

ABSTRACT
HIV-1 must overcome multiple innate antiviral mechanisms to replicate in CD4+ T lymphocytes and macrophages. Previous studies have demonstrated that the apolipoprotein B mRNA editing enzyme polypeptide-like 3 (APOBEC3, A3) family of proteins (at least A3D, A3F, A3G, and stable A3H haplotypes) contribute to HIV-1 restriction in CD4+ T lymphocytes. Virus-encoded virion infectivity factor (Vif) counteracts this antiviral activity by degrading A3 enzymes allowing HIV-1 replication in infected cells. In addition to A3 proteins, Vif also targets other cellular proteins in CD4+ T lymphocytes, including PPP2R5 proteins. However, whether Vif primarily degrades only A3 proteins during viral replication is currently unknown. Herein, we describe the development and characterization of A3F-, A3F/A3G-, and A3A-to-A3G-null THP-1 cells. In comparison to Vif-proficient HIV-1, Vif-deficient viruses have substantially reduced infectivity in parental and A3F-null THP-1 cells, and a more modest decrease in infectivity in A3F/A3G-null cells. Remarkably, disruption of A3A–A3G protein expression completely restores the infectivity of Vif-deficient viruses in THP-1 cells. These results indicate that the primary function of Vif during infectious HIV-1 production from THP-1 cells is the targeting and degradation of A3 enzymes.
IMPORTANCE
HIV-1 Vif neutralizes the HIV-1 restriction activity of A3 proteins. However, it is currently unclear whether Vif has additional essential cellular targets. To address this question, we disrupted A3A to A3G genes in the THP-1 myeloid cell line using CRISPR and compared the infectivity of wild-type HIV-1 and Vif mutants with the selective A3 neutralization activities. Our results demonstrate that the infectivity of Vif-deficient HIV-1 and the other Vif mutants is fully restored by ablating the expression of cellular A3A to A3G proteins. These results indicate that A3 proteins are the only essential target of Vif that is required for fully infectious HIV-1 production from THP-1 cells.
KEYWORDS: HIV-1, APOBEC3, G-to-A mutations, deaminase-dependent mechanism, deaminase-independent mechanism, Vif
INTRODUCTION
The A3 family of proteins comprise seven single-strand DNA cytosine deaminases (A3A–A3D and A3F–A3H) in humans (1 - 3). A3 enzymes have broad and essential roles in innate antiviral immunity against parasitic DNA-based elements (4 - 6). Retroviruses are sensitive to A3 enzyme activity due to the obligate step of reverse transcription during viral replication that produces single-stranded cDNA intermediates. These viral cDNA intermediates can act as substrates for A3 enzymes, as demonstrated by C-to-U deamination resulting in G-to-A mutations in the genomic strand. To date, the best-characterized substrate of A3 enzymes is human immunodeficiency virus type 1 (HIV-1). In CD4+ T lymphocytes, four A3 proteins (A3D, A3F, A3G, and stable A3H haplotypes) restrict HIV-1 replication by mutating viral cDNA intermediates and by physically blocking reverse transcription (7 - 14). A3 enzymes have a preference for specific dinucleotide motifs (5´-CC for A3G and 5´-TC for other A3 enzymes) at target cytosine bases, which appear as 5´-AG or 5´-AA mutations in the genomic strand (7, 8, 15, 16).
Virus-encoded virion infectivity factor (Vif) functions in disrupting the activity of A3 enzymes. Vif forms an E3 ubiquitin ligase complex that degrades A3 enzymes through a proteasome-mediated pathway (2, 3, 17, 18). The central domain of this complex is a Vif heterodimer with the transcription factor, core binding factor subunit β (CBF-β), which stabilizes Vif during disruption of A3 protein activity (19, 20). Vif also suppresses the transcription of A3 enzymes by hijacking RUNX/CBF-β complex (21). In addition to these Vif-dependent mechanisms, HIV-1 reverse transcriptase and protease have been shown to disrupt the activity of A3 enzymes via Vif-independent mechanisms (22, 23). Recently, functional proteomic analyses have demonstrated that Vif has several target proteins, including the PPP2R5 family of proteins, in CD4+ T cell lines and lymphocytes (24, 25). These findings indicate that Vif may have additional essential target proteins during HIV-1 infection.
We previously reported that endogenous A3G protein contributes to HIV-1 restriction in a deaminase-dependent manner in THP-1 cells (26). Although disruption of the A3G gene nearly eliminates viral G-to-A mutations, Vif-deficient HIV-1 virions have 50% lower infectivity than wild-type HIV-1 or mutants selectively lacking A3G degradation activity (26). These results indicated that Vif-mediated inhibition of A3G and at least one additional A3 proteins is required for efficient infectious HIV-1 production.
In the present study, we evaluate the effects of other A3 proteins on HIV-1 infectivity by developing and characterizing A3F-, A3F/A3G-, and A3A-to-A3G-null THP-1 cells using HIV-1 Vif mutants with selective A3 neutralization activities. In comparison to wild-type HIV-1, Vif-deficient HIV-1 infectivity is strongly inhibited in A3F-null THP-1 cells and modestly inhibited in A3F/A3G-null THP-1 cells. In contrast, an HIV-1 Vif mutant selectively lacking A3F degradation activity had comparable infectivity to wild-type HIV-1 in A3F-null THP-1 cells and 50% infectivity in parental THP-1 cells, indicating that A3F protein contributes to HIV-1 restriction in THP-1 cells. Furthermore, Vif-deficient HIV-1 infectivity is comparable to wild-type HIV-1 in A3A-to-A3G-null THP-1 cells. These results demonstrate that A3 proteins are the primary target of HIV-1 Vif during infectious virus production from THP-1 cells.
RESULTS
Endogenous A3H protein is not involved in HIV-1 restriction in THP-1 cells
THP-1 cells express significant levels of A3B, A3C, A3F, A3G, and A3H mRNA (26). The results of our previous study indicated that A3G and at least one additional A3 proteins are involved in HIV-1 restriction in THP-1 cells (26). Variations in the amino acid sequence of A3 family proteins are known to influence HIV-1 restriction activity (27), and the A3H gene is the most polymorphic of all human A3 genes (10, 22, 28, 29). The A3H allele is grouped into stable and unstable haplotypes according to the combination of amino acid residues at positions 15, 18, 105, 121, and 178 (10, 22, 28, 29). Stable A3H haplotypes are active against HIV-1, whereas unstable A3H haplotypes have absent or minimal activity as they encode proteins with low stability (9, 10, 22, 29, 30). To determine A3H genotypes, we sequenced A3H cDNA from THP-1 cells. Sequencing data identified an unstable haplotype in the THP-1 genome, termed A3H hapI gene (Fig. 1A). These data suggest that endogenous A3H protein has minimal restriction activity against Vif-deficient HIV-1 in THP-1 cells.
Fig 1.

Endogenous A3H protein does not inhibit HIV-1 in THP-1 cells. (A) A3H haplotypes in THP-1 cells. The indicated positions are key amino acid residues that determine the expression of unstable (hapI) or stable (hapII) A3H protein. (B) Schematic of the susceptibility of HIV-1 Vif mutants to antiviral activity by stable A3H haplotypes. Key amino acid residues of Vif that determine the susceptibility of HIV-1 IIIB to restriction by stable A3H haplotypes. −, full resistance; +, partial resistance; +++, sensitivity. (C) Schematic depiction of the pseudo-single cycle infectivity assay. For details, see the main text and the “Pseudo-single cycle infectivity assays” section in Materials and Methods. (D) Representative infectivity of HIV-1 mutants with hyper- and hypo-functional Vif produced from THP-1 cells. Top panels show the infectivity of hyper-Vif, hypo-Vif, IIIB Vif, and Vif-deficient HIV-1 mutants produced in THP-1 cells. The amounts of produced viruses used to infect TZM-bl cells were normalized to p24 levels. Each bar shows the average of four independent experiments with standard deviation (SD). Data are represented as relative infectivity compared to hyper-Vif HIV-1. Statistical significance was determined using the two-sided paired t test. *P < 0.05 compared with the infectivity of hyper-Vif HIV-1. The bottom panels are representative western blots of three independent experiments. The levels of viral and cellular proteins in viral particles and whole-cell lysates are shown. p24 and HSP90 were used as loading controls. (E) G-to-A mutations. Average number of G-to-A mutations in the 564 bp pol gene after infection with hyper-Vif, hypo-Vif, IIIB Vif, or Vif-deficient HIV-1 produced from THP-1. Each bar depicts the average of three independent experiments with SD. (F) G-to-A mutation profile. Dinucleotide sequence contexts of G-to-A mutations in the 564 bp pol gene after infection with the indicated viruses produced from indicated cell lines. Each vertical line indicates the location of the dinucleotide sequence contexts described in the legend within the 564 bp amplicon (horizontal line).
The A3H hapI protein results in expression of an unstable protein that has weak anti-HIV-1 activity (28, 29, 31). However, this protein is enzymatically active and has an HIV-1 restriction phenotype similar to the stable A3H haplotype, A3H hapII protein, when both proteins are expressed at the same levels (31). In addition, A3H protein expression levels are upregulated during HIV-1 infection (10, 22), and A3H hapI protein is resistant to Vif-mediated degradation (32). Accordingly, we evaluated whether the expression of A3H hapI protein is associated with HIV-1 restriction in THP-1 cells. To address this question, we utilized HIV-1 Vif mutants that selectively degrade stable A3H protein (hyper-functional Vif; hyper-Vif) or lack stable A3H degradation (hypo-functional Vif; hypo-Vif) (10) (Fig. 1B). IIIB Vif displays an intermediate phenotype (Fig. 1B). Of note, hyper-Vif, hypo-Vif, and IIIB Vif have full neutralization activity against A3D, A3F, and A3G proteins (10).
To allow efficient and equivalent delivery of HIV-1 to THP-1 and its derivatives, vesicular stomatitis virus G glycoprotein (VSV-G) pseudotyped full-length HIV-1 and its Vif mutants were produced from 293T cells and titrated with CEM-GXR cells for determination of the multiplicity of infection (MOI) as described (23, 26) (Fig. 1C). Then, these VSV-G pseudotyped infectious viruses were used to infect SupT11 (MOI = 0.05) and THP-1 (MOI = 0.25) cells, thereby creating virus-producing cells (Fig. 1C). The MOI calculations are based on titers determined by CEM-GXR cells and these MOIs therefore may not reflect the true MOI on SupT11 or THP-1 cells. The resultant infectious viruses were used to measure viral infectivity in TZM-bl cells, evaluate the packaging of A3 proteins by western blotting, and analyze the frequency of G-to-A mutations (Fig. 1C). Hereafter, this assay is referred to as pseudo-single cycle infectivity assay (see the “Pseudo-single cycle infectivity assays” section in the Materials and Methods for details).
The susceptibility of hyper-Vif and hypo-Vif HIV-1 to stable A3H hapII protein was validated in SupT11 cell lines (Fig. S1A). In SupT11-vector cells, hyper-Vif, hypo-Vif, IIIB Vif, and Vif-deficient HIV-1 had comparable infectivity with TZM-bl cells (Fig. S1A, top panel). As expected, the infectivity of hypo-Vif and Vif-deficient HIV-1 was restricted in SupT11-A3H hapII cells because these mutants are unable to degrade A3H hapII protein, leading to packaging of A3H hapII protein into viral particles (Fig. S1A, bottom panel). The partial degradation of A3H hapII protein by IIIB Vif resulted in moderate inhibition of IIIB Vif HIV-1 infectivity (Fig. S1A). The infection of hyper-Vif HIV-1 resulted in A3H hapII degradation, where stable A3H hapII protein is undetectable in the viral particles and unable to inhibit the Vif mutant (Fig. S1A). Next, to determine whether G-to-A mutations were introduced into proviral DNA, we recovered proviral DNA from SupT11 cells after infection with each HIV-1 mutant produced from either SupT11-vector or SupT11-A3H hapII cells and sequenced the pol region. Hypo-Vif and Vif-deficient HIV-1 showed G-to-A mutations preferred by A3H protein (GA-to-AA signature motif; hypo-Vif: 2.8 ± 0.7 mutations/kb and ∆Vif: 3.2 ± 0.8 mutations/kb, respectively) in proviral DNA (Fig. S1B and C). These results are consistent with previous reports demonstrating the susceptibility of Vif mutants to A3H hapII protein (10).
As shown in Fig. 1D (top panel), hyper-Vif HIV-1, hypo-Vif HIV-1, and IIIB Vif HIV-1 (IIIB) produced in THP-1 cells had similar viral infectivity. While Vif did not degrade A3H protein in THP-1 cells, it was not packaged into viral particles (Fig. 1D, bottom panel). To examine whether G-to-A mutations were introduced into proviral DNA, we sequenced the pol region of the proviruses from SupT11 cells after infection with each HIV-1 mutant produced from THP-1 cells. Sequencing data demonstrated that hyper-Vif HIV-1, hypo-Vif HIV-1, and IIIB Vif HIV-1 had minimal G-to-A mutations preferred by A3H protein (GA-to-AA signature motif) in proviral DNA (Fig. 1E and F), indicating that endogenous A3H protein expressed in THP-1 cells is not involved in HIV-1 restriction. In contrast, the infectivity of Vif-null HIV-1 was restricted in THP-1 cells and A3G protein was packaged into viral particles, thereby inducing profound G-to-A mutations (10.3 ± 3.5 mutations/kb) (Fig. 1D through F). Most of mutations were in the GG-to-AG signature motif preferred by A3G protein (80 ± 10%) in proviral DNA (Fig. 1E and F). Taken together, these results indicate that A3G and perhaps other A3 proteins, but unlikely A3H protein, contribute to HIV-1 restriction in THP-1 cells.
Development of A3F-, A3F/A3G-, and A3A-to-A3G-null THP-1 cells
A3F protein has a restrictive effect on HIV-1 among A3 family members and is a target of Vif, in addition to A3G protein, in CD4+ T cell lines and lymphocytes (7, 33 - 35). To determine whether A3F protein also reduces HIV-1 infectivity in THP-1 cells, we used CRISPR to create A3F and A3F/A3G gene knockout cell lines. Two independent subclones of A3F and A3F/A3G-null THP-1 cells were obtained, as evidenced by the results of genomic DNA sequencing and western blotting (Fig. S2).
A3 proteins include single- and double-domain deaminases and are phylogenetically classified into three groups: Z1, Z2, and Z3 domains (3, 4) (Fig. 2A represented in green, orange, and blue, respectively). A3A, A3B carboxy-terminal domain (CTD), and A3G CTD proteins are classified as Z1 domains (Fig. 2A, represented in green). Of note, exon 4 of the A3A gene, exon 7 of the A3B gene, and exon 7 of A3G gene are highly conserved at the nucleotide level (A3A exon 4 and A3B exon 7 have 95% identity; A3A exon 4 and A3G exon 7 have >99% identity; and A3B exon 7 and A3G exon 7 have 95% identity, respectively). Interestingly, each of these exons has an identical sequence (5´-GAG TGG GAG GCT GCG GGC CA). We therefore designed a guide RNA (gRNA) homologous to this sequence and used it to delete the entire 125 kbp interval spanning A3A to A3G genes in THP-1 cells (Fig. 2A, represented in arrows, and Fig. S3). We predicted that successful Cas9-mediated cleavage would cause one of the following three scenarios: (i) fusion of exon 4 of the A3A gene with exon 7 of the A3B gene (30 kbp deletion); (ii) fusion of exon 7 of the A3B gene with exon 7 of the A3G gene (95 kbp deletion); or (iii) fusion of exon 4 of the A3A gene with exon 7 of the A3G gene (125 kbp deletion; Fig. 2A). To obtain THP-1 cells lacking expression of A3A to A3G proteins, a lentiviral vector expressing gRNA against the target sequence was transduced into THP-1 cells. Finally, two independent subclones (THP-1#11-4 and THP-1#11-7) were obtained, with whole-genome sequencing (WGS) analysis demonstrating an extensive deletion between A3A exon 4 and A3G exon 7 at the A3 gene locus (Fig. 2B). In THP-1#11-4, six alleles of the fusion of A3A exon 4 with A3G exon 7 are observed, and each A3A/A3G hybrid exon had six different insertions or deletions (indels) (Fig. S3). THP-1#11-7 harbors three alleles of A3A exon 4 and A3G exon 7 fusions (one may be A3A exon 4) with three different deletions (Fig. S3). Although more than 20 potential off-target sites with two or three nucleotides mismatched with the designed gRNA were predicted, a significant deletion was only found downstream of the predicted A3G pseudogene harboring 2 bp mismatched with the target sequence (Fig. S4; potential target sequence in an orange box and deletions indicated by green dotted lines). In comparison to parental THP-1 cells, these subclones had similar growth capacities under normal cell culture conditions. Reverse transcription-quantitative PCR (RT-qPCR) analyses demonstrated that A3B to A3G mRNAs are not detectable in either clone (Fig. 2C). However, A3A mRNA expression remained detectable in parental THP-1 cells and the two subclones as the A3A promoter remains intact and potentially functional (Fig. 2A through C). A3A mRNA expression is known to be upregulated 100- to 1,000-fold in THP-1 cells treated with type I interferon (IFN) (36). To confirm the expression of A3A mRNA and protein in THP-1 cells, parental THP-1 cell and the respective subclones were cultured in the presence of type I IFN for 6 h, and A3 mRNA and protein expression levels were then analyzed by RT-qPCR and western blotting, respectively. In parental THP-1 cells, A3A, A3B, A3F, and A3G mRNA and protein expression levels were increased following IFN treatment (Fig. 2C and D). In the THP-1#11-4 subclone, A3A mRNA expression is increased following IFN treatment; however, A3A, A3B, A3C, A3F, and A3G proteins are not detectable, even after IFN treatment (Fig. 2C and D). Furthermore, A3A to A3G proteins are not detectable in the THP-1#11-7 subclone under normal cell culture conditions (Fig. 2D). Interestingly, low levels of a protein with comparable size to A3A protein are detected in the THP-1#11-7 subclone after IFN treatment (Fig. 2D). Sanger sequence analyses indicated that this protein was an A3A and A3G hybrid with a 3 bp deletion (Fig. S3). Collectively, these data indicate that the THP-1#11-4 and THP-1#11-7 subclones lack expression of A3A to A3G proteins under normal cell culture conditions and that clone THP-1#11-4 is a clean knockout that fails to express functional versions of any of these proteins.
Fig 2.

Disruption of the A3A to A3G genes in THP-1 cells. (A) Schematic of the A3 gene at the A3 locus. The A3 family of genes comprises seven members with one or two Z domains (single- or double-domain deaminases) which belong to three phylogenetically distinct groups shown in green, orange, and blue. Three sites with an identical sequence (5´-GAG TGG GAG GCT GCG GGC CA) in exon 4 of the A3A gene, exon 7 of the A3B gene, and exon 7 of the A3G gene are targeted by gRNA, as indicated by arrows. The three predicted scenarios are shown. Bar represents 15,000 bp. (B) Mapping of WGS data to the A3 locus. Genomic DNAs from parental THP-1, THP-1#11-4, and #11-7 cells were subjected to WGS analysis, with an extensive deletion including the A3A–A3G genes observed in THP-1#11-4 and #11-7 clones. (C) RT-qPCR data. Parental THP-1, THP-1#11-4, and #11-7 cells were treated with 500 units/mL type I IFN. Total RNA was isolated after 6 h. A3 mRNA expression levels were quantified by RT-qPCR and are normalized to TATA-binding protein (TBP) mRNA levels. Each bar represents the average of three independent experiments with SD. Statistical significance was determined using the two-sided paired t test. *, P < 0.05 compared to untreated cells. (D) Representative western blots of three independent experiments. Levels of indicated A3 proteins in whole-cell lysates from cells treated with or without type I IFN are shown. HSP90 was used as a loading control.
Disruption of A3A to A3G protein expression fully restores the infectivity of Vif-deficient HIV-1 in THP-1 cells
We next determined whether endogenous A3F protein is degraded by Vif in addition to A3G protein. HIV-1 Vif mutants with selective A3 neutralization activities were used for pseudo-single cycle infectivity assays as mentioned above. For example, a Vif4A mutant harboring 14AKTK17 substitutions (14DRMR17 in IIIB) is susceptible to HIV-1 restriction activity by A3D and A3F proteins but resistant to the restriction by A3G protein (37 - 39) (Fig. 3A). We examined the ability of Vif4A to counteract the restriction activity of A3D and A3F proteins, although A3D protein could not be detected by western blotting (anti-A3D antibodies are unavailable) and it may be inconsequential because its mRNA expression levels are relatively low in this cell line (26) (Fig. 2C). As our group and others have shown previously (26, 37, 38, 40), HIV-1 with Vif5A containing five alanine substitutions (40YRHHY44 to 40AAAAA44) is sensitive to the HIV-1 restriction activity of A3G protein but not A3D and A3F proteins (Fig. 3A). HIV-1 harboring Vif4A5A is susceptible to inhibition by A3D, A3F and A3G proteins (37) (Fig. 3A). VSV-G pseudotyped HIV-1 and these Vif mutants were used to infect SupT11 derivatives and engineered A3F-null THP-1 cells. First, the susceptibilities of these Vif mutants to A3F and A3G proteins were validated in SupT11 cell lines (Fig. S5A). In SupT11-vector cells, Vif-proficient HIV-1 and all Vif mutants had comparable infectivity in TZM-bl cells (Fig. S5A). As expected, the infectivity of Vif-deficient HIV-1 and the Vif4A and 4A5A mutants was reduced in SupT11-A3F cells as these mutants are unable to degrade A3F protein, thereby leading to packaging of A3F protein in viral particles (Fig. S5A). Further, infection with Vif-deficient HIV-1 or the Vif5A and Vif4A5A mutants resulted in packaging of A3G protein in viral particles from SupT11-A3G cells in addition to reduced infectivity of these Vif mutants (Fig. S5A). These results are consistent with previous reports demonstrating the susceptibilities of Vif mutants to A3 proteins (26, 37 - 40).
Fig 3.
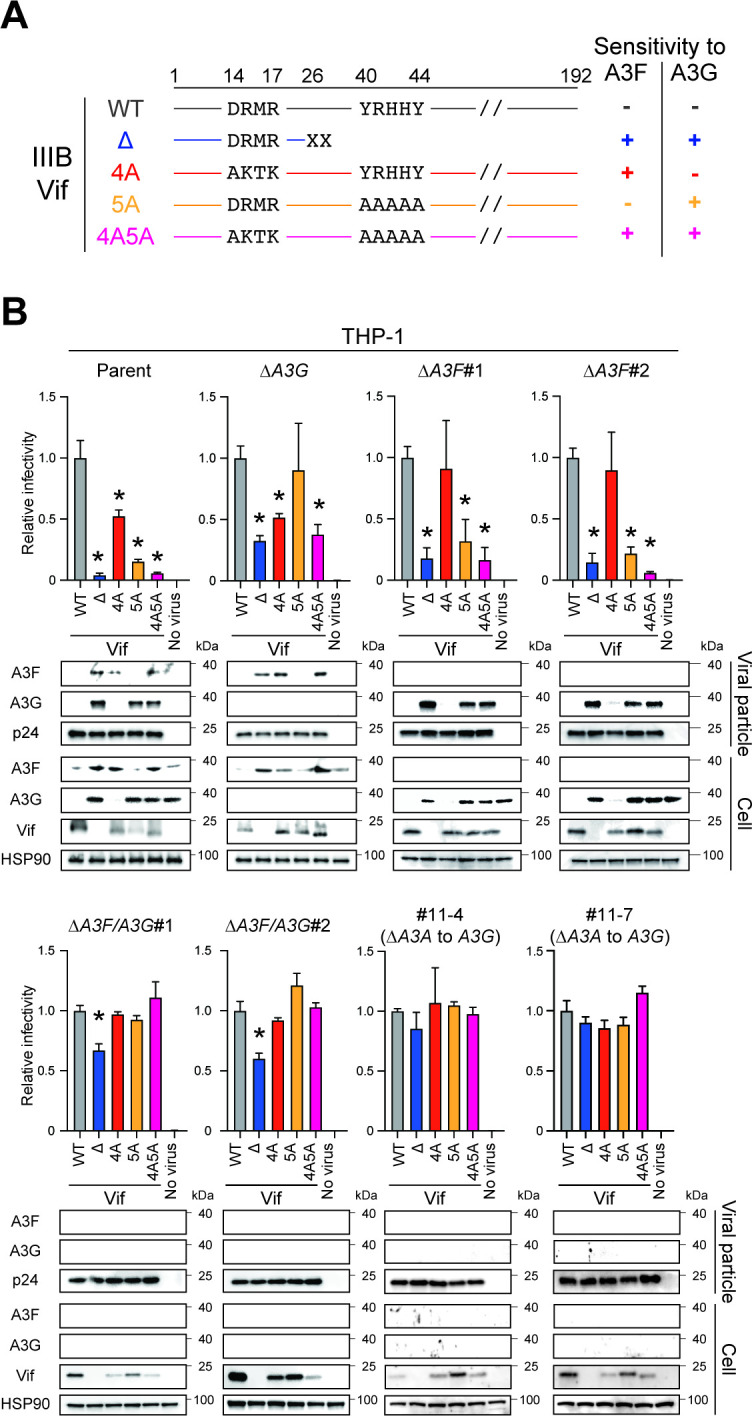
Pseudo-single cycle infectivity assays for each HIV-1 mutant in A3-null THP-1 cells. (A) Schematic of the susceptibility of HIV-1 Vif mutants to HIV-1 restriction activity by A3F and A3G proteins. Key amino acid residues of Vif that determine the susceptibility of HIV-1 IIIB to restriction by A3F and A3G proteins. −, resistance; +, sensitivity. (B) Representative infectivity of Vif-proficient, Vif-deficient, Vif4A, Vif5A, and Vif4A5A HIV-1 mutants produced from parental or A3-null THP-1 cells. Top panels show the infectivity of indicated HIV-1 mutants produced in parental or A3-null THP-1 cells. The amounts of produced viruses used to infect TZM-bl cells were normalized to p24 levels. Each bar represents the average of four independent experiments with SD. Data are presented as infectivity relative to Vif-proficient HIV-1 (WT). Statistical significance was assessed using the two-sided paired t test. *P < 0.05 compared to Vif-proficient HIV-1. Bottom panels are representative western blots of three independent experiments. Levels of indicated viral and cellular proteins in viral particles and whole-cell lysates are shown. p24 and HSP90 were used as loading controls.
Pseudo-single cycle infectivity assays were then performed in parental THP-1, A3G-null, and A3F-null cells using these Vif mutants. Vif-proficient HIV-1 degraded A3F and A3G proteins in THP-1 cells, and lower amounts of these A3 proteins were packaged into viral particles (Fig. 3B; THP-1 parent). In contrast, Vif-deficient HIV-1 was unable to degrade A3F and A3G proteins, thereby leading to reduced viral infectivity compared to Vif-proficient HIV-1 (Fig. 3B; THP-1 parent). The infectivity of A3F-susceptible Vif mutants, Vif4A and Vif4A5A, was lower than that of Vif-proficient HIV-1, indicating that endogenous A3F protein contributes to Vif-deficient HIV-1 restriction in THP-1 cells (Fig. 3B; THP-1 parent). This finding was supported by results in A3G-null THP-1 cells where Vif4A mutants are restricted, as observed in parental THP-1 cells (Fig. 3B; THP-1 ∆A3G). The involvement of endogenous A3G protein in HIV-1 restriction was confirmed in A3G-null THP-1 cells, as reported (26) (Fig. 3B; THP-1 ∆A3G). To determine whether endogenous A3F protein contributes to HIV-1 restriction in THP-1 cells, pseudo-single cycle infectivity assays were performed according to the methods described above in two independent A3F-null THP-1 clones (Fig. 1C; Fig. S2A and B). Vif-deficient HIV-1 and the Vif5A and Vif4A5A mutants had reduced infectivity in A3F-null subclones due to the inhibitory effect of A3G protein (Fig. 3B; THP-1 ∆A3F#1 and #2). However, the infectivity of the Vif4A mutant was restored to near wild-type levels following disruption of A3F gene expression in THP-1 cells. These data demonstrate that endogenous A3F and potentially A3D proteins contribute to Vif-deficient HIV-1 restriction in THP-1 cells, and that Vif degrades A3F protein and thereby prevents packaging and restriction upon target cell infection.
A3F and A3G proteins are involved in Vif-deficient HIV-1 restriction in THP-1 cells and are degraded by Vif (26) (Fig. 3B). However, it is unclear whether only these A3 proteins are associated with Vif-deficient HIV-1 restriction in THP-1 cells. To address this issue, we performed pseudo-single cycle infectivity assays in A3F/A3G-null THP-1 cells using separation-of-function Vif mutants. Although Vif-deficient HIV-1 had greater infectivity defects in parental, A3G-null, and A3F-null THP-1 cells compared to wild-type HIV-1 (parent: <10% infectivity, ∆A3G: 30% to 40% infectivity, and ∆A3F: 20% infectivity, respectively), the infectivity of Vif-deficient HIV-1 was 30% lower in A3F/A3G-null THP-1 cells (Fig. 3B; THP-1 parent, ∆A3G, ∆A3F#1 and #2, and ∆A3F/A3G#1 and #2). On the other hand, the Vif4A, Vif5A, and Vif4A5A mutants had similar infectivity to wild-type HIV-1 in A3F/A3G-null THP-1 cells (Fig. 3B; THP-1 ∆A3F/A3G#1 and #2). These data indicate that other A3 proteins, in addition to A3F and A3G proteins, contribute to Vif-deficient HIV-1 restriction in THP-1 cells or that Vif disrupts an additional essential target during infectious virus production from THP-1 cells.
The universally recognized primary target of Vif is the A3 family of proteins (2, 3, 17, 18). However, Vif-mediated A3 degradation may mask an additional A3-independent Vif function required for fully infectious virus production. To address this issue, we constructed two independent A3A-to-A3G-null THP-1 clones (Fig. 2) and characterized HIV-1 infection using pseudo-single cycle infectivity assays with Vif mutants. As mentioned above, the disruption of A3F and A3G protein expression results in Vif-deficient HIV-1 having 70% of wild-type HIV-1 infectivity in THP-1 cells (Fig. 3B; THP-1 ∆A3F/A3G#1 and #2). Remarkably, Vif-deficient HIV-1 and the other Vif mutants have comparable infectivity to Vif-proficient HIV-1 lacking expression of A3A to A3G proteins in THP-1 cells (Fig. 3B; THP-1#11-4 and #11-7). These results indicate that A3 degradation is the only function of Vif required for fully infectious virus production from THP-1 cells.
A3 proteins restrict HIV-1 infectivity via deaminase-dependent and deaminase-independent mechanisms in THP-1 cells
Our previous results indicated that A3G protein is the primary source of A3 mutagenesis in THP-1 cells (26). To further investigate the G-to-A mutation spectra in each A3-null THP-1 subclone, the pol region was cloned and sequenced from the proviruses used in the aforementioned infectivity assays. As expected, GG-to-AG mutations are observed in the proviral DNA of Vif mutants lacking A3G neutralization activity (Vif-deficient HIV-1 and Vif5A and Vif4A5A mutants) produced from SupT11-A3G cells (Fig. S5B and C; SupT11-A3G). Consistent with a previous report (26), THP-1 expresses A3G protein capable of mutating A3G-susceptible Vif mutants, including Vif-deficient HIV-1 and Vif5A and Vif4A5A mutants, as seen in parental THP-1 cells. These GG-to-AG mutations are not observed in A3G-null THP-1 cells (Fig. 4A and B; THP-1 parent and ∆A3G). Similarly, GG-to-AG mutations preferred by A3G protein were seen in the proviruses of the A3G-susceptible Vif mutants produced from two independent A3F-null THP-1 cells, with disruption of A3G protein nearly completely eliminating these mutations in THP-1 cells (Fig. 4A and B; THP-1 ∆A3F#1 and #2, ∆A3F/A3G#1 and #2, #11-4, and #11-7). These data indicate that A3G protein is the primary source of GG-to-AG mutations in HIV-1 proviruses produced by THP-1 cells.
Fig 4.

A3 proteins inhibit Vif-deficient HIV-1 by both deaminase-dependent and -independent mechanisms in THP-1 cells. (A) G-to-A mutations. Average number of G-to-A mutations in the 564 bp pol gene after infection with hyper-Vif, hypo-Vif, IIIB Vif, or Vif-deficient HIV-1 produced from parental or A3-null THP-1 cells. Each bar depicts the average of three independent experiments with SD. (B) G-to-A mutation profile. Dinucleotide sequence contexts of G-to-A mutations in the 564 bp pol gene after infection with the indicated viruses produced from indicated cell lines. Each vertical line indicates the location of the dinucleotide sequence contexts described in the legend within the 564 bp amplicon (horizontal line). (C) Representative LRT quantification data for Vif-proficient, Vif-deficient, Vif4A, Vif5A, and Vif4A5A HIV-1 mutants produced from each A3-null THP-1 subclone. Data show LRT products of the indicated HIV-1 mutants produced in parental or indicated A3-null THP-1 cells. The amount of produced viruses used to infect SupT11 cells was normalized to p24 levels. LRT products were measured by qPCR. Each bar represents the average of four independent experiments with SD. LRT products were normalized to the quantity of the CCR5 gene relative to Vif-proficient HIV-1 (WT). Statistical significance was assessed using the two-sided paired t test. *P < 0.05 compared to Vif-proficient HIV-1 LRT products.
Although the Vif mutants lacking A3F neutralization activity (Vif-deficient HIV-1 and Vif4A and Vif4A5A mutants) produced from SupT11-A3F cells have a relatively low number of G-to-A mutations, the observed G-to-A mutations are predominantly within the GA-to-AA sequence motif preferred by A3F protein (Fig. S5B and C; SupT11-A3F). However, A3F-preferred GA-to-AA mutations are not observed in proviruses of A3F-susceptible Vif mutants produced from parental or A3G-null THP-1 cells, in support of prior observations (26) (Fig. 4A and B; THP-1 parent and ∆A3G). In addition, fewer GA-to-AA mutations are observed in THP-1 cells, even after disruption of A3F protein expression (Fig. 4A and B; THP-1 ∆A3F#1 and #2, ∆A3F/A3G#1 and #2, #11-4, and #11-7). Accordingly, these results combine to indicate that A3F protein in THP-1 cells is involved in Vif-deficient HIV-1 restriction via a deaminase-independent mechanism.
A3F protein has been shown to inhibit the accumulation of reverse transcription (RT) products (14). To investigate a potential effect on RT, SupT11 cells were infected with viruses from the pseudo-single cycle infectivity assays described above, and late RT (LRT) products were examined by quantitative PCR (qPCR). As expected, all Vif mutants were decreased in LRT products in comparison to wild-type virus when these mutants were produced in parental THP-1 cells and used to infect SupT11 cells (Fig. 4C; THP-1 parent). LRT products of Vif5A and Vif4A mutants were restored to levels comparable to Vif-proficient HIV-1 following the disruption of A3G or A3F protein expression in THP-1 cells (Fig. 4C; THP-1 ∆A3G and ∆A3F#1 and #2), indicating that both A3G and A3F proteins inhibit HIV-1 via a deaminase-independent mechanism. However, double knockout of A3G and A3F proteins in THP-1 cells did not increase the LRT products of Vif-deficient HIV-1 compared to those of Vif-proficient virus (Fig. 4C; THP-1 ∆A3F/A3G#1 and #2), indicating that other A3 proteins, in addition to A3F and A3G proteins, may contribute to the restriction of HIV-1 in THP-1 cells via a deaminase-independent mechanism or that a separate protein targeted by Vif blocks the accumulation of RT products. To test this hypothesis, we measured LRT products by infecting SupT11 cells with HIV-1 Vif mutants produced in A3A-to-A3G-null clones. Consistent with the results of the pseudo-single cycle infectivity assays (Fig. 3B), Vif-deficient HIV-1 and other Vif mutants had comparable levels of LRT products to Vif-proficient HIV-1 lacking expression of A3A to A3G proteins in THP-1 cells (Fig. 4C; THP-1#11-4 and #11-7). These data indicate that Vif-mediated A3 degradation is required for fully infectious virus production from THP-1 to counteract deaminase-dependent and -independent HIV-1 restriction by A3 proteins.
Transmitted/founder (TF) HIV-1 Vif also only targets A3 family proteins to enable fully infectious virus production from THP-1 cells
As an additional experiment, we examined whether the A3-dependent function of Vif was present in TF viruses. To address this issue, Vif-proficient and deficient versions of the CH58 TF virus were produced from parental THP-1 and A3A-to-A3G-null cells, with viral infectivity measured in TZM-bl cells (Fig. 5). Similar to the results observed with IIIB viruses, Vif-deficient CH58 virus was restricted in parental THP-1 cells; however, this restriction is completely abolished by disruption of the A3A to A3G genes (Fig. 5). These data indicate that TF viruses also utilize a primarily A3-dependent function of Vif during infectious HIV-1 production from THP-1 cells.
Fig 5.
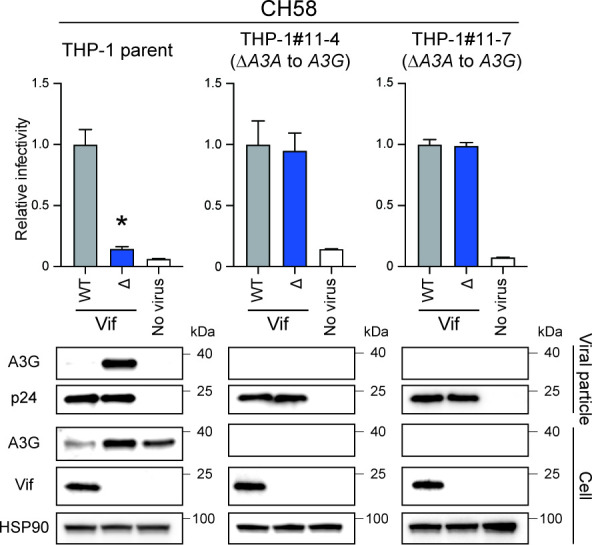
Pseudo-single cycle infectivity assays of TF virus molecular clone in A3A-to-A3G-null THP-1 cells. Infectivity of Vif-proficient and Vif-deficient CH58 viruses. Top panels show the infectivity of Vif-proficient and Vif-deficient HIV-1 produced from parental THP-1, THP-1#11-4, or THP-1#11-7 cells. The amounts of produced viruses used to infect TZM-bl cells were normalized to p24 levels. Each bar represents the average of four independent experiments with SD. Data are represented as relative to Vif-proficient HIV-1 (WT). Statistical significance was assessed using the two-sided paired t test. *P < 0.05 compared to Vif-proficient HIV-1. The bottom panels are representative western blots of three independent experiments. The levels of indicated viral and cellular proteins in viral particles and whole-cell lysates are shown. p24 and HSP90 were used as loading controls.
DISCUSSION
Vif-mediated A3 degradation is critical for HIV-1 replication in CD4+ T lymphocytes and myeloid cells (2, 3, 17, 18). In CD4+ T lymphocytes, at least A3D, A3F, A3G, and A3H (only stable haplotypes) proteins are involved in Vif-deficient HIV-1 restriction, and Vif is required to degrade A3 enzymes and allow efficient viral replication (2, 3, 17, 18). However, the degradation of A3 enzymes by Vif during infectious HIV-1 production from myeloid lineage cells has yet to be fully elucidated. We previously reported that A3G protein contributes to Vif-deficient HIV-1 restriction in a deaminase-dependent manner in THP-1 cells (26). Herein, we demonstrate that A3F protein also inhibits Vif-deficient HIV-1 in a largely deaminase-independent manner and that Vif avoids this HIV-1 restriction mechanism by degrading A3F protein (Fig. 3 and 4). Importantly, the results of pseudo-single cycle infectivity assays demonstrate that the disruption of A3A to A3G proteins confers comparable infectivity to wild-type HIV-1 in a Vif-deficient lab-adapted virus (IIIB) and TF virus (CH58) (Fig. 3 to 5). These results indicate that Vif-mediated A3 degradation is the primary function of Vif during infectious HIV-1 production from THP-1 cells.
Our results demonstrate that A3F and A3G but not A3H proteins restrict Vif-deficient HIV-1 via deaminase-dependent and -independent mechanisms in THP-1 cells (Fig. 1, 3, and 4). In addition to A3F and A3G proteins, our findings indicate that at least one additional A3 protein is involved in Vif-deficient HIV-1 restriction via a deaminase-independent mechanism (Fig. 3 and 4). Accordingly, the remaining four A3 proteins (A3A, A3B, A3C, and A3D) may contribute to Vif-deficient HIV-1 restriction in a deaminase-independent manner in THP-1 cells (Fig. 4). However, A3A and A3B proteins are highly unlikely to contribute in this manner as A3A mRNA and protein expression levels are very low or undetectable in THP-1 cells without IFN treatment (Fig. 2C and D). Further, both A3A and A3B proteins are resistant to degradation by HIV-1 Vif (7, 34, 41 - 43). It is therefore plausible that A3C and A3D proteins contribute to Vif-deficient HIV-1 restriction in THP-1 cells. An A3C-isoleucine 188 variant is reportedly more active against HIV-1 than a serine 188 variant (44, 45). To ask which A3C variant is expressed by THP-1 cells, we determined the A3C genotypes of THP-1 cells using cDNA sequencing. These results demonstrated that the amino acid residue of A3C protein at position 188 is serine. This result indicates that A3C protein may have a modest effect on Vif-deficient HIV-1 restriction via a deaminase-independent mechanism in THP-1 cells, as shown in prior studies (44, 45). Similarly, the results of previous studies indicate that A3D protein has a weak effect on Vif-deficient HIV-1 restriction in 293, SupT11, and CEM2n cells (7, 8, 37, 46, 47). Nevertheless, the fact that Vif-deficient HIV-1 has 20% lower infectivity indicates that a synergistic mechanism may enhance the effect of A3 proteins on HIV-1 infectivity (48, 49). Further studies are required to fully elucidate the mechanisms underlying the effect of A3 proteins on HIV-1 infectivity.
Similar to CD4+ T lymphocytes, HIV-1 can also target myeloid cells such as monocytes and macrophages, and these infections are associated with viral dissemination, persistence, and latency (50, 51). Accordingly, it is important to understand the role of restriction factors, including A3 proteins, in myeloid cells. In monocytes, A3A mRNA levels are 10 to 1,000 times higher than other A3 mRNA expression levels, and A3A mRNA expression is reduced by 10- to 100-fold after differentiation into monocyte-derived macrophages (MDMs) (52 - 54). In contrast, A3G mRNA expression levels are reduced approximately 10-fold lower after differentiation of monocytes into MDMs (52, 53). A3F mRNA expression levels are less variable during the differentiation of monocytes into MDMs (52). Interestingly, suppression of A3A and A3G protein levels by siRNA reportedly leads to a four- to fivefold increase in p24 production by HIV-1-infected monocytes (53). As MDMs are generally more sensitive to HIV-1 infection than monocytes, it is highly likely that A3A and A3G proteins contribute to the lack of susceptibility of monocytes to HIV-1 infection. However, as previous studies have reported that A3A protein is less active against HIV-1 in 293T and SupT11 cell lines (7, 34, 55), further studies are required to determine the contribution of A3A protein to HIV-1 restriction in monocytes.
In addition to A3A and A3G proteins, A3F and A3H proteins may be involved in HIV-1 restriction in monocytes. Although A3F mRNA expression levels are essentially unchanged during differentiation from monocytes into MDMs (53), A3F mRNA expression levels are comparable to A3G mRNA expression levels (53, 54), indicating that A3F protein likely contributes to HIV-1 restriction in monocytes. It is possible that only stable A3H haplotypes and the A3C-I188 variant are associated with HIV-1 restriction in monocytes. According to previous observations in 293, SupT11, and CEM2n cells (7, 8, 37, 46, 47), A3D protein may modestly contribute to HIV-1 restriction in monocytes. As A3B mRNA expression levels are relatively low, it is unlikely that this A3B protein inhibits HIV-1 in monocytes. However, the contribution of A3 proteins other than A3A and A3G proteins to HIV-1 suppression in monocytes remains unclear, and the antiviral activities of these A3 proteins warrant further investigation.
In MDMs, A3A protein appears to be associated with anti-HIV-1 activity as increasing HIV-1 infectivity has been reported following siRNA knockdown of A3A gene (53, 54). In addition, HIV-1 replication assays in MDMs using HIV-1 Vif4A and Vif5A mutants demonstrated that the replication kinetics of both mutants were slower than that of the Vif-proficient HIV-1, indicating that A3D, A3F, and A3G proteins contribute to HIV-1 restriction in MDMs (39). However, the effects of A3D and A3F proteins on HIV-1 replication are donor-dependent, likely due to their respective expression levels (39). As the antiviral activity of A3B, A3C, and A3H proteins has not been reported in MDMs, further studies are required to address these issues.
Vif is required for HIV-1 replication in CD4+ T lymphocytes and macrophages (2, 3, 17, 18). In the absence of Vif, HIV-1 is attacked by A3 proteins in CD4+ T lymphocytes, macrophages, monocytes, dendritic cells, and CD4+ T cell lines, and massive G-to-A mutations accumulate in HIV-1 proviral DNA (7, 8, 10, 15, 23, 26, 39, 56, 57). HIV-1 Vif recruits A3 proteins into an E3 ubiquitin ligase complex, thereby avoiding the antiviral activity of these proteins by promoting their degradation through a proteasome-mediated pathway (2, 3, 17, 18). The primary function of Vif has long been posited to be the suppression of the antiviral activity of A3 proteins. On the other hand, Vif causes G2/M cell cycle arrest (58 - 60). As the amino acid residues of Vif responsible for G2/M cell cycle arrest do not completely match with the amino acid residues required for Vif-mediated A3 degradation, these functions of Vif may be independent of each other (61 - 63). In 2016, a functional proteomic analysis identified the PPP2R5 family of proteins, which function as regulators of protein phosphatase 2A, as novel targets of Vif (25). Subsequently, studies revealed that Vif induces G2/M arrest by degrading PPP2R5 proteins (60, 64, 65). Vif-induced G2/M arrest has been observed in many cell types, including 293T, SupT11, CEM-SS, and THP-1 cell lines, and primary CD4+ T lymphocytes (25, 61, 63). However, Vif-mediated G2/M arrest is not required for HIV-1 infection, supporting our findings that A3 family proteins are the sole essential substrate of Vif during infectious virus production from THP-1 cells under normal cell culture conditions (Fig. 3 to 5). It has recently been reported that fragile X mental retardation 1 and diphthamide biosynthesis 7 are degraded by Vif in CD4+ T lymphocytes (24). Further studies are required to determine whether a substrate of Vif other than A3 proteins is required for fully infectious HIV-1 production in vivo.
In this study, we revealed an A3-dependent Vif function required for fully infectious HIV-1 production from THP-1 cells using only one lab-adapted virus (IIIB) and one TF virus (CH58) (Fig. 3 to 5). However, it is likely that Vif plays additional roles beyond A3 antagonism in some HIV-1 strains. Furthermore, the results of our pseudo-single cycle infectivity assays do not exclude the possibility that Vif may target non-A3 proteins required for HIV-1 replication. Moreover, we could not exclude the possibility that immunomodulatory effects may induce additional Vif targets other than A3 proteins in HIV-1-infected individuals.
In summary, the findings of the studies here demonstrate that the primary target of Vif is the A3 family of proteins during infectious HIV-1 production from THP-1 cells (i.e., A3G, A3F, and potentially A3C and/or A3D proteins; unlikely A3A, A3B, or A3H hapI protein). Whether this observation is applicable to primary CD4+ T lymphocytes and myeloid cells, such as monocytes and macrophages, is important for the development of antiviral therapies targeting the A3-Vif axis. Such studies may contribute to a functional cure for HIV-1 by manipulating A3 mutagenesis.
MATERIALS AND METHODS
Cell lines and culture conditions
293T (CRL-3216) was obtained from American Type Culture Collection. TZM-bl (#8129) (66) was obtained from the NIH AIDS Reagent Program (NARP). The creation and characterization of the permissive T cell line SupT11 and the SupT11 single clones stably expressing untagged A3 (SupT11-vector, SupT11-A3F, SupT11-A3G, and SupT11-A3H hapII high) have been reported (10, 33). CEM-GXR (CEM-GFP expressing CCR5) was provided by Dr. Todd Allen (Harvard University, USA) (67). THP-1 was provided by Dr. Andrea Cimarelli (INSERM, France) (53). The generation and characterization of THP-1 ΔA3G#1 was reported (26). Adherent cells were cultured in DMEM (Wako, Cat# 044-29765) supplemented with 10% fetal bovine serum (FBS) (Nichirei, Cat#175012) and 1% penicillin/streptomycin (P/S) (Wako, Cat# 168-23191). Suspension cells were maintained in Roswell Park Memorial Institute Medium (RPMI) (Thermo Fisher Scientific, Cat# C11875500BT) with 10% FBS and 1% P/S.
Genotyping of A3C and A3H genes
Total RNA was isolated from THP-1 by RNA Premium Kit (NIPPON Genetics, Cat# FG-81250). Then, cDNA was synthesized by Transcriptor Reverse Transcriptase (Roche, Cat# 03531287001) and used to amplify A3C or A3H gene with the following primers: [A3C outer primers: (5´-GCG CTT CAG AAA AGA GTG GG) and (5´-GGA GAC AGA CCA TGA GGC); A3C inner primers: (5´-ACA TGA ATC CAC AGA TCA GAA A) and (5´-CCC CTC ACT GGA GAC TCT CC); A3H outer primers: (5´-CCA GAA GCA CAG ATC AGA AAC ACG AT) and (5´-GAC CAG CAG GCT ATG AGG CAA); A3H inner primers: (5´-TGT TAA CAG CCG AAA CAT TCC) and (5´-TCT TGA GTT GCT TCT TGA TAA T)]. The amplified fragments were cloned into the pJET cloning vector (Thermo Fisher Scientific, Cat# K1231). At least 10 independent clones were subjected to Sanger sequencing (Azenta) and sequence data were analyzed by Sequencher v5.4.6 (Gene Codes Corporation).
Construction of pLentiCRISPR-Blast
The pLentiCRISPR1000 system was previously described (68). pLentiCRISPR1000-Blast was generated by restriction digest with BmtI and MluI to excise the P2A-puromycin cassette. An oligo containing a P2A-blasticidin cassette was purchased from IDT (5´-AGC GGA GCT ACT AAC TTC AGC CTG CTG AAG CAG GCT GGC GAC GTG GAG GAG AAC CCT GGA CCT ACC GGT ATG GCC AAG CCA CTG TCC CAA GAA GAG TCA ACT CTG ATC GAG AGG GCC ACT GCA ACC ATT AAT AGC ATT CCC ATC TCT GAA GAC TAT AGC GTA GCT AGT GCC GCA CTC AGC TCT GAT GGA CGC ATA TTC ACC GGC GTT AAT GTC TAC CAC TTC ACC GGC GGA CCC TGC GCC GAA CTG GTC GTG CTG GGG ACC GCA GCC GCC GCG GCT GCC GGG AAT TTG ACG TGC ATT GTT GCA ATA GGC AAC GAG AAT AGG GGC ATC CTG TCA CCT TGC GGC CGG TGT CGG CAA GTG CTG CTG GAC CTG CAC CCC GGC ATC AAG GCC ATA GTC AAG GAT AGT GAT GGC CAG CCG ACC GCC GTT GGG ATT CGA GAA CTT CTG CCT TCT GGG TAC GTC TGG GAA GGC TAG) and amplified with the primers (5´-CAA GAC TAG TGG AAG CGG AGC TAC TAA CTT CAG CCT GCT GAA GCA GGC TGG CGA CGT GGA GGA and 5´-NNN NAC GCG TCT AGC CTT CCC AGA CGT ACC C) using high-fidelity Phusion polymerase (NEB, Cat# M0530S). The PCR fragment was digested with BmtI and MluI, and ligated into the cut pLentiCRISPR1000, producing pLentiCRISPR1000-Blast.
Creation of THP-1 cells disrupting A3 genes
An A3F specific guide for exon 3 was designed (Fig. S2A and C) and evaluated manually for specificity to the A3F target sequence via an alignment with the most related members of the A3 family as described previously (26). Oligos with ends compatible with the Esp3I sites in pLentiCRISPR1000-Blast were purchased from IDT [ΔA3F gRNA: (5´-CAC CGG TAG TAG TAG AGG CGG GCG G) and (5´-CCA TCA TCA TCT CCG CCC GCC CAA G)]. The targeting construct was generated by annealing oligos and cloned by Golden Gate ligation into pLentiCRISPR1000-Blast. A guide with a common sequence among A3A exon 4, A3B exon 7, and A3G exon 7 was designed (Fig. 2A) and oligos with ends compatible with the Esp3I sites in pLentiCRISPR1000 (68) were purchased from IDT [PanZ1 gRNA: (5´-CAC CGT GGC CCG CAG CCT CCC ACT C) and (5´-GAA CGA GTG GGA GGC TGC GGG CCA C)]. The targeting construct was generated by annealing oligos and cloned by Golden Gate ligation into pLentiCRISPR1000 (68). All constructs were confirmed by Sanger sequencing (Azenta) and sequence data were analyzed by Sequencher v5.4.6 (Gene Codes Corporation).
For transduction, VSV-G pseudotyped virus was generated by transfecting 2.5 µg of the pLentiCRISPR1000 or pLentiCRISPR1000-Blast targeting construct along with 1.67 µg of pΔ-NRF (HIV-1 gag, pol, rev, tat genes) (69) and 0.83 µg of pMD.G (VSV-G) expression vectors using TransIT-LT1 (Takara, Cat# MIR2306) into 293T cells. At 48 h post-transfection, viral supernatants were harvested, filtered with 0.45 µm filters (Merck, Cat# SLHVR33RB), and concentrated by centrifugation (26,200 × g, 4°C, 2 h). Then, viral pellets were resuspended in 10% FBS/RPMI and incubated with cells for 48 h. Forty-eight hours later, cells were placed under drug selection in 10% FBS/RPMI containing 1 µg/mL puromycin (InvivoGen, Cat# ant-pr) or 6 ng/mL blasticidin (InvivoGen, Cat# ant-bl). Single-cell clones were isolated by the limiting dilution of the drug-resistant cell pool and expanded. The expression levels of A3F protein in THP-1 ΔA3F#1 and #2 and THP-1ΔA3F/A3G#1 and #2 cells were confirmed by western blotting (see “Western blot” section). To confirm indels in the A3F target sequence of the selected clones, genomic DNA was isolated by DNeasy Blood & Tissue Kits (Qiagen, Cat# 69504) and amplified with Choice-Taq DNA polymerase (Denville Scientific, Cat# CB4050-2) using primers (5´-GCT GAA GTC GCC CTT GAA TAA ACA CGC and 5´-TGT CAG TGC TGG CCC CG). The amplified PCR products were cloned into the pJET cloning vector (Thermo Fisher Scientific, Cat# K1231) and subjected to Sanger sequencing (Azenta). To confirm indels in the A3A, A3B, and A3G target sequences of the selected clones (THP-1#11-4 and #11-7), genomic DNA was isolated by DNeasy Blood & Tissue Kits (Qiagen, Cat# 69504) and subjected to whole-genome sequencing (Macrogen). The WGS data were enrolled in the NCBI BioSample database and the respective accession numbers are SAMN35719796 for parental THP-1, SAMN35719797 for THP-1#11-4, and SAMN35719798 for THP-1#11-7. The sequencing data were aligned by Isaac aligner (iSAAC-04.18.11.09). Off-target sites were analyzed by Cas-OFFinder (http://www.rgenome.net/cas-offinder/). For further analysis of indels between A3A and A3G genes, genomic DNAs from THP-1#11-4 and #11-7 were amplified using primers (5´-GGG GCT TTC TGA AAG AAT GAG AAC TGG GC and 5´-CAG CTG GAG ATG GTG GTG AAC AGC C). The amplified PCR products were cloned into the pJET cloning vector (Thermo Fisher Scientific, Cat# K1231) and subjected to Sanger sequencing (Azenta). All sequence data were analyzed by Sequencher v5.4.6 (Gene Codes Corporation). To assess the expression levels of A3 mRNAs and proteins, THP-1 parent, #11-4, and #11-7 were incubated in 10% FBS/RPMI including 500 units/mL IFN (R&D Systems, Cat# 11200-2) for 6 h. Then, cells were harvested and subjected to RT-qPCR (see “RT-qPCR” section) (Fig. 2C) and western blotting (see “Western blot” section) (Fig. 2D).
Pseudo-single cycle infectivity assays
Vif-proficient and Vif-deficient (X26 and X27) HIV-1 IIIB C200 proviral expression constructs have been reported (70). HIV-1 IIIB C200 mutants with hyper- (H48 and 60EKGE63) and hypo-functional (V39) Vifs have been reported (10). An HIV-1 IIIB C200 Vif 5A mutant (40AAAAA44) has been described (26). HIV-1 IIIB C200 Vif 4A (14AKTK18) and 4A5A (14AKTK18 and 40AAAAA44) mutants were created by digesting pNLCSFV3-4A, and −4A5A proviral DNA construct [(37); kindly provided by Dr. Kei Sato, University of Tokyo, Japan] at SwaI and SalI sites and cloned into pIIIB C200 proviral construct. The proviral expression vector encoding full-length TF virus, CH58 (NARP, #11856) was obtained from the NARP. The creation of Vif-deficient CH58 mutant has been described previously (71).
HIV-1 single-cycle assays using VSV-G pseudotyped viruses were performed as described previously (23, 26) (Fig. 1C). 293T cells were cotransfected with 2.4 µg of proviral DNA construct and 0.6 µg of VSV-G expression vector using TransIT-LT1 reagent (Takara, Cat# MIR2306) into 293T cells (3 × 106). Forty-eight hours later, supernatants were harvested, filtered (0.45 µm filters, Merck, Cat# SLHVR33RB), and used to titrate on 2.5 × 104 CEM-GXR reporter cells for MOI determinations. GFP+ cells were measured using a FACS Canto II (BD Biosciences) and the data were analyzed using FlowJo software v10.7.1 (BD Biosciences). One or 5 × 106 target cells were infected with an MOI of 0.05 (for SupT11 derivatives) or 0.25 (for THP-1 derivatives) and washed with phosphate-buffered saline (PBS) twice at 24 h postinfection and then incubated for an additional 24 h. After 24 h, supernatants were collected and filtered. The resulting viral particles were quantified by p24 ELISA (ZeptoMetrix, Cat# 0801008) and used to infect 1 × 104 TZM-bl cells (1 or 2 ng of p24). At 48 h postinfection, the infected cells were lysed with a Bright-Glo luciferase assay system (Promega, Cat# E2650) and the intracellular luciferase activity was measured by a Synergy H1 microplate reader (BioTek) or Centro XS3 LB960 microplate luminometer (Berthold Technologies).
Quantification of LRT products
Viruses were produced by infecting VSV-G pseudotyped virus into THP-1 cells as described above (see “Pseudo-single cycle infectivity assays” section) and the resulting viral particles were quantified by p24 ELISA (ZeptoMetrix, Cat# 0801008). The viral supernatants including 20 ng of p24 antigen were used for infection into SupT11 cells. At 12 h postinfection, cells were harvested and washed with PBS twice. Then, total DNA was isolated by DNeasy Blood & Tissue Kits (Qiagen, Cat# 69504) and treated with RNase A (Qiagen, Cat# 19101) according to the manufacturer’s instruction. Following DpnI digestion, 50 ng of DNA was used to amplify LRT products and CCR5 gene with the following primers: LRT forward: (5´-CGT CTG TTG TGT GAC TCT GG) and LRT reverse: (5´-TTT TGG CGT ACT CAC CAG TCG); CCR5 forward: (5´-CCA GAA GAG CTG AGA CAT CCG) and CCR5 reverse (5´-GCC AAG CAG CTG AGA GGT TAC T). qPCR was performed using Power SYBR Green PCR Master Mix (Thermo Fisher Scientific, Cat# 4367659) and fluorescent signals from resulting PCR products were acquired using a Thermal Cycler Dice Real Time System III (Takara). Finally, each LRT product was represented as values normalized by the quantity of the CCR5 gene (Fig. 4C).
RT-qPCR
Cells were harvested and washed with PBS twice. Then, total RNA was isolated by RNA Premium Kit (NIPPON Genetics, Cat# FG-81250) and cDNA was synthesized by Transcriptor Reverse Transcriptase (Roche, Cat# 03531287001) with random hexamer. RT-qPCR was performed using Power SYBR Green PCR Master Mix (Thermo Fisher Scientific, Cat# 4367659). Primers for each A3 mRNA have been reported previously (72, 73): A3A forward: (5´-GAG AAG GGA CAA GCA CAT GG) and A3A reverse: (5´-TGG ATC CAT CAA GTG TCT GG); A3B forward: (5´-GAC CCT TTG GTC CTT CGA C) and A3B reverse: (5´-GCA CAG CCC CAG GAG AAG); A3C forward: (5´-AGC GCT TCA GAA AAG AGT GG) and A3C reverse: (5´-AAG TTT CGT TCC GAT CGT TG); A3D forward: (5´-ACC CAA ACG TCA GTC GAA TC) and A3D reverse: (5´-CAC ATT TCT GCG TGG TTC TC); A3F forward: (5´-CCG TTT GGA CGC AAA GAT) and A3F reverse: (5´-CCA GGT GAT CTG GAA ACA CTT); A3G forward: (5´-CCG AGG ACC CGA AGG TTA C) and A3G reverse: (5´-TCC AAC AGT GCT GAA ATT CG); A3H forward: (5´-AGC TGT GGC CAG AAG CAC) and A3H reverse: (5´-CGG AAT GTT TCG GCT GTT); TATA-binding protein (TBP) forward: (5´-CCC ATG ACT CCC ATG ACC) and TBP reverse: (5´-TTT ACA ACC AAG ATT CAC TGT GG). Fluorescent signals from resulting PCR products were acquired using a Thermal Cycler Dice Real Time System III (Takara). Finally, each A3 mRNA expression level was represented as values normalized by TBP mRNA expression levels (Fig. 2C).
Hypermutation analyses
Hypermutation analyses were performed as previously described (23, 26, 45). Genomic DNAs containing HIV-1 proviruses were recovered by infecting viruses produced in derivatives of THP-1 or SupT11 cells into SupT11 using DNeasy Blood & Tissue Kits (Qiagen, Cat# 69504). Following DpnI digestion, the viral pol region was amplified by nested PCR with outer primers (876 bp) [(5´-TCC ART ATT TRC CAT AAA RAA AAA) and (5´-TTY AGA TTT TTA AAT GGY TYT TGA)] and inner primers (564 bp) [(5´-AAT ATT CCA RTR TAR CAT RAC AAA AAT) and (5´-AAT GGY TYT TGA TAA ATT TGA TAT GT)]. The resulting 564 bp amplicon was subjected to pJET cloning. At least 10 independent clones were Sanger sequenced (Azenta) for each condition and analyzed by the HIV sequence database (https://www.hiv.lanl.gov/content/sequence/HYPERMUT/hypermut.html). Clones with identical mutations were eliminated.
Western blot
Western blotting for cell and viral lysates was performed as described previously (23, 26, 74). Cells were harvested, washed with PBS twice, and lysed in lysis buffer [25 mM HEPES (pH7.2), 20% glycerol, 125 mM NaCl, 1% Nonidet P40 (NP40) substitute (Nacalai Tesque, Cat# 18558-54)]. After quantification of total protein by protein assay dye (Bio-Rad, Cat# 5000006), lysates were diluted with 2× SDS sample buffer [100 mM Tris-HCl (pH 6.8), 4% SDS, 12% β-mercaptoethanol, 20% glycerol, 0.05% bromophenol blue] and boiled for 10 min. Virions were dissolved in 2× SDS sample buffer and boiled for 10 min after pelleting down using 20% sucrose (26,200 × g, 4°C, 2 h). Then, the quantity of p24 antigen was measured by p24 ELISA (ZeptoMetrix, Cat# 0801008).
Proteins in the cell and viral lysates (5 µg of total protein and 10 ng of p24 antigen) were separated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Cat# IPVH00010). Membranes were blocked with 5% milk in PBS containing 0.1% Tween 20 (0.1% PBST) and incubated in 4% milk/0.1% PBST containing primary antibodies: mouse anti-HSP90 (BD Transduction Laboratories, Cat# 610418, 1:5,000); rabbit anti-A3B (5210-87-13, 1:1,000) (75); rabbit anti-A3C (Proteintech, Cat# 105911-1-AP, 1:1,000); rabbit anti-A3F (675, 1:1,000) (76); rabbit anti-A3G (NARP, #10201, 1:2,500); rabbit anti-A3H (Novus Biologicals, NBP1-91682, 1:5,000): mouse anti-Vif (NARP, #6459, 1:2,000); mouse anti-p24 (NARP, #1513, 1:2,000). Subsequently, the membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies: donkey anti-rabbit IgG-HRP (Jackson ImmunoResearch, 711-035-152, 1:5,000); donkey anti-mouse IgG-HRP (Jackson ImmunoResearch, 715-035-150, 1:5,000). SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, Cat# 34095) or Super signal atto (Thermo Fisher Scientific, Cat# A38555) was used for HRP detection. Bands were visualized by the Amersham Imager 600 (Amersham).
Statistical analyses
Statistical significance was performed using a two-sided paired t test (Fig. 1D, 2C, 3B, 4C, 5, S1A, and S5A). GraphPad Prism software v8.4.3 was used for these statistical tests.
ACKNOWLEDGMENTS
We would like to thank Haruyo Hasebe, Kimiko Ichihara, Kazuko Kitazato, Otowa Takahashi, and all Ikeda lab members for technical assistance. We also would like to thank Drs. Todd Allen, Andrea Cimarelli, and Kei Sato for sharing reagents.
This study was supported in part by AMED Research Program on Emerging and Re-emerging Infectious Diseases (JP21fk0108574, to Hesham Nasser; JP21fk0108494 to Terumasa Ikeda); AMED Research Program on HIV/AIDS (JP22fk0410055, to Terumasa Ikeda); JSPS KAKENHI Grant-in-Aid for Scientific Research C (22K07103, to Terumasa Ikeda); JSPS KAKENHI Grant-in-Aid for Early-Career Scientists (22K16375, to Hesham Nasser); JSPS Leading Initiative for Excellent Young Researchers (LEADER) (to Terumasa Ikeda); Takeda Science Foundation (to Terumasa Ikeda); Mochida Memorial Foundation for Medical and Pharmaceutical Research (to Terumasa Ikeda); The Naito Foundation (to Terumasa Ikeda); Shin-Nihon Foundation of Advanced Medical Research (to Terumasa Ikeda); Waksman Foundation of Japan (to Terumasa Ikeda); an intramural grant from Kumamoto University COVID-19 Research Projects (AMABIE) (to Terumasa Ikeda); Intercontinental Research and Educational Platform Aiming for Eradication of HIV/AIDS (to Terumasa Ikeda); International Joint Research Project of the Institute of Medical Science, the University of Tokyo (to Terumasa Ikeda); SPP1923 and the Heisenberg Program of the German Research Foundation (SA 2676/3-1; SA 2676/1-2) (to Daniel Sauter); as well as the Canon Foundation Europe (to Daniel Sauter).
Contributions from the Harris lab were supported by NIAID R37-AI064046 and a Recruitment of Established Investigators Award from the Cancer Prevention and Research Institute of Texas (CPRIT RR220053). R.S.H. is an Investigator of the Howard Hughes Medical Institute and the Ewing Halsell President’s Council Distinguished Chair.
The authors declare that they have no competing interests.
Contributor Information
Terumasa Ikeda, Email: ikedat@kumamoto-u.ac.jp.
Stephen P. Goff, Columbia University Medical Center, New York, New York, USA
Stuart J. Neil, King's College London, London, United Kingdom
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/mbio.00782-23.
Pseudo-single cycle infectivity assays for each HIV-1 mutant in SupT11 cells stably expressing stable A3H haplotype.
Legends to Fig. S1 to S5.
Development of A3F and A3F/A3G-null THP-1 cells.
Sequence analysis of flanking region targeted by gRNA in THP-1#11-4 and #11-7.
Deletions around predicted A3G pseudogene.
Pseudo-single cycle infectivity assays for each HIV-1 mutant in SupT11 cells stably expressing A3.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Ikeda T, Yue Y, Shimizu R, Nasser H. 2021. Potential utilization of APOBEC3-mediated mutagenesis for an HIV-1 functional cure. Front Microbiol 12:686357. doi: 10.3389/fmicb.2021.686357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Desimmie BA, Delviks-Frankenberrry KA, Burdick RC, Qi D, Izumi T, Pathak VK. 2014. Multiple APOBEC3 restriction factors for HIV-1 and one Vif to rule them all. J Mol Biol 426:1220–1245. doi: 10.1016/j.jmb.2013.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harris RS, Dudley JP. 2015. APOBECs and virus restriction. Virology 479–480:131–145. doi: 10.1016/j.virol.2015.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Koito A, Ikeda T. 2012. Apolipoprotein B mRNA-editing, catalytic polypeptide cytidine deaminases and retroviral restriction. Wiley Interdiscip Rev RNA 3:529–541. doi: 10.1002/wrna.1117 [DOI] [PubMed] [Google Scholar]
- 5. Cheng AZ, Moraes SN, Shaban NM, Fanunza E, Bierle CJ, Southern PJ, Bresnahan WA, Rice SA, Harris RS. 2021. APOBECs and herpesviruses. Viruses 13:390. doi: 10.3390/v13030390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Holmes RK, Malim MH, Bishop KN. 2007. APOBEC-mediated viral restriction: not simply editing? Trends Biochem Sci 32:118–128. doi: 10.1016/j.tibs.2007.01.004 [DOI] [PubMed] [Google Scholar]
- 7. Hultquist JF, Lengyel JA, Refsland EW, LaRue RS, Lackey L, Brown WL, Harris RS. 2011. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J Virol 85:11220–11234. doi: 10.1128/JVI.05238-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Refsland EW, Hultquist JF, Harris RS. 2012. Endogenous origins of HIV-1 G-to-A hypermutation and restriction in the nonpermissive T cell line CEM2n. PLoS Pathog 8:e1002800. doi: 10.1371/journal.ppat.1002800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ooms M, Brayton B, Letko M, Maio SM, Pilcher CD, Hecht FM, Barbour JD, Simon V. 2013. HIV-1 Vif adaptation to human APOBEC3H haplotypes. Cell Host Microbe 14:411–421. doi: 10.1016/j.chom.2013.09.006 [DOI] [PubMed] [Google Scholar]
- 10. Refsland EW, Hultquist JF, Luengas EM, Ikeda T, Shaban NM, Law EK, Brown WL, Reilly C, Emerman M, Harris RS. 2014. Natural polymorphisms in human APOBEC3H and HIV-1 Vif combine in primary T lymphocytes to affect viral G-to-A mutation levels and infectivity. PLoS Genet 10:e1004761. doi: 10.1371/journal.pgen.1004761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Newman ENC, Holmes RK, Craig HM, Klein KC, Lingappa JR, Malim MH, Sheehy AM. 2005. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr Biol 15:166–170. doi: 10.1016/j.cub.2004.12.068 [DOI] [PubMed] [Google Scholar]
- 12. Wang X, Ao Z, Chen L, Kobinger G, Peng J, Yao X. 2012. The cellular antiviral protein APOBEC3G interacts with HIV-1 reverse transcriptase and inhibits its function during viral replication. J Virol 86:3777–3786. doi: 10.1128/JVI.06594-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pollpeter D, Parsons M, Sobala AE, Coxhead S, Lang RD, Bruns AM, Papaioannou S, McDonnell JM, Apolonia L, Chowdhury JA, Horvath CM, Malim MH. 2018. Deep sequencing of HIV-1 reverse transcripts reveals the multifaceted antiviral functions of APOBEC3G. Nat Microbiol 3:220–233. doi: 10.1038/s41564-017-0063-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Holmes RK, Koning FA, Bishop KN, Malim MH. 2007. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. comparisons with APOBEC3G. J Biol Chem 282:2587–2595. doi: 10.1074/jbc.M607298200 [DOI] [PubMed] [Google Scholar]
- 15. Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH. 2003. DNA deamination mediates innate immunity to retroviral infection. Cell 113:803–809. doi: 10.1016/s0092-8674(03)00423-9 [DOI] [PubMed] [Google Scholar]
- 16. Rathore A, Carpenter MA, Demir Ö, Ikeda T, Li M, Shaban NM, Law EK, Anokhin D, Brown WL, Amaro RE, Harris RS. 2013. The local dinucleotide preference of APOBEC3G can be altered from 5'-CC to 5'-TC by a single amino acid substitution. J Mol Biol 425:4442–4454. doi: 10.1016/j.jmb.2013.07.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Salamango DJ, Harris RS. 2020. Dual functionality of HIV-1 Vif in APOBEC3 counteraction and cell cycle arrest. Front Microbiol 11:622012. doi: 10.3389/fmicb.2020.622012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Takaori-Kondo A, Shindo K. 2013. HIV-1 Vif: a guardian of the virus that opens up a new era in the research field of restriction factors. Front Microbiol 4:34. doi: 10.3389/fmicb.2013.00034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jäger S, Kim DY, Hultquist JF, Shindo K, LaRue RS, Kwon E, Li M, Anderson BD, Yen L, Stanley D, Mahon C, Kane J, Franks-Skiba K, Cimermancic P, Burlingame A, Sali A, Craik CS, Harris RS, Gross JD, Krogan NJ. 2011. Vif hijacks CBF-β to degrade APOBEC3G and promote HIV-1 infection. Nature 481:371–375. doi: 10.1038/nature10693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang W, Du J, Evans SL, Yu Y, Yu XF. 2011. T-cell differentiation factor CBF-β regulates HIV-1 Vif-mediated evasion of host restriction. Nature 481:376–379. doi: 10.1038/nature10718 [DOI] [PubMed] [Google Scholar]
- 21. Anderson BD, Harris RS. 2015. Transcriptional regulation of APOBEC3 antiviral immunity through the CBF-β/RUNX axis. Sci Adv 1:e1500296. doi: 10.1126/sciadv.1500296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ebrahimi D, Richards CM, Carpenter MA, Wang J, Ikeda T, Becker JT, Cheng AZ, McCann JL, Shaban NM, Salamango DJ, Starrett GJ, Lingappa JR, Yong J, Brown WL, Harris RS. 2018. Genetic and mechanistic basis for APOBEC3H alternative splicing, retrovirus restriction, and counteraction by HIV-1 protease. Nat Commun 9:4137. doi: 10.1038/s41467-018-06594-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ikeda T, Symeonides M, Albin JS, Li M, Thali M, Harris RS, Silvestri G. 2018. HIV-1 adaptation studies reveal a novel Env-mediated homeostasis mechanism for evading lethal hypermutation by APOBEC3G. PLoS Pathog 14:e1007010. doi: 10.1371/journal.ppat.1007010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Naamati A, Williamson JC, Greenwood EJ, Marelli S, Lehner PJ, Matheson NJ. 2019. Functional proteomic atlas of HIV infection in primary human CD4+ T cells. Elife 8:e41431. doi: 10.7554/eLife.41431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Greenwood EJ, Matheson NJ, Wals K, van den Boomen DJ, Antrobus R, Williamson JC, Lehner PJ. 2016. Temporal proteomic analysis of HIV infection reveals remodelling of the host phosphoproteome by lentiviral Vif variants. Elife 5:e18296. doi: 10.7554/eLife.18296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ikeda T, Molan AM, Jarvis MC, Carpenter MA, Salamango DJ, Brown WL, Harris RS. 2019. HIV-1 restriction by endogenous APOBEC3G in the myeloid cell line THP-1. J Gen Virol 100:1140–1152. doi: 10.1099/jgv.0.001276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sadeghpour S, Khodaee S, Rahnama M, Rahimi H, Ebrahimi D. 2021. Human APOBEC3 variations and viral infection. Viruses 13:1366. doi: 10.3390/v13071366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. OhAinle M, Kerns JA, Li MMH, Malik HS, Emerman M. 2008. Antiretroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host Microbe 4:249–259. doi: 10.1016/j.chom.2008.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang X, Abudu A, Son S, Dang Y, Venta PJ, Zheng Y-H. 2011. Analysis of human APOBEC3H haplotypes and anti-human immunodeficiency virus type 1 activity. J Virol 85:3142–3152. doi: 10.1128/JVI.02049-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nakano Y, Misawa N, Juarez-Fernandez G, Moriwaki M, Nakaoka S, Funo T, Yamada E, Soper A, Yoshikawa R, Ebrahimi D, Tachiki Y, Iwami S, Harris RS, Koyanagi Y, Sato K, Cullen BR. 2017. HIV-1 competition experiments in humanized mice show that APOBEC3H imposes selective pressure and promotes virus adaptation. PLoS Pathog 13:e1006606. doi: 10.1371/journal.ppat.1006606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Starrett GJ, Luengas EM, McCann JL, Ebrahimi D, Temiz NA, Love RP, Feng Y, Adolph MB, Chelico L, Law EK, Carpenter MA, Harris RS. 2016. The DNA cytosine deaminase APOBEC3H haplotype I likely contributes to breast and lung cancer mutagenesis. Nat Commun 7:12918. doi: 10.1038/ncomms12918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhen A, Wang T, Zhao K, Xiong Y, Yu X-F. 2010. A single amino acid difference in human APOBEC3H variants determines HIV-1 Vif sensitivity. J Virol 84:1902–1911. doi: 10.1128/JVI.01509-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Albin JS, Brown WL, Harris RS. 2014. Catalytic activity of APOBEC3F is required for efficient restriction of Vif-deficient human immunodeficiency virus. Virology 450–451:49–54. doi: 10.1016/j.virol.2013.11.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho S-J, Malim MH. 2004. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr Biol 14:1392–1396. doi: 10.1016/j.cub.2004.06.057 [DOI] [PubMed] [Google Scholar]
- 35. Zheng Y-H, Irwin D, Kurosu T, Tokunaga K, Sata T, Peterlin BM. 2004. Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. J Virol 78:6073–6076. doi: 10.1128/JVI.78.11.6073-6076.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Land AM, Law EK, Carpenter MA, Lackey L, Brown WL, Harris RS. 2013. Endogenous APOBEC3A DNA cytosine deaminase is cytoplasmic and nongenotoxic. J Biol Chem 288:17253–17260. doi: 10.1074/jbc.M113.458661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sato K, Takeuchi JS, Misawa N, Izumi T, Kobayashi T, Kimura Y, Iwami S, Takaori-Kondo A, Hu W-S, Aihara K, Ito M, An DS, Pathak VK, Koyanagi Y. 2014. APOBEC3D and APOBEC3F potently promote HIV-1 diversification and evolution in humanized mouse model. PLoS Pathog 10:e1004453. doi: 10.1371/journal.ppat.1004453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Russell RA, Smith J, Barr R, Bhattacharyya D, Pathak VK. 2009. Distinct domains within APOBEC3G and APOBEC3F interact with separate regions of human immunodeficiency virus type 1 Vif. J Virol 83:1992–2003. doi: 10.1128/JVI.01621-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chaipan C, Smith JL, Hu W-S, Pathak VK. 2013. APOBEC3G restricts HIV-1 to a greater extent than APOBEC3F and APOBEC3De in human primary CD4+ T cells and macrophages. J Virol 87:444–453. doi: 10.1128/JVI.00676-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Smith JL, Pathak VK. 2010. Identification of specific determinants of human APOBEC3F, APOBEC3C, and APOBEC3De and African green monkey APOBEC3F that interact with HIV-1 Vif. J Virol 84:12599–12608. doi: 10.1128/JVI.01437-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yu Q, Chen D, König R, Mariani R, Unutmaz D, Landau NR. 2004. APOBEC3B and APOBEC3C are potent inhibitors of simian immunodeficiency virus replication. J Biol Chem 279:53379–53386. doi: 10.1074/jbc.M408802200 [DOI] [PubMed] [Google Scholar]
- 42. Doehle BP, Schäfer A, Cullen BR. 2005. Human APOBEC3B is a potent inhibitor of HIV-1 infectivity and is resistant to HIV-1 Vif. Virology 339:281–288. doi: 10.1016/j.virol.2005.06.005 [DOI] [PubMed] [Google Scholar]
- 43. Goila-Gaur R, Khan MA, Miyagi E, Kao S, Strebel K. 2007. Targeting APOBEC3A to the viral nucleoprotein complex confers antiviral activity. Retrovirology 4:61. doi: 10.1186/1742-4690-4-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wittkopp CJ, Adolph MB, Wu LI, Chelico L, Emerman M. 2016. A single nucleotide polymorphism in human APOBEC3C enhances restriction of lentiviruses. PLoS Pathog 12:e1005865. doi: 10.1371/journal.ppat.1005865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Anderson BD, Ikeda T, Moghadasi SA, Martin AS, Brown WL, Harris RS. 2018. Natural APOBEC3C variants can elicit differential HIV-1 restriction activity. Retrovirology 15:78. doi: 10.1186/s12977-018-0459-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dang Y, Wang X, Esselman WJ, Zheng Y-H. 2006. Identification of APOBEC3De as another antiretroviral factor from the human APOBEC family. J Virol 80:10522–10533. doi: 10.1128/JVI.01123-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Takei H, Fukuda H, Pan G, Yamazaki H, Matsumoto T, Kazuma Y, Fujii M, Nakayama S, Kobayashi IS, Shindo K, Yamashita R, Shirakawa K, Takaori-Kondo A, Kobayashi SS. 2020. Alternative splicing of APOBEC3D generates functional diversity and its role as a DNA mutator. Int J Hematol 112:395–408. doi: 10.1007/s12185-020-02904-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Desimmie BA, Burdick RC, Izumi T, Doi H, Shao W, Alvord WG, Sato K, Koyanagi Y, Jones S, Wilson E, Hill S, Maldarelli F, Hu W-S, Pathak VK. 2016. APOBEC3 proteins can copackage and comutate HIV-1 genomes. Nucleic Acids Res 44:7848–7865. doi: 10.1093/nar/gkw653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ara A, Love RP, Follack TB, Ahmed KA, Adolph MB, Chelico L. 2017. Mechanism of enhanced HIV restriction by virion coencapsidated cytidine deaminases APOBEC3F and APOBEC3G. J Virol 91:e02230-16. doi: 10.1128/JVI.02230-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hendricks CM, Cordeiro T, Gomes AP, Stevenson M. 2021. The interplay of HIV-1 and macrophages in viral persistence. Front Microbiol 12:646447. doi: 10.3389/fmicb.2021.646447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Herskovitz J, Gendelman HE. 2019. HIV and the macrophage: from cell reservoirs to drug delivery to viral eradication. J Neuroimmune Pharmacol 14:52–67. doi: 10.1007/s11481-018-9785-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Peng G, Lei KJ, Jin W, Greenwell-Wild T, Wahl SM. 2006. Induction of APOBEC3 family proteins, a defensive maneuver underlying interferon-induced anti-HIV-1 activity. J Exp Med 203:41–46. doi: 10.1084/jem.20051512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Berger G, Durand S, Fargier G, Nguyen X-N, Cordeil S, Bouaziz S, Muriaux D, Darlix J-L, Cimarelli A, Emerman M. 2011. APOBEC3A is a specific inhibitor of the early phases of HIV-1 infection in myeloid cells. PLoS Pathog 7:e1002221. doi: 10.1371/journal.ppat.1002221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Koning FA, Newman ENC, Kim E-Y, Kunstman KJ, Wolinsky SM, Malim MH. 2009. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J Virol 83:9474–9485. doi: 10.1128/JVI.01089-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Aguiar RS, Lovsin N, Tanuri A, Peterlin BM. 2008. Vpr.A3A chimera inhibits HIV replication. J Biol Chem 283:2518–2525. doi: 10.1074/jbc.M706436200 [DOI] [PubMed] [Google Scholar]
- 56. Koning FA, Goujon C, Bauby H, Malim MH. 2011. Target cell-mediated editing of HIV-1 cDNA by APOBEC3 proteins in human macrophages. J Virol 85:13448–13452. doi: 10.1128/JVI.00775-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pion M, Granelli-Piperno A, Mangeat B, Stalder R, Correa R, Steinman RM, Piguet V. 2006. APOBEC3G/3F mediates intrinsic resistance of monocyte-derived dendritic cells to HIV-1 infection. J Exp Med 203:2887–2893. doi: 10.1084/jem.20061519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sakai K, Dimas J, Lenardo MJ. 2006. The Vif and Vpr accessory proteins independently cause HIV-1-induced T cell cytopathicity and cell cycle arrest. Proc Natl Acad Sci U S A 103:3369–3374. doi: 10.1073/pnas.0509417103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang J, Shackelford JM, Casella CR, Shivers DK, Rapaport EL, Liu B, Yu X-F, Finkel TH. 2007. The Vif accessory protein alters the cell cycle of human immunodeficiency virus type 1 infected cells. Virology 359:243–252. doi: 10.1016/j.virol.2006.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Salamango DJ, Ikeda T, Moghadasi SA, Wang J, McCann JL, Serebrenik AA, Ebrahimi D, Jarvis MC, Brown WL, Harris RS. 2019. HIV-1 Vif triggers cell cycle arrest by degrading cellular PPP2R5 phospho-regulators. Cell Rep 29:1057–1065. doi: 10.1016/j.celrep.2019.09.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. DeHart JL, Bosque A, Harris RS, Planelles V. 2008. Human immunodeficiency virus type 1 Vif induces cell cycle delay via recruitment of the same E3 ubiquitin ligase complex that targets APOBEC3 proteins for degradation. J Virol 82:9265–9272. doi: 10.1128/JVI.00377-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhao K, Du J, Rui Y, Zheng W, Kang J, Hou J, Wang K, Zhang W, Simon VA, Yu X-F. 2015. Evolutionarily conserved pressure for the existence of distinct G2/M cell cycle arrest and A3H inactivation functions in HIV-1 Vif. Cell Cycle 14:838–847. doi: 10.1080/15384101.2014.1000212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Du J, Rui Y, Zheng W, Li P, Kang J, Zhao K, Sun T, Yu X-F. 2019. Vif-CBFβ interaction is essential for Vif-induced cell cycle arrest. Biochem Biophys Res Commun 511:910–915. doi: 10.1016/j.bbrc.2019.02.136 [DOI] [PubMed] [Google Scholar]
- 64. Marelli S, Williamson JC, Protasio AV, Naamati A, Greenwood EJ, Deane JE, Lehner PJ, Matheson NJ. 2020. Antagonism of PP2A is an independent and conserved function of HIV-1 Vif and causes cell cycle arrest. Elife 9:e53036. doi: 10.7554/eLife.53036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nagata K, Shindo K, Matsui Y, Shirakawa K, Takaori-Kondo A. 2020. Critical role of PP2A-B56 family protein degradation in HIV-1 Vif mediated G2 cell cycle arrest. Biochem Biophys Res Commun 527:257–263. doi: 10.1016/j.bbrc.2020.04.123 [DOI] [PubMed] [Google Scholar]
- 66. Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother 46:1896–1905. doi: 10.1128/AAC.46.6.1896-1905.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Brockman MA, Tanzi GO, Walker BD, Allen TM. 2006. Use of a novel GFP reporter cell line to examine replication capacity of CXCR4- and CCR5-tropic HIV-1 by flow cytometry. J Virol Methods 131:134–142. doi: 10.1016/j.jviromet.2005.08.003 [DOI] [PubMed] [Google Scholar]
- 68. Carpenter MA, Law EK, Serebrenik A, Brown WL, Harris RS. 2019. A lentivirus-based system for Cas9/gRNA expression and subsequent removal by cre-mediated recombination. Methods 156:79–84. doi: 10.1016/j.ymeth.2018.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. 1996. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272:263–267. doi: 10.1126/science.272.5259.263 [DOI] [PubMed] [Google Scholar]
- 70. Haché G, Shindo K, Albin JS, Harris RS. 2008. Evolution of HIV-1 isolates that use a novel Vif-independent mechanism to resist restriction by human APOBEC23G. Curr Biol 18:819–824. doi: 10.1016/j.cub.2008.04.073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hopfensperger K, Richard J, Stürzel CM, Bibollet-Ruche F, Apps R, Leoz M, Plantier J-C, Hahn BH, Finzi A, Kirchhoff F, Sauter D, Brockman MA, Smithgall TE. 2020. Convergent evolution of HLA-C downmodulation in HIV-1 and HIV-2. mBio 11. doi: 10.1128/mBio.00782-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Refsland EW, Stenglein MD, Shindo K, Albin JS, Brown WL, Harris RS. 2010. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res 38:4274–4284. doi: 10.1093/nar/gkq174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, Refsland EW, Kotandeniya D, Tretyakova N, Nikas JB, Yee D, Temiz NA, Donohue DE, McDougle RM, Brown WL, Law EK, Harris RS. 2013. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 494:366–370. doi: 10.1038/nature11881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Nasser H, Shimizu R, Ito J, Genotype to Phenotype Japan (G2P-Japan) Consortium, Saito A, Sato K, Ikeda T. 2022. Monitoring fusion kinetics of viral and target cell membranes in living cells using a SARS-CoV-2 spike-protein-mediated membrane fusion assay. STAR Protoc 3:101773. doi: 10.1016/j.xpro.2022.101773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Brown WL, Law EK, Argyris PP, Carpenter MA, Levin-Klein R, Ranum AN, Molan AM, Forster CL, Anderson BD, Lackey L, Harris RS. 2019. A rabbit monoclonal antibody against the antiviral and cancer genomic DNA mutating enzyme APOBEC3B. Antibodies (Basel) 8:47. doi: 10.3390/antib8030047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang J, Shaban NM, Land AM, Brown WL, Harris RS. 2018. Simian immunodeficiency virus Vif and human APOBEC3B interactions resemble those between HIV-1 Vif and human APOBEC3G. J Virol 92:e00447-18. doi: 10.1128/JVI.00447-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Pseudo-single cycle infectivity assays for each HIV-1 mutant in SupT11 cells stably expressing stable A3H haplotype.
Legends to Fig. S1 to S5.
Development of A3F and A3F/A3G-null THP-1 cells.
Sequence analysis of flanking region targeted by gRNA in THP-1#11-4 and #11-7.
Deletions around predicted A3G pseudogene.
Pseudo-single cycle infectivity assays for each HIV-1 mutant in SupT11 cells stably expressing A3.