

ABSTRACT
Despite its name, the current diagnosis of acute kidney injury (AKI) still depends on markers of decreased kidney function and not on markers of injury. This results in a delayed diagnosis: AKI is diagnosed based on serum creatinine criteria only when the severity of injury is enough to decrease glomerular filtration rate. Moreover, by the time AKI is diagnosed, the insult may have already ceased, and even appropriate therapy targeted at the specific insult and its associated pathogenic pathways may no longer be effective. Biomarkers of injury are needed that allow the diagnosis of AKI based on injury criteria. At least three commercially available immunoassays assessing urinary or plasma neutrophil gelatinase-associated lipocalin and urinary tissue inhibitor of metalloproteinases-2 × insulin-like growth factor-binding protein-7 ([TIMP2]*[IGFBP7]) (NephroCheck®) have generated promising data regarding prediction and early diagnosis of AKI, although their relative performance may depend on clinical context. Recently, a urinary peptidomics classifier (PeptAKI) was reported to predict AKI better than current biomarkers. Focusing on [TIMP2]*[IGFBP7], the cellular origin of urinary TIMP2 and IGFBP7 remains unclear, especially under the most common predisposing condition for AKI, i.e. chronic kidney disease. We now discuss novel data on the kidney cell expression of TIMP2 and IGFBP7 and its clinical implications.
Keywords: acute kidney injury, biomarkers, chronic kidney disease, IGFBP7, NephroCheck, NGAL, TIMP2, urinary proteomics
BACKGROUND
Acute kidney injury (AKI) is common among hospitalized patients, occurs more frequently in those with preexistent chronic kidney disease (CKD) and is associated with high mortality, as recently confirmed during the coronavirus disease 2019 (COVID-19) pandemic [1, 2]. Currently, there is no treatment for AKI beyond kidney replacement therapy (KRT) and hoping for a spontaneous recovery, with some exceptions in which a cause with specific treatment is identified (e.g. crescentic glomerulonephritis). Furthermore, AKI preventive measures rely mainly on avoiding malpractice, such a maintaining adequate kidney perfusion or avoiding nephrotoxic drugs, rather than on modulating molecular drivers of kidney injury, and despite this, compliance was poor in clinical trials [3].
SHORTCOMINGS OF THE CURRENT DIAGNOSTIC CRITERIA FOR AKI
A suboptimal definition may partly explain unmet clinical needs in the treatment and outcomes of AKI. Despite its name, its current definition still depends on markers of decreased kidney function and not on markers of injury. This results in a delayed diagnosis: AKI is diagnosed based on serum creatinine criteria only when the severity of injury is enough to decrease the estimated glomerular filtration rate (eGFR). However, the magnitude of the decrease of eGFR associated with a 0.3 mg/dL increase in serum creatinine may vary from ≥1 to ≥36 mL/min/1.73 m2, depending on baseline eGFR (Fig. 1) [4]. This very broad range of functional deterioration encompassed under the umbrella term of AKI also illustrates the need for biomarkers of injury that allow a diagnosis of AKI in patients with milder functional deterioration. Given these limitations, the 2021 American Association for Clinical Chemistry (AACC) Guidance Document on Laboratory Investigation of AKI proposed to amend the KDIGO serum creatinine thresholds for diagnosis of AKI to an increase of +0.20 mg/dL or +20% (whichever is greater) [5]. Furthermore, the AACC guidelines recommend the use of creatinine assays with intra-laboratory analytical variability ≤3.4% [5]. This would prevent a diagnosis of AKI in patients with preexisting advanced CKD who display small changes in serum creatinine.
Figure 1:

Decrease in eGFR associated with an increase in serum creatinine of 0.3 mg/dL or more in individuals with different levels on baseline serum creatinine. Baseline eGFR and eGFR after serum creatinine increase by 0.3 mg/dL was estimated in females aged 20 years using the 2009 CKD Epidemiology Collaboration equation without using race correction. For the calculations, it was assumed that following the increase in serum creatinine that allowed the diagnosis of AKI, serum creatinine remained stable thereafter for at least a week.
To complicate matters further, by the time AKI is diagnosed, the insult may have already ceased, and even appropriate therapy targeted at the specific insult and its associated pathogenic pathways may no longer be effective. The combination of delayed diagnosis, lack of information on ongoing injury or on active pathophysiological mechanisms, and a serum creatinine–based definition of AKI that encompasses decrements in eGFR ranging from 1 to >100 mL/min/1.73 m2 makes it especially difficult to design clinical trials that recruit a homogenous patient population characterized by active ongoing kidney injury and to define appropriate endpoints that also reflect a homogeneous assessment of outcome. Biomarkers of injury are needed that allow the diagnosis of AKI or risk of AKI based on injury criteria. This may allow the recruitment of more homogenous populations for early treatment clinical trials and, eventually, early intervention in clinical practice.
KEY NOVEL BIOMARKERS FOR AKI DIAGNOSIS AND RISK CATEGORIZATION
Among the multiple emerging biomarkers in the field of AKI, urinary tissue inhibitor of metalloproteinases-2 × insulin-like growth factor-binding protein-7 ([TIMP2]*[IGFBP7]) and urinary or plasma neutrophil gelatinase-associated lipocalin (NGAL) are supported by more evidence. At least three commercially available immunoassays are marketed to enable earlier identification of AKI or short-term risk of AKI based on these biomarkers, thus facilitating the early adoption of therapeutic measures, and potentially leading to improved outcomes. NephroCheck® (Astute Medical, Inc., San Diego, CA, USA) assesses urinary [TIMP2]*[IGFBP7]. Additionally, urine NGAL (ARCHITECT®, Abbott Laboratories, Abbott Park, IL, USA) and urine and plasma NGAL (BioPorto Diagnostics A/S, Hellerup, Denmark) can also be assessed [6]. However, a 2022 systematic review and meta-analysis found no studies addressing the clinical impact of the use of these biomarkers on patient outcomes compared with standard care, and concluded that it was not possible to determine a plausible base-case incremental cost-effectiveness ratio for the tests, again compared with standard care [6]. In this regard, urinary [TIMP2]*[IGFBP7] was not yet recommended by the AACC for routine risk assessment of AKI due to the lack of evidence of benefit shown in outcome studies, high false-positive rate, and limited performance studies outside of the intensive care unit (ICU) or perioperative setting [5].
In addition to these immunoassays, serum cystatin A and a point-of-care test for plasma proenkephalin A 119–159 have been proposed as more precise measures of GFR than serum creatinine to be used in patients with AKI (penKid, Sphingotec GmbH, Hennigsdorf, Germany) [7], and the Nephroclear CCL14 Test assessing urinary C-C motif chemokine ligand 14 (CCL4) predicted persistent severe AKI [8, 9]. The 2021 AACC Guidance Document on Laboratory Investigation of AKI indicates that serum cystatin C may be helpful in predicting renal recovery earlier than serum creatinine among hospitalized patients with AKI. However, the assay was not universally recommended due to poor standardization, the lack of availability from most vendors and high cost (in comparison with creatinine) [5]. Additionally, the 2021 AACC document recommended the use of a urine sediment scoring system based on the number of granular casts and renal tubular epithelial cells per high-power field to differentially diagnose AKI [5].
[TIMP2]*[IGFBP7], NephroCheck AND AKIRisk
NephroCheck is a bedside test that provides a numeric result, labeled AKIRisk, obtained from calculating the product of urinary TIMP2 and IGFBP7 concentrations ([TIMP2]*[IGFBP7]) [10]. In 2014, the US Food and Drug Administration approved the use of [TIMP2]*[IGFBP7] to aid in the early prediction of AKI [10]. In four clinical settings [ischemia–reperfusion injury (IRI), cardiac failure, severe AKI and chemotherapy-induced kidney injury], AKIRisk increased rapidly prior to any change in serum creatinine. Moreover, a rapid decline in AKIRisk was associated with the restoration of kidney function, whereas a persistent increase was associated with kidney failure. However, the dynamics may differ, depending on the cause and the extent of injury [11].
NephroCheck is based on studies going back 10 years [12] (Fig. 2). In 2013, Kashani et al. identified and validated urinary [TIMP2]*[IGFBP7] as a predictor of moderate to severe AKI (KDIGO stage 2–3) within 12 h of sample collection among over 1200 participants from three observational studies that enrolled critically ill patients divided into discovery and validation cohorts [12]. The area under the receiver operating characteristic curve (AUC) for AKI KDIGO stage 2–3 was 0.80 and was superior to urine or plasma NGAL, or urine kidney injury molecule-1 (KIM-1), interleukin (IL)-18, Glutathione S-transferase pi (GST-pi) or liver-type fatty acid-binding protein (L-FABP), none of which achieved an AUC >0.72. However, the combination added little overall information over that provided by urinary TIMP2 alone [AUC 0.79 with overlapping 95% confidence intervals (CIs)], although this may vary with clinical setting, and this was likely the reason for combining both biomarkers into a single numeric result. The best performance was observed in patients with sepsis (AUC 0.82) and post-surgery (AUC 0.85). Differential performances according to clinical setting were also suggested for other biomarkers: urine and plasma NGAL performed better than urine KIM-1 for sepsis, while the reverse was true post-surgery [12]. Furthermore, [TIMP2]*[IGFBP7] significantly improved risk stratification for AKI when added to a nine-variable clinical model and predicted major adverse kidney events (death, dialysis or persistent renal dysfunction) within 30 days [12]. Over the next year, cut-off points were established for clinical use in the prediction of AKI. The relative risk (95% CI) of AKI within 12 h for subjects with AKIRisk™ score values of >0.3–2.0 (ng/mL)2/1000 was 4.4–4.7 higher than for those with values ≤0.3 (ng/mL)2/1000. For AKIRisk™ scores >2.0 (ng/mL)2/1000, the relative risk was 12–18 higher than for those with lower scores. Sensitivity was 89% and 42%–43% and specificity 50%–53% and 90%–95%, respectively for each cut-off point [13]. Moreover, a positive test also identified patients with AKI at increased risk for mortality or need for KRT over the next 9 months [14]. A rapid decline in [TIMP2]*[IGFBP7] also predicted renal recovery in patients with AKI [15]. In 2019, an expert panel representing Europe and North America suggested that patients undergoing major surgery (both cardiac and non-cardiac), those who were hemodynamically unstable or those with sepsis, may be tested for kidney stress using [TIMP2]*[IGFBP7] and categorized into those at high risk and those candidates for “fast-track” protocols [16].
Figure 2:
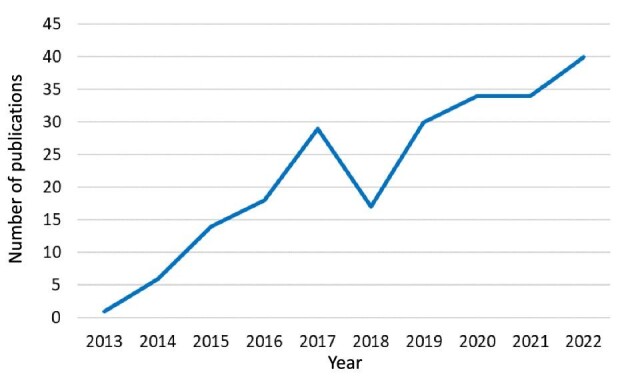
Number of publications in PubMed retrieved by the query “(nephrocheck OR TIMP2 OR IGFBP7) AND acute kidney injury” on 15 February 2023.
In additional studies, serial urinary [TIMP2]*[IGFBP7] values at baseline, 12 and 24 h, and up through 3 days were prognostic for the occurrence of AKI stage 2/3 over the course of critical illness [17]. Three consecutive negative values [≤0.3 (ng/mL)2/1000] were associated with very low (13.0%) incidence of AKI stage 2/3 over the course of 7 days. Conversely, strongly positive results [>2.0 (ng/mL)2/1000], emerging or persistent, predict very high incidence rates (up to 94.4%) of AKI stage 2/3. There was a low rate of test results between 0.3 and 2.0 (ng/mL)2/1000, where the primary endpoint was observed in a third of cases [17].
The PrevAKI-Multicenter Randomized Controlled Trial examined the adherence to the KDIGO bundle (primary endpoint) consisting of optimization of volume status and hemodynamics, functional hemodynamic monitoring, avoidance of nephrotoxic drugs and prevention of hyperglycemia in high-risk patients identified by urinary [TIMP2]*[IGFBP7] after cardiac surgery. This pilot study analyzed 278 patients, found that the protocol was feasible, although compliance was poor (65%) and observed a reduction in moderate to severe AKI [secondary endpoint: 14.0% vs 23.9%; absolute risk reduction 10.0% (95% CI, 0.9–19.1); P = .034] in the intervention group, despite no differences in the overall incidence of AKI [3]. In further studies, the combination of urinary [TIMP2]*[IGFBP7] with serum creatinine, but not with cystatin C or proenkephalin, improved risk stratification for the development of AKI stage 2/3 over [TIMP2]*[IGFBP7] alone [18].
Urinary [TIMP2]*[IGFBP7] was also reported to respond to kidney protective therapy. Patients with acute decompensated heart failure with or without diabetes were randomized to empagliflozin 10 mg or placebo for 30 days. Empagliflozin significantly reduced urinary [TIMP2]*[IGFBP7], which became significant after 3 days of treatment [placebo: 1.1 ± 1.1 (ng/mL)2/1000; empagliflozin: 0.3 ± 0.2 (ng/mL)2/1000; P = .02] and remained significant at the 7-day time point [placebo: 2.5 ± 3.8 (ng/mL)2/1000; empagliflozin: 0.3 ± 0.2 (ng/mL)2/1000; P = .003] [19]. Thus, an intervention improved urinary [TIMP2]*[IGFBP7] values, but since sodium-glucose cotransporter 2 (SGLT2) inhibition is both kidney protective and heart protective [20], a pathophysiological interpretation of the data is not straightforward.
Interestingly, following a marathon race, serum creatinine increased by 40%, urinary TIMP2 by 555% and urinary IGFBP7 by 1094% over the course of the race, but in the limited number of participants who supplied samples 24 h post-race, values were similar to baseline for all variables. The study suggests that beyond functional changes in kidney function, marathon running causes kidney stress and potential injury [21]. Unfortunately, given the low number of participants with follow-up data, no firm conclusions can be reached on the relative dynamics of serum creatinine vs urinary [TIMP2]*[IGFBP7] values.
However, not all datasets support a superiority of urinary [TIMP2]*[IGFBP7] over other biomarkers for AKI prediction. As an example, in a military operation scenario, urine NGAL dipstick clearly outperformed NephroCheck to predict KRT or death in combat casualties [22]. Moreover, a November 2022 systematic review and meta-analysis of 110 studies with 38 725 patients concluded that biomarkers containing NGAL had the best predictive accuracy for the occurrence of AKI, regardless of whether the values were adjusted by urinary creatinine [23]. However, differences were found according to setting. In surgical patients, urinary NGAL/creatinine was the most accurate biomarker. Differences between biomarkers were larger for non-critical patients while serum NGAL and urinary NGAL/creatinine, NGAL, KIM-1, L-FABP, IL-18 and [TIMP2]*[IGFBP7] had similar predictive performance in critically ill patients [23]. Additional subtle differences were found for different contexts of use, but in none was [TIMP2]*[IGFBP7] the best option.
EXPRESSION AND FUNCTION OF TIMP2 AND IGFBP7 IN KIDNEY INJURY
Both TIMP2 and IGFBP7 have been reported to induce G1 cell cycle arrest, mainly in non-kidney studies, and to contribute to the severity of AKI. Indeed, IGFBP7 overexpression induced cell cycle arrest at the G1–G0 phases and apoptosis in HK-2 human proximal tubular cells [24]. Mechanistically, IGFBP7 bound to poly(ADP-ribose) polymerase 1 (PARP1) and inhibited its degradation by antagonizing the E3 ubiquitin ligase ring finger protein 4 (RNF4) [25]. Global or tubular knockout of IGFBP7 prevented AKI induced by cisplatin, kidney ischemia–reperfusion, lipopolysaccharide (LPS) or bacterial sepsis in mice [25, 26]. TIMP2 overexpression induced endoplasmic reticulum stress and apoptosis, and amplified the inflammatory response to LPS in HK-2 cells [27, 28]. Kidney- or tubule-specific deficiency of TIMP2 prevented LPS or sepsis-induced AKI as well as kidney fibrosis induced by ureteral obstruction [27–29]. Supporting a tubular cell origin of the biomarkers, renal vein levels of IGFBP7 and TIMP2 were increased in patients with renal artery stenosis, and baseline IGFBP7 correlated inversely with pre-stent cortical blood flow [30]. Moreover, both proteins are expressed and secreted by primary cultures of human tubules: IGFBP7 is equally expressed across tubule cell types yet preferentially secreted by cells of proximal tubule origin, while TIMP2 is both expressed and secreted preferentially by cells of distal tubule origin [31]. In human kidney tissue, strong staining of IGFBP7 was seen in the luminal brush-border region of a subset of proximal tubule cells, and TIMP2 stained intracellularly in distal tubules. An in vitro model of ischemia–reperfusion demonstrated enhanced secretion of both markers early after reperfusion [31]. Thus, there is clear evidence that both tubular TIMP2 and IGFBP7 contribute to the pathogenesis experimental kidney injury. Moreover, these studies point to extracellular TIMP2 and IGFBP7 as bioactive contributors to the severity of AKI, which at least in mice, are secreted by tubular cells.
However, some reports have questioned the tubular origin of urinary TIMP2 and IGFBP7 or, at least, that their de novo synthesis accounts for the increased urinary levels.
In murine AKI induced by rhabdomyolysis (glycerol), maleate or ischemia–reperfusion, increased urinary TIMP2 and IGFBP7 proteins were observed within 4–18 h, however this occurred in the absence of upregulation of kidney mRNA expression, arguing against de novo gene transcription in tubular cells as the source of the urinary biomarkers [32]. Additionally, a progressive decrease in TIMP2/IGFBP7 immunostaining was observed in injured proximal tubule cells and evidence was generated supporting that TIMP2/IGFBP7 are reabsorbed by healthy proximal tubular cells. In this regard, their low molecular weights (≈24 and ≈30 kDa, respectively) suggest that they may undergo glomerular filtration and proximal tubular reabsorption. These findings are compatible with preexistent TIMP2/IGFBP7 inside tubular cells leaking into urine during AKI, decreased reabsorption by injured proximal tubular cells of molecules physiologically filtered by glomeruli or a combination of both mechanisms [32]. If these molecules are filtered by normal glomeruli, a further source of urinary TIMP2 and/or IGFBP7 during AKI or severe illness should be contemplated, i.e. massive production and release into the circulation from organs stressed during conditions such as surgery, sepsis or marathon running (Fig. 3). For example, heart injury or failure increase the local production and circulating levels of IGFBP7 [33, 34], while TIMP2 is increased in heart tissue surrounding a myocardial infarction [35].
Figure 3:

Potential sources of urinary TIMP2 and IGFBP7. Urinary [TIMP2]*[IGFBP7] values may be the end-result of multiple simultaneous or sequential processes. (1) Both molecules may be released from injured organs outside the kidneys. For example, this has been shown for the injured heart. (2) Circulating TIMP2 and IGFBP7 of non-renal origin may be filtered by normal glomeruli, given that they are small proteins. Increased non-renal production with subsequent increased glomerular filtration may contribute to higher urinary [TIMP2]*[IGFBP7] levels and to their association with adverse outcomes if urinary [TIMP2]*[IGFBP7] represents more severe non-renal injury. (3) As for other small proteins, proximal tubular cells could reabsorb both TIMP2 and IGFBP7. Injured proximal tubular cells may fail to reabsorb TIMP2 and IGFBP7 and this may also contribute to higher urinary [TIMP2]*[IGFBP7] levels and to their association with adverse outcomes. (4) Additionally, both TIMP2 and IGFBP7 can directly injure proximal tubular cells. The fact that both molecules may cause tubular cell injury may also contribute to explain the association between higher urinary [TIMP2]*[IGFBP7] levels and adverse outcome. (5) Immunostaining of human kidney tissue has localized IGFBP7 to tubular cells, mainly proximal tubular cells. Gene expression data and the fact that IGFBP7 immunostaining is not limited to proximal tubular cells support that proximal tubular cells are indeed a source of IGFBP7 beyond any immunostaining resulting from reabsorbed proteins. Increased IGFBP7 release from injured proximal tubular cells may contribute to higher urinary [TIMP2]*[IGFBP7] levels and to their association with adverse outcome. (6) Finally, immunostaining of human kidney tissue has localized TIMP2 to distal tubular cells, suggesting that they may be a source of urinary TIMP2. TIMP2 release from injured distal tubular cells may contribute to higher urinary [TIMP2]*[IGFBP7] levels and to their association with adverse outcomes. (7) Overall, urinary [TIMP2]*[IGFBP7] may be the end result of multiple processes, each of which may be independently associated with more severe kidney injury.
Timing of sampling may explain some of the potential discrepancies observed. A clear increase in tubular IGFBP7 immunostaining was observed in human AKI and at 24 h in murine cisplatin- and IRI-induced AKI, in the latter associated with increased kidney mRNA expression [25]. In sepsis, the kidney gene and protein expression of TIMP2 increased and correlated with the severity of AKI [28]. However, although kidney TIMP2 protein was observed in control kidneys, increased kidney TIMP2 was only observed from 12 h onwards, i.e. later than expected for the urinary pattern of TIMP2/IGFBP7 in at least some clinical scenarios. In murine unilateral ureteric obstruction, kidney Timp2 mRNA did not increase until aft the chronic phase (after 14 days following IRI), while tubular TIMP2 remained unchanged or increased after 48 h [36, 37]. These findings are consistent with a scenario in which high urinary [TIMP2]*[IGFBP7] may to some extent reflect preexistent kidney injury, a hypothesis supported by a recent manuscript in CKJ that presents human kidney biopsy data [38]. In this regard, CKD is a key risk factor for AKI and a decreased eGFR is a late consequence (and diagnostic criterion) of CKD.
CELL CYCLE ARREST IN AKI
[TIMP2]*[IGFBP7] are thought to be markers of tubular cell cycle arrest. However, cell cycle arrest may have diverse roles in AKI [38, 39]. On one hand, a transient, protective G1 cell cycle arrest may be triggered by ischemic preconditioning and contribute to kidney protection [38]. In this regard, cell cycle arrest can protect cells from the disastrous consequences of entering cell division with damaged DNA or insufficient bioenergetic resources during injury or stress [39]. On the other hand, epithelial cell arrest in G1 or G2 favors a hypertrophic and fibrotic phenotype [39]. Thus, biomarkers of cell cycle arrest may theoretically reflect aspects of these two faces of cell cycle arrest. In this regard, the kidney expression of both Timp2 and Igfbp7 mRNA expression increased transiently in reversible ischemia–reperfusion AKI in mice, while both increased progressively, peaking at last follow-up at 14 days in mice with irreversible unilateral ureteral obstruction [40]. However, the site of expression differed: fibroblasts, myofibroblasts and failed-repair proximal tubule cells expressed the highest levels of Igfbp7, while fibroblasts, myofibroblasts and acute injury proximal tubule cells expressed the highest levels of Timp2 [40].
TIMP2 AND IGFBP7 IMMUNOSTAINING IN HUMAN CKD
In this issue of CKJ, Schanz et al. analyzed 37 kidney biopsies of patients with kidney disease (19 with a clinical diagnosis of AKI) and 10 non-diseased control biopsies for TIMP2 and IGFBP7 expression using immunohistochemistry and assessed urinary [TIMP2]*[IGFBP7] at the time of biopsy using NephroCheck [41]. The kidney expression of TIMP2 and IGFBP7 was increased in diseased kidney biopsies as compared with control tissue and staining was prominent in the tubular compartment: IGFBP7 was located to proximal and distal tubules while TIMP2 was predominantly localized in the collecting ducts. This location was consistent with prior cell culture and human kidney data [31].
Tubular TIMP2 and IGFBP7 staining intensity and urinary [TIMP2]*[IGFBP7] correlated with the tubular injury score, again consistent with the fact that urinary [TIMP2]*[IGFBP7] is a marker of early AKI or short-term risk of AKI [41]. However, tubular expression and urinary findings did not correlate. To some extent, it is expected that different markers of kidney injury assessed in the same small piece of tissue corresponding to a kidney biopsy are correlated. However, the lack of correlation between immunohistochemistry and urinary results is more difficult to interpret, given that immunohistochemistry is not quantitative, and the biopsy sample may not be fully representative of the whole kidney parenchyma in both kidneys. Despite these limitations, urinary biomarkers have frequently correlated to histological assessment of the protein [42] or with overall severity of kidney injury [43]. Thus, the lack of correlation between urinary and tissue [TIMP2]*[IGFBP7] may reflect a non-tubular origin of these urinary biomarkers or an impact of reabsorption or secretion dynamics [32–35].
In Schanz’s study, patients with AKI had more often and more severe preexistent CKD as assessed by low baseline eGFR [23 (13–29) mL/min/1.73 m2] and evidence of chronicity in the kidney biopsy [41]. However, urinary [TIMP2]*[IGFBP7] was high [>0.3 (ng/mL)2/1000] in a majority of participants, without significant differences between participants with and without AKI in either TIMP2 or IGFBP7 immunostaining or urinary [TIMP2]*[IGFBP7]. Interestingly, an increase in both glomerular and tubular TIMP2 or IGFBP7 immunostaining versus controls was observed in very diverse nephropathies, ranging from membranous nephropathy to ANCA-associated vasculitis to a miscellaneous group of glomerular and interstitial nephropathies. Consistent with these results, Schanz et al. concluded that TIMP2 and IGFBP7 are unspecific markers of renal injury. This may explain some of the discrepant results regarding the information provided by urinary [TIMP2]*[IGFBP7] in different clinical contexts. The frequent coexistence of AKI and CKD in this cohort and the lack of data on severity or timing of AKI limit the conclusions to be drawn on urinary [TIMP2]*[IGFBP7] values in patients with CKD. Nevertheless, the data call for a more thorough evaluation of the performance of urinary [TIMP2]*[IGFBP7] across patients with a wide range of baseline eGFR, albuminuria/proteinuria values and causes of CKD.
NEW KIDS ON THE BLOCK
In 2022, Piedrafita et al. studied derivation and validation cohorts totalling 1170 major cardiac bypass surgery patients and an external cohort of 1569 ICU patients to develop a urinary peptidomics-based score predictive of AKI (7-day KDIGO classification) termed PeptAKI [44]. PeptAKI is composed of 204 urinary peptides derived from 48 proteins related to fibrosis, hemolysis, inflammation, immune cells, and cell growth and survival. A positive PeptAKI score conferred an odds ratio of 6.1 for AKI, which was almost double that associated with clinical risk scores, urinary [TIMP2]*[IGFBP7] and urinary NGAL among cardiac surgery patients with AUCs of 0.78. 0.70, 0.76 and 0.70, respectively for the different biomarkers. The performance of PeptAKI was confirmed in the ICU cohort in which it also predicted death. In the ICU setting, the overall PeptAKI performance was closer to that of urinary NGAL [44]. However, in post-operative AKI, the PeptAKI AUC (0.86) compared favorably with the AUC obtained in the derivation and internal validation cohorts. While these results are promising, there is currently no point-of-care device that allows timely assessment of urinary peptide biomarkers to guide rapid clinical decision making, contrary to the situation with [TIMP2]*[IGFBP7] or NGAL.
CONCLUSION
In conclusion, after 10 years of clinical development, urinary [TIMP2]*[IGFBP7] as assessed by the point-of-care test NephroCheck is already in clinical use, although it remains costly and its impact on clinical outcomes has not been conclusively demonstrated. In this regard, no major clinical guidance documents by scientific societies provide recommendations for context of use and actions to be implemented in response to [TIMP2]*[IGFBP7] values. However, it remains an attractive biomarker for future outcomes-focused clinical trials that use NephroCheck results as a port of entry for enrollment, clinical decision-making and treatment prescription. The recent data by Schanz et al. suggest widespread upregulation of kidney tissue TIMP2 and IGFBP7 in human CKD, without further upregulation in AKI, and an unclear role of urinary [TIMP2]*[IGFBP7] in providing information on kidney histology in presence of AKI and/or CKD. Further studies should carefully characterize baseline urinary [TIMP2]*[IGFBP7] values in patients with preexistent CKD of different causes and severity. Similar issues mar NGAL, which may be released by inflammatory cells addition to damaged tubular cells, and levels can increase in other chronic and acute inflammatory conditions, including CKD and urinary tract infections [45]. In this regard, the urinary biomarkers field keeps evolving and multiparametric panels potentially including non-kidney-derived and kidney-derived markers such as urinary peptidomics may provide the major breakthrough in the AKI prediction field not yet provided by current individual biomarkers, although the technology should be adapted to point-of-care testing.
Contributor Information
Catalina Martin-Cleary, Department of Nephrology and Hypertension, IIS-Fundacion Jimenez Diaz UAM, Madrid, Spain; RICORS2040, Madrid, Spain; Departamento de Medicina, Facultad de Medicina, Universidad Autónoma de Madrid, Madrid, Spain.
Ana Belen Sanz, Department of Nephrology and Hypertension, IIS-Fundacion Jimenez Diaz UAM, Madrid, Spain; RICORS2040, Madrid, Spain; Departamento de Medicina, Facultad de Medicina, Universidad Autónoma de Madrid, Madrid, Spain.
Alejandro Avello, Department of Nephrology and Hypertension, IIS-Fundacion Jimenez Diaz UAM, Madrid, Spain; RICORS2040, Madrid, Spain; Departamento de Medicina, Facultad de Medicina, Universidad Autónoma de Madrid, Madrid, Spain.
Maria Dolores Sanchez-Niño, Department of Nephrology and Hypertension, IIS-Fundacion Jimenez Diaz UAM, Madrid, Spain; RICORS2040, Madrid, Spain; Departamento de Farmacología, Facultad de Medicina, Universidad Autónoma de Madrid, Madrid, Spain.
Alberto Ortiz, Department of Nephrology and Hypertension, IIS-Fundacion Jimenez Diaz UAM, Madrid, Spain; RICORS2040, Madrid, Spain; Departamento de Medicina, Facultad de Medicina, Universidad Autónoma de Madrid, Madrid, Spain.
FUNDING
FIS/Fondos FEDER (PI22/00 469, PI22/00 050, PI21/00 251), ERA-PerMed-JTC2018 (KIDNEY ATTACK AC18/00 064, ISCIII-RETIC REDinREN RD016/0009), Sociedad Española de Nefrología, Sociedad Madrileña de Nefrología (SOMANE), FRIAT, Comunidad de Madrid en Biomedicina P2022/BMD-7223, CIFRA_COR-CM. Instituto de Salud Carlos III (ISCIII) RICORS program to RICORS2040 (RD21/0005/0001) funded by European Union—NextGenerationEU, Mecanismo para la Recuperación y la Resiliencia (MRR) and SPACKDc PMP21/00 109, FEDER funds. This publication is based upon work from COST Action PERMEDIK CA21165, supported by COST (European Cooperation in Science and Technology).
CONFLICT OF INTEREST STATEMENT
A.O. has received grants from Sanofi and consultancy or speaker fees or travel support from Adviccene, Alexion, Astellas, AstraZeneca, Amicus, Amgen, Boehringer Ingelheim, Fresenius Medical Care, GSK, Bayer, Sanofi-Genzyme, Menarini, Mundipharma, Kyowa Kirin, Lilly, Freeline, Idorsia, Chiesi, Otsuka, Novo-Nordisk, Sysmex and Vifor Fresenius Medical Care Renal Pharma, and is Director of the Catedra Mundipharma-UAM of diabetic kidney disease and the Catedra AstraZeneca-UAM of chronic kidney disease and electrolytes. He has stock in Telara Farma and is a previous CKJ Editor-in-Chief.
REFERENCES
- 1. Chawla LS, Eggers PW, Star RAet al. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med 2014;371:58–66. 10.1056/NEJMra1214243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Roushani J, Thomas D, Oliver MJet al. Acute kidney injury requiring renal replacement therapy in people with COVID-19 disease in Ontario, Canada: a prospective analysis of risk factors and outcomes. Clin Kidney J 2022;15:507–16. 10.1093/ckj/sfab237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zarbock A, Küllmar M, Ostermann Met al. Prevention of cardiac surgery-associated acute kidney injury by implementing the KDIGO guidelines in high-risk patients identified by biomarkers: the PrevAKI-multicenter randomized controlled trial. Anesth Analg 2021;133:292–302. 10.1213/ANE.0000000000005458 [DOI] [PubMed] [Google Scholar]
- 4. Levey AS, Stevens LA, Schmid CHet al. A new equation to estimate glomerular filtration rate. Ann Intern Med 2009;150:604–12. 10.7326/0003-4819-150-9-200905050-00006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. El-Khoury JM, Hoenig MP, Jones GRDet al. AACC guidance document on laboratory investigation of acute kidney injury. J Appl Lab Med 2021;6:1316–37. [DOI] [PubMed] [Google Scholar]
- 6. Brazzelli M, Aucott L, Aceves-Martins Met al. Biomarkers for assessing acute kidney injury for people who are being considered for admission to critical care: a systematic review and cost-effectiveness analysis. Health Technol Assess 2022;26:1–286. 10.3310/UGEZ4120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. von Groote T, Albert F, Meersch Met al. Proenkephalin A 119-159 predicts early and successful liberation from renal replacement therapy in critically ill patients with acute kidney injury: a post hoc analysis of the ELAIN trial. Crit Care 2022;26:333. 10.1186/s13054-022-04217-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Koyner JL, Chawla LS, Bihorac Aet al. Performance of a standardized clinical assay for urinary C-C motif chemokine ligand 14 (CCL14) for persistent severe acute kidney injury. Kidney360 2022;3:1158–68. 10.34067/KID.0008002021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bagshaw SM, Al-Khafaji A, Artigas Aet al. External validation of urinary C-C motif chemokine ligand 14 (CCL14) for prediction of persistent acute kidney injury. Crit Care 2021;25:185. 10.1186/s13054-021-03618-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ilaria G, Kianoush K, Ruxandra Bet al. Clinical adoption of Nephrocheck® in the early detection of acute kidney injury. Ann Clin Biochem 2021;58:6–15. 10.1177/0004563220970032 [DOI] [PubMed] [Google Scholar]
- 11. Pajenda S, Ilhan-Mutlu A, Preusser Met al. NephroCheck data compared to serum creatinine in various clinical settings. BMC Nephrol 2015;16:206. 10.1186/s12882-015-0203-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kashani K, Al-Khafaji A, Ardiles Tet al. Discovery and validation of cell cycle arrest biomarkers in human acute kidney injury. Crit Care 2013;17:R25. 10.1186/cc12503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hoste EA, McCullough PA, Kashani Ket al. Derivation and validation of cutoffs for clinical use of cell cycle arrest biomarkers. Nephrol Dial Transplant 2014;29:2054–61. 10.1093/ndt/gfu292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koyner JL, Shaw AD, Chawla LSet al. Tissue inhibitor metalloproteinase-2 (TIMP-2)⋅IGF-binding protein-7 (IGFBP7) levels are associated with adverse long-term outcomes in patients with AKI. J Am Soc Nephrol 2015;26:1747–54. 10.1681/ASN.2014060556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dewitte A, Joannès-Boyau O, Sidobre Cet al. Kinetic eGFR and novel AKI biomarkers to predict renal recovery. Clin J Am Soc Nephrol 2015;10:1900–10. 10.2215/CJN.12651214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guzzi LM, Bergler T, Binnall Bet al. Clinical use of [TIMP-2]•[IGFBP7] biomarker testing to assess risk of acute kidney injury in critical care: guidance from an expert panel. Crit Care 2019;23:225. 10.1186/s13054-019-2504-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McCullough PA, Ostermann M, Forni LGet al. Serial urinary tissue inhibitor of metalloproteinase-2 and insulin-like growth factor-binding protein 7 and the prognosis for acute kidney injury over the course of critical illness. Cardiorenal Med 2019;9:358–69. 10.1159/000502837 [DOI] [PubMed] [Google Scholar]
- 18. Forni LG, Joannidis M, Artigas Aet al. Characterising acute kidney injury: the complementary roles of biomarkers of renal stress and renal function. J Crit Care 2022;71:154066. 10.1016/j.jcrc.2022.154066 [DOI] [PubMed] [Google Scholar]
- 19. Thiele K, Rau M, Hartmann NKet al. Empagliflozin reduces markers of acute kidney injury in patients with acute decompensated heart failure. ESC Heart Fail 2022;9:2233–8. 10.1002/ehf2.13955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fernandez-Fernandez B, Sarafidis P, Kanbay Met al. SGLT2 inhibitors for non-diabetic kidney disease: drugs to treat CKD that also improve glycaemia. Clin Kidney J 2020;13:728–33. 10.1093/ckj/sfaa198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Leckie T, Fitzpatrick D, Richardson AJet al. Marathon running and cell-cycle arrest biomarkers of acute kidney injury. J Sci Med Sport 2023;26:14–8. 10.1016/j.jsams.2022.10.012 [DOI] [PubMed] [Google Scholar]
- 22. Beyer CA, Burmeister DM, Gómez BIet al. Point-of-care urinary biomarker testing for risk prediction in critically injured combat casualties. J Am Coll Surg 2019;229:508–15.e501. 10.1016/j.jamcollsurg.2019.07.003 [DOI] [PubMed] [Google Scholar]
- 23. Pan HC, Yang SY, Chiou TTet al. Comparative accuracy of biomarkers for the prediction of hospital-acquired acute kidney injury: a systematic review and meta-analysis. Crit Care 2022;26:349. 10.1186/s13054-022-04223-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang X, Ma T, Wan Xet al. IGFBP7 regulates sepsis-induced acute kidney injury through ERK1/2 signaling. J Cell Biochem 2019;120:7602–11. [DOI] [PubMed] [Google Scholar]
- 25. Yu JT, Hu XW, Yang Qet al. Insulin-like growth factor binding protein 7 promotes acute kidney injury by alleviating poly ADP ribose polymerase 1 degradation. Kidney Int 2022;102:828–44. 10.1016/j.kint.2022.05.026 [DOI] [PubMed] [Google Scholar]
- 26. Wang X, Ma T, Wan Xet al. IGFBP7 regulates sepsis-induced acute kidney injury through ERK1/2 signaling. J Cell Biochem 2019;120:7602–11. 10.1002/jcb.28035 [DOI] [PubMed] [Google Scholar]
- 27. Jiang N, Huang R, Zhang Jet al. TIMP2 mediates endoplasmic reticulum stress contributing to sepsis-induced acute kidney injury. FASEB J 2022;36:e22228. 10.1096/fj.202101555RR [DOI] [PubMed] [Google Scholar]
- 28. Li YM, Zhang J, Su LJet al. Downregulation of TIMP2 attenuates sepsis-induced AKI through the NF-κb pathway. Biochim Biophys Acta Mol Basis Dis 2019;1865:558–69. 10.1016/j.bbadis.2018.10.041 [DOI] [PubMed] [Google Scholar]
- 29. Wang Z, Famulski K, Lee Jet al. TIMP2 and TIMP3 have divergent roles in early renal tubulointerstitial injury. Kidney Int 2014;85:82–93. 10.1038/ki.2013.225 [DOI] [PubMed] [Google Scholar]
- 30. Saad A, Wang W, Herrmann SMet al. Atherosclerotic renal artery stenosis is associated with elevated cell cycle arrest markers related to reduced renal blood flow and postcontrast hypoxia. Nephrol Dial Transplant 2016;31:1855–63. 10.1093/ndt/gfw265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Emlet DR, Pastor-Soler N, Marciszyn Aet al. Insulin-like growth factor binding protein 7 and tissue inhibitor of metalloproteinases-2: differential expression and secretion in human kidney tubule cells. Am J Physiol Renal Physiol 2017;312:F284–96. 10.1152/ajprenal.00271.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Johnson ACM, Zager RA.. Mechanisms underlying increased TIMP2 and IGFBP7 urinary excretion in experimental AKI. J Am Soc Nephrol 2018;29:2157–67. 10.1681/ASN.2018030265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ko T, Nomura S, Yamada Set al. Cardiac fibroblasts regulate the development of heart failure via Htra3-TGF-β-IGFBP7 axis. Nat Commun 2022;13:3275. 10.1038/s41467-022-30630-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chugh S, Ouzounian M, Lu Zet al. Pilot study identifying myosin heavy chain 7, desmin, insulin-like growth factor 7, and annexin A2 as circulating biomarkers of human heart failure. Proteomics 2013;13:2324–34. 10.1002/pmic.201200455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wilson EM, Moainie SL, Baskin JMet al. Region- and type-specific induction of matrix metalloproteinases in post-myocardial infarction remodeling. Circulation 2003;107:2857–63. 10.1161/01.CIR.0000068375.40887.FA [DOI] [PubMed] [Google Scholar]
- 36. Kirita Y, Wu H, Uchimura Ket al. Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. Proc Natl Acad Sci USA 2020;117:15874–83. 10.1073/pnas.2005477117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dixon EE, Wu H, Muto Yet al. Spatially resolved transcriptomic analysis of acute kidney injury in a female murine model. J Am Soc Nephrol 2022;33:279–89. 10.1681/ASN.2021081150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rossaint J, Meersch M, Thomas Ket al. Remote ischemic preconditioning causes transient cell cycle arrest and renal protection by a NF-κb-dependent Sema5B pathway. JCI Insight 2022;7:e158523. 10.1172/jci.insight.158523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kellum JA, Chawla LS.. Cell-cycle arrest and acute kidney injury: the light and the dark sides. Nephrol Dial Transplant 2016;31:16–22. 10.1093/ndt/gfv130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li H, Dixon EE, Wu Het al. Comprehensive single-cell transcriptional profiling defines shared and unique epithelial injury responses during kidney fibrosis. Cell Metab 2022;34:1977–98.e9. 10.1016/j.cmet.2022.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schanz M, Kimmel M, Alscher MDet al. TIMP2 and IGFBP7 in human kidney biopsies in renal disease. Clin Kidney J 2023. 10.1093/ckj/sfad010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Perez-Gomez MV, Pizarro-Sanchez S, Gracia-Iguacel Cet al. Urinary Growth Differentiation Factor-15 (GDF15) levels as a biomarker of adverse outcomes and biopsy findings in chronic kidney disease. J Nephrol 2021;34:1819–32. 10.1007/s40620-021-01020-2 [DOI] [PubMed] [Google Scholar]
- 43. Magalhães P, Pejchinovski M, Markoska Ket al. Association of kidney fibrosis with urinary peptides: a path towards non-invasive liquid biopsies? Sci Rep 2017;7:16915. 10.1038/s41598-017-17083-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Piedrafita A, Siwy J, Klein Jet al. A universal predictive and mechanistic urinary peptide signature in acute kidney injury. Crit Care 2022;26:344. 10.1186/s13054-022-04193-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mårtensson J, Bellomo R.. The rise and fall of NGAL in acute kidney injury. Blood Purif 2014;37:304–10. 10.1159/000364937 [DOI] [PubMed] [Google Scholar]