

Abstract
Objective:
Succinic semialdehyde dehydrogenase deficiency (SSADHD) is a rare inherited metabolic disorder caused by a defect of ɣ-aminobutyrate (GABA) catabolism. Despite the resultant hyperGABAergic environment facilitated by the metabolic defect, individuals with this disorder have a paradoxically high prevalence of epilepsy. We aimed to study the characteristics of epilepsy in SSADHD and its concordance with GABA-related metabolites and neurophysiologic markers of cortical excitation.
Methods:
Subjects in an international natural history study of SSADHD underwent clinical assessments, electroencephalography, transcranial magnetic stimulation (TMS), magnetic resonance spectroscopy for GABA/NAA quantification, and plasma GABA-related metabolite measurements.
Results:
A total of 61 subjects with SSADHD and 42 healthy controls were included in the study. Epilepsy was present in 49% of the SSADHD cohort. Over time, there was an increase in severity in 33% of the subjects with seizures. The presence of seizures was associated with increasing age (p=0.001) and lower levels of GABA (p=0.002), γ-hydroxybutyrate (GHB) (p=0.004), and γ-guanidinobutyrate (GBA) (p=0.003). Seizure severity was associated with increasing age and lower levels of GABA-related metabolites as well as lower TMS-derived resting motor thresholds (rMT) (p=0.04). The cut-off values with the highest discriminative ability to predict seizures were age >9.2 years (p=0.001), GABA<2.57μM/L (p=0.002), GHB<143.6 (p=0.004), and GBA<0.075 (p=0.007). A prediction model for seizures in SSADHD was comprised of the additive effect of older age, lower plasma GABA, GHB, and GBA (area under the ROC of 0.798, p=0.008).
Significance:
Epilepsy is highly prevalent in SSADHD, and its onset and severity correlate with an age-related decline in GABA and GABA-related metabolite levels as well as TMS markers of reduced cortical inhibition. The reduction of GABAergic activity in this otherwise hyperGABAergic disorder demonstrates a concordance between epileptogenesis and compensatory responses. These findings may furthermore inform the timing of molecular interventions for SSADHD.
Keywords: Seizures, Epileptogenesis, Excitation, Inhibition, Pathomechanism
Introduction
ɣ-aminobutyrate (GABA), the major cortical inhibitory neurotransmitter, normally converts via the enzyme GABA-transaminase to the intermediate metabolite succinic semialdehyde (SSA), which is rapidly converted to succinate by succinic semialdehyde dehydrogenase (SSADH). Succinate then enters the tricarboxylic cycle, generating ATP and α-ketoglutarate. SSADH deficiency (SSADHD) (OMIM #271980) is an autosomal recessive disorder driven by biallelic loss of function mutations of the ALDH5A1 gene 1. SSADHD results in SSA and GABA accumulation, and conversion of GABA into γ-hydroxybutyrate (GHB). The disorder results in a broad spectrum of encephalopathic manifestations, including seizures in up to 50% of patients with SSADHD 2.
Previous studies in the ALDH5A1−/− null mouse3 and in patients with SSADHD 4 have suggested that seizures in a hyperGABAergic environment occur because of heightened compensatory activity, likely associated with downregulation of GABAA and GABAB receptors leading to impaired GABA signaling 5–7. Additionally, the elevation of GABA and GHB (in blood and hair) observed in newborn dried blood spots or in young patients decreases over the first decade 8. We employed TMS, MRS imaging with GABA editing, and GABA-related metabolite quantification to test the hypothesis that seizure onset or exacerbation in SSADHD occurs with reduced cortical inhibition or GABA-related metabolite levels.
Materials and Methods
Study design and population
This is an analysis of data collected in an ongoing natural history study of SSADHD (ClinicalTrials.gov ID: NCT03758521) by investigators of the SSADH Deficiency Consortium, between March 2019-December 2022. Participants were recruited at three clinical sites: Boston Children’s Hospital (BCH) in the US, University Children’s Hospital Heidelberg (UCHH) in Germany, and Hospital Sant Joan Déu Barcelona Children’s Hospital (UDB) in Spain. Remote subjects unable to attend the on-site procedures constituted a “standard of care” (SOC) cohort that sent relevant medical reports to BCH. Study oversight was provided by BCH (clinical), Washington State University (laboratory), and the Health Informatics Institute at University of South Florida (data management). The study was approved by the Institutional Review Board (IRB) of each participating site under the umbrella of a single IRB at BCH (IRB #P00029917). Some of the data used in this study were reported at the half-way point of the SSADHD natural history study, a time at which only 28 individuals were enrolled 9. Study participants included individuals with confirmed molecular diagnosis of SSADHD (n = 61), and healthy individuals (controls, n = 42) recruited among the patients’ family members (parents and siblings) and unrelated individuals. Participants completed procedures including neurological and neuropsychological assessments, electroencephalography (EEG), magnetic resonance imaging (MRI), magnetic resonance spectroscopy ([1H]MRS), and blood collection for GABA, GHB, and γ-guanidinobutyrate (GBA) measurements, all of which are elevated in SSADHD 10. Seizures were assessed for age of onset, frequency, duration, and responsiveness to medications. Transcranial magnetic stimulation (TMS) studies were completed by those enrolled in the BCH site.
Seizure-Related Definitions
Seizure types and epilepsy were classified accordingly to the International League Against Epilepsy (ILAE) criteria 11; 12. “An increase in seizure severity” was defined when there was an increase in seizure frequency or duration of at least 2-fold over time, and [per the definition of drug-resistant epilepsy (DRE) 13] when two tolerated and appropriately chosen antiseizure medications failed to control the seizures.
Neurophysiology, Neuroimaging, and Biochemical Measurements
EEGs- ~30-minute EEGs were done as 21-channel studies with electrodes placed per international standards, recording awake and sleep states.
Brain MRS- Brain GABA/NAA ratio was measured using 1H-NMR spectroscopy. Data were acquired using both single-voxel methods (MEGA PRESS) with a voxel size of 30 mm isotropic and multi-voxel methods using two-dimensional chemical shift imaging 14.
Neuronavigated TMS- was applied using the Nexstim 5.1.1 system (Nexstim, Finland). The use of an anatomical T1-weighted MRI sequence for each participant was required for co-registration and frameless-stereotaxy. Each participant’s primary motor cortex was identified using a Figure-of-eight coil, while motor evoked potentials were recorded from the contralateral abductor pollicis brevis (APB) muscle 15. EMG was recorded at 3 kHz with a bandpass filter from 10-500 Hz. Resting motor threshold (rMT) was operationally defined as the minimum machine output required to elicit an MEP ≥50 μV from the target muscle at rest in >50% of trials. Cortical silent period (CSP) was calculated as the duration from stimulation at 150% rMT intensity to the spontaneous return of voluntary muscle activity in the EMG of the right APB. Long-interval cortical inhibition (LICI), a paired-pulse metric of intracortical inhibition, was collected by pairs of stimulations delivered at 120% rMT with 100 millisecond interpulse intervals. LICI was reported as the log transformation of the test:conditioning right APB peak-to-peak amplitude ratio (peak-to-peak amplitude of the resultant MEP from the second pulse divided by the peak-to-peak amplitude of the resultant MEP from the first pulse). EMG signals were analyzed using LabChart v8.1.17 to extract CSP durations and LICI ratios.
Biochemical tests- Total plasma quantitation of GABA (done after hydrolysis with 6N HCl using isotope dilution methodology 16), GHB, and GBA was performed 17.
Statistical Analysis
Data were analyzed with SPSS Statistics (IBM SPSS Statistics, Version 28, 2021, IBM Corp, Armonk, NY, USA). Descriptive statistics methods were used due to the sample size and the study’s objectives. Categorical variables were reported by their relative frequencies and compared by a Pearson Chi-square test or a Fisher exact test. The normality of distributions of continuous variables was tested using a histogram and the Kolmogorov–Smirnov test. Continuous variables following a normal distribution were described by their means and standard deviations (SD), and those not following a normal distribution by medians and interquartile ranges (IQR). Associations between categorical and normally distributed continuous variables were compared using independent sample t tests and skewed variables through a Mann-Whitney test. Pearson’s correlation coefficient was applied to study the association between two continuous variables. Discrimination ability of potential predictors of seizures was evaluated by an area under the receiver operating characteristic (ROC) curve. The maximal Youden index 18 was used to identify the threshold value with the highest discriminative ability for these variables. Positive and negative predictive values (PPV and NPV) of the best cutoffs identified for the predicting variables were also calculated. Variables with the highest predicting significance were applied to examine the best-compounded prediction model for seizures in SSADHD. A p value ≤ 0.05 was considered significant for all analyses. The correlations between age and GABA, GHB and TMS were analyzed using Statgraphics Centurion 18, Version 18.1.14 (Statgraphics Technologies, Inc., The Plains, VA, USA) and the Excel Solver function. Regression models describing the best fit of the data to age were chosen based on the highest correlation coefficients and R-squared values and the lowest sum of the squared difference between observed and fitted data. Inflection points were determined graphically as the age at which the parameters of interest (GABA, GHB, TMS) were half their value at birth.
Results
A total of 103 individuals were included in the study; 61 (54% female) with confirmed SSADHD (29 from BCH, 14 from UCHH, 10 from UDB, and 7 from the standard of care cohort), and 42 healthy controls. The median (IQR) age at enrollment was 9.6 (5.4-14.7) years, and 38 (62%) were <12 years old. No participant was born prematurely, had a birth weight lower than 2000 grams, or sustained major postnatal complications. Thirty-eight SSADHD individuals completed an MRI, of which 10 (26%) were determined as abnormal, with cortical and mild cerebellar volume loss, T2-weighted hyperintensities in the cerebellar dentate nucleus and globus pallidi, and mild to moderate diffuse white matter changes. Genetic information showed 57 subjects who were either homozygous or compound heterozygous for 32 different ALDH5A1 variants (Fig 1).
Fig 1.
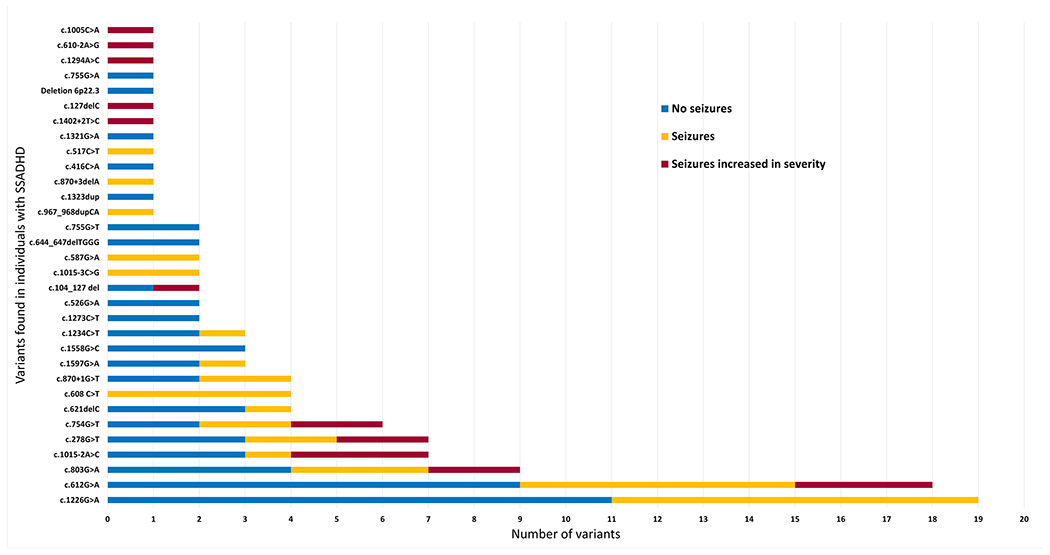
Display of the distribution of frequency, seizure occurrence, and seizure severity of all the ALDH5A1 variants found in individuals with SSADHD in the study group.
Plasma GABA and GHB concentrations were inversely correlated with age (GABA: r=−0.540, p<0.001; GHB r=−0.430, p=0.01), showing a rapid decline in values up to 10-15 years of age and reaching a plateau thereafter (Fig 2). TMS-measured rMT and CSP decreased with age (r=−0.659, p=0.004 for rMT, and r=−0.524, p=0.01 for CSP), reaching a plateau similar to what was observed for GABA and GHB.
Fig 2.
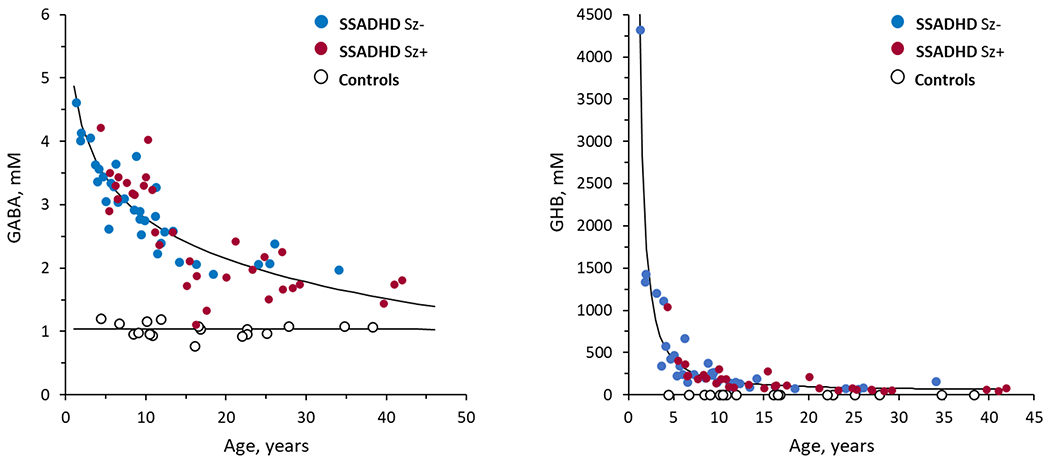
Relationship between plasma concentrations of GABA (left), and GHB (right) to age in SSADHD individuals with (Sz+, red dots) and without (Sz−, blue dots) seizures, and controls. The lines represent the best fits [Equation 1: GABA = 4.85 – (0.90 * LN(Age), p < 0.001; Equation 2: GHB = (0.36 +(70.3/Age)2, p<0.001; NS for controls].
Compared to healthy controls, participants of the SSADHD group were significantly younger [median (IQR) of 9.6 (5.4-14.7) vs. 15.8 (10.2-23.3) years, (p=0.006)]. However, the age-adjusted estimated marginal means [standard errors, (SE)] of the SSADHD group were significantly higher for plasma GABA [2.85 (0.14) vs. 1.19 (0.22) μM/L, p<0.001], plasma GHB [390.67 (101.53) vs. 59.41 (140.31) μM/L, p = 0.002], MRS-measured GABA/NAA ratio [0.20 (0.007) vs. 0.07 (0.008), p<0.001], and TMS-measured rMT [median [66.17 (23.16) vs. 47.33 (17.55) MO%, p<0.001] and CSP [200.54 (12.45) vs. 122.14 (14.79) ms, p<0.001], and significantly lower for TMS-measured LICI [−0.11 (0.41) vs. 2.80 (0.59) log MEPtest/MEPcondition, p = 0.002].
Correlates of Epilepsy
Thirty (49%) individuals with SSADHD had seizures, with a median (IQR) age of seizure onset of 9.0 (5.25-12.0) years and a full range of 4 months to 19 years. Only three (5%) individuals from the study group had seizures as part of their initial diagnosis. Eleven (18%) subjects had more than one seizure type, and the overall number of seizure types were generalized tonic clonic (GTC) [n= 18 (60%)]; absence [n= 7 (23%)]; myoclonic [n=5 (17%)]; atonic [n= 4 (13%)]; focal with impaired awareness [n=3 (10%)]; epileptic spasms [n=2 (7%)], and other types [n=4 (13%)]. Episodes of status epilepticus were experienced by 2 (3%) individuals. The most common type of seizure in childhood (≤12 years) was absence, and in adolescence and adulthood GTC.
Seizures were significantly more prevalent in individuals who were older [median (IQR) of 13.3 (9.1-20.3) vs. 7.8 (4.2-11.1), p =0.001], had lower overall adaptive scores (p=0.01) and motor ability scores (p=0.03), lower values of plasma GABA (μM/L) (mean ± SD of 2.48 ± 1.08 vs. 3.41 ± 1.22, p=0.002), plasma GBA (μM/L) (mean ± SD of 0.62 ± 0.21 vs. 0.92 ± 0.29, p=0.003), and plasma GHB (μM/L) [median (IQR) of 114.3 (67.6-226.8) vs. 270.6 (154.0-624.5), p=0.004] (Table 1). The cut-off values with a significant discriminative ability to predict seizures were age above 9.22 years, and plasma GABA, GBA and GHB levels (μM/L) below 2.57, 0.075, and 143.65, respectively (Table 2). The additive effect of the variables with the highest discriminative ability (older age, lower plasma GABA, GBA, and GHB) yielded a prediction model for seizures in SSADHD with an area under the ROC of 0.798 (p=0.008), sensitivity of 82%, specificity of 78%, Youden index of 0.596, and PPV of 81% (Table 2, Fig 3). There were no significant differences between individuals with and without seizures in terms of sex, overall IQ, presence of abnormalities of EEG, MRS peak GABA/NAA, and TMS measurements (Table 1).
Table 1.
Demographic and clinical characteristics of SSADHD individuals with and without seizures, and with an unchanged vs. increased seizure severity over time.
| Characteristic | Presence of Seizures | p value | Seizure severity* | p value | ||
|---|---|---|---|---|---|---|
|
|
|
|||||
| Without seizures N=31 (%) |
With seizures N=30 (%) |
Unchanged N=20 (%) |
Increased N=10 (%) |
|||
|
| ||||||
| Sex | ||||||
| Males | 14 (45) | 14 (47) | 0.90 | 10 (50) | 4 (40) | 0.60 |
| Females | 17 (55) | 16 (53) | 10 (50) | 6 (60) | ||
|
| ||||||
| Age, years, median (IQR) | 7.8 (4.2-11.1) | 13.3 (9.1-20.3) | 0.001 | 10.9 (7.0-18.3) | 15.6 (12.3-24.2) | 0.04 |
|
| ||||||
| Intelligence quotient (N=27) | (N=13) 57.15 ± 17.11 | (N=14) 50.79 ± 10.79 | 0.26 | (N=8) 50.88 ± 11.05 | (N=6) 50.67 ± 11.48 | 0.973 |
| Adaptive function scores (N=31) | (N=15) 67.33 ± 13.08 | N=16) 55.56 ± 11.78 | 0.01 | (N=10) 56.20 ± 12.48 | (N=6) 54.50 ± 10.84 | 0.791 |
| Motor ability scores (N=26) | (N=15) 72.07 ± 7.66 | (N=11) 64.73 ± 8.28 | 0.03 | (N=7) 64.57 ± 9.53 | (N=4) 65.00 ± 6.83 | 0.939 |
|
| ||||||
| Abnormal EEG (N=54) | (N=26) 17 (65) | (N=28) 22 (79) | 0.28 | (N=18) 12 (66) | (N=10) 10 (100) | 0.03 |
|
| ||||||
| Plasma GABA , μM/L, mean ± SD (N=45) | (N=23) 3.41 ± 1.22 | (N=22) 2.48 ± 1.08 | 0.002 | (N=15) 2.82 ± 1.11 | (N=7) 1.71 ± 0.45 | 0.02 |
| Plasma GBA, μM/L, mean ± SD (N=31) | (N=15) 0.92 ± 0.29 | (N=16) 0.62 ± 0.21 | 0.003 | (N=10) 0.68 ± 0.02 | (N=6) 0.53 ± 0.01 | 0.18 |
| Plasma GHB, μM/L, median (IQR) (N=34) | (N=17) 270.6 (154.0-624.5) | (N=17) 114.3 (67.6-226.8) | 0.004 | (N=11) 208.6 (78.2-302.0) | (N=6) 68.5 (52.7-109.8) | 0.03 |
|
| ||||||
| MRS peak GABA/NAA, mean ± SD (N=19) | (N=8) 0.20 ± 0.04 | (N=11) 0.19 ± 0.03 | 0.57 | (N=5) 0.19 ± 0.02 | (N=6) 0.19 ± 0.03 | 0.80 |
|
| ||||||
| TMS, median (IQR) | ||||||
| rMT, %MO (N=23) | (N=11) 72.7 (55.0-100.0) | (N=12) 56.0 (44.0-73.0) | 0.19 | (N=6) 68.5 (57.0-79.7) | (N=6) 44.0 (33.5-62.5) | 0.04 |
| CSP, ms (N=21) | (N=9) 209.0 (175.0-255.0) | (N=12) 210.5 (144.0-253.4) | 0.82 | (N=7) 238.0 (163.0-254.2) | (N=5) 159.0 (131.0-223.7) | 0.29 |
| LICI, log (MEPtest/MEPcondition) (N=18) | (N=7) 0.0004 (−0.08-0.51) | (N=11) 0.037 (−0.739-0.56) | 0.55 | (N=6) 0.01 (−0.54-0.54) | (N=5) 0.33 (−0.76-0.59) | 0.71 |
CSP- cortical silent period; EEG- electroencephalography; GABA- γ-aminobutyric acid; GBA- γ-guanidinobutyrate; IQR- interquartile range; LICI- long-interval intracortical inhibition; MEP- motor evoked potential; MO- machine output; MRS- Magnetic resonance spectroscopy; NAA- N-acetyl aspartate; SD- Standard deviation; TMS- transcranial magnetic stimulation; Bold indicates the significance of the p-value.
As defined by an increase in seizure frequency or duration of at least 2-fold over time, and when two tolerated and appropriately chosen antiseizure medications failed to control the seizures.
Table 2.
Cut-off values with the highest discriminative ability of variables associated with seizures in SSADHD individuals.
| Parameter | Cut-off point | Sensitivity | Specificity | Youden index | PPV | NPV | AUC | 95% CI of AUC | p value |
|---|---|---|---|---|---|---|---|---|---|
| 1. Older age, years (N=61) | 9.22 | 77% | 65% | 0.412 | 70% | 75% | 0.745 | 0.620-0.869 | 0.001 |
| 2. MRS GABA/NAA (N=19) | 0.232 | 91% | 38% | 0.284 | 67% | 75% | 0.551 | 0.263-0.840 | 0.710 |
| 3. Lower plasma GABA, μM/L (N=45) | 2.57 | 68% | 78% | 0.465 | 75% | 72% | 0.775 | 0.635-0.914 | 0.002 |
| 4. Lower plasma GBA, μM/L (N=31) | 0.075 | 75% | 80% | 0.550 | 100% | 75% | 0.783 | 0.613-0.953 | 0.007 |
| 5. Lower plasma GHB, μM/L (N=34) | 143.65 | 65% | 82% | 0.471 | 79% | 70% | 0.785 | 0.632-0.939 | 0.004 |
| 6. Lower rMT, %MO (N=23) | 54.5 | 50% | 82% | 0.318 | 100% | 60% | 0.659 | 0.433-0.886 | 0.19 |
| 7. Lower CSP, ms (N=21) | 124 | 17% | 100% | 0.167 | 100% | 47% | 0.579 | 0.328-0.830 | 0.54 |
| 8. Higher LICI, log (MEPtest/MEPcondition) (N=18) | 0.51 | 36% | 86% | 0.221 | 100% | 36% | 0.532 | 0.255-0.810 | 0.82 |
| Combination of variables 1, 3, 4, 5 | - | 82% | 78% | 0.596 | 81% | 73% | 0.798 | 0.588-1.000 | 0.008 |
AUC- area under the curve; CI- confidence interval; CSP- cortical silent period; EEG- electroencephalography; GABA- γ-aminobutyric acid; GBA- γ-guanidinobutyrate; LICI- long-interval intracortical inhibition; MEP- motor evoked potential; MO- machine output; MRS- Magnetic resonance spectroscopy; NAA- N-acetyl aspartate; NPV- negative predictive value; PPV- positive predictive value; rMT- resting motor threshold; SSADHD- succinic semialdehyde dehydrogenase deficiency; TMS- transcranial magnetic stimulation; Bold indicates the significance of the p-value.
Fig 3.
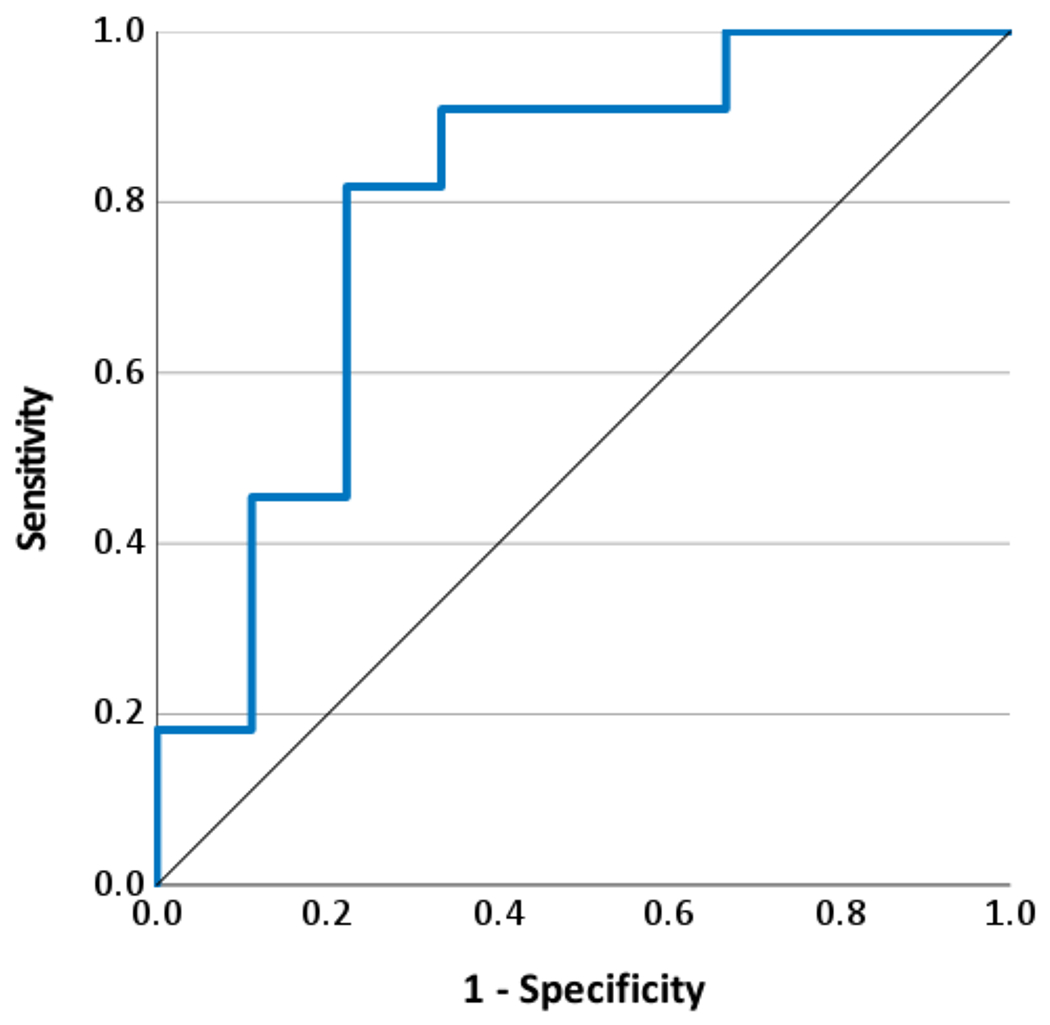
Area under the receiver operating characteristic (ROC) curve reflecting the predictive compounded model of seizures in SSADHD subjects. The model is composed of the additive effect of four independent seizure-predicting variables that are relevant for this condition: an older age, and lower plasma ɣ-aminobutyrate (GABA), γ-guanidinobutyrate (GBA) and γ-hydroxybutyrate (GHB) levels.
At baseline, a rare (≤1 per year) seizure frequency was noted in 15 (50%) subjects, and the median (IQR) seizure frequency of the other affected subjects was 2.0 (1.0-3.0) seizures per month. Seizure duration was ≤1 minute in 8 (27%) subjects, and the overall average seizure duration was 1-5 minutes. Overtime, out of 30 individuals with SSADHD who had epilepsy, the seizure frequency or duration of 10 (33%) had increased by more than 2-fold. The epilepsy was considered as drug-resistant in all of the individuals in whom seizure severity increased. An increase in seizure severity was significantly associated with an older age [median (IQR) of 15.6 (12.3-24.2) vs. 10.9 (7.0-18.3) years (p=0.04)], an abnormal EEG (p=0.03), lower plasma levels of GABA (mean ± SD of 1.71 ± 0.45 vs. 2.82 ± 1.11 μM/L, p=0.02), and GHB [median (IQR) of 68.5 (52.7-109.8) vs. 208.6 (78.2-302.0) μM/L, p=0.03), and a lower value of TMS-measured rMT [median (IQR) of 44.0 (33.5-62.5) vs. 68.5 (57.0-79.7) MO, p=0.04] (Table 1). No significant differences were found between individuals in whom seizure severity was unchanged vs. those in whom it was increased, in sex, FSIQ, test scores of adaptive function and motor ability, plasma GBA levels, MRS-measured peak GABA/NAA, and TMS-measured CSP and LICI. The distribution of variants of individuals with seizures of unchanged and increased severity is presented in Fig 1. None of the variants were found to be significantly associated with seizures that increased in severity.
Neurophysiologic Findings
Fifty-four subjects (89%) with SSADHD had undergone EEG recordings, which were abnormal in 39 (73%). Twenty-two (56%) individuals with an abnormal EEG had clinical seizures. Diffuse background slowing was seen in 24 (45%) subjects and epileptiform discharges in 14 (26%) of those who underwent EEGs. Focal epileptiform discharges were mainly in centrotemporal or frontal regions. The posterior dominant rhythm was absent or deceased in frequency, in relation to age, in nine (17%) subjects. Table 3 demonstrates the age distribution of these EEG abnormalities. Older individuals were significantly more prone to EEG abnormalities coexisting with clinical seizures (p=0.02).
Table 3.
EEG abnormalities of individuals with SSADHD
| Age range, years | Abnormal EEG N=39 (%) |
Diffuse background slowing N=24 (%) |
Interictal epileptiform activity N=14 (%) |
Ictal activity N=3 (100%) |
Absent / Age-inappropriate posterior dominant rhythm N=9 (%) |
|---|---|---|---|---|---|
| 0-5 | 9 (17) | 6 (25) | 4 (28) | 3 (100) | 0 (0) |
| 6-12 | 18 (49) | 9 (37) | 7 (50) | 0 (0) | 4 (44) |
| 12-17 | 8 (17) | 5 (21) | 3 (21) | 0 (0) | 3 (33) |
| ≥18 | 4 (8) | 4 (12) | 0 (0) | 0 (0) | 2 (22) |
EEG- electroencephalography; SSADHD- succinic semialdehyde dehydrogenase deficiency
Response to Anti-Seizure Medications (ASMs)
A total of 52 trials using 13 standard ASMs were documented for SSADHD subjects with seizures. The mean number of ASMs used by those with DRE was 2.7 ± 1.5. Nine (30%) individuals with seizures did not require any ASM and 4 (13%) required ≥ 3 ASMs. The most common ASMs that were used were levetiracetam [n=13 (43%)] and lamotrigine [n=8 (27%)]. ASMs given as monotherapy that led to seizure reduction were lamotrigine [n=3 (10%)], levetiracetam [n=2 (7%)], and oxcarbazepine [n=1 (3%)]. The combined therapy of lamotrigine and levetiracetam [n=2 (7%)], lamotrigine with clobazam, oxcarbazepine, and clobazam, and levetiracetam and perampanel [n=1 (3%)] in each case, were also effective at seizure control. Vigabatrin was not clinically efficacious for the three (10%) individuals who were treated with it.
Discussion
This prospective, international, longitudinal study of the largest reported cohort of SSADHD subjects showed that the onset and severity of epilepsy increased at the end of the first decade, in concordance with reductions in GABA and its primary metabolites, GHB and GBA, which are otherwise elevated in this condition 10. In addition, TMS derived measurements of the cortical excitatory: inhibitory ratio showed evidence of decreased inhibition at this time. These findings are consistent with earlier reports of worsening epilepsy in SSADHD during adolescence 2; 19. Also consistent with prior reports, half of our cohort had epilepsy, and nearly ¾ had abnormal EEGs, predominantly manifesting diffuse slowing and focal epileptiform discharges 2; 19–22. EEG abnormalities, together with clinical seizures, were significantly more prevalent in older individuals. Four of six individuals in our study who had an abnormal EEG but no seizures at baseline developed seizures with time. None of the genetic variants was specifically associated with epilepsy or an increase of seizure severity (Fig 1). However, since some of the study participants are still young, and seizures may occur later in their lives, examining this genotype to seizure-phenotype correlation in the future may be informative.
The most suitable ASM for treating seizures in SSADHD is still uncertain. This was a noninterventional study that did not systematically study the patients’ ASMs. Therefore, we cannot draw clear-cut conclusions from any of the findings seen in this context. However, according to the latest reports 23–25 and the observations of this study, there seems to be some affirmation that seizure control is best achieved with lamotrigine or levetiracetam, either as monotherapy or in combination with other ASMs as clobazam, oxcarbazepine, and perampanel. Although valproate is generally not recommended due to its ability to inhibit any residual enzymatic activity 26; 27, valproate was given to several individuals in the study, and was not proven to be beneficial. Likewise, vigabatrin, an irreversible inhibitor of GABA transaminase, did not lead to seizure control in the three individuals from our study who received it, adding to the conflicting evidence of its clinical yield 28–34.
Elevated GABA levels, representing a general decrease in cortical excitability, are paradoxical given the high prevalence of seizures in SSADHD. To discern this discrepancy, we performed analyses aimed to reflect GABA-ergic influences. These showed significantly lower GABA, GBA, and GHB levels in SSADHD individuals with epilepsy, and even lower GABA and GHB levels in subjects in whom seizure severity increased over time (Table 1). TMS measurements indicated that rMT directly correlated with GABA and GHB levels, as it was reduced in SSADHD patients with epilepsy, and was significantly reduced in those with increased seizure severity. This is in accordance with rMT being a measure of aggregate cortical excitability that is increased in epilepsy 35. Similar to neurotransmitter levels, both the rMT and CSP values significantly declined with age, reaching a plateau around 10-15 years. Taken together, the TMS data supports increased cortical excitability toward the end of the first decade in SSADHD.
The neurobiological explanation of seemingly paradoxical reduced cerebral inhibition in individuals with SSADHD, a hyper-GABAergic state, may be multifactorial. Gene expression studies have suggested that GABAA receptors remain largely immature in ALDH5A1 null mice and remain excitatory, consistent with altered chloride channel activity 24. There may be persistence of the postnatal excitatory qualities of the GABAA receptor past the postnatal period. With age, a persistent hyperGABAergic state appears to facilitate the down-regulation of GABA receptors, as demonstrated by flumazenil-PET and paired pulse TMS studies 36–38. Another factor that may influence the E:I ratio is the accumulation of GHB, a neuroactive metabolite of GABA. In normal concentrations, GHB mainly activates GHB receptors, but in high concentrations, it activates postsynaptic GABAB receptors 39. Presynaptically, GABAB receptors induce the release of GABA by curbing the neuronal calcium conductance, and postsynaptically, they lead to neuronal hyperpolarization 40; 41. In SSADHD, the persistently elevated GHB has been shown to lead to the downregulation of GABAB receptors, further disrupting inhibition 38; 42. The reduction of GABA and GHB levels that occurs with increasing age 8, in the presence of impaired GABAA and GABAB receptors signaling and delayed upregulation of these receptors, likely further exacerbates the E:I ratio, resulting in a lower seizure threshold, neuropsychiatric decline 25; 43, and behavioral worsening 44; 45.
We showed that the decline in GABA, GHB, and TMS measured rMT and CSP correlate with age. The reduction of these essential markers that represent decreased cortical inhibition occurs slightly before ten years and reaches a plateau around 10-15 years (Fig 2). This finding is supported by the threshold age of > 9.22 years, which had a high discriminative ability for seizure onset (Table 2).
Recent efforts to develop a gene therapy for SSADHD are based on a novel SSADHD mouse model termed ALDH5A1lox-STOP that can restore SSADH “on-demand” 46. One of the challenging aspects of this treatment approach is predicting the correct time at which SSADH is optimally restored. In the presence of downregulated GABAA receptors, fast SSADH restoration may inadvertently result in a hypoGABAergic state, which may intensify seizures and neurodevelopmental deficits 46. Therefore, these findings are relevant to the timing of genetically targeted intervention 7. Additionally, this study’s findings, providing a comprehensive mapping of clinical, biochemical, and neurophysiological correlative features of seizures in SSADHD, set the stage for future mechanistic investigations determining the elements that regulate the maturational trajectory of GABA and related metabolites, leading to seizures. Moreover, utilizing the biomarkers we have identified, future studies could be replicated in SSADHD and in other disease states of GABA metabolism, ranging from GABA transaminase deficiency to the numerous disorders of GABA receptors and transporters, to pharmacological hyperGABAergic states, such as those induced by vigabatrin, an irreversible inhibitor of GABA transaminase.
Conclusions
Seizure onset and severity in SSADHD, a hyper-GABAergic metabolic epilepsy, coincide with an age-related decline in inhibitory neurotransmitter levels and TMS-derived motor thresholds and cortical silent period, markers of increased cortical excitation. Cut-off values of age > 9 years, plasma GABA levels <2.57μM/L, plasma GHB levels < 143.65μM/L, and plasma GBA < 0.075μM/L have a high discriminative value for prediction of seizure onset or exacerbation. These findings are furthermore informative for the timing of targeted gene therapy or enzyme replacement therapy in SSADHD.
Key points.
Individuals with SSADHD, a rare inherited disorder of GABA catabolism, have a high prevalence of seizures which are characterized by absence and later generalized tonic-clonic semiology, and which typically have onset or increased severity at the end of the first decade.
Decline in plasma GABA, GABA-related metabolites, and TMS-derived indices of cortical inhibition correlate with seizure onset and severity in SSADHD.
Patient age and inhibitory neurotransmitter levels have the highest discriminative ability for prediction of seizure onset and provide information for the timing of molecularly targeted treatment.
Compensatory downregulation of GABAergic activity may explain the pathophysiology of epilepsy in hyperGABAergic states.
Funding statement
The SSADHD Natural History study is funded by the National Institutes of Health (1R01HD091142). The content presented in this study is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Conflict of interest disclosure
None of the authors has any conflict of interest to disclose.
Epilepsia Ethical Publication Statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Ethics approval statement
The study was approved by the Institutional Review Board (IRB) of each participating site under the umbrella of a single IRB at Boston Children’s Hospital (IRB #P00029917).
Patient consent statement
All the patients and healthy controls the (or their parents/legal guardians in cases of children and adolescents under the age of 18 years) who were included in the study signed a written formal informed consent form.
Permission to reproduce material from other sources
Non-applicable
Clinical trial registration
Non-applicable
Data availability statement
The data that support the findings of this study are available on reasonable request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
References
- 1.Pearl PL, Parviz M, Vogel K, et al. Inherited disorders of gamma-aminobutyric acid metabolism and advances in ALDH5A1 mutation identification. Dev. Med. Child Neurol 2015;57:611–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pearl PL, Gibson KM, Acosta MT, et al. Clinical spectrum of succinic semialdehyde dehydrogenase deficiency. Neurology 2003;60:1413–1417 [DOI] [PubMed] [Google Scholar]
- 3.Gupta M, Hogema BM, Grompe M, et al. Murine succinate semialdehyde dehydrogenase deficiency. Ann. Neurol 2003;54 Suppl 6:S81–90 [DOI] [PubMed] [Google Scholar]
- 4.Pearl PL, Gibson KM, Cortez MA, et al. Succinic semialdehyde dehydrogenase deficiency: lessons from mice and men. J. Inherit. Metab. Dis 2009;32:343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jansen EE, Struys E, Jakobs C, et al. Neurotransmitter alterations in embryonic succinate semialdehyde dehydrogenase (SSADH) deficiency suggest a heightened excitatory state during development. BMC Dev. Biol 2008;8:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim KJ, Pearl PL, Jensen K, et al. Succinic semialdehyde dehydrogenase: biochemical-molecular-clinical disease mechanisms, redox regulation, and functional significance. Antioxid Redox Signal 2011;15:691–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee HHC, McGinty GE, Pearl PL, et al. Understanding the Molecular Mechanisms of Succinic Semialdehyde Dehydrogenase Deficiency (SSADHD): Towards the Development of SSADH-Targeted Medicine. Int J Mol Sci 2022;23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jansen EE, Vogel KR, Salomons GS, et al. Correlation of blood biomarkers with age informs pathomechanisms in succinic semialdehyde dehydrogenase deficiency (SSADHD), a disorder of GABA metabolism. J. Inherit. Metab. Dis 2016;39:795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pearl PL, DiBacco ML, Papadelis C, et al. Succinic Semialdehyde Dehydrogenase Deficiency: Review of the Natural History Study. J. Child Neurol 2021;36:1153–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kirby T, Walters DC, Brown M, et al. Post-mortem tissue analyses in a patient with succinic semialdehyde dehydrogenase deficiency (SSADHD). I. Metabolomic outcomes. Metab. Brain Dis 2020;35:601–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fisher RS, Cross JH, French JA, et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58:522–530 [DOI] [PubMed] [Google Scholar]
- 12.Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58:512–521.5386840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kwan P, Arzimanoglou A, Berg AT, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 2010;51:1069–1077 [DOI] [PubMed] [Google Scholar]
- 14.Afacan O, Yang E, Lin AP, et al. Magnetic Resonance Imaging (MRI) and Spectroscopy in Succinic Semialdehyde Dehydrogenase Deficiency. J. Child Neurol 2021;36:1162–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsuboyama M, Kaye HL, Rotenberg A. Review of Transcranial Magnetic Stimulation in Epilepsy. Clin. Ther 2020;42:1155–1168 [DOI] [PubMed] [Google Scholar]
- 16.Arning E, Wasek B, Bottiglieri T. Quantitation of γ-Aminobutyric Acid in Cerebrospinal Fluid Using Liquid Chromatography-Electrospray-Tandem Mass Spectrometry Clinical Applications of Mass Spectrometry in Biomolecular Analysis, Springer; 2022:165–174. [DOI] [PubMed] [Google Scholar]
- 17.Jansen EE, Verhoeven NM, Jakobs C, et al. Increased guanidino species in murine and human succinate semialdehyde dehydrogenase (SSADH) deficiency. Biochim. Biophys. Acta 2006;1762:494–498 [DOI] [PubMed] [Google Scholar]
- 18.Fluss R, Faraggi D, Reiser B. Estimation of the Youden Index and its associated cutoff point. Biom J 2005;47:458–472 [DOI] [PubMed] [Google Scholar]
- 19.Pearl PL, Shukla L, Theodore WH, et al. Epilepsy in succinic semialdehyde dehydrogenase deficiency, a disorder of GABA metabolism. Brain Dev. 2011;33:796–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DiBacco ML, Roullet JB, Kapur K, et al. Age-related phenotype and biomarker changes in SSADH deficiency. Ann Clin Transl Neurol 2019;6:114–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cortes-Saladelafont E, Molero-Luis M, Cuadras D, et al. Gamma-aminobutyric acid levels in cerebrospinal fluid in neuropaediatric disorders. Dev. Med. Child Neurol 2018;60:780–792 [DOI] [PubMed] [Google Scholar]
- 22.Cortez MA, Wu Y, Gibson KM, et al. Absence seizures in succinic semialdehyde dehydrogenase deficient mice: a model of juvenile absence epilepsy. Pharmacol. Biochem. Behav 2004;79:547–553 [DOI] [PubMed] [Google Scholar]
- 23.Didiasova M, Banning A, Brennenstuhl H, et al. Succinic Semialdehyde Dehydrogenase Deficiency: An Update. Cells 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vogel KR, Ainslie GR, Walters DC, et al. Succinic semialdehyde dehydrogenase deficiency, a disorder of GABA metabolism: an update on pharmacological and enzyme-replacement therapeutic strategies. J. Inherit. Metab. Dis 2018;41:699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parviz M, Vogel K, Gibson KM, et al. Disorders of GABA metabolism: SSADH and GABA-transaminase deficiencies. J Pediatr Epilepsy 2014;3:217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shinka T, Ohfu M, Hirose S, et al. Effect of valproic acid on the urinary metabolic profile of a patient with succinic semialdehyde dehydrogenase deficiency. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci 2003;792:99–106 [DOI] [PubMed] [Google Scholar]
- 27.Vanadia E, Gibson KM, Pearl PL, et al. Therapeutic efficacy of magnesium valproate in succinic semialdehyde dehydrogenase deficiency. JIMD Rep 2013;8:133–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Casarano M, Alessandri MG, Salomons GS, et al. Efficacy of vigabatrin intervention in a mild phenotypic expression of succinic semialdehyde dehydrogenase deficiency. JIMD Rep 2012;2:119–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gibson KM, DeVivo DC, Jakobs C. Vigabatrin therapy in patient with succinic semialdehyde dehydrogenase deficiency. Lancet 1989;2:1105–1106 [DOI] [PubMed] [Google Scholar]
- 30.Gibson KM, Jakobs C, Ogier H, et al. Vigabatrin therapy in six patients with succinic semialdehyde dehydrogenase deficiency. J. Inherit. Metab. Dis 1995;18:143–146 [DOI] [PubMed] [Google Scholar]
- 31.Gropman A. Vigabatrin and newer interventions in succinic semialdehyde dehydrogenase deficiency. Ann. Neurol 2003;54 Suppl 6:S66–72 [DOI] [PubMed] [Google Scholar]
- 32.Horvath GA, Hukin J, Stockler-Ipsiroglu SG, et al. Eye Findings on Vigabatrin and Taurine Treatment in Two Patients with Succinic Semialdehyde Dehydrogenase Deficiency. Neuropediatrics 2016;47:263–267 [DOI] [PubMed] [Google Scholar]
- 33.Matern D, Lehnert W, Gibson KM, et al. Seizures in a boy with succinic semialdehyde dehydrogenase deficiency treated with vigabatrin (gamma-vinyl-GABA). J. Inherit. Metab. Dis 1996;19:313–318 [DOI] [PubMed] [Google Scholar]
- 34.Vogel KR, Pearl PL, Theodore WH, et al. Thirty years beyond discovery--clinical trials in succinic semialdehyde dehydrogenase deficiency, a disorder of GABA metabolism. J. Inherit. Metab. Dis 2013;36:401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsuboyama M, Lee Kaye H, Rotenberg A. Biomarkers obtained by transcranial magnetic stimulation of the motor cortex in epilepsy. Front. Integr. Neurosci 2019;13:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pearl PL, Gibson KM, Quezado Z, et al. Decreased GABA-A binding on FMZ-PET in succinic semialdehyde dehydrogenase deficiency. Neurology 2009;73:423–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Macdonald RL, Kang JQ. Molecular pathology of genetic epilepsies associated with GABAA receptor subunit mutations. Epilepsy Curr. 2009;9:18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reis J, Cohen LG, Pearl PL, et al. GABAB-ergic motor cortex dysfunction in SSADH deficiency. Neurology 2012;79:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carter LP, Koek W, France CP. Behavioral analyses of GHB: receptor mechanisms. Pharmacol. Ther 2009;121:100–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pinard A, Seddik R, Bettler B. GABAB receptors: physiological functions and mechanisms of diversity. Adv. Pharmacol 2010;58:231–255 [DOI] [PubMed] [Google Scholar]
- 41.Bettler B, Kaupmann K, Mosbacher J, et al. Molecular structure and physiological functions of GABA(B) receptors. Physiol. Rev 2004;84:835–867 [DOI] [PubMed] [Google Scholar]
- 42.Buzzi A, Wu Y, Frantseva MV, et al. Succinic semialdehyde dehydrogenase deficiency: GABAB receptor-mediated function. Brain Res. 2006;1090:15–22 [DOI] [PubMed] [Google Scholar]
- 43.Malaspina P, Roullet JB, Pearl PL, et al. Succinic semialdehyde dehydrogenase deficiency (SSADHD): Pathophysiological complexity and multifactorial trait associations in a rare monogenic disorder of GABA metabolism. Neurochem. Int 2016;99:72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Knerr I, Gibson KM, Murdoch G, et al. Neuropathology in succinic semialdehyde dehydrogenase deficiency. Pediatr. Neurol 2010;42:255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bjork JM, Moeller FG, Kramer GL, et al. Plasma GABA levels correlate with aggressiveness in relatives of patients with unipolar depressive disorder. Psychiatry Res. 2001;101:131–136 [DOI] [PubMed] [Google Scholar]
- 46.Lee HHC, Pearl PL, Rotenberg A. Enzyme Replacement Therapy for Succinic Semialdehyde Dehydrogenase Deficiency: Relevance in gamma-Aminobutyric Acid Plasticity. J. Child Neurol 2021;36:1200–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available on reasonable request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.