

Abstract
Radiation-induced intestinal injury (RIII), a common gastrointestinal complication caused by radiotherapy on pelvic, abdominal and retroperitoneal tumors, seriously affects the life quality of patients and may result in termination of radiotherapy. At present, the pathogenesis of RIII has not been fully understood. Herein, we demonstrated that ferroptosis played a critical role in RIII occurrence. The RNA sequencing analysis strongly hinted ferroptosis was involved in RIII mice. In line with this, the levels of 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA), markers of lipid peroxidation, remarkably increased in RIII mice. And the ferroptosis inhibitor, Ferrostatin-1 (Fer-1), improved the mice survival and alleviated intestinal fibrosis in vivo. Moreover, our results revealed that arachidonic acid (AA) enhanced ferroptosis in cultured intestinal epithelial cells (IECs) and organoids in vitro after irradiation, and AA gavage aggravated RIII in mice. Mechanistic studies revealed the level of ACSL4 protein significantly increased in mouse jejunums and IECs after irradiation. Radiation-induced ferroptosis in IECs was also prevented following ACSL4 knockdown or with the function inhibitor of ACSL4. Furthermore, we found that transcription of ACSL4 induced by irradiation was regulated by STAT1/IRF1 axis, and AMPK activation triggered by AA negatively regulated radiation-induced ferroptosis. Taken together, our results suggest that ferroptosis mediates RIII and reducing dietary AA intake as well as targeting the STAT1-IRF1-ACSL4 axis or AMPK may be the potential approaches to alleviate RIII.
Keywords: Radiation-induced intestinal injury, Ferroptosis, Lipid peroxidation, Arachidonic acid, ACSL4
Graphical abstract
1. Introduction
Radiation-induced intestinal injury (RIII) is a common gastrointestinal complication caused by radiotherapy on pelvic, abdominal, and retroperitoneal tumors. Up to 90% of patients develop acute gastrointestinal symptoms (such as abdominal pain, diarrhea, bloating, etc.) within three months after abdominopelvic irradiation and at least 50% of the patients experience chronic gastrointestinal symptoms (such as fibrosis, obliterating vasculitis, atrophy, etc.) which seriously affect their quality of life [[1], [2], [3], [4]]. Although pathogenesis of RIII is still not fully understood, various studies reported that epithelial injury, impaired vascular systems as well as the disordered gut immune and microbiota are involved in RIII [5,6]. At present, no unified and effective methods are available for clinical prevention and treatment of RIII.
Ferroptosis is a recently discovered form of regulated necrosis driven by iron-dependent phospholipid peroxidation [7]. Accumulation of iron, increased reactive oxygen species (ROS) production, shrunken mitochondria, and condensed mitochondrial membranes are typical features of ferroptosis [8]. Ferroptosis is regulated by multiple cellular metabolic events, including redox homeostasis, metabolism of iron, lipids and amino acids [9]. Recently, ferroptosis has been implicated in multiple diseases, including ischemic heart disease, stroke, kidney failure, liver damage, neurodegenerative diseases and inflammatory bowel disease [[9], [10], [11]]. Moreover, ferroptosis also functions as a tumor suppression mechanism via promoting the death of cancer cells and activating anti-tumor immunity [12]. Intriguingly, emerging evidences suggest that ferroptosis facilitates death of cancer cells in radiotherapy but at the same time mediates radiation-induced organs injuries [13,14]. Despite having been mentioned in some studies [15,16], the exact role of ferroptosis in RIII development, and the mechanism underlying ferroptosis induced by ionizing radiation (IR) in intestines are still not clear.
Ferroptosis is controlled by numerous metabolism-related proteins. Acyl-CoA synthetase long chain family member 4 (ACSL4) is one of the critical drivers of ferroptosis. ACSL4 ligates polyunsaturated fatty acids (PUFAs) especially arachidonic acid (AA) with coenzyme A, thus facilitating the esterification of PUFAs-CoA into phospholipids, and ultimately promotes formation of phospholipid hydroperoxides (PLOOHs) which act as major executioners of ferroptosis [17,18]. ACSL4 has been shown to contribute to multiple ferroptosis-related diseases and tumor suppression [[19], [20], [21]]. Moreover, IR induces expression of ACSL4, resulting in ferroptosis of cancer cells. Depletion of ACSL4 largely suppresses radiation-induced ferroptosis and promotes radioresistance [13]. However, how ACSL4 is upregulated by IR has not been revealed. In addition, owing to the differences between cancer and normal cells, the role of ACSL4 in damages of organs induced by IR still also remains unknown.
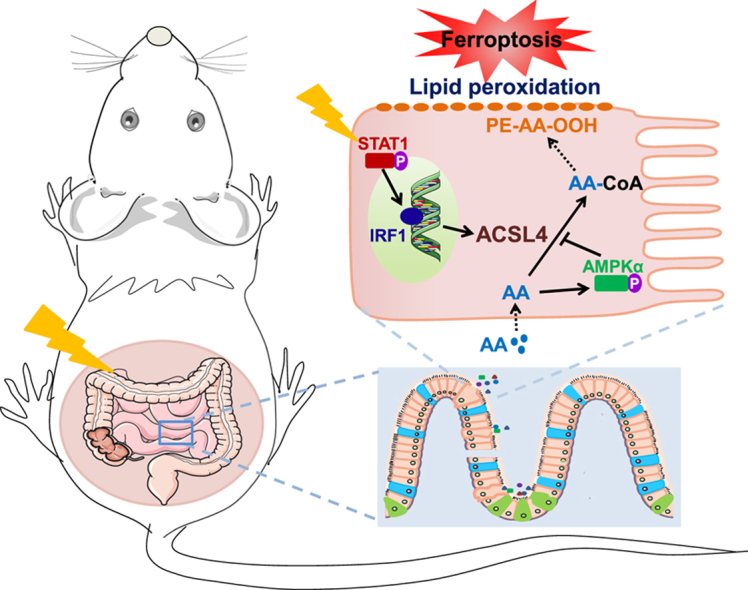
In the present study, we found that IR induced ferroptosis in the jejunums of mice. Moreover, intestinal injury and IECs damage caused by IR were distinctly aggravated by the additional AA, and were alleviated through inhibiting ferroptosis, indicating that AA-enhanced ferroptosis was involved in RIII. Mechanistic studies revealed that IR induced ferroptosis via activating STAT1-IRF1-ACSL4 pathway and AMPK activation triggered by AA negatively regulated the radiation-induced ferroptosis. These results provide some hints that ferroptosis and STAT1-IRF1-ACSL4 axis or AMPK are potential targets to alleviate RIII clinically, and limiting excessive dietary intake of AA would be beneficial.
2. Materials and methods
2.1. Animals
Male C57BL/6J mice aged 6-8 weeks (21-25 g) were purchased from GemPharmatech Co. Ltd (Nanjing, China). Mice were housed under specific-pathogen-free environment with a 12/12-h light/dark cycle, and were provided with free access to water and food. All animal studies were conducted according to protocols approved by the Ethical Committee of Experimental Animals of Hefei Institutes of Physical Science, Chinese Academy of Sciences (Code of Ethics: SWYX-DW-2021-64).
2.2. Cell culture
The human intestinal epithelial cell line FHs74Int, purchased from American Type Culture Collection (Manassas, VA, USA), was cultured in RPMI 1640 medium (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Gibco), 100 μg/mL streptomycin (Gibco) and 100 U/mL penicillin (Gibco). Cells were maintained at 37 °C in a humidified incubator with 5% CO2 and 95% air.
2.3. Mouse intestinal organoids culture
Mouse crypt isolation and organoid establishment were performed as previously described [22]. Briefly, the jejunum fragments were incubated in EDTA (2 mM) with PBS for 30 min on ice. After removing EDTA solution, the jejunum fragments were gently washed with cold PBS. The crypt-villus was collected through scraping the inner side of intestine and resuspended in PBS. After standing for 5 min, the sediment was retained and passed through a 70 μm cell strainer (Biosharp, Hefei, China). The isolated crypts were centrifuged at 65×g for 5 min and resuspended in matrigel (Corning, Glendale, AZ, USA) for culture and formation of organoids. For irradiation treatment, organoids were seeded into 48-well plates at a density of ∼100 organoids/20 μL Matrigel per well.
Organoid survival assays were performed as described previously [23]. Briefly, the number of live/dead organoids in each group was counted on day 9 after IR, and the ratio of organoid survival was calculated. Live organoids fulfil the following criteria: the diameter of organoid >30 μm, with solid or cystic structure. The dead organoids were identified by the characteristics, the structural collapse and the presence of dispersed cells or dark debris where the organoid was previously located. The sizes of live organoids were measured with the software ImageJ (National Institutes of Health).
2.4. Irradiation treatment
Mice were anesthetized and irradiated with Elekta Infinity linear accelerator (Elekta, Stockholm, Sweden) at a dose rate of 6 Gy/min. For RNA sequencing (RNAseq) analysis, mice were irradiated with a non-lethal dose of 11 Gy total abdominal irradiation (ABI). For survival and intestinal fibrosis measurements, mice were irradiated with doses of 15/17 Gy or 14 Gy ABI, respectively. To inhibit ferroptosis in vivo, mice were intraperitoneal injected with Ferrostatin-1 (Fer-1; MCE, Shanghai, China) at a dose of 5 mg/kg body weight per day. Fer-1 administration was started at 24 h before ABI and lasted for 14 days post ABI. The corresponding control mice were intraperitoneally injected with Vehicle solution (DMSO + PEG300+Tween-80+saline). To verify the effect of AA (MCE) intake on RIII, AA gavage at a dose of 10 mg/mice per day was started at 24 h before 15 Gy ABI and lasted for 14 days post ABI [24]. The survival analysis of mice was performed with the software GraphPad Prism 7.0 (GraphPad Software, La Jolla, CA, USA) using Kaplan-Meier statistics with a log-rank test.
Cells and mouse intestinal organoids were irradiated with an XHA600D X-ray irradiator (SHINVA, Zibo, Shandong, China) at a dose rate of 0.189 Gy/min in the presence or absence of AA, α-Linolenic acid (αLA; MCE), Linoleic acid (LA; MCE), Docosahexaenoic acid (DHA; MCE), Fer-1 or RSL3 (MCE). Cells or organoids were pretreated with PUFAs, Fer-1 or RSL3 for 24 h before IR.
2.5. Histological and immunohistochemical analysis
The jejunums of mice were fixed with 10% formalin and embedded in paraffin. Sections of jejunums (3 μm) were deparaffinized and rehydrated, and the tissue slides were then stained with hematoxylin and eosin. Collagen fibers were visualized with Masson's trichrome staining. For immunohistochemical analysis, tissue slides were incubated with anti-4-Hydroxynonenal (4-HNE) antibody (0.1 μg/mL; R&D Systems, Minneapolis, MN, USA) at 4 °C overnight followed by incubation with HRP-conjugated goat anti-mouse IgG H&L (1:2000; Abcam, Cambridge, MA, USA). After incubating with streptavidin peroxidase, slides were visualized using DAB substrate (Beyotime Biotechnology, Shanghai, China). Images were captured with Pannoramic Scan (3DHISTECH Ltd., Budapest, Hungary). The villus height, crypt depth and the thickness of the jejunal submucosa fibrous layer were measured with the ImageJ software (National Institutes of Health, Bethesda, MD, USA). The level of 4-HNE staining was quantified with ImagePro Plus (Media Cybernetics, Rockville, MD, USA).
2.6. RNA sequencing analysis
Three paired jejunum samples cut from RIII and control mice were collected at 4 days after ABI. The total RNA extraction, integrity checking, amplification, labeling, purification as well as gene expression sequencing analysis were carried out by SeqHealth Tech (Wuhan, China). The differential expression genes (DEG) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment were analyzed as described in Ref.[25].
2.7. MDA measurement
Jejunums were homogenized in Cell lysis buffer for western blotting (Beyotime Biotechnology), and the supernatant was harvested for protein concentration and MDA measurements after centrifugation (12,000×g, 10 min). The content of MDA in jejunums of mice was measured with MDA Detection Kit (Beyotime Biotechnology) according to the manufacturer's instructions. Briefly, the supernatant was mixed with the MDA detection solution and heated at 100 °C for 15 min. After cooling, the mixture was centrifuged and the supernatant was harvested for the measurement of optical density (OD) value at 532 nm with a microplate reader (Thermo Scientific, Waltham, MA, USA). The absolute content of MDA was calculated according to a standard curve.
2.8. Quantification of AA
Jejunums were cut into small pieces and homogenized with pre-cooled PBS (adding 9 mL PBS to every gram of tissue). The homogenate was further broken with ultrasound and centrifuged at 5000×g for 10 min. The supernatant of homogenate was subsequently collected for AA detection with an enzyme-linked immunosorbent assay (ELISA), following the manufacturer's instructions (Sangon Biotech, Shanghai, China).
2.9. Cell death assays
The dead cells were detected with propidium iodide (PI; Beyotime Biotechnology) staining and flow cytometry at 120 h post IR. For PI staining, cells were collected after trypsinization and resuspended in PBS containing PI (5 μg/mL). After incubation for 20 min at room temperature, the cells were washed with PBS and analyzed with a flow cytometer (Accuri C6, BD Biosciences, San Jose, CA, USA).
2.10. Detection of lipid peroxidation with BODIPY-C11 staining
The level of lipid peroxidation was measured with BODIPY-C11 staining at 72 h post IR. Cells were collected after trypsinization and resuspended in PBS containing BODIPY 581/591C11 dye (5 μM, Invitrogen, Waltham, MA, USA). After incubation for 30 min at 37 °C in a humidified incubator, the cells were washed with PBS and analyzed with a flow cytometer (Accuri C6, BD Biosciences).
2.11. Western blotting
The whole proteins from jejunums of mice and FHs74Int cells were extracted with RIPA lysis buffer (Beyotime Biotechnology). BCA protein assay kit (Beyotime Biotechnology) was used to determine the concentration of protein. Proteins were separated with SDS-PAGE and transferred to PVDF membranes (Merck Millipore, Darmstadt, Germany) followed by blocking with 5% skim milk (BD/Difco, Sparks, MD, USA). The various primary antibodies used in the experiments were as follows: anti-GPX4 (1:1000; Protein Tech Group, Wuhan, China), anti-ACSL4 (1:1000; ABclonal, Wuhan, China), anti-pSTAT1 (Tyr701) (1:1000; Cell Signaling Technology, Beverly, MA, USA), anti-STAT1 (1:1000; Protein Tech Group), anti-IRF1 (1:1000; ABclonal), anti-AMPKα (1:1000; Cell Signaling Technology), anti-pAMPKα (Thr172) (1:1000; Cell Signaling Technology) and anti-β-actin (1:1000; Protein Tech Group). After incubation with primary antibodies and washing with TBST, membranes were incubated with IRDye-conjugated secondary antibodies (1:10000; Li-COR Biosciences, Lincoln, NE, USA). Images of immunoreactive bands were captured with Odyssey CLx Infrared Imaging system (Li-COR Biosciences) and quantified with ImageJ software (National Institutes of Health).
2.12. qRT-PCR
The total RNA from jejunums of mice and FHs74Int cells was extracted with HiPure Total RNA Mini Kit (Magen, Guangzhou, China) and subjected to cDNA synthesis with Hifair II 1st Strand cDNA Synthesis SuperMix for qPCR (gDNA digester plus) kit (Yeasen Biotechnology, Shanghai, China). The qPCR was performed with Hifair qPCR SYBR Green Master Mix (No Rox) kit (Yeasen Biotechnology) on a Roche 480 Light Cycler. The primers used for PCR amplification are shown as follows: 5′-ACTGGCCGACCTAAGGGAG-3′, 5′-GCCAAAGGCAAGTAGCCAATA-3′ (ACSL4) and 5′-CTGGGACGACATGGAGAAAA-3′, 5′-AAGGAAGGCTGGAAGAGTGC-3′ (ACTB). ACTB was used as a normalizing control. The fold changes of mRNA were calculated with the 2−ΔΔCt method.
2.13. Small interfering RNA transfection
Cells were transfected with negative control RNA sequence (siNC) or small interfering RNA targeted to ACSL4 or STAT1 (GenePharma, Shanghai, China) with Lipofectamine 2000 Transfection Reagent (Invitrogen). The target sequences of siRNA are as follows: GGGAGTGATGATGCATCATAGCAAT (ACSL4); GCACCTGCAATTGAAAGAA (STAT1); AGACCAGAGCAGGAACAAG (IRF1) and ATGATGTCAGATGGTGAATTT (AMPKα). Cells were used for the further experiments at 48 h after transfection.
2.14. Statistical analysis
Data were presented as mean ± SD. Differences were evaluated by using Student's t-test with SPSS v21.0 software (SPSS, USA), and p < 0.05 was considered a statistically significant difference.
3. Results
3.1. Ferroptosis occurs in jejunum of mice after ABI
Recently, multiple studies suggested that ROS production and lipid peroxidation induced by IR played important roles in RIII occurrence [[26], [27], [28]]. Considering that ROS accumulation and excessive lipid peroxidation were critical factors triggering ferroptosis, we hypothesized that ferroptosis was related to RIII. A mouse RIII model with obvious destruction of jejunum architecture and significant reduction in villus height to crypt depth ratio was established following ABI (Fig. 1A and B), and the jejunum of mice was collected to perform RNAseq analysis. The DEG analysis results showed that 2108 genes were up-regulated while 2715 genes were down-regulated in jejunum on the fourth day (D4) after ABI (Fig. 1C). Moreover, KEGG pathway enrichment analysis showed that partial DEGs were enriched in ferroptosis (Fig. 1D), which supported our hypothesis that ferroptosis was related to RIII. Accordingly, the gene expression profile showed that ferroptosis-promoting genes such as ACSL4 and heme oxygenase 1 (HMOX1) were remarkably upregulated, whereas ferroptosis-inhibiting genes such as glutathione peroxidase 4 (GPX4) and solute carrier family 3 member 2 (SLC3A2) were significantly downregulated in RIII specimens (Fig. 1E). Moreover, we found significant decrease of GPX4 protein level in jejunum after IR, which was consistent with the results of gene expression analysis (Fig. 1F and G). Taken together, these results indicate that ferroptosis may be involved in RIII.
Fig. 1.

IR induces ferroptosis in mice jejunum. (A) Representative images of H&E staining of jejunum sections from control and irradiated mice at D4 after 11 Gy ABI. Scale bar, 100 μm. (B) Radio of the villus height to crypt depth in jejunum sections at D4 after 11 Gy ABI (n = 3). (C) Differentially expressed genes in jejunum samples harvested from control and RIII mice. (D) KEGG pathway enrichment analysis enriched by DEGs. (E) The heatmap of ferroptosis-related genes expression in jejunum samples from control and RIII mice (n = 3). (F) Western blot of GPX4 in the jejunum samples from control mice and irradiated mice at the indicated times post 11 Gy of ABI. Quantification of GPX4 expression is depicted in (G). (H) Representative immunohistochemistry images of 4-HNE in the jejunum sections of control and irradiated mice at D4 after 11 Gy of ABI. Scale bar, 50 μm. Quantification of the 4-HNE staining is depicted in (I), n = 3. (J) MDA levels in the jejunum samples from control and irradiated mice at D2 after 11 Gy of ABI (n = 3). *: p < 0.05, **: p < 0.01.
We next detected the accumulation of lipid peroxidation products such as 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA), which were characteristics of ferroptosis, to confirm that ferroptosis was involved in RIII. As shown in Fig. 1H-J, the RIII group displayed significantly increased 4-HNE and MDA levels compared with the control group. It was also noticed that the positive staining of 4-HNE was predominately in the intestinal epithelial cells (IECs) of jejunum (Fig. 1H). According to the findings described above, we concluded that ferroptosis was induced in the jejunum of mice after ABI.
3.2. Inhibition of ferroptosis alleviates RIII in mice
To clarify the role of ferroptosis in RIII, we used Fer-1, a selective small-molecule inhibitor of ferroptosis, to treat mice before IR and assessed the survival and intestinal fibrosis (Fig. 2A). In the survival experiment, mice were exposed to 17 Gy ABI and the results showed that Fer-1 significantly delayed the death of mice (Fig. 2B). Moreover, at 12 weeks after exposure to 14 Gy ABI, we observed typical morphological features of intestinal fibrosis, fibrotic submucosa thickening and hyperplasia of muscular layer (Fig. 2C). Fer-1 treatment significantly alleviated the radiation-induced thickening of submucosa fibrous layer, which indicated that ferroptosis promoted intestinal fibrosis induced by IR (Fig. 2C and D). Taken together, these results suggested that ferroptosis played an important role in RIII and inhibiting ferroptosis could alleviate RIII.
Fig. 2.

Fer-1 alleviates RIII in mice. (A) Treatment schema of mice with intraperitoneal injection (i.p.) of Fer-1 and ABI. (B) The percent survival of mice after 17 Gy ABI. (C) Representative images of Masson staining of the jejunum sections from Vehicle- and Fer-1-treated mice at 12 weeks post 14 Gy ABI. Scale bar: 50 μm. Quantification of the thickness of submucosa fibrous layer are depicted in (D), n = 3. *: p < 0.05.
3.3. AA enhances radiation-induced ferroptosis and RIII
Since IECs function as a physical and biochemical barrier that separates host tissue from gut microbiome to maintain intestinal homeostasis, dysfunction of IECs caused by IR was closely associated with RIII [5]. In the present study, immunohistochemical analysis showed the increase of 4-HNE level in IECs following ABI, indicating IR may induce ferroptosis in IECs which contributes to RIII (Fig. 1H). To test this hypothesis, we attempted to clarify the effect of ferroptosis on IECs survival after IR in vitro. Unexpectedly, the death of IECs induced by IR alone could not be rescued by Fer-1 (Fig. 3A and B), which suggested IR alone did not trigger ferroptosis in IECs in vitro. We noticed that the cell culture medium lacked PUFAs, which were abundant in gut and critical to ferroptosis induction in the in vitro cell culture system [29]. To better mimic the gut environment and to trigger ferroptosis in vitro, we supplemented the culture medium with various types of common PUFAs (Fig. 3A-C). Interestingly, only AA, but not other PUFAs, enhanced IR-induced cell death in FHs74Int cells. Moreover, the enhanced part of cell death with AA after IR was completely rescued by Fer-1 (Fig. 3B and C). Correspondingly, the level of lipid peroxidation in FHs74Int cells following IR was also remarkably enhanced with AA treatment and significantly decreased with Fer-1 treatment in the presence of AA (Fig. 3D). We also found that radiation-induced cell death and lipid peroxidation were significantly enhanced by treatment with RSL3, a well-known inducer of ferroptosis, in the presence of AA but not in the absence of AA (Fig. 3E and F). These results indicate that AA is necessary for IR-induced ferroptosis in IECs in vitro.
Fig. 3.

Radiation induces ferroptosis in the presence of AA. (A–C) Percentage of dead FHs74Int cells, treated with indicated PUFAs (20 or 40 μM) or/and Fer-1 (5 μM), detected at 120 h after irradiation. αLA: α-Linolenic acid; LA: Linoleic acid; DHA: Docosahexaenoic acid; AA: Arachidonic acid. (D) Lipid peroxidation levels in FHs74Int cells, in the absence or presence of AA (40 μM) and Fer-1 (5 μM), detected at 72 h after 10 Gy irradiation. (E) Percentage of dead FHs74Int cells, treated with AA (40 μM) or/and RSL3 (0.05 μM), detected at 120 h after 10 Gy irradiation. (F) Lipid peroxidation levels in FHs74Int cells, in the absence or presence of AA (40 μM) and RSL3 (0.05 μM), detected at 72 h after 10 Gy irradiation. (G) Representative images of intestinal crypt organoids in indicated groups captured at D9 post IR in the presence or absence of AA (100 μM). Organoids were pretreated with Fer-1 (10 μM) or/and AA for 24 h followed by 6 Gy irradiation, and the culture medium was replaced every 24 h. Scale bar: 50 μm. Quantification of survived organoids are depicted in (H). (I) Schema of AA gavage and irradiation treatment. (J) AA levels in jejunums from Ctrl and AA gavage mice measured by ELISA at D4 (n = 3). (K) The percent survival of mice after 15 Gy ABI in the presence or absence of AA. *: p < 0.05, **: p < 0.01, ***: p < 0.001, NS: no significance.
Since the intestinal stem cells (ISCs) located in the crypts are critical to maintain intestinal integrity via differentiating into functional IECs after IR, we next employed the model of intestinal organoid to investigate the effect of ferroptosis on IECs regeneration. As shown in Fig. 3G&H, the survival fraction of organoids in AA + IR group was significantly less than that in only IR group, while Fer-1 treatment effectively only attenuated the organoids death induced by AA + IR but not IR, and this also indicates that AA enhances ferroptosis following IR to impair the survival of ISCs.
Furthermore, we also identified the effect of AA on RIII in vivo (Fig. 3I). As shown in Fig. 3J&K, AA gavage remarkably elevated AA levels in jejunums of mice and significantly aggravated the death of mice after ABI. Taken together, radiation-induced ferroptosis in the presence of AA not only mediated death of IECs but also impaired their regeneration, and excessive intake of AA might be a risk factor for RIII.
3.4. Radiation induces ferroptosis in IECs via ACSL4
We next explored the mechanism underlying radiation-induced ferroptosis in IECs. Interestingly, we found that radiation remarkably upregulated the expression of ACSL4 proteins in both jejunums of mice and FHs74Int cells (Fig. 4A-C). Moreover, the mRNA level of ACSL4 significantly increased following IR (Fig. 4D), consistent with the RNAseq analysis results shown in Fig. 1E, suggesting that irradiation might upregulate ACSL4 expression via activating its transcription. Addition of AA did not increase the protein expression of ACSL4 after irradiation (Fig. 4C). ACSL4 is one of ferroptosis-promoting proteins, which functions as regulating PUFA-phospholipids biosynthesis and preferentially utilizes AA as its substrate. To identify whether ACSL4 is a critical enzyme to mediate radiation-induced ferroptosis, we employed siRNA to interfere ACSL4 expression in FHs74Int cells (Fig. 4E). The results revealed that radiation-induced cell death was attenuated after ACSL4 interference (Fig. 4F). In line with the change of cell death, radiation-induced lipid peroxidation in IECs also decreased in the group of ACSL4 interference (Fig. 4G). Notably, the effect of ACSL4 interference on inhibiting radiation-induced cell death and lipid peroxidation is similar to that of Fer-1 treatment, suggesting that IR induced ferroptosis in IECs mainly through ACSL4 (Fig. 4F and G). Moreover, we found that rosiglitazone, an enzymatic inhibitor of ACSL4, effectively attenuated radiation-induced cell death and lipid peroxidation of FHs74Int cells in a concentration-dependent manner in the presence of AA (Fig. 4H and I). Taken together, these results indicate that radiation induces ferroptosis in the presence of AA in IECs via upregulating ACSL4.
Fig. 4.

Radiation induces ferroptosis via upregulating ACSL4 expression. (A) Western blot of ACSL4 in the jejunum samples from control and irradiated mice at 48 h post 11 Gy ABI. Quantification of ACSL4 expression is depicted in (B). (C) Western blot of ACSL4 in FHs74Int cells at 48 h post 10 or 20 Gy irradiation with or without AA (40 μM) treatment. (D) Quantitative PCR detection of the relative expression of ACSL4 transcripts in FHs74Int cells at indicated time points post 10 Gy irradiation in the presence of AA (40 μM). (E) Western blot of ACSL4 in FHs74Int cells at 48 h following siRNA transfection. (F) Percentage of dead NC or siACSL4 FHs74Int cells, treated with DMSO or Fer-1 (5 μM), detected at 120 h after IR in the presence of AA (40 μM). (G) Lipid peroxidation levels in NC or siACSL4 FHs74Int cells, treated with DMSO or Fer-1 (5 μM), detected at 72 h after IR in the presence of AA (40 μM). (H) Percentage of dead FHs74Int cells, treated with indicated concentrations of rosiglitazone, detected at 120 h after 10 Gy irradiation in the presence of AA (40 μM). (I) Lipid peroxidation levels in FHs74Int cells, treated with indicated concentrations of rosiglitazone, detected at 72 h after 10 Gy irradiation in the presence of AA (40 μM). *: p < 0.05, **: p < 0.01, ***: p < 0.001.
3.5. Radiation stimulates ACSL4 expression via STAT1/IRF1 axis
Signal transducers and activators of transcription 1 (STAT1), one major transcription factor regulating gene expression in response to IFN-γ, is frequently activated by irradiation in both tumor and normal tissue [[30], [31], [32]]. Recently, STAT1 signaling pathway was demonstrated to mediate ACSL4 expression upon IFN-γ stimulation [21]. Herein, we explored whether the radiation-induced ACSL4 expression was regulated by STAT1 signaling pathway. As shown in Fig. 5A, the phosphorylation level of STAT1 was elevated after irradiation, indicating that irradiation activated STAT1 signaling in IECs. In order to verify the connection between radiation-induced activation of STAT1 and ACSL4 expression, we knocked down STAT1 with siRNA interference. As shown in Fig. 5B, the expression of ACSL4 protein remarkably decreased after STAT1 siRNA interference. Consistent with the change of ACSL4 protein, the transcription of ACSL4 induced by IR was also effectively inhibited with STAT1 siRNA interference (Fig. 5C). As expected, the cell death and production of lipid peroxidation induced by IR was significantly decreased following STAT1 siRNA interference (Fig. 5D and E). Moreover, we found that the expression of IRF1 was inhibited by silencing STAT1 with siRNA, which suggests that IRF1 works as a downstream of STAT1 (Fig. 5F). Given IRF1 has five binding sites (BS1-5) in the promoter region of ACSL4, we next investigated the possible role of IRF1 in ferroptosis mediated by ACSL4 after irradiation. As shown in Fig. 5G&H, the protein and mRNA expression levels of ACSL4 after irradiation remarkably decreased after IRF1 siRNA interference. Moreover, the cell death and production of lipid peroxidation induced by irradiation also significantly decreased following IRF1 siRNA interference (Fig. 5I and J). Taken together, these data suggest that STAT1/IRF1 axis is essential to the expression of ACSL4 induced by irradiation.
Fig. 5.

Radiation upregulates ACSL4 expression via STAT1/IRF1 axis. (A) Protein expression level of pSTAT1/STAT1 in FHs74Int cells detected at 48 h post 10 or 20 Gy irradiation in the presence of AA (40 μM). (B) Protein expression level of ACSL4 and pSTAT1/STAT1 in NC or siSTAT1 FHs74Int cells detected at 48 h post 10 Gy irradiation in the presence of AA (40 μM). (C) Quantitative PCR detection of the relative expression of ACSL4 transcripts in NC or siSTAT1 FHs74Int cells at 48 h post irradiation. (D) Percentage of dead NC or siSTAT1 FHs74Int cells, treated with DMSO or Fer-1 (5 μM), detected at 120 h after irradiation in the presence of AA (40 μM). (E) Lipid peroxidation levels in NC or siSTAT1 FHs74Int cells, treated with DMSO or Fer-1 (5 μM), detected at 72 h after irradiation in the presence of AA (40 μM). (F) Protein expression level of IRF1 and pSTAT1/STAT1 in NC or siSTAT1 FHs74Int cells detected at 48 h post irradiation in the presence of AA (40 μM). (G) Protein expression level of ACSL4 and IRF1 in NC and siIRF1 FHs74Int cells detected at 48 h post irradiation in the presence of AA (40 μM). (H) Quantitative PCR detection of the relative expression of ACSL4 transcripts in NC and siIRF1 FHs74Int cells at 48 h post irradiation. (I) Percentage of dead NC and siIRF1 FHs74Int cells, treated with DMSO or Fer-1 (5 μM), detected at 120 h after irradiation in the presence of AA (40 μM). (J) Lipid peroxidation levels in NC and siIRF1 FHs74Int cells, treated with DMSO or Fer-1 (5 μM), detected at 72 h after irradiation in the presence of AA (40 μM). *: p < 0.05, **: p < 0.01, ***: p < 0.001.
3.6. AMPK activation triggered by AA negatively regulates radiation-induced ferroptosis
Upon the intake of AA, the cellular energy status and fatty acid metabolism are likely to be altered. AMP-activated protein kinase (AMPK) is a crucial sensor of cellular energy status, and plays a critical role in maintaining the homeostasis of cellular energy and fatty acid metabolism [33]. The potential role of AMPK activation in responding to AA stimulation prompted us to explore the possible role of AMPK in ferroptosis induced by AA + IR. Interestingly, AMPKα was remarkably activated by AA but not irradiation (Fig. 6A). As expected, the expression of ACSL4 induced by IR was not affected with AMPKα siRNA interference (Fig. 6B). However, we found the cell death and production of lipid peroxidation induced by AA + IR were significantly enhanced following AMPKα siRNA interference, which could be rescued by Fer-1 (Fig. 6C and D). Taken together, these results suggest that AMPK activation exerts negative regulation on ferroptosis induced by AA + IR.
Fig. 6.

AMPK activation inhibits ferroptosis induced by AA + IR. (A) Protein expression level of pAMPKα/AMPKα in FHs74Int cells detected at 48 h post irradiation in the absence or presence of AA (40 μM). (B) Protein expression level of ACSL4 and AMPKα in FHs74Int cells detected at 48 h post irradiation in the presence of AA (40 μM). (C) Percentage of dead NC or siAMPKα FHs74Int cells, treated with DMSO or Fer-1 (5 μM), detected at 120 h after irradiation in the presence of AA (40 μM). (D) Lipid peroxidation levels in NC or siAMPKα FHs74Int cells, treated with DMSO or Fer-1 (5 μM), detected at 72 h after irradiation in the presence of AA (40 μM). (E) Schematic of ferroptosis-mediated RIII. *: p < 0.05, **: p < 0.01.
4. Discussion
Recently, growing evidences show that multiple programmed cell deaths (PCDs) including apoptosis, pyroptosis, autophagy and necroptosis are involved in RIII [[34], [35], [36]]. Here, we found that ferroptosis, a novel form of PCD, also played an important role in RIII. Moreover, we revealed that IR induced ferroptosis in IECs via activating STAT1-IRF1-ACSL4 pathway.
Ferroptosis occurs on the basis of excess ferrous iron and lipid peroxidation regulated by complex metabolic pathways. Our results of RNAseq analysis showed that differentially expressed genes were enriched in ferroptosis in jejunum specimens of RIII mice. Among these genes, the expression of ACSL4 is essential for ferroptosis execution [17]. Genetically or pharmacologically inhibiting ACSL4 is a viable therapeutic approach to prevent ferroptosis-related diseases. Tuo et al. revealed that cerebral ischemia/reperfusion injury was prevented by performing AAV-assisted ACSL4 knockout or using ACSL4 inhibitors (triacsin C and pioglitazone) [37]. In the present study, we also found the survival of IECs increased significantly after irradiation following ACSL4 silence or treatment with rosiglitazone, an enzymatic inhibitor of ACSL4. Given the importance of IECs in maintaining intestinal integrity, targeted inhibition of ACSL4 would be a promising approach to mitigate RIII.
It is worth noting that ferroptosis is not only triggered by the ferroptosis promoting mechanisms, but also regulated by the anti-ferroptosis mechanisms. We noticed that GPX4 expression was remarkably reduced in RIII mice. GPX4 is the first known central anti-ferroptosis enzyme utilizing glutathione (GSH) to protect cells from ferroptosis by eliminating phospholipid peroxides and cholesterol hydroperoxides [38]. The loss function of GPX4 results in ferroptosis-related diseases such as neurodegeneration and ischemia/reperfusion injury in kidney and liver [9]. However, whether GPX4 expression reduction plays a key role in RIII needs further studies.
AA, an essential ω-6 PUFA, can be obtained from diet or synthesized through desaturation and chain elongation of linoleic acid [39]. As the preferential substrate for ACSL4, AA plays a vital role in ferroptosis. It is reported that AA triggers ferroptosis in GPX4-restricted IECs accompanied with the increased transcription of inflammatory cytokines and causes enteritis resembling Crohn's disease [24]. Furthermore, the addition of exogenous AA is essential for IFN-γ-induced ferroptosis in tumor cells and enhances the sensitivity of mouse embryonic fibroblasts to ferroptosis triggered by RSL3 in vitro [18,21]. Consistent with these studies, we found exogenous AA significantly promoted ferroptosis in IECs and intestinal organoids after IR in vitro. For the lack of AA in the cell culture medium, supplementation of exogenous AA may enhance ferroptosis via providing sufficient substrate for ACSL4 in vitro. Previous studies showed that excessive dietary intake and mucosal accumulation of AA was closely correlated with the risk of developing inflammatory bowel disease (IBD) [40,41]. Our results suggest that AA is also a risk factor for RIII and controlling the dietary intake of AA may be beneficial in alleviating RIII.
Interestingly, our results further revealed that STAT1-IRF1 axis was involved in IR + AA-induced ferroptosis. STAT1, an important transcription factor, is essential for IFN signaling. Activated by multiple stimulators such as cytokines, growth factors and hormones, the phosphorylated STAT1 translocates into the nucleus and binds to specific promoters of target genes to promote gene expressions [42]. Numerous studies identified the activation of STAT1 signaling as an important risk factor involved in inflammatory diseases, such as asthma, rheumatoid arthritis, ulcerative colitis and Crohn's disease [43]. STAT1 cascade is involved in different pathologies correlated to the inflammatory process by regulating inflammation-related gene expression, such as inducible nitric oxide synthase (iNOS), cyclooxygenase (COX), vascular cell adhesion molecules (VCAM) and intercellular cell adhesion molecules (ICAM) [44]. In the current study, our data demonstrated that phosphorylation of STAT1 induced by IR + AA contributed to ferroptosis in IECs via promoting ACSL4 expression. However, the ENCODE chromatin immunoprecipitation (ChIP)-seq database suggests the existence of the binding sites for IRF1, a well-known downstream target of STAT1 signaling, are at the promoter region of ACSL4 but not for STAT1 [21]. Therefore, STAT1 activation might induce the transcription of ACSL4 via IRF1, which has been identified as a critical factor in mediating inflammatory cell death. The hypothesis was supported by our results that silencing IRF1 effectively inhibited IR-induced ACSL4 expression and ferroptosis. Hence, targeting STAT1-IRF1 axis may be a previously unappreciated approach to alleviate RIII. Nevertheless, the regulatory mechanism underlying STAT1 activation following IR needs further study.
In addition to the positive regulatory mechanism of ACSL4-mediated ferroptosis, we also identified AMPK activation as a negative feedback pathway for ferroptosis upon AA treatment. Once having sensed AA in cells, AMPK was activated. It was reported that AMPK activation suppressed the biosynthesis of AA-containing phosphatidylethanolamines (PEs), the direct triggers for ferroptosis, via inducing phosphorylation of acetyl-CoA carboxylase [45]. It is reasonable to infer that AMPK agonists may also have the potential to relieve RIII.
In summary, the present study shows that ferroptosis via activating STAT1-IRF1-ACSL4 pathway plays a critical role in RIII, and AMPK activation functions as a negative feedback mechanism to inhibit ferroptosis induced by IR + AA (Fig. 6E). Additionally, reducing dietary intake of AA, which functions as a risk factor for RIII, and targeting STAT1-IRF1-ACSL4 signaling pathway or AMPK might be promising therapeutic approaches to alleviate RIII.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This research was supported by the Chinese National Natural Science Foundation (grant nos. 82102842, 81974484 and U20A20372), CASHIPS Director's Fund (grant nos. YZJJ2021QN39 and YZJJ2022QN47), Key Research and Development Project of Anhui Province (202104j07020009) and project funded by Hefei Comprehensive National Science Center, the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) and Jiangsu Provincial Key Laboratory of Radiation Medicine and Protection.
References
- 1.Andreyev J. Gastrointestinal complications of pelvic radiotherapy: are they of any importance? Gut. 2005;54:1051–1054. doi: 10.1136/gut.2004.062596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hauer-Jensen M., Denham J.W., Andreyev H.J. Radiation enteropathy--pathogenesis, treatment and prevention. Nat. Rev. Gastroenterol. Hepatol. 2014;11:470–479. doi: 10.1038/nrgastro.2014.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abayomi J., Kirwan J., Hackett A. The prevalence of chronic radiation enteritis following radiotherapy for cervical or endometrial cancer and its impact on quality of life. Eur. J. Oncol. Nurs. 2009;13:262–267. doi: 10.1016/j.ejon.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 4.Andreyev J. Gastrointestinal symptoms after pelvic radiotherapy: a new understanding to improve management of symptomatic patients. Lancet Oncol. 2007;8:1007–1017. doi: 10.1016/S1470-2045(07)70341-8. [DOI] [PubMed] [Google Scholar]
- 5.Kim C.K., Yang V.W., Bialkowska A.B. The role of intestinal stem cells in epithelial regeneration following radiation-induced gut injury. Curr Stem Cell Rep. 2017;3:320–332. doi: 10.1007/s40778-017-0103-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumagai T., Rahman F., Smith A.M. The microbiome and radiation induced-bowel injury: evidence for potential mechanistic role in disease pathogenesis. Nutrients. 2018;10:1045. doi: 10.3390/nu10101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang X., Stockwell B.R. Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021;22:266–282. doi: 10.1038/s41580-020-00324-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dixon S.J., Lemberg K.M., Lamprecht M.R., Skouta R., Zaitsev E.M., Gleason C.E., et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stockwell B.R., Jiang X., Gu W. Emerging mechanisms and disease relevance of ferroptosis. Trends Cell Biol. 2020;30:478–490. doi: 10.1016/j.tcb.2020.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qiu Y., Cao Y., Cao W., Jia Y., Lu N. The application of ferroptosis in diseases. Pharmacol. Res. 2020;159 doi: 10.1016/j.phrs.2020.104919. [DOI] [PubMed] [Google Scholar]
- 11.Xu M., Tao J., Yang Y., Tan S., Liu H., Jiang J., et al. Ferroptosis involves in intestinal epithelial cell death in ulcerative colitis. Cell Death Dis. 2020;11:86. doi: 10.1038/s41419-020-2299-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun L.L., Linghu D.L., Hung M.C. Ferroptosis: a promising target for cancer immunotherapy. Am. J. Cancer Res. 2021;11:5856–5863. [PMC free article] [PubMed] [Google Scholar]
- 13.Lei G., Zhang Y. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020;30:146–162. doi: 10.1038/s41422-019-0263-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li X., Duan L., Yuan S., Zhuang X., Qiao T., He J. Ferroptosis inhibitor alleviates Radiation-induced lung fibrosis (RILF) via down-regulation of TGF-β1. J. Inflamm. 2019;16:11. doi: 10.1186/s12950-019-0216-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie L.W., Cai S., Zhao T.S., Li M., Tian Y. Green tea derivative (-)-epigallocatechin-3-gallate (EGCG) confers protection against ionizing radiation-induced intestinal epithelial cell death both in vitro and in vivo. Free Radic. Biol. Med. 2020;161:175–186. doi: 10.1016/j.freeradbiomed.2020.10.012. [DOI] [PubMed] [Google Scholar]
- 16.Wang L., Wang A., Fu Q., Shi Z., Chen X., Wang Y., et al. Ferroptosis plays an important role in promoting ionizing radiation-induced intestinal injuries. Biochem. Biophys. Res. Commun. 2022;595:7–13. doi: 10.1016/j.bbrc.2022.01.068. [DOI] [PubMed] [Google Scholar]
- 17.Doll S., Proneth B., Tyurina Y.Y., Panzilius E., Kobayashi S., Ingold I., et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017;13:91–98. doi: 10.1038/nchembio.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kagan V.E., Mao G., Qu F., Angeli J.P., Doll S., Croix C.S., et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017;13:81–90. doi: 10.1038/nchembio.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cui Y., Zhang Y., Zhao X., Shao L., Liu G., Sun C., et al. ACSL4 exacerbates ischemic stroke by promoting ferroptosis-induced brain injury and neuroinflammation. Brain Behav. Immun. 2021;93:312–321. doi: 10.1016/j.bbi.2021.01.003. [DOI] [PubMed] [Google Scholar]
- 20.Li Y., Feng D., Wang Z., Zhao Y., Sun R., Tian D., et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 2019;26:2284–2299. doi: 10.1038/s41418-019-0299-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liao P., Wang W., Wang W., Kryczek I., Li X., Bian Y., et al. CD8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell. 2022;40 doi: 10.1016/j.ccell.2022.02.003. 365-78.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sato T., Vries R.G., Snippert H.J., van de Wetering M., Barker N., Stange D.E., et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 23.Fu G., Chen S., Liang L., Li X., Tang P., Rao X., et al. SIRT1 inhibitors mitigate radiation-induced GI syndrome by enhancing intestinal-stem-cell survival. Cancer Lett. 2021;501:20–30. doi: 10.1016/j.canlet.2020.12.034. [DOI] [PubMed] [Google Scholar]
- 24.Mayr L., Grabherr F., Schwärzler J., Reitmeier I., Sommer F. Dietary lipids fuel GPX4-restricted enteritis resembling Crohn's disease. Nat. Commun. 2020;11:1775. doi: 10.1038/s41467-020-15646-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hou J., Wang F., Kong P., Yu P.K., Wang H., Han W. Gene profiling characteristics of radioadaptive response in AG01522 normal human fibroblasts. PLoS One. 2015;10 doi: 10.1371/journal.pone.0123316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dutta A., Gupta M.L., Verma S. Podophyllotoxin and rutin in combination prevents oxidative stress mediated cell death and advances revival of mice gastrointestine following lethal radiation injury. Free Radic. Res. 2018;52:103–117. doi: 10.1080/10715762.2017.1418982. [DOI] [PubMed] [Google Scholar]
- 27.Li M., Gu M.M., Lang Y., Shi J., Chen B.P.C., Guan H., et al. The vanillin derivative VND3207 protects intestine against radiation injury by modulating p53/NOXA signaling pathway and restoring the balance of gut microbiota. Free Radic. Biol. Med. 2019;145:223–236. doi: 10.1016/j.freeradbiomed.2019.09.035. [DOI] [PubMed] [Google Scholar]
- 28.Mohamed H.A., Said R.S. Coenzyme Q10 attenuates inflammation and fibrosis implicated in radiation enteropathy through suppression of NF-kB/TGF-β/MMP-9 pathways. Int. Immunopharm. 2021;92 doi: 10.1016/j.intimp.2020.107347. [DOI] [PubMed] [Google Scholar]
- 29.Pearl D.S., Masoodi M., Eiden M., Brümmer J., Gullick D., McKeever T.M., et al. Altered colonic mucosal availability of n-3 and n-6 polyunsaturated fatty acids in ulcerative colitis and the relationship to disease activity. J Crohns Colitis. 2014;8:70–79. doi: 10.1016/j.crohns.2013.03.013. [DOI] [PubMed] [Google Scholar]
- 30.Raspaglio G., Buttarelli M., Filippetti F., Battaglia A., Buzzonetti A., Scambia G., et al. Stat1 confers sensitivity to radiation in cervical cancer cells by controlling Parp1 levels: a new perspective for Parp1 inhibition. Cell Death Dis. 2021;12:933. doi: 10.1038/s41419-021-04229-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Du J., Kageyama S.I., Yamashita R., Hirata H., Hakozaki Y., Okumura M., et al. Impacts of the STING-IFNAR1-STAT1-IRF1 pathway on the cellular immune reaction induced by fractionated irradiation. Cancer Sci. 2022;113:1352–1361. doi: 10.1111/cas.15297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Philipp J., Sievert W., Azimzadeh O., von Toerne C., Metzger F., Posch A., et al. Data independent acquisition mass spectrometry of irradiated mouse lung endothelial cells reveals a STAT-associated inflammatory response. Int. J. Radiat. Biol. 2020;96:642–650. doi: 10.1080/09553002.2020.1712492. [DOI] [PubMed] [Google Scholar]
- 33.Ren Y., Shen H.M. Critical role of AMPK in redox regulation under glucose starvation. Redox Biol. 2019;25 doi: 10.1016/j.redox.2019.101154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu D., Han R., Deng S., Liu T., Zhang T., Xie H., et al. Protective effects of flagellin A N/C against radiation-induced NLR pyrin domain containing 3 inflammasome-dependent pyroptosis in intestinal cells. Int. J. Radiat. Oncol. Biol. Phys. 2018;101:107–117. doi: 10.1016/j.ijrobp.2018.01.035. [DOI] [PubMed] [Google Scholar]
- 35.Wu D.M., Li J., Shen R., Li J., Yu Y., Li L., et al. Autophagy induced by micheliolide alleviates acute irradiation-induced intestinal injury via inhibition of the NLRP3 inflammasome. Front. Pharmacol. 2021;12 doi: 10.3389/fphar.2021.773150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu Y., Tu W., Sun D., Chen X., Ge Y., Yao S., et al. Nrf2 alleviates radiation-induced rectal injury by inhibiting of necroptosis. Biochem. Biophys. Res. Commun. 2021;554:49–55. doi: 10.1016/j.bbrc.2021.03.004. [DOI] [PubMed] [Google Scholar]
- 37.Tuo Q.Z., Liu Y., Xiang Z., Yan H.F., Zou T., Shu Y., et al. Thrombin induces ACSL4-dependent ferroptosis during cerebral ischemia/reperfusion. Signal Transduct. Targeted Ther. 2022;7:59. doi: 10.1038/s41392-022-00917-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seibt T.M., Proneth B., Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic. Biol. Med. 2019;133:144–152. doi: 10.1016/j.freeradbiomed.2018.09.014. [DOI] [PubMed] [Google Scholar]
- 39.Tallima H., El Ridi R. Arachidonic acid: physiological roles and potential health benefits - a review. J. Adv. Res. 2018;11:33–41. doi: 10.1016/j.jare.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Silva P.S., Olsen A., Christensen J., Schmidt E.B., Overvaad K., Tjonneland A., et al. An association between dietary arachidonic acid, measured in adipose tissue, and ulcerative colitis. Gastroenterology. 2010;139:1912–1917. doi: 10.1053/j.gastro.2010.07.065. [DOI] [PubMed] [Google Scholar]
- 41.Nishida T., Miwa H., Shigematsu A., Yamamoto M., Iida M., Fujishima M. Increased arachidonic acid composition of phospholipids in colonic mucosa from patients with active ulcerative colitis. Gut. 1987;28:1002–1007. doi: 10.1136/gut.28.8.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim H.S., Lee M.S. STAT1 as a key modulator of cell death. Cell. Signal. 2007;19:454–465. doi: 10.1016/j.cellsig.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 43.Yu Y.L., Chen M., Zhu H., Zhuo M.X., Chen P., Mao Y.J., et al. STAT1 epigenetically regulates LCP2 and TNFAIP2 by recruiting EP300 to contribute to the pathogenesis of inflammatory bowel disease. Clin. Epigenet. 2021;13:127. doi: 10.1186/s13148-021-01101-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Prati A.C., Ciampa A.R., Cavalieri E., Zaffini R., Darra E., Menegazzi M., et al. STAT1 as a new molecular target of anti-inflammatory treatment. Curr. Med. Chem. 2005;12:1819–1828. doi: 10.2174/0929867054546645. [DOI] [PubMed] [Google Scholar]
- 45.Lee H., Zandkarimi F., Zhang Y. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 2020;22:225–234. doi: 10.1038/s41556-020-0461-8. [DOI] [PMC free article] [PubMed] [Google Scholar]