

Summary
We performed a study to present a phenotypic and genotypic characterization of a patient clinically diagnosed with carbonic anhydrase II (CAII) deficiency syndrome. Medical records were reviewed, and oral examination was performed. Sanger sequencing was undertaken for molecular diagnosis. The patient presented with osteopetrosis, renal tubular acidosis, cerebral calcification, blindness, deafness, and development delay. The oral manifestations included anterior open bite, posterior crossbite, tooth eruption impairment, and hypoplastic amelogenesis imperfecta (AI). Molecular analysis revealed a CA2 homozygous deletion (c.753delG, p.Asn252Thrfs*14) and confirmed the clinical diagnosis. This study suggests that AI can be another feature of CAII deficiency syndrome. For the first time, a CA2 disease-causing variant is reported to be associated with syndromic AI.
Keywords: amelogenesis imperfecta, osteopetrosis, renal tubular acidosis, carbonic anhydrase II
Carbonic anhydrase II (CAII) deficiency syndrome (#OMIM 259730), also known as marble brain disease or Guibaud-Vainsel syndrome, is an autosomal recessive disease characterized by an increased bone density and metabolic acidosis that cause osteopetrosis, renal tubular acidosis and cerebral calcification (1). Affected individuals usually present with cranial nerve compression causing intellectual disability, blindness and deafness, recurrent bone fractures, and nephrocalcinosis. CAII is a cytoplasmic enzyme encoded by the CA2 gene, mainly expressed in bone, brain, distal renal tubules, and erythrocytes, regulating intracellular pH by catalyzing the conversion of carbonic acid (H2CO3) to bicarbonate (HCO3-) and hydrogen ion (H+). CAII is involved in osteoclast differentiation, bone resorption, and in the acid-base physiology of the kidneys (2).
In recent years, experimental studies in mice have also demonstrated that ameloblasts express CAII during amelogenesis, especially in the transition and maturation stage (3-5). In mice, this protein plays a role in intracellular pH control, which is essential for normal enamel development. However, the enamel phenotype description has not been the focus of the clinical reports. Tooth eruption disturbances, severe dental caries, crowded teeth, and dental malocclusion are the most common oral manifestations reported (6-8).
Amelogenesis imperfecta (AI) comprises a heterogeneous group of inherited development defects of enamel (DDE) (9). AI can be an isolated or syndromic trait that affects all or almost all teeth in both dentitions. The mode of inheritance can be autosomal recessive, autosomal dominant, or X-linked. In hypoplastic AI, the enamel thickness or shape is altered; in hypomaturation and hypocalcified AI, the enamel matrix mineralization is incomplete, and enamel hardness is reduced.
The present study aimed to perform a phenotypic and genotypic characterization in a current 26-year-old female Brazilian patient (III:1) clinically diagnosed with CAII deficiency at age nine years old.
The study was approved by the Research Ethics Committee, Faculty of Health Sciences, University of Brasilia (certificate of presentation for ethical appreciation number 43064320.3.0000.0030) in accordance with the Declaration of Helsinki. Informed consent was obtained from the participants.
The proband was the only affected member in the family (Figure 1, A). Parents reported no consanguinity, but both were born in a small village in the State of Maranhão, Brazil. She was an only child, delivered from an uncomplicated pregnancy. She presented with osteopetrosis, microcephaly, cerebral calcifications, developmental delay, short stature, limb malformations, and recurrent fractures of the femur, tibia, and fibula (Figure 1, C-E). Furthermore, she presented right optic nerve atrophy with visual impairment, strabismus, horizontal nystagmus, and conductive hearing loss. At age nine years old, she presented with low values of venous blood pH (7.21), low HCO3 (16.7 mEq/L) and low base excess (-10.5 mmol/L); high urinary pH (6.0); high urinary excretion of calcium (5.4 mg/kg/24 h), and low excretion of citrate (0.32 mmol/24 h). She was diagnosed with distal renal tubular acidosis, metabolic acidosis, hypercalciuria, and hypocitraturia.
Figure 1.
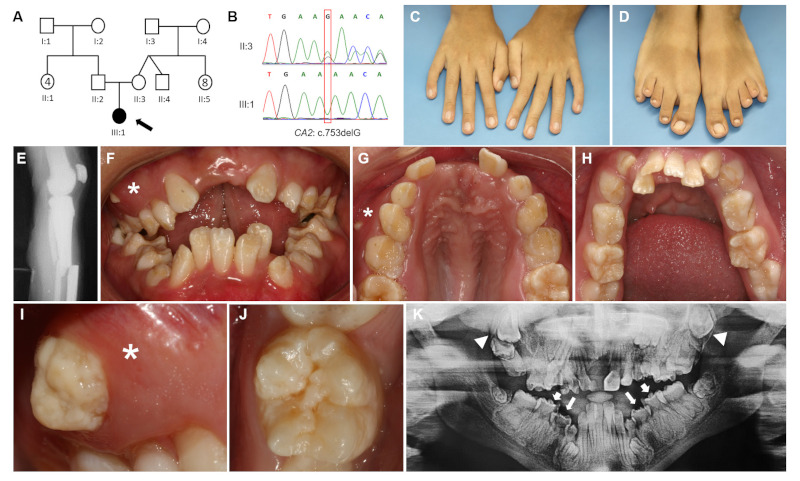
Genetic, clinical, and radiographic findings of this study. (A) Pedigree illustrates the absence of other affected members in the family or parental consanguinity. (B) Chromatograms show mother heterozygosity and proband homozygosity for the CA2 variant. (C) Physical extraoral examination detected slender fingers and bilateral 5th finger clinodactyly in upper limbs. (D) It was also observed hammertoe bilaterally (1st toes), sandal gap, and short 3rd/4th toes in lower limbs. (E) Radiography of knee and leg showing tibial and fibular fractures. (F) Intraoral examination at age 17: anterior open bite, posterior crossbite with crowded teeth, and ectopic eruption (*). (G) The occlusal view shows a high-arched palate with maxillary atresia. (H) Crowded teeth in the mandible and primary second molars retention. (I) Ectopic eruption of maxillary first premolar with hypoplastic pitting enamel (*). (J) Approximate view of a lower permanent first molar with hypoplastic pitting enamel. (K) Dental panoramic radiography at age 26 revealed retention of primary molars, eruption failure of upper permanent molars (white triangles), and abnormal enamel thickness in primary and permanent molars (white arrows).
At age 9, she was referred to the Oral Care Center for Inherited Diseases, University Hospital of Brasilia, University of Brasilia, and a complete oral examination was performed. She presented mixed dentition with generalized enamel hypoplasia, high-arched and ogival palate, anterior open bite, posterior crossbite, crowded teeth, ectopic eruption, and altered tooth shape. Later, at age 17, she presented with retained primary and impacted permanent teeth. All primary and permanent teeth presented with hypoplastic pits and grooves resembling hypoplastic AI (Figure 1, F-J). She also presented with gingival inflammation in areas of crowded teeth due to difficult access for oral hygiene. Dental panoramic radiography showed tooth eruption impairment and impacted teeth (Figure 1, K).
Venous blood was collected from the proband and the mother for DNA isolation. CA2 gene was amplified using specific pairs of primers (available upon request), and PCR products were sequenced. Sanger sequencing results were compared with CA2 reference sequences (http://www.ensembl.org) and revealed that the proband was homozygous and the mother (II:3) was heterozygous for a single base pair deletion resulting in a frameshift mutation in exon 7 (CA2: c.753delG, p.Asn252Thrfs*14) (Figure 1, B). The variant was predicted to be likely pathogenic (PM2, PVS1, PP4) according to the American College of Medical Genetics and Genomics (ACMG) criteria (10). The father (II:2) was not available for the genetic exam. The variant nomenclature was checked on the Mutalyzer Website (https://mutalyzer.nl) and registered in the Leiden Open Variation Database (LOVD).
In this study, the patient presented with the classic features of CAII deficiency syndrome, except for the absence of nephrocalcinosis, and the presence of hypoplastic AI, which had never been previously reported in this syndrome. The same variant detected in our patient had already been reported in two Mexican siblings (11), but the orodental features were not described.
CAII deficiency cases present a particular geographic distribution and have been mostly reported in the Mediterranean region and the Middle East (6,12), i.e. patients of Arab descent, probably due to the high occurrence of consanguineous marriages in this population. In the present family, no consanguinity was reported, but once the parents are natives from a small village with a reduced population, the possibility of familial genetic similarities between them may be considered to explain the homozygosity detected in the proband.
We performed a literature search in 3 databases (Medline, PubMed, and Web of Science) until July 2023, covering the topics: "CAII deficiency OR carbonic anhydrase 2" AND "oral manifestations OR enamel". After the removal of duplicates and reports in foreign languages, a total of 309 studies were retrieved. Abstracts were screened, and 60 records were eligible for the next phase. After reading the complete studies, we identified only four case reports (13-16) and one series case report (6) that observed enamel hypoplasia in some patients. Among them, only one case report confirmed the molecular etiology and described the presence of enamel hypoplasia in three patients from two unrelated families (13). None of them mentioned generalized DDE resembling AI.
The renal phenotype of Car2(-/-) mice are similar to the observed in CAII deficiency patients (17), i.e., metabolic acidosis and impaired urine acidification, but there is no transgenic Ca2 animal models study with an orodental approach in the literature. Our patient presented AI in both primary and permanent dentition. As the enamel development of primary dentition occurs during prenatal life, when the placenta provides the fetal homeostasis for amelogenesis, it may be assumed that DDE in primary teeth are unlikely to be caused by systemic disturbances but by an inherited condition in patients born from uncomplicated pregnancies (18). DDE may also be caused by other factors such as local infections, dental trauma, environmental and systemic disturbances. Thus, a secondary effect of the metabolic acidosis found in our patient cannot be discharged once the ameloblasts are sensitive to pH changes during amelogenesis.
The CAII role during amelogenesis has only recently been described (3-5), and it may explain why dental enamel phenotype has not been carefully characterized in previous studies. Furthermore, in the past, AI used to be classified only as an isolated trait caused by variants in genes that encoded for enamel matrix proteins or proteases. Advances in molecular diagnosis techniques have identified disease-causing variants in at least 26 different genes that cause 19 types of syndromic AI (9), including five autosomal recessive renal diseases linked to CLDN16 (#OMIM 248250), CLDN19 (#OMIM 248190), FAM20A (#OMIM 204690), KCNJ1 (#OMIM 241200) and SLC4A4 genes (#OMIM 604278). All these genes are functionally expressed in both ameloblasts and kidney cells.
In conclusion, the generalized enamel hypoplasia observed in the primary and permanent teeth in the present case suggests that AI can be part of the phenotypic spectrum of CAII deficiency syndrome. Additionally, we suggest that this syndrome should be included in the hall of syndromic AI and, more specifically, in the group of autosomal recessive diseases with renal and dental enamel involvement. These results reinforce the importance of referring patients with autosomal recessive renal diseases for oral examination. Likewise, AI patients should be referred for nephrological evaluation.
Acknowledgements
We thank the study participants for their kind cooperation.
Funding
This study was supported by CAPES/ COFECUB, National Council for Scientific and Technological Development (CNPq), and Decanato de Pesquisa e Inovação, University of Brasilia.
Conflict of Interest
The authors have no conflicts of interest to disclose.
References
- 1. Whyte MP. Carbonic anhydrase II deficiency. Bone. 2023; 169:116684. [DOI] [PubMed] [Google Scholar]
- 2. Borthwick KJ, Kandemir N, Topaloglu R, Kornak U, Bakkaloglu A, Yordam N, Ozen S, Mocan H, Shah GN, Sly WS, Karet FE. A phenocopy of CAII deficiency: A novel genetic explanation for inherited infantile osteopetrosis with distal renal tubular acidosis. J Med Genet. 2003; 40:115-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bori E, Guo J, Rácz R, Burghardt B, Földes A, Kerémi B, Harada H, Steward MC, Den Besten P, Bronckers AL, Varga G. Evidence for bicarbonate secretion by ameloblasts in a novel cellular model. J Dent Res. 2016; 95:588-596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yin K, Paine ML. Bicarbonate transport during enamel maturation. Calcif Tissue Int. 2017; 101:457-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lin HM, Nakamura H, Noda T, Ozawa H. Localization of H+-ATPase and carbonic anhydrase II in ameloblasts at maturation. Calcif Tissue Int. 1994; 55:38-45. [DOI] [PubMed] [Google Scholar]
- 6. Awad M, Al-Ashwal AA, Sakati N, Al-Abbad AA, Bin- Abbas BS. Long-term follow up of carbonic anhydrase II deficiency syndrome. Saudi Med J. 2002; 23:25-29. [PubMed] [Google Scholar]
- 7. Pang Q, Qi X, Jiang Y, Wang O, Li M, Xing X, Dong J, Xia W. Two novel CAII mutations causing carbonic anhydrase II deficiency syndrome in two unrelated Chinese families. Metab Brain Dis. 2015; 30:989-997. [DOI] [PubMed] [Google Scholar]
- 8. Alsemari A, Alsuhaibani M, Alhathlool R, Ali BM. Potential oligogenic disease of mental retardation, short stature, spastic paraparesis, and osteopetrosis. Appl Clin Genet. 2018; 11:129-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dong J, Ruan W, Duan X. Molecular-based phenotype variations in amelogenesis imperfecta. Oral Dis. 2023; doi: 10.1111/odi.14599. [DOI] [PubMed] [Google Scholar]
- 10. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17:405-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hu P, Lim E, Ciccolella J, Strisciuglio P, Sly W. Seven novel mutations in carbonic anhydrase II deficiency syndrome identified by SSCP and direct sequencing analysis. Hum Mutat. 1997; 9:383-387. [DOI] [PubMed] [Google Scholar]
- 12. Sly WS, Whyte MP, Sundaram V, et al. Carbonic anhydrase II deficiency in 12 families with the autosomal recessive syndrome of osteopetrosis with renal tubular acidosis and cerebral calcification. N Engl J Med. 1985; 313:139-145. [DOI] [PubMed] [Google Scholar]
- 13. Ismail EA, Abul Saad S, Sabry MA. Nephrocalcinosis and urolithiasis in carbonic anhydrase II deficiency syndrome. Eur J Pediatr. 1997; 156:957-962. [DOI] [PubMed] [Google Scholar]
- 14. Ohlsson A, Stark G, Sakati N. Marble brain disease: Recessive osteopetrosis, renal tubular acidosis and cerebral calcification in three Saudi Arabian families. Dev Med Child Neurol. 1980; 22:72-84. [DOI] [PubMed] [Google Scholar]
- 15. Strisciuglio P, Sartorio R, Pecoraro C, Lotito F, Sly WS. Variable clinical presentation of carbonic anhydrase deficiency: Evidence for heterogeneity? Eur J Pediatr. 1990; 149:337-340. [DOI] [PubMed] [Google Scholar]
- 16. Nagai R, Kooh SW, Balfe JW, Fenton T, Halperin ML. Renal tubular acidosis and osteopetrosis with carbonic anhydrase II deficiency: Pathogenesis of impaired acidification. Pediatr Nephrol. 1997; 11:633-636. [DOI] [PubMed] [Google Scholar]
- 17. Hains DS, Chen X, Saxena V, Barr-Beare E, Flemming W, Easterling R, Becknell B, Schwartz GJ, Schwaderer AL. Carbonic anhydrase 2 deficiency leads to increased pyelonephritis susceptibility. Am J Physiol Renal Physiol. 2014; 307:F869-F880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koch MJ, Bührer R, Pioch T, Schärer K. Enamel hypoplasia of primary teeth in chronic renal failure. Pediatr Nephrol. 1999; 13:68-72. [DOI] [PubMed] [Google Scholar]