

Abstract
Transposable elements (TEs) comprise about half of the mammalian genome. TEs often contain sequences capable of recruiting the host transcription machinery, which they use to express their own products and promote transposition. However, the regulatory sequences carried by TEs may affect host transcription long after the TEs have lost the ability to transpose. Recent advances in genome analysis and engineering have facilitated systematic interrogation of the regulatory activities of TEs. In this Review, we discuss diverse mechanisms by which TEs contribute to transcription regulation. Notably, TEs can donate enhancer and promoter sequences that influence the expression of host genes, modify 3D chromatin architecture and give rise to novel regulatory genes, including non-coding RNAs and transcription factors. We discuss how TEs spur regulatory evolution and facilitate the emergence of genetic novelties in mammalian physiology and development. By virtue of their repetitive and interspersed nature, TEs offer unique opportunities to dissect the effects of mutation and genomic context on the function and evolution of cis-regulatory elements. We argue that TE-centric studies hold the key to unlocking general principles of transcription regulation and evolution.
From protists to mammals, almost all eukaryotic genomes are populated with transposable elements (TEs)1. TEs are parasitic mobile DNA elements that are diverse in their genetic structure and transposition mechanisms1 (FIG. 1). As they propagate in the genome, TEs are potent insertional mutagens. When landing within exons, they are usually disruptive, whereas TE insertions in non-coding sequences, within or between genes, can have immediate or latent effects on host gene expression in cis or in trans. As for any mutational process, the effects of TE insertions on host gene expression may range from deleterious to adaptive. Over evolutionary timescales, mutations in their sequences can render TEs incapable of transposing. However, the regulatory sequences of TEs can be co-opted for host regulatory functions regardless of their transposition competence. Mechanistically, TEs can affect gene expression transcriptionally2,3, post-transcriptionally4–6 or in trans through their encoded products, which include both non-coding RNAs (ncRNAs) and proteins5,7.
Fig. 1 |. Major types of TEs in mammalian genomes.
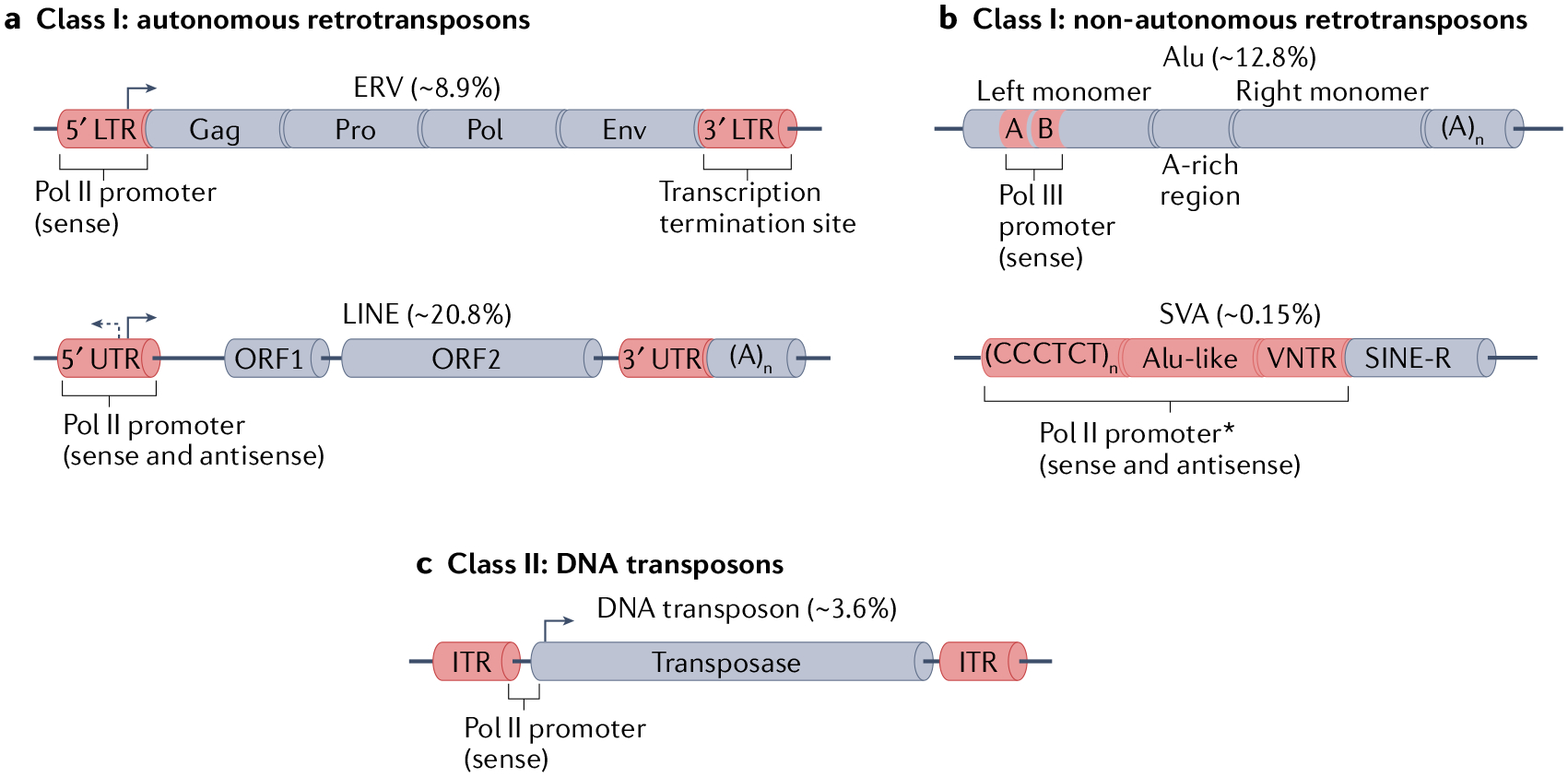
Transposable elements (TEs) are divided into Class I and Class II depending on their transposition mechanism1. Class I elements are called retrotransposons because they use RNA as an intermediate that is reverse transcribed into DNA and integrated in the genome (not shown). a | Autonomous retrotransposons encode all the required proteins for their retrotransposition. Endogenous retroviruses (ERVs) consist of two long terminal repeats (LTRs) flanking the open reading frames (ORFs) that encode the viral proteins. During evolution, ERVs are often reduced to a single LTR, or ‘solo LTR’, which renders them incapable of retrotransposition. Long interspersed nuclear elements (LINEs), such as L1, contain two ORFs that encode proteins required for their retrotransposition, which are flanked by untranslated regions (UTRs). At the 3′ end, they possess an adenines tail of variable length. b | Non-autonomous retrotransposons comprise those TEs that require the machinery encoded by autonomous retrotransposons to be mobilized (the TEs depicted here use the L1 machinery). Alu elements are primate-specific short interspersed nuclear elements (SINEs), and their structure consists of two monomers derived from the 7SL non-coding RNA, flanking an adenine (A)-rich region. At the 3′ end, they possess an adenines tail of variable length. On the left monomer, boxes A and B indicate a bipartite promoter for RNA polymerase III (Pol III). SINE-VNTR-Alu (SVA) elements are hominoid-specific composite non-autonomous retrotransposons comprised of a 5′ region of a variable number of repeats of the hexamer CCCTCT, followed by an Alu-like region, a variable number of tandem repeats (VNTR), and a 3′ ‘SINE-R’ region derived from the LTR of human endogenous retrovirus K. c | Class II DNA transposons encode a transposase that is required for their excision and insertion through a ‘cut-and-paste’ mechanism. The transposase ORF is flanked by two inverted terminal repeats (ITRs). Asterisk indicates that the precise localization of the cis-regulatory feature within the delimited region is unknown. Numbers in brackets indicate the percentage of the human genome (chm13-v1.0 (REF.17)) comprised by the specific subfamily. env, envelope; pol, reverse-transcriptase; pro, protease.
Co-opted.
Co-opted or exapted refers to the process by which transposable element (TE) sequences are repurposed for host function. Classically, this term implies that the process conferred adaptive changes that increased organismal fitness. in the context of gene regulation, the term is also used to describe the process by which TEs give rise to novel cis-regulatory elements that cause demonstrable changes in host gene expression.
In this Review, we discuss recent literature illuminating how TEs influence mammalian gene regulation at the transcriptional level through cis and trans mechanisms. These studies have been enabled by technological advances in genome editing (TABLE 1; BOX 1), which have opened new frontiers for the identification and characterization of TE-derived cis-regulatory elements (CREs), and for their functional validation in embryonic development and adult tissues8–11. We also discuss challenges in demonstrating and interpreting the adaptive relevance of gene expression changes driven by TEs. We surmise that, although many TEs exert measurable effects on gene expression or cause regulatory divergence between species, only a minority of these effects contribute to phenotypic changes at the organismal level. Finally, we argue that TE-derived CREs provide a unique model system for interrogating the mammalian cis-regulatory lexicon owing to their distinctive features compared with non-TE CREs.
Table 1 |.
Genome editing-driven advances in the study of TEs
| Technique | Brief description | Adaptations for TEs | Examples of key findings |
|---|---|---|---|
| Targeting individual TEs by nucleases: TALEs and CRISPR–Cas9 | Highly versatile genome-editing technologies based on the generation of double-strand breaks at a desired genomic locus for the introduction of genetic modifications8 | Target the unique flanking regions of the TE sequence to limit off target effects on other TEs of the same family | An MTC retrotransposon drives the expression of an oocyte-specific Dicer isoform49; LINE1 transcription is required for mouse embryo pre-implantation127; MER41 elements have been co-opted as innate immunity-responsive enhancers64; mouse liver LTR and LINE1 elements are required for proper gene function208; the regulatory capacity of ERVs is buffered by HSP90 (REF209); a HERV-K polymorphism regulates RASGRF2, a gene-related to dopaminergic activity194; specific insertions of the RLTR13 and RLTR9 subfamilies of TEs function as enhancers in mESCs and mTSCs45; HERV-H demarcates TADs in hESCs107 |
| Large-scale TE perturbations with CRISPRi or CRISPRa | Variation of the CRISPR–Cas9 technology that uses dCas9 fused to transcription activators or repressors to modify the functional status of a target region | The guide RNA must bind to a sequence that is common to all copies of the targeted TE subfamily; successful perturbation might require the combined action of multiple guide RNAs and molecules of dCas9-effector at the target region | LTR5HS insertions function as ape-specific early embryonic enhancers35; the RLTR13D6 TEs minimally contribute to gene regulation despite containing histone modifications characteristic of enhancers45; LTR5HS and SVA TEs function as enhancers in naive hESCs34; the HERV-K envelope protein promotes neurodegeneration210; ZNF417 and ZNF587 repress LTR5HS and SVA elements in neurons211; RLTR10 elements function as enhancers during mouse spermatogenesis47; HERV-K activation impairs cortical neuron differentiation212 |
CRISPRa, CRISPR activation; CRISPRi, CRISPR inhibition; dCas9, catalytic mutant of Cas9; ERVs, endogenous retroviruses; HERV-K: human endogenous retrovirus type K; hESCs, human embryonic stem cells; HSP90, heat-shock protein 90; LINE1, long interspersed nuclear element 1; LTR, long terminal repeat; LTR5HS, long terminal repeat 5 of HERV-K; MER41, medium reiterated sequence 41; mESCs, mouse embryonic stem cells; MTC, mouse transcript family type C; mTSCs, mouse trophoblast stem cells; SVA, SINE-VNTR-Alu; TAD, topologically associating domain; TALEs, transcriptional activator-like effectors; TEs, transposable elements.
Box 1 |. Functional assessment of transposon-derived cis-regulatory elements.
Genetic manipulation methods (see the figure, parts a–d) and modalities of reporter assays (see the figure, parts e–h) permit the functional validation of cis-regulatory elements (CREs). the transposable element (TE)-derived CRE of interest can be removed from the genome using CRISPR–Cas9 by designing pairs of guide RNAs flanking its sequence. Loss of enhancer activity is evaluated by measuring RNA expression of the putative CRE target gene (see the figure, part a). the TE-derived CRE or TE family of interest can be epigenetically repressed by a repressor protein domain (for example, KRAB) fused to a catalytic mutant Cas9 (dCas9). A single guide RNA or multiple guide RNAs can be designed to target the TE-derived CREs. Loss of enhancer activity is evaluated by measuring RNA expression of the putative CRE target genes (see the figure, part b). The TE-derived CRE or TE family can be epigenetically activated by an activator protein domain (for example, VPR or p300) fused to dCas9. increase of enhancer activity is evaluated by measuring the RNA expression of the target genes (see the figure, part c). When performing the experiment in vivo, the manipulation may or may not result in an organism-level phenotype (see the figure, part d). TE sequences can be tested for their functionality as a CRE one by one or in a massively parallel manner using methods like STARR-seq224 (see the figure, part e). reporter assays can be carried out with the sequences cloned into an episomal plasmid or integrated into the genome. the latter provides a chromatin environment that might be required for obtaining a reporter signal (see the figure, part f). To test the necessity of a transcription factor to activate a TE-derived CRE, the reporter assay can include sequences with mutant versions of the transcription factor binding sites (TFBSs). if the analysed transcription factor is involved in gene activation, the reporter activity should decrease with the introduction of the mutations (see the figure, part g). when performing the reporter assay in vivo, the model organism can display a positive signal or no signal (see the figure, part h).

How TEs influence gene transcription
In their prescient ‘gene-battery’ model, Roy Britten and Eric Davidson were among the first to realize the potential of interspersed DNA repeats as raw material for the wiring of cis-regulatory circuits12,13 (reviewed in REFS2,14–16). We now appreciate that these repeats are mostly derived from the accumulation of diverse TE families that can account for a substantial fraction of the genome. For instance, at least half of the human genome is comprised of TEs and their remnants17,18 (FIG. 1). A body of literature has now emerged to support the contribution of specific TE families to the dispersion of transcription factor binding sites (TFBSs) and CREs throughout mammalian genomes14,15,19–24 (reviewed in REF.25). In fact, it has been estimated that the majority of CREs newly evolved during primate evolution are directly derived from TEs26,27.
Gene-battery model.
Theory developed by britten and Davidson postulating that repetitive sequences are a driver of the evolution of protein expression control with a limited number of effectors.
Nevertheless, the mechanisms by which TEs influence host gene-regulatory networks are far more diverse than initially envisioned and can broadly be divided into five classes, discussed in this section (FIG. 2): (1) introduction of TFBSs, promoters and enhancers, (2) modification of 3D chromatin architecture, (3) production of regulatory ncRNAs, (4) co-option of TE-derived coding sequences as new transcriptional effector proteins, and (5) collateral effects of TE silencing mechanisms.
Fig. 2 |. Overview of mechanisms by which TEs influence host transcription regulation.

a | Transposable elements (TEs) can introduce new enhancers or promoters of cellular genes. A TE is depicted providing a transcription factor binding site (TFBS), which influences the transcription of the gene. b | TEs modulate 3D chromatin structure. A TE is depicted providing a TFBS for CCCTC-binding factor (CTCF), thereby demarcating a new boundary between two topologically associating domains (TADs; represented as dark-red triangles in a Hi-C map). c | TEs give rise to novel nuclear long non-coding RNAs (lncRNAs). A TE-derived non-coding RNA serves as a scaffold for a transcription factor that modulates the expression of a nearby gene in cis or of another gene in trans. d | TEs generate new transcription factors by fusion of DNA-binding domains of their transposase (TPase) with host transcription factor domains. A transposase-transcription factor fusion is depicted modulating two genes as a result of its binding to cognate TE sequences in the genome. e | Collateral effects of TE silencing on host gene transcription. A TE is bound by a host repressive transcription factor, thereby causing the silencing of nearby gene.
TEs function as cis-regulatory elements
In order to promote their parasitic existence, many TEs contain regulatory sequences that facilitate their own transcription. Over evolutionary timescales, TEs are typically silenced by the host and accumulate mutations, eventually leading to their immobilization, after which they no longer retain the biochemical activities necessary to promote their own spread. However, remnants of their ancestral regulatory sequences often persist and can be repurposed for the transcriptional control of host genes (FIG. 2a).
An early survey of genome-wide binding patterns of transcription factors in human and mouse revealed that a particular family of TEs is often over-represented in the set of binding sites for a given transcription factor20. This correlation suggests that the progenitor of a TE family frequently contains and disperses sequences that can attract host transcription factors. As a result, the process of TE amplification spreads large amounts of TFBSs throughout the genome. A seminal study of 26 transcription factors in human and mouse demonstrated that between 2% and 40% of their binding sites were derived from TEs21. In human embryonic stem cells (hESCs) and mouse embryonic stem cells (mESCs), ~19% of various TFBSs of pluripotency factors are located in TEs22,23. It is worth noting that TEs frequently harbour modules of multiple TFBSs, which may expand their regulatory potential and transcriptional activity to a wide range of cell types, tissues and developmental stages19,23,25,28–31.
TEs have remarkable cis-regulatory activities in stem cells.
New TE insertions must occur in the germline to be vertically transmitted. Thus, one would expect TEs to possess promoters that support their transcription in the germline or its progenitors to successfully propagate. Indeed, the transcriptional activity of TEs is frequently elevated in totipotent and pluripotent embryonic stem cells, and early germ cells, which share many transcriptional features, rely on a partially overlapping set of transcription factors, and have broadly permissive chromatin landscapes that may further facilitate TE activation32. As a result, one might predict that recently active TE families would have a propensity for being co-opted as early embryonic CREs since such sequences would be pre-existing within the TEs and readily available upon their insertion. Consistent with this prediction, multiple examples of young TE families having deposited CREs that are active in early embryonic cells have now been documented in humans and mice (FIG. 3). For example, the long terminal repeat 5 (LTR5HS) of the human endogenous retrovirus type-K (HERV-K) functions as an enhancer upon activation by the pluripotency transcription factors octamer-binding transcription factor 4 (OCT4), Krüppel-like factor 4 (KLF4) and KLF17 (REFS33,34). As these elements were inserted after the split of Old World monkeys from hominoids (apes), they might have contributed to hominoid-specific embryonic gene expression patterns33–35.
Fig. 3 |. Examples of families of TEs in humans and mice that may have cis-regulatory functions based on the binding of specific TFs or on functional experiments.
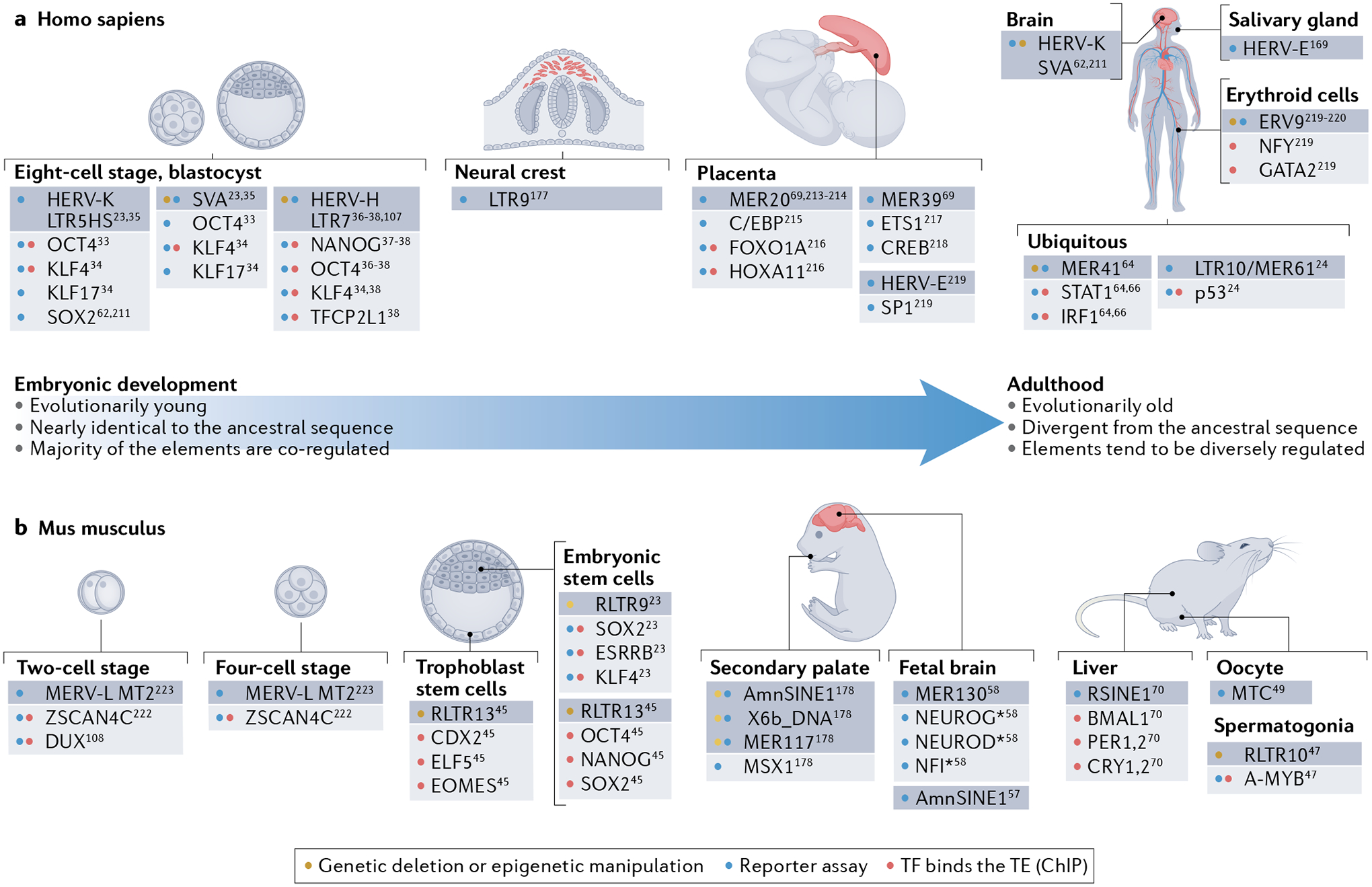
a | During human embryonic development and in human differentiated tissues24,33–38,62,64,66,69,107,169,177,211,213–221. b | During mouse embryonic development and in mouse differentiated tissues23,24,45,47,49,57,58,70,108,178,222,223. The figure shows how the transposable elements (TEs; dark grey boxes) that exhibit cis-regulatory activity at different stages of the life of an organism (namely humans and mice) differ in their characteristics. TEs that are active during early embryonic development belong to evolutionarily young families, whose sequences are nearly identical to their ancestral TE sequence. As a consequence, these elements share transcription factor (TF) binding sites and are often co-regulated. As development proceeds, the TEs that become active belong to evolutionarily older families and their DNA sequences frequently diverge from the ancestral sequence. In contrast to the co-regulation of the young TEs, only specific copies of these older TEs become active in the different tissues. Asterisk indicates that the specific member of the family of TFs that activates the reporter was not specified. Only the cis-regulatory activity of TEs and TFs demonstrated by genetic manipulation, reporter assay or chromatin immunoprecipitation (ChIP), is depicted. CDX2, caudal type homeobox 2; ELF5, E74-like factor 5; EOMES, eomesodermin; FOXO1A, Forkhead box O1; HERV-K: human endogenous retrovirus type K; KLF4, Krüppel-like factor 4; LTR, long terminal repeat; LTR5HS, long terminal repeat 5 human specific; MER41, medium reiterated sequence 41; MTC, mouse transcript family type C; OCT4, octamer-binding transcription factor 4; SOX2, SRY-box transcription factor 2; SVA, SINE-VNTR-Alu; TFCP2L1, transcription factor CP2-like 1.
Another remarkable example of this phenomenon is the primate-specific LTR7 of HERV-H. As an endogenous retrovirus (ERV), HERV-H copies are flanked by two identical-upon-insertion long terminal repeats (LTRs) that contain promoter sequences that drive transcription of the ERV (FIG. 1a). Members of this family are bound by the pluripotency transcription factors NANOG, OCT4, KLF4 and transcription factor CP2-like 1 (TFCP2L1), and they have been shown to function as enhancers in hESCs34,36,37 and as promoters of naive stem cell-specific chimeric and alternative transcripts38 (FIG. 3a). Interestingly, different subfamilies of HERV-H LTRs (LTR7Y, LTR7B, etc.) are transcriptionally active at distinct stages of zygote-to-blastocyst development, indicating that differences in the LTR DNA sequence promote precise stage-specific activation39. Activation of HERV-H has also been observed in human primordial germ cells, although this activation involves a different subset of HERV-H insertions than those observed in hESCs40. As reported in a recent preprint, dissection of the LTR7 family revealed that there are eight distinct subfamilies of LTR7. These subfamilies arose as the result of a complex evolutionary pattern of recombination and duplication events. A particular subfamily that is highly active in hESCs (LTR7up) has a binding site for SRY-box transcription factor 2 (SOX2) and SOX3, which is essential for promoter activity in vitro41. Thus, even within a single TE family, one can observe an extensive diversification of cis-regulatory activities. This diversification may be driven by the need to colonize new embryonic cell types in order to evade the emergence of stage-specific host repressors42–44.
Chimeric and alternative transcripts.
Transcripts that result from joining by splicing of RNAs that are not part of the same region of origin. They may comprise exons of different genes or combinations of sequences of TE and non-TE origin.
In mice, two families of LTR elements, RLTR9 and RLTR13, are bound by SOX2, OCT4, NANOG, oestrogen-related receptor-β (ESRRB), and KLF4 and have enhancer activity in mESCs23 (FIG. 3b). Importantly, the sequence motifs recognized by each of these transcription factors were present in the TE ancestral sequences, which may explain how these elements were able to successfully colonize the mouse genome23. The developing placenta also appears to be a niche repeatedly targeted by some TEs. In mouse trophoblast stem cells, RLTR13B and RLTR13D5 elements are bound by caudal type homeobox 2 (CDX2), E74-like factor 5 (ELF5), and eomesodermin (EOMES) and make a substantial contribution to the enhancer repertoire of mouse trophoblast stem cells45,46.
Some TEs also recruit transcription factors to directly infiltrate germ cells. As a result, these elements can deposit CREs that may be co-opted to control sex-specific gene expression. For example, studies in mice spermatogenesis demonstrated that RLTR10 elements act as enhancers of male germline genes upon binding of A-MYB, a crucial regulator of male meiosis47,48 (FIG. 3b). In female mice, a mouse transcript family type C (MTC) retrotransposon drives the oocyte-specific expression of a Dicer isoform that is essential for fertility49.
Altogether, these examples illustrate that young TE families — often LTR elements with embryonic TFBSs in their ancestral sequence — exhibit exquisitely specific patterns of transcription during early development. In developmental biology, the hourglass model posits that the embryonic morphology (and its underlying gene expression) of species belonging to the same phylum (for example, vertebrates) is divergent at the earlier and later steps of embryogenesis whereas, in the mid-embryonic stages, the different embryos have similar morphology, pointing to a common anterior–posterior body plan50,51. We speculate that the species-specific features that characterize early embryogenesis reflect the accelerated transcriptional divergence driven by recent waves of TE colonization, which have introduced large numbers of CREs that are active in different subpopulations of totipotent and pluripotent embryonic cells3,23,45.
TEs contribute to cis-regulatory gene networks in somatic cells.
In contrast to early embryos and germ cells, there is no intuitive reason for TE families to possess cis-regulatory activity in somatic tissues. From an evolutionary perspective, somatic activity should be selected against since it seemingly offers no advantage for TE propagation but creates an additional process by which new insertions can cause detrimental changes to their host52. Despite these theoretical predictions, many reports have now confirmed that certain TEs do in fact readily transpose in somatic cells53 and some have been co-opted as CREs in somatic tissues19,31,34,46,54–60 (FIG. 3). We posit that this process can occur through one or more of the following mechanisms: (1) overlap between cis-regulatory programmes of stem cells and somatic cells, (2) retroviral hijacking of transcription factors expressed in immune cell types, or (3) gain of somatic regulatory activity through mutations in the TE sequence that occur after genomic insertion.
As outlined above, TE-derived cis-regulatory activity in somatic tissues may be explained by the presence of ancestral binding sites for transcription activators that are also expressed in germ cells or embryonic stem cells. This overlap between embryonic and somatic cis-regulatory programmes may be explained by the promiscuity of expression of pluripotency transcription factors. Indeed, it is known that pluripotency transcription factors, such as SOX2, OCT4, NANOG, ESRRB and KLF4, are also individually expressed in various differentiated tissues61. Thus, binding of these transcription factors to TE families containing their cognate motifs could trigger regulatory activity of these young TEs in somatic cells. For example, upon binding of SOX2, a HERV-K element acts as an enhancer of PRODH, a schizophrenia-linked gene, in human neuronal cell lines62. Similarly, SINE-VNTR-Alu (SVA) elements, which are active in the early human embryo, are also reactivated in the fetal and adult brain and liver34,63 (FIG. 3a).
In a mechanism restricted to ERVs, somatic CRE activity may reflect the hijacking of innate immunity transcription factors by their exogenous retroviral ancestors. This is exemplified by MER41, a family of primate-specific LTR elements that are frequently bound by signal transducer and activator of transcription 1 (STAT1) and act as interferon-inducible enhancers for several innate immunity genes64–66 (FIG. 3a). Remarkably, MER41 enhancers are often located distal to the genes they activate but are in contact with their target promoters prior to interferon induction65, a finding consistent with the function of stress-responsive enhancers observed in other systems65. Because MER41 is of retroviral origin, it is tempting to speculate that its hijacking of immunity transcription factors is a relic of strategies evolved prior to its endogenization to promote viral replication in immune cells, similar to a strategy adopted by contemporary exogenous viruses such as HIV1 (REFS64,67).
Finally, an alternative explanation for TE cis-regulatory activity in somatic cells is that it emerged following genomic insertion through mutation of the TE sequence, which led to gain of new TFBSs. Consistent with this model, TE-derived enhancers active in somatic tissues are predominantly observed in members of older TE families, which also tend to be present across more species68 than the young ERV families that participate in early embryonic regulatory programmes. For instance, in human fetal brain, several members of the ultraconserved element 29 family of DNA transposons (UCON29) are hypomethylated, marked as enhancers by mono-methylation of histone H3 Lys4 (H3K4me1), and located near genes that show fetal brain-specific activation54. LTR77-derived CREs are preferentially active in blood, CRE activity of the MER121 family is enriched in mesodermal tissues, and CRE activity of several old short interspersed elements (SINE) families is enriched in fetal and epithelial tissues54. Neural development in mammals is partially regulated by a set of lineage-specific enhancers that are enriched in MER130 DNA transposons (an old repeat family that contains key binding sites for neurodevelopmental transcription factors, including neural differentiation factors, neurogenins and neurofibromin 1) and in deeply conserved insertions of the Amniotes-clade SINE1 (AmnSINE1) family of retrotransposons57,58. Importantly, all these TE families have been inactive for at least 100 million years and their ancestral (consensus) sequences did not originally contain the lineage-specific TFBSs responsible for the cis-regulatory activity observed today. These observations suggest that these older TEs acquired CRE activity by recurrent post-insertional mutations of individual copies rather than by dispersion of built-in TFBS modules such as those observed in younger LTR elements. It seems that the ancestral sequence of these TE families contained ‘proto-motifs’ for specific transcription factors that matured into bona fide binding sites upon mutation. Thus, the proto-motif content and abundance of certain TE families may predispose them to serve as a substrate for evolving some but not other TFBSs69,70. Additionally, their nucleotide composition and location in the genome may influence their mutational trajectory, notably due to the presence of hypermutable methyl-Cpg sites.
Methyl-CpG sites.
The cytosine in 5′-Cpg-3′ dinucleotides (C-phosphate-g) can be methylated to form 5-methylcytosine, which frequently triggers a C-to-T mutation through deamination.
TE-derived promoters drive transcription of chimeric RNAs and protein isoforms.
Besides acting as enhancers that modulate gene expression, TEs can also introduce functional promoters in new genomic locations, thereby creating new host gene transcripts71,72 (FIG. 2a). These TE-driven transcripts may encode protein isoforms (for example, truncated) with novel biochemical properties and functions73 or new patterns of expression49. One of the most well-characterized examples of this process is the antisense promoter activity of long interspersed nuclear elements (LINE1), which generates chimeric transcripts with human genes74–76. The LTRs of ERVs are also a common source of alternative promoters that drive the production of chimeric transcripts in a tissue-specific or developmental-specific fashion71,77–79. For example, a recent study shows that the mouse MT2B2 LTR promoter generates an embryonic-specific RNA isoform of cyclin-dependent kinase 2-associated protein 1, which encodes a truncated protein essential for pre-implantation development80. Although the vast majority of LINE1 and LTR promoters are repressed in normal cells, they may be reactivated in disease states — notably in tumours and transformed cells81. This reactivation may result not only in expression of their own proteins, which can drive cancer-specific transposition82, but also in aberrant host gene transcription. Both mechanisms can directly contribute to oncogenesis83 (reviewed in REF.84). Widespread de-repression of latent TE promoters can be experimentally induced with epigenetic drugs such as histone deacetylase or DNA-methyltransferase inhibitors, which may be harnessed for cancer treatment by driving the expression of tumour-specific antigens, or as nucleic acid adjuvants that potentiate immunotherapies85–88. Thus, the activity and proper control of TE-derived CREs are integral to cellular homeostasis.
TEs influence 3D chromatin architecture
The relationship between 3D organization of the genome and gene expression has been a subject of intense investigation (reviewed in REFS89–95). The development of chromatin conformation capture methods for measuring the contact frequency between distant regions of DNA revealed that, in the interphase nucleus, the genome partitions into topologically associating domains (TADs) in the megabase scale89,96–98. TADs are proposed to have a key role in specifying CRE function by restricting enhancers to only activate promoters within the same TAD91,99. However, the precise role of TADs in gene regulation remains controversial. Deletions of individual TAD boundary elements can result in dysregulation of the expression of adjacent genes, in some cases with clear phenotypic consequences100–104. Paradoxically, acute global perturbations of TAD-organizing proteins, such as the cohesin complex and CCCTC-binding factor (CTCF), apparently have a relatively minor effect on transcription despite global loss of chromatin loop domains104.
Topologically associating domain.
(TAD). large architectural domains of chromatin demarcated by insulator proteins that generally restrict the space in which three-dimensional DNA contacts and enhancer–promoter interactions are favoured.
Chromatin loop domain.
Folding of chromatin that causes two regions of the genome that are separated by (a large) linear space along the DNA to come into close three-dimensional proximity.
The contribution of TEs to 3D genome architecture has just begun to be explored. Several TE families are specifically enriched at TAD boundaries or harbour insulator activity105,106, and manipulative experiments suggest that some TEs have a direct influence on the folding of chromosomes (FIG. 2b). Notably, HERV-H elements are enriched at TAD boundaries that are transiently established in hESCs prior to their in vitro differentiation, and CRISPR–Cas-mediated deletion of two individual HERV-H elements leads to loss of their cognate boundaries107. Interestingly, this boundary function appears to be dependent on the transcription of the elements, which may suggest that they drive the formation of transient TAD boundaries during stages of embryonic development in which HERV-H is specifically expressed107. Similarly, in mice, widespread hypomethylation and transcriptional activation of the murine endogenous retrovirus type-L (MERVL) elements at the two-cell stage of embryonic development77,108 results in the establishment of stage-specific TAD boundaries at these elements (reported in a preprint109). It remains to be seen whether TE-derived TAD boundaries established at these early embryonic stages and the spatial genomic remodelling they engender have functional consequences for development.
The zinc finger protein CTCF has been demonstrated to demarcate many TAD boundaries, to mediate chromatin loop formation, and to be generally crucial for proper 3D genome organization110. Early studies reported that a large fraction of CTCF binding sites in various mammalian genomes derive from several classes of TEs20,21. For example, in humans, CTCF binding sites often derive from primate-specific ERVs whereas, in the mouse genome, B2 SINEs are a major source of murine-specific CTCF binding sites20,111. The observations that TEs have dispersed so many lineage-specific CTCF sites in mammalian genomes have posed a conundrum because TAD boundaries are highly conserved between human and mouse and they often correspond to regions that are deeply syntenic across metazoan genomes97,112. How are genomes protected from the formation of new TADs introduced by TE-driven CTCF sites and from a widespread remodelling of genome organization? Recent work has provided two solutions to this conundrum. First, TAD boundaries have been identified where one CTCF site introduced by a lineage-specific TE has functionally replaced an adjacent ancestral CTCF site, resulting in CTCF anchor turnover without a major change in 3D genome organization113. Another mechanism mitigates the widespread remodelling of TAD boundaries through CTCF sites dispersed by B2 SINEs, which are preferentially bound by the ChAHP complex (abbreviation for CHD4, ADNP and HP1 complex) that recognizes a motif nearly identical to that bound by CTCF but with higher affinity114. Thus, ChAHP binding tends to outcompete CTCF binding at B2 SINEs and prevent the establishment of ectopic TAD boundaries at these sites114.
TEs can also influence host chromatin structure in a CTCF-independent way by acting as insulator elements. In the developing mouse embryo, a B2 SINE ensures proper spatiotemporal activation of the growth hormone locus by blocking the spread of repressive chromatin. This insulator activity is dependent on the transcription of the B2 element by both RNA polymerase II (Pol II) and Pol III, which is surprising because such tRNA-derived SINEs are not naturally transcribed by Pol II115. By leveraging the polymorphic nature of B2 SINEs between lab mouse strains, a recent study identified a polymorphic B2 insertion that constrains the spread of histone H3 Lys9 acetylation (H3K9ac) and DNA hypomethylation in the strain where it is present116. This particular element was bound by CTCF; however, ChIP-seq data revealed that B2 SINEs are typically enriched at the boundaries of active-chromatin marks, and that the majority of these elements are not bound by CTCF116. The RNA expression levels of B2 SINEs are relatively low in the tissues where these boundary elements were observed, which argues against the model that the expression of these elements is universally required for their boundary function. A parallel situation exists in humans, where mammalian-wide interspersed repeats (MIR) are enriched at CTCF-independent boundaries between active and repressive chromatin features. These SINEs are also bound by Pol III105, suggesting that, in mammalian cells, one or more components of the Pol III machinery may facilitate insulator function as has been proposed for TFIIIC in yeast117.
Gene regulation by TE-derived ncRNA
Long non-coding RNAs (lncRNAs) contain a much higher density of TE-derived sequences than protein-coding genes, with TEs comprising ~30% of the total lncRNA sequence in human and mouse118,119. As they do for protein-coding genes, TEs provide crucial signals for the biogenesis of lncRNAs. For example, the expression of ~10% of human lncRNAs is driven by LTR-derived promoters118. In addition, TE sequences transcribed as part of lncRNA species can affect gene expression through a variety of mechanisms120,121 (FIG. 2c).
Several TE-derived lncRNAs have been implicated in embryonic development. Notably, some are highly expressed in pluripotent stem cells, where the transcriptome has been reported to be 30% more complex than those of differentiated cells, presumably due to the pronounced transcriptional activation of TEs122. Depletions of lncRNA transcripts derived from HERV-H in hESCs and from ERV-K and mammalian apparent LTR retrotransposon (MaLR) elements in mESCs suggest that they are essential for the maintenance of pluripotency in vitro36,38,122,123. In another example, the MER41-containing lncRNA BRAF-activated non-protein coding RNA (BANCR) stimulates the migration of ESC-derived human and non-human primate cardiomyocytes and cardiac enlargement in a mouse model124. The molecular mechanisms by which these ERV-derived lncRNAs modulate development remain murky but may involve a combination of transcriptional36,38,107 and post-transcriptional123,125,126 processes.
LINE1-derived RNAs have been reported to have a role in mouse embryonic pre-implantation development127 and in mESC self-renewal128. LINE1 RNA is highly expressed in the early mouse embryo, peaking at the two-cell stage, where it accumulates predominantly in the nucleus. Perturbation of LINE1 transcription at those early stages using designer transcription activator-like effectors (TALEs) fused to either activating or repressive transcription factor domains revealed that the precise temporal pattern of LINE1 transcription is required to avoid developmental arrest127. Interestingly, this phenotype cannot be rescued by the addition of exogenous LINE1 RNA in trans, which might indicate that it is the nascent LINE1 RNA or the process of LINE1 transcription that is required for development. Despite the dramatic phenotypic consequences of LINE1 perturbation to embryonic development, only modest changes in host gene expression were observed in these experiments127. Furthermore, another study reported that LINE1 nuclear RNA is essential to safeguard mESC identity by repressing the two-cell stage gene expression programme128. This conclusion was drawn by depleting LINE1-derived nuclear RNA using antisense oligonucleotides, suggesting that stable expression of LINE1 RNA is required for this regulatory activity. Further experimentation is required to explore the potential role of LINE1 expression in development as it remains unclear which LINE1 insertions or RNA species these different perturbation techniques were able to target.
Another example of regulatory activities of a TE-derived ncRNA is provided by the mouse B2 SINE RNAs upon heat shock. The heat shock transcriptional response is characterized by robust induction of a small set of heat-responsive genes alongside modest repression of many other genes129. B2 SINE RNAs bind to the stress-responsive genes and repress their transcription in basal conditions but, upon heat-shock, the Polycomb protein enhancer of zeste homologue 2 (EZH2) is recruited and accelerates the degradation of B2 RNA and de-repression of the stress-responsive genes130. B2 RNA degradation occurs through the activity of a self-cleaving ribozyme, which is greatly stimulated by EZH2 (REF.131). B2 SINE RNA has also been implicated in repressing the transcription of non-induced genes after heat shock132,133. Additionally, a recent report revealed that B2 SINE RNA cleavage can also be stimulated by heat shock transcription factor 1 (HSF1; a transcription factor that has a major role in the heat-shock response) in a mouse hippocampal neuronal cell line134. These results further strengthen the evidence of a trans-regulatory role for B2 SINE RNAs in this type of transcriptional stress response. It is possible that these regulatory functions may extend to other SINE RNAs because Alu transcripts also accumulate during and modulate the heat-shock response in human cells135 and Alu RNA possesses EZH2-stimulated self-cleaving RNA activity131.
These examples illustrate how ncRNAs derived from TEs act as regulators of transcription through a variety of mechanisms. In recent years, the number of molecular techniques available for the study of TE-derived ncRNAs has increased exponentially, so we foresee that many new and exciting roles of TE-derived ncRNAs are yet to be discovered.
Transposase-derived transcription factors
While it has long been speculated that the repeat sequences that TEs spread throughout genomes provide a fodder for the dispersion of TFBSs that facilitate the emergence of new gene regulatory networks12,13, it has more recently come to be appreciated that the proteins encoded by TEs themselves provide complementary pathways to achieve this outcome. Transposase proteins encoded by DNA transposons contain DNA-binding domains that recognize and bind their cognate TE sequences dispersed in the genome. Thus, it is easy to envision how the fusion of a transposase DNA-binding domain to a transcription-regulation domain could form a new transcription factor instantaneously capable of recognizing a ready-made network of binding sites distributed across the genome by its parent TE family (FIG. 2d). The fact that well-characterized transcription factors, such as paired box (PAX) proteins, possess DNA-binding domains that appear to have originated from transposases provided a hint that this process of transposase capture may be a recurrent theme in the birth of transcription factors (reviewed in REF.16). However, the ancient origin of most transposase-derived transcription factors has made it difficult to trace the steps by which these proteins and their cis-regulatory network were assembled.
A recent study has shed new light on the prevalence and modalities by which transposases supply protein domains for the emergence of new transcription factors in vertebrate evolution. A survey of ~600 tetrapod (limbed vertebrates) genomes suggests that fusion of transposase domains with host regulatory domains is a recurrent mechanism for the assembly of novel transcription factors during evolution136. This study identified 94 independent events across the phylogeny of transposase domains fused to a variety of host domains, with an apparent proclivity for KRAB domains. A mariner-transposase-KRAB fusion gene dubbed KRABINER, which evolved in the vespertilionid bat lineage, was further characterized. Reporter assays, loss-of-function and rescue experiments in bat cells show that KRABINER behaves as a canonical sequence-dependent transcription factor that binds thousands of genomic sites, including hundreds of cognate mariner transposons, and modulates the transcription of a large set of genes and CREs136. Together with previously characterized host-transposase fusion proteins137–141, these new findings demonstrate that the fusion of host and transposase domains can give rise to a wide variety of proteins with novel architectures and functions, including transcription factors. Because the emergence of new transcription factors is often pivotal to the emergence of major evolutionary novelties (for example, PAX6 in the evolution of eyes141), it is tempting to speculate that transposase capture has had a profound impact on phenotypic diversification.
KRAB domains.
Repressive transcription factor domain, characterizing KRAB containing zinc-finger repressor proteins.
Collateral effects of TE silencing
Organisms have evolved strategies to tame the activity of TEs142,143, and there are also a few examples that describe how TEs have evolved mechanisms to temper host defenses144. One of the major silencing mechanisms of TEs in mammals is DNA methylation145,146, which can lead to long-term, heritable transcription silencing (reviewed in REF.147). The need for DNA methylation to suppress TEs is underscored by the observation that, when some TEs are demethylated, their enhancer potential is unmasked, leading to aberrant activation of nearby genes54,148.
A classic case of the interplay between DNA methylation and the activity of TE-derived CREs are the Agouti metastable epialleles. These elements confer a range of yellow coloured coating to the brown-furred C57BL/6J mice in a manner dependent on the DNA methylation levels of the cryptic promoter that controls the transcription of intracisternal A-particle (IAP) retrotransposons149–154. In mice, IAP retrotransposons with disparate levels of CpG methylation across individuals (that is, with metastable epiallele features) mostly belong to the youngest and most polymorphic IAP subtypes155,156. Current data suggest that, at the implantation–gastrulation transition, the developing embryo may be exposed to environmental cues that modulate the activity of methylation maintenance complexes, giving rise to these differential methylation patterns157. Following their establishment in early development, these DNA methylation patterns are maintained throughout the organism157. Recent work demonstrates that DNA methylation at metastable epialleles is highly dependent on genetic background158. Additionally, studies in humans have revealed that metastable epialleles overlap with ERVs and LINE1s, suggesting that these elements contribute to human epigenetic variation159.
Metastable epialleles.
Alleles that are differentially epigenetically regulated and maintained throughout the organism’s lifetime and, in some cases, also across generations. They typically arise due to the variable levels of DNA methylation established during early development and can respond to environmental stressors and potentially drive phenotypic variation.
Another process through which TEs are silenced is the targeted deposition of repressive histone modifications. For example, in mESCs, TEs are silenced through tri-methylation of histone H3 Lys9 (H3K9me3) by SET domain bifurcated histone lysine methyltransferase 1 (SETDB1), which is recruited to TEs by KRAB zinc-finger proteins (KZFPs) through interaction with tripartite motif containing 28 (TRIM28; also known as KAP1)43. SETDB1 knockout leads to widespread de-repression of class I and class II ERV elements and transcription of chimeric RNAs, suggesting that repression of these elements not only prevents mutagenic transposition but also deleterious cis-regulatory effects160. An elegant experiment using trans-chromosomic mESCs demonstrated that a subset of SVA and LINE1 retrotransposons on the transferred human chromosome were activated in the non-primate nuclear environment because mice lack the transcription repressors ZNF91 and ZNF93 (REF.42). ZNF91 and ZNF93 are two KZFPs that have rapidly evolved in the primate lineage to adapt their DNA binding domains to specifically repress these retrotransposons, but the youngest LINE1HS elements have escaped repression through a deletion of the ZNF93 binding site42. Interestingly, interplay between TEs and KZFPs has also been described in naive hESCs, human primordial germ cells and, recently, in cancer34,40,161. In naive hESCs, accessible chromatin regions marked by H3K27ac and KLF4 binding are enriched in young TEs such as LTR5HS of HERV-K, LTR7 of HERV-H, and SVA elements. Curiously, these TEs, which are upregulated during pre-implantation development, function as enhancers of young KZFPs that, in return, target them for repression34. Thus, TEs and KZFPs appear to form complex regulatory loops that modulate the expression of developmental genes in a cell type-specific fashion. Interestingly, many KZFPs are expressed in somatic tissues, where they may mitigate the deleterious cis-regulatory effects of the TEs they target. By comparison, lack of KZFP expression in the germline may facilitate the spread of TEs and their fixation in the population. Thus, rather than a defence system against transposition, the KZFP system may actually enable the genomic accumulation of TEs with strong CREs (such as LTR elements), which increases the likelihood of these elements being subsequently co-opted for host functions.
Trans-chromosomic mESCs.
Mouse embryonic stem cell (mesC) clones to which a human chromosome has been transferred, resulting in aneuploid cells in which the effects of a non-primate cellular context on a primate chromosome can be assayed.
An additional H3K9me3-related repression mechanism is the silencing of retrotransposons by the human silencing hub (HUSH) complex, which was first identified in humans as a mediator of position-effect variegation162. A genome-wide screen of proteins affecting LINE1 retrotransposition in human cells revealed that the ATPase MORC2 and HUSH interfere with the mobilization and mutagenic potential of these elements by promoting the deposition of H3K9me3 on evolutionarily young LINE1 elements that reside in euchromatin163. In naive mESCs, TRIM28 and HUSH also co-repress young LINE1s and a subset of ERVs164. As many of these LINE1s are located within the introns of active host genes, their silencing by HUSH has a collateral repressive effect on the genes that harbour them, even when the silenced LINE1s are located a long distance from the gene promoters163,164. Host-gene silencing is likely mediated by a decrease in Pol II elongation rates caused by the presence of intragenic islands of H3K9me3 heterochromatin165. Recently, it has also been reported that HUSH repression of LINE1s safeguards the cells from inflammation triggered by the sensing of LINE1 and ERV RNAs in the cytoplasm by the double-stranded RNA sensors melanoma differentiation-associated protein 5 (MDA5) and retinoic acid-inducible gene I (RIG-1)166. These examples reveal that TE silencing not only affects TE activity but can also have collateral effects on the regulation of host-gene transcription (FIG.2e).
Position-effect variegation.
Phenomenon that defines the distinct expression levels that a gene or transgene exhibits when located at different positions in the genome, usually in correlation with the activated or repressed status of the neighbouring chromatin.
Phenotypic and evolutionary effects
The complex relationship between genotype and phenotype is challenging to decipher, yet it forms the foundation of our understanding of how genomic variation underlies the diversity of living organisms. In this section, we discuss the phenotypic consequences of gene regulatory changes triggered by TEs in mammals. TE insertions are thought to be generally deleterious in nature, yet TEs have colonized mammalian genomes to an impressive extent. This accumulation is likely driven by the small effective population size of mammalian species, which promotes the fixation of mildly deleterious TE insertions through genetic drift167. Although the majority of TEs in mammalian genomes have long lost the ability to transpose, their accumulation constitutes a vast amount of DNA with pre-existing functionalities, including CREs, which can provide fodder for phenotypic evolution.
TE-driven phenotypic change
Mammalian TE sequences can be co-opted to assemble new protein-coding genes (reviewed in REF.7) or contribute in non-coding capacity to the rewiring of gene regulatory networks. Some studies estimate that about 20% of CREs in the human genome may have been co-opted from TEs168. The extent to which exaptation of TEs drives phenotypic changes during evolution remains largely unknown, but there are some documented cases of TE-derived regulatory innovations with clear adaptive consequences.
A paradigmatic example is the evolution of the salivary-specific expression of the digestive enzyme amylase, which emerged during hominoid evolution through the insertion of an ERV-derived CRE upstream of the gene AMY1C (encoding α-amylase 1C)169,170. Another adaptation of mammals driven by a TE-derived CRE is the expression of prolactin in the endometrium, which is required for embryonic implantation171. In a striking case of convergent evolution, different mammal species utilize different TEs as endometrial promoters of prolactin: MER39 and MER20 in primates, MER77 in mice, and the LINE L1–2_LA in elephants172. Multiple members of the MER20 and MER121 families might also have had important roles in the evolution of pregnancy by dispersing numerous TFBSs for the transcription factors progesterone receptor (PGR) or Fork head box O1 (FOXO1A), thus potentially contributing to endometrial decidualization173,174. The placenta is another evolutionary novelty of mammals where TEs appear to have made repeated contributions through cis-regulatory innovations (FIG. 3a). Notably, insertion of a primate-specific transposon-like human element 1B (THE1B) controls the expression of corticotropin-releasing hormone (CRH), which modu lates gestational length175, and another TE controls the expression of placensin, a hormone that promotes placental cell invasiveness176.
TE-derived CREs that spur phenotypic novelties are not restricted to physiological or reproductive processes but may have also affected anatomical innovations. Enrichment in specific families of ERVs, such as LTR9, has been detected in neural crest enhancers with divergent activity between human and chimp177. These evolutionary gains in enhancer activity in neural crest cells have been linked to changes in the TFBS content of the enhancers, notably within a 17-bp sequence motif termed Coordinator. This motif was apparently not present in the progenitor sequence of LTR9 elements; thus, repeated mutations in a Coordinator-like proto-motif might have contributed to the adoption of some of these LTRs for neural crest enhancer function during recent human evolution177. Consistent with progressive gain of TEs and cooperative action of transcription factors91, a Wnt family member 5A (WNT5A)-dependent enhancer that regulates mammalian secondary palate development originated from concerted co-option of three distinct TEs (AmnSINE1, X6b_DNA and MER117)178.
As examples continue to aggregate in the literature, it is becoming increasingly apparent that TE-derived sequences have provided a fertile ground for the emergence of CREs that drove adaptive changes in gene expression during mammalian evolution. Nonetheless, we lack systematic studies that delete large numbers of TE-derived CREs to test their regulatory activity in animal models or even in cells. The scarcity of loss-of-function experiments in mammalian model organisms such as mice (but see REF.80 for a recent example), often precludes the demonstration of the direct involvement of TEs in driving organism-level phenotypes. In humans, these experiments introduce additional challenges. For example, many human enhancer regions do not have an orthologous sequence in the mouse179, and even if the mouse orthologous sequence exists, its spatiotemporal pattern of activity or the strength of it may not be conserved; thus, deletion of the mouse sequence may lead to much smaller gene expression changes than those seen for the human enhancer180.
Impact of TE insertion polymorphisms
Thus far, all known examples of human TEs with adaptive CRE activity come from insertions that have long been fixed in the genome. However, could TE insertion polymorphisms contribute to human phenotypic variation, including to disease susceptibility? In humans, only LINE1, Alu and SVA elements retain the ability to transpose and, as such, they represent the vast majority of TE insertion polymorphisms between and within populations181,182. De novo insertions of these elements can disrupt proper gene function leading to highly penetrant phenotypes and monogenic diseases181,183,184. There is also growing evidence that TE insertion polymorphisms that segregate in non-coding regions of the human genome can have subtler regulatory effects that may contribute to Mendelian or polygenic diseases or to the emergence of complex traits (FIG. 4). Recent studies suggest that these TE insertion variants can be mapped as cis-expression quantitative trait loci with substantial effects on gene expression, especially at loci involved in immune response and cognitive function185–188. Decrease in chromatin accessibility at or near TE insertion sites seems to underlie gene expression differences associated with most polymorphic TE insertions188. For instance, insertion of an SVA into a cell type-specific enhancer leads to a decrease in expression of the B4GALT1 gene (encoding β−1,4-galactosyltransferase 1), which has been associated by genome-wide association studies (GWAS) with susceptibility to autoimmune disease187. Another study identified 44 polymorphic Alu insertions in non-coding regions that are in linkage disequilibrium with GWAS single-nucleotide polymorphisms associated with various complex diseases, suggesting that these elements may contribute to the disease phenotypes through cis-regulatory effects189.
Fig. 4 |. Transposable element insertional polymorphisms that drive cis-regulatory changes and their phenotypic association in GWAS.
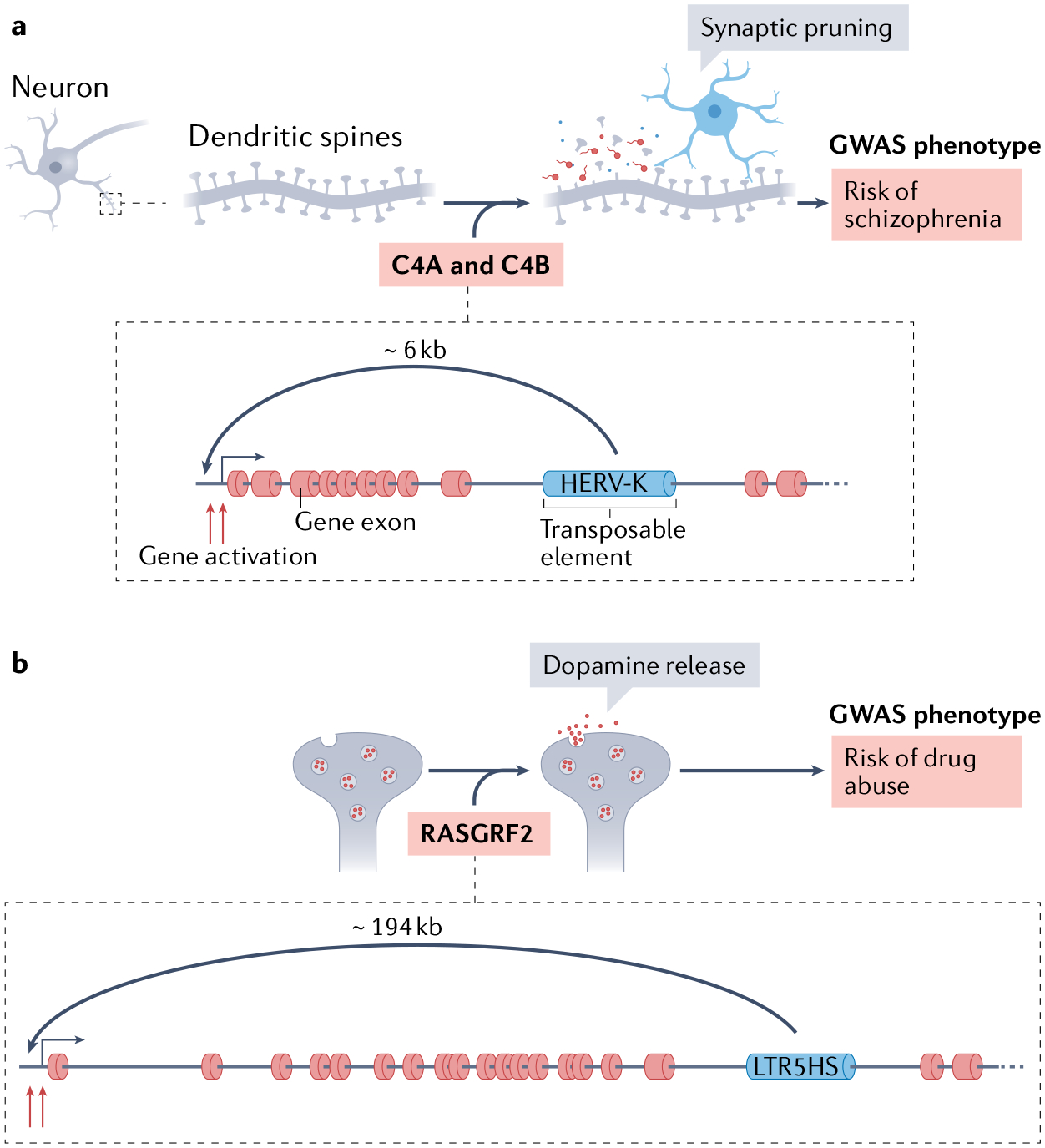
a | In neurons, a human endogenous retrovirus type-K (HERV-K) polymorphic insertion located between exons 9 and 10 of the gene encoding the C4A and C4B complement components promotes its transcriptional upregulation. The complement component signalling pathway is associated with synaptic pruning, and the HERV-K-containing C4A and C4B gene variants have been associated with schizophrenia193. b | Also in neurons, a sole long terminal repeat (LTR5HS) of HERV-K located at the RASGRF2 gene (encoding Ras-specific guanine nucleotide releasing factor 2) ~194 kb away from the promoter, leads to an increase in RASGRF2 expression. RASGRF2 is required for dopamine release, and this LTR5HS polymorphism has been associated with drug abuse194. GWAS, genome-wide association studies.
TE insertion polymorphism.
A TE insertion allele that is not yet fixed but segregating in the population; emerges from transpositions that occur in the germline and may be transmitted to the offspring, thereby increasing TE frequency in the population.
Expression quantitative trait loci.
Loci that explain expression variation among individuals in a population.
Genome-wide association studies.
(GWAS). a population genetics method of linking genetic variants present within the population with various traits and diseases.
Although no ERV is known to actively transpose in humans, the HERV-K family was recently active (~250,000 years ago) and seeded several insertions that remain unfixed in the human population190,191. Analysis of data from the 1000 Genomes and Human Genome Diversity projects revealed associations between HERV-K polymorphisms and human traits and diseases, and notably with neurological and immune afflictions192. For example, a polymorphic HERV-K insertion increases the expression of the gene encoding the C4A and C4B complement factors and has been correlated with higher schizophrenia risk193 (FIG. 4a). Another polymorphic HERV-K insertion has been associated with increased drug abuse due to an upregulation of the addiction-related gene RASGRF2 (encoding Ras-specific guanine nucleotide releasing factor 2)194 (FIG. 4b). These findings are concordant with the enhancer function of HERV-K in several cellular contexts34,35 and beg for further functional experiments to characterize the effects of HERV-K insertion polymorphisms on gene expression variation.
Evolutionary sequence conservation, or purifying selection, is a hallmark of functional constraint. Thus, it can be used as a proxy to quantify the fraction of TEs that have undergone co-option for organismal function. It has been estimated that ~6% of TEs annotated in the human genome harbour non-exonic sequences that have evolved under functional constraint throughout mammalian or primate evolution, suggesting they have assumed some regulatory functions168. However, this benchmark is not always appropriate in defining TE co-option or, more broadly, the functionality of non-coding regions. Enhancers are sometimes poorly conserved at the sequence level, and TEs that have given rise to enhancers are often clade specific and, as such, have not had sufficient time to exhibit measurable sequence conservation when closely related species are compared.
Very few cases have been described of human TEs co-opted as CREs that give rise to gene expression changes that lead to adaptive phenotypic changes at the organism level. It is likely that many TE-driven regulatory changes have resulted in subtler but still adaptive effects that are difficult to capture pheno-typically. Notably, redundancy is a common feature of gene-regulatory networks (at the level of both regulatory elements195 and protein-coding genes196). This phenomenon has been noted previously by a study that found that many enhancers have ‘shadow enhancers’ or redundant regulatory elements that may provide regulatory buffering and protect the enhancers against harmful changes195–197. TE-driven regulatory changes likely contribute to this adaptive redundancy and, in this case, their deletion would not lead to obvious phenotypic changes or such changes would only reveal themselves in certain, suboptimal environmental conditions198.
Using TEs to decipher transcription
TEs have several characteristics that enable unique lines of investigation into broader mechanistic aspects of transcription regulation. First, TE sequences from the same family are essentially identical upon their insertion. The very high genomic copy number (hundreds to thousands) of some TEs provides considerable advantages when investigating their regulatory activity. This repetitive nature has been a boon for the design of large-scale perturbative experiments utilizing sequence-guided catalytic mutant Cas9 (dCas9)-based synthetic transcriptional effectors (TABLE 1) because a large copy-number of elements can be targeted simultaneously using relatively few guide RNAs34,35,45. This efficiency allows the interrogation and comparison of the activity of hundreds to thousands of CREs in a single experiment (FIG. 5a). Second, although the different insertions of a specific TE family are identical or highly similar in sequence, they are integrated into distinct chromosomal environments, allowing for probing of the effect of the local chromatin context on CRE function. Furthermore, TE families are not evenly distributed in the genome but show distinct biases for insertion and retention in different genomic locales1. For example, in the human genome, Alu elements are enriched in gene-dense regions, whereas LINE1s are depleted from these regions18. These patterns enable contrasting the effects of distinct genomic environments on cis-regulatory activity. Third, just as the current sequences of the different TE families have permitted the reconstruction of their ancestral sequences10,18, it is possible to finely assess how mutations in TE-derived regulatory elements that occurred after insertion have modulated their activity (FIG. 5b). This property also enables the examination of how the unique genomic context of otherwise identical regulatory elements has influenced their mutational trajectory towards the gain or loss of CRE activity during evolution (FIG. 5c). Finally, TEs often have exquisitely specific patterns of expression, which enable inferences to be made regarding the role of sequence motifs — and by extension of transcription factors that recognize them — in a given cell type or tissue. Due to these special characteristics, TEs can be thought of as a model system within the genome that can bolster our general understanding of mechanisms governing the control of gene expression.
Fig. 5 |. Use of TEs as a model system for studying transcription regulation.
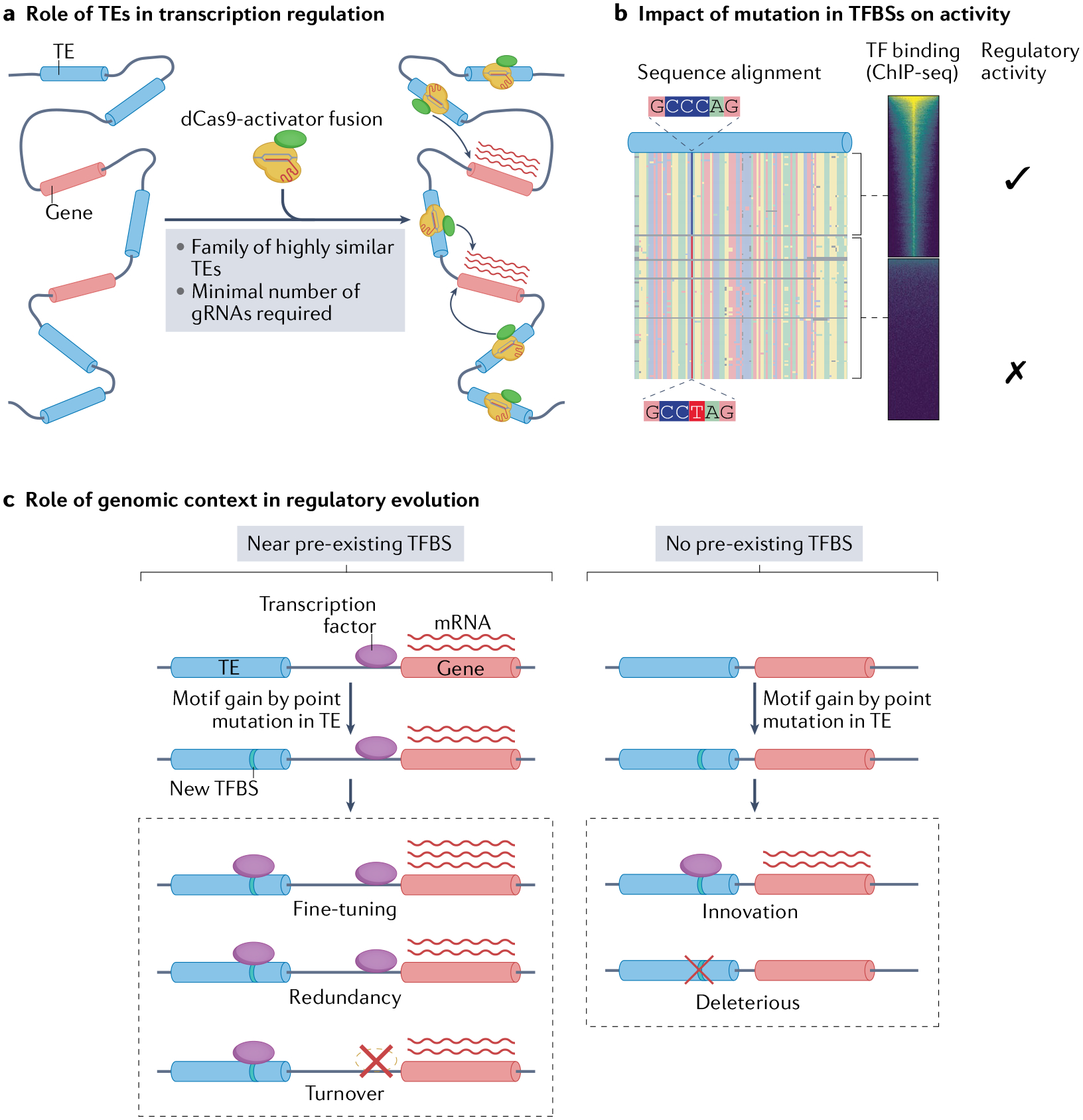
a | The dispersed, repetitive nature of transposable elements (TEs) enables large-scale sequence-specific transcriptional alterations using catalytically dead Cas9 (dCas9) fused to a transcription factor (domain). In the case of dCas9 fusion to a transcription activator, a minimal number of guide RNAs (gRNAs) complementary to a highly repetitive TE family can enable simultaneous transcriptional perturbation of many individual TE loci. b | The repetitive nature of TE families allows discrimination between the effects of subtle changes in sequence on transcription factor binding and regulatory activity. c | TEs enable the study of how the genomic context of a regulatory element influences its activity and evolution. A TE that inserted near a pre-existing transcription factor binding site (TFBS) gains an additional TFBS for the same transcription factor by point mutation. This process can result in either cis-regulatory fine-tuning, owing to the addition of a second binding site with similar activity; cis-regulatory redundancy, which can buffer against harmful regulatory changes introduced by future mutations; or cis-regulatory turnover, where introduction of the second TFBS relaxes selection on the pre-existing TFBS, which then decays by mutation and leaves the new nearby TE-derived TFBS in its place. A TE that gains a TFBS with no pre-existing TFBS nearby has two outcomes: the introduction of a cis-regulatory innovation, where new activity arises de novo, or the activity is deleterious and is purged by selection. ChIP, chromatin immunoprecipitation.
Consequences of TFBS mutation
TEs often harbour clusters of TFBSs, and their ancestral sequence can be approximated by generating phylogenetic trees or consensus sequences10,18 (taking into account that a consensus does not necessarily represent the actual ancestral sequence as selective forces can introduce bias in mutational patterns). Accordingly, a particular TE family can be used to measure the effects of mutations in TFBSs on their function as CREs. For example, if an LTR element containing a Pol II promoter and a specific combination of TFBSs is dispersed in many sites throughout the genome, one could investigate, in a highly granular manner, the effect of point mutations in each of these different TFBSs on the transcriptional output of the associated promoter. For example, HERV-K LTR5HS is the youngest ERV in the human genome and, consequently, the sequences of its 697 LTRs are >90% identical18,35. Despite this sequence homogeneity, LTR5HS copies display heterogeneous levels of the enhancer-associated modification H3K27ac in NCCIT embryonic carcinoma cells, with approximately 30% of the copies exhibiting high levels of H3K27ac while the remaining fraction shows low or negligible levels of the modification35. Likewise, copies of the RLTR13D6 and RTLR9 families in the mouse genome are also highly similar in sequence but exhibit varying levels of H3K27ac in mESCs and have disparate effects on gene expression45. These examples highlight the power of using TEs to study gene regulation despite having highly similar sequences; members of the same family may possess different activities, offering the opportunity to evaluate whether variation in regulatory activity is caused by mutations that led to gain or loss of TFBSs (FIG. 5b) or by local genomic features unique to each TE insertion site. Currently, the presence of suboptimal TFBSs or the vari able spacing between them are being uncovered as key determinants of enhancer function199. We speculate that the multi-copy nature of TEs and their variable regu lation will boost the exploration of the cis-regulatory grammar in the coming years.
Effects of genomic environment
Another interesting question to ask is whether the genomic context of a TE insertion can influence the element’s propensity to evolve regulatory activity. Although most examples of TE-derived TFBSs involve an intact transcription-factor binding motif pre-existing in their ancestral sequence, there are also instances where the TE gained a new TFBS through a mutation introduced after its genomic insertion. For example, a recent study revealed that RSINE1 elements in mouse spread proto-motifs for circadian transcription factors throughout the genome that, in many distinct cases, matured into bona fide TFBSs through CpG deamination70, which is a common mutation in methylated TEs (reviewed in REF.200). A similar mutational process has been observed previously within human Alu elements, which contain proto-motifs for p53, PAX6, and MYC and can generate different combinations of TFBSs through differential CpG deamination201. Interestingly, RSINE1 elements inserted nearby pre-existing circadian TFBSs were more likely to acquire circadian TFBSs through CpG deamination in proto-motifs. One possible explanat ion for this pattern is that it is driven by natural selection for mutations that introduce cis-regulatory redundancy, an idea that has been explored previously197. Epistatic interactions with other TFBSs located outside the TE but near its insertion site may also facilitate the acquisition of new TFBSs within the TE, a mechanism dubbed ‘epistatic capture’69. Using TEs to measure the effects of genomic context on the evolutionary trajectory of regulatory elements has the potential to unlock many of the secrets of the non-coding genome (FIG. 5c).
How do distinct local epigenomic features or the 3D structure of chromatin shape the regulatory activity of TEs? Several reports modelling the features that predict regulatory potential of enhancer–promoter pairs have recently been published202,203. The strength of the CRE, the distance to the cognate promoters, the underlying TFBSs and the 3D topology of chromatin have emerged as the main features influencing a CRE’s regulatory potential. Additionally, 4Tran65 (a technique akin to capture Hi-C204 but centred around transposons) has revealed that TEs interact with genomic regions that are located in the same compartment and the same TAD, which is consistent with the general behaviour of enhancer–promoter connections65. Further study of TE-derived CREs leveraging the experimental and analytical advantages described above may solidify our understanding of how 3D chromatin structure influences transcription regulation.
Tissue-specific transcription factors
Because TEs contain various TFBSs and often have highly specific patterns of expression across different cell types31, knowing which TEs are expressed in a given cell type might provide a way to infer which transcription factors may be important for transcription activation in that environment. Rapid diversification of the cis-regulatory activity of a TE family — driven by the need to colonize new cell lineages — can cause a group of highly related TE subfamilies to exhibit highly specific activation profiles across different cell types39 and developmental cell lineages41. Careful examination of the sequence motif content of these highly similar subfamilies may facilitate the identification of previously unrecognized transcription factors governing cell type identity41. This analysis could even be performed with more granularity by cross-referencing the TFBS content of all expressed TEs for common occurrence of TFBSs and might reveal different transcription factor specificity in distinct yet related cell types. By using TEs as a proxy for transcription factor affinity across cell lineages, we believe that the degree of overlap between developmental programmes in terms of the combination of transcription factors required to generate a particular cell type could also be examined in detail. The advent of single-cell methodologies is likely to greatly enhance this type of analyses. Indeed, recent forays into single-cell RNA sequencing analysis of TE expression have revealed that certain TE families behave as precise markers of cell lineages during vertebrate development41,205–207.
Conclusions and future perspective
Technological advances in genomics and systems biology over the past few years are ushering in a golden era of TE research. Methods such as CRISPR–Cas9 have empowered the systematic dissection of TE-driven cis-regulatory evolution in mammalian cells. These experiments have revealed that, although many TEs bear the biochemical hallmarks of CREs, only a subset of these elements significantly contribute to gene regulation in their native chromosomal context. As the number of functionally validated TE-derived CREs continues to grow, it will soon become possible to explain their potential redundancies, synergies and interactions with each other and with non-TE regulatory elements. Empirical demonstration of TE co-option for adaptive cis-regulatory changes remains challenging as regulatory changes driven by TEs may result in subtle phenotypes. However, methodological improvements in TE-centric analyses and the ease and precision with which genomes can now be functionally profiled and modified have elevated TEs from components that were once recalcitrant to genomic analyses to an emerging platform for understanding general principles of gene regulation. Unique features of TEs offer powerful tools to decipher the lexicon of mammalian transcription regulation and probe its evolutionary dynamics with unprecedented depth. These advances open exciting avenues for improving our understanding of gene regulation in health, disease and evolution.
Acknowledgements
We apologize to our colleagues whose work we were unable to cite due to space limitations. This work was supported by awards R35-GM122550, U01-HG009391 and R01-CA260691 from the National Institutes of Health to C.F., and R35-GM131757 and HHMI investigator award to J.W. J.J. was supported by NHGRI fellowship F31-HG010820. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. R.F. was funded by an EMBO long-term fellowship and a Cancer Research Institute/Bristol-Myers Squibb postdoctoral fellowship.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Wells JN & Feschotte C A field guide to eukaryotic transposable elements. Annu. Rev. Genet 54, 539–561 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chuong EB, Elde NC & Feschotte C Regulatory activities of transposable elements: from conflicts to benefits. Nat. Rev. Genet 18, 71–86 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sundaram V & Wysocka J Transposable elements as a potent source of diverse cis-regulatory sequences in mammalian genomes. Philos. Trans. R. Soc. B Biol. Sci 375, 20190347 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drongitis D, Aniello F, Fucci L & Donizetti A Roles of transposable elements in the different layers of gene expression regulation. Int. J. Mol. Sci 20, 5755 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fort V, Khelifi G & Hussein SMI Long non-coding RNAs and transposable elements: a functional relationship. Biochim. Biophys. Acta Mol. Cell Res 1868, 118837 (2021). [DOI] [PubMed] [Google Scholar]
- 6.Ozata DM, Gainetdinov I, Zoch A, O’Carroll D & Zamore PD PIWI-interacting RNAs: small RNAs with big functions. Nat. Rev. Genet 20, 89–108 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Jangam D, Feschotte C & Betrán E Transposable element domestication as an adaptation to evolutionary conflicts. Trends Genet 33, 817–831 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pickar-Oliver A & Gersbach CA The next generation of CRISPR–Cas technologies and applications. Nat. Rev. Mol. Cell Biol 20, 490–507 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goodwin S, McPherson JD & McCombie WR Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet 17, 333–351 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goerner-Potvin P & Bourque G Computational tools to unmask transposable elements. Nat. Rev. Genet 19, 688–704 (2018). [DOI] [PubMed] [Google Scholar]
- 11.McGuire AL et al. The road ahead in genetics and genomics. Nat. Rev. Genet 21, 581–596 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Britten RJ & Davidson EH Gene regulation for higher cells: a theory. Science 165, 349 (1969). [DOI] [PubMed] [Google Scholar]
- 13.Davidson EH & Britten RJ Regulation of gene expression: possible role of repetitive sequences. Science 204, 1052–1059 (1979). [DOI] [PubMed] [Google Scholar]
- 14.Sundaram V & Wang T Transposable element mediated innovation in gene regulatory landscapes of cells: re-visiting the “Gene-Battery” model. BioEssays 40, 1700155 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Enriquez-Gasca R, Gould PA & Rowe HM Host gene regulation by transposable elements: the new, the old and the ugly. Viruses 12, 1089 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feschotte C Transposable elements and the evolution of regulatory networks. Nat. Rev. Genet 9, 397–405 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoyt SJ et al. From telomere to telomere: the transcriptional and epigenetic state of human repeat elements. bioRxiv 10.1101/2021.07.12.451456v1 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lander ES et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001). [DOI] [PubMed] [Google Scholar]
- 19.Pehrsson EC, Choudhary MNK, Sundaram V & Wang T The epigenomic landscape of transposable elements across normal human development and anatomy. Nat. Commun 10, 5640 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bourque G et al. Evolution of the mammalian transcription factor binding repertoire via transposable elements. Genome Res 18, 1752–1762 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sundaram V et al. Widespread contribution of transposable elements to the innovation of gene regulatory networks. Genome Res 24, 1963–1976 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kunarso G et al. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat. Genet 42, 631–634 (2010). [DOI] [PubMed] [Google Scholar]
- 23.Sundaram V Functional cis -regulatory modules encoded by mouse-specific endogenous retrovirus. Nat. Commun 8, 14550 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang T et al. Species-specific endogenous retroviruses shape the transcriptional network of the human tumor suppressor protein p53. Proc. Natl Acad. Sci. USA 104, 18613–18618 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hermant C & Torres-Padilla M-E TFs for TEs: the transcription factor repertoire of mammalian transposable elements. Genes Dev 35, 22–39 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacques P-É, Jeyakani J & Bourque G The majority of primate-specific regulatory sequences are derived from transposable elements. PLoS Genet 9, e1003504 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trizzino M et al. Transposable elements are the primary source of novelty in primate gene regulation. Genome Res 27, 1623–1633 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ito J et al. Systematic identification and characterization of regulatory elements derived from human endogenous retroviruses. PLoS Genet 13, e1006883 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun X et al. Transcription factor profiling reveals molecular choreography and key regulators of human retrotransposon expression. Proc. Natl Acad. Sci. USA 115, E5526–E5535 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Faulkner GJ et al. The regulated retrotransposon transcriptome of mammalian cells. Nat. Genet 41, 563–571 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Miao B et al. Tissue-specific usage of transposable element-derived promoters in mouse development. Genome Biol 21, 255 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodriguez-Terrones D & Torres-Padilla M-E Nimble and ready to mingle: transposon outbursts of early development. Trends Genet 34, 806–820 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Grow EJ et al. Intrinsic retroviral reactivation in human preimplantation embryos and pluripotent cells. Nature 522, 221–225 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pontis J et al. Hominoid-specific transposable elements and KZFPs facilitate human embryonic genome activation and control transcription in naive human ESCs. Cell Stem Cell 24, 724–735.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fuentes DR, Swigut T & Wysocka J Systematic perturbation of retroviral LTRs reveals widespread long-range effects on human gene regulation. eLife 7, e35989 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu X et al. The retrovirus HERVH is a long noncoding RNA required for human embryonic stem cell identity. Nat. Struct. Mol. Biol 21, 423–425 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Barakat TS et al. Functional dissection of the enhancer repertoire in human embryonic stem cells. Cell Stem Cell 23, 276–288.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang J et al. Primate-specific endogenous retrovirus-driven transcription defines naive-like stem cells. Nature 516, 405–409 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Göke J et al. Dynamic transcription of distinct classes of endogenous retroviral elements marks specific populations of early human embryonic cells. Cell Stem Cell 16, 135–141 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Tang WWC et al. A unique gene regulatory network resets the human germline epigenome for development. Cell 161, 1453–1467 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carter TA et al. Mosaic cis-regulatory evolution drives transcriptional partitioning of HERVH endogenous retrovirus in the human embryo. bioRxiv 10.1101/2021.07.08.451617v1 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jacobs FM et al. An evolutionary arms race between KRAB zinc finger genes 91/93 and SVA/L1 retrotransposons. Nature 516, 242–245 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Imbeault M, Helleboid P-Y & Trono D KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature 543, 550–554 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Bruno M, Mahgoub M & Macfarlan TS The arms race between KRAB–zinc finger proteins and endogenous retroelements and its impact on mammals. Annu. Rev. Genet 53, 393–416 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Todd CD, Deniz Ö, Taylor D & Branco MR Functional evaluation of transposable elements as enhancers in mouse embryonic and trophoblast stem cells. eLife 8, e44344 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chuong EB, Rumi MAK, Soares MJ & Baker JC Endogenous retroviruses function as species-specific enhancer elements in the placenta. Nat. Genet 45, 325–329 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sakashita A et al. Endogenous retroviruses drive species-specific germline transcriptomes in mammals. Nat. Struct. Mol. Biol 27, 967–977 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bolcun-Filas E et al. A-MYB (MYBL1) transcription factor is a master regulator of male meiosis. Development 138, 3319–3330 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Flemr M et al. A retrotransposon-driven dicer isoform directs endogenous small interfering RNA production in mouse oocytes. Cell 155, 807–816 (2013). [DOI] [PubMed] [Google Scholar]
- 50.Duboule D Temporal colinearity and the phylotypic progression: a basis for the stability of a vertebrate Bauplan and the evolution of morphologies through heterochrony. Dev. Suppl 1994, 135–142 (1994). [PubMed] [Google Scholar]
- 51.Irie N & Kuratani S The developmental hourglass model: a predictor of the basic body plan? Development 141, 4649–4655 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Haig D Transposable elements: self-seekers of the germline, team-players of the soma. BioEssays 38, 1158–1166 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Loreto ELS & Pereira CM Somatizing the transposons action. Mob. Genet. Elem 7, 1–9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xie M et al. DNA hypomethylation within specific transposable element families associates with tissue-specific enhancer landscape. Nat. Genet 45, 836–841 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Villar D et al. Enhancer evolution across 20 mammalian species. Cell 160, 554–566 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Okhovat M et al. Co-option of the lineage-specific LAVA retrotransposon in the gibbon genome. Proc. Natl Acad. Sci. USA 117, 19328–19338 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sasaki T et al. Possible involvement of SINEs in mammalian-specific brain formation. Proc. Natl Acad. Sci. USA 105, 4220–4225 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Notwell JH, Chung T, Heavner W & Bejerano G A family of transposable elements co-opted into developmental enhancers in the mouse neocortex. Nat. Commun 6, 6644 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ye M et al. Specific subfamilies of transposable elements contribute to different domains of T lymphocyte enhancers. Proc. Natl Acad. Sci. USA 117, 7905–7916 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pavlicev M, Hiratsuka K, Swaggart KA, Dunn C & Muglia L Detecting endogenous retrovirus-driven tissue-specific gene transcription. Genome Biol. Evol 7, 1082–1097 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Uhlén M et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 62.Suntsova M et al. Human-specific endogenous retroviral insert serves as an enhancer for the schizophrenia-linked gene PRODH. Proc. Natl Acad. Sci. USA 110, 19472–19477 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Trizzino M, Kapusta A & Brown CD Transposable elements generate regulatory novelty in a tissue-specific fashion. BMC Genomics 19, 468 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chuong EB, Elde NC & Feschotte C Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 351, 1083–1087 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Raviram R et al. Analysis of 3D genomic interactions identifies candidate host genes that transposable elements potentially regulate. Genome Biol 19, 216 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schmid CD & Bucher P MER41 repeat sequences contain inducible STAT1 binding sites. PLoS One 5, e11425 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sgarbanti M et al. IRF-1 is required for full NF-kappaB transcriptional activity at the human immunodeficiency virus type 1 long terminal repeat enhancer. J. Virol 82, 3632–3641 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Simonti CN, Pavlicev M & Capra JA Transposable element exaptation into regulatory regions is rare, influenced by evolutionary age, and subject to pleiotropic constraints. Mol. Biol. Evol 34, 2856–2869 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Emera D & Wagner GP Transformation of a transposon into a derived prolactin promoter with function during human pregnancy. Proc. Natl Acad. Sci. USA 109, 11246–11251 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Judd J, Sanderson H & Feschotte C Evolution of mouse circadian enhancers from transposable elements. Genome Biol 22, 193 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cohen CJ, Lock WM & Mager DL Endogenous retroviral LTRs as promoters for human genes: a critical assessment. Gene 448, 105–114 (2009). [DOI] [PubMed] [Google Scholar]
- 72.Lanciano S & Cristofari G Measuring and interpreting transposable element expression. Nat. Rev. Genet 21, 721–736 (2020). [DOI] [PubMed] [Google Scholar]
- 73.Etchegaray E, Naville M, Volff J-N & Haftek-Terreau Z Transposable element-derived sequences in vertebrate development. Mob. DNA 12, 1 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nigumann P, Redik K, Mätlik K & Speek M Many human genes are transcribed from the antisense promoter of L1 retrotransposon. Genomics 79, 628–634 (2002). [DOI] [PubMed] [Google Scholar]
- 75.Mätlik K, Redik K & Speek M L1 antisense promoter drives tissue-specific transcription of human genes. J. Biomed. Biotechnol 2006, 71753 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Criscione SW et al. Genome-wide characterization of human L1 antisense promoter-driven transcripts. BMC Genomics 17, 463 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Macfarlan TS et al. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature 487, 57–63 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang F et al. DUX-miR-344-ZMYM2-mediated activation of MERVL LTRs induces a totipotent 2C-like state. Cell Stem Cell 26, 234–250.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Peaston AE et al. Retrotransposons regulate host genes in mouse oocytes and preimplantation embryos. Dev. Cell 7, 597–606 (2004). [DOI] [PubMed] [Google Scholar]
- 80.Modzelewski AJ et al. A mouse-specific retrotransposon drives a conserved Cdk2ap1 isoform essential for development. Cell 184, 5541–5558 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ardeljan D, Taylor MS, Ting DT & Burns KH The human long interspersed element-1 retrotransposon: an emerging biomarker of neoplasia. Clin. Chem 63, 816–822 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Burns KH Transposable elements in cancer. Nat. Rev. Cancer 17, 415–424 (2017). [DOI] [PubMed] [Google Scholar]
- 83.Jang HS et al. Transposable elements drive widespread expression of oncogenes in human cancers. Nat. Genet 51, 611–617 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Babaian A & Mager DL Endogenous retroviral promoter exaptation in human cancer. Mob. DNA 7, 24 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Roulois D DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 162, 961–973 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chiappinelli KB Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 162, 974–986 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brocks D et al. DNMT and HDAC inhibitors induce cryptic transcription start sites encoded in long terminal repeats. Nat. Genet 49, 1052–1060 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ishak CA & De Carvalho DD Reactivation of endogenous retroelements in cancer development and therapy. Annu. Rev. Cancer Biol 4, 159–176 (2020). [Google Scholar]
- 89.Rowley MJ & Corces VG Organizational principles of 3D genome architecture. Nat. Rev. Genet 19, 789–800 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Robson MI, Ringel AR & Mundlos S Regulatory landscaping: how enhancer-promoter communication is sculpted in 3D. Mol. Cell 74, 1110–1122 (2019). [DOI] [PubMed] [Google Scholar]
- 91.Long HK, Prescott SL & Wysocka J Ever-changing landscapes: transcriptional enhancers in development and evolution. Cell 167, 1170–1187 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bonev B & Cavalli G Organization and function of the 3D genome. Nat. Rev. Genet 17, 772 (2016). [DOI] [PubMed] [Google Scholar]
- 93.Rada-Iglesias A, Grosveld FG & Papantonis A Forces driving the three-dimensional folding of eukaryotic genomes. Mol. Syst. Biol 14, e8214 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zheng H & Xie W The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol 20, 535–550 (2019). [DOI] [PubMed] [Google Scholar]
- 95.Oudelaar AM & Higgs DR The relationship between genome structure and function. Nat. Rev. Genet 22, 154–168 (2020). [DOI] [PubMed] [Google Scholar]
- 96.Lieberman-Aiden E et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dixon JR et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hildebrand EM & Dekker J Mechanisms and functions of chromosome compartmentalization. Trends Biochem. Sci 45, 385–396 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schoenfelder S & Fraser P Long-range enhancer-promoter contacts in gene expression control. Nat. Rev. Genet 20, 437–455 (2019). [DOI] [PubMed] [Google Scholar]
- 100.Finn EH & Misteli T Molecular basis and biological function of variability in spatial genome organization. Science 365, eaaw9498 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mir M, Bickmore W, Furlong EEM & Narlikar G Chromatin topology, condensates and gene regulation: shifting paradigms or just a phase? Development 146, dev182766 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ghavi-Helm Y Functional consequences of chromosomal rearrangements on gene expression: not so deleterious after all? J. Mol. Biol 432, 665–675 (2020). [DOI] [PubMed] [Google Scholar]
- 103.Nora EP et al. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell 169, 930–944.e22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rao SSP et al. Cohesin loss eliminates all loop domains. Cell 171, 305–320.e24 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang J et al. MIR retrotransposon sequences provide insulators to the human genome. Proc. Natl Acad. Sci. USA 112, E4428–E4437 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cournac A, Koszul R & Mozziconacci J The 3D folding of metazoan genomes correlates with the association of similar repetitive elements. Nucleic Acids Res 44, 245–255 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang Y et al. Transcriptionally active HERV-H retrotransposons demarcate topologically associating domains in human pluripotent stem cells. Nat. Genet 51, 1380–1388 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hendrickson PG et al. Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL/HERVL retrotransposons. Nat. Genet 49, 925–934 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kruse K et al. Transposable elements drive reorganisation of 3D chromatin during early embryogenesis. bioRxiv 10.1101/523712 (2019). [DOI] [Google Scholar]
- 110.Ghirlando R & Felsenfeld G CTCF: making the right connections. Genes Dev 30, 881–891 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schmidt D et al. Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Cell 148, 335–348 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Harmston N et al. Topologically associating domains are ancient features that coincide with Metazoan clusters of extreme noncoding conservation. Nat. Commun 8, 441 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Choudhary MN et al. Co-opted transposons help perpetuate conserved higher-order chromosomal structures. Genome Biol 21, 16 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kaaij LJT, Mohn F, van der Weide RH, de Wit E & Bühler M The ChAHP complex counteracts chromatin looping at CTCF sites that emerged from SINE expansions in mouse. Cell 178, 1437–1451.e14 (2019). [DOI] [PubMed] [Google Scholar]
- 115.Lunyak VV et al. Developmentally regulated activation of a SINE B2 repeat as a domain boundary in organogenesis. Science 317, 248–251 (2007). [DOI] [PubMed] [Google Scholar]
- 116.Ichiyanagi T et al. B2 SINE copies serve as a transposable boundary of DNA methylation and histone modifications in the mouse. Mol. Biol. Evol 38, 2380–2395 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kirkland JG, Raab JR & Kamakaka RT TFIIIC bound DNA elements in nuclear organization and insulation. Biochim. Biophys. Acta 1829, 418–424 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kapusta A et al. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS Genet 9, e1003470 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kelley D & Rinn J Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol 13, R107 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Johnson R & Guigó R The RIDL hypothesis: transposable elements as functional domains of long noncoding RNAs. RNA 20, 959–976 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kapusta A & Feschotte C Volatile evolution of long noncoding RNA repertoires: mechanisms and biological implications. Trends Genet 30, 439–452 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fort A et al. Deep transcriptome profiling of mammalian stem cells supports a regulatory role for retrotransposons in pluripotency maintenance. Nat. Genet 46, 558–566 (2014). [DOI] [PubMed] [Google Scholar]
- 123.Durruthy-Durruthy J et al. The primate-specific noncoding RNA HPAT5 regulates pluripotency during human preimplantation development and nuclear reprogramming. Nat. Genet 48, 44–52 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wilson KD et al. Endogenous retrovirus-derived lncRNA BANCR promotes cardiomyocyte migration in humans and non-human primates. Dev. Cell 54, 694–709.e9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wang Y et al. Endogenous miRNA sponge lincRNA-RoR regulates Oct4, Nanog, and Sox2 in human embryonic stem cell self-renewal. Dev. Cell 25, 69–80 (2013). [DOI] [PubMed] [Google Scholar]
- 126.Ohnuki M et al. Dynamic regulation of human endogenous retroviruses mediates factor-induced reprogramming and differentiation potential. Proc. Natl Acad. Sci. USA 111, 12426–12431 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jachowicz JW et al. LINE-1 activation after fertilization regulates global chromatin accessibility in the early mouse embryo. Nat. Genet 49, 1502–1510 (2017). [DOI] [PubMed] [Google Scholar]
- 128.Percharde M et al. A LINE1-nucleolin partnership regulates early development and ESC identity. Cell 174, 391–405.e19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Vihervaara A, Duarte FM & Lis JT Molecular mechanisms driving transcriptional stress responses. Nat. Rev. Genet 19, 385–397 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zovoilis A, Cifuentes-Rojas C, Chu H-P, Hernandez AJ & Lee JT Destabilization of B2 RNA by EZH2 activates the stress response. Cell 167, 1788–1802.e13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hernandez AJ et al. B2 and ALU retrotransposons are self-cleaving ribozymes whose activity is enhanced by EZH2. Proc. Natl Acad. Sci. USA 117, 415–425 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Allen TA, Von Kaenel S, Goodrich JA & Kugel JF The SINE-encoded mouse B2 RNA represses mRNA transcription in response to heat shock. Nat. Struct. Mol. Biol 11, 816–821 (2004). [DOI] [PubMed] [Google Scholar]
- 133.Espinoza CA, Allen TA, Hieb AR, Kugel JF & Goodrich JA B2 RNA binds directly to RNA polymerase II to repress transcript synthesis. Nat. Struct. Mol. Biol 11, 822–829 (2004). [DOI] [PubMed] [Google Scholar]
- 134.Cheng Y et al. Increased processing of SINE B2 ncRNAs unveils a novel type of transcriptome deregulation in amyloid beta neuropathology. eLife 9, e61265 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mariner PD et al. Human Alu RNA is a modular transacting repressor of mRNA transcription during heat shock. Mol. Cell 29, 499–509 (2008). [DOI] [PubMed] [Google Scholar]
- 136.Cosby RL et al. Recurrent evolution of vertebrate transcription factors by transposase capture. Science 371, eabc6405 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cordaux R, Udit S, Batzer MA & Feschotte C Birth of a chimeric primate gene by capture of the transposase gene from a mobile element. Proc. Natl Acad. Sci. USA 103, 8101–8106 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lee S-H et al. The SET domain protein Metnase mediates foreign DNA integration and links integration to nonhomologous end-joining repair. Proc. Natl Acad. Sci. USA 102, 18075–18080 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tellier M & Chalmers R Human SETMAR is a DNA sequence-specific histone-methylase with a broad effect on the transcriptome. Nucleic Acids Res 47, 122–133 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bailey AD et al. The conserved Cockayne syndrome B-piggyBac fusion protein (CSB-PGBD3) affects DNA repair and induces both interferon-like and innate antiviral responses in CSB-null cells. DNA Repair 11, 488–501 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Breitling R & Gerber JK Origin of the paired domain. Dev. Genes Evol 210, 644–650 (2000). [DOI] [PubMed] [Google Scholar]
- 142.Goodier JL Restricting retrotransposons: a review. Mob. DNA 7, 16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Molaro A & Malik HS Hide and seek: how chromatin-based pathways silence retroelements in the mammalian germline. Curr. Opin. Genet. Dev 37, 51–58 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cosby RL, Chang N-C & Feschotte C Host-transposon interactions: conflict, cooperation, and cooption. Genes Dev 33, 1098–1116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Smith ZD et al. DNA methylation dynamics of the human preimplantation embryo. Nature 511, 611–615 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Deniz Ö, Frost JM & Branco MR Regulation of transposable elements by DNA modifications. Nat. Rev. Genet 20, 417–431 (2019). [DOI] [PubMed] [Google Scholar]
- 147.Miranda TB & Jones PA DNA methylation: the nuts and bolts of repression. J. Cell. Physiol 213, 384–390 (2007). [DOI] [PubMed] [Google Scholar]
- 148.De la Rica L et al. TET-dependent regulation of retrotransposable elements in mouse embryonic stem cells. Genome Biol 17, 234 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Elmer JL & Ferguson-Smith AC Strain-specific epigenetic regulation of endogenous retroviruses: the role of trans-acting modifiers. Viruses 12, E810 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Waterland RA & Jirtle RL Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol. Cell. Biol 23, 5293–5300 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Whitelaw E & Martin DI Retrotransposons as epigenetic mediators of phenotypic variation in mammals. Nat. Genet 27, 361–365 (2001). [DOI] [PubMed] [Google Scholar]
- 152.Bertozzi TM & Ferguson-Smith AC Metastable epialleles and their contribution to epigenetic inheritance in mammals. Semin. Cell Dev. Biol 97, 93–105 (2020). [DOI] [PubMed] [Google Scholar]
- 153.Duhl DMJ, Vrieling H, Miller KA, Wolff GL & Barsh GS Neomorphic agouti mutations in obese yellow mice. Nat. Genet 8, 59–65 (1994). [DOI] [PubMed] [Google Scholar]
- 154.Dickies MM A new viable yellow mutation in the house mouse. J. Hered 53, 84–86 (1962). [DOI] [PubMed] [Google Scholar]
- 155.Faulk C, Barks A & Dolinoy DC Phylogenetic and DNA methylation analysis reveal novel regions of variable methylation in the mouse IAP class of transposons. BMC Genomics 14, 48 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kazachenka A et al. Identification, characterization, and heritability of murine metastable epialleles: implications for non-genetic inheritance. Cell 175, 1259–1271 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kessler NJ, Waterland RA, Prentice AM & Silver MJ Establishment of environmentally sensitive DNA methylation states in the very early human embryo. Sci. Adv 4, eaat2624 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bertozzi TM, Elmer JL, Macfarlan TS & Ferguson-Smith AC KRAB zinc finger protein diversification drives mammalian interindividual methylation variability. Proc. Natl Acad. Sci. USA 117, 31290–31300 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Silver MJ et al. Independent genomewide screens identify the tumor suppressor VTRNA2–1 as a human epiallele responsive to periconceptional environment. Genome Biol 16, 118 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Karimi MM et al. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell 8, 676–687 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ito J et al. Endogenous retroviruses drive KRAB zinc-finger protein family expression for tumor suppression. Sci. Adv 6, abc3020 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Timms RT, Tchasovnikarova IA & Lehner PJ Position-effect variegation revisited: HUSHing up heterochromatin in human cells. BioEssays 38, 333–343 (2016). [DOI] [PubMed] [Google Scholar]
- 163.Liu N et al. Selective silencing of euchromatic L1s revealed by genome-wide screens for L1 regulators. Nature 553, 228–232 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Robbez-Masson L et al. The HUSH complex cooperates with TRIM28 to repress young retrotransposons and new genes. Genome Res 28, 836–845 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Saint-André V, Batsché E, Rachez C & Muchardt C Histone H3 lysine 9 trimethylation and HP1γ favor inclusion of alternative exons. Nat. Struct. Mol. Biol 18, 337–344 (2011). [DOI] [PubMed] [Google Scholar]
- 166.Tunbak H et al. The HUSH complex is a gatekeeper of type I interferon through epigenetic regulation of LINE-1s. Nat. Commun 11, 5387 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lynch M The Origins of Genome Architecture (Sinauer Associates Inc., 2007). [Google Scholar]
- 168.Lowe CB & Haussler D 29 mammalian genomes reveal novel exaptations of mobile elements for likely regulatory functions in the human genome. PLoS One 7, e43128 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ting CN, Rosenberg MP, Snow CM, Samuelson LC & Meisler MH Endogenous retroviral sequences are required for tissue-specific expression of a human salivary amylase gene. Genes Dev 6, 1457–1465 (1992). [DOI] [PubMed] [Google Scholar]
- 170.Samuelson LC, Phillips RS & Swanberg LJ Amylase gene structures in primates: retroposon insertions and promoter evolution. Mol. Biol. Evol 13, 767–779 (1996). [DOI] [PubMed] [Google Scholar]
- 171.Gerlo S, Davis JRE, Mager DL & Kooijman R Prolactin in man: a tale of two promoters. BioEssays 28, 1051–1055 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Emera D et al. Convergent evolution of endometrial prolactin expression in primates, mice, and elephants through the independent recruitment of transposable elements. Mol. Biol. Evol 29, 239–247 (2012). [DOI] [PubMed] [Google Scholar]
- 173.Lynch VJ Ancient transposable elements transformed the uterine regulatory landscape and transcriptome during the evolution of mammalian pregnancy. Cell Rep 10, 551–561 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lynch VJ, Leclerc RD, May G & Wagner GP Transposon-mediated rewiring of gene regulatory networks contributed to the evolution of pregnancy in mammals. Nat. Genet 43, 1154–1159 (2011). [DOI] [PubMed] [Google Scholar]
- 175.Dunn-Fletcher CE et al. Anthropoid primate-specific retroviral element THE1B controls expression of CRH in placenta and alters gestation length. PLoS Biol 16, e2006337 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sun M et al. Endogenous retroviruses drive lineage-specific regulatory evolution across primate and rodent placentae. Mol. Biol. Evol 38, 4992–5004 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Prescott SL et al. Enhancer divergence and cis-regulatory evolution in the human and chimp neural crest. Cell 163, 68–83 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Nishihara H Coordinately co-opted multiple transposable elements constitute an enhancer for wnt5a expression in the mammalian secondary palate. PLoS Genet 12, 1006380 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.ENCODE Project Consortium. et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Long HK et al. Loss of extreme long-range enhancers in human neural crest drives a craniofacial disorder. Cell Stem Cell 27, 765–783.e14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Payer LM & Burns KH Transposable elements in human genetic disease. Nat. Rev. Genet 20, 760–772 (2019). [DOI] [PubMed] [Google Scholar]
- 182.Sudmant PH An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Tam OH, Ostrow LW & Gale Hammell M Diseases of the nERVous system: retrotransposon activity in neurodegenerative disease. Mob. DNA 10, 32 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hancks DC & Kazazian HH Roles for retrotransposon insertions in human disease. Mob. DNA 7, 9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Gymrek M et al. Abundant contribution of short tandem repeats to gene expression variation in humans. Nat. Genet 48, 22–29 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Spirito G, Mangoni D, Sanges R & Gustincich S Impact of polymorphic transposable elements on transcription in lymphoblastoid cell lines from public data. BMC Bioinform 20, 495 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wang L, Norris ET & Jordan IK Human retrotransposon insertion polymorphisms are associated with health and disease via gene regulatory phenotypes. Front. Microbiol 8, 1418 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Goubert C, Zevallos NA & Feschotte C Contribution of unfixed transposable element insertions to human regulatory variation. Philos. Trans. R. Soc. B Biol. Sci 375, 20190331 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Payer LM Structural variants caused by Alu insertions are associated with risks for many human diseases. Proc. Natl Acad. Sci. USA 114, 3984–3992 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Marchi E, Kanapin A, Magiorkinis G & Belshaw R Unfixed endogenous retroviral insertions in the human population. J. Virol 88, 9529–9537 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Turner G Insertional polymorphisms of full-length endogenous retroviruses in humans. Curr. Biol 11, 1531–1535 (2001). [DOI] [PubMed] [Google Scholar]
- 192.Wallace AD et al. To ERV is human: a phenotype-wide scan linking polymorphic human endogenous retrovirus-K insertions to complex phenotypes. Front. Genet 9, 298 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Sekar A Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Karamitros T Human endogenous retrovirus-K HML-2 integration within RASGRF2 is associated with intravenous drug abuse and modulates transcription in a cell-line model. Proc. Natl Acad. Sci. USA 115, 10434–10439 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Osterwalder M et al. Enhancer redundancy provides phenotypic robustness in mammalian development. Nature 554, 239–243 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Nowak MA, Boerlijst MC, Cooke J & Smith JM Evolution of genetic redundancy. Nature 388, 167–171 (1997). [DOI] [PubMed] [Google Scholar]
- 197.Cannavò E et al. Shadow enhancers are pervasive features of developmental regulatory networks. Curr. Biol 26, 38–51 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Frankel N et al. Phenotypic robustness conferred by apparently redundant transcriptional enhancers. Nature 466, 490–493 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Farley EK et al. Suboptimization of developmental enhancers. Science 350, 325–328 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Greenberg MVC & Bourc’his D The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol 20, 590–607 (2019). [DOI] [PubMed] [Google Scholar]
- 201.Zemojtel T et al. CpG deamination creates transcription factor-binding sites with high efficiency. Genome Biol. Evol 3, 1304–1311 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Gasperini M et al. A genome-wide framework for mapping gene regulation via cellular genetic screens. Cell 176, 377–390 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Fulco CP et al. Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat. Genet 51, 1664–1669 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Mifsud B et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat. Genet 47, 598–606 (2015). [DOI] [PubMed] [Google Scholar]
- 205.He J et al. Identifying transposable element expression dynamics and heterogeneity during development at the single-cell level with a processing pipeline scTE. Nat. Commun 12, 1456 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Shao W & Wang T Transcript assembly improves expression quantification of transposable elements in single-cell RNA-seq data. Genome Res 31, 88–100 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Chang N-C, Rovira Q, Wells JN, Feschotte C & Vaquerizas JM A genomic portrait of zebrafish transposable elements and their spatiotemporal embryonic expression. Genome Res 10.1101/gr.275655.121 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Du J et al. Chromatin variation associated with liver metabolism is mediated by transposable elements. Epigenetics Chromatin 9, 28 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Hummel B The evolutionary capacitor HSP90 buffers the regulatory effects of mammalian endogenous retroviruses. Nat. Struct. Mol. Biol 24, 234–242 (2017). [DOI] [PubMed] [Google Scholar]
- 210.Li W et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci. Transl. Med 7, 307ra153 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Turelli P et al. Primate-restricted KRAB zinc finger proteins and target retrotransposons control gene expression in human neurons. Sci. Adv 6, eaba3200 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Padmanabhan Nair V et al. Activation of HERV-K(HML-2) disrupts cortical patterning and neuronal differentiation by increasing NTRK3. Cell Stem Cell 28, 1566–1581.e8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Berwaer M, Martial JA & Davis JR Characterization of an up-stream promoter directing extrapituitary expression of the human prolactin gene. Mol. Endocrinol 8, 635–642 (1994). [DOI] [PubMed] [Google Scholar]
- 214.Gellersen B, Kempf R, Telgmann R & DiMattia GE Nonpituitary human prolactin gene transcription is independent of Pit-1 and differentially controlled in lymphocytes and in endometrial stroma. Mol. Endocrinol 8, 356–373 (1994). [DOI] [PubMed] [Google Scholar]
- 215.Jacob KK & Stanley FM CCAAT/enhancer-binding protein alpha is a physiological regulator of prolactin gene expression. Endocrinology 140, 4542–4550 (1999). [DOI] [PubMed] [Google Scholar]
- 216.Lynch VJ, Brayer K, Gellersen B & Wagner GP HoxA-11 and FOXO1A cooperate to regulate decidual prolactin expression: towards inferring the core transcriptional regulators of decidual genes. PLoS One 4, e6845 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Brar AK, Kessler CA & Handwerger S An Ets motif in the proximal decidual prolactin promoter is essential for basal gene expression. J. Mol. Endocrinol 29, 99–112 (2002). [DOI] [PubMed] [Google Scholar]
- 218.Ishida M et al. Involvement of cAMP response element-binding protein in the regulation of cell proliferation and the prolactin promoter of lactotrophs in primary culture. Am. J. Physiol. Endocrinol. Metab 293, E1529–E1537 (2007). [DOI] [PubMed] [Google Scholar]
- 219.Schulte AM et al. Influence of the human endogenous retrovirus-like element HERV-E.PTN on the expression of growth factor pleiotrophin: a critical role of a retroviral Sp1-binding site. Oncogene 19, 3988–3998 (2000). [DOI] [PubMed] [Google Scholar]
- 220.Ling J et al. The solitary long terminal repeats of ERV-9 endogenous retrovirus are conserved during primate evolution and possess enhancer activities in embryonic and hematopoietic cells. J. Virol 76, 2410–2423 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Pi W et al. Long-range function of an intergenic retrotransposon. Proc. Natl Acad. Sci. USA 107, 12992–12997 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Zhang W et al. Zscan4c activates endogenous retrovirus MERVL and cleavage embryo genes. Nucleic Acids Res 47, 8485–8501 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Macfarlan TS et al. Endogenous retroviruses and neighboring genes are coordinately repressed by LSD1/KDM1A. Genes Dev 25, 594–607 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Arnold CD et al. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 10.1126/science.1232542 (2013). [DOI] [PubMed] [Google Scholar]