

Abstract
Fusions involving CRAF (RAF1) are infrequent oncogenic drivers in pediatric low‐grade gliomas, rarely identified in tumors bearing features of pilocytic astrocytoma, and involving a limited number of known fusion partners. We describe recurrent TRAK1::RAF1 fusions, previously unreported in brain tumors, in three pediatric patients with low‐grade glial‐glioneuronal tumors. We present the associated clinical, histopathologic and molecular features. Patients were all female, aged 8 years, 15 months, and 10 months at diagnosis. All tumors were located in the cerebral hemispheres and predominantly cortical, with leptomeningeal involvement in 2/3 patients. Similar to previously described activating RAF1 fusions, the breakpoints in RAF1 all occurred 5′ of the kinase domain, while the breakpoints in the 3′ partner preserved the N‐terminal kinesin‐interacting domain and coiled‐coil motifs of TRAK1. Two of the three cases demonstrated methylation profiles (v12.5) compatible with desmoplastic infantile ganglioglioma (DIG)/desmoplastic infantile astrocytoma (DIA) and have remained clinically stable and without disease progression/recurrence after resection. The remaining tumor was non‐classifiable; with focal recurrence 14 months after initial resection; the patient remains symptom free and without further recurrence/progression (5 months post re‐resection and 19 months from initial diagnosis). Our report expands the landscape of oncogenic RAF1 fusions in pediatric gliomas, which will help to further refine tumor classification and guide management of patients with these alterations.
Keywords: brain pathology, CRAF, glioneuronal tumors, low‐grade gliomas, RAF1
We describe recurrent TRAK1::RAF1 fusions, previously unreported in brain tumors, in three pediatric patients with low‐grade glial‐glioneuronal tumors. We present the associated clinical, histopathologic and molecular features.
1. INTRODUCTION
The 2021 Classification of Central Nervous System Tumors has taken a new approach to the classification gliomas, glioneuronal and neuronal tumors, driven in large part by our increasing understanding of differences in the genetic features and largely indolent nature of low‐grade glial and glioneuronal tumors that present in childhood as compared to their adult counterparts. Although a number of recurrent RAS–mitogen‐activated protein kinase (RAS/MAPK) pathway alterations have been implicated as a hallmark of pediatric low‐grade gliomas, they remain challenging to diagnose and manage. Due to frequent overlap of histologic features and specific genetic alterations among pediatric low‐grade glial and glioneuronal tumors, careful morphologic assessment and comprehensive molecular characterization may not always result in a definitive diagnosis [1, 2]. This is particularly true in cases with uncommon genetic features.
Fusions involving RAF1 are infrequent drivers in pediatric low‐grade gliomas, rarely identified in tumors bearing features of pilocytic astrocytoma, and involving a number of fusion partners, and involving a number of fusion partners, namely QKI, FYCO, TRIM33, SRGAP3, NF1A, and ATG7 [3, 4, 5, 6, 7, 8, 9, 10, 11]. RAF1 has been implicated in cell proliferation and survival. Yet while RAF1 fusions have generally been shown to activate the RAS/MAPK pathway, their clinical implication in the context of pediatric brain tumors remains unclear [7, 8, 12].
Here we describe recurrent TRAK1::RAF1 fusions, previously unreported in brain tumors, in three pediatric patients with low‐grade glial‐glioneuronal tumors. Patients were all female, aged 8 years, 15 months, and 10 months at diagnosis. All tumors were supratentorial and peripherally located with leptomeningeal involvement in 2/3 cases. We present the associated clinical and histopathologic features and results of DNA methylation profiling.
2. MATERIALS AND METHODS
This retrospective study was conducted with the approval of respective Institutional Review Boards.
2.1. Histopathology
Case materials were collected by the authors in the course of their hospital and consultative practices. Formalin‐fixed and paraffin‐embedded (FFPE) tissues were employed for all immunohistochemical and molecular diagnostic assays. The streptavidin–biotin peroxidase complex method was utilized for immunohistochemical studies as described, with antibodies as follows: Ki‐67 (MIB‐1; Ventana, monoclonal), CD34 (Ventana, monoclonal), synaptophysin (SYN; Biogenex, monoclonal), neuronal nuclear protein (NeuN; Chemicon, monoclonal), glial fibrillary acidic protein (GFAP; Cell Marque, monoclonal), oligodendrocyte Transcription Factor 2 (OLIG2; Abcam, monoclonal).
2.2. Mutational profiling
All cases underwent clinical targeted sequencing analysis using the validated platform available at the treating institution(s) at the time of diagnosis. Testing panels covered mutational hotspots in 3 genes (Case 1), 50 genes (Case 2), and 505 genes (Case 3) respectively, as previously described [13, 14, 15]. Complete gene lists are included in Supplemental 1.
2.3. Fusion transcript detection
Anchored Multiplex PCR (AMP™) technology for detection of fusion transcripts (Archer FusionPlex™) was performed on total RNA extracted from formalin‐fixed, paraffin‐embedded (FFPE) material as previously described [16]. Briefly, unidirectional gene‐specific primers (GSPs) were used to target a custom panel of several exons in 123 cancer‐related genes frequently involved in chromosomal rearrangements. GSPs in combination with adapter‐specific primers amplify known and novel fusion transcripts. Enriched amplicons are sequenced on an Illumina MiSeq instrument.
2.4. Genome‐wide DNA methylation profiling
Cases were analyzed with the Infinium MethylationEPIC (850 K) platform, which interrogates >850,000 CpG methylation sites across the genome, as previously described [17]. Briefly, 250 ng of input genomic DNA, extracted from FFPE tissue (Chemagic DNA Tissue kit, PerkinElmer chemagen Technologie, GmbH, Baesweiler, Germany), was used for bisulfite conversion (EZ DNA Methylation Kit; Zymo Research; catalog number D5002), followed by FFPE restoration (Infinium HD FFPE DNA Restore Kit; Illumina; catalog number WG‐321‐1002). Samples were processed and scanned according to manufacturer's recommended protocol using the Infinium 850 k array and Illumina iScan. Cases were assigned methylation‐based classes using version 0.1.124 of the random forest‐based mnp.v11b4 R package obtained from the German Cancer Research Center (DKFZ) [18]. Dimensionality reduction with principal component analysis and t‐distributed stochastic neighbor embedding was performed and overlaid on a reference cohort composed of select tumors of known methylation classes from the previously published DKFZ study using version 0.15 of the Rtsne package with the following non‐default parameters: initial_dims = 100, max_iter = 1500, and theta = 0. Cross‐reactive, sex‐chromosome, and failed probes were excluded from these analyses. Analyses were performed using the mnp.v11b6 package in R version 3.6.1; as previously described [15].
3. RESULTS
Key features of all three cases are summarized in Table 1.
TABLE 1.
Summary of histopathologic and clinical features of TRAK1::RAF1 fusion positive pediatric low grade glial‐glioneuronal tumors
| Age at Diagnosis | Sex | Presenting symptoms | Tumor Location | Surgery* | Tumor Ki‐67 proliferative index | Tumor Mitoses | Histopathologic Diagnosis* | v12.5 Methylation Class (Score) | Fusion | Additional tumor cytogenetic and molecular features | Recurrence/progression | Follow‐up (months) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Case 1 | 8 years | Female | Seizure | Left frontal cortex | GTR | elevated (5‐10%) | Rare | LGG/LGNT, NOS | Pilocytic astrocytoma, hemispheric ( 0.6) | TRAK1::RAF1 | broad loss of Chromosome 3p | focal recurrent tumor noted on 14 month surveillance MRI; treated by complete re‐resection (NED at 5 month follow‐up) | 19 |
| Case 2 | 9 months | Female | Decreased motor activity (arm/leg) | Left fronto‐parietal leptomeninges | GTR | low (< 5%) | Absent | DIG | Desmoplastic infantile ganglioglioma / desmoplastic infantile astrocytoma (1.0 ) | TRAK1::RAF1 | (none detected; germline Chr 15p deletion) | none | 53 |
| Case 3 | 15 months | Female | Seizure | Right parietal/occipital leptomeninges | STR | elevated (>10%) | Rare | DLGNT | Desmoplastic infantile ganglioglioma / desmoplastic infantile astrocytoma (1.0 ) | TRAK1::RAF1 | (none detected) | none | 15 |
*Abbreviations: DLGNT = diffuse leptomeningeal glioneuronal tumor; GTR = gross total resection; LGG‐LGNT, NOS = low grade glial‐glioneuronal tumor, not other specified; NED = no evidence of disease; STR+ subtotal resection.
3.1. Clinical histories
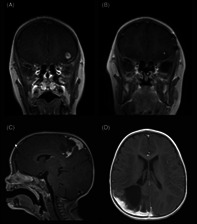
Case 1: An 8‐year‐old otherwise healthy female presented with new onset seizures. Magnetic resonance imaging (MRI) studies demonstrated a ~1 cm well‐circumscribed ovoid T2‐hypointense, T1‐slightly hyperintense lesion with heterogenous rim‐like enhancement. The lesion was superficially located in the cortex/subcortical white matter of the left inferolateral frontal lobe, with a small amount of adjacent signal hyperintensity, compatible with edema (Figure 1A). Findings remained stable after 4‐week interval MRI. The patient underwent craniotomy and gross total resection and has remained seizure‐free. Surveillance MRI 14 months post resection demonstrated a new area of nodular enhancement measuring up to 5 mm in association with the prior resection bed, suspicious for tumor recurrence (Figure 1B). The patient underwent re‐resection and remains without further tumor recurrence/progression (5 months post re‐resection and 19 months from the original resection).
FIGURE 1.
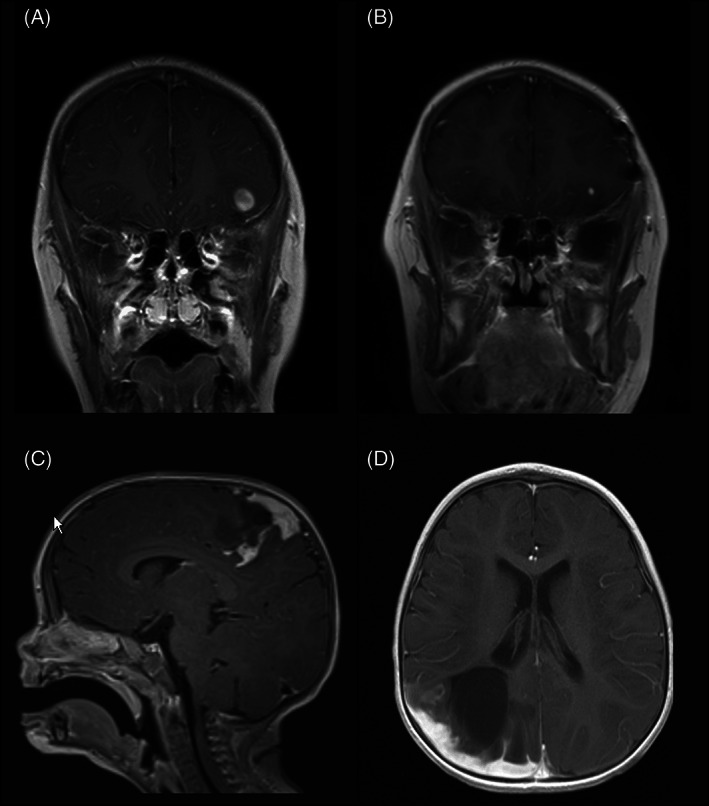
MRI findings in TRAK1::RAF1 fusion positive gliomas: T1‐weighted, post‐contrast MRI demonstrates a hypointense, well‐circumscribed 1 cm ovoid lesion with heterogenous rim‐like enhancement, cortically‐based in the left inferotemporal lobe in Case 1 (A); after gross total resection, an area of focal nodular enhancement subjacent to the surgical site was noted on 14‐month surveillance scan (B). Tumors in Case 2 (C) and Case 3 (D) involved both brain and leptomeninges. T1‐weighted, post‐contrast MRI in Case 2 demonstrated a 3 cm solid and cystic enhancing mass with significant surrounding edema (C), while T1‐weighted, post‐contrast MRI in Case 3 demonstrated an infiltrative lesion in the R parieto‐occipital lobe with thick nodular leptomeningeal enhancement and ~4 cm non‐enhancing cystic‐solid component (D).
Case 2: A 9‐month‐old female presented with decreased right arm and right leg movement. Past medical history was notable for birth at 39 weeks requiring NICU admission caused by omphalocele (status post repair) and known constitutional deletion of chromosome 15q11.2, consistent with Prader Willi/Angelman syndrome. Head CT revealed a brain mass and subsequent MRI confirmed a ~3 cm solid and cystic left fronto‐parietal lobe mass, abutting/involving the leptomeninges, with significant surrounding edema (Figure 1C). The patient underwent a craniotomy and gross total resection, without further tumor recurrence or progression of disease after several years of follow‐up (~53 months).
Case 3: A 15‐month‐old previously healthy female presented with seizures. Head CT demonstrated 2.8 cm right parietal hypodensity with associated edema. MRI studies confirmed an infiltrative lesion in the R parietal/occipital lobe with 1 cm thickness nodular leptomeningeal enhancement and associated non‐enhancing cystic‐solid lesion spanning up to 4 cm (Figure 1D). MRI spine was negative. A subtotal resection was performed. Postoperatively, the patient recovered fully without neurologic impairment or additional seizures, and no further clinical or MRI progression (follow up ~15 months).
3.2. Light microscopic features
Case 1
The tumor in both the original and re‐resection samples consisted of a cortical‐based (left inferotemporal lobe) glial proliferation, comprised predominately of astrocytes with prominent eosinophilic cytoplasm and ovoid nuclei, with occasional perinuclear halos (Figure 2A). While generally well‐circumscribed, a slightly infiltrative border was noted (Figure 2B). Additional features included scattered eosinophilic granular bodies and patchy, mild perivascular lymphocytic infiltrate. Ki‐67 proliferative index was focally moderately elevated (up to 5%–10%) and a rare mitotic figure was present (Figure 2G). No increased mitotic/proliferative activity or other high‐grade features were present in the re‐resection sample. Immunohistochemical stain for GFAP highlighted most tumor cells, with patchy‐scattered OLIG2 staining, while NeuN and synaptophysin staining were negative. CD34 demonstrated focal aberrant (ramified astrocytic) expression. A desmoplastic component was not present, as supported by trichrome and reticulin stains. The findings were felt to be non‐specific, but most compatible with a low‐grade glial‐glioneuronal tumor.
Case 2
The tumor was predominately based in the leptomeninges with focal infiltration into the superficial cortex, creating a biphasic appearance. Tumor cells consisted of intermixed nests of gemistocytic astrocytes displaying plump, eosinophilic cytoplasm, and occasional ganglion‐like morphology (Figure 2C). A second population of more primitive‐appearing neurocyte‐like cells was present, particularly at the interface of tumor an adjacent brain parenchyma. The tumor was predominately composed of a desmoplastic, dense collagenous matrix with storiform spindled growth pattern (Figure 2D). Rare mitoses were present. Only the astrocytic component expressed GFAP, with scattered clusters of OLIG2‐positive cells, while NeuN and synaptophysin showed scattered positive expression in ganglionic and primitive cells. CD34 was negative and Ki‐67 proliferative index was low (<1%). The histologic findings were most compatible with desmoplastic infantile ganglioglioma, WHO grade 1.
Case 3
The tumor was located in the brain and leptomeninges with desmoplastic infiltration of the overlying dura (Figure 2E). Infiltrating tumor cells demonstrated bland rounded nuclei, with frequent perinuclear halos. Focally, nests of primitive‐appearing neurocyte‐like cells are noted (Figure 2F). GFAP staining was weak to negative, with scattered OLIG2‐positive cells, and patchy expression of synaptophysin. Rare mitoses were present and Ki‐67 proliferative index was focally elevated (>10%, Figure 2E). The findings were most compatible with low‐grade glioneuronal tumor with features of diffuse leptomeningeal glioneuronal tumor (DLGNT) without dissemination.
FIGURE 2.
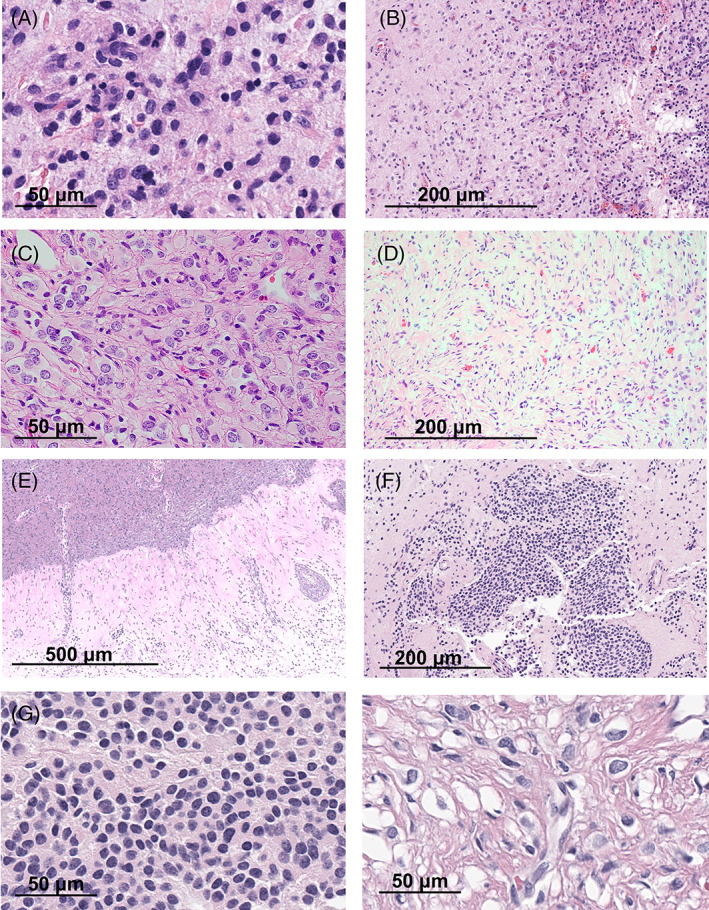
Histologic features of TRAK1::RAF1 fusion positive gliomas: The tumor in Case 1 demonstrated features compatible with a low grade glial‐glioneuronal tumor, comprised of bland astrocytic cells with ovoid nuclei and occasional perinuclear halos (A) . While generally well‐circumscribed, a slightly infiltrative border was noted (B). The tumors in Cases 2 and 3 demonstrated features compatible with diffuse leptomeningeal glioneuronal tumor, involving both leptomeninges and superficial cortex. In Case 2, tumor cells consisted of intermixed nests of gemistoytic astrocytes displaying plump, eosinophilic cytoplasm, and occasional ganglion‐like morphology (C). Tumors in Cases 2 and 3 demonstrated a desmoplastic, dense collagenous matrix with storiform spindled growth pattern (D [Case 2]; E [Case 3]), and in both cases a second population of more primitive‐appearing neurocyte‐like cells was present (F). Ki‐67 proliferative index was moderately elevated in Cases 1 (G) and 3 (H).
3.3. TRAK1 :: RAF1 fusions retain RAF1 kinase domain and TRAK1 coiled‐coiled motifs
RNA‐seq‐based methodology (ArcherDx) to identify fusion transcripts demonstrated an intrachromosomal rearrangement on chromosome 3p involving TRAK1 (3p22.1) and RAF1 (3p25.2) in samples from all three cases (Figure 3). Similar to previously described activating RAF1 fusions, the breakpoints in RAF1 (NM_002880) all occurred 5′ of the kinase domain (exons 7, 10 and 8 in Cases 1, 2, and 3, respectively). The breakpoint in exon 5 of the 3′ partner gene TRAK1 (NM_001042646), seen in Case 3 was previously reported in melanoma [19]. Fusion partner genes identified in RAF1 fusions across a number of cancer types frequently contain a coiled–coil dimerization motif leading to RAF1 activation [20]. TRAK1, a kinesin adaptor protein important in intracellular trafficking and transport, contains an N‐terminal kinesin‐interacting conserved region which contains three predicted coiled‐coil domains, reported to facilitate homodimerization [21, 22, 23, 24, 25]. These regions are also preserved in TRAK1 breakpoints demonstrated in Cases 1 and 2 (exon 15 [NM_001042646] and exon 8 [NM_014965.4]).
FIGURE 3.
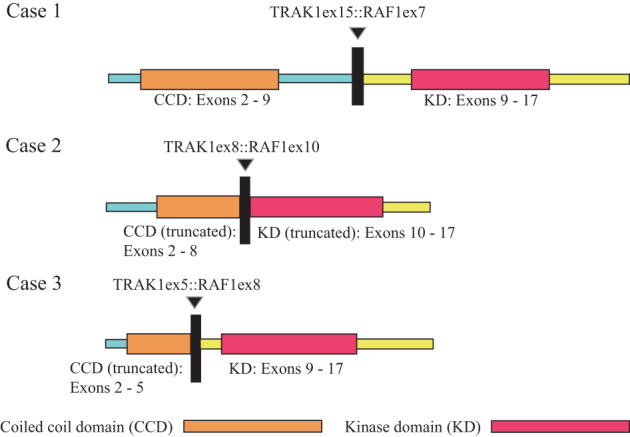
Features of recurrent TRAK1::RAF1 fusions in pediatric low grade glial‐glioneuronal tumors: Schematics showing the intrachromosomal rearrangement event on chromosome 3p leading to fusions TRAK1 (3p22.1) and RAF1 (3p25.2) in all 3 cases. Breakpoints in RAF1 (NM_002880) all occurred 5′ of the kinase domain (exons 7, 10 and 8 in Cases 1, 2, and 3 respectively). TRAK1 contains an N‐terminal kinesin‐interacting conserved region with three coiled‐coil domains, expected to facilitate homodimerization, and preserved by the TRAK1 breakpoints identified here (exon 15 [NM_001042646], exon 8 [NM_014965.4], and exon 5[NM_001042646]).
3.4. TRAK1 :: RAF1 fusion bearing tumors do not all segregate with a unifying pediatric low‐grade neuroepithelial tumor methylation class
Using the DNA methylation–based CNS tumor classification version available at the time of the original clinical assessment (v11b4), none of the tumors met the generally accepted calibrated score threshold for valid classification (0.84–0.9). Case 1 demonstrated the highest probability result (class score: 0.6805) for methylation class “low grade glioma, subclass hemispheric pilocytic astrocytoma and ganglioglioma (LGG, PA/GG ST).” For both Cases 2 and 3, the highest methylation class scores were for “low grade glioma, desmoplastic infantile astrocytoma/ganglioglioma (LGG, DIG/DIA)” (0.6889 and 0.2696, respectively).
As the Heidelberg classifier undergoes continuous modification with the refinement of classes and addition of newly recognized tumor entities, the samples were reanalyzed using the most recently updated version available at the time of this report (v12.5). While a similar result was obtained for Case 1 (highest probability match to methylation class “pilocytic astrocytoma, hemispheric” with sub‐threshold calibrated score of 0.60205), Cases 2 and 3 both demonstrated high probability matches to methylation class “Desmoplastic infantile ganglioglioma/desmoplastic infantile astrocytoma” with calibrated scores well exceeding threshold for valid classification (0.99999 and 0.90719, respectively). A focused t‐SNE analysis with select reference comparison groups was conducted, results are demonstrated in Figure 4.
FIGURE 4.
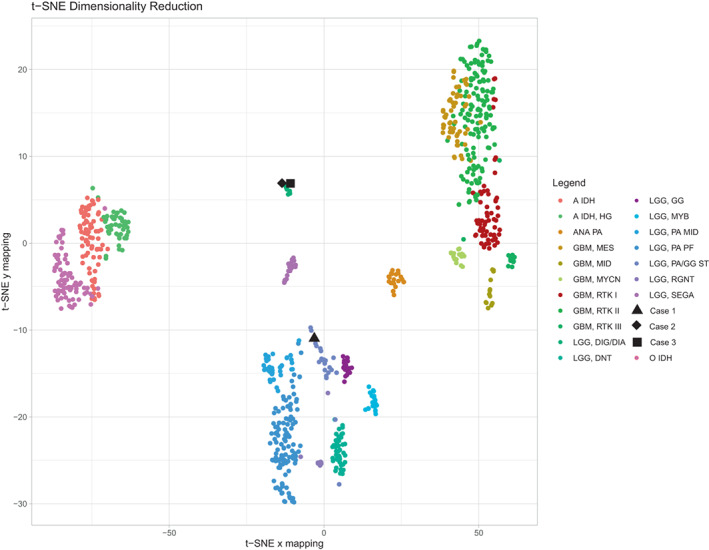
DNA methylation‐based classification of TRAK1::RAF1 fusion positive pediatric low grade glial‐glioneuronal tumors: T‐SNE dimensionality reduction was performed on the tumors from Case 1 (▲), Case 2 (♦), Case 3 (■), and a subset of CNS tumors from the DKFZ reference set (color coded according to methylation cluster assignation) with the Rtsne package version 0.15. Analyses were performed using the mnp.v11b6 package in R version 3.6.1; as previously described (PMID: 29539639).
3.5. Additional molecular and cytogenetic features of TRAK1 :: RAF1 Fusion bearing tumors
Analysis of copy number profiles derived from DNA methylation data from all three tumors demonstrated relatively few alterations (Figure S1). Case 1 showed only a broad low‐level loss on chromosome 3p. Both Cases 2 and 3 showed relatively flat copy number profiles (excluding the known constitutional 15p deletion in Case 2). While sequencing results are limited by variation among testing platforms, no additional tumor variants were detected.
4. DISCUSSION
We describe recurrent TRAK1::RAF1 fusions, previously unreported in brain tumors, in three glial/glioneuronal tumors occurring in pediatric female patients. Two of the three cases demonstrated methylation profiles (v12.5), compatible with desmoplastic infantile ganglioglioma/astrocytoma (DIG/DIA), while one case (Case 1), remains difficult to classify. In keeping with other MAPK‐driven low grade glial/glioneuronal tumors, a range of histologic features was present. The patients in Cases 2 and 3 have remained clinically stable and without disease progression/recurrence after resection with no adjuvant therapy. In cases of DIG/DIA, no relapses have been reported in cases treated with by complete resection, even after extensive follow‐up [26, 27, 28].
While Cases 2 and 3 suggest that TRAK1::RAF1 fusions may be enriched among tumors classifiable as DIG/DIA, the tumor described in Case 1 underscores that specific genetic alterations, including TRAK1::RAF1 fusions, may represent a shared molecular feature across several, possibly related, tumor types or subtypes. Our findings underscore the evolving robustness of methylome profiling, particularly with regard to the classification of low‐grade glial and glioneuronal tumors. RAF1 fusions with a number of 3′ fusion partner genes have been previously identified in pediatric low‐grade glial and glioneuronal tumors, including those diagnosed as pilocytic astrocytoma, DIG/DIA, and DLGNT [3, 6, 29, 30]. The potential for morphologic overlap among pediatric low grade glial and glioneuronal tumor entities is well‐recognized [2, 31]. At initial diagnosis, the differential diagnoses for each of these tumors was broad. The histologic features among many glial and glioneuronal tumors overlap, making them difficult to distinguish on morphologic grounds. For example, in Case 3, the extensive leptomeningeal component with oligodendrocyte‐like cells both in the intraparenchymal and leptomeningeal component and extensive desmoplastic changes, initially favored a diagnosis of DLGNT. However, these features are also often characteristic of DIG/DIA. Both entities have also been described to evidence a variable astrocytic or ganglionic component, and foci of primitive neuronal/embryonal cells. Ahead of clinical validation of the v12.5 classifier, the histologic and molecular data suggest that these tumors are best regarded as low grade glial/glioneuronal tumors, RAF1‐fusion positive. However, future studies benefitting from increased sample diversity and molecular characterization with multiple methodologies are required to further refine the precise classification of TRAK1::RAF1 fusion‐ bearing brain tumors, and further studies with long‐term follow‐up are required to determine the prognostic implications of TRAK1::RAF1 fusions in the context of other histologic, molecular and clinical data.
RAF1 fusions have also been previously identified in numerous other solid tumors [9, 30]. Specifically, activating RAF1 fusions characterize a subset of melanoma, including fusions of TRAK1::RAF1 [32, 33, 34, 35], where they have been demonstrated to show common breakpoints 5′ of the region encoding the kinase domain and result in enhanced MAPK signaling [34]. These TRAK1::RAF1 fusions of melanoma share structural similarity with those reported here, for the first time in brain tumors.
While the full mutational profile of all tumors was not able to be interrogated with whole exome/genome sequencing, targeted sequencing results from Case 3 suggest that, as commonly seen in other MAPK‐altered low‐grade tumors, the TRAK1::RAF1 fusion represents the sole detectable alteration [3, 30, 36]. As with all previously described RAF1 fusions in pediatric CNS tumors, the cases described here share preserved coiled‐coil dimerization domains of TRK1 (as the N‐terminal partner), suggesting that dimerization of the resultant fusion protein is required for oncogenesis [9]. However, caused by the comparative rarity of RAF1 fusions, their contribution to tumor biology, and their potential as a therapeutic target remains an area of active investigation.
It has been reported that, like BRAF fusions, RAF1 fusions aberrantly activate the MAPK signaling pathways, but additionally activate phosphoinositide‐3 kinase/mammalian target of rapamycin (PI3K/mTOR). Unlike BRAF fusions, RAF1 fusions are unresponsive to first and second‐generation RAF inhibitors [9, 12, 37]. While no clinical studies are available examining RAF1 fusions as a therapeutic target, there are anecdotal reports of clinical benefit from trametinib, including in one pediatric patient with LGG [38, 39]. It has been suggested that a combination of RAF inhibition together with blockade of dimerization of N‐terminal fusion may be an effective therapeutic strategy [9]. As all patients in this study have been treated with resection alone by the time of this report, we are not able to comment on a role for additional therapy.
Future studies are reliant on the increased awareness of and the ability to detect the full spectrum of RAF1 fusions. Our report expands the landscape of oncogenic RAF1 fusions in pediatric gliomas. Increased recognition of RAF1 fusions in brain tumors will assist to further refine tumor classification and hopefully guide management of patients with tumors bearing these alterations.
AUTHOR CONTRIBUTIONS
Jamal K. Benhamida, Hannah J. Harmsen, Deqin Ma, Christopher M. William, Bryan K. Li, Michael C. Dewan, and Matthias A. Karajannis compiled the clinical records and imaging. Hannah J. Harmsen, Deqin Ma, Christopher M. William, Matija Snuderl, David Zagzag, Marc K. Rosenblum, and Tejus A. Bale performed histopathologic assessment. Jamal K. Benhamida, Liliana Villafania, Purvil Sukhadia, Kerry A. Mullaney, Efsevia Vakiani, Marc Ladanyi, and Tejus A. Bale conducted molecular studies. Jamal K. Benhamida and Tejus A. Bale drafted the manuscript. All authors reviewed the manuscript.
FUNDING INFORMATION
This work has been supported in part by the Marie‐Josée and Henry R. Kravis Center for Molecular Oncology and the National Cancer Institute Cancer Center Core Grant No. P30‐CA008748.
CONFLICT OF INTEREST STATEMENT
David Zagzag and Marc K. Rosenblum are members of the Editorial Board of Brain Pathology and were not involved in peer review of this submission.
Supporting information
Data S1. Supporting Information.
Figure S1: Copy number profiles derived from DNA methylation data from all three tumors demonstrated relatively few alterations. Case 1 showed only a broad low‐level loss on chromosome 3q. Both Cases 2 and 3 showed flat copy number profiles, excepting the known constitutional 15p deletion in Case 2.
{kind=link}
ACKNOWLEDGMENTS
We gratefully acknowledge the members of the Molecular Diagnostics Service in the Department of Pathology at MSK. We would also like to thank Ms. Shadia Carlo for administrative assistance in preparation of this manuscript.
Benhamida JK, Harmsen HJ, Ma D, William CM, Li BK, Villafania L, et al. Recurrent TRAK1::RAF1 Fusions in pediatric low‐grade gliomas. Brain Pathology. 2023;33(5):e13185. 10.1111/bpa.13185
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Ellison DW, Hawkins C, Jones DTW, Onar‐Thomas A, Pfister SM, Reifenberger G, et al. cIMPACT‐NOW update 4: diffuse gliomas characterized by MYB, MYBL1, or FGFR1 alterations or BRAF(V600E) mutation. Acta Neuropathol. 2019;137(4):683–7. [DOI] [PubMed] [Google Scholar]
- 2. Ryall S, Tabori U, Hawkins C. Pediatric low‐grade glioma in the era of molecular diagnostics. Acta Neuropathol Commun. 2020;8(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B, et al. Whole‐genome sequencing identifies genetic alterations in pediatric low‐grade gliomas. Nat Genet. 2013;45(6):602–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114(2):97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lind KT, Chatwin HV, DeSisto J, Coleman P, Sanford B, Donson AM, et al. Novel RAF fusions in pediatric low‐grade gliomas demonstrate MAPK pathway activation. J Neuropathol Exp Neurol. 2021;80(12):1099–107. [DOI] [PubMed] [Google Scholar]
- 6. Deng MY, Sill M, Chiang J, Schittenhelm J, Ebinger M, Schuhmann MU, et al. Molecularly defined diffuse leptomeningeal glioneuronal tumor (DLGNT) comprises two subgroups with distinct clinical and genetic features. Acta Neuropathol. 2018;136(2):239–53. [DOI] [PubMed] [Google Scholar]
- 7. Jones DT, Ichimura K, Liu L, Pearson DM, Plant K, Collins VP. Genomic analysis of pilocytic astrocytomas at 0.97 Mb resolution shows an increasing tendency toward chromosomal copy number change with age. J Neuropathol Exp Neurol. 2006;65(11):1049–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jones DT, Kocialkowski S, Liu L, Pearson DM, Backlund LM, Ichimura K, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008;68(21):8673–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Roosen M, Ode Z, Bunt J, Kool M. The oncogenic fusion landscape in pediatric CNS neoplasms. Acta Neuropathol. 2022;143(4):427–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Johnson A, Severson E, Gay L, Vergilio JA, Elvin J, Suh J, et al. Comprehensive genomic profiling of 282 pediatric low‐ and high‐grade gliomas reveals genomic drivers, tumor mutational burden, and hypermutation signatures. Oncologist. 2017;22(12):1478–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jones DT, Gronych J, Lichter P, Witt O, Pfister SM. MAPK pathway activation in pilocytic astrocytoma. Cell Mol Life Sci. 2012;69(11):1799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jain P, Fierst TM, Han HJ, Smith TE, Vakil A, Storm PB, et al. CRAF gene fusions in pediatric low‐grade gliomas define a distinct drug response based on dimerization profiles. Oncogene. 2017;36(45):6348–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dias‐Santagata D, Akhavanfard S, David SS, Vernovsky K, Kuhlmann G, Boisvert SL, et al. Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO Mol Med. 2010;2(5):146–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sande CM, Chang B, Monga V, Bossler AD, Ma D. Biallelic TP53 gain of function mutations in rapidly progressing solid tumors. Cancer Genet. 2018;222‐223:20–4. [DOI] [PubMed] [Google Scholar]
- 15. Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering‐integrated mutation profiling of actionable cancer targets (MSK‐IMPACT): a hybridization capture‐based next‐generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17(3):251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zheng Z, Liebers M, Zhelyazkova B, Cao Y, Panditi D, Lynch KD, et al. Anchored multiplex PCR for targeted next‐generation sequencing. Nat Med. 2014;20(12):1479–84. [DOI] [PubMed] [Google Scholar]
- 17. Benhamida JK, Hechtman JF, Nafa K, Villafania L, Sadowska J, Wang J, et al. Reliable clinical MLH1 promoter Hypermethylation assessment using a high‐throughput genome‐wide methylation Array platform. J Mol Diagn. 2020;22(3):368–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, et al. DNA methylation‐based classification of central nervous system tumours. Nature. 2018;555(7697):469–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Donati M, Martinek P, Kastnerova L, Persichetti P, Michal M, Kazakov DV. RAF1 gene fusions as a possible driver mechanism in rare BAP1‐inactivated melanocytic tumors: a report of 2 cases. Am J Dermatopathol. 2020;42(12):961–6. [DOI] [PubMed] [Google Scholar]
- 20. Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun. 2014;5:4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Webber E, Li L, Chin LS. Hypertonia‐associated protein Trak1 is a novel regulator of endosome‐to‐lysosome trafficking. J Mol Biol. 2008;382(3):638–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pekkurnaz G, Trinidad JC, Wang X, Kong D, Schwarz TL. Glucose regulates mitochondrial motility via Milton modification by O‐GlcNAc transferase. Cell. 2014;158(1):54–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Glater EE, Megeath LJ, Stowers RS, Schwarz TL. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol. 2006;173(4):545–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stowers RS, Megeath LJ, Gorska‐Andrzejak J, Meinertzhagen IA, Schwarz TL. Axonal transport of mitochondria to synapses depends on milton, a novel drosophila protein. Neuron. 2002;36(6):1063–77. [DOI] [PubMed] [Google Scholar]
- 25. Lee CA, Chin LS, Li L. Hypertonia‐linked protein Trak1 functions with mitofusins to promote mitochondrial tethering and fusion. Protein Cell. 2018;9(8):693–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Taratuto AL, Monges J, Lylyk P, Leiguarda R. Superficial cerebral astrocytoma attached to dura. Report of six cases in infants. Cancer. 1984;54(11):2505–12. [DOI] [PubMed] [Google Scholar]
- 27. VandenBerg SR. Desmoplastic infantile ganglioglioma and desmoplastic cerebral astrocytoma of infancy. Brain Pathol. 1993;3(3):275–81. [DOI] [PubMed] [Google Scholar]
- 28. Bianchi F, Tamburrini G, Massimi L, Caldarelli M. Supratentorial tumors typical of the infantile age: desmoplastic infantile ganglioglioma (DIG) and astrocytoma (DIA). A Review Childs Nerv Syst. 2016;32(10):1833–8. [DOI] [PubMed] [Google Scholar]
- 29. Jones DT, Kocialkowski S, Liu L, Pearson DM, Ichimura K, Collins VP. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene. 2009;28(20):2119–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ryall S, Zapotocky M, Fukuoka K, Nobre L, Guerreiro Stucklin A, Bennett J, et al. Integrated molecular and clinical analysis of 1,000 pediatric low‐grade gliomas. Cancer Cell. 2020;37(4):569–83 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bale TA, Rosenblum MK. The 2021 WHO classification of tumors of the central nervous system: an update on pediatric low‐grade gliomas and glioneuronal tumors. Brain Pathol. 2022;32(4):e13060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yoshihara K, Wang Q, Torres‐Garcia W, Zheng S, Vegesna R, Kim H, et al. The landscape and therapeutic relevance of cancer‐associated transcript fusions. Oncogene. 2015;34(37):4845–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cancer Genome Atlas N . Genomic classification of cutaneous melanoma. Cell. 2015;161(7):1681–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li J, Lu H, Ng PK, Pantazi A, Ip CKM, Jeong KJ, et al. A functional genomic approach to actionable gene fusions for precision oncology. Sci Adv. 2022;8(6):eabm2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Williams EA, Shah N, Montesion M, Sharaf R, Pavlick DC, Sokol ES, et al. Melanomas with activating RAF1 fusions: clinical, histopathologic, and molecular profiles. Mod Pathol. 2020;33(8):1466–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Qaddoumi I, Orisme W, Wen J, Santiago T, Gupta K, Dalton JD, et al. Genetic alterations in uncommon low‐grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol. 2016;131(6):833–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sievert AJ, Lang SS, Boucher KL, Madsen PJ, Slaunwhite E, Choudhari N, et al. Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas. Proc Natl Acad Sci U S A. 2013;110(15):5957–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yde CW, Sehested A, Mateu‐Regue A, Ostrup O, Scheie D, Nysom K, et al. A new NFIA:RAF1 fusion activating the MAPK pathway in pilocytic astrocytoma. Cancer Genet. 2016;209(10):440–4. [DOI] [PubMed] [Google Scholar]
- 39. Rankin A, Johnson A, Roos A, Kannan G, Knipstein J, Britt N, et al. Targetable BRAF and RAF1 alterations in advanced pediatric cancers. Oncologist. 2021;26(1):e153–e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting Information.
Figure S1: Copy number profiles derived from DNA methylation data from all three tumors demonstrated relatively few alterations. Case 1 showed only a broad low‐level loss on chromosome 3q. Both Cases 2 and 3 showed flat copy number profiles, excepting the known constitutional 15p deletion in Case 2.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.