

Summary
Recent studies suggest that infection reprograms hematopoietic stem and progenitor cells (HSPCs) to enhance innate immune responses upon secondary infectious challenge, a process called “trained immunity.” However, the specificity and cell types responsible for this response remain poorly defined. We established a model of trained immunity in mice in response to Mycobacterium avium infection. scRNA-seq analysis revealed that HSPCs activate interferon gamma-response genes heterogeneously upon primary challenge, while rare cell populations expand. Macrophages derived from trained HSPCs demonstrated enhanced bacterial killing and metabolism, and a single dose of recombinant interferon gamma exposure was sufficient to induce similar training. Mice transplanted with influenza-trained HSPCs displayed enhanced immunity against M. avium challenge and vice versa, demonstrating cross protection against antigenically distinct pathogens. Together, these results indicate that heterogeneous responses to infection by HSPCs can lead to long-term production of bone marrow derived macrophages with enhanced function and confer cross-protection against alternative pathogens.
Subject areas: Cell biology, Immunology, Microbiology, Stem cells research, Transcriptomics
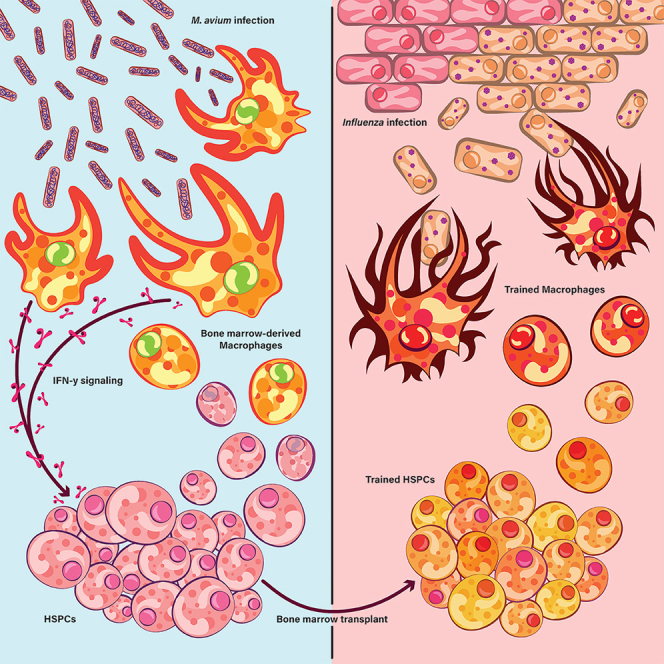
Graphical abstract
Highlights
-
•
Mycobacterium avium exposure induces HSPC-encoded trained immunity
-
•
Transcriptional responses to infection are heterogeneous among HSPCs
-
•
rIFNγ HSPC training induces increased BMDM killing and lasting metabolic rewiring
-
•
HSPC-encoded M. avium trained immunity cross-protects against influenza
Cell biology; Immunology; Microbiology; Stem cells research; Transcriptomics
Introduction
Over the past decade, innate immune cells have been shown to have immunological memory, mounting enhanced responses to infectious pathogens upon secondary challenge. This phenomenon, termed innate immune memory, has been observed in monocytes, macrophages, natural killer (NK) cells, dendritic cells (DCs) and neutrophils.1 Functional phenotypes affiliated with trained immunity include increased pathogen clearance and cytokine production upon secondary challenge.1,2
Innate immune memory likely underlies nonspecific cross-protection from vaccines, as documented in many human studies.3,4,5,6 For example, children who have been vaccinated with Bacillus Calmette–Guérin (BCG), MMR, or polio vaccines have significantly reduced morbidity and mortality from unrelated respiratory pathogens.3 Despite this phenomenon’s importance, much remains to be discovered about which cells encode immunological memory, the mechanisms underlying memory, and the specificity of the trained immune response.
Although innate immune memory phenotypes were originally described in terminally differentiated innate immune cells, these cells are typically short-lived, prompting the hypothesis that hematopoietic stem and progenitor cells (HSPCs) serve as the reservoir for long term innate immune memory.1,7 Recent studies confirm that HSPCs may encode trained immunity and that epigenetically reprogramming HSPCs can affect myelopoiesis and myeloid cell function. In one study, transferring trained HSPCs into naive mice was sufficient to confer immunity against Mycobacterium tuberculosis via an interferon gamma (IFNγ) signaling-dependent process.8 Trained immunity encoded by stem and progenitor cells in the bone marrow is now defined as “central trained immunity” to distinguish it from “peripheral trained immunity,” which encompasses enhanced immune function by innate effector cells at the infection site.9
Mechanistic studies suggest that trained immunity phenotypes are attributable to epigenetic reprogramming in hematopoietic cells after primary stimulation.7,10 In particular, lineage-negative, Sca1-positive, cKit-positive (LSK) cells—which represent a diverse amalgam of short and long-term hematopoietic stem cells and multipotent progenitors—have been shown to accumulate H3K9me3, H3K9me, and H3K27ac in promoter and enhancer regions of immunity-related genes. These activating histone modifications lead to heightened immune responses upon secondary challenge.10,11 However, much remains to be learned about how specific stem cell and multipotent progenitor populations contribute to protective trained immunity.
We previously investigated how persistent inflammation—specifically, persistent IFNγ signaling produced during chronic Mycobacterium avium infection—leads to changes in HSC quiescence, self-renewal, and differentiation.12,13 Chronic infection causes HSPCs to differentiate with a myeloid bias and lose self-renewal capacity.12,14,15,16 Given IFNγ signaling’s importance in trained immunity, we used our M. avium infection model to study outstanding questions in the trained immunity field. To evaluate the cellular basis for central trained immunity, we surveyed IFNγ-mediated transcriptional responses among HSPCs using scRNA-seq analysis. We tested whether transient IFNγ is sufficient to induce enhanced HSPC-encoded immunity and tested the specificity of M. avium-induced trained immunity by challenging chimeric mice with influenza A, a common respiratory pathogen. Altogether, we show that subsets of multiple HSPC compartments respond transcriptionally to M. avium infection without a dominant responding cell type. We provide in vivo evidence that epigenetic and transcriptomic changes in HSPCs in response to IFNγ alone are sufficient to induce lasting changes in macrophage killing and metabolism, and that training by one pathogen can be cross-protective against an unrelated secondary pathogen.
Results
Transplanting HSPCs exposed to M. avium confers in vivo protection to recipient mice up to one year post training stimulus
Prior studies have demonstrated that HSPCs exposed to BCG confer protection against a subsequent M. tuberculosis infection via an IFNγ-dependent process.8 To evaluate whether HSPCs previously exposed to systemic M. avium infection protect recipient mice from subsequent infectious challenge, we isolated c-Kit+ HSPCs from mice systemically infected or “trained” with M. avium one month prior and transplanted them into naive sub-lethally irradiated recipient mice. A separate control mouse cohort received c-Kit+ cells from uninfected mice (Figure 1A). To minimize transmission of the primary M. avium infection, we treated donor cells with clarithromycin prior to transplant; there were no signs of infection in the recipient unchallenged mice at the experimental endpoint (e.g., they retained normal spleen size) (Figures 1C and S1C). Twelve weeks after transplant, engraftment was slightly lower in mice receiving trained versus untrained cells, consistent with a known engraftment defect in HSPCs from M. avium-infected mice (Figures S1A and S1B).14 We then mock PBS challenged (“control”) or challenged (“control infected”) the mice with M. avium by intravenous injection—including an additional set of control mice that did not undergo bone marrow transplant—and assessed for disease outcome four weeks later (Figure 1A). The primary outcome was bacterial load in the spleen, the central reservoir for infection in systemically infected mice. Transplanting untrained cells did not affect splenic bacterial load compared to controls; however, transferring trained HSPCs significantly decreased splenic bacterial loads and overall splenomegaly (Figures 1B, 1C, and S1C). H&E-stained splenic sections comparing disease histopathology between experimental groups revealed a significant reduction in inflammation, destruction of germinal centers, and total granulomas in spleens in animals receiving M. avium-trained HSPCs, which correlated with reduced overall spleen size (Figures 1D and 1E).
Figure 1.

Transplant of M. avium-exposed HSPCs confers immunity to recipient mice following M. avium challenge
(A) Chimeric mouse model of M. avium trained immunity experiments.
(B) Splenic bacterial load in experimental mice one month post M. avium challenge. Results representative of 3 independent experiments, n = 5–9 per experimental group.
(C) Representative image of spleens one-month post-challenge.
(D) Quantification of splenic granulomas per stitched 10x image of longitudinal spleen section. Results representative of 2 independent experiments, n = 3–5 per group.
(E) Splenic histology one month post M. avium infection. 10x magnification, Images representative of 3 independent experiments, scale bar 210μm.
(F–H) Serum cytokine levels of transplant recipients one month post M. avium challenge.
(I–K) Whole bone flow cytometry of transplant, challenged experiments. (I) Percentage of CD45.2 LT-HSCs (Lin−,ckit+,CD150+,CD48-,Flk2-,CD34-), (J) CD45.2 MPP2 (Lin−,ckit+, CD150+,CD48+,Flk2-,CD34+), and (K) CD45.2 MPP3 (Lin−, ckit+, CD150-,CD48+,Flk2-,CD34+) in transplanted recipients post M. avium challenge.
(L) Representative image of spleens harvested from naive and challenged experimental mice rested for six months post-transplant.
(M) Splenic bacterial load in experimental mice one month post M. avium challenge in trained M. avium HSPC recipients that had been rested for six months post-transplant. n = 4–6 per experimental group.
(N) Representative image of spleens harvested from challenged experimental mice rested for 1 year post-transplant.
(O) Splenic bacterial load in experimental M. avium trained or untrained mice recipients that had been rested for 1 year post-transplant. n = 3–4 per experimental group.
(P) Volcano plot representation of differentially expressed genes of CD45.2 untrained vs. trained HSPCs (LK CD150+, CD48−) recovered from transplanted recipients 1 month following M. avium infection.
(Q) Gene ontology analysis of genes upregulated in challenged M. avium trained HSPCs, showing an enrichment of metabolism related pathways.
(R) Gene Set Enrichment Analysis (GSEA) results for Hallmark pathways comparing gene expression in untrained and trained HSPCs. For comparisons of two groups, statistics were calculated using Student’s t test. For comparisons of more than two groups, one-way ANOVA with Tukey’s multiple comparisons was completed. ∗p < 0.05, ∗∗p < 0.01 ∗∗∗p < 0.001 ∗∗∗∗p < 0.0001. Analysis, cutoff for bulk RNA-seq analysis was FDR <0.1 for a log-fold change of < -1.5 or >1.5.
Mice that received trained HSPCs had reduced systemic inflammation in both serum cytokine levels and circulating immune cells. Whereas IFNγ, TNFα, and IL-6 serum levels were elevated in control mice and mice that received untrained HSPCs (Figures 1F and 1H), these inflammatory cytokine levels were near normal in mice that received trained HSPCs. Mice receiving trained HSPCs also demonstrated normal or near-normal peripheral blood cell distribution compared to mice receiving untrained HSPCs or control infected animals (Figures S1D–S1G). Mice receiving trained HSPCs had significantly increased donor-derived LT-HSCs, MPP2, and MPP3 cells but no difference in ST-HSCs (MPP1) or lymphoid biased MPP4 progenitors (Figures 1I, 1K, S1H, and S1I), which is consistent with reports in other trained immunity models.8,10,17,18
In mice that received trained HSPCs, the percentage of Ly6c+ inflammatory monocytes in the bone marrow was significantly reduced, B cell levels were increased, and T cells were unchanged (Figures S1J–S1L). When we used NIH Ag85240-254 tetramer to assess M. avium-specific splenic T cells via flow cytometry, we found a significant reduction in tetramer-positive CD4+ T cells in both percentage and number in trained HSPC recipients (Figures S1M–S1O). Of the tetramer-positive T cells, most were Foxp3+ whereas few were Tbet+ or IFNγ-expressing T cells (Figures S1P–S1R). Altogether, these findings suggest that the reduction in bacterial load in mice receiving trained HSPCs was not principally attributable to an adaptive immune response.
To test M. avium induced HSPC trained immunity’s longevity, we rested transplanted chimeric mice for up to one year post transplantation and then challenged them with M. avium infection. M. avium trained HSPC recipients exhibited a significant reduction in bacterial burden at both six months and one year and a reduction in overall splenomegaly (Figures 1L and 1O). These results demonstrate that transferring cKit+ cells from M. avium-infected mice confers enhanced immunity to M. avium in transplant recipients and are consistent with a trained immunity phenotype that is encoded within the HSPC compartment and durable up to 1-year post transplant.
M. avium exposure durably alters transcription of genes related to immunity and metabolism
Prior studies investigating trained immunity in mycobacterial and fungal infection settings have identified epigenetic modifications and corresponding transcriptional changes in trained macrophages.10,17 To evaluate how prior training affects transcriptional responses, we re-isolated CD45.2 untrained vs. trained HSCs (LK CD150+CD48–) from transplanted mice one month after M. avium challenge to generate bulk RNA-Seq libraries. Comparing the transcriptional response of trained versus untrained CD45.2 HSCs after challenge revealed global transcriptional changes, many more genes (670) were upregulated upon secondary infection in the trained versus untrained population (255) (Figure 1P). Strikingly, gene set enrichment analysis (GSEA) of the genes uniquely upregulated upon infectious challenge in the trained HSCs aligned with GCPR signaling and metabolic processes, which are reportedly rewired in macrophages in other trained immunity models (Figure 1Q). Specifically, oxidative phosphorylation, adipogenesis, fatty acid oxidation, and glycolysis were enhanced in the trained HSCs (Figure 1R). Together, these analyses reveal that, even after transplant into a naive host, trained HSCs have altered responses to infection at one month post-challenge compared to untrained controls and significantly upregulated metabolic pathway genes.
M. avium exposure induces heterogeneous responses in HSPCs which are conserved in innate immune cells
Having confirmed that transferring HSPCs is sufficient to confer enhanced immunity to naive recipient mice, we sought to understand the transcriptional landscape of HSPCs after infection to gain mechanistic insight. We therefore performed single-cell RNA sequencing on HSPCs from M. avium-infected and control mice (Figure 2A). To ensure adequate representation of rare cell types, we sorted phenotypically defined LT-HSCs, CD41+ HSCs, MPP3, GMP, inflammatory macrophages, neutrophils, and B cells from M. avium- or mock-infected mice’s bone marrow according to the definitions listed in Figure S2A. CD41+ HSCs are a subset of HSCs that respond rapidly to inflammatory stimulation and differentiate with a myeloid bias (Gekas, 2013). While this sorting approach may obscure shifts in cell populations, it enables us to robustly evaluate transcriptional responses in progenitor and myeloid marrow components, even if they are rare. Cells were clustered and assigned identities based on manually curated gene lists and the scCATCH package (Figure 2B). UMAP plots showed that primitive HSC populations clustered together while MPP3 and myeloid progenitors were situated between the HSCs and differentiated myeloid cells as expected (Figure 2B). Cells from all primitive cell populations heterogeneously expressed IFNγ-response genes known to be induced upon M. avium challenge, like Batf2, Cxcl9, and Ccl5, and a large subset of cells showed no expression while a small subset showed high expression (Figures 2C, 2E, and S2B). Responsive cells were scattered across all progenitor populations indicating that no particular cell identity was poised to express these genes. The total percentage of IFNγ-responsive cells ranged from 10 to 25% among LT-HSCs, CD41+ HSCs, and ST-HSCs (Figure 2F). By analyzing Xist expression, we confirmed that the heterogeneity observed upon M. avium stimulation was not due to sex bias and that both males and females were represented in the pooled cell populations as expected across the single cell dataset (Figures S2C and S2D).
Figure 2.

M. avium exposure induces heterogeneous responses within HSPCs
(A) Experimental schematic of single cell RNAseq library generation from control or M. avium exposed bone marrow one month post infection. Cells were isolated by FACS sorting and pooled for sequencing using the 10x genomics platform.
(B) UMAP plot representing cell populations as identified by characteristic gene expression using scCATCH and curated gene lists.
(C) UMAP plots of gene expression in naive or M. avium infected samples. Relative gene expression is represented by corresponding color scale.
(D and E) Raincloud diagrams showing “non-responder” vs. “responder” single cell gene expression. (D) Batf2 expression by cell type (E) Cxcl9 expression by cell type.
(F) Percentages of cells expressing IFNγ response genes Batf2, Cxcl9, and Ccl5 by cell type.
Like the HSC subsets, MPP3 myeloid-biased progenitors and terminally differentiated innate immune cells demonstrated significant heterogeneity in gene expression (Figure 2C). Indeed, Cxcl9, Batf2, and Ccl5 were strongly induced in a subset of monocyte/macrophages and neutrophils but not in mature B cells (Figures 2D, 2F, and S2B), indicating that the expression pattern in myeloid cells mirrored the bimodal transcriptional patterns observed in HSPCs. Differential expression analysis of naive and WT infected HSC populations revealed gene enrichment related to antigen processing and presentation, innate immune responses, and response to viruses as in previously reported bulk RNAseq data (Figures S2E–S2Q).
We generated an interferon score based on expression of genes that are typically upregulated during M. avium infection to assess responder cell percentage across populations (Figure S2R). We found a low correlation between the interferon response score and expression of cytokine receptors Ifngr1 or Tnfrsf1a (Ifngr1: R = 0.4, Tnfrsf1a: R = 0.3) (Figures S2S and S2T), suggesting that heterogeneous responses to infection were not attributable to differences in cytokine receptor expression. Together, these results suggest that transcriptional responses to M. avium infection, particularly IFNγ response genes, are heterogeneously induced across HSCs, MPPs, and terminally differentiated innate immune cells of the myeloid lineage. These findings imply that only a subset of the cells transferred in trained immunity transplant experiments may be responsible for the protective phenotype. Furthermore, they suggest that HSPC compartment training does not necessarily lead to changes in all the cells.
LT-HSCs do not transmit protective trained immune responses, but rare HSPC subpopulations expand during infection
It has been proposed that HSPCs are the key cells that encode a lasting and effective trained immune response for several months following training exposure.19 To test whether the long-term HSCs (LT-HSCs) that sit at the top of the hematopoietic hierarchy are sufficient to confer trained immunity, we transplanted 250 LT-HSCs (LK CD150+ CD48− CD34− ePCR+) from untrained or M. avium-trained mouse bone marrow one month post infection into naive recipient mice (Figure 3A). This number of LT-HSCs approximates the number that would have been contained among the 2x105 cKit+ cells transferred in the experiments shown in Figure 1. After confirming engraftment 12 weeks post-transplant (Figure S3A), we challenged the transplanted mice with M. avium and assessed disease morbidity. There was no difference in splenomegaly, splenic weight, or bacterial burden between untrained and trained LT-HSC recipient mice (Figure 3B, 3C, and S3B). Further, serum cytokine profiling of trained LT-HSC recipient mice showed no significant reduction in TNFα or IFNγ or improvement in granuloma burden compared to untrained controls (Figures S3C–S3E). These results suggest that LT-HSCs alone are not sufficient to confer trained immunity in this model.
Figure 3.

HSCs alone do not confer immune protection, but M. avium infection results in the emergence of a subpopulation of infection-activated HSCs (IA-HSCs)
(A) Mosaic model of M. avium trained LT-HSC transplant.
(B) Representative images showing splenomegaly in untrained and M. avium trained HSC recipients post M. avium challenge. Data are representative of two independent experiments with n = 6–12.
(C) Splenic bacterial load in untrained and M. avium trained HSC transplant recipients post M. avium challenge. n = 6–12, Statistics: Unpaired t-test.
(D) UMAP plots of scRNAseq data from BM cells of naive versus M. avium-infected mice.
(E) Top gene occupancy of genes expressed by emerging infection activated HSCs (IA-HSCs).
(F) UMAP representation of pseudotime analysis of combined naive and M. avium infected scRNA-seq datasets. Primitive cells with stem like properties are represented in red, while terminally differentiated immune cells are represented in blue. IA-HSCs circled in black.
(G) Mosaic mouse model of IA-HSC transplant recipients and experimental schematic.
(H) Engraftment percentage of CD45.2 donor cells at specific time points twelve weeks post-transplant. n = 6–10 biological replicates per group, statistics: One-way ANOVA with Tukey’s multiple comparisons.
(I) Lineage distribution of trained SLAM HSC and IA-HSC recipients. n = 6–10 biological replicates per group, statistics: One-way ANOVA with Tukey’s multiple comparisons. ∗p < 0.05, ∗∗p < 0.01 ∗∗∗p < 0.001 ∗∗∗∗p < 0.0001.
Notably, when comparing single-cell transcriptome data UMAP plots from control versus M. avium infected mice, we noted a cell population that expanded near the LT-HSC population (Figures 3D and 3E). This cell set expressed not only standard HSC markers (Sca, cKit, Slamf1/CD150) but also markers related to activation and immune responses to infection CD81,20 CD9,21 Coro1a, CD93,22,23,24 Ptprc,25 and markers associated with B cells CD24a26 and CD7227 (Figures 3F and S4A). CD81 and CD9 are tetraspanins associated with HSC re-entry to quiescence after stress,20,21,28 and Ptprc is involved in cell motility.29 This cell population identified in the scRNA-seq dataset comprised both male and female cells, indicating that these cells were not a single mouse’s artifact (Figures S3F and S3G). Interestingly, however, there were more cells of female origin than male, inverse to the input population. To reflect their unique gene expression pattern, we refer to these cells as infection-activated HSCs (IA-HSCs).
Pseudotime analysis of the single cell libraries showed IA-HSCs in closest proximity to LT-HSCs as opposed to terminally differentiated immune cells, which suggests highly overlapping gene expression profiles (Figures 3G, S3H, and S3I). Additionally, arranging cells into 7 lineage slings originating from primitive LT-HSCs showed that IA-HSCs reside only within the first lineage sling (Figures S3J and S3K). In a separate experiment, we performed flow cytometric analysis of WBM from M. avium-infected mice and found IA-HSCs (Lin− Sca+ CD24a+ CD81+ CD9+ to exist as 0.004% of nucleated WBM cells (standard deviation 6.8 x 10−6; n = 6) (Figure S4B). Only 0.7% of these IA-HSCs additionally met strict HSC criteria (CD150+, CD48−, CD34−, Flk2-). To confirm IA-HSCs’ stemness and self-renewal capacity, we transplanted 500 phenotypically defined IA-HSCs (Lin− c-kit+ Sca+ CD24a+ CD81+ CD9+) (Figure S3L) or SLAM HSCs (LK CD150+ CD48−) from control or M. avium-infected mice with CD45.1 rescue whole bone marrow (WBM) into lethally irradiated CD45.1 mice (Figure 3H). IA-HSCs demonstrated trilineage engraftment into donor mice at 12 weeks following transplantation (Figures 3I, 3J, and S3M–S3O). Thus, this rare population which expands in the bone marrow upon M. avium infection demonstrates long term engraftment capacity and trilineage multipotency. Future studies will be required to determine these cells’ significance and function.
M. avium trained HSPCs generate bone marrow derived macrophages with increased bacterial clearance ex vivo
To assess whether HSPC M. avium training leads to the production of macrophages with enhanced bacterial clearance, we isolated lineage-ckit+ HSPCs from control and M. avium trained CD45.2 mice and cultured bone marrow derived macrophages (BMDMs), verifying surface marker expression via flow cytometry (Figures 4A and S5A). Upon bacterial challenge, BMDMs derived from trained HSPCS demonstrated a trend toward increased bacterial load at 4 h post infection, suggesting normal to increased bacterial engulfment compared to untrained controls (Figure 4B). Importantly, BMDMs derived from M. avium trained HSPCs had significantly reduced bacterial load at 3 days post challenge, indicating increased bacterial killing capacity compared to untrained controls (Figure 4C). These results indicate that M. avium-trained BMDMs are capable of enhanced immune function independent of the adaptive immune system.
Figure 4.

M. avium trained HSPCS differentiate into macrophages that exhibit increased bacterial killing and metabolism
(A) Experimental scheme for generation of bone marrow derived macrophages (BMDM) from HSPCs (LK).
(B) M. avium bacterial CFU counts in BMDMs 4 h post M. avium challenge.
(C) M. avium bacterial CFU counts in BMDMs 3 days post M. avium challenge. n = 4 per group.
(D–F) Seahorse Mito Stress test of naive vs. M. avium trained BMDMs. n = 4–5 per group, data are representative of 2 independent experiments. (D) Metabolic trace of naive and M. avium trained BMDMs at baseline. (E) Basal respiration of naive vs. M. avium-trained BMDMs. (F) Maximal respiration of naive vs. M. avium trained BMDMs.
(G) Experimental scheme of rIFNγ training and BMDM generation from whole bone marrow (WBM). Mice were treated with rIFNγ 24 h prior to harvest of WBM for culture.
(H) M. avium bacterial CFU counts in BMDMs 4 h post challenge.
(I) M. avium bacterial CFU counts in BMDMs 3 days post challenge. n = 6; data are representative of three independent experiments.
(J–M) Seahorse Mito Stress test of naive vs. rIFNγ trained BMDMs. n = 6; data are representative of two independent experiments. (J) Metabolic trace of naive and rIFNγ-trained BMDMs at baseline. (K) Basal respiration of naive vs. rIFNγ-trained BMDMs. (L) Maximal respiration of naive vs. rIFNγ-trained BMDMs. (M) ATP production of naive vs. rIFNγ-trained BMDMs.
(N) Experimental scheme: Mice were trained for 24 h with rIFNγ and rested for seven days prior to harvest of WBM for BMDM culture.
(O) M. avium bacterial CFU counts in BMDMs 3 days post challenge. n = 3 per group; data are representative of two independent experiments. Statistics were calculated using Student’s t test for two groups or one-way ANOVA with Tukey’s multiple comparisons. ∗p < 0.05, ∗∗p < 0.01 ∗∗∗p < 0.001 ∗∗∗∗p < 0.0001.
Because trained immunity functional phenotypes are often affiliated with metabolic rewiring, we investigated whether macrophages derived from untrained and M. avium trained hematopoietic progenitor cells exhibited different metabolic phenotypes. Seahorse Mito stress tests of control and M. avium trained BMDMs demonstrated that M. avium-trained BMDMs had significantly increased basal respiration and maximal respiration, increased ATP production, and a likelihood of increased proton leak compared with untrained BMDMs (Figures 4D, 4F, and S5B–S5D). These results indicate increased metabolic function of trained BMDMs at baseline.
rIFNγ induces increased bacterial killing and metabolic rewiring of bone marrow-derived macrophages (BMDMs)
A 2018 study showed that systemic BCG infection induces trained immunity in hematopoietic progenitors via an IFNγ-dependent mechanism.8 Since IFNγ mediates many of systemic M. avium infection’s effects on HSCs, we evaluated whether prior exposure to IFNγ alone is sufficient to increase HSPC immune responses against M. avium infection. We injected mice with rIFNγ intravenously at a dose sufficient to induce cell cycle and gene expression changes in HSCs.12,14 Twenty-four hours later, we isolated whole bone marrow and cultivated BMDMs ex vivo (Figure 4G) and confirmed BMDM identity using surface marker expression. Challenging BMDMs from rIFNγ-treated mice with M. avium revealed similar bacterial uptake at 4 h (Figure 4H) but reduced CFU after 3 days compared to BMDMs from mice treated with PBS alone (Figure 4I), which points to enhanced bacterial killing. Basal respiration, maximal respiration, total ATP production, and spare respiratory capacity were significantly enhanced in BMDMs derived from IFNγ-treated mice compared to controls, indicating increased metabolism (Figures 4J–4M, S5E, and S5F). We saw similarly improved bacterial clearance when mice were rested for 7 days after IFNγ stimulation and prior to BM collection for BMDM culture, suggesting that the phenotype was not a result of acute activation alone (Figures 4N and 4O). Together, these data demonstrate that a single dose of systemic IFNγ is sufficient to functionally alter hematopoietic progenitors and aspects of their downstream immune progeny.
To test whether the functional differences induced by rIFNγ training were sufficient to confer host protection in vivo, we transplanted sub-lethally irradiated mice with HSPCs from mice that had been injected with a single dose of rIFNγ or PBS control. Twelve weeks after transplant, we challenged these mice with M. avium (Figure S5G). One dose of pro-inflammatory cytokine stimulus was insufficient to protect mice from subsequent M. avium challenge in terms of bacterial load or splenomegaly (Figures S5H–S5J). However, we detected increased cellular respiration and ATP production in BMDMs derived from rIFNγ trained M. avium challenged mice compared to controls even four months after rIFNγ training and transplant (Figures S5K–S5N). These results indicate that a single dose of IFNγ alone was insufficient to protect against subsequent infectious challenge, but it was sufficient to confer long lasting changes in metabolism in BMDMs.
Influenza training of HSPCs leads to subtle cross-protection against M. avium infection
While β-glucan can induce cross-protective trained immunity against TB and BCG vaccine can protect against influenza, much remains to be discovered about the extent to which training innate immune cells provides cross-protective immunity.30,31,32 Given that IFNγ alone is sufficient to induce metabolic characteristics of trained immunity, we hypothesized that Th1-inducing pathogens may generate trained immunity against other Th1-inducing pathogens. Thus, we tested whether influenza A infection training can confer cross-protection against M. avium infection. We assessed cytokine production and bacterial killing in BMDMs derived from the whole bone marrow of control or influenza A (H1N1 PR8) infected mice (Figure 5A). BMDMs from influenza trained bone marrow had increased bacterial engulfment and cytokine production including IL-4, TNFα, and IL-12 4 h post-infection, which indicates sustained effects (Figures 5B–5E). Unlike our other training models, however, there was no increase in bacterial killing at three days post infection (Figure S6A).
Figure 5.

Influenza training leads to increased pro-inflammatory responses in BMDMs and mild cross-protection in vivo
(A) Experimental schematic for influenza trained WBM-derived BMDMs.
(B) M. avium bacterial CFU engulfed per BMDM 4 h post M. avium challenge.
(C–E) BMDM supernatant cytokine concentration from BMDM cultures 4 h post M. avium challenge for: (C) IL-4 (D) IL-12p70 (E) TNFα Statistics: two-way ANOVA with Tukey’s multiple comparisons, n = 3–4 per experimental group.
(F) Mosaic model of H1N1 PR8-trained HSPC transplants followed by M. avium challenge.
(G) Representative images of spleens from control and challenged transplant recipients one month post M. avium infection.
(H) M. avium bacterial load in challenged recipients of untrained versus trained HSPCs. Data are pooled from two independent experiments, n = 7–17 per experimental group. Significant by two-way ANOVA with Tukey’s multiple comparisons.
(I) Representative histology of spleens of transplanted challenged recipients at 10x magnification, 210 μm scale bar.
(J) Splenic granuloma quantification from one longitudinally cut complete spleen section at 10x magnification. p values calculated by one-way ANOVA, n = 3 per experimental group.
(K–M) Serum cytokine from transplant recipient mice 1 month after M. avium challenge. (K) IL-6 (L) CXCL9 and (M) CXCL10 serum cytokine levels were elevated in H1N1 PR8-trained recipients post challenge compared to untrained controls. Statistics: Unpaired t-test, n = 6–10 per group. ∗p < 0.05, ∗∗p < 0.01 ∗∗∗p < 0.001 ∗∗∗∗p < 0.0001.
Next, we generated chimeric mice transplanted with control or influenza-trained HSPCs, challenged the mice with M. avium, and assessed their disease morbidity one month post infection (Figure 5F). Although there was no difference in splenomegaly or splenic histology (Figures 5G–5J), there was a subtle reduction in bacterial load and enhanced IL-6, CXCL9, and CXCL10 serum cytokine levels in mice transplanted with influenza-trained HSPCs (Figures 5H–5M), which suggests an enhanced immune response. We also found that BMDMs generated from these trained M. avium challenged mice had higher MHC II and iNOS expression but significantly decreased Ly6c expression (Figures S6D–S6F). These results indicate that influenza training can induce lasting cellular changes that lead to enhanced BMDM functionality, cytokine production, and pathogen clearance against an unrelated Th1-inducing bacterial pathogen.
M. avium training induces cross-protection against acute influenza infection
Reciprocally, we tested whether M. avium training can confer cross-protection against influenza A infection. We created mosaic mice by transplanting HSPCs from M. avium-infected or mock-infected animals into naive recipients and challenged them 12 weeks later with influenza A H1N1 PR8 (20pfu). We assessed disease morbidity three days post viral challenge, when lungs have a high viral load and the innate immune system is very active (Figure 6A). Mice receiving M. avium trained HSPCs demonstrated enhanced Th1-type cytokine responses in the serum, specifically IFNγ, CXCL5, and CXCL9 (Figures 6B–6D). These mice also had reduced lung pathology (Figure 6E), reduced immune cell infiltration (Figure 6F), and significantly reduced viral copy numbers in the lung (Figure 6G). Myeloid-biased progenitors — specifically, donor-derived MPP1, MPP3, GMP and Ly6c− monocytes — were enhanced in the animals receiving trained cells, whereas LT-HSCs, MPP2s, MPP4s, and Ly6c+ monocytes were not affected (Figures 6H–6K and S7A–S7D). CD3+ T and B cells were decreased in the M. avium-trained HSPC recipients (Figures S7E and S7F) while donor-derived megakaryocyte and erythrocyte precursors were increased (Figures S7G–S7J). To test whether M. avium training induced protection against lethal influenza infection, we exposed the chimeric mice to 150 pfu (5xLD50) influenza and monitored disease morbidity and mortality for 21 days post infection. Mice receiving M. avium trained HSPCs had a small but statistically significant weight and survival advantage upon challenge with a lethal influenza dose (Figures 6L and S7K). Altogether, these data show that M. avium-induced HSPC training is sufficient to reduce morbidity and improve survival from influenza and, therefore, that IFNγ-dependent central trained immunity can cross-protect against alternative pathogens that share a Th1-type immunological response.
Figure 6.

M. avium training induces cross-protection against acute influenza infection
(A) Mosaic mouse model of M. avium trained HSPC recipients and H1N1 influenza challenge.
(B–D) Serum cytokine levels in transplant recipients three days post H1N1 PR8 challenge for: (B) IFNγ, (C) CXCL5, and (D) CXCL9. n = 5–6 per experimental group. Statistics: unpaired t-test.
(E–G) H&E-stained lung sections from untrained and trained challenged mice. From left to right: 2x and 5x magnification: whole lung sections representing global tissue damage. 10x magnification: representative section of bronchioles, untrained HSPC recipients have more immune infiltration within the bronchiole space compared to trained HSPC recipients. 40x: representative section of alveolar sacs (F) Nucleated immune cells per mm2 of lung tissue. n = 4 per group (G) Viral titer 3 days post influenza infection quantified by influenza nucleoprotein (NP) qPCR from lung supernatants, n = 6 mice per group, statistics:unpaired t-test.
(H–K) Flow cytometry analysis of bone marrow leukocytes from challenged transplant recipients three days post H1N1 PR8 challenge. (H) CD45.2 ST-HSC/MPP1 (LK, CD150+, CD48−, Flk2-, CD34+), (I) CD45.2 myeloid biased progenitors (MPP3) (LK, CD150-, CD48+, Flk2-, CD34+), (J) CD45.2 GMP (LK, CD150-, CD16/32+), and (K) CD45.2 Ly6c− monocytes (CD11b+, Ly6g−, SSC-A low, Ly6c−) were increased in M. avium trained HSPC recipients compared to untrained controls. n = 5–6 per group, Statistics: Unpaired t test.
(L) Kaplan Meier survival curve of transplanted mice challenged with 5xLD50 (150 pfu) influenza H1N1 PR8. Results were pooled from two independent experiments, n = 11–12 per group per experiment. ∗p < 0.05, ∗∗p < 0.01 ∗∗∗p < 0.001 ∗∗∗∗p < 0.0001.
Discussion
Here we established an M. avium training and infection chimeric mouse model to investigate the cellular basis of central trained immunity. We found that systemic M. avium infection induces memory in ckit+ HSPCs, conferring a protective effect on transplant recipients 12 weeks after transplant. BMDMs derived from trained HSCs demonstrated increased bacterial killing and enhanced metabolism. This model enabled us to investigate two major questions in trained immunity. First, is there a particular subset of HSPCs that is responsible for trained immunity? We found that M. avium infection induced heterogeneous transcriptional effects in HSPCs, that a subset of HSCs and MPPs demonstrated transcriptional responses to infection, and the expansion of a distinct HSC subpopulation with an activated phenotype (IA-HSCs). Importantly, transferring LT-HSCs alone did not result in improved immunity, suggesting that the protective immune response is likely dependent on other HSPC-compartment cells. Second, can training with a pathogen or IFNγ alone induce cross-protection against other pathogens? IFNγ exposure alone was sufficient to induce long-term changes in metabolism of HSPC-derived BMDMs even after transplant, and transplanting HSCs from influenza-trained mice protected against subsequent M. avium infection and vice versa, demonstrating cross-protective effects. Together, these data provide strong evidence that HSPC changes upon inflammatory stress do generate lasting effects that impact downstream innate immune function.
The chimeric transplant model used in this study is unique because we utilized sublethal irradiation to make space for the trained c-kit+ cells to engraft in the bone marrow, leading to relatively low engraftment levels (Figure S1A). Further, single cell transcriptional profiling suggests that only a portion of HSPCs are transcriptionally active in response to systemic infection. Collectively, our data show that only a fraction of HSPC cells require training to confer a protective effect. Trained HSPC-derived BMDMs were more metabolically active than untrained cells and had enhanced bacterial killing properties. These results align with previously established reprogramming models within the trained immunity field.33,34
We observed persistent changes in BMDMs after a single dose of rIFNγ alone, with increased pathogen killing and metabolic profiles immediately following training exposure that lasted for four months after transplantation and infectious challenge. HSPCs induced by M. avium infection were sufficient to confer protection against influenza-associated morbidity and mortality in transplant recipients. This cross-protection indicates that IFNγ-induced trained immunity can protect against antigenically unrelated pathogens affecting distant organ systems. Our results suggest that pathogens that elicit similar Th1 immune responses can provide cross-protection and that these protective effects may be induced via systemic signaling generated in response to the training pathogen. Determining the ideal combination of cytokines and other signaling factors could allow for HSPC training in the absence of an infectious pathogen in vivo or ex vivo. This strategy could serve as a translational platform to protect vulnerable patients like bone marrow transplant recipients from life-threatening infections.
Collectively, these studies—which integrate datasets at the single cell and bulk level—form an initial in-depth analysis of transcriptional trained immune responses in HSPCs. Whereas some HSPCs showed strong immune response gene induction following M. avium infection, others were unresponsive in our single cell analysis. Notably, response heterogeneity within the HSPC pool was not correlated with IFNγ receptor expression. Heterogeneity in responses may be due to local differences in pathogen or cytokine exposure and leaves open the possibility that alternate HSPC subsets could encode diverse responses to a variety of pathogens or immune challenges. Because our immune systems are constantly sampling the environment, there is an evolutionary advantage to cell specific circumstances that allow for training following specific inflammatory stressors. It is possible that certain cells within the HSC pool are more susceptible to training by some pathogens and resistant to others, allowing for the preservation of the stem cell pool throughout the host’s lifetime. For example, LT-HSCs were not sufficient to confer trained immunity in our M. avium infection model. The heterogeneity we observed within our single cell dataset suggests that multiple cell types may work together within the HSPC pool to execute trained immune responses, particularly HSC subsets and myeloid biased MPPs and GMPs. The factors that influence whether an HSPC is susceptible to training remain to be discovered but may include location and localized cytokine differences within the bone marrow niche,35,36 prior exposure to training stimuli, and age.
Limitations of the study
It is important to note that the work we present here cannot entirely rule out a contribution from the adaptive immune system. However, the demonstration of persistent changes in bacterial killing and metabolism in macrophages derived from HSPCs in vitro indicates that HSPCs and their innate immune cell progeny are persistently altered upon training. Our study does not diminish training’s potential effects on T cell function, and in our tetramer data we observed significant decreases in antigen specific T cell percentage and number.37 Additionally, using sorted cell populations to perform scRNA-seq analysis without sample hash-tagging potentially could have obscured shifts in cell populations. Future work will be required to understand the role of IA-HSCs, sex-based differences in their expansion, and whether they emerge in a variety of infectious contexts.
Finally, while our work indicates that M. avium induces cross-protection against influenza, further research is necessary to understand this protection’s breadth. Training using B-glucan and LPS also reprograms HSPCs and influences downstream immune function.11,17,38,39,40 While it is likely that each individual immune signal has diverse cellular targets and functional effects, the degree to which different stimulants work in parallel or potentially reverse preexisting programming in the same cell remains unknown. Future work in this area will have important biological implications for long-term immunity, vaccine efficacy, metabolic function, and bone marrow transplantation outcomes.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Mouse CD45.1 (PB conjugated, clone A20) | Biolegend | Cat#110721, RRID:AB_492867 |
| Anti-Mouse CD45.1 (APC conjugated, clone A20) | ebioscience | Cat#17-0453-8, RRID:AB_469397 |
| Anti-Mouse CD45.2 (BV 605 conjugated, clone 104) | Biolegend | Cat#109841, RRID:AB_2563485 |
| Anti-Mouse CD45.2 (PE conjugated, clone A20) | ebioscience | Cat#12-0453-81, RRID:AB_465674 |
| Anti-Mouse CD45.2 (BV711 conjugated, clone 104) | Biolegend | 109847, RRID: AB_2616859 |
| Anti-Mouse c-kit/CD117 (APC-Cy7 conjugated, clone 2B8) | ebioscience | Cat#25-1171-82, RRID:AB_469644 |
| Anti-Mouse CD3 (AF 700 conjugated, clone 17A2) | ebioscience | 100215, RRID: AB_2815745 |
| Anti-Mouse CD4 (PE-Cy5 conjugated, clone GK 1.5) | ebioscience | Cat#15-0041-82, AB_468695 |
| Anti-Mouse CD4 (FITC conjugated, clone GK1.5) | ebioscience | Cat#11-0041-82, RRID:AB_464892 |
| Anti-Mouse CD4 (PE conjugated, clone GK1.5) | ebioscience | 12-0041-82, RRID: AB_465506 |
| Anti-Mouse CD8 (PE-Cy5 conjugated, clone 53-6.7) | ebioscience | Cat#15-0081-82, RRID:AB_468706 |
| Anti-Mouse CD8 (FITC conjugated, clone 53-6.7) | ebioscience | Cat#11-0081-82, RRID:AB_464915 |
| Anti Mouse NK 1.1 (BV605 conjugated, clone PK136) | Biolegend | 108753 RRID: AB_2686977 |
| Anti-Mouse CD45R/B220 (PE-Cy5 conjugated, clone RA3-682) | ebioscience | Cat#15-0452-82, RRID:AB_468755 |
| Anti-Mouse CD45R/B220 (PE-Cy7 conjugated, clone RA3-6B2) | ebioscience | Cat#25-0452-82, RRID:AB_469627 |
| Anti-Mouse/Human CD45R/B220 (FITC conjugated, clone RA3-6B2) | ebioscience | Cat#11-0452-82, RRID:AB_465054 |
| Anti-Mouse Ly-6G/Gr1 (PE-Cy5 conjugated, clone RB6-8C5) | ebioscience | Cat#15-5931-82, RRID:AB_468813 |
| Anti-Mouse Ly-6G/Gr1 (PE-Cy7 conjugated, clone RB6-8C5) | ebioscience | Cat#25-5931-82, RRID:AB_469663 |
| Anti-Mouse CD11b/Mac1 (PE-Cy5 conjugated, clone M1/70) | ebioscience | Cat#15-0112-82, RRID:AB_468714 |
| Anti-Mouse CD11b/Mac1 (PE-Cy7 conjugated, clone M1/70) | ebioscience | Cat#25-0112-81, RRID:AB_469587 |
| Anti-Mouse CD11b/Mac1 (PE conjugated, clone M1/70) | ebioscience | 12-0112-82, RRID: AB_2734869 |
| Anti-Mouse CD117 (APC-Cy7 conjugated, clone 2B8) | ebioscience | 47-1171-82, RRID: AB_1272177 |
| Anti-Mouse Ter119 (PE-Cy5 conjugated, clone M1/70) | ebioscience | Cat#15-5921-82, RRID:AB_468810 |
| Anti-Mouse CD150/SLAM (PE-Cy7 conjugated, clone TC15-12F12.2) | Biolegend | Cat#115914, RRID:AB_439797 |
| Anti-Mouse CD150/SLAM (APC conjugated, clone TC15-12F12.2) | Biolegend | 115910, RRID: AB_493460 |
| Anti-Mouse CD48 (APC conjugated, clone HM48-1) | ebioscience | Cat#17-0481-82, RRID: AB_469408 |
| Anti-Mouse CD48 (FITC conjugated, clone HM48-1) | ebioscience | Cat#11-0481-82, RRID: AB_465077 |
| Anti-Mouse CD34 (FITC conjugated, clone RAM34) | ebioscience | 11-0341-82, RRID:AB_465021 |
| Anti-Mouse Flk2/Flt3/CD135 (PE conjugated, clone A2F10) | ebioscience | Cat#12-1351-82, RRID:AB_465859 |
| Anti-Mouse CD41 (PE-Cy7 conjugated, clone MWReg30) | abcam | ab95726, RRID: AB_10679742 |
| Anti-Mouse CD16/32 (PE conjugated, clone 93) | ebioscience | 12-0161-82, RRID: AB_465568 |
| Anti-Mouse CD105 (FITC conjugated, clone MJ7/18) | Biolegend | 120406, RRID: AB_961053 |
| Anti-Mouse Ki67 (PE conjugated, clone SolA15) | ebioscience | Cat#12-5698-82, RRID: AB_11150954 |
| Anti-Mouse Ki67 (FITC conjugated, clone SolA15) | eBioscience | 11-5698-82, RRID: AB_11151330 |
| Anti-Mouse Annexin V (PE conjugated) | BD Pharmingen | Cat#556421: RRID: AB_2869071 |
| Anti-Mouse I-A/I-E, MHCII (BV421 conjugated, clone M5/114) | BD Horizon | 562564, RRID:AB_2716857 |
| Anti-Mouse iNOS (PE-Cy7 conjugated, clone CXNFT) | ebioscience | 25-5920-82 |
| Anti-Mouse CD86 (BV785 conjugated, clone GL-1) | Biolegend | 105043, RRID:AB_2566722 |
| Anti-Mouse CD80 (BV711 conjugated, clone 16-10A1) | Biolegend | 104743, RRID:AB_2810338 |
| Anti-Mouse CD11c (APC-Cy7 conjugated, APC-Vio770) | Miltenyi | 130-110-841, RRID:AB_2654715 |
| Anti-Mouse Ly6c (APC conjugated, clone HK1.4) | Biolegend | 128016, RRID: AB_1732076 |
| Anti-Mouse Ly6g (FITC conjugated, clone 1A8) | Biolegend | 127605, RRID: AB_1236488 |
| Anti-Mouse CD9 (PE conjugated, clone KMC8) | eBioscience | 12-0091-81, RRID: AB_891498 |
| Anti-Mouse CD81 (FITC conjugated, clone Eat2) | Invitrogen | MA5-17939, RRID:AB_2539323 |
| Anti-Mouse CD24 (PE-Cy7 conjugated, clone M1/69) | BD Pharmingen | 560536, RRID:AB_1727452 |
| DAPI | Life technologies | Cat#D1306 |
| Fc block | MACS Miltenyi Biotec | Cat#130-046-702 |
| Ag85240-254-loaded PE-conjugated I-Ab tetramer | NIH Tetramer Core | NA |
| Bacterial and virus strains | ||
| Mycobacterium avium | SmT 2151 | |
| Influenza | Dr. Silke Paust | H1N1 PR8 |
| Cell lines | ||
| MDCK.2 cell line | ATCC | CRL-2936 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Mouse IFNy | ebioscience | Cat#BMS326 |
| Mouse CD117 Micro Beads | MACS Miltenyi Biotec | Cat#130-091-224 |
| Mouse Biotin Micro Beads | MACS Miltenyi Biotec | Cat# 130-090-485 |
| iTaq Universal SYBR Green Supermix | BioRad | 172-5121 |
| Tn5 Transposase | Lucigen | TNP92110 |
| Digitonin | Promega | G9441 |
| NP-40 | Roche | 11332473001 |
| Tween 20 | Sigma-Aldrich | P1379 |
| Murine recombinant M-CSF | Peprotech | 315-02-50UG |
| Murine recombinant GM-CSF | Peprotech | 10780-710 |
| Murine recombinant IL-3 | Peprotech | 213-1310ug |
| Murine recombinant SCF | Peprotech | 10773-124 |
| Critical commercial assays | ||
| NucleoSpin RNA Plus XS kit | Macherey-Nagel | Cat# 740990 |
| SMARTer® Stranded Total RNA-Seq Kit v2 - Pico Input Mammalian | Takara | Cat# 634411 |
| RNAqueous Kit | Ambion | Cat#AM1912 |
| SuperScript III First-Strand Synthesis Supermix | Invitrogen | Cat#18080051 |
| iTaq Universal SYBR Green Supermix | BioRad | Cat#172-5121 |
| Annexin V Binding Buffer, 10X concentrate | BD Pharmingen | Cat#556422 |
| Seahorse XFp Mito Stress test kit | Agilent Technologies | 103010-100 |
| Mouse Cytokine Array/Chemokine array 31-Plex | Eve Technologies | MD31 |
| Mouse Cytokine Array Proinflammatory Focused 10-plex | Eve Technologies | MDF10 |
| Deposited data | ||
| RNAseq_bulk untrained vs trained HSPCs and WGBS comparisons | GEO | GSE197294 |
| Whole Genome Bisulfite data WT naïve vs. M. avium infected | GEO | GSE197274 |
| Single cell RNA-seq data WT naïve vs, M. avium infected | GEO | GSE197407 |
| Code Availability for scRNA analysis | Github | https://github.com/KingLabBCM/TrainedImmunity |
| Experimental models: Organisms/strains | ||
| Mice: CD45.1 | CCM core - BCM | NA |
| Mice: CD45.2 | CCM core - BCM | NA |
| Mice: CD45.1/CD45.2 chimeric transplant model | CCM core- BCM | NA |
| Mice: Ifngr1-/- | The Jackson Laboratory | Stock No: 025394 |
| Oligonucleotides | ||
| Mycobacterium avium 16s probe -forward (CCTCAAGACGCATGTCTTC) | IDT | NA |
| Mycobacterium avium 16s probe -reverse (ACCTACCGTCAATCCGAGAA) | IDT | NA |
| PR8NP F1 primer (GGGTGAGAATGGACGAAAAAC) | IDT | NA |
| PR8NP R1 primer (GATCCATCATTGCTTTTTGTGCA) | IDT | NA |
| Software and algorithms | ||
| Prism 7 | https://www.graphpad.com/scientific-software/prism/ | |
| Flow Jo | https://www.flowjo.com/solutions/flowjo | |
| Aivia | https://www.aivia-software.com/ | |
| Image J | https://imagej.nih.gov/ij/ | |
| R Project for Statistical Computing | https://www.r-project.org | |
| R studio | https://www.rstudio.com/products/rstudio/download/#download | |
| HTStream | https://github.com/ibest/HTStream | |
| STAR | https://github.com/alexdobin/STAR | |
| Biomart –Genome annotations- | http://uswest.ensembl.org/biomart/martview/f7089c3f574fcc3ee405faed118b166e | |
| Differential Expression with Limma-Voom | https://ucdavis-bioinformatics-training.github.io/2018-June-RNA-Seq-Workshop/thursday/DE.html | |
| Samtools | https://samtools.github.io/samtools/samtools.html | |
| Loupe Cell Browser | https://www.10xgenomics.com/products/loupe-browser | |
| Seurat | https://satijalab.org/seurat/ | |
| Other | ||
| Seahorse XFp Cell culture miniplates | Agilent technologies | 103025-100 |
| Cell-Tak | Corning | 354240 |
| AMPURE XP | Beckman Coulter | A63880 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Katherine King (kyk@bcm.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and study participant details
Mice
For trained immunity transplant experiments we used 8-10 week-old wild-type C57BL/6 (CD45.2) and C57BL/6.SJL (CD45.1) mice. For BMDM ex vivo functional assessment we used 8-16 week-old CD45.2 mice. Mice of both sexes were used with equal ratios across experimental groups. All mice were maintained at an AALAC-accredited, specific pathogen-free animal facility at Baylor College of Medicine. Baylor College of Medicine’s IACUC approved all experiments under protocol AN-4802.
In vivo infections
For chronic bacterial infections, we infected mice intravenously (IV) with 2 x 106 colony-forming units (CFU) Mycobacterium avium (strain SmT 2151) as described41 and detected M. avium by growth on Middlebrook agar or qPCR (Park et al., 2000). Dr. Silke Paust graciously provided the H1N1 PR8 stocks. We completed virus titering via plaque assay and calculated median lethal dose (LD50) estimations using mice of the same genotype, age, and sex. We infected mice with influenza viral suspension intranasally under isoflurane anesthesia and monitored them 1-2 times a day for 21 days or until they exhibited disease recovery. Mice that lost over 20% body weight were euthanized in accordance with the animal protocol.
Chimeric mouse generation
We intravenously infected CD45.2 donor mice with 2 x 106 CFU of M. avium as described.41 Four weeks after infection, we isolated whole bone marrow from the donors’ tibias, femurs, and hips. To isolate HSPCs, we performed RBC lysis and magnetic c-kit+ cell separation using CD117-conjugated magnetic beads (Miltenyi). cKit+ cells were incubated with clarithromycin for four hours at 4C, washed, and re-suspended in HBSS supplemented with penicillin-streptomycin. We transplanted HSPCs via IV injection of 2 × 105 CD45.2 donor c-kit+ cells from M. avium infected mice or naive controls following one 5.25 Gy dose of sublethal irradiation. Non-transplanted control mice were subject to the same dose of irradiation but did not receive donor cells. Four weeks following transplant, we checked mice for donor cell engraftment via retro-orbital bleeding. Twelve weeks after transplant, we checked mice for M. avium transmission via qPCR of peripheral blood prior to infectious challenge. In IFNγ experiments, we injected mice with 100 ug of murine rIFNγ (eBioscience). For influenza experiments, we intranasally infected mice with 150 pfu of H1N1PR8. Four weeks after infection, we isolated c-kit+ cells from surviving donor mice and transplanted them as previously described for the M. avium trained transplants. Infection timepoints remained the same for all challenge experiments.
For long-term hematopoietic stem cell (LT-HSC) transplants, we isolated LT-HSCs from infected mice or naïve control CD45.2 donors using ckit enrichment and sorted lineage- CD150+ CD48- CD34- ePCR+ cells via FACS on a Sony cell sorter SH800. We transplanted 250 LT-HSCs with 2 x 105 CD45.1 competitor whole bone marrow into 2-month-old CD45.1 recipients following a split dose of 10.5 Gy irradiation. We checked engraftment in the peripheral blood every four weeks for 8 weeks prior to M. avium challenge and completed CD45.2 IA-HSC (Lin- ckit-enriched CD24α+ CD81+ CD9+) and SLAM-HSC (Lin- ckit-enriched CD150+ CD48-) experiments in a similar fashion, using 500 cells transplanted with 2 x 105 CD45.1 competitor whole bone marrow into CD45.1 recipients.
Method details
Flow cytometry
Murine peripheral complete blood counts (CBCs) were analyzed with a Heska element HT5. WBM cells were isolated from femurs, tibias, and pelvises. For antibody staining, cells were suspended at a concentration of 108 cells/mL and incubated at 4°C for 30 minutes with the desired antibody cocktail at a staining ratio of 1:100. For FACS sorting, magnetic enrichment was performed with anti-c-Kit microbeads (eBioscience, San Diego, CA) on manual flow-through columns (Miltenyi Biotec, Germany). Post-enrichment, the positive cell fraction was labeled with antibodies to identify HSCs (Lineage- CD150+ CD48- CD34- Flk2-). Cell sorting was performed on an Aria II (BD Biosciences, San Jose, CA) or SH800 (Sony Biotechnology), and analysis was performed on an LSR Fortessa (BD Biosciences, San Jose, CA). Antibodies used for flow cytometry and cell sorting are listed in the key resources table. Flow cytometric analysis was completed using FlowJo (BD Biosciences, San Jose, CA).
Intracellular staining
Murine cells stained with cell surface markers were fixed, permeabilized and refixed using buffers from BD Biosciences BrdU Flow Kit (BD Biosciences, San Jose, CA) according to the manufacturer’s instructions. After fixation, cells were stained for 30 mins on ice with desired intracellular markers (eBioSciences) and washed prior to flow cytometric assessment.
M. avium transmission check using qPCR
For M. avium transmission checks in transplanted mice, we isolated RNA or DNA using an RNAeasy or DNeasy kit, respectively (Qiagen). RNA was reverse transcribed with random hexamer primers using SuperScript IV (Invitrogen) following the manufacturer’s protocol. We standardized cDNA input and performed RT-PCR using iTaq Universal SYBR Green Supermix (BioRad) with 16S rRNA probes (F:CCTCAAGACGCATGTCTTC and R: ACCTACCGTCAATCCGAGAA,42 of M. avium for 40 cycles with an Applied Biosystems StepOnePlus Real-Time PCR System. M. avium copy number level per sample was calculated using a standard curve from M. avium stock samples. Primers used in RT-PCR reactions are in Table S4.
Bacterial CFU counting
Spleens from naïve and infected experimental mice were isolated and weighed prior to being divided longitudinally for histopathology or bacterial CFU counting. For bacterial CFU counting, spleen segments were re-weighed, crushed, and filtered in 15 mL conical tubes. Spleen suspensions were serially diluted in sterile 1 x PBS and plated in duplicate on 7H10 agar plates as previously described.43 Plates were incubated at 37°C in a biohazard incubator for 7-10 days prior to colony counting. Plates with 30-250 colonies were used for the calculation of bacterial CFU/g spleen.
Assessment of organ histopathology
For spleen histology, spleen segments were stored in 10% formalin prior to being submitted at the BCM Pathology and Histology Core. Each spleen was parafilm embedded, sectioned, and hematoxylin and eosin (H&E) stained. For lung histology, lung lobes were isolated and inflated with 1 x PBS prior to being preserved in 10% formalin for processing. Images of spleen and lung histopathology were captured using a Leica Upright fluorescent microscope or an Echo Revolve microscope (San Diego, CA). Scale bars for each image were embedded using Image J or Evo technologies software, respectively. Granulomas visualized via H&E staining were counted following the stitching of 10x magnification images via Aivia technology and quantified using ImageJ.
Tetramer specific T cell flow cytometry staining
Spleens were collected from naïve and infected WT and Dnmt3a-/- mice. Spleens were weighed, crushed in 8-10 mL HBSS with 1x Brefeldin A (Invitrogen B7450), and filtered in 15 mL conical tubes. RBCs were removed using Biolegend RBC lysis buffer (Cat# 420301). For tetramer staining, Ag85240-254-loaded PE-conjugated I-Ab tetramer (NIH Core) was used to stain cells for 1 hour at 37°C. Ag85-specific cells were enriched using magnetic cell separation with CD117+ microbeads on MACS columns (Miltenyi Biotec, Germany). Positive and negative cell fragments were stained with desired antibodies at a concentration of 108 cells/mL and incubated at 4C for 30 minutes.
Bulk RNA-Seq library generation and analysis of trained immunity transplant HSPCs
10,000-50,000 HSCs (CD45.1/CD45.2 KL CD150+ CD48-) into HBSS from naive or 1-month infected WT/transplanted mice (n=10-12 per group). DNA and RNA were isolated with the NucleoSpin ® RNA Plus XS kit (Macherey Nagel). RNA-seq libraries were prepared using SMARTer® Stranded Total RNA-Seq Kit v2 - Pico Input Mammalian (Takara Bio Usa). Illumina NovaSeq SP was used for sequencing with a paired-end sequencing length of 10bp. Samples were sequenced at Admera Health using an Illumina HiSeq 2x150 at sequencing depth of ∼40 million reads. FASTQ files were preprocessed using HTS stream (https://github.com/ibest/HTStream) and the clean FASTQ file were aligned using STAR. Differential expression (DE) analysis of gene expression was performed using Limma-Voom. False discovery rate (FDR)<0.05 was considered statistically significant. Further analysis was completed using Illumina Basespace packages and programs (deSeq2) We performed gene ontology analysis for differentially expressed genes with q value of <0.05. Gene set enrichment analysis (GSEA, Broad Institute and UC San Diego) was completed using normalized gene counts as previously described (.44 Results were visualized using Tidyverse packages in R. Comparison of differentially expressed gene lists and generation of Venn diagrams were generated using the GeneVenn webtool. Biological process and molecular function gene ontology analysis of differentially expressed genes found in bulk RNA-seq datasets were completed using GENEONTOLOGY of the Mus musculus database.45,46
Whole genome bisulfite sequencing (WGBS) data analysis
A previously published WGBS dataset47 was mined for protein encoding genes that were differentially methylated with a DMR.padj of p<0.1. GO analysis of this gene list was conducted as described above in bulk RNA-seq analysis methods.
Cytokine bead arrays
Peripheral blood was collected via cardiac puncture, and serum was isolated by centrifugation using BD microtainers and frozen at –80°C until further use. Samples were shipped overnight on dry ice to Eve technologies (Calgary, Canada) and assessed using the Mouse Cytokine Array/ Chemokine array 31-Plex (MD31) for murine serum samples and the Mouse Cytokine Array Proinflammatory Focused 10-plex (MDF10) for cytokine release assessment of murine primary ex vivo macrophages. Analyte concentrations (pg/mL) were determined based on standard curves generated by Eve technologies.
Bone marrow derived macrophage (BMDM) generation from either whole bone marrow or lineage-negative, ckit-enriched (LK) cells
For generation of BMDM from whole bone marrow (WBM), WBM was isolated from tibias, femurs, and pelvic bones of experimental or control mice by flushing. Flushed marrow was resuspended in BMDM media (Dulbecco’s Modified Eagle Medium with F12 Glutamax, 10% Fetal Bovine Serum, 10% Penicilin and Streptomycin), treated with murine macrophage-colony stimulating factor (M-CSF, Peprotech, 40ng/mL), and incubated in petri dishes and placed in CO2 incubators at 37°C and 5% CO2 for six days. Cultures were washed three times with sterile 1X PBS and isolated by mechanical scraping. The cellular phenotype (CD11b+, F4/80+) was confirmed using flow cytometry. Cells were then seeded for further BMDM phenotype analysis.
For the generation of BMDMs from lineage-negative, ckit-enriched cells (LK cells), WBM was isolated from the tibias, femurs, and pelvic bones of experimental or control mice after crushing. Homogenates were then incubated in RBC lysis buffer (Biolegend, cat: 420301, diluted to 1X in deionized H2O) for five minutes on ice. After RBC lysis, cells are washed and filtered into new conical tubes and spun at 400 RCF for six minutes. Cells were then incubated and enriched for cKit (CD117) via magnetic antibody cell separation (MACS) (Miltenyi, 130-097-146). cKit+ and lineage negative cells were isolated via flow activated cell sorting (FACS) on a Sony Sorter SH800. LK cells were seeded at 500,000 cells in six-well plates with the same BMDM media used in WBM BMDM generation. Cells were treated with M-CSF (Peprotech, 50ng/mL) and murine interleukin 3 (IL-3, Peprotech, 50ng/mL) for the first four days. Cells were split and supplemented with additional BMDM media and cytokines every second day to facilitate expansion. After day four, cells were treated with only M-CSF (50ng/mL) for an additional six days. Once cells reached >90% confluency, cells were purified and seeded as described in the above WBM method.
BMDM M. avium ex vivo killing assays
Purified BMDMs were seeded into 6- or 12-well plates at concentration of 500,000 -1,000,000 cells/well or 200,000 cells/well, respectively. The following day, cells were either left untreated or infected with an MOI of 5 M. avium/seeded BMDM. Cells were incubated with bacteria for four hours or 3 days (protocol modified from Kaufman et al. 2018). At harvest, supernatant from each well was collected and stored at -80°C for cytokine analysis. Cells were washed with 1 x PBS, scraped, and resuspended into Eppendorf tubes. Following manual counting using a hematocytometer, cells were centrifuged at 13,000 rpm for ten minutes and resuspended in sterile dH2O to lyse eukaryotic cells. Cell suspensions were then either plated for CFU counting (as previously described) or frozen at -80°C for pre-amplification of DNA for qPCR.
A standard curve to determine M. avium concentration was generated using our laboratory stock of M. avium. An aliquot of bacteria was spun down at 13,000 RPM in a table-top centrifuge, resuspended in sterile dH2O, and boiled at 95°C for 10 minutes to generate a stock of 16s DNA. Following boiling, DNA was quantified using a Nanodrop Microvolume UV-Vis Spectrophotometer (ThermoFisher Scientific) and aliquoted into serial dilutions ranging from undiluted DNA – 10-6. Standard curve samples were pre-amplified using Pre-Amp Mastermix (Fluidigm, San Francisco, CA) according to manufacturer’s instructions. Diluted pre-amplifcation product was used to complete RT-PCR using iTaq Universal SYBR Green Supermix (BioRad) and 16s M. avium primers. Ct values generated by these samples were used to quantify M. avium copy number in samples from BMDM killing assays. BMDM killing assay samples were processed and run using the same protocol as standard curve generation.
BMDM seahorse metabolic assays
Seahorse XFp Cell culture miniplates (Agilent technologies) were coated in a CellTak suspension containing 6 mM NaOH and 100 mM NaHCO3. Wells were coated with 25uL of solution and incubated for 20 minutes, then washed with sterile dH2O twice prior to storage at 4°C until further use. One day before seahorse assay, sensor cartridges were hydrated and placed in a 37°C incubator with no CO2 to equilibrate overnight. BMDMs were harvested from culture plates and spun down at 1200 rpm for 5 minutes at 4°C in a fixed angle centrifuge. Cells were resuspended in XFp Basal media kept at 37°C (Agilent) supplemented with 25mM glucose, 2mM Na-Pyruvate, 2mM L-glutamine and washed twice prior to being resuspended in seahorse media and counting (Cellometer Auto 2000, Nexcelom, Lawrence, MA). Cells were seeded in 6-well seahorse plates at a concentration of 90,000-100,000 cells/well. Cells were adhered to the plate by centrifuging for one minute with no brake. Drugs at a concentration of 1.5uM Oligomycin, 0.75 uM FCCP, and 0.5uM AA/Rotenone were loaded into the cartridge and allowed to equilibrate in the 37°C incubator.48 Once cells were adhered, proper drug calculations were completed using the Seahorse XFp manual. Prior to the assay, images of each well were captured for post assay-normalization with cell counts. Mitochondrial function was assessed on a Seahorse XFp analyzer in real time following the injection of oligomycin, FCCP, and AA/Rotenone according to the manufacturer’s instructions. Specific mitochondrial parameters were assessed as previously described.
Single cell RNA-seq library generation
CD45.2 mice were infected with 2 x 106 CFU M. avium for one month prior to bone marrow harvesting. One month post infection, experimental mice were euthanized and infection was confirmed by measuring splenic weight and size. WBM from the hips, tibias, and femurs of each mouse was extracted through crushing with a mortar and pestle. The suspension was then RBC lysed and CD117 (c-kit)+ and cKit- cells were separated using a manual magnetic separation system (Miltenyi). Experimental samples were stained with desired antibodies (listed in key resources table) and sorted using a BD FACS Aria cell sorter. The following cell types were isolated according to the following phenotypic definitions:
| Hematopoietic cell type | Cellular fraction | Cellular Phenotype |
|---|---|---|
| LT-HSCs | c-kit + | Lin- c-kit+ CD48- CD150+ Flk2- CD34- |
| MPP3 | c-kit + | Lin- c-kit+ CD48+ CD150- Flk2- CD34+ |
| GMP | c-kit + | Lin- c-kit+ CD41- CD16/CD32+ |
| CD41+ HSCs | c-kit + | Lin- c-kit+ CD48- CD150+ CD41+ (Gekas and Graf Blood 2013) |
| Inflammatory Macrophages | c-kit - | B220- Gr1+ Mac-1+ CD3- CD11c- Ly6g- Ly6c+ |
| Neutrophils | c-kit - | B220- Gr1+ Mac-1+ CD3- CD11c- Ly6g+ |
| B cells | c-kit - | B220+ Gr1- Mac-1- CD3- |
The cellular purity and count of each sample was confirmed prior to pooling for submission to the BCM Single Cell Core. Depending on the rarity of the isolated population, 20,000-50,000 sorted cells were combined to a concentration of ∼7200 cells/uL. Samples were loaded into the Chromium controller system and generated 3’ 10x scRNAseq barcoded libraries for each sample. Prior to sequencing, quality control of the libraries was completed using a Bioanalyzer (Agilent, Santa Clara, CA) and Nanodrop system. Samples were sequenced on a NovaSeq at a sequencing depth of 300M reads at the BCM Genomic and RNA Profiling (GARP) core. After sequencing, fastq files for each sample were generated (R1, R2, and I1 files) to demultiplex the data using the CellRanger pipeline. Initial analysis through the Cell Ranger pipeline was completed prior to additional bioinformatic analysis described below.
Single cell RNA-seq statistical analysis
For the statistical analysis of the single cell RNA-seq data, we first performed quality control by removing low-quality cells with <500 or >6500 unique genes, <6000 Unique Molecular Identifiers, and >10% reads that mapped to the mitochondrial genome. To account for cell variations due to cell cycle effect, we applied cell cycle scaling on each sample group individually by predicting the cell cycle phase of each cell using canonical gene markers and regressing out the predicted score. To remove batch effects and allow for comparative analyses, we then integrated the datasets for each of the sample groups using the 4000 most variable features. We reduced the feature space of the integrated dataset for analysis using Uniform Manifold Approximation and Projection (UMAP) on the first 30 principal components of a Principal Components Analysis (PCA). The UMAP dimension reduction was calculated using the cosine metric with 50 neighboring points and a minimum distance of 0.5. We then clustered the cells using the FindNeighbors function with default parameters and the FindCluster functions using the Louvain algorithm with multilevel refinement and a resolution of 0.4, resulting in 22 clusters. For each possible cell type in the data, we plotted the gene expression values across the clusters using a self-curated list of known gene markers. We classified the clusters to the cell types that matched the corresponding gene signatures, which we additionally verified by applying the scCATCH R package. We then found gene markers for the Activated HSCs by performing differential expression analysis on the cell type groupings using the default log fold change cut-off of 0.25 with adjusted P-value <0.001. Using the slingshot R package (v1.4.0) we performed pseudotime analysis on the cell type groupings using a PCA dimension reduction and specifying LT-HSCs as the starting cell type and approximating the curves using 100 points. All analyses were performed in R (v3.2.3) using the Seurat R package (v3.6.1) unless otherwise specified.
NP standard curve and H1N1 PR8 tittering
An NP standard curve was generated using our laboratory stock of H1N1 PR8 and forward and reverse primers49 were adjusted for optimized Tm and amplicon and size (PR8NP F1 GGGTGAGAATGGACGAAAAAC and PR8NP R1 GATCCATCATTGCTTTTTGTGCA). To generate our RNA from our H1N1PR8 stock, we isolated RNA using an RNAeasy kit (Qiagen). RNA was reverse transcribed with random hexamer primers using SuperScript IV (Invitrogen) following the manufacturer’s protocol. Serial diluted stocks of influenza cDNA were generated and amplified using the following thermocycling steps:
| Step | Temperature (C) | Time (minutes) |
|---|---|---|
| Initial Denaturation | 95 | 2 |
| Denaturation | 95 | 0.5-1 |
| Annealing | 50 | 0.5-1 |
| Extension | 72 | 0.5 |
| Final Extension | 72 | 5 |
For experimental samples, virus from infected lungs were concentrated using Amicon Ultra 0.5 mL centrifugal filters (Millipore-sigma). Samples were processed similarly to the influenza stock to develop the standard curve and cDNA was normalized to the same concentration prior to completing RT-PCR using iTaq Universal SYBR Green Supermix (BioRad) and H1N1 PR8 primers. Ct values compared to stock standards were used to calculate influenza copy number. Copy number was normalized according to the weight of the starting lung material.
Quantification and statistical analysis
Statistical analysis of experimental results was conducted using GraphPad Prism. All datapoints were assessed for outliers using the ROUT method (<5%) and assessed for normality and lognormality using a Shapiro-Wilk test. Depending on distribution, comparisons between two groups were completed using two-tailed unpaired t-tests or a non-parametric test. For comparisons of three or more groups, ordinary one-way ANOVAs or Kruskal-Wallis tests with Tukey’s multiple comparisons were completed. For assessing survival, the Kaplan-Meyer curve analysis was utilized. For Seahorse assay and influenza weight loss analysis requiring comparisons between multiple timepoints, a repeated measure t-test was completed.
Acknowledgments
The authors would like to thank Meghan Kisiel, Trisha Wathan, Hannah Yan, Hilda Chan, Camryn Lam, and Mara Willis for their technical assistance and all members of the King Lab for their thoughtful suggestions. We wish to acknowledge Catherine Gillespie and Emily Schaffer for help with editing, Courtney Hodges for assistance with genomics related experimental design, and Alexandre Carisey at the Center of Human Immunobiology for technical assistance with microscopy and image analysis. This project depended on the support of the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the CPRIT Core Facility Support Award (CPRIT-RP180672), the NIH (CA125123 and RR024574), and the assistance of Joel M. Sederstrom. Single cell genomic libraries were generated in collaboration with the BCM Single Cell Genomics core and was partially supported by NIH shared instrument grants (S10OD023469, S10OD025240) and P30EY002520. This project was also aided by the Flow Cytometry core at Texas Children’s Hospital, the Center for Human Immunobiology at Texas Children’s Hospital, and the Genomic and RNA Profiling Core at BCM. This project was supported by NIH grants Hematology Training Grant T32DK60445 (BK, LM), T32AI053831 Immunology Training Grant (BK), Cell and Gene Therapy Training Grant T32HL2332 (BT), F31 HL164287 (BT), R01s DK115454 (AC), HL134880, AI141716, and HL136333, P01 CA265748, and R35 HL155672 (KYK). RJ was supported by The Polish National Science Centre grant 2016/23/D/ST7/03665. DEMM is supported by the Howard Hughes Medical Institute (HHMI) Gilliam Fellowship for Advanced Study. Figures were generated using Biorender software.
Author contributions
B.K. and K.Y.K. designed the chimeric trained immunity transplant model, planned and conducted the major experiments, and wrote the paper. P.L. and C.S. conducted the single cell RNA-seq analysis, generated a webtool, and provided expertise related to statistical analysis of single cell datasets. B.T.T. designed the LT-HSC transplant model and optimized LK-cell BMDM differentiation method, planned and performed experiments, and contributed to figure design. R.C. and S.K. assisted with the BMDM functionality experiments’ design, experimentation, and analysis. D.L. and M.F. conducted BMDM functional experiments including killing assays and flow cytometric assays. D.E.M.M. completed the bacterial CFU plating for each trained immunity transplant experiment. L.M. and A.C. provided guidance for experimental design and Seahorse assay execution. J.T. and H.Y. assisted with generating the transplant model and single cell datasets. R.J. analyzed the WGBS data and generated featured plots of differential methylation.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: August 9, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107596.
Supplemental information
Data and code availability
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Original code used for single cell data analysis is available at: https://github.com/KingLabBCM/TrainedImmunity. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Bekkering S., Domínguez-Andrés J., Joosten L.A.B., Riksen N.P., Netea M.G. Trained Immunity: Reprogramming Innate Immunity in Health and Disease. Annu. Rev. Immunol. 2021;39:667–693. doi: 10.1146/annurev-immunol-102119-073855. [DOI] [PubMed] [Google Scholar]
- 2.Domínguez-Andrés J., Joosten L.A., Netea M.G. Induction of innate immune memory: the role of cellular metabolism. Curr. Opin. Immunol. 2019;56:10–16. doi: 10.1016/j.coi.2018.09.001. [DOI] [PubMed] [Google Scholar]
- 3.Butkeviciute E., Jones C.E., Smith S.G. Heterologous effects of infant BCG vaccination: Potential mechanisms of immunity. Future Microbiol. 2018;13:1193–1208. doi: 10.2217/fmb-2018-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arts R.J.W., Moorlag S.J.C.F.M., Novakovic B., Li Y., Wang S.Y., Oosting M., Kumar V., Xavier R.J., Wijmenga C., Joosten L.A.B., et al. BCG Vaccination Protects against Experimental Viral Infection in Humans through the Induction of Cytokines Associated with Trained Immunity. Cell Host Microbe. 2018;23:89–100.e5. doi: 10.1016/j.chom.2017.12.010. [DOI] [PubMed] [Google Scholar]
- 5.Berendsen M.L., van Gijzel S.W., Smits J., de Mast Q., Aaby P., Benn C.S., Netea M.G., van der Ven A.J. BCG vaccination is associated with reduced malaria prevalence in children under the age of 5 years in sub-Saharan Africa. BMJ Glob. Health. 2019;4:e001862. doi: 10.1136/bmjgh-2019-001862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walk J., de Bree L.C.J., Graumans W., Stoter R., van Gemert G.J., van de Vegte-Bolmer M., Teelen K., Hermsen C.C., Arts R.J.W., Behet M.C., et al. Outcomes of controlled human malaria infection after BCG vaccination. Nat. Commun. 2019;10:874. doi: 10.1038/s41467-019-08659-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fanucchi S., Mhlanga M.M. Lnc-ing Trained Immunity to Chromatin Architecture. Front. Cell Dev. Biol. 2019;7:2–7. doi: 10.3389/fcell.2019.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaufmann E., Sanz J., Dunn J.L., Khan N., Mendonça L.E., Pacis A., Tzelepis F., Pernet E., Dumaine A., Grenier J.C., et al. BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis. Cell. 2018;172:176–190.e19. doi: 10.1016/j.cell.2017.12.031. [DOI] [PubMed] [Google Scholar]
- 9.Divangahi M., Aaby P., Khader S.A., Barreiro L.B., Bekkering S., Chavakis T., van Crevel R., Curtis N., DiNardo A.R., Dominguez-Andres J., et al. Trained immunity, tolerance, priming and differentiation: distinct immunological processes. Nat. Immunol. 2021;22:2–6. doi: 10.1038/s41590-020-00845-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Laval B., Maurizio J., Kandalla P.K., Brisou G., Simonnet L., Huber C., Gimenez G., Matcovitch-Natan O., Reinhardt S., David E., et al. C/EBPβ-Dependent Epigenetic Memory Induces Trained Immunity in Hematopoietic Stem Cells. Cell Stem Cell. 2020;26:657–674.e8. doi: 10.1016/j.stem.2020.01.017. [DOI] [PubMed] [Google Scholar]
- 11.Saeed S., Quintin J., Kerstens H.H.D., Rao N.A., Aghajanirefah A., Matarese F., Cheng S.C., Ratter J., Berentsen K., Van Der Ent M.A., et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. 2014;345:1251086. doi: 10.1126/science.1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baldridge M.T., King K.Y., Boles N.C., Weksberg D.C., Goodell M.A. Quiescent haematopoietic stem cells are activated by IFN-γ in response to chronic infection. Nature. 2010;465:793–797. doi: 10.1038/nature09135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.King K.Y., Baldridge M.T., Weksberg D.C., Chambers S.M., Lukov G.L., Wu S., Boles N.C., Jung S.Y., Qin J., Liu D., et al. Irgm1 protects hematopoietic stem cells by negative regulation of IFN signaling. Blood. 2011;118:1525–1533. doi: 10.1182/blood-2011-01-328682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matatall K.A., Jeong M., Chen S., Sun D., Chen F., Mo Q., Kimmel M., King K.Y. Chronic Infection Depletes Hematopoietic Stem Cells through Stress-Induced Terminal Differentiation. Cell Rep. 2016;17:2584–2595. doi: 10.1016/j.celrep.2016.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matatall K.A., Shen C.C., Challen G.A., King K.Y. Type II interferon promotes differentiation of myeloid-biased hematopoietic stem cells. Stem Cell. 2014;32:3023–3030. doi: 10.1002/stem.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morales-Mantilla D.E., Kain B., Le D., Flores A.R., Paust S., King K.Y. Hematopoietic stem and progenitor cells improve survival from sepsis by boosting immunomodulatory cells. Elife. 2022;11:e74561. doi: 10.7554/eLife.74561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moorlag S.J.C.F.M., Khan N., Novakovic B., Kaufmann E., Jansen T., van Crevel R., Divangahi M., Netea M.G. β-Glucan Induces Protective Trained Immunity against Mycobacterium tuberculosis Infection: A Key Role for IL-1. Cell Rep. 2020;31:107634. doi: 10.1016/j.celrep.2020.107634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitroulis I., Ruppova K., Wang B., Chen L.S., Grzybek M., Grinenko T., Eugster A., Troullinaki M., Palladini A., Kourtzelis I., et al. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell. 2018;172:147–161.e12. doi: 10.1016/j.cell.2017.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Zuani M., Frič J. Train the Trainer: Hematopoietic Stem Cell Control of Trained Immunity. Front. Immunol. 2022;13:827250. doi: 10.3389/fimmu.2022.827250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin K.K., Rossi L., Boles N.C., Hall B.E., George T.C., Goodell M.A. CD81 is essential for the re-entry of hematopoietic stem cells to quiescence following stress-induced proliferation via deactivation of the Akt pathway. PLoS Biol. 2011;9:e1001148. doi: 10.1371/journal.pbio.1001148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balise V.D., Saito-Reis C.A., Gillette J.M. Tetraspanin Scaffold Proteins Function as Key Regulators of Hematopoietic Stem Cells. Front. Cell Dev. Biol. 2020;8:598. doi: 10.3389/fcell.2020.00598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grosche L., Knippertz I., König C., Royzman D., Wild A.B., Zinser E., Sticht H., Muller Y.A., Steinkasserer A., Lechmann M. The CD83 Molecule - An Important Immune Checkpoint. Front. Immunol. 2020;11:721. doi: 10.3389/fimmu.2020.00721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim J.W., Park H.Y., Lee M.J., Jang M.J., Lee S.Y., Park Y.M., Son D.H., Chang Y.C., Bae Y.S., Kwak J.Y. Phosphatidic acid and tumor necrosis factor-alpha induce the expression of CD83 through mitogen activated protein kinase pathway in a CD34+ hematopoietic progenitor cell line. Int. Immunopharmacol. 2004;4:1603–1613. doi: 10.1016/j.intimp.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 24.Nakasone H., Kikuchi M., Kawamura K., Akahoshi Y., Sato M., Kawamura S., Yoshino N., Takeshita J., Yoshimura K., Misaki Y., et al. Increased CD83 expression of CD34-positive monocytes in donors during peripheral blood stem cell mobilization in humans. Sci. Rep. 2019;9:16499. doi: 10.1038/s41598-019-53020-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quarmyne M., Doan P.L., Himburg H.A., Yan X., Nakamura M., Zhao L., Chao N.J., Chute J.P. Protein tyrosine phosphatase-sigma regulates hematopoietic stem cell-repopulating capacity. J. Clin. Invest. 2015;125:177–182. doi: 10.1172/JCI77866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mensah F.F.K., Armstrong C.W., Reddy V., Bansal A.S., Berkovitz S., Leandro M.J., Cambridge G. CD24 Expression and B Cell Maturation Shows a Novel Link With Energy Metabolism: Potential Implications for Patients With Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Front. Immunol. 2018;9:2421. doi: 10.3389/fimmu.2018.02421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adachi T., Wakabayashi C., Nakayama T., Yakura H., Tsubata T. CD72 negatively regulates signaling through the antigen receptor of B cells. J. Immunol. 2000;164:1223–1229. doi: 10.4049/jimmunol.164.3.1223. [DOI] [PubMed] [Google Scholar]
- 28.Brosseau C., Colas L., Magnan A., Brouard S. CD9 Tetraspanin: A New Pathway for the Regulation of Inflammation? Front. Immunol. 2018;9:2316. doi: 10.3389/fimmu.2018.02316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williamson A.J.K., Pierce A., Jaworska E., Zhou C., Aspinall-O'Dea M., Lancashire L., Unwin R.D., Abraham S.A., Walker M.J., Cadecco S., et al. A specific PTPRC/CD45 phosphorylation event governed by stem cell chemokine CXCL12 regulates primitive hematopoietic cell motility. Mol. Cell. Proteomics. 2013;12:3319–3329. doi: 10.1074/mcp.M112.024604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leentjens J., Kox M., Stokman R., Gerretsen J., Diavatopoulos D.A., Van Crevel R., Rimmelzwaan G.F., Pickkers P., Netea M.G. BCG vaccination enhances the immunogenicity of subsequent influenza vaccination in healthy volunteers: A randomized, placebo-controlled pilot study. J. Infect. Dis. 2015;212:1930–1938. doi: 10.1093/infdis/jiv332. [DOI] [PubMed] [Google Scholar]
- 31.de Bree L.C.J., Marijnissen R.J., Kel J.M., Rosendahl Huber S.K., Aaby P., Benn C.S., Wijnands M.V.W., Diavatopoulos D.A., van Crevel R., Joosten L.A.B., et al. Bacillus Calmette-Guérin-Induced trained immunity is not protective for experimental influenza A/Anhui/1/2013 (H7N9) infection in mice. Front. Immunol. 2018;9:869. doi: 10.3389/fimmu.2018.00869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaufmann E., Khan N., Tran K.A., Ulndreaj A., Pernet E., Fontes G., Lupien A., Desmeules P., McIntosh F., Abow A., et al. BCG vaccination provides protection against IAV but not SARS-CoV-2. Cell Rep. 2022;38:110502. doi: 10.1016/j.celrep.2022.110502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferreira A.V., Domiguéz-Andrés J., Netea M.G. The Role of Cell Metabolism in Innate Immune Memory. J. Innate Immun. 2022;14:42–50. doi: 10.1159/000512280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.del Fresno C., García-Rio F., Gómez-Piña V., Soares-Schanoski A., Fernández-Ruíz I., Jurado T., Kajiji T., Shu C., Marín E., Gutierrez del Arroyo A., et al. Potent phagocytic activity with impaired antigen presentation identifying lipopolysaccharide-tolerant human monocytes: demonstration in isolated monocytes from cystic fibrosis patients. J. Immunol. 2009;182:6494–6507. doi: 10.4049/jimmunol.0803350. [DOI] [PubMed] [Google Scholar]
- 35.Bowers E., Slaughter A., Frenette P.S., Kuick R., Pello O.M., Lucas D. Granulocyte-derived TNFalpha promotes vascular and hematopoietic regeneration in the bone marrow. Nat. Med. 2018;24:95–102. doi: 10.1038/nm.4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.May M., Slaughter A., Lucas D. Dynamic regulation of hematopoietic stem cells by bone marrow niches. Curr. Stem Cell Rep. 2018;4:201–208. doi: 10.1007/s40778-018-0132-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zebley C.C., Abdelsamed H.A., Ghoneim H.E., Alli S., Brown C., Haydar D., Mi T., Harris T., McGargill M.A., Krenciute G., Youngblood B. Proinflammatory cytokines promote TET2-mediated DNA demethylation during CD8 T cell effector differentiation. Cell Rep. 2021;37:109796. doi: 10.1016/j.celrep.2021.109796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng S.C., Quintin J., Cramer R.A., Shepardson K.M., Saeed S., Kumar V., Giamarellos-Bourboulis E.J., Martens J.H.A., Rao N.A., Aghajanirefah A., et al. MTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345:1250684. doi: 10.1126/science.1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Librán-Pérez M., Costa M.M., Figueras A., Novoa B. β-glucan administration induces metabolic changes and differential survival rates after bacterial or viral infection in turbot (Scophthalmus maximus) Fish Shellfish Immunol. 2018;82:173–182. doi: 10.1016/j.fsi.2018.08.005. [DOI] [PubMed] [Google Scholar]
- 40.Quintin J., Saeed S., Martens J.H.A., Giamarellos-Bourboulis E.J., Ifrim D.C., Logie C., Jacobs L., Jansen T., Kullberg B.J., Wijmenga C., et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. 2012;12:223–232. doi: 10.1016/j.chom.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feng C.G., Weksberg D.C., Taylor G.A., Sher A., Goodell M.A. The p47 GTPase Lrg-47 (Irgm1) links host defense and hematopoietic stem cell proliferation. Cell Stem Cell. 2008;2:83–89. doi: 10.1016/j.stem.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keerthirathne T.P., Magana-Arachchi D.N., Madegedara D., Sooriyapathirana S.S. Real time PCR for the rapid identification and drug susceptibility of Mycobacteria present in Bronchial washings. BMC Infect. Dis. 2016;16:607. doi: 10.1186/s12879-016-1943-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rojony R., Martin M., Campeau A., Wozniak J.M., Gonzalez D.J., Jaiswal P., Danelishvili L., Bermudez L.E. Quantitative analysis of Mycobacterium avium subsp. hominissuis proteome in response to antibiotics and during exposure to different environmental conditions. Clin. Proteomics. 2019;16:39. doi: 10.1186/s12014-019-9260-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gene Ontology Consortium The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 2021;49:D325–D334. doi: 10.1093/nar/gkaa1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hormaechea-Agulla D., Matatall K.A., Le D.T., Kain B., Long X., Kus P., Jaksik R., Challen G.A., Kimmel M., King K.Y. Chronic infection drives Dnmt3a-loss-of-function clonal hematopoiesis via IFNgamma signaling. Cell Stem Cell. 2021;28:1428–1442.e6. doi: 10.1016/j.stem.2021.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jessop F., Buntyn R., Schwarz B., Wehrly T., Scott D., Bosio C.M. Interferon Gamma Reprograms Host Mitochondrial Metabolism through Inhibition of Complex II To Control Intracellular Bacterial Replication. Infect. Immun. 2020;88:e00744-19. doi: 10.1128/IAI.00744-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Er J.Z., Koean R.A.G., Ding J.L. Loss of T-bet confers survival advantage to influenza-bacterial superinfection. EMBO J. 2019;38:e99176. doi: 10.15252/embj.201899176. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Original code used for single cell data analysis is available at: https://github.com/KingLabBCM/TrainedImmunity. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.