cells transplanted in NSG mice. (G) Survival curve for MOLM-13 transplanted mice treated with DMSO or 50 mg/kg DEG-35. n=10 mice for each group. (H,I) (H) Spleen weight and (I) liver weights of mice from (G). Data shown is the means +/− SEM of the indicated samples tested. (J) Survival curve of NSG mice transplanted with PDX AML#2 and treated with DMSO (n=10) or DEG-35 (n=5). For statistics in (E, G, and J), log rank test was used *p < 0.05, ***p < 0.001 and for results in (H and I) Student’s t test was used, *p < 0.05. See also Figure S6.
DEG-35 treatment also delayed leukemia progression in BL6 mice transplanted with MLL-AF9 CrbnI391V cells compared to DMSO treated mice without overt weight loss at 50 mg/kg (Figures 6D–E and S6F). Reduction of CK1α and IKZF2 was observed by immunoblot analysis in splenic leukemic cells in DEG-35 treated mice (Figure S6G). Similarly, DEG-35 treatment significantly prolonged survival in the MOLM-13 transplanted mice (Figures 6F–G) without overt weight loss (Figure S6H). Furthermore, DEG-35 treated mice demonstrated reduced disease burden shown by decreased spleen and liver weights (Figures 6H–I). Immunoblot analysis of sorted MOLM-13 cells from the spleen of moribund mice at the endpoint showed reduced CK1α but not IKZF2 abundance (Figure S6I). These data suggest potential resistance of leukemic cells to degradation of IKZF2 in vivo over time. Additionally, we found reduced engraftment of two AML PDXs at 2 weeks and 3 weeks after transplant (Figures S6J–K). Although AML PDX#1 successfully engrafted, it did not cause leukemia-induced death. DEG-35 treatment led to significant prolonged survival and reduced spleen burden in mice transplanted with AML PDX#2 without reducing body weight (Figures 6J, S6L–M).
7. DEG-77, an analog of DEG-35, has comparable activities in vitro.
To improve in vivo efficacy, we performed another round of structural optimization to enhance the solubility of DEG-35 while maintaining the dual degradation of IKZF2 and CK1α. Permutation of the naphthalene in DEG-35 with a sp3-hybridized carbon and oxygen yielded DEG-77 (Figure 7A). Importantly, DEG-77 exhibited a 10-fold increase in solubility in DMSO compared to that of DEG-35 and maintained comparable activity against IKZF2 and CK1α in MOLM-13 cells at 24 h. Similar potency in degradation, myeloid differentiation, apoptosis, and cytotoxicity with comparable IC50 values were also observed (IC50 DEG-35 = 5 nM, IC50 DEG-77 = 4 nM, Figures 7B–F). Additionally, DEG-77 had increased selectivity against IKZF2 (DC50 = 15.3 nM) and CK1α (DC50 = 10 nM) over other known neosubstrates (GSPT1, IKZF1, IKZF3, Figures S7A–B). As expected, degradation of IKZF2 and CK1α by DEG-77 was CRBN-dependent and occurred post-translationally (Figure S7C–E). DEG-77 additionally reduced abundance of HOXA9 and c-MYC in MOLM-13 cells (Figure 7G). Furthermore, DEG-77 had similar activity and kinetics as DEG-35 resulting in increased apoptosis, myeloid differentiation, and induction of the TP53 pathway, and comparable preferential sensitivity to leukemia versus healthy cells (Figures 7H–O and Figures S7F–L).
Figure 7. DEG-77, an analog of DEG-35 has similar potency in killing AML cells.
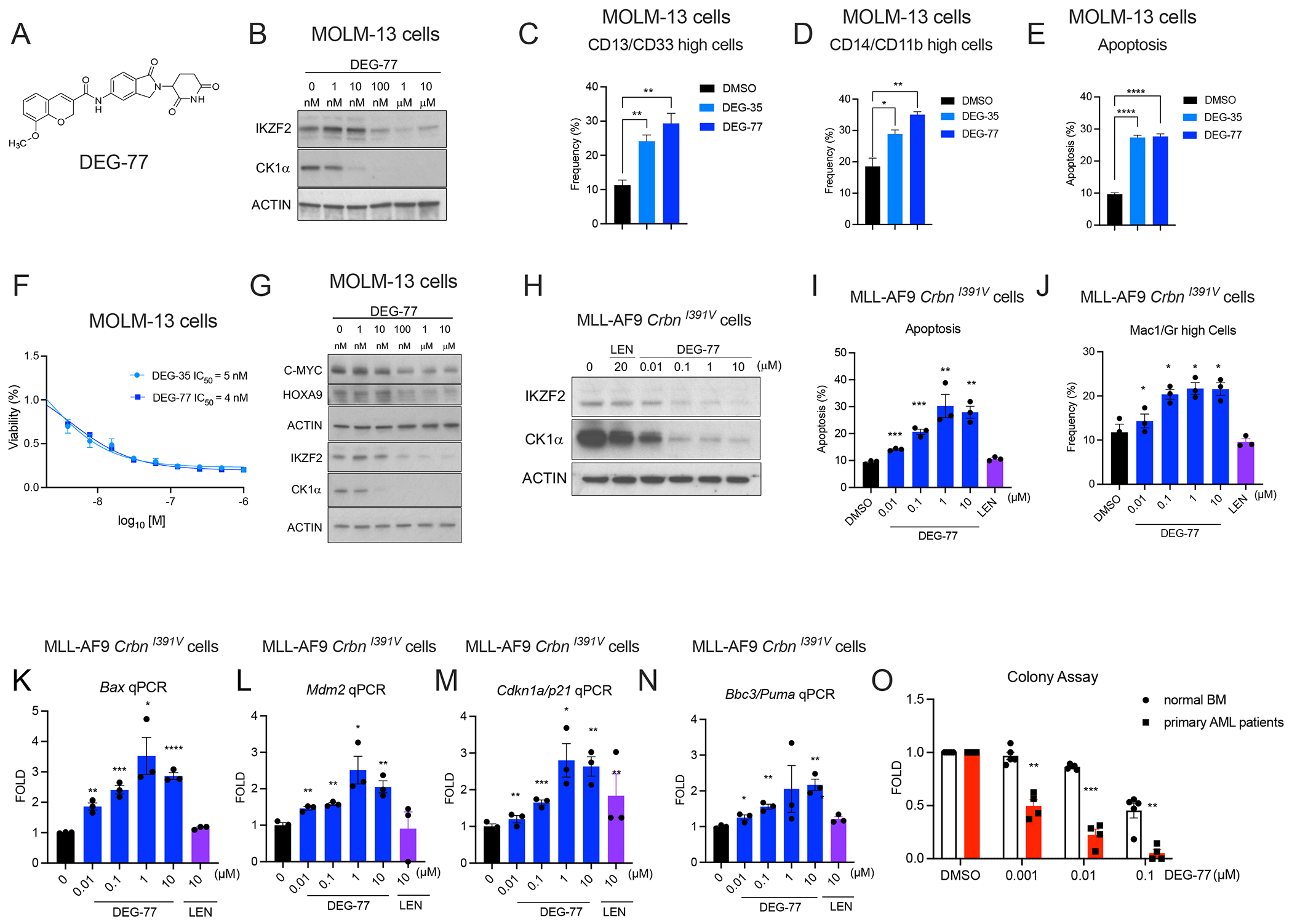
(A) Chemical structure of DEG-77. (B) Western blot analysis of IKZF2 and CK1α in MOLM-13 cells treated with indicated concentrations of DEG-77 for 24 h. (C,D) MOLM-13 cells were stained with myeloid markers (C) CD13/CD33 and (D) CD14/CD11b and then measured by flow cytometry for differentiation at day 2 post-treatment with 100 nM DEG-35 or DEG-77. (E) Apoptosis was measured by flow cytometry using Annexin V and DAPI as apoptotic marker. (F) Cell viability assay was performed in MOLM-13 cells treated with DEG-35 and DEG-77 for 3 days to obtain IC50 values. Data shown is the representative result of three independent experiments. Mean ± SEM of triplicates. (G) Western blot analysis showing reduction of HOXA9 and MYC by DEG-77 treatment at 24 h post-treatment in MOLM-13 cells. (H) Western blot analysis showing degradation of CK1α and IKZF2 in MLL-AF9 CrbnI391V by different concentration of DEG-77 after 24 h. (I,J) (I) Apoptosis and (J) differentiation were measured by flow cytometry of cells stained with Annexin V and myeloid markers Mac1/Gr1 respectively. Flow cytometry was performed 24 h post-treatment with indicated concentrations of DEG-35 or 10 μM lenalidomide. (K-N) mRNA levels of p53 targets, (K) Bax, (L) Mdm2, (M) Cdkn1a/p21, (N) Bbc3/Puma measured by qPCR analysis in MLL-AF9 CrbnI391V cells treated with DEG-77 and lenalidomide for 24 h. (O) Normalized colony assay of four primary AML patient cells and five normal bone marrow/CD34+ HSPC cord blood cells treated with DEG-35 plated in H443 methylcellulose for 14 days. For statistics used in (C–E, I–J, K–N, O), results shown were combined from at least three independent experiments, Mean ± SEM. Student’s t test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. See also Figure S7.
8. DEG-77 has improved efficacy in vivo and can block leukemic stem cell activity
To measure the efficacy of DEG-77 in vivo, we performed pharmacokinetic and pharmacodynamic experiments (Figures 8A–C). The pharmacokinetic study revealed that a single treatment of DEG-77 at 223 mg/kg in CrbnI391V mice had a half-life of 16.9 h (Figure 8A). In the pharmacodynamic experiment, one injection of DEG-77 in BL6 mice transplanted with MLL-AF9 CrbnI391V cells led to the degradation of IKZF2 and CK1α but not GSPT1 in bone marrow and splenic leukemic cells at 24, 48, and 72 h (Figure 8C and S8A). After 24 h following DEG-77 injection, leukemic cells in blood, bone marrow, and spleen underwent increased apoptosis and myeloid differentiation, and leukemic cells had reduced colony forming ability (Figures 8D and S8B–C).
Figure 8. DEG-77, an analog of DEG-35 has better efficacy and can block leukemic stem cell activity.
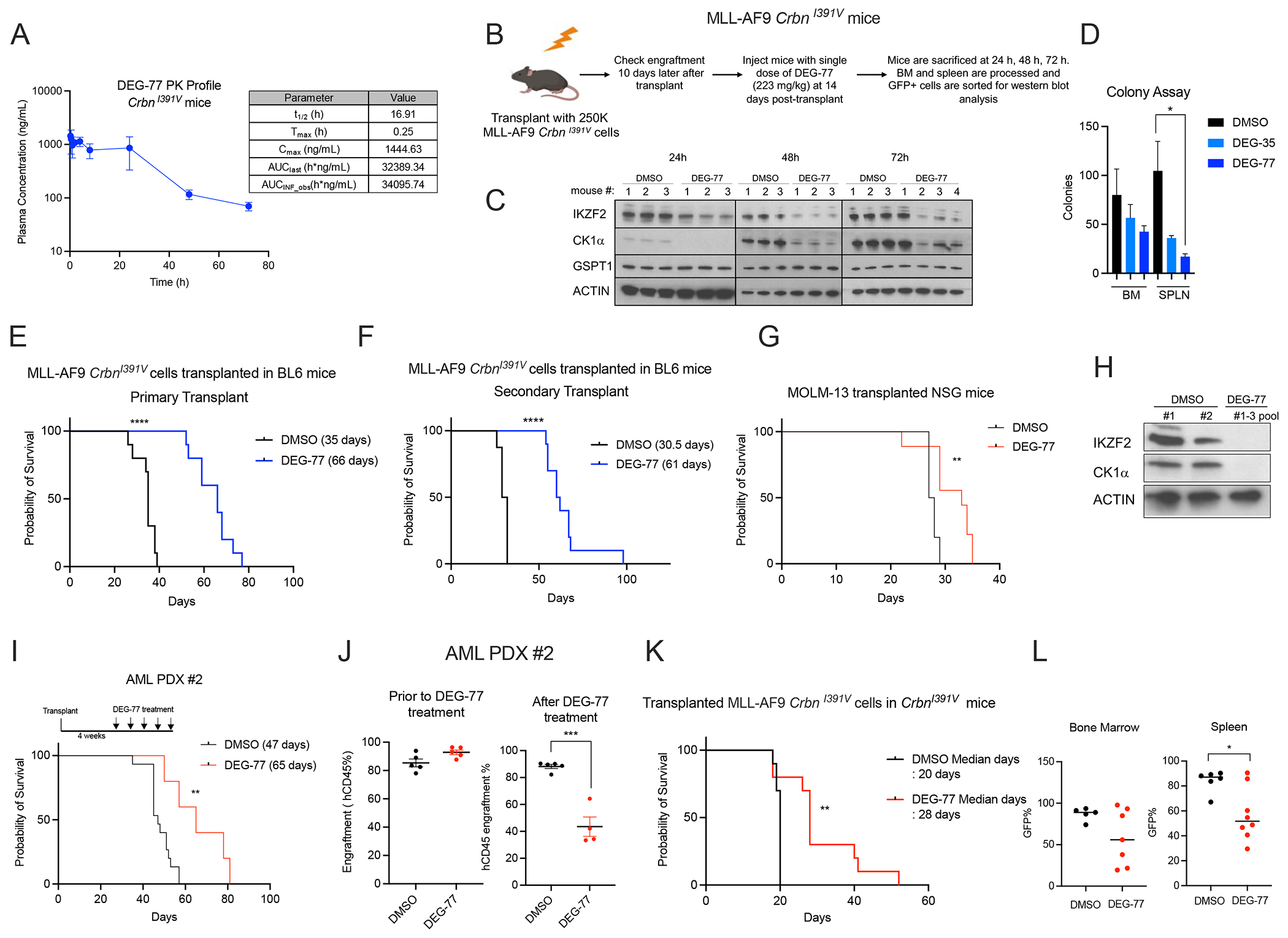
(A) Pharmacokinetic study showing plasma concentration of DEG-77 over a 72 h period in mice after a single intraperitoneal injection of DEG-35 at 223 mg/kg. The plasma concentrations represent the means +/− SEM from three mice per time point. (B) Scheme showing strategy for analyzing pharmacodynamics of targets of DEG-77 in the MLL-AF9 CrbnI391V mice model. (C) Western blot analysis of targets IKZF2, CK1α and GSPT1 of leukemic bone marrow cells from mice treated with DEG-77 at 223 mg/kg, at indicated time point. (D) Colony assay of leukemic cells from bone marrow and spleen of mice treated with DMSO or DEG-77 24 h post treatment. Data shown is the means +/− SEM of n=3 samples for each condition. Student’s t test, *p < 0.05. (E, F) Survival curves of BL/6 mice transplanted with (E) MLL-AF9 CrbnI391V cells treated with DMSO or DEG-77 at dose of 223mg/kg weekly or (F) secondarily transplanted. ****p < 0.0001 log-rank test. n=10 mice for each group. (G) Survival curve of NSG mice transplanted with MOLM-13 cells and treated with DMSO (n=10) or DEG-77 (n=9) treated at 223mg/kg weekly. **p < 0.01 log-rank test. (H) Western blot analysis of MOLM-13 cells sorted from bone marrow of NSG mice 24h after drug treatment. (I) Survival curve of NSG mice transplanted with AML PDX#2 treated with DMSO (n=15) or DEG-77 (n=5) at dose of 223mg/kg, weekly from 4 weeks post-transplant. **p < 0.01 log-rank test. (J) Engraftment frequency of AML PDX cells measured by flow cytometry by hCD45 using bone marrow aspirates at 4 weeks post-transplant (left panel) before and (right panel) one week after a single dose of DEG-77 treatment. Data shown is means +/− SEM of the indicated samples as shown. Student’s t test, ***p < 0.001. (K) Survival curve of non-irradiated CrbnI391V mice transplanted with MLL-AF9 CrbnI391V cells treated with DMSO or DEG-77 at dose of 223 mg/kg weekly. **p < 0.01 log-rank test. n=10 mice for each group. (L) Disease burden shown by frequency of GFP positive cells (MLL-AF9 CrbnI391V cells) in bone marrow or spleen at time of sacrifice. Data shown is means +/− SEM of the indicated samples as shown. Student’s t test, *p < 0.05. See also Figure S8.
DEG-77-treated mice had significantly prolonged survival of 66 days compared to 35 days of vehicle-treated mice (Figure 8E) without loss of total body weight (Figure S8D). Importantly, the secondary transplant of DMSO and DEG-77 treated mice maintained a significant survival difference, where cells from DEG-77 treated mice in the primary had a pronounced delay in leukemia progression, indicating that DEG-77 can also reduce leukemic stem cell activity (Figure 8F).
DEG-77 treatment also increased survival and reduced leukemia burden in bone marrow of MOLM-13 transplanted mice without gross weight loss (Figures 8G and S8E–F). Both IKZF2 and CK1α were degraded in bone marrow leukemic cells after DEG-77 treatment (Figure 8H). In the PDX AML model, PDX cells were allowed to engraft for 4 weeks and then treated with DMSO or DEG-77 weekly. This resulted in a significant extension of survival and reduced disease burden for the animals treated with DEG-77 (Figure 8I and S8G–H). PDX cells exhibited >85% engraftment prior to DEG-77 treatment in the bone marrow and only around 40% engraftment after a single injection (Figure 8J) indicating that DEG-77 can reduce leukemic cells in the bone marrow.
Toxicity was determined by testing CrbnI391V mice with four rounds of DMSO or DEG-77 resulting in reduced HSC and MPP1 frequency in the stem cell compartment and CMP and MEP in the progenitor compartment (Figures S8I–K). However, we did not observe significant changes in absolute number of LSK and HSC in the DEG-77 treated mice (Figures S8L–M). We found a decrease in B cells and increase in myeloid cells which resulted in a modest reduction of WBC counts (Figures S8N–Q). Importantly, platelets and RBC counts remained unchanged without a major reduction in total body weight (Figures S8R–T).
To further test the therapeutic index, we found a significant increase in survival and reduction of leukemic cell burden in the spleen when DEG-77 was administered to CrbnI391V mice transplanted with MLL-AF9 CrbnI391V cells (Figures 8K–L). These data suggest that DEG-77 has efficiency in selectively killing leukemic cells in an in vivo model where all cells are sensitive to the degrader.
Discussion
We report the development of dual degraders DEG-35 and DEG-77 that have selectivity for IKZF2 among other Ikaros family members and efficiently target CK1α. Utilizing large cell-based screens and unbiased proteomics, we discovered the substrates and a driving mechanism for DEG-35 function. Through a PRISM screen, we showed that DEG-35 activates the CK1α–p53 pathway in various cell lines. Unfortunately, we did not see CK1α and IKZF2 in the correlations of PRISM data with the CRISPR knockout screen because CK1α, an essential gene, was not included in the screen and IKZF2 has thus far been validated to be important only in AML cells. We demonstrate that DEG-35 mediates its effects mainly through dual activity on the CK1α–TP53 axis and IKZF2 inhibition.
Targeting CK1α could have potential liabilities for normal cells considering its reported function in HSCs.28 Although we observed changes to the normal hematopoietic system including a reduction of B cells and increased myeloid cells, we found overall tolerability in the stem and progenitor compartment. Furthermore, treatment of DEG-77 in an AML model in CrbnI391V mice demonstrated a significant delay in latency of disease suggesting a therapeutic index.
Targeting CK1α could be especially beneficial in MDS or AML patients who have lost one copy of CK1α.29 Furthermore, in addition to the TP53 pathway and for patients that have wildtype p53, we have shown that DEG-35 can reduce HOXA9 and MYC abundance. HOXA9 is overexpressed in more than 70% of AMLs and is associated with poor prognosis. 30,31 HOXA9 and MYC are known essential factors for survival of AML cells that act downstream in AML lineages with various upstream genetic alterations, including MLL-translocations, NUP98-fusions, NPM1c mutations, CDX dysregulation, and MOZ-fusions.32 We have tested primary AML patient samples that harbor mutations or genetic alterations such as DNMT3A, NPM1, FLT3, CEBPA, MLL-fusions, del-5q, IDH1 and SF3B1 and found that these cells are sensitive to dual degradation of IKZF2 and CK1α. Thus, DEG-35 may be beneficial for AML patients with different cytogenetic abnormalities.
Consistent with our results, several studies have shown that drugs targeting the TP53 pathway such as nutlin have additive or synergistic effects when used in combination with midostaurin or cytarabine.33,34 Previous studies have shown that resistance to lenalidomide arises through abnormalities that directly affect CRBN abundance the resurgence of specific populations of cancer cells, or alteration in downstream pathways including TP53 mutagenesis.35–37 We would expect to potentially observe similar resistance mechanisms in response to DEG-35 or DEG-77, in addition to other mechanisms.
Importantly, the degradation mechanism enables this beneficial activity against multiple substrates and in multiple contexts. Interestingly, IKZF2 and IKZF4 are highly expressed in regulatory T cells and their loss leads to a reversal of immune suppressive activity converting T regs into effector T cells.38 This approach was recently demonstrated with another small molecule degrader ALV2 that targeted IKZF2 in T regs, reducing the suppressive activity of T reg cells.10 Furthermore, DKY709, an IKZF2 degrader, is under investigation in a Phase 1 clinical trial for advanced solid cancers as a monotherapy and in combination with PD1 inhibitors. 39 It will be important to test how dual blockade of intrinsic and extrinsic mechanisms would improve immunotherapy. As additional CRBN ligands and degraders are developed, it will also be important to understand the optimal set of substrates for targeting specific cancer subtypes and the potential immunotherapeutic contribution. DEG-35 and DEG-77 are thus selective and potent tool compounds that demonstrate that dual degradation of IKZF2 and CK1α is a viable therapeutic strategy. Overall, our study provides a framework for the development of other degraders with an expanded set of substrates to enhance their function and activity.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Christina Woo (cwoo@chemistry.harvard.edu).
Materials Availability
Plasmids generated in this study have been deposited to Addgene.
Data and code availability
RNA-seq data has been deposited in GEO (GSE220707).
All proteomics datasets have been deposited to the PRIDE repository with the dataset identifier PXD039474.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines and Primary patient cells
All cell lines were grown at 37 °C with 5% CO2. MOLM-13 (male), MOLM-14 (male), OCI-AML3 (male), Kasumi-1 (male), NB4 (female), KG-1A (male), NOMO-1 (female) cells were cultured in RPMI media (Cell gro) supplemented with 10% FBS, glutamine, and penicillin/streptomycin. OCI-AML5 (male) was grown in MEM media (Cell gro) supplemented with 20% FBS, MEM, glutamine, penicillin/streptomycin and 10ng/ml of GM-CSF. All human AML cell lines were authenticated from the Integrated Genomics Operation core at MSKCC using STR analysis. HEK293T cells (female) were cultured in DMEM (Lonza) supplemented with 10% FBS, glutamine, and 10% penicillin/streptomycin.
Culture of all primary AML patient samples, unless otherwise noted, were collected under the Biospecimen collection/banking study 09-141. The use of the samples for research purposes is covered under the Biospecimen research protocol 16-354. Frozen AML patient cells or AML PDX cells were thawed in warm RPMI containing 20% FBS and penicillin/streptomycin, washed, counted, and cultured in StemSpan SFEMII containing StemSpan CD34+ expansion supplement (Stem Cell tech) at 1X106 cells/ml. Cytogenetic information of patient cells is available in Table S7.
Mouse leukemic bone marrow cells from MLL-AF9 wt and MLL-AF9 CrbnI391V mice (female) were grown in RPMI (Cellgro) medium containing 10% FBS, 6 ng/ml IL-3, 10 ng/ml SCF, 10 ng/ml IL-6 and 10 ng/ml GM-CSF.
Animal Models
6-8 weeks old female C57BL/6 or 6-10 weeks old female NSG mice purchased from Jackson Laboratories respectively, were used as recipients for transplant experiments. CrbnI391V mice were purchased from Jackson Laboratories and bred in the mouse facility in Memorial Sloan Kettering Cancer Center. 6-8 weeks old female mice were used for experiments. All animal procedures were approved by the Institutional Animal Care and Use Committee at Memorial Sloan Kettering Cancer Center.
METHOD DETAILS
Transduction of human leukemia cell lines
Cells were transduced with virus expressing empty vector, CK1α wt, CK1α G40N, murine or human IKZF2 wt or IKZF2 mutants by spinning in RPMI with 10% FBS together with viral supernatant containing 10 ug/ml polybrene at 1600 RPM for 1 h. Media was changed 24 h later, and after 72 h post-transduction cells were sorted for either GFP or RFP and used for experiments.
Transduction of HEK293T cells for fluorescence assays
HEK293T cells were cultured in a 10 cm plate in antibiotic-free DMEM supplemented with 10% FBS and grown to between 70-90% confluence. VSV-G (4 μg), Gag-Pol (6 μg), desired vector (IKZF2-GFP, IKZF1-GFP, GFP-CK1α, 10 μg), and TransitPro (30 μL) were diluted into OptiMEM to a final volume of 1 mL. After tapping to mix and incubation at room temperature (15 min), the DNA/OptiMEM solution was added dropwise to HEK293T cells and swirled gently to mix. Cells were incubated at 37 °C and 5% CO2 for 5-8 h. Media was replaced with antibiotic-free DMEM supplemented with 10% FBS (10 mL). Cells were incubated for an additional 24-48 h prior to harvesting virus. To harvest, media was collected and centrifuged (500 x g, 5 min). The supernatant was removed, passed through a 0.45 μm filter, and aliquoted prior to being snap frozen in liquid nitrogen and stored at − 80 °C prior to use.
An aliquot of lentivirus (1 mL) was thawed and used to create a 6-well viral dilution plate containing 0, 1:3, 1:5, 1:10, 1:50, and 1:100 viral dilutions in antibiotic-free DMEM supplemented with 10% FBS and polybrene (10 μg/mL) for a final volume of 500 μL per well. 1 x 105 HEK293T cells in antibiotic-free DMEM (1 mL) supplemented with 10% FBS and polybrene (10 μg/mL) were added to each well and rocked gently to mix. Cells were incubated with virus at 37 °C and 5% CO2 for 48 h. The media was changed and cells were incubated for an additional 24 h. To begin the selection, media was changed to DMEM supplemented with 10% FBS, 10% penicillin/streptomycin, and puromycin (2 μg/mL). Media was periodically refreshed until cells in control well (no virus) appeared dead, generally about 4-5 days after beginning selection. Healthy cells from all wells of virus were harvested and sorted for GFP expression using fluorescence-activated cell sorting (FACS) on the Fortessa flow cytometer at the Harvard Bauer Core and binned into “high”, “medium”, and “low” expression levels as compared to a non-transduced HEK293T cell line. Each cell pool was then evaluated for POI-GFP degradation via Western blot and flow cytometry and subsequently used for experiments.
Site-directed mutagenesis of plasmids
Site-directed mutagenesis of pMSCV-IRES-RFP15-mouse IKZF2, pHFUW-human IKZF2-IRES-GFP and pLC-Flag-CSNK1A1 constructs was performed to obtain the mutants. QuikChange Lightning and Multi Site-Directed Mutagenesis Kits (#210513 and #210518, Agilent Technologies) were used. The designed primers are listed in the (Table S8).
Isolation of cord blood-derived CD34+ HSPC cells
Five mixed cord blood units (each unit is from a healthy donor) were used for purifying cord blood CD34+ HSPCs. Mononuclear cells were purified from cord blood by using Hespan and Ficoll-Hypaque Plus density centrifugation, followed by positive selection using the Auto MACS Pro Separator and isolation Kit (Miltenyi).
Generation of MLL-AF9 CrbnI391V leukemic cells
Lin-Sca+Kit+ (LSK) cells were isolated from bone marrow cells of wildtype or CrbnI391V mice that were 6 to 8 weeks old. Briefly, bone marrow cells were enriched for c-Kit positive cells by incubating with CD117/c-Kit microbeads and then processed through the autoMACS Pro Separator (Miltenyi Biotec), following the manufacturer’s instructions. C-Kit enriched cells were stained with Lineage antibody cocktail (CD3, CD4, CD8, Gr1, B220, CD19, TER119 conjugated with PeCy5), Sca1-Pac Blue, CD34-FITC, SLAM-APC, CD48-PE, and c-KIT-APC-Cy7. After staining, cells were sorted for LSK cells using a BD FACS Aria II instrument. Sorted LSK cells were incubated overnight in Stemspan SFEM medium (Stem Cell Technologies) with 10 ng/ml IL-3, 10 ng/ml IL-6, 50 ng/ml SCF, 10 ng/ml thrombopoietin (TPO), and 20 ng/ml FLT-3 ligand.
Cells were plated on retronectin-coated plates together with supernatant containing retrovirus expressing MLL-AF9 together with GFP (a gift from Scott Armstrong, Dana Farber Cancer Institute) and spun for 1h at 2500 RPM. After two cycles of spinfection, the cells were grown in M3434 methylcellulose medium (Stem Cell Technologies) for a week. Cells were sorted for GFP positivity and sorted 200,000 GFP positive cells were injected retro-orbitally with 250,000 bone marrow support cells into lethally irradiated 6-week-old female C57BL/6 mice.
Western blot analysis
1 × 106 human AML cell line or murine leukemic cells were counted and treated with stock solutions of drugs in DMSO at the indicated concentrations for the indicated time points. Cells that were treated with MG132 or MLN4924 were pre-incubated for 1 h. Cells were pelleted (500 × g), washed with PBS (2 × 1 mL), and lysed in Laemmli buffer (150 μL) by vigorous vortexing for 10 min. Lysates were boiled at 95 °C for 5 min. Boiled lysates were processed through Qia Shredder and electrophoresed on 4%–15% gradient SDS-PAGE gels. Proteins were transferred onto the PVDF membrane and blotted with the specified antibodies.
Viability Assay
10,000 cells per well (AML PDX cells, AML cell lines, MLL-AF9 leukemic cells or normal BM cells) were plated onto flat-bottom 96-well plates in media with increasing concentration of DEG-35, DEG-9 or Lenalidomide up to 100 μM. Cells were cultured for 72 h at 37 °C in a 5% CO2 incubator. For measuring viability, Cell-Titer Glo kit (Promega) or an MTT assay were used following manufacturer’s instructions.
For Cell-Titer Glo assay, cells were cooled to room temperature for 20–30 min, 100 μL of the cultured cells were transferred to opaque-white bottom 96-well plates and mixed with 100 μL of Cell-Titer Glo reagent. The mixture was incubated for 15 min at room temperature and read using a Synergy H1 Hybrid reader (BioTek) for luminescence. Data was normalized as percentage viability and graphed by non-linear regression curves in Graph Pad PRISM 7.0.
For the MTT assay, 10 μL of a 5 mg/mL solution of MTT in PBS was pipetted into each well containing cells and/or media. Plates were spun down at 500 x g for 30 seconds and incubated for 3 h. Plates were removed from incubation and 100 μL of a solution of 10% w/v SDS + 0.01 M HCl in diH2O were pipetted into each well containing cells and/or media. Plates were incubated overnight and absorbance of each well was measured at 570 nM using a plate reader (Molecular Devices SpectraMax ID5). The three wells containing media only were annotated as blanks and their average absorbance was used as a background value that was subtracted from the other values during data analysis. Background normalized absorbance values were normalized to the average absorbance of the three DMSO wells per plate and multiplied by 100% to get cell viability percentage values.
Flow cytometry for apoptosis and differentiation
For measuring differentiation, MOLM-13 cells treated with DEG-35 or DEG-77 for 48 h were stained with anti-CD13-FITC, anti-CD33-APC, anti-CD14-FITC or anti-CD11b/Mac1-APC. For murine MLL-AF9 leukemic cells, cells treated with DEG-35 for 24 h were stained with anti-Mac1-PB, anti-Ly-6G(Gr1)-PE, anti-CD115-APC and anti-F4/80-Pe-Cy7. Samples were stained with 2%FBS RPMI with diluted antibodies for 20 min in the dark, washed and analyzed with LSRII or LSR Fortessa (BD Biosciences) and data was graphed by using FlowJo™ version 10.4. For apoptosis assessment, MOLM-13 cells treated with DEG-35 or DEG-77 for 48 h or MLL-AF9 cells treated with DEG-35 for 24 h, were washed with cold PBS and incubated with anti-Annexin V-APC (BD Biosciences) in the Annexin-V binding buffer (10 mM HEPES, pH 7.4, 140 mM NaCl, 4 mM KCl, 0.75 mM MgCl2, 1 mM CaCl2) together with DAPI in the reaction volume of 100 ml_ for 15 min as recommended by the manufacturer. For co-staining of myeloid markers and Annexin-V, cells were first stained with myeloid markers, CD11b or CD13 and later washed and further stained with Annexin-V in Annexin-V binding buffer. Cells were washed and analyzed using flow cytometry.
Flow cytometry for degradation
3.2 x 104 HEK293T GFP-CK1α cells were seeded in a U-bottom 96-well plate in 200 μL of complete DMEM (10% FBS + P/S) and allowed to grow for 24 h. Non-transduced HEK293T cells were seeded as a negative control. The cells were treated with a 100× stock solution of compound/DMSO (2 μL, 1× final concentration) and incubated at 37 °C for 5 h. The media was removed and 0.5% trypsin-EDTA (100 μL) was added to each well and incubated at 37 °C for 3-5 min. Media (100 μL) and propidium iodide/PBS (0.5 mg/mL, 4 μL) were added to each well and analyzed with a BD LSR Fortessa instrument.
Cell Cycle Analysis
For cell cycle analysis, MOLM-13 cells treated with 1μM DEG-35 or DEG-77 were washed with PBS and fixed in ice-cold 70% ethanol for few hours. Fixed cells were pelleted and washed twice with PBS and incubated in PBS containing 3.8 mM Sodium Citrate, propidium iodide (50μg/ml) and RNase A (100μg/ml) for 30 min at 4 °C in the dark. Nuclei were then analyzed by running at low flow rate in LSRII for 10,000 events.
Wright-Giemsa Staining
50,000-100,000 MOLM-13 cells were cytospun onto poly-lysine coated glass slides. Slides were then processed for Wright-Giemsa Staining by dipping slides into fixative, Solution A and Solution B of the Three-Step Stain Kit (Richard-Allan Scientific) for 30 seconds each. Dried slides were mounted with cover slides.
Colony forming Unit Assay
Colony Assay for human CD34+ HSPCs, normal bone marrow cells or AML patient cells were performed by plating 5000 cells on methylcellulose (MethoCult H4434 Classic-Stem Cell Technologies). Colonies were assessed and counted 10 to14 days after seeding. 500 sorted murine LSK cells from Wt or Crbn I391V mice and 1000 bone marrow cells from MLL-AF9 Wt or Crbn I391V mice were plated on MethoCult M3434 and counted 5 -10 days after plating.
Quantitative global proteomics
MOLM-13 cells were seeded in a 6-well plate at a density of ~1 × 106 cells/well and treated with 50 nM DEG-35 or DMSO (four replicates each) and incubated for 2 h at 37 °C. The cells were pelleted by centrifugation (500 × g) for 3 min at room temperature and then washed with PBS (2 × 1 mL). The washed cells were lysed with 5% SDS/50 mM TEAB, pH 7.55 (100 μL) and sonicated on ice using a probe tip sonicator (10% amplitude, 5 sec on, 3 sec off, 10 sec total). Cell lysate was cleared by centrifugation (13,000 × g) for 10 min at 4 °C and the soluble protein concentration was determined by BCA assay. Protein concentrations were adjusted to 0.23 mg/mL in lysis buffer. Lysates (100 μL) were reduced with 20 mM dithiothreitol (DTT) for 30 min at 24 °C and alkylated with 36 mM iodoacetamide for 30 min at 24 °C in the dark. The samples were acidified with 12% phosphoric acid/water (13.75 μL) and diluted with 90% methanol, 10% water, 100 mM TEAB, pH 7.1 (900 μL). The samples were transferred to an S-Trap micro attached to a vacuum line and the supernatant was drained. 10% water, 100 mM TEAB, pH 7.1 (400 μL) was used to transfer any remaining sample. The samples were washed with 10% water, 100 mM TEAB, pH 7.1 (3 × 200 μL) and removed from the vacuum line. Sequencing-grade trypsin (4 μg) in 50 mM TEAB (40 μL) was added to the S-Trap and incubated for 1 h at 47 °C. The digested peptides were eluted sequentially with 50 mM TEAB (40 μL), water (40 μL), and 0.1% formic acid/50% acetonitrile/water (35 μL). The elutions were combined and dried by vacufuge. The dried samples were resuspended in water (25 μL). TMT reagent (10 μL) was added and the samples were incubated for 1 h at 24 °C. Excess TMT reagent was quenched by addition of 5% hydroxylamine (1.2 μL) followed by incubation for 15 min at 24 °C. The labeled samples were combined and then dried by vacufuge. The dried TMT-labeled samples were resuspended in 0.1% TFA/water (300 μL) and 100 μL was fractionated into 20 fractions using a high pH reversed-phase peptide fraction kit.
Mass spectrometry data analysis
Analysis was performed in Thermo Scientific Proteome Discoverer version 2.4.1.15. The raw data was searched against a protein sequence database including all entries from the Human Uniprot database (August 19, 2016; 20,156 entries, SwissProt) and a list of common contaminant proteins. Search parameters included a 10-ppm precursor ion tolerance and 0.02 Da (HCD) or 0.6 Da (CID) fragment ion tolerance, full tryptic protease specificity with up to two missed cleavages, a static modification of TMTpro 16-plex at lysine residues and N-termini (+304.2071 Da), static carboxyamidomethylation of cysteine residues (+57.0214 Da), and variable oxidation on methionine residues (+15.9949 Da). Peptide spectral matches (PSMs) were filtered for a false discovery rate (FDR) of 1% using Percolator. TMT reporter ions were quantified using the Reporter Ions Quantifier node and the total intensity was normalized for each TMT channel. Quantified proteins were required to have at least three unique peptides and four separate PSMs. All TMT-based experiments were performed with four replicates.
RT-qPCR
MOLM-13 cells were seeded in a 6-well plate at a density of ×1 × 106 cells/well and treated with 50 nM DEG-35 or DMSO and incubated for 24 h at 37 °C. MLL-AF9 Crbn I391V cells were treated with various doses of DEG-35 or DEG-77 for 24h and processed. The cells were pelleted by centrifugation (500 × g) for 3 min at room temperature and then washed with PBS (2 × 1 mL). The Monarch Total RNA Miniprep Kit was used to isolate and purify RNA from the cells. Resulting RNA extracts were diluted to 10 ng/μL and the Luna Universal One-Step RT-qPCR Kit was used to prepare the samples for RT-qPCR. 20 μL of combined RNA/enzyme/primer mixes were aliquoted into a 96-well plate and centrifuged (1000 × g) for 1 min at room temperature. RT-qPCR was performed in a BioRad iQ5 Real-Time PCR Detection System and results were exported to Microsoft Excel for analysis.
In cellulo co-immunoprecipitation
HEK-293T cells stably transfected with Flag-tagged CRBN were plated in a 10 cm dish and allowed to grow to full confluency. Cells were treated with 10 μM MG132 and incubated for 1 h at 37 °C. The cells were then treated with either 10 μM deg35 or DMSO and incubated for 3 h at 37 °C. Cells were washed with PBS (2 × 10 mL) and Cell Lysis Buffer (CST, 400 μL) containing MG132 (10 μM) and 1× protease inhibitors was added to each plate. The cells were incubated for 5 min on ice and then harvested by scraping. Lysate was incubated on ice for 5 min and then cleared by centrifugation (21,130 × g) for 10 min at 4 °C. Solubilized protein concentration was determined by BCA assay and adjusted to 1 mg/mL in lysis buffer. Adjusted lysate (200 μL) was added to anti-Flag M2 magnetic beads (30 μL) previously washed with TBS (3 × 300 μL) and incubated for 1 h at 4 °C. The supernatant was removed, and the beads were washed with lysis buffer (3 × 300 μL). 1× Laemmli sample buffer (50 μL) was added to the beads and boiled for 5 min at 95 °C. Results were analyzed by Western blot.
AlphaScreen
3× AlphaScreen buffer (150 mM HEPES pH 7.4, 450 mM NaCl, 0.3% w/v BSA, v/v 0.03% Tween 20) was prepared fresh for each experiment. A 3× stock solution of DEG-35 and lenalidomide (3% DMSO/1× AlphaScreen buffer) and a series of 2-fold serial dilutions in 3% DMSO/1× AlphaScreen buffer were also prepared fresh for each experiment. A solution of 600 nM streptactin-CK1α/1× AlphaScreen buffer (5 μL) and a solution of 500 nM CULT/1× AlphaScreen buffer (5 uL) were added into each well of a 384-well Optiplate. Compound (5 μL) was added to each well with each concentration assayed in triplicate. The plate was sealed with TopSeal-A PLUS, centrifuged (200 rcf, 25 °C, 1 min) and incubated at 25 °C for 1 h. The seal was removed and under low light, AlphaScreen streptactin acceptor beads in 1× AlphaScreen buffer (60 μg/mL, 5 μL) and AlphaLISA nickel chelate donor beads in 1× AlphaScreen buffer (60 μg/mL, 5 μL) were added to each well. The plate was sealed with TopSeal-A PLUS, centrifuged (200 rcf, 25 °C, 1 min), wrapped in foil, and incubated at 25 °C for 1 h. The seal was removed and the plate was analyzed.
HTR-FRET
PerkinElmer’s Cereblon binding assay kit was used. A series of 4-fold serial dilutions of lenalidomide, DEG-35, and DEG-77 in 4% DMSO/dilution buffer were prepared fresh for each experiment. Compound (5 μL) was added to each well of a 384-well microplate. A solution of CRBN/1× PROTAC binding buffer (5 μL) and a solution of premixed thalidomide red glutathione-europium cryptate antibody/1× PROTAC binding buffer (10 μL) were added to each well. The plate was sealed with TopSeal-A PLUS and incubated at 25 °C for 3 h. The seal was removed, and the plate was analyzed.
In vivo drug administration in MLL-AF9 and PDX mouse models
A total of 250,000 of MLL-AF9 BM secondary mouse leukemia cells were injected retro-orbitally into 6-8 weeks-old female C57BL/6 recipient mice that have been sublethally irradiated at 450 cGy. For the AML PDX model, 1 million PDX cells were transplanted by intravenous injection into 8-10 weeks old female NSG mice that had been sublethally irradiated at 200 rads 24 h before transplant. For both models, 3 days after transplant, DEG-35 or vehicle was administered by intraperitoneal injections at 50 mg/kg in DMSO in 50ul volume for in vivo long-term studies. DEG-77 was treated at 223 mg/kg in DMSO in 50ul volume. Mice weight was monitored every day to check for toxicity. Mice were sacrificed when moribund, and organ weights were measured for disease burden. Spleen and bone marrow cells were isolated and processed for western blot analysis. All animal studies were performed on animal protocols approved by the Institutional Animal Care and Use Committee (IACUC) at Memorial Sloan Kettering Cancer Center.
In the case for Pharmacodynamic experiments, DEG-35 or DEG-77 were administered 2 weeks post-transplant of MLL-AF9 BM secondary mouse leukemia cells in C57BL/6 recipient mice. DEG-35, DEG-77 or DMSO was administered by intraperitoneal injections at 50 mg/kg or 223 mg/kg. After single dose treatment of DEG-35 or DEG-77, mice were sacrificed at either 24 h, 48 h or 72 h later and, spleen and bone marrow cells were processed for analysis.
For Pharmacokinetics experiments, a single dose of DEG-35 at 5 mg/kg or 50 mg/kg in DMSO was administered intraperitoneally into male CD1 mice. DEG-77 was injected intraperitoneally at 223 mg/kg in DMSO into female CrbnI391V mice. Plasma was extracted from each mouse after indicated time and DEG-35 or DEG-77 in plasma was measured using HPLC.
For examining the toxicity of DEG-77 in the hematopoietic system of Crbn I391V mice, four rounds of DMSO or 223mg/kg DEG-77 was treated in 8-12 weeks old female Crbn I391V mice. Mice were injected once a week and were sacrificed 4 weeks later for blood analysis using the Element HT5 Hematology Analyzer (HESKA) and analysis of the hematopoietic cells by flow cytometry.
RNA sequencing
MOLM-13 cells treated with DMSO (n = 3) or 50nM DEG-35 (n = 3) for 24 h, were processed for RNA extraction using TRIZOL and RNeasy RNA extraction kit. High purity mRNAs were enriched from total RNAs using Dynabeads mRNA purification kit (Thermo Fisher). After PicoGreen quantification and quality control by Agilent BioAnalyzer, mRNA input was used for library preparation (TrueSeq Stranded mRNA LT Sample Prep Kit). Libraries were run on a HiSeq 4000 in a 100bp/100bp paired end run, using the HiSeq 3000/4000 SBS Kit (Illumina). The average number of read pairs per sample was 100 million. Sequence data were aligned using STAR aligner to human reference genome (version hg19). Fragments Per Kilobase of transcript per Million mapped reads (FPKM) were calculated and differential expression analysis was conducted using the DE Seq software package. Differentially expressed genes were identified as those with FPKM greater than 1 showing differential expression greater than twofold (up or down) with an adjustment p value less than 0.05. GSEA was performed using the rank list of differentially expressed genes from the MOLM-13 cells treated with DMSO and DEG-35 on all curated gene sets in the Molecular Signatures Database (MSigDB, https://www.broadinstitute.org/msigdb; 3,256 gene sets)(Liberzon et al., 2011) combined with an additional set of relevant gene sets (92 gene sets from our experimentally derived or published hematopoietic self-renewal and differentiation signatures, CK1 KO keratinocyte signature) (Table S4 and 5).
General Chemical Methods
All reactions were performed in a flame-dried, single neck, round bottom flask fitted with a rubber septum under argon unless otherwise noted. N,N-dimethylformamide was vigorously purged with argon for 1 h followed by passage under argon pressure through two packed columns of neutral alumina (Pure Process Technology). Organic solutions were concentrated by rotary evaporation at 30–33 °C. Normal phase flash-column chromatography was performed with silica gel (60 Å, 40–63 μM particle size) purchased from Silicycle (Quebec, Canada). Analytical thin-layer chromatography (TLC) was performed using glass plates pre-coated with silica gel (0.25 μM, 60 Å pore size) impregnated with a fluorescent indicator. TLC plates were visualized by exposure to ultraviolet (UV) light.
Chemical Instrumentation
Proton nuclear magnetic resonance spectra (1H NMR) and proton-decoupled carbon nuclear magnetic resonance spectra (13C NMR) experiments were performed on a Bruker 400 MHz, JEOL 400 MHz, Varian 500 MHz, or Agilent 600 MHz instruments at 25 °C unless otherwise noted. Chemical shifts are expressed in parts per million (ppm, δ scale) and are referenced to residual solvent (DMSO-d6, δ 2.50 ppm for 1H and δ 39.52 ppm for 13C; DMF-d7, δ 2.75 ppm for 1H and δ 29.76 ppm for 13C; Acetone-d6, δ 2.05 ppm for 1H and δ 29.84 ppm for 13C). The following abbreviations were used to express multiplicities: s = singlet, d = doublet, t = triplet dd = doublet of doublets, m = multiplet. Infrared (IR) spectra experiments were performed on a Bruker ALPHA FT-IR instrument. Peaks are expressed as frequency of absorption (cm−1) and intensity of absorption (s = strong, m = medium, w = weak, br = broad). Small molecule high-resolution mass spectra (HRMS) was performed at the Harvard Center for Mass Spectrometry using a Thermo q-Exactive Plus. Preparative HPLC was performed on a Waters Prep 150 LC system and Agilent Infinity II system with a XBridge BEH C18 OBD prep column (130 Å, 5 μm, 19 mm × 100 mm) and an Agilent C18 prep column (100 Å, 5 μm, 30 mm × 100 mm).
Compound Synthesis and Characterization
All associated NMR and IR spectra can be found in Figure S9.

N-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-5-yl)-2-naphthamide (DEG-9)
HATU (110.0 mg, 0.29 mmol, 3.00 equiv), and diisopropylamine (50 μL, 0.29 mmol, 3.00 equiv) were added sequentially to a solution of 2-naphthoic acid (24.9 mg, 0.14 mmol, 1.50 equiv) and 5-NH2-lenalidomide (25.0 mg, 0.10 mmol, 1.00 equiv) in dimethylformamide (964 μL, 0.1 M) and stirred for 18 h at 24 °C. The product mixture was diluted with brine (5 mL) and transferred to a separatory funnel. The aqueous layer was extracted with ethyl acetate (2 × 5 mL) and the organic layers were combined. The combined organic layers were dried over sodium sulfate and filtered through cotton. The filtrate was concentrated by rotary evaporation. The residue obtained was purified by flash column chromatography (SiO2, 2%–4% methanol-dichloromethane, 1 step) and further purified by high performance liquid chromatography to afford DEG-9 as a white solid (7.3 mg, 18% yield).
Rf = 0.50 (10% methanol–dichloromethane, UV). 1H NMR (400 MHz, DMSO-d6) δ 11.01 (s, 1H, H1), 10.76 (s, 1H, H9), 8.62 (s, 1H, H16), 8.20 (s, 1H, H6),8.13–8.01 (m, 4H, H10/H11/H12/H15), 7.89 (dd, J = 8.3, 1.6 Hz, 1H, H7), 7.74 (d, J = 8.3 Hz, 1H, H8), 7.69–7.62 (m, 2H, H13/H14), 5.12 (dd, J = 13.3, 5.1 Hz, 1H, H4), 4.5 (d, J = 17.3 Hz, 1H, H5), 3.35 (d, J = 17.3 Hz, 1H, H5), 2.98–2.88 (m, 1H, H2), 2.65–2.58 (m, 1H, H2), 2.46–2.33 (m, 1H, H3), 2.05–1.98 (m, 1H, H3). 13C NMR (101 MHz, DMSO-d6) δ 172.96 (C), 171.17 (C), 167.87 (C), 166.06 (C), 143.18 (C), 142.55 (C), 134.40 (C), 132.06 (C), 131.99 (C), 129.04 (CH), 128.24 (CH), 128.15 (CH), 128.03 (CH), 127.74 (CH), 126.98 (CH), 126.80 (C), 124.49 (CH), 123.59 (CH), 119.97 (CH), 114.43 (CH), 51.58 (CH), 47.22 (CH2), 31.26 (CH2), 22.57 (CH2). IR (ATR-FTIR), cm−1: 1715 (m), 1672 (s), 1539 (m), 1342 (m), 1181 (m). HRMS-ESI (m/z): [M+H]+ calculated for C24H20N3O4, 414.1448; found, 414.1448.
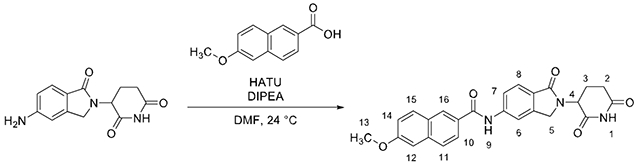
N-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-5-yl)-6-methoxy-2-naphthamide (DEG-35)
HATU (88.0 mg, 0.23 mmol, 3.00 equiv), and diisopropylamine (40 μL, 0.23 mmol, 3.00 equiv) were added sequentially to a solution of 6-methoxy-2-naphthoic acid (23.4 mg, 0.12 mmol, 1.50 equiv) and 5-NH2-lenalidomide (20.0 mg, 0.07 mg, 1.00 equiv) in dimethylformamide (771 μL, 0.1 M) and stirred for 18 h at 24 °C. The product mixture was diluted with brine (5 mL) and transferred to a separatory funnel. The aqueous layer was extracted with ethyl acetate (2 × 5 mL) and the organic layers were combined. The combined organic layers were dried over sodium sulfate and filtered through cotton. The filtrate was concentrated by rotary evaporation. The residue obtained was purified by flash column chromatography (SiO2, 1%–4% methanol–dichloromethane, 3 steps) and further purified by high performance liquid chromatography to afford DEG-35 as a white solid (7.3 mg, 21% yield).
Rf = 0.25 (5% methanol–dichloromethane, UV). 1H NMR (400 MHz, DMSO-d6): δ 10.99 (s, 1H, H1), 10.65 (s, 1H, H9), 8.55 (s, 1H, H16), 8.19 (s, 1H, H6), 8.02–7.94 (m, 3H, H10/H11/H15), 7.89 (d, J = 9.7 Hz, 1H, H7), 7.73 (d, J = 8.3 Hz, 1H, H8), 7.43 (d, J = 2.3 Hz, 1H, H12), 7.27 (dd, J = 8.94, 2.46 Hz, 1H, H14), 5.11 (dd, J = 13.24, 5.08 Hz, 1H, H4), 4.49 (d, J = 17.3 Hz, 1H, H5), 4.35 (d, J = 17.3 Hz, 1H, H5), 3.92 (s, 3H, H13), 2.98–2.87 (m, 1H, H2), 2.61 (d, J = 16.4 Hz, 1H, H2), 2.46–2.31 (m, 1H, H3), 2.05–1.98 (m, 1H, H3). 13C NMR (101 MHz, DMSO-d6): δ 172.90 (C), 171.12 (C), 167.85 (C), 166.03 (C), 158.83 (C), 143.13 (C), 142.64 (C), 136.11 (C), 130.61 (CH), 129.58 (C), 128.12 (CH), 127.36 (C), 126.87 (CH), 126.64 (C), 124.99 (CH), 123.52 (CH), 119.88 (CH), 119.57 (CH), 114.32 (CH), 105.96 (CH), 55.38 (CH3), 51.56 (CH), 47.18 (CH2), 31.23 (CH2), 22.54 (CH2). IR (ATR-FTIR), cm−1: 2924 (m), 2854 (w), 1672 (s), 1204 (m). HRMS-ESI (m/z): [M+H]+ calculated for C25H22N3O5, 444.1554; found, 444.1554.

3-(5-(((6-methoxynaphthalen-2-yl)methyl)amino)-1-oxoisoindolin-2-yl)piperidine-2,6-dione (CH2-DEG-35)
Sodium cyanoborohydride (19.7 mg, 314 μmol, 3.00 equiv) was added to a solution of 5-NH2-lenalidomide (27.1 mg, 105 μmol, 1.00 equiv), 6-methoxy-2-naphthaldehyde (58.4 mg, 314 μmol, 3.00 equiv) in DMF (784 μL, 0.13 M). Acetic acid (17.9 μL, 314 μmol, 3.00 equiv) was added to the solution and stirred at 24 °C for 16 h. The reaction mixture was diluted with aqueous sodium bicarbonate (5 mL) and transferred to a separatory funnel. The aqueous layer was extracted with ethyl acetate (3 × 5 mL). The combined organic layers were dried over sodium sulfate and filtered through cotton. The filtrate was concentrated by rotary evaporation. The resulting residue was purified by flash column chromatography (SiO2, 1%–4% methanol–dichloromethane, 3 steps) and high-performance liquid chromatography afforded CH2-DEG-35 as a white solid (9.2 mg, 20% yield).
Rf = 0.37 (5% methanol–dichloromethane, UV). 1H NMR (400 MHz, DMSO-d6) δ 10.90 (s, 1H, H1), 7.81–7.74 (m, 3H, H12/H16/H17), 7.46 (d, J = 8.3 Hz, 1H, H11), 7.38 (d, J = 8.3 Hz, 1H, H8), 7.29 (d, J = 2.4 Hz, 1H, H13), 7.13 (dd, J = 9.0, 2.5 Hz, 1H, H15), 7.06 (t, J = 5.9 Hz, 1H, H9), 6.73 (d, J = 8.8 Hz, 1H, H7), 6.69 (s, 1H, H6), 4.99 (dd, J = 13.3, 5.0 Hz, 1H, H4), 4.48 (d, J = 5.6 Hz, 2H, H10), 4.23 (d, J = 16.7 Hz, 1H, H5), 4.09 (d, J = 16.8 Hz, 1H, H5), 3.86 (s, 3H, H14), 2.93–2.82 (m, 1H, H2), 2.56 (d, J = 17.6 Hz, 1H, H2), 2.30 (qd, J = 13.2, 4.4 Hz, 1H, H3), 1.94–1.87 (m, 1H, H3). 13C NMR (101 MHz, DMSO-d6) 172.93 (C), 171.37 (C), 168.60 (C), 157.03(C), 152.09 (C), 144.33 (C), 134.56 (C), 133.42 (C), 129.02 (CH), 128.36 (C), 126.94 (CH), 126.24 (CH), 125.20 (CH), 123.92 (CH), 119.16 (C), 118.64 (CH), 112.72 (CH), 105.84 (CH), 104.76 (CH), 55.14 (CH3), 51.25 (CH), 46.74 (CH2), 46.26 (CH2), 31.25 (CH2), 22.61 (CH2). IR (ATR-FTIR), cm−1: 3297 (w), 1659 (s), 1233 (s), 824 (m). HRMS-ESI (m/z): [M+H]+ calculated for C25H24N3O4, 430.1761; found, 430.1765.
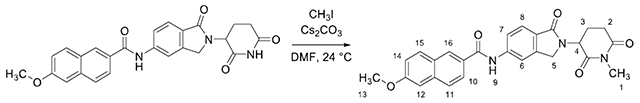
6-methoxy-N-(2-(1-methyl-2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-5-yl)-2-naphthamide (Me-DEG-35)
Methyl iodide (8.0 μL 0.12 mmol, 1.00 equiv) was added to a solution of DEG-35 (50.0 mg, 0.113 mmol, 1.00 equiv) and cesium carbonate (40.0 mg, 0.12 mmol, 1.00 equiv) in DMF (1.13 ml_, 0.1 M) under argon and stirred for 5 h at 24 °C. The reaction mixture was diluted with water (5 mL) and transferred to a separatory funnel. The solution was diluted with brine (30 mL) and the aqueous layer was extracted with ethyl acetate (4 × 20 mL). The combined organic layers were dried over sodium sulfate and filtered through cotton. The filtrate was concentrated by rotary evaporation. The residue obtained was purified by high performance liquid chromatography (reverse phase, 95% to 5% water/acetonitrile) to afford Me-DEG-35 as a white solid (41.4 mg, 80% yield).
Rf = 0.53 (5% methanol-dichloromethane, UV). 1H NMR (400 MHz, DMSO-d6) δ 10.67 (s, 1H, H9), 8.54 (s, 1H, H16), 8.19 (s, 1H, H6), 8.03 – 7.94 (m, 3H, H10/H11/H15), 7.88 (d, J = 9.0 Hz, 1H, H7), 7.74 (d, J = 8.3 Hz, 1H, H8), 7.43 (d, J = 2.1 Hz, 1H, H12), 7.27 (dd, J = 8.9, 2.4 Hz, 1H, H14), 5.18 (dd, J = 13.4, 5.0 Hz, 1H, H4), 4.49 (d, J = 17.2 Hz, 1H, H5), 4.33 (d, J = 17.1 Hz, 1H, H5), 3.92 (s, 3H, H13), 3.05 – 2.94 (m, 4H, H1/H2), 2.80 – 2.73 (m, 1H, H2), 2.47 – 2.33 (m, 1H, H3), 2.07 – 1.98 (m, 1H, H3). 13C NMR (101 MHz, DMSO-d6) δ 171.98 (C), 170.81 (C), 167.90 (C), 166.06 (C), 158.85 (C), 143.17 (C), 142.70 (C), 136.13 (C), 130.64 (CH), 129.60 (C), 128.15 (CH), 127.38 (C), 126.90 (CH), 126.64 (C), 125.02 (CH), 123.60 (CH), 119.92 (CH), 119.61 (CH), 114.33 (CH), 105.96 (CH), 55.40 (CH3), 52.09 (CH), 47.20 (CH2), 31.40 (CH2), 26.61 (CH3), 21.81 (CH2). IR (ATR-FTIR), cm−1: 3357 (w), 1431, (m), 1333 (s), 860 (s). HRMS-ESI (m/z): [M+H]+ calculated for C26H24N3O5, 458.1710; found, 458.1713.

N-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-5-yl)-8-methoxy-2H-chromene-3-carboxamide (DEG-77)
HATU (154.0 mg, 0.40 mmol, 3.00 equiv), and diisopropylamine (71 μL, 0.40 mmol, 3.00 equiv) were added sequentially to a solution of 8-methoxy-2H-chromene-3-carboxylic acid (55.7 mg, 0.27 mmol, 2.00 equiv) and 5-NH2-lenalidomide (35.0 mg, 0.13 mg, 1.00 equiv) in dimethylformamide (1.35 mL, 0.1 M) and stirred for 18 h at 24 °C. The product mixture was diluted with water (5 mL) and transferred to a separatory funnel. The aqueous layer was extracted with ethyl acetate (2 × 5 mL) and the organic layers were combined. The combined organic layers were dried over sodium sulfate and filtered through cotton. The filtrate was concentrated by rotary evaporation. The residue obtained was purified by flash column chromatography (SiO2, 1%–4% methanol–dichloromethane, 3 steps) and further purified by high performance liquid chromatography to afford DEG-77 as a yellow solid (27.9 mg, 46% yield).
Rf = 0.52 (5% methanol–dichloromethane, UV). 1H NMR (400 MHz, DMF-d7): δ 10.96 (s, 1H, H1), 10.44 (s, 1H, H9), 8.20 (s, 1H, H6), 7.89 (dd, J = 8.3, 1.5 Hz, 1H, H7), 7.74 (d, J = 8.3 Hz, 1H, H8), 7.61 (s, 1H, H15), 7.09 (dd, J = 8.0, 1.3 Hz, 1H, H12), 6.97 (t, J = 7.8 Hz, 1H, H13), 6.91 (dd, J = 7.6, 1.3 Hz, 1H, H14), 5.24 (dd, J = 13.3, 5.1 Hz, 1H, H4), 5.08 (d, J = 1.0 Hz, 1H, H10), 4.57 (d, J = 17.0 Hz, 1H, H5), 4.45 (d, J = 17.0 Hz, 1H, H5), 3.87 (s, 3H, H11), 3.10–3.00 (m, 1H, H2), 2.78–2.72 (m, 1H, H2), 2.61–2.50 (m, 1H, He), 2.22–2.14 (m, 1H, H3). 13C NMR (101 MHz, DMF-d7) δ 173.08 (C), 171.48 (C), 168.47 (C), 164.28 (C), 148.54 (C), 144.19 (C), 143.89 (C), 143.09 (C), 129.17 (CH), 127.47 (C), 127.31 (C), 123.88 (CH), 122.46 (C), 121.98 (CH), 120.90 (CH), 120.00 (CH), 115.16 (CH), 114.51 (CH), 64.77 (CH2), 55.97 (CH3), 52.33 (CH), 47.66 (CH2), 31.81 (CH2), 23.35 (CH2). IR (ATR-FTIR), cm−1: 1692 (m), 1531 (s), 1422 (s), 1224 (m). HRMS-ESI (m/z): [M+H]+ calculated for C24H22N3O6, 448.1503; found, 448.1504.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters for each data are reported in Figure Legends. The 2-tailed Student’s t test was used for significance testing in the bar graphs, except where stated otherwise. P values less than 0.05 were considered significant. Error bars reflect the SEM, except where stated otherwise. Survival probabilities were estimated using the Kaplan-Meier method and compared with the log-rank test. All statistical analyses were carried out using GraphPad Prism 7.0 and the R statistical environment.
Supplementary Material
Table S1, Related to Figure 1. Mutations in AML cell lines
Table S2, Related to Figure 2. Proteomics data for MOLM-13 cells treated with DEG-35.
Table S2, Related to Figure 2. Mass spectrometry data for MOLM-13 cells treated with DEG-35.
Table S3, Related to Figure 3. Correlation of CRISPR screen data to DEG-35 PRISM cell line sensitivity signature.
Table S3, Related to Figure 3. Correlation of Repurposed drug database to DEG-35 PRISM cell line sensitivity signature.
Table S3, Related to Figure 3. Correlation of mutations to DEG-35 PRISM cell line sensitivity signature.
Table S3, Related to Figure 3. Correlation of Copy Number Aberrations to DEG-35 PRISM cell line sensitivity signature.
Table S3, Related to Figure 3. Correlation of Gene expression to DEG-35 PRISM cell line sensitivity signature.
Table S3, Related to Figure 3. Correlation of CRISPR screen to Nutlin-3a PRISM cell line sensitivity signature.
Table S4, Related to Figure 4. Enrichr analysis (TF perturbation) of ShRNA positive correlation (Coef>0.2, qvalue <0.05)
Table S4, Related to Figure 4. Enrichr analysis (ENCODE and ChEA) of Gene expression positive correlation (Coef>0.2, qvalue <0.05)
Table S4, Related to Figure 4. Enrichr analysis (ChEA 2016) of Gene expression positive correlation (Coef>0.2, qvalue <0.05)
Table S5, Related to Figure 4. List of Differentially up-regulated genes (log 2-fold change >1, p adjusted <0.05)
Table S5, Related to Figure 4. List of Differentially down-regulated genes (log 2-fold change >1, p adjusted <0.05)
Table S6, Related to Figure 4. Rank list for GSEA for the RNA-seq DEG-35 vs DMSO in MOLM-13 cells.
Table S6, Related to Figure 4. Rank list for GSEA for the RNA-seq Control versus IKZF2 knockout primary MLL-AF9 c-Kit high cells
Table S6, Related to Figure 4. GSEA positively enriched using DEG35 regulated genes as Rank list.
Table S6, Related to Figure 4. GSEA negatively enriched using DEG35 regulated genes as Rank list.
Table S7, Related to Star Methods. List of AML patient characteristics.
Table S8, Related to Star Methods. List of primer sequences.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-Helios | Cell Signaling Technology | Cat#42427; RRID:AB_2799221 |
| Goat polyclonal anti-Helios | Santa Cruz Biotechnology | Cat#sc-9864; RRID: AB_2124965 |
| Rabbit monoclonal anti-Ikaros | Cell Signaling Technology | Cat#9034; RRID: AB_2797691 |
| Rabbit monoclonal anti-Aiolos | Cell Signaling Technology | Cat#15103; RRID: AB_2744524 |
| Rabbit polyclonal anti-IKZF4 | Abcam | Cat#ab220329; RRID: NA |
| Rabbit polyclonal anti-CMYC | Cell Signaling Technology | Cat# 5605S; RRID: 1903938 |
| Rabbit polyclonal anti-HOXA9 | Abcam | Cat# ab140631; RRID:NA |
| Rabbit monoclonal anti-p21 Waf1/Cip1 | Cell Signaling Technology | Cat#2947; RRID:823586 |
| Rabbit monoclonal anti-p53 | Cell Signaling Technology | Cat#2527; RRID: AB_10695803 |
| Rabbit monoclonal anti-MDM2 | Cell Signaling Technology | Cat#86934; RRID: AB_2784534 |
| Rabbit monoclonal anti-casein kinase 1 alpha | Abcam | Cat#ab206652; RRID: AB_2925161 |
| Rabbit polyclonal anti-eRF3a/GSPT1 | Proteintech | Cat#10763-1-AP; RRID: AB_2115506 |
| Rabbit polyclonal anti-CRBN | Sigma-Aldrich | Cat#HPA045910; RRID: AB_10960409 |
| Mouse monoclonal anti-β actin | Santa Cruz Biotechnology | Cat#sc-47778; RRID: AB_2714189 |
| Goat anti-rabbit IgG secondary antibody peroxidase conjugated | Rockland | Cat#611-1302; RRID: AB_219720 |
| Goat IRDye 800CW anti-mouse IgG secondary antibody | LI-COR | Cat#926-32210; RRID: AB_621842 |
| Human monoclonal anti-CD14, PE | BD Biosciences | Cat#555398; RRID: AB_395799 |
| Human monoclonal anti-CD11b, APC | Life Technologies | Cat#CD11B05; RRID: AB_1464195 |
| Human monoclonal anti-CD13, APC | Life Technologies | Cat#MHCD1305; RRID: AB_1464860 |
| Human monoclonal anti-CD33, PE | BD Pharmingen | Cat#555450; RRID: AB_395843 |
| Human monoclonal anti-CD45, PerCP-PB | Invitrogen | Cat#45-9459-42; RRID: AB_1548697 |
| Human/mouse monoclonal anti-CD11b, PB | BioLegend | Cat#101224; RRID: AB_755986 |
| Mouse monoclonal Ly-6G/Ly-6C, PeCy5 | Invitrogen | Cat#15-5931-82; RRID: AB_468813 |
| Mouse monoclonal anti-CD115, APC | BioLegend | Cat#135510; RRID: AB_2085221 |
| Mouse monoclonal anti-F4/80, PeCy7 | BioLegend | Cat#123114; RRID: AB_1626278 |
| Mouse monoclonal anti-CD45 | BD Pharmingen | Cat#559864; RRID: AB_398672 |
| Anti-Annexin V, APC | BD Biosciences | Cat#550475; RRID: AB_2868885 |
| Mouse monoclonal anti-CD3, PeCy5 | Invitrogen | Cat#15-0031-83; RRID: AB_468691 |
| Mouse monoclonal anti-CD4, PeCy5 | Invitrogen | Cat#15-0041-83; RRID: AB_468696 |
| Mouse monoclonal anti-CD8a, PeCy5 | Invitrogen | Cat#15-0081-83 RRID: AB_468707 |
| Mouse monoclonal anti-B220, PeCy5 | Invitrogen | Cat#15-0452-83; RRID: AB_468756 |
| Mouse monoclonal anti-TER-119, PeCy5 | Invitrogen | Cat#15-5921-83; RRID: AB_468811 |
| Mouse monoclonal anti-Ly-6A/E (Sca-1) | BioLegend | Cat#122520; RRID: AB_2143237 |
| Mouse monoclonal anti-Ly-6G/Ly-6C, FITC | BD Biosciences | Cat#553127; RRID: AB_394643 |
| Mouse monoclonal anti-Ly-6G/Ly-6C, APC-Cy7 | BioLegend | Cat#108424; RRID: AB_2137485 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Patient-derived xenographs (PDX) | Hematologic Oncology Tissue Bank at Memorial Sloan Kettering Cancer Center | N/A |
| Primary AML patient cells | Hematologic Oncology Tissue Bank at Memorial Sloan Kettering Cancer Center | N/A |
| Normal bone marrow cells | Hematologic Oncology Tissue Bank at Memorial Sloan Kettering Cancer Center | N/A |
| Cord blood | National Cord Blood Program, NY Blood Center | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| MG132 | Selleck Chemicals | Cat#S2619; CAS 1211877-36-9 |
| MLN4924 | Selleck Chemicals | Cat#S7109; CAS 905579-51-3 |
| CC-885 | Selleck Chemicals | Cat#S7109; CAS 1010100-07-8 |
| Lenalidomide | Sigma-Aldrich | Cat#SML2283; CAS 191732-72-6 |
| Pomalidomide | Sigma-Aldrich | Cat#P0018; CAS 19171-19-8 |
| complete, EDTA-free protease inhibitor cocktail | Sigma-Aldrich | Cat#11873580001 |
| MTT | Sigma-Aldrich | Cat#475989; CAS 298-93-1 |
| Cell lysis buffer | Cell Signaling Technology | Cat#9803S |
| Sequencing-grade trypsin | Promega | Cat#V5111 |
| Midostaurin | Selleck Chemicals | Cat#S8064; CAS 120685-11-2 |
| Cytarabine (U-19920A) | Selleck Chemicals | Cat#S1648; CAS 147-94-4 |
| Venetoclax | Selleck Chemicals | Cat#S8048; CAS 1257044-40-8 |
| Methothrexate | Selleck Chemicals | Cat#S1210; CAS 59-05-2 |
| Propium Iodide | Sigma-Aldrich | Cat#P4170; CAS 25535-16-4 |
| Murine SCF | Peprotech | Cat# 250-03 |
| Murine IL-3 | Peprotech | Cat# 213-13 |
| Murine IL-6 | Peprotech | Cat# 216-16 |
| Murine GM-CSF | Peprotech | Cat# 315-03 |
| MethoCult M3434 | Stem Cell Technologies | Cat# 03434 |
| MethoCult H4434 | Stem Cell Technologies | Cat# 04434 |
| Critical commercial assays | ||
| Luna Universal One-Step RT-qPCR Kit | New England BioLabs | Cat#E3005L |
| RNeasy Mini Kit | Qiagen | Cat#74104 |
| HTRF Human Cereblon Binding Kit | PerkinElmer | Cat#64BDCRBNPEG |
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat#G7570 |
| QuikChange Lightning Multi Site-Directed Mutagenesis Kit | Agilent | Cat#210513 |
| QuikChange Lightning Site-Directed Mutagenesis Kit | Agilent | Cat#210518 |
| Dynabeads mRNA Purification Kit | Thermo Fisher Scientific | Cat#61006 |
| Monarch Total RNA Miniprep Kit | New England BioLabs | Cat#T2010S |
| TMTpro 16plex Label Reagent Set | Thermo Fisher Scientific | Cat#A44520 |
| Strep-Tactin Alpha Donor Beads | PerkinElmer | Cat#AS106D |
| AlphaLisa Nickel Chelate Acceptor Beads | PerkinElmer | Cat#AL108M |
| Richard-Allan Scientific Three Step Stain Set | Thermo Fisher Scientific | Cat#3300 |
| Deposited data | ||
| RNA-seq Data | GEO | GSE220707 |
| Proteomics Data | PRIDE Repository | PXD039474 |
| Experimental models: Cell lines | ||
| Human: MOLM-13 cells | Gary Gilliland Lab | RRID: CVCL_2119 |
| Human: MOLM-14 cells | Gary Gilliland Lab | RRID: CVCL_7916 |
| Human: OCI-AML3 cells | Gary Gilliland Lab | RRID: CVCL_1844 |
| Human: OCI-AML5 cells | Uli Steidl Lab | RRID: CVCL_1620 |
| Human: Kasumi-1 cells | Stephen Nimer Lab | RRID: CVCL_0589 |
| Human: NB-4 cells | Gary Gilliland Lab | RRID: CVCL_0005 |
| Human: KG-1A cells | Gary Gilliland Lab | RRID: CVCL_1824 |
| Human: NOMO-1 cells | Gary Gilliland Lab | RRID: CVCL_1609 |
| Human: HEK293T cells | ATCC | RRID: CVCL_0063 |
| Murine MLL-AF9 Crbn I391V leukemic cells | This paper | |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6-Crbntm1.1Ble/J | The Jackson Laboratory | Stock No: 032487 |
| Mouse: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) | The Jackson Laboratory | Stock No: 005557 |
| Mouse: C57BL/6J | The Jackson Laboratory | Stock No: 000664 |
| Oligonucleotides | ||
| Primers for RT-qPCR, See Table S7 | This paper | N/A |
| sgRNA 1: TP53 GATCCACTCACAGTTTCCAT | This paper | N/A |
| sgRNA 2: TP53 GAGCGCTGCTCAGATAGCGA | This paper | N/A |
| Recombinant DNA | ||
| pLC-Flag-CSNK1A1 | (Patil et al., 2018) | Addgene #123319 |
| pMSCV-IRES-RFP(MIR)-mouse IKZF2 | (Park et al., 2019) | |
| pHFUW-human IKZF2-IRES-GFP | This paper | N/A |
| Software and algorithms | ||
| Proteome Discoverer version 2.4.1.15 | Thermo Fisher Scientific | https://www.thermofisher.com/us/en/home/industrial/mass-spectrometry/liquid-chromatography-mass-spectrometry-lc-ms/lc-ms-software/multi-omics-data-analysis/proteome-discoverer-software.html |
| ImageJ | https://imagej.nih.gov/ij/ | |
| ChemDraw Professional 16.0 | PerkinElmer | https://perkinelmerinformatics.com/products/research/chemdraw |
| Mnova x64 | Mestrelab Research | https://mestrelab.com/download/mnova/ |
| Molecular Operating Environment | Chemical Computing Group | https://www.chemcomp.com/index.htm |
| Prism 7.0 | GraphPad | https://www.graphpad.com/ |
| Enrichr | Chen et al. | https://maayanlab.cloud/Enrichr/ |
| FlowJo 10.8.1 | Becton Dickinson & Company (BD) | https://www.flowjo.com/ |
| Other | ||
HIGHLIGHTS:
DEG-35 and DEG-77 are cereblon-dependent dual degraders of IKZF2 and CK1α
IKZF2 and CK1α degradation induces cell cycle arrest, apoptosis and differentiation
DEG-35 and DEG-77 are selective against leukemic cells and prolong survival in vivo
Acknowledgements
We thank members of the Kharas and Woo laboratories for helpful advice and suggestions. We thank the Harvard University Mass Spectrometry and Proteomics Resource Laboratory for mass spectrometry support, the Cancer Program at the Broad Institute for assistance with the PRISM screen, and the Antitumor Assessment Core in MSKCC for their help with bone marrow aspirates, PK study and advice on drug treatment in mice. We also thank Dr. Daniel Heller and Chen Chen for helpful discussions on formulation. CK1α was a generous gift from the Fischer lab. Support from the Blavatnik Accelerator at Harvard University (C.M.W.), Starr Cancer Consortium Translational Commercialization Fund at the Broad Institute (C.M.W., M.G.K.), and Anna Fuller Fund (M.G.K.) is gratefully acknowledged. M.G.K. is a Scholar of the Leukemia and Lymphoma Society and supported by NIDDK NIH R01-DK101989-01A1, NCI 1R01CA193842-01, NCI 1R01CA193842-06A1,5R01CA186702-07, 1R01DK1010989-06A1, R01HL135564, and R01CA 225231-01; The Starr Cancer Consortium; the Alex’s Lemonade Stand A Award. MSK core facilities are supported by P30CA008748.
Declaration of Interests
Harvard University filed PCT patent applications on April 29, 2021 and September 14, 2022 covering the chemical structures described in this article and their use. C.M.W.,D.M., S.P., and M.G.K. are listed as inventors on these patents. The Woo laboratory receives support from Ono Pharmaceuticals and Merck. M.G.K is on the scientific advisory board of 858 Therapeutics and has received honorarium from Kumquat Biosciences and Astrazeneca.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Estey E, and Dohner H (2006). Acute myeloid leukaemia. Lancet 368, 1894–1907. 10.1016/S0140-6736(06)69780-8. [DOI] [PubMed] [Google Scholar]
- 2.Suva ML, Riggi N, and Bernstein BE (2013). Epigenetic reprogramming in cancer. Science 339, 1567–1570. 10.1126/science.1230184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dohner H, Wei AH, and Lowenberg B (2021). Towards precision medicine for AML. Nat Rev Clin Oncol 18, 577–590. 10.1038/s41571-021-00509-w. [DOI] [PubMed] [Google Scholar]
- 4.Pirozzi CJ, and Yan H (2021). The implications of IDH mutations for cancer development and therapy. Nat Rev Clin Oncol 18, 645–661. 10.1038/s41571-021-00521-0. [DOI] [PubMed] [Google Scholar]
- 5.Park SM, Cho H, Thornton AM, Barlowe TS, Chou T, Chhangawala S, Fairchild L, Taggart J, Chow A, Schurer A, et al. (2019). IKZF2 Drives Leukemia Stem Cell Self-Renewal and Inhibits Myeloid Differentiation. Cell stem cell 24, 153–165 e157. 10.1016/j.stem.2018.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kronke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, Svinkina T, Heckl D, Comer E, Li X, et al. (2014). Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305. 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, Wong KK, Bradner JE, and Kaelin WG Jr. (2014). The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 343, 305–309. 10.1126/science.1244917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sievers QL, Petzold G, Bunker RD, Renneville A, Slabicki M, Liddicoat BJ, Abdulrahman W, Mikkelsen T, Ebert BL, and Thoma NH (2018). Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 362. 10.1126/science.aat0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beckwith REJ, Bonazzi Simone, Cernijenko Artiom, Visser Michael Scott (2020) 3-(1-Oxo-5-(piperidin-4-yl)piperidine-2,6-dion Derivatives and Uses Therof. United States of America patent application WO 2020/16583 A1.
- 10.Wang ES, Verano AL, Nowak RP, Yuan JC, Donovan KA, Eleuteri NA, Yue H, Ngo KH, Lizotte PH, Gokhale PC, et al. (2021). Acute pharmacological degradation of Helios destabilizes regulatory T cells. Nat Chem Biol 17, 711–717. 10.1038/s41589-021-00802-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fink EC, McConkey M, Adams DN, Haldar SD, Kennedy JA, Guirguis AA, Udeshi ND, Mani DR, Chen M, Liddicoat B, et al. (2018). Crbn (I391V) is sufficient to confer in vivo sensitivity to thalidomide and its derivatives in mice. Blood 132, 1535–1544. 10.1182/blood-2018-05-852798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krönke J, Fink EC, Hollenbach PW, MacBeth KJ, Hurst SN, Udeshi ND, Chamberlain PP, Mani D, Man HW, and Gandhi AK (2015). Lenalidomide induces ubiquitination and degradation of CK1α in del (5q) MDS. Nature 523, 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin Z, Amako Y, Kabir F, Flaxman HA, Budnik B, and Woo CM (2022). Development of Photolenalidomide for Cellular Target Identification. J Am Chem Soc. 10.1021/jacs.1c11920. [DOI] [PubMed] [Google Scholar]
- 14.Ishoey M, Chorn S, Singh N, Jaeger MG, Brand M, Paulk J, Bauer S, Erb MA, Parapatics K, Muller AC, et al. (2018). Translation Termination Factor GSPT1 Is a Phenotypically Relevant Off-Target of Heterobifunctional Phthalimide Degraders. ACS Chem Biol 13, 553–560. 10.1021/acschembio.7b00969. [DOI] [PubMed] [Google Scholar]
- 15.Corsello SM, Nagari RT, Spangler RD, Rossen J, Kocak M, Bryan JG, Humeidi R, Peck D, Wu X, Tang AA, et al. (2020). Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat Cancer 1, 235–248. 10.1038/S43018-019-0018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu C, Mannan AM, Yvone GM, Ross KN, Zhang YL, Marton MA, Taylor BR, Crenshaw A, Gould JZ, Tamayo P, et al. (2016). High-throughput identification of genotype-specific cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nat Biotechnol 34, 419–423. 10.1038/nbt.3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen L, Li C, Pan Y, and Chen J (2005). Regulation of p53-MDMX interaction by casein kinase 1 alpha. Mol Cell Biol 25, 6509–6520. 10.1128/MCB.25.15.6509-6520.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huart A-S, MacLaine NJ, Meek DW, and Hupp TR (2009). CK1α Plays a Central Role in Mediating MDM2 Control of p53 and E2F-1 Protein Stability. Journal of Biological Chemistry 284, 32384–32394. 10.1074/jbc.M109.052647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patil A, Manzano M, and Gottwein E (2018). CK1alpha and IRF4 are essential and independent effectors of immunomodulatory drugs in primary effusion lymphoma. Blood 132, 577–586. 10.1182/blood-2018-01-828418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu S, Chen L, Becker A, Schonbrunn E, and Chen J (2012). Casein kinase 1alpha regulates an MDMX intramolecular interaction to stimulate p53 binding. Mol Cell Biol 32, 4821–4832. 10.1128/MCB.00851-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jakobsen JS, Laursen LG, Schuster MB, Pundhir S, Schoof E, Ge Y, d’Altri T, Vitting-Seerup K, Rapin N, Gentil C, et al. (2019). Mutant CEBPA directly drives the expression of the targetable tumor-promoting factor CD73 in AML. Sci Adv 5, eaaw4304. 10.1126/sciadv.aaw4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Juan TS, Wilson DR, Wilde MD, and Darlington GJ (1993). Participation of the transcription factor C/EBP delta in the acute-phase regulation of the human gene for complement component C3. Proc Natl Acad Sci U S A 90, 2584–2588. 10.1073/pnas.90.7.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rojo R, Pridans C, Langlais D, and Hume DA (2017). Transcriptional mechanisms that control expression of the macrophage colony-stimulating factor receptor locus. Clin Sci (Lond) 131, 2161–2182. 10.1042/CS20170238. [DOI] [PubMed] [Google Scholar]
- 25.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, and Mesirov JP (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cruciat CM (2014). Casein kinase 1 and Wnt/beta-catenin signaling. Curr Opin Cell Biol 31, 46–55. 10.1016/j.ceb.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 27.Grzanka J, Leveson-Gower D, Golab K, Wang XJ, Marek-Trzonkowska N, Krzystyniak A, Wardowska A, Mills JM, Trzonkowski P, and Witkowski P (2013). FoxP3, Helios, and SATB1: roles and relationships in regulatory T cells. Int Immunopharmacol 16, 343–347. 10.1016/j.intimp.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 28.Schneider RK, Adema V, Heckl D, Jaras M, Mallo M, Lord AM, Chu LP, McConkey ME, Kramann R, Mullally A, et al. (2014). Role of casein kinase 1A1 in the biology and targeted therapy of del(5q) MDS. Cancer Cell 26, 509–520. 10.1016/j.ccr.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jaras M, Miller PG, Chu LP, Puram RV, Fink EC, Schneider RK, Al-Shahrour F, Pena P, Breyfogle LJ, Hartwell KA, et al. (2014). Csnk1a1 inhibition has p53-dependent therapeutic efficacy in acute myeloid leukemia. J Exp Med 211, 605–612. 10.1084/jem.20131033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, et al. (1999). Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 286, 531–537. 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 31.Lambert M, Alioui M, Jambon S, Depauw S, Van Seuningen I, and David-Cordonnier MH (2019). Direct and Indirect Targeting of HOXA9 Transcription Factor in Acute Myeloid Leukemia. Cancers (Basel) 11. 10.3390/cancers11060837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collins CT, and Hess JL (2016). Role of HOXA9 in leukemia: dysregulation, cofactors and essential targets. Oncogene 35, 1090–1098. 10.1038/onc.2015.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zauli G, Celeghini C, Melloni E, Voltan R, Ongari M, Tiribelli M, di Iasio MG, Lanza F, and Secchiero P (2012). The sorafenib plus nutlin-3 combination promotes synergistic cytotoxicity in acute myeloid leukemic cells irrespectively of FLT3 and p53 status. Haematologica 97, 1722–1730. 10.3324/haematol.2012.062083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shabashvili DE, Feng Y, Kaur P, Venugopal K, and Guryanova OA (2022). Combination strategies to promote sensitivity to cytarabine-induced replication stress in acute myeloid leukemia with and without DNMT3A mutations. Exp Hematol 110, 20–27. 10.1016/j.exphem.2022.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martinez-Hoyer S, and Karsan A (2020). Mechanisms of lenalidomide sensitivity and resistance. Exp Hematol 91, 22–31. 10.1016/j.exphem.2020.09.196. [DOI] [PubMed] [Google Scholar]
- 36.Sperling AS, Guerra VA, Kennedy JA, Yan Y, Hsu JI, Wang F, Nguyen AT, Miller PG, McConkey ME, Quevedo Barrios VA, et al. (2022). Lenalidomide promotes the development of TP53-mutated therapy-related myeloid neoplasms. Blood 140, 1753–1763. 10.1182/blood.2021014956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu YX, Shi CX, Bruins LA, Wang X, Riggs DL, Porter B, Ahmann JM, de Campos CB, Braggio E, Bergsagel PL, and Stewart AK (2019). Identification of lenalidomide resistance pathways in myeloma and targeted resensitization using cereblon replacement, inhibition of STAT3 or targeting of IRF4. Blood Cancer J 9, 19. 10.1038/s41408-019-0173-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakagawa H, Sido JM, Reyes EE, Kiers V, Cantor H, and Kim HJ (2016). Instability of Helios-deficient Tregs is associated with conversion to a T-effector phenotype and enhanced antitumor immunity. Proc Natl Acad Sci U S A 113, 6248–6253. 10.1073/pnas.1604765113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mullard A. (2021). Targeted protein degraders crowd into the clinic. Nat Rev Drug Discov 20, 247–250. 10.1038/d41573-021-00052-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, Related to Figure 1. Mutations in AML cell lines
Table S2, Related to Figure 2. Proteomics data for MOLM-13 cells treated with DEG-35.
Table S2, Related to Figure 2. Mass spectrometry data for MOLM-13 cells treated with DEG-35.
Table S3, Related to Figure 3. Correlation of CRISPR screen data to DEG-35 PRISM cell line sensitivity signature.
Table S3, Related to Figure 3. Correlation of Repurposed drug database to DEG-35 PRISM cell line sensitivity signature.
Table S3, Related to Figure 3. Correlation of mutations to DEG-35 PRISM cell line sensitivity signature.
Table S3, Related to Figure 3. Correlation of Copy Number Aberrations to DEG-35 PRISM cell line sensitivity signature.
Table S3, Related to Figure 3. Correlation of Gene expression to DEG-35 PRISM cell line sensitivity signature.
Table S3, Related to Figure 3. Correlation of CRISPR screen to Nutlin-3a PRISM cell line sensitivity signature.
Table S4, Related to Figure 4. Enrichr analysis (TF perturbation) of ShRNA positive correlation (Coef>0.2, qvalue <0.05)
Table S4, Related to Figure 4. Enrichr analysis (ENCODE and ChEA) of Gene expression positive correlation (Coef>0.2, qvalue <0.05)
Table S4, Related to Figure 4. Enrichr analysis (ChEA 2016) of Gene expression positive correlation (Coef>0.2, qvalue <0.05)
Table S5, Related to Figure 4. List of Differentially up-regulated genes (log 2-fold change >1, p adjusted <0.05)
Table S5, Related to Figure 4. List of Differentially down-regulated genes (log 2-fold change >1, p adjusted <0.05)
Table S6, Related to Figure 4. Rank list for GSEA for the RNA-seq DEG-35 vs DMSO in MOLM-13 cells.
Table S6, Related to Figure 4. Rank list for GSEA for the RNA-seq Control versus IKZF2 knockout primary MLL-AF9 c-Kit high cells
Table S6, Related to Figure 4. GSEA positively enriched using DEG35 regulated genes as Rank list.
Table S6, Related to Figure 4. GSEA negatively enriched using DEG35 regulated genes as Rank list.
Table S7, Related to Star Methods. List of AML patient characteristics.
Table S8, Related to Star Methods. List of primer sequences.
Data Availability Statement
RNA-seq data has been deposited in GEO (GSE220707).
All proteomics datasets have been deposited to the PRIDE repository with the dataset identifier PXD039474.