

Abstract
Single-cell sequencing have been widely used to characterize cellular heterogeneity. Sample multiplexing where multiple samples are pooled together for single-cell experiments, attracts wide attention due to its benefits of increasing capacity, reducing costs, and minimizing batch effects. To analyze multiplexed data, the first crucial step is to demultiplex, the process of assigning cells to individual samples. Inaccurate demultiplexing will create false cell types and result in misleading characterization. We propose scDemultiplex, which models hashtag oligo (HTO) counts with beta-binomial distribution and uses an iterative strategy for further refinement. Compared with seven existing demultiplexing approaches, scDemultiplex achieved great performance in both high-quality and low-quality data. Additionally, scDemultiplex can be combined with other approaches to improve their performance.
Keywords: Single-cell sequencing, Hashtag oligo (HTO), Demultiplexing, Beta-binomial
Graphical Abstract

1. Introduction
Single-cell sequencing provides an unprecedent scale for investigating cellular heterogeneity systematically [1], [13], [14], [20], [21], [25], [28]. Sample multiplexing where multiple samples are pooled and sequenced together is often used in single-cell experiments to increase capacity and reduce costs, and most importantly, minimize batch effects [5]. There are two main methods used for sample multiplexing: barcode-based and single nucleotide polymorphism (SNP)-based. Barcode-based multiplexing labels samples with unique DNA barcodes (also termed hashtag oligos, HTO) using either an antibody tagged with a DNA barcode that targets the cell surface protein [19] or nucleus pore complex [6], or lipid/cholesterol-modified oligonucleotides that tag the cell membrane [15], [16]. SNP-based multiplexing distinguishes multiplexed samples based on their natural genetic landscapes [10], [27], [7], [9], but it can only be applied to genetically distinct samples, not those that share the same genetic background, such as samples from the same individuals at different developmental/lineage/experimental stages. Here, we focus solely on barcode-based sample multiplexing.
While barcoded-based multiplexing is extremely useful, it introduces artifacts due to cross- contamination in library construction and sequencing errors, giving rise to the potential that cells are not labelled only by one barcode but varying degrees of other barcodes [15], [16], [18]. In order to assign cells to each individual sample, demultiplexing approaches are aimed to classify cells into negatives (not true cells), singlets (cells from one sample), and multiplets (cells from two or more samples). Accurate demultiplexing is to recall as many singlets as possible and at the same time not to misrecognize negatives and multiplets as singlets. After cells are hash tagged with barcoded oligos and then pooled and processed for single-cell RNA sequencing, reads are aligned to generate gene-by-cell and HTO-by-cell count matrices. The next step is demultiplexing based on the HTO count matrix, which is critical for the downstream cell type identification and characterization. Misrecognition of negatives and multiplets as singlets will result in false cell types and misleading characterization.
Several demultiplexing approaches have been developed (Table 1). HTODemux uses k-medoids clustering to find negative clusters and then determines thresholds for classifying cells as positives or negatives by fitting a negative binomial distribution to the negative clusters [19]. MULTIseqDemux determines cutoffs based on Gaussian kernel density to classify cells [15], [16]. hashedDrops identifies singlets and doublets by the log-fold changes between the most abundant HTO, the second most abundant HTO, and ambient contamination[12]. Bimodal Flexible Fitting (BFF), including BFF_raw and BFF_cluster, first determines thresholds based on a simple assumption that count distribution is bimodal. Then BFF_raw and BFF_cluster use determined thresholds to identify negatives, singlets and multiplets [3]. Instead of determining thresholds, GMM-Demux assumes HTO counts come from two separate sources, background and the real sample. GMM-Demux then fits the HTO counts of each sample into a Gaussian mixture model and computes the posterior probability of being singlets or multiplets [26]. Demuxmix uses two-component mixture models like GMM-Demux. Different from GMM-Demux modeling centered log ratio (CLR)-transformed counts by Gaussian distribution, demuxmix models raw HTO counts by negative binomial distribution [11] (Table 1). Overcoming the limitations of threshold-based approaches, GMM-Demux and Demuxmix achieved better performance in identifying singlets and removing multiplets [11], [26], [8].
Table 1.
Summary of eight demultiplexing methods.
| Method | Models | Description |
|---|---|---|
| HTODemux [19] |
|
|
| MULTIseqDemux[15], [16] |
|
|
| hashedDrops[12] |
|
|
| BFF_raw [3] |
|
|
| BFF_cluster [3] |
|
|
| GMM-Demux [26] |
|
|
| Demuxmix [11] |
|
|
| scDemultiplex |
|
|
Here, we present scDemultiplex, which models HTO counts of each sample with beta-binomial distribution and calculates the probability of one cell being from the sample. Additionally, scDemultiplex uses an iterative strategy to further refine the model and the classifying result (Table 1). Compared to GMM-Demux using Gaussian mixture distribution, negative-binomial distribution in demuxmix and beta-binomial distribution fit the count nature of the HTO data better. Compared to MULTIseqDemux determining HTO-specific thresholds by an iterative strategy [15], [16], scDemultiplex fits HTO counts (specific and non-specific) with beta-binomial distributions (Table 1). Therefore, sample classification in scDemultiplex is determined not only by HTO-specific but also HTO-nonspecific counts. We benchmarked scDemultiplex against seven existing demultiplexing approaches, hashedDrops, HTODemux, GMM-Demux, MULTIseqDemux, BFF_raw, BFF_cluster, and demuxmix using five real HTO datasets. The evaluation demonstrated that scDemultiplex achieved high performance in both high-quality and low-quality data. The iterative strategy makes it easy to combine scDemultiplex with other approaches, which obtained higher performance than using those approaches alone.
2. Materials and methods
2.1. scDemultiplex
Suppose the data have m cells pooled from n samples, i.e, labeled with n HTOs. The HTO count matrix xij contains the count of the HTO i in the cell j, where i = 1, 2, …, n and j = 1, 2,., m. scDemultiplex is designed to classify the cell j as a negative, a singlet, or a multiplet based on the HTO count distributions of the cell, denoted as Xj= (x1j ,x2j, …, xnj ).
2.2. Parameter estimation of beta-binomial distribution
scDemultiplex models HTO counts of each sample with beta-binomial distribution. That is, the corresponding HTO counts Xj from the sample i follow beta-binomial distribution with parameter BetaBin(nj, αi, βi). where nj is the total counts of the cell j, . αi and βi are estimated from the data.
To estimate the parameters αi and βi for the sample i, scDemultiplex uses Gaussian mixture model to perform an initial classifying. First, HTO raw count xij is normalized by centered log-ratio (CLR) transformation, where the normalized HTO values is the natural log-transformed of the counts divided by the geometric mean of a specified HTO [19]:
For a given sample i, the normalized HTO values of the cell j, Yj= ( y1j ,y2j, …, ynj ) follow a mixture of two Gaussian distributions, one is a “negative” distribution coming from background and the other is a “positive” distribution deriving from the sample i. Let Zj[k] indicates that the cell j comes from the component k, Zj[k] ϵ {0, 1}, k = 1, 2. Zj[1] = 1 if the cell from the component 1, otherwise 0; and Zj[2]= 1 if the cell is from the component 2, otherwise 0. Let πk be the mixture proportions, the probability density function (pdf) for the mixture model is
where the k-th component follows N(μik, σik). To assist in identification of “negative” and “positive” cells, scDemultiplex applied the Expectation–maximization (EM) algorithm to estimate the parameters in the Gaussian mixture model. The fitted mixed model is then used to calculate the threshold Ti that discriminate the cell j into two groups [22]. Given a cell j, if the normalized HTO value yij greater than the threshold Ti, it is a “positive” cell; otherwise, it is a “negative” cell.
scDemultiplex models each sample with Gaussian mixture model and assign cells to every sample. If the cell j is identified as negative in all samples, that is, , this cell is negative; if the cell j is recognized as positive in only one sample, e.g., ykj greater than Tk, that is, , the cell belongs to the sample k (singlet); otherwise the cell j is a multiplet, . The formula is laid out below:
where I() is an indicator function. The cell assignment based on Gaussian mixture model is used to estimate the parameters αi and βi in the BetaBin(nj, αi, βi) for the sample i. scDemultiplex first fits HTO raw counts with Dirichlet-multinomial distribution DirMult(nj, αi’). Condition on the total HTO counts, the raw count for each HTO can be modeled with Dirichlet-multinomial distribution, which is justified by the study [12]. The parameters αi’= (αi1’, αi2’, …, αin’) are estimated by the method described in Lun et al. [12] using R dirmult package [23]. To simplify the calculation, scDemultiplex replaces Dirichlet-multinomial distribution with beta-binomial distribution. To fit beta-binomial distribution, scDemultiplex aggregates the n-dimensional vector Xj= (x1j ,x2j, …, xnj ) into a two-dimensional vector with counts coming from the sample i (xij) and sum of counts from other samples . scDemultiplex estimates αi by taking and .
2.3. Demultiplexing
The probability mass function (pmf) of beta-binomial distribution of a given sample i for the cell j follows
where nj is the total counts of the cell j, . The probability of the cell j coming from a given sample i, denoted by , is estimated to be the tail cumulative probability p(x > =. The probability is then adjusted by controlling the false discovery rate (FDR) using the Benjamini-Hochberg (BH) method [2]. A specified FDR threshold is set at 0.1%. The cell j is identified as negative if the cell is not assigned to any samples, singlet if the cell is only assigned to one sample, multiplets if others.
2.4. Iterative refinement of beta-binomial models and reclassifying
scDemultiplex repeats beta-binomial parameter estimations and the probability calculations n times (default: n = 10) to further refine the model and the classification. Specifically, the classifying result from the previous iteration is used to estimate parameters α and β for every sample and then the estimated parameters are utilized to calculate the probabilities of each cell belonging to each sample and classify each cell to negatives, singlets or multiplets in the next iteration.
The procedure will terminate earlier if there are no reassignment or multiple types of singlets reclassified to another type of singlet (default: >= 3) during one iteration. Multiple reclassifications suggest something might go awry in the remodeling. This stopping criterion not only increases the speed in high-quality data, but also helps prevent wrong models in poor-quality data, where different types of singlets, negatives, and multiplets are indistinct from each other.
2.5. Real HTO datasets
We used five real HTO datasets to evaluate the performance of scDemultiplex, named as batch1, batch2, batch3, Barnyard, and PBMC8.
The three batches were utilized in a recent study benchmarking single-cell demultiplexing methods [8]. They contain 24 genetically distinct samples of bronchoalveolar lavage fluid. Each batch contains two captures pooled from 8 samples. Each sample was tagged with different Totalseq-A antibody-derived tag (ADT). Batch1 consists of 11,900 and 12,923 cells, batch2 consists of 24,905 and 25,763 cells, and batch 3 contains 32,886 and 31,956 cells, respectively. All the datasets have been demultiplexed using genetic variants from the RNA by Vireo [8], [9], which are used as “ground truth” to evaluate the accuracy of the HTO demultiplexing methods. As indicated in the study [8], cells in the batch1 are well labeled (high-quality), while those in the batch2 and batch3 are hash-tagged poorly (low-quality), highlighting demultiplexing in these two batches is more challenging.
The Barnyard dataset was downloaded from a previous study [15], [16]. Briefly, by combining lipid- and cholesterol-modified oligonucleotides (LMOs, CMOs) with three separate cell lines, including Human Embryonic Kidney (HEK) cells, Jurkat T cells, and Mouse Embryonic Fibroblast (MEF) cells, twelve samples consisting of 5877 cells were multiplexed and sequenced. The ‘ground truth’ was obtained from the original study [15], [16], which was generated by marker gene expression analysis.
The PBMC8 dataset was downloaded from a previous study [19]. The dataset comprised 15,113 cells extracted from PBMC in eight different human donors. The “ground truth” was obtained by genetic-based demultiplexing using Vireo [9].
2.6. Performance evaluation
We first evaluated the performance of demultiplexing by visual inspection of low dimensional embeddings of HTO profiles. The low dimensional embeddings was generated by the Seurat RunUMAP function using HTO counts as input [4]. In the low-dimensional embeddings, we expect to find cells from the same sample form a distinct cluster (singlet-cluster), while negative cells generally spread and/or loosely connect to clusters from singlets, and multiplets locate at the edge of singlet-clusters or form a separate cluster between two singlet-clusters. The visual distribution is useful but subjective and lack a quantitative measure for robust comparisons.
Additionally, we used two quantitative metrics to evaluate the performance, the adjusted rand index (ARI) and F-score based on the “ground truth”. ARI measures the agreement between the HTO classification and the “ground truth”. The higher ARI value means the better agreement, indicating more accuracy of HTO classification. ARI was calculated by the mclust package using adjustedRandIndex function [17]. The F-score is the harmonic mean of precision and recall, defined as TP/(TP+1/2(FP+FN))[8]. The higher F-score suggests better performance.
2.6.1. Implementation and Code availability
We implemented our algorithm in an R package scDemultiplex, which is publicly available at Github (https://github.com/shengqh/scDemultiplex). The code for analyzing the three batches, Barnyard, and PBMC8 datasets by the eight demultiplexing approaches is also provided at the Github repository (https://github.com/shengqh/scDemultiplex_analysis). A web server running scDemultiplex is available at https://bioinfo.vanderbilt.edu/scdemult/.
3. Results
3.1. Application on the batch1, Barnyard, and PBMC8 datasets with high-quality
We applied scDemultiplex and seven existing demultiplexing approaches on the batch1 datasets with two captures (batch1_c1 and batch1_c2). We obtained similar results for the two captures within the batch1. The UMAP plots labeled with demultiplexing results from each method showed similar patterns (Fig. 1 and Supplementary Fig. S1). There were eight distinct and dense clusters representing eight samples, which were identified as singlets by every method (singlet-cluster). The distinct singlet clusters indicates high-quality data [8]. Negatives spread and/or loosely connected to singlet clusters, and multiplets located at the edge of singlet-clusters or formed a separate cluster between two singlet-clusters.
Fig. 1.

UMAP plots of the batch1_c1 dataset labeled with demultiplexing results from scDemultiplex, HTODemux, MULTIseqDemux, GMM-Demux, BFF_raw, BFF_cluster, demuxmix, and hashedDrops.
We further investigated negatives and multiplets identified by each method (Fig. 2 and Supplementary Fig. S2). The visual inspection found most methods obtained reasonable results, which had negatives spreading and loosely connecting to singlet-clusters, and multiplets locating at the edge of singlet-clusters. MULTIseqDemux and BFF_raw, however, misrecognized many singlets as negatives. MULTIseqDemux and BFF_raw had the greatest number of negatives identified (Supplementary Tables S1-S2). The negatives identified by MULTIseqDemux and BFF_raw extended to the center of singlet-clusters (Fig. 2 and Supplementary Fig. S2).
Fig. 2.

UMAP plots of the batch1_c1 dataset labeled with singlets, negatives, and multiplets classified by scDemultiplex, HTODemux, MULTIseqDemux, GMM-Demux, BFF_raw, BFF_cluster, demuxmix, and hashedDrops.
Table 2 listed the ARI and F-score based on the genetic “ground truth”. BFF_cluster achieved the highest ARI and F-score in both two captures, followed by scDemultiplex. MULTIseqDemux, in contrast, had the lowest ARI and F-score due to its misrecognizing many singlets as negatives, especially for the BAL_02 and BAL_08 samples (Supplementary Tables S3 and S4). Other methods, such as HTODemux, GMM_Demux, demuxmix, and hashedDrops performed comparably well in the batch1.
Table 2.
Adjusted rand index and F-score of eight demultiplex methods in the batch1 datasets.
 |
Note: The highest ARI and F-score among the eight approaches is highlighted in red while the second highest ARI and F-score is highlighted in blue.
In addition, we applied scDemultiplex and seven existing demultiplexing approaches on the Barnyard and the PBMC8 datasets. Both datasets showed distinct singlet clusters, indicating their high quality (Supplementary Figs. S3-S6). Similar to the results from the batch1, most methods performed well when data quality is high. BFF_cluster achieved the highest ARI and F-score, followed by scDemultiplex in the Barnyard dataset (Supplementary Tables S5-S7), while scDemultiplex achieved the highest ARI and F-score, followed by BFF_raw and demuxmix in the PBMC8 dataset (Supplementary Tables S8-S10). In contrast, hashedDrops and MULTIseqDemux misrecognized many singlets as negatives, and HTODemux misclassified many singlets as multiplets in the Barnyard dataset (Supplementary Fig. S4), resulting in poor performance with low ARI and F-scores (Supplementary Tables S5-S7). In the PBMC8 dataset, MULTIseqDemux also had the lowest ARI and F-score due to its misrecognizing many singlets as negatives (Supplementary Tables S8-S10).
3.2. Application on the batch2 datasets with low-quality
We applied the eight demultiplexing approaches on the batch2 datasets with two captures (batch2_c1 and batch2_c2). We obtained similar results for the two captures within the batch2. The UMAP plots labeled with demultiplexing results from each method showed ambiguous patterns (Fig. 3 and Supplementary Fig. S7). Although most methods identified eight clusters, they were not that distinct from each other compared to the batch1. There was no clear separation between singlet-clusters, negatives, or multiplets, suggesting poor quality of the batch2. Notably, BFF_raw and BFF_cluster only identified five HTO clusters in the batch2_c1 and six in the batch2_c2. BFF_raw failed to recognize BAL-10 and BAL-14, while BFF_cluster missed BAL-15 and BAL-16 in both two captures (Supplementary Tables S11 and S12).
Fig. 3.

UMAP plots of the batch2_c1 dataset labeled with demultiplexing results from scDemultiplex, HTODemux, MULTIseqDemux, GMM-Demux, BFF_raw, BFF_cluster, demuxmix and hashedDrops.
We further investigated negatives and multiplets identified by each method (Fig. 4 and Supplementary Fig. S8). scDemultiplex obtained the most reasonable results, where most negatives either spread in the middle or at the edge of singlet-clusters, and most multiplets located between singlet-clusters or at the edge of singlet-clusters. In comparison, MULTIseqDemux, hashedDrops, and BFF_raw misclassified many singlets as negatives, while HTODemux, GMM_Demux, BFF_cluster, and demuxmix classified numerous cells as multiplets (Supplementary Tables S11 and S12). Those misclassified negatives and multiplets extended to the center of singlet-clusters or even dominated the single-cluster.
Fig. 4.

UMAP plots of the batch2_c1 dataset labeled with singlets, negatives, and multiplets classified by scDemultiplex, HTODemux, MULTIseqDemux, GMM-Demux, BFF_raw, BFF_cluster, demuxmix and hashedDrops.
Consistent with the UMAP visualization, scDemultiplex achieved the highest/second highest ARI and highest F-score (Table 3) (ARI=0.671 and 0.633; F_score=0.842 and 0.836), demonstrating its high performance in the low-quality data (Supplementary Table S13 and S14). HTODemux, GMM_Demux, and demuxmix showed decent performance. Notably, compared to great performance in the high-quality batch1 datasets, BFF_cluster had the lowest F-score in the batch2, especially for the BAL-15 and BAL-16 samples (Supplementary Table S13 and S14). This result is consistent with a recent benchmark study [8], reporting that methods that assume a bimodal count distribution perform poorly on low-quality data.
Table 3.
Adjusted rand index and F-score of eight demultiplex methods in the batch2 datasets.
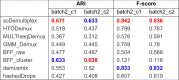 |
Note: The highest ARI and F-score among the eight approaches is highlighted in red while the second highest ARI and F-score is highlighted in blue.
3.3. Application on the batch3 datasets with low-quality
We applied the eight demultiplexing approaches on the batch3 datasets with two captures (batch3_c1 and batch3_c2). We obtained similar results for the two captures within the batch3. Similar to the results in the batch2, the UMAP plots labeled with demultiplexing results from each method showed ambiguous patterns (Fig. 5 and Supplementary Fig. S9). Although most methods identified eight clusters, they were not that distinct from each other. Different singlet-clusters were fused into each other, suggesting even worse quality of the batch3 than the batch2. Notably, BFF_raw and BFF_cluster failed to recognize several HTO clusters in both two captures (Supplementary Tables S15 and S16).
Fig. 5.

UMAP plots of the batch3_c1 dataset labeled with demultiplexing results from scDemultiplex, HTODemux, MULTIseqDemux, GMM-Demux, BFF_raw, BFF_cluster, demuxmix and hashedDrops.
We further investigated negatives and multiplets identified by each method (Fig. 6 and Supplementary Fig. S10). scDemultiplex obtained the most reasonable results, where most negatives either spread in the middle or at the edge of singlet-clusters, and most multiplets located between singlet-clusters or at the edge of singlet-clusters. In comparison, MULTIseqDemux, hashedDrops, and BFF_raw misclassified many singlets as negatives, while HTODemux, GMM_Demux, BFF_cluster, and demuxmix classified numerous cells as multiplets (Supplementary Tables S15 and S16). Those misclassified negatives and multiplets extended to the center of singlet-clusters or even dominated singlet-clusters.
Fig. 6.

UMAP plots of the batch3_c1 dataset labeled with singlets, negatives, and multiplets classified by scDemultiplex, HTODemux, MULTIseqDemux, GMM-Demux, BFF_raw, BFF_cluster, demuxmix and hashedDrops.
Consistently, scDemultiplex achieved the highest ARI and F-score (Table 4) (ARI=0.483 and 0.535; F-score=0.767 and 0.791), demonstrating its high performance in low-quality data. Similar to the results in the batch2, BFF_cluster performed poorly in the low-quality data, while HTODemux, GMM_Demux, and demuxmix had acceptable performance (Table 4 and Supplementary Tables 17 and 18).
Table 4.
Adjusted rand index and F-score of eight demultiplex methods in the batch3 datasets.
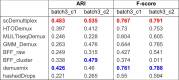 |
Note: The highest ARI and F-score among the eight approaches is highlighted in red while the second highest ARI and F-score is highlighted in blue.
In summary, scDemultiplex performs consistently well in both high-quality and low-quality data. In the high-quality data (batch1, Barnyard, and PBMC8), most approaches performed well and scDemultiplex ranked either the best or the second-best. In the low-quality data (batch2 and batch3), performance of demultiplexing approaches differed a lot. Notably, scDemultiplex achieved the highest performance when data quality was poor, demonstrating its power and consistency.
3.4. Integration with other approaches
scDemultiplex uses a two-step strategy, where the first step is to find a starting point and the second step is to fit the beta-binomial model based on the starting point and to refine the model. The two-step strategy makes it very easy to combine other approaches with scDemultiplex by using their results as the starting point.
We found combining approaches with scDemultiplex achieved better performance than using those approaches alone. In all the five datasets, the integrative approaches obtained higher ARI (Table 5) and F-scores (Table 6) except BFF_cluster in the batch1, GMM_Demux in the batch2_c2. We did not combine BFF_cluster and BFF_raw with scDemultiplex in the batch2 and batch3 since they failed to recognize several HTO clusters completely. The improved performance demonstrated that scDemultiplex was able to refine the result from each individual approach, indicating beta-binomial distribution is helpful to model raw HTO counts accurately.
Table 5.
ARI obtained by integrative approaches (each individual approach followed by scDemultiplex) in the three batches.
 |
Note: the number in brackets is the result using the approach alone. Red/blue indicates increased/decreased values after scDemultiplex refinement, and black means no change.
Table 6.
F-score obtained by integrative approaches (each individual approach followed by scDemultiplex) in the three batches.
Note: the number in brackets is from the result using the approach alone. Red/blue indicates increased/decreased values after scDemultiplex refinement, and black means no change.
4. Discussion
The experimental design of pooling single cells from multiple samples together with computational demultiplexing is not only cost-effective but also beneficial to the downstream analysis with minimized batch effects. Here, we present scDemultiplex, a novel barcoded-based demultiplexing method. Compared to most methods using Gaussian distribution to model log-transformed count, scDemultiplex directly models raw count by beta-binomial distribution, which fits the nature of the data better. Sample classification based on beta-binomial distribution calculates the probability of cells being from one sample not only by HTO-specific but also HTO-nonspecific counts, which is better than using HTO-specific counts alone. In addition, scDemultiplex uses an iterative strategy to further refine the model and the classification. We evaluated the performance of scDemultiplex by visual inspection, ARI, and F-score based on the “ground truth” obtained from other studies. scDemultiplex not only had great performance in high-quality data, but also performed the best in low-quality data. Since low-quality data is critical to distinguish performance of demultiplexing methods, the best achievement in those challenging datasets demonstrated the power and ability of scDemultiplex.
scDemultiplex uses CLR to normalize the HTO counts. The selection of different normalization methods would affect demultiplexing performance. For example, we found the log-transformed library size normalization worsened the performance of scDemultiplex in most cases (Supplementary Table S19), which is possibly because its assumption that each cell has the same number of total HTO counts might not hold true.
scDemultiplex uses an iterative strategy to refine the model. The maximum number of iterations is a tunable parameter to balance between the performance and speed (default=10). The accuracy over the iteration showed a substantial improvement in the first several iterations and slight increase or no change after that in both high-quality and low-quality datasets (Supplementary Fig. S11-S18), which suggested the default 10 times is appropriate. The procedure even stopped earlier since there were no reassignments or multiple misclassifications during one iteration in all the evaluation datasets.
Generally demultiplexing approaches performed comparably on good quality data with a clear bimodal distribution [3]. When the data quality is low and bimodal distribution is ambiguous, demultiplexing approaches have difficulty in determine the thresholds for classifying negatives, singlets, and multiplets and fitting the model. For example, although BFF_cluster achieved the best performance in the high-quality data, it even misrecognized several groups of singlets as multiplets in the low-quality data due to the failure to determine the thresholds when counts did not follow bimodal distribution. With the two-step strategy, scDemultiplex allows users to choose a start point manually, which is very useful for analyzing poor-quality data. Users can start with a stringent cutoff and then fit and refine the model in the second step. Additionally, users can use demultiplexing results of other methods as the starting point and then followed by scDemultiplex for refinement, which has been demonstrated to gain improved performance.
Besides barcoded-based demultiplexing, a number of SNP-based tools have been developed to separate samples with different genetic signatures, such as Demuxlet [10], souporcell [7], Vireo [9], and scSplit [27]. In this study, we used SNP-based demultiplexing as the ground truth, which would not be “gold standard” since SNP-based tools have their own limitations, such as high recall and low precision and reduced performance in high proportions of ambient RNA [24]. Those methods would not work when pooled samples have the same genetic background, but they are very useful in the cross-donor or cross-species design. If samples from different genetic background are barcoded and pooled together, an approach to combine scDemultiplex with other SNP-based tools holds great promise to improve the demultiplexing accuracy.
CRediT authorship contribution statement
Li-Ching Huang, Qi Liu and Quanhu Sheng designed the study, implemented the package, analyzed the data, and wrote the manuscript. Hua-Chang Chen implemented web service. Lindsey K Stolze tested the package and drafted the manuscript. Alexander Gelbard and Yu Shyr reviewed and edited the manuscript. All authors read and approved the final manuscript.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported in part by the National Institutes of Health (NIH) P01AI139449 (Q.L.) and R01HL146401 (A.G.), National Cancer Institute U2CCA233291, U54CA217450, P01CA229123, U54CA274367 (Y.S. and Q.L.) and Cancer Center Support Grant P30CA068485 (Y.S., Q.L. and Q.S.), and Patient-Centered Outcomes Research Institute (PCORI) 1409-22214 (A.G.). We also thank Dr. Zev Gartner for providing the ground truth of the PBMC8 dataset.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.csbj.2023.08.013.
Contributor Information
Qi Liu, Email: qi.liu@vumc.org.
Quanhu Sheng, Email: quanhu.sheng.1@vumc.org.
Appendix A. Supplementary material
Supplementary material.
.
References
- 1.Adil A., et al. Single-cell transcriptomics: current methods and challenges in data acquisition and analysis. Front Neurosci. 2021;15 doi: 10.3389/fnins.2021.591122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc. 1995;57:289–300. [Google Scholar]
- 3.Boggy G.J., et al. BFF and cellhashR: analysis tools for accurate demultiplexing of cell hashing data. Bioinformatics. 2022;38(10):2791–2801. doi: 10.1093/bioinformatics/btac213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Butler A., et al. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36(5):411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng J., et al. Multiplexing methods for simultaneous large-scale transcriptomic profiling of samples at single-cell resolution. Adv Sci (Weinh) 2021;8(17) doi: 10.1002/advs.202101229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gaublomme J.T., et al. Nuclei multiplexing with barcoded antibodies for single-nucleus genomics. Nat Commun. 2019;10(1):2907. doi: 10.1038/s41467-019-10756-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heaton H., et al. Souporcell: robust clustering of single-cell RNA-seq data by genotype without reference genotypes. Nat Methods. 2020;17(6):615–620. doi: 10.1038/s41592-020-0820-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Howitt, G., et al. Benchmarking single-cell hashtag oligo demultiplexing methods. bioRxiv [Preprint] 2022:2022.2012.2020.521313. [DOI] [PMC free article] [PubMed]
- 9.Huang Y., McCarthy D.J., Stegle O. Vireo: Bayesian demultiplexing of pooled single-cell RNA-seq data without genotype reference. Genome Biol. 2019;20(1):273. doi: 10.1186/s13059-019-1865-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kang H.M., et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat Biotechnol. 2018;36(1):89–94. doi: 10.1038/nbt.4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klein, H.-U. demuxmix: Demultiplexing oligonucleotide-barcoded single-cell RNA sequencing data with regression mixture models. bioRxiv [Preprint] 2023:2023.2001.2027.525961. [DOI] [PMC free article] [PubMed]
- 12.Lun A.T.L., et al. EmptyDrops: distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biol. 2019;20(1):63. doi: 10.1186/s13059-019-1662-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma P., et al. Bacterial droplet-based single-cell RNA-seq reveals antibiotic-associated heterogeneous cellular states. Cell. 2023;186(4):877–891. doi: 10.1016/j.cell.2023.01.002. e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McFarland J.M., et al. Multiplexed single-cell transcriptional response profiling to define cancer vulnerabilities and therapeutic mechanism of action. Nat Commun. 2020;11(1):4296. doi: 10.1038/s41467-020-17440-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McGinnis C.S., Murrow L.M., Gartner Z.J. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 2019;8(4):329–337. doi: 10.1016/j.cels.2019.03.003. e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McGinnis C.S., et al. MULTI-seq: sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nat Methods. 2019;16(7):619–626. doi: 10.1038/s41592-019-0433-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scrucca L., et al. mclust 5: clustering, classification and density estimation using Gaussian finite mixture models. R J. 2016;8(1):289–317. [PMC free article] [PubMed] [Google Scholar]
- 18.Shin D., et al. Multiplexed single-cell RNA-seq via transient barcoding for simultaneous expression profiling of various drug perturbations. Sci Adv. 2019;5(5):eaav2249. doi: 10.1126/sciadv.aav2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stoeckius M., et al. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 2018;19(1):224. doi: 10.1186/s13059-018-1603-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tabula Sapiens C., et al. The Tabula Sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science. 2022;376(6594) doi: 10.1126/science.abl4896. eabl4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tietscher S., et al. A comprehensive single-cell map of T cell exhaustion-associated immune environments in human breast cancer. Nat Commun. 2023;14(1):98. doi: 10.1038/s41467-022-35238-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trang N.V., et al. Determination of cut-off cycle threshold values in routine RT-PCR assays to assist differential diagnosis of norovirus in children hospitalized for acute gastroenteritis. Epidemiol Infect. 2015;143(15):3292–3299. doi: 10.1017/S095026881500059X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tvedebrink T. Overdispersion in allelic counts and theta-correction in forensic genetics. Theor Popul Biol. 2010;78(3):200–210. doi: 10.1016/j.tpb.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 24.Weber L.M., et al. Genetic demultiplexing of pooled single-cell RNA-sequencing samples in cancer facilitates effective experimental design. GigaScience. 2021;10:9. doi: 10.1093/gigascience/giab062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Winkler E.A., et al. A single-cell atlas of the normal and malformed human brain vasculature. Science. 2022;375(6584):eabi7377. doi: 10.1126/science.abi7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xin H., et al. GMM-Demux: sample demultiplexing, multiplet detection, experiment planning, and novel cell-type verification in single cell sequencing. Genome Biol. 2020;21(1):188. doi: 10.1186/s13059-020-02084-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu J., et al. Genotype-free demultiplexing of pooled single-cell RNA-seq. Genome Biol. 2019;20(1):290. doi: 10.1186/s13059-019-1852-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ziffra R.S., et al. Single-cell epigenomics reveals mechanisms of human cortical development. Nature. 2021;598(7879):205–213. doi: 10.1038/s41586-021-03209-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material.