

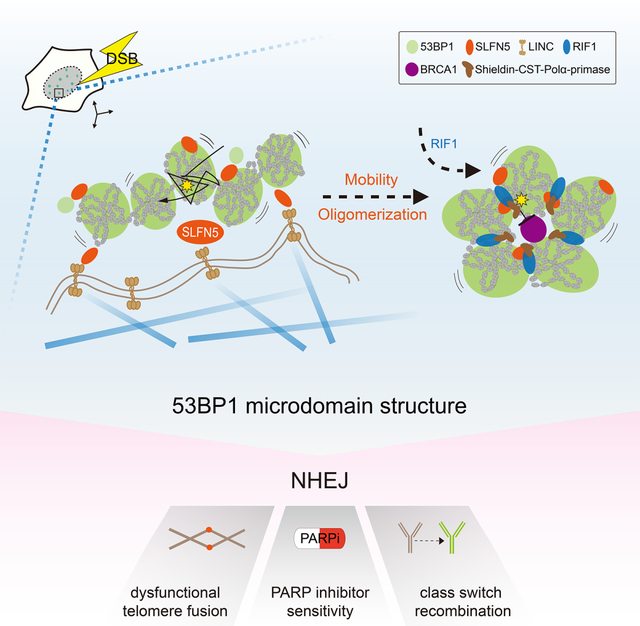
SUMMARY
Repair of DNA double-strand-breaks (DSBs) elicits three-dimensional (3D) chromatin topological changes. A recent finding reveals that 53BP1 assembles into a 3D chromatin topology pattern around DSBs. How this formation of a higher-order structure is configured and regulated remains enigmatic. Here, we report that SLFN5 is a critical factor for 53BP1 topological arrangement at DSBs. Using super-resolution imaging, we find that SLFN5 binds to 53BP1 chromatin domains to assemble a higher-order microdomain architecture by driving damaged chromatin dynamics at both DSBs and deprotected telomeres. Mechanistically, we propose that 53BP1 topology is shaped by two processes: (1) chromatin mobility driven by the SLFN5-LINC-microtubule axis and (2) the assembly of 53BP1 oligomers mediated by SLFN5. In mammals, SLFN5 deficiency disrupts the DSB repair topology and impairs non-homologous end joining, telomere fusions, class switch recombination, and sensitivity to poly (ADP-ribose) polymerase inhibitor. We establish a molecular mechanism that shapes higher-order chromatin topologies to safeguard genomic stability.
Keywords: Non-homologous end joining, Chromatin topology, Chromatin mobility, 53BP1, SLFN5, Telomere fusions, Class switch recombination, PARP inhibitor sensitivity, Super-resolution microscopy
Graphical Abstract
INTRODUCTION
In response to DNA double-strand-breaks (DSBs), mammalian cells use two main repair pathways, homologous recombination (HR) and non-homologous end joining (NHEJ), to safeguard genome integrity1. NHEJ rejoins broken ends, whereas HR requires end resection, which is restricted to the S and G2 phases1–3. DSB repair pathway choice is orchestrated by chromatin-proximal factors that couple DNA repair events with chromatin topological changes1,4,5. BRCA1 and 53BP1, two key factors for HR and NHEJ, respectively, are observed to form unique and mutually exclusive topological domains on damaged chromatin5–7. Recent work indicates that 53BP1 can form nanodomains (NDs) that highly overlap with topologically associated domains (TADs), which then mature into a higher-order topologically organized microdomain (MD)7 to restrict BRCA1 physically and functionally. The downstream effector RIF1 subsequently binds to the 53BP1-ND boundaries and stabilizes the topological architecture7. Importantly, the loss of higher-order 53BP1-MD structure leads to BRCA1 spreading, which may cause excessive resection and genomic instability7–9. However, how 53BP1 organizes into an MD after the induction of DSBs and whether this is an actively regulated process remain unclear.
The function of 53BP1 in DSB repair has been implicated in NHEJ, telomere fusion, class switch recombination (CSR), and poly (ADP-ribose) polymerase inhibitor (PARPi) response4,10. These functions for 53BP1 are mediated both by its role in protecting the ends of DSBs and by its role in regulating chromatin dynamics in response to DSBs4,11–15. 53BP1 governs DSB end protection by recruiting the RIF1-Shieldin complex, which in turn promotes CST-Polα-primase dependent fill-in or ASTE1-dependent cleavage processing to antagonize ssDNA resection4,16–22. On the other hand, 53BP1 is required for increased mobility of damaged chromatin which is linked to the microtubule-LINC complex dynamics14,15. However, the main factor proposed to mediate the mobility of DSB ends remains unknown4,15. In addition, the relationship between 53BP1-mediated chromatin mobility and 53BP1 topological reorganization is unclear.
Schlafen (SLFN) family proteins have been implicated in immune cell proliferation, differentiation, activation, and antiviral restriction23–27. All SLFNs contain an SLFN domain, and the largest subgroup III harbors an additional domain, which is homologous to the DNA/RNA helicase superfamily I27,28. As a SLFN subgroup III member, SLFN5 has three paralogs (SLFN11, SLFN13, and SLFN14) in humans27–29. The SLFN family has poorly described roles in genome stability, with SLFN11 implicated in regulating HR30 and stressed replication forks31. SLFN5 acts as a transcriptional co-repressor in the interferon response32, viral transcription25, and epithelial–mesenchymal transition-driving factor33,34 but has no known function in DNA repair.
In this study, we report that SLFN5 is a critical factor in shaping 53BP1 chromatin topology at DSBs. Our data suggest that SLFN5 links 53BP1 and microtubule-LINC to drive 53BP1-MD formation. Similar to 53BP1, SLFN5 deficiency impairs NHEJ, interferes with telomere maintenance and CSR, and causes genomic instability. Our findings establish the SLFN5-microtubule-LINC axis as a key regulator of damaged chromatin mobility and 53BP1 topological rearrangement while also connecting these two processes together.
RESULTS
SLFN5 facilities deprotected telomere fusions
Recent proximity-based proteomics of DNA repair networks (PROX-NET dataset) reveal 53BP1, BRCA1 and MDC1 interaction neighborhoods16. The UpSet plot showed shared or unique interactions among the above baits (Figure 1A; Table S1). SLFN5 was among the high-confident hits in the same 53BP1-enriched intersection node as SHLD3, a recently identified Shieldin complex component that promotes DSB end jointing16–20,35. SLFN5 and 53BP1 interacted reciprocally, and the interaction network of SLFN5 extended to other NHEJ factors, including RIF1, PTIP and the Shieldin complex subunits (Figures 1B and S1A). No interaction was detected between SLFN5 and BRCA1 or CtIP. We also verified this association using proximity ligation assays between SLFN5 and 53BP1 in cells following DNA damage (Figures S1B and S1C). Interestingly, analyses of the weighted genomic integrity index (WGII) from the cancer genome atlas (TCGA), an indicative signature of genomic instability, showed that patients with SLFN5-deficiency harbored higher genomic instability (Figure S1D). These hints prompted us to determine a connection between SLFN5 and 53BP1-dependent pathway.
Figure 1. SLFN5 promotes telomere-mediated fusions.
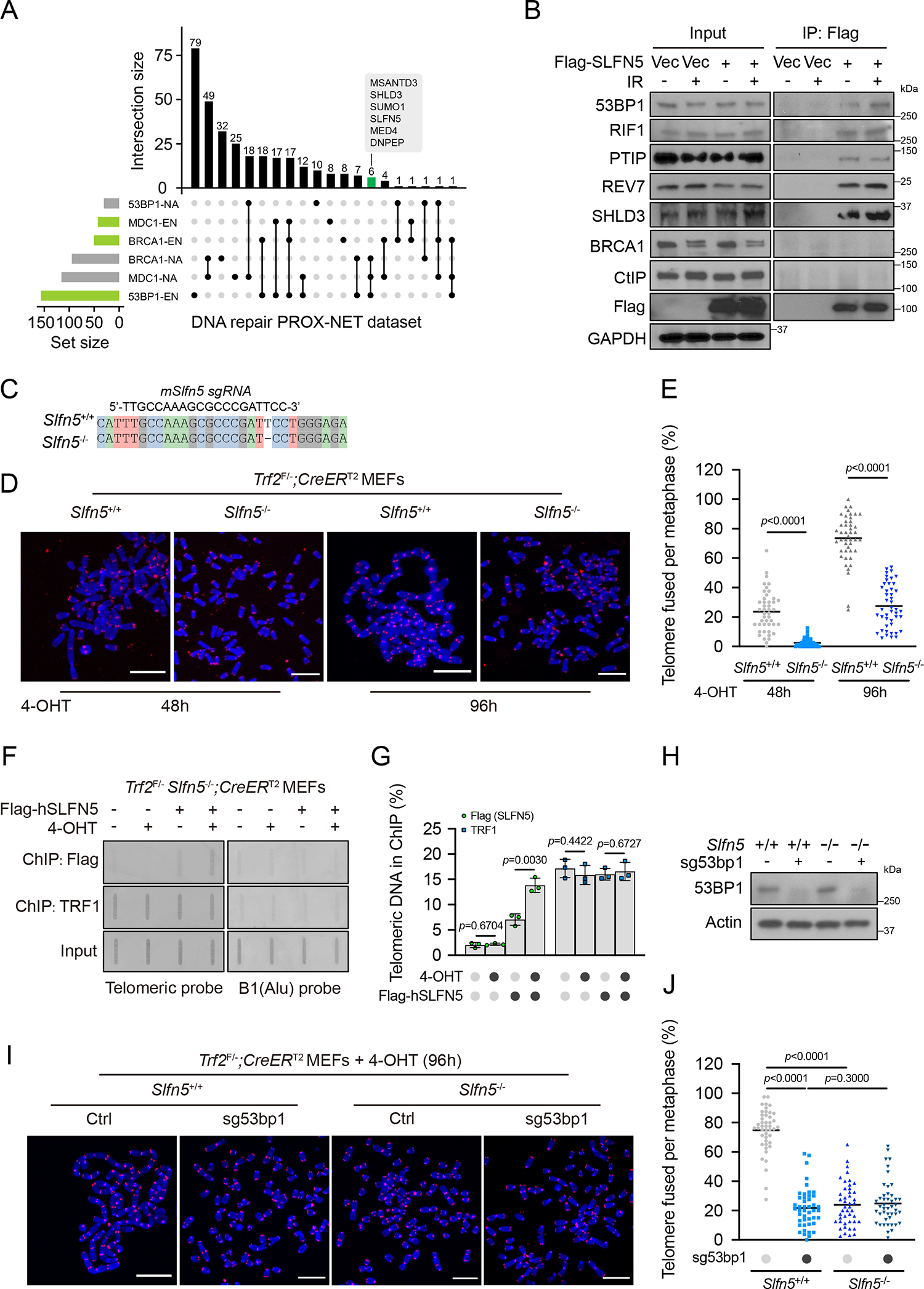
(A) UpSet plot shows the intersection of significant proximal proteins in the PROX-NET dataset of 53BP1, BRCA1, and MDC116. Horizontal bars show the set of proteins with (EN, the log2[bait/control] was ≥ 1 in six independent replicates) or without (NA, not detected in six independent replicates) enrichment of 53BP1, BRCA1, and MDC1 neighborhood networks. Connected lines represent shared proteins between sets. Bar graphs show intersection sizes. A full list is provided in Table S1.
(B) HEK293T cells transfected with empty vector (Vec) or Flag-SLFN5 were untreated or irradiated (5 Gy, 1 h). Cell lysates were immunoprecipitated with Flag beads and blotted with indicated antibodies.
(C) DNA sequencing showed that Slfn5 KO Trf2F/−; CreERT2 MEFs have a frameshift mutation at the CRISPR-Cas9 targeted site.
(D) The indicated Trf2F/−; CreERT2 MEFs were treated with 4-hydroxytamoxifen (4-OHT) for the indicated times. Metaphase spreads were stained by telomeric fluorescence in situ hybridization (FISH) and assessed for telomere fusion. Telomeres and DNA were stained with PNA probe (red) and DAPI (blue), respectively. Representative images are shown in (D). Scale bars, 10 μm.
(E) Telomere fusion quantification from (D). Each dot represents an individual metaphase cell, and the center line indicates the mean; per condition. Data are representative of three independent experiments.
(F) Trf2F/− Slfn5−/−; CreERT2 MEFs stably expressing Vec or Flag-human SLFN5 were treated with MeOH or 4-OHT for 70 h. Flag-SLFN5 and TRF1 accumulation at deprotected telomeres was detected by ChIP. Input, 12.5% of the input DNA. Slot blots were hybridized with telomeric or B1 probes.
(G) Telomere ChIP quantification from (F). Signals were normalized to input. Error bars indicate the mean ± SD of four independent experiments.
(H-J) The indicated Trf2F/−; CreERT2 MEFs were treated with 4-OHT for 96 h. Metaphase spreads were assessed for telomere fusion by telomeric FISH. Telomeres and DNA were stained with PNA probe (red) and DAPI (blue), respectively. The expression level of the indicated proteins was assessed by western blot (H). Representative images are shown in (I). Telomere fusion quantification is shown in (J). Each dot represents an individual metaphase cell, and the center line indicates the mean; per condition. Data are representative of three independent experiments. Scale bars, 10 μm.
p values calculated with two-tailed unpaired t tests.
53BP1 promotes the joining of dysfunctional telomeres through NHEJ14,15. To test whether SLFN5 is involved in the regulation of damaged telomeres, we used conditional Trf2-knockout (KO) mouse embryonic fibroblasts (MEFs) that show telomere uncapping and NHEJ-dependent end-to-end fusions after the deletion of TRF214. Like 53BP1, SLFN5 deficiency resulted in a dramatic suppression of end-to-end telomere fusions induced by Trf2-KO (Figures 1C–1E). Following telomere uncapping, SLFN5 accumulated at the dysfunctional telomeres (Figures 1F and 1G). These results suggest that SLFN5 regulates NHEJ at telomeres that lack TRF2 protection. Consistent with this, we observed genetic epistasis between Slfn5 and 53bp1 in modulating telomere fusions in Trf2-KO MEFs (Figures 1H–1J), which suggests that SLFN5 and 53BP1 act in the same genetic pathway at damaged telomeres.
Slfn5−/− mice exhibit genomic instability
To investigate the function of SLFN5 in vivo, we generated a mouse model in which a “knockout” allele was inserted in the Slfn5 gene (Figures S1E and S1F). MEFs derived from Slfn5−/− embryos showed more chromosomal abnormalities than those of wild-type (WT) controls (Figures S1G and S1H). In agreement, a significant accumulation of γH2AX signals was observed in Slfn5−/− splenocytes (Figures S1I and S1J). In addition, we found that mouse normochromic erythrocytes had a markedly increased frequency of micronuclei compared to WT controls (Figures S1K and S1L). These data suggest that Slfn5−/− mice have genomic instability.
SLFN5 is important for Ig CSR
53BP1 and its downstream effectors RIF1 and REV7 have been proposed to be important for DSB repair during CSR4,36. We asked whether SLFN5 also regulates CSR. We first characterized the levels of serum immunoglobulin (Ig) and found Slfn5−/− mice had normal IgM but reduced titers of IgG1, IgG2a, IgG2b, IgG3, and IgA (Figure 2A). In agreement with this, the KO of Slfn5 impeded class switching to IgG1, IgE, IgG2b, and IgG3 in splenic B lymphocytes (Figures 2B and 2C), indicating that SLFN5 deficiency results in CSR failure. We also observed that Slfn5 and 53bp1 were epistatic (Figure 2D), supporting that they function within the same pathway during CSR. Slfn5−/− mice displayed normal lymphocyte development in the spleen, bone marrow, and thymus (Figures S2A–S2N). We also examined the thymus and spleen weight and found that Slfn5−/− mice had normal spleen and thymus development (Figures S2O and S2P). These data suggest that SLFN5 promotes CSR without affecting lymphocyte development.
Figure 2. Slfn5−/− mice exhibit defective CSR.
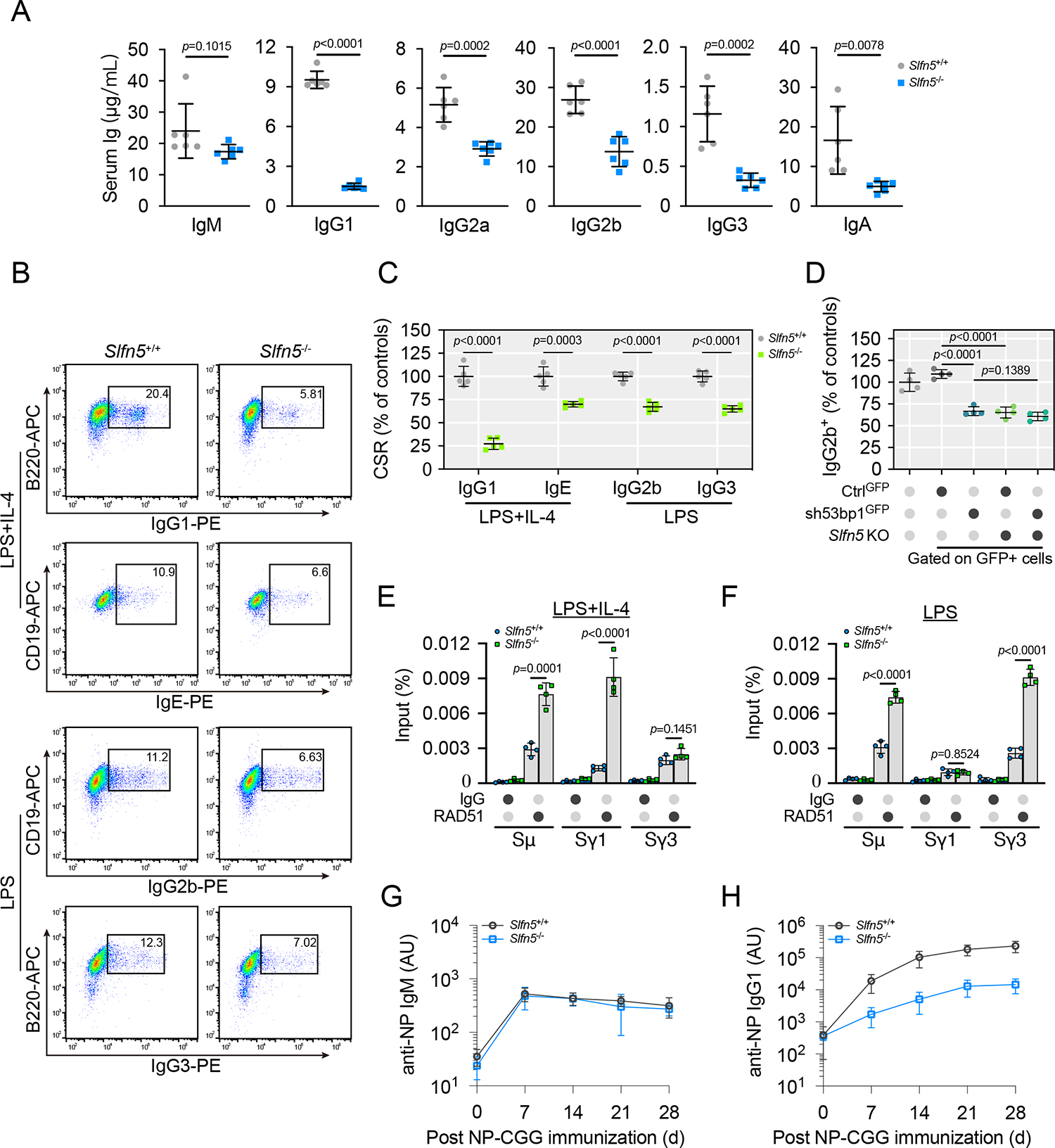
(A) Quantification of serum immunoglobulin from Slfn5+/+ and Slfn5−/− mice. mice per genotype. Error bars indicate mean ± SD.
(B and C) Slfn5+/+ and Slfn5−/− splenic B cells were cultured with the indicated stimuli for 96 h and stained for surface IgG1, IgE, IgG2b, and IgG3. Representative plots of flow cytometry (FACS) are shown in (B). Quantification of CSR percentage for indicated isotypes is shown in (C). mice per genotype. Error bars indicate mean ± SD.
(D) Slfn5+/+ and Slfn5−/− splenic B cells transduced with control-GFP (CtrlGFP) or 53bp1 shRNA-GFP (sh53bp1GFP) were cultured in the presence of lipopolysaccharides (LPS) for 96 h. Transduced cells were stained for IgG2b and analyzed by FACS. IgG2b quantification is shown in (D). Slfn5+/+ splenic B cell were included as a control. mice per genotype. Error bars indicate mean ± SD.
(E and F) Slfn5+/+ and Slfn5−/− splenic B cells were cultured in the presence of LPS + IL-4 (E) or LPS (F) for 96 h. RAD51 accumulation at the CSR-targeted S region DNA sites was detected by ChIP-qPCR. Error bars indicate the mean ± SD of four independent experiments.
(G and H) Slfn5+/+ and Slfn5−/− mice were immunized with 4-hydroxy-3-nitrophenyl)acetyl-chicken γ-globulin (NP-CGG), and NP-specific serum IgM (G) and IgG1 (H) were measured at indicated times after immunization. mice per genotype. Error bars indicate mean ± SD.
p values calculated with two-tailed unpaired t tests.
During CSR, cytidine deamination and DNA cleavage on opposite strands generate DSBs37. These DSBs at the switch region are repaired by components of the NHEJ pathway38,39. We found that SLFN5 deficiency resulted in RAD51 accumulation at the Sμ and Sγ regions (Figures 2E and 2F), without affecting the cell cycle (Figures S2Q–S2T). These findings suggest that SLFN5 loss shifts NHEJ to HR during CSR.
Finally, we tested the role of SLFN5 in CSR in vivo. To do so, Slfn5−/− mice and WT littermates were immunized with the antigen NP-CGG and monitored for the production of NP-specific Ig isotypes. We observed that serum concentrations of NP-specific IgG1 in Slfn5−/− mice were significantly lower than those of Slfn5+/+ mice, whereas NP-specific IgM levels were comparable between groups (Figures 2G and 2H). These data further support that SLFN5 functions in the 53BP1 axis.
Loss of SLFN5 restores HR in BRCA1-deficient cells
53BP1 plays an important role in PARPi sensitivity by protecting DSB ends4. The concurrent loss of 53BP1 in BRCA1-deficient cells has been shown to restore HR, leading to PARPi resistance17,20,40. Therefore, we hypothesized that SLFN5 depletion might modulate PARPi sensitivity. Like 53BP1, loss of SLFN5 conferred resistance to the PARPi olaparib in BRCA1-depleted U2OS cells (Figures 3A and 3B). Consistently, Slfn5−/− MEF cells with BRCA1 deficiency also showed marked resistance to olaparib or cisplatin (Figures 3C–3E). Furthermore, in BRCA1-deficient cells, SLFN5 depletion restored HR, similar to the effect in cells lacking 53BP1 (Figure 3F). Ionizing radiation (IR)-induced focus formation of RAD51 and RPA2 were also restored in the absence of both SLFN5 and BRCA1 (Figures 3G–3I), implying the reactivation of DNA end resection. In addition, the HR deficiency (HRD) score, which describes the sum of three metrics of chromosomal level aberration (loss of heterozygosity [LOH], telomeric allelic imbalance [TAI], and large-scale transitions [LSTs]), was analyzed in triple-negative breast cancer samples from TCGA. We found that SLFN5 deficiency had lower HRD scores in BRCA1-deficient breast tumors (Figure 3J), consistent with the effect of SLFN5 on HR in BRCA1-deficient cells. Taken together, these data suggest that, like 53BP1, loss of SLFN5 confers PARPi resistance in BRCA1-deficient cancers through the restoration of HR.
Figure 3. SLFN5 loss promotes PARPi resistance in BRCA1-deficient tumors.
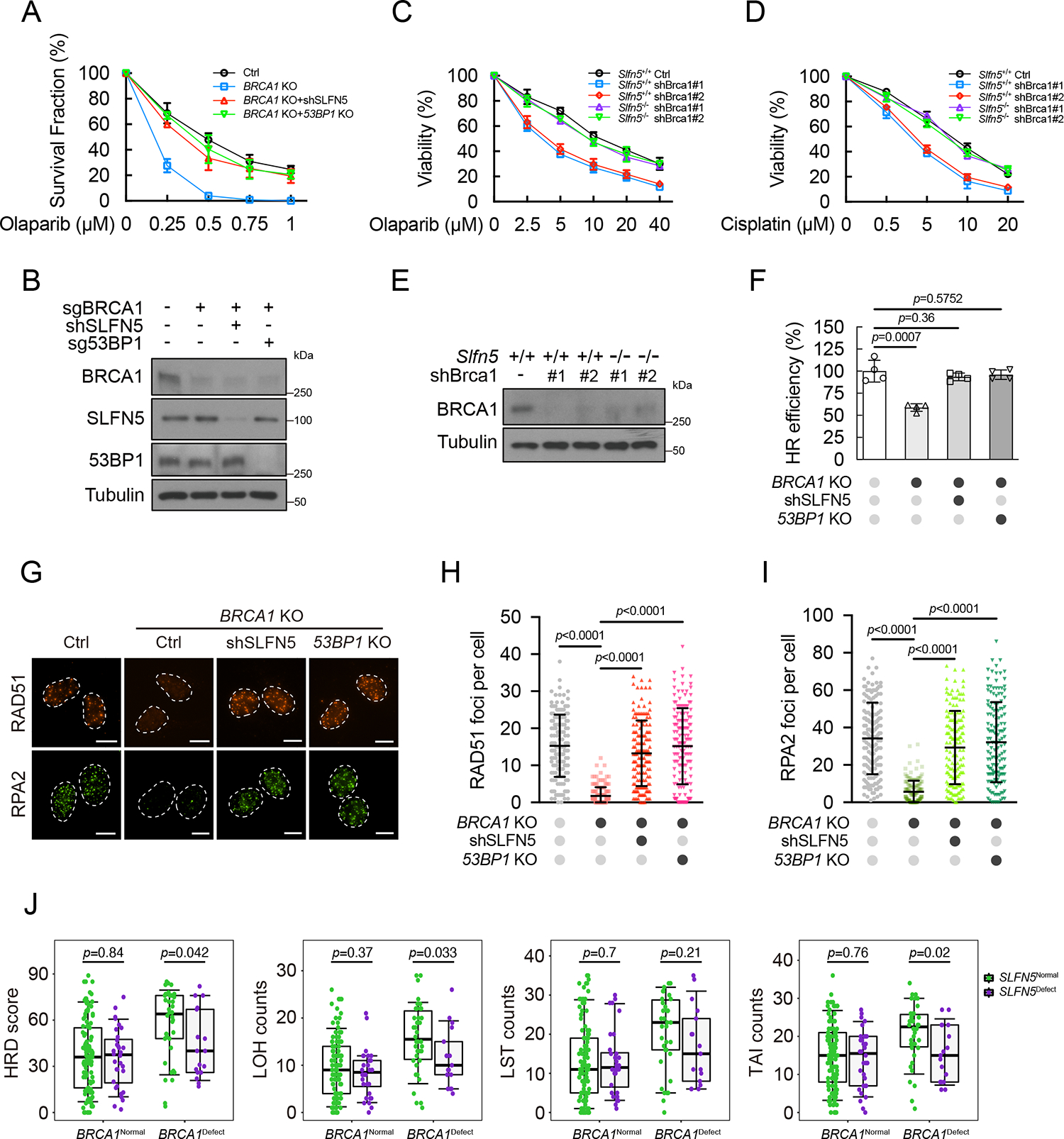
(A and B) The indicated U2OS cells were subjected to colony formation assays to assess olaparib sensitivity (A). Error bars indicate the mean ± SD of three independent experiments. The expression level of the indicated proteins was assessed by western blot (B).
(C-E) Survival assays for Slfn5+/+ and Slfn5−/− MEFs (with or without Brca1 knockdown) exposed to olaparib (C) or cisplatin (D). Error bars indicate the mean ± SD of six independent experiments. The expression level of the indicated proteins was assessed by western blot (E).
(F) Cells from (A) were subjected to a reporter assay to assess the efficiency of HR. Error bars indicate the mean ± SD of four independent experiments.
(G-I) Cells from (A) were exposed to irradiation (2 Gy, 4 h), and the indicated foci were detected by immunofluorescence. Representative images are shown in (G). Quantification of focus numbers is shown in (H and I). Each dot represents a single cell; per condition. Error bars indicate mean ± SD. Data are representative of three independent experiments. Scale bars, 10 μm.
(J) Analysis of HR deficiency (HRD) scores and loss of heterozygosity (LOH), large-scale transitions (LSTs), and telomeric allelic imbalance (TAI) counts in triple-negative breast cancer samples from TCGA. Each dot represents an individual sample, the center line represents the median, the box limits at 25th and 75th centiles, and the whiskers indicate ± 1.5× interquartile range; distinct patients.
p values calculated with two-tailed unpaired t tests (F, H, and I) or two-tailed Mann-Whitney U tests (J).
SLFN5 accumulates at DSB sites and functions downstream of 53BP1
The observed effects of SLFN5 on telomere fusions, CSR, and PARPi response suggest a role of SLFN5 in regulating 53BP1-dependent DSB repair. Indeed, SLFN5 deficiency resulted in enhanced γH2AX foci at late time points (Figures 4A and S3A–S3E), implying that SLFN5 loss impairs DSB repair in cells. Consistently, increased comet tail moments, an indicator for unrepaired DSB lesions, were also observed in SLFN5-depleted cells at late time points (Figures 4B and S3F–S3H). Using dual reporter assays, SLFN5 deficiency led to an increase in HR but a decrease in NHEJ without affecting the cell cycle (Figures S3I–S3M), suggesting a role for SLFN5 in regulating the balance of HR versus NHEJ. These data are consistent with SLFN5 acting in three specialized NHEJ contexts described above (see Figures 1, 2, and 3), establishing SLFN5 as a bona fide regulator of NHEJ in the 53BP1 pathway.
Figure 4. SLFN5 accumulates at DSB sites and functions downstream of 53BP1.
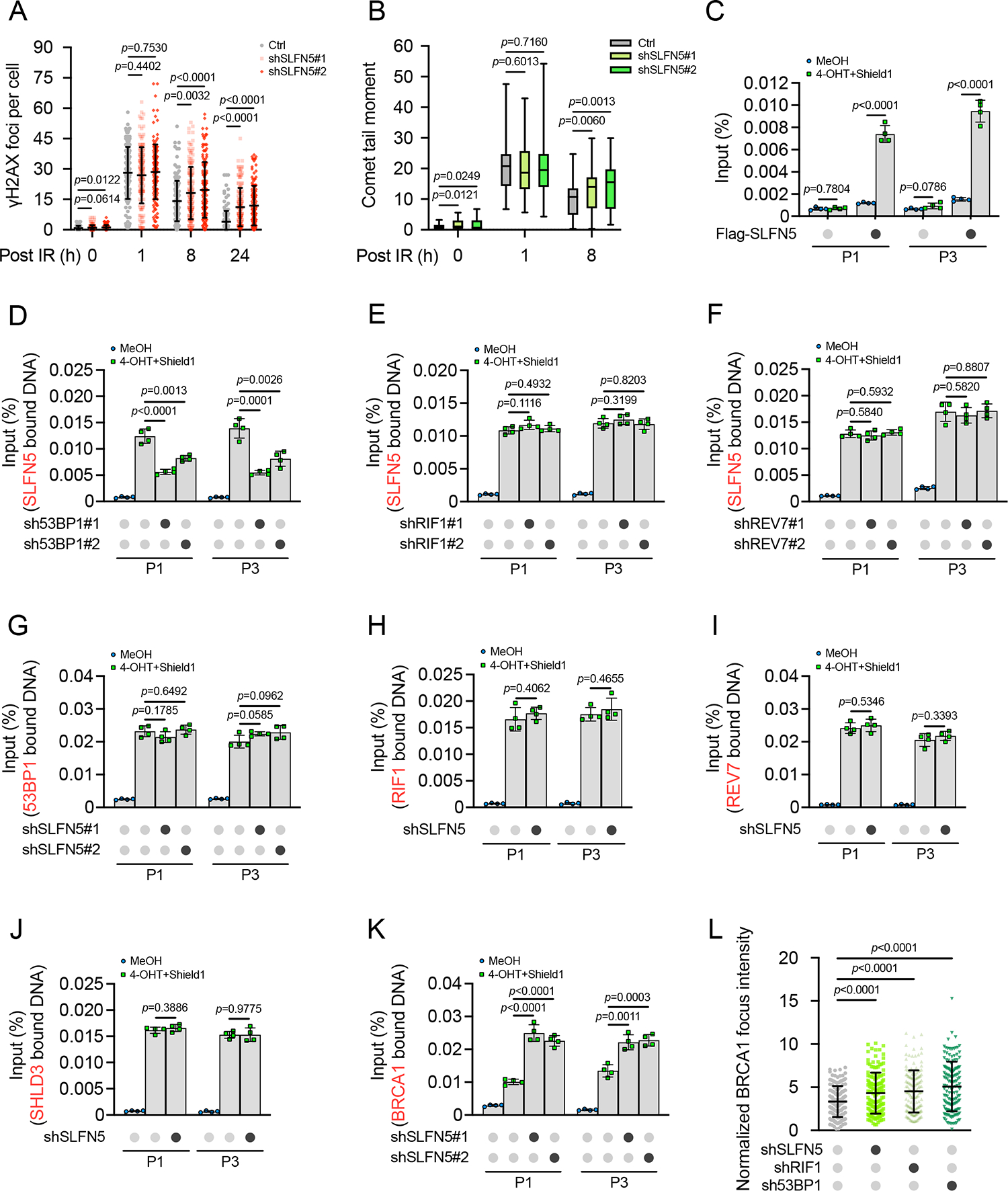
(A) Control (Ctrl) or SLFN5 knockdown U2OS cells were irradiated (2 Gy) for the indicated times. γH2AX foci were detected by immunofluorescence. Quantification of γH2AX focus numbers is shown in (A). Each dot represents a single cell; per condition. Error bars indicate mean ± SD. Data are representative of three independent experiments. Representative images are shown in Figure S3A.
(B) Quantification of tail moments from ctrl or SLFN5 knockdown U2OS cells that were irradiated (5 Gy) for the indicated times. Cells were harvested and subjected to neutral comet assay. The center line represents the median, the box limits at 25th and 75th centiles, and the whiskers indicate the minimum and maximum values; per condition. Data are representative of three independent experiments. Representative images are shown in Figure S3F.
(C) U2OS-FokI cells transfected with Vec or Flag-SLFN5 were treated with 4-OHT and Shield1 for 4 h. Flag-SLFN5 accumulation at DSB sites generated by FokI was detected by ChIP-qPCR. Error bars indicate the mean ± SD of four independent experiments.
(D-F) Ctrl or 53BP1 (D), RIF1 (E), or REV7 (F) knockdown U2OS-FokI cells transfected with Flag-SLFN5 were treated with MeOH or 4-OHT + Shield1 for 4 h. Flag-SLFN5 accumulation at DSB sites generated by FokI was detected by ChIP-qPCR. Error bars indicate the mean ± SD of four independent experiments.
(G) Ctrl or SLFN5 knockdown U2OS-FokI cells were treated with MeOH or 4-OHT + Shield1 for 4 h. 53BP1 accumulation at DSB sites generated by FokI was detected by ChIP-qPCR. Error bars indicate the mean ± SD of four independent experiments.
(H-J) Ctrl or SLFN5 knockdown U2OS-FokI cells transfected with Flag-RIF1 (H), SFB-REV7 (I), or SFB-SHLD3 (J) were treated with MeOH or 4-OHT + Shield1 for 4 h. Flag-RIF1 (H), SFB-REV7 (I), or SFB-SHLD3 (J) accumulation at DSB sites generated by FokI was detected by ChIP-qPCR. Error bars indicate the mean ± SD of four independent experiments.
(K) Ctrl or SLFN5 knockdown U2OS-FokI cells were treated with MeOH or 4-OHT + Shield1 for 4 h. BRCA1 accumulation at DSB sites generated by FokI was detected by ChIP-qPCR. Error bars indicate the mean ± SD of four independent experiments.
(L) The indicated U2OS cells were irradiated (2 Gy, 1 h), and the BRCA1 foci were detected by immunofluorescence. Each dot represents a single cell; per condition. Error bars indicate mean ± SD. Data are representative of three independent experiments. Representative images are shown in Figure S4B.
p values calculated with two-tailed unpaired t tests.
To address how SLFN5 promotes NHEJ, we next assessed its localization. We examined SLFN5 localization using an inducible mCherry-LacI-FokI nuclease to generate DSBs with local transcription silencing41,42. SLFN5 was localized at FokI-generated DSB sites, as revealed by co-localization with mCherry-FokI focus formation and quantifying its enrichment by chromatin immunoprecipitation qPCR (ChIP-qPCR) (Figures 4C and S3N).
We next determined whether SLFN5 localized to DSBs in a 53BP1-dependent manner. As shown in Figures 4D–4F and S3O–S3Q, the recruitment of SLFN5 to DSB sites was dramatically decreased in 53BP1-depleted cells, but not in RIF1/REV7-depleted cells, suggesting that SLFN5 recruitment to DSBs requires 53BP1. On the other hand, SLFN5 depletion did not affect the recruitment of 53BP1, RIF1 and Shieldin complex to DSBs (Figures 4G–4J), placing SLFN5 functions in a different functional module than RIF1-Shieldin. Conversely, the loss of SLFN5 significantly promoted BRCA1 recruitment at DSBs (Figure 4K). Consistently, BRCA1 focus intensity at DNA damage sites was enhanced upon SLFN5 depletion (Figures 4L, S4A, and S4B). Considering that SLFN5 and 53BP1 promote NHEJ while inhibiting HR, it is likely that the higher BRCA1 intensity in SLFN5-depleted cells is due to a defect in the 53BP1 pathway.
SLFN5 modulates the higher-order topological arrangement of 53BP1
Previous studies indicate that BRCA1 and 53BP1 spatially organize into defined and mutually exclusive territories at sites of damage chromatin6. Recent super-resolution imaging of DNA damage foci reveals a more complex chromatin topology with different domains occupied by BRCA1 and 53BP17. To characterize how SLFN5 affects BRCA1 recruitment in more detail, we set out to visualize the three-dimensional (3D) organization of BRCA1/53BP1 foci. We found that BRCA1 largely localized to focal compartments, whereas 53BP1 exhibited a high-order, ring-shaped pattern form (Figure 5A). This observation is consistent with a recent report suggesting that the 53BP1 repair focus consists of several nanodomains (NDs), which then assemble into a higher-ordered microdomain (MD)7. Notably, as shown in Figure 5A, the knockdown of SLFN5 enhanced BRCA1 focal appearance and spatial distribution, consistent with the brighter BRCA1 focus intensity observed under conventional microscopy. Interestingly, we found that the depletion of SLFN5 led to the disruption of 53BP1-MD architecture, which appeared as aberrantly disordered 53BP1-NDs (Figures 5A and S4F). Quantification of the topological textures7 of 53BP1-MDs also showed the knockdown of SLFN5 significantly increased the mean breadth and the aberrant distribution of 53BP1-MDs (Figures 5A–5C and S4C–S4G). In addition, the topologic arrangements of 53BP1-MDs affected by SLFN5 deficiency were cell cycle independent (Figures S4H–S4J). SLFN5 deficiency also impaired γH2AX-MD architecture that is predominately marked by 53BP1-MD7 (Figures S4K and S4L), indicating a role for SLFN5 in maintaining damaged chromatin topology. These results suggest that SLFN5 is required for the topological arrangement of 53BP1-MDs.
Figure 5. SLFN5 regulates the higher-order chromatin topology of 53BP1.
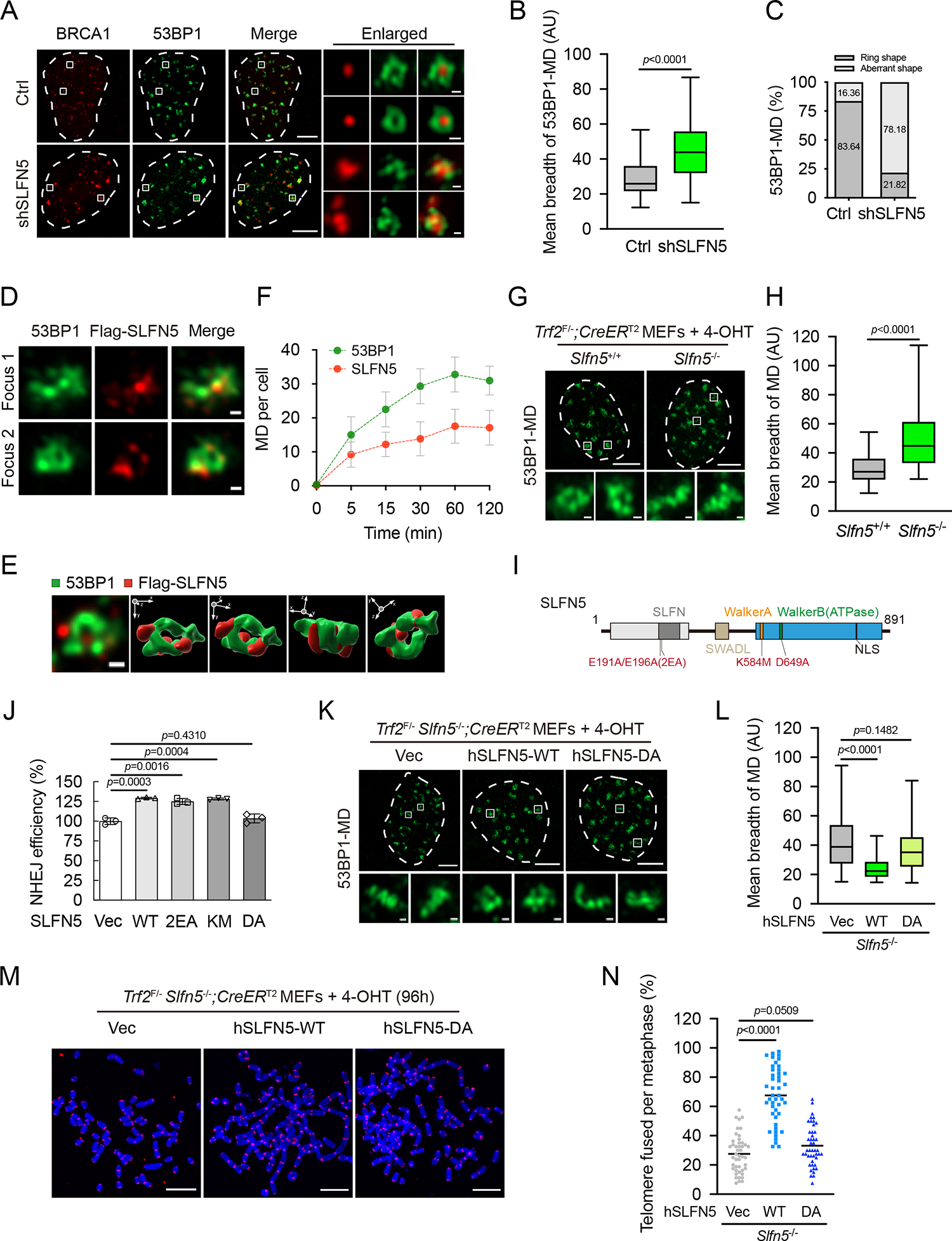
(A) Ctrl or SLFN5 knockdown U2OS cells were irradiated (2 Gy, 1 h), and the indicated foci were detected by Airyscan2 super-resolution microscope (Airyscan2-SR). Scale bars, 5 μm and 200 nm (insets).
(B) Quantification of the mean breadth of 53BP1 microdomains (53BP1-MDs) from (A) was performed by QUANTEX7. The center line represents the median, the box limits at 25th and 75th centiles, and the whiskers indicate the minimum and maximum values; per condition. Data are representative of three independent experiments.
(C) Quantification of the percentage of ring distribution patterns versus aberrant patterns of 53BP1-MDs from (A). per condition.
(D) U2OS cells stably expressing Flag-SLFN5 were irradiated (2 Gy, 1 h) and subjected to Airyscan2-SR with the indicated antibodies. Foci 1 and 2 are magnified from a large-field-of-view image in Figure S5A. Scale bars, 200 nm.
(E) A representative 3D view of an arrangement of 53BP1-MD with Flag-SLFN5. The 3D image is processed under maximal intensity projection. 3D opacity view is displayed in three orientations indicated by arrows.
(F) U2OS cells stably expressing Flag-SLFN5 were irradiated (2 Gy) for the indicated times and subjected to Airyscan2-SR with the indicated antibodies. Quantification of recruitment of 53BP1 and Flag-SLFN5 to DSB sites is shown in (F). per condition. Error bars indicate mean ± SD. Data are representative of two independent experiments. Representative images for the indicated times are shown in Figure S5.
(G and H) The indicated Trf2F/−; CreERT2 MEFs were treated with 4-OHT for 70 h. The 53BP1-MD signals were detected by Airyscan2-SR. Representative images are shown in (G). Quantification of mean breadth of 53BP1-MDs is shown in (H). The center line represents the median, the box limits at 25th and 75th centiles, and the whiskers indicate the minimum and maximum values; per condition. Data are representative of three independent experiments. Scale bars, 5 μm and 200 nm (insets).
(I) A schematic depiction of SLFN5 wild-type (WT) and its mutants.
(J) HEK293T cells were transfected with Vec, WT, or the indicated mutants of SLFN5 and were subjected to a reporter assay to assess the efficiency of NHEJ. Error bars indicate the mean ± SD of three independent experiments.
(K and L) Trf2F/− Slfn5−/−; CreERT2 MEFs stably expressing Vec, WT, or DA Flag-human SLFN5 were treated with 4-OHT for 70 h. The 53BP1-MD signals were detected by Airyscan2-SR. Representative images are shown in (K). Quantification of the mean breadth of 53BP1-MDs is shown in (L). The center line represents the median, the box limits at 25th and 75th centiles, and the whiskers indicate the minimum and maximum values; per condition. Data are representative of three independent experiments. Scale bars, 5 μm and 200 nm (insets).
(M and N) Trf2F/− Slfn5−/−; CreERT2 MEFs stably expressing Vec or WT or DA Flag-human SLFN5 were treated with 4-OHT for 96 h. Metaphase spreads were stained with Telomeric FISH and assessed for telomere fusion. Telomeres and DNA were stained with PNA probe (red) and DAPI (blue), respectively. Representative images are shown in (M). Quantification of the percentage of telomere fusions is shown in (N). Each dot represents an individual metaphase cell, and the center line indicates the mean; per condition. p values calculated with two-tailed unpaired t tests. Data are representative of three independent experiments. Scale bars, 10 μm.
p values calculated with two-tailed Mann-Whitney U tests (B, H, and L) or two-tailed unpaired t tests (J and N).
In support of this hypothesis, we found that SLFN5 localized to the 53BP1-MD, crossing the interfaces and borders of the ring pattern (Figures 5D, 5E, and S5A). Importantly, the kinetic recruitment of SLFN5 was similar to that of 53BP1, which was detectable early after DNA breakage and rose to its peak at around the subsequent 60 min (Figures 5F, S5A, and S5B). These data suggest that SLFN5 regulates the initial step of 53BP1-MD assembly. Interestingly, we observed that 53BP1-decorated dysfunctional telomere formed a higher-order topological structure (Figure 5G), which is consistent with observations at DSB ends, indicating that 53BP1-MDs present a general response in both DSB ends and deprotected telomeres. SLFN5 deficiency caused the topological disruption of 53BP1-MDs at dysfunctional telomeres (Figures 5G and 5H). We conclude that SLFN5 functions in higher-order chromatin assembly of 53BP1-MDs at both DSBs and deprotected telomeres.
The ATPase activity is required for SLFN5-mediated 53BP1-MD topology
Protein sequence analysis revealed that SLFN5 has an N-terminal nuclease domain, which is highly conserved with SLFN13’s RNase domain43, and the C-terminal domain contains a Walker A motif and Walker B ATPase motif29,44 (Figure 5I). To explore the role of SLFN5’s catalytic activity on 53BP1 regulation, we generated mutants at the putative RNase domain (E191A/E196A, 2EA), Walker A domain (K584M, KM), or Walker B domain (D649A, DA), respectively (Figures 5I and S6A). We found that only the DA mutant abolished SLFN5’s function in suppressing HR and promoting NHEJ (Figures 5J, S6B, and S6C). As expected, recombinant WT SLFN5 displayed ATPase activity in vitro, while the DA mutation abolished the ATPase activity of SLFN5 (Figures S6D and S6E). Furthermore, this ATPase activity was enhanced following DNA damage and this activation of ATPase activity was significantly suppressed by ATM inhibition (Figures S6F–S6I), suggesting that the ATPase activity of SLFN5 is regulated by the DNA damage response (DDR). To test the functional significance of SLFN5’s ATPase activity, we reconstituted SLFN5-WT or the DA mutant into cells in which endogenous SLFN5 had been depleted. SLFN5-WT rescued the topologically disordered 53BP1-MDs in SLFN5-deficient cells, whereas the DA mutant had no effect (Figures 5K, 5L, S6J, and S6K). In agreement with this data, human SLFN5-WT, rather than SLFN5-DA, could promote end-to-end telomere fusions induced by Trf2-KO (Figures 5M and 5N). Altogether, these results suggest that the ATPase activity of SLFN5 is necessary for the topologic arrangement of 53BP1.
SLFN5 regulates damaged chromatin mobility through 53BP1
To study how SLFN5 cooperates with 53BP1 to mediate its function, we set out to investigate which domain(s) of 53BP1 interacts with SLFN5. First, we found that SLFN5 interacted with the 53BP1 N-terminus (residues 1–1302), but not the Tudor or BRCT domain (Figures S7A–S7C). We then mapped the SLFN5-interacting region of 53BP1 using a panel of deletion and substitution mutants of the 53BP1 N-terminus (Figure 6A). We found that mutation of all 28 N-terminal S/TQ sites (Δ28) inhibited SLFN5 binding (Figures 6B and S7D). These mutations completely abolish its phosphorylation by ATM following DNA damage38. The deletion of the C-terminal BRCT domain (DB) had no effect. We further used the following mutations15,45 to pinpoint which S/TQ sites are required: a mutant harboring the first 15 N-terminal S/TQ sites (ΔPro), a mutant impairing 53BP1-mediated chromatin mobility (ΔMob), a mutant spanning the PTIP binding motif (ΔPTIP), and a mutant compromising RIF1 binding (ΔRIF1). We also included a mutant with impaired oligomerization of 53BP1 (ΔCore)45. Notably, 53BP1-ΔMob and 53BP1-ΔCore significantly compromised the interaction between 53BP1 and SLFN5 (Figures 6B and S7D). The Mob motif has previously been shown to regulate DSB/chromatin movement following DNA damage15. These studies suggest that SLFN5 interacts with 53BP1 functional modules that mediate DNA damage-dependent chromatin mobility and oligomerization.
Figure 6. SLFN5 promotes 53BP1-dependent DSB dynamics.
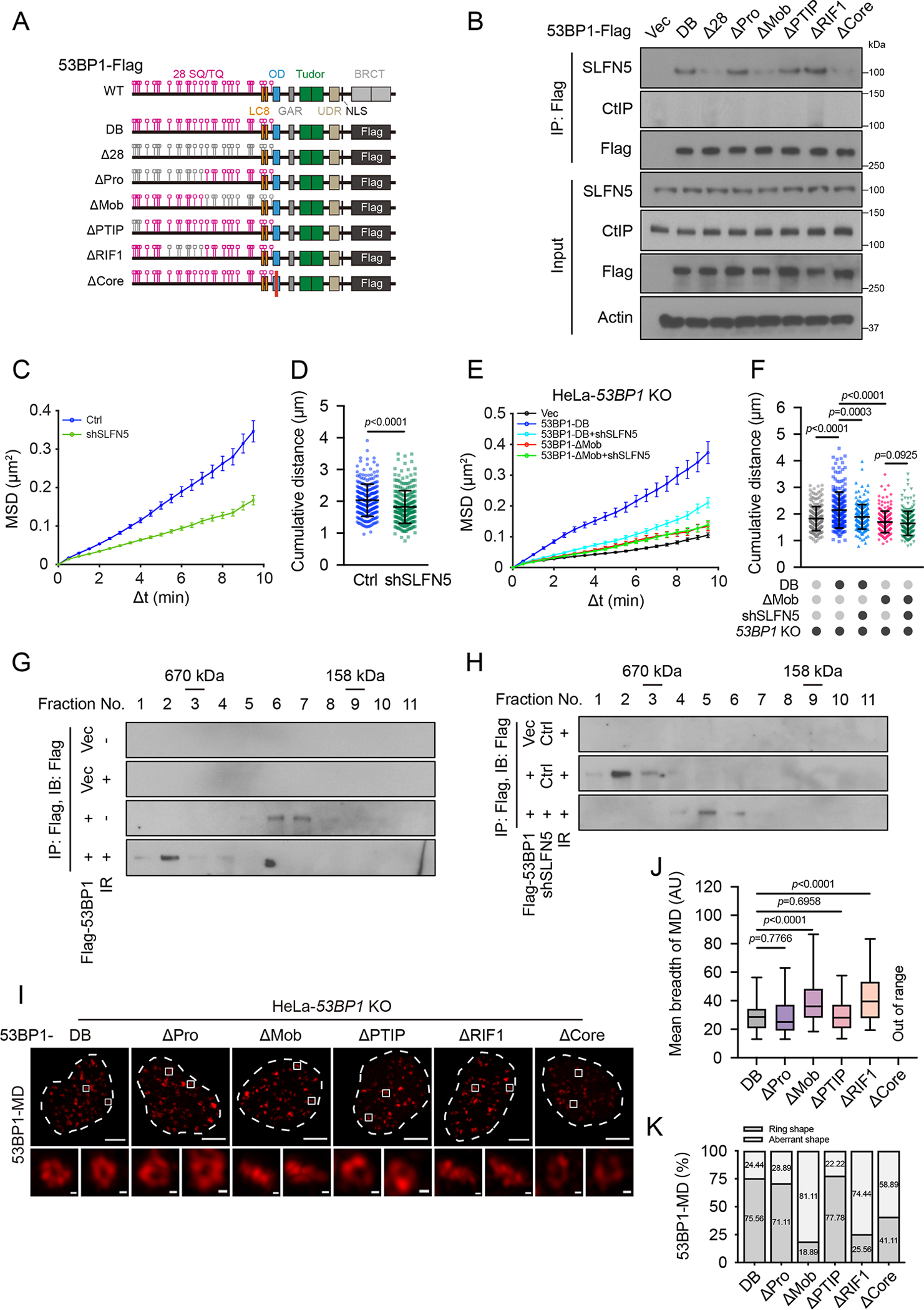
(A) A schematic depiction of 53BP1 mutants.
(B) 53BP1 knockout (KO) 293A cells transfected with the indicated 53BP1-Flag constructs were irradiated (5 Gy, 1 h). Cell lysates were immunoprecipitated with Flag beads and blotted with the indicated antibodies.
(C) Ctrl or SLFN5 knockdown U2OS cells expressing mCherry-BP1-2 were irradiated (2 Gy, 10 min). mCherry-BP1-2 foci were then traced over 10 min via live-cell imaging. Mean square displacement (MSD) analysis of mCherry-BP1-2 foci is shown in (C). Δt, time interval. and 267. Error bars indicate mean ± weighted SD. Data are representative of three independent experiments.
(D) Median cumulative distance traveled by mCherry-BP1-2 foci from (C). Error bars indicate mean ± SD. and 267. Data are representative of three independent experiments.
€ The indicated HeLa cells expressing mCherry-BP1-2 were irradiated (2 Gy, 10 min). mCherry-BP1-2 foci were then traced over 10 min via live-cell imaging. MSD analysis of mCherry-BP1-2 foci is shown in €. Δt, time interval. , 230, 205, 211, and 208. Error bars indicate mean ± weighted SD. Data are representative of three independent experiments.
(F) Median cumulative distance traveled by mCherry-BP1-2 foci from (E). Error bars indicate mean ± SD. , 230, 205, 211, and 208. Data are representative of three independent experiments.
(G) Flag-53BP1 was purified from HEK293T cells untreated or irradiated (5 Gy, 1 h). Samples were fractionated by fast protein liquid chromatography (FPLC) and then immunoblotted with an anti-Flag antibody.
(H) Flag-53BP1 was purified from ctrl or SLFN5 knockdown HEK293T cells after irradiation (5 Gy, 1 h). Samples were fractionated by FPLC and immunoblotted with an anti-Flag antibody.
(I and J) The indicated HeLa cells were exposed to irradiation (2 Gy, 1 h). 53BP1-MD signals were detected by Airyscan2-SR. Representative images are shown in (I). Quantification of the mean breadth of 53BP1-MDs is shown in (J). The center line represents the median, the box limits at 25th and 75th centiles, and the whiskers indicate the minimum and maximum values; per condition. Data are representative of three independent experiments. Scale bars, 5 μm and 200 nm (insets). Out of Range, focus intensity of 53BP1-ΔCore mutant is too weak/discrete (non-continuous) to detect the successive signals.
(K) Quantification of the percentage of ring distribution patterns versus aberrant patterns of 53BP1-MDs from (I). per condition.
p values calculated with two-tailed Mann-Whitney U tests.
To test this possibility, we first investigated whether SLFN5 influences chromatin mobility during DSB repair. We visualized DSB chromatin movements in cells stably expressing an mCherry-53BP1 fusion protein (mCherry-BP1-2), a widely used indicator for monitoring DSB chromatin mobility without affecting endogenous 53BP1 function14,15,46. Mean-square displacement (MSD) analysis of mCherry-BP1-2 foci in U2OS cells revealed a diffusion coefficient of 1.008 × 10−4 μm2 s−1, which is in the range observed by previous reports15,47,48 (Figures 6C, S8A, and S8K; Video S1). Calculation of the anomalous diffusion coefficient (α) showed values of all groups were consistent with sub-diffusive motion (Figure S8K). Quantification of DSB tracks indicated that mCherry-BP1-2 foci in U2OS cells explored a mean cumulative distance of 2.04 μm over 10 min (Figure 6D). Accordingly, we investigated DSB movements by tracking mCherry-BP1-2 foci and calculating the above motion properties. Notably, SLFN5 deficiency significantly decreased mCherry-BP1-2 foci diffusion coefficient from 1.008 × 10−4 μm2 s−1 to 0.5633 × 10−4 μm2 s−1 (Figure 6C; Video S1). Likewise, the distance traveled by mCherry-BP1-2 foci was diminished in SLFN5-depleted cells (Figures 6D and S8K). These data suggest that SLFN5 loss impairs damaged chromatin mobility. Next, we asked whether the dynamic behavior of damaged chromatin is dependent on the 53BP1-SLFN5 interaction. We reconstituted 53BP1-KO HeLa cells with 53BP1-DB and 53BP1-ΔMob, respectively. Unlike 53BP1-DB, 53BP1-ΔMob was unable to restore damaged chromatin movement, as previously reported15. We found that SLFN5 knockdown did not affect the DSB mobility or the total traveled distance in 53BP1-KO cells expressing 53BP1-ΔMob (Figures 6E, 6F, S8B, and S8K; Video S2), suggesting that SLFN5 regulates DSB chromatin movements through its interaction with 53BP1. In addition, SLFN5-DA, but not SLFN5-WT, failed to rescue impaired damaged chromatin movements in SLFN5-deficient cells (Figures S8C–S8E and S8K; Video S3). These results suggest that SLFN5-mediated motion of damaged chromatin requires its interaction with 53BP1 and its ATPase activity.
To further test this possibility, we assessed the impact of SLFN5 on the movement of dysfunctional telomeres. Motion analysis of mCherry-BP1-2 foci at dysfunctional telomeres revealed a diffusion coefficient of 1.614 × 10−4 μm2 s−1, a mean cumulative distance of 2.34 μm over 10 min, and the α-coefficient indicated sub-diffusive motion (Figure S8K). Notably, the depletion of SLFN5 significantly decreased the mobility of deprotected telomeres (Figures S8F, S8G, and S8K; Video S4) and this limited telomeric mobility could not be restored by human SLFN5-DA mutant (Figures S8H–S8K; Video S5). These results suggest that SLFN5 promotes damaged telomere dynamics, as it does at DSB ends.
Because SLFN5 also interacts with the 53BP1 oligomerization domain, we examined whether SLFN5 regulates 53BP1 oligomerization. We utilized fast protein liquid chromatography (FPLC) to analyze the 53BP1 complex purified from cell lysates upon DNA damage. We observed, following DNA damage, 53BP1 showed an increase in higher-molecular-weight complexes (Figure 6G). However, SLFN5 deficiency decreased the sizes of 53BP1 complexes and redistributed it from larger to smaller ones (Figure 6H), suggesting that SLFN5 may play a role in promoting 53BP1 oligomerization.
Taken together, our data suggest that the SLFN5–53BP1 interaction is critical for 53BP1-mediated damaged chromatin mobility and 53BP1 oligomerization. The regulation of damaged chromatin dynamics by SLFN5 and its interaction with the 53BP1 mobility domain is very similar to a Protein X proposed to promote 53BP1-dependent mobility of DSB ends4,15.
53BP1 Mob and Core domains are required for its topological arrangement
Based on the results shown above, we hypothesized that the regulation of 53BP1 and damaged chromatin movement might be an underlying mechanism by which SLFN5 regulates the topological arrangement of 53BP1. To this end, we reconstituted 53BP1-KO HeLa cells with a panel of deletion and substitution mutants of 53BP1. The 53BP1-ΔRIF1 mutant disrupted 53BP1 topology, consistent with a previous report7, which suggests that RIF1 stabilizes the formation of chromatin topology. Importantly, the 53BP1-ΔMob mutant failed to mature into a higher-order topological formation of 53BP1-MD, phenocopying the topological disruptions in SLFN5-depleted cells (Figures 6I–6K). The 53BP1-MD signals were also weaker and discontinuous in the 53BP1-ΔCore mutant (Figures 6I–6K). These data suggest that 53BP1-dependent mobility of damaged chromatin and 53BP1 oligomerization favor the topological arrangement of 53BP1-MDs, and SLFN5 appears to have a unique role in promoting this process.
Our data suggest that both 53BP1 Mob and Core domains are important for 53BP1-MD formation. We next studied their relationship. We found that the 53BP1-ΔCore mutants did not affect damaged chromatin mobility and the 53BP1-ΔMob mutant did not affect 53BP1 oligomerization, suggesting that the 53BP1-ΔMob and 53BP1-ΔCore mutants are separation-of-function mutations4,49 (Figures S8K and S9A–S9C). Given the single mutant (ΔMob or ΔCore) suppresses higher-order assembly of 53BP1-MD (see Figure 6) yet has a modest effect on DNA end resection15,49, we therefore evaluated the ability of double mutants (ΔMob/ΔCore) to suppress end resection in cells lacking both 53BP1 and BRCA1. Single mutants of 53BP1 suppressed IR-induced RAD51 focus formation (Figures S9D–S9G), though they are modestly less effective than 53BP1-DB. By contrast, the 53BP1-ΔMob/ΔCore mutant almost abolished the inhibition of RAD51 foci (Figures S9F and S9G). These results suggest that higher-order assembly of 53BP1-MD may be important for its role in blocking resection. In support of this, we found that the double-mutant 53BP1 restored HR to a greater extent (Figure S9H). These data suggest that the higher-order topological formation of 53BP1-MD is important for 53BP1 activity that suppresses HR.
A previous study suggests that dynamic microtubule-LINC complex contributes to 53BP1-mediated DSB mobility15, although how the LINC complex is connected to 53BP1 is not clear, and there might exist an X factor in between. We hypothesized that SLFN5 is said factor that connects the LINC complex to 53BP1 to promote chromatin movement and high order structure of 53BP1. Consistent with this hypothesis, we found that chemical perturbation of microtubule dynamics but not actin polymerization led to the topological disorder of 53BP1-MDs (Figures 7A and 7B). Moreover, the LINC complex subunits SUN1 and SUN2 strongly interacted with SLFN5 (Figures 7C and S7E). The knockdown of SUN1/2 disrupted the 53BP1 topological structure, phenocopying SLFN5 depletion (Figures 7D–7F). Consistently, a mutant lacking the nucleoplasmic domain of SUN2 failed to rescue the topologically disordered 53BP1-MDs as well as the interaction with SLFN5 in SUN1/2-depleted cells (Figures 7G–7I and S7F), indicating that SLFN5 acts through interaction with SUN. These results suggest that the microtubule-LINC complex promotes DSB chromatin mobility and drives the topological arrangement of 53BP1-MDs, and SLFN5 acts as a potential link between 53BP1 function and dynamic microtubule-LINC complex in this highly dynamic process.
Figure 7. Microtubule-LINC complex mediates the topological arrangement of 53BP1-MDs.
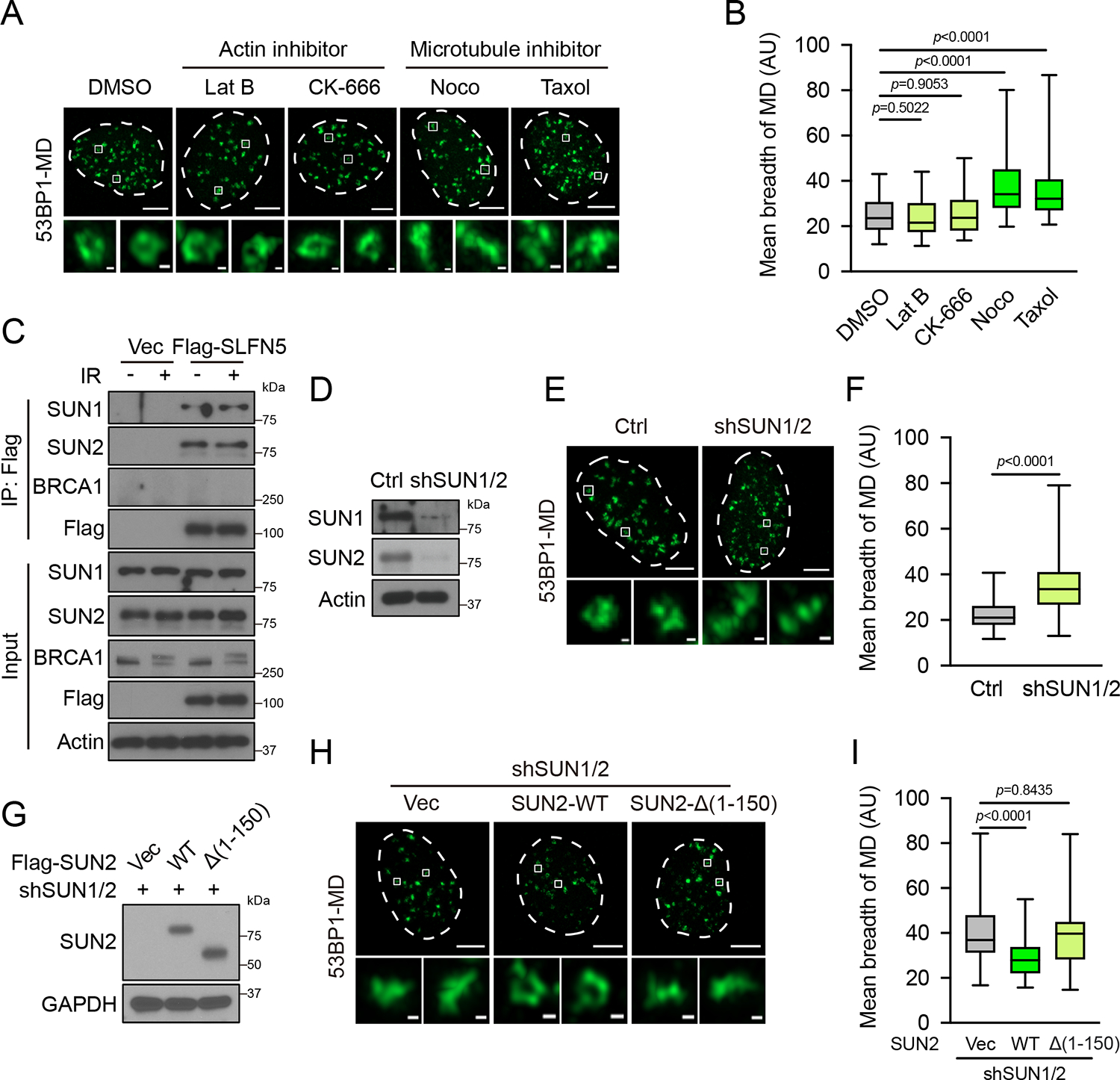
(A and B) U2OS cells were irradiated (2 Gy, 1 h) with Latrunculin B (4 μM), CK-666 (100 μM), Nocodazole (1 μg/mL), or Taxol (20 μM). The 53BP1-MD signals were detected by Airyscan2-SR. Representative images are shown in (A). Quantification of the mean breadth of 53BP1-MDs (B). The center line represents the median, the box limits at 25th and 75th centiles, and the whiskers indicate the minimum and maximum values; per condition. Data are representative of three independent experiments. Scale bars, 5 μm and 200 nm (insets).
(C) HEK293T cells transfected with Vec or Flag-SLFN5 were untreated or irradiated (5 Gy, 1 h). Cell lysates were immunoprecipitated with Flag beads and blotted with the indicated antibodies.
(D-F) Ctrl or SUN1/2 knockdown U2OS cells were irradiated (2 Gy, 1 h). The 53BP1-MD signals were detected by Airyscan2-SR. Immunoblots of the indicated proteins are shown in (D). Representative images are shown in (E). Quantification of the mean breadth of 53BP1-MDs is shown in (F). The center line represents the median, the box limits at 25th and 75th centiles, and the whiskers indicate the minimum and maximum values; per condition. Data are representative of three independent experiments. Scale bars, 5 μm and 200 nm (insets).
(G-I) SUN1/2 knockdown U2OS cells stably expressing Vec, WT, or nucleoplasmic domain mutant Flag-SUN2 were irradiated (2 Gy, 1 h). Immunoblots of the indicated proteins are shown in (G). Representative images are shown in (H). Quantification of the mean breadth of 53BP1-MDs is shown in (I). The center line represents the median, the box limits at 25th and 75th centiles, and the whiskers indicate the minimum and maximum values; per condition. Data are representative of three independent experiments. Scale bars, 5 μm and 200 nm (insets).
p values calculated with two-tailed Mann-Whitney U tests.
Collectively, our findings reveal that SLFN5 is a key effector that mediates 53BP1-dependent DSB chromatin mobility and 53BP1 oligomerization, and the formation of a functional 53BP1 topological module.
DISCUSSION
Our results reveal an important role of SLFN5 in the topological organization of 53BP1. We propose a model in which SLFN5 interacts with 53BP1 to promote damaged chromatin mobility and 53BP1 oligomerization. When DSBs trigger 53BP1 chromatin decoration to form NDs, SLFN5 might enable damaged chromatin mobility and oligomerization of 53BP1 to prompt the formation of a higher-order 53BP1 MD. RIF1 subsequently reinforces this topology7. The depletion of SLFN5 or RIF17 leads to the topological disruption of 53BP1-MDs, causing BRCA1 spreading, excessive DSB resection, and genomic instability. Our observations suggest a temporal difference between SLFN5 and RIF1 in the 53BP1 spatial events. In contrast to RIF1, which is recruited later and located at the borders between 53BP1-NDs7, SLFN5 recruitment occurred simultaneously with 53BP1 and was largely localized in spots or boundaries of 53BP1-MD. Thus, we propose that the recruitment of SLFN5 at DSBs contributes to modulating the initial step of chromatin arrangement of 53BP1-MD topology, which RIF1 helps to stabilize once it forms.
Interestingly, SLFN5 interacts with the Mob and Core domains of 53BP1, implying that SLFN5 has a unique role in the 53BP1 functional modules. The topological disruption of 53BP1-MD was reproduced by the 53BP1 mutant that abrogates its regulation on DSB mobility15, supporting the notion that DSB or damaged chromatin mobility is linked to 53BP1-mediated topology arrangement. More broadly, we note that such 53BP1 spatial events are also common at dysfunctional telomeres, suggesting that a higher-order assembly of 53BP1-MD is a central feature of its function to protect DNA ends. The present model extends our mechanical insight into the mobility of damaged chromatin and the formation of 53BP1 higher-order topology. The requirement of SLFN5’s ATPase in 53BP1 topological rearrangement further suggests that SLFN5 plays a unique role in the formation of higher-order structure, apart from the action of other well-known 53BP1 effectors such as RIF1, PTIP, and so on. A plausible scenario is that once SLFN5 accumulates at damaged chromatin regions, SLFN5 may catalyze the ATP hydrolysis process, which provides the energy that is necessary for the change of chromatin dynamics and topology50,51. We suggest that DSB dynamics prompt DSB-flanking chromatin to form a favorable topology to protect DNA ends. Both chromatin topology and chromatin mobility are proposed to be important for genomic stability4,5,7,14,15,52–the discovery here potentially links the two processes through SLFN5. However, the temporal order of these two processes is not precisely determined and should be further investigated in the future.
The fact that SLFN5 mediates 53BP1-dependent DSB dynamic behavior is reminiscent of an unidentified factor linking 53BP1 and microtubule-LINC complex during DSB mobility15. We found that SLFN5 interacted with the nucleoplasmic region of SUN proteins, the inner components of the LINC complex. Importantly, interfering with microtubule dynamics or depletion of SUN proteins phenocopied the topological disruption of 53BP1 by SLFN5 depletion. This study therefore establishes a link between microtubule forces and the arrangement of 53BP1 topology to potentiate the NHEJ process. It is interesting that nuclear F-actin facilitates HR repair of DSBs53,54 or replication stress55, whereas microtubule forces potentiate NHEJ through the arrangement of 53BP1/damaged chromatin topology. As the LINC complexes are localized at the nuclear envelope (NE), one interpretation is that SLFN5 as a connector brings the repair complexes to the NE. Consistent with this, recent live 3D imaging studies showed that global chromatin organization is fairly close to the NE56,57, providing another likely scenario that most DSBs are peripheral, and the repair complexes assemble near the NE. The LINC complexes and microtubule force they transduced might serve to position and organize 53BP1 complexes at the inner face of the NE through SLFN5. These issues warrant attention in future studies. Other studies also suggest that chromatin mobility at sites of DSBs not only stems from external cytoskeletal forces4,15 but also arises from internal alteration of chromatin structure52. The latter model proposed INO80-C dependent histone degradation drives damaged chromatin mobility52,58. INO80 dependent chromatin remodeling involves ATPase activity. It will be interesting to test whether this process sculpts the 3D chromatin architecture and whether SLFN5 has an epistatic role.
The idea that SLFN5 is important to facilitate the topological arrangement of 53BP1 and that loss of SLFN5 is sufficient to restore HR and confer PARPi resistance in the face of BRCA1 deficiency for the first time establishes the clinical significance of 53BP1 topology. Furthermore, our findings suggest a possibility that perturbation of microtubule dynamics might endow HR-deficient cells with the ability to tolerate PARPi treatment. Another SLFN family protein, SLFN11 has been reported to inhibit HR by promoting the disassociation of RPA-ssDNA complex30, and the loss of SLFN11 has also been shown to induce PARPi resistance59. Although the evolutionary distance between SLFN5 and SLFN11 supports their classification into distinct phylogenetic groups and the evolution of SLFN5 is more conserved across species27, whether SLFN11 contributes to the regulation of chromatin dynamics and topology is a noteworthy possibility to be explored. Our analysis hints that the remaining human SLFN proteins may function in genome integrity and supports future efforts to study the precise mechanisms SLFN members play in genome instability or human disease.
In conclusion, we present evidence that SLFN5 is an important regulator of 53BP1 chromatin topology in DSB repair. Analogous to 53BP1, SLFN5 regulates NHEJ-dependent physiological processes including telomere fusions, CSR, and PARPi sensitivity. We propose a model where SLFN5 promotes the dynamic behavior of DSBs and induces a 53BP1 higher-order topological arrangement, to promote NHEJ, end protection, telomere fusion, and genome stability.
Limitations of the Study
Many of our conclusions are based on observations under super-resolution imaging. However, the work cannot completely rule out the possibility that SLFN5 also affects basal chromatin architecture in the absence of DSB induction. Whether SLFN5 depletion disrupts other chromatin topology/repair factories remains unclear. We analyzed DSB motion and 53BP1 topology, a 3D process, using 2D-maximum-intensity projections. The nuclei we studied are intrinsically flat, so 2D motion can largely capture the 3D mobility15,53. Furthermore, follow-up studies are required to mechanistically study how SLFN5 mediates 53BP1 function. For example, our results show that the ATPase domain of SLFN5 is important, but its catalytic function during 53BP1-MD formation remains unclear. Because we propose that SLFN5 connects microtubule-LINC and 53BP1, the relationship and signaling among 53BP1, SLFN5, LINC, NE, and the nuclear and cytoskeletal forces merit further testing. It also will be of interest to test whether SLFN5 directly connects the LINC complex to DDR foci or regulates other factor(s) that does. Finally, although we evaluated the impact of disabling the 53BP1 mobility module in 53BP1 topology, we did not assess the DSB movements/53BP1 topology in HR-deficient strain with and without SLFN5. The temporal correlation between 53BP1-mediated mobility and the formation of 53BP1-MD remains to be determined.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Zhenkun Lou (Lou.Zhenkun@mayo.edu).
Materials Availability
Plasmids and cell lines generated in this study will be available upon request from the lead contact.
Data and Code Availability
Original imaging data (including microscopy, gels and western blots) have been deposited to Mendeley Data (http://dx.doi.org/10.17632/9n4g54vcdh.1).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Slfn5 knockout mice
Slfn5 knockout (KO) mice (C57BL/6J strain) were generated at Cyagen Biosciences. Briefly, Slfn5 KO mice were generated using the CRISPR–Cas9 systems. The guide RNA1 (sense: TCCTTCTTGTTCTAGTCTAATGG) and guide RNA2 (antisense: TTTCAGCAACGGCCTTCGAAAGG) were used to target the region between exon 3 and exon 6 of Slfn5. The size of effective KO region is 6331 base pair (bp). The guide RNAs were synthesized and co-injected with Cas9 mRNA into fertilized eggs from C57BL/6J females (3–4-week-old). Surviving embryos were transferred into the oviducts of pseudo-pregnant females (8–10-week-old). Genotyping was performed by PCR analyses of tail DNA using three primers: forward1 (TTTACAGATGACCCGAGAGACTTT), forward2 (CTATGATTTCAGGGTGAGTCCAG), reverse (AGTTTCAGAGAAGCCGAGCGTGG). The PCR products are 516 bp for the wild-type allele and 584 bp for the KO allele. All the pups were screened by genotyping with primers flanking the targeting sites followed by sequencing of the PCR products. Both male and female mice were used in this study. All the animal procedures were approved by Mayo Clinic/Tongji University Institutional Animal Care and Use Committee.
Cell lines
Cell lines used in this study are listed in the Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-SLFN5 | Novus Biologicals | Cat#NBP1-81178; RRID:AB_11003398 |
| Rabbit polyclonal anti-53BP1 | Novus Biologicals | Cat#NB100-304; RRID:AB_10003037 |
| Mouse monoclonal anti-BRCA1 | Santa Cruz | Cat#sc-6954; RRID:AB_626761 |
| Mouse monoclonal anti-BRCA1 | GeneTex | Cat#GTX70111; RRID:AB_368627 |
| Mouse monoclonal anti-phospho-Histone H2A.X (Ser139) | Millipore | Cat#05-636; RRID:AB_309864 |
| Rabbit polyclonal anti-RIF1 | Bethyl Laboratories | Cat#A300-569A; RRID:AB_669804 |
| Rabbit polyclonal anti-PTIP | Thermo Fisher Scientific | Cat#A300-370A; RRID:AB_2160127 |
| Mouse monoclonal anti- REV7/MAD2L2 | Santa Cruz | Cat#sc-135977; RRID:AB_2139534 |
| Rabbit polyclonal anti-SHLD3 | Novus Biologicals | Cat#NBP2-49564 |
| Mouse monoclonal anti-CtIP | Active Motif | Cat#61141; RRID:AB_2714164 |
| Goat polyclonal anti-MCM2 | Bethyl Laboratories | Cat#A300-122A; RRID:AB_155897 |
| Rabbit polyclonal anti-RAD51 | GeneTex | Cat#GTX100469; RRID:AB_1951602 |
| Rabbit polyclonal anti-RAD51 | Abcam | Cat#ab176458; RRID:AB_2665405 |
| Mouse monoclonal anti-RPA2 | Santa Cruz | Cat#sc-56770; RRID:AB_785534 |
| Rabbit polyclonal anti-SUN1 | Sigma-Aldrich | Cat#HPA008346; RRID:AB_1080462 |
| Rabbit polyclonal anti-SUN2 | Sigma-Aldrich | Cat#HPA001209; RRID:AB_1080465 |
| Rabbit monoclonal anti-phospho--Chk2 (Thr68) | Cell Signaling | Cat#2197; RRID:AB_2080501 |
| Rabbit polyclonal anti-mouse TRF1(#1449) | Gift from Dr. Titia de Lange; Lottersberger et al.15 | N/A |
| Mouse monoclonal anti-FLAG | Sigma-Aldrich | Cat#F1804; RRID:AB_262044 |
| Mouse monoclonal anti-HA | Sigma-Aldrich | Cat#H9658; RRID:AB_260092 |
| Mouse monoclonal anti-GFP | Santa Cruz | Cat#sc-9996; RRID:AB_627695 |
| Mouse monoclonal anti-β-Actin | Sigma-Aldrich | Cat#A2228; RRID:AB_476697 |
| Mouse monoclonal anti-α-Tubulin | Sigma-Aldrich | Cat#T5168; RRID:AB_477579 |
| Mouse monoclonal anti-GAPDH | Proteintech | Cat#60004-1-Ig; RRID:AB_2107436 |
| Rabbit IgG (ChIP grade) | Abcam | Cat#ab171870; RRID:AB_2687657 |
| Normal rabbit IgG | Millipore | Cat#12-370; RRID:AB_145841 |
| APC mouse anti-phospho-Histone H2A.X (Ser139) | BioLegend | Cat#613416; RRID:AB_2629534 |
| APC rat anti-mouse CD71 | BioLegend | Cat#113820; RRID:AB_2728135 |
| FITC rat anti-mouse CD19 | BD Biosciences | Cat#557398; RRID:AB_396681 |
| APC rat anti-mouse CD19 | BD Biosciences | Cat#550992; RRID:AB_398483 |
| APC rat anti-mouse B220 | BioLegend | Cat#103212; RRID:AB_312997; |
| PE rat anti-mouse IgG1 | BD Biosciences | Cat#562027; RRID:AB_10894761 |
| PE rat anti-mouse IgG2b | BioLegend | Cat#406708; RRID:AB_2563381 |
| PE rat anti-mouse IgE | BioLegend | Cat#406907; RRID:AB_493291 |
| Rat anti-mouse IgG3 | BD Biosciences | Cat#553401; RRID:AB_394838 |
| FITC rat anti-mouse CD45 | BioLegend | Cat#103108; RRID:AB_312973 |
| APC rat anti-mouse CD4 | BD Biosciences | Cat#553051; RRID:AB_398528 |
| PE rat anti-mouse CD4 | BioLegend | Cat#100512; RRID:AB_312715 |
| APC rat anti-mouse CD8a | BioLegend | Cat#100712; RRID:AB_312751 |
| AP-conjugated goat anti-mouse IgM | SouthernBiotech | Cat#1020-04; RRID:AB_2794200 |
| AP-conjugated goat anti-mouse IgG1 | SouthernBiotech | Cat#1070-04; RRID:AB_2794411 |
| Alexa Fluor 488-labeled goat anti-mouse IgG (H+L) | Jackson ImmunoResearch | Cat#115-545-062; RRID:AB_2338845 |
| Alexa Fluor 488-labeled goat anti-rabbit IgG (H+L) | Jackson ImmunoResearch | Cat#111-545-045; RRID:AB_2338049 |
| Alexa Fluor 488- labeled donkey anti-rabbit IgG (H+L) | Jackson ImmunoResearch | Cat#711-545-152; RRID:AB_2313584 |
| Rhodamine Red-X-labeled goat anti-mouse IgG (H+L) | Jackson ImmunoResearch | Cat#115-295-146; RRID:AB_2338766 |
| Rhodamine Red-X-labeled goat anti-rabbit IgG (H+L) | Jackson ImmunoResearch | Cat#111-295-144; RRID:AB_2338028 |
| Alexa Fluor 647-labeled donkey anti-goat IgG (H+L) | Jackson ImmunoResearch | Cat#705-605-147; RRID:AB_2340437 |
| HRP goat anti-mouse IgG (H+L) | Jackson ImmunoResearch | Cat#115-035-146; RRID:AB_2307392 |
| HRP goat anti-rabbit IgG (H+L) | Jackson ImmunoResearch | Cat#111-035-144; RRID:AB_2307391 |
| Bacterial and virus strains | ||
| Bacteria: DH5α competent cells | NEB | Cat#C2987H |
| Bacteria: BL21(DE3) competent cells | Thermo Fisher Scientific | Cat#EC0114 |
| Chemicals, peptides, and recombinant proteins | ||
| (Z)-4-Hydroxytamoxifen (4-OHT) | Sigma-Aldrich | Cat#H7904 |
| Shield-1 ligand | AOBIOUS | Cat#AOB1848 |
| TransIT-X2 | Mirus | Cat#MIR6006 |
| Anti-FLAG M2 Affinity Gel | Sigma-Aldrich | Cat#A2220 |
| Anti-HA Affinity Gel | Sigma-Aldrich | Cat#E6779 |
| 3× Flag peptide | Sigma-Aldrich | Cat#F4799 |
| Protein A/G Magnetic Beads | Thermo Fisher Scientific | Cat#88803 |
| Gel Filtration Standard | Bio-Rad | Cat#1511901 |
| Isopropyl β-D-1-thiogalactopyranoside (IPTG) | Thermo Fisher Scientific | Cat#15529019 |
| Protease Inhibitor Cocktail | Roche | Cat#4693159001 |
| Glutathione Sepharose 4B | Millipore | Cat#GE17-0756-01 |
| Olaparib (AZD2281) | LC labs | Cat#O-9201 |
| Cisplatin | MedChemExpress | Cat#HY-17394 |
| ATM kinase inhibitor KU55933 | Abcam | Cat#ab120637 |
| Latrunculin B (Lat B) | Sigma-Aldrich | Cat#L5288 |
| CK-666 | Sigma-Aldrich | Cat#SML0006 |
| Nocodazole (Noco) | Sigma-Aldrich | Cat#M1404 |
| Paclitaxel (Taxol) | Sigma-Aldrich | Cat#T7402 |
| CCK-8 solution | MesGen Biotech | Cat#MG6432 |
| Giemsa solution | Sigma-Aldrich | Cat#GS500 |
| Colcemid | Thermo Fisher Scientific | Cat#15210040 |
| Heparin | Calbiochem | Cat#375095 |
| PerfectHyb Plus hybridization buffer | Sigma-Aldrich | Cat#H7033 |
| Red blood cell lysis buffer | Sigma-Aldrich | Cat#R7757 |
| LPS | Sigma-Aldrich | Cat#L7770 |
| IL4 | R&D Systems | Cat#404-ML-050 |
| NP-BSA | Biosearch Technologies | Cat#5050H |
| NP-CGG | Biosearch Technologies | Cat#5055C |
| Imject Alum adjuvant | Thermo Fisher Scientific | Cat#77161 |
| PI/RNase Staining Solution | Thermo Fisher Scientific | Cat#F10797 |
| DAPI | Thermo Fisher Scientific | Cat#D1306 |
| SYBR Gold | Thermo Fisher Scientific | Cat#S11494 |
| Streptavidin PE Conjugate | Thermo Fisher Scientific | Cat#12-4317-87 |
| Phosphatase substrate | Sigma-Aldrich | Cat#P4744 |
| Critical commercial assays | ||
| Telomere PNA FISH Kit/Cy3 | Agilent | Cat#K532611-8 |
| QuikChange Site-Directed Mutagenesis Kit | Agilent | Cat#200518 |
| Duo-link in situ PLA Kit | Sigma-Aldrich | Cat#DUO92101 |
| Biotin Chromogenic Detection Kit | Thermo Fisher Scientific | Cat#K0661 |
| ATPase/GTPase Activity Assay Kit | Sigma-Aldrich | Cat#MAK113 |
| Single-Cell Gel Electrophoresis Assay Kit | R&D Systems | Cat#4250-050-K |
| Simple ChIP Enzymatic Chromatin IP Kit | Cell Signaling | Cat#9004 |
| PerfectStart Green qPCR SuperMix | TransGen Biotech | Cat#AQ601-01 |
| Mouse Immunoglobulin Isotyping Kit | BioLegend | Cat#740492 |
| EasySep Mouse B Cell Isolation Kit | Stemcell Tech | Cat#19854 |
| Deposited data | ||
| Mendeley dataset | This paper | http://dx.doi.org/10.17632/9n4g54vcdh.1 |
| Experimental models: Cell lines | ||
| Human: HEK293T | ATCC | Cat#CRL-11268; RRID:CVCL_1926 |
| Human: Phoenix-AMPHO | ATCC | Cat#CRL-3213; RRID:CVCL_H716 |
| Human: U2OS | ATCC | Cat#HTB-96; RRID:CVCL_0042 |
| Human: HeLa-53BP1 KO | Gift from Dr. Junjie Chen | N/A |
| Human: 293A-53BP1 KO | Gift from Dr. Junjie Chen | N/A |
| Human: ER-mCherry-LacI-FokI-DD U2OS | Gift from Dr. Roger A. Greenberg; Tang et al.42 | N/A |
| Human: U2OS-BRCA1 KO | Zhao et al.21 | N/A |
| Human: U2OS-BRCA1/53BP1 double KO | Zhao et al.21 | N/A |
| MEF: Trf2F/−; CreERT2 | ATCC | Cat#CRL-3317; RRID:CVCL_UE13 |
| MEF: Trf2F/− Slfn5−/−; CreERT2 | This paper | N/A |
| MEF: Slfn5+/+ | This paper | N/A |
| MEF: Slfn5−/− | This paper | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: Slfn5+/+. C57BL/6J | This paper | N/A |
| Mouse: Slfn5−/−. C57BL/6J | This paper | N/A |
| Oligonucleotides | ||
| Primer for genotyping Slfn5 allele: Forward#1, 5’-TTTACAGATGACCCGAGAGACTTT-3’; | This paper | N/A |
| Primer for genotyping Slfn5 allele: Forward#2, 5’-CTATGATTTCAGGGTGAGTCCAG-3’ | This paper | N/A |
| Primer for genotyping Slfn5 allele: Reverse, 5’-AGTTTCAGAGAAGCCGAGCGTGG-3’ | This paper | N/A |
| Telomeric probe: Biotin-100 bp of repeated TTAGGG | This paper | N/A |
| B1 probe: Biotin-TAATCCCAGCACTTGGGAGGC | This paper | N/A |
| Primer for U2OS-DSB-reporter locus qPCR: P1: Forward, 5’-GGAAGATGTCCCTTGTATCACCAT-3’ | Tang et al.42 | N/A |
| Primer for U2OS-DSB-reporter locus qPCR: P1: Reverse, 5’-TGGTTGTCAACAGAGTAGAAAGTGAA-3’ | Tang et al.42 | N/A |
| Primer for U2OS-DSB-reporter locus qPCR: P3: Forward, 5’-GGCATTTCAGTCAGTTGCTCAA-3’ | Tang et al.42 | N/A |
| Primer for U2OS-DSB-reporter locus qPCR: P3: Reverse, 5’-TTGGCCGATTCATTAATGCA-3’ | Tang et al.42 | N/A |
| Primer for Sμ region: Forward, 5’-GCTAAACTGAGGTGATTACTCTGAGGTAAG-3’ | Zan et al.60 | N/A |
| Primer for Sμ region: Reverse, 5’-GTTTAGCTTAGCGGCCCAGCTCATTCCAGT-3’ | Zan et al.60 | N/A |
| Primer for Sγ1 region: Forward, 5’-ATAAGTAGTAGTTGGGGATTC-3’ | Zan et al.60 | N/A |
| Primer for Sγ1 region: Reverse, 5’-CTCAGCCTGGTACCTTATACA-3’ | Zan et al.60 | N/A |
| Primer for Sγ3 region: Forward, 5’-AATCTACAGAGAGCCAGGTGG-3’ | Zan et al.60 | N/A |
| Primer for Sγ3 region: Reverse, 5’-TGGTTTTCCATGTTCCCACTT-3’ | Zan et al.60 | N/A |
| Recombinant DNA | ||
| pLVX3-FLAG-SLFN5 | This paper | N/A |
| pLVX3-FLAG-SLFN5 (E191A/E196A) | This paper | N/A |
| pLVX3-FLAG-SLFN5 (K584M) | This paper | N/A |
| pLVX3-FLAG-SLFN5 (D649A) | This paper | N/A |
| pGEX-4T-2-SLFN5 | This paper | N/A |
| pGEX-4T-2-SLFN5 (D649A) | This paper | N/A |
| pMX-53BP1-DB-FLAG | Gift from Dr. Titia de Lange; Lottersberger et al.15 | N/A |
| pMX-53BP1-DB-Δ28-FLAG | Gift from Dr. Titia de Lange; Lottersberger et al.15 | N/A |
| pMX-53BP1-DB-ΔPTIP-FLAG | Gift from Dr. Titia de Lange; Lottersberger et al.15 | N/A |
| pMX-53BP1-DB-ΔMob-FLAG | Gift from Dr. Titia de Lange; Lottersberger et al.15 | N/A |
| pMX-53BP1-DB-ΔPro-FLAG | Gift from Dr. Michela Di Virgilio; Sundaravinayagam et al.45 | N/A |
| pMX-53BP1-DB-ΔRIF1-FLAG | Gift from Dr. Michela Di Virgilio; Sundaravinayagam et al.45 | N/A |
| pMX-53BP1-DB-ΔCore-FLAG | Gift from Dr. Michela Di Virgilio; Sundaravinayagam et al.45 | N/A |
| pMX-53BP1-DB-ΔMob/ΔCore-FLAG | This paper | N/A |
| HA-53BP1 | Gift from Dr. Junjie Chen | N/A |
| HA-53BP1-Δ(1–1051) | Gift from Dr. Junjie Chen | N/A |
| HA-53BP1-Δ(1052–1302) | Gift from Dr. Junjie Chen | N/A |
| HA-53BP1-ΔTudor | Gift from Dr. Junjie Chen | N/A |
| HA-53BP1-ΔBRCT | Gift from Dr. Junjie Chen | N/A |
| pLVX2-HA-53BP1 | This paper | N/A |
| pLVX2-HA-53BP1-ΔTudor/ΔBRCT | This paper | N/A |
| FLAG-RIF1 | Gift from Dr. Dongyi Xu | N/A |
| SFB-REV7 | Gift from Dr. Jun Huang | N/A |
| SFB-SHLD3 | Gift from Dr. Jun Huang | N/A |
| pLVX3-FLAG-SUN2 | This paper | N/A |
| pLVX3-FLAG-SUN2-Δ(1–150) | This paper | N/A |
| mCherry-BP1-2 pLPC-Puro | Addgene | Cat#19835 |
| shRNA#1 targeting SLFN5 | Sigma-Aldrich | Cat#TRCN0000154288 |
| shRNA#2 targeting SLFN5 | Sigma-Aldrich | Cat#TRCN0000157669 |
| shRNA#1 targeting 53BP1 | Sigma-Aldrich | Cat#TRCN0000218999 |
| shRNA#2 targeting 53BP1 | Sigma-Aldrich | Cat#TRCN0000018865 |
| shRNA#1 targeting RIF1 | Sigma-Aldrich | Cat#TRCN0000155022 |
| shRNA#2 targeting RIF1 | Sigma-Aldrich | Cat#TRCN0000220017 |
| shRNA#1 targeting REV7 | Sigma-Aldrich | Cat#TRCN0000006573 |
| shRNA#2 targeting REV7 | Sigma-Aldrich | Cat#TRCN0000006570 |
| shRNA targeting SUN1 | Sigma-Aldrich | Cat#TRCN0000134596 |
| shRNA targeting SUN2 | Sigma-Aldrich | Cat#TRCN0000141514 |
| shRNA#1 targeting Brca1 | Addgene | Cat#44594 |
| shRNA#2 targeting Brca1 | Addgene | Cat#44595 |
| sgRNA targeting 53bp1 | Santa Cruz | Cat#sc-424212- KO-2 |
| pLent-U6-CMV-copGFP-P2A-puro-shRNA targeting 53bp1 (sense: GCTATTGTGGAGATTGTGTTT) | WZ Biosciences; Xu et al.61 | N/A |
| lentiCRISPRv2-sgRNA targeting Slfn5 (sense: TTGCCAAAGCGCCCGATTCC) | Genscript | N/A |
| Software and algorithms | ||
| ZEN Blue | Zeiss | https://www.zeiss.com/microscopy/en/products/software/zeiss-zen-lite |
| Fiji | Open source | https://imagej.net/software/fiji/ |
| Prism 9 | GraphPad | https://www.graphpad.com/ |
| FlowJo 10.1 | FlowJo LLC | https://www.flowjo.com/ |
| Matlab R2019b (9.7.0) | MathWorks | https://uk.mathworks.com |
| Python 3.0 | Python | https://www.python.org/ |
Primary Slfn5+/+ and Slfn5−/− MEFs were isolated from E13.5 embryos produced by crossing heterozygous Slfn5 mice and immortalized by serial passaging. The Trf2F/− Slfn5−/−; CreERT2 MEF cell line was generated from Trf2F/−; CreERT2 MEF using lentiCRISPRv2-sgSlfn5, followed by single clone isolation. HEK293T, HeLa and MEFs were cultured in Dulbecco’s Modified Eagle Medium (DMEM) medium with 10% fetal bovine serum (FBS). U2OS was cultured in McCoy’s 5A with 10% FBS. To induce the Trf2 knockout allele in Trf2F/−; CreERT2 and Trf2F/− Slfn5−/−; CreERT2 MEFs, cells were treated with 1 μM (Z)-4-Hydroxytamoxifen (4-OHT) dissolved in methanol (MeOH) for 2–4 d14. To induce site-specific DSBs by FokI in ER-mCherry-LacI-FokI-DD U2OS, cells were treated with 1 μM Shield-1 and 1 μM 4-OHT for 4 h42.
METHOD DETAILS
Plasmids, transfection and lentiviral infection
SLFN5 was cloned into pLVX3-CMV-puro (3×Flag at N-terminus) and pGEX-4T-2 vectors. SLFN5 E191A/E196A (2EA), SLFN5 K584M, and SLFN5 D649A mutants were generated using a QuikChange site-directed mutagenesis kit (Agilent). HA-53BP1, HA-53BP1-Δ(1–1051), HA-53BP1-Δ(1052–1302), HA-53BP1-ΔTudor and HA-53BP1-ΔBRCT plasmids were provided by Dr. Junjie Chen. 53BP1-DB-Flag, 53BP1-DB-Δ28-Flag, 53BP1-DB-ΔPTIP-Flag and 53BP1-DB-ΔMob-Flag plasmids were gifts of Dr. Titia de Lange. 53BP1-DB-ΔPro-Flag, 53BP1-DB-ΔRIF1-Flag and 53BP1-DB-ΔCore-Flag plasmids were gifts from Dr. Michela Di Virgilio. The Flag-RIF1 plasmid was a gift of Dr. Dongyi Xu. SFB-REV7 and SFB-SHLD3 were gifts from Dr. Jun Huang. SUN2 and SUN2-Δ(1–150) mutant were cloned into pLVX3-CMV-puro. The HA-53BP1 plasmid was subcloned into pLVX2-CMV-puro with a HA cassette. The 53BP1-ΔTudor/ΔBRCT mutant was generated by PCR based on HA-53BP1-ΔTudor backbone and cloned into pLVX2-CMV-puro with a HA cassette. The 53BP1-DB-ΔMob/ΔCore mutant was generated by site-directed mutagenesis from 53BP1-DB-ΔMob-Flag using QuikChange (Agilent). The mCherry-BP1-2 pLPC-Puro plasmid was obtained from Addgene (Plasmid #19835).
SLFN5 shRNA (NM_144975), 53BP1 shRNA (NM_005657), RIF1 shRNA (NM_018151), REV7 shRNA (NM_006341), SUN1 shRNA (XM_379766) and SUN2 shRNA (NM_015374) were purchased from Sigma-Aldrich. The shRNAs targeting mouse Brca1 were obtained from Addgene (Plasmids #44594, #44595). The shRNA targeting mouse 53bp1 (sense: GCTATTGTGGAGATTGTGTTT)61 was clone by WZ Biosciences into pLent-U6-shRNA-CMV-copGFP-P2A-puro. The mouse Slfn5 guide RNA (sense: TTGCCAAAGCGCCCGATTCC) cloned in lentiCRISPRv2 vector was generated at Genscript. The mouse 53bp1 CRISPR/Cas9 KO Plasmid was obtained from Santa Cruz. Cells were transfected with TransIT-X2 (Mirus) according to the manufacturer’s instructions. Lentiviruses and retroviruses were packaged in HEK293T and Phoenix-AMPHO cells, respectively. Viral infection of cells was performed as described previously62.
Immunofluorescence
Cells were grown on coverslips 24 h before experiments and treated with 2 Gy irradiation followed by recovery for the indicated times. Cells were rinsed in ice-cold PBS and stained dependent on the foci. For γH2AX, 53BP1, BRCA1 and RAD51 staining, cells were pre-extracted using ice-cold 0.25% Triton-X in PBS for 1 min and fixed with 4% paraformaldehyde (PFA) for 10 min at RT. For RPA2 staining, incubation at −20 °C in methanol for 15 min was used as a fixative. Primary and secondary antibodies were diluted in Tris-buffered saline with 0.1% Tween-20 (TBST) containing 1% BSA. Cells were incubated with primary antibody at RT for 1h and subsequently incubated with secondary antibody at RT for 30 min. Nuclei were stained with DAPI. The coverslips were mounted onto glass slides with an anti-fade solution and visualized using an ImageXpress Micro Confocal system (Molecular Devices).
For super-resolution immunofluorescence, cells were grown on coverslips, treated with irradiation (2 Gy, 1 h), rinsed in PBS, pre-extracted using ice-cold 0.25% Triton-X in PBS for 1 min and fixed with 4% PFA for 10 min at RT. Cells were then incubated with primary antibody and subsequently incubated with the corresponding secondary antibody. The coverslips were mounted onto glass slides with an anti-fade solution and visualized using a Zeiss LSM 980 Airyscan2 super-resolution microscope.
Super-resolution imaging
Super-resolution imaging was carried out on a Zeiss LSM 980 AxioObserver.Z1/7 microscope fitted with an Airyscan2 detector using a Plan-Apochromat 63×/1.40 Oil DIC M27 objective using the Zeiss algorithm (ZEN Blue). Cells were imaged using 0.8% excitation power of 488 nm laser, 1% excitation power of 561 nm laser, and 1% excitation power of 639 nm laser, with 1.7× scan zoom for all laser conditions combined with appropriate filter sets. Z-stacks of 50–70 xy planes at an interval of 0.125 μm were captured with frame scanning, SR mode and bidirectional scanning. Super-resolution images were generated using the Airyscan processing with ZEN Blue.
Live-cell imaging
The indicated cells were seeded on 35-mm glass-bottom microwell dishes (MatTek). Before imaging, cells were changed into DMEM without phenol red medium (Thermo Fisher) containing 10% FBS and subjected to irradiation (2 Gy, 10 min). Images were acquired on a Zeiss LSM 980 microscope equipped with a Plan-Apochromat 63×/1.40 Oil DIC M27 objective using a heated, humidified chamber with 5% CO2 atmosphere. Focus movements were examined by collecting z-stacks at 0.5 μm intervals with frame scanning, multiplex (MPLX) mode and bidirectional scanning, throughout the entire nucleus every 30 s for 10 min.
Proximity ligation assay (PLA)
Cells were rinsed in ice-cold PBS, fixed with 4% PFA for 10 min on ice, permeabilized with 0.25% Triton-X for 5 min on ice. PLA was performed by a Duo-link in situ PLA kit (Sigma) according to the manufacturer’s protocol. Briefly, samples were blocked in blocking solution at 37 °C for 1 h and incubated with the mixture of primary antibodies (1:1000) at 4 °C overnight. Then probes were incubated at 37 °C for 1 h, followed by hybridization, ligation, amplification, and detection. Nuclei were stained with DAPI. Coverslips were mounted onto glass slides with an anti-fade solution and visualized using an ImageXpress Micro Confocal system (Molecular Devices).
Neutral comet assay
Comet assay was performed using a Single-Cell Gel Electrophoresis Assay Kit (R&D Systems) according to the manufacturer’s instruction. Briefly, the indicated cells were left untreated or irradiated (5 Gy) and recovered for indicated times at 37 °C. 2 × 105 cells were combined with 1% low melting agarose at 37 °C at the ratio of 1:10 (v/v) and pipetted onto slides. Slides were immersed in the lysis buffer at 4 °C overnight, subjected to electrophoresis at 31 V for 45 min and stained with SYBR Gold (Invitrogen, 1: 20000) for 20 min. Images were captured on a Nikon Eclipse 80i fluorescence microscope. Comet tail data were analyzed using the OpenComent plugin63 for Fiji/ImageJ.
Chromatin immunoprecipitation (ChIP)
ChIP was performed by a Simple ChIP Enzymatic Chromatin IP Kit (Cell Signaling) according to the manufacturer’s instruction. In brief, the indicated cells were crosslinked with formaldehyde and neutralized with glycine. The cross-linked nuclear lysates were digested with micrococcal nuclease and then sonicated to yield genomic DNA fragments of approximately 150–900 bp. Digested chromatins were immunoprecipitated with the indicated primary antibody at 4 °C overnight. The immunocomplexes were pulled down using magnetic beads, reverse crosslinked at 65 °C for 30 min and digested with proteinase K overnight. DNA samples were purified using Miniprep columns. qPCR was performed with ABI PRISM 7500 using PerfectStart Green qPCR SuperMix (TransGen Biotech). Primers for ChIP-qPCR were as follows: P1 (forward: GGAAGATGTCCCTTGTATCACCAT, reverse: TGGTTGTCAACAGAGTAGAAAGTGAA), P3 (forward: GGCATTTCAGTCAGTTGCTCAA, reverse: TTGGCCGATTCATTAATGCA), Sμ (forward: GCTAAACTGAGGTGATTACTCTGAGGTAAG, reverse: GTTTAGCTTAGCGGCCCAGCTCATTCCAGT), Sγ1 (forward: ATAAGTAGTAGTTGGGGATTC, reverse, CTCAGCCTGGTACCTTATACA) and Sγ3 (forward: AATCTACAGAGAGCCAGGTGG, reverse: TGGTTTTCCATGTTCCCACTT).
Immunoprecipitation and western blot
Cells were lysed with NETN buffer (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40) supplemented with 50 mM β-glycerophosphate, 10 mM NaF and 1 mg mL−1 each of pepstatin A and aprotinin. Whole-cell lysates were centrifuged at 12,000 rpm for 10 min. The supernatants were incubated with Anti-Flag M2 Affinity Gel (Sigma) or Anti-HA Affinity Gel (Sigma) for 2 h or at 4 °C overnight. The immunocomplexes were washed with NETN buffer and separated by SDS–PAGE. Western blotting was done using standard procedures.
Fast protein liquid chromatography (FPLC)
The indicated HEK-293T cells were left untreated or treated with irradiation (5 Gy, 1 h). Cells were harvested and subjected to immunoprecipitation. The supernatants were incubated with Anti-Flag M2 Affinity Gel (Sigma) and eluted with 3× Flag peptide (Sigma). The immunocomplexes were run on an AKTA Purifier with a Superdex 200 gel filtration column (GE Healthcare) to separate the different oligomeric fractions. Gel Filtration Standard (Bio-Rad) was loaded first to indicate the protein size. Collected fractionations were subjected to western blotting.
Recombinant protein expression
The recombinant GST-SLFN5 and SLFN5 D649A were induced in BL21 cells with 0.2 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at 18.5 °C for 20 h. Cells were then lysed using lysis buffer (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40, 1% Triton-X) supplemented with Protease Inhibitor Cocktail (Roche). Proteins were purified by binding to glutathione agarose beads (Millipore) for 30 min at 4 °C. Beads were washed with lysis buffer twice and bound proteins were subsequently eluted using elution buffer (100 mM Tris-HCl, pH 8.0, 150 mM NaCl, 20 mM reduced glutathione). Samples were dialyzed against buffer containing 50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 10% glycerol before storage at −80 °C.
ATPase assay
ATPase assays were performed using an ATPase Activity Assay Kit (Sigma) according to the manufacturer’s instruction. In brief, equimolar amounts (300 nM) of protein were incubated in 1× assay buffer with 1 mM ATP in a final volume of 40 μL at RT for the indicated times. The reaction was terminated by the addition of 200 μL Malachite green solution and incubated for an additional 30 min. The optical density at 620 nm was detected using an Epoch2 microplate reader (BioTek). The concentration of free phosphate (Pi) was calculated from the phosphate standard curve and plotted.
Clonogenic and cell viability assays
For colony formation, 800–1000 cells were plated in triplicate in each well of 6 well plates. 16 h later, cells were treated with olaparib (LC labs) and left for 10–14 d at 37 °C to allow colony formation. Colonies were stained with Giemsa solution (Sigma) and counted. Data were normalized to plating efficiencies.
For the cell viability assay, 2000 cells were plated in triplicate in each well of 96 well plates overnight. Cells were treated with olaparib (LC labs) or cisplatin (MedChemExpress) and left at 37 °C for 72 h. Cells were subsequently incubated with CCK-8 solution (MesGen Biotech) at 37 °C for 2 h. The optical density at 450 nm was detected using a SpectraMax M5 microplate reader (Molecular Devices).
Cell cycle analysis
The indicated cells were harvested and fixed in 70% ice-cold ethanol overnight. Fixed cells were washed with PBS and resuspended in PI/RNase solution (Thermo Fisher) at RT for 30 min. Samples were analyzed on CytoFLEX flow cytometry (Beckman) and processed using ModFit LT.
HR and NHEJ reporter assays
To quantify the repair of I-SceI-generated DSBs by HR, the indicated HEK293T cells were transfected with DR-GFP, pCBA-I-SecI and pCherry. The HR efficiency was determined 48 h later, and GFP-positive cells were quantified by an Attune NxT flow cytometry (Thermo Fisher). To quantify the repair of I-SecI-generated DSBs by NHEJ, cells were transfected with EJ5-GFP, pCBA-I-SecI and pCherry. The NHEJ efficiency was determined and quantified as described above.
Metaphase spreads
MEFs were incubated with 20 ng mL−1 colcemid (Thermo Fisher) at 37 °C for 2 h. Cells were harvested and swollen in prewarmed 75 mM KCl at 37 °C for 25 min. After centrifugation, cells were fixed with Carnoy’s buffer (methanol: acetic acid in 3:1 ratio) at RT for 10 min. Fixed cells were centrifuged and then resuspended in Carnoy’s buffer twice. The final supernatant was dropped onto slides and air-dried. Slides were stained with Giemsa solution (Sigma).
Telomere fusion assay
The indicated Trf2F/−; CreERT2 MEFs were treated with 1 μM 4-OHT for 48 or 96 h. Cells were collected and subjected to metaphase spread. Then the telomeric fluorescence in situ hybridization (FISH) was performed using a Telomere PNA FISH Kit (Agilent) following the manufacturer’s instruction. DNA was stained with DAPI. Images were acquired using a Zeiss LSM780 laser-scanning confocal microscope.
Telomeric ChIP
Telomeric ChIP was performed as described previously with slight modification64,65. Briefly, the indicated Trf2F/−; CreERT2 MEFs were treated with MeOH or 4-OHT for 70 h. Cells were collected and subjected to ChIP. The purified DNA was denatured in 2× SSC (Sigma) at 100 °C for 10 min and blotted on Biodyne B Nylon Membrane (Thermo Fisher) using a slot blot manifold (Hoefer). Membranes were incubated at 42 °C overnight in PerfectHyb Plus hybridization buffer (Sigma) containing denatured biotin-labeled telomeric, 100 bp of repeated TTAGGG probes or denatured biotin-labeled murine B1 probe (the Alu-equivalent in human, TAATCCCAGCACTTGGGAGGC). The ChIP signals were measured using a Biotin Chromogenic Detection Kit (Thermo Fisher) according to the manufacturer’s protocol. The signal intensity was normalized to the signals of input DNA on the same blot.
Micronucleus assay
The micronucleus assay was performed as described previously66. Briefly, mouse blood (10–12-week-old) was mixed with 100 μL PBS containing 1000 U mL−1 of heparin (Calbiochem). Blood suspension was added to 1 mL of ice-cold methanol and stored at −80 °C overnight until further processing. Fixed blood cells were washed with bicarbonate buffer (0.9% NaCl, 5.3 mM NaHCO3). Cells were suspended in 100 μL of bicarbonate buffer with 1 μL of APC-conjugated CD71 antibodies at 4 °C for 45 min. After centrifugation, pellets were washed with bicarbonate buffer and resuspended in 5 μg mL−1 PI/RNase Staining Solution. Samples were analyzed on a CytoFLEX flow cytometry (Beckman) and processed using FlowJo.
Flow cytometric analyses of lymphocyte
Single-cell suspensions of lymphocyte were prepared from Slfn5−/− mice or wild-type littermates (10–12-week-old) and analyzed by flow cytometry. Briefly, mouse spleen, bone marrow and thymus were disrupted in PBS and the aggregates and debris were removed using a 70 μm mesh nylon strainer. The suspension was centrifuged, resuspended in red blood cell lysis buffer at RT for 10 min, and washed twice with PBS. The final cell suspension was stained with the indicated antibodies at 4 °C for 45 min. Data were collected on a CytoFLEX flow cytometry (Beckman) and analyzed with FlowJo.
Flow cytometric analyses of serum immunoglobulin
Mouse serum immunoglobulins were examined using a Mouse Immunoglobulin Isotyping Kit (BioLegend) according to the manufacturer’s instructions. Briefly, mouse blood samples (10–12-week-old, both male and female) were clotted at least 30 min and centrifuge at 1000 g. for 10 min. Serum was diluted at 50000-fold with assay buffer. Dilutes serum was incubated with 25 μL Ig capture beads at RT for 2 h and then added 25 μL detection antibodies for 1 h. Samples were then added 25 μL SA-PE and incubated in dark at RT for 30 min. Finally, the beads were washed three times with wash buffer and then analyzed on a CytoFLEX flow cytometry (Beckman) and processed using LEGENDplex data analysis software (BioLegend).
in vitro class switch recombination assay
Mouse splenic B cells were purified from Slfn5−/− mice (10–12-week-old) and age- and sex-matched littermate controls using an EasySep Mouse B Cell Isolation Kit (Stemcell Tech) according to the manufacturer’s instructions. Purified B cells were cultured in RPMI 1640 supplemented with 10% FBS and 50 μM β-mercaptoethanol. 2 × 106 cells were stimulated with 10 μg mL−1 LPS (Sigma) or 10 μg mL−1 LPS plus 10 ng mL−1 mouse recombinant IL-4 (R&D systems) for 96 h. Stimulated B cells were analyzed on a CytoFLEX flow cytometry (Beckman) and processed using FlowJo.
Immunizations
Slfn5−/− mice and wild-type littermates were immunized intraperitoneally with 200 μg of NP-CGG (Biosearch Technologies) in an emulsion with Imject Alum adjuvant (Thermo Fisher). Mouse blood samples were collected from the tail vein at 0, 7, 14, 21 and 28 days after immunization.
Enzyme-linked immunosorbent assay (ELISA)
ELISA was used to quantify the production of NP-specific antibodies in mice serum and performed as described previously19. Briefly, polycarbonate 96 well plates (Nunc MaxiSorp) were coated with 1 μg mL−1 NP-BSA (Biosearch Technologies) in bicarbonate buffer at 4 °C overnight, blocked with 5% milk in PBS at 37 °C for 1h and incubated with serial dilutions of serum collected at the indicated times from immunized mice. Plates were then probed with alkaline phosphatase-coupled antibodies against mouse IgM or IgG1 (Southern Biotech). phosphatase substrate (Sigma) was used for detection and the optical density was measured at 405 nm. For IgG1, pooled blood from post-immunization wild-type littermates was used as a standard and serially diluted into a standard curve. The first dilution was established as 1000 arbitrary units. For IgM, pooled blood from day 7 was used as a standard.
Analysis of 53BP1-MD topological textures
53BP1-MD texture analysis followed the approach recently described7. Imaging data were Airyscan processed in the ZEN Blue software and imported into Fiji/ImageJ to generate a TIF Image Sequence format. The QUANTEX software was used to quantify the morphology, texture and geometry features of 53BP1-MD objects following the software user manual. For segmentation of cell nuclei, z-stacks were clipped to the minimum number of slices, smoothened by Gaussian filter bullring, and then underwent automated weighted nuclear Otsu-based segmentation. Then, the segmentation of 53BP1-MDs was conducted using the following parameters: nuclear background subtraction (Rolling ball, size 3), automated Otsu segmentation (Threshold scale factor 1.0), morphology filtering-1 (Min object size 10). The parameter output of object features was exported as .xlsm document. The mean breadth of QUANTEX feature was used to analyze the geometrical change of 53BP1-MD.
Analysis of damaged chromatin mobility
Damaged chromatin mobility was visualized using mCherry-BP1-2 and analyzed as described previously53. Imaging data were Airyscan processed and a maximum-intensity projection of each z-stack was generated using the ZEN Blue software. Images were imported to Fiji/ImageJ and converted to 32-bit tiffs. T-stacks were registered using the StackReg plugin67 to normalize cell movements. Cells that underwent large-scale deformations or expansions were discarded. Individual focus particle was tracked using the TrackMate plugin68 for Fiji/ImageJ. ImageJ output data were converted to meters by the formula, 1 pixel = 0.049 μm, based on the characteristics of the objective. All foci in a cell that were continuously tracked at least 19 out of 20 frames were analyzed. Focus trajectories were subsequently transferred into MATLAB (MathWorks) and analyzed using the class @msdanalyzer69. The MSD of DNA damage foci plots the average squared distance travelled by foci at increasing time intervals, whereas the diffusion coefficient D(t) is approximated through the linear weighted fit of the initial mean MSD curve69. The MSD values were calculated using the formula,, where reflects focus position and t is the time in minutes. The error bars for each data point represent the weighted SD over all MSD curves. The diffusion coefficient was calculated from the linear fit of the first 25% of each MSD curve. The anomalous diffusion coefficient α was derived using MATLAB through log-log fitting of the power law, . The cumulative distance traveled in 10 min by each of the foci was calculated using Python by the formula, , where and refer to the and coordinates of the focus at time .
WGII signature analysis
Segmented copy number data and SLFN (SLFN5, SLFN11, SLFN12, SLFN13 and SLFN14) RNA expression data as log transformed TPM in TCGA triple-negative breast cancer cohort ( samples) were downloaded from UCSC xenabrowser70. The chromatin instability signature WGII for all samples was recalculated on the basis of Andrea et al.71. Samples were subgrouped based on the SLFN levels. SLFN deficiency was defined the lowest 20% expression or at least heterozygous deletion. Statistical significance was tested by two-tailed Mann-Whitney U tests.
HRD score analysis
TCGA breast cancer cohort HRD scores and the three subscores (LOH, LST and TAI) were published previously and were downloaded from Genomic Data Commons (GDC)72,73. Expression data as log-transformed transcripts per million (TPM) were downloaded from UCSC xenabrowser70. The analysis was performed on triple-negative breast cancer ( samples) as defined by immunohistochemical stain of ER, PR and HER2 proteins. Samples were subgrouped based on BRCA1 and SLFN5 levels. BRCA1 and SLFN5 deficiency was defined the lowest 20% expression or at least heterozygous deletion. Statistical significance was tested by two-tailed Mann-Whitney U tests.
QUANTIFICATION AND STATISTICAL ANALYSIS
All experiments were repeated at least 3 independent times otherwise stated in the figure legends. The replicate number, mean and error bars are explained in the figure legends. The statistical tests we used and resulting p values are indicated in the figure panels and/or figure legends. Statistical analyses were performed using GraphPad Prism software.
Supplementary Material
Table S1. A list of the UpSet plot, Related to Figure 1A
Video S1. SLFN5 deficiency impairs damaged chromatin mobility, Related to Figures 6C and 6D
Representative videos of mCherry-BP1-2 foci in ctrl (a) or SLFN5 knockdown (b) U2OS cells. Scale bar, 10 μm.
Video S2. SLFN5 promotes 53BP1-dependent damaged chromatin mobility, Related to Figures 6E and 6F
Representative videos of mCherry-BP1-2 foci in 53BP1-KO HeLa cells expressing Vec (a), 53BP1-DB (b), 53BP1-DB with SLFN5 knockdown (c), 53BP1-ΔMob (d), or 53BP1-ΔMob with SLFN5 knockdown (e). Scale bar, 10 μm.
Video S3. SLFN5-mediated motion of damaged chromatin requires its ATPase activity, Related to Figures 6, S8C and S8D
Representative videos of mCherry-BP1-2 foci in SLFN5-depleted U2OS cells expressing Vec (a), SLFN5-WT (b) or SLFN5-DA (c). Scale bar, 10 μm.
Video S4. SLFN5 deficiency impairs dysfunctional telomeric mobility, Related to Figures 6, S8F and S8G
Representative videos of mCherry-BP1-2 foci in Slfn5+/+ (a) or Slfn5−/− (b) Trf2F/−; CreERT2 MEFs. Scale bar, 10 μm.
Video S5. SLFN5-mediated motion of dysfunctional telomeres requires its ATPase activity, Related to Figures 6, S8H and S8I
Representative videos of mCherry-BP1-2 foci in Trf2F/− Slfn5−/−; CreERT2 MEFs cells expressing Vec (a), human SLFN5-WT (b), or human SLFN5-DA (c). Scale bar, 10 μm.
ACKNOWLEDGEMENTS
We thank members of the Lou lab for comments and discussion throughout the project. We also thank Junjie Chen for his generous sharing of the 53BP1-KO cell lines and Roger A. Greenberg for the ER-mCherry-LacI-FokI-DD U2OS cell line. We thank Titia de Lange, Jiri Lukas, Michela Di Virgilio and Junjie Chen for providing 53BP1 constructs. We thank Dongyi Xu for providing the Flag-RIF1 plasmid and Jun Huang for SFB-REV7 and SFB-SHLD3 plasmids. We are grateful to Jiri Lukas for the help of QUANTEX analysis. We thank Mayo Clinic Microscopy and Cell Analysis Core for assistance with super-resolution microscope. This work was supported in part by a grant from the Mayo Foundation (Z.L.) and the Mayo Edward C. Kendall Fellowship in Biochemistry Award (J.H.). J.A.K was supported by the NIH T32GM065841.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Scully R, Panday A, Elango R, and Willis NA (2019). DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol 20, 698–714. 10.1038/s41580-019-0152-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Panier S, and Boulton SJ (2014). Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Bio 15, 7–18. 10.1038/nrm3719. [DOI] [PubMed] [Google Scholar]
- 3.Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing MT, Young JTF, Tkac J, Cook MA, Rosebrock AP, Munro M, Canny MD, et al. (2013). A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Molecular Cell 49, 872–883. 10.1016/j.molcel.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Mirman Z, and de Lange T (2020). 53BP1: a DSB escort. Gene Dev 34, 7–23. 10.1101/gad.333237.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lou J, Scipioni L, Wright BK, Bartolec TK, Zhang J, Masamsetti VP, Gaus K, Gratton E, Cesare AJ, and Hinde E (2019). Phasor histone FLIM-FRET microscopy quantifies spatiotemporal rearrangement of chromatin architecture during the DNA damage response. Proc Natl Acad Sci U S A 116, 7323–7332. 10.1073/pnas.1814965116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chapman JR, Sossick AJ, Boulton SJ, and Jackson SP (2012). BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J Cell Sci 125, 3529–3534. 10.1242/jcs.105353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ochs F, Karemore G, Miron E, Brown J, Sedlackova H, Rask MB, Lampe M, Buckle V, Schermelleh L, Lukas J, and Lukas C (2019). Stabilization of chromatin topology safeguards genome integrity. Nature 574, 571-+. 10.1038/s41586-019-1659-4. [DOI] [PubMed] [Google Scholar]
- 8.Ghodke I, and Soutoglou E (2019). 53BP1-RIF1: sculpting the DNA repair focus in 3D. Nature Structural & Molecular Biology 26, 1087–1088. 10.1038/s41594-019-0348-1. [DOI] [PubMed] [Google Scholar]
- 9.Caron P, and Polo SE (2020). Reshaping Chromatin Architecture around DNA Breaks. Trends Biochem Sci 45, 177–179. 10.1016/j.tibs.2019.12.001. [DOI] [PubMed] [Google Scholar]
- 10.Zhao BL, Rothenberg E, Ramsden DA, and Lieber MR (2020). The molecular basis and disease relevance of non-homologous DNA end joining. Nat Rev Mol Cell Bio 21, 765–781. 10.1038/s41580-020-00297-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, and de Lange T (2013). 53BP1 Regulates DSB Repair Using Rif1 to Control 5 ‘ End Resection. Science 339, 700–704. 10.1126/science.1231573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paiano J, Zolnerowich N, Wu W, Pavani R, Wang C, Li HZ, Zheng L, Shen BH, Sleckman BP, Chen BR, and Nussenzweig A (2021). Role of 53BP1 in end protection and DNA synthesis at DNA breaks. Gene Dev 35, 1356–1367. 10.1101/gad.348667.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, Sartori AA, Adams IR, Batista FD, and Boulton SJ (2013). RIF1 Is Essential for 53BP1-Dependent Nonhomologous End Joining and Suppression of DNA Double-Strand Break Resection. Molecular Cell 49, 858–871. 10.1016/j.molcel.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dimitrova N, Chen YCM, Spector DL, and de Lange T (2008). 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 456, 524–U551. 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lottersberger F, Karssemeijer RA, Dimitrova N, and de Lange T (2015). 53BP1 and the LINC Complex Promote Microtubule-Dependent DSB Mobility and DNA Repair. Cell 163, 880–893. 10.1016/j.cell.2015.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gupta R, Somyajit K, Narita T, Maskey E, Stanlie A, Kremer M, Typas D, Lammers M, Mailand N, Nussenzweig A, et al. (2018). DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell 173, 972-+. 10.1016/j.cell.2018.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Noordermeer SM, Adam S, Setiaputra D, Barazas M, Pettitt SJ, Ling AK, Olivieri M, Alvarez-Quilon A, Moatti N, Zimmermann M, et al. (2018). The shieldin complex mediates 53BP1-dependent DNA repair. Nature 560, 117-+. 10.1038/s41586-018-0340-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mirman Z, Lottersberger F, Takai H, Kibe T, Gong Y, Takai K, Bianchi A, Zimmermann M, Durocher D, and de Lange T (2018). 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Pol alpha-dependent fill-in. Nature 560, 112-+. 10.1038/s41586-018-0324-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghezraoui H, Oliveira C, Becker JR, Bilham K, Moralli D, Anzilotti C, Fischer R, Deobagkar-Lele M, Sanchiz-Calvo M, Fueyo-Marcos E, et al. (2018). 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature 560, 122-+. 10.1038/s41586-018-0362-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dev H, Chiang TWW, Lescale C, de Krijger I, Martin AG, Pilger D, Coates J, Sczaniecka-Clift M, Wei WM, Ostermaier M, et al. (2018). Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat Cell Biol 20, 954-+. 10.1038/s41556-018-0140-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao F, Kim W, Gao HY, Liu C, Zhang Y, Chen YP, Deng M, Zhou Q, Huang JZ, Hu Q, et al. (2021). ASTE1 promotes shieldin-complex-mediated DNA repair by attenuating end resection. Nat Cell Biol 23, 894-+. 10.1038/s41556-021-00723-9. [DOI] [PubMed] [Google Scholar]
- 22.Mirman Z, Sasi NK, King A, Chapman JR, and de Lange T (2022). 53BP1-shieldin-dependent DSB processing in BRCA1-deficient cells requires CST-Polalpha-primase fill-in synthesis. Nat Cell Biol 24, 51–61. 10.1038/s41556-021-00812-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu F, Zhou P, Wang Q, Zhang M, and Li D (2018). The Schlafen family: complex roles in different cell types and virus replication. Cell Biol Int 42, 2–8. 10.1002/cbin.10778. [DOI] [PubMed] [Google Scholar]
- 24.Yue T, Zhan X, Zhang D, Jain R, Wang KW, Choi JH, Misawa T, Su L, Quan J, Hildebrand S, et al. (2021). SLFN2 protection of tRNAs from stress-induced cleavage is essential for T cell-mediated immunity. Science 372. 10.1126/science.aba4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim ET, Dybas JM, Kulej K, Reyes ED, Price AM, Akhtar LN, Orr A, Garcia BA, Boutell C, and Weitzman MD (2021). Comparative proteomics identifies Schlafen 5 (SLFN5) as a herpes simplex virus restriction factor that suppresses viral transcription. Nat Microbiol 6, 234–245. 10.1038/s41564-020-00826-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li M, Kao E, Gao X, Sandig H, Limmer K, Pavon-Eternod M, Jones TE, Landry S, Pan T, Weitzman MD, and David M (2012). Codon-usage-based inhibition of HIV protein synthesis by human schlafen 11. Nature 491, 125–128. 10.1038/nature11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bustos O, Naik S, Ayers G, Casola C, Perez-Lamigueiro MA, Chippindale PT, Pritham EJ, and de la Casa-Esperon E (2009). Evolution of the Schlafen genes, a gene family associated with embryonic lethality, meiotic drive, immune processes and orthopoxvirus virulence. Gene 447, 1–11. 10.1016/j.gene.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neumann B, Zhao L, Murphy K, and Gonda TJ (2008). Subcellular localization of the Schlafen protein family. Biochem Bioph Res Co 370, 62–66. 10.1016/j.bbrc.2008.03.032. [DOI] [PubMed] [Google Scholar]
- 29.Geserick P, Kaiser F, Klemm U, Kaufmann SHE, and Zerrahn J (2004). Modulation of T cell development and activation by novel members of the Schlafen (slfn) gene family harbouring an RNA helicase-like motif. Int Immunol 16, 1535–1548. 10.1093/intimm/dxh155. [DOI] [PubMed] [Google Scholar]
- 30.Mu Y, Lou J, Srivastava M, Zhao B, Feng XH, Liu T, Chen J, and Huang J (2016). SLFN11 inhibits checkpoint maintenance and homologous recombination repair. Embo Rep 17, 94–109. 10.15252/embr.201540964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murai J, Tang SW, Leo E, Baechler SA, Redon CE, Zhang H, Al Abo M, Rajapakse VN, Nakamura E, Jenkins LMM, et al. (2018). SLFN11 Blocks Stressed Replication Forks Independently of ATR. Mol Cell 69, 371–384 e376. 10.1016/j.molcel.2018.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arslan AD, Sassano A, Saleiro D, Lisowski P, Kosciuczuk EM, Fischietti M, Eckerdt F, Fish EN, and Platanias LC (2017). Human SLFN5 is a transcriptional co-repressor of STAT1-mediated interferon responses and promotes the malignant phenotype in glioblastoma. Oncogene 36, 6006–6019. 10.1038/onc.2017.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wan G, Zhu J, Gu X, Yang Y, Liu Y, Wang Z, Zhao Y, Wu H, Huang G, and Lu C (2020). Human Schlafen 5 regulates reversible epithelial and mesenchymal transitions in breast cancer by suppression of ZEB1 transcription. Br J Cancer 123, 633–643. 10.1038/s41416-020-0873-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gu X, Wan G, Yang Y, Liu Y, Yang X, Zheng Y, Jiang L, Zhang P, Liu D, Zhao W, et al. (2020). SLFN5 influences proliferation and apoptosis by upregulating PTEN transcription via ZEB1 and inhibits the purine metabolic pathway in breast cancer. Am J Cancer Res 10, 2832–2850. [PMC free article] [PubMed] [Google Scholar]
- 35.Setiaputra D, and Durocher D (2019). Shieldin - the protector of DNA ends. Embo Rep 20. ARTN e47560. 10.15252/embr.201847560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manis JP, Morales JC, Xia ZF, Kutok JL, Alt FW, and Carpenter PB (2004). 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat Immunol 5, 481–487. 10.1038/ni1067. [DOI] [PubMed] [Google Scholar]
- 37.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, and Honjo T (2000). Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102, 553–563. Doi 10.1016/S0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 38.Bothmer A, Robbiani DF, Di Virgilio M, Bunting SF, Klein IA, Feldhahn N, Barlow J, Chen HT, Bosque D, Callen E, et al. (2011). Regulation of DNA End Joining, Resection, and Immunoglobulin Class Switch Recombination by 53BP1. Molecular Cell 42, 319–329. 10.1016/j.molcel.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manis JP, Tian M, and Alt FW (2002). Mechanism and control of class-switch recombination. Trends Immunol 23, 31–39. Doi 10.1016/S1471-4906(01)02111-1. [DOI] [PubMed] [Google Scholar]
- 40.Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. (2010). 53BP1 Inhibits Homologous Recombination in Brca1-Deficient Cells by Blocking Resection of DNA Breaks. Cell 141, 243–254. 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shanbhag NM, Rafalska-Metcalf IU, Balane-Bolivar C, Janicki SM, and Greenberg RA (2010). ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 141, 970–981. 10.1016/j.cell.2010.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang J, Cho NW, Cui G, Manion EM, Shanbhag NM, Botuyan MV, Mer G, and Greenberg RA (2013). Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat Struct Mol Biol 20, 317–325. 10.1038/nsmb.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang JY, Deng XY, Li YS, Ma XC, Feng JX, Yu B, Chen Y, Luo YL, Wang X, Chen ML, et al. (2018). Structure of Schlafen13 reveals a new class of tRNA/rRNA- targeting RNase engaged in translational control. Nature Communications 9. ARTN 1165. 10.1038/s41467-018-03544-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murai J, Thomas A, Miettinen M, and Pommier Y (2019). Schlafen 11 (SLFN11), a restriction factor for replicative stress induced by DNA-targeting anti-cancer therapies. Pharmacol Therapeut 201, 94–102. 10.1016/j.pharmthera.2019.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sundaravinayagam D, Rahjouei A, Andreani M, Tupina D, Balasubramanian S, Saha T, Delgado-Benito V, Coralluzzo V, Daumke O, and Di Virgilio M (2019). 53BP1 Supports Immunoglobulin Class Switch Recombination Independently of Its DNA Double-Strand Break End Protection Function. Cell Rep 28, 1389-+. 10.1016/j.celrep.2019.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li W, Bai XZ, Li J, Zhao YC, Liu JY, Zhao HY, Liu L, Ding M, Wang QS, Shi FY, et al. (2019). The nucleoskeleton protein IFFO1 immobilizes broken DNA and suppresses chromosome translocation during tumorigenesis. Nat Cell Biol 21, 1273-+. 10.1038/s41556-019-0388-0. [DOI] [PubMed] [Google Scholar]
- 47.Amitai A, Seeber A, Gasser SM, and Holcman D (2017). Visualization of Chromatin Decompaction and Break Site Extrusion as Predicted by Statistical Polymer Modeling of Single-Locus Trajectories. Cell Rep 18, 1200–1214. 10.1016/j.celrep.2017.01.018. [DOI] [PubMed] [Google Scholar]
- 48.Shukron O, Seeber A, Amitai A, and Holcman D (2019). Advances Using Single-Particle Trajectories to Reconstruct Chromatin Organization and Dynamics. Trends Genet 35, 685–705. 10.1016/j.tig.2019.06.007. [DOI] [PubMed] [Google Scholar]
- 49.Lottersberger F, Bothmer A, Robbiani DF, Nussenzweig MC, and de Lange T (2013). Role of 53BP1 oligomerization in regulating double-strand break repair. P Natl Acad Sci USA 110, 2146–2151. 10.1073/pnas.1222617110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wright RHG, Le Dily F, and Beato M (2019). ATP, Mg(2+), Nuclear Phase Separation, and Genome Accessibility. Trends Biochem Sci 44, 565–574. 10.1016/j.tibs.2019.03.001. [DOI] [PubMed] [Google Scholar]
- 51.Kruhlak MJ, Celeste A, Dellaire G, Fernandez-Capetillo O, Muller WG, McNally JG, Bazett-Jones DP, and Nussenzweig A (2006). Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J Cell Biol 172, 823–834. 10.1083/jcb.200510015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hauer MH, and Gasser SM (2017). Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev 31, 2204–2221. 10.1101/gad.307702.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schrank BR, Aparicio T, Li Y, Chang W, Chait BT, Gundersen GG, Gottesman ME, and Gautier J (2018). Nuclear ARP2/3 drives DNA break clustering for homology-directed repair. Nature 559, 61–66. 10.1038/s41586-018-0237-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Caridi CP, D’Agostino C, Ryu T, Zapotoczny G, Delabaere L, Li X, Khodaverdian VY, Amaral N, Lin E, Rau AR, and Chiolo I (2018). Nuclear F-actin and myosins drive relocalization of heterochromatic breaks. Nature 559, 54–60. 10.1038/s41586-018-0242-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lamm N, Read MN, Nobis M, Van Ly D, Page SG, Masamsetti VP, Timpson P, Biro M, and Cesare AJ (2020). Nuclear F-actin counteracts nuclear deformation and promotes fork repair during replication stress. Nat Cell Biol 22, 1460–1470. 10.1038/s41556-020-00605-6. [DOI] [PubMed] [Google Scholar]
- 56.Amiad-Pavlov D, Lorber D, Bajpai G, Reuveny A, Roncato F, Alon R, Safran S, and Volk T (2021). Live imaging of chromatin distribution reveals novel principles of nuclear architecture and chromatin compartmentalization. Sci Adv 7. ARTN eabf6251. 10.1126/sciadv.abf6251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bajpai G, Amiad Pavlov D, Lorber D, Volk T, and Safran S (2021). Mesoscale phase separation of chromatin in the nucleus. Elife 10. 10.7554/eLife.63976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hauer MH, Seeber A, Singh V, Thierry R, Sack R, Amitai A, Kryzhanovska M, Eglinger J, Holcman D, Owen-Hughes T, and Gasser SM (2017). Histone degradation in response to DNA damage enhances chromatin dynamics and recombination rates. Nat Struct Mol Biol 24, 99–107. 10.1038/nsmb.3347. [DOI] [PubMed] [Google Scholar]
- 59.Lok BH, Gardner EE, Schneeberger VE, Ni A, Desmeules P, Rekhtman N, de Stanchina E, Teicher BA, Riaz N, Powell SN, et al. (2017). PARP Inhibitor Activity Correlates with SLFN11 Expression and Demonstrates Synergy with Temozolomide in Small Cell Lung Cancer. Clin Cancer Res 23, 523–535. 10.1158/1078-0432.CCR-16-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zan H, Tat C, Qiu Z, Taylor JR, Guerrero JA, Shen T, and Casali P (2017). Rad52 competes with Ku70/Ku86 for binding to S-region DSB ends to modulate antibody class-switch DNA recombination. Nat Commun 8, 14244. 10.1038/ncomms14244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, Bartkova J, Gogola E, Warmerdam D, Barazas M, et al. (2015). REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 521, 541–544. 10.1038/nature14328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang JZ, Chen M, Chen, Gao XC, Zhu S, Huang H, Hu M, Zhu H., and Yan GR. (2017). A Peptide Encoded by a Putative lncRNA HOXB-AS3 Suppresses Colon Cancer Growth. Mol Cell 68, 171–184 e176. 10.1016/j.molcel.2017.09.015. [DOI] [PubMed] [Google Scholar]
- 63.Gyori BM, Venkatachalam G, Thiagarajan PS, Hsu D, and Clement MV (2014). OpenComet: an automated tool for comet assay image analysis. Redox Biol 2, 457–465. 10.1016/j.redox.2013.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Frescas D, and de Lange T (2014). TRF2-Tethered TIN2 Can Mediate Telomere Protection by TPP1/POT1. Mol Cell Biol 34, 1349–1362. 10.1128/Mcb.01052-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Simonet T, Zaragosi LE, Philippe C, Lebrigand K, Schouteden C, Augereau A, Bauwens S, Ye J, Santagostino M, Giulotto E, et al. (2011). The human TTAGGG repeat factors 1 and 2 bind to a subset of interstitial telomeric sequences and satellite repeats. Cell Res 21, 1028–1038. 10.1038/cr.2011.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang J, Zhou Q, Gao M, Nowsheen S, Zhao F, Kim W, Zhu Q, Kojima Y, Yin P, Zhang Y, et al. (2020). Tandem Deubiquitination and Acetylation of SPRTN Promotes DNA-Protein Crosslink Repair and Protects against Aging. Mol Cell 79, 824–835 e825. 10.1016/j.molcel.2020.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thevenaz P, Ruttimann UE, and Unser M (1998). A pyramid approach to subpixel registration based on intensity. IEEE Trans Image Process 7, 27–41. 10.1109/83.650848. [DOI] [PubMed] [Google Scholar]
- 68.Tinevez JY, Perry N, Schindelin J, Hoopes GM, Reynolds GD, Laplantine E, Bednarek SY, Shorte SL, and Eliceiri KW (2017). TrackMate: An open and extensible platform for single-particle tracking. Methods 115, 80–90. 10.1016/j.ymeth.2016.09.016. [DOI] [PubMed] [Google Scholar]
- 69.Tarantino N, Tinevez JY, Crowell EF, Boisson B, Henriques R, Mhlanga M, Agou F, Israel A, and Laplantine E (2014). TNF and IL-1 exhibit distinct ubiquitin requirements for inducing NEMO-IKK supramolecular structures. J Cell Biol 204, 231–245. 10.1083/jcb.201307172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Goldman M, Craft D, Kamath A, Brooks A, Zhu J, and Haussler D (2018). The UCSC Xena Platform for cancer genomics data visualization and interpretation. bioRxiv. 10.1101/326470. [DOI] [Google Scholar]
- 71.Marquard AM, Eklund AC, Joshi T, Krzystanek M, Favero F, Wang ZC, Richardson AL, Silver DP, Szallasi Z, and Birkbak NJ (2015). Pan-cancer analysis of genomic scar signatures associated with homologous recombination deficiency suggests novel indications for existing cancer drugs. Biomark Res 3, 9. 10.1186/s40364-015-0033-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, Fan H, Shen H, Way GP, Greene CS, et al. (2018). Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep 23, 239–254 e236. 10.1016/j.celrep.2018.03.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Telli ML, Timms KM, Reid J, Hennessy B, Mills GB, Jensen KC, Szallasi Z, Barry WT, Winer EP, Tung NM, et al. (2016). Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin Cancer Res 22, 3764–3773. 10.1158/1078-0432.CCR-15-2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. A list of the UpSet plot, Related to Figure 1A
Video S1. SLFN5 deficiency impairs damaged chromatin mobility, Related to Figures 6C and 6D
Representative videos of mCherry-BP1-2 foci in ctrl (a) or SLFN5 knockdown (b) U2OS cells. Scale bar, 10 μm.
Video S2. SLFN5 promotes 53BP1-dependent damaged chromatin mobility, Related to Figures 6E and 6F
Representative videos of mCherry-BP1-2 foci in 53BP1-KO HeLa cells expressing Vec (a), 53BP1-DB (b), 53BP1-DB with SLFN5 knockdown (c), 53BP1-ΔMob (d), or 53BP1-ΔMob with SLFN5 knockdown (e). Scale bar, 10 μm.
Video S3. SLFN5-mediated motion of damaged chromatin requires its ATPase activity, Related to Figures 6, S8C and S8D
Representative videos of mCherry-BP1-2 foci in SLFN5-depleted U2OS cells expressing Vec (a), SLFN5-WT (b) or SLFN5-DA (c). Scale bar, 10 μm.
Video S4. SLFN5 deficiency impairs dysfunctional telomeric mobility, Related to Figures 6, S8F and S8G
Representative videos of mCherry-BP1-2 foci in Slfn5+/+ (a) or Slfn5−/− (b) Trf2F/−; CreERT2 MEFs. Scale bar, 10 μm.
Video S5. SLFN5-mediated motion of dysfunctional telomeres requires its ATPase activity, Related to Figures 6, S8H and S8I
Representative videos of mCherry-BP1-2 foci in Trf2F/− Slfn5−/−; CreERT2 MEFs cells expressing Vec (a), human SLFN5-WT (b), or human SLFN5-DA (c). Scale bar, 10 μm.
Data Availability Statement
Original imaging data (including microscopy, gels and western blots) have been deposited to Mendeley Data (http://dx.doi.org/10.17632/9n4g54vcdh.1).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.