

Abstract
Purpose of Review
Tumor-like brain lesions are rare and commonly suggest a neoplastic etiology. Failure to rapidly identify non-neoplastic causes can lead to increased morbidity and mortality. In this review, we describe 10 patients who presented with atypical, non-neoplastic tumor-like brain lesions in which brain biopsy was essential for a correct diagnosis and treatment.
Recent Findings
There has been increasing recognition of autoimmune conditions affecting the nervous system, and many of those diseases can cause tumor-like brain lesions. Currently available reports of non-neoplastic tumor-like brain lesions are scarce. Most case series focus on tumefactive demyelinating lesions, and a comprehensive review including other neuroimmunological conditions such as CNS vasculitis, neurosarcoidosis, histiocytic and infectious etiologies is lacking.
Summary
We review the literature on tumor-like brain lesions intending to increase the awareness and differential diagnosis of non-neoplastic brain tumor mimics. We advocate for earlier brain biopsies, which, in our case series, significantly changed diagnosis, management, and outcomes.
Introduction
Tumor-like brain lesions are defined as lesions that measure more than 2 cm in size on MRI, potentially causing edema and mass effect.1 The most common cause of tumor-like brain abnormalities include primary CNS neoplasm or metastatic disease; however, up to 13% of patients with tumor-like brain lesions consulting with neuro-oncologists have non-neoplastic tumor mimics with heterogeneous etiologies.2 MRI of the brain may have unique findings on some of these entities and can help guide the initial workup and treatment. However, those findings can be nonspecific, preventing the correct identification of non-neoplastic tumor-like lesions, leading to delay or incorrect treatment and worse outcomes. Our case series aims to describe 10 patients who presented with tumor-like brain lesions mimicking neoplasms, and MRI brain findings were not enough to differentiate from neoplastic causes; hence, brain biopsies were obtained.
Case Descriptions
Demyelinating Disease
Tumefactive Lesions Causing Uncal Herniation in a Patient on Fingolimod
Figure 1, A–D: Patient 1 is a 31-year-old man with multiple sclerosis on fingolimod who presented with dysarthria and encephalopathy for 1 week despite medication adherence. MRI (Figure 1, A and B) revealed an 8 × 5 centimeters (cm) incomplete enhancing right temporal lobe lesion with 1.3 cm uncal herniation treated with intravenous (IV) steroids x 3 weeks. Steroids were weaned, and 2 months later, he received the Pfizer mRNACovid-19 vaccine. One week after the second immunization, patient returned with expressive aphasia and MRI revealed a new enhancing lesion with targetoid appearance in the left centrum semiovale (Figure 1, C and D). Patient underwent biopsy to rule out T-cell lymphoproliferative process, which has been reported with fingolimod and can have a more heterogenous appearance in setting of immunosuppression. Biopsy revealed a macrophage-rich demyelinating lesion with sheets of CD163-positive foamy macrophages and CD3-positive T lymphocytes. He improved with plasmapheresis followed by 7 sessions of cyclophosphamide.
Figure 1. Demyelinating Diseases.

(A–D) Patient 1, tumefactive multiple sclerosis. (E–I) Patient 2, neuromyelitis optica. (J–M) Patient 3, myelin oligodendrocyte glycoprotein antibody–associated disease (MOGAD).
Tumefactive demyelinating lesions (TDL) represent a heterogeneous group of inflammatory CNS demyelinating diseases. They most commonly occur in the second to third decade of life; only 4% of the patients are younger than 18 years, and 4% are older than 65 years.1 The incidence of TDL has been estimated to be around 0.3/100.000 people, and the prevalence is 1–3/1000 cases of MS.2 The ratio of women to men differs across series; some have found higher prevalence in women, while others have found a ratio close to 1:1.1,3 In 46% of the patients with TDL, there is a known history of MS; however, tumefactive demyelination can be the first neurologic event, and 30%–70% of patients with TDL will develop MS over time.3,4 In patients evolving to MS, the time to second relapse is significantly longer after a tumefactive lesion when compared with a clinically isolated syndrome (CIS) and has been estimated to be 4.8 years.3 Sphingosine-1 phosphate receptor (S1P) inhibitors, natalizumab, and alemtuzumab have been associated with tumefactive demyelination in patients with MS.2 Balo's concentric sclerosis (BCS) is a form of tumefactive demyelinating disease, characterized by alternating rings of demyelinated and preserved myelin layers with a venule oftentimes at the center.5,6
CSF studies may be normal or may reveal pleocytosis and elevated proteins. Oligoclonal bands are present in 33% of patients with TDL, and 35% will have an elevated IgG index, which is less frequent than in typical multiple sclerosis.1,3 Patients with these markers are more likely to develop MS, although oligoclonal bands are not specific for demyelination. For example, oligoclonal bands are present in 27% of CNS lymphoma cases, which can lead to diagnostic uncertainity.7
MRI of the brain may differentiate demyelinating tumefactive lesions from brain tumors, with an estimated sensitivity of 89% and specificity of 94%.1 Obtaining serial MRIs is suggested because TDLs tend to decrease in size or even disappear on interval imaging several weeks later, while neoplasm will tend to increase in size.2 TDL are commonly focal and supratentorial, located in the cerebral bihemispheric white matter of the frontal and parietal lobes but can also be found elsewhere in the CNS, including the gray matter.1 Multifocal tumefactive lesions can occur, and almost all tumefactive demyelinating lesions will have gadolinium enhancement with heterogeneous patterns.1,8 Important imaging findings for demyelination include an open ring or incomplete ring of enhancement pointing toward the gray matter, which is present in approximately 35–70% of the cases, with a specificity of 98–100%.8,9 The enhancing ring area represents the leading edge of demyelination favoring the white matter and is associated with infiltration of macrophages and angiogenesis at the inflammatory border.10 Closed ring enhancement is frequently seen in high-grade neoplasms, but it can also be seen in approximately 18% of demyelinating lesions.4 There may be a perilesional T2 hypointense and T1 hypointense ring in 48% of the patients in the same area of ring enhancement.2,8 Central diffusion restriction mimicking abscess can occur in lesions with an intense inflammatory core.1 TDLs classically show high ADC in the center of the lesion due to vasogenic edema and myelin destruction and low ADC peripherally due to inflammatory cell infiltrates.4 Finally, a butterfly configuration through the corpus callosum mimicking glioblastoma could be seen in up to 12% of patients with TDL.3
Magnetic resonance spectroscopy (MRS) has a high sensitivity but low specificity for demyelination because it can demonstrate increased choline (Cho) and reduced N-acetylaspartate (NAA), with a resultant increased Cho/NAA ratio; however, this finding may also be seen in non-necrotic high-grade brain tumors such as anaplastic astrocytoma, GBM, and primary CNS lymphoma.11 MR perfusion images can help differentiate TDL from brain tumors by measuring the mean relative cerebral blood volume, which will be significantly less in demyelination as compared with neoplasms.2 However, perfusion images can be inconclusive given the variability of tumor types, and primary CNS lymphoma may have decreased cerebral blood volume mimicking demyelination.2 Nuclear medicine studies such as brain single photon emission computed tomography (SPECT) or PET can be helpful in the differentiation of intracranial neoplasms, with delayed increased accumulation in SPECT being more commonly seen in lymphoma and melanoma. The value of these studies in distinguishing TDL from malignancy is uncertain because TDL can demonstrate increased accumulations of the tracer and high retention in both early and delayed sequences in SPECT. TDL tend to be hypometabolic in PET scan but could demonstrate hypermetabolism mimicking lymphoma as well (Table 1).16,17 Interestingly, hypoattenuated areas on computed tomography scan (CT) that correspond to areas of enhancement on MRI occur at a higher rate in tumefactive demyelination than in tumor (93% vs 4%).1 Hence, CT plus MRI have a stronger diagnostic accuracy than MRI alone, with a sensitivity of 87% and specificity of 100%.4
Table 1.
Diagnostic Tests for Differentiating Tumefactive Brain Demyelinating Lesions vs Malignancy

Brain biopsy is the gold standard for the diagnosis and is particularly beneficial when there is absence of other lesions in the brain and spinal cord compatible with demyelination. Stains for myelin, axons, and macrophages can help differentiate demyelination from CNS lymphoma or glial neoplasm. Features of demyelination include relative axonal sparing, foamy macrophages that contain myelin in the absence of coagulative necrosis, reactive astrocytes, and perivascular lymphocytes (Figure 2A).4,18 Creuztfelt cells are a nonspecific feature of demyelination that appear as reactive astrocytes with karyorrhexis.2 The diagnostic yield of brain biopsy is 95.7% in brain tumors; however, in TDL and other tumor-like brain lesions, the exact yield is brain biopsy is unknown.19
Figure 2. Examples of Pathologic Findings.
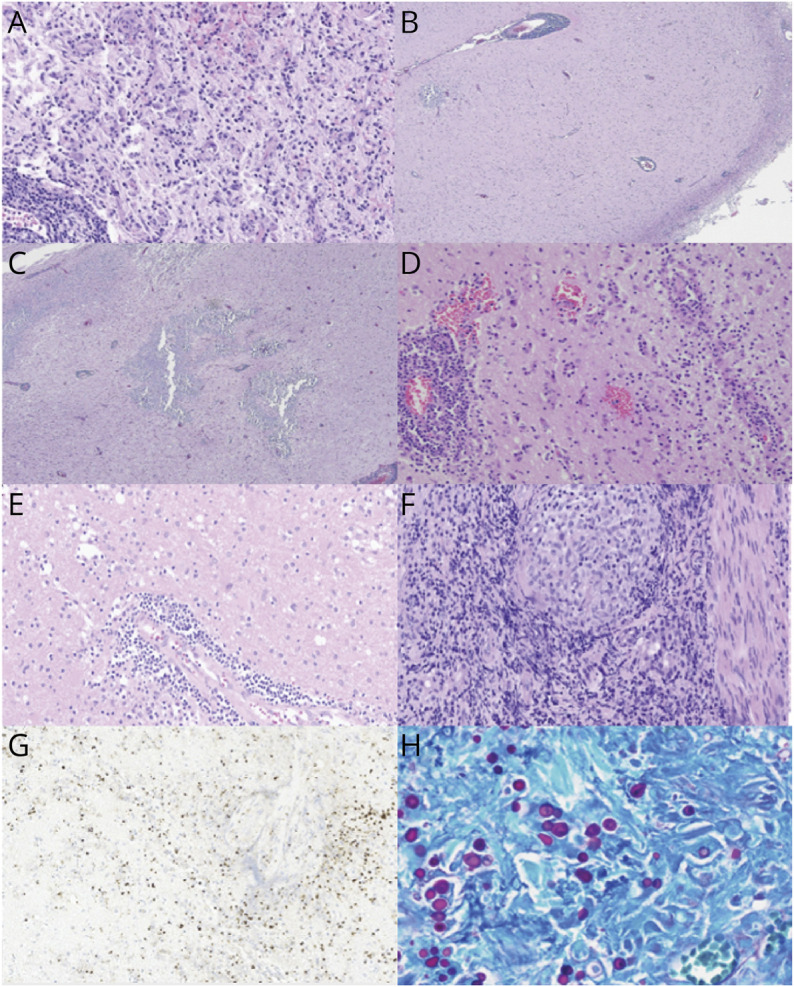
(A) Shows typical histopathologic features of a demyelinating lesion, with perivascular lymphocytes (lower left), reactive astrocytosis, and macrophage accumulation, in viable neuropil (relative axonal preservation) (magnification = ×20 before enlargement). (B, C) Images correspond to patient 2. Shows dense perivascular lymphocytic infiltrates in the meninges and the cortex, geographic areas of dystrophic calcifications and gliosis in the cortex and degenerative change in the white matter with macrophages aggregates and cavitary lesion. (D) Demonstrates histopathologic features of vasculitis, with transmural and perivascular infiltrates of mononuclear inflammatory cells affecting the medium-sized and small sized vessels (magnification = ×20 before enlargement). (E) Displays features of vasculitis with perivascular infiltrates of mononuclear inflammatory cells and intraparenchymal macrophages (magnification = ×20 before enlargement). (F) Shows features of neurosarcoidosis with well-formed non-necrotizing granuloma composed of epithelioid histiocytes (magnification = 20X before enlargement). (G) Images correspond to patient 9. Immunohistochemical stain using antibodies to Toxoplasma gondii, demonstrating tachyzoites in cerebral toxoplasmosis (magnifications = 60X before enlargement). (H) Images correspond to patient 10. Periodic acid-Schiff (PAS) special stain highlights numerous yeast-form fungal organisms with broad-based budding, demonstrating cerebral blastomycosis (magnifications = 40X before enlargement).
Most patients with tumefactive demyelination improve after IV corticosteroids. If there is an incomplete response, plasmapheresis may be warranted. In severe cases, rituximab or cyclophosphamide could be tried.2
Tumefactive NMO in a Patient With Mitochondrial Disorder
Figure 1, E–I: Patient 2 is a 23-year-old man who presented with a focal onset seizure with impaired awareness at age 12 years. MRI showed a right temporal mass which was resected (Figure 1, E–G). Low-grade glioneuronal tumor was questioned; however, the pathology did not show neoplastic proliferation. Brain tissue revealed perivascular lymphocytic infiltrates in the meninges and cortex, with areas of dystrophic calcifications and gliosis (Figure 2, B and C). At age 18 years, he developed headaches, recurrent multifocal seizures, and subclinical status epilepticus. Brain MRI showed extensive enhancing periventricular and corpus callosum T2 hyperintense lesions. MR spectroscopy demonstrated double lactate peak suggestive of mitochondrial disease. Gene testing showed an abnormality in MT-ND4 gene encoding for NADH dehydrogenase 4 in mitochondrial complex 1. At age 19 years, he had left vision loss, serum aquaporin-4 antibody was positive 1:10000, and he was diagnosed with NMOSD. The plan was to start rituximab, but the patient was lost in follow-up. At age 21 years, he had recurrence of left optic neuritis with vision loss treated with IV solumedrol, plasmapheresis, and rituximab. He developed mucormycotic pneumonia with cavitation requiring thoracotomy precluding further use of rituximab and other immunosuppressants. At age 22 years, he had right hemiparesis, a new left subcortical white matter hyperintensity with negative infectious workup (Figure 1, H–I). He developed multiple infections due to neutropenia and leukopenia prompting antiepileptic modification. He started prednisone 20 mg every other day to treat NMO, monthly IVIG and filgrastim for immune support, and carnitine, riboflavin, thiamine, and arginine to treat mitochondrial disorder. The patient has been stable without relapses or infections since.
Neuromyelitis Optica Spectrum Disorder (NMOSD) may present with brain lesions in 24%–89% of the patients and typically follow the distribution of the aquaporin-4 (AQP4) channel, including the subpia, periependymal region, brainstem, chiasm, hypothalamus, area postrema, and corpus callosum.20 A distinctive feature in NMOSD is the involvement of corticospinal tracts, including the posterior limb of the internal capsule.20 Typical patterns in NMOSD include patchy enhancement with ill-defined margins and thin linear ependymal enhancement. Leptomeningeal and nodular enhancement can rarely occur. Tumefactive lesions are more common in MS but can also occur in 3% of NMOSD cases, usually with positive AQP4 IgG.20,21 In a series of 30 patients with Balo's Concentric Sclerosis and NMOSD, all patients were found to have extensive AQP4 loss in all actively demyelinating lesions.5 The associated vasogenic edema may be due to deficient water elimination associated with AQP4 antibodies.22 Pathology reveals demyelination and/or necrosis, which may progress to cystic cavitation.23 Rescue treatment includes high doses of IV corticosteroids and plasmapheresis. Satralizumab, inebilizumab, and eculizumab are FDA-approved medications for long-term immunotherapy, which is necessary to prevent relapses. Rituximab has been used off-label for many years for this condition, as well as mycophenolate, azathioprine, and methotrexate, with the latter 3 being less efficacious.24
MOGAD With Features of Vasculitis
Figure 1, J–M: Patient 3 is a 29-year-old woman who presented with progressive aphasia and right hemiparesis over months. She was found to have a left parietal mass with vasogenic edema and heterogeneous enhancement. Brain biopsy revealed perivascular lymphocytes with abundant macrophages and patches of necrosis. Reticulin staining showed infiltration of several scattered vessels by inflammatory cells; demyelination was not excluded. Malignancy was ruled out. Special stains to detect microorganisms were negative. Serum MOG IgG titer was positive at 1:40, and serum AQP4 IgG was negative. She received IV solumedrol for 5 days, and on follow-up MRI, lesion size and edema decreased significantly. Symptoms have not recurred in 3 years without additional treatment. Arguably, without a significant MOG titer, this might not be considered true myelin oligodendrocyte glycoprotein antibody–associated disease (MOGAD) by some experts; however, vasculitis has been reported with MOGAD.
MOGAD can present with brain lesions in less than 50% of adults,25,26 which are usually few, bilateral, with ill-defined margins,26 and a fluffy appearance.27 MOGAD can be a monophasic or a relapsing condition and has a relapse risk of 32% within 4 years, such risk is higher in young adults between 18 and 40 years who had optic neuritis at onset, which is the most common relapsing phenotype.28 Locations of brain lesions include the thalamus and the pons. MOGAD can present with an encephalitis pattern resembling acute disseminated encephalomyelitis (ADEM), involving gray and white matter.26 Encephalopathy, headaches, and seizures occur more commonly in MOGAD as compared with NMO and MS.21,28 Active lesions may demonstrate patchy enhancement and tumefactive demyelinating lesions occur in 22% of the cases.21,26,29 On follow-up MRI, complete or near-complete resolution of the lesion favors MOGAD or ADEM diagnosis, as compared with MS and NMOSD, in which lesions tend to persist, albeit may shrink over time.26 Higher MOG antibody levels and persistent seropositivity had been associated with a higher risk of a relapsing disease course; however, recent series have not suggested prognostic value in persistent positive titers in adults.30 Use of prednisolone, azathioprine, mycophenolate, and methotrexate individually or in combination has been associated with decreased risk of relapse of approximately 50%.28 In a multicenter retrospective study, the annualized relapse rate (ARR) was lower with IVIG (20%), as compared with rituximab (61%), mycophenolate (74%), and azathioprine (59%), although only 10 patients were treated with IVIG; hence, definitive conclusions cannot be drawn from these data.31 Despite MOGAD being considered an antibody-mediated disorder, rituximab does not seem to be as effective in MOGAD as it is in MS and NMO, raising the question of whether MOG IgG is itself pathogenic or a biomarker of disease.32,33
Vasculitis
CNS Vasculitis Appearing as Glial Neoplasm
Patient 4 is a 23-year-old man who presented with headache and seizures. He was found to have a left frontal enhancing mass, suspicious for primary glial neoplasm favoring anaplastic astrocytoma (Figure 3, A and B). Brain biopsy identified predominantly lymphocytic infiltration and vessel wall necrosis consistent with vasculitis. He was started on prednisone 1 mg/kg daily with gradual steroid taper and transition to mycophenolate with good response without relapse for 2.8 years.
Figure 3. CNS Vasculitis.

(A, B) Patient 4. (C–H) Patient 5.
CNS vasculitis is an idiopathic vascular inflammatory disease limited to the CNS that occurs in all age groups and affects parenchymal, leptomeningeal medium, and small blood vessels. Spinal cord involvement can occur in 5% of the patients, and tumor-like brain lesions occur in approximately 4% of the cases.34-36 The presentation of primary CNS angiitis is variable and nonspecific, both clinically and radiologically, with the most common symptoms being encephalopathy, headache, and seizures.36 Brain imaging is always abnormal, and lesions may mimic demyelination, neoplasm, infarction, infection, and others. Such lesions can be faint, diffuse, and coalescent; and the presentation may include infarcts and hemorrhage paired with multifocal stenosis on vascular imaging.34,36 High resolution vessel wall imaging (ie MRA black blood protocol) may be normal or may show inflammation of the vessel wall and enhancement that is typically concentric, as opposed to atherosclerotic plaque which is eccentric.37 Parenchymal and meningeal biopsy is required for definite diagnosis of this condition and demonstrates inflammation of the vessel wall, which can be granulomatous, lymphocytic, or necrotizing (Figure 2, D–E).36 Acute treatment involves IV corticosteroids, with a gradual oral taper over months. Induction with either cyclophosphamide or rituximab can also be given. Long-term immunotherapy may be warranted, with mycophenolate mofetil and azathioprine being commonly used.36
Neuro-Behcet Mimicking Infectious Encephalitis
Patient 5 is a 41-year-old woman with latent tuberculosis and a history of ulcerating oral and urogenital lesions who presented with fevers and altered mental status. Imaging demonstrated an enhancing lesion centered at the left basal ganglia extending into the pons and cerebellum (Figure 3, C–H). CSF showed neutrophilic pleocytosis, WBC 283/mm3 (PMN 56%), and elevated protein 105 mg/dL. Infectious workup was negative. HLA-B51 was present. Brain biopsy showed reactive gliosis and was negative for neoplasm. The patient was treated for neuro-Behcet, given the constellation of meningoencephalitis and urogenital ulcerations. She improved with high-dose steroids and mycophenolate. She had a relapse 5 years later while off mycophenolate.
Behcet disease is a chronic, relapsing-remitting, multisystemic vasculitic disease, prevalent in Mediterranean countries and more common in men.38 HLA-B51 susceptibility gene is present in 50%–80% of the patients within endemic geographic regions, but its presence is not required for the diagnosis of this condition.39 The characteristic clinical findings include mucocutaneous and genital ulcers, uveitis, and pathergy. CNS involvement is variable within different ethnicities, and cerebral mass lesions are rare.38 Mesodiencephalic location is a typical finding for Behcet, presenting with enhancing lesions and surrounding edema centered in the midbrain with contiguous involvement of the rest of the brainstem, basal ganglia, and internal capsule.20,40 Spinal cord involvement can occur, and fever is present in 22% of patients.38,41 Frequent pathologic findings include intense parenchymal inflammatory infiltration of lymphocytes, eosinophils, and macrophages with areas of necrosis and neuronal loss. Fibrinoid necrosis of blood vessels does not occur. Treatment involves chronic immune suppression similar to other vasculitides.38
Granulomatous, Histiocytic, and Plasma Cell Disorders
Neurosarcoidosis Presenting as a Solitary Enhancing Mass
Patient 6 is a 48-year-old woman who presented with headaches and right temporal field vision loss. She was found to have T2 hyperintense left periventricular, parietal, occipital, and temporal lobe hyperintensities (Figure 4, A–D). CSF revealed 29 WBC/mm3, protein of 50 mg/dL, and no oligoclonal bands. Her brain biopsy showed small noncaseating granulomas without significant involvement or destruction of blood vessels. She was diagnosed with neurosarcoidosis, started on infliximab and methotrexate with resolution of enhancement and improvement of T2/FLAIR hyperintensity.
Figure 4. Granulomatous, Histiocytic, and Plasma Cell Disorders.

(A–D) Patient 6, neurosarcoidosis. (E–H) Patient 7, IgG-4–related disease (IGG4RD). (I–L) Patient 8, Langerhans histiocytosis.
Sarcoidosis is an immune-mediated granulomatous disorder that can affect every organ system. Neurosarcoidosis occurs in 5%–10% of patients with systemic sarcoidosis; however, recent studies have found higher numbers, up to 34%.42 Incidence varies among different ethnicities, 10/100,000 in White patients, and as high as 80/100,000 in Black patients.42 It can affect any part of the nervous system, and isolated CNS involvement without systemic findings can occur in up to 20% of the patients.42 Brain parenchyma infiltration resembling a cerebral mass can occur.43 Pathogenesis is believed to be an exaggerated granulomatous reaction after exposure to unidentified antigens in genetically susceptible patients, and brain biopsy demonstrates well-formed, non-necrotizing granulomas (Figure 2F).44 Sarcoidosis is a remarkably steroid-responsive condition, which represents standard of treatment, with long and slow tapers being typically required.42 Steroid sparing agents can be used as well, depending on the severity of the disease. Given the important role of TNF alpha in initiation and perpetuation of granulomatous response, TNF alpha inhibitors are frequently used. Methotrexate, mycophenolate, and azathioprine can be used as well.42
IgG4-Related Disease Presenting as a Cerebral Mass With Leptomeningeal Enhancement
Patient 7 is a 42-year-old, right-handed man who presented with progressive right hemiparesis, hemisensory loss, and focal motor seizures with secondary generalization. MRI brain (Figure 4, E and F) showed extensive left parietal vasogenic edema with abnormal leptomeningeal and dura enhancement in the left frontoparietal and occipital regions. CSF showed 5 WBC, protein 62, high IgG 9 (reference 0.0 to 8.1), high IgG index 1.21, and >5 CSF-specific oligoclonal bands. CSF cytology was negative for cancerous cells. CSF infectious workup was negative. Extensive rheumatologic workup was also unrevealing. CT chest/abdomen/pelvis and whole-body PET scan were normal. Serum IgG panel showed high IgG4 (98 mg/dL, normal value 2–96 mg/dL) and high IgG4/IgG ratio (15%, normal value 5%). Biopsy showed marked chronic inflammatory infiltrate with increased IgG4 plasma cells per high power field (HPF) (>50) and increased IgG4/IgG ratio involving leptomeninges and overlying dura. The inflammatory infiltrate extended superficially in the underlying cortex with dominant perivascular distribution. A repeat MRI brain (Figure 4, G and H) 2 months later showed significant improvement in mass size, leptomeningeal enhancement, and edema. He was treated with prolonged oral prednisone taper, with relapse on discontinuation. Rituximab was added, with good clinical and radiologic response.
IgG4-RD is an immune-mediated, fibroinflammatory disorder of unknown cause characterized by unique pathologic features involving a wide variety of organs.45 It can mimic any other condition including neoplastic, infectious, and other inflammatory diseases. Although uncommon, brain parenchymal involvement and tumor-like presentation are possible.45,46 Pathologic findings include infiltration of lymphocytes and plasmacytes with demonstration of IgG4 plasma cells (ratio of IgG4/IgG cell >40% and >10 plasma cell/HPF), fibrosis that is organized in a storiform pattern, and obliterative phlebitis.46 Corticosteroids are the first-line treatment. Relapse rates during glucocorticoid taper or maintenance therapy are high, and in such case, B-cell depletion is recommended.46 Untreated IgG4RD often progresses from lymphoplasmacytic inflammation to extensive fibrosis, and at this stage, patients are less likely to respond to treatment. Hence, recognition is important to avoid permanent organ dysfunction and disability.45
Langerhans Cell Histiocytosis Presenting as a Suprasellar Mass
Patient 8 is a 42-year-old woman with a history of DM-2, hypothyroidism, skin and vaginal ulcers, and psoriatic arthritis who presented with progressive cognitive decline over 1 year and diabetes insipidus. MRI brain (Figure 4, I–K) showed a T2 hyperintense mass-like lesion with enhancement involving the hypothalamus and optic chiasm extending along the tuber cinereum and optic tracts. CSF showed 7 WBC and high protein. Serum AQP-4 and MOG antibodies were negative. Biopsy of the mass showed granulomatous features and heavy histiocytic-lymphocytic infiltration. Clinical condition worsened in 2 months with visual hallucinations and delusions. Repeat MRI brain (Figure 4L) revealed increase in size of the lesion and involvement of the right medial temporal lobe. Whole-body PET scan showed a hypermetabolic mass in the suprasellar area. Bone marrow biopsy was normal. Re-evaluation of pathology slides revealed fragments of neuropil with perivascular and intraparenchymal aggregates of histiocytes, eosinophils, scattered giant cells, admixed lymphocytes, plasma cells, and reactive astrocytes. Stain for CD1a, langerin, and BRAF V600E mutant protein was positive, suggestive of Langerhans cell histiocytosis.
Histiocytoses are rare heterogeneous hematopoietic conditions, with the most recognized being Langerhans cell histiocytosis (LCH), Erdheim-Chester disease (ECD), and Rosai-Dorfman-Destombes disease (RDD). These diseases are characterized by the accumulation of CD68 cells with various inflammatory infiltrates and fibrosis.47 LCH is more common in children but can also be present in young adults and is characterized by the presence of BRAF V600E mutation. Constitutional symptoms such as fever and weight loss can occur, as well as bone pain, mucosal or cutaneous lesions, cytopenias, and diabetes insipidus.47 Brain lesions are rare, can have a tumor-like appearance, and are most frequently located in the pituitary stalk, pineal gland, and other circumventricular regions.48 Pathology demonstrates positive CD1a and Langerin in LCH and tissue infiltration of CD68 (+) cells in LHC and ECD. S100 positivity and VRAF V600E mutations can be present in LCH and ECD.48 Therapy directed against myeloid precursors with activated MAPK signaling may be effective for LCH.49
Infections
Toxoplasmosis in a Patient With Waldenstrom Macroglobulinemia
Patient 9 is a 62-year-old man with Waldenstrom macroglobulinemia treated with bendamustine and rituximab who presented with impulsive behavior, spontaneous irregular left arm movements, left-sided numbness, and gait ataxia. MRI revealed an enhancing lesion in the right thalamus, midbrain, and pons with surrounding vasogenic edema (Figure 5, A–D). Serum studies showed a CD4 count of 181, positive toxoplasma IgG and negative IgM. CSF revealed lymphocytic pleocytosis with WBC 31/mm3 (85% lymphocytes) and protein 123 mg/dL, otherwise unremarkable. Biopsy revealed a macrophage-rich lesion with scattered mononuclear lymphoid cells, primarily CD3-positive T cells, without evidence of glioma or lymphoma, and negative fungal and acid-fast bacilli stains (Figure 2G). Next-generation sequencing of biopsy was positive for toxoplasmosis. He was treated with pyrimethamine, sulfadiazine, and clindamycin for 6 weeks with clinical and radiographic improvement.
Figure 5. Infections.

(A–D) Patient 9, toxoplasmosis. (E–L) Patient 10, blastomycosis.
Toxoplasma gondii is a ubiquitous protozoan parasite that can cause opportunistic cerebral infection due to the reactivation of latent brain cysts in the setting of decreased T-cell activity.50,51 Cerebral toxoplasmosis most commonly presents with multiple ring-enhancing lesions predominantly in the basal ganglia, which may demonstrate a small eccentric enhancing nodule along the lesion wall, known as the eccentric target sign, which is seen in <30% of the cases.50 The concentric target sign has also been described and represents concentric alternating zones of hyperintensities and hypointensities on T2. Nodular enhancing or expansile nonenhancing brain mass lesions may also occur.50 Treatment consists of at least 4–6 weeks of pyrimethamine with sulfadiazine or clindamycin with concurrent administration of folinic acid; antimicrobial course may be longer in immunocompromised patients until resolution of all signs and symptoms.52
Blastomycosis in a Patient With Medication-Induced Lymphopenia
Patient 10 is a 68-year-old woman with a history of multiple sclerosis, HTN, and DM type II who developed grade III lymphopenia while on dimethyl fumarate (ALC 200/mm3). She presented with numbness, weakness, and spasticity in her right arm and leg. MRI of the brain revealed a right cerebellar and left thalamic enhancing lesion containing several small cysts with surrounding edema (Figure 5, E-H). CT chest showed a right lung mass. She received 3 days of IV corticosteroids as was believed to have sarcoidosis. Brain biopsy showed granulomatous inflammation with multinucleated histiocytes, lymphocytes, and plasma cells with numerous thick-walled yeast-form fungal organisms, positive for periodic acid-Schiff (PAS), Gomori methenamine silver (GMS) stains, and negative for AFB (Figure 2H). Bronchoalveolar lavage (BAL) revealed Blastomyces dermatitidis; hence, the patient completed induction therapy with IV amphotericin B and 2 years of voriconazole treatment for both CNS and pulmonary blastomycosis. Her symptoms greatly resolved with antifungal therapy, and her brain lesion improved significantly, with only mild residual enhancement in the left thalamus on follow-up imaging 1 year after therapy completion (Figure 5, I–L).
Blastomycosis is caused by Blastomyces dermatitidis, which is endemic in the Midwest and Southeast in North America. The host typically contracts the infection by inhalation of the spores, which can disseminate through blood and lymphatics to the CNS.53 Fungal infections affecting the CNS most commonly occur in immunocompromised patients and are frequently located at the gray-white junction and perforated arterial zones.54 CNS blastomycosis is rare, affects 5–10% of patients with disseminated disease, and can present with parenchymal abscesses or as meningitis, with a wide range of symptoms and severity.55 Important MRI findings include heterogeneous diffusion restriction seen in DWI that typically precedes enhancement, because of the high viscosity of fungal pus, as well as a thin rim of peripheral enhancement known as a weak ring.54,55 Spinal fluid studies can be nondiagnostic because cultures may be negative for this infection.55 Tissue samples or cytologic material can reveal broad-based budding yeast forms of B. dermatitidis, with positive GMS and PAS stains.56 Recommended antimicrobial treatment consists of induction therapy with amphotericin B and maintenance with an azole agent for at least 12 months until there is resolution of infection.53
Discussion
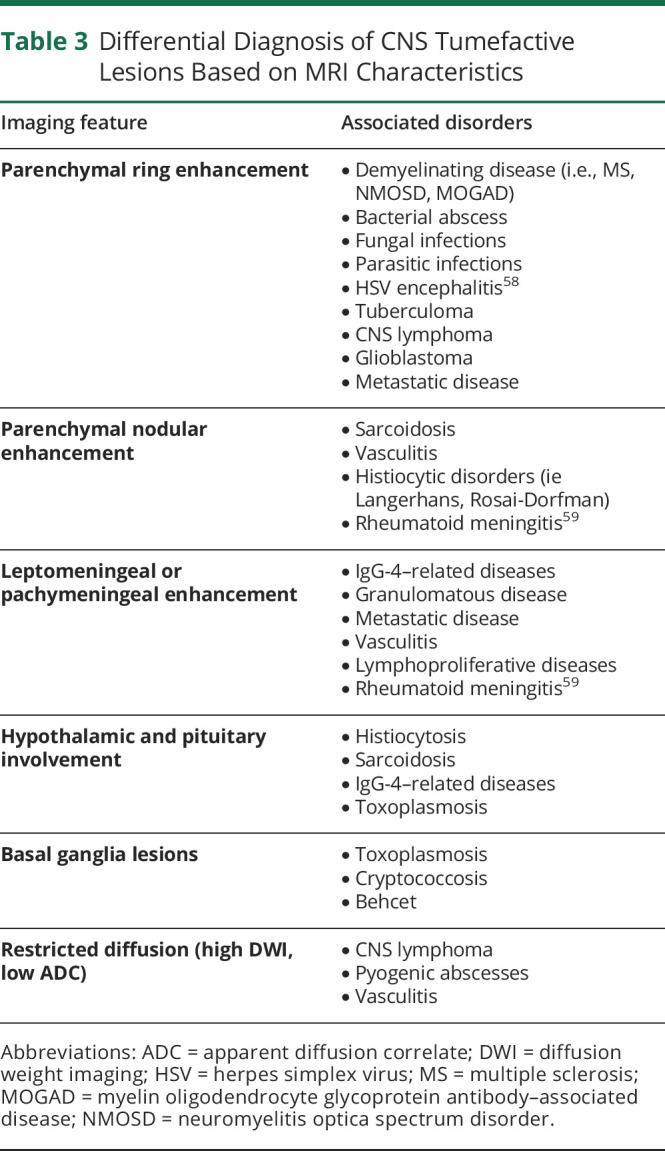
Tumor-like brain lesions have heterogeneous etiologies and can be challenging to diagnose (Table 2). Erroneous diagnosis and use of empiric treatment can lead to significant morbidity because chemotherapy and/or radiation can worsen these conditions and can decrease the yield of subsequent biopsies, even further contributing to accumulating disability. The appearance of such lesions on conventional imaging can be similar (Table 3), and advanced diagnostic aids such as MR spectroscopy, SPECT, PET, and perfusion images may be inconclusive. Fortunately, stereotactic brain biopsy has a high diagnostic yield and relatively benign risk, a recent meta-analysis noted mortality rates from 0.7% to 4% and morbidity ranged from 3% to 13%.60
Table 2.
Causes of Tumor-like Brain Lesions Mimicking Neoplasms, Pertinent MRI Findings, Pathology and Treatment

Table 3.
Differential Diagnosis of CNS Tumefactive Lesions Based on MRI Characteristics
Our case series and review of literature illustrate a potentially broad range of inflammatory/infectious etiologies for tumor-like brain lesions, which required brain biopsy for a correct diagnosis, and support the need for consideration of brain biopsy early in the diagnostic process for lesions with uncertain etiology because pathology-directed therapy improves clinical outcomes.
Tumor-like brain lesions represent a diagnostic challenge and may be neoplastic or non-neoplastic. The wide range of infectious and inflammatory potential etiologies of non-neoplastic tumor-like brain lesions and the relatively low risk of biopsy support early confirmation of pathology to guide appropriate therapeutic decisions.
Appendix. Authors

Footnotes
Editorial, page e200183
Study Funding
The authors report no targeted funding.
Disclosure
G.S. Perez reports no disclosures relevant to the manuscript; L. Singer reports no disclosures relevant to the manuscript; T. Cao reports no disclosures relevant to the manuscript; P. Jamshidi reports no disclosures relevant to the manuscript; K. Dixit reports no disclosures relevant to the manuscript; M. Kontzialis reports no disclosures relevant to the manuscript; R. Castellani reports no disclosures relevant to the manuscript; P. Pytel reports no disclosures relevant to the manuscript; N. Anadani reports no disclosures relevant to the manuscript; C. Bevan has participated in an ad board for Genentech; E. Grebenciucova reports no disclosures relevant to the manuscript; R. Balabanov received honoraria from Biogen, Sanofi, Alexion, and Teva Pharmaceutical and research support from the National Multiple Sclerosis Society, National Institute of Health, Nextcure, and Biogen; B. Cohen reports no disclosures relevant to the manuscript. E. Graham received consulting and advisory board fees from Novartis, Atara Biotherapeutics, Tavistock Life Sciences, Horizon Therapeutics, Roche Genentech. She receives research support from F. Hoffman-La Roche Ltd. She received compensation for question writing from ACP MKSAP. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/cp.
References
- 1.Frederick MC, Cameron MH. Tumefactive demyelinating lesions in multiple sclerosis and associated disorders. Curr Neurol Neurosci Rep. 2016;16(3):1-7. doi: 10.1007/s11910-016-0626-9 [DOI] [PubMed] [Google Scholar]
- 2.Algahtani H, Shirah B, Alassiri A. Tumefactive demyelinating lesions: a comprehensive review. Mult Scler Relat Disord. 2017;14:72-79. doi: 10.1016/j.msard.2017.04.003 [DOI] [PubMed] [Google Scholar]
- 3.Lucchinetti CF, Gavrilova RH, Metz I, et al. Clinical and radiographic spectrum of pathologically confirmed tumefactive multiple sclerosis. Brain. 2008;131(7):1759-1775. doi: 10.1093/brain/awn098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakayama M, Naganawa S, Ouyang M, et al. A review of clinical and imaging findings in tumefactive demyelination. Am J Roentgenol. 2021;217(1):186-197. doi: 10.2214/AJR.20.23226 [DOI] [PubMed] [Google Scholar]
- 5.Matsuoka T, Suzuki SO, Iwaki T, Tabira T, Ordinario AT, Kira JI. Aquaporin-4 astrocytopathy in Baló’s disease. Acta Neuropathol. 2010;120(5):651-660. doi: 10.1007/s00401-010-0733-7 [DOI] [PubMed] [Google Scholar]
- 6.Karaarslan E, Altintas A, Senol U, et al. Baló’s concentric sclerosis: clinical and radiologic features of five cases. Am J Neuroradiol; 2001:22(7):1362-7. [PMC free article] [PubMed] [Google Scholar]
- 7.Husseini L, Saleh A, Reifenberger G, Hartung HP, Kieseier BC. Inflammatory demyelinating brain lesions heralding primary CNS lymphoma. Can J Neurol Sci. 2012;39(1):6-10. doi: 10.1017/S0317167100012610 [DOI] [PubMed] [Google Scholar]
- 8.Suh CH, Kim HS, Jung SC, Choi CG, Kim SJ. MRI findings in tumefactive demyelinating lesions: a systematic review and meta-analysis. Am J Neuroradiol. 2018;39(9):1643-1649. doi: 10.3174/ajnr.A5775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xia L, Lin S, Wang ZC, et al. Tumefactive demyelinating lesions: nine cases and a review of the literature. Neurosurg Rev. 2009;32(2):171-179. doi: 10.1007/s10143-009-0185-5 [DOI] [PubMed] [Google Scholar]
- 10.Kobayashi M, Shimizu Y, Shibata N, Uchiyama S. Gadolinium enhancement patterns of tumefactive demyelinating lesions: correlations with brain biopsy findings and pathophysiology. J Neurol. 2014;261(10):1902-1910. doi: 10.1007/s00415-014-7437-1 [DOI] [PubMed] [Google Scholar]
- 11.Horská A, Barker PB. Imaging of brain tumors: MR spectroscopy and metabolic imaging. Neuroimaging Clin N Am. 2010;20(3): 293-310. doi.doi: 10.1016/j.nic.2010.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alharbi B, Alammar H, Alkhaibary A, et al. Primary spinal cord glioblastoma: a rare cause of paraplegia. Surg Neurol Int. 2022;13:160. doi: 10.25259/SNI_135_2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campello C, Tabouret E, Chinot O. Challenges in diagnosis and management of adult spinal cord gliomas. Rev Neurol (Paris). 2021;177(5):515-523. doi: 10.1016/j.neurol.2021.02.384 [DOI] [PubMed] [Google Scholar]
- 14.Kawai N, Miyake K, Yamamoto Y, Nishiyama Y, Tamiya T. 18F-FDG PET in the diagnosis and treatment of primary central nervous system lymphoma. Biomed Res Int. 2013;2013:247152. doi: 10.1155/2013/247152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elavarasi A, Dash D, Warrier AR, et al. Spinal cord involvement in primary CNS lymphoma. J Clin Neurosci. 2018;47:145-148. doi: 10.1016/j.jocn.2017.10.027 [DOI] [PubMed] [Google Scholar]
- 16.Sagiuchi T, Oka H, Utsuki S, et al. Increased accumulations of n-isopropyi-p-[123I]-iodoamphetamine related to tumefactive multiple sclerosis. Ann Nucl Med. 2005. Oct;19(7):603-6. doi: 10.1007/BF02985054. [DOI] [PubMed] [Google Scholar]
- 17.Biswas S, Nagaraj C, Mangalore S, Gupta AK. Tumefactive demyelination versus primary central nervous system lymphoma on 18F-fluorodeoxyglucose positron emission tomography magnetic resonance imaging: a twist in the tale. Indian J Nucl Med. 2019;34(3):237-240. doi: 10.4103/ijnm.IJNM_31_19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuhlmann T, Lassmann H, Brück W. Diagnosis of inflammatory demyelination in biopsy specimens: a practical approach. Acta Neuropathol. 2008;115(3):275-287. doi: 10.1007/s00401-007-0320-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.di Bonaventura R, Montano N, Giordano M, et al. Reassessing the role of brain tumor biopsy in the era of advanced surgical, molecular, and imaging techniques—a single-center experience with long-term follow-up. J Pers Med. 2021;11(9). doi: 10.3390/jpm11090909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dutra BG, da Rocha AJ, Nunes RH, Maia ACM. Neuromyelitis optica spectrum disorders: spectrum of MR imaging findings and their differential diagnosis. Radiographics. 2018;38(1):169-193. doi: 10.1148/rg.2018170141 [DOI] [PubMed] [Google Scholar]
- 21.Cacciaguerra L, Morris P, Tobin WO, et al. Tumefactive demyelination in MOG Ab–associated disease, multiple sclerosis, and AQP-4-IgG–positive neuromyelitis optica spectrum disorder. Neurology. 2023;100(13):e1418-e1432. doi: 10.1212/WNL.0000000000206820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saiki S, Ueno Y, Moritani T, et al. Extensive hemispheric lesions with radiological evidence of blood-brain barrier integrity in a patient with neuromyelitis optica. J Neurol Sci. 2009;284(1-2):217-219. doi: 10.1016/j.jns.2009.05.022 [DOI] [PubMed] [Google Scholar]
- 23.Mandler RN, Davis LE, Jeffery DR, Kornfeld M. Devic's neuromyelitis optica: a clinicopathological study of 8 patients. Ann Neurol. 1993;34(2):162-168. doi: 10.1002/ana.410340211 [DOI] [PubMed] [Google Scholar]
- 24.Loda E, Arellano G, Perez-Giraldo G, Miller SD, Balabanov R. Can immune tolerance be Re-established in neuromyelitis optica? Front Neurol. 2021;12. doi: 10.3389/fneur.2021.783304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramanathan S, Mohammad S, Tantsis E, et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J Neurol Neurosurg Psychiatry. 2018;89(2):127-137. doi: 10.1136/jnnp-2017-316880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shahriari M, Sotirchos ES, Newsome SD, Yousem DM. MOGAD: how it differs from and resembles other neuroinflammatory disorders. Am J Roentgenol. 2021;216(4):1031-1039. doi: 10.2214/AJR.20.24061 [DOI] [PubMed] [Google Scholar]
- 27.Wynford-Thomas R, Jacob A, Tomassini V. Neurological update: MOG antibody disease. J Neurol. 2019;266(5):1280-1286. doi: 10.1007/s00415-018-9122-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Satukijchai C, Mariano R, Messina S, et al. Factors associated with relapse and treatment of myelin oligodendrocyte glycoprotein antibody-associated disease in the United Kingdom. JAMA Netw Open. 2022;5(1):e2142780. doi: 10.1001/jamanetworkopen.2021.42780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ataka T, Kimura N, Matsubara E. A case of myelin oligodendrocyte glycoprotein-antibody-associated disease presenting with tumefactive demyelinating lesion. Mult Scler Relat Disord. 2020;43:102191. doi: 10.1016/j.msard.2020.102191 [DOI] [PubMed] [Google Scholar]
- 30.Cobo-Calvo A, Ruiz A, Rollot F, et al. Clinical features and risk of relapse in children and adults with myelin oligodendrocyte glycoprotei antibody-associated disease. Ann Neurol. 2021;89(1):30-41. doi. 10.1002/ana.25909 [DOI] [PubMed] [Google Scholar]
- 31.Chen JJ, Flanagan EP, Bhatti MT, et al. Steroid-sparing maintenance immunotherapy for MOG-IgG associated disorder. Neurology. 2020;95(2):E111-E120. doi: 10.1212/WNL.0000000000009758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Durozard P, Rico A, Boutiere C, et al. Comparison of the response to rituximab between myelin oligodendrocyte glycoprotein and aquaporin-4 antibody diseases. Ann Neurol. 2020;87(2):256-266. doi: 10.1002/ana.25648 [DOI] [PubMed] [Google Scholar]
- 33.Barreras P, Vasileiou ES, Filippatou AG, et al. Long-term effectiveness and safety of rituximab in neuromyelitis optica spectrum disorder and MOG antibody disease. Neurology. 2022;99(22):e2504-e2516. doi: 10.1212/wnl.0000000000201260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee JS, Jung TY, Lee KH, Kim SK. Primary central nervous system vasculitis mimicking a cortical brain tumor: a case report. Brain Tumor Res Treat. 2017;5(1):30. doi: 10.14791/btrt.2017.5.1.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salvarani C, Brown RD, Calamia KT, et al. Primary CNS vasculitis with spinal cord involvement. Neurology. 2008;70(24 Part 2):2394-2400. doi: 10.1212/01.wnl.0000314687.69681.24 [DOI] [PubMed] [Google Scholar]
- 36.Kraemer M, Berlit P. Primary central nervous system vasculitis – an update on diagnosis, differential diagnosis and treatment. J Neurol Sci. 2021;424:117422. doi: 10.1016/j.jns.2021.117422 [DOI] [PubMed] [Google Scholar]
- 37.Mandell DM, Mossa-Basha M, Qiao Y, et al. Intracranial vessel wall MRI: principles and expert consensus recommendations of the American Society of Neuroradiology. Am J Neuroradiol. 2017;38(2):218-229. doi: 10.3174/ajnr.A4893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Al-Araji A, Kidd DP. Neuro-Behçet’s disease: epidemiology, clinical characteristics, and management. Lancet Neurol. 2009;8(2):192-204. doi: 10.1016/S1474-4422(09)70015-8 [DOI] [PubMed] [Google Scholar]
- 39.Takeno M. The association of Behçet’s syndrome with HLA-B51 as understood in 2021. Curr Opin Rheumatol. 2022;34(1):4-9. doi: 10.1097/BOR.0000000000000846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yasar Bilge NS, Saylisoy S, Kasifoglu T, Korkmaz C. Mass-like lesions as a rare form of neuro-Behçet’s disease: a case report and review of the literature. Europen J Rheumatol. 2014;1(1):34-38. doi: 10.5152/eurjrheum.2014.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seyahi E, Karaaslan H, Ugurlu S, Yazici H. Fever in Behçet’s syndrome. Clin Exp Rheumatol. 2013;31(3 suppl 77):64-67. [PubMed] [Google Scholar]
- 42.Bradshaw MJ, Pawate S, Koth LL, Cho TA, Gelfand JM. Neurosarcoidosis: pathophysiology, diagnosis, and treatment. Neurol Neuroimmunol Neuroinflamm. 2021;8:e1084. doi: 10.1212/nxi.0000000000001084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stern BJ, Royal W, Gelfand JM, et al. Definition and consensus diagnostic criteria for neurosarcoidosis: from the neurosarcoidosis consortium consensus group. JAMA Neurol. 2018;75(12):1546-1553. doi: 10.1001/jamaneurol.2018.2295 [DOI] [PubMed] [Google Scholar]
- 44.Valeyre D, Prasse A, Nunes H, Uzunhan Y, Brillet PY, Müller-Quernheim J. Sarcoidosis. Lancet. 2014;383(9923):1155-1167. doi: 10.1016/S0140-6736(13)60680-7 [DOI] [PubMed] [Google Scholar]
- 45.Saitakis G, Chwalisz BK. The neurology of IGG4-related disease. J Neurol Sci. 2021;424:117420. doi: 10.1016/j.jns.2021.117420 [DOI] [PubMed] [Google Scholar]
- 46.Baptista B, Casian A, Mrcp MA, Rice CM. Neurological manifestations of IgG4-related disease. Curr Treat Options Neurol. 2017;19(4):14. doi. 10.1007/s11940-017-0450-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cohen Aubart F, Idbaih A, Emile JF, et al. Histiocytosis and the nervous system: from diagnosis to targeted therapies. Neuro Oncol. 2021;23(9):1433-1446. doi: 10.1093/neuonc/noab107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goyal G, Young JR, Koster MJ, et al. The Mayo Clinic Histiocytosis Working Group consensus statement for the diagnosis and evaluation of adult patients with histiocytic neoplasms: Erdheim-Chester disease, langerhans cell histiocytosis, and rosai-dorfman disease. Mayo Clin Proc. 2019;94(10):2054-2071. doi: 10.1016/j.mayocp.2019.02.023 [DOI] [PubMed] [Google Scholar]
- 49.McClain KL, Picarsic J, Chakraborty R, et al. CNS Langerhans cell histiocytosis: common hematopoietic origin for LCH-associated neurodegeneration and mass lesions. Cancer. 2018;124(12):2607-2620. doi: 10.1002/cncr.31348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vidal JE. HIV-related cerebral toxoplasmosis revisited: current concepts and controversies of an old disease. J Int Assoc Provid AIDS Care. 2019;18:2325958219867315. doi: 10.1177/2325958219867315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Donahue JH, Patel SH, Fadul CE, Mukherjee S. Imaging mimics of brain tumors. Radiol Clin North Am. 2021;59(3):457-470. doi: 10.1016/j.rcl.2021.02.003 [DOI] [PubMed] [Google Scholar]
- 52.Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet. 2004;363(9425):1965-1976. doi. 10.1016/S0140-6736(04)16412-X [DOI] [PubMed] [Google Scholar]
- 53.Ta M, Flowers SA, Rogers D. The role of voriconazole in the treatment of central nervous system blastomycosis. Ann Pharmacother. 2009;43(10):1696-1700. doi. 10.1345/aph.1M010 [DOI] [PubMed] [Google Scholar]
- 54.Starkey J, Moritani T, Kirby P. MRI of CNS fungal infections: review of aspergillosis to histoplasmosis and everything in between. Clin Neuroradiol. 2014;24(3):217-230. doi: 10.1007/s00062-014-0305-7 [DOI] [PubMed] [Google Scholar]
- 55.Majdick K, Kaye K, Shorman MA. Central nervous system blastomycosis clinical characteristics and outcomes. Med Mycol. 2021;59(1):87-92. doi: 10.1093/mmy/myaa041 [DOI] [PubMed] [Google Scholar]
- 56.Patel AJ, Gattuso P, Reddy VB. Diagnosis of Blastomycosis in Surgical Pathology and Cytopathology: Correlation With Microbiologic Culture; 2010. ajsp.com [DOI] [PubMed] [Google Scholar]
- 57.Ayrignac X, Carra-Dallière C, Marelli C, Taïeb G, Labauge P. Adult-onset genetic central nervous system disorders masquerading as acquired neuroinflammatory disorders. JAMA Neurol. 2022;79(10):1069-1078. doi: 10.1001/jamaneurol.2022.2141 [DOI] [PubMed] [Google Scholar]
- 58.Das B, Goyal MK, Modi M, et al. A rare cause of temporal lobe ring-enhancing lesion. Neurol Clin Pract. 2015;5(6):538-539. doi: 10.1212/CPJ.0000000000000195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qin Z, Kim J, Valencia D, et al. Rheumatoid meningitis. Neurol Clin Pract. 2020;10(1):73-83. doi: 10.1212/cpj.0000000000000678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Riche M, Amelot A, Peyre M, Capelle L, Carpentier A, Mathon B. Complications after frame-based stereotactic brain biopsy: a systematic review. Neurosurg Rev. 2021;44(1):301-307. doi. 10.1007/s10143-019-01234-w [DOI] [PubMed] [Google Scholar]