

Abstract
Complement overactivation mediates microglial synapse elimination in neurological diseases such as Alzheimer’s disease (AD) and frontotemporal dementia (FTD), but how complement activity is regulated in the brain remains largely unknown. We identified that the secreted neuronal pentraxin Nptx2 binds complement C1q and thereby regulates its activity in the brain. Nptx2-deficient mice show increased complement activity, C1q-dependent microglial synapse engulfment, and loss of excitatory synapses. In a neuroinflammation culture model and in aged TauP301S mice, adeno-associated virus (AAV)–mediated neuronal overexpression of Nptx2 was sufficient to restrain complement activity and ameliorate microglia-mediated synapse loss. Analysis of human cerebrospinal fluid (CSF) samples from a genetic FTD cohort revealed reduced concentrations of Nptx2 and Nptx2-C1q protein complexes in symptomatic patients, which correlated with elevated C1q and activated C3. Together, these results show that Nptx2 regulates complement activity and microglial synapse elimination in the brain and that diminished Nptx2 concentrations might exacerbate complement-mediated neurodegeneration in patients with FTD.
INTRODUCTION
Tight regulation of synapse formation and removal, as well as maintenance of appropriate connections, is important for the assembly of neuronal circuits in the mature brain. Aberrant synapse function and aberrant elimination is a key pathomechanism in neuropsychiatric and neurodegenerative diseases like schizophrenia and Alzheimer’s disease (AD) (1–3). Innate immune molecules, including the classical complement pathway (CCP), play a role in synapse removal during developmental circuit refinement and in pathological conditions (4–6). C1q, the initiating factor of the CCP, binds to synapses and, upon activation of the downstream cascade, leads to engulfment of the complement-opsonized synapses by microglia (4, 7, 8). This process is overactivated in several central nervous system (CNS) diseases, including AD, virus-induced synapse loss, frontotemporal dementia (FTD), Huntington’s disease, and perhaps schizophrenia, leading to synapse loss and subsequent cognitive decline (6–12). Blocking C1q binding to neurons is sufficient to attenuate synapse loss in mouse models of AD, FTD, and Huntington’s disease (7, 11, 13).
Despite the pivotal role of C1q in neurodegeneration and synapse loss, it is not well understood how C1q and downstream complement activity are regulated in the brain. Recently, the secreted neuronal sushi repeat protein X-linked 2 (SRPX2) has been shown to bind C1q and block complement-mediated synapse elimination during development of the visual system and somatosensory cortex (14). However, loss of SPRX2 had no effect on complement activity and synapse density after the developmental period (14), suggesting that SPRX2 is not involved in complement regulation in the adult brain. Apolipoprotein E (ApoE), a major risk factor for AD, has been shown to interact with C1q and thereby modulate CCP activity (15).
In the periphery, several proteins have been described to bind C1q and other complement factors to regulate complement activation. Members of the pentraxin family directly bind C1q and thereby regulate peripheral complement activity (16). Long pentraxins are characterized by their C-terminal pentraxin domain and sequence homology among family members. In the CNS, three pentraxins are primarily expressed by excitatory neurons, neuronal pentraxin 1 and 2 (Nptx1 and Nptx2) and the neuronal pentraxin receptor (Nptxr). NPTXs form disulfide-linked heterooligomers that remain tethered to the plasma membrane by the transmembrane domain of Nptxr (17). NPTX oligomers bind and promote the clustering of postsynaptic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) type glutamate receptors (AMPA-R), thereby contributing to different forms of synaptic plasticity (18–23). Moreover, Nptxr-containing complexes can be shed from the synapse upon enzymatic cleavage of Nptxr (19). In vivo imaging of Nptx2 indicates that the Nptx1/Nptx2/Nptxr complex is exocytosed at excitatory synapses on parvalbumin interneurons (PV-INs) during periods of behavioral activity; as much as 40% of the complex is later shed on a diurnal and circadian schedule during sleep, presumably in the process of circuit refinement (24). The shedding process can be monitored in humans by measuring Nptx2 in cerebrospinal fluid (CSF) and is disrupted by sleep deprivation in both mice and humans, highlighting its dynamic physiological regulation (24).
Nptx2 is expressed as an immediate early gene by excitatory neurons and is particularly important for activity-dependent strengthening of GluA4 AMPA-R–containing excitatory synapses on PV-INs (20, 23, 25). Ocular dominance plasticity requires Nptx2 (26), and monocular deprivation results in rapid shedding of Nptx2 expressed by pyramidal neurons at excitatory synapses on PV-INs in the deprived cortex. Coincidently, excitatory synapses that couple pyramidal neurons with PV-INs disconnect (27). In an AD amyloidosis mouse model, Nptx2 deletion amplifies the effect of amyloid-beta to reduce inhibitory circuit function required for gamma rhythmicity (25). Nptx2 in CSF serves as a biomarker in genetic FTD (28), AD (25, 29), Down syndrome (30), dementia with Lewy bodies (31), schizophrenia (24), and normal aging (32) and consistently correlates with cognitive measures. The reduction of Nptx2 in CSF may reflect inhibitory circuit dysfunction with associated excitatory synapse damage or loss, a pathomechanism common between neurodegenerative and neuropsychiatric diseases (24, 25, 33).
Here, we provide evidence that NPTX proteins bind C1q and thereby regulate CCP activity. Nptx2 is critical to target the Nptx1/Nptx2/Nptxr complex to presynaptic sites (25), and we found that deletion of Nptx2 leads to increased activation of the CCP and microglia-mediated elimination of excitatory synapses in the adult mouse brain. These consequences of Nptx2 knockout (Nptx2KO) are prevented by genetic deletion of C1q or by C1q-blocking antibodies, consistent with the notion that the natural function of Nptx2 is to inhibit C1q and CCP activation in the brain. Nptx2 also inhibits the cytotoxic action of activated microglia in a coculture system. Moreover, in TauP301S mice, a FTD and AD model characterized by neuroinflammation and complement-dependent neuronal damage (7–9), adeno-associated virus (AAV)–mediated overexpression of Nptx2 decreased complement activation and associated synapse loss. Last, we show that Nptx2 interacts with C1q in human CSF and that the fraction of Nptx2-bound C1q is decreased in symptomatic carriers of genetic FTD, whereas complement activity is increased compared with healthy noncarriers and presymptomatic carriers.
RESULTS
NPTXs bind C1q and inhibit CCP activity in vitro
C1q binds peripheral pentraxins with high affinity (34). To determine whether C1q interacts with NPTXs, we immobilized purified NPTXs, or immunoglobulin M (IgM) as a positive control, onto microtiter plate wells and measured C1q binding. C1q bound to all three NPTXs (Fig. 1A). Binding to immobilized Nptxr was protein concentration dependent and saturable (fig. S1A). The interaction was C1q specific, because NPTXs did not bind to immobilized complement C2, C3, or C4 (Fig. 1B). To visualize the C1q-Nptx2 complex, we analyzed purified Nptx2 and C1q by negative staining electron microscopy. As expected, C1q showed a characteristic radial spoke structure with six globular heads connected by collagen-like stalks. Consistent with prior biochemical studies (17), Nptx2 formed a hexameric ring about 10 nm across (Fig. 1C). When incubated together, the resulting C1q-Nptx2 complex showed Nptx2 bound to the core of the C1q molecule in between the globular heads (Fig. 1C). Peripheral pentraxins bind C1q with their pentraxin domain (34). To test whether C1q binds to the pentraxin domain of NPTXs, we expressed and purified Nptx2 pentraxin domain (X2-PD) and immobilized it to cyanogen bromide–activated Sepharose beads. X2-PD–coated beads, but not control beads, pulled down C1q (fig. S1B). To independently confirm the C1q-NPTX interaction, we performed cell surface binding assays with a stable doxycycline-inducible CHO cell line that coexpressed Nptxr and Nptx2. Upon addition of C1q to fresh cell medium, essentially all CHO cells with doxycycline-induced Nptxr/Nptx2 expression were decorated with C1q, whereas C1q did not bind to the CHO cell lines in the absence of tetracycline (and without detectable expression of Nptxr and Nptx2) (Fig. 1D). To estimate the C1q-Nptxr/Nptx2 binding affinity, we incubated Nptxr/Nptx2-expressing CHO cells with increasing concentrations of C1q, fixed the cells, and quantified the amount of bound C1q on the surface using an anti-C1q antibody. We measured a saturable interaction with a dissociation constant (KD) of about 2 nM for C1q and Nptxr/Nptx2 (Fig. 1E).
Fig. 1. NPTXs bind C1q and inhibit CCP activity in vitro.
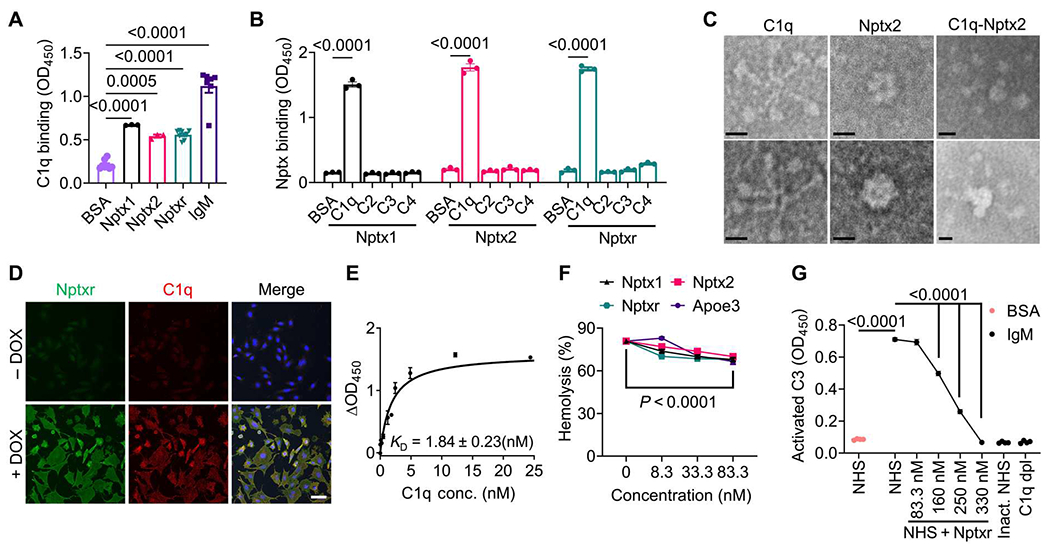
(A) Microtiter wells were coated with BSA, IgM, or NPTXs (Nptx1, Nptx2, and Nptxr) as indicated and incubated with C1q. Binding of C1q to the wells was detected using an anti-C1q antibody. P values were determined by one-way ANOVA followed by Šidák post hoc test. (B) Microtiter wells were coated with BSA or complement proteins (C1q, C2, C3, and C4) as indicated. Binding of His-tagged NPTXs was monitored using an anti-His antibody. P values were determined by two-way ANOVA followed by Šidák post hoc test. (C) Shown are representative images of negative staining electron microscopy of purified C1q, Nptx2, and C1q-Nptx2 complexes. Scale bars, 10 nm. (D) C1q cell surface binding assays were conducted with a stable tetracycline-inducible CHO cell line that coexpressed Nptxr and Nptx2. C1q proteins were added to the fresh culture medium, and bound C1q was visualized by immunofluorescence labeling (red). Expression of Nptxr was detected with a Nptxr-specific antibody (green). Images are shown of cells treated with (+DOX) or without (−DOX) doxycycline. Scale bar, 50 μm. (E) Cell surface binding of C1q to Nptxr- and Nptx2-expressing CHO cells was quantified. Stable-inducible CHO cells expressing Nptxr and Nptx2 were incubated with C1q at indicated concentrations. Bound C1q was detected with an anti-C1q antibody. The signal shown was subtracted from the background in CHO cells without induced Nptxr expression (ΔOD450). (F) The ability of NPTX to inhibit hemolysis of sheep erythrocytes was tested. Purified Nptx1, Nptx2, Nptxr, or ApoE3 was incubated in NHS, which was activated using CCP-specific GVB++ buffer, and lysis of sheep erythrocytes was analyzed by measuring released hemoglobin at 415 nm. P values were determined by two-way ANOVA followed by Šidák post hoc test. (G) The ability of Nptxr to inhibit CCP and activated C3 deposition was measured. NHS (1%) supplemented with recombinant Nptxr was added to IgM-coated microtiter plates, and activated C3 deposition was measured using specific antibodies. Heat-inactivated (Inact.) NHS and C1q-depleted (dpl) human serum were used as negative controls. P values were determined by one-way ANOVA followed by Šidák post hoc test. Data are presented as means ± SEM. Each dot represents data from individual samples. OD450, optical density at 450 nm.
On the basis of the interaction data, we tested whether NPTXs modulate CCP activity. In a hemolytic complement activity assay, addition of normal human serum (NHS), which contains all CCP proteins, leads to the lysis of erythrocytes (fig. S1C). Similar to ApoE, which was previously shown to inhibit CCP (15), addition of nanomolar amounts of Nptx1, Nptx2, or Nptxr decreased complement-mediated hemolysis in a dose-dependent manner (Fig. 1F). Addition of glutathione S-transferase (GST)–tagged Nptxr pentraxin domain (XR-PD), but not GST alone, efficiently inhibited CCP-mediated hemolysis in a dose-dependent manner (fig. S1D). Furthermore, in a CCP-mediated Escherichia coli killing assay, bacteria cells remained viable in the presence of XR-PD in a dose-dependent manner (fig. S1E). Last, we used a plate-based assay in which we immobilized IgM onto microtiter plate wells. C1q binds with high affinity to aggregated antibodies, resulting in the activation of the CCP; addition of NHS with CCP-activating buffer showed potent C1q-dependent deposition of cleaved C3 in IgM-coated wells, reflecting CCP activation. Addition of recombinant Nptxr inhibited CCP activity in a dose-dependent manner (Fig. 1G). Together, these results show that all three NPTXs can bind C1q and inhibit the CCP, likely by blocking C1q.
Nptx2 deletion increases CCP activity and leads to loss of excitatory synapses in vivo
In AD, Nptx2 is decreased in postmortem brain and in patient CSF, where its concentration correlates with disease status, cognitive performance, and disease progression (25). Nptx2 concentrations are also reduced in CSF from patients with schizophrenia, and Nptx2KO mice show schizophrenia-related behavioral deficits (24). Given the identification of Nptx2 as a regulator of complement, we next examined whether Nptx2KO mouse brains exhibited dysregulated complement. We first immunostained C1q in 2- to 3-month-old Nptx2KO mouse brains; C1q immunofluorescence intensity was similar in Nptx2KO compared with wild-type (WT) brains (Fig. 2A), which we further validated by C1q enzyme-linked immunosorbent assay (ELISA) (Fig. 2B). Whereas full-length protein concentration of the downstream C4 was also unchanged in Nptx2KO brains (Fig. 2C), the amount of cleaved activated C4 (C4b) was significantly (P = 0.0036) increased in Nptx2KO brains (Fig. 2D), which is consistent with increased processing of C4 resulting from increased CCP activity in NPTX2KO brains.
Fig. 2. Nptx2 regulates complement activity in vivo.
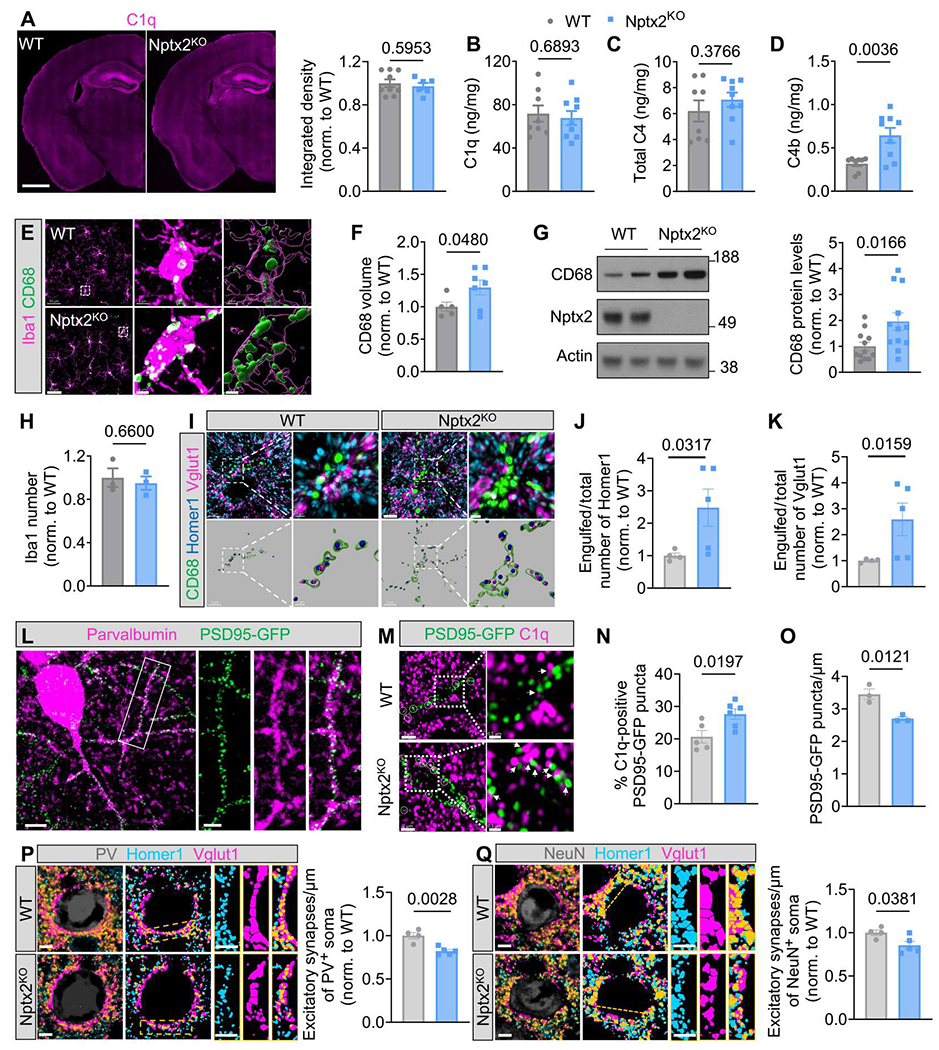
(A) Shown on the left are representative images of C1q immunostaining in WT and Nptx2KO mice brains. Scale bar, 1000 μm. The right shows quantification of C1q immunofluorescence in WT and Nptx2KO brains. P values were determined by unpaired t test. n = 9 for WT; n = 6 for Nptx2KO. Norm., normalized. (B to D) C1q (B), C4 (C), and C4b (D) concentration in WT and Nptx2KO cortices was measured by ELISA. P values were determined by unpaired t test. n = 8 for WT; n = 9 for Nptx2KO. (E) Shown are representative confocal images of CD68 (green) and Iba1 (magenta) immunostaining in WT and Nptx2KO cortical layers II to V. Scale bars, 30 μm and 2 μm (inset). (F) CD68 volume was quantified in WT and Nptx2KO cortical layers II to V. P values were determined by unpaired t test. n = 5 for WT; n = 7 for Nptx2KO. (G) Representative Western blots and quantification show CD68 abundance in hippocampi from WT and Nptx2KO cortices. P values were determined by unpaired t test. n = 12 for WT; n = 12 for Nptx2KO. (H) The number of Iba1+ microglia in WT and Nptx2KO whole brains was quantified. P values were determined by unpaired t test. n = 3 for WT; n = 3 for Nptx2KO. (I to K) Shown are representative confocal images and three-dimensional (3D) surface rendering of immunostained CD68 (green), Vglut1 (magenta), and Homer1 (blue) in WT and Nptx2KO cortical layers II to V (I). Scale bars, 5 μm and 1 μm (inset). Homer1 (J) and Vglut1 (K) puncta inside CD68+ lysosomes were quantified. P values were determined by unpaired t test. n = 4 for WT; n = 5 for Nptx2KO. (L) Representative confocal images of PSD95-GFP and immunostained parvalbumin are shown in samples from PSD95-GFPf/f:PV-Cre WT cortical layers II to V. Scale bars, 10 μm and 4 μm (inset). (M) Shown are representative confocal images of immunostained C1q (magenta) and PSD95-GFP (green) in PSD95-GFPf/f:PV-Cre:WT and Nptx2KO hippocampus CA1 strata radiata. White circles and white arrowheads indicate C1q colocalized with PSD95-GFP. Scale bars, 2 μm and 0.5 μm (inset). (N) The percentage of C1q + PSD95-GFP puncta in WT and Nptx2KO hippocampi was quantified. P values were determined by unpaired t test. n = 5 for WT; n = 6 for Nptx2KO. (O) PSD95-GFP puncta density was quantified in PSD95-GFPf/f:PV-Cre:WT and Nptx2KO hippocampi. P values were determined by unpaired t test. n = 3 for WT; n = 3 for Nptx2KO. (P) Shown on the left are representative confocal images and mask of immunostained PV (gray), Homer1 (blue), and Vglut1 (magenta) in WT and Nptx2KO cortical layers II to V. The bar graph on the right shows excitatory synapse density around somas of PV+ cells. P values were determined by unpaired t test. n = 4 for WT; n = 5 for Nptx2KO. Scale bars, 5 μm. (Q) Shown on the left are representative confocal images and mask of immunostained NeuN (gray), Homer1 (blue), and Vglut1 (magenta) in WT and Nptx2KO cortical layers II to V. Scale bars, 5 μm. The bar graph on the right shows normalized number of excitatory synapses around NeuN+ soma. P values were determined by unpaired t test. n = 4 for WT; n = 5 for Nptx2KO. Scale bars, 5 μm. Data are presented as means ± SEM. Each dot represents data from one mouse.
Microglia are the cellular effectors of complement-mediated synapse pruning. Given the increased complement activity, we wondered whether microglia show a phagocytic phenotype in Nptx2KO brains. By immunohistochemistry (IHC) staining and immunoblotting, we found that immunoreactivity of the phagolysosomal microglial marker CD68 was significantly (P = 0.048) increased in the cortices from Nptx2KO mice (Fig. 2, E to G). The number of microglia was unchanged in the cortices of Nptx2KO brains (Fig. 2H). By IHC, we confirmed that Nptx2 is expressed by excitatory neurons (fig. S2, A and B), and we detected synaptic Nptx2 puncta in the vicinity of excitatory synapse marker proteins vesicular glutamate transporter 1 (Vglut1) and synaptophysin, but not the inhibitory synapse marker protein gephyrin (fig. S2, C and D). Thus, we wondered whether loss of Nptx2 might render synapses more vulnerable to complement-dependent synapse loss through microglial phagocytosis. By coimmunostaining of excitatory postsynaptic marker Homer1 and presynaptic marker Vglut1 with microglial CD68+ lysosomes (Fig. 2I), we found significantly more Homer1 (P = 0.0317) and Vglut1 (P = 0.0159) immunoreactivity inside CD68+ lysosomes in the cortices from Nptx2KO mice compared with WT brains (Fig. 2, J and K), suggesting that microglial phagocytosis of excitatory synapses is increased in Nptx2KO brains. Given that Nptx2 concentration is particularly high on dendrites of PV-INs (20), we hypothesized that excitatory synapses on PV-INs might be especially vulnerable to complement-mediated pruning in Nptx2KO mice. Using mice that selectively express green fluorescent protein (GFP)–tagged postsynaptic density protein 95 (PSD95) in PV-INs (Fig. 2L), we found a significant (P = 0.0197) increase in C1q-labeled PSD95-GFP puncta in Nptx2KO mice (Fig. 2, M and N). The increase in C1q and synapses correlated with decreased PSD95-GFP density along dendrites in Nptx2KO brains (Fig. 2O). We speculated that microglia eliminate the excitatory synapses on PV-INs; however, we failed to identify GFP fluorescence in microglial lysosomes, possibly because GFP fluorescence is pH sensitive. Instead, using a PV-Cre reporter mouse that expresses the pH-stable tdTomato selectively in PV-INs, we found more tdTomato+ structures within CD68+ microglial lysosomes in NPTX2KO versus WT brains (fig. S2E). To validate the loss of excitatory synapses on PV-INs, we measured the density of colocalized postsynaptic Homer1 and presynaptic Vglut1 puncta around PV somas. Excitatory synapse density around PV somas was significantly (P = 0.0028) decreased in Nptx2KO brains by about 20% (Fig. 2P). PV cell density was unaffected, suggesting that microglia remove synapses (and possibly other structures) from PV-INs without inducing PV neuronal loss in Nptx2KO brains (fig. S2F). To test whether excitatory synapses are more globally affected in Nptx2KO brains, we measured excitatory synapse density around NeuN+ somas (a panneuronal marker). Intriguingly, excitatory synapse density was significantly (P = 0.0381) decreased by about 15% around NeuN+ somas (Fig. 2Q), suggesting that loss of Nptx2 leads to loss of excitatory synapses from additional neuronal subtypes beyond PV-INs.
Microglia have been shown to prune inhibitory synapses under physiological and pathological conditions through a complement-dependent mechanism (8, 35, 36). To determine whether loss of Nptx2 has an impact on inhibitory synapse density, we immunostained the inhibitory postsynaptic marker gephyrin and presynaptic marker glutamic acid decarboxylase 67 (GAD67) and measured inhibitory synapse density around PV+ and NeuN+ somas (fig. S3, A to D). Inhibitory synapse density was unchanged in Nptx2KO versus WT brains (fig. S3, B and D). The amount of gephyrin puncta inside microglial lysosomes was not significantly (P = 0.1561) different between WT and Nptx2KO brains, whereas GAD67 content was slightly but significantly (P = 0.007) elevated in Nptx2KO microglial lysosomes (fig. S3, E and F). Together, our results show that microglia remove excitatory synapses in the Nptx2KO cortex, whereas inhibitory synapses are largely unaffected by Nptx2 deficiency.
Complement inhibition rescues synapse loss in Nptx2KO brains
To test whether synapse loss in Nptx2KO brains is complement dependent, we compared animals that were deficient in C1q, Nptx2, or both (Nptx2KO;C1qKO double knockout mice). Given that the volume of microglial CD68+ lysosomes was increased in Nptx2KO brains, we first analyzed whether this microglial phenotype is C1q dependent. CD68 volume in C1qKO brains was unchanged compared with WT mice, but C1q deletion prevented the CD68 increase in Nptx2KO brains (fig. S4A). Concordantly, microglial excitatory synapse engulfment, as measured by density of Homer1 and Vglut1 puncta inside CD68+ lysosomes, was unchanged in C1qKO compared with WT brains but was significantly (Homer1, P = 0.0053; Vglut1, P = 0.0006) decreased in Nptx2KOC1qKO versus Nptx2KO brains (Fig. 3, A to C). To measure whether C1q deletion in Nptx2KO brains resulted in increased synapse density, we measured excitatory synapse density around NeuN+ and PV+ somas. At 10 weeks of age, C1qKO mice had a comparable synapse density to WT mice around NeuN+ and PV+ somas, suggesting that C1q is not essential for synapse pruning in the adult cortex (Fig. 3, D to G). The decreased synapse density seen in Nptx2KO mice was rescued by C1q deletion, because Nptx2KOC1qKO mice had a similar synapse density to WT mice (Fig. 3, D to G). Because in all these IHC readouts C1qKO and Nptx2KOC1qKO brains were not statistically different, the data imply that C1q is required in Nptx2KO brains to induce microglia activation and excitatory synapse elimination.
Fig. 3. Complement inhibition rescues synapse loss in Nptx2KO brains.
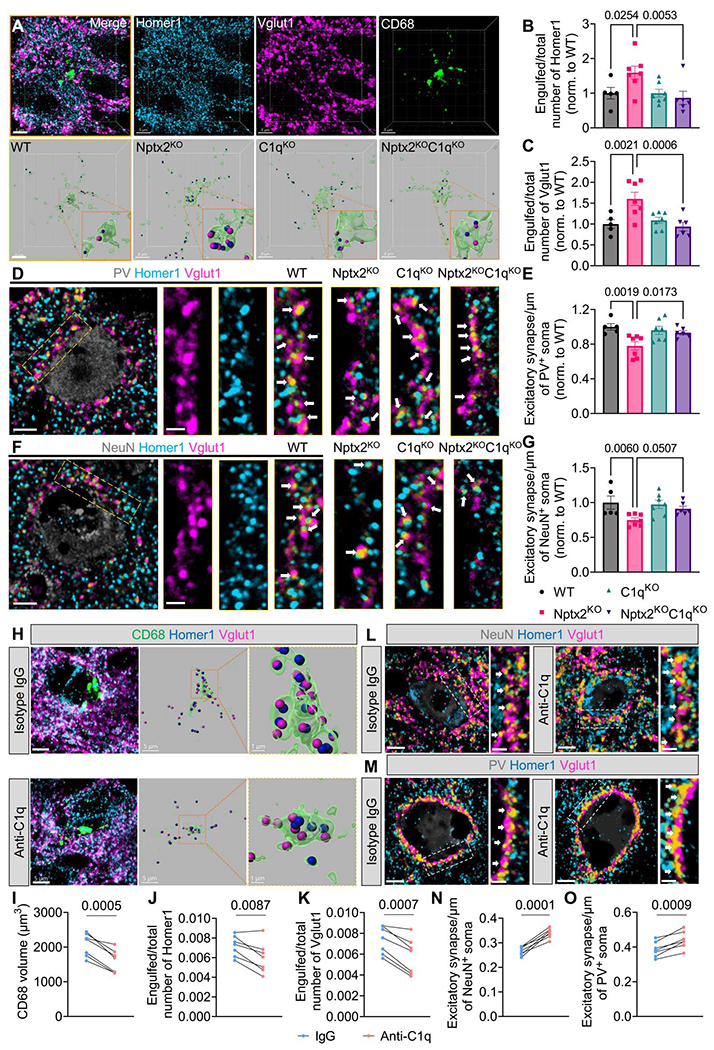
(A) Shown are representative confocal images and 3D surface reconstructions of Homer1 (blue), Vglut1 (magenta), and CD68 (green) in mouse cortical layers II to IV. Scale bars, 5 μm. (B and C) The relative amount of Homer1 (B) and Vglut1 (C) puncta within CD68+ microglial lysosomes was quantified. P values were determined by one-way ANOVA followed by Šidák post hoc test. n = 5 for WT; n = 7 for Nptx2KO; n = 7 for C1qKO; n = 6 for Nptx2KO;C1qKO. (D and E) Shown are representative confocal images (D) and quantification (E) of excitatory synapse density around somas of PV+ cells in the cortical layers II to V. White arrows indicate synapses, which were identified by colocalization of Homer1 and Vglut1 puncta. P values were determined by one-way ANOVA followed by Šidák post hoc test. n = 5 for WT; n = 7 for Nptx2KO; n = 7 for C1qKO; n = 6 for Nptx2KO;C1qKO. Scale bars, 5 μm and 2 μm (inset). (F and G) Shown are representative confocal images (F) and quantification (G) of excitatory synapse density around somas of NeuN+ cells in cortical layers II to V. White arrows indicate synapses, which were identified by colocalization of Homer1 and Vglut1 puncta. P values were determined by one-way ANOVA followed by Šidák post hoc test. n = 5 for WT; n = 7 for Nptx2KO; n = 7 for C1qKO; n = 6 for Nptx2KO;C1qKO. Scale bars, 5 μm and 2 μm (inset). (H) Shown are representative confocal images and 3D surface reconstructions of CD68 (green), Vglut1 (magenta), and Homer1 (blue) in control IgG and C1q-blocking antibody-injected Nptx2KO cortices. Scale bars, 5 μm and 1 μm (inset). (I) Volume of CD68+ microglial lysosomes in the cortices of Nptx2KO mice injected with control IgG and C1q-blocking antibody was quantified. P value was determined by paired t test. n = 7. (J and K) The relative amount of Homer1 (J) and Vglut1 (K) puncta within CD68+ microglial lysosomes was quantified in the cortices of Nptx2KO mice injected with isotype IgG antibody or C1q-blocking antibody. P values were determined by paired t test. n = 7. (L) Shown are representative confocal images of immunostained NeuN (gray), Homer1 (blue), and Vglut1 (magenta) in the cortical layers II-V of Nptx2KO mice injected with control IgG or C1q-blocking antibody. White arrows indicate synapses, which were identified by colocalization of Homer1 and Vglut1. P value was determined by paired t test. n = 7; Scale bars, 5 μm and 2 μm (inset). (M) Shown are representative confocal images of immunostained PV (gray), Homer1 (blue), and Vglut1 (magenta) in the cortical layers II to V of Nptx2KO mice injected with control IgG or C1q-blocking antibody. White arrows indicate synapses, which were identified by colocalized Homer1 and Vglut1 puncta. Scale bars, 5 μm and 2 μm (inset). (N) Excitatory synapse density around somas of NeuN+ cells was quantified. P value was determined by paired t test. n = 7. (O) Excitatory synapse density around somas of PV+ cells was quantified. P value was determined by paired t test. n = 7. Data are presented as means ± SEM. Each dot represents data from one mouse.
Fig. 4. Neuronal Nptx2 overexpression limits complement-mediated neurotoxicity in a neuroinflammation neuron-microglia coculture model.
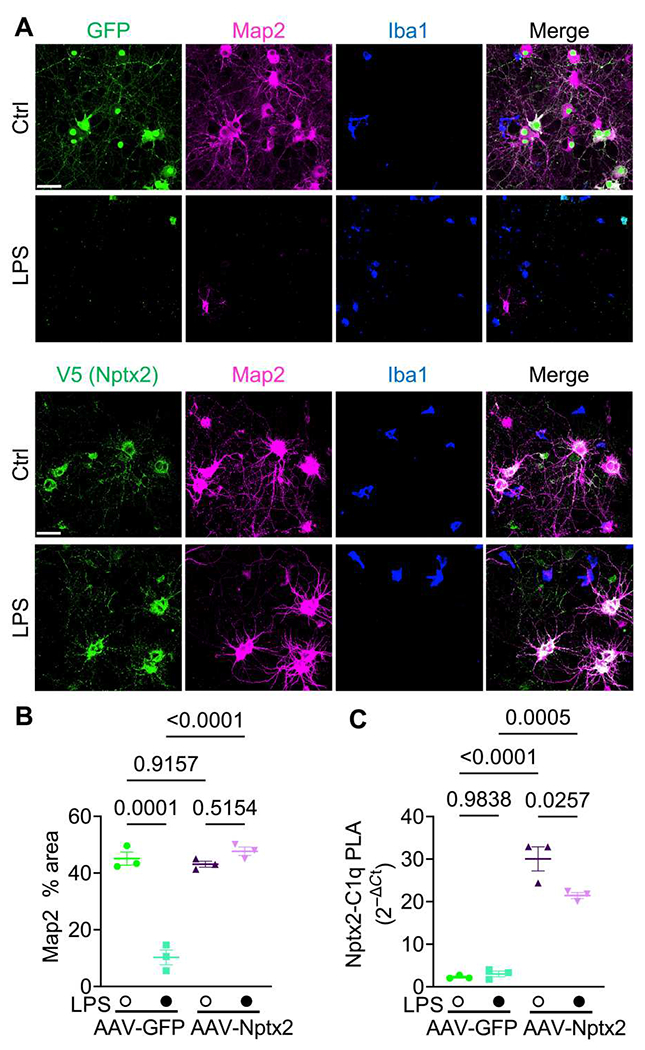
(A) Shown are representative images of immunostained Map2, Iba1, and Nptx2-V5 or GFP fluorescence in neuron-microglia cocultures treated with LPS or vehicle (Ctrl) for 24 hours. Neurons were infected with AAV-GFP or AAV-Nptx2-V5. Scale bars, 50 μm. (B) The percentage of Map2+ area in indicated cultures was quantified. Three independent neuron-microglia cocultures were used per condition. P values were determined by two-way ANOVA followed by Tukey post hoc test. (C) Nptx2-C1q complex abundance in culture medium was analyzed by proximity ligation assay (PLA). Media from three independent neuron-microglia cocultures were used. P values were determined by two-way ANOVA followed by Tukey post hoc test.
We showed previously that C1q-blocking antibodies rescued synapse loss in neuron-microglia cocultures and in P301S mice (7). To examine whether acute inhibition of C1q is sufficient to reduce microglial synapse elimination and rescue synapse density, we stereotactically injected C1q-blocking antibodies that potently inhibit CCP activity in vitro (fig. S4B), or isotype control IgGs into opposite somatosensory cortical hemispheres of 2- to 3-month-old Nptx2KO mice, and analyzed the brains 6 days after injection (fig. S4, C and D). Although the control IgG was diffusely distributed in the hippocampus and cortex, the anti-C1q antibody was strongly enriched in the hippocampus and cortex and showed a punctate staining that is characteristic for C1q distribution in the brain (fig. S4E) (7). By IHC, we found a significant (P = 0.0005) decrease in CD68 volume in the C1q-blocking antibody-injected cortex compared with the contralateral control IgG-injected cortex (Fig. 3, H and I). Furthermore, microglial engulfment of the excitatory synapse marker proteins Homer1 and Vglut1 was significantly (Homer1, P = 0.0087; Vglut1, P = 0.0007) decreased in C1q-blocking versus control IgG-injected cortical hemispheres (Fig. 3, H, J, and K). To determine whether decreased synapse engulfment resulted in increased synapse density, we measured Homer1-Vglut1–colocalized puncta around NeuN+ and PV+ somas 6 days after antibody injection. A single dose of C1q-blocking antibody was sufficient to increase synapse density around NeuN+ and PV+ somas compared with IgG-injected contralateral cortical hemispheres in all Nptx2KO mice used in the experiment (Fig. 3, L to O). Together, these findings reveal that Nptx2 deletion leads to C1q/CCP-dependent synapse elimination through microglial pruning in the adult mouse brain.
Neuronal Nptx2 overexpression limits complement-mediated neurotoxicity in a neuroinflammation neuron-microglia coculture model
Activated microglia can mediate neurodegeneration in culture. Addition of the bacterial endotoxin lipopolysaccharide (LPS) to neuron-microglia cocultures induces a strong proinflammatory and phagocytic microglia state and increased expression of complement components (37, 38), leading to notable loss of dendrites and neurons as measured by microtubule-associated protein 2 (Map2) immunostaining (fig. S5, A and B). It has been shown previously that blocking C1q is sufficient to ameliorate neurotoxicity mediated by activated microglia in cultures (38). Notably, when we added C1q-blocking antibodies together with LPS to neuron-microglia cocultures, neuronal loss was completely prevented (fig. S5, A and B). Thus, C1q-mediated activation of CCP is essential for LPS-induced neurotoxicity in those cocultures. Given that Nptx2 binds C1q and blocks CCP activation, we wondered whether increasing Nptx2 quantities would be sufficient to prevent LPS-induced neurotoxicity in neuron-microglia cocultures. We added recombinant Nptx2 protein (rNptx2) or bovine serum albumin (BSA) as a negative control together with LPS to the cultures (fig. S5C). Addition of rNptx2 prevented neurotoxicity induced by LPS (fig. S5, C and D). To test whether enhanced Nptx2 expression by neurons would be sufficient to ameliorate neurotoxicity, we infected neuronal cultures with AAVs overexpressing V5-tagged Nptx2 or GFP as a control for 3 days before the addition of microglia to the cultures. AAV-Nptx2 resulted in increased expression and secretion of Nptx2 into the medium (fig. S5E). Overexpression of Nptx2, but not GFP, prevented neuronal killing by LPS-activated microglia (Fig. 4, A and B). We speculated that secreted Nptx2 bound and blocked extracellular C1q, thereby inhibiting CCP activation. To test this hypothesis, we established a proximity ligation assay (PLA) using DNA-labeled antibodies, followed by ligation of the two DNA tails, amplification, and detection by quantitative polymerase chain reaction (qPCR) to detect and quantify amounts of C1q-Nptx2 complex in the culture medium. AAV-mediated overexpression of Nptx2 or the addition of rNptx2 strongly increased the abundance of C1q-Nptx2 complexes as compared with the respective controls in coculture media (Fig. 4C and fig. S5F). These results show that, analogously to C1q-blocking antibodies, secreted Nptx2 can bind and block C1q, thereby preventing neuronal loss in microglia-neuron cocultures.
AAV-induced Nptx2 overexpression ameliorates synapse loss in P301S mice
We showed previously in the TauP301S mouse model of FTD/AD (termed P301S mice hereafter) that C1q labels synapses for microglial elimination and that a C1q-blocking antibody or genetic deletion of C1q or the downstream C3 is sufficient to ameliorate synapse loss (7–9). C1q immunoreactivity was elevated in all layers of the frontal cortex and was even stronger in hippocampal regions of 9-month-old P301S mice compared with WT mice (fig. S6, A to D). However, Nptx2 immunoreactivity was comparable across cortical layers and hippocampal regions in 9-month-old P301S versus WT brains, with the exception of the stratum lucidum of the hippocampus, where Nptx2 immunoreactivity was slightly but significantly (P = 0.0467) reduced in P301S brains (fig. S6, E to H). C1q and Nptx2 immunoreactivity in the frontal cortex and hippocampus was comparable between P301S and WT mice at 3 months (fig. S6). This suggests that at 9 months, an age when synapse loss is detectable in P301S mice (7, 8), there is a C1q-Nptx2 imbalance, which might contribute to CCP overactivation. Therefore, we considered whether overexpression of Nptx2 is sufficient to blunt CCP activity and ameliorate complement-mediated synapse loss in P301S brains. AAVs overexpressing V5-tagged Nptx2 or GFP as a control were bilaterally injected into the hippocampi of 9-month-old P301S mice (Fig. 5, A and B). Three weeks after injection, we measured the protein concentration of complement proteins by ELISA. C1q and C4 concentrations were comparable in GFP- and Nptx2-overexpressing hippocampi (Fig. 5, C and D). However, C4b, the activated cleaved form of C4, was significantly (P = 0.015) lower in Nptx2-versus GFP-overexpressing hippocampi (Fig. 5E). Mean concentrations of total C3 and cleaved C3 (C3b) were reduced in Nptx2- compared with GFP-overexpressing hippocampi by about 35 and 50%, respectively, but this was not statistically significant (total C3, P = 0.144; C3b, P = 0.205) (Fig. 5, F and G). As expected, the concentration of all measured complement proteins was elevated in P301S hippocampi relative to WT hippocampi of the same age (fig. S7A). These results imply that overexpression of Nptx2 is sufficient to limit CCP overactivation in P301S brains. To test whether Nptx2-mediated complement inhibition is sufficient to ameliorate synapse loss in P301S mice, we injected AAV-Nptx2 or AAV-GFP into the ipsilateral and contralateral hippocampal hemispheres of 8.5-month-old P301S mice and analyzed the brains 6 weeks after injection by IHC (Fig. 5H and fig. S7B). As expected at this age, P301S brains were characterized by robust phospho-Tau staining (measured by AT8 immunoreactivity) as well as microgliosis and astrogliosis, as measured by ionized calcium–binding adaptor molecule 1 (Iba1) and glial fibrillary acidic protein (GFAP) immunoreactivity, respectively (fig. S7, C to F); all those hallmarks were most strongly detectable in the hippocampus as previously described (fig. S7C) (7, 9). Nptx2 overexpression did not affect AT8, Iba1, or GFAP immunoreactivity (fig. S7, G to I). In line with our ELISA results (Fig. 5C), C1q immunoreactivity was also unaffected by Nptx2 overexpression (fig. S7J). However, overexpression of Nptx2 reduced the percentage of C3-labeled synapses in the hippocampus, whereas the total number of C3 structures in the hippocampus was not changed (Fig. 5, I and J, and fig. S7K). Consistent with restrained CCP activity, the volume of CD68+ microglial lysosomes was significantly (P = 0.0126) smaller (Fig. 5, K and L), and microglial engulfment of excitatory synapses was significantly (P = 0.0165) reduced in Nptx2-versus GFP-overexpressing hippocampal regions (Fig. 5, K and M). This correlated with an increase in synapse density in Nptx2-overexpressing hippocampi, as measured by number of Homer1 puncta (Fig. 5N). Thus, Nptx2 overexpression is sufficient to ameliorate CCP-mediated microglial synapse pruning in P301S mice without affecting AT8 immunostaining or gliosis, implying that Nptx2 and complement act downstream of Tau pathology and microglial activation as measured here.
Fig. 5. AAV-induced Nptx2 overexpression ameliorates synapse loss in P301S mice.
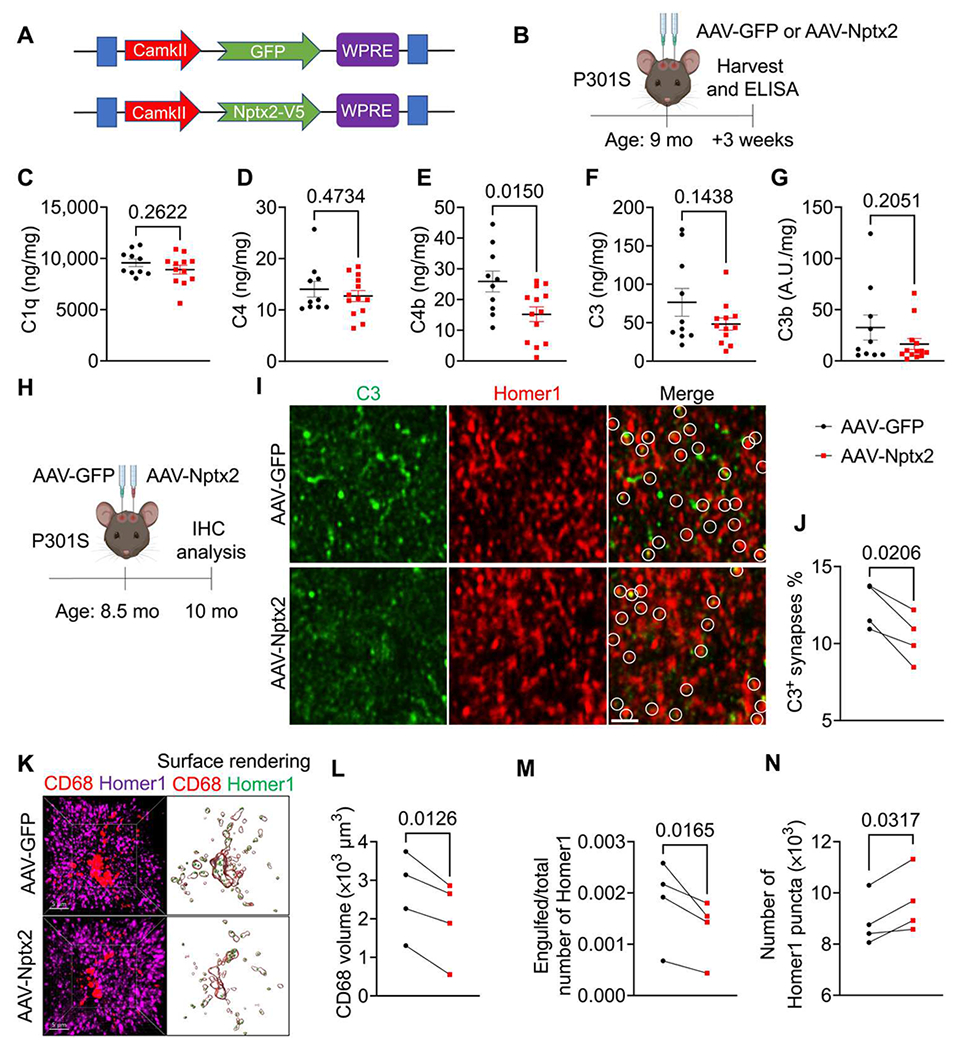
(A) Shown is a schematic representation of AAV-CamKII-GFP and AAV-CamKII-Nptx2-V5 constructs used for in vivo studies. (B) Shown is a schematic illustration of the workflow and data analysis. AAVs were injected bilaterally into hippocampi of 9-month-old male and female P301S mice and analyzed by ELISA 3 weeks later. (C to G) Concentrations of C1q (C), C4 (D), C4b (E), C3 (F), and C3b (G) were measured in hippocampus lysates from AAV-GFP– and AAV-NPTX2–injected P301S mice. Protein concentrations were measured by ELISA and are expressed as ng or A.U. (arbitrary unit)/mg of total protein. P values were determined by unpaired t test. AAV-GFP, n = 10; AAV-Nptx2, n = 13 (n = 12 for C1q and C3 ELISA because of low protein yield in one sample). (H) Shown is a schematic illustration of AAV injection into hippocampi of 8.5-month-old P301S mice and IHC analysis 6 weeks later. (I) Shown are representative confocal images of immunostained C3 (green) and Homer1 (red) in AAV-GFP– and AAV-Nptx2–injected hippocampal regions of P301S mice. White circles indicate C3 colocalized with Homer1. Scale bar, 2 μm. (J) The percentage of C3-labeled Homer1 puncta in AAV-GFP– and AAV-Nptx2–injected hippocampi from P301S mice was quantified. P value was determined by paired t test. n = 4. (K) Shown are representative confocal images and 3D surface renderings of Homer1 and CD68+ microglial lysosomes in AAV-GFP– and AAV-Nptx2–injected hippocampi of P301S mice. (L) The volume of CD68+ structures in AAV-GFP– and AAV-Nptx2–injected hippocampi of P301S mice was quantified. P value was determined by paired t test. n = 4. (M) The fraction of Homer1 puncta within CD68+ lysosomes in AAV-GFP– and AAV-NPTX2–injected hippocampi of P301S mice was quantified. P value was determined by paired t test. n = 4. (N) The relative spot number of Homer1 in AAV-GFP– and AAV-NPTX2–injected hippocampi of P301S mice was quantified. P value was determined by paired t test. n = 4. Data are presented as means ± SEM. Each dot represents data from individual samples.
Nptx2, C1q, and Nptx2-C1q complex concentrations are altered in genetic FTD CSF
Complement is recognized as a major driver of synapse loss and neuronal damage in many neurodegenerative diseases, and we reported previously that total and CCP activation–induced cleavage of C3 and C4 are increased in CSF from patients with AD (8, 9). In a previously characterized cohort of patients with genetic FTD, symptomatic carriers of pathogenic mutations in GRN or C9orf72 had lower Nptx2 concentrations in CSF, and decreased amounts of Nptx1 and Nptxr, compared with presymptomatic carriers or noncarrier controls (Fig. 6A) (28). In a subset of this cohort, we measured CSF C1q concentrations by ELISA (Fig. 6B). Whereas C1q concentrations were similar in CSF samples from noncarriers and presymptomatic carriers, they were significantly (P = 0.0002) elevated, on average, in CSF from symptomatic patients (Fig. 6B). Next, we wanted to determine whether C1q and Nptx2 present in the CSF were biochemically associated and whether Nptx2-C1q complex amounts were changed in FTD CSF. We used the PLA that we previously used to measure Nptx2-C1q complex concentration in culture medium. The specificity of this assay in complex samples was validated using Nptx2KO and C1qKO mouse brain lysates (fig. S8A). Furthermore, we monitored whether Nptx2 and C1q would form new protein complexes in solution by coincubating Nptx2KO and C1qKO lysates. However, we detected only background signal compared with lysates from WT brains (fig. S8A), suggesting that we primarily detected Nptx2-C1q complexes that preexisted in vivo. In addition, we confirmed that the assay requires the presence of both oligo-labeled C1q and Nptx2 antibodies in WT brain lysates (fig. S8B). Last, compared with NHS, we measured only background Nptx2-C1q PLA signal in C1q-depleted human serum (fig. S8C). Using the C1q-Nptx2 PLA assay, we measured robust signal in the CSF, suggesting that C1q and Nptx2 are present in a complex in the CSF (Fig. 6C). The Nptx2-C1q PLA signal, corresponding to the amount of the protein complexes containing these two proteins, was decreased in symptomatic carrier CSF versus both presymptomatic and noncarrier CSF (Fig. 6C). To test whether downstream complement activity is changed in FTD CSF, we measured the concentration of the C3 cleavage product C3b by ELISA. CSF C3b concentration strongly correlated with C1q (fig. S8D). Compared with those in presymptomatic carriers and noncarriers, C3b concentration was significantly (presymptomatic carriers, P = 0.0001; noncarriers, P = 0.0001) increased in symptomatic CSF (Fig. 6D). By comparison, CSF concentration of factor B, a component of the alternative complement pathway, was similar between the three patient groups (Fig. 6E). Thus, in CSF from patients with symptomatic genetic FTD, Nptx2 and Nptx2-C1q complex concentrations are lowered, whereas CCP activity (C3b) is increased. These findings support the role of Nptx2 as a potential regulator of CCP activity in the brain.
Fig. 6. Nptx2, C1q, and Nptx2-C1q complex concentrations are altered in genetic FTD.
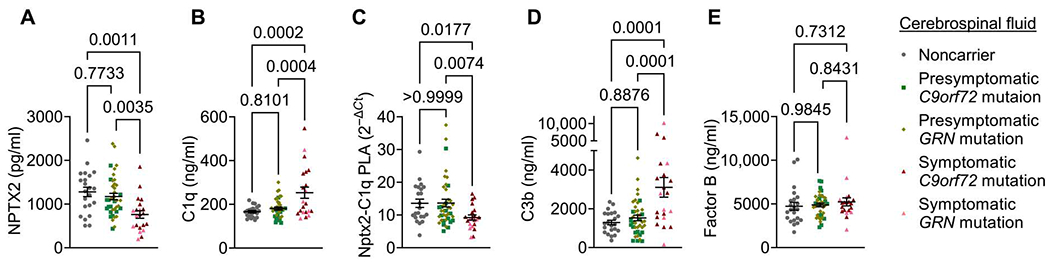
(A and B) Concentrations of Nptx2 (A) and C1q (B) were quantified in cerebrospinal fluid (CSF) from noncarriers as well as presymptomatic and symptomatic carriers of C9orf72 or GRN mutations. Protein concentrations were measured by ELISA. Nptx2 concentrations were measured and reported previously (28). P values were determined by one-way ANOVA test followed by Šidák post hoc test. (C) NPTX2-C1q complex abundance in CSF samples was analyzed by PLA. Quantitative qPCR data are represented using 2−ΔCt. P values were determined by one-way ANOVA test followed by Šidák post hoc test. (D and E) Concentrations of C3b (D) and factor B (E) were quantified in CSF from noncarriers as well as presymptomatic and symptomatic carriers of C9orf72 or GRN mutations. Protein concentrations were measured by ELISA. P values were determined by one-way ANOVA test followed by Šidák post hoc test. All data are presented as means ± SEM. Each dot represents data from one individual. Noncarriers, n = 22; presymptomatic GRN mutation, n = 25; presymptomatic C9orf72, n = 15; symptomatic GRN mutation, n = 7; symptomatic C9orf72, n = 14 (n = 13 for factor B ELISA).
DISCUSSION
We report that Nptx2 binds C1q and thereby regulates CCP activity in the brain under physiological and pathological conditions. The CCP is triggered by binding of C1q to a wide range of receptors and subsequent activation of the downstream pathway. Although other neuronal CCP inhibitors have been identified to play a role in synapse sculpting during neurodevelopment, including SRPX2 and CUB and sushi multiple domains 1 (CSMD1) (14, 39), to our knowledge, Nptx2 is the first neuronal protein that regulates CCP activity in the adult brain. Deletion of Nptx2 in mice led to increased CCP activity, resulting in microglia-mediated removal of excitatory synapses in the adult brain. Consistent with the hypothesis that Nptx2 acts as a CCP inhibitor, overexpression of Nptx2 was sufficient to decrease CCP activity and ameliorate neural damage in a neuroinflammation cell model and in P301S mice. One important finding of our study is the diminished concentrations of Nptx2-C1q complexes and elevated C3b in the CSF from individuals with symptomatic genetic FTD, which is indicative of elevated CCP activity in the brain at a stage of disease when cognitive failure is present.
A recent study reported that Nptx2 interacts with C1q’s globular head domain with a binding affinity in the nanomolar range (40), which independently validates our protein interaction analysis. Because Nptx2 binds to the head region of C1q, the domain by which C1q interacts with targets like antigen-bound synapses or IgGs, the interaction blocks downstream CCP activation. A similar mode of action has been described for a therapeutical C1q-neutralizing antibody that binds to the head domain of C1q and prevents the interaction of C1q with diverse ligands (41, 42), including synapses (7), and is currently in clinical trials for Huntington’s disease and amyotrophic lateral sclerosis.
Without a “stressor” and subsequent glial cell activation and expression of downstream complement factors, C1q accumulation alone is likely insufficient to trigger an aberrant innate immune reaction and widespread neuronal damage (9, 43). Although C1q is relatively abundant in the brain, expression of downstream CCP genes, including C1s and C1r which, together with C1q, form the CCP-initiating C1 complex, is very low and only induced under pathological conditions (9). Our data suggest that in the absence of Nptx2, basal CCP activity is elevated enough to increase microglia-mediated excitatory synapse elimination without inducing more severe neuronal damage. This is consistent with previous work showing that mice deficient in the complement inhibitors SRPX2 or CSMD1 are characterized by loss of excitatory synapses without frank neurodegeneration early in brain development (14, 39). Genetic variations of the complement component 4 (C4) gene C4A, which lead to increased expression of the C4a protein, are among the strongest common variant risk factors associated with schizophrenia (44, 45). Increased expression of C4 is sufficient to elevate microglia-mediated elimination of excitatory synapses, resulting in aberrant behavior in mouse models (10, 46). Nptx2KO mice parallel the phenotype of C4-overexpressing mice; we found elevated concentration of cleaved C4b in brains from Nptx2KO mice, and we previously reported that Nptx2KO mice exhibit increased vulnerability to social isolation stress, with emergent neuropsychiatric behavioral deficits (24). The present finding extends the notion that Nptx2 loss of function spans the complex genetics of schizophrenia as a shared common pathway (24).
In contrast to modestly raised CCP activity and moderate loss of excitatory synapses in Nptx2KO or C4-overexpressing mice, C1q and downstream complement components are highly up-regulated in many chronic neurodegenerative diseases, including AD, FTD, and Huntington’s disease (7–9, 11, 13, 36, 47). The marked increase in CCP activity in these diseases correlates with exacerbated synapse loss and severe neurodegeneration, and genetic or pharmacological inhibition of CCP or downstream complement components is sufficient to ameliorate neuronal damage (7–9, 11, 13, 36, 47–49). Thus, the degree of CCP activity correlates with the severity of neuronal damage across multiple diseases with different etiologies and genetic risk factors. At least in our neuroinflammation neuron-microglia coculture model, C1q activity is required for microglia-mediated neurodegeneration. In this context, it is remarkable that neuronal overexpression of Nptx2 in neuron-microglia cultures or in P301S mice at an age when Tau pathology and neuroinflammation are present limited CCP activity, complement tagging, and subsequent microglial removal of synapses. Our genetic FTD CSF analysis and recently published data (50, 51) identify that CCP activity is elevated in brains of symptomatic patients, potentially driven by the increased amount of C1q that is not regulated by Nptx2. The CSF results support the idea that, similar to our data in neuron-microglia cocultures and P301S mice, Nptx2 binds C1q and thereby constrains CCP activity. Notably, CSF Nptx2 concentration changes are detectable before CSF C1q and C3b concentrations increase at a later disease stage (51). Together with decreased amounts of C1q-Nptx2 complexes that we measured in CSF from symptomatic GRN or C9orf72 mutation carriers, the data support the idea that decreased Nptx2 concentration might unleash detrimental CCP activity. We hypothesize that beyond genetic FTD, CSF C1q-Nptx2 complex concentration might be a disease-relevant biomarker across other CNS diseases characterized by aberrant CCP activity.
Our study has limitations. NPTXs bind postsynaptic AMPA receptors through a transsynaptic interaction, and, at least in neuronal cultures, this interaction appears to be sufficient to induce postsynaptic specializations (17, 18). Although we cannot exclude the possibility that Nptx2 might modulate synapse stability or density independent of complement through binding to AMPA receptors or other (synaptic) interaction partners, the decreased synapse density in Nptx2KO mice was C1q dependent. The dynamic redistribution of synaptic Nptx2 in response to behavior, sleep, circadian rhythm, and afferent activity (24, 27) indicates that Nptx2-C1q might contribute to the precise refinement of circuits that mediate homeostasis of excitability, rhythmicity, and capacity for information processing. Another limitation is that we did not analyze whether rescue of synapses by Nptx2 overexpression in P301S mice results in functional benefits. Last, there is no human genetics evidence so far that NPTX2 is associated with neurodegeneration.
Overall, our study identifies Nptx2 as a neuronal synaptic CCP inhibitor. Elevating Nptx2 expression might be a viable therapeutic strategy to curb complement overactivation and subsequent neuronal damage in FTD, AD, and other neuroinflammatory diseases.
MATERIALS AND METHODS
Study design
This study aimed to test whether NPTX Nptx2 binds complement C1q and thereby regulates complement activity. To this end, we performed in vitro and in vivo experiments and analyzed CSF samples from patients with FTD. Sample sizes were determined on the basis of previous experience for each experiment. No statistical methods were used to predetermine sample size. No data were excluded, and no outlier analysis was performed. Mice were randomly assigned to experimental groups whenever possible. All experiments were performed and analyzed blinded to the genotype, group, and treatment. In vivo experiments were performed in biological replicates as indicated by n values in the figure legends. For in vitro cell assays, data were collected from at least three independent cultures, as indicated in the figure legends.
Statistical analysis
All raw, individual-level data for experiments where n < 20 are presented in data file S1. The number of samples is shown in figures and described in figure legends. All values are presented as means ± SEM unless otherwise stated. Student’s t test was performed for comparison between two groups. Data were tested for normality using Shapiro-Wilk test. One-way analysis of variance (ANOVA) or two-way ANOVA with Šidák’s or Tukey’s post hoc test was performed to analyze more than two groups, as indicated in the figure legends. Statistical analysis was performed using GraphPad Prism 9, and specific statistical tests are defined in the figure legends. For all experiments, statistical significance is defined as follows: not significant (n.s.), *P < 0.05, **P < 0.01, and ***P < 0.001.
Supplementary Material
Acknowledgments:
We thank the Janelia’s Virus Tool facility for helping prepare the AAV. We thank R. Huganir’s laboratory (Johns Hopkins University) for providing the PSD95-GFPf/f mice. We thank T.-M. Wang and D. Hackos for help with creating the Nptxr/Nptx2 CHO cell lines. We also thank K. Chan and B. Deverman for helping with AAV design. We thank the members of the Worley’s laboratory, especially W. Zhang and L. Yang, for insightful discussion of this project. We thank R. Lin for insightful discussion of this project and valuable suggestions for the manuscript writing and editing.
Funding:
This study was supported by funding from the NIH (grant no. R35NS097966 and U19 AG065169 to P.F.W.) and the Stanley Center for Psychiatric Research (to M.S.).
Footnotes
Supplementary Materials
This PDF file includes:
Other Supplementary Material for this manuscript includes the following:
MDAR Reproducibility Checklist
Competing interests: M.S. is a scientific cofounder and senior advisory board member of Neumora Therapeutics and a senior advisory board member of Biogen, Vanqua Bio, ArcLight Therapeutics, and Cerevel Therapeutics. P.F.W. and M.-F.X. are cofounders of CogNext. J.E.H. is employed by Genentech. The other authors declare that they have no competing interests.
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. All biological resources, antibodies, and model organisms and tools are either available through commercial sources or may be requested from P.F.W. and can be shared with the scientific community through a completed materials transfer agreement (MTA). The stable CHO cell line may be requested from Genentech by completing an MTA (www.gene.com/scientists/mta).
REFERENCES AND NOTES
- 1.Volk L, Chiu S-L, Sharma K, Huganir RL, Glutamate synapses in human cognitive disorders. Annu. Rev. Neurosci 38, 127–149 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Wilton DK, Dissing-Olesen L, Stevens B, Neuron-glia signaling in synapse elimination. Annu. Rev. Neurosci 42, 107–127 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Neniskyte U, Gross CT, Errant gardeners: Glial-cell-dependent synaptic pruning and neurodevelopmental disorders. Nat. Rev. Neurosci 18, 658–670 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SWM, Barres BA, The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B, Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bohlen CJ, Friedman BA, Dejanovic B, Sheng M, Microglia in brain development, homeostasis, and neurodegeneration. Annu. Rev. Genet 53, 263–288(2019). [DOI] [PubMed] [Google Scholar]
- 7.Dejanovic B, Huntley MA, Mazière AD, Meilandt WJ, Wu T, Srinivasan K, Jiang Z, Gandham V, Friedman BA, Ngu H, Foreman O, Carano RAD, Chih B, Klumperman J, Bakalarski C, Hanson JE, Sheng M, Changes in the synaptic proteome in tauopathy and rescue of tau-induced synapse loss by C1q antibodies. Neuron 100, 1322–1336.e7 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Dejanovic B, Wu T, Tsai M-C, Graykowski D, Gandham VD, Rose CM, Bakalarski CE, Ngu H, Wang Y, Pandey S, Rezzonico MG, Friedman BA, Edmonds R, Mazière AD, Rakosi-Schmidt R, Singh T, Klumperman J, Foreman O, Chang MC, Xie L, Sheng M, Hanson JE, Complement C1q-dependent excitatory and inhibitory synapse elimination by astrocytes and microglia in Alzheimer’s disease mouse models. Nat. Aging 2, 837–850 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu T, Dejanovic B, Gandham VD, Gogineni A, Edmonds R, Schauer S, Srinivasan K, Huntley MA, Wang Y, Wang T-M, Hedehus M, Barck KH, Stark M, Ngu H, Foreman O, Meilandt WJ, Elstrott J, Chang MC, Hansen DV, Carano RAD, Sheng M, Hanson JE, Complement C3 is activated in human AD Brain and is required for neurodegeneration in mouse models of amyloidosis and tauopathy. Cell Rep. 28, 2111–2123.e6 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Comer AL, Jinadasa T, Sriram B, Phadke RA, Kretsge LN, Nguyen TPH, Antognetti G, Gilbert JP, Lee J, Newmark ER, Hausmann FS, Rosenthal S, Kot KL, Liu Y, Yen WW, Dejanovic B, Cruz-Martín A, Increased expression of schizophrenia-associated gene C4 leads to hypoconnectivity of prefrontal cortex and reduced social interaction. PLOS Biol. 18, e3000604 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B, Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, Soung A, Yu J, Perez-Torres C, Frouin A, Wilton DK, Funk K, DeMasters BK, Jiang X, Bowen JR, Mennerick S, Robinson JK, Garbow JR, Tyler KL, Suthar MS, Schmidt RE, Stevens B, Klein RS, A complement–Microglial axis drives synapse loss during virus-induced memory impairment. Nature 534, 538–543 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilton DK, Mastro K, Heller MD, Gergits FW, Willing CR, Frouin A, Daggett A, Gu X, Kim AY, Faull R, Jayadev S, Yednock T, Yang XW, Stevens B, Microglia mediate early corticostriatal synapse loss and cognitive dysfunction in Huntington’s disease through complement-dependent mechanisms. bioRxiv 471180.4 December 2021. 10.1101/2021.12.03.471180. [DOI] [Google Scholar]
- 14.Cong Q, Soteros BM, Wollet M, Kim JH, Sia G-M, The endogenous neuronal complement inhibitor SRPX2 protects against complement-mediated synapse elimination during development. Nat. Neurosci 23, 1067–1078(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yin C, Ackermann S, Ma Z, Mohanta SK, Zhang C, Li Y, Nietzsche S, Westermann M, Peng L, Hu D, Bontha SV, Srikakulapu P, Beer M, Megens RTA, Steffens S, Hildner M, Halder LD, Eckstein H-H, Pelisek J, Herms J, Roeber S, Arzberger T, Borodovsky A, Habenicht L, Binder CJ, Weber C, Zipfel PF, Skerka C, Habenicht AJR, ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat. Med 25, 496–506 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nauta AJ, Bottazzi B, Mantovani A, Salvatori G, Kishore U, Schwaeble WJ, Gingras AR, Tzima S, Vivanco F, Egido J, Tijsma O, Hack EC, Daha MR, Roos A, Biochemical and functional characterization of the interaction between pentraxin 3 and C1q. Eur. J. Immunol 33, 465–473 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Xu D, Hopf C, Reddy R, Cho RW, Guo L, Lanahan A, Petralia RS, Wenthold RJ, O’Brien RJ, Worley P, Narp and NP1 form heterocomplexes that function in developmental and activity-dependent synaptic plasticity. Neuron 39, 513–528 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Lee S-J, Wei M, Zhang C, Maxeiner S, Pak C, Botelho SC, Trotter J, Sterky FH, Südhof TC, Presynaptic neuronal pentraxin receptor organizes excitatory and inhibitory synapses. J. Neurosci 37, 1062–1080 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cho RW, Park JM, Wolff SBE, Xu D, Hopf C, Kim J, Reddy RC, Petralia RS, Perin MS, Linden DJ, Worley PF, mGluR1/5-dependent long-term depression requires the regulated ectodomain cleavage of neuronal pentraxin NPR by TACE. Neuron 57, 858–871 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang MC, Park JM, Pelkey KA, Grabenstatter HL, Xu D, Linden DJ, Sutula TP, McBain CJ, Worley PF, Narp regulates homeostatic scaling of excitatory synapses on parvalbumin-expressing interneurons. Nat. Neurosci 13, 1090–1097 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pelkey KA, Barksdale E, Craig MT, Yuan X, Sukumaran M, Vargish GA, Mitchell RM, Wyeth MS, Petralia RS, Chittajallu R, Karlsson R-M, Cameron HA, Murata Y, Colonnese MT, Worley PF, McBain CJ, Pentraxins coordinate excitatory synapse maturation and circuit integration of parvalbumin interneurons. Neuron 85, 1257–1272 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Brien RJ, Xu D, Petralia RS, Steward O, Huganir RL, Worley P, Synaptic clustering of AMPA receptors by the extracellular immediate-early gene product Narp. Neuron 23, 309–323 (1999). [DOI] [PubMed] [Google Scholar]
- 23.Sia G-M, Béïque J-C, Rumbaugh G, Cho R, Worley PF, Huganir RL, Interaction of the N-Terminal domain of the AMPA receptor GluR4 subunit with the neuronal pentraxin NP1 mediates GluR4 synaptic recruitment. Neuron 55, 87–102 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Xiao M-F, Roh S-E, Zhou J, Chien C-C, Lucey BP, Craig MT, Hayes LN, Coughlin JM, Leweke FM, Jia M, Xu D,Zhou W, Talbot CC Jr., Arnold DB, Staley M, Jiang C, Reti IM, Sawa A, Pelkey KA, McBain CJ, Savonenko A, Worley PF, A biomarker-authenticated model of schizophrenia implicating NPTX2 loss of function. Sci. Adv 7, eabf6935 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao M-F, Xu D, Craig MT, Pelkey KA, Chien C-C, Shi Y, Zhang J, Resnick S, Pletnikova O, Salmon D, Brewer J, Edland S, Wegiel J, Tycko B, Savonenko A, Reeves RH, Troncoso JC, McBain CJ, Galasko D, Worley PF, NPTX2 and cognitive dysfunction in Alzheimer’s disease. eLife 6, e23798 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gu Y, Huang S, Chang MC, Worley P, Kirkwood A, Quinlan EM, Obligatory role for the immediate early gene NARP in critical period plasticity. Neuron 79, 335–346 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Severin D, Hong SZ, Roh S-E, Huang S, Zhou J, Bridi MCD, Hong I, Murase S, Robertson S, Haberman RP, Huganir RL, Gallagher M, Quinlan EM, Worley P, Kirkwood A, All-or-none disconnection of pyramidal inputs onto parvalbumin-positive interneurons gates ocular dominance plasticity. Proc. Nat. Acad. Sci. U.S.A 118, e2105388118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van der Ende EL, Xiao M, Xu D, Poos JM, Panman JL, Jiskoot LC, Meeter LH, Dopper EG, Papma JM, Heller C, Convery R, Moore K, Bocchetta M, Neason M, Peakman G, Cash DM, Teunissen CE, Graff C, Synofzik M, Moreno F, Finger E, Sánchez-Valle R, Vandenberghe R, Jr RL, Masellis M, Tartaglia MC, Rowe JB, Butler CR, Ducharme S, Gerhard A, Danek A, Levin J, Pijnenburg YA, Otto M, Borroni B, Tagliavini F, de Mendonca A, Santana I, Galimberti D, Seelaar H, Rohrer JD, Worley PF, van Swieten JC; Genetic Frontotemporal Dementia Initiative (GENFI), Neuronal pentraxin 2: A synapse-derived CSF biomarker in genetic frontotemporal dementia. J. Neuro. Neurosurg. Psychiatry 91, 612–621 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Galasko D, Xiao M, Xu D, Smirnov D, Salmon DP, Dewit N, Vanbrabant J, Jacobs D, Vanderstichele H, Vanmechelen E; Alzheimer’s Disease Neuroimaging Initiative (ADNI), P. Worley, Synaptic biomarkers in CSF aid in diagnosis, correlate with cognition and predict progression in MCI and Alzheimer’s disease. Alzheimer’s Dementia Transl. Res. Clin. Interventions 5, 871–882 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belbin O, Xiao M-F, Xu D, Carmona-Iragui M, Pegueroles J, Benejam B, Videla L, Fernández S, Barroeta I, Nuñez-Llaves R, Montal V, Vilaplana E, Altuna M, Clarimón J, Alcolea D, Blesa R, Lleó A, Worley PF, Fortea J, Cerebrospinal fluid profile of NPTX2 supports role of Alzheimer’s disease-related inhibitory circuit dysfunction in adults with Down syndrome. Mol. Neurodegener 15, 46 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Steenoven I, Koel-Simmelink MJA, Vergouw LJM, Tijms BM, Piersma SR, Pham TV, Bridel C, Ferri G-L, Cocco C, Noli B, Worley PF, Xiao M-F, Xu D, Oeckl P, Otto M, van der Flier WM, de Jong FJ, Jimenez CR, Lemstra AW, Teunissen CE, Identification of novel cerebrospinal fluid biomarker candidates for dementia with Lewy bodies: A proteomic approach. Mol. Neurodegener 15, 36 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soldan A, Moghekar A, Walker KA, Pettigrew C, Hou X, Lu H, Miller MI, Alfini A, Albert M, Xu D, Xiao M-F, Worley P, Csernansky J, Holtzman D, Knopman D, Kukull W, Grimm K, Hsiao J, Ryan L, Lyketsos C, Pardo C, Schellenberg G, Shaw L, Thambisetty M, Trojanowski J, Resting-state functional connectivity is associated with cerebrospinal fluid levels of the synaptic protein NPTX2 in non-demented older adults. Front. Aging Neurosci 11, 132 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Styr B, Slutsky I, Imbalance between firing homeostasis and synaptic plasticity drives early-phase Alzheimer’s disease. Nat. Neurosci 21, 463–473 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Inforzato A, Peri G, Doni A, Garlanda C, Mantovani A, Bastone A, Carpentieri A, Amoresano A, Pucci P, Roos A, Daha MR, Vincenti S, Gallo G, Carminati P, Santis RD, Salvatori G, Structure and function of the long pentraxin PTX3 glycosidic moiety: Finetuning of the interaction with C1q and complement activation. Biochemistry 45, 11540–11551 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Favuzzi E, Huang S, Saldi GA, Binan L, Ibrahim LA, Fernández-Otero M, Cao Y, Zeine A, Sefah A, Zheng K, Xu Q, Khlestova E, Farhi SL, Bonneau R, Datta SR, Stevens B, Fishell G, GABA-receptive microglia selectively sculpt developing inhibitory circuits. Cell 184, 5686 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lui H, Zhang J, Makinson SR, Cahill MK, Kelley KW, Huang H-Y, Shang Y, Oldham MC, Martens LH, Gao F, Coppola G, Sloan SA, Hsieh CL, Kim CC, Bigio EH, Weintraub S, Mesulam M-M, Rademakers R, Mackenzie IR, Seeley WW, Karydas A, Miller BL, Borroni B, Ghidoni R, Farese RV, Paz JT, Barres BA, Huang EJ, Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation. Cell 165, 921–935 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pulido-Salgado M, Vidal-Taboada JM, Barriga GG-D, Solà C, Saura J, RNA-Seq transcriptomic profiling of primary murine microglia treated with LPS or LPS + IFNγ. Sci. Rep 8, 16096 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ndoja A, Reja R, Lee S-H, Webster JD, Ngu H, Rose CM, Kirkpatrick DS, Modrusan Z, Chen Y-JJ, Dugger DL, Gandham V, Xie L, Newton K, Dixit VM, Ubiquitin ligase COP1 suppresses neuroinflammation by degrading c/EBPβ in microglia. Cell 182, 1156–1169.e12 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Baum ML, Wilton DK, Muthukumar A, Fox RG, Carey A, Crotty W, Scott-Hewitt N, Bien E, Sabatini DA, Lanser T, Frouin A, Gergits F, Håvik B, Gialeli C, Nacu E, Blom AM, Eggan K, Johnson MB, McCarroll SA, Stevens B, CUB and sushi multiple domains 1 (CSMD1) opposes the complement cascade in neural tissues. bioRxiv 291427. 12 September 2020. 10.1101/2020.09.11.291427. [DOI] [Google Scholar]
- 40.Vadászi H, Kiss B, Micsonai A, Schlosser G, Szaniszló T, Kovács RÁ, Györffy BA, Kékesi KA, Goto Y, Uzonyi B, Liliom K, Kardos J, Competitive inhibition of the classical complement pathway using exogenous single-chain C1q recognition proteins. J. Biol. Chem 298, 102113 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vukojicic A, Delestrée N, Fletcher EV, Pagiazitis JG, Sankaranarayanan S, Yednock TA, Barres BA, Mentis GZ, The classical complement pathway mediates microglia-dependent remodeling of spinal motor circuits during development and in SMA. Cell Rep. 29, 3087–3100.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lansita JA, Mease KM, Qiu H, Yednock T, Sankaranarayanan S, Kramer S, Nonclinical development of ANX005: A humanized anti-C1q antibody for treatment of autoimmune and neurodegenerative diseases. Int. J. Toxicol 36, 449–462 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Stephan AH, Madison DV, Mateos JM, Fraser DA, Lovelett EA, Coutellier L, Kim L, Tsai H-H, Huang EJ, Rowitch DH, Berns DS, Tenner AJ, Shamloo M, Barres BA, A dramatic increase of C1q protein in the CNS during normal aging. J. Neurosci 33, 13460–13474 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kamitaki N, Sekar A, Handsaker RE, de Rivera H, Tooley K, Morris DL, Taylor KE, Whelan CW, Tombleson P, Loohuis LMO; Schizophrenia Working Group of the Psychiatric Genomics Consortium, Kimberly RP, Kaufman KM, Harley JB, Langefeld CD, Seidman CE, Pato MT, Pato CN, Ophoff RA, Graham RR, Criswell LA, Vyse TJ, McCarroll SA, Complement genes contribute sex-biased vulnerability in diverse disorders. Nature 582, 577–581 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, Tooley K, Presumey J, Baum M, Doren VV, Genovese G, Rose SA, Handsaker RE, Daly MJ, Carroll MC, Stevens B, McCarroll SA; Schizophrenia Working Group of the Psychiatric Genomics Consortium, Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yilmaz M, Yalcin E, Presumey J, Aw E, Ma M, Whelan CW, Stevens B, McCarroll SA, Carroll MC, Overexpression of schizophrenia susceptibility factor human complement C4A promotes excessive synaptic loss and behavioral changes in mice. Nat. Neurosci 24, 214–224 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shi Q, Chowdhury S, Ma R, Le KX, Hong S, Caldarone BJ, Stevens B, Lemere CA, Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med 9, eaaf6295 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hernandez MX, Jiang S, Cole TA, Chu S-H, Fonseca MI, Fang MJ, Hohsfield LA, Torres MD, Green KN, Wetsel RA, Mortazavi A, Tenner AJ, Prevention of C5aR1 signaling delays microglial inflammatory polarization, favors clearance pathways and suppresses cognitive loss. Mol. Neurodegener 12, 66 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carpanini SM, Torvell M, Bevan RJ, Byrne RAJ, Daskoulidou N, Saito T, Saido TC, Taylor PR, Hughes TR, Zelek WM, Morgan BP, Terminal complement pathway activation drives synaptic loss in Alzheimer’s disease models. Acta Neuropathologica Commun. 10, 99 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van der Ende EL, Heller C, Sogorb-Esteve A, Swift IJ, McFall D, Peakman G, Bouzigues A, Poos JM, Jiskoot LC, Panman JL, Papma JM, Meeter LH, Dopper EGP, Bocchetta M, Todd E, Cash D, Graff C, Synofzik M, Moreno F, Finger E, Sanchez-Valle R, Vandenberghe R, Laforce R Jr, Masellis M, Tartaglia MC, Rowe JB, Butler C, Ducharme S, Gerhard A, Danek A, Levin J, Pijnenburg YAL, Otto M, Borroni B, Tagliavini F, de Mendonça A, Santana I, Galimberti D, Sorbi S, Zetterberg H, Huang E, van Swieten JC, Rohrer JD, Seelaar H; Genetic Frontotemporal Dementia Initiative (GENFI), Elevated CSF and plasma complement proteins in genetic frontotemporal dementia: Results from the GENFI study. J. Neuroinflamm 19, 217 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van der Ende EL, Bron EE, Poos JM, Jiskoot LC, Panman JL, Papma JM, Meeter LH, Dopper EGP, Wilke C, Synofzik M, Heller C, Swift IJ, Sogorb-Esteve A, Bouzigues A, Borroni B, Sanchez-Valle R, Moreno F, Graff C, Laforce R, Galimberti D, Masellis M, Tartaglia MC, Finger E, Vandenberghe R, Rowe JB, de Mendonça A, Tagliavini F, Santana I, Ducharme S, Butler CR, Gerhard A, Levin J, Danek A, Otto M, Pijnenburg YAL, Sorbi S, Zetterberg H, Niessen WJ, Rohrer JD, Klein S, van Swieten JC, Venkatraghavan V, Seelaar H; GENFI consortium A data-driven disease progression model of fluid biomarkers in genetic frontotemporal dementia. Brain 145, 1805–1817 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. All biological resources, antibodies, and model organisms and tools are either available through commercial sources or may be requested from P.F.W. and can be shared with the scientific community through a completed materials transfer agreement (MTA). The stable CHO cell line may be requested from Genentech by completing an MTA (www.gene.com/scientists/mta).