

The authors characterize a library of engineered chimeric cytokine receptors (CCR) in the context of CAR T-cell therapy. These CCRs hold the potential for tailoring cytokine signaling to the type of adoptive cell therapy being developed.
Abstract
Adoptive T-cell therapy aims to achieve lasting tumor clearance, requiring enhanced engraftment and survival of the immune cells. Cytokines are paramount modulators of T-cell survival and proliferation. Cytokine receptors signal via ligand-induced dimerization, and this principle has been hijacked utilizing nonnative dimerization domains. A major limitation of current technologies resides in the absence of a module that recapitulates the natural cytokine receptor heterodimeric pairing. To circumvent this, we created a new engineered cytokine receptor able to constitutively recreate receptor-heterodimer utilizing the heterodimerization domain derived from the IgG1 antibody (dFab_CCR). We found that the signal delivered by the dFab_CCR-IL2 proficiently mimicked the cytokine receptor heterodimerization, with transcriptomic signatures like those obtained by activation of the native IL2 receptor. Moreover, we found that this dimerization structure was agnostic, efficiently activating signaling through four cytokine receptor families. Using a combination of in vivo and in vitro screening approaches, we characterized a library of 18 dFab_CCRs coexpressed with a clinically relevant solid tumor–specific GD2-specific chimeric antigen receptor (CAR). Based on this characterization, we suggest that the coexpression of either the common β-chain GMCSF or the IL18 dFab_CCRs is optimal to improve CAR T-cell expansion, engraftment, and efficacy. Our results demonstrate how Fab dimerization is efficient and versatile in recapitulating a cytokine receptor heterodimerization signal. This module could be applied for the enhancement of adoptive T-cell therapies, as well as therapies based on other immune cell types. Furthermore, these results provide a choice of cytokine signal to incorporate with adoptive T-cell therapies.
Introduction
The full activation and proliferation of adoptively transferred immune cells [e.g., tumor-infiltrating lymphocytes (TIL), T-cell receptor (TCR) T cells, and chimeric antigen receptor (CAR) T cells] require multiple immunologic signals (1). In addition to antigen receptor signaling, cytokine receptor activation directs and amplifies T-cell differentiation and expansion (2). However, within the tumor microenvironment, immunostimulatory cytokines are limited (3). Furthermore, in conditions of minimal residual disease or prolonged trafficking to sites of disease, the scarcity of antigen receptor activation limits T-cell auto- and paracrine cytokine production, additionally restricting T-cell activation by cytokines (4).
Several approaches have been explored in adoptive T-cell therapy to overcome this lack of endogenous immunostimulatory cytokines. Cytokines such as IL2 can be given systemically; however, side-effects are common and prolonged administration is impractical (5, 6). T cells can be engineered to secrete cytokines, yet this approach is also associated with systemic adverse effects (7, 8).
Alternative approaches have focused on engineering cytokine receptors to deliver cytokine signals in the absence of cytokines. Some engineered cytokine receptors signal in response to a pharmaceutical agent (9), and others signal in response to ubiquitous and inhibitory microenvironmental factors (10, 11). In other approaches, cytokine receptors are engineered to be constitutively active without the requirement for exogenous stimuli (12, 13).
With this report, we add to the literature of constitutively active cytokine receptors with a format that forces cytokine receptor heterodimerization by replacing cytokine receptor ectodomains with antibody kappa constant light chain (KCL) and constant heavy 1 (CH1) domain (dFab_CCR). When expressed in human T cells, we found that the dFab_CCR design was highly versatile, being able to transmit any major cytokine receptor family signal. Such flexibility allowed for the characterization of several dFab_CCRs that enhanced CAR T-cell effector function and persistence both in vitro and in vivo. We identified IL18 and GMCSF dFab_CCR as multifaceted modulators of CAR T-cell effector function by improving persistence, enhancing cytokine secretion, and augmenting aerobic metabolism. This translated into enhanced antitumor activity in several animal models.
Materials and Methods
Retroviral and plasmid constructs
Molecular cloning was performed by Golden Gate assembly using Bsa I (NEB, R3733L), BsmB I (NEB, R0739L), and Bbs I (NEB, R0539L) restriction enzymes depending on the DNA sequence. G-blocks encoding for cytokine receptor sequences, constant heavy chain 1, constant light kappa chain were purchased from IDT. Information regarding the cytokine receptor sequences and all other key resources and reagents can be found in Supplementary Table S1. Codon optimization was performed using GeneArt Codon Optimization Tool that strove to keep GC content at 60% and eliminate cryptic splicing, hairpins, literal repeats, and any possible cis-acting sequences. Each open reading frame was cloned into the splicing oncoretroviral vector SFG as polycistronic nucleotide sequences separated by interposed foot-and-mouth disease (FMD)-2A TaV motifs, codon wobbled to prevent retroviral recombination. The human GD2-specific CAR comprised the Huk666 scFv (14), a CD8 alpha stalk and transmembrane domain along with a 41bb-CD3ζ endodomain. The human CD19-specific CAR comprised the fmc63 scFv (15), a CD8 alpha stalk and a transmembrane domain along with a 41bb-CD3ζ endodomain. The PSMA CAR was composed of the 10C11 scFv (in house), a CD8 alpha stalk and a transmembrane domain along with a 41bb-CD3ζ endodomain.
The dFab_CCRs were cloned utilizing the CH1 region and the Constant Light region of the human IgG antibody. The transmembrane and endodomain were derived from the indicated cytokine receptor. The murine dFab_CCRs were generated utilizing the indicated murine cytokine receptor transmembrane and endodomain. All human constructs were coexpressed with RQR8 (16) as a surrogate of CAR gene expression.
The murine GD2-specific CAR comprised the muk666 scFv, a murine CD8 alpha stalk and transmembrane domain along with a murine CD28 and CD3ζ endodomains (17). The murine constructs were coexpressed with THY1.1 as a surrogate of CAR expression.
Cell lines
HEK-293T (2018, ATCC, CRL-3216, RRID:CVCL_0063), Phoenix ECO (2019, ATCC, SD-3444, RRID:CVCL_H717), SKOV-3 GD2+, SKOV-3 mKATE/CD19+, and CHLA-255-FFluc cells were cultured in I10 medium consisting of Iscove's modified Dulbecco's medium (IMDM, Gibco, 12440053) supplemented with 10% fetal bovine serum (FBS, BIOSERA, FB-1101/100) and 2 mmol/L GlutaMAX (Sigma, 35050061). SupT1 NT (2019, ATCC, CRL-1942, RRID:CVCL_1714), SupT1 GD2+, CT26 NT (2018, ATCC, CRL-263, RRID:CVCL_7256), CT26 GD2+, B16F10 NT (2018, ATCC, CRL-6475, RRID:CVCL_0159), B16F10 GD2+, NALM6 (2018, DSMZ, ACC-128, RRID:CVCL_0092), and NALM6-CD19KO cell lines were cultured in R10 medium consisting of RPMI (RPMI1640, Gibco, 21875034) supplemented with 10% FBS (BIOSERA, FB-1101/100) and 2 mmol/L GlutaMAX (Sigma, 35050061). HEK-293 T, Phoenix Eco, SupT1, CT26, SKOV3, and B16F10 lines were obtained from the American Type Culture Collection. NALM6 cells were obtained from the DSMZ, German Collection of Microorganisms and Cell Cultures GmbH. CHLA-255 cells were kindly donated by the COG Cell Culture Core + Xenograft Repository, Texas Tech Uni HSC Cancer in 2017.
NALM6-CD19KO cells were generated in-house using CRISPR/Cas9 genome engineering. Guide RNAs (gRNA) were designed targeting exon 2 of the CD19 gene, choosing the guides with the top quality and least off-target score; gRNA: CGCUGUGCUGCAGUGCCUCA.SupT1, SKOV3, CT26, and B16F10 were transduced with an SFG retroviral vector to express human GD2. SKOV3 were transduced with an SFG vector to express human CD19 and red fluorescent protein mKATE. CHLA-255 and NALM6 were transduced with an SFG vector to express firefly luciferase. Briefly, the cells were plated at a density of 0.3 × 106 cells (0.5 mL) in retronectin-coated (Takara, T100B) 24-well plates with 1.5 mL of retroviral supernatant. The plates were centrifuged at 1,000 × g for 40 minutes. At 24 hours after spinoculation, the cells were harvested and rechallenged with fresh viral supernatant.
All the cells utilized in the article were between passage 5 and passage 10 and Mycoplasma tested utilizing MycoAlert Mycoplasma Detection Kit (Lonza, LT07–318). Each cell line was archived with unique serial number, and thereafter, not reauthenticated. The transduced cells were analyzed prior to each assay via flowcytometry for the expression of the respective transgene.
Retroviral transduction of primary human T cells
RD114-pseudotyped γ-retroviral supernatants were generated by transient transfection of HEK-293T cells with an SFG transfer vector plasmid (4.7 μg), an RD114 envelope expression plasmid (RDF, a gift from M. Collins and Y. Takeuchi, University College London, 3.2 μg), and a packaging plasmid expressing MoMLV Gag-pol (a gift from E. Vanin, Baylor College of Medicine, 4.7 μg). Transfection was facilitated using GeneJuice (Merck Millipore, 70967), following the manufacturer's instructions.
Leucocyte cones of healthy donors were purchased from National Health Service Blood and Transplant (NHSBT, United Kingdom). Whole blood was extracted from each cone and diluted to 50 mL with sterile PBS. Peripheral blood mononuclear cells (PBMC) were isolated by Ficoll gradient centrifugation using SepMate 50 (StemCell, 85450) layering 25 mL of whole blood mixture to each SepMate 50. The cells were centrifuged at 1,200 × g for 20 minutes. The buffy coat was extracted and washed twice with sterile PBS. Isolated PBMCs cells were resuspended at 1 × 106 cells/mL in R10 and stimulated with 50 ng anti-CD3 (Miltenyi Biotec, 170–076–124) and 50 ng anti-CD28 (Miltenyi Biotec, 170–076–117) per 1 × 106 cells. A total of 100 iU/mL human IL2 (GenScript, Z0036) was added following overnight stimulation. Twenty-four hours after IL2 supplementation, cells were collected, plated at a density of 1 × 106 cells per well (1 mL) on retronectin-coated (Takara, T100B) 6-well plates with 3 mL of retroviral supernatant in the presence of 100 iU/mL of human IL2, and centrifuged at 1,000 × g for 40 minutes. At 24 hours after spinoculation, the T cells were harvested and replated in complete R10 media supplemented with 100 iU human IL2. Transduction efficiency was determined on day 5 after transduction and further experiments were commenced on days 5 to 9 after transduction.
Retroviral transduction of murine T cells
Ecotropic virus was prepared by transiently transfecting Phoenix ECO adherent packaging cells with an SFG vector (4.7 μg) and pseudotyped with ecotropic envelope vector pCL-ECO (2.68 μg, PhEco, RRID:Addgene_12371). Transfection was facilitated using GeneJuice (Merck Millipore, 70967), following the manufacturer's instructions.
Splenocytes were extracted as follows: Spleens were pulped and passed through a 70 μm cell strainer. Red blood cells were lysed using ACK red blood cell lysis buffer (Gibco, A1049201) for 5 minutes at room temperature. The reaction was stopped with 15 mL of Hank's Balanced Salt Solution (HBSS, Gibco, J67763.K2). After the cells were pelleted, the splenocytes were passed through a 40-μm cell strainer to obtain a single-cell suspension. Isolated splenocytes were activated with 2 μg Concanavalin A (Sigma, C0412–5MG) and 1 ng murine IL7 (Miltenyi Biotec, 130–094–066) per 1.5 × 106 cells for 24 hours. Cells (5 × 106) were plated on retronectin-coated (Takara, T100B) 6-well plates in 2 mL of retroviral supernatant and spun 800 × g for 90 minutes. The wells were supplemented with 4 mL of complete media R10, incubated for 72 hours at 37°C in 5% CO2, in the presence of human IL2 (100 iU/mL; GenScript, Z00368).
Transduction efficiency evaluation
Staining steps to evaluate transduction efficiency were performed at room temperature for 12 minutes, with PBS washes between steps. Cells were costained with either eFluor 780 (eBioscience, 65–0865–15) or eFluor450 (eBioscience, 65–0863–18) fixable viability dye depending on the experimental condition. Transduced human T cells were detected by staining for the RQR8 marker, utilizing anti-CD34 (R&D system, FAB7227G, RRID:AB_10973657). Murine CAR T-cell transduction was evaluated by staining for THY1.1 marker, utilizing anti-Thy1.1 (BioLegend, 202522, RRID:AB_1595477). The samples were stained in 96-well plates and resuspended in 100 μL of PBS. Flow cytometry was performed using the MacsQuantX flow cytometer (Miltenyi Biotec), running 50 μL of cell suspension. Data analysis was conducted using FlowJo v10 (Treestar, RRID:SCR_008520).
T-cell immunophenotyping
Staining steps were performed at room temperature for 12 minutes, with PBS washes between steps. Transduced cells were identified by RQR8 positivity utilizing a CD34-specific antibody, and live cells selected via 7AAD (BioLegend, 420404) live dead cell dye exclusion. Memory, exhaustion, and activation phenotypes in CD4+ and CD8+ CAR T cells were based on the expression of CD45RA (Miltenyi Biotec, 130–117–747, RRID:AB_2658333), CCR7 (Miltenyi Biotec, 130–119–583, RRID:AB_2655953), PD-1 (Miltenyi Biotec, 130–120–382, RRID:AB_2661364), LAG-3 (Miltenyi Biotec, 130–120–012, RRID:AB_2656417), TIM-3 (Miltenyi Biotec, 130–121–334, RRID:AB_2784166), and KLRG-1 (Miltenyi Biotec, 130–120–423, RRID:AB_2727835). Information regarding the antibodies can be found in Supplementary Table S1.
The samples were stained in 96-well plates and resuspended in 100 μL of PBS. Flow cytometry was performed using the MacsQuantX flow cytometer (Miltenyi Biotec), running 50 μL of cell suspension. Data analysis was conducted using FlowJo v10 (Treestar, RRID:SCR_008520).
Animals study approval
All animal procedures in this study gained the approval of The Animal Welfare and Ethical Review Body and the United Kingdom Home Office (Autolus PPL No. P244BBE6B). All procedures are performed in accordance with the United Kingdom Home Office Animals (Scientific Procedures) Act 1986 and in adherence to Imperial College London or Autolus SOPs. This study is necessary and justifiable with due consideration to the “3Rs” (the reduction, refinement, and replacement of animals in research).
Establishment of subcutaneous syngeneic colon carcinoma mouse model
Six- to 8-week-old Balb/c female mice (strain code 028, Charles Rivers Laboratories) were subcutaneously injected with 1 × 106 (100 μL) of CT26 colon carcinoma cells modified to express GD2. Nine days after tumor engraftment, the mice were sublethally irritated with 4.5 Gy TBI. All mice were analyzed to measure tumor size on D-1 (related to CAR T-cell injection) and ranked in order of tumor volume, after which cohorts were randomly selected with a similar total group volume average. Twenty-four hours after TBI, 1 × 106 transduced CAR T cells were injected intravenously (100 μL). Tumor growth was monitored twice a week via caliper measurements. Peripheral blood via tail cut was taken every 7 days. Mice were euthanized when the tumor reached a maximum length or breadth of 1 cm3 or sudden body weight loss ≥ 20%. Animals were sacrificed by isoflurane intoxication followed by cervical dislocation. Spleen were collected at the time of euthanization for CAR T-cell tracking.
Establishment of subcutaneous syngeneic melanoma mouse model
Six- to 8-week-old female C57Bl/6J mice (strain code 632, Charles River Laboratories) were subcutaneously injected with 1 × 105 (100 μL) of B16F10 melanoma cells modified to express GD2. Seven days after tumor engraftment, the mice were sublethally irritated with 4.5 Gy TBI. All mice were analyzed to measure tumor size on D-1 (related to CAR T-cell injection) and ranked in order of tumor volume, after which cohorts are randomly selected with a similar total group volume average. Twenty-four hours after TBI, 3 × 106 transduced T cells were injected intravenously (100 μL). Tumor growth was monitored twice a week via caliper measurements. Peripheral blood via tail cut was taken every 7 days. Mice were euthanized when the tumor reached a maximum length or breadth of 1 cm3 or sudden body weight loss ≥ 20%. Animals were sacrificed by isoflurane intoxication followed by cervical dislocation. Bone marrow and spleen were collected at the time of euthanization for CAR T-cell tracking.
Establishment of metastatic neuroblastoma xenograft mouse model
Ten- to 14-week-old female NOD.Cg-Prkdc scid Il2rg tm1Wjl /SzJ (NSG, The Jackson Laboratory, #005557) were intravenously injected with 1 × 106 Firefly-luciferase expressing GD2+ CHLA-255 cells (CHLA-255 FLluc, in house; 100 μL). All mice were scanned to measure BLI counts on D-1 (related to CAR T-cell injection) and ranked in order of total flux, after which cohorts are randomly selected with a similar total group BLI average. Fifteen days later, mice were injected intravenously with 1 × 106 transduced CAR T cells (100 μL).
Tumor growth was indirectly assessed by biweekly bioluminescent imaging. The mice were imaged for bioluminescence signal from tumor cells using the IVIS system (IVIS, Xenogen Corporation) 10 to 15 minutes after 150 mg/kg D-luciferin (Promega, P1043) per mouse was injected intraperitoneally. Mice were euthanized when the sudden body weight loss ≥ 20%. Animals were sacrificed by isoflurane intoxication followed by cervical dislocation. Bone marrow and spleen were collected at the time of euthanization for CAR T-cell tracking.
Establishment of human acute lymphoblastic leukemia xenograft mouse model
Ten- to 14-week-old female NOD.Cg-Prkdc scid Il2rg tm1Wjl /SzJ (NSG, The Jackson Laboratory, #005557) were intravenously injected with 1 × 106 Firefly-luciferase expressing NALM6 cells (NALM6 FLuc, in house; 100 μL). Four days later, mice were injected intravenously with 2.5 × 106 transduced CAR T cells (100 μL). Tumor growth was indirectly assessed by biweekly bioluminescent imaging. The mice were imaged for bioluminescence signal from tumor cells using the IVIS system (IVIS, Xenogen Corporation) 10 to 15 minutes after 150 mg/kg D-luciferin (Promega, P1043) per mouse was injected intraperitoneally. Mice were euthanized when body weight loss ≥20%. Animals were sacrificed by isoflurane intoxication followed by cervical dislocation. Bone marrow and spleen were collected at the time of euthanization for CAR T-cell tracking.
Animal sample phenotyping
Day 10 post murine CAR T-cell injection, Balb/c mice (see Subcutaneous syngeneic colon carcinoma mouse model) were sacrificed by isoflurane intoxication followed by cervical dislocation. Spleens were processed as described above (see Retroviral transduction of murine T cells). TILs were enriched from tumor biopsies by using Tumor Dissociation Kit, murine (Miltenyi Biotec, 130–096–730) and processed utilizing gentleMACS Dissociator (Miltenyi Biotec). Erythrocytes were lysed using 500 μL of ACK lysis buffer (Sigma, A1049201) for 5 minutes at room temperature. Cells were strained twice, first utilizing a 70-μm cell strainer then a 40 μm cell strainer. Cells (2 × 106) were aliquoted for phenotyping by flow cytometry. First, the Fc receptor was blocked using anti-CD32/CD16 (BioLegend, 156604, RRID: AB_2783138) to avoid nonspecific binding. Transduced CAR T-cell populations were identified based on the expression of CD45 (BioLegend, 103132, RIID: AB_893340), CD3 (BioLegend, 100236, RRID:AB_2561456), CD4 (BioLegend, 116004, RRID:AB_313688), THY1.1 (BioLegend, 202522, RRID:AB_1595477), and the absence of CD11b (BioLegend, 101216, RRID, AB_312799). The identification of murine T-cell memory subpopulation was performed by the detection of CD62 L (BioLegend, 104441, RRID AB_2561537) and CD44 (BioLegend, 163609, RRID AB_2924492). The myeloid population was defined by the expression of CD45 and CD11b. Monocytes were identified by the expression of F4/80 (BioLegend, 157304, RRID AB_2832547). Dendritic cells (DC) were identified by the expression of CD11b. Information regarding the antibodies can be found in Supplementary Table S1.
The samples were stained in 96-well plates and resuspended in 100 μL of PBS. Flow cytometry was performed using the MacsQuantX flow cytometer (Miltenyi Biotec), running 50 μL of cell suspension. Data analysis was conducted using FlowJo v10 (Treestar, RRID:SCR_008520).
Intracellular staining
Between 1 × 105 and 5 × 104 human CAR T cells or nontransduced human T cells/well were pelleted by centrifugation at 1,000 × g for 2 minutes at room temperature. The cells were stained with eFluor450 fixable viability dye, CD3 (BioLegend), and CD34 (R&D Systems) for 10 minutes at room temperature. The cells were washed once with PBS and pelleted 1,000 × g for 2 minutes. The cells were fixed and permeabilized according to the manufacturer's protocol using BD Cytofix/Cytoperm Fixation/Permeabilization Solution Kit (BD Biosciences, 554714). The cells were then stained with anti-Human Kappa Light Chain Biotin (Thermo, MA5–12114, RRID:AB_10984749) in permeabilization/washing solution for 12 minutes at room temperature, washed once with permeabilization/washing solution, and pelleted by centrifugation at 1,000 × g for 2 minutes. The cells washed once more with 200 μL of permeabilization/washing solution and pelleted as before. The cells were then stained with PE-conjugated Streptavidin (BioLegend, 405204) in permeabilization/washing solution for 12 minutes at room temperature. The samples were stained in 96-well plates and resuspended in 100 μL of PBS. Flow cytometry was performed using the MacsQuantX flow cytometer (Miltenyi Biotec), running 50 μL of cell suspension. Data analysis was conducted using FlowJo v10 (Treestar, RRID:SCR_008520).
Flow cytometry–based assay of human CAR T-cell cytotoxicity
CAR transduced human T cells were normalized based on RQR8 expression by dilution with nontransduced human T cells. SupT1 NT, SupT1 GD2+, NALM6, NALM6-CD19KO targets, and effector cells were resuspended in R10 media and plated in a 96-well plate flat bottom at the desired volume to achieve the desired effector:target ratio. Target cell number was kept constant within each experiment, at 5 × 104 cells. After 48 hours, the plate was centrifuged at 1,000 × g for 2 minutes, 100 μL of supernatant was removed for cytokine analysis (see Analysis of cytokine production), and CountBright beads (Thermo Fisher Scientific, C36950) were added to allow normalization of cell numbers acquired. T cells were distinguished from target cells by positivity for CD2 (BioLegend, 300214, RRID: AB_10900259) and CD3. Gating on single live target cells was performed according to exclusion of fixable viability dye eFluor780. Flow cytometry was performed using the MacsQuantX flow cytometer (Miltenyi Biotec). Data analysis was conducted using FlowJo v10 (Treestar, RRID:SCR_008520). Percentage of live cells was calculated relative to the number of live target cells after coculture with nontransduced T cells.
 |
Flow cytometry–based assay for murine CAR T-cell cytotoxicity
B16F10, B16F10-GD2+, CT26, and CT26-GD2+ target cells were plated 24 hours prior to the assay at a density of 2.5 × 104 cells/well in a 96-well, flat bottom plate. Murine CAR transduced T cells were normalized based on THY-expression by dilution with nontransduced T cells and plated over the target cells to achieve the desired effector:target (E:T) ratio. After 48 hours, the plate was centrifuged at 1,000 × g for 2 minutes, supernatant was removed for cytokine analysis (see Analysis of cytokine production), and CountBright beads (Thermo Fisher Scientific) were added to allow normalization of cells numbers acquired. Target cells were detached from the plate using 50 μL of Trypsin (Gibco, 25200056) for 5 minutes at 37°C. The reaction was stopped with 100 μL of R10, and the cells were collected in a new 96-well plate. Following a wash with PBS, the cells were ready to stain. Murine T cells were distinguished from target cells by positivity for CD3 and CD45. Gating on single live target cells was performed according to exclusion of fixable viability dye eFluor780. Flow cytometry was performed using the MacsQuantX flow cytometer (Miltenyi Biotec). Data analysis was conducted using FlowJo v10 (Treestar, RRID:SCR_008520). Percentage of live cells was calculated relative to the number of live target cells after coculture with nontransduced T cells.
 |
Cytokine starvation assay
Human CAR T cells were prelabeled with Cell Trace Violet (CTV, Thermo Fisher Scientific, C34557) according to the manufacturer's instructions and seeded at a density of 1 × 105 cells without the addition of exogenous cytokines in a 96-well, flat bottom plate. If ruxolitinib was used, human Fab_CCR-IL2 or nontransduced T cells were preincubated with either 1 μmol/L or 10 μmol/L of ruxolitinib (Stem Cell Technologies, 73402) for 4 hours, then washed to remove excess ruxolitinib. The cells were then seeded at a density of 1 × 105 cells without the addition of exogenous cytokines in a 96-well, flat bottom plate. Murine CAR-positive, CAR/dFab_CCR-positive, and nontransduced T cells were plated at 1 × 105 cells in a 96-well, flat bottom plate, without CTV prelabeling and exogenous cytokines 4 or 7 days in R10 media. The proliferative potential was analyzed either 4 or 7 days after, depending on the experiment.
CTV mean fluoresced value was calculated, and gating on single live target cells was performed according to exclusion of fixable viability dye eFluor780 and positivity for RQR8 (human CAR T cells) or THY1.1 (murine CAR T cells). Flow cytometry was performed using the MacsQuantX flow cytometer (Miltenyi Biotec). Data analysis was conducted using FlowJo v10 (Treestar, RRID:SCR_008520). The T fold expansion was calculated as follows:
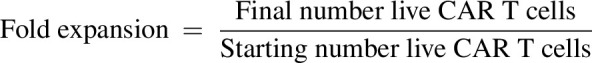 |
Serial tumor rechallenge assay
A total of 1 × 106 human GD2-specific or CD19-specific CAR T cells and 1 × 105 of either SKOV-3-GD2+ cells or SKOV-3-CD19+ cells, E:T ratio of 10:1, were cocultured in cytokine-free R10 media for 7 days in a 24-well plate. After 7 days, the CAR T cells were carefully harvested, and the viable CAR T cells were enumerated utilizing NucleoCounter NC-250 (Chemometec) automated cell analyzer together with AO•DAPI Solution 18 Staining Reagent (Chemometec, 910–3018) for live/dead discrimination. Live CAR T cells (1 × 106) were replated over fresh 1 × 106 SKOV-3-GD2+ cells or SKOV-3-CD19+ cells to reach the E:T ratio of 10:1. The coculture was incubated for a further 7 days. The cycle of stimulations was performed for a total of three rechallenges.
Serial starvation assay
Human CAR T cells (0.5 × 106) were cultured in cytokine-free R10 media for 7 days in a 24-well plate. After 7 days, the CAR T cells were carefully harvested, and the viable T cells were enumerated utilizing NucleoCounter NC-250 (Chemometec) automated cell analyzer together with AO•DAPI Solution 18 Staining Reagent (Chemometec, 910–3018) for live/dead discrimination. Up to 0.5 × 106 CAR T cells were replated and cultured for a further 7 days. The cycle of starvation was continued until the number of CAR T cells at the day of harvest fell below the counter limit of detection (0.05 × 106). The data are plotted as 106 cumulative numbers, calculated as the sum of 106 viable CAR T cells every 7 days.
Activation of GD2-specific and CD19-specific CAR T cells with scFv anti-idiotype antibodies
Anti-huk666 idiotype and anti-fmc63 idiotype were generated in house from expi-CHO cells (Thermo Fisher Scientific, A29127) and purified using mAb Select SuRe protein A column (Cytiva, 11003494) couple with Akta Pure system (see Supplementary Fig. S1 for flow cytometry characterization).
Ten micrograms of anti-huk666 idiotype (for GD2-specific CAR T Cells) or 1 μg of anti-fmc63 idiotype (for CD19-specific CAR T cells) was coated on non-tissue culture–treated 24-well plates for 16 hours at 4°C. Plates were washed twice before plating T cells (1 × 106 cells/well). Plates were centrifuged at 500 × g for 5 minutes and incubated at 37°C for 24 to 48 hours.
Analysis of cytokine production
Human IFNγ (BioLegend, 430115), human IL2 (BioLegend, 431815), and mouse IFNγ (BioLegend, 430804) were detected utilizing the indicated BioLegend ElisaMax Kits, following the manufacturer's instructions. For human CAR T-cell coculture supernatants, Cytokine bead array (BioLegend, Legend Plex custom-made) was performed according to the manufacturer's protocol. For blood serum from NSG mice [see Establishment of human acute lymphoblastic leukemia xenograft mouse model], Cytokine bead array (BioLegend, LEGENDplex Human CD8/NK Panel, 13-plex, 741065) was performed according to the manufacturer's protocol. For blood serum from day 10 euthanized Balb-c mice (see Establishment of subcutaneous syngeneic colon carcinoma mouse model), Cytokine bead array (BioLegend, LEGENDplex MU Cytokine Release Syndrome Panel, 13-plex, 741024) was performed according to the manufacturer's protocol. The samples were detected using Fortessa LRS X20 (BD Biosciences) and analyzed using LEGENDplex Data Analysis Software Suite (BioLegend).
RNA sequencing
The dFab_CCR-IL2 positive and nontransduced human T cells from 3 individual donors were isolated using CD34 microbeads (Miltenyi Biotec, 130–046–702) and rested for 24 hours in R10 media. Subsequently, the T cells were seeded at 1 × 106 cells/mL with or without 100,000 iU/mL of human IL2. After 72 hours, rHuIL2 was replenished. After 7 days, the cells were lysed, and the mRNA was extracted with the illustra RNAspin Mini RNA Isolation Kit (GE Healthcare, 12183018A). The RNA was quantified with NanoDrop (Thermo Fisher Scientific). The quality of the RNA was assessed with Agilent TapeStation (Agilent, RRID:SCR_019547). Poly-A selection was performed to remove ribosomal RNA. Roughly, 40 million paired end reads (150 bp) were sequenced on Illumina Hiseq sequencer (GeneWiz) for each individual CAR T-cell sample with different CCR modules.
Fastq reads were mapped to the human genome (B37.3) and gene model GenCode (V19) with Omicsoft Aligner 4 (18). Gene count data was normalized by logGenometric mean values. Differential gene expression analysis was carried out using DeSeq2 General Linear Model test (RRID:SCR_015687; ref. 19). Gene set enrichment analysis (Subramanian) Pre-ranked tool (GSEAPreranked) was used to conduct the pathway and functional analysis on the differentially expressed genes.
NanoString
Total mRNA from activated or nonactivated human GD2-specific CAR T cells was collected using the PureLink RNA Mini Kit (Thermo Fisher Scientific, 12183018A). Gene expression analysis was performed using the CAR T cell Panel version 1 (NanoString) RNA Profiling Core. Briefly, 50 ng of total mRNA, Reporter CodeSet, and Capture ProbeSet were hybridized for 16 hours at 65°C in a thermal cycler. The samples were then loaded in the nCounter Analysis System. Data were analyzed using nSolver 4.0 software (NanoString) and the Advanced Analysis software utilizing the standard analysis parameters.
Analysis of metabolic parameters
Mitochondrial function was assessed on both human GD2-specific CAR T cells and human CD19-specific CAR T cells with the extracellular flux analyzer SeaHorse (Agilent). Individual wells of the XF96 cell-culture microplates were coated with CellTak (Corning, 354240) following the manufacturer's instructions. The matrix was adsorbed overnight at 37°C. Mitochondrial function was assessed 24 hours postactivation or 24 hours poststarvation. Regarding the GD2-specific CAR T cells, 1.25 × 105 cells were resuspended in XF assay medium (non-buffered RPMI1640, Agilent, 103681–100) containing 5.5 mmol/L glucose (Agilent, 103577–100), 2 mmol/L L-glutamine (Agilent, 103579–100), and 1 mmol/L sodium pyruvate (Agilent, 103578–100). Regarding the CD19-specific CAR T-cell experiments, 5 × 104 cells were resuspended in XF assay medium containing 5.5 mmol/L glucose, 2 mmol/L L-glutamine, and 1 mmol/L sodium pyruvate. The microplate was centrifuged at 1,000 × g for 5 minutes and incubated in standard culture conditions for 60 minutes. During instrument calibration (30 minutes), the XF96 assay cartridges (Agilent) were calibrated in accordance with the manufacturer's instructions. Cellular oxygen consumption rates (OCR) were measured under basal conditions and, following treatment with 1.5 mmol/L oligomycin, 1.5 mmol/L FCCP, and 40 nmol/L rotenone, with 1 mmol/L antimycin A (XF Cell Mito Stress Kit, Agilent, 103015–100).
Generation of the DNA barcoded dFab_CCR library
Each individual DNA barcode cassette was created as DNA g-block and ordered from IDT. Each g-block was digested with MluI (NEB, R3198L)/ClaI (NEB, R0197L), and inserted in a linearized SFG retroviral vector encoding for specific human dFab_CCRs/GD2-CAR creating individually barcoded vectors. Human T cells were transduced (see Retroviral transduction of primary human T cells), the transduction efficiency evaluated (see Transduction efficiency evaluation) and individual human GD2-dFab_CCRs barcoded T cells were mixed to obtain equivalent numbers of transduced cells for each sample.
Barcode analysis
Genomic DNA from human GD2/dFab_CCRs barcoded pooled CAR T cells, bone marrow, spleen, and liver harvested from euthanized NSG mice (see Establishment of metastatic neuroblastoma xenograft mouse model) was isolated using PureLink Genomic DNA Mini Kit (Thermo Fisher Scientific, K182002). Barcode sequence was amplified in a 50 μL PCR, utilizing Q5 High-Fidelity DNA Polymerase (NEB, M0491L), deoxynucleotide (dNTP) Mix (NEB, N0447S), and 2 μmol/L each of the forward primer (ACACTCTTTCCCTACACGACGCTCTTCCGATCTCCGCCAACGCGGTCGCAC) and the reverse primer (GACTGGAGTTCAGACGTGTGCTCTTCCGATCTGATGAGAACAGTATCGATTAGGGTTGACGGC) at 65°C amplification temperature. PCR products were isolated from a 3% agarose gel. The purified PCR products were sequenced using Amplicon-EZ next generation sequencing service (GeneWiz). Roughly 50,000 reads per sample were fully sequenced with 2 × 250 bp sequencing. The contigs of paired-end reads were created and Fastq quality performed to exclude disastrous reads. The pair-end reads were aligned and the barcode sequence reads counted. Values were normalized to the total reads obtained in each sample.
Phosphorylated protein immunoblot analysis
A total of 1 × 106 human dFab_CCR transduced and nontransduced T cells were harvested and pelleted at 5,000 × g for 2 minutes. The samples were washed with ice-cold PBS, and the cells were pelleted at 5,000 × g for 2 minutes. The pellet was lysed in 50 μL of 1× RIPA buffer (Millipore, 20–188) supplemented with 1× protease and phosphatase inhibitor (Abcam, ab201119). The cells were incubated on ice for 15 minutes. The tubes were then vortexed and incubated for a further 15 minutes. The lysate was clarified by centrifuging the cells at maximum speed for 10 minutes at 4°C. The lysate was then transferred in PCR tube strips with 12.5 μL of NuPAGE LDS Sample Buffer (4×; Invitrogen, NP0007). The sample was then boiled at 95°C for 5 minutes. Eight microliters of each sample was run on a premade 4%–20% Mini-PROTEAN TGX Precast Gel (Bio-Rad, 4561043), at 180V, 500 mA for 37 minutes on constant voltage mode. Five microliters of protein ladder (Bio-Rad, 1610374) was run as a size marker. Postseparation, SDS-PAGE gel was transferred to Trans-Blot Turbo Midi 0.2 μm PVDF membrane (Abcam, 1704157) using Trans-Blot Turbo Midi by semi-dry transfer (Abcam). Following transfer, membranes were blocked with 1× TBST (Sigma, 28360) supplemented with 5% BSA for 24 hours at 4°C. Membrane staining was undertaken with specific antibodies (Supplementary Table S1) diluted to the appropriate concentration in 1× TBST supplemented with 5% BSA for 1 hour on orbital shaker, 480 rpm, at room temperature. The membranes were then washed 3 times with 1× TBST for 10 minutes each time. The secondary staining (Supplementary Table S1) was performed as per primary staining. Following the last TBST wash, the membrane was washed with distilled water; then, the membrane was developed by incubation with Pierce ECL Plus Western Blotting Substrate (Bio-Rad, 1705061), according to the manufacturer's instructions.
Statistical analysis
Data were presented as mean ± SEM unless indicated otherwise. Graphs and statistics were generated using Prism 9.0 software for Windows (GraphPad Software Inc., RRID:SCR_000306). The differences between means were calculated using two-tailed unpaired t test, one-way ANOVA, and two-way ANOVA. Tukey correction for multiple comparisons was used to calculate adjusted P value when appropriate.
Specific statistical tests used for analyzing the data for each figure are described in the corresponding figure legends. Survival determined from the time of tumor cell injection was analyzed by the Kaplan–Meier method, and differences in survival between groups were compared by the log-rank test.
P values: ns P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001.
Data availability
The data generated in this study are available within the article and its supplementary data files or upon request from the corresponding author. The transcriptomic data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE227161 and GSE227312.
Results
The IL2 receptor endodomains can be constitutively activated by Ig Fab domain-induced heterodimerization
We sought to engineer a constitutively active IL2 receptor by approximation of the IL2 receptor β chain (IL2Rβ) and the common γ chain (CγC). To achieve this, the transmembrane and intracellular domains of IL2Rβ and CγC were fused with the human Ig CH1 and ΚLC, respectively (dFab_CCR-IL2). dFab_CCR-IL2 chains and the marker gene RQR8 were coexpressed in a single retroviral vector (Fig. 1A). Flow cytometric analysis of dFab_CCR-IL2 transduced human T cells via detection of KLC (Fig. 1B) or CH1 chain (Supplementary Fig. S2A) showed that both dFab components were expressed, although mainly detected intracellularly. When cultured under conditions of cytokine starvation, dFab_CCR-IL2–expressing T cells increased in numbers (Fig. 1C, right), and diluted CTV dye (Fig. 1C, left), suggesting that some cytokine receptor signaling was recapitulated.
Figure 1.

Constitutive heterodimeric dFab_CCR-IL2 recapitulates a proliferative IL2 signal in primary human T cells. A, Schematic of the dFab_CCR-IL2 format composed of CH1 fused to the IL2 receptor β chain. The IL2 receptor CγC was fused to the constant light κ chain (dFabκ_CCR-IL2). The illustration was created with BioRender.com (RRID:SCR_018361). B, Flow cytometric analysis of the constant light κ chain expression and RQR8 (CD34) in dFab_CCR-IL2–expressing T cells evaluated by surface staining or intracellular staining after fixation/permeabilization. C, Quantitated proliferation of dFab_CCR-IL2 T cells cultured for 96 hours in cytokine starvation. Proliferation expressed as CTV MFI dilution (left), and fold expansion (right) of RQR8+CD3+ T cells (n = 6, Student t test; ****, P ≤ 0.0001); data, mean ± SEM. D, PCA plot analyzing the variation in gene expression profile driven by the donors and by the two conditions. E, Volcano plot representing the differential gene expression between dFab_CCR-IL2 T cells and nontransduced T cells stimulated with 100,000 iU/mL IL2 (n = 3). F, Venn Diagram of the top 50 gene set enrichment analysis (GSEA) between dFab_CCR-IL2 T cells and T cells stimulated with 100,000 iU/mL IL2 (n = 3).
We next determined that dFab_CCR-IL2 signal could be modulated by truncating the receptor endodomain (20, 21) from either the CγC (Supplementary Fig. S3A and S3B) or IL2Rβ (Supplementary Fig. S3C and S3D). Alternatively, dFab_CCR-IL2 signal was controlled by exposure to the off-the-shelf FDA-approved JAK inhibitor ruxolitinib (Supplementary Fig. S4A; ref. 22).
These data suggested that Fab-based cytokine receptors are a versatile way of achieving constitutive cytokine receptor function.
dFab_CCR-IL2 induces functional effects similar to those of the native IL2 receptor
To determine whether differences existed in signaling between the dFab_CCR and the native cytokine receptor, we compared human T cells transduced with the dFab_CCR-IL2 with nontransduced T cells cultured in the presence of 100,000 iU/mL exogenous IL2. Both resulted in T-cell proliferation with T cells transduced with the dFab_CCR-IL2 expanding more than nontransduced T cells in 100,000 iU/mL human IL2 (Supplementary Fig. S5A), and both exogenous IL2 stimulation and dFab-CCR_IL2 expression resulting in phosphorylated STAT5 (Supplementary Fig. S5B).
We next performed a transcriptomic analysis following 7 days of cytokine starvation of human T cells expressing dFab_CCR-IL2 compared with nontransduced human T cells exposed to exogenous cytokine or not (Supplementary Fig. S5C). Principal component analysis (PCA) revealed no influence from the donors tested (n = 3) but showed a degree of separation between the three conditions (Fig. 1D; Supplementary Fig. S5D). Nonetheless, differential gene expression analysis showed no global transcriptomic shift between the signaling delivered by the dFab_CCR-IL2 and exogenous IL2 receptor (Fig. 1E).
Given the similarity in the overall gene expression patterns, we interrogated differences in specific pathways activated by the dFab_CCR-IL2 and the exogenous IL2 stimulation. Forty-four of the top 50 upregulated pathways (Supplementary Table S1) were shared between the two conditions (Fig. 1F). Furthermore, the false discovery rate (FDR) q-value of the top 10 shared pathways revealed a more pronounced proliferative profile from the dFab_CCR-IL2 transduced T cells with lower overall values than for the nontransduced T cells stimulated with exogenous IL2 (Supplementary Table S2).
Taken together, these data indicated that dFab_CCR-IL2 provided a functional cytokine-like signal.
The dFab_CCR format can be used to activate multiple cytokine receptor families
A broad range of cytokine dFab-CCR formats were generated (Fig. 2A) and grouped by respective cytokine receptor family: CγC receptor, the IL12 family, the IL1 family, and the IL10 and IL17 family (Supplementary Fig. S6A). Surface and intracellular Fab expression (light Kappa %) and Fab density levels (light Kappa MFI) detected by flow cytometry on transduced dFab_CCRs T cells (Supplementary Fig. S6B) varied between receptor families (Supplementary Fig. S6C and S6D), but were comparable within the same receptor family. Functional effects in primary human T cells were then explored.
Figure 2.

The dFab-CCR heterodimerization format activates a broad range of cytokine receptors families in primary human T cells. A, Schematic of the main cytokine receptors family members and the viral vector design selected for the generation of the dFab_CCR library. The illustration was created with BioRender.com (RRID:SCR_018361). B, Quantitated proliferation of T cells engineered with either IL2 or IL7 dFab_CCRs, expressed as CTV MFI dilution (left), and fold expansion (right) of RQR8+CD3+ T cells (n = 4, one-way ANOVA; ns P > 0.05, ****, P ≤ 0.0001). C, Immunoblot analysis of STAT5 phosphorylation, Y694. Nontransduced T cells were cultured in cytokine starvation for 24 hours. dFab_CCR-IL2 or -IL7 were cytokine starved for 96 hours. GAPDH was used as loading control. Data are representative of three independent experiments. D, Immunoblot analysis of in vitro STAT6 phosphorylation, Y641. Nontransduced and dFab_CCR-IL4 T cells were cultured in cytokine starvation for 24 hours. GAPDH was used as loading control in the same membrane. Data are representative of three independent experiments. E, Immunoblot analysis of in vitro STAT3 phosphorylation, Y705. Nontransduced, IL9, and IL21 dFab_CCR T cells were cultured in cytokine starvation for 24 hours. GAPDH was used as loading control in the same membrane. Data are representative of three independent experiments. F, IFNγ secreted by IL2, IL12, IL23, and IL27 dFab_CCR transduced T cells cultured for either 24 or 48 hours in cytokine starvation (n = 3, one-way ANOVA; ns P > 0.05; **, P ≤ 0.01; ***, P ≤ 0.001). G, Immunoblot analysis of in vitro STAT1 and STAT3 phosphorylation. Nontransduced, dFab_CCR-IL12, -IL23, and -IL27 T cells were cytokine starved for 24 hours. GAPDH was used as loading control in the same membrane. Data are representative of three independent experiments. H, IFNγ secreted by IL2 and IL18 dFab_CCR T cells cultured for either 24 or 48 hours in cytokine starvation (n = 3, one-way ANOVA; ns P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01). I, Immunoblot analysis of in vitro ERK1/2 phosphorylation, T202 and Y204. Nontransduced, IL1, IL18, and IL33 dFab_CCR T cells were cytokine starved for 24 hours. GAPDH was used as loading control in the same membrane. Data are representative of three independent experiments. J, Quantitated in vitro proliferation of T cells engineered with either -IL3, -IL5 and -GMCSF dFab_CCRs, expressed as CTV MFI dilution (left), and fold expansion (right) of RQR8+/CD3+ T cells (n = 4, one-way ANOVA; ns P > 0.05; *, P ≤ 0.05). K, Immunoblot analysis of in vitro STAT3 and STAT5 phosphorylation. Nontransduced T cells were cytokine starved for 24 hours. IL3, IL5, and GMCSF dFab_CCRs T cells were cytokine starved for 96 hours. GAPDH was used as loading control in the same membrane. Data are representative of three independent experiments. All data are presented as mean ± SEM.
Western blot analysis of the dFab_CCR CγC receptor family demonstrated the correct activation of the respective STAT molecules (Fig. 2C–E), yet only the dFab_CCR-IL7 was able to induce T-cell proliferation (Fig. 2B; Supplementary Fig. S7A–S7C).
The IL12 family members (IL12 and IL23) and IL18 from the IL1 receptor family dFab_CCRs showed increased baseline IFNγ release both at 24 and 48 hours (Fig. 2F and H), as expected (23, 24). Furthermore, all the IL12 receptor family (Fig. 2G, STAT1 and STAT3), IL1 family (Fig. 2I, ERK1/2), and both IL10 and IL17A (Supplementary Fig. S7D and S7E) dFab_CCRs, correctly activated their respective signaling molecules.
Finally, we determined whether dFab_CCR could be applied beyond lymphoid cytokine receptors by testing myeloid cell–specific common β-chain: dFab_CCR-IL3, -IL5 and -GMCSF (Supplementary Fig. S6A). T cells engineered with all the CβC dFab_CCRs displayed significant proliferation (Fig. 2J) and resulted in the phosphorylation of either STAT3 or STAT5 proteins (Fig. 2K).
Taken together, these results demonstrated that the dFab format could be applied to a broad spectrum of cytokine receptor families.
dFab_CCR-IL2 and CAR can function when coexpressed in a single vector
We initially tested whether RQR8/dFab_CCR-IL2 could be coexpressed with a 2nd generation 41bbζ GD2-specific CAR (Fig. 3A). The expected RQR8 and dFab_CCR-IL2 ratio was maintained (Supplementary Fig. S8A), and coexpressing the dFab_CCR-IL2 provided a proliferative advantage (Supplementary Fig. S8B and S8C), while killing and cytokine secretion induced via the GD2-specific CAR was largely comparable (Supplementary Fig. S8D–S8F).
Figure 3.

Coexpression of the dFab_CCRs with GD2 CAR in primary human T cells reveal differential effects on CAR T-cell functionality. A, Schematic of the of the tetra-cistronic γ-RV vectors coexpressing RQR8, 2nd generation 41bbζ GD2-specific CAR and dFab_CCR. The illustration was created with BioRender.com (RRID:SCR_018361). B, Day 7 quantification of in vitro proliferation of GD2-specific CAR T cells engineered with the library of dFab_CCRs, expressed as fold expansion of RQR8+CD3+ GD2 CAR T cells (n = 11, GD2CAR + dFab_CCR-IL7 n = 4, one-way, ANOVA; *, P ≤ 0.05; ***, P ≤ 0.001; ****, P ≤ 0.0001). C, Killing of SupT1-NT (black) or SupT1-GD2+ (red) after 48-hour coculture with CAR-T cells coexpressing the library of dFab_CCRs at a 1:4 effector:target ratio. Data show mean percentage (± SD) of live cells compared with nontransduced (NT) control (n = 11, GD2CAR + dFab_CCR-IL7 n = 4, two-way ANOVA). D, Extended in vitro persistence of IL2, IL7, IL3, IL5, or GMCSF dFab_CCR coexpressing GD2-specific CAR T cells cultured in cytokine-free complete cell culture media. Live cells were counted weekly using DAPI/acridine orange (AO)I staining. Data show mean 106 live cells (± SD; n = 6). E, Killing of SupT1-GD2+ after 48-hour coculture with GD2 CAR-T cells recovered after 7 days of cytokine starvation at 1:2 and 1:4 effector:target ratio. Data shows mean percentage (± SD) of live cells compared with NT control (n = 4, two-way ANOVA; **, P ≤ 0.01; ****, P ≤ 0.0001). All data are presented as mean ± SEM.
Given that these preliminary data indicated that dFab_CCR-IL2 may have a functional benefit when coexpressed with a CAR, we proceeded to test 18 dFab_CCRs receptors, representing different cytokine receptor families, coexpressed with the same GD2-specfic CAR. Coexpression of dFab_CCR-IL2 or -GMCSF with the GD2-specific CAR resulted in the strongest antigen-independent proliferation, with a fold expansion between 5 and 7 (Fig. 3B). Looking at the CAR effector function, the expression of most of the CγC dFab_CCRs (IL2, IL7), CβC (GMCSF), and the proinflammatory cytokine receptors (IL1, IL18) preserved CAR T-cell cytotoxicity (Fig. 3C). The remaining dFab_CCRs library impacted GD2-specific CAR T-cell cytotoxicity with different magnitudes (Fig. 3C).
Most of the dFab_CCRs tested did not alter CAR-mediated IFNγ secretion; however, IL18 and IL12 dFab_CCRs increased IFNγ secretion in response to cognate antigen (Supplementary Fig. S9A). In addition, dFab_CCR-IL18 expression enhanced IL2 secretion (Supplementary Fig. S9B). Similar results with respect to proliferation, cytotoxicity, and cytokine secretion were obtained with the dFab_CCR IL2, IL7, GMCSF and IL18 coexpressed with a second-generation CD19 CAR (Supplementary Fig. S10A and S10B). In addition, dFab_CCR-IL18 demonstrated increased IFNγ and IL2 secretion when coexpressed with the second-generation CD19 CAR (Supplementary Fig. S10C and S10D). Finally, coexpression of dFab_CCRs with an irrelevant second-generation CAR did not mediate nonspecific tumor cell killing or autonomous cytokine secretion despite demonstrating the expected proliferation (Supplementary Fig. S11).
dFab_CCRs prolong CAR T-cell expansion over time
Constitutive cytokine signaling could cause uncontrolled T-cell proliferation. To address this, we challenged a selection of dFab_CCRs to continuous antigen and cytokine starvation. Nontransduced T cells died within the first week in culture, whereas CAR alone sustained T cells for two weeks (Fig. 3D). All the dFab_CCRs CAR T cells expanded over the first 21 days (Fig. 3D). After day 21 the proliferative potency of the CβC dFab_CCRs and the CγC dFab_CCRs diverged. The CβC dFab_CCRs exhibited a gradual reduction in live cell number until day 49. Conversely, the CγC dFab_CCRs (IL2 and IL7) maintained a steady cell growth peaking at day 49, and a steady contraction until day 63 (Fig. 3D).
We then assessed whether cytokine withdrawal would affect CAR T-cell cytotoxicity. Seven day–starved GD2-specific CAR T cells were cocultured with GD2+ target cells (SupT1 GD2+). At both E:T ratios tested, the selected dFab_CCRs tested retained a strong CAR T-cell cytolytic capacity (Fig. 3E). Concordantly, dFab_CCR coexpression with the CAR sustained elevated IFNγ secretion (Supplementary Fig. S9C). Expression of the CAR alone failed to protect both cytotoxicity and IFNγ secretion.
These results demonstrated that the selection of dFab_CCRs showed a prolonged but finite proliferative potential.
dFab_CCRs influence CAR T-cell performance after serial antigen exposure
In vitro chronic antigen exposure is a relevant way to evaluate T-cell functional exhaustion (25). We therefore subjected dFab_CCR CAR T cells to weekly exposure of fresh antigen-positive tumor cells (Fig. 4A). In contrast to dFab_CCR-IL12 GD2-specific CAR T cells, The IL2, IL12, IL7, IL18, and GMCSF dFab_CCR CAR T cells expanded in cell number following 3 rounds of antigen stimulation, with the GMCSF dFab_CCR inducing the strongest expansion, to a greater degree compared to GD2-specific CAR alone (Fig. 4B). Chronic stimulation did not interfere with dFab_CCR signaling. Expression of IL2 or GMCSF dFab_CCRs resulted in 3- to 4-fold CAR T-cell expansion compared with CAR T alone, while the IL7 dFab_CCR conferred more modest proliferation (Fig. 4C). The remaining dFab_CCRs showed differential functionality, with the CβC CCRs mediating sustained expansion (Supplementary Fig. S12). Repeated stimulations reduced the cytolytic ability of GD2-specific CAR T cells (Fig. 4D). No improvement in cytolytic ability was observed with the co-expression of either IL7 or IL18 dFab_CCR with the CAR. In contrast, IL2 and IL12 dFab_CCR showed a trend of improved CAR T-cell cytotoxicity (Fig. 4D), with dFab_CCR-GMCSF showing significant enhancement. Coexpression of the dFab_CCRs with the GD2-specific CAR resulted in increased levels of IFNγ (Supplementary Fig. S13), with IL2, IL7, GMCSF, and IL18 dFab_CCRs doubling the amount of IFNγ secreted, whilst dFab_CCR-IL12 improved IFNγ secretion to approximately 4-fold compared with GD2-specific CAR alone.
Figure 4.

dFab_CCR coexpression sustains GD2-specific human CAR T-cell function after chronic antigen exposure. A, Chronic antigen stimulation experimental design. The first coculture was initiated with 1 × 106 GD2-specific CAR T cells together with 0.1 × 106 SKOV3 GD2+ cells for 7 days, in the absence of exogenous cytokines. For the second and third cocultures, T cells were harvested from the previous coculture and then replated in new culture medium with fresh tumor cells at the same 10:1 E:T ratio. The illustration was created with BioRender.com (RRID:SCR_018361). B, Cumulative expansion of selected GD2-specific CAR T cells from A after three rounds of coculture (n = 11, n = 4 GD2CAR + dFab_CCR-IL7, one-way ANOVA; ns P > 0.05; ***, P ≤ 0.001). C, Quantitated in vitro proliferation of GD2-specific CAR T cells recovered after three rounds of coculture (B) and cultured for 7 days in cytokine starvation condition. Proliferation expressed as fold expansion of RQR8+CD3+ GD2-specific CAR T cells (n = 11; GD2CAR + dFab_CCR-IL7 n = 4; one-way ANOVA; **, P ≤ 0.01; ****, P ≤ 0.0001). All data are presented as mean ± SEM. D, Killing of SupT1 nontransduced (NT; black) and GD2+ (red) after 48-hour coculture with GD2-specific CAR T cells recovered after three rounds of coculture (B) at 1:4 E:T ratio. Data show mean percentage (± SD) of live cells compared with NT control (n = 11; GD2CAR + dFab_CCR-IL7 n = 4, GD2CAR + dFab_CCR-IL7; two-way ANOVA; **, P ≤ 0.01). E, Exhaustion phenotype of CAR T cells after three rounds of chronic antigen exposure. Exhaustion evaluated by the expression of TIM3, LAG3, PD-1, or KLRG1 in either CD4+ (left) or CD8+ (right) T cells. Stacked bars show percentage of cells expressing 0, 1, 2, 3, or 4 markers per individual donors (n = 11; GD2CAR + dFab_CCR-IL7 n = 4). F, Memory phenotype of CAR T cells after three rounds of chronic antigen exposure. Memory phenotype evaluated by the expression of CD45RA and/or CCR7 in either CD4+ (left) or CD8+ (right) T cells. Memory phenotype defined as: naïve T cells (TN, CD45RA+, CCR7+), central memory (TCM) T cells (CD45RA−, CCR7+), effector memory (TEM) T cells (CD45RA−, CCR7), and terminally differentiated (TEMRA) T cells (CD45RA+, CCR7−). Stacked bars show percentage of cells in each population markers per individual donors (n = 11; GD2CAR + dFab_CCR-IL7 n = 4). All data are presented as mean ± SEM.
When coexpressed with a CD19-specific CAR, repeated stimulation resulted in similar expansion of T cells expressing both the CAR and the selected dFab_CCRs (Supplementary Fig. S14A). However, chronic stimulation did not impair CD19-specific CAR T-cell cytotoxicity, regardless of the presence or absence of the dFab_CCRs (Supplementary Fig. S14B). Here, the dFab_CCR-18 was able improve IFNγ secretion 3-fold compared with CD19 CAR alone (Supplementary Fig. S14C), while dFab_CCR-IL2 improved IL2 production (Supplementary Fig. S14D)
dFab_CCR coexpression maintains a less differentiated and a nonexhausted CAR T-cell phenotype
We next sought to determine the effect of each of the dFab_CCRs on CAR T-cell memory differentiation and exhaustion (Fig. 4E and F; Supplementary Fig. S15). After three rounds of antigen exposure, coexpression of the GD2-specific CAR with the IL2, IL7, GMCSF, or IL12 dFab_CCRs showed an increased percentage of cells expressing multiple exhaustion markers compared with the CAR alone (Fig. 4E), with a more prominent effect in the CD8+ T-cell population than the CD4+ T-cell population.
The IL18 dFab_CCR, despite enhancing IFNγ and IL2 secretion, maintained a similar exhaustion marker expression profile as the CAR alone (Fig. 4E). The majority of the dFab_CCRs expanded the central memory population in both the CD4+ and CD8+ T-cell compartments (Supplementary Fig. S15B). Moreover, GD2-specific CAR T cells coexpressing the IL2, IL7, GMCSF, or IL18 dFab_CCRs demonstrated a less differentiated phenotype (Fig. 4F). Others, (e.g., the IL9, IL21, and IL12 dFab_CCRs) increased the percentage of terminally differentiated effector cells and effector memory population, with a more pronounced effect in the CD8+ T-cell compartment (Supplementary Fig. S15B). Similarly, coexpression of selected dFab_CCRs with the CD19-specific CAR promoted lower exhaustion marker expression and mediated a less differentiated phenotype despite the enhanced functionality (Supplementary Fig. S16A and S16B).
dFab_CCR-IL18 improves human CAR T-cell persistence in a neuroblastoma xenograft model
Only a small panel of dFab CCRs can be easily screened in parallel in vivo due to the low-throughput characteristic of standard models. To assess the persistence of the dFab_CCRs in vivo, we employed a pooled DNA library of barcoded GD2-specific CAR dFab_CCRs constructs, which allowed assessment of the proliferative capacity of each individual CCR (Supplementary Fig. S17). We inoculated T cells expressing the library into NSG mice engrafted with GD2-positive CHLA255 tumors (Fig. 5A and B). Flow cytometric analysis showed higher CAR T-cell engraftment in tumor-bearing mice (Fig. 5C and E), compared to tumor-free mice (Fig. 5D and F). The analysis of barcode frequencies from both tumor-free and tumor-bearing NSG mice showed expansion of GD2-specific CAR T cells coexpressing the IL2, IL4, and IL18 dFab_CCRs compared with their relative abundance at the day of injection (Fig. 5E and F). The greatest degree of expansion was shown by those coexpressing the dFab_CCR-IL18, which represented 40% of the total CAR T-cell population recovered (Fig. 5F) in vivo.
Figure 5.

The pooled DNA-barcoded dFab_CCR library identifies prosurvival dFab_CCRs in vivo. A, Experimental set-up the xenograph in vivo model of the barcoded GD2CAR coexpressing dFab_CCRs pooled library. The illustration was created with BioRender.com (RRID:SCR_018361). B, NOD.Cg-Prkdc scid Il2rg tm1Wjl /SzJ (NSG) mice were either engrafted with the GD2-expressing CHLA neuroblastoma cell line or left tumor free. The illustration was created with BioRender.com (RRID:SCR_018361). C and D, Flow cytometry analysis of barcoded human CAR T cells pooled population, 14 days after injection. Graphs represent the percentage of CD45+CD3+ human T cells from CHLA255 neuroblastoma xenograft (C) or PBS control (D) NSG mice. All data are presented as mean ± SEM. E and F, Quantitated representation of individual barcoded frequency within the pooled barcode population 14 days postinfusion in CHLA255 neuroblastoma xenograft (E) or PBS control (F) NSG mice (red: bone marrow; green: spleen; purple: liver; n = 5 mice per group, one-way ANOVA; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001). All data are presented as mean ± SEM.
dFab_CCR-IL18 and GMCSF coexpression promote a distinctive T-cell effector profile
To understand better the in vivo effects of either the dFab_CCR-IL18 or dFab_CCR-GMCSF, transcriptomic, cytokine array and metabolomic analysis were performed on a selection of CAR T cells coexpressing dFab_CCRs. First, NanoString was utilized in resting conditions or following GD2-specific CAR activation (Supplementary Fig. S18A). PCA indicated that the presence of the dFab_CCR-IL18 promoted a distinctive transcriptomic profile following GD2-specific CAR activation (Fig. 6A), clustering distinctly from CAR alone or the other dFab_CCRs tested (IL2, IL7, GMCSF). IL2 and IL7 dFab_CCRs colocalized in both activated and nonactivated CAR T cells (Fig. 6A). Finally, dFab_CCR-GMCSF segregated more closely with the IL7 and IL2 dFab_CCR, although retained a degree of separation (Fig. 6A).
Figure 6.

IL18 and GMCSF dFab_CCRs deliver functionally and transcriptomically different output to GD2-specific human CAR T cells. A, PCA analysis of each individual dFab_CCR transcriptome in activated CAR T cells (left) or nonactivated (resting) CAR T cells (right). B, Volcano plot representing the comparison of dFab_CCR-IL18 CAR T cells against either dFab_CCR-IL7, dFab_CCR-GMCSF, or CAR alone in activated CAR T cells. C, Volcano plot representing the comparison of dFab_CCR-IL18 CAR T cells against either dFab_CCR-IL7 or dFab_CCR-GMCSF or CAR alone in nonactivated CAR T cells. D and E, The oxygen consumption rates (OCR) and the individual metabolic parameter were analyzed either after 24 hours of antigen starvation (nonactivated CAR T cells, D) or after 24 hours of activation (activated CAR T cells, F; n = 5, one-way ANOVA; ***, P ≤ 0.001). All data are presented as mean ± SEM.
Differential gene expression analysis showed that dFab_CCR-IL18 coexpression with the GD2-specific CAR uniquely influenced the expression of interleukin- and chemokine-related genes such as CXCL9 and CXCL10, cytokines such as the Th2 cytokine IL5, and the IL17 and IL1 genes, suggesting a more profound proinflammatory state (Fig. 6B: activated T cells, 6C: resting T cells). In accordance with the PCA data, differential gene expression confirmed the overlapping similarities of IL7 and IL2 dFab_CCRs (Supplementary Fig. S18B activated T cells, S17C resting T cells), while dFab_CCR-GMCSF coexpression induced more pronounced differences in genes expression (Supplementary Fig. S18B and S18C).
We then tested the secretion of Th1 and Th2 cytokines and chemokines. When coexpressed with either the GD2-specific or CD19-specific CAR, dFab_CCR-IL18 CAR T cells demonstrated a higher trend for chemokine secretion compared to CAR alone, despite not reaching statistical significance (Supplementary Fig. S19). Concurring with findings from Lange and colleagues (26), dFab_CCR-IL18–expressing CAR T cells demonstrated increased levels of Th2 cytokines IL5 and IL13, particularly when coexpressed with a CD19-specific CAR (Supplementary Fig. S19A and S19B). However, dFab_CCR-GMCSF coexpressing CAR T cells exhibited a reduction in the secretion of both chemokines and cytokines (Supplementary Fig. S19).
Finally, regardless of the CAR, dFab_CCR-IL18 influenced the CAR T-cell metabolic profile with higher respiratory capacity mainly in resting conditions (Fig. 6D: resting T cells, 6E: activated T cells). IL18 and IL2 dFab_CCRs drove a greater maximal respiratory capacity and ATP production in nonactivated T cells (Supplementary Fig. S20). Furthermore, dFab_CCR-IL18 induced higher basal respiration in both resting and activated conditions (Supplementary Fig. S20). Despite not reaching statistical significance, dFab_CCR-IL18 showed higher maximal respiratory capacity and ATP production compared with the other dFab_CCRs in activated CAR T cells (Supplementary Fig. S20).
Overall, these data indicated that dFab_CCR-IL18 created a distinct profile characterized by proinflammatory transcriptomic and metabolic state representative of a less differentiated T-cell population (27).
GMCSFmu and IL18mu dFab_CCR coexpression enhances in vivo murine GD2-specific CAR T-cell antitumor activity
To test the efficacy of selected dFab_CCRs on human CAR T-cell products in vivo, we tested dFab_CCRs/CD19-specific CAR T cells in a xenograft model of B-ALL (Supplementary Fig. S21A). Coexpression of either dFab_CCR-GMCSF or dFab_CCR-IL7 induced peripheral expansion of the CD19-specific CAR T cells (Supplementary Fig. S21B) and complete tumor control by day 14 (Supplementary Figs. S21C and S22A). However, early onset of graft-versus-host disease (GVHD) was observed, hampering the longevity of the experiment. In contrast, dFab_CCR-18 promoted comparable antitumor activity, with complete tumor control until the end of the model (day 24), with no GVHD observed. Moreover, none of the dFab_CCRs coexpressed with the irrelevant CAR promoted antitumor activity or GVHD toxicity (Supplementary Fig. S21C and S22B). Blood serum inflammatory cytokines were also quantified (Supplementary Fig. S21D).
We then designed murine equivalents of the CCRs and confirmed function in murine T cells (Supplementary Fig. S23) prior to assessing function in a syngeneic, immunocompetent CT26 colon carcinoma in vivo model (Supplementary Fig. S24A). In vitro, GD2-specific CAR T cells alone were unable to mediate tumor rejection (Fig. 7A; Supplementary Fig. S24B), resulting in no mice surviving past day 24 (Fig. 7B). In contrast, coexpression of the dFab_CCR_IL18mu with the CAR completely controlled tumor growth in 3 of 10 mice, up to day 50, and delayed tumor progression in a further 3 mice (Fig. 7A; Supplementary Fig. S24B), with 50% of the mice alive at day 50 post CAR T-cell injection (Fig. 7B). Similarly, dFab_CCR-GMCSFmu promoted a delay in tumor growth in 4 of 8 mice, unpalpable tumor in 1 of 8 mice by day 31 (Fig. 7A; Supplementary Fig. S24B), and improved mice survival by more than 15 days compared with the CAR alone (Fig. 7B). The enhanced tumor control displayed by dFab_CCR-IL18mu and dFab_CCR-GMCSFmu was accompanied by increased GD2-specific CAR T-cell engraftment, with a higher peak at day 10 after CAR T-cell infusion. In addition, both dFab_CCRs sustained prolonged peripheral persistence of the transduced CAR T cells over time (Fig. 7C; Supplementary Fig. S24C). The use of dFab_CCR-IL18mu was accompanied by weight loss in treated mice, resulting in the culling of 2 of 9 mice by day 10 posttreatment; however, the remaining mice recovered weight to baseline (Supplementary Fig. S24D).
Figure 7.

IL18 and GMCSF dFab_CCRs enhances adoptive GD2-specific mouse CAR T-cell immunotherapy against colon carcinoma immunocompetent animal models. A, Tumor volume quantification over time (n = 8). B, Kaplan–Meier survival curve (*, P ≤ 0.05, calculated via log-rank or Gehan–Breslow–Wilcoxon tests and Bonferroni-corrected α value). C, GD2 CAR T cells peripheral engraftment quantitated via flow cytometry by the expression of murine CD45, murine CD3 and THY1.1 (n = 8, day 10 one-way ANOVA; **, P ≤ 0.01; ****, P ≤ 0.0001). D, Day10 GD2-specific CAR T cells tumor trafficking measured as number of THY1.1+ CAR T cells every 1 × 106 CD3+CD45+CD11b− lymphoid cells (n-8). E, Memory phenotype of murine THY1.1 CAR T cells in the tumor. Memory phenotype evaluated by the expression of CD62 L and CD44. Naïve T cells (TN, CD44−, CD62L+), effector memory (TEM) T cells (CD44+, CD62L+), central memory (TCM) T cells (CD44+, CD62L+) and terminally differentiated (TEMRA) T cells CD44−, CD62L−). Stacked bars show percentage of cells in each population markers per individual donor (n = 8). F, Myeloid cells quantification in spleen, bone marrow, and tumor defines by the expression of total CD11b (myeloid), CD11b/CD11c (dendritic cells), CD11b/F4–80 (macrophages), every 1 × 106 CD45+CD11b− cells (n = 8). All data are presented as mean ± SEM.
Enhanced functionality of GD2-specific CAR T cells expressing dFab_CCRs was also observed in a highly aggressive B16F10 melanoma subcutaneous model (Supplementary Fig. S25A). T cells expressing the CAR alone failed to control tumor growth with a comparable overall survival with nontransduced T cells (Supplementary Fig. S25B). Coexpression with the -IL7mu, -IL18mu, and -GMCSFmu dFab_CCRs improved antitumor efficacy with prolonged survival up to days 31, 35, and 39, respectively (Supplementary Fig. S25C), without apparent toxicity manifesting as weight loss (Supplementary Fig. S25D). In addition, dFab_CCR-GMCSFmu–treated mice showed significantly greater T-cell engraftment (Supplementary Fig. S25E).
dFab_CCR-IL18 influences the recruitment of immune cells in the tumor microenvironment
To investigate the mechanisms underlying the improved antitumor efficacy in the syngeneic colon carcinoma and melanoma models, we evaluated the effect of dFab_CCR expression on the trafficking and phenotype of the murine GD2-specific CAR T cells and their interaction with the host immune milieu (Supplementary Fig. S26A). dFab_CCR-IL18 CAR T cell–treated mice demonstrated lower tumor volume (Supplementary Fig. S26B) and improved trafficking within the tumor microenvironment (Fig. 7D), maintaining a less differentiated phenotype with a higher proportion of naïve and central memory T cells (Fig. 7E), and a higher proportion of central memory T cells in both bone marrow and spleen (Supplementary Fig. S26D). In addition, dFab_CCR-18 expression induced prominent recruitment of myeloid cells, especially DCs and macrophage in both bone marrow and spleen (Fig. 7F), while within the tumor, IL7 and GMCSF dFab_CCRs promoted higher myeloid-cell recruitment (Supplementary Fig. S26C). Finally, dFab_CCR-18 GD2–specific CAR T cells induced significantly higher IFNγ levels and displayed a trend to increased levels of CXCL10 chemokine, TNFα, and IL6, albeit without reaching statistical significance (Supplementary Fig. S26E).
Altogether, these data indicated that IL18 dFab_CCR promoted the strongest interaction with the host immune system, likely caused by its complex chemokine and cytokine secretion.
Discussion
In this study, we describe a new format of constitutively active cytokine receptor (dFab_CCR) that is broadly generalizable across different cytokine receptors families. We tested the utility of coexpressing dFab_CCRs in CAR T cells and demonstrated an enhancement of CAR T-cell activity. We found that IL18 and GMCSF dFab_CCRs conferred particular and differing functional benefits to CAR T cells.
Given that cytokine receptors usually require endodomain heterodimerization to signal, we explored using the Ig CH1/Light chain constant region interaction to induce cytokine receptor heterodimerization. CH1 and kappa or lambda light chain constant (KLC or LLC) form a densely packed and stable heterodimeric interface (28, 29) and have been previously used for heterodimeric assembly of recombinant proteins (30, 31). We hypothesized that we could induce constitutive cytokine receptor signaling by replacing cytokine receptor ectodomains with CH1 and the KLC region. Initial exploration of this strategy utilizing the IL2 receptor endodomains in primary human T cells demonstrated that an IL2 signal could be recapitulated by functional and transcriptional analysis.
Adoptive immunotherapy has frequently been combined with systemic administration of cytokines to enhance T-cell activity; however, this often causes systemic toxicity (6). Strategies have been developed to engineer therapeutic immune cells to avoid systemic administration of cytokines. The simplest strategy is to engineer T cells to constitutively secrete a particular cytokine (7, 8). This approach still risks systemic cytokine toxicity (32) and is susceptible to downregulation of the cognate cytokine receptor (33). Further strategies were devised that overcame these limitations. Early solutions included chimeric cytokine receptors that could be activated by pharmaceuticals such erythropoietin (34) or small molecules such as rapamycin (9). Alternative approaches were also developed to engineer constitutively active receptors, such as IL7 by cysteine and/or proline insertions in the IL7Rα transmembrane (CR7; ref. 13). Despite being efficacious, this strategy is not applicable to cytokine receptors that function as homodimers, such as IL2 β-chain (20, 35) or IL12Rβ2 homodimers (36).
Alternative methods include a membrane-tethered IL15 receptor generated by fusing the IL15 to the IL15Rα by a flexible linker (12). However, this approach might result in unwanted activation of neighboring cells. Furthermore, such formats could be restricted by γ-chain availability due to their interaction with other native common γ-chain receptor partners (2).
We add to this literature with the dFab_CCR approach. Following initial proof-of-concept with IL2, truncations of either the IL2Rβ chain or common γ-chain endodomains were demonstrated to potentially enable “tuning” of the signal intensity and the degree of T-cell proliferation.
Given the similar architecture of the entire class I cytokine receptor family, the versatility of the dFab_CCR was explored. Six cytokine receptor families comprising of a total of 18 different individual dFab_CCRs were investigated spanning from T cell– to myeloid cell–specific receptors. The dFab_CCR approach resulted in activation of downstream signaling molecules from all the 18 cytokine receptors tested. The IL12 family (IL12, IL23) and IL18 dFab_CCRs were functionally active resulting in high basal level of IFNγ secretion, possibly due to their higher expression density. Interestingly, despite being myeloid cell–specific, common β-chain dFab_CCRs (e.g., GMCSF) promoted strong proliferation in T cells through the activation of both STAT3 and STAT5.
Having established the versatility of dFab_CCR, the utility in the setting of adoptive immunotherapy was then investigated. Given the success of CAR T cells in lymphoid malignancies (37–39) and early promise in solid cancers (14), dFab_CCRs were tested in combination with both a GD2-specific and a CD19-specific second generation 41bbζ CARs. We explored whether the 18 individual dFab_CCRs conferred a differential functional advantage to CAR T cells. Using individualized in vitro dFab_CCR testing as well as a pooled barcoded in vitro and in vivo approach, the majority of dFab_CCRs maintained CAR T-cell antigen-specific cytotoxicity and cytokine secretion. This remained true when a selection of dFab_CCRs was coexpressed with an irrelevant CAR.
Under conditions of chronic antigen stimulation, IL2 and the common β-chain dFab_CCRs sustained CAR T-cell cytolytic activity upon serial rechallenge with fresh target cells, compared to the CAR alone. Importantly, IL2, IL7, and the common β-chain dFab_CCRs maintained enhanced CAR T-cell expansion in the absence of exogenous cytokines, before and after repeated antigen stimulation, regardless of the coexpressed CAR partner. In addition, these dFab_CCR–expressing CAR T cells displayed a higher proportion of the central memory T-cell population suggesting a less differentiated state. When coexpressed with the GD2-specific CAR, dFab_CCRs based upon immunostimulatory cytokines such as IL23 and IL27 (24) reduced cytolytic activity, cytokine secretion and hampered proliferation upon chronic antigen stimulation.
These experimental approaches highlighted distinct effects of dFab_CCR-IL18 on CAR T-cell function. IL18 is a proinflammatory cytokine belonging to the IL1 superfamily. In T cells, IL18 facilitates CD4+ T helper 1 (Th1) polarization in conjunction with IL12 but Th2 polarization together with IL2 (40). In preclinical models, intratumoral production of IL18 showed preferable safety profile compared with IL12 (41–43). The IL18 receptor signals via MYD88 (44), which plays a critical role in innate immunity (45). MYD88 has been used in adoptive cell therapy as an alternative CAR costimulatory domain, showing improved CAR T-cell effector function in the setting of chronic antigen stimulation (46–48). Furthermore, several approaches have been characterized by delivering an engineered IL18 cytokine or an IL18 receptor signal within the tumor microenvironment, showing increased chemokine and Th1 cytokine secretion (26, 49, 50).
In this study, dFab_CCR-IL18 increased CAR T-cell persistence and antitumor responses in two syngeneic animal models, enhanced survival in an NSG neuroblastoma model, and enhanced antitumor control in a CD19 NSG model. Furthermore, in the syngeneic colon carcinoma model, a less differentiated phenotype of dFab_CCR-IL18 expressing CAR T cells was observed accompanied by an increased recruitment of the host lymphoid and myeloid cells.
Furthermore, transcriptomic and metabolomic in vitro analysis revealed that dFab_CCR-IL18 promoted a pronounced proinflammatory state, and a higher aerobic metabolism characteristic of less differentiated CAR T cells (27). Recently, Gottschalk and colleagues presented an elegant receptor switch that redirects GMCSF secreted upon CAR T-cell activation, to an IL18 signal in CAR T cells (26). Concordantly, they also reported an increase in Th2 cytokine secretion together with improved Th1 effector function and better in vivo tumor control (26).
This toolkit of a range of constitutively active CCRs displayed various ways could enhance CAR T-cell therapy: (i) improvement of proliferation; (ii) maintenance of effector function; (iii) enhancement of cytokine secretion influencing the host immune system; (iv) improved aerobic metabolism; and (v) acceptable safety profile. Gene therapies based upon the expression of the common γ-chain have been associated with malignant transformation in the context of hematopoietic stem cell SCID therapy (51). However, such toxicities have been related to viral construct design (52, 53), as subsequent clinical studies using self-inactivating lentivirus reported no malignant transformation (54). Here, we demonstrate that the use of common γ-chain dFab_CCRs in mature T cells showed enhanced but time-limited cell expansion, both in vitro and in vivo. Accordingly, expression of CR7 in CAR T cells exhibited time-limited cell proliferation (13). In addition, a single incubation with FDA-approved JAK inhibitor ruxolitinib inhibited dFab_CCR-IL2 signaling.
dFab_CCR is a new architecture for constitutive cytokine receptors. The dFab_CCR architecture is versatile and can be used to induce constitutive signaling from a range of cytokine receptors. Certain dFab_CCRs, for instance, those derived from GM-CSF or IL18 receptors, may convey advantages for enhanced CAR T-cell functions. Future clinical exploration may determine the utility of this and other forms of cytokine signaling, particularly in the setting of adoptive immunotherapy for solid cancers.
Supplementary Material
Supplementary Figure S1-S26
Supplementary Table 1: Key Resources
Supplementary Table S2. Top 50 up-regulated GSEA pathways.
Acknowledgments
All work was supported by Autolus Therapeutics.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Immunology Research Online (http://cancerimmunolres.aacrjournals.org/).
Authors' Disclosures
M. Righi reports a patent for GB20190011187 issued and is an employee and shareholder of Autolus Ltd. I. Gannon is an employee and shareholder of Autolus Ltd. E. Kokalaki is an Autolus employee and owns Autolus shares. J. Sillibourne reports a patent for GB20190011187 issued. S. Cordoba reports a patent for WO 2021/023987 A1 pending. S. Thomas reports a patent #GB20190011187 issued. M. Pule reports other support from Autolus Therapeutics during the conduct of the study, as well as a patent for GB20190011187 pending to Autolus Therapeutics. No disclosures were reported by the other authors.
Authors' Contributions
M. Righi: Conceptualization, data curation, formal analysis, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing. I. Gannon: Investigation, seahorse metabolomic analysis. M. Robson: Investigation, animal model generation, animal model follow-up and takedown. S. Srivastava: Investigation, animal model take down and processing. E. Kokalaki: Investigation. T. Grothier: Investigation, animal model take down and processing, immunoblot. F. Nannini: Data curation, investigation, rna sequencing analysis, dna barcode library analysis. C. Allen: Investigation. Y.V. Bai: Data curation, formal analysis. J. Sillibourne: Validation, investigation, plasmid dna design and generation. S. Cordoba: Conceptualization, data curation, supervision, validation, visualization, writing–original draft, project administration. S. Thomas: Data curation, validation, visualization, writing–review and editing. M. Pule: Conceptualization, data curation, supervision, funding acquisition, writing–review and editing.
References
- 1. Kershaw MH, Westwood JA, Darcy PK. Gene-engineered T cells for cancer therapy. Nat Rev Cancer 2013;13:525–41. [DOI] [PubMed] [Google Scholar]
- 2. Dwyer CJ, Knochelmann HM, Smith AS, Wyatt MM, Rangel Rivera GO, Arhontoulis DC, et al. Fueling cancer immunotherapy with common gamma chain cytokines. Front Immunol 2019;10:263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008;27:5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol 2020;17:147–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, et al. Effects of single-dose interleukin-12 exposure on interleukin-12–associated toxicity and interferon-γ production. Blood 1997;90:2541–8. [PubMed] [Google Scholar]
- 6. Rosenberg SA, Yannelli JR, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, et al. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J Natl Cancer Inst 1994;86:1159–66. [DOI] [PubMed] [Google Scholar]
- 7. Chen Y, Sun C, Landoni E, Metelitsa L, Dotti G, Savoldo B. Eradication of neuroblastoma by T cells redirected with an optimized GD2-specific chimeric antigen receptor and interleukin-15. Clin Cancer Res 2019;25:2915–24. [DOI] [PubMed] [Google Scholar]
- 8. Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia 2010;24:1160–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ogawa K, Kawahara M, Nagamune T. Construction of unnatural heterodimeric receptors based on IL-2 and IL-6 receptor subunits. Biotechnol Progr 2013;29:1512–8. [DOI] [PubMed] [Google Scholar]
- 10. Wang Y, Jiang H, Luo H, Sun Y, Shi B, Sun R, et al. An IL-4/21 inverted cytokine receptor improving CAR-T cell potency in immunosuppressive solid-tumor microenvironment. Front Immunol 2019;10:1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wilkie S, Burbridge SE, Chiapero-Stanke L, Pereira ACP, Cleary S, van der Stegen SJC, et al. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J Biol Chem 2010;285:25538–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hurton LV, Singh H, Najjar AM, Switzer KC, Mi T, Maiti S, et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc Natl Acad Sci U S A 2016;113:E7788–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shum T, Omer B, Tashiro H, Kruse RL, Wagner DL, Parikh K, et al. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Discov 2017;7:1238–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Straathof K, Flutter B, Wallace R, Jain N, Loka T, Depani S, et al. Antitumor activity without on-target off-tumor toxicity of GD2–chimeric antigen receptor T cells in patients with neuroblastoma. Sci Transl Med 2020;12:eabd6169. [DOI] [PubMed] [Google Scholar]
- 15. Nicholson IC, Lenton KA, Little DJ, Decorso T, Lee FT, Scott AM, et al. Construction and characterisation of a functional CD19 specific single chain Fv fragment for immunotherapy of B lineage leukaemia and lymphoma. Mol Immunol 1997;34:1157–65. [DOI] [PubMed] [Google Scholar]
- 16. Philip B, Kokalaki E, Mekkaoui L, Thomas S, Straathof K, Flutter B, et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood 2014;124:1277–87. [DOI] [PubMed] [Google Scholar]
- 17. Thomas S, Straathof K, Himoudi N, Anderson J, Pule M. An optimized GD2-targeting retroviral cassette for more potent and safer cellular therapy of neuroblastoma and other cancers. PLoS One 2016;11:e0152196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hu J, Ge H, Newman M, Liu K. OSA: a fast and accurate alignment tool for RNA-Seq. Bioinformatics 2012;28:1933–4. [DOI] [PubMed] [Google Scholar]
- 19. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nelson BH, Lord JD, Greenberg PD. Cytoplasmic domains of the interleukin-2 receptor beta and gamma chains mediate the signal for T-cell proliferation. Nature 1994;369:333–6. [DOI] [PubMed] [Google Scholar]
- 21. Usacheva A, Sandoval R, Domanski P, Kotenko SV, Nelms K, Goldsmith MA, et al. Contribution of the Box 1 and Box 2 motifs of cytokine receptors to Jak1 association and activation. J Biol Chem 2002;277:48220–6. [DOI] [PubMed] [Google Scholar]
- 22. Kenderian SS, Ruella M, Shestova O, Kim MY, Klichinsky M, Chen F, et al. Ruxolitinib prevents cytokine release syndrome after CART cell therapy without impairing the anti-tumor effect in a xenograft model. Blood 2016;128:652. [Google Scholar]
- 23. Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev 2018;281:8–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vignali DAA, Kuchroo VK. IL-12 family cytokines: immunological playmakers. Nat Immunol 2012;13:722–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bucks CM, Norton JA, Boesteanu AC, Mueller YM, Katsikis PD. Chronic antigen stimulation alone is sufficient to drive CD8+ T cell exhaustion. J Immunol 2009;182:6697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lange S, Sand LG, Bell M, Patil SL, Langfitt D, Gottschalk S. A chimeric GM-CSF/IL18 receptor to sustain CAR T-cell function. Cancer Discov 2021;11:1661–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Geltink RI Klein, Kyle RL, Pearce EL. Unraveling the complex interplay between T cell metabolism and function. Annu Rev Immunol 2018;36:461–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Feige MJ, Groscurth S, Marcinowski M, Shimizu Y, Kessler H, Hendershot LM, et al. An unfolded CH1 domain controls the assembly and secretion of IgG antibodies. Mol Cell 2009;34:569–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Röthlisberger D, Honegger A, Plückthun A. Domain interactions in the Fab fragment: a comparative evaluation of the single-chain Fv and Fab format engineered with variable domains of different stability. J Mol Biol 2005;347:773–89. [DOI] [PubMed] [Google Scholar]
- 30. Hust M, Jostock T, Menzel C, Voedisch B, Mohr A, Brenneis M, et al. Single chain Fab (scFab) fragment. BMC Biotech 2007;7:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Klein C, Schaefer W, Regula JT. The use of CrossMAb technology for the generation of bi- and multispecific antibodies. MAbs 2016;8:1010–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang L, Morgan RA, Beane JD, Zheng Z, Dudley ME, Kassim SH, et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin Cancer Res 2015;21:2278–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cendrowski J, Mamińska A, Miaczynska M. Endocytic regulation of cytokine receptor signaling. Cytokine Growth Factor Rev 2016;32:63–73. [DOI] [PubMed] [Google Scholar]
- 34. Moraga I, Wernig G, Wilmes S, Gryshkova V, Richter CP, Hong W-J, et al. Tuning cytokine receptor signaling by re-orienting dimer geometry with surrogate ligands. Cell 2015;160:1196–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nakamura Y, Russell SM, Mess SA, Friedmann M, Erdos M, Francois C, et al. Heterodimerization of the IL-2 receptor beta- and gamma-chain cytoplasmic domains is required for signalling. Nature 1994;369:330–3. [DOI] [PubMed] [Google Scholar]
- 36. Floss DM, Schönberg M, Franke M, Horstmeier FC, Engelowski E, Schneider A, et al. IL-6/IL-12 cytokine receptor shuffling of extra- and intracellular domains reveals canonical STAT activation via synthetic IL-35 and IL-39 signaling. Sci Rep 2017;7:15172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol 2019;20:31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med 2018;378:439–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med 2019;380:1726–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nakanishi K. Unique action of interleukin-18 on T cells and other immune cells. Front Immunol 2018;9:763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kunert A, Chmielewski M, Wijers R, Berrevoets C, Abken H, Debets R. Intra-tumoral production of IL18, but not IL12, by TCR-engineered T cells is non-toxic and counteracts immune evasion of solid tumors. OncoImmunology 2018;7:e1378842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chmielewski M, Abken H. CAR T cells releasing IL-18 convert to T-Bethigh FoxO1low effectors that exhibit augmented activity against advanced solid tumors. Cell Rep 2017;21:3205–19. [DOI] [PubMed] [Google Scholar]
- 43. Hu B, Ren J, Luo Y, Keith B, Young RM, Scholler J, et al. Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL-18. Cell Rep 2017;20:3025–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fields JK, Günther S, Sundberg EJ. Structural basis of IL-1 family cytokine signaling. Front Immunol 2019;10:1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Warner N, Núñez G. MyD88: a critical adaptor protein in innate immunity signal transduction. J Immunol 2013;190:3–4. [DOI] [PubMed] [Google Scholar]
- 46. Mata M, Gerken C, Nguyen P, Krenciute G, Spencer DM, Gottschalk S. Inducible activation of MyD88 and CD40 in CAR T cells results in controllable and potent antitumor activity in preclinical solid tumor models. Cancer Discov 2017;7:1306–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Prinzing B, Schreiner P, Bell M, Fan Y, Krenciute G, Gottschalk S. MyD88/CD40 signaling retains CAR T cells in a less differentiated state. JCI Insight 2020;5:136093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Glienke W, Dragon AC, Zimmermann K, Martyniszyn-Eiben A, Mertens M, Abken H, et al. GMP-compliant manufacturing of TRUCKs: CAR T Cells targeting GD2 and releasing inducible IL-18. Front Immunol 2022;13:839783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Blokon-Kogan D, Levi-Mann M, Malka-Levy L, Itzhaki O, Besser MJ, Shiftan Y, et al. Membrane anchored IL-18 linked to constitutively active TLR4 and CD40 improves human T cell antitumor capacities for adoptive cell therapy. J Immunother Cancer 2022;10:e001544. [Google Scholar]
- 50. Zhou T, Damsky W, Weizman O-E, McGeary MK, Hartmann KP, Rosen CE, et al. IL-18BP is a secreted immune checkpoint and barrier to IL-18 immunotherapy. Nature 2020;583:609–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, Gross F, Yvon E, Nusbaum P, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science 2000;288:669–72. [DOI] [PubMed] [Google Scholar]
- 52. Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003;302:415–9. [DOI] [PubMed] [Google Scholar]
- 53. Ruggero K, Al-Assar O, Chambers JS, Codrington R, Brend T, Rabbitts TH. LMO2 and IL2RG synergize in thymocytes to mimic the evolution of SCID-X1 gene therapy-associated T-cell leukaemia. Leukemia 2016;30:1959–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhou S, Mody D, DeRavin SS, Hauer J, Lu T, Ma Z, et al. A self-inactivating lentiviral vector for SCID-X1 gene therapy that does not activate LMO2 expression in human T cells. Blood 2010;116:900–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1-S26
Supplementary Table 1: Key Resources
Supplementary Table S2. Top 50 up-regulated GSEA pathways.
Data Availability Statement
The data generated in this study are available within the article and its supplementary data files or upon request from the corresponding author. The transcriptomic data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE227161 and GSE227312.