

Abstract
Nonalcoholic fatty liver disease (NAFLD) is one of the etiologies that contribute to hepatocellular carcinoma (HCC), and chronic inflammation is one of the proposed mediators of HCC. Because necroptosis is a cell death pathway that induces inflammation, we tested whether necroptosis-induced inflammation contributes to the progression of NAFLD to HCC in a mouse model of diet-induced HCC. Male and female wild-type (WT) mice and mouse models where necroptosis is blocked (Ripk3−/− or Mlkl−/− mice) were fed either a control diet, choline-deficient low-fat diet or choline-deficient high-fat diet. Blocking necroptosis reduced markers of inflammation [proinflammatory cytokines (TNFα, IL6, and IL1β), F4/80+ve macrophages, CCR2+ve infiltrating monocytes], inflammation-associated oncogenic pathways (JNK, PD-L1/PD-1, β-catenin), and HCC in male mice. We demonstrate that hepatic necroptosis promotes recruitment and activation of liver macrophages leading to chronic inflammation, which in turn trigger oncogenic pathways leading to the progression of NAFLD to HCC in male mice. Whereas in female mice, blocking necroptosis reduced HCC independent of inflammation. Our data show a sex-specific difference in the development of inflammation, fibrosis, and HCC in WT mice. However, blocking necroptosis reduced HCC in both males and females without altering liver fibrosis. Thus, our study suggests that necroptosis is a valid therapeutic target for NAFLD-mediated HCC.
Implications:
Necroptosis is a major contributor to hepatic inflammation that drives the progression of NAFLD to HCC and therefore represents a valid target for NAFLD-mediated HCC.
Visual Overview
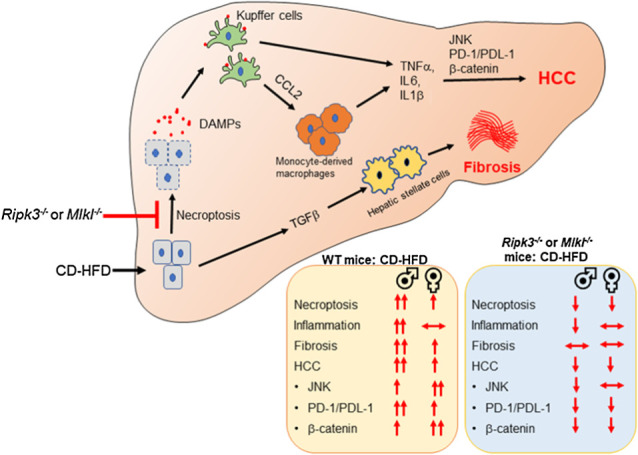
Introduction
Hepatocellular carcinoma (HCC), the primary form of liver cancer, is the fourth cancer-related cause of death worldwide with limited therapeutic options and poor survival (1). Nonalcoholic fatty liver disease (NAFLD) is one of the etiologies that contribute to HCC. NAFLD covers a spectrum of diseases ranging from fat deposition in the liver (nonalcoholic fatty liver, NAFL) to nonalcoholic steatohepatitis (NASH) that is characterized by steatosis, increased hepatic inflammation, fibrosis, and hepatocyte death. Nearly 30% of people with NAFL progress to NASH, and from these, 2%–13% progress to HCC (2). Despite the strong association between NAFLD and HCC, the pathway(s) that cause the progression of NAFLD to HCC are not clearly understood.
Nonresolving chronic inflammation is believed to be a contributor to the development and progression of HCC. One of the pathways that trigger a persistent inflammatory response is the activation of immune cells by damage-associated molecular patterns (DAMP; refs. 3, 4). DAMPs such as high mobility group box protein 1 (HMGB1) and mitochondrial DNA released from hepatocytes are proposed mediators of chronic inflammation in NASH, and levels of these DAMPs are increased in mouse models and patients with NASH (5). One of the pathways that release DAMPs from cells is necroptosis, a regulated form of inflammatory cell death (6). Necroptosis is initiated when necroptotic stimuli (e.g., TNFα, oxidative stress, lipotoxicity) sequentially activate receptor-interacting protein kinase 1 (RIPK1), RIPK3, and the pseudokinase mixed lineage kinase domain-like (MLKL) protein. Phosphorylation of MLKL leads to its oligomerization and membrane attachment, which results in permeabilization of the cell membrane and DAMP release (7). Necroptosis has been reported to be increased in the livers of NAFLD and NASH patients and in mouse models of NASH (8, 9).
Previously, we reported that inhibiting necroptosis using necrostatin-1s (Nec-1s) in a mouse model of spontaneous HCC (mice deficient in the antioxidant enzyme Cu/Zn superoxide dismutase, Sod1−/− mice) reduced hepatic inflammation, fibrosis, and pathways associated with HCC development (10). We also found that inhibiting necroptosis reduced hepatic inflammation and fibrosis in old wild-type (WT) mice that exhibit NASH pathology (11). On the basis of these findings, we hypothesized that necroptosis-mediated inflammation contributes to the progression of NAFLD to HCC. Our data show that WT male mice fed a choline-deficient amino acid–defined high-fat diet (CD-HFD) have increased inflammation, fibrosis, and HCC relative to WT female mice, and blocking necroptosis using either Ripk3−/− or Mlkl−/− mice reduced HCC in both males and females without altering liver fibrosis.
Materials and Methods
Animals and diet feeding
All procedures were approved by the Institutional Animal Care and Use Committee at the University of Oklahoma Health Sciences Center (OUHSC). Colonies of Ripk3−/− (12), Ripk3+/+, Mlkl−/− (13), and Mlkl+/+ mice were generated by breeding male and female Ripk3+/− or Mlkl+/− mice. The WT mice used in the study are a mix of littermates from both Ripk3 and Mlkl crossings, as they do not show any difference in the parameters measured. The mice were group housed in ventilated cages at 20°C ± 2°C, 12-hour dark/light cycle. Starting at 2 months of age, male and female mice (n = 7–10/group) were fed a choline-deficient amino acid–defined diet containing 60% fat by calories (CD-HFD; A06071302i, Research Diets) for a period of 6 months at the OUHSC animal care facility. Normal chow diet (NC; 5053 Pico Lab, Purina Mills) or choline-deficient amino acid–defined diet containing 10% fat by calories (CD-LFD; A06071324i, Research diets) were used as controls (Supplementary Fig. S1A). The livers were examined for the presence of visible tumors and classified on the basis of the size and the numbers of tumors per mouse.
Western blotting
Western blotting was performed and quantified using ImageJ software (U.S. NIH) as described previously (11). The primary antibodies used were: MLKL (#MABC604) and α-smooth muscle actin (α-SMA, #A5228; Millipore Sigma), RIPK3 (#NBP1-77299, Novus Biologicals), β-tubulin (#T5201, Sigma-Aldrich), desmin (#PA5-16705, Invitrogen), P-SAPK/JNK (T183/Y185; #4668P), SAPK/JNK (#9252), P-p38 MAPK (T180/Y182; #9211S), p38 MAPK (#9212), P-p44/42 MAPK (ERK1/2; #9101S), p44/42 MAPK (ERK1/2; #4695S), β-catenin (#8480), Cyclin D1 (#2978), P-AKT (T308; #4056), AKT2 (#3063S), P-S6 ribosomal protein (S235/236; #4857), S6 ribosomal Protein (#2217), cleaved caspase‐3 (#9664), and caspase-3 (#9662) from Cell Signaling Technology, p62 (#MAB8028-SP) and PD-L1 (#AF 1019) from R&D Systems. In the representative Western blots, each feeding condition per experimental group is represented by 2 independent mice. Graphical representation of the quantified Western blots for 4–6 animals/group is shown alongside the blots. In the graphs, each dot represents a mouse.
Detection of MLKL oligomers
MLKL oligomers were detected as described previously (14). Protein samples were prepared by heating with Laemmli buffer at 37°C for 15 minutes and loaded into 8% polyacrylamide gel without SDS. MLKL oligomers were detected as bands above 250 kDa using MLKL antibody.
qRT-PCR
Briefly, total RNA was isolated using RNeasy kit (# 74106, Qiagen), first-strand cDNA was synthesized using High capacity cDNA reverse transcription kit (# 4368813, Thermo Fisher Scientific), and RT-PCR using Power SYBR Green PCR Master Mix (#4368708, Thermo Fisher Scientific) in a Quantstudio 12K Flex real-time PCR system (Applied Biosystems). β-microglobulin and hypoxanthine phosphoribosyl transferase 1 (HPRT) were used as controls, as described previously (10). The list of primers used is given in Supplementary Table S3.
Characterization of immune cells by flow cytometry
Immune cell populations were analyzed by flow cytometry as described previously (15) with modifications. Briefly, mice were euthanized and liver was perfused with perfusion buffer followed by incubation in digestion buffer containing 0.075 mg/mL Liberase (#05401127001, Sigma-Aldrich). The reaction was stopped using blocking buffer, and the suspension was filtered through 70 μm cell strainer and centrifuged. The supernatant containing the non-parenchymal cells (NPC) was collected, centrifuged at 600 × g for 10 minutes, and pelleted NPC was subjected to differential centrifugation using OptiPrep density gradient media (#AXS-1114542, Cosmo Bio USA) at 1,500 × g for 30 minutes. The interphase containing NPC was collected and pelleted at 800 × g for 5 minutes. The cells were incubated with Live/Dead fixable violet dead cell stain (#L34955, Thermo Fisher Scientific) for the live cell gating and with the following antibodies from BioLegend: CD16/32 (#101302), CD45-APC/Cy7 (#103116), CD11b-PE/Cy7 (#101216), Ly6C-APC (#128016), F4/80-PE (#157304), CCR2-BV605 (#150615). Data were collected using Stratedigm 4-Laser flow cytometer, and analyzed using Flow Jo software (BD Biosciences).
Histologic analysis of liver sections
Paraffin-embedded liver sections were stained with hematoxylin & eosin (H&E) as per standardized protocol at the Stephenson Cancer Center Tissue Pathology core. Images were acquired using a Nikon Ti Eclipse microscope (Nikon) at 200× magnification for three random nonoverlapping fields per sample. Kupffer cell (KC) clusters were identified as groups of 10–12 cells clustered together and counted manually in a blinded fashion. The number of KC clusters/10 mm2 was quantified and represented graphically.
IHC
IHC was done as described previously (16). Paraffin-embedded liver sections were incubated with primary antibodies against Glypican 3 (#MA5-17083, Thermo Fisher Scientific), Ki-67 (#ab15580, Abcam), Cleaved Caspase-3 (Cell Signaling Technology), phospho-MLKL (#ab196436, Abcam), phospho-RIPK3 (#ab205421, Abcam) overnight at 4°C. Diaminobenzidine-based colorimetric method was used for the detection of target proteins in the sections. Nuclei were counter stained with Mayer's Hematoxylin (#MHS16, Sigma-Aldrich). Images were taken using a Nikon Ti Eclipse microscope (Nikon) for three random fields per sample. Staining intensity was quantified using Image J software.
TUNEL staining
TUNEL staining was performed using paraffin-embedded liver sections (5 μm) by using the DeadEnd Colorimetric TUNEL System (#G7130, Promega) following the manufacturer's instructions.
Picrosirius red staining
Picrosirius red staining was performed with paraffin-embedded liver sections (5 μm) by following a standardized protocol at the Imaging Core facility at the Oklahoma Medical Research Foundation. Images were acquired using Nikon Ti Eclipse microscope (Nikon) for three random nonoverlapping fields per sample at 200× magnification and quantified using Image J software (U.S. NIH).
ELISA
The following ELISA kits were used to determine plasma levels of the following as per manufacturer's instructions. TNFα mouse high sensitivity ELISA kit (#BMS607-3) and IL6 mouse high sensitivity ELISA kit (#BMS603HS; Thermo Fisher Scientific), mouse CCL2/JE/MCP-1 quantikine ELISA kit (#MJE00B), mouse alpha-fetoprotein (AFP) quantikine ELISA kit (#MAFP00; R&D Systems), mouse HMGB1 ELISA Kit (#E-EL-M0676, Elabscience).
Alanine transaminase colorimetric activity assay
The levels of alanine transaminase (ALT) in plasma were measured using ALT colorimetric activity assay kit from Cayman Chemical Company (#700260) as per manufacturer's instructions.
Hydroxyproline assay
The collagen content in the liver was measured by hydroxyproline (OHP) content as described previously (17). The absorbance values at 558 nm were converted into μg units using the 4-parameter standard curve generated using the standards and expressed as μg hydroxyproline/g of tissue.
RNA sequencing and data processing
Total RNA was isolated from liver tissue using RNeasy kit (Qiagen) and library preparation was done using NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (New England Biolabs; ref. 18). Paired-end 150 bp read sequencing was performed, in four to six biological replicates per diet by mouse type condition, on an Illumina NextSeq 500 sequencing platform. Raw reads, in a FASTQ format, were trimmed of residual adaptor sequences using the Scythe software. Low-quality bases at the beginning and end of reads were removed using Sickle, then the quality of remaining sequences was confirmed with FastQC. Trimmed quality reads were aligned to the Mus musculus genome reference (GRCm39/mm39) using STAR v2.4.0h (19). Gene-level read counts were determined using HTSeq v0.5.3p9 (20) with the GENCODE Release M29 (GRCm39) annotation. Read-count normalization and differentially expression analyses were performed using the edgeR package from Bioconductor, following the widely used limma/voom workflow (21). Differentially expressed genes were organized in expression profile sets following similar phenotype variation patterns observed in tumor incidence. Functional analysis, identifying sets of genes sharing the same functionality (Gene Ontology, Kyoto Encyclopedia of Genes and Genomes pathways), overrepresented among the differentially expressed genes, was performed with specialized packages from Bioconductor. Ingenuity Pathway Analysis (IPA, QIAGEN; https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/?cmpid=QDI_GA_Comp&gclid=Cj0KCQjw_5unBhCMARIsACZyzS2JMPkfj3taUIlBU-laIzPAshbtyOLSmBI6nkqSYovvLHDGKwfR8bsaApXzEALw_wcB) was used further for discovery and interactive exploration of significantly impacted static and causal gene networks, pathways, disease, upstream regulators, and regulatory effects.
Statistical analysis
All data are represented as mean ± SEM. Two‐way ANOVA with Tukey post hoc test was used to analyze data using GraphPad Prism. P < 0.05 is considered statistically significant. The symbols used for statistical comparison between groups are described in the figure legend.
Data availability
The data generated in this study are available within the article and its Supplementary Data. Analytic methods, and study materials will be made available to other researchers on request. Complete RNA-sequencing (RNA-seq) data are available as GSE200923 on Gene Expression Omnibus.
Results
CD-HFD increased necroptosis markers in the liver
The level of MLKL oligomers, a marker of necroptosis, were significantly increased in CD-HFD fed male (6-fold) and female WT mice (4-fold) relative to mice fed either NC or CD-LFD, and deleting Ripk3 or Mlkl significantly reduced MLKL oligomerization (Fig. 1A). Consistent with this, levels of phospho-MLKL (22, 23) and phospho-RIPK3 were increased in the livers of mice fed CD-HFD and absence of Ripk3 or Mlkl reduced the expression (Fig. 1B; Supplementary Fig. S1B). The increase in MLKL protein expression in WT and Ripk3−/− mice could be attributed to increased Mlkl transcript levels (Fig. 1A; Supplementary Fig. S1C). CD-HFD feeding increased RIPK3 protein levels in WT male mice; however, the absence of Mlkl blunted this effect in male mice (Fig. 1A). Transcript levels of Ripk3 remained unaltered in the absence of Mlkl in male and female mice (Supplementary Fig. S1C). In addition, blocking necroptosis significantly reduced the elevated levels of circulating HMGB1 in male mice fed CD-HFD, though not statistically significant in females (Fig. 1C). Thus, increased MLKL phosphorylation and oligomerization in response to CD-HFD in WT male mice parallels elevated HMGB1 levels.
Figure 1.

CD-HFD increased necroptosis in liver and deleting either Ripk3 or Mlkl reduced necroptosis. A, Left: Immunoblots of liver tissue extracts for MLKL oligomers, MLKL, RIPK3, and β-tubulin from WT, Ripk3−/−, and Mlkl−/− male (top) and female (bottom) mice fed NC (C), CD-LFD (L) or CD-HFD (H). Right: Graphical representation of quantified blots normalized to β-tubulin. Males: NC (white), CD-LFD (light blue) or CD-HFD (dark blue). Females: NC (white), CD-LFD (light orange) or CD-HFD (dark orange). B, IHC staining for P-MLKL in liver sections from WT, Ripk3−/−, and Mlkl−/− male mice fed NC or CD-LFD or CD-HFD. The intensity of staining was quantified and represented. Scale bar: 25 μm. C, HMGB1 in plasma of WT, Ripk3−/−, and Mlkl−/− male (left) and female (right) mice. D, Top: IHC staining for cleaved caspase-3 in liver sections from male mice. Bottom: Graphical representation of number of cleaved caspase-3 positive cells per high-power field (HPF). Scale bar: 50 μm. E, Top: TUNEL staining in liver sections from male mice. Bottom: Graphical representation of number of TUNEL positive cells per HPF. Scale bar: 50 μm. Arrowheads indicate positive cells. For Figs. 1–7, data are represented as mean ± SEM, n = 4–6 per group, Two-way ANOVA P < 0.05. a-NC versus CD-LFD, b-NC versus CD-HFD, c-CD-LFD versus CD-HFD. Between groups: j-WT NC versus Ripk3−/− NC, k-WT NC versus Mlkl−/− NC, m-Ripk3−/− NC versus Mlkl−/− NC, g-WT CD-LFD versus Ripk3−/− CD-LFD, h-WT CD-LFD versus Mlkl−/− CD-LFD, i-Ripk3−/− CD-LFD versus Mlkl−/− CD-LFD, d-WT CD-HFD versus Ripk3−/− CD-HFD, e-WT CD-HFD versus Mlkl−/− CD-HFD, f-Ripk3−/− CD-HFD versus Mlkl−/− CD-HFD.
Expression of cleaved caspase-3, a marker of apoptosis, was increased in WT male mice in response to CD-LFD and CD-HFD (Fig. 1D; Supplementary Fig. S1D). Absence of Ripk3 further increased cleaved caspase-3 levels whereas absence of Mlkl reduced its expression as shown by IHC as well as by Western blotting (Fig. 1D; Supplementary Fig. S1D). Though, CD-HFD increased the expression of cleaved caspase-3 in WT female mice, it was approximately 2-fold lower than in males and showed no change in Ripk3−/− or Mlkl−/− females (Supplementary Fig. S1D). To further assess cell death, TUNEL assay was performed. WT mice fed a CD-LFD or CD-HFD resulted in 3- or 6-fold increase in TUNEL positive cells, relative to WT mice fed normal chow diet. In contrast, Ripk3−/− and Mlkl−/− mice fed CD-HFD had significantly reduced TUNEL positive cells, indicating reduced cell death in the absence of Ripk3 or Mlkl (Fig. 1E). Unlike the sex-specific differences in body weight gain, no such difference was observed in liver weight in response to the diets (Supplementary Fig. S2A and S2B).
Deleting Ripk3 or Mlkl reduced CD-HFD–induced inflammation in male mice
Transcript levels of IL6, TNFα, and IL1β, as well as circulating IL6, were significantly increased in WT male mice fed CD-HFD and were significantly reduced in Ripk3−/− and Mlkl−/− mice, whereas no such effect was observed in female mice (Fig. 2A–C; Supplementary Fig. S2C). Circulating TNFα was elevated in both male and female WT mice fed CD-HFD, where the levels were reduced in knockout male mice, but not in female mice (Fig. 2D). KC clusters, a measure of the proinflammatory status of the liver (24), was significantly elevated in response to CD-LFD and CD-HFD in WT male mice, whereas blocking necroptosis reduced KC clusters in the knockout CD-HFD group (Fig. 2E). In WT females, though KC clusters were significantly elevated while fed with CD-HFD, this was approximately 1.5-fold lower than in male mice, and a significant reduction was seen only in Ripk3−/− group (Supplementary Fig. S2D).
Figure 2.

Deleting Ripk3 or Mlkl reduced CD-HFD induced inflammation in male mice. Transcript levels of hepatic IL6 (A), circulating IL6 (B), transcript levels of hepatic TNFα (C), and circulating TNFα (D) in male (left) and female (right) mice. E, Left: Images of H&E stained sections. Black dotted circles represent KC clusters. Scale bar: 50 μm. Right: Graphical representation of the number of KC clusters in each group. Males: NC (white), CD-LFD (light blue) or CD-HFD (dark blue). Females: NC (white), CD-LFD (light orange) or CD-HFD (dark orange).
The elevated levels of total immune cell population (CD45+ve cells) and macrophages (F4/80+ve cells), the major immune cell type mediating hepatic inflammation in response to DAMPs, increased in WT male mice fed CD-HFD and was significantly reduced in knockout male mice fed the same diet (Fig. 3A and B; Supplementary Fig. S3A). The gating strategy that was followed for the analysis is shown in Supplementary Fig S3B. In contrast to male mice, while WT females fed CD-HFD showed a significant reduction in the CD45+ve population, no significant change in F4/80+ve population was observed (Fig. 3A and B; Supplementary Fig. S3A). Levels of proinflammatory macrophages derived from infiltrating monocytes (CCR2+ve cells; ref. 25) showed a significant increase in male WT CD-HFD group and was significantly reduced in Ripk3−/− and Mlkl−/− mice (Fig. 3C and D). Though CD-HFD fed WT female mice also had increased CCR2+ve cells, their levels were approximately 2-fold lower than in males, and blocking necroptosis had no effect on these cells in females (Fig. 3C and D).
Figure 3.

Deleting Ripk3 or Mlkl reduced CD-HFD–induced CCL2 and CCR2+ve macrophages. Left: Flow cytometric analysis of percentage of F4/80+ve cell population (A) and CCR2+ve cell population (C) in the livers of male (left) and female (right) mice. Right: Graphical representation of the percentage population of F4/80+ve cells gated on liver CD45+ve cell subset (B) and CCR2+ve cells, gated on liver CD45+, CD11b+, F4/80low, Ly6C+, Ly6G− cell subsets (D) in male (top) and female (bottom) mice. Transcript levels of hepatic CCL2 (E) and circulating CCL2 (F). Males: NC (white), CD-LFD (light blue) or CD-HFD (dark blue). Females: NC (white), CD-LFD (light orange) or CD-HFD (dark orange).
Transcript levels of chemokine (C-C motif) ligand 2 (CCL2), a chemokine that recruits monocytes to the sites of injury (25), were significantly increased in WT mice in response to CD-HFD and these levels were significantly reduced in Ripk3−/− and Mlkl−/− mice (Fig. 3E). Similarly, circulating CCL2 was elevated in response to CD-HFD in WT male and female mice; however, CCL2 levels were significantly reduced in Ripk3−/− and Mlkl−/− males, but not in females (Fig. 3F). Also, the markers of proinflammatory M1 macrophages, CD68 and TLR4, elevated in WT male mice fed CD-HFD were significantly reduced in knockout groups (Supplementary Fig. S3C).
Blocking necroptosis did not alter CD-HFD–induced liver fibrosis
CD-HFD feeding resulted in severe fibrosis as seen by the increased intensity in PicroSirius red (PSR) staining of liver tissue which was not reduced when necroptosis was blocked. Moreover, the staining intensity was higher in males (∼3-fold) than in females (Fig. 4A). The increased transcript levels of Col3α1, Acta2, and Col1α1 in WT males fed with CD-HFD (Fig. 4B and C; Supplementary Fig. S4A) was reduced when Ripk3 was absent. Whereas, only Col1α1 showed a significant reduction in Mlkl−/− mice fed CD-HFD, in males and females (Fig. 4B and C; Supplementary Fig. S4A). Assessment of hepatic stellate cell (HSC) markers showed that desmin and α-SMA increased in WT CD-HFD were reduced only in male Ripk3−/− mice (Fig. 4D; Supplementary Fig. S4B). TGFβ transcript levels were significantly upregulated in WT mice fed CD-HFD and blocking necroptosis had no effect on TGFβ levels in male or female mice (Fig. 4E; Supplementary Fig. S4C). Similarly, OHP which was significantly elevated in the livers of WT male mice fed CD-LFD and CD-HFD were not reduced in knockout males (Fig. 4F). Circulating levels of ALT, a biomarker of hepatocellular damage, were also significantly elevated in WT male mice in response to CD-LFD and CD-HFD and the levels did not change significantly in the knockout groups (Fig. 4G).
Figure 4.

Deleting either Ripk3 or Mlkl did not alter liver fibrosis. A, Left: PSR staining of liver sections of male (left) and female (right) mice. Scale bar: 50 μm. Right: Graphical representation of PSR quantification. Transcript levels of Col3α1 (B) and Acta2 (C) in male (left) and female mice (right). D, Left: Immunoblots of liver tissue extracts for desmin, α-SMA, and β-tubulin from male mice fed NC (C) or CD-LFD (L) or CD-HFD (H). Right: Graphical representation of quantified blots normalized to β-tubulin. Transcript levels of hepatic TGFβ normalized to β-microglobulin (E), liver hydroxyproline content (F), and serum ALT levels (G). Males: NC (white), CD-LFD (light blue) or CD-HFD (dark blue). Females: NC (white), CD-LFD (light orange) or CD-HFD (dark orange).
Deleting either Ripk3 or Mlkl significantly reduced HCC development in response to CD-HFD
WT male mice fed CD-HFD developed liver nodules (2–25 nodules/mouse) while no or few liver nodules were detected in mice fed NC or CD-LFD (Fig. 5A; Supplementary Fig. S5A). While a similar effect was observed in female mice, the number of liver nodules was lower (∼2- to 3-fold) relative to males (Fig. 5A). Importantly, the number and size of liver nodules were significantly reduced in both Ripk3−/− and Mlkl−/− male and female mice fed CD-HFD (Fig. 5A and B; Table 1). The tumor (T) region of liver sections from CD-HFD fed WT mice showed strong positive staining for glypican-3, a marker for HCC, (Fig. 5C) as well as the cell proliferation marker, Ki-67, compared with non-tumor (NT) region and this was reduced in both Ripk3−/− and Mlkl−/− mice (Fig. 5D). The elevated levels of serum AFP, a biomarker of HCC, in WT mice fed CD-HFD were significantly reduced in the knockout groups (Fig. 5E). Transcript levels of liver AFP also showed a similar trend for males, but not for females (Supplementary Fig. S5B).
Figure 5.
![Figure 5. Deleting either Ripk3 or Mlkl significantly reduced HCC in response to a CD-HFD. A, Tumor nodules in the livers of WT, Ripk3−/−, and Mlkl−/− male mice (left) and female mice (right). B, Graphical representation of large (>5 mm, yellow), medium (2–4 mm, green), and small (<1 mm, white) liver nodules in male (left) and female (right) mice [aWT small tumor (ST) vs. WT medium tumor (MT), P < 0.0001; bWT ST vs. WT large tumor (LT), P < 0.0001; cMlkl−/− ST vs. Mlkl−/− MT, P < 0.01; dMlkl−/− ST vs. Mlkl−/− LT, P < 0.01; eWT ST vs. Ripk3−/− ST, P < 0.0001; f WT ST vs. Mlkl−/− ST, P < 0.03]. IHC staining for glypican-3 (C) and Ki-67 (D) in the non-tumor (NT) and tumor (T) of liver sections from male mice. Dark red color in tumor (T) region indicate positive staining for glypican-3 and dark brown spots in tumor region indicate positive staining for Ki-67. Scale bar: 50 μm. E, Circulating AFP levels male (left) and female (right) mice. Males: NC (white), CD-LFD (light blue) or CD-HFD (dark blue). Females: NC (white), CD-LFD (light orange) or CD-HFD (dark orange).](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=10472095_933fig5.jpg)
Deleting either Ripk3 or Mlkl significantly reduced HCC in response to a CD-HFD. A, Tumor nodules in the livers of WT, Ripk3−/−, and Mlkl−/− male mice (left) and female mice (right). B, Graphical representation of large (>5 mm, yellow), medium (2–4 mm, green), and small (<1 mm, white) liver nodules in male (left) and female (right) mice [aWT small tumor (ST) vs. WT medium tumor (MT), P < 0.0001; bWT ST vs. WT large tumor (LT), P < 0.0001; cMlkl−/− ST vs. Mlkl−/− MT, P < 0.01; dMlkl−/− ST vs. Mlkl−/− LT, P < 0.01; eWT ST vs. Ripk3−/− ST, P < 0.0001; f WT ST vs. Mlkl−/− ST, P < 0.03]. IHC staining for glypican-3 (C) and Ki-67 (D) in the non-tumor (NT) and tumor (T) of liver sections from male mice. Dark red color in tumor (T) region indicate positive staining for glypican-3 and dark brown spots in tumor region indicate positive staining for Ki-67. Scale bar: 50 μm. E, Circulating AFP levels male (left) and female (right) mice. Males: NC (white), CD-LFD (light blue) or CD-HFD (dark blue). Females: NC (white), CD-LFD (light orange) or CD-HFD (dark orange).
Table 1.
Tumor incidence and size-based indication of tumors in the experimental groups.
| Males | Females | |||||
|---|---|---|---|---|---|---|
| WT | Ripk3 −/− | Mlkl −/− | WT | Ripk3 −/− | Mlkl −/− | |
| Tumor incidence | 100% (10/10) | 75% (6/8) | 100% (7/7) | 87.5% (7/8) | 80% (4/5) | 75% (3/4) |
| Small sized tumors: <0.1 mm | 10.36 ± 1.86, | 2.86 ± 1.01, | 6.14 ± 1.32, | 4.42 ± 0.66, | 2.2 ± 0.66, | 1.25 ± 0.48, |
| (number and percentage) | 68% | 80% | 87% | 90% | 92% | 64% |
| Medium sized tumors: 2–4 mm | 3 ± 1.27, | 0.625 ± 0.42, | 0.33 ± 0.21, | 0.42 ± 0.23, | 0.2 ± 0.2, | 0.75 ± 0.7, |
| (number and percentage) | 20% | 20% | 5% | 9% | 8% | 36% |
| Large sized tumors: >5 mm | 1.88 ± 1.28, | 0 | 0.57 ± 0.297, | 0.1 ± 0.1, | 0,0 | 0,0 |
| (number and percentage) | 12% | 0 | 8% | 2% | ||
Gene expression modulation in response to CD-LFD or CD-HFD was attenuated when necroptosis is blocked
To determine the effect of blocking necroptosis on the liver transcriptome of male mice, RNA-seq was performed. The transcriptomic changes in WT mice induced by CD-LFD and CD-HFD feeding was largely prevented in the Ripk3−/− and Mlkl−/− groups (Fig. 6A). A list of the transcripts that significantly changed is given in Supplementary Tables S1 and S2. To validate the RNA-seq data, we performed qRT-PCR analysis of selected genes associated with tumorigenesis that were altered in WT mice fed CD-HFD: Tonsl (26), Dppa2 (27), and Pogz (28). The mRNA levels of these genes were significantly increased in WT mice fed CD-HFD and blocking necroptosis reduced their expression (Fig. 6B).
Figure 6.

Genes upregulated or downregulated in response to CD-LFD or CD-HFD in WT, Ripk3−/− or Mlkl−/− mice. A, Schematic representation of transcriptome analysis of liver tissue extracts. Red color represents genes that are significantly upregulated and blue color represents genes that are significantly downregulated (1: NC, 2: CD-LFD, 3: CD-HFD). B, Transcript levels of Tonsl, Dppa2, and Pogz normalized to β-microglobulin in male mice. C, Graphical representation of canonical pathways which are significantly upregulated (red) and downregulated (blue). D, Transcript levels of p16, p21, and p53 in male (top) and female (bottom). E, Upstream regulators that are significantly upregulated (red) and downregulated (blue) in male mice. Males: NC (white), CD-LFD (light blue) or CD-HFD (dark blue). Females: NC (white), CD-LFD (light orange) or CD-HFD (dark orange).
Further analysis by IPA identified 24 pathways that were significantly altered (Supplementary Fig. S6A) and of these, pathways that showed positive (necroptosis signaling pathway and senescence pathway), and negative (sirtuin signaling pathway and CLEAR signaling pathway) z-score are showed in Fig. 6C. WT male mice fed CD-HFD showed increase in markers of senescence, p16, p21 and p53, compared with control. p21 was significantly reduced in Ripk3−/− and Mlkl−/− male mice fed CD-HFD, whereas p16 and p53 was significantly reduced only in Ripk3−/− group (Fig. 6D). In WT female mice, p16 and p21, but not p53, were significantly elevated in response to CD-HFD and blocking necroptosis reduced their expression in Mlkl−/− mice, but not in Ripk3−/− mice (Fig. 6D). IPA further identified 15 upstream regulators of the pathways (Supplementary Fig. S6B), of which one showed a positive z-score (STAT5) and two showed negative z-score (LEP and PPARα; Fig. 6E).
Oncogenic pathways induced by CD-HFD is attenuated when necroptosis is blocked
To identify pathways mediating HCC development in response to CD-HFD, the activation of HCC-associated pathways was assessed. Male mice were used for the initial studies and the pathways that were found to be activated in males were then validated in females. We assessed MAPK pathways that are modulated by inflammation and are involved in HCC, for example, JNK, ERK and p38 pathways, by assessing phosphorylation levels of these proteins which represent the active form. Phosphorylation of JNK was increased in the livers of CD-HFD fed WT mice, and was significantly reduced in knockout male mice, but not in females (Fig. 7A). The AKT/mTOR and p38 pathways were not activated in WT male mice fed CD-HFD, whereas ERK phosphorylation was reduced and remained unaltered in knockout mice (Supplementary Fig. S7A and S7B).
Figure 7.

Changes in pathways involved in HCC. A, Left: Immunoblots of liver tissue extracts for P-JNK and JNK male (top) and female (bottom) mice fed NC (C), CD-LFD (L) or CD-HFD (H). Right: Graphical representation of quantified phosphoprotein normalized to total protein. B, Immunoblots of liver tissue extracts for β-catenin, cyclin-D1 and β-tubulin (left) from male mice. Right: Graphical representation of quantified blots normalized to β-tubulin. C, Transcript levels of c-Myc (left) and EpCAM (right). D, Left: Immunoblots of liver tissue extracts for PD-L1 and β-tubulin. Right: Graphical representation of quantified blots normalized to β-tubulin. Transcript levels of (E) hepatic PD-L1 (top) and PD-1 (bottom), and (F) PPARγ normalized to β-microglobulin. Males: NC (white), CD-LFD (light blue) or CD-HFD (dark blue). Females: NC (white), CD-LFD (light orange) or CD-HFD (dark orange).
The elevated levels of β-catenin and its downstream target, cyclin D1, in CD-HFD fed WT male mice were significantly reduced in knockout groups (Fig. 7B). Similarly, blocking necroptosis reduced β-catenin expression in females in response to CD-HFD (Supplementary Fig. S7C). Also, transcript levels of c-Myc and EpCAM, the downstream targets of the JNK and β-catenin pathways, elevated in WT male mice fed CD-HFD was significantly reduced in knockout groups (Fig. 7C). Interestingly, the increased expression of PD-L1, the immune checkpoint protein, which was significantly upregulated in the livers of WT male mice fed CD-HFD was reduced in both knockout groups fed CD-HFD with a greater reduction in Mlkl−/− group (Fig. 7D). Similarly, transcript levels of PD-L1 and PD-1 in the liver were increased in male and female WT mice in response to CD-HFD, and were significantly reduced in all knockout groups fed CD-HFD (Fig. 7E). Furthermore, similar to previous reports (29), we found PPARγ to be significantly upregulated in Ripk3−/− and Mlkl−/− mice fed CD-HFD compared with WT group, thereby underlining the role of necroptotic proteins in PPARγ modulation (Fig. 7F). Also, p62, a marker of autophagy that is elevated in HCC, was significantly upregulated in WT male mice fed CD-HFD with no change seen in the knockout groups (Supplementary Fig. S7D).
Discussion
This study addressed the role of two key proteins in the necroptosis pathway, RIPK3 and MLKL, on hepatic inflammation and HCC development in a NASH-induced HCC mouse model; and the sex-specific effects of deleting Ripk3 or Mlkl on inflammation, liver fibrosis, and HCC. The major findings of the study are that deleting either Ripk3 or Mlkl ameliorated hepatic inflammation and HCC in male mice and HCC only in female mice. Liver fibrosis was however not improved in either male or female mice. By showing that both Ripk3 and Mlkl knockout mice had similar effects on reducing inflammation and/or HCC in response to CD-HFD, we propose that necroptosis plays a role in HCC induction in the CD-HFD mouse model.
Our study agrees with previous reports showing increased necroptosis in the livers of male mice fed NAFLD or NASH inducing diets (8, 29, 30). While the absence of Mlkl abolished MLKL oligomer levels, deletion of Ripk3 reduced MLKL oligomer levels, suggesting a role of TAM (Tyro3, Axl, and Mer) kinases in MLKL phosphorylation and oligomerization (31). Liver macrophages, in particular the infiltrating monocyte-derived macrophages, are the major mediators of the inflammatory response in NASH and HCC (32). Our finding that blocking necroptosis reduced total liver macrophages and infiltrating monocytes suggests a role for liver macrophages in necroptosis-mediated inflammation in male WT mice, not female mice. The female sex hormone, estrogen, has been shown to inhibit production of proinflammatory cytokines by KC (33), and male mice is reported to accumulate more macrophages in the adipose tissue than females leading to elevated levels of inflammatory cytokines (34). Although female mice fed CD-HFD had increased levels of necroptosis markers, neither HMGB1 nor proinflammatory cytokines were elevated, suggesting that plasma membrane rupture, the terminal step in necroptosis is inhibited in females. Future studies will determine whether MLKL oligomers are ubiquitinated to block the cytotoxic potential in female mice (35).
Oncogenic pathways regulated by inflammatory cytokines are increased in the livers of WT male mice fed CD-HFD, that is, JNK, PD-L1/PD-1, and β-catenin pathways (36–38). Deleting either Ripk3 or Mlkl reduced JNK, PD-1/PD-L1 signaling, and β-catenin pathways in male mice, supporting a role of necroptosis-mediated inflammation in HCC in male mice. These pathways were also elevated in WT female mice fed CD-HFD, but blocking necroptosis reduced only PD-1/PD-L1 signaling and β-catenin pathways, suggesting other factors in addition to necroptosis-mediated inflammation can modulate these pathways. Our study also confirms the role of RIPK3 in NASH progression to HCC, where the expression of RIPK3 in human NASH subjects negatively correlates with PPARγ expression (29), thereby suggesting the role of necroptotic proteins in controlling the hepatic lipid metabolism, and hence NASH incidence.
Absence of Ripk3 or Mlkl reduced the number and size of liver tumor nodules in male and female mice. Even though WT female mice developed HCC in response to CD-HFD, the number of tumor nodules were approximately 2.5-fold lower than in WT male mice, consistent with the fact that HCC incidence in humans is >2-fold higher in males than females (39). Similarly, diethylnitrosamine-induced hepatocarcinogenesis model had shown higher incidence of HCC in males relative to females (33). Surprisingly, though blocking necroptosis reduced liver tumor nodules in females, the reduction occurred in the absence of any significant decrease in inflammation. This finding suggests the existence of alternate pathways regulated by both Ripk3 and Mlkl in HCC development in females.
Chronic liver inflammation is a well-known mediator of liver fibrosis (40). Even though the increase in hepatic inflammation was paralleled by increased liver fibrosis in male mice fed CD-HFD, deleting neither Ripk3 nor Mlkl had any effect on liver fibrosis suggesting that necroptosis-mediated inflammation does not play a role in liver fibrosis. Contrary to our findings, deleting Ripk3 reduced hepatic inflammation and liver fibrosis in male mice fed methionine- and choline-deficient diet or choline-deficient diet (8, 29). Also, feeding Ripk3−/− mice with HFD exacerbated hepatic inflammation and liver fibrosis (22, 41). Similarly, Ripk3 deletion is protective, whereas Ripk1 inhibition aggravated disease and hastened animal death in ConA-induced autoimmune hepatitis (42). It is possible that discrepancies in dietary fat sources as well as the composition in diets or compounds used to induce chronic liver disease is the cause of different outcomes. Our data show that deleting neither Ripk3 nor Mlkl had any effect on the elevated levels of TGFβ, the fibrogenic factor involved in the activation of HSCs. Similar to humans, liver fibrosis was observed to be higher in male mice relative to females (43). A role for the female sex hormone, estrogen, has been proposed for the gender differences in liver fibrosis and HCC based on the finding that the degree of liver fibrosis and HCC are higher in postmenopausal women than in premenopausal women (44).
Our study identified a novel cross-talk between necroptosis and other pathways that regulate tumorigenesis, for example, apoptosis and cell senescence. Deletion of Ripk3 elevated apoptosis markers as reported previously (29), whereas Mlkl deficiency reduced apoptosis, suggesting that reduction of HCC is independent of apoptosis. Recently, we reported that inhibiting necroptosis using Nec-1s reduced markers of cell senescence in the livers of Sod1−/− mice, a mouse model of spontaneous HCC, as well as in the livers of old WT mice (11, 45), further supporting a role of necroptosis in cell senescence. As cell senescence also regulates inflammation, it is possible that the observed outcome of HCC inhibition in both Ripk3−/− and Mlkl−/− mice could also be due to inhibition of both necroptosis and cellular senescence (46).
The novel aspects of our study are: (i) To our knowledge, this is the first study showing a protective role of MLKL deficiency on HCC in response to CD-HFD. Even though a previous study has reported that absence of Ripk3 could reduce HCC (29), the study used a choline-deficient amino acid–defined diet that lacks high fat. (ii) Our study shows that reduced incidence of HCC due to Ripk3 or Mlkl deficiency is independent of fibrosis. (iii) RIPK3 and MLKL regulate cellular processes other than necroptosis: RIPK3 modulates apoptosis, NLRP3 activation, and lipid metabolism (29, 47), whereas MLKL regulates autophagy and endosomal trafficking (48, 49). Therefore, similar outcomes using both Ripk3−/− and Mlkl−/− mouse models in our study suggest involvement of necroptosis. Ours is the only study that tested effect of Ripk3 or Mlkl deletion in males and females simultaneously to show a sex-specific difference.
In summary, our data provide strong support for necroptosis playing a role in HCC in a mouse model of NASH-induced HCC. In addition, our study offers insight into sex-specific differences in the progression of NASH to HCC, demonstrating the need for using both males and females for studies, especially for developing therapeutic strategies for treating HCC. As HCC incidence is higher in postmenopausal women relative to premenopausal women (50), the study also highlights the importance of using estrogen-deficient female mice for studying HCC.
Supplementary Material
List of Differentially Expressed Genes from RNA Seq analysis
List of significantly altered transcripts from RNA Seq analysis
List of primers used for quantitative real time PCR
Figure S1 shows the experimental plan of the study, IHC staining for Ripk3, Transcript levels of Mlkl and Ripk3, Western blots for Cleaved and Total Caspase 3
Figure S2 shows body weight changes, liver weight changes, Transcript levels of Il1b and H&E staining for showing KC population
Fig S3 shows flow cytometry data for CD45, gating strategy and transcript levels of CD68 and TLR4
Fig S4 shows transcript levels of Col1a1, western blots for Desmin, a-SMA and b-tubulin, Transcript levels of TGFb
Figure S5 shows images of liver nodules, transcript levels of AFP
Fig S6 shows ingenuity pathway analysis for canonical pathways and upstream regulators
Figure S7 shows western blots for p-AKT/AKT, p-S6/S6, b tubulin, p-ERK/ERK, P-p-38/p-38, b catenin, p62 and b tubulin
Supplementary Figures 1 -7 and Supplementary Tables 1 - 3 legends
Acknowledgments
The efforts of authors were supported by NIH grants R01AG059718 (S.S. Deepa), R03 CA262044 (S.S. Deepa), P30AG050911(W.M. Freeman, J.D. Wren, and B.F. Miller), R01 AG064951 (B.F. Miller), R56 AG067754 (B.F. Miller), R21 AR077387 (B.F. Miller), P20 GM139763 (B.F. Miller), GeroOncology Pilot Grant (R. Janknecht and S.S. Deepa) from the University of Oklahoma Health Sciences Center, Oklahoma Center for Adult Stem Cell Research Grant (OCASCR; S.S. Deepa) and Oklahoma Center for the Advancement of Science and Technology (OCAST) Postdoctoral grant HF21-009 (S. Mohammed).
The authors would like to thank Stephenson Cancer Center Tissue Pathology Core for performing H&E staining, the Imaging Core facility at the Oklahoma Medical Research Foundation for performing Picrosirius red, cleaved caspase-3 and TUNEL staining, and the Laboratory for Molecular Biology and Cytometry Research at the University of Oklahoma Health Sciences Center for providing the facilities for the flow cytometry experiments.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/).
Authors' Disclosures
No disclosures were reported.
Authors' Contributions
S. Mohammed: Data curation, formal analysis, investigation, methodology, writing–review and editing. N. Thadathil: Data curation, formal analysis, investigation, methodology, writing–review and editing. P. Ohene-Marfo: Formal analysis, investigation, methodology, writing–review and editing. A.L. Tran: Formal analysis, investigation, methodology, writing–review and editing. M. Van Der Veldt: Formal analysis, investigation. C. Georgescu: Formal analysis, investigation, writing–review and editing. S. Oh: Formal analysis, writing–review and editing. E.H. Nicklas: Formal analysis, investigation, writing–review and editing. D. Wang: Formal analysis, investigation. N.H. Haritha: Formal analysis, investigation, writing–review and editing. W. Luo: Formal analysis, writing–review and editing. R. Janknecht: Formal analysis, writing–review and editing. B.F. Miller: Formal analysis, writing–review and editing. J.D. Wren: Formal analysis, writing–review and editing. W.M. Freeman: Formal analysis, writing–review and editing. S.S. Deepa: Conceptualization, formal analysis, supervision, funding acquisition, writing–original draft, project administration, writing–review and editing.
References
- 1. Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, et al. Hepatocellular carcinoma. Nat Rev Dis Prim 2021;7:6. [DOI] [PubMed] [Google Scholar]
- 2. Huang DQ, El-Serag HB, Loomba R. Global epidemiology of NAFLD-related HCC: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 2021;18:223–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yu LX, Ling Y, Wang HY. Role of nonresolving inflammation in hepatocellular carcinoma development and progression. NPJ Precis Oncol 2018;2:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mihm S. Danger-associated molecular patterns (DAMPs): molecular triggers for sterile inflammation in the liver. Int J Mol Sci 2018;19:3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gaskell H, Ge X, Nieto N. High-mobility group box-1 and liver disease. Hepatol Commun 2018;2:1005–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Newton K, Manning G. Necroptosis and inflammation. Annu Rev Biochem 2016;85:743–63. [DOI] [PubMed] [Google Scholar]
- 7. Samson AL, Zhang Y, Geoghegan ND, Gavin XJ, Davies KA, Mlodzianoski MJ, et al. MLKL trafficking and accumulation at the plasma membrane control the kinetics and threshold for necroptosis. Nat Commun 2020;11:3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gautheron J, Vucur M, Reisinger F, Cardenas DV, Roderburg C, Koppe C, et al. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol Med 2014;6:1062–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gautheron J, Vucur M, Schneider AT, Severi I, Roderburg C, Roy S, et al. The necroptosis-inducing kinase RIPK3 dampens adipose tissue inflammation and glucose intolerance. Nat Commun 2016;7:11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mohammed S, Nicklas EH, Thadathil N, Selvarani R, Royce GH, Kinter M, et al. Role of necroptosis in chronic hepatic inflammation and fibrosis in a mouse model of increased oxidative stress. Free Radic Biol Med 2021;164:315–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mohammed S, Thadathil N, Selvarani R, Nicklas EH, Wang D, Miller BF, et al. Necroptosis contributes to chronic inflammation and fibrosis in aging liver. Aging Cell 2021;20:e13512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Newton K, Sun X, Dixit VM. Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol 2004;24:1464–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 2013;39:443–53. [DOI] [PubMed] [Google Scholar]
- 14. Miyata T, Wu X, Fan X, Huang E, Sanz-Garcia C, Cajigas-Du Ross CK, et al. Differential role of MLKL in alcohol-associated and non-alcohol-associated fatty liver diseases in mice and humans. JCI insight 2021;6:e140180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stijlemans B, Sparkes A, Abels C, Keirsse J, Brys L, Elkrim Y, et al. Murine liver myeloid cell isolation protocol. Bio-Protocol 2015;5:e1471. [Google Scholar]
- 16. Lankadasari MB, Aparna JS, Mohammed S, James S, Aoki K, Binu VS, et al. Targeting S1PR1/STAT3 loop abrogates desmoplasia and chemosensitizes pancreatic cancer to gemcitabine. Theranostics 2018;8:3824–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Smith LR, Hammers DW, Sweeney HL, Barton ER. Increased collagen cross-linking is a signature of dystrophin-deficient muscle. Muscle Nerve 2016;54:71–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ocañas SR, Pham KD, Blankenship HE, Machalinski AH, Chucair-Elliott AJ, Freeman WM. Minimizing the ex vivo confounds of cell-isolation techniques on transcriptomic and translatomic profiles of purified microglia. eNeuro 2022;9:ENEURO.0348-21.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anders S, Pyl PT, Huber W. HTSeq–a python framework to work with high-throughput sequencing data. Bioinformatics 2015;31:166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Law CW, Chen Y, Shi W, Smyth GK. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 2014;15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Roychowdhury S, McCullough RL, Sanz-Garcia C, Saikia P, Alkhouri N, Matloob A, et al. Receptor interacting protein 3 protects mice from high-fat diet-induced liver injury. Hepatology 2016;64:1518–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu X, Nagy LE. MLKL contributes to western diet-induced liver injury through inhibiting autophagy. Autophagy 2020;16:1351–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Woltman AM, Boonstra A, Naito M, Leenen PJM. Kupffer cells in health and disease. Macrophages: biology and role in the pathology of diseases. New York (NY): Springer; 2014. p. 217–47. [Google Scholar]
- 25. Zimmermann HW, Trautwein C, Tacke F. Functional role of monocytes and macrophages for the inflammatory response in acute liver injury. Front Physiol 2012;3:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yu B, Ding Y, Liao X, Wang C, Wang B, Chen X. Overexpression of TONSL might be an independent unfavorable prognostic indicator in hepatocellular carcinoma. Pathol Res Pract 2019;215:939–45. [DOI] [PubMed] [Google Scholar]
- 27. Monk M, Hitchins M, Hawes S. Differential expression of the embryo/cancer gene ECSA(DPPA2), the cancer/testis gene BORIS and the pluripotency structural gene OCT4, in human preimplantation development. Mol Hum Reprod 2008;14:347–55. [DOI] [PubMed] [Google Scholar]
- 28. Zheng S, Liu Y, Sun H, Jia J, Wu T, Ding R, et al. Identification of abnormally high expression of POGZ as a new biomarker associated with a poor prognosis in osteosarcoma. Eur J Histochem 2021;65:3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Afonso MB, Rodrigues PM, Mateus-Pinheiro M, Simão AL, Gaspar MM, Majdi A, et al. RIPK3 acts as a lipid metabolism regulator contributing to inflammation and carcinogenesis in non-alcoholic fatty liver disease. Gut 2021;70:2359–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Afonso MB, Rodrigues PM, Carvalho T, Caridade M, Borralho P, Cortez-Pinto H, et al. Necroptosis is a key pathogenic event in human and experimental murine models of non-alcoholic steatohepatitis. Clin Sci 2015;129:721–39. [DOI] [PubMed] [Google Scholar]
- 31. Najafov A, Mookhtiar AK, Luu HS, Ordureau A, Pan H, Amin PP, et al. TAM kinases promote necroptosis by regulating oligomerization of MLKL. Mol Cell 2019;75:457–68. [DOI] [PubMed] [Google Scholar]
- 32. Daemen S, Schilling JD. The interplay between tissue niche and macrophage cellular metabolism in obesity. Front Immunol 2019;10:3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Naugler WE, Sakurai T, Kim S, Maeda S, Kim KH, Elsharkawy AM, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 2007;317:121–4. [DOI] [PubMed] [Google Scholar]
- 34. Chen K-HE, Lainez NM, Coss D. Sex differences in macrophage responses to obesity-mediated changes determine migratory and inflammatory traits. J Immunol 2021;206:141–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu Z, Dagley LF, Shield-Artin K, Young SN, Bankovacki A, Wang X, et al. Oligomerization-driven MLKL ubiquitylation antagonizes necroptosis. EMBO J 2021;40:e103718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kuntzen C, Sonuc N, De Toni EN, Opelz C, Mucha SR, Gerbes AL, et al. Inhibition of c-Jun-N-terminal-kinase sensitizes tumor cells to CD95-induced apoptosis and induces G2–M cell cycle arrest. Cancer Res 2005;65:6780–8. [DOI] [PubMed] [Google Scholar]
- 37. Khalaf AM, Fuentes D, Morshid AI, Burke MR, Kaseb AO, Hassan M, et al. Role of wnt/β-catenin signaling in hepatocellular carcinoma, pathogenesis, and clinical significance. J Hepatocell Carcinoma 2018;5:61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Singh V, Khurana A, Allawadhi P, Banothu AK, Bharani KK, Weiskirchen R. Emerging role of PD-1/PD-L1 inhibitors in chronic liver diseases. Front Pharmacol 2021;12:790963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wu EM, Wong LL, Hernandez BY, Ji JF, Jia W, Kwee SA, et al. Gender differences in hepatocellular cancer: disparities in nonalcoholic fatty liver disease/steatohepatitis and liver transplantation. Hepatoma Res 2018;4:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest 2017;127:55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Saeed WK, Jun DW, Jang K, Ahn SB, Oh JH, Chae YJ, et al. Mismatched effects of receptor interacting protein kinase-3 on hepatic steatosis and inflammation in non-alcoholic fatty liver disease. World J Gastroenterol 2018;24:5477–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Deutsch M, Graffeo CS, Rokosh R, Pansari M, Ochi A, Levie EM, et al. Divergent effects of RIP1 or RIP3 blockade in murine models of acute liver injury. Cell Death Dis 2015;6:e1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ballestri S, Nascimbeni F, Baldelli E, Marrazzo A, Romagnoli D, Lonardo A. NAFLD as a sexual dimorphic disease: role of gender and reproductive status in the development and progression of nonalcoholic fatty liver disease and inherent cardiovascular risk. Adv Ther 2017;34:1291–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ascha MS, Hanouneh IA, Lopez R, Tamimi TAR, Feldstein AF, Zein NN. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology 2010;51:1972–8. [DOI] [PubMed] [Google Scholar]
- 45. Thadathil N, Selvarani R, Mohammed S, Nicklas EH, Tran AL, Kamal M, et al. Senolytic treatment reduces cell senescence and necroptosis in Sod1 knockout mice that is associated with reduced inflammation and hepatocellular carcinoma. Aging Cell 2022;21:e13676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Giannakoulis VG, Dubovan P, Papoutsi E, Kataki A, Koskinas J. Senescence in HBV-, HCV- and NAFLD- mediated hepatocellular carcinoma and senotherapeutics: current evidence and future perspective. Cancers 2021;13:4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Moriwaki K, Chan FKM. The inflammatory signal adaptor RIPK3: functions beyond necroptosis. Int Rev Cell Mol Biol 2017;328:253–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wu X, Poulsen KL, Sanz-Garcia C, Huang E, McMullen MR, Roychowdhury S, et al. MLKL-dependent signaling regulates autophagic flux in a murine model of non-alcoholic fatty liver disease. J Hepatol 2020;73:616–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yoon S, Kovalenko A, Bogdanov K, Wallach D. MLKL, the protein that mediates necroptosis, also regulates endosomal trafficking and extracellular vesicle generation. Immunity 2017;47:51–65. [DOI] [PubMed] [Google Scholar]
- 50. Zhang W, Liu F, Huang J, Guo X, Dong W, Wei S, et al. Effect of menopausal status on the survival and recurrence of sex-classified hepatocellular carcinoma after liver resection: a case-matched study with propensity score matching. Aging 2020;12:25895–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of Differentially Expressed Genes from RNA Seq analysis
List of significantly altered transcripts from RNA Seq analysis
List of primers used for quantitative real time PCR
Figure S1 shows the experimental plan of the study, IHC staining for Ripk3, Transcript levels of Mlkl and Ripk3, Western blots for Cleaved and Total Caspase 3
Figure S2 shows body weight changes, liver weight changes, Transcript levels of Il1b and H&E staining for showing KC population
Fig S3 shows flow cytometry data for CD45, gating strategy and transcript levels of CD68 and TLR4
Fig S4 shows transcript levels of Col1a1, western blots for Desmin, a-SMA and b-tubulin, Transcript levels of TGFb
Figure S5 shows images of liver nodules, transcript levels of AFP
Fig S6 shows ingenuity pathway analysis for canonical pathways and upstream regulators
Figure S7 shows western blots for p-AKT/AKT, p-S6/S6, b tubulin, p-ERK/ERK, P-p-38/p-38, b catenin, p62 and b tubulin
Supplementary Figures 1 -7 and Supplementary Tables 1 - 3 legends
Data Availability Statement
The data generated in this study are available within the article and its Supplementary Data. Analytic methods, and study materials will be made available to other researchers on request. Complete RNA-sequencing (RNA-seq) data are available as GSE200923 on Gene Expression Omnibus.