

Abstract
Alpha‐synuclein (αsyn) aggregates are pathological features of several neurodegenerative conditions including Parkinson disease (PD), dementia with Lewy bodies, and multiple system atrophy (MSA). Accumulating evidence suggests that mitochondrial dysfunction and impairments of the autophagic‐lysosomal system can contribute to the deposition of αsyn, which in turn may interfere with health and function of these organelles in a potentially vicious cycle. Here we investigated a potential convergence of αsyn with the PINK1‐PRKN‐mediated mitochondrial autophagy pathway in cell models, αsyn transgenic mice, and human autopsy brain. PINK1 and PRKN identify and selectively label damaged mitochondria with phosphorylated ubiquitin (pS65‐Ub) to mark them for degradation (mitophagy). We found that disease‐causing multiplications of αsyn resulted in accumulation of the ubiquitin ligase PRKN in cells. This effect could be normalized by starvation‐induced autophagy activation and by CRISPR/Cas9‐mediated αsyn knockout. Upon acute mitochondrial damage, the increased levels of PRKN protein contributed to an enhanced pS65‐Ub response. We further confirmed increased pS65‐Ub‐immunopositive signals in mouse brain with αsyn overexpression and in postmortem human disease brain. Of note, increased pS65‐Ub was associated with neuronal Lewy body‐type αsyn pathology, but not glial cytoplasmic inclusions of αsyn as seen in MSA. While our results add another layer of complexity to the crosstalk between αsyn and the PINK1‐PRKN pathway, distinct mechanisms may underlie in cells and brain tissue despite similar outcomes. Notwithstanding, our finding suggests that pS65‐Ub may be useful as a biomarker to discriminate different synucleinopathies and may serve as a potential therapeutic target for Lewy body disease.
Keywords: alpha‐synuclein, autophagy, Lewy body disease, mitochondria, mitophagy, multiple system atrophy, Parkinson disease, phosphorylated ubiquitin, PINK1, PRKN
The increase of the selective mitophagy marker pS65‐Ub in human autopsy brain is associated with neuronal Lewy body‐type, but not glial cytoplasmic inclusions of alpha‐synuclein pathology.
1. INTRODUCTION
Parkinson disease (PD) is an incurable neurodegenerative disorder which is characterized by slow progression of the cardinal motor symptoms and the occurrence of comorbid, nonmotor symptoms as the disease develops [1]. Aside from significant loss of dopaminergic neurons and projections, the main pathological hallmark of PD is the abnormal accumulation of alpha‐synuclein (αsyn) in the form of Lewy bodies (LBs) and Lewy neurites within the surviving neurons [2]. αsyn inclusions are also features of other neurodegenerative disorders, including, but not limited to, dementia with Lewy bodies and multiple system atrophy (MSA). While these diseases are collectively referred to as synucleinopathies, the affected brain regions and cell types as well as the distribution and morphology of αsyn aggregates vary considerably [3].
Mitochondrial dysfunction is believed to be a major contributor to PD and has also been linked to MSA as the resulting energy deficits, oxidative stress, and impaired signaling are all known to promote the aggregation of αsyn [4, 5]. Moreover, accumulation of αsyn is likely also a consequence of impaired autophagic‐lysosomal degradation [6, 7]. The main pathway for selective elimination of damaged mitochondria by autophagy (mitophagy) is orchestrated by the ubiquitin (Ub) kinase PINK1 and the Ub ligase PRKN [8]. Complete loss of either gene function causes early‐onset PD and it is thought that the concomitant buildup of damaged mitochondria eventually leads to cell death. PINK1 continuously surveils mitochondrial health and accumulates only on depolarized mitochondrial membranes where it phosphorylates serine‐65 of Ub (pS65‐Ub). Binding to pS65‐Ub recruits and activates the Ub ligase PRKN which provides additional substrates for PINK1 and thus amplifies the mitophagy signal [8]. Damaged mitochondria decorated with pS65‐Ub are then subjected to the autophagic‐lysosomal system for degradation.
A functional relationship between αsyn aggregation and mitophagy alterations is emerging, but greater insights into this likely complex crosstalk and the underlying mechanisms have remained elusive [9]. It has been shown that loss of either PINK1 or PRKN can aggravate αsyn‐induced phenotypes [10, 11, 12, 13], while overexpression of each enzyme can ameliorate those [14, 15, 16, 17, 18]. Furthermore, αsyn was shown to impact mitochondria, autophagy, and lysosomes and thus could alter the mitophagy process at different steps, but perhaps also in different directions [7, 9, 19, 20, 21, 22, 23, 24]. Cells and transgenic mice expressing A53T mutant αsyn showed marked accumulation of mitochondria‐containing autophagic vesicles [25, 26]. Certain αsyn species were shown to strongly bind to the mitochondrial import receptor TOMM20, which could lead to mitophagy defects [27]. Another conformationally distinct, nonfibrillar form of αsyn induced structural and functional mitochondrial damage leading to increased fission and enhanced mitophagy [28]. Altogether, this suggests that alterations of mitophagy may well play a key role in the αsyn‐related pathogenesis.
Since both lysosomal storage disorders and mitochondrial diseases appear disproportionately vulnerable to the deposition of LBs, the process of LB formation may be the consequence of dysfunctions in either of these organelles [29]. Indeed, LBs were recently shown to contain membranous components and organellar remnants, including mitochondria and lysosomes [30]. Previously we have already demonstrated the accumulation of the mitophagy marker pS65‐Ub in Lewy body disease (LBD) cases, where the punctate structures colocalized with αsyn signals and closely decorated the surface of LBs [31]. We here set out to further explore the complex interrelation between αsyn and mitophagy, the potential underlying molecular mechanisms, and the pathological implications in different cell models, αsyn transgenic mice, and in autopsy brain from cases with LBD or MSA.
2. MATERIALS AND METHODS
2.1. Collection, culture, and genetic or pharmacological manipulation of patient fibroblasts
Primary human dermal fibroblasts were collected from two SNCA triplication (SNCAx3) carriers, one SNCA duplication (SNCAx2) carrier, and their respective healthy siblings that served as controls. The collection, processing, and analyses of primary dermal fibroblast were approved by the Institutional Review Board at Mayo Clinic. All fibroblasts were cultured in Dulbecco's modified Eagle medium (Thermo Fisher, 11965118) supplemented with 10% fetal bovine serum (Neuromics, FBS001800112), 1% PenStrep (Thermo Fisher, 15140122), and 1% nonessential amino acids (Thermo Fisher, 11140050). Fibroblasts were grown at 37°C, 5% CO2:air in humidified atmosphere. αsyn knockout fibroblasts were generated by transducing cells with LentiCRISPRv2 vector (Addgene, Plasmid #52961) containing a single guide RNA targeting the SNCA start codon (TCCTTTCATGAATACATCCA) and followed by antibiotic selection. PRKN knockdown fibroblasts were generated by transfecting cells with siRNA (Qiagen, SI02661267) using nucleofection (Lonza, V4XP‐2024). 20 μg/mL cycloheximide (CHX, Sigma–Aldrich, C1988), 200 nM bafilomycin A1 (Cayman Chemicals, 11038), 1 μM epoxomicin (Sigma, E3652), HBSS (Thermo Fisher, 14025076), 0.5 μM valinomycin (Enzo Life Science, BML‐KC140‐0025), or vehicle DMSO (Sigma–Aldrich, D4540) were used for cell treatments.
2.2. Direct conversion of fibroblasts to induced neurons
The trans‐differentiation of fibroblasts into induced neurons (iNeurons) was performed as previously described with modifications [32, 33]. Briefly, primary patient fibroblasts were seeded in 6‐well plates and transduced with lentiviral particles containing human PTB shRNA for 48 h in the presence of 5 μg/mL Polybrene (Sigma–Aldrich, TR‐1003‐G) at 32°C. Transduced cells were selected with 2 μg/mL puromycin (Thermo Fisher, A1138‐03) starting 2 days after transduction. Two days later, 10 ng/mL basic fibroblast growth factor (GenScript, Z02734) was added to the fibroblast media and cultivation continued for additional 2 days. From day 7 on, cells were maintained in differentiation medium containing DMEM/F12 (Thermo Fisher, 11320‐–082), 5% fetal bovine serum (Neuromics, FBS001800112; further reduced to 2% after 2 days), 1% PenStrep (Thermo Fisher, 15140122), 25 μg/mL insulin (Sigma–Aldrich, I9278), 100 nM putrescine (Sigma–Aldrich, P5780), 50 μg/mL transferrin (Sigma–Aldrich, T8158), 30 nM sodium selenite (Sigma–Aldrich, S5261), and 15 ng/mL basic fibroblast growth factor. After 6 days, 2% B27 supplement without antioxidants (Thermo Fisher, 10889‐038) and 10 ng/mL each of brain‐derived neurotrophic factor (R&D Systems, 248‐BD‐025), glial cell‐derived neurotrophic factor (R&D Systems, 212‐GD‐050), neurotensin‐3 (Peprotech, AF‐450‐03), and ciliary neurotrophic factor (Peprotech, 450‐13) were added to the differentiation medium. The cells were used for experiments 2 days later. Neuronal differentiation was confirmed by expression of the neuronal marker tubulin beta 3 class III (TUBB3).
2.3. Maintenance and expression regulation of H4 cells
Stable human H4 neuroglioma cells expressing αsyn fusion proteins under a tetracycline‐controlled transcriptional activation system [34] were provided by Dr. McLean (Mayo Clinic, Jacksonville, USA). H4 cells were grown in Opti‐MEM reduced serum growth medium (Thermo Fisher, 51985091) supplemented with 10% fetal bovine serum, 200 μg/mL of G418 (Thermo Fisher, 10131‐035), 200 μg/mL of hygromycin (Thermo Fisher, 10687010), and 1 μg/mL of tetracycline (Sigma–Aldrich, T7660) at 37°C, 5% CO2:air in humidified atmosphere. To induce αsyn expression in H4 cells, tetracycline was removed from the culture medium.
2.4. RNA extraction and real‐time quantitative PCR
RNA was extracted from fibroblast cell pellets using a RNeasy Mini Kit (Qiagen, 74106). Real‐time quantitative PCR was performed using iTaq Universal SYBR Green One‐Step Kit (Bio‐Rad, #1725150). Specifically, 50 ng of RNA was mixed with primers for the targeted genes (Table S1), SYBR Green, and iScript reverse transcriptase in a 5 μL reaction. The PCR was executed using a 384‐well block on a LightCycler 480 system (Roche, Switzerland). Relative transcript levels for targeted genes were calculated with 2‐ΔΔCT method using RPL27 as housekeeping gene and normalized to the relative expression level of the control [35].
2.5. Western blot
Cells were lysed in RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 0.1% SDS, 0.5% deoxycholate, 1% NP‐40) containing protease inhibitor cocktail and phosphatase inhibitors (Sigma–Aldrich, 11697498001 and 04906837001). Cell lysates were cleared for 15 min, 4°C at 20,817 × g and protein concentrations were determined by BCA assay (Thermo Fisher, 23225). Cell lysates containing 15 μg of protein were diluted in Laemmli buffer (62.5 mM Tris, pH 6.8, 1.5% SDS, 8.33% glycerol, 1.5% β‐Mercaptoethanol, 0.005% bromophenol blue) and boiled at 95°C for 5 min before running on Tris‐Glycine gels (Invitrogen, EC60485BOX). Post transfer of protein onto PVDF membranes (Millipore Sigma, IPVH00010), membranes were blocked in 5% skim milk (Genesee, 20‐241) and incubated with primary antibodies against αsyn (BD Biosciences, 610787; 1:2000), PRKN (Millipore, MAB5512; 1:5000), PINK1 (Cell Signaling Technology, 6946; 1:1000), TUBB3 (Cell Signaling Technology, 5568; 1:5000), LC3 (Novus Biologicals, NB100‐2220; 1:10,000), GFP (Takara Bio, 632381; 1:10,000), cyclin dependent kinase inhibitor 1A (CDKN1A/P21) (Cell Signaling Technology, 2947; 1:2000), pS65‐Ub (in‐house, 1:10,000) [33], mitofusin 2 (MFN2) (Abcam, ab56889; 1:2000), GAPDH (Meridian Life Sciences, H86504M; 1:500,000), and vinculin (VCL) (Sigma–Aldrich, V9131; 1:500,000) overnight at 4°C, followed by secondary HRP‐conjugated antibodies (Jackson ImmunoResearch, 711‐035‐152, 715‐035‐150, 115‐035‐207, 115‐035‐205; 1:10,000) for 1 h at room temperature. For αsyn immunoblotting, PVDF membranes were incubated with 4% paraformaldehyde (Sigma–Aldrich, 441244) containing 0.01% glutaraldehyde (Electron Microscopy Science, 16020) for 30 min at room temperature prior to blocking [36, 37]. Protein bands were visualized using Immobilon Western Chemiluminescent HRP Substrate (Millipore Sigma, WBKLS0500) and Blue Devil Lite X‐ray films (Genesee Scientific, 30‐810L).
2.6. Immunofluorescence staining of iNeurons
To examine the neuronal structure, iNeurons were also differentiated on glass coverslips that were coated with growth‐factor reduced matrigel (Millipore, CLS356252, 1:1000 in PBS). Cells were fixed with 4% paraformaldehyde, permeabilized with 1% Triton X‐100, blocked with 10% goat serum, and incubated with primary antibody against TUBB3 (Millipore, AB9354; 1:250) followed by incubation with secondary antibody (Invitrogen, A‐11034; 1:1000) and Hoechst 33342 (Invitrogen, H21492; 1:5000). Coverslips were mounted onto microscope slides using fluorescent mounting medium (Dako, S302380‐2). High‐resolution confocal fluorescent images were taken with an AxioObserver microscope equipped with an ApoTome Imaging System (Zeiss, Oberkochen, Germany).
2.7. Immunofluorescence staining of fibroblasts and high content imaging
To quantify pS65‐Ub levels using automated high‐content imaging, fibroblasts were seeded in 96‐well imaging plates (Fisher Scientific, 08772225) and allowed to attach overnight. Cells were then treated for 0, 4, 8, or 24 h with 1 μM valinomycin and fixed in 4% paraformaldehyde after one wash with PBS. Fibroblasts were immunostained with primary antibodies against pS65‐Ub (Cell Signaling Technology, 62802; 1:1250) and HSP60 (arigo Biolaboratories, ARG10757; 1:2000) followed by incubation with secondary antibody (Invitrogen, A‐11034 and A‐11041; 1:1000) and Hoechst 33342 (Invitrogen, H21492; 1:5000). Plates were imaged on a BD Pathway 855 (BD Biosciences, San Jose, CA, USA) with a 20× objective using a 2 × 2 montage (no gaps) with laser autofocus every second frame as previously described [33]. Raw images were processed using the built‐in AttoVision V1.6 software. Regions of interest were defined as nucleus and cytoplasm using the built‐in “RING‐2 outputs” segmentation for the Hoechst channel after applying a shading algorithm. Values were normalized to 0 h and 24 h valinomycin treated control cells as 0% and 100%, respectively.
2.8. Animal cohort and tissue collection
All procedures involving animals were in accordance with the ethical standards and approved by the Institutional Animal Care and Use Committee at Mayo Clinic. Brain tissue from 9 to 18 months old Line D mice (n = 18) that overexpress wild‐type human αsyn from the platelet‐derived growth factor‐β promoter [38] and their nontransgenic littermate controls (n = 11) were used. Mice were euthanized and transcardially perfused with PBS and then brains were quickly removed and fixed in 4% paraformaldehyde overnight. Fixed brain was then embedded in paraffin wax for sectioning.
2.9. Human autopsy brain
All brain samples are from autopsies performed after approval by the legal next‐of‐kin. Research on de‐identified postmortem brain tissue is considered exempt from human subjects' regulations by the Mayo Clinic Institutional Review Board. In the current study, autopsy brains of neurologically normal controls (n = 15), LBD cases without known familial risk factors (n = 9), LBD cases with SNCA multiplications or missense mutations (LBDmut, n = 6), and MSA‐parkinsonian type (MSA‐P) cases (n = 15) from non‐Hispanic Caucasians were retrieved for immunohistochemical analyses from the Mayo Clinic Florida Brain Bank. All brains were examined in a systematic and standardized manner by a single neuropathologist, as previously described [39]. Available neuropathological information included age at death, sex, Braak neurofibrillary tangle stage (0–VI), and Thal amyloid phase (0–5) (see Table S2 and S3).
2.10. Immunohistochemistry
Paraffin embedded brain tissue was cut into 5 μm sections and allowed to dry overnight at 60°C. Slides were deparaffinized and rehydrated, followed by antigen retrieval in steaming deionized water for 30 min. After blocking with 0.03% hydrogen peroxide and 5% normal goat serum (Invitrogen, 16210072), sections were incubated with primary antibodies against pS65‐Ub (in‐house, 1:650) [31], αsyn (for human brain: NACP [40], Mayo Clinic Jacksonville, 1:3000; for mouse brain: Thermo Scientific, MA1‐90346, 4B12, 1:5000), or phospho‐tau (CP13, gift from the late Dr. Peter Davies, Feinstein Institute, North Shore Hospital, NY, 1:1000), followed by rabbit‐ or mouse‐labeled polymer HRP (Agilent, K4011 and K4007) at room temperature. For NACP staining, slides were pretreated with 98% formic acid for 30 min before steaming to retrieve the antigen. Peroxidase labeling was visualized with the chromogen solution 3,3′‐diaminobenzidine (Agilent, K4011 and K4007). The sections were then counterstained with Lerner 1 hematoxylin (Fisher Scientific, CS400‐1D) and coverslipped with Cytoseal mounting medium (Thermo Scientific, 8310). After immunohistochemical staining, all sections were scanned with an Aperio AT2 digital pathology scanner (Leica Biosystems, Wetzlar, Germany) and then traced and quantified using optimized Aperio algorithms to measure the total signal or count the positive cell number followed by manual quality control [31].
2.11. Immunofluorescence and imaging human brain tissue
Following target retrieval and blocking, brain sections were incubated in primary antibodies against pS65‐Ub (in‐house, 1:650) and pS129‐αsyn (Wako Chemicals USA, 015‐25191; 1:3000) at 4°C overnight and in secondary antibodies (Invitrogen, A‐11034 and A‐11004; 1:1000) with DAPI (Sigma–Aldrich, D9542; 1:1000) at room temperature for 1.5 h. 3% Sudan black (SPI Supplies, 02560‐AB) was used to quench autofluorescence before slides were coverslipped in fluorescence mounting medium (Agilent, S302380). After immunofluorescence staining, super‐resolution confocal (Airyscan) images were taken with a LSM 880 microscope (Zeiss, Oberkochen, Germany) with z‐stack.
2.12. Statistical analysis
Statistical analysis of cell experiments was performed with one‐way ANOVA. For measures in animals, unpaired t tests were used. For human cohorts, given that most measures were not normally distributed and had differing variances between groups, nonparametric tests (Kruskal–Wallis and Mann–Whitney U tests followed by adjustment with Bonferroni correction) were used. Statistical analyses were performed using GraphPad Prism (GraphPad Software; version 9).
3. RESULTS
3.1. Elevated αsyn expression leads to an increase in PRKN protein across different cell types
To examine potential αsyn‐associated alterations in PINK1‐PRKN‐mediated mitophagy, we first assessed their gene expression in primary fibroblasts from three siblings, two of which carry a triplication of the SNCA locus. As expected, mRNA levels of SNCA were significantly increased in both triplication cell lines, albeit to different extent (Figure 1A). Neither PINK1 nor PRKN transcription was changed in any of the cells. While it is known that fibroblasts generally have very low expression of SNCA [41, 42], both triplication cells showed more than 5–10 fold increased αsyn protein levels compared to the sibling control (Figure 1B,C), consistent with their relative increase in mRNA. PINK1 protein was not detectable as expected in the absence of mitochondrial stress. However, PRKN protein levels were notably elevated in both triplication cell lines. A similar trend for elevated PRKN levels, though less pronounced, was observed when analyzing an additional, independent set of fibroblasts from a SNCAx2 carrier and the sibling control (Figure S1A,B).
FIGURE 1.
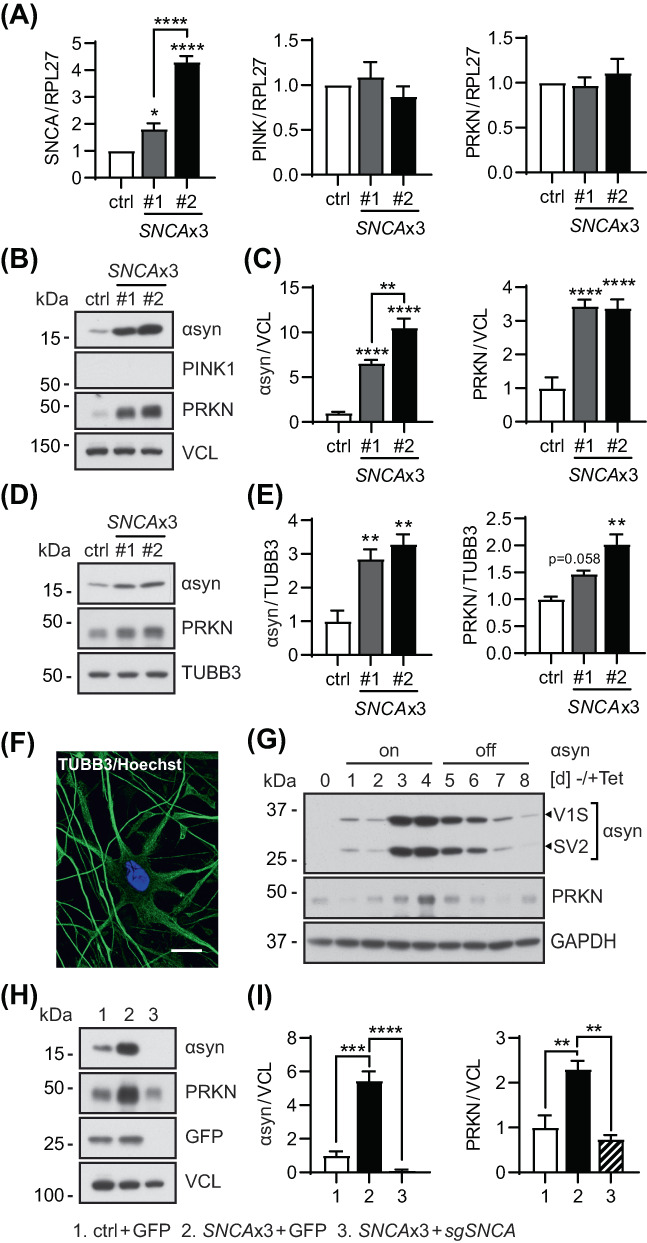
PRKN protein levels are consistently increased in cells with higher αsyn expression. (A) Reverse transcriptase qPCR shows a significant increase of SNCA mRNA levels in both SNCAx3 fibroblasts compared to a sibling control (p = 0.016 for #1 and p < 0.0001 for #2; p < 0.0001 for #1 vs. #2), while PINK1 and PRKN mRNA levels remain unchanged among all three cell lines. n = 4–5 independent experiments. (B) Representative western blot of control and two SNCAx3 fibroblast lines. (C) Immunoblot quantification shows significantly increased protein levels of αsyn (p < 0.0001 for both lines; p = 0.0011 for #1 vs. #2) and PRKN (p < 0.0001 for both lines) in two SNCAx3 fibroblasts compared to control. n = 6 independent experiments. (D) Representative western blot of control and two SNCAx3 iNeurons. (E) Immunoblot quantification shows significantly increased levels of αsyn (p = 0.0091 for #1, p = 0.0032 for #2) and PRKN (p = 0.058 for #1, p = 0.0016 for #2) in two SNCAx3 cells compared to control. Induction of the neuronal marker TUBB3 confirms the successful trans‐differentiation of fibroblasts to iNeurons. n = 3 independent differentiation experiments. (F) Representative immunofluorescence images of iNeurons stained with TUBB3 (green) and Hoechst (blue) confirmed the neuronal conversion. Scale bar: 20 μm. (G) Representative western blot of H4 cells shows an increase and then a decrease in PRKN protein when αsyn expression was turned on (Tet −) for 4 days and then turned off (Tet +) for another 4 days. Tet, tetracycline. Note the H4 cell line is for bimolecular fluorescence complementation assays and expresses two split venus YFP tagged forms of αsyn [Venus1‐αsyn (V1S) and αsyn‐Venus2 (SV2)] from a bidirectional promoter. (H) Representative western blot of fibroblasts transduced with GFP (ctrl + GFP and SNCAx3 + GFP) and SNCAx3 fibroblasts with CRISPR/Cas9 knockdown of SNCA (SNCAx3 + sgSNCA) are shown. (I) Western blot quantification shows significantly decreased αsyn (p < 0.0001) and PRKN (p = 0.0026) levels in SNCAx3 fibroblasts transduced with sgSNCA compared to the GFP control. n = 3 independent experiments. Quantifications are shown as fold change with the control set to 1. Data are shown as mean with standard error. One‐way ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 when compared to controls. Significance level between two groups as indicated by brackets.
To validate our observations in a different cell type, we directly converted the patient fibroblasts to iNeurons. Consistent to the fibroblast results, the relative increases of both αsyn and PRKN proteins in the respective SNCAx3 compared to the control cells were maintained in iNeurons (Figure 1D,E). Successful neuronal conversion and morphology of the cells was confirmed by the expression of the neuronal marker TUBB3 in western blot and immunofluorescence staining (Figure 1D,F). To further corroborate our findings in an independent model, we employed a H4 neuroglioma cell line with a stable inducible expression system of split venus‐tagged αsyn [Venus1‐αsyn (V1S) and αsyn‐Venus2 (SV2)] [34]. Tetracycline was initially removed from the culture media to allow the expression of αsyn for 4 days and was then added back in to shut off αsyn transcription for another 4 days. Consistently, we found an increase of αsyn protein upon induction followed by a decline over the second half of the time course (Figure 1G). Of note, PRKN protein levels followed a similar trend, gradually increasing over the first 4 days, peaking with the maximal expression of αsyn, and then decreasing again over the last 4 days.
To confirm that elevated PRKN protein levels were indeed caused by the increased expression of αsyn, we knocked‐out αsyn in the triplication fibroblasts using CRISPR/Cas9. Patient cells were transduced with a lentivirus expressing GFP as a control or a single guide RNA targeting the start codon of SNCA (sgSNCA). This strategy effectively abolished αsyn expression from the triplication fibroblasts compared to the GFP control transduction (Figure 1H,I). Of note, elimination of αsyn significantly reduced PRKN protein to levels seen in the control fibroblast line. Altogether, the analyses of different cell lines and rescue experiments highlight a close relationship between enhanced αsyn and PRKN expression at the protein level.
3.2. PRKN accumulation in SNCAx3 fibroblasts is linked to altered, basal autophagic flux
To explore possible mechanisms underlying PRKN protein accumulation in SNCAx3 fibroblasts, we first followed its half‐life after protein synthesis inhibition via cycloheximide (CHX) treatment (Figure 2A,B). Typically, PRKN requires activation by PINK1 to release its auto‐inhibited conformation, but otherwise can be quite stable under basal, nonstress conditions. While no significant change in PRKN turnover rate was found in either of the three fibroblast lines over the duration of the treatment, slightly more PRKN protein was turned over after 48 h CHX treatment in the control fibroblast line when compared to each of the SNCAx3 fibroblasts (65% vs. 49% and 56%), suggesting a slightly slower degradation rate that may partially contribute to PRKN accumulation over time in the presence of elevated αsyn expression.
FIGURE 2.
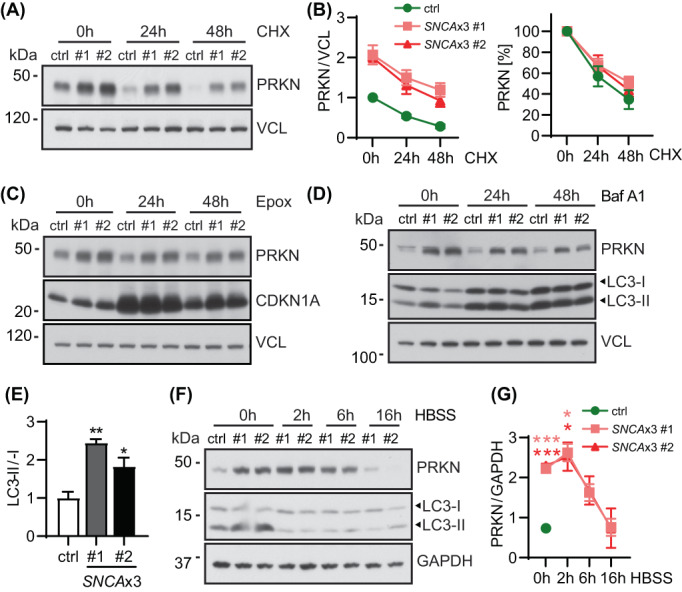
Activation of general autophagy reduces PRKN accumulation in SNCAx3 fibroblasts. (A) Representative western blot of control and SNCAx3 fibroblasts treated with cycloheximide (CHX) for the indicated timepoints. (B) Western blot quantification of PRKN normalized with the value of the control line set to 1 (left) or using each value at 0 h as 100% (right). Results show comparable PRKN turnover rates in all three fibroblast lines. n = 4 independent experiments. (C) Proteosome inhibition does not affect PRKN levels in control or SNCAx3 fibroblasts. Representative western blot of PRKN, CDKN1A, and VCL from control and SNCAx3 fibroblasts after 1 μM epoxomicin (Epox) treatment for 0, 24, and 48 h. (D) Inhibition of autophagic flux does not further increase PRKN levels in SNCAx3 fibroblasts. Representative western blot of PRKN, LC3, and VCL from control and SNCAx3 fibroblasts after 200 nM bafilomycin A1 (Baf A1) treatment for 0, 24, and 48 h. (E) Immunoblot quantification shows significant increases of lipidated to unlipidated LC3 ratio (LC3‐II/I) in two SNCAx3 fibroblasts compared to control (p = 0.0022 for #1, p = 0.031 for #2). Quantifications are shown as fold change with the control set to 1. n = 3 independent experiments. (F) Representative western blot of control and SNCAx3 fibroblasts treated by HBSS for indicated timepoints. (G) Western blot quantification shows that HBSS‐induced autophagy activation is able to lower PRKN levels in SNCAx3 fibroblasts to the control level after 16 h of starvation. n = 2 independent experiments. Data shown as mean with standard error. One‐way ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001 in pink/red for the corresponding SNCAx3 cells when compared to control at the same timepoint.
In general, two major proteolytic pathways, the ubiquitin‐proteasome and the autophagy‐lysosome system, mediate cellular degradation, and αsyn is known to interfere with both routes [43]. To explore the contribution of either, we first inhibited proteasome function in the control and SNCAx3 fibroblasts with epoxomicin (Epox) (Figure 2C). Successful proteasome inhibition was confirmed by the accumulation of CDKN1A after 24 h of treatment. However, no obvious change in PRKN levels was found in any of the three fibroblast lines. Next, we treated fibroblasts with the autophagy inhibitor bafilomycin A1 (Baf A1) (Figure 2D). Successful inhibition of autophagic flux was confirmed by a strong upregulation of LC3 across all lines, yet no additional increase of PRKN protein was noted. However, a significant increase in the LC3‐II/I ratio in both SNCAx3 lines compared to the control was apparent at basal conditions, that is, in the absence of stress (Figure 2E). While this can be interpreted as either, an induction of autophagosome formation or a decrease in degradation rates, αsyn has been previously linked to impairments of autophagic flux [19]. To ascertain this effect, we stimulated general autophagy in SNCAx3 fibroblasts by starvation. This led to a time‐dependent decrease of PRKN protein to similar levels seen in the control fibroblast over a period of just 16 h (Figure 2F,G). Taken together, accumulation of PRKN protein in SNCAx3 fibroblasts was likely associated with an alteration of basal autophagy but could be effectively cleared and normalized by starvation‐induced autophagy activation.
3.3. Abnormally enhanced PRKN levels modify the mitophagy response in SNCAx3 fibroblasts
To assess potential functional consequences of the increased PRKN levels in cells, we next studied mitophagy upon acute mitochondrial stress. For this, fibroblasts were treated with mitochondrial stressor valinomycin (Val) and levels of PINK1 and PRKN as well as the mitophagy marker pS65‐Ub and the PRKN substrate MFN2 were studied by western blot at different time points (Figure 3A,B). Induction of mitophagy led to an immediate and significant stabilization of PINK1 over time that was comparable across all three cell lines. In contrast, levels of the Ub ligase PRKN typically decrease over time once activated by PINK1. As expected, PRKN levels were significantly higher in both triplication lines, but the signal declined over time at a rate similar to the consanguineous control cells (data not shown). Yet, levels of the mitophagy tag pS65‐Ub were also significantly elevated in the SNCAx3 cells compared to the control. Consistently, Ub modification of MFN2 (MFN2‐Ub) that is observed as a 8 kDa shifted band was also markedly increased in SNCAx3 cells (especially at early time points). The significant boost of the pS65‐Ub signal was additionally confirmed by immunofluorescence staining of fibroblasts followed by quantification with high content imaging (Figure 3C–E). Over time, both SNCAx3 cells showed much greater accumulation of pS65‐Ub‐positsive mitochondria than the control line. Similar findings were obtained from the SNCAx2 fibroblasts that also showed elevated pS65‐Ub levels compared to the sibling control upon 24 h of Val treatment (Figure S1C).
FIGURE 3.
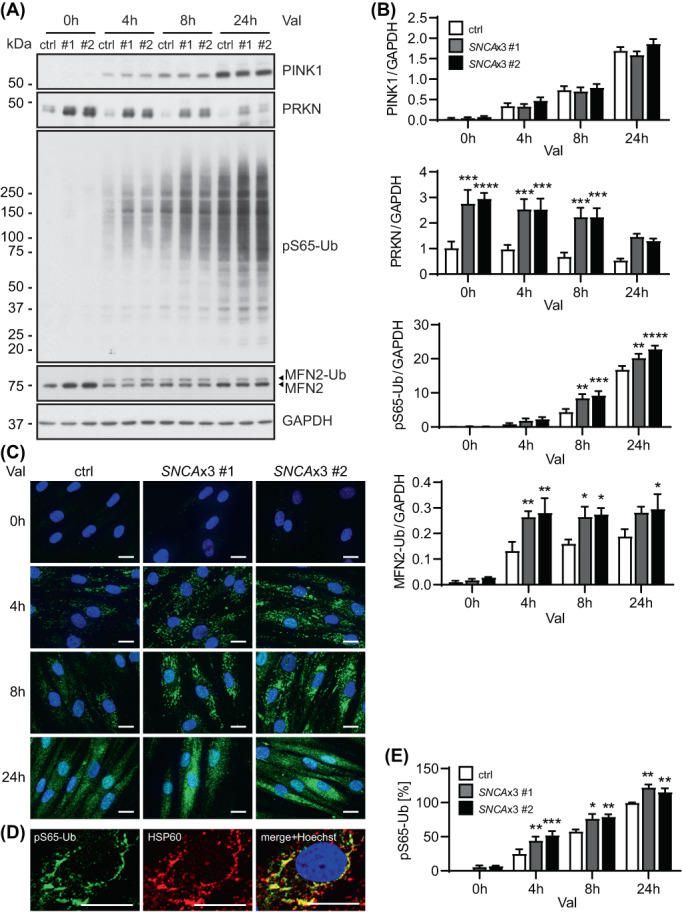
Increased mitophagy response upon acute stress in SNCAx3 fibroblasts. (A) Representative western blot of control and two SNCAx3 fibroblasts lines upon valinomycin (Val) treatment for the indicated time points. (B) Western blot quantification shows similar stabilization of PINK1, but significantly increased levels of PRKN, pS65‐Ub, and ubiquitinated MFN2 (MFN2‐Ub) in SNCAx3 fibroblasts compared to the control with the same treatment (for PRKN, p < 0.001 for both lines at 0, 4, 8 h; for pS65‐Ub, p < 0.01 for both lines at 8, 24 h; for MFN2‐Ub, p < 0.05 for #1 at 4, 8 h and for #2 at 4, 8, and 24 h). n = 6 independent experiments. (C) Representative images of pS65‐Ub immunofluorescence staining (green) in fibroblasts upon Val treatment. Scale bar: 20 μm. (D) Representative immunofluorescence images of fibroblasts stained with pS65‐Ub (green) and the mitochondrial marker HSP60 (red) showing colocalization of pS65‐Ub on the mitochondria. Scale bar: 20 μm. (E) High content imaging quantification confirms the significant increase in pS65‐Ub‐positive signal in SNCAx3 fibroblasts compared to the control with the same treatment (p < 0.05 for #1 and p < 0.01 for #2 at 4, 8, 24 h). n = 4 independent experiments. Data shown as mean with standard error. One‐way ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 when compared to the corresponding control at the same time point.
Given that activated PRKN provides additional substrates for PINK1 and thereby typically amplifies the pS65‐Ub response, we asked whether the abnormally increased levels of the E3 Ub ligase PRKN were causal to the elevated mitophagy signaling observed in SNCAx3 fibroblasts. To test this hypothesis, we knocked down PRKN expression by siRNA to similar levels seen in the control fibroblast (Figure 4A,B). We then assessed mitophagy signaling again in response to Val treatment. While PINK1 stabilization over time remained similar, knockdown of PRKN reduced the pS65‐Ub response to levels comparable to the control cell line. Taken together, our results suggest a primary role for elevated PRKN protein in exacerbating the pS65‐Ub response in SNCAx3 fibroblasts. While PRKN protein seems to accumulate due to altered basal autophagy, the Ub ligase can be activated and its increased levels amplify the mitophagy response upon mitochondrial stress in SNCAx3 fibroblasts.
FIGURE 4.
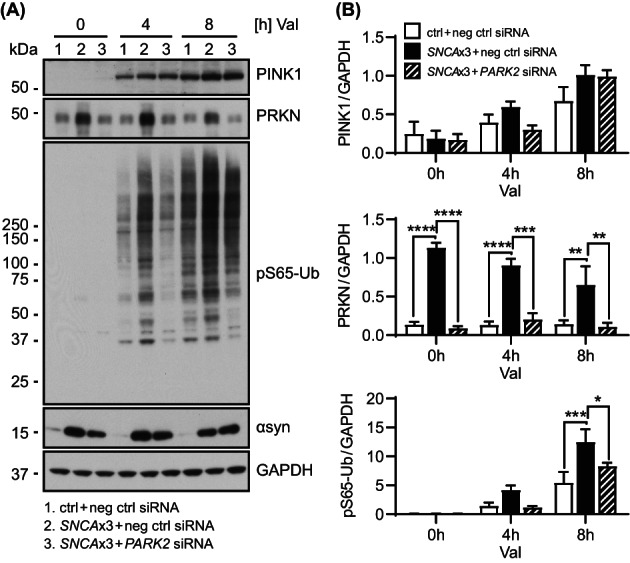
PRKN knockdown in SNCAx3 fibroblasts normalizes the mitophagy response. (A) Representative western blot of control and SNCAx3 fibroblasts transfected with negative control siRNA (ctrl + neg ctrl siRNA and SNCAx3 + neg ctrl siRNA) and SNCAx3 fibroblasts transfected with PARK2 siRNA (SNCAx3 + PARK2 siRNA). (B) Western blot quantification shows that PRKN knockdown in SNCAx3 fibroblasts significantly reduces PRKN (p < 0.01 at 0, 4, 8 h) and pS65‐Ub (p = 0.019 at 8 h) levels but does not affect PINK1 and αsyn levels when compared to negative control siRNA transfected SNCAx3 fibroblasts with same duration of valinomycin (Val) treatment. n = 3 independent experiments. Data shown as mean with standard error. One‐way ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
3.4. The mitophagy marker pS65‐Ub correlates with the pathological αsyn load in LBD brain
To evaluate the disease relevance of findings from cell culture, we turned to human autopsy brain comparing LBD cases with different burden of αsyn pathology to neurologically normal individuals. Given the lack of tools to reliably detect PINK1 and PRKN in tissue, we chose to focus on their joint product pS65‐Ub to assess mitophagy alterations by immunohistochemistry [31, 33, 44]. Compared to our previous work, here we examined pS65‐Ub levels in LBD cases with SNCA mutations or multiplications (LBDmut group) that generally show more severe αsyn pathology at younger age [45, 46]. Age‐ and sex‐matched neurologically normal controls and sporadic LBD cases (LBD group) were also included in the analysis. For a summary and the detailed characteristics of all subjects studied, see Tables S2 and S3, respectively. Significant pathological burden, as determined by αsyn (NACP) immunostaining, was detected in the substantia nigra (SN) and hippocampus (Figure S2A) as well as in the amygdala, nucleus basalis of Meynert (nbM), and putamen (Figure 5A,B).
FIGURE 5.
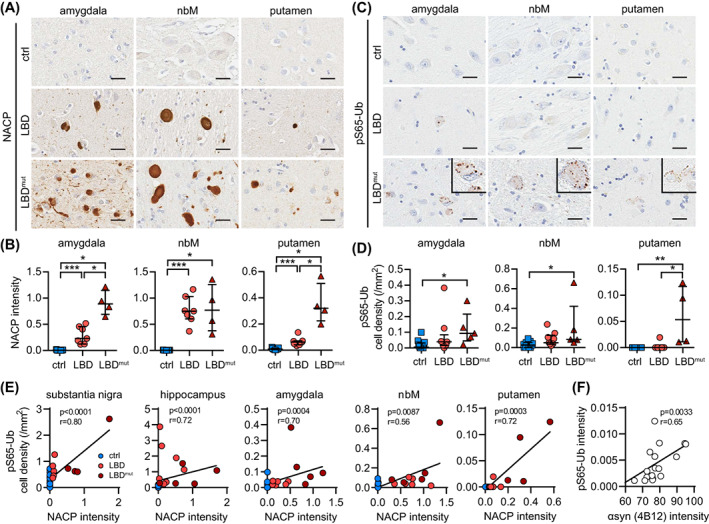
The mitophagy marker pS65‐Ub is increased across regions in human autopsy LBD brains with SNCA missense mutations or multiplications. Representative images of (A) αsyn (stained by NACP) and (C) pS65‐Ub immunostaining in the indicated brain regions from control, LBD, and LBDmut groups. Insets show magnified views of pS65‐Ub positive cells in all three regions. Scale bar: 25 μm. (B) NACP intensity is significantly increased in LBD and increased further in the LBDmut group in the amygdala, nbM, and putamen compared to the age‐matched controls (p < 0.0001 for LBD, p = 0.0015 for LBDmut, p = 0.0061 for LBD vs. LBDmut). (D) pS65‐Ub‐positive cell density is significantly increased only in the LBDmut group in the amygdala (p = 0.004), nbM (p = 0.0053), and putamen (p = 0.0005) compared to age‐matched controls. Kruskal–Wallis and Mann–Whitney U tests followed by adjustment with Bonferroni correction, *p < 0.0167 (i.e., the statistical significance threshold after Bonferroni correction), **p < 0.001, ***p < 0.0001. Data is shown as median with interquartile range. (E) pS65‐Ub positive cell density and NACP intensity are strongly correlated in the substantia nigra, hippocampus, amygdala, nbM, and putamen. Spearman's test of correlation, significance threshold: p < 0.01. n = 11–15 for controls, n = 6–9 for LBD group, n = 4–5 for LBDmut group. nbM, nucleus basalis of Meynert. (F) pS65‐Ub and human αsyn levels are strongly correlated in brains of Line D mice. Spearman's test of correlation, significance threshold: p < 0.05. n = 18.
As expected, the LBDmut group showed more widespread and more severe αsyn pathology compared to the LBD group in most of the regions examined. In line with these and our prior findings [31], significant increases of the pS65‐Ub were restricted to the SN and hippocampus in the LBD cases (Figure S2B) but were observed in all studied brain regions in the LBDmut group (Figure 5C,D). The increased pS65‐Ub level was particularly prominent in the SNCA triplication case compared to SNCA duplication or A53T mutation cases, in agreement with their respective αsyn burden. The increase of pS65‐Ub was independent of the comorbid tau pathology as there was no significant difference in the density of CP13 (phospho‐tau) positive cells between LBD and LBDmut groups (Figures S2C and S3). However, pS65‐Ub significantly correlated with an increased burden of neuropathological αsyn across all brain regions studied (Figure 5E). To further validate this finding, we immunostained and quantified pS65‐Ub in a mouse model overexpressing wild‐type human αsyn (Line D) [38]. Transgenic mice expressing high levels of human αsyn showed significant increases of pS65‐Ub immunoreactive signal in the brain compared to the respective nontransgenic controls (Figure S4). Consistent with results from human brain, pS65‐Ub levels also significantly correlated with the αsyn burden in brains of transgenic mice (Figure 5F).
3.5. pS65‐Ub accumulates early on during the development of and in proximity to the αsyn pathology
To investigate the spatial relationship between mitophagy changes and αsyn pathology, we immunostained human tissue for pS65‐Ub and phosphorylated‐αsyn (pS129‐αsyn), the pathognomonic form of αsyn, and used super‐resolution microscopy for colocalization studies (Figure 6A). We first categorized pS65‐Ub‐positive cells in the SN based on their respective pS129‐αsyn immunoreactivity: no pS129‐αsyn, diffuse pS129‐αsyn, or aggregated pS129‐αsyn staining. While only a small percentage of pS65‐Ub‐positive cells were completely devoid of pS129‐αsyn immunoreactivity, their number was slightly greater in the LBDmut cases compared to LBD cases (Figure 6B). The LBDmut group also contained more pS65‐Ub‐positive cells with diffuse pS129‐αsyn staining particularly in the SNCA triplication case (the highest data point). The number of pS65‐Ub positive cells with aggregated pS129‐αsyn in the form of either cortical‐type or classical, brainstem‐type LBs were rather similar between both disease groups. In either case, the majority of pS65‐Ub positive structures did not necessarily colocalize with the pS129‐αsyn signal but were especially abundant in cells with smaller, pathological αsyn aggregates (Figure 6C). pS65‐Ub positive puncta were frequently found adjacent to or closely decorated the surface of αsyn aggregates that resembled premature LBs in neurons in the studied regions (Figure 6D). However, in neurons with rather mature LBs, pS65‐Ub granules were less prominent, suggesting that mitophagy changes occur early on but may not be detectable during late‐stage pathology.
FIGURE 6.
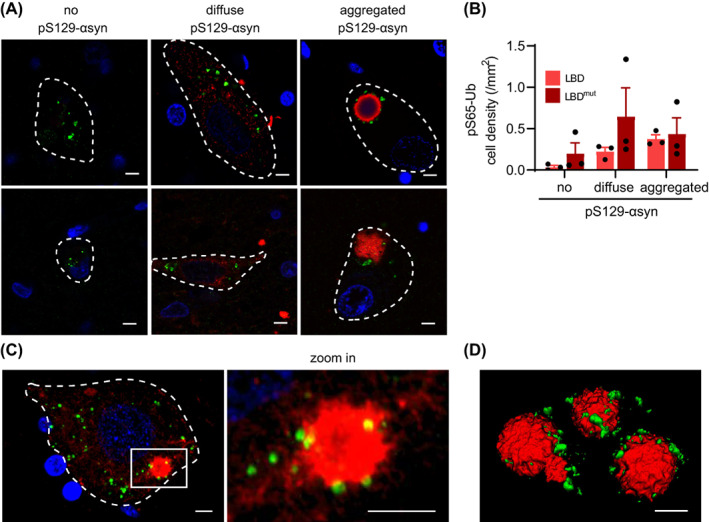
Spatial relationship between pS65‐Ub and αsyn pathology in LBD. (A) Representative images of pS65‐Ub positive cells with different pS129‐αsyn immunoreactive signals. Classical (top right image) and cortical (bottom right image) LBs with double immunostaining of pS65‐Ub (green) and pS129‐αsyn (red) are shown. (B) The quantification of pS65‐Ub‐positive cell density that was categorized based on their respective pS129‐αsyn immunoreactivity in the substantia nigra of three LBD and three LBDmut cases. (C) Representative immunofluorescence images in 3D maximum projection rendering of one cell containing small pS129‐αsyn‐positive aggregation is shown. A magnified image of the boxed area is shown to the right. pS65‐Ub‐positive granules (green) surround the pS129‐αsyn‐positive aggregation (red). (D) The spatial relationship of pS65‐Ub‐ and pS129‐αsyn‐positive LB is shown in 3D surface rendering.
3.6. pS65‐Ub accumulates alongside neuropathological αsyn in LBD, but not in MSA brain
LBD and MSA are both synucleinopathies but are pathologically characterized by distinct intracellular αsyn inclusions, namely LB‐type deposits and glial cytoplasmic inclusions (GCIs), respectively [3, 47]. To explore the effect of different αsyn inclusions on mitophagy, we examined αsyn burden and compared pS65‐Ub levels in brains of LBD and MSA patients along with their age‐ and sex‐matched neurologically normal controls. We chose to examine the amygdala, nbM, and putamen here to cover brain regions with low, intermediate, or high αsyn burden in both types of diseases (Figure 7A). While both LBD and LBDmut groups contained high αsyn burden in the amygdala, αsyn levels in LBDmut cases in this region were similar to αsyn levels in MSA cases in the putamen and thus this group was used here for comparison with MSA. αsyn (NACP) immunostaining confirmed the strongest αsyn burden in the amygdala in LBDmut and in the putamen in MSA with comparable levels seen in the nbM in both diseases (Figure 7B,C). pS65‐Ub seemed to track exclusively with the LB pathology found in LBDmut as levels of pS65‐Ub were also the highest in the amygdala, somewhat decreased in the nbM, and the lowest in the putamen (Figure 7D). On the contrary, pS65‐Ub signals were not elevated in MSA cases compared to controls, even in the nbM or the putamen with higher αsyn burden and despite the additional tau pathology (Figure 7E).
FIGURE 7.
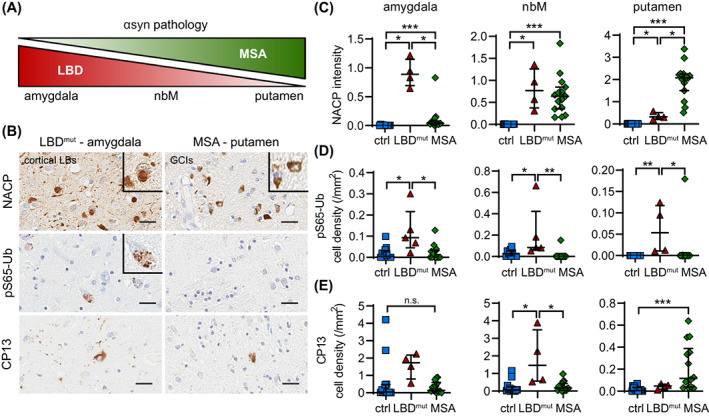
The increase of the mitophagy marker pS65‐Ub in human autopsy brain is associated with LB‐, but not GCI‐type of αsyn pathology. (A) The schematic shows that αsyn pathology is predominantly found in the amygdala and to a lesser extent in the nbM and putamen in LBD, while it is predominantly found in the putamen and to a lesser extent in the amygdala and nbM in MSA. (B) Representative images of αsyn (NACP, top), pS65‐Ub (middle), and phospho‐tau (CP13, bottom) immunostaining in the amygdala and putamen of LBDmut and MSA cases, respectively. Scale bar: 25 μm. Insets show magnified views of cortical type LBs in LBD, GCIs in MSA, and pS65‐Ub positive cells. (C) Consistent with the schematic, NACP intensity is differentially increased in the amygdala, nbM, and putamen of LBDmut and MSA cases compared to age‐matched controls (p = 0.0015 for LBDmut, p < 0.0001 for MSA, p < 0.01 for LBDmut vs. MSA). (D) pS65‐Ub positive cell density is increased in all three regions, but only in the LBDmut group compared to age‐matched controls (p = 0.004, 0.0053, and 0.0005 in the amygdala, nbM, and putamen, respectively) and MSA group (p = 0.0035, 0.0003, and 0.0013 in the amygdala, nbM, and putamen, respectively). (E) CP13 positive cell density is selectively increased in the nbM of the LBDmut group (p = 0.0097 for LBDmut, p = 0.0062 for LBDmut vs. MSA) and in the putamen of MSA cases (p < 0.0001) compared to age‐matched controls. n = 11–15 for controls, n = 4–5 for LBDmut group, n = 14–15 for MSA group. Kruskal–Wallis and Mann–Whitney U tests followed by adjustment with Bonferroni correction, *p < 0.0167 (i.e., the statistical significance threshold after Bonferroni correction), **p < 0.001, ***p < 0.0001, n.s., not significant. Data is shown as median with interquartile range. GCI, glial cytoplasmic inclusion; LB, Lewy body; nbM, nucleus basalis of Meynert.
Collectively our new data confirm and extend previous findings to additional brain regions from overall younger, but more severe cases of LBD that show more advanced and more widespread neuropathology. In subjects with disease‐causing missense mutations or multiplications of SNCA, pS65‐Ub levels were significantly increased and strongly correlated with the αsyn pathology in all regions analyzed and independently of the comorbid tau pathology. We further discovered that despite comparable neuropathological load, the observed mitophagy alterations were specific to LB‐type deposits of αsyn rather than GCIs seen in MSA.
4. DISCUSSION
Characterization of primary fibroblasts obtained from siblings with or without SNCA triplications showed that PRKN protein levels increase in cells with higher αsyn expression. We confirmed this effect in independent sets of patients' cells and different cell types including directly differentiated neurons and an inducible αsyn overexpression model. Moreover, SNCA knockout by CRISPR/Cas9 caused a reduction of PRKN protein to control levels. PINK1 and PRKN mRNA however remained unchanged, arguing for a posttranslational mechanism rather than a compensatory transcriptional effect. While αsyn has been proposed to impair mitochondrial function directly or indirectly through a plethora of mechanisms [25, 26, 27, 28, 48, 49, 50, 51], we could not observe stabilization of PINK1 protein in SNCAx3 cells as one would expect if increased expression of αsyn caused mitochondrial damage. Likewise, at baseline we did not detect an increase of pS65‐Ub, the most sensitive readout for activation of the PINK1‐PRKN pathway. Instead, we found elevated levels of αsyn to be associated with an altered basal autophagy, which likely contributed to the accumulation of PRKN protein.
The impairment of autophagy through mutant or aggregated αsyn is well documented in the literature (reviewed in [6]), but our current study suggests that even increased levels of soluble αsyn can alter basal autophagic flux. While these effects were subtle and largely reversible in fibroblasts, increased soluble αsyn was recently identified as a shared feature of induced pluripotent stem cells (iPSC)‐derived dopamine neurons from young‐onset PD patients [52]. The mechanism(s) by which soluble αsyn can alter basal flux are unresolved, but may include both direct and indirect effects on autophagy or lysosomes that are probably also context dependent with regard to cell type and actual αsyn species. Regardless, stalled basal autophagy may not only contribute to spreading and propagation of αsyn, but partial degradation can lead to further aberrant processing of αsyn which may produce forms that are even more aggregation‐prone and neurotoxic [28, 53, 54, 55]. The resulting pathologic αsyn species then preferentially bind to and impair mitochondria and contribute to a viscous cycle that directly affects the PINK1‐PRKN‐mediated mitophagy [27, 28, 49, 50, 51, 56, 57].
We hypothesize that PRKN most likely accumulates in its auto‐inhibited form as PINK1‐dependent recruitment and activation results in its own degradation alongside damaged mitochondria. Consistently, PRKN protein accrues in PINK1 knockout cells and tissue due to lack of prerequisite activation (Watzlawik et al., in submission). Notwithstanding, the increased pool of PRKN in SNCAx3 fibroblasts was activatable and, due to the known feed‐forward loop in which products of PRKN become additional substrates for PINK1 [8], strongly amplified the pS65‐Ub response to acute mitochondrial insult. A boosted pS65‐Ub signal may lead to additional buildup of mitophagosomes or, in case of sufficient degradative capacities, to excessive mitophagy, both of which have been described in the context of αsyn overexpression [25, 26, 48]. Yet, levels of the E3 Ub ligase PRKN, once activated, eventually declined at similar rates in control and SNCAx3 fibroblasts, suggesting a comparable turnover of damaged mitochondria among three fibroblast lines. Although pharmacological block of neither autophagy nor proteasome system alone further increased levels of PRKN, induction of general autophagy by nutrient starvation in SNCAx3 cells effectively cleared the elevated pool of PRKN over time.
In human LBD autopsy brain, we previously identified an age‐dependent increase of the mitophagy marker pS65‐Ub as well as independent αsyn‐ and tau‐mediated associations in select vulnerable brain regions including the SN and the hippocampus [31, 33]. Compared to these prior studies, we here expanded our analyses to younger and more severe cases of LBD including individuals with SNCA missense mutations or gene multiplications (LBDmut group). We observed significant increases of pS65‐Ub in all brain regions analyzed, including amygdala, nbM, and putamen, areas that are typically affected later than the SN or in more aggressive forms of LBD. Since tau can independently drive pS65‐Ub in both human brain and transgenic mice [44], we adjusted for the comorbid phospho‐tau pathology and identified a strong correlation of pS65‐Ub with the more abundant αsyn pathology. Consistently, pS65‐Ub also strongly correlated with αsyn levels in a transgenic mouse model overexpressing human αsyn in neurons [38]. However, comparison of LBD to MSA revealed a significant pS65‐Ub increase only in the context of LBs, but not GCIs, despite a similar or higher burden of pathologic αsyn in vulnerable regions, and regardless of greater neuronal loss seen in some, but not all MSA cases. While it is known that pathological αsyn from LBs and GCIs is conformationally and biologically distinct [58, 59, 60], it remains unclear whether our findings primarily reflect a cell type specific mitophagy effect (neurons vs. oligodendrocytes), a difference in the respective αsyn aggregate, or the diverse structures and other molecular content of the inclusions.
While loss of PINK1 or PRKN causes EOPD and impedes the initial steps of mitophagy, that is, the identification and labeling of damaged mitochondria with pS65‐Ub, the age‐dependent decline in degradative capacities may impair the pathway at later steps contributing to sporadic late‐onset PD. However, loss of PINK1 or PRKN not only results in the accumulation of dysfunctional mitochondria, but also contributes to αsyn aggregation in various cell and animal models [10, 61, 62, 63, 64]. Thus, a functional relationship and convergence of both αsyn and PINK1‐PRKN‐mediated mitophagy is further emerging [9]. In line with our previous study [31], the results herein suggest that mitophagy alterations in LBD brain may happen early on and prior to prominent αsyn aggregation and maturation into LBs. Such overarching concept is also compatible with the earlier notion that PRKN activity might be required for LB formation, in part because mutant cases seem to mostly lack LBs [65, 66, 67, 68] as well as more recent findings that clusters of membranous compartments derived from mitochondria and lysosomes accumulate in LBs [30, 69]. Furthermore, it was suggested that the process of LB formation may be the consequence of dysfunctions in either of these organelles [29], and the major driver of neurodegeneration [70, 71].
With the ongoing discussion about the biological origin of synucleinopathies and what triggers the disease relevant events [72, 73], our study further emphasizes the need to determine the specific bioactivity of the various forms of αsyn. We found similar outcomes associated with higher levels of αsyn in cells and in brain tissue (i.e., higher levels of the mitophagy tag pS65‐Ub), but the underlying mechanisms might be distinct. Although pS65‐Ub is a direct readout of both PINK1 and PRKN enzymatic activities as well as a quantitative measure of mitochondrial damage, both increased activation of mitophagy as well as reduced flux through the autophagy‐lysosome system can lead to pS65‐Ub accumulation. The current detailed analyses in human brain tissue mostly focused on LBs and not on GCIs. Going forward, additional analyses in iPSC‐derived cultures, in novel organoid models [74], and in in vivo animal models are needed to confirm our findings in LBs vs. GCIs and to further dissect effects of individual αsyn species on different organelles and aspects involved in mitophagy. High resolution imaging of fixed tissue should help to at least approximate the most likely primary defects leading to the increase in pS65‐Ub [44]. Yet, this seems to strongly depend on the respective brain region and cell type as well as the nature and progression of the neuropathology. Nevertheless, our current findings together with the identification of pS65‐Ub in patients' biofluids [75] may help facilitate a differential diagnosis between synucleinopathies despite the similar extrapyramidal symptoms seen in PD and MSA.
AUTHOR CONTRIBUTIONS
Conceptualization: Wolfdieter Springer, Xu Hou. Methodology: Xu Hou, Fabienne C. Fiesel. Formal analysis: Xu Hou. Investigation: Xu Hou, Taylor Hsuan‐Yu Chen, Jenny M. Bredenberg, Shunsuke Koga. Resources: Shunsuke Koga, Ayman H. Faroqi, Marion Delenclos, Guojun Bu, Zbigniew K. Wszolek, Jonathan A. Carr, Owen A. Ross, Pamela J. McLean, Melissa E. Murray, Dennis W. Dickson. Writing—original draft: Xu Hou, Wolfdieter Springer. Visualization: Xu Hou. Funding acquisition: Wolfdieter Springer.
FUNDING INFORMATION
W.S., O.A.R., P.J.M., and D.W.D. are members of the American Parkinson Disease Association (APDA) Center for Advanced Research at Mayo Clinic Florida and are further supported by the National Institute of Neurological Disorders and Stroke (NIH/NINDS) Lewy Body Dementia Center Without Walls (U54 NS110435). W.S. is additionally supported by NIH (R01 NS085070, R01 NS110085, and R56 AG062556), the Department of Defense Congressionally Directed Medical Research Programs (CDMRP) (W81XWH‐17‐1‐0248), the Michael J. Fox Foundation for Parkinson's Research (MJFF), the Ted Nash Long Life Foundation, Mayo Clinic Foundation, the Center for Biomedical Discovery (CBD), and the Robert and Arlene Kogod Center on Aging. X.H. is supported by a pilot grant and a developmental project award from the Mayo Clinic Alzheimer Disease Research Center (ADRC, P30 AG062677) and fellowships awarded by the APDA and Alzheimer's Association (AARF‐22‐973152). F.C.F. is the recipient of fellowships from the Younkin Scholar Program and the APDA and is supported in part by the Florida Department of Health—Ed and Ethel Moore Alzheimer's Disease Research Program (22A07), the MJFF, a Gerstner Family Career Development Award from the Center for Individualized Medicine (CIM) and an auxiliary award from the CBD at Mayo Clinic. Z.K.W. is partially supported by the NIH (U19 AG063911), Mayo Clinic Center for Regenerative Medicine, gifts from the Donald G. and Jodi P. Heeringa Family, the Haworth Family Professorship in Neurodegenerative Diseases fund, and the Albertson Parkinson's Research Foundation. A.H.F., M.D., P.J.M. are partially supported by Mayo Clinic APDA Center for Advanced Research grant and American Brain Foundation (ABF) Cure One Cure All grant. O.A.R. is supported in part by NIH (P50 NS072187, R01 NS078086, U54 NS100693), Department of Defense (W81XWH‐17‐1‐0249), the ABF, the MJFF, the Little Family Foundation, and the CIM at Mayo Clinic. D.W.D. is further supported by the Mangurian Foundation Lewy Body Dementia Program at Mayo Clinic.
CONFLICT OF INTEREST STATEMENT
Mayo Clinic, F.C.F., and W.S. have filed a patent related to PRKN activators. Z.K.W. serves as an external advisory board member for Vigil Neuroscience, Inc. Additional funding sources to disclose but not pertinent to the current study include grants from Biohaven Pharmaceuticals, Inc. (BHV4157‐206 and BHV3241‐301 to Z.K.W.), Neuraly, Inc. (NLY01‐PD‐1 to Z.K.W.), Vigil Neuroscience, Inc. (VGL101‐01.001 and VGL101‐01.002 to Z.K.W.), and Amazentis SA (to W.S.). All other authors declare they have no competing interests. This research was conducted in compliance with Mayo Clinic conflict of interest policies.
ETHICS STATEMENT
The collection, processing, and analyses of primary dermal fibroblast were approved by the Institutional Review Board at Mayo Clinic. All procedures involving animals were in accordance with the ethical standards and approved by the Institutional Animal Care and Use Committee at Mayo Clinic. All brain samples are from autopsies performed after approval by the legal next‐of‐kin. Research on de‐identified postmortem brain tissue is considered exempt from human subjects' regulations by the Mayo Clinic Institutional Review Board.
Supporting information
Data S1. Supporting Information
Data S2. Supporting Information
Data S3. Supporting Information
ACKNOWLEDGMENTS
We are grateful to the patients and their families who made this study possible. We thank Monica Castanedes‐Casey and Virginia R. Phillips from the Neuropathology Laboratory for processing human postmortem tissues and the excellent technical support. We thank the Center for Regenerative Medicine and the Neuroregeneration Laboratory for biobanking of patient cells at Mayo Clinic Florida. We thank Audrey J. Strongosky for coordinating clinical research studies and help with obtaining patient specimens. We also thank the late Dr. Peter Davies from the Feinstein Institute for his generous contribution of CP13 antibodies.
Hou X, Chen TH‐Y, Koga S, Bredenberg JM, Faroqi AH, Delenclos M, et al. Alpha‐synuclein‐associated changes in PINK1‐PRKN‐mediated mitophagy are disease context dependent. Brain Pathology. 2023;33(5):e13175. 10.1111/bpa.13175
DATA AVAILABILITY STATEMENT
The datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.
REFERENCES
- 1. Lees AJ, Hardy J, Revesz T. Parkinson's disease. Lancet. 2009;373:2055–66. [DOI] [PubMed] [Google Scholar]
- 2. Dickson DW, Braak H, Duda JE, Duyckaerts C, Gasser T, Halliday GM, et al. Neuropathological assessment of Parkinson's disease: refining the diagnostic criteria. Lancet Neurol. 2009;8:1150–7. [DOI] [PubMed] [Google Scholar]
- 3. McCann H, Stevens CH, Cartwright H, Halliday GM. Alpha‐synucleinopathy phenotypes. Parkinsonism Relat Disord. 2014;20(Suppl 1):S62–7. [DOI] [PubMed] [Google Scholar]
- 4. Esteves AR, Arduino DM, Silva DF, Oliveira CR, Cardoso SM. Mitochondrial dysfunction: the road to alpha‐synuclein oligomerization in PD. Parkinsons Dis. 2011;2011:693761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Monzio Compagnoni G, Di Fonzo A. Understanding the pathogenesis of multiple system atrophy: state of the art and future perspectives. Acta Neuropathol Commun. 2019;7:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hou X, Watzlawik JO, Fiesel FC, Springer W. Autophagy in Parkinson's disease. J Mol Biol. 2020;432:2651–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Moors T, Paciotti S, Chiasserini D, Calabresi P, Parnetti L, Beccari T, et al. Lysosomal dysfunction and alpha‐synuclein aggregation in Parkinson's disease: diagnostic links. Mov Disord. 2016;31:791–801. [DOI] [PubMed] [Google Scholar]
- 8. Truban D, Hou X, Caulfield TR, Fiesel FC, Springer W. PINK1, parkin, and mitochondrial quality control: what can we learn about Parkinson's disease pathobiology? J Parkinsons Dis. 2017;7:13–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ryan BJ, Hoek S, Fon EA, Wade‐Martins R. Mitochondrial dysfunction and mitophagy in Parkinson's: from familial to sporadic disease. Trends Biochem Sci. 2015;40:200–10. [DOI] [PubMed] [Google Scholar]
- 10. Gispert S, Brehm N, Weil J, Seidel K, Rub U, Kern B, et al. Potentiation of neurotoxicity in double‐mutant mice with Pink1 ablation and A53T‐SNCA overexpression. Hum Mol Genet. 2015;24:1061–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stichel CC, Zhu XR, Bader V, Linnartz B, Schmidt S, Lubbert H. Mono‐ and double‐mutant mouse models of Parkinson's disease display severe mitochondrial damage. Hum Mol Genet. 2007;16:2377–93. [DOI] [PubMed] [Google Scholar]
- 12. Creed RB, Goldberg MS. Analysis of alpha‐synuclein pathology in PINK1 knockout rat brains. Front Neurosci. 2018;12:1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Creed RB, Goldberg MS. Enhanced susceptibility of PINK1 knockout rats to alpha‐synuclein fibrils. Neuroscience. 2020;437:64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Todd AM, Staveley BE. Pink1 suppresses alpha‐synuclein‐induced phenotypes in a drosophila model of Parkinson's disease. Genome. 2008;51:1040–6. [DOI] [PubMed] [Google Scholar]
- 15. Yasuda T, Miyachi S, Kitagawa R, Wada K, Nihira T, Ren YR, et al. Neuronal specificity of alpha‐synuclein toxicity and effect of parkin co‐expression in primates. Neuroscience. 2007;144:743–53. [DOI] [PubMed] [Google Scholar]
- 16. Lo Bianco C, Schneider BL, Bauer M, Sajadi A, Brice A, Iwatsubo T, et al. Lentiviral vector delivery of parkin prevents dopaminergic degeneration in an alpha‐synuclein rat model of Parkinson's disease. Proc Natl Acad Sci U S A. 2004;101:17510–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Krzystek TJ, Banerjee R, Thurston L, Huang J, Swinter K, Rahman SN, et al. Differential mitochondrial roles for alpha‐synuclein in DRP1‐dependent fission and PINK1/parkin‐mediated oxidation. Cell Death Dis. 2021;12:796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chung E, Choi Y, Park J, Nah W, Park J, Jung Y, et al. Intracellular delivery of parkin rescues neurons from accumulation of damaged mitochondria and pathological alpha‐synuclein. Sci Adv. 2020;6:eaba1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xilouri M, Brekk OR, Stefanis L. Autophagy and alpha‐synuclein: relevance to Parkinson's disease and related synucleopathies. Mov Disord. 2016;31:178–92. [DOI] [PubMed] [Google Scholar]
- 20. Ordonez DG, Lee MK, Feany MB. Alpha‐synuclein induces mitochondrial dysfunction through Spectrin and the Actin cytoskeleton. Neuron. 2018;97:108–124.e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bido S, Soria FN, Fan RZ, Bezard E, Tieu K. Mitochondrial division inhibitor‐1 is neuroprotective in the A53T‐alpha‐synuclein rat model of Parkinson's disease. Sci Rep. 2017;7:7495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Paillusson S, Gomez‐Suaga P, Stoica R, Little D, Gissen P, Devine MJ, et al. Alpha‐synuclein binds to the ER‐mitochondria tethering protein VAPB to disrupt Ca(2+) homeostasis and mitochondrial ATP production. Acta Neuropathol. 2017;134:129–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Choi ML, Chappard A, Singh BP, Maclachlan C, Rodrigues M, Fedotova EI, et al. Pathological structural conversion of alpha‐synuclein at the mitochondria induces neuronal toxicity. Nat Neurosci. 2022;25:1134–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Creed RB, Memon AA, Komaragiri SP, Barodia SK, Goldberg MS. Analysis of hemisphere‐dependent effects of unilateral intrastriatal injection of alpha‐synuclein pre‐formed fibrils on mitochondrial protein levels, dynamics, and function. Acta Neuropathol Commun. 2022;10:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Choubey V, Safiulina D, Vaarmann A, Cagalinec M, Wareski P, Kuum M, et al. Mutant A53T alpha‐synuclein induces neuronal death by increasing mitochondrial autophagy. J Biol Chem. 2011;286:10814–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen L, Xie Z, Turkson S, Zhuang X. A53T human alpha‐synuclein overexpression in transgenic mice induces pervasive mitochondria macroautophagy defects preceding dopamine neuron degeneration. J Neurosci. 2015;35:890–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Di Maio R, Barrett PJ, Hoffman EK, Barrett CW, Zharikov A, Borah A, et al. Alpha‐synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson's disease. Sci Transl Med. 2016;8:342ra378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Grassi D, Howard S, Zhou M, Diaz‐Perez N, Urban NT, Guerrero‐Given D, et al. Identification of a highly neurotoxic alpha‐synuclein species inducing mitochondrial damage and mitophagy in Parkinson's disease. Proc Natl Acad Sci U S A. 2018;115:E2634–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Erskine D, Koss D, Korolchuk VI, Outeiro TF, Attems J, McKeith I. Lipids, lysosomes and mitochondria: insights into Lewy body formation from rare monogenic disorders. Acta Neuropathol. 2021;141:511–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shahmoradian SH, Lewis AJ, Genoud C, Hench J, Moors TE, Navarro PP, et al. Lewy pathology in Parkinson's disease consists of crowded organelles and lipid membranes. Nat Neurosci. 2019;22:1099–109. [DOI] [PubMed] [Google Scholar]
- 31. Hou X, Fiesel FC, Truban D, Castanedes Casey M, Lin WL, Soto AI, et al. Age‐ and disease‐dependent increase of the mitophagy marker phospho‐ubiquitin in normal aging and Lewy body disease. Autophagy. 2018;14:1404–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xue Y, Ouyang K, Huang J, Zhou Y, Ouyang H, Li H, et al. Direct conversion of fibroblasts to neurons by reprogramming PTB‐regulated microRNA circuits. Cell. 2013;152:82–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fiesel FC, Ando M, Hudec R, Hill AR, Castanedes‐Casey M, Caulfield TR, et al. (Patho‐)physiological relevance of PINK1‐dependent ubiquitin phosphorylation. EMBO Rep. 2015;16:1114–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Moussaud S, Malany S, Mehta A, Vasile S, Smith LH, McLean PJ. Targeting alpha‐synuclein oligomers by protein‐fragment complementation for drug discovery in synucleinopathies. Expert Opin Ther Targets. 2015;19:589–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) method. Methods. 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- 36. Sasaki A, Arawaka S, Sato H, Kato T. Sensitive western blotting for detection of endogenous Ser129‐phosphorylated alpha‐synuclein in intracellular and extracellular spaces. Sci Rep. 2015;5:14211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee BR, Kamitani T. Improved immunodetection of endogenous alpha‐synuclein. PLoS One. 2011;6:e23939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, et al. Dopaminergic loss and inclusion body formation in alpha‐synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–9. [DOI] [PubMed] [Google Scholar]
- 39. Koga S, Ikeda A, Dickson DW. Deep learning‐based model for diagnosing Alzheimer's disease and tauopathies. Neuropathol Appl Neurobiol. 2022;48:e12759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gwinn‐Hardy K, Mehta ND, Farrer M, Maraganore D, Muenter M, Yen SH, et al. Distinctive neuropathology revealed by alpha‐synuclein antibodies in hereditary parkinsonism and dementia linked to chromosome 4p. Acta Neuropathol. 2000;99:663–72. [DOI] [PubMed] [Google Scholar]
- 41. Ambrosi G, Ghezzi C, Sepe S, Milanese C, Payan‐Gomez C, Bombardieri CR, et al. Bioenergetic and proteolytic defects in fibroblasts from patients with sporadic Parkinson's disease. Biochim Biophys Acta. 2014;1842:1385–94. [DOI] [PubMed] [Google Scholar]
- 42. Devine MJ, Ryten M, Vodicka P, Thomson AJ, Burdon T, Houlden H, et al. Parkinson's disease induced pluripotent stem cells with triplication of the alpha‐synuclein locus. Nat Commun. 2011;2:440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xilouri M, Brekk OR, Stefanis L. Alpha‐synuclein and protein degradation systems: a reciprocal relationship. Mol Neurobiol. 2013;47:537–51. [DOI] [PubMed] [Google Scholar]
- 44. Hou X, Watzlawik JO, Cook C, Liu CC, Kang SS, Lin WL, et al. Mitophagy alterations in Alzheimer's disease are associated with granulovacuolar degeneration and early tau pathology. Alzheimers Dement. 2020;17:417–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fujishiro H, Imamura AY, Lin WL, Uchikado H, Mark MH, Golbe LI, et al. Diversity of pathological features other than Lewy bodies in familial Parkinson's disease due to SNCA mutations. Am J Neurodegener Dis. 2013;2:266–75. [PMC free article] [PubMed] [Google Scholar]
- 46. Konno T, Ross OA, Puschmann A, Dickson DW, Wszolek ZK. Autosomal dominant Parkinson's disease caused by SNCA duplications. Parkinsonism Relat Disord. 2016;22(Suppl 1):S1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Koga S, Sekiya H, Kondru N, Ross OA, Dickson DW. Neuropathology and molecular diagnosis of synucleinopathies. Mol Neurodegener. 2021;16:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sarkar S, Olsen AL, Sygnecka K, Lohr KM, Feany MB. Alpha‐synuclein impairs autophagosome maturation through abnormal Actin stabilization. PLoS Genet. 2021;17:e1009359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nakamura K, Nemani VM, Azarbal F, Skibinski G, Levy JM, Egami K, et al. Direct membrane association drives mitochondrial fission by the Parkinson disease‐associated protein alpha‐synuclein. J Biol Chem. 2011;286:20710–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Park JH, Burgess JD, Faroqi AH, DeMeo NN, Fiesel FC, Springer W, et al. Alpha‐synuclein‐induced mitochondrial dysfunction is mediated via a sirtuin 3‐dependent pathway. Mol Neurodegener. 2020;15:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang X, Becker K, Levine N, Zhang M, Lieberman AP, Moore DJ, et al. Pathogenic alpha‐synuclein aggregates preferentially bind to mitochondria and affect cellular respiration. Acta Neuropathol Commun. 2019;7:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Laperle AH, Sances S, Yucer N, Dardov VJ, Garcia VJ, Ho R, et al. iPSC modeling of young‐onset Parkinson's disease reveals a molecular signature of disease and novel therapeutic candidates. Nat Med. 2020;26:289–99. [DOI] [PubMed] [Google Scholar]
- 53. Ejlerskov P, Rasmussen I, Nielsen TT, Bergstrom AL, Tohyama Y, Jensen PH, et al. Tubulin polymerization‐promoting protein (TPPP/p25alpha) promotes unconventional secretion of alpha‐synuclein through exophagy by impairing autophagosome‐lysosome fusion. J Biol Chem. 2013;288:17313–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Minakaki G, Menges S, Kittel A, Emmanouilidou E, Schaeffner I, Barkovits K, et al. Autophagy inhibition promotes SNCA/alpha‐synuclein release and transfer via extracellular vesicles with a hybrid autophagosome‐exosome‐like phenotype. Autophagy. 2018;14:98–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lee HJ, Cho ED, Lee KW, Kim JH, Cho SG, Lee SJ. Autophagic failure promotes the exocytosis and intercellular transfer of alpha‐synuclein. Exp Mol Med. 2013;45:e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, et al. Parkinson's disease alpha‐synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J Neurosci. 2006;26:41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shaltouki A, Hsieh CH, Kim MJ, Wang X. Alpha‐synuclein delays mitophagy and targeting Miro rescues neuron loss in Parkinson's models. Acta Neuropathol. 2018;136:607–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Peng C, Gathagan RJ, Covell DJ, Medellin C, Stieber A, Robinson JL, et al. Cellular milieu imparts distinct pathological alpha‐synuclein strains in alpha‐synucleinopathies. Nature. 2018;557:558–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Van der Perren A, Gelders G, Fenyi A, Bousset L, Brito F, Peelaerts W, et al. The structural differences between patient‐derived alpha‐synuclein strains dictate characteristics of Parkinson's disease, multiple system atrophy and dementia with Lewy bodies. Acta Neuropathol. 2020;139:977–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, et al. Alpha‐synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. 2015;522:340–4. [DOI] [PubMed] [Google Scholar]
- 61. Imaizumi Y, Okada Y, Akamatsu W, Koike M, Kuzumaki N, Hayakawa H, et al. Mitochondrial dysfunction associated with increased oxidative stress and alpha‐synuclein accumulation in PARK2 iPSC‐derived neurons and postmortem brain tissue. Mol Brain. 2012;5:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Oliveras‐Salva M, Macchi F, Coessens V, Deleersnijder A, Gerard M, Van der Perren A, et al. Alpha‐synuclein‐induced neurodegeneration is exacerbated in PINK1 knockout mice. Neurobiol Aging. 2014;35:2625–36. [DOI] [PubMed] [Google Scholar]
- 63. Norris KL, Hao R, Chen LF, Lai CH, Kapur M, Shaughnessy PJ, et al. Convergence of parkin, PINK1, and alpha‐synuclein on stress‐induced mitochondrial morphological remodeling. J Biol Chem. 2015;290:13862–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chung SY, Kishinevsky S, Mazzulli JR, Graziotto J, Mrejeru A, Mosharov EV, et al. Parkin and PINK1 patient iPSC‐derived midbrain dopamine neurons exhibit mitochondrial dysfunction and alpha‐synuclein accumulation. Stem Cell Rep. 2016;7:664–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Madsen DA, Schmidt SI, Blaabjerg M, Meyer M. Interaction between parkin and alpha‐synuclein in PARK2‐mediated Parkinson's disease. Cell. 2021;10:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lim KL, Chew KC, Tan JM, Wang C, Chung KK, Zhang Y, et al. Parkin mediates nonclassical, proteasomal‐independent ubiquitination of synphilin‐1: implications for Lewy body formation. J Neurosci. 2005;25:2002–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chung KK, Zhang Y, Lim KL, Tanaka Y, Huang H, Gao J, et al. Parkin ubiquitinates the alpha‐synuclein‐interacting protein, synphilin‐1: implications for Lewy‐body formation in Parkinson disease. Nat Med. 2001;7:1144–50. [DOI] [PubMed] [Google Scholar]
- 68. Schlossmacher MG, Frosch MP, Gai WP, Medina M, Sharma N, Forno L, et al. Parkin localizes to the Lewy bodies of Parkinson disease and dementia with Lewy bodies. Am J Pathol. 2002;160:1655–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fanning S, Selkoe D, Dettmer U. Parkinson's disease: proteinopathy or lipidopathy? NPJ Parkinsons Dis. 2020;6:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sanderson JB, De S, Jiang H, Rovere M, Jin M, Zaccagnini L, et al. Analysis of alpha‐synuclein species enriched from cerebral cortex of humans with sporadic dementia with Lewy bodies. Brain Commun. 2020;2:fcaa010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mahul‐Mellier AL, Burtscher J, Maharjan N, Weerens L, Croisier M, Kuttler F, et al. The process of Lewy body formation, rather than simply alpha‐synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc Natl Acad Sci U S A. 2020;117:4971–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Killinger BA, Melki R, Brundin P, Kordower JH. Endogenous alpha‐synuclein monomers, oligomers and resulting pathology: let's talk about the lipids in the room. NPJ Parkinsons Dis. 2019;5:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ghanem SS, Majbour NK, Vaikath NN, Ardah MT, Erskine D, Jensen NM, et al. Alpha‐synuclein phosphorylation at serine 129 occurs after initial protein deposition and inhibits seeded fibril formation and toxicity. Proc Natl Acad Sci U S A. 2022;119:e2109617119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mohamed NV, Sirois J, Ramamurthy J, Mathur M, Lepine P, Deneault E, et al. Midbrain organoids with an SNCA gene triplication model key features of synucleinopathy. Brain Commun. 2021;3:fcab223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Watzlawik JO, Hou X, Fricova D, Ramnarine C, Barodia SK, Gendron TF, et al. Sensitive ELISA‐based detection method for the mitophagy marker p‐S65‐Ub in human cells, autopsy brain, and blood samples. Autophagy. 2021;17:2613–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting Information
Data S2. Supporting Information
Data S3. Supporting Information
Data Availability Statement
The datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.