

Abstract
Tricyclic bridgehead carbon centers (TBCCs) are a synthetically challenging substructure found in many complex natural products. Here we review the syntheses of ten representative families of TBCC-containing isolates, with the goal of outlining the strategies and tactics used to install these centers, including a discussion of the evolution of the successful synthetic design. We provide a summary of common strategies to inform future synthetic endeavors.
1. Introduction
1.1. Significance of tricyclic bridgehead carbon centers (TBCCs)
The syntheses of isolates containing quaternary carbon centers are challenging and have been the subject of numerous reviews.1–10 Tricyclic bridgehead carbon centers (TBCCs hereafter) are quaternary carbon centers located at a bridgehead position and embedded within three fused rings. This additional skeletal complexity creates considerable challenges for synthesis. Beyond the realm of natural product chemistry, TBCCs have emerged in drug discovery campaigns as their three-dimensionality11 is aligned with a renewed focus on sp3-rich molecules in industry.12
Here we systematically review syntheses of natural product targets bearing TBCCs, with an emphasis on the evolution of the successful synthetic approach and the final pathway that provided access to these structures. We close with a summary of general strategies to construct TBCCs, which may be applicable to a broader array of natural products and drug-like molecules. We envision that this review will benefit the synthetic community by aggregating and organizing the literature around the successful strategies that have been developed to access TBCCs. This review is meant to present representative examples and is not intended to be comprehensive. The reader is directed to earlier reviews of quaternary carbon centers for further discussion and examples from the literature.1–10
1.2. Trends in design strategies for syntheses of TBCCs: syntheses of enmein diterpenoids as an example
Early approaches to TBCCs are characterized by iterative acyclic bond-forming events, followed by ring-closing reactions. Subsequently, there was a major shift to cycloaddition reactions, which allow stereocontrolled formation of one or more TBCCs with concomitant generation of one or more rings. Advances in synthetic methods over the past ten years have provided highly efficient, stereoselective methods, such as asymmetric catalysis, photochemical transformations, and skeletal rearrangement enabled by newly invented promoters or catalysts, that can be used to access TBCCs. The total syntheses of enmein (1) by the Fujita group in 197413 and the Dong group in 201814 serve as illustrative examples that bookend the ends of this synthetic spectrum.
(−)-Enmein (1) is an ent-kaurene diterpenoid with a highly oxidized pentacyclic system (Scheme 1). (−)-Enmein (1) was first isolated from I. japonica by Natsume and co-workers in 1958.15,16 (−)-Enmein (1) derives biosynthetically from C6–C7 bond cleavage of the ent-kaurene skeleton (Scheme 1).17 Its structure was elucidated by chemical degradation,16 X-ray analysis of its heavy atom derivative,15 and chemical conversion to the known natural product, (−)-kaurene.18 (−)-Enmein (1) contains two TBCCs, C6 and C7.
Scheme 1.
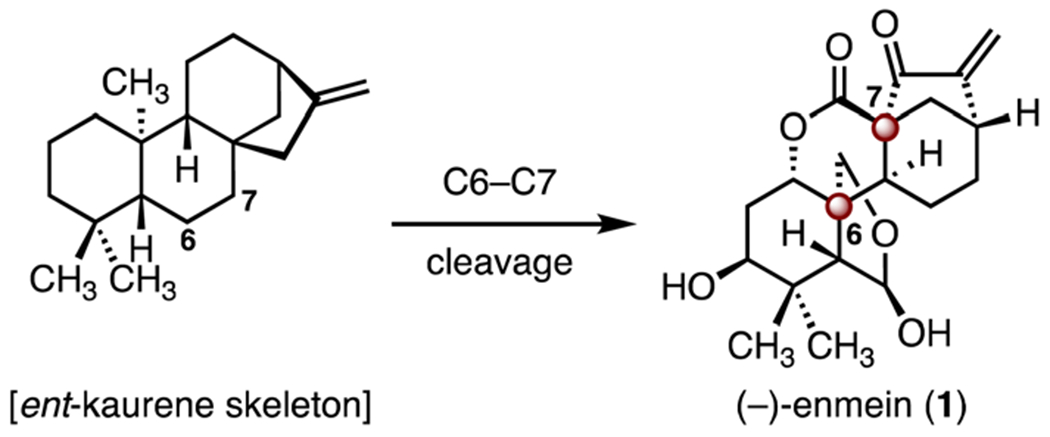
Structure of (−)-enmein (1) and its biosynthetic origin.
The Fujita group employed a relay strategy for the synthesis of (±)-enmein (1, Scheme 2).13,19 A Robinson annulation employing 5-chloro-pentane-3-one and the β-ketoester 2, promoted by p-toluenesulfonic acid, provided the tricycle (±)-3, which bears the C6 quaternary stereogenic center of the target (marked with a blue sphere; 59%).20 A six-step sequence then generated the ketal 4 (22% overall). Birch reduction (lithium, ammonia),222 followed by acid-catalyzed hydrolysis and hemiacetal formation (hydrochloric acid; p-toluenesulfonic acid, methanol) formed the cyclohexanone 5, with the C6 TBCC established (marked with a red sphere; 64%, three steps). A six-step sequence involving alkylation at the more-hindered α-position of the ketone 5 (C7) provided the aldehyde 6, which bears the C7 quaternary stereogenic center. An intramolecular aldol reaction (sodium methoxide) generated the cyclic hydroxy ketone 7, which bears the C7 TBCC (23%, seven steps). The hydroxy ketone 7 was elaborated to the alkene 9 through the relay intermediate 8 (nine steps, 1.6% overall). Oxidative cleavage of the alkene 9 (ozone), followed by oxidative lactonization (Jones’ reagent) and methylation (diazomethane), provided the lactone 10 (13%, three steps). A seven-step sequence was developed to transform the lactone 10 to the carboxylic acid 11 (10% overall). Lactonization (boron trifluoride diethyl etherate complex) formed the second lactone in 12, as a single diastereomer (72%). The total synthesis of (±)-enmein (1) was completed by a ten-step sequence (0.45% overall). In sum, the route provided access to the target in forty-seven steps and <0.0001% overall yield.
Scheme 2.
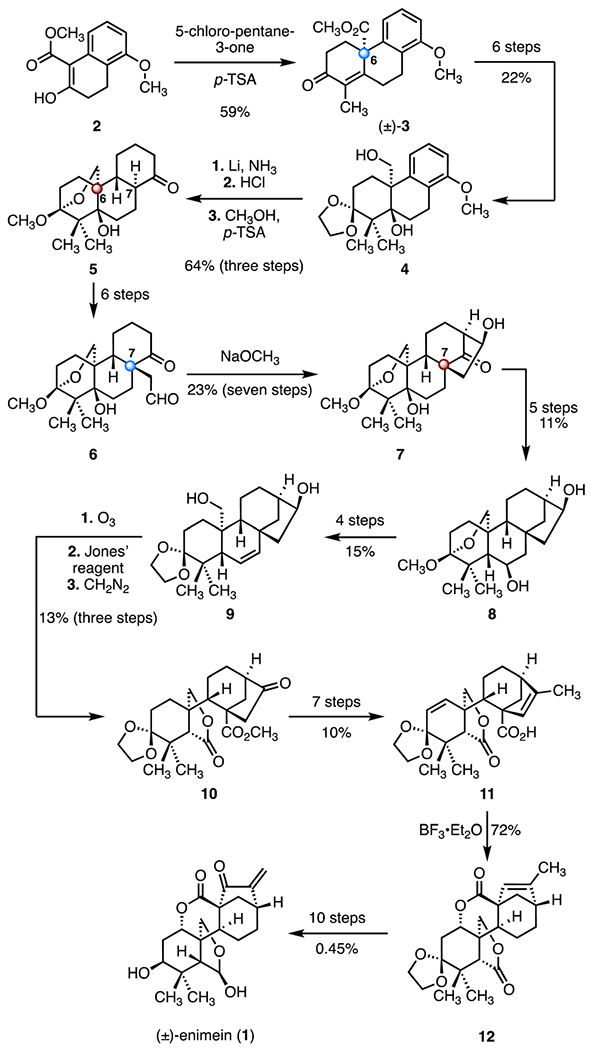
Synthesis of (±)-enmein (1) by Fujita and co-workers.13,19 p-TSA = p-toluenesulfonic acid.
Dong and co-workers developed a convergent approach to (−)-enmein (1) that featured a cycloaddition and a reductive alkenylation as key steps (Scheme 3).14 The synthesis began with a Diels–Alder cycloaddition between the chiral Danishefsky diene derivative 13 (two steps from (S)-1-phenylethanol, 80%) and 3-(3-methoxyphenyl)furan-2,5-dione21 (14; butylated hydroxytoluene, 100 °C; then, hydrochloric acid, 52%, 14% de, 97% ee), which afforded the tricyclic cyclohexanone (−)-15 bearing the C6 quaternary stereogenic center. The cyclohexanone (−)-15 was converted to the enone 16 by a five-step sequence (42% overall). A one-flask acylation–alkylation–lactonization cascade (lithium bis(trimethylsilyl)amide, Mander’s reagent; then, 17, sodium iodide; then, hydrochloric acid, 55%) generated the tricyclic lactone 18, which contains the C6 TBCC (>10 : 1 dr at C7). The alkylation conditions were extensively optimized to suppress competitive O-alkylation (not shown). Conjugate reduction of the enone 18 (L-selectride) produced the lithium enolate 19. An intramolecular enolate vinylation was achieved by treatment with tetrakis(triphenylphosphine) palladium(0) and cesium carbonate22 to furnish the ketone 20, which contains the D/E ring system of the target and the C7 TBCC (53%). A three-step sequence then formed the alcohol 21 (80% overall). Inversion of the C3 alcohol in 21, followed by allylic oxidation and methyl ether cleavage (29%, four steps), provided (−)-enmein (1). The target was obtained in seventeen steps with 1.2% overall yield.
Scheme 3.
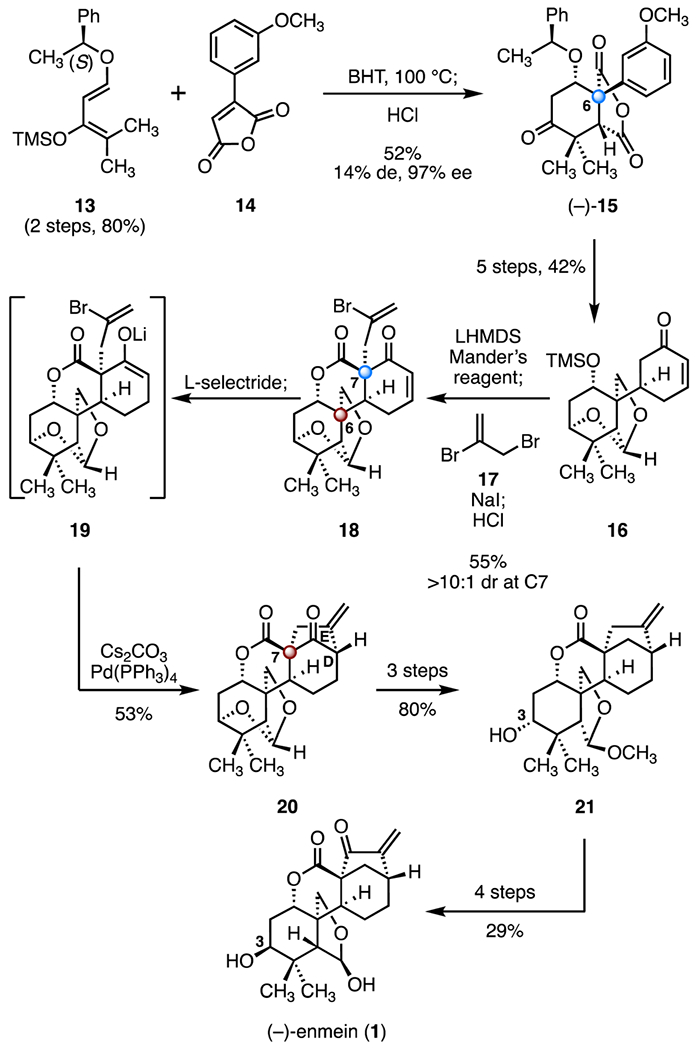
Synthesis of (−)-enmein (1) by Dong and co-workers.14 BHT = butylated hydroxytoluene, LHMDS = lithium bis(trimethylsilyl)amide.
These two syntheses of enmein (1) illustrate the dramatic increase in efficiency of synthetic approaches to targets containing TBCCs over the past fifty years. Replacement of one-bond forming cyclization events by two-bond forming cycloadditions greatly expedited construction of the polycyclic system. A fragment coupling strategy (13 + 14 → 15) increased convergence and avoided late-stage oxidation state manipulations. Palladium-mediated enolate vinylation methodology (18 → 20) facilitated construction of the carbon skeleton while minimizing lateral transformations. Additional syntheses of ent-kaurene and Isodon diterpenoids containing TBCCs have been reported; and we direct readers to more specialized recent reviews on this topic for additional illustrative examples.17,23,24
2. Case studies
2.1. Ingenol: TBCC embedded in a highly strained terpenoid ring system
Ingenanes are complex polycyclic diterpenoids derived from the tigliane skeleton (Scheme 4).25 Ingenanes possess a unique 5/7/7/3 tetracyclic system containing an in,out-bridgehead bicyclo [4.4.1]undecane core. The first known member of this family, (+)-ingenol (23), was isolated in 1968 from Euphorbia ingens by Hecker and co-workers.26 The structure of (+)-ingenol was elucidated in 1970 by X-ray crystallography.27 Many natural ingenol derivatives are known, and some possess antiviral and anticancer activities.28–30 The semisynthetic derivative ingenol mebutate (Picato®, 24) was approved for the treatment of actinic keratosis by the US FDA in 2013.31 From a structural perspective, (+)-ingenol (23) contains a C10 TBCC and two highly oxidized rings (A and B rings).32 The in,out-bicyclo[4.4.1]undecane scaffold is calculated to be 10.3 kcal mol−1 higher in energy than the out,out-diastereomer, due to torsional strain.33 Numerous research groups have published synthetic studies toward (+)-ingenol (23).34 Here we discuss syntheses of ingenol (23) by the Winkler,35 Tanino–Kuwajima,36 Wood,37 and Baran groups.38
Scheme 4.
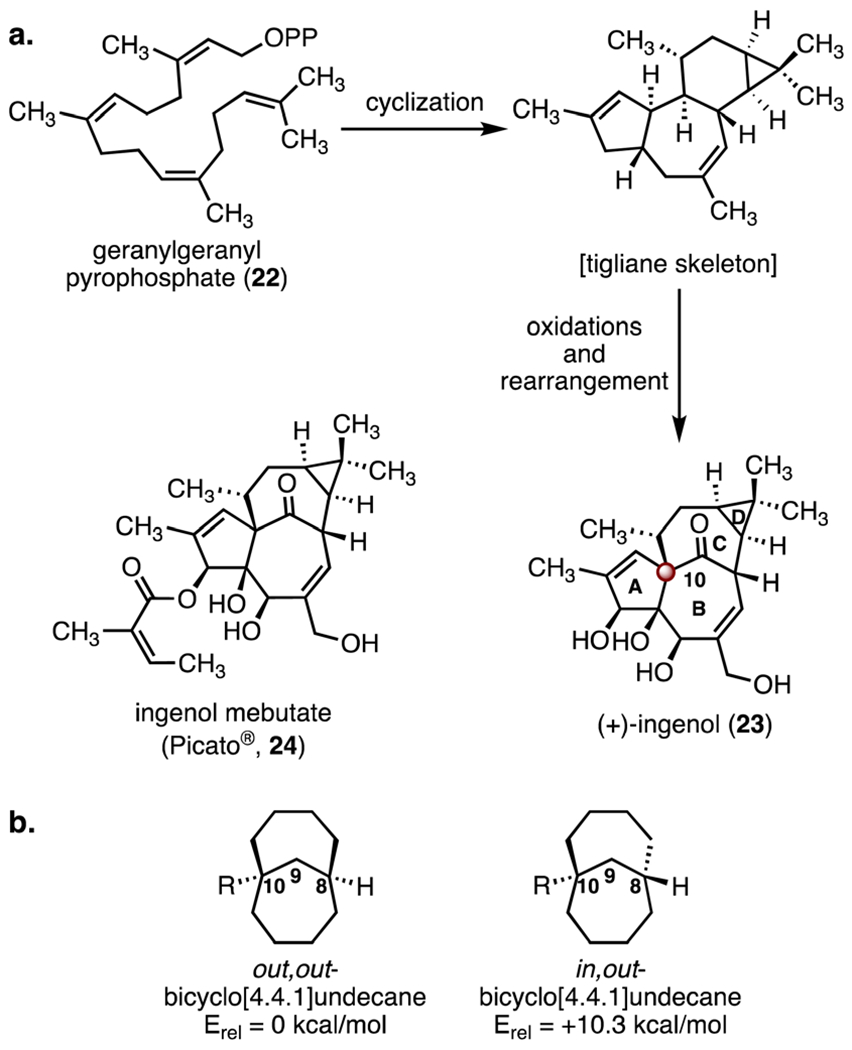
(a) Structure and biosynthesis of (+)-ingenol (23), and the structure of ingenol mebutate (Picato®, 24). (b) Calculated relative energies of out,out- and in,out-bicyclo[4.4.1]undecane diastereomers.
In 2002, Winkler and co-workers reported the first total synthesis of (±)-ingenol (23), employing a tandem [2 + 2] cycloaddition-ring opening as a key step (Scheme 5).35 Their synthesis began with reductive coupling of the cyclopentenone 25 and methyl crotonate.39 Conjugate reduction (lithium, ammonia) of the cyclopentanone 25, followed by a chelation-controlled 1,4-addition of the resulting enolate (26) to methyl crotonate, provided the methyl ester (±)-27 as a 14 : 1 mixture of diastereomers at C11. The C10 all-carbon quaternary center was established with full stereocontrol. The enoxysilane 28 was prepared by treatment of 27 with tert-butyldimethylsilyl trifluoromethanesulfonate and triethylamine (69%, two steps). This product was elaborated to the chlorodiene 29 in nine steps and as a ~1 : 1 mixture of C14 diastereomers (34% overall). Intramolecular photocycloaddition of the diene 29 (mercury lamp, 200–400 nm) provided the cyclobutane 30, which contains the C10 TBCC and the desired (8R) configuration. Unexpectedly, the product of 1,2-migration of the chlorine atom (not shown) was also formed. These two products were not separable on preparative scales (60% combined yield, 5 : 2 ratio of 30 and the chlorine migration product). It is noteworthy that the same photocycloaddition of the C14 hydroxyl analog of 29 (not shown) proceeded in only 16% yield.40 Cleavage of the lactone (potassium carbonate, methanol) promoted a Grob fragmentation of the C9–C6 bond; a three-step sequence then provided the ketone 31 (35%, four steps). This approach established the in,out-BC ring system. The gem-dimethylcyclopropane was constructed by addition of dibromocarbene (bromoform, sodium hydroxide, benzyl triethylammonium chloride) to 31, followed by reductive methylation (methyl lithium, copper(i) thiocyanate, iodomethane, 72%, two steps).41 The tetracyclic product 32 contains the complete skeleton of ingenol (23). The triol 33 was synthesized from 32 in twelve steps by sequential oxidations at C3, C4, C5, and C6 (13% overall). The triol 33 was then advanced to the alkene 34 by a nine-step sequence (23% overall). Finally, installation of the C2 methyl substituent, followed by oxidation state manipulations and removal of the protecting groups, completed the synthesis (six steps, 6.4% overall). In sum, the first synthesis of (±)-ingenol (23) was completed in forty-five steps and 0.0068% overall yield.
Scheme 5.
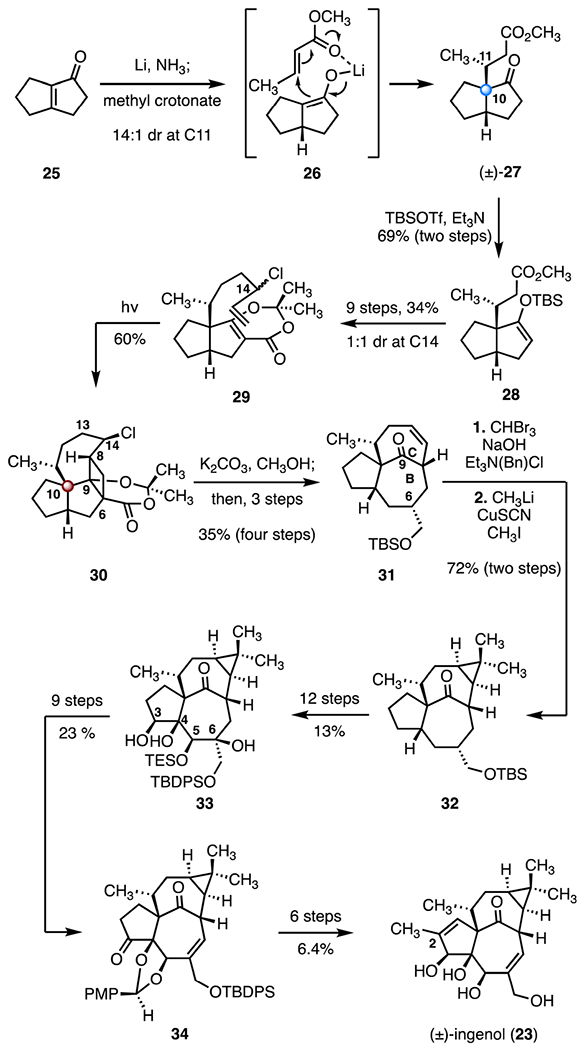
Synthesis of (±)-ingenol (23) by Winkler and co-workers.35 TBSOTf = tert-butyldimethylsilyl trifluoromethanesulfonate.
In 2003, Tanino, Kuwajima and co-workers disclosed a total synthesis of (±)-ingenol (23), employing a tandem cyclization-rearrangement strategy (Scheme 6).36 The enone 36 was prepared in six steps and 17% overall yield from the alcohol (±)-35. A diastereoselective aldol addition (tert-butyl acetate, lithium diisopropylamide, lithium bromide), followed by a chelation-controlled alkylation (trimethylaluminum, lithium diisopropylamide) and acidic workup (hydrochloric acid) provided the trans-decalin 37 as a single diastereomer (60%, two steps). The cobalt complex 38 was synthesized in seven steps from the ester 37 (59% overall). Treatment of the cobalt complex 38 with the bulky Lewis acid 39 triggered a cyclization reaction that formed the C ring bearing the C11 α-methyl group (not shown); reductive demetalation (lithium, ammonia) afforded the diene 40 (67%, two steps). A two-step sequence was used to introduce the gem-dimethyl cyclopropane (bromoform, sodium hydroxide, benzyl triethylammonium chloride; lithium dimethyl cuprate methyl lithium complex, iodomethane, 67% overall). In a key reaction, directed epoxidation of the allylic alcohol 41 (tert-butyl hydroperoxide, titanium(IV) isopropoxide), followed by Lewis-acid promoted ring-opening rearrangement (trimethylaluminum), provided the tetracycle 43 (76%, two steps). In this approach, the configuration of the C9 hydroxy group was used to direct the formation of the C10 TBCC. The tetracycle 43 was elaborated to the tetraol 44 in twelve steps (35% overall). A ten-step sequence was developed to convert the tetraol 44 to the epoxide 45 (4.7% overall). Finally, reductive cleavage of the bromoepoxide 45 (zinc, ammonium chloride) followed by deprotection (potassium hydroxide, 81%, two steps) generated (±)-ingenol (23). The synthesis was completed in forty-five steps and 0.027% overall yield.
Scheme 6.
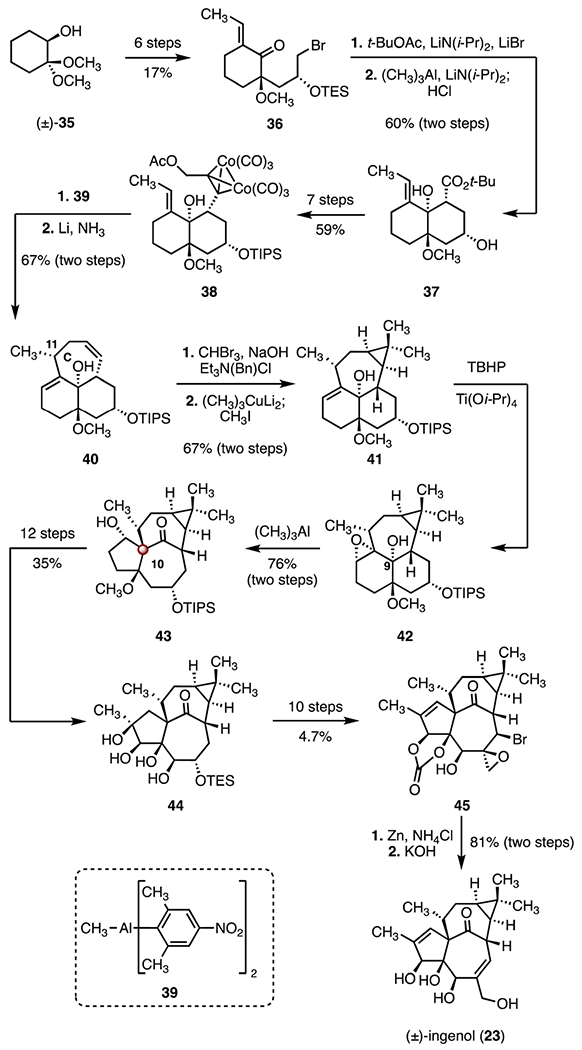
Synthesis of (±)-ingenol (23) by Tanino and co-workers.36 TBHP = tert-butyl hydroperoxide.
In 2004, Wood and co-workers disclosed a synthetic route to (+)-ingenol (23) that featured a ring-opening metathesis (ROM)–ring-closing metathesis (RCM) cascade (Scheme 7).37 Their synthesis began with (+)-3-carene (46), which was converted in four steps42 to the bicyclic cycloheptenone 47 (29% overall). 1,4-Addition of lithium cyanomethyl cuprate to the ester 47 provided the ketoester 48 as a 9.5 : 6.6 : 4.5 : 1 mixture of C10 and C11 diastereomers (86%). The mixture of keto esters 48 was converted to the exocyclic alkene 49 in five steps (70% overall).43 A substrate-controlled Diels–Alder addition between 49 and cyclopentadiene, promoted by boron trifluoride diethyl etherate complex, afforded the spirocyclic alkene 50, which bears the C10 quaternary center (59%). Ring-opening metathesis (ROM; Grubbs I catalyst, ethylene) furnished the diene 51 (98%).44 Selective functionalization of the C3–C3a alkene provided the acetal 52 (three steps, 73% overall). Deprotonation of 52 (potassium hydride) in the presence of the allylic chloride 53 then formed the cyclization precursor 54, as a single C8 diastereomer (98%). In a key transformation, 54 was converted to the tetracycle 55 by ring-closing metathesis (RCM) promoted by the second-generation Hoveyda–Grubbs catalyst (76%). The high conversion of this reaction was attributed to the formation of a more stable trisubstituted alkene in the product 55, which disfavors the reverse ROM reaction. Functionalization of the A-ring (eight steps, 18% overall) then provided the allylic alcohol 56. Directed epoxidation (vanadium(iv) oxide acetylacetonate, tert-butyl hydroperoxide, 73%) furnished the epoxide 57. An eleven-step sequence (5.2% overall) then completed the total synthesis of (+)-ingenol (23). The synthesis was completed in thirty-seven steps and 0.038% overall yield.
Scheme 7.
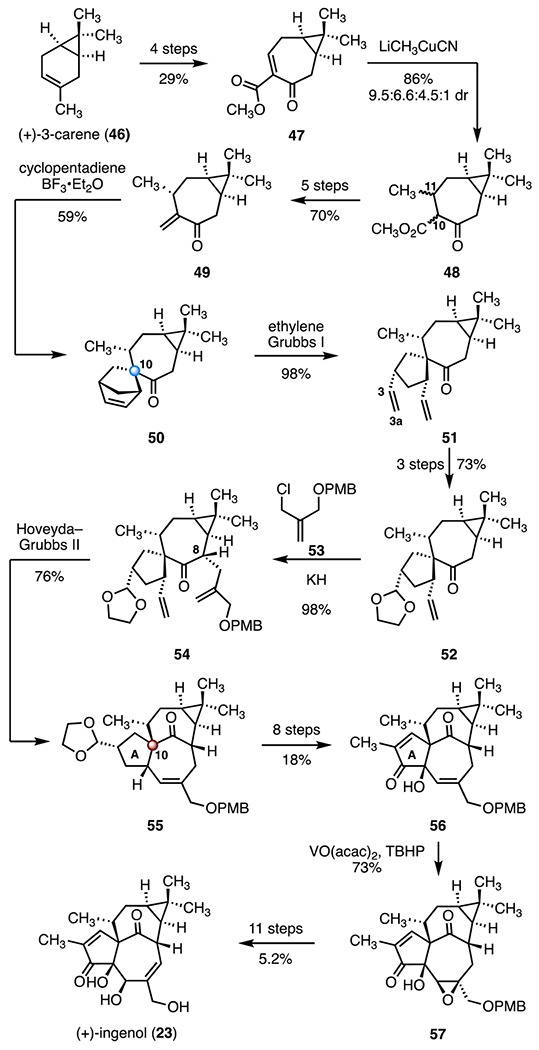
Synthesis of (+)-ingenol (23) by Wood and co-workers.37 TBHP = tert-butyl hydroperoxide, VO(acac)2 = vanadium(IV) oxide acetylacetonate.
In 2013, Baran and co-workers reported a synthesis of (+)-ingenol (23). They employed a Pauson–Khand cyclization and an allylic pinacol rearrangement as key steps (Scheme 8).38 Their synthesis began with a diastereoselective C11 chlorination (N-chlorosuccinimide, 4-dimethylaminopyridine) of (+)-3-carene (46), followed by oxidative cleavage of the resulting exocyclic alkene (ozone; then, thiourea, 48%, two steps). A one-flask diastereoselective reductive methylation–aldol addition (lithium naphthalenide, iodomethane; then, lithium bis(trimethylsilyl)amide, 59)45 provided the allene 60 as a single diastereomer with the requisite (8R)-configuration (44%). The allene 60 was advanced to the allene-yne 61 in two steps (52% overall). A rhodium-catalyzed Pauson–Khand reaction46 (ruthenium(I) chloride bis(carbonyl) dimer, carbon monoxide) then afforded the cyclopentenone 62 (72%). A three-step sequence provided the tertiary allylic alcohol 63 (54% overall). Exposure of the allylic alcohol 63 to boron trifluoride diethyl etherate complex triggered an allylic pinacol rearrangement, furnishing the ketone 64 with the C10 TBCC (80%). This approach to the ingenane core differs from the work of Tanino and Kuwajima,36 in that a tertiary allylic alcohol, rather than an epoxide, was used as the ionizable functional group. The temperature of this reaction was carefully optimized (−78 to −40 °C) to suppress premature elimination of the C2 alcohol. A three-step sequence was devised to convert the rearrangement product to the triol 65 (43% overall). The synthesis was completed by an allylic oxidation47 of the triol 65 (selenium(IV) oxide, formic acid, 76%). Overall, this synthesis of (+)-ingenol (23) proceeded in fourteen steps and 1.1% overall yield.
Scheme 8.
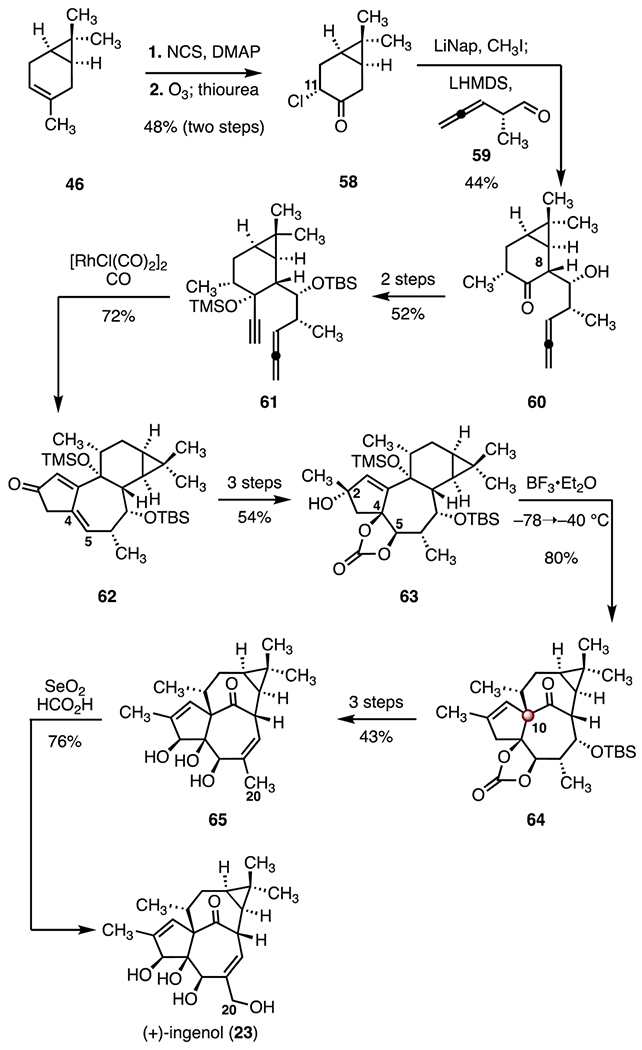
Synthesis of (+)-ingenol (23) by Baran and co-workers.38 NCS = N-chlorosuccinimide, DMAP = 4-dimethylaminopyridine, LiNap = lithium naphthalenide.
The C10 TBCC of ingenol (23) serves as the fusion point of the five-membered A ring and the highly strained in,out-bicyclo [4.4.1]undecane (BC rings). Two strategies were developed to access the TBCC within this ring system. The Winkler35 and Wood37 groups constructed the C10 center early in their synthetic routes (see substrates 27 and 50) through a 1,4-addition–Michael addition or a [4 + 2] cycloaddition, respectively. The corresponding cyclization events (29 → 30, 54 → 55) required harsher than usual conditions due to the strained BC ring junction. Tanino, Kuwajima and co-workers,36 as well as Baran38 and co-workers, took advantage of cationic rearrangement strategies to efficiently access this strained system. This allowed them to accomplish the cyclization events by construction of simpler fused tetracyclic ring systems (42 and 63). The stereochemistry at C8 was established early in each route, and the C10 configuration was derived from the C9 tertiary alcohol, which proved to be easier to install with stereocontrol. All four syntheses employed late-stage oxidation sequences, where the incorporation of highly unsaturated synthons (e.g. 59) improved the efficiency of the oxidation sequences.
2.2. Bilobalide: vicinal TBCCs embedded in a highly oxidized terpenoid ring system
(−)-Bilobalide (67) is a fifteen-carbon tetracyclic trilactone that is derived from biological degradation of the diterpenoid ginkgolide (66, Scheme 9). (−)-Bilobalide (67) was isolated in 1971 from Ginkgo biloba by Nakanishi and co-workers. Its structure was elucidated by NMR spectroscopy.48 A recent study reported that bilobalide (67) inhibits recombinant γ-amino-butyric acid receptors (GABAARs), with an IC50 of 4.6 μM (α1β2γ2L receptor, Xenopus laevis oocytes).49 Additionally, bilobalide (67) exhibits beneficial effects on cognition.50 Structurally, 67 features three adjacent γ-lactones fused to a cyclopentane ring. The C5 and C9 TBCCs are in a vicinal relationship, which further complicates synthetic planning. Here we review the syntheses of bilobalide (67) by the Corey,51 Crimmins,52 and Shenvi laboratories.53
Scheme 9.
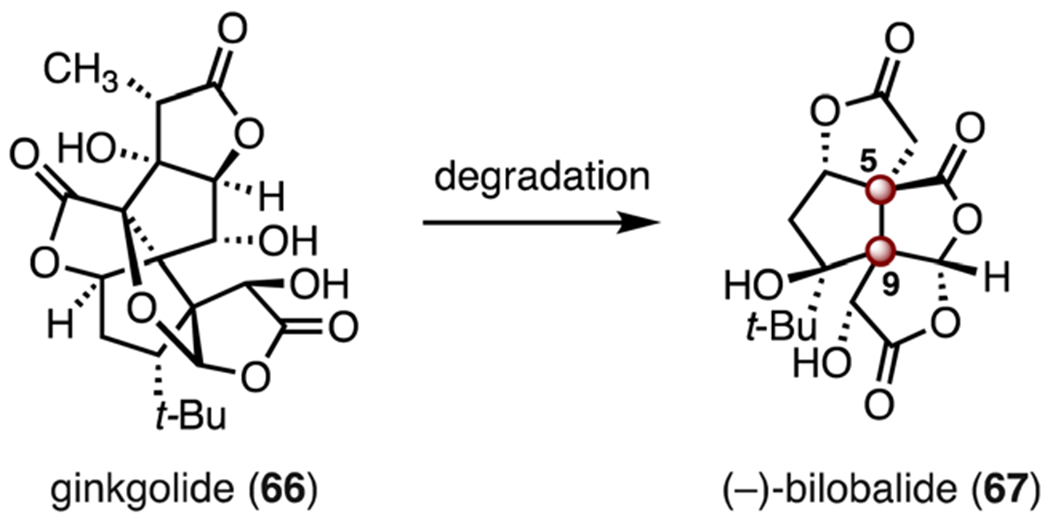
Structures of (−)-bilobalide (67) and ginkgolide (66).
In 1987, Corey and co-workers reported the first total synthesis of (±)-bilobalide (67). The route is notable for its rapid construction of the tetracyclic skeleton (Scheme 10).51 Exposure of a mixture of the ynone 68 and the diester (±)-69 to lithium diisopropylamide triggered a tandem acylation–cyclization reaction, furnishing the enone 70 (72%). This first step provided stereocontrolled construction of the C5 and C9 vicinal quaternary centers. Further investigation revealed that this step likely proceeds through a ketene intermediate.54 Reduction of the C6 carbonyl (sodium borohydride); selective ozonolysis of C4–C12 alkene (ozone; then, dimethyl sulfide), and hemiacetal formation (trimethyl orthoformate) provided the hemiacetal 71 (43%, three steps). A five-step sequence was developed to advance the hemiacetal 71 to the lactone 72, which possesses the full carbon skeleton of the target (72% overall). Dehydration of the hemiacetal (methanesulfonyl chloride, triethylamine; N,N-diisopropylethylamine, 83%, two steps) provided the enol ether 73. Two-fold diastereoselective epoxidation of the enol ether 73 was accomplished by treatment of 73 with 3,5-dinitroperoxybenzoic acid, to furnish the bis(epoxide) 74 (95%); the latter was elaborated to the tris(lactone) 75 in five steps (79% overall). The C7–C8 epoxide was reduced by heating of 75 with a large excess of triethylsilane at elevated temperatures (300 °C), to form the C7–C8 alkene 76 (90%). Finally, dihydroxylation of the C7–C8 alkene of 76 (osmium tetroxide) followed by a selective deoxygenation of the less-hindered C7 alcohol (oxalyl chloride, N,N-diisopropylethylamine; tri-n-butyltin hydride) and deacetylation (hydrochloric acid) completed the synthesis (four steps, 39%). The synthesis of (±)-bilobalide (67) was accomplished in twenty-two steps and 4.9% overall yield.
Scheme 10.
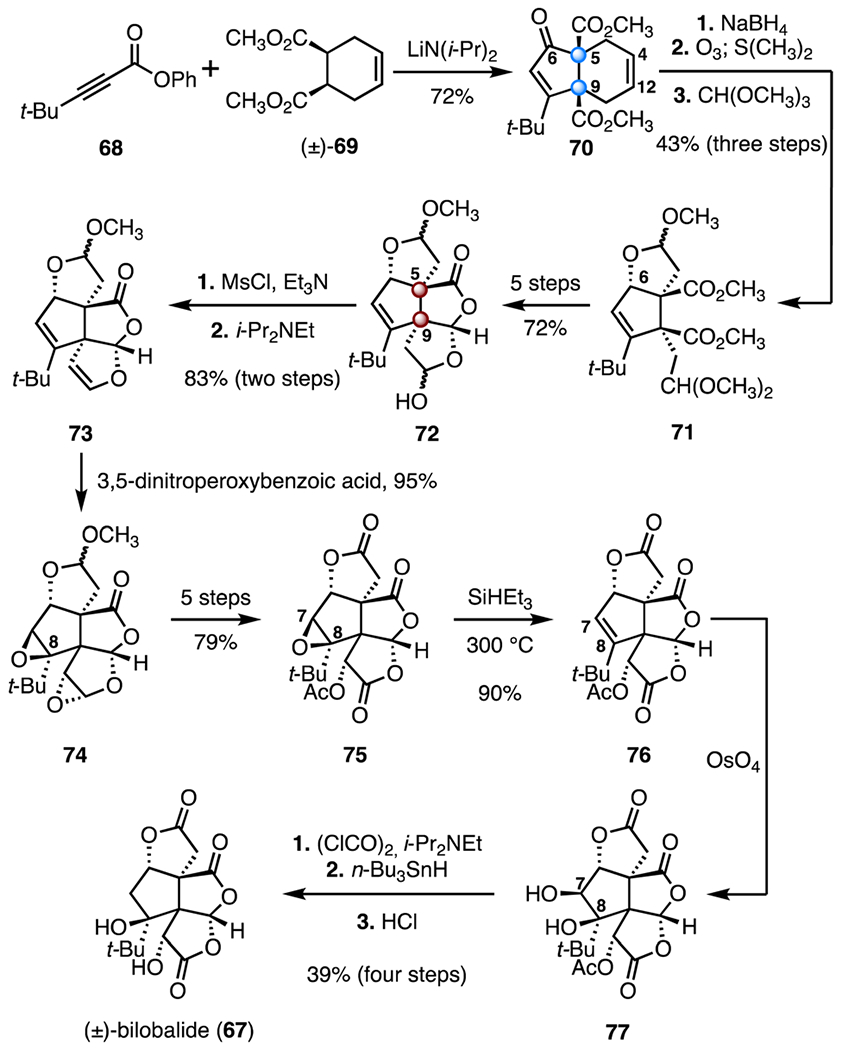
Synthesis of (±)-bilobalide (67) by Corey and co-workers.51 MsCl = methanesulfonyl chloride.
In 1993, Crimmins and co-workers reported a synthesis of (±)-bilobalide (67) that employed an intramolecular [2 + 2] addition to establish the C5 and C9 TBCCs (Scheme 11).52 3-Furaldehyde (78) was converted to the aldehyde (±)-79 by a four-step sequence (48% overall). Fragment coupling by addition of lithium diisopropylamide to a mixture of the aldehyde (±)-79 and the enone 80 generated the diol 81 in 85% yield and as a 1 : 1 mixture of diastereomers at C7. A three-step sequence was developed to convert the mixture of diols 81 to the rearranged enone 82 (50% overall). An intramolecular [2 + 2] cycloaddition was performed by irradiation of 82, to provide a regioisomeric mixture of the cyclobutanes 83 (25%) and 84 (50%). The stereoselectivity of this addition was thought to derive from cyclization of the conformer shown for 82, wherein the tert-butyl and the trimethylsilyloxy substituents occupy pseudoequatorial orientations. A three-step sequence then generated the ketal 85 (68% overall). Reduction (lithium aluminum hydride) followed by oxidative cleavage (lead(IV) acetate) afforded the cyclobutanone 86 (80%, two steps). Baeyer–Villiger oxidation of the cyclobutanone 86 (m-chloroperoxybenzoic acid) provided the lactone 87 (95%). Finally, the bis(lactone) 88, formed by oxidation of 87 (Jones’ reagent, 96%), was subjected to sequential oxidations (dimethyldioxirane; Jones’ reagent, 86%, two steps) to furnish the target. The synthesis of (±)-bilobalide (67) was completed in eighteen steps with 4.4% overall yield.
Scheme 11.
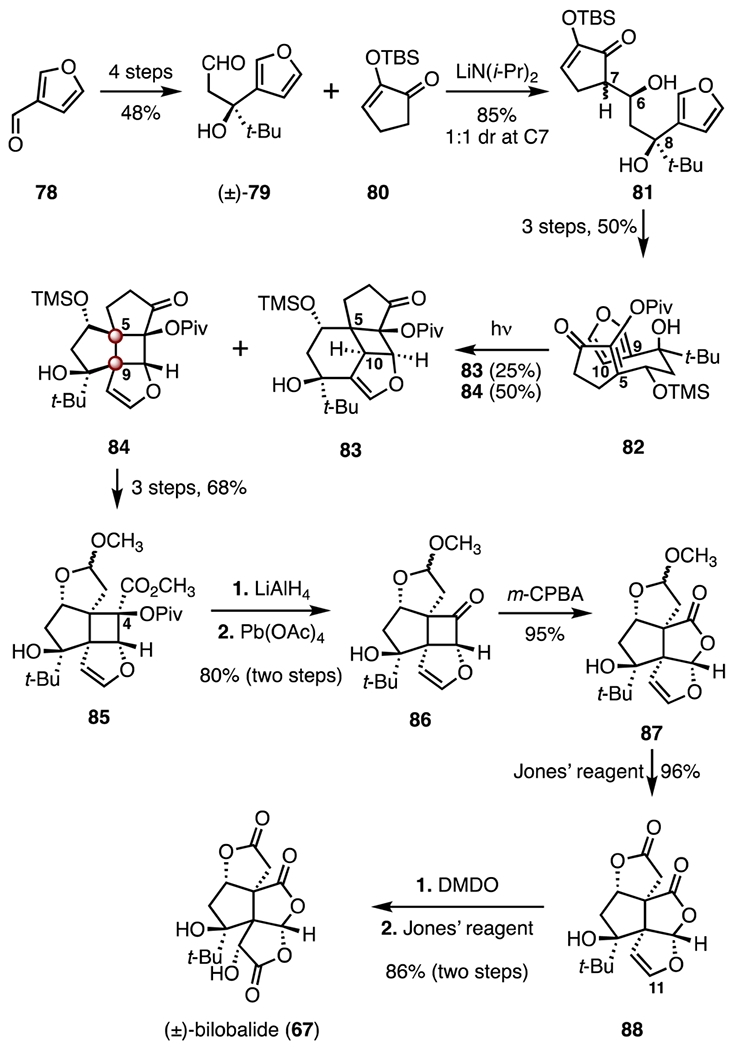
Synthesis of (±)-bilobalide (67) by Crimmins and co-workers.52 m-CPBA = m-chloroperoxybenzoic acid, DMDO = dimethyldioxirane.
In 2019, Shenvi and co-workers disclosed an enantioselective total synthesis of (−)-bilobalide (67) employing an asymmetric Reformatsky addition, a solvent-controlled Mukaiyama hydration and a selective alkyne oxidation as key steps (Scheme 12).53 The synthesis began with an asymmetric vinylogous Reformatsky addition of the allylic bromide 90 (prepared in two steps from benzyl 2-(triphenylphosphaneylidene)acetate) to the aldehyde 89 (prepared in one step from pinacolone, 70%) promoted by diethylzinc and the “indabox” ligand (−)-91.55,56 The addition product (−)-92 was obtained in 44% yield, 94% ee, and with 2.3 : 1 diastereoselectivity at C6. Stereoselective reductive cyclization (tri-n-butyltin hydride, azobisisobutyronitrile) provided the cyclopentene 93 (60%, 20 : 1 dr at C9), which contains the desired C9 all-carbon quaternary center. Mukaiyama hydration (tris(2,2,6,6-tetramethyl-3,5-heptanedionato)manganese(III), isopropoxy(phenyl)silane, oxygen, 67%) of the cyclopentene 93 provided the hindered tertiary C8 alcohol 94 (3 : 1 dr at C8).57 Treatment of the alcohol 94 with the chiral phosphoric acid derivative (−)-95 generated the oxetanyl acetal 96 with 4.5 : 1 diastereoselectivity at C1 (87%). Oxidation of the C6 hydroxyl of 96 (2-iodoxybenzoic acid), followed by C5 alkynylation using trimethylsilyl ethynylbenziodoxolone,58 and reduction of the C6 ketone (samarium(II) iodide), furnished the alkyne 97 (60% over three steps, 20 : 1 dr). In this three-step procedure, the C5 quaternary center was established by electrophilic alkynylation. Treatment of the alkyne 97 with lithium bis(trimethylsilyl)amide and trimethyl borate, followed by addition of m-chloroperoxybenzoic acid, provided the lactone 98 (55%). Removal of the benzyl ester protecting groups (palladium on carbon, hydrogen; then, hydrochloric acid), followed by a benzoate ester-assisted one-pot translactonization–α-hydroxylation cascade through the spirocyclic intermediate 99 (benzoyl anhydride, 4-dimethylaminopyridine; then, potassium bis(trimethylsilyl)amide, Davis’ oxaziridine; then, hydrochloric acid) accomplished the final oxidations and skeletal rearrangement (44%, two steps). The total synthesis of (−)-bilobalide (67) was completed in twelve steps and 1.7% overall yield.
Scheme 12.
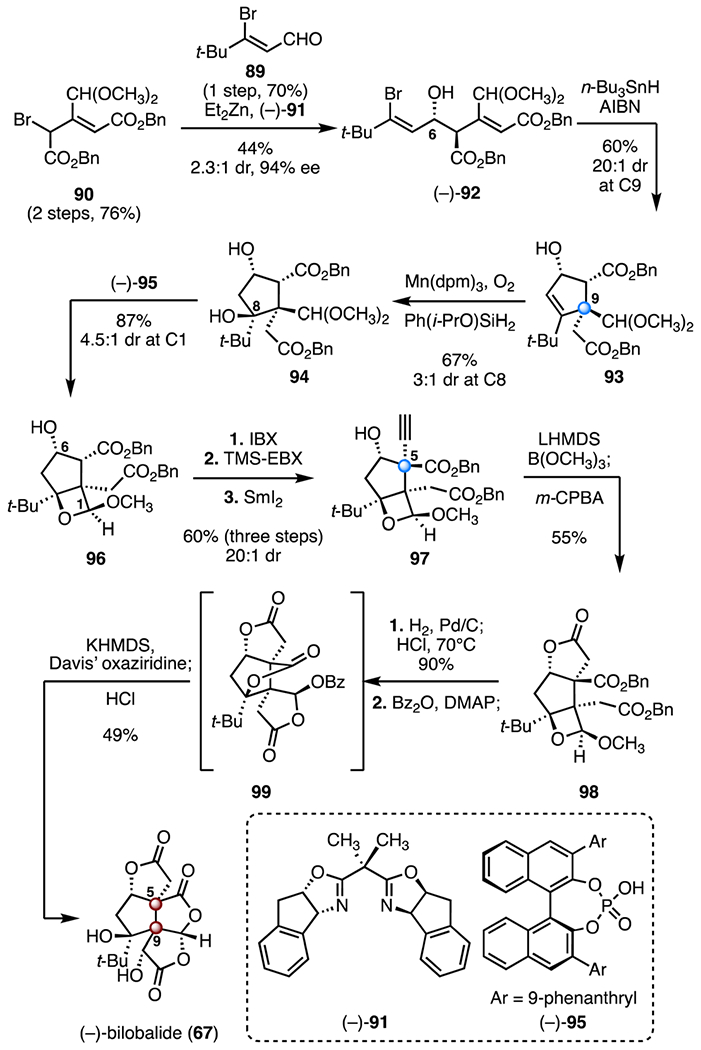
Synthesis of (−)-bilobalide (67) by Shenvi and co-workers.53 AIBN = azobisisobutyronitrile, Mn(dpm)3 = tris(2,2,6,6-tetramethyl-3,5-heptanedionato)manganese(III), IBX = 2-iodoxybenzoic acid, TMS-EBX = trimethhylsilyl ethynylbenziodoxolone, LHMDS = lithium bis(trimethylsilyl)amide, m-CPBA = m-chloroperoxybenzoic acid, DMAP = 4-dimethylaminopyridine, KHMDS = potassium bis(trimethylsilyl)amide.
The vicinal C5 and C9 TBCCs in (−)-bilobalide (67) are the fusion points of a cyclopentane ring and three γ-lactones. A common element of all three synthetic routes is installation of the quaternary centers before lactone formations, by CγO bond construction. The synthesis by Corey and co-workers51 generated the vicinal quaternary centers by an acylation–cyclization reaction (68 + 69 → 70), while Crimmins and co-workers52 introduced these centers through a [2 + 2] cycloaddition (82 → 84). By comparison, Shenvi and co-workers53 constructed these centers in a stepwise fashion (92 → 93, 96 → 97). Corey and co-workers51 formed the C6 and C8 stereocenters after introducing C5 and C9 (70 → 71, 73 → 74). In contrast, the Crimmins synthesis52 introduced them in the linear cyclization precursor (82) early in the route. The Shenvi synthesis53 constructed these centers through two novel reactions before (89 + 90 → 92) and after (93 → 94) introduction of the all-carbon five-membered ring.
2.3. Waihoensene: vicinal TBCCs within a terpenoid ring system bearing sparse heteroatom functionality
The tetracyclic diterpene (+)-waihoensene (100) was isolated in 1997 by Weavers and co-workers from the New Zealand podocarp Podocarpus totara var. waihoensis. Its structure was elucidated by NMR spectroscopy (Scheme 13).59 Like (−)-bilobalide (67), (+)-waihoensene (100) possess two vicinal TBCCs, at C3a and C9a. Instead of lactones, however, the ring system of (+)-waihoensene (100) contains only carbon atoms. The absence of functional group handles in the target creates challenges for synthetic planning. To date, five total syntheses of waihoensene (100) have been reported; each will be discussed here.60–64
Scheme 13.
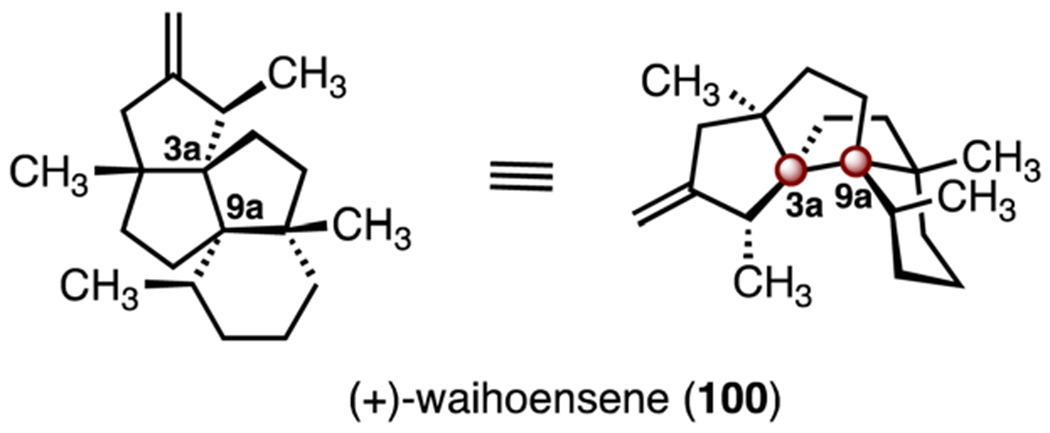
Structure of (+)-waihoensene (100).
In 2017, Lee and co-workers disclosed a total synthesis of (±)-waihoensene (100) that employed a cycloaddition–fragmentation–cyclization cascade to construct the carbon skeleton (Scheme 14).60 The allene 102 was prepared in twelve steps and 35% yield from the known ketoester (±)-101.65 The hydrazone 103 was obtained from the allene 102 by condensation (p-toluenesulfonyl hydrazide). Treatment with sodium hydride provided the diazoalkane 104, which underwent an intramolecular [3 + 2] cycloaddition, extrusion of dinitrogen, and cyclization of the resulting trimethylenemethane diyl intermediate 106, to furnish the tetracyclic alkene 107, which bears the C3a and C9a TBCCs (83%, 3.3 : 1 dr, two steps). A similar strategy was employed in the synthesis of (−)-crinipellin A by the same group.66 A four-step sequence was developed to transform the alkene 107 to the enone 108 (13% overall). Stereoselective installation of the C3 and C11a methyl substituents and ketone methylenylation provided (±)-waihoensene (100; 16% over three steps). The synthesis of (±)-waihoensene (100) was completed in twenty-one steps and 0.60% overall yield.
Scheme 14.
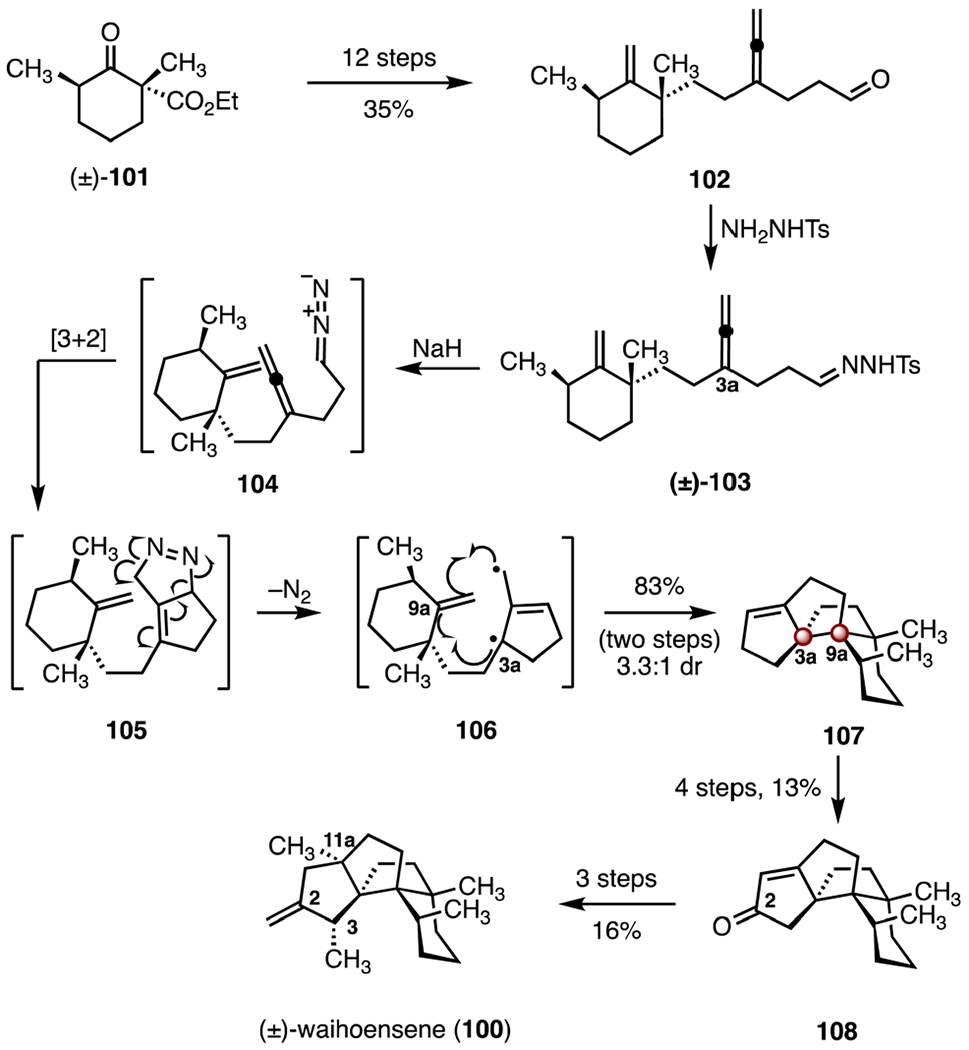
Synthesis of (±)-waihoensene (100) by Lee and co-workers.60 NH2NHTs = p-toluenesulfonyl hydrazide.
In 2020, Yang and co-workers reported the first enantioselective synthesis of (+)-waihoensene (100). Their synthetic route employed Conia-ene and Pauson–Khand reactions to construct the carbon skeleton (Scheme 15).61 The synthesis began with addition of the Grignard reagent 110 to 3-ethoxycyclohexenone (109), followed by hydrolysis of the 1,2-addition product (hydrochloric acid). Enantioselective, copper-catalyzed 1,4-addition of trimethylaluminum (copper(I) thiophene-2-carboxylate, (R,R)-111) generated a stable aluminum enolate with the established C5a quaternary center (not shown). Addition of N-(butylmethoxy)-N-ethylethanamine, followed by oxidative elimination (m-chloroperoxybenzoic acid), produced the enantioenriched enone (−)-112 (60%, three steps, 91% ee).67 A three-step sequence was developed to convert the enone (−)-112 to the diyne 113 (42% overall, 1.5 : 1 mixture of diastereomers at C9a). A base-promoted Conia-ene reaction (potassium tert-butoxide) furnished the enyne 114, which bears the quaternary stereogenic center at C9a (83%). A Pauson–Khand annulation (dicobalt octacarbonyl, nitrous oxide) then produced the enone 115 (59%).68 By this approach, the configuration of the vicinal C3a and C9a TBCCs were established by a stereochemical relay from C9a. A four-step sequence then provided the alkene 116 (51% overall). Diastereoselective hydrogenation of the C9–C15 alkene of 116 (iron(III) acetylacetonate, phenylsilane, 75%) delivered the ketone 117.69 A two-step sequence was developed to convert the ketone 117 to (+)-waihoensene (100; 81% overall). The synthesis of (+)-waihoensene (100) was completed in fifteen steps and 3.8% overall yield.
Scheme 15.
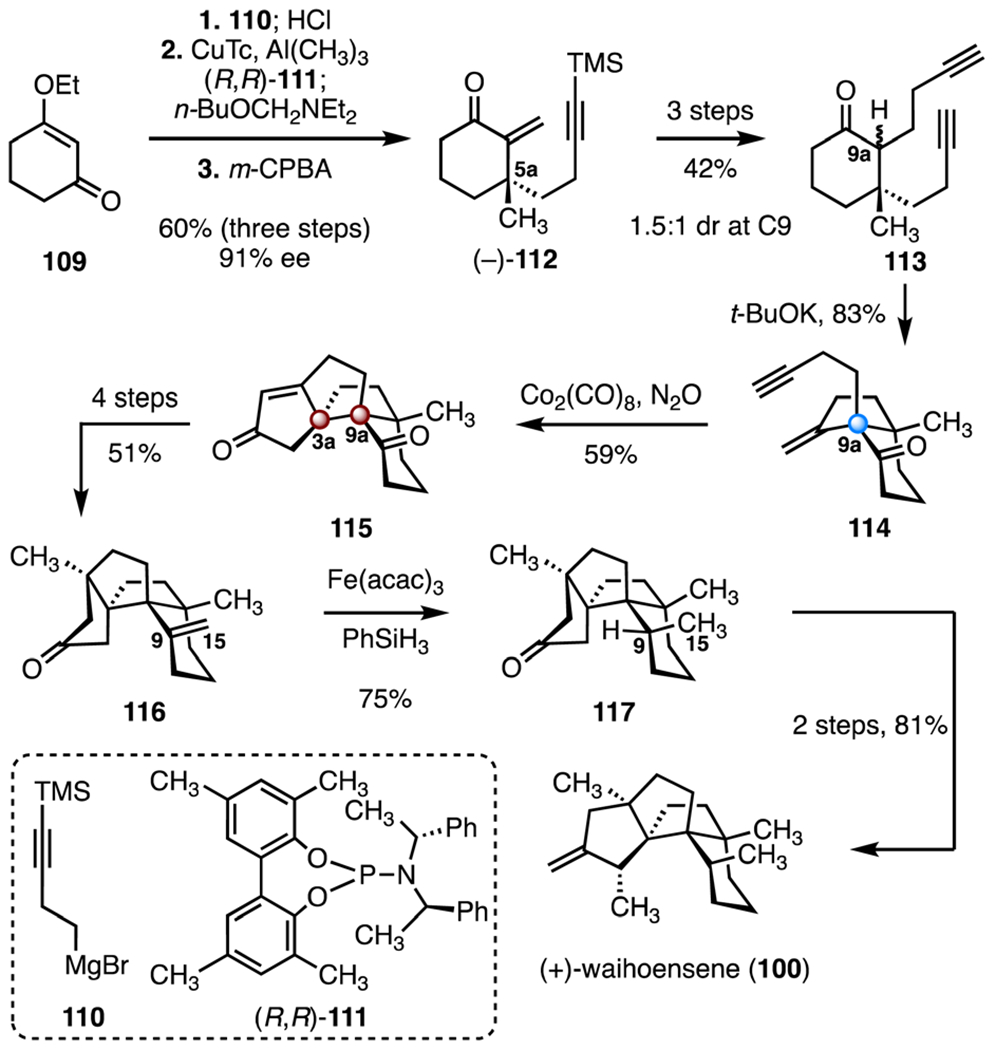
Synthesis of (+)-waihoensene (100) by Yang and co-workers.61 CuTc = copper(I) thiophene-2-carboxylate, m-CPBA = m-chloroperoxybenzoic acid, Fe(acac)3 = iron(III) acetylacetonate.
In the same and the subsequent years, two additional enantioselective syntheses of (+)-waihoensene (100) were disclosed by Snyder and Gaich.62,63 Snyder’s synthesis also relied on Conia-ene and Pauson–Khand reactions to form the carbon skeleton, though the implementation of the transformations was distinct (Scheme 16). The Snyder synthesis began with addition of the alkyl Grignard reagent 119 to 3-methoxycyclopentenone (118). Hydrolysis of the 1,2-addition product was achieved by treatment with hydrochloric acid on workup (product not shown). An enantioselective conjugate addition of trimethylaluminum was promoted by the N-heterocyclic carbene complex 120 and copper(n) trifluoromethanesulfonate, to provide the ketone (−)-121 (44%, two steps, 92% ee).70 Deprotonation of the ketone within 121 (sodium bis(trimethylsilyl) amide), followed by addition of Mander’s reagent, formed the ketoester 122 as a 1 : 1 mixture of C9a diastereomers (56%), along with a 1 : 1 diastereomeric mixture of its constitutional isomer (31%, not shown). Removal of the silyl protecting group (tetra-n-butylammonium fluoride), followed by a gold-catalyzed Conia-ene reaction (triphenylphosphine gold(I) chloride, silver(I) trifluoromethanesulfonate) generated the hydrindane 123, which bears the C9a quaternary stereogenic center (94%, two steps).71 An eight-step sequence was developed to advance the bicycle 123 to the enyne 124 (18% overall). A Pauson–Khand annulation (dicobalt octacarbonyl, carbon monoxide) then provided the tetracycle 108, an intermediate in the Lee synthesis (50%).60 This reaction required unusually high temperatures (160 °C), likely due to the steric congestion of the substrate. The tetracycle 108 was advanced to (+)-waihoensene (100) in three steps (36% overall). The synthesis was completed in seventeen steps and 0.75% overall yield.
Scheme 16.
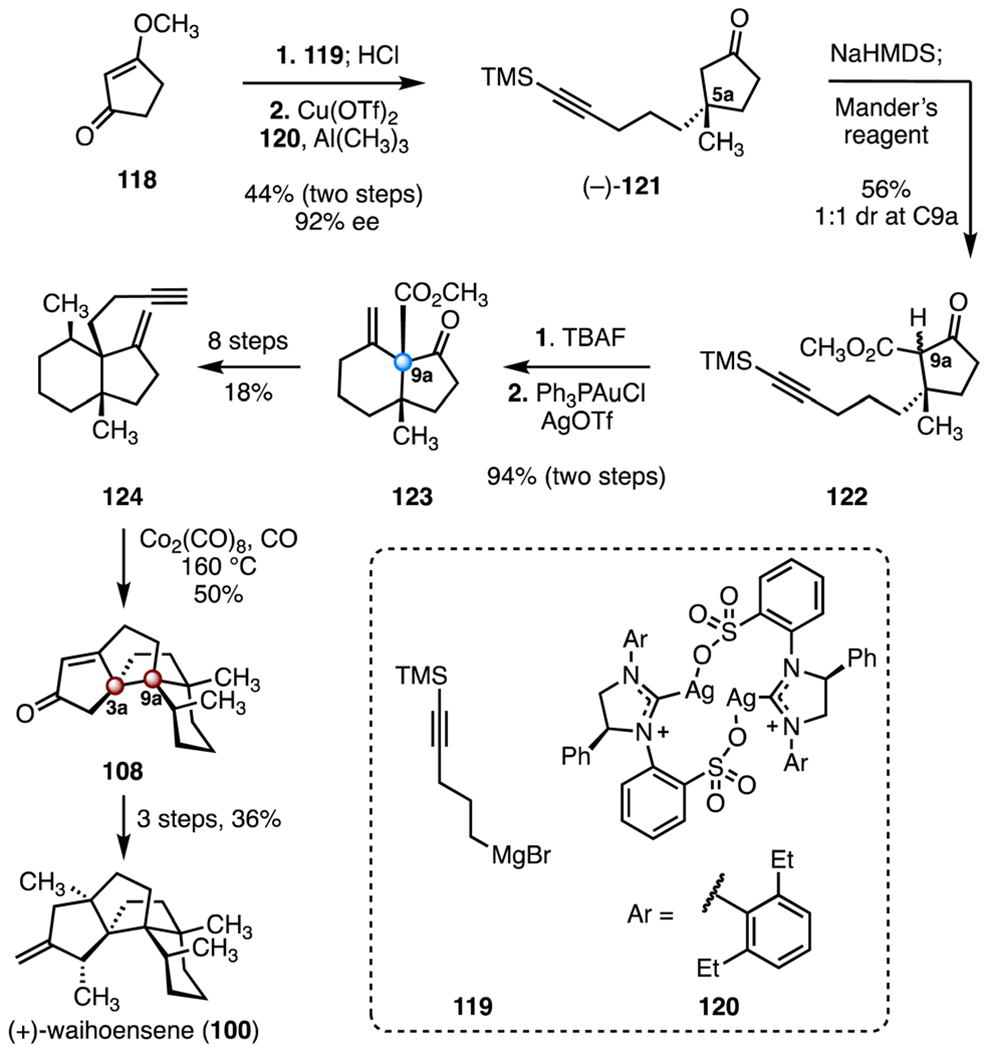
Synthesis of (+)-waihoensene (100) by Snyder and co-workers.62 Cu(OTf)2 = copper(II) trifluoromethanesulfonate, NaHMDS = sodium bis(trimethylsilyl)amide, TBAF = tetra-n-butylammonium fluoride, AgOTf = silver(I) trifluoromethanesulfonate.
The Gaich laboratory also employed a Pauson–Khand annulation as a key step, but these researchers developed a diastereoselective radical cyclization rather than a Conia-ene reaction to establish the C9a center (Scheme 17). The synthesis began with an α-methylation of the β-ketoester (±)-125 (sodium hydride, iodomethane), followed by an enantioselective decarboxylative allylic alkylation promoted by the chiral ligand (S,S)-126 and tris(dibenzylideneacetone) dipalladium, which provided the enantioenriched alkene (+)-127 (72% over two steps, 96% ee).72 A five-step sequence was developed to transform the alkene (+)-127 to the alkyne 128 (46% overall). A radical cyclization (tri-n-butyltin hydride, azobisisobutyronitrile), destannylation (pyridinium p-toluenesulfonate), and α-epimerization (sodium hydroxide, methanol) then formed the bicyclic ketone 129, which contains the C9a quaternary center (84% over three steps, 33.3 : 1 dr at C9). The C8 carbonyl was reduced by a two-step sequence (58% overall) to provide the bicycle 130, which was converted to the enyne 124 (59%, three steps). Pauson–Khand annulation (dicobalt octacarbonyl) provided the enone 108 (46%). A three-step sequence was developed to obtain (+)-waihoensene (100) (40% overall). Thus, the route to (+)-waihoensene (100) was completed in nineteen steps and 1.8% overall yield.
Scheme 17.
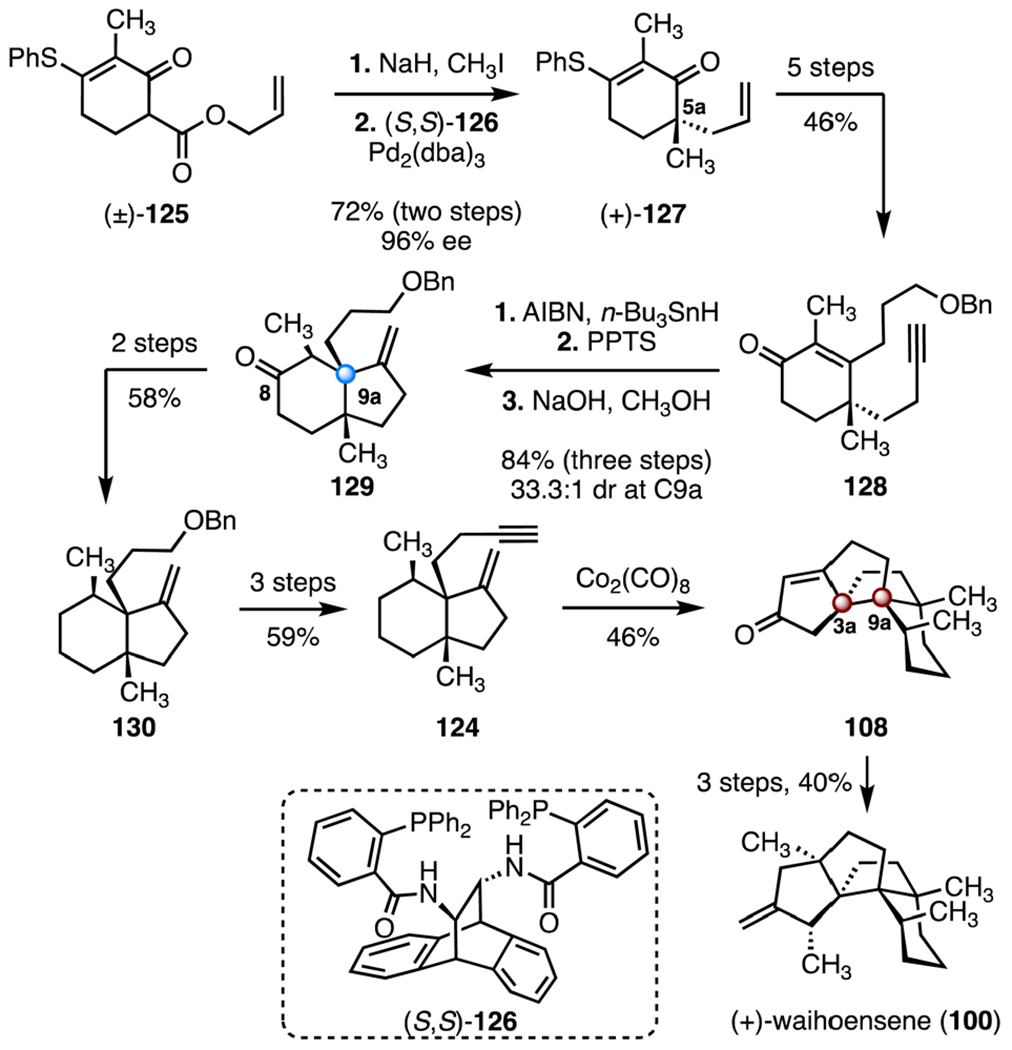
Synthesis of (+)-waihoensene (100) by Gaich and co-workers.63 Pd2(dba)3 = tris(dibenzylideneacetone) dipalladium, AIBN = azobisisobutyronitrile, PPTS = pyridinium p-toluenesulfonate.
In 2022, Tu and co-workers reported a total synthesis of (±)-waihoensene (100) that employed a Nazarov cyclization and a double ring expansion cascade as key steps (Scheme 18).64 The synthesis began with a Horner–Wadsworth–Emmons olefination of the silylated cyclobutanone (±)-131 (132, sodium hydride). Ester hydrolysis followed by a Dieckmann condensation (lithium hydroxide; oxalyl chloride; lithium diisopropylamide, methyl acetate) then furnished the β-ketoester 133 (82%, three steps). Titanium-mediated aldol dehydration using cyclobutanone as electrophile furnished the bis(enone) precursor 134 (73%). In a remarkable transformation, treatment of 134 with indium(III) hexafluoroantimonate provided the angular tricycle 139 in 81% yield and 7 : 1 dr at C2. This cascade reaction was proposed to comprise a Nazarov cyclization (through 135) to afford the cationic intermediate 136, sequential 1,2-migrations (136 → 137, 137 → 138), proton elimination, and isomerization (138 → 139). The enone 139 bears the C3a TBCC. The diene 140 was then accessed by a five-step sequence (22% overall). Irradiation of the diene 140 (350 nm) induced a [2 + 2] cycloaddition to provide the pentacyclic intermediate 141, which bears the C9a TBCC. Subsequent reductive ring-opening (samarium(II) iodide) and ketone reduction (diisobutylaluminum hydride) delivered the tetracycle 142 (47%, two steps). A six-step procedure was developed to advance the tetracycle 142 to the target (29% overall). The total synthesis of (±)-waihoensene (100) was completed in eighteen steps and 1.5% overall yield.
Scheme 18.
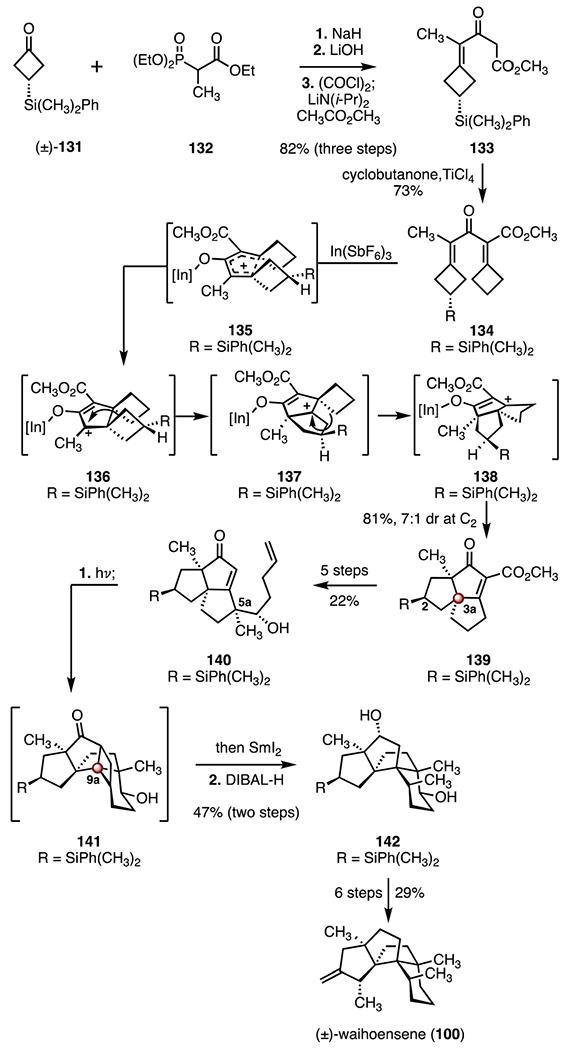
Synthesis of (±)-waihoensene (100) by Tu and co-workers.64 DIABL-H = diisobutylaluminum hydride.
In the syntheses discussed above, the contiguous stereocenters C5a, C9a, C3a, C11a, including the C9a and C3a TBCCs, were constructed by three approaches. In the Yang,61 Snyder,62 and Gaich63 syntheses, the C5a quaternary center was introduced by stereoselective 1,4-addition (109 → 112, 118 → 121) or decarboxylative allylic alkylation (125 → 127), followed by Pauson–Khand cyclizations (114 → 115, 124 → 108), to form the C3a and C9a TBCCs. The configuration of C5a was relayed to the C9a and C3a TBCCs. Both the Lee60 and the Tu64 syntheses took advantage of novel cyclization strategies to construct these centers. In the Lee synthesis,60 a linear precursor 102 was first prepared. Tandem cycloaddition of this precursor was used to construct the C3a and C9a TBCCs from planar carbon centers. In the Tu synthesis,64 the cyclization cascade produced the tricyclic structure 139 and the C3a TBCC. Stereoselective installation of the C5a quaternary center by similar carbonyl chemistry occurred before the generation of the C9a TBCC by a [2 + 2] cycloaddition (140 → 141).
2.4. Epicolactone: congested cluster of TBCCs within a highly unsaturated polyketide ring system
(−)-Epicolactone (145) is a secondary metabolite isolated from sugarcane by Marsaioli and co-workers in 2012.73 Biosynthetically, (−)-epicolactone (145) is derived from the oxidative coupling of epicoccone B (144) and epicoccine (143, Scheme 19).74 (−)-Epicolactone (145) possesses antifungal and antimicrobial activities.75 Structurally, (−)-epicolactone (145) contains three TBCCs at C1, C5 and C9. The syntheses of epicolactone (145) by the Trauner76 and Carreira77 laboratories are discussed below.
Scheme 19.
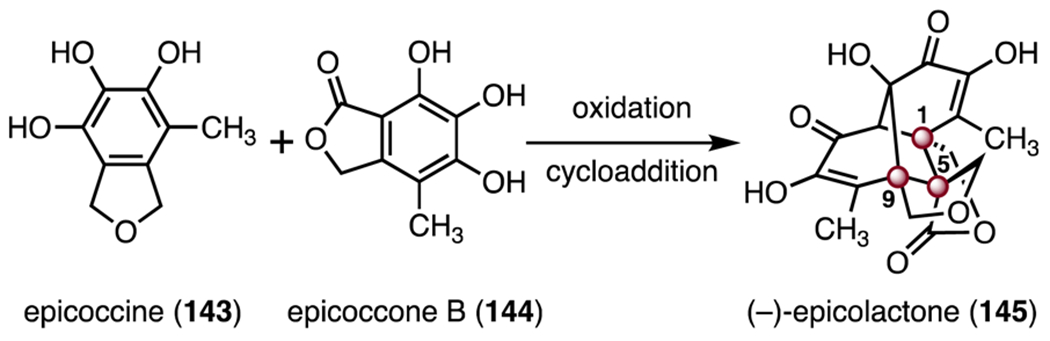
Structure of (−)-epicolactone (145) and its biosynthetic precursors.
The Trauner group developed a biomimetic route to (±)-epicolactone (145, Scheme 20).76 The synthesis began with preparation of the catechol 148 as a synthetic surrogate of the natural precursor epicoccone B (144). A four-step sequence was developed to convert 4-(hydroxymethyl)-2-methoxyphenol (146) to the aryl bromide 147 (38% overall).78 Substitution of the bromide function by hydroxide (copper, sodium hydroxide), followed by aldehyde reduction (sodium borohydride), provided the catechol 148 (59%, two steps). Treatment of a mixture of the catechol 148 and epicoccine (143)79 with potassium ferrocyanide initiated a biomimetic cyclization-rearrangement cascade. It was proposed that the substrates were oxidized to the ortho-quinones 149 and 150; a spontaneous intermolecular [5 + 2] cycloaddition then generated the tetracyclic intermediate 151, which contains the C1 quaternary center and the C5 TBCC. Intramolecular translactonization formed the dienol 152. Finally, an intramolecular aldol addition afforded the bis(enone) 153. Cleavage of the methyl ether (magnesium iodide, quinoline) then provided (±)-epicolactone (145; 75%). By this biomimetic approach, (±)-epicolactone (145) was obtained in eight steps and 7.1% overall yield.
Scheme 20.
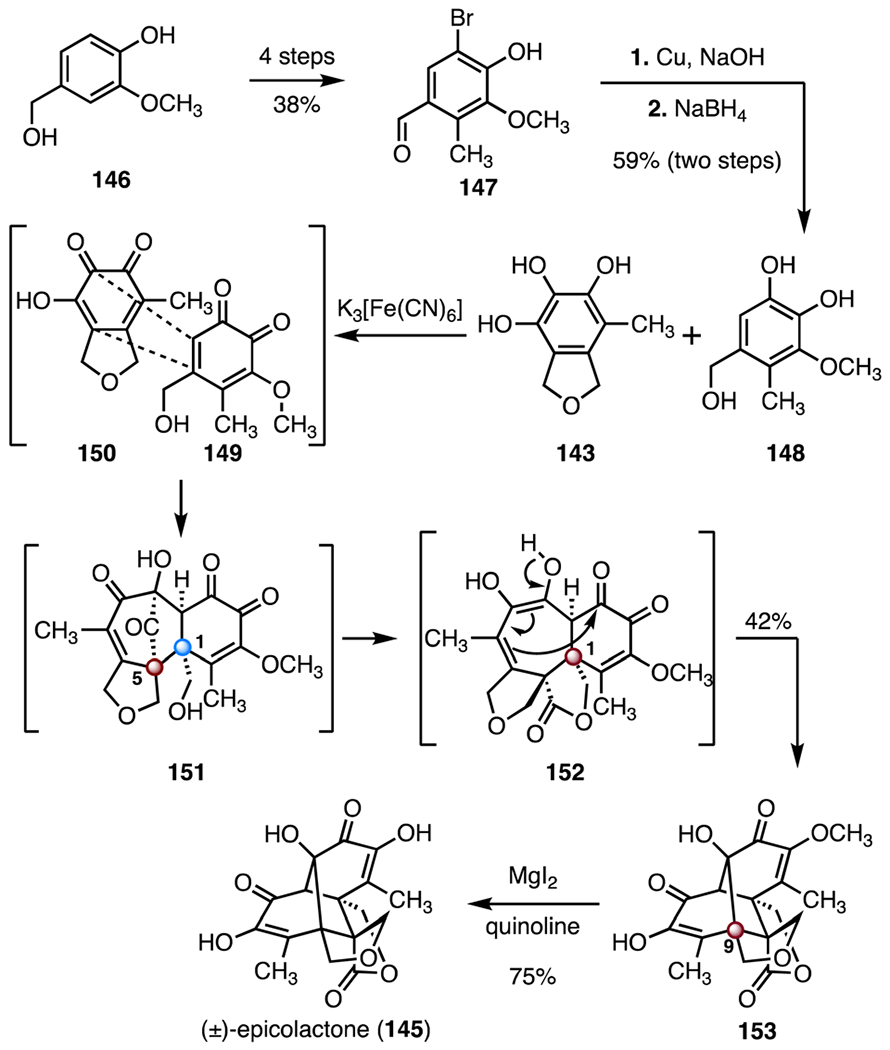
Synthesis of (±)-epicolactone (145) by Trauner and co-workers.76
In 2018, Carreira and co-workers reported a synthesis of (±)-epicolactone (145) that employed a [2 + 2] cycloaddition–ring expansion cascade (Scheme 21).77 The synthesis began with a Stille coupling between the stannane 154 (two steps from 2,3,4,6-tetramethoxybenzaldehyde, 78%) and the vinyl bromide 155 80 using bis(triphenylphosphine)palladium(II) chloride as precatalyst. Oxidative demethylation (ceric ammonium nitrate) then provided the ether-linked bicycle 156 (42%, two steps). Irradiation of the bicycle 156 using 400–450 nm light promoted an intramolecular [2 + 2] cycloaddition,81 to provide the cyclic ether (±)-157 (76%). The C1 quaternary center and the C5 and C9 TBCCs were established in this single transformation. Oxidative cleavage of the terminal olefin (osmium(VII) oxide; then, lead(IV) acetate) followed by a 1,2-alkyl shift (boron trifluoride acetic acid complex) produced the methyl ketone 158 (54%, two steps).82 In this step, a Lewis acid-promoted demethylation of the C14 ether triggered the C14–C1 bond migration. The methyl ketone 158 was advanced to the enol ether 159 by a four-step sequence (39% overall). A Prins cyclization (trifluoroacetic acid, >99%) produced the alcohol 160, which bears the C1 TBCC. A two-step sequence was then developed to convert the alcohol 160 to the iodomethyl silyl ether 161 (76% overall). Treatment of the silyl ether 161 with samarium(II)83 followed by exposure to hydrochloric acid provided the β-methylenone 162 (90%). The synthesis was completed by removal of the ethylene unit in 162 (three steps, 56%). The synthesis of (±)-epicolactone (145) was completed in eighteen steps and 2.0% overall yield.
Scheme 21.
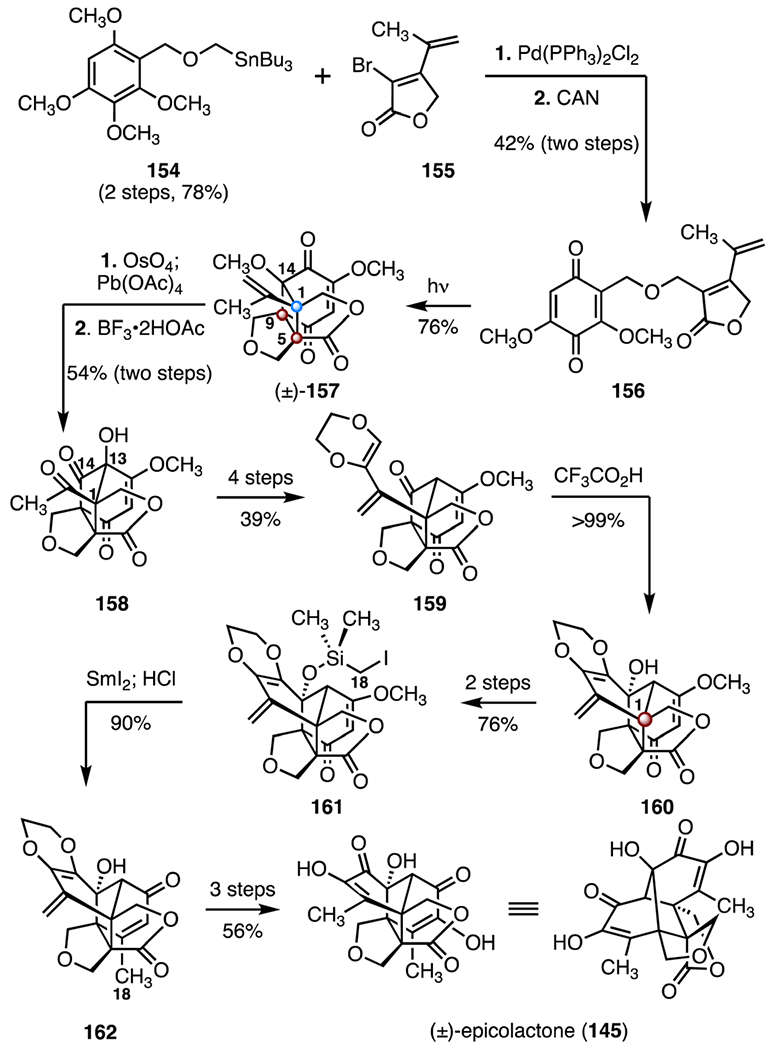
Synthesis of (±)-epicolactone (145) by Carreira and co-workers.77 CAN = ceric ammonium nitrate.
(−)-Epicolactone (145) possesses three contiguous C1, C5, and C9 TBCCs. Both the Trauner76 and Carreira77 syntheses employed cycloaddition reactions to construct the congested stereocenters. The Trauner synthesis76 employed a bimolecular [5 + 2] cycloaddition (149 + 150 → 151) to form the C1 quaternary center and C5 TBCC, followed by a biomimetic cascade to introduce the C9 TBCC. The Carreira synthesis77 utilized the tethered precursor 156 to achieve a unimolecular [2 + 2] cyclization, which formed the C1 quaternary center, as well as the C5 and C9 TBCCs in a single step. The C1-fused all-carbon ring was installed late-stage by a Prins cyclization (159 → 160).
2.5. Hetisine alkaloids: TBCCs within a complex polycyclic terpenoid alkaloid
Hetisine natural products are a family of C20-diterpenoid alkaloids isolated from the Aconitum, Consolida, Delphinium, Rumex, and Spiraea genera. They display a diverse spectrum of biological activities including antiarrhythmic, antitumor, insecticidal, antimicrobial and antiviral activities.84 Hetisines contain a heptacyclic carbon skeleton comprised of a bicyclo [2.2.2]octane core and N–C6 and C14–C20 linkages (Scheme 22). The skeleton contains TBCCs at C8 and C10. Here we discuss syntheses of (±)-nominine (175) by Muratake and Natsume85,86 and Gin.87 Additionally we discuss syntheses of (+)-cossonidine (197) and (+)-spirasines IV (208) and XI (209) by the Sarpong88 and Zhang laboratories.89
Scheme 22.
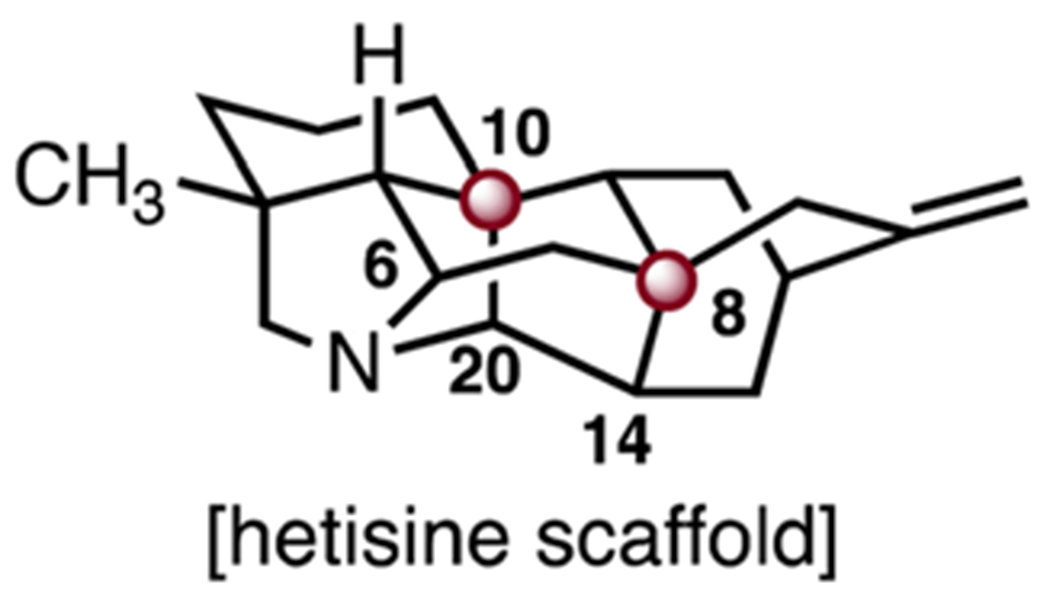
Structure of the hetisine scaffold.
(+)-Nominine (175) was first isolated as “Nomi-base I” from Aconitum sanyoense Nakai by Ochiai and co-workers in 1956.90 It was later named “nominine” by Sakai and co-workers. Its structure was established by chemical correlation with kobusine, whose structure was elucidated by X-ray crystallography.91 In 2004, Muratake and Natsume accomplished the first synthesis of (±)-nominine (175).85,86 Their approach relied primarily on single bond-forming events to construct the complex carbon skeleton (Scheme 23). The synthesis began with a five-step sequence to convert 2-bromo-5-methoxyphenethyl iodide (163) to the aldehyde (±)-164 as a 1 : 1 diastereomeric mixture at C11 (39% overall). The aldehyde 164 was then transformed to the benzylic aldehyde 165, which contains the C10 quaternary center, by a palladium-catalyzed α-arylation (bis(triphenylphosphine)palladium(II) chloride, cesium carbonate, 65%, 4.2 : 1 dr at C11). A four-step sequence was developed to advance the tetracyclic aldehyde 165 to the allylic alcohol 166 as a single diastereomer (16% overall). Eschenmoser–Claisen rearrangement, using acetal 167 as reagent, provided the amide 168, which contains the C8 quaternary stereogenic center (68%).92 The amide 168 was elaborated to the pivalate 169 by a two-step sequence (92% overall). An acetal–ene reaction (boron trifluoride diethyl etherate complex; then, p-toluenesulfonic acid) then formed the tetracycle 170, with the C14–C20 bond and the C10 TBCC constructed.93 A nine-step sequence afforded the tertiary cyanide 171 (4.1% overall). Nitrile reduction and protecting group manipulations generated the pyrrolidine 172 (lithium diisopropylamide, trimethylsilyl chloride; lithium aluminum hydride; triethylamine, benzyl chloroformate, 63%, three steps). The enyne 173 was obtained from the pyrrolidine 172 by a four-step sequence (71% overall). A radical cyclization (tri-n-butyltin hydride, azobisisobutyronitrile), followed by protodestannylation (hydrochloric acid), then formed the alkene 174 (57%). This reaction generated the bicyclo[2.2.2]octane scaffold of the target with the C8 TBCC in place. Ultimately, the alkene 174 was converted to (±)-nominine (175) by a nine-step sequence (46% overall). The total synthesis of (±)-nominine (175) was completed in forty steps with an overall yield of 0.0081%.
Scheme 23.
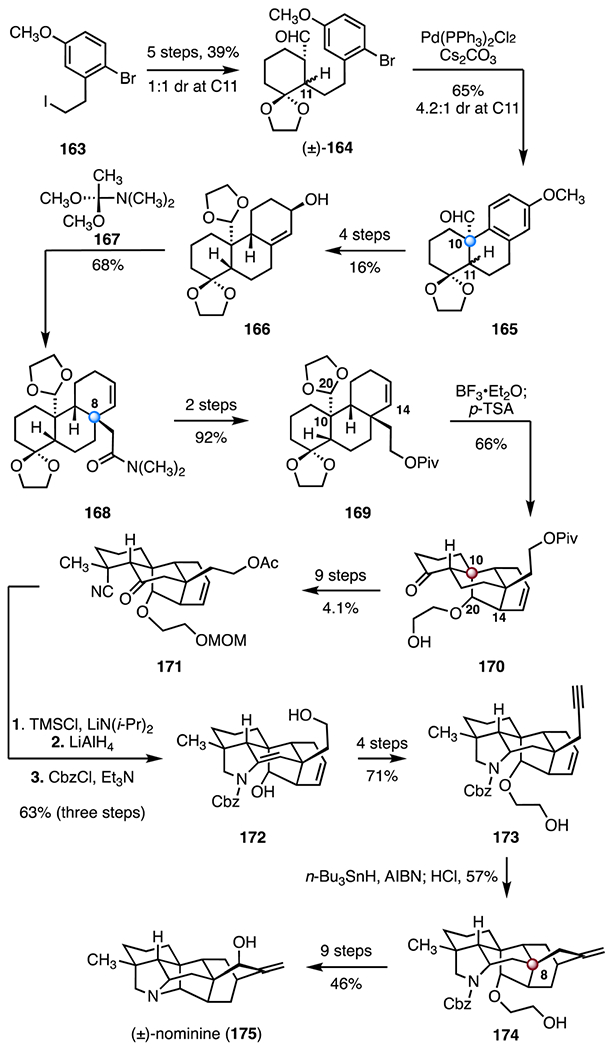
Synthesis of (±)-nominine (175) by Muratake and Natsume.85,86 p-TSA = p-toluenesulfonic acid, TMSCl = trimethylsilyl chloride, CbzCl = benzyl chloroformate, AIBN = azobisisobutyronitrile.
In 2006, Gin and co-workers disclosed an alternative approach to (±)-nominine (175).87 The synthesis employed an aza-1,3-dipolar cycloaddition and an intramolecular Diels–Alder (IMDA) reaction as key steps (Scheme 24). The synthesis began with the preparation of the azide 177 (three steps from the methyl ether 176, 49% overall) and the aldehyde (±)-179 (three steps from the enone 178, 63% overall). A reductive fragment coupling (tri-n-butyl phosphine, sodium triacetoxyborohydride) provided the tethered bicyclic amine 180 (79%). Acid-catalyzed (trifluoroacetic acid) elimination of methanol and isomerization furnished the 4-oxido-isoquinolinium betaine 181 (93%). Heating of 181 to 180 °C promoted an intramolecular [3 + 2] cycloaddition, to provide a 3.6 : 1 mixture of the regioisomeric cycloaddition products 182 and 183 (97% conversion). The cycloaddition was found to be reversible, allowing for recycling of the undesired cycloadduct 182. This transformation generated the C10 quaternary center in the target. The nitrile 183 was converted to the alkene 184 by a six-step sequence (54% overall). Exposure of the alkene 184 to pyrrolidine affected an IMDA reaction, providing the cycloaddition product 186, likely through the enamine 185. The adduct 186 possesses the full carbon skeleton of the target, including the C10 and C8 TBCCs. The adduct 186 was transformed to (±)-nominine (175) in two steps (51% overall). The total synthesis was completed in fifteen steps (LLS) and 1.6% overall yield.
Scheme 24.
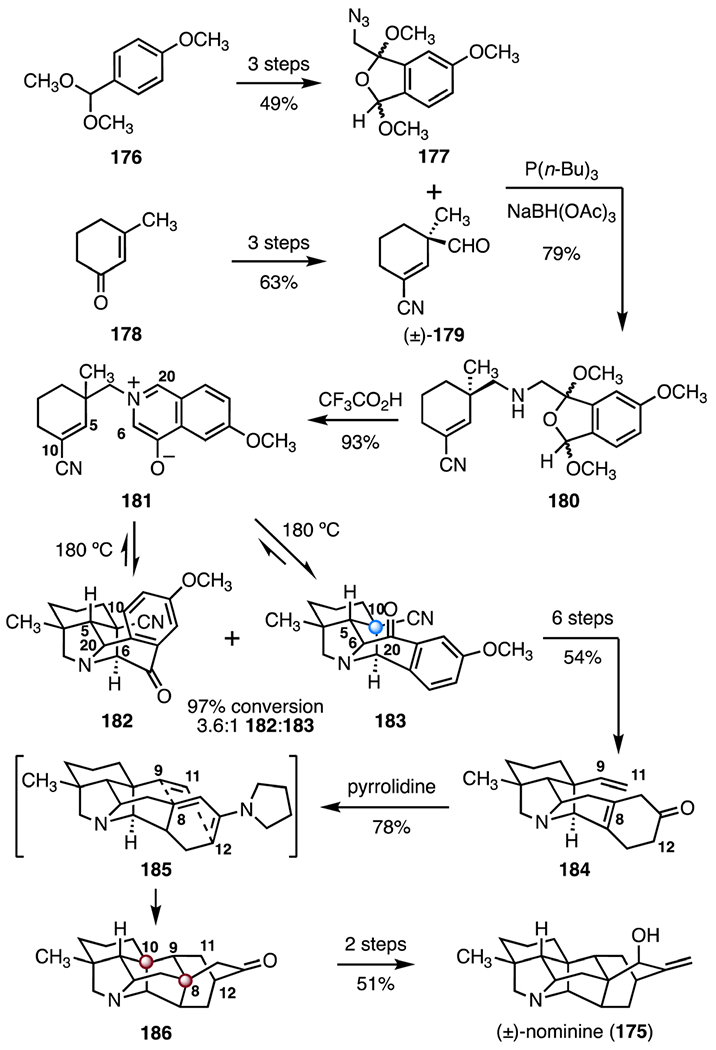
Synthesis of (±)-nominine (175) by Gin and co-workers.87
(+)-Cossonidine (197) was isolated in 1996 by Fuente and co-workers. Its structure was elucidated by NMR spectroscopy.94 In 2018, Sarpong and co-worker disclosed a synthesis of (±)-cossonidine (197) that employed a [4 + 2] cycloaddition, alkylation and acylation of a benzyne intermediate, and a double amination to facilitate disconnections of maximally bridged ring systems (Scheme 25).88 The route began with an intermolecular Diels–Alder cycloaddition between the diene 187 and the dienophile 188 (110 °C). Following catalytic hydrogenation (palladium on carbon, dihydrogen), the bicycle (±)-189 was obtained in 70% overall yield.95,96 The bicycle (±)-189 was converted to the β-ketoester 190 by a four-step sequence (62% overall). Treatment of the β-ketoester 190 with cesium fluoride in the presence of the aryl bromide 191 promoted formation of the cycloheptanone 192 through acylation and alkylation of a benzyne intermediate (45%).97 A five-step sequence was developed to convert the cycloheptanone 192 to the nitrile 193 (39% overall). Reduction of the nitrile, followed by reductive amination (cobalt boride, tert-butylamine boron complex; lithium aluminum hydride), provided the diene 194. A photochemical hydro-amination delivered the pyrrolidine 195 (n-butyl lithium, 320–1100 nm tungsten halogen lamp, isopropylamine; 71% over three steps).98 A Birch reduction (sodium, ammonia; then, hydrochloric acid), followed by a pyrrolidine-mediated IMDA, produced the cycloadduct 196, which contains the full carbon skeleton of cossonidine (197) as well as the C8 and C10 TBCCs (54%, two steps). The cycloadduct 196 was advanced to (±)-cossonidine (197) in six steps (15% overall). The synthesis was completed in twenty-three steps and 0.44% overall yield.
Scheme 25.
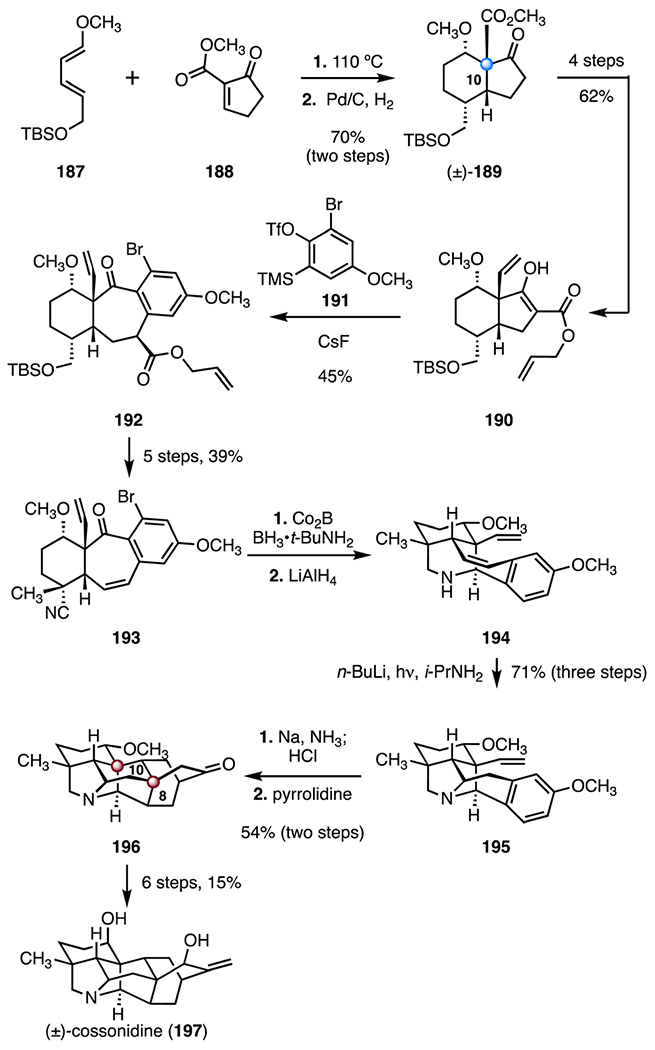
Synthesis of (±)-cossonidine (197) by Sarpong and co-workers.88
(+)-Spirasines IV (208) and XI (209) are hetisine alkaloids isolated from Spiraea japonica L. f. var. fortunei by Sun and Yu in 1985. Their structures were elucidated by NMR spectroscopy.99 In 2018, Zhang and co-workers reported a synthesis of (±)-208 and (±)-209 that employed an intramolecular 1,3-dipolar cycloaddition to construct the carbon skeleton (Scheme 26).89 The synthesis began with the ester 198,100 which was converted to the aldehyde (±)-199 by a six-step sequence (40% overall). Treatment of the aldehyde (±)-199 with the phosphinimine 200 and silver(I) acetate promoted an intramolecular [3 + 2] cycloaddition, through the aza-Wittig product 201, to provide the ketone 202, which bears the C10 TBCC (68%, 7 : 1 dr at C20). The tetracycle 202 was elaborated to the diol 203 by a three-step sequence (68% overall). Sulfonylation of the hydroxyl substituents in 203 (methanesulfonyl chloride, pyridine), followed by a three-step debenzylation–reductive amination–ester reduction–reductive deoxygenation sequence (palladium on active carbon, hydrogen; lithium triethylborohydride) provided the amine 204 (63%, three steps). Conversion of the primary alcohol to a halide proceeded in low yields (product not shown), resulting in exploration of alternative radical precursors. Ultimately, the thiocarbonate 205 was accessed from 204 (phenyl chlorothionocarbonate, 4-dimethylaminopyridine, 87%). Treatment of the thiocarbonate 205 with samarium(II) iodide promoted a reductive cyclization, to provide the diene 206 (61%). The C8 TBCC was installed in this step. The diene 206 was advanced to the alcohol 207 in three steps (37% overall). Finally, a three-step sequence was developed to convert the alcohol 207 to (±)-spirasine IV (208, 43% overall). Stereo-controlled reduction of 208 (L-selectride) provided (±)-spirasine XI (209; 83%, >20 : 1 dr). Overall, the synthesis of (±)-spirasine IV (208) was completed in twenty-two steps and 0.98% overall yield and the synthesis of (±)-spirasine XI (209) was completed in twenty-three steps and 0.82% overall yield.
Scheme 26.
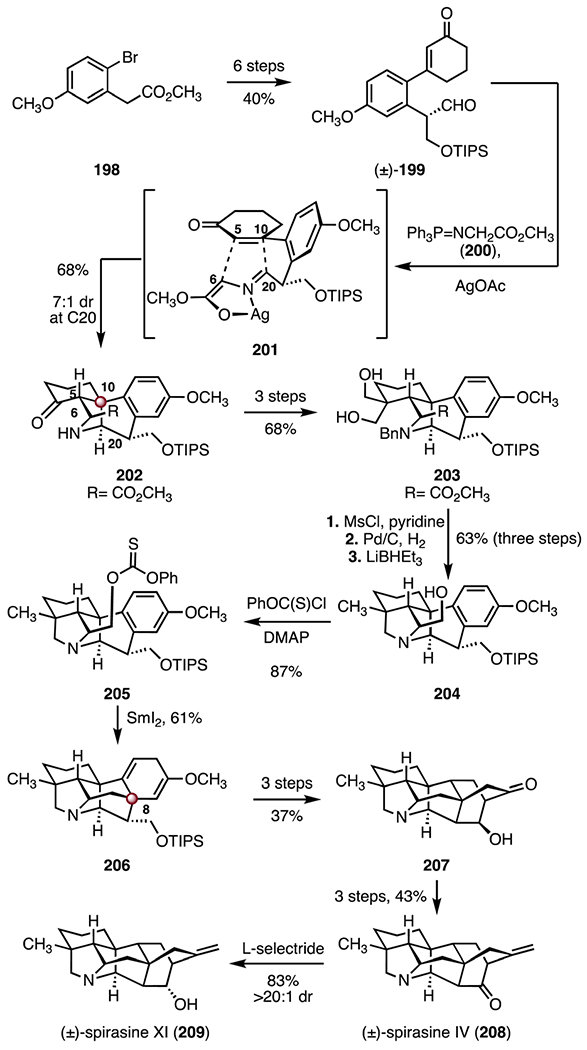
Synthesis of (±)-spirasine IV (208) and (±)-spirasine XI (209) by Zhang and co-workers.89 MsCl = methanesulfonyl chloride, DMAP = 4-dimethylaminopyridine.
The syntheses outlined above highlight the evolution of strategies from iterative individual bond constructions to concerted cycloaddition reactions in which the C8 and C10 TBCCs are constructed in a single step. In Natsume’s synthesis,85,86 the C10 quaternary center was formed early in the route by an α-arylation reaction (164 → 165). The C8 quaternary center was later introduced by an Eschenmoser–Claisen rearrangement (166 → 168). Single bond cyclization events about the two quaternary centers were then used to construct the full carbon skeleton. Gin’s synthesis87 constructed the C10 quaternary center by an aza-1,3-dipolar cycloaddition (181 → 183), while Sarpong and co-workers88 established this center by a [4 + 2] cycloaddition (187 + 188 → 189). Both Gin87 and Sarpong88 employed a base-mediated [4 + 2] cycloaddition (184 → 186, 195 → 196) to construct the bicyclo[2.2.2]octane core bearing the C8 TBCC. The stereochemical outcome of this reaction was relayed from the pre-established C10 quaternary center. Zhang’s synthesis89 employed a unique intramolecular 1,3-dipolar cycloaddition of an azamethine ylide to direct construct the C10 TBCC (199 → 202). The C8 TBCC was accessed by a reductive cyclization (205 → 206).
2.6. Arcutine alkaloids and atropurpuran: dispersed TBCCs within a rearranged hetisine core
The arcutine diterpenoids are characterized by a complex tetracyclo[5.3.3.04,9.04,12]tridecane framework that possesses TBCCs at C5 and C8 (Scheme 27). (−)-Atropurpuran (210) was isolated by Wang and co-workers from Aconitum hemslyanum var. atropurpureum in 2009, and its structure was elucidated by X-ray crystallography.101 (−)-Arcutinine (211) was first isolated by Saidkhodzhaeva and co-workers from Aconitum arcuatum in 2000. (−)-Arcutinidine (212) is the product of saponification of (−)-arcutinine (211). Their structures were elucidated by X-ray crystallography.102 Biosynthetically, the arcutine terpenoid alkaloids are proposed to arise from rearrangements of hetidine or hetisine alkaloids.103 The first total synthesis of atropurpuran (210) was reported in 2016 by Qin and co-workers,104 and a second synthesis was reported by Xu and co-workers in 2019.105 Total syntheses of arcutinine (211) and arcutinidine (212) have been reported by the Qin, Sarpong, and Li groups.106–108
Scheme 27.
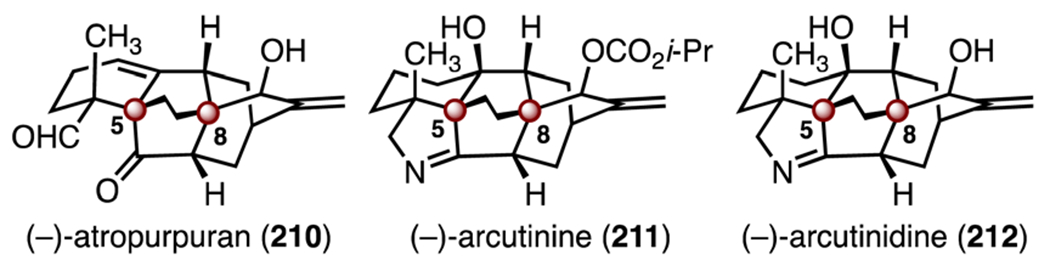
Structure of (−)-atropurpuran (210), (−)-arcutinine (211) and (−)-arcutinidine (212).
In 2016, Qin and co-workers disclosed the synthesis of (±)-atropurpuran (210, Scheme 28).104 Their approach to the complex tetracyclic [5.3.3.04,9.04,12]tridecane system relied on a late-stage ketyl–olefin cyclization and an aldol addition. The synthesis began with the aryl aldehyde 213,109 which was transformed to the acrylate 214 in four steps (42% overall). Oxidative dearomatization of 214 (di(acetoxyiodo)benzene, methanol) followed by a thermal IMDA (150 °C) provided the bicyclo[2.2.2]octane derivative (±)-215, which contains the C8 TBCC (72%, two steps). The cyclization product (±)-215 was converted to the thioester 216 by a four-step sequence (49% overall). A reductive Knoevenagel condensation between the thioester 216 and 217 (218, l-proline; then tert-butyl(chloro) diphenylsilane, triethylamine) generated the β-silyloxy enone 219 (82%).110 Reduction of the thioester (palladium on active carbon, triethylsilane), followed by a stereoselective Mukaiyama aldol addition (tetra-n-butylammonium fluoride) generated the alcohol 220, which contains the C5 quaternary stereogenic center (80%, >20 : 1 dr, two steps). A silyl ether was introduced (tert-butyldimethylsilyl trifluoromethanesulfonate, 2,6-lutidine, 90%) to bias the conformation of 221 towards a boat-like conformation. This conformation brings the C10 carbonyl and the C9 alkene in closer proximity than the corresponding chair conformer. Treatment of the silyl ether 221 with samarium(II) iodide promoted a ketyl–olefin cyclization to provide the tertiary alcohol 222, which contains the C5 TBCC (95%). The remaining functionality was then introduced by an eleven-step sequence (4.9% overall). The total synthesis of (±)-atropurpuran (210) was completed in twenty-six steps and 0.41% overall yield.
Scheme 28.
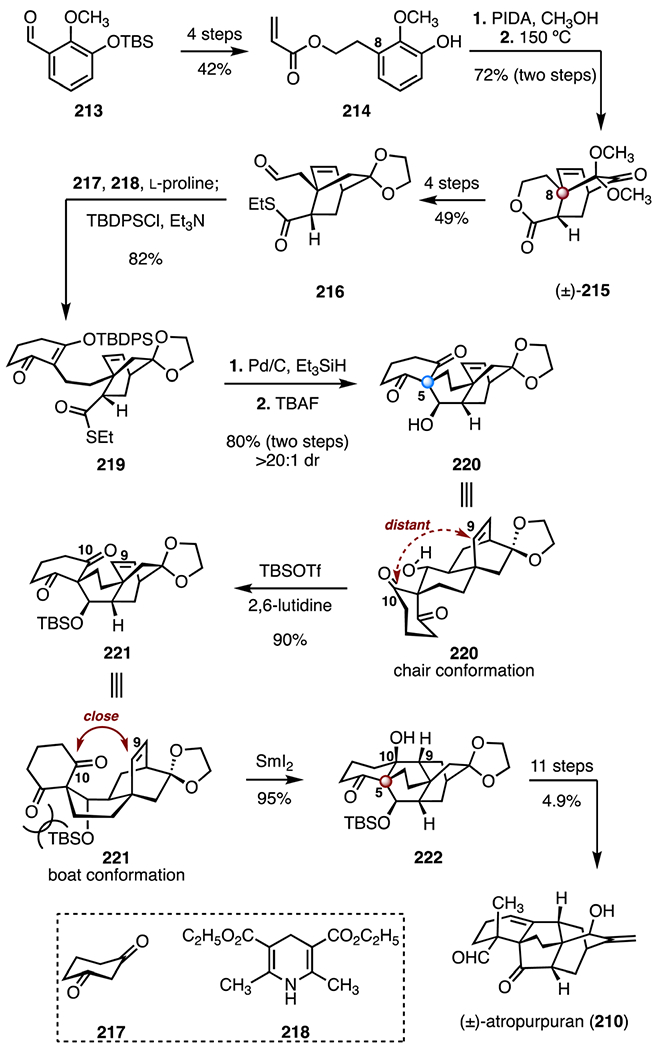
Synthesis of (±)-atropurpuran (210) by Qin and co-workers.104 PIDA = di(acetoxyiodo)benzene, TBDPSCl = tert-butyl(chloro)diphenylsilane, TBSOTf = tert-butyldimethylsilyl trifluoromethanesulfonate.
In 2019, Xu and co-workers completed the synthesis of (±)-atropurpuran (210) using a strategy that relied on generating both contiguous bicyclo[2.2.2]octane systems by a dearomatization–IMDA sequence (Scheme 29).105 Their route began with the tetralone 223. Treatment of 223 with lithium bis(trimethylsilyl) amide and the pent-4-enoyl chloride (224) followed by addition of tetra-n-butylammonium fluoride and trimethylsilyl ethynylbenziodoxolone (TMS-EBX) provided the alkynyl diketone (±)-225, which bears the C5 quaternary center (74%, two steps). Ring-closing enyne metathesis (Grubbs II catalyst), followed by cleavage of the methyl ether (boron tribromide) and diastereoselective reduction of the C4 and the C20 ketones (trimethylaluminum, lithium aluminum hydride), generated the spirocyclic diol 226 (50%). Oxidative dearomatization of the diol 226 (di(acetoxyiodo)benzene, methanol) followed by IMDA (160 °C) formed the tetracyclo[5.3.3.04,9.04,12] tridecane 227 (55%). Both the C5 and C8 TBCCs were established in this step. A five-step sequence then provided the aldehyde 228 (20% overall). The final C4 quaternary stereocenter was introduced by methylation (potassium tert-butoxide, iodomethane, 42%, 3 : 1 dr). The methylated product 229 was elaborated to (±)-atropurpuran (210) by a three-step sequence (41% overall). The total synthesis of (±)-atropurpuran (210) was completed in thirteen steps and 0.70% overall yield.
Scheme 29.
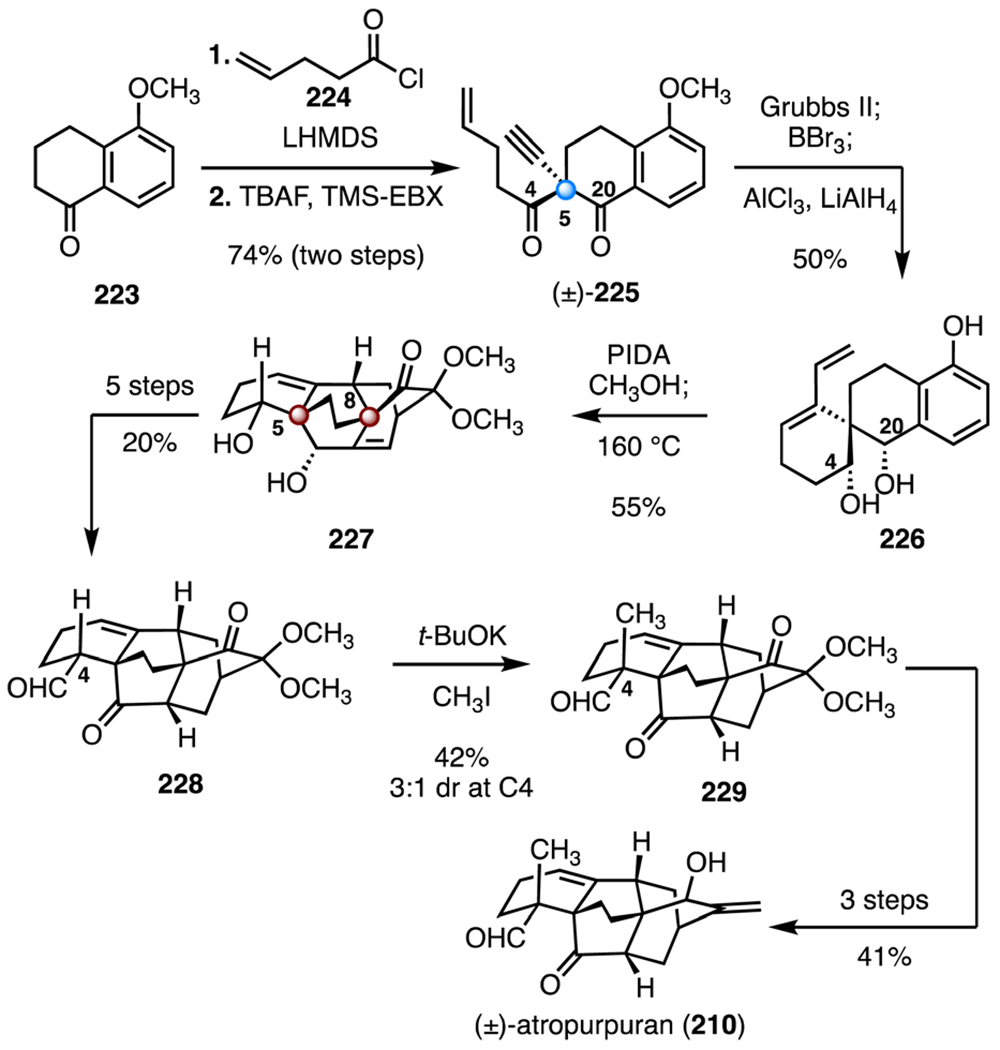
Synthesis of (±)-atropurpuran (210) by Xu and co-workers.105 LHMDS = lithium bis(trimethylsilyl) amide, TBAF = tetra-n-butylammonium fluoride, TMS-EBX = trimethylsilyl ethynylbenziodoxolone, PIDA = di(acetoxyiodo)benzene.
In 2019, Qin and co-workers reported a synthesis of (−)-arcutinine (211) and (−)-arcutinidine (212).106 Their synthetic strategy relied on an aza-Wacker reaction followed by IMDA and reductive cyclization to form the carbon skeleton (Scheme 30). Their synthesis began with an enantioselective 1,4-addition67 of trimethylaluminum to the enone 230 (copper(I) thiophene-2-carboxylate (S,S)-111), followed by an aldol addition to the aldehyde 231 in situ (45%, 92% ee).111 The resulting mixture of inseparable C5 and C6 diastereomers of the ketone 232 was transformed to the tertiary nitrile 233 by a six-step sequence (33% overall, 1 : 1.5 dr at C5). Cleavage of the methoxymethyl ether (p-toluenesulfonic acid), followed by two-fold enoxysilane formation (trimethylsilyl chloride, lithium iodide, bis(trimethylsilyl) amine) and O-allylic alkylation (methyl lithium; then, allyl chloroformate), formed the bis(allyl carbonate) 234 (43%, three steps). A palladium-catalyzed decarboxylative allylic alkylation of 234 (tetrakis(triphenylphosphine) palladium(0)) and reintroduction of the aryl methoxymethyl ether protecting group (methoxymethyl chloride, N,N-diisopropylethylamine) provided the α-allyl ketone (+)-235 (55%).112 The ketone (+)-235 was transformed to the silyl ether 236 by a four-step sequence (60% overall). An aza-Wacker reaction (palladium(II) acetate, 237, dioxygen) generated the pyrrolidine 238 (76%, 2.5 : 1 dr at C20).113 Removal of the silyl ether (tetra-n-butylammonium fluoride), oxidation (Dess–Martin periodinane), and methoxymethyl ether cleavage (trifluoroacetic acid) furnished the expected phenol (not shown). Oxidative dearomatization (di(acetoxyiodo)benzene, methanol) followed by an IMDA (65 °C) furnished the cycloadduct 239 (59%, four steps), which bears the C5 and C8 TBCCs. The synthesis of the carbon framework was completed by a samarium(II) iodide-mediated ketyl–olefin cyclization, to generate the tertiary alcohol 240 (85%). The alcohol 240 was transformed to (−)-arcutinine (211) in five steps (29% overall), which after saponification (sodium methoxide, 70%), provided (−)-arcutinindine (212). The synthesis of (−)-arcutinine (211) was completed in twenty-six steps and 0.23% overall yield. The synthesis of (−)-arcutinindine (212) was completed in twenty-seven steps and 0.16% yield.
Scheme 30.
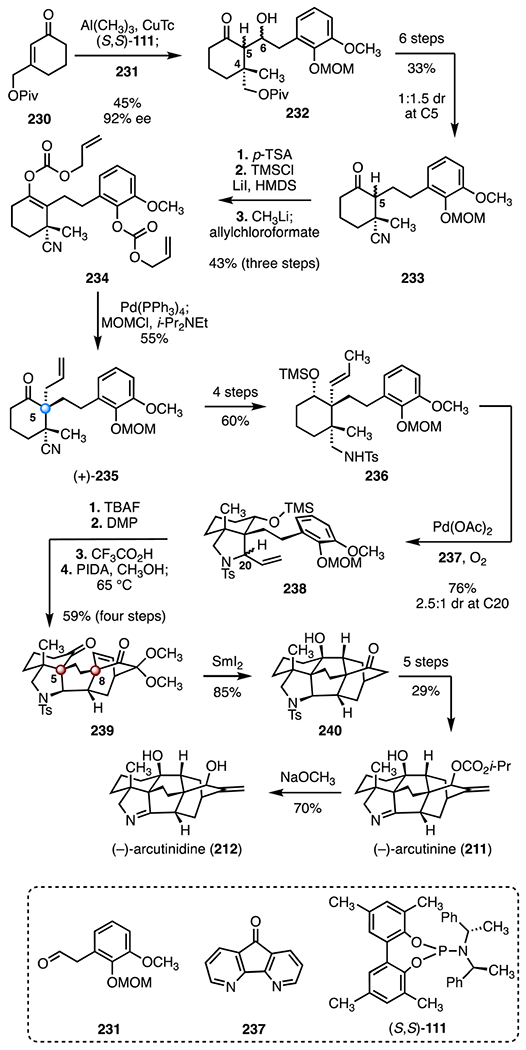
Synthesis of (−)-arcutinine (211) and (−)-arcutinidine (212) by Qin and co-workers.106 CuTc = copper(I) thiophene-2-carboxylate, p-TsOH = p-toluenesulfonic acid, TMSCl = trimethylsilyl chloride, HMDS = bis(trimethylsilyl) amine, MOMCl = methoxymethyl chloride, TBAF = tetra-n-butylammonium fluoride, DMP = Dess–Martin periodinane, PIDA = di(acetoxyiodo)benzene.
In 2019, Sarpong and co-workers disclosed a synthesis of (±)-arcutinidine (212).107 Their synthetic strategy employed two [4 + 2] cycloadditions to construct the carbon skeleton (Scheme 31). The route began with conversion of citracomimide (241)114,115 to the tethered bicyclic ether 242 (52%, two steps), which was then advanced to the tricyclic hemiaminal (±)-243 by a Friedel–Crafts cyclization (trifluoromethanesulfonic acid, 89%). Treatment of (±)-243 with the diene 244 in the presence of aluminum chloride furnished the tetracycle 245, through generation of an oxopyrrolium dienophile (78%). This first [4 + 2] cycloaddition constructed the vicinal C4 quaternary center and C5 TBCC in a single transformation. A two-step sequence was developed to elaborate 245 to the amide 246 (78% overall). The amide 246 was transformed to the phenol 247 by reduction (lithium aluminum hydride), acylation (acetic anhydride, pyridine) and demethylation (trimethylsilyl iodide, 65%, three steps). Exposure of the phenol 247 to lead(IV) acrylate (generated in situ from acrylic acid and lead(iv) acetate) provided the α-keto vinyl acrylate intermediate 248, which then underwent a second [4 + 2] cycloaddition upon heating to 110 °C, to form the cycloadduct 249 bearing the C8 TBCC (60%, two steps). The adduct 249 was transformed to the diketone 250 in three steps. A pinacol coupling (samarium(II) iodide) furnished the vicinal diol 251 (25%, four steps), which was converted to (±)-arcutinidine (212) in nine steps (0.75% overall). The synthesis of (±)-arcutinidine (212) was completed in twenty-four steps and 0.021% overall yield.
Scheme 31.
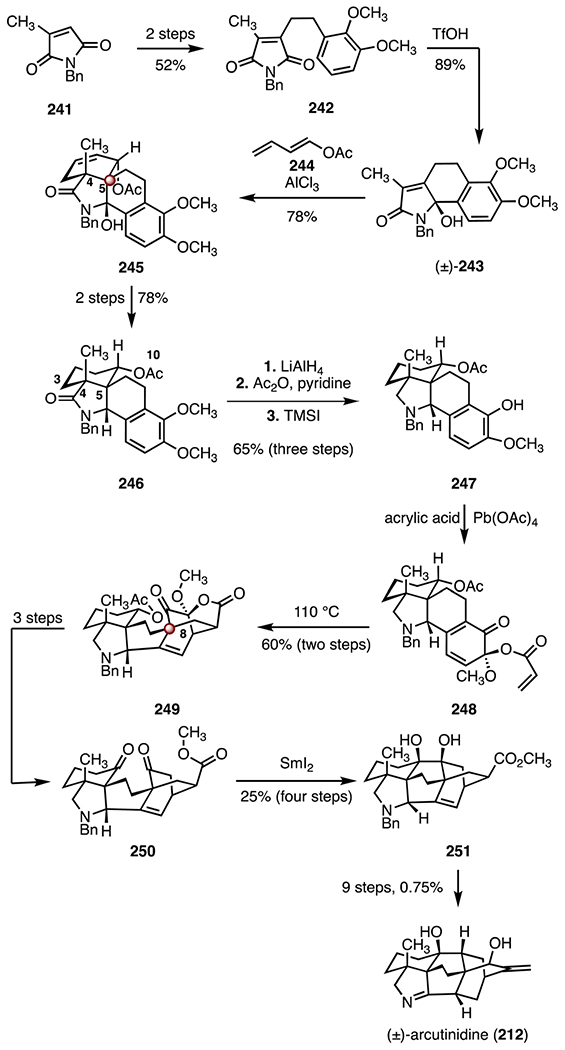
Synthesis of (±)-arcutinidine (212) by Sarpong and co-workers.107 TfOH = trifluoromethanesulfonic acid.
In 2019, Li and co-workers disclosed a synthesis of (−)-arcutinine (211) and (−)-arcutinidine (212).108 Li’s strategy employed two [4 + 2] cycloadditions, a biomimetic cationic rearrangement, and a late-stage reductive amination (Scheme 32). The synthesis began with the enantioenriched alcohol (−)-252 (>99% ee),116 which was converted to the enal 253 by a two-step sequence (85% overall). Lewis acid-catalyzed [4 + 2] cycloaddition (boron trifluoride diethyl etherate complex) between the enal 253 and the diene 254 then provided the tricyclic alkene 255 (68%). Stereoselective 1,2-addition (vinyllithum), protection of the resulting alcohol (methoxymethyl chloride) and ketal removal (perchloric acid) with concomitant alkene isomerization furnished the enone 256 (61%, two steps). An anionic [4 + 2] cycloaddition was achieved by treatment of the enone 256 with lithium bis(trimethylsilyl)amide (through 257); cleavage of the silyl ether (tetra-n-butylammonium fluoride) in situ then delivered the ketone 258 (85%).117 The alkene 259 was accessed by formal dehydration of 258 (thionyl chloride, pyridine, 76%). Exposure of the alkene 259 to tin(IV) chloride furnished the cyclic ether 262 (63%). This transformation was hypothesized to proceed by ionization of the methoxymethyl ether (259 → 260), Prins cyclization (260 → 261), 1,2-alkyl shift, and proton elimination (261 → 262). The ketone 262 was elaborated to the aldehyde 263 by a seven-step sequence (17% overall). Oxime formation and enone reduction (hydroxylamine hydrochloride, sodium acetate; then, sodium borohydride), followed by selective reduction (titanium(III) chloride, sodium cyanoborohydride) and imine formation furnished (−)-arcutinidine (212; 46%, two steps).118 Acylation of 212 (4-(dimethylamino)pyridine, isobutyric acid, N,N′-dicyclohexylcarbodiimide, 77%) provided (−)-arcutinine (211). The syntheses of (−)-arcutinidine (212) and (−)-arcutinine (211) were achieved in seventeen steps and 1.1% overall yield, and eighteen steps and 0.86% overall yield, respectively.
Scheme 32.
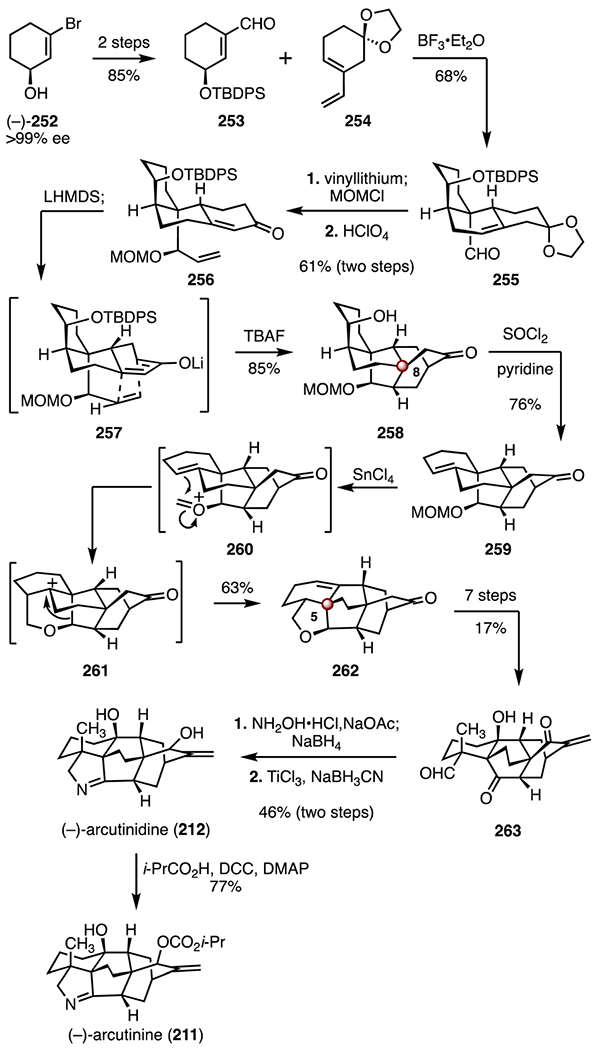
Synthesis of (−)-arcutinidine (212) and (−)-arcutinine (211) by Li and co-workers.108 MOMCl = methoxymethyl chloride, LHMDS = lithium bis(trimethylsilyl)amide, TBAF = tetra-n-butylammonium fluoride, DCC = N,N′-dicyclohexylcarbodiimide, DMAP = 4-dimethylaminopyridine.
The syntheses discussed all employed two-bond disconnections by [4 + 2] cycloaddition reactions. Qin and co-workers104 employed an oxidative–dearomatization IMDA early in the route to form the C8 quaternary carbon (214 → 215) in their synthesis of (±)-atropurpuran (211). This in turn allowed them to construct the C5 TBCC by an aldol addition (219 → 220) and a ketyl–olefin cyclization (221 → 222). The Xu group105 also utilized a dearomative [4 + 2] cycloaddition to establish the tetracyclo[5.3.3.04,9.04,12]tridecane system (226 → 227). The C5 center was accessed by asymmetric alkylation (223 → 225) and enyne metathesis (225 → 226). The Qin group106 formed the C5 quaternary center of the arcutine-type alkaloids by a decarboxylative allylic alkylation (234 → 235), followed by a dearomatization–IMDA sequence to install the C8 TBCC (238 → 239). The Sarpong synthesis107 employed a late-stage IMDA reaction to form the bicyclo[2.2.2]octane (248 → 249) and the C8 TBCC, while constructing the vicinal C4 and C5 quaternary centers by a second [4 + 2] cycloaddition involving a novel oxopyrrolium dienophile (243 + 244 → 245). The Li group108 took advantage of a biomimetic cationic rearrangement to construct the C5 TBCC (259 → 262). An anionic [4 + 2] cycloaddition enabled the C8 TBCC formation (256 → 258). The conformational rigidity of caged polycyclic structures results in reactivity driven by well-defined conformations. The reductive cyclization (221 → 222) in Qin’s synthesis of (±)-atropurpuran (210)104 illustrated the strategic use of protecting groups to manipulate the conformation of a molecule and to enable desired reactivity.
2.7. Bukittinggine alkaloids: congested cluster of TBCCs within a terpenoid alkaloid scaffold
The daphniphyllum alkaloids have attracted much attention due to their complex structures.119,120 One of the most synthetically challenging members of this family are the bukittinggine alkaloids, which contain a heptacyclic ring system bearing TBCCs at C5, C8 and C10 (Scheme 33). Related daphniphyllum alkaloids have been the subject of many synthetic studies.121,122 By comparison, only two total syntheses of bukittinggine-type alkaloids have been disclosed. (±)-Bukittinggine (264) was synthesized by Heathcock and co-workers in 1992.123 (−)-Caldaphnidine O (265) was synthesized by Xu and co-workers in 2019.124 (−)-Bukittinggine (264) was isolated in 1990 from Sapium baccatum by Cannon and co-workers. Its structure was determined through X-ray analysis of its hydrobromide salt.125 (−)-Bukittinggine (264) possesses anti-inflammatory activities.126 (−)-Caldaphnidine O (265) was isolated from the twigs of Daphniphyllum calycinum by Yue and co-workers in 2008. Its structure was determined by NMR spectroscopy.127
Scheme 33.
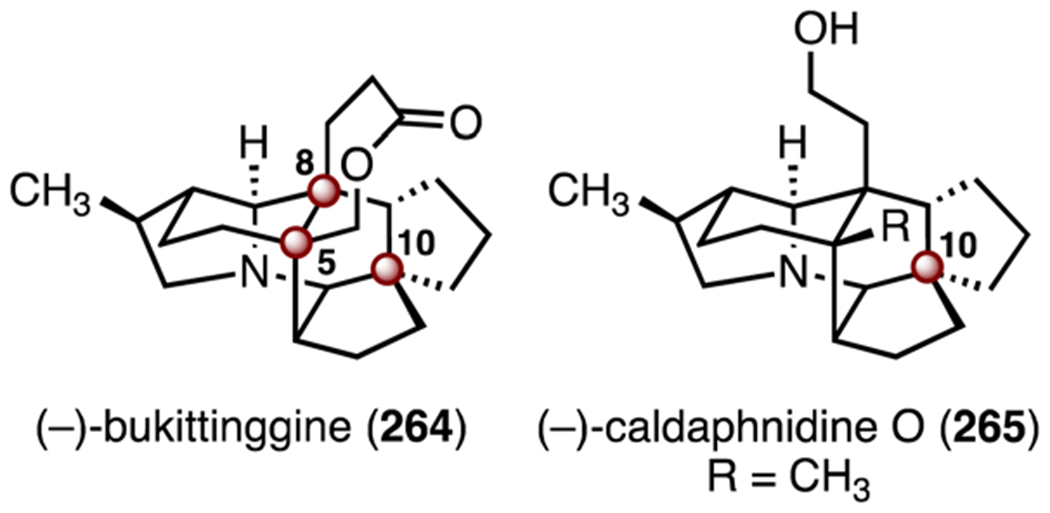
Structures of (−)-bukittinggine (264) and (−)-caldaphnidine O (265).
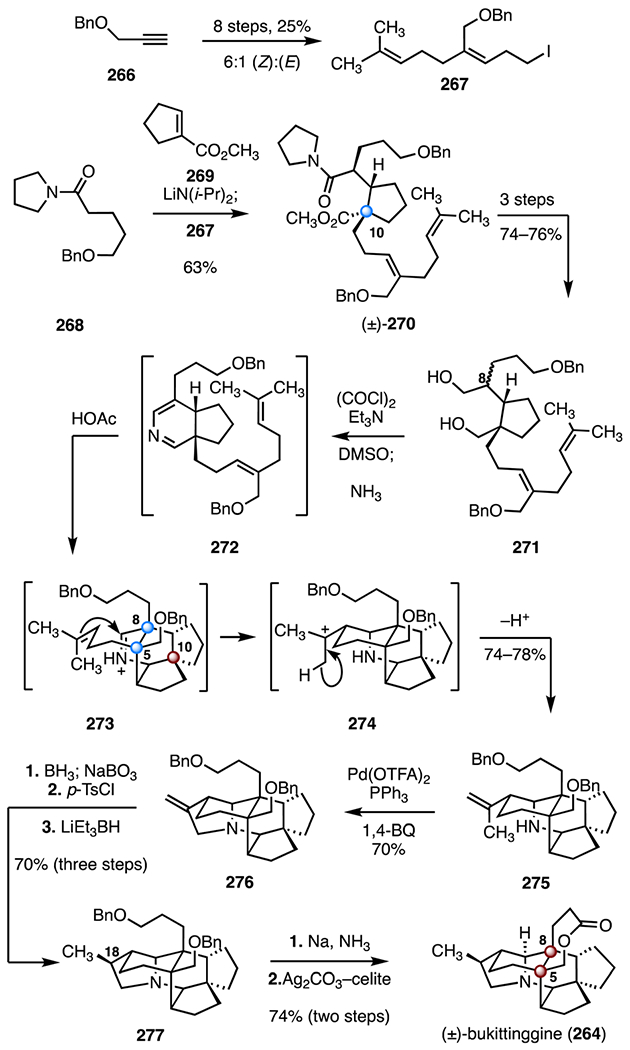
Heathcock and co-workers employed a [4 + 2] cycloaddition–aza-Prins cascade to form the tetracyclic core of bukittinggine (264; Scheme 34).123 The diene 267 was prepared from the alkynyl benzyl ether 266 in eight steps and as a 6 : 1 mixture of (Z)- and (E)-diastereomers (25% overall). The methyl ester (±)-270, which contains the C10 quaternary center, was prepared in a convergent fashion by a one-flask Michael addition–alkylation between the diene 267, the amide 268, and the ester 269 (lithium diisopropylamide, 63%). A three-step sequence then provided the diol 271, as an inconsequential C8 diastereomeric mixture (74–76% overall). Swern oxidation of the diol (oxalyl chloride, triethylamine, dimethylsulfoxide) followed by sequential addition of ammonia and acetic acid, provided the polycyclic amine 275 (74–78%). This transformation was thought to proceed by an intramolecular inverse-electron-demand Diels–Alder reaction, initiated by protonation of the 2-aza diene intermediate 272 (272 → 273), an aza-Prins cyclization (273 → 274) and an elimination (274 → 275). Three rings, the C5 and C8 quaternary stereogenic centers, and the C10 TBCC were generated in this transformation. Palladium-mediated cyclization of the amine 275 (palladium(II) trifluoroacetate, triphenylphosphine, 1,4-benzoquinone, 70%) delivered the pyrrolidine 276.128
Scheme 34.
Synthesis of (±)-bukittinggine (264) by Heathcock and co-workers.123 DMSO = dimethylsulfoxide, p-TsCl = p-toluenesulfonic chloride, 1,4-BQ = 1,4-benzoquinone, TFA = trifluoroacetate.
Hydroboration–oxidation of the pyrrolidine 276 (borane, sodium perborate), sulfonylation of the resulting alcohol (p-toluenesulfonic chloride), and reduction (lithium triethylborohydride) furnished the amine 277 (70%, three steps). A debenzylation–oxidation sequence (sodium, ammonia; silver(I) carbonate on Celite, 74%, two steps) was developed to obtain (±)-bukittinggine (264). (±)-Bukittinggine (264) was synthesized in nineteen steps and 3.1–3.4% overall yield.
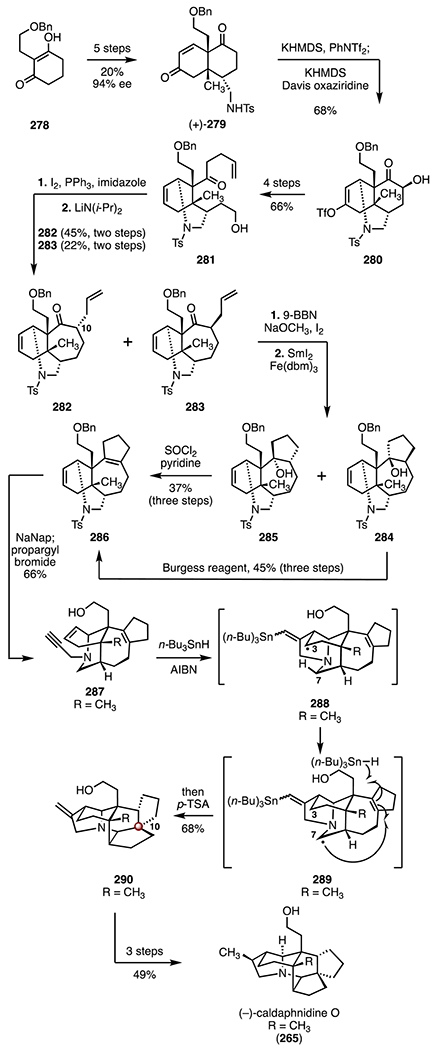
Xu and co-workers developed a late-stage radical cyclization cascade to form the C10 TBCC in (−)-caldaphnidine O (265; Scheme 35).124 The synthesis began with the benzyl ether 278,129 which was advanced in five steps to the enantio-enriched bicyclic enone (+)-279 (20% overall, 94% ee).130 Intramolecular aza-Michael addition (IMAM) and trifluoromethanesulfonylation of the resulting enolate (potassium bis(trimethylsilyl)amide, bis(trifluoromethanesulfonyl)aniline), followed by α-hydroxylation of the ketone (potassium bis(trimethylsilyl)amide, Davis oxaziridine), provided the hydroxy ketone 280 as a single diastereomer (68%). A four-step sequence was developed to convert the α-hydroxy ketone 280 to the homoallylic ketone 281 (66% overall). Deoxyiodination (iodine, triphenylphosphine, imidazole), followed by an intramolecular alkylation (lithium diisopropylamide) furnished a mixture of the diastereomeric cycloheptanones 282 (45%, two steps) and 283 (22%, two steps). Hydroboration–iodination (9-borabicyclo[3.3.1]nonane; then, sodium methoxide, iodine) followed by reductive coupling (samarium(II) iodide, iron tris(diisobutyrylmethane)) provided the tricyclic alcohols 284 and 285.131–133 Dehydration of either diastereomer (thionyl chloride, pyridine for 285, 37%, three steps; Burgess reagent for 284, 45%, three steps) then formed the cyclopentene 286. Reductive removal of the sulfonamide and benzyl ether protecting groups (sodium naphthalenide), followed by in situ N-propargylation (propargyl bromide), furnished the enyne 287 (66%). Exposure of the enyne 287 to azobisisobutyronitrile and tri-n-butyltin hydride, followed by addition of p-toluenesulfonic acid, yielded the alkene 290, which bears the C10 TBCC (68%). This reaction was proposed to proceed through sequential 5-exo-trig cyclization (287 → 288), 1,5-hydrogen atom transfer (288 → 289), transannular 5-exo-trig cyclization, reduction of the C9 radical, and destannylation (289 → 290). A three-step sequence then provided (−)-caldaphnidine O (265, 49% overall). The synthesis of (−)-caldaphnidine O (265) was completed in twenty-one steps and 0.48% overall yield.
Scheme 35.
Synthesis of (−)-caldaphnidine O (265) by Xu and co-workers.124 KHMDS = potassium bis(trimethylsilyl)amide, PhNTf2 = bis(trifluoromethanesulfonyl)aniline, 9-BBN = 9-borabicyclo[3.3.1]nonane, Fe(dbm)3 = iron tris(diisobutyrylmethane), NaNap = sodium naphthalenide, AIBN = azobisisobutyronitrile, p-TSA = p-toluenesulfonic acid.
The syntheses discussed above illustrate two distinct approaches to these alkaloid targets. Heathcock and co-workers123 employed a Michael addition–alkylation early in the synthesis to construct the C10 quaternary center (268 → 270). A carefully orchestrated inverse-electron-demand Diels–Alder (IEDDA)/aza-Prins cyclization cascade furnished the C5 and C8 quaternary stereogenic centers and the C10 TBCC in a single step (271 → 275). The Xu group124 employed a distinct, late-stage transannular skeletal cyclization cascade (287 → 290) to access the carbon skeleton and the C10 TBCC.
2.8. Akuammiline alkaloids: an indole-containing isolate bearing a TBCC at the 3-position of the indole
The akuammiline alkaloids are indole diterpenoid alkaloids isolated from Africa, India, and Southeast Asia.134 They possess opioid, cytotoxic, and glycine receptor antagonist activities.135 The akuammiline alkaloids contain a pentacyclic skeleton comprised of a tricyclic furoindoline residue (Scheme 36). The skeleton contains one TBCC at C7. Numerous syntheses and synthetic studies toward these isolates have been disclosed.136 Here, we discuss syntheses of two representative isolates, (−)-aspidophylline A (291)137–139 and (+)-strictamine (292).140–142
Scheme 36.
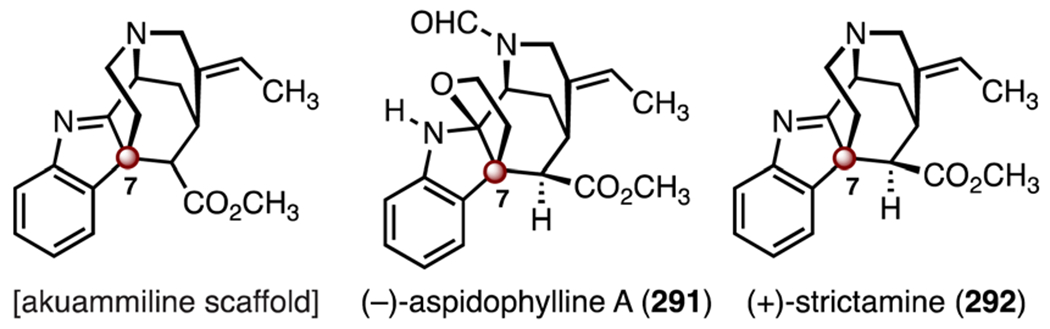
Structures of the akuammiline alkaloid scaffold, (−)-aspidophylline A (291), and (+)-strictamine (292).
(−)-Aspidophylline A (291) was isolated by Kam and co-workers in 2007. Its structure was determined by NMR spectroscopy and mass spectrometry. In the same study, (−)-aspidophylline A (291) was found to reverse drug resistance in cancer cells (KB).143 (+)-Strictamine (292) was first isolated by the Ganguli group from Rhazya stricta Decaisne in 1966. Its structure was initially elucidated by NMR spectroscopy and MS,144 and later confirmed by X-ray crystallographic analysis.145
In 2014, Ma and co-workers disclosed a synthesis of (±)-aspidophylline A (291) that employed an oxidative coupling reaction to form the C7 TBCC (Scheme 37).137 Their synthesis began with the silyl ether 293, which was converted to the azide (±)-294 by a four-step sequence (23% overall). An intramolecular oxidative coupling of (±)-294 (lithium bis(trimethylsilyl)amide, iodine, 54%, 2 : 1 dr at C3) provided a diastereomeric mixture of the ethers 296 with the C7 TBCC established.146 It was hypothesized that the free primary hydroxyl group in (±)-294 promoted formation of the chelated intermediate 295, which facilitated C7–C16 bond formation. The unsaturated ester 297 was prepared in four steps from the ether 296 (22% overall). A Staudinger reduction (triphenyl-phosphine), followed by N-alkylation (298, cesium carbonate) and N-formylation (formic acid, N,N′-diisopropylcarbodiimide, 4-dimethylaminopyridine), produced the vinyl iodide 299 (51%, three steps). Reductive cyclization (bis(cyclooctadiene) nickel(0), triethylamine) then furnished the pentacycle 300 (58%).147 Finally, removal of the tert-butyloxycarbonyl protecting group (trimethylsilyl trifluoromethanesulfonate, 95%) delivered (±)-aspidophylline A (291). The synthesis was completed in fourteen steps and 0.77% overall yield.
Scheme 37.
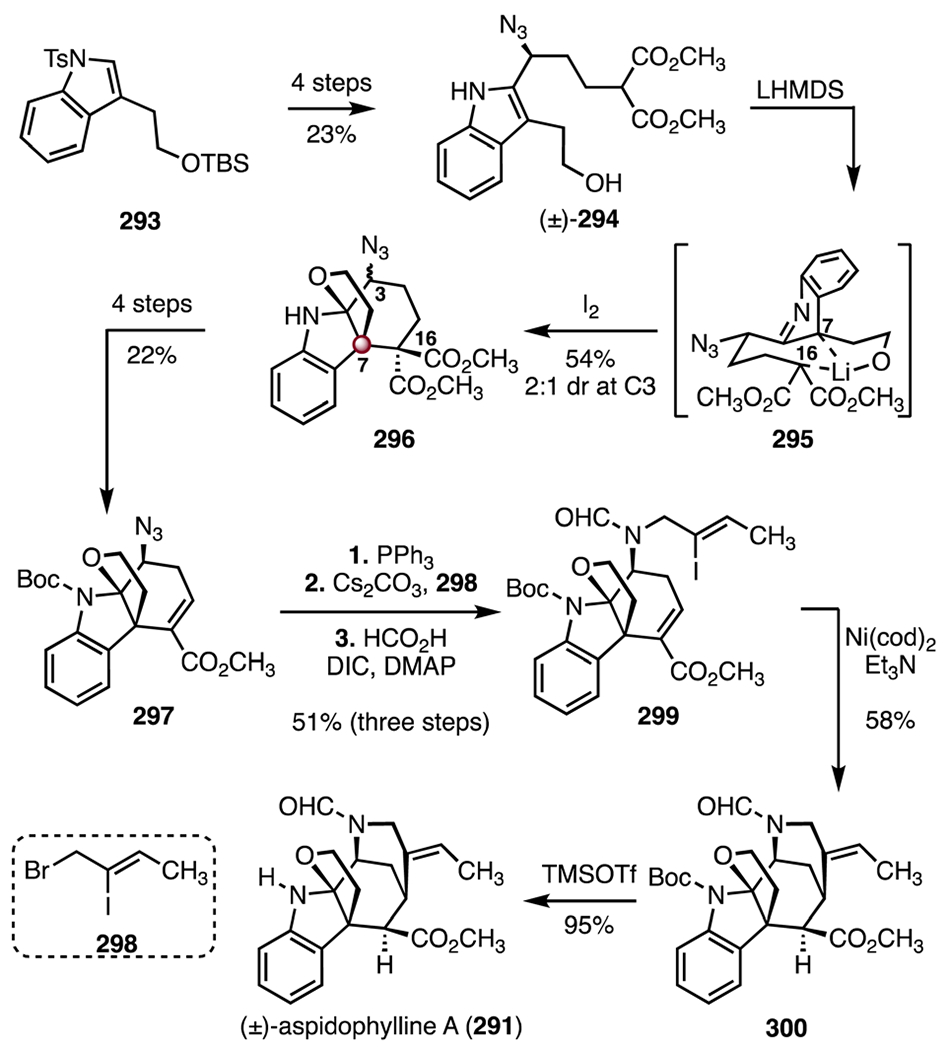
Synthesis of (±)-aspidophylline A (291) by Ma and co-workers.137 LHMDS = lithium bis(trimethylsilyl)amide, DIC = N,N′-diisopropylcarbodiimide, DMAP = 4-dimethylaminopyridine, Ni(cod)2 = bis(cyclooctadiene) nickel(0), TMSOTf = trimethylsilyl trifluoromethanesulfonate.
In 2014, Zhu and co-workers disclosed a total synthesis of (±)-aspidophylline A (291) that employed an oxidative azidoalkoxylation reaction to form the C3–N bond (Scheme 38).138 The known diketone (±)-301 was prepared in three steps from cyclohexane-1,3-dione (217, 53% overall). Here, the C7 quaternary center was established by SNAr and allylic alkylation reactions.148 A two-step sequence was developed to transform 301 to the vinyl trifluoromethanesulfonate 302 (trimethylsilyl chloride; bis(trifluoromethanesulfonyl)aniline, 55%, two steps). Reduction of the nitro group (titanium(iii) chloride, ammonium acetate), followed by in situ imine formation, provided the indolenine 303 (80%). Acylation of the indolenine 303 (methylchloroformate, sodium hydride) followed by oxidative cleavage and aldehyde reduction (osmium(viii) oxide, N-methylmorpholine N-oxide; then, sodium periodate; sodium borohydride; then, hydrochloric acid) generated the hemiaminal 304 (60%, three steps). The C7 TBCC was established in this step by hemiaminal formation. A two-step sequence was developed to convert the hemiaminal 304 to the enamine 305. In a key transformation, the enamine 305 was treated with ceric ammonium nitrate and sodium azide, resulting in production of the alkyl azide 307 as a single diastereomer after separation (49%, three steps, 1:1 dr at C3).149 This reaction was hypothesized to proceed through the radical cation intermediate 306. A two-step sequence was developed to convert the azide 307 to the vinyl iodide 308 (64% overall). An intramolecular 1,4-addition (tert-butyl lithium, trimethylsilyl chloride) then furnished the pentacycle 309 (51%). (±)-Aspidophylline A (291) was obtained by a two-step sequence (53% overall). The synthesis was completed in seventeen steps and 1.2% overall yield.
Scheme 38.
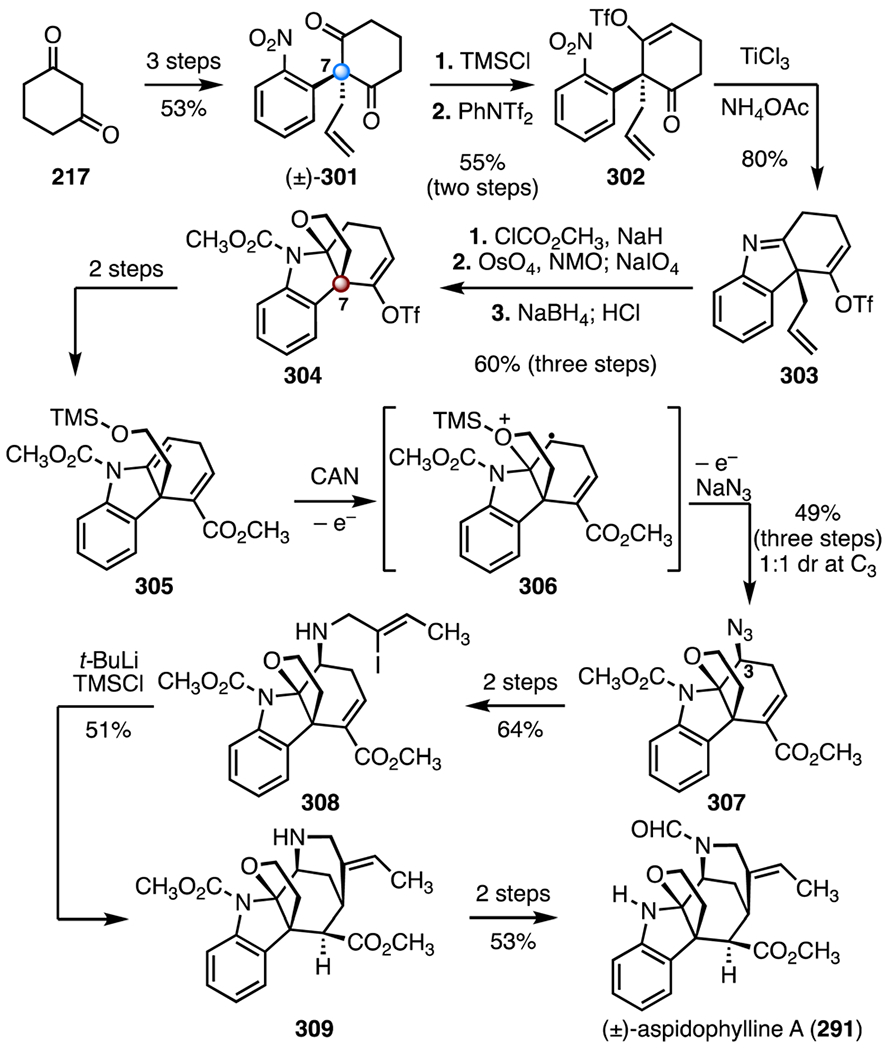
Synthesis of (±)-aspidophylline A (291) by Zhu and co-workers.138 TMSCl = trimethylsilyl chloride, PhNTf2 = bis(trifluoromethanesulfonyl) aniline, NMO = N-methylmorpholine N-oxide, CAN = ceric ammonium nitrate.
In 2016, the Yang group reported an enantioselective synthesis of (−)-aspidophylline A (291) that employed an enantioselective iridium-catalyzed iminium ion cyclization to form the C7 TBCC (Scheme 39).139 The synthesis began with a Jiao–Bach alkylation of the indole 310 (palladium(ii) chloride, norbornene, ethyl 4-bromobutyrate, potassium carbonate, air), to form the ester 311 (85%).150,151 The ester 311 was converted to the allylic alcohol (±)-312 in four steps (51% overall). In a key reaction, the allylic alcohol (±)-312 underwent an enantioselective iridium-catalyzed cyclization (chloro(1,5-cyclooctadiene)iridium(I) dimer, (S)-313, zinc(II) trifluoromethanesulfonate) to the cyclic aminal (−)-315, through the iminium ion intermediate 314 (75%, 5 : 1 dr at C16, 98% ee). The aminal (−)-315 was elaborated to the unsaturated ester 316 in four steps (40% overall). An oxidative azidoalkoxylation (trimethylsilyl trifluoromethanesulfonate, 2,6-lutidine, sodium azide, ceric ammonium nitrate)138 provided the azide 317 (42%, 1:1 dr at C3). A three-step sequence was developed to transform the azide 317 to the vinyl iodide 318 (65% overall). Finally, a reductive cyclization, promoted by bis(cyclooctadiene) nickel(0) and triethylamine, followed by removal of the acetamide (sodium methoxide, 32%, two steps), delivered (−)-aspidophylline A (291). The route was completed in sixteen steps and 1.1% overall yield.
Scheme 39.
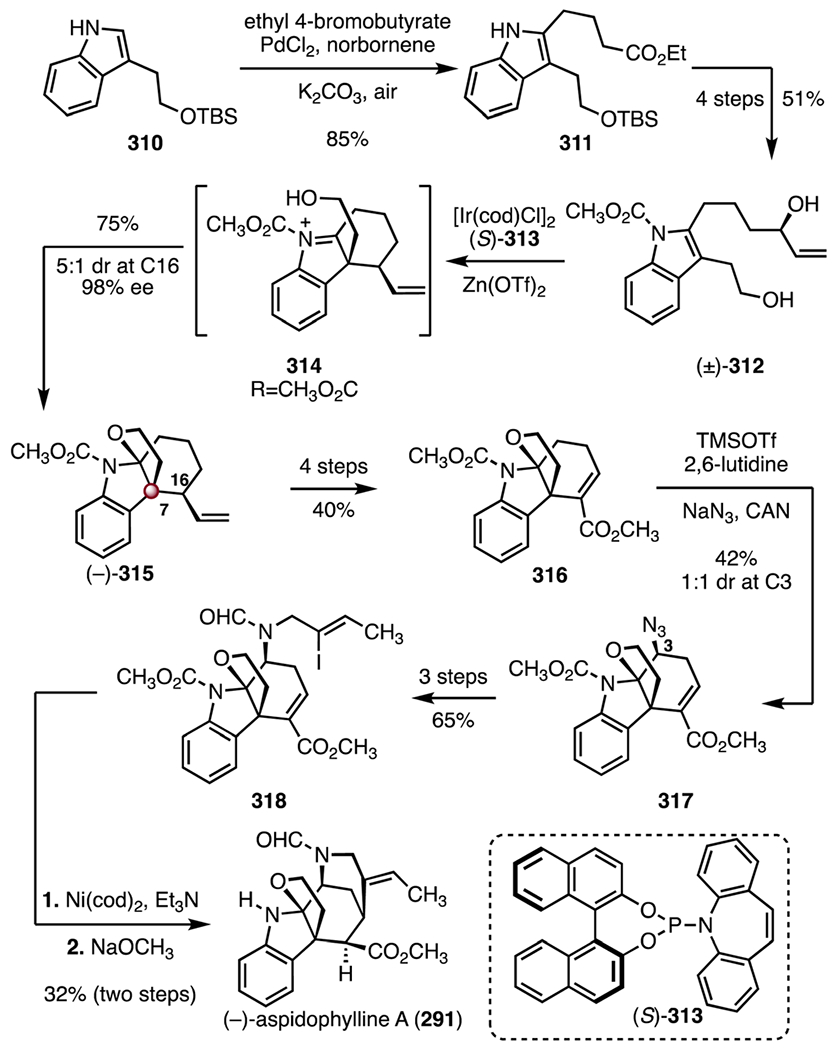
Synthesis of (−)-aspidophylline A (291) by Yang and co-workers.139 [Ir(cod)Cl]2 = chloro(1,5-cyclooctadiene)iridium(I) dimer, Zn(OTf)2 = zinc(II) trifluoromethanesulfonate, TMSOTf = trimethylsilyl trifluoromethanesulfonate, CAN = ceric ammonium nitrate, Ni(cod)2 = bis(cyclooctadiene) nickel(0).
In 2016, Garg and co-workers reported the first synthesis of (+)-strictamine (292).140,141 The route employed an interrupted Fischer indolization as a key step (Scheme 40).152 The synthesis began with an enantioselective desymmetrization153 of the bis(benzoate) (±)-319 (320, allylpalladium(II) chloride dimer, (R,R)-321, cesium carbonate); hydrolysis of the benzoyl group (lithium hydroxide) provided the allylic alcohol (−)-322 (89%, 96% ee, two steps). A two-step sequence was developed to convert the allylic alcohol (−)-322 to the enoxysilane 323 (83% overall). A gold-catalyzed cyclization of the enoxysilane 323 (chloro(trimethylphosphine) gold(I), silver trifluoromethanesulfonate, tert-butyl alcohol; then, p-toluenesulfonic acid)154 delivered the bicyclic enone 324, which was converted to the hydroxyl enal 325 by epoxidation (sodium perborate tetrahydrate) and Wittig olefination ((methoxymethyl)triphenylphosphonium chloride, 49%, three steps). The lactone 326 was obtained by a seven-step sequence (28% overall). In a key reaction, treatment of the lactone 326 with phenylhydrazine and trifluoroacetic acid, followed by addition of triethylsilane, provided the pentacycle 330, which bears the C7 TBCC (83%). The reaction was proposed to proceed by [3,3]-sigmatropic rearrangement (327 → 328), imine formation (328 → 329), and silane reduction (329 → 330). The pentacycle 330 was converted to the alkyl chloride 331 in seven steps (20% overall). Finally, oxidation of the alkyl chloride 331 (pyridinium chlorochromate), followed by deprotection and N-alkylation (MetSThiol®)155 provided (+)-strictamine (292; >99%, two steps). The synthesis was completed in twenty-four steps and 1.7% overall yield.
Scheme 40.
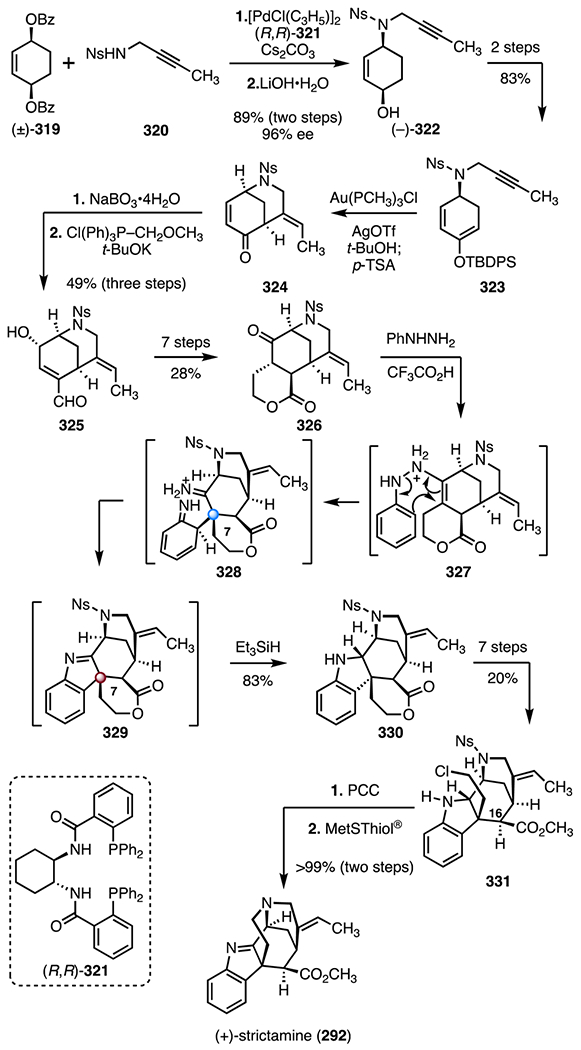
Synthesis of (+)-strictamine (292) by Garg and co-workers.140,141 AgOTf = silver(I) trifluoromethanesulfonate, p-TSA = p-toluenesulfonic acid, PCC = pyridinium chlorochromate.
In 2019, the Qin group disclosed an enantioselective synthesis of (−)-strictamine (292) that employed a photocatalytic radical cyclization as a key step (Scheme 41).142 Enantioselective Michael addition of tert-butyl methyl malonate to the enal 332, promoted by the chiral pyrrolidine derivative (S)-333, followed by an acid-mediated deprotection and decarboxylation (trifluoroacetic acid; then, triethylamine), generated the aldehyde (+)-334 (46%, 96% ee, two steps).156 Exposure of the aldehyde (+)-334 to 4-methoxybenzylamine, followed by nitro reduction (zinc) and sulfonylation (p-toluenesulfonyl chloride, pyridine), provided the enamide 335 (47%, three steps). In a key transformation, irradiation of a mixture of the enamide 335, the iridium catalyst 336, and acrolein with blue LEDs (350 nm) generated the aldehyde 338, likely by formation of the N-sulfonyl aminal radical 337, 5-exo-trig cyclization and radical 1,4-addition to acrolein. Addition of vinyl magnesium bromide to the aldehyde intermediate 338 followed by removal of the N-sulfonyl substituent (sodium naphthalenide) provided the indoline 339 as an inseparable mixture of diastereomers at C16 (47%, 1.15 :1 dr). A two-step sequence was developed to convert the indoline 339 to the indole 340 (83% overall, 1.15 : 1 dr at C16). Tsuji–Trost cyclization (C7–C16 bond formation; tetra-kis(triphenylphosphine) palladium(0)) provided the alkene 341, which bears the C7 TBCC (74%, 5:1 dr at C16). Amide ethanolysis (sodium hydroxide, ethanol) and reduction (lithium aluminum hydride) generated the tricyclic alcohol 342 (60%, two steps). A seven-step sequence was developed to convert the alcohol 342 to the diol 343 (25% overall). A nickel-catalyzed reductive cyclization (bis(cyclooctadiene) nickel(0), triethylamine) provided the exocyclic alkene 344 (61%). (−)-Strictamine (292) was then obtained by a five-step sequence (22% overall). The synthesis was completed in twenty-four steps and 0.13% yield.
Scheme 41.
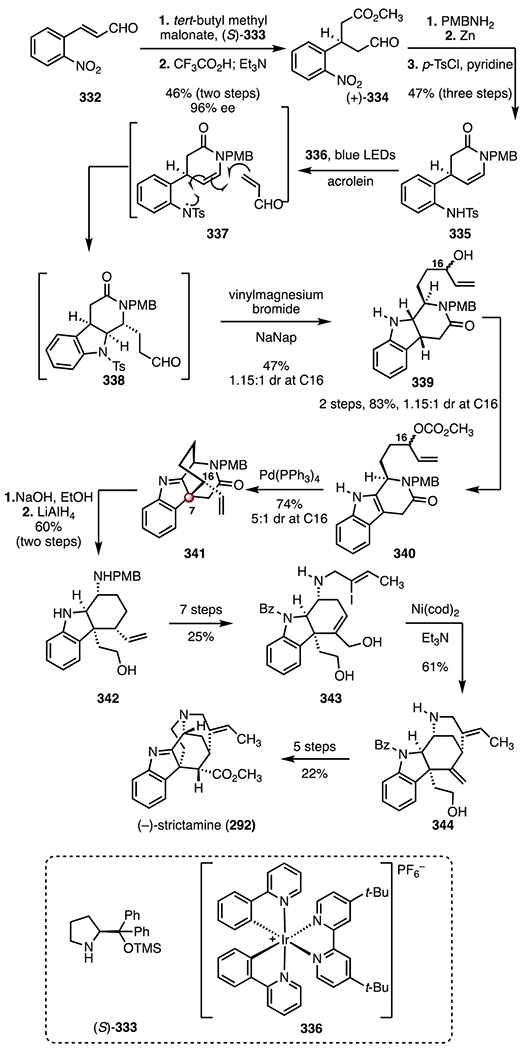
Synthesis of (−)-strictamine (292) by Qin and co-workers.142 PMBNH2 = 4-methoxybenzylamine, p-TsCl = p-toluenesulfonyl chloride, NaNap = sodium naphthalenide, Ni(cod)2 = bis(cyclooctadiene) nickel(0).
All syntheses except the enolate-based strategy of Zhu and co-workers138 (217 → 301) constructed the C7 quaternary center by nucleophilic C–C bond formation from a sp2-hybridized precursor. Ma and co-workers137 utilized a chelation-controlled intramolecular oxidative coupling to form the C7–C16 bond (294 → 296). Yang and co-workers139 developed an iridium-catalyzed cyclization to construct the C7 TBCC with control of absolute stereochemistry (312 → 315). Qin and co-workers142 used a Tsuji–Trost reaction (340 → 341) to form the C7–C16 bond in a diastereoselective fashion. Garg and co-workers140,141 employed an interrupted Fischer indolization reaction to simultaneously form the C7 TBCC and the AB rings (326 → 330).
2.9. Kopsia alkaloids: congested cluster of TBCCs within an indole alkaloid
Kopsia indole alkaloids have been isolated from multiple Kopsia species157 and exhibit a diverse range of biological activities, including anticancer,158–160 antileishmania,161 antimanic,162 antitussive,163 antibacterial, and antifungal164 activities. Structurally, kopsia alkaloids are categorized into four classes (Scheme 42), all possessing polycyclic structures with TBCCs at C7 and C20. Over ten members of the kopsia alkaloid family have been synthesized to date;165 selected syntheses are discussed here.166–171
Scheme 42.
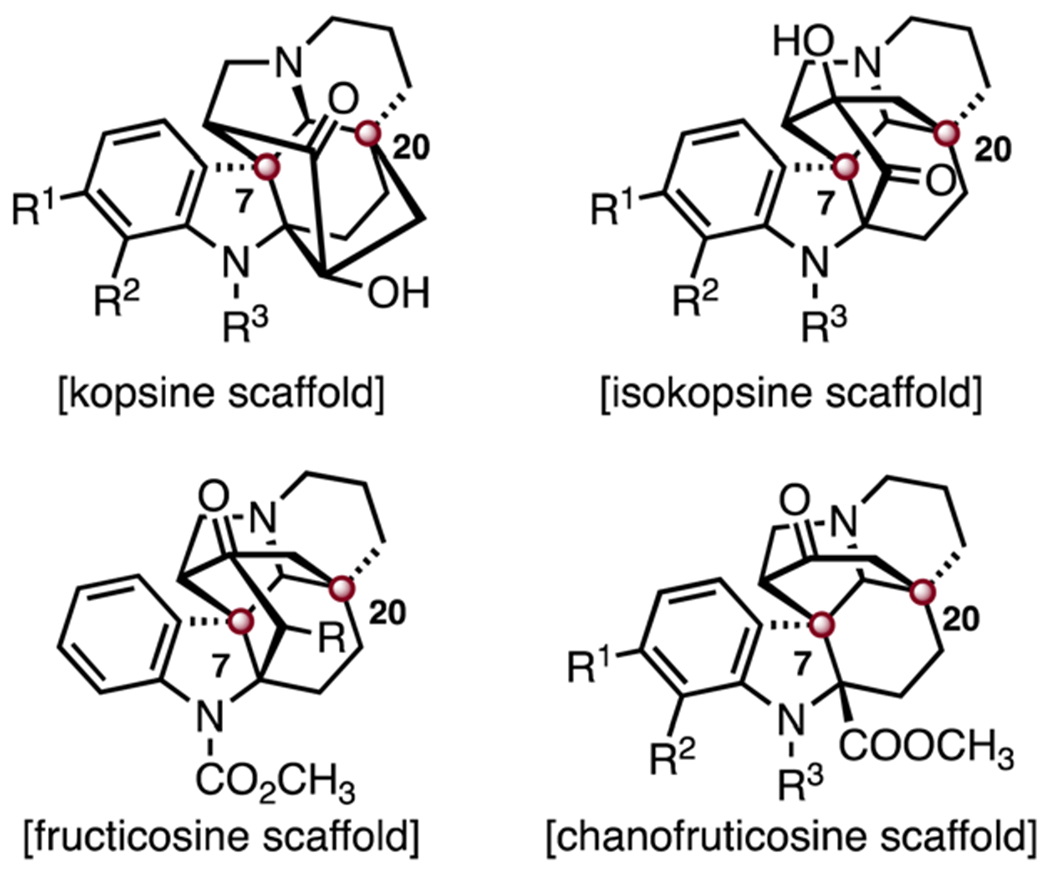
The common scaffolds of the four categories of kopsia alkaloids: kopsine, isokopsine, fructicosine, and chanofruticosine.
(+)-10,22-Dioxokopsane (357) was isolated from Pleiocarpa mutica in 1965 by Biemann and co-workers. Its structure was elucidated by mass spectrometry and NMR spectroscopy.172 (−)-Kopsanone (358) was isolated from Aspidosperma verbascifolium in 1966 by Durham and co-workers. Its structure was first elucidated by NMR spectroscopy and later confirmed by X-ray crystallographic analysis.173,174
In 1983, Magnus and co-workers disclosed a synthesis of (±)-kopsanone (358) and (±)-10,22-dioxokopsane (357; Scheme 43).166 The synthesis began with condensation of the aldehyde 345 and the amine 346 to form the imine 347 (>98%). Exposure of 347 to the mixed anhydride 348 and N,N-diisopropylethylamine promoted a diene translocation by N-acylation (347 → 349). Thermal [4 + 2] cycloaddition then provided the tetracycle (±)-350 as a single diastereomer (50%). A two-step sequence was developed to convert the tetracycle 350 to the sulfoxide 351 (82% overall). Alkylation of the indole by a Pummerer-like pathway, followed by hydrogen chloride elimination (trifluoroacetic anhydride; then, 130 °C), furnished the pentacyclic diene 352 containing the C7 TBCC (78%). An α-alkylation (potassium bis(trimethylsilyl) amide, allyl bromide) provided the triene 353 as a single diastereomer. Heating to 100 °C promoted a second cycloaddition to generate the alkene 354 (78%, two steps), which bears the C20 TBCC. A four-step sequence was developed to transform 354 to the 1,3-diketone 355 (49% overall). The 1,3-diketone 355 was converted to (±)-10,22-dioxokopsane (357, 66%, two steps) or (±)-kopsanone (358, 46%, two steps) by reduction and Moffatt oxidation (lithium aluminum hydride; 356 or lithium, ammonia; 356). The synthetic routes to (±)-kopsanone (357) and (±)-kopsanone (358) were completed both in thirteen steps and 8.1% or 5.6% overall yields, respectively.
Scheme 43.
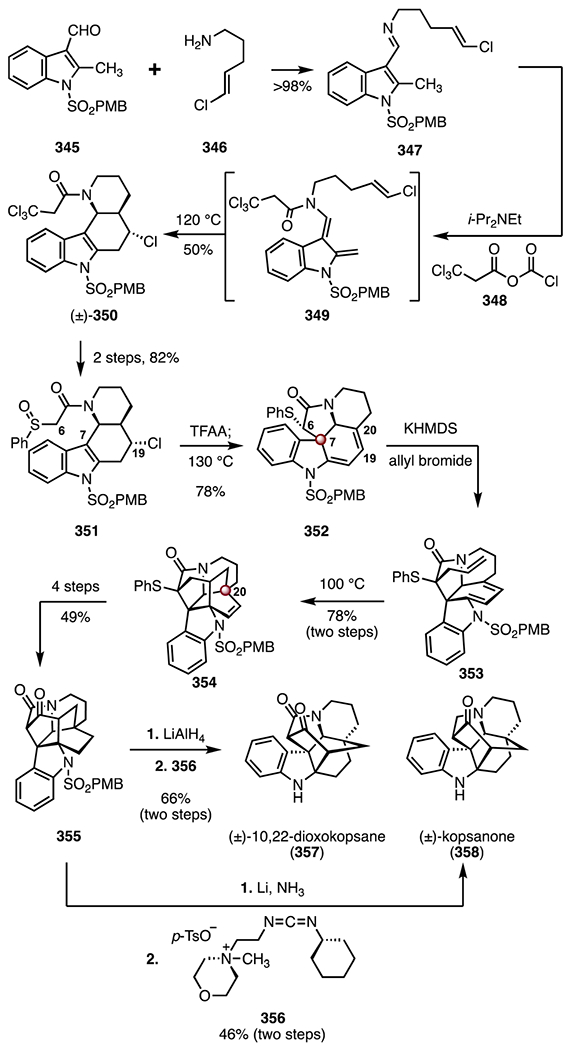
Synthesis of (±)-kopsanone (357) and (±)-10,22-dioxokopsane (358) by Magnus and co-workers.166 TFAA = trifluoroacetic anhydride, KHMDS = potassium bis(trimethylsilyl)amide.
(−)-Kopsinine (370) was isolated from Kopsia longiflora in 1955 by Michael and co-workers. Its structure was elucidated by NMR spectroscopy.175 In 2011, the MacMillan laboratory disclosed a synthesis of (−)-kopsanone (358) and (−)-kopsinine (370) that employed an organocatalytic cyclization cascade reaction as a key step (Scheme 44).167 The synthesis began with the selenide 360, which was prepared from the indole derivative 359 by a three-step sequence (63% overall). Coupling with the alkynyl aldehyde 361, mediated by the imidazolidinone catalyst (S,S)-362, provided the aldehyde (+)-366 (83%, 97% ee).176 Condensation of 361 with (S,S)-362 formed an iminium intermediate (not shown), which underwent an enantioselective [4 + 2] cycloaddition with the selenide 360, to provide the skipped diene intermediate 363. Elimination of the selenide substituent (363 → 364), intramolecular 1,4-addition (364 → 365), and imine hydrolysis furnished (+)-366, which bears the C7 TBCC. A six-step sequence was developed to convert the aldehyde (+)-366 to the pentacyclic diene 367 (40% overall). Thermal [4 + 2] cycloaddition between the diene 367 and phenyl vinyl sulfone (368) generated the alkene 369 as a single diastereomer (86%). The intermediate 369 contains the complete carbon skeleton of the targets, including the C20 TBCC. Sulfone reduction and alkene hydrogenation (RANEY® nickel) delivered (−)-kopsinine (370, 83%). The synthetic route to (−)-kopsinine (370) was completed in twelve steps and 15% overall yield. Hydrolysis of 370 (hydrochloric acid), followed by a thermolytic rearrangement (130 → 200 °C), provided (−)-kopsanone (357; 74%). (−)-Kopsanone (357) was completed in fourteen steps and 11% overall yield.
Scheme 44.
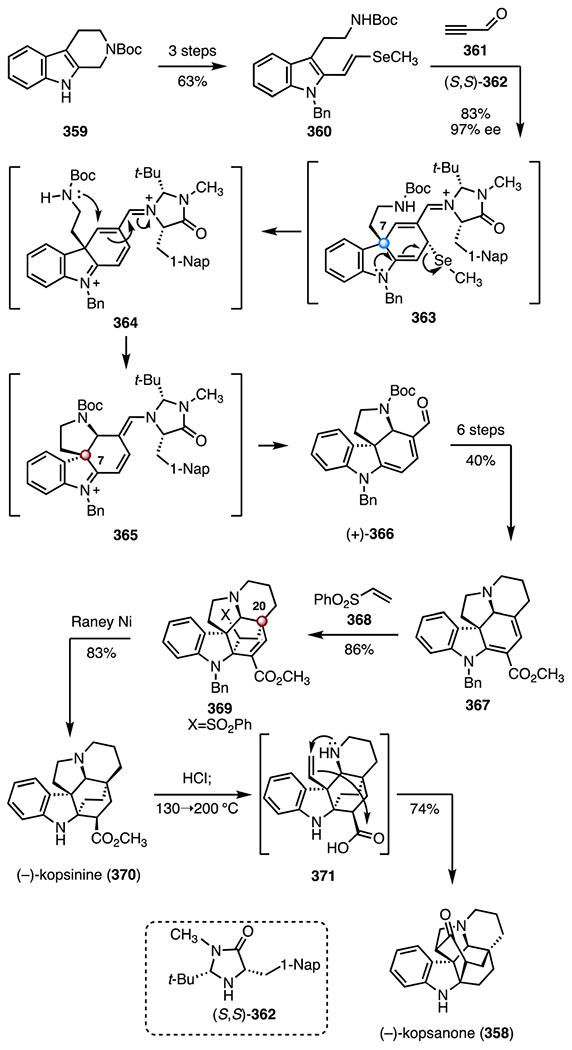
Synthesis of (−)-kopsanone (357) and (−)-kopsinine (370) by MacMillan and co-workers.167
(+)-Methyl N-decarbomethoxychanofruticosinate (379) was isolated from Kopsia officinalis in 1981 by Breitmaier and co-workers. Its structure was elucidated by correlation of the NMR spectrum to methyl 6,7-methylene dioxychanofruticosinate, whose structure was determined earlier by X-ray crystallography.177 In 2013, Ma and co-workers reported a synthesis of (+)-methyl N-decarbomethoxychanofruticosinate (379; Scheme 45).168 The synthesis began with an enantioselective, palladium-catalyzed, decarboxylative allylic alkylation (tris(dibenzylideneacetone) dipalladium(0), (R)-373), of the carbazolone (±)-372 (prepared in three steps and 53% yield from tetrahydro-4-oxocarbazole), to provide the allylic nitrile (−)-374 (90%, 94% ee).178 An eight-step sequence was developed to convert 374 to the bromide 375 (54% overall). Reductive cyclization (samarium(II) iodide) provided the β-hydroxy ketone 376 as an inconsequential mixture of diastereomers at C16 (78%, 1.4:1 dr).179 A two-step sequence then provided the ketone 377 (74% overall). Oxidative coupling (lithium bis(trimethylsilyl)amide; then, iodine) generated the imine 378 (77%). The imine 378 was converted to the target in three steps (43% overall). The route was completed in nineteen steps and 4.9% overall yield.
Scheme 45.
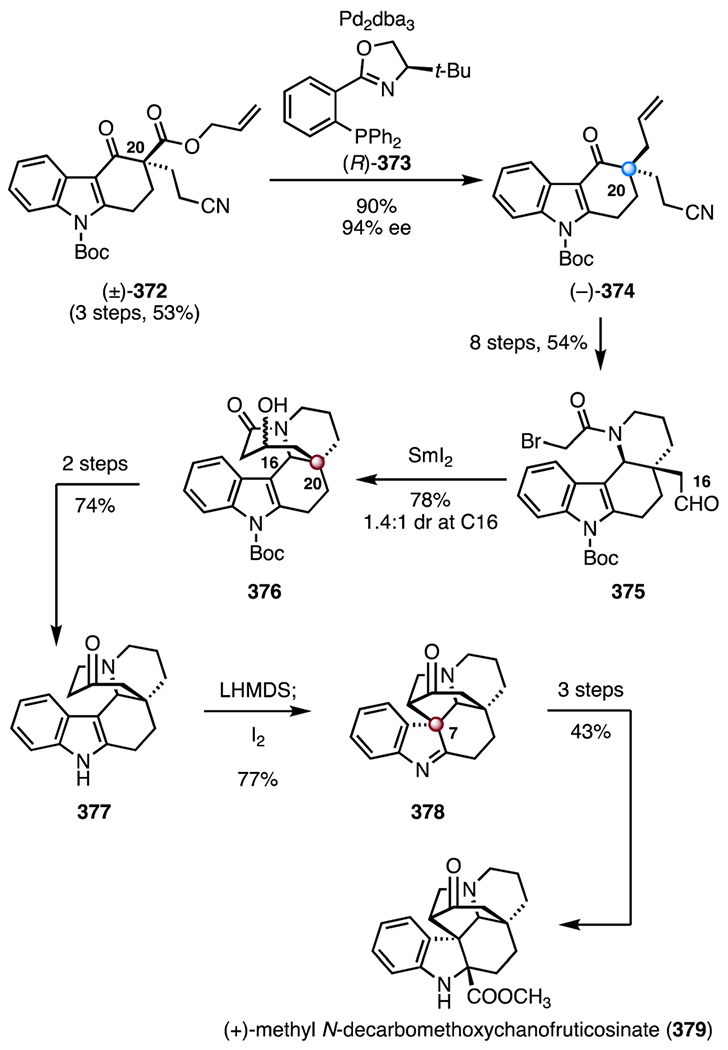
Synthesis of (+)-methyl N-decarbomethoxychanofruticosinate (379) by Ma and co-workers.168 Pd2(dba)3 = tris(dibenzylideneacetone) dipalladium, LHMDS = lithium bis(trimethylsilyl) amide.
(+)-Kopsinidine C (390) was isolated from Kopsia officinalis in 2017 by Li and co-workers. The structure of 390 was elucidated by NMR spectroscopy.180 Demethoxycarbonylkopsin (389) was isolated from Kopsia fruticose in 1962 by Schmid and co-workers. The structure of 389 was elucidated by NMR spectroscopy.181 In 2018, Ma and co-workers reported an enantioselective synthesis of (+)-10,22-dioxokopsane(357), (+)-kopsinidine C (390) and (−)-demethoxycarbonylkopsin (389, Scheme 46).169 The synthesis began with an enantioselective 1,4-addition of the carbazole (±)-380 to α-chloroacrylonitrile (381) catalyzed by the thiourea catalyst (R,R)-382. In situ removal of the chloride substituent (zinc, acetic acid) provided the ketoamide (−)-383 (93%, 97% ee), which bears the C20 quaternary center.182 Nitrile reduction (RANEY® nickel, hydrogen) generated the imine 384 (75%). The imine 384 was transformed to the aldehyde 385 by a six-step sequence (25% overall). Treatment of the aldehyde 385 with samarium iodide promoted a reductive cyclization to deliver, after oxidation of the resulting secondary alcohol (Dess–Martin periodinane), the β-ketoamide 386, which contains the C20 TBCC (53%, two steps). An oxidative cyclization was achieved by treating the β-ketoamide 386 with manganese(III) acetate dihydrate, copper(II) acetate, and methanol, providing a mixture of the methyl ether 388 (76%) and the hemiacetal 387 (10%), both of which bear the C7 TBCC. A six-step sequence was developed to convert the methyl ether 388 to (+)-10,22-dioxokopsane (357; 22% overall). The synthesis of 357 was completed in seventeen steps and 1.5% overall yield. Alternatively, the methyl ether 388 was converted to the hemiacetal 387 by treatment with hydrochloric acid (94%). The hemiacetal 387 was advanced to (−)-demethoxycarbonylkopsin (389) by a four-step sequence (44% overall). Reduction of (−)-demethoxycarbonylkopsin (389) (lithium aluminum hydride) delivered (+)-kopsinidine C (390; 40%). The syntheses of (+)-demethoxycarbonylkopsin (389) and (+)-kopsinidine C (390) were completed in sixteen and seventeen steps, and 3.3% and 1.3% overall yield, respectively.
Scheme 46.
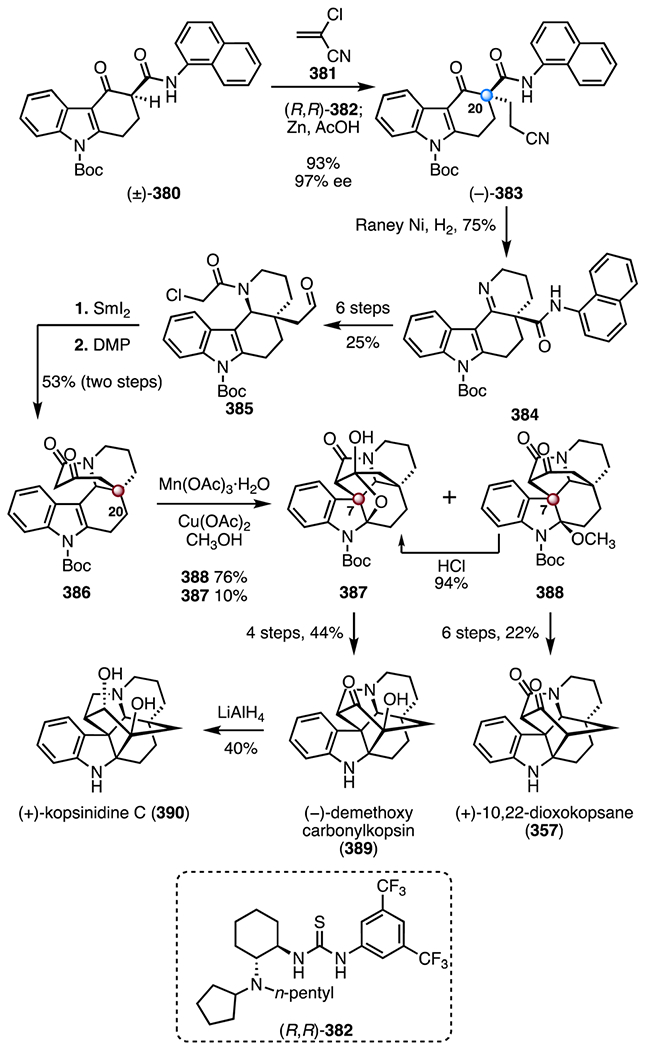
Synthesis of (+)-10,22-dioxokopsane (358), (+)-kopsinidine C (390) and (−)-demethoxycarbonylkopsin (389) by Ma and co-workers.169 DMP = Dess–Martin periodinane.
(−)-Isokopsine (402) and (+)-methyl chanofruticosinate (403) were isolated by Schmid and co-workers in 1963 and 1966, respectively, from Kopsia fruticosa.183,184 (−)-Kopsine (401) was isolated from Kopsia fruticosa by Bose and co-workers in 1949.185 (−)-Fruticosine (404) was isolated by Gregory and co-workers from Kopsia fruticosa in 1963.186 The structures of all of these isolates were elucidated by NMR spectroscopy. In 2017, the Qin laboratory disclosed a collective synthesis of (−)-isokopsine (402), (+)-methyl chanofruticosinate (403), (−)-kopsine (401), (−)-fruticosine (404), and kopsanone (358).170 Their synthetic route employed cyclopropanation to form the C7 and C20 TBCCs as a key step (Scheme 47). Treatment of the allylic ester (±)-391 (three steps from tetrahydro-4-oxocarbazole, 68%) with tris(dibenzylideneacetone)dipalladium(0) and the chiral ligand (S)-373 187 provided the decarboxylative allylation product (+)-392 in 91% yield and 94% ee. The allylation product 392 contains the C20 quaternary stereogenic center in the target. A six-step sequence was developed to transform the ketone (+)-392 to the amine 393 (64% overall). Reductive ring-opening (iron(II) chloride) and diazo transfer (394) generated the α-diazo ketone 395 (91%, two steps). An intramolecular cyclopropanation, promoted by copper(II) hexafluoroacetylacetonate, provided the cyclopropane 396, which bears the C7 and C20 TBCCs (52%). A four-step sequence was developed to access the bis(cyanide) 397 (41% overall). An intramolecular acyloin condensation (samarium(II) iodide) then provided the α-hydroxy ketone 398 (74%). A three-step sequence was developed to transform the α-hydroxy ketone 398 to the advanced intermediates 399 (24% overall) and 400 (29% overall). The hydroxy ketone 399 was converted to (−)-kopsine (401) by treatment with triphosgene and pyridine (42%), or to (−)-kopsanone (358) through a two-step deoxygenation sequence (26% overall). The syntheses of these targets were completed in twenty-two steps and 0.84% overall yield (for 401), and twenty-three steps and 0.52% overall yield (for 358). Intermediate 400 was elaborated to (−)-isokopsine (402) by a two-step sequence (64% overall). The synthesis of 402 was completed in twenty-three steps and 1.6% overall yield. (−)-Isokopsine (402) was converted to (+)-methyl chanofruticosinate (403) by treatment with lead(IV) acetate and methanol (65%), or, alternatively to (−)-fruticosine (404) by a three-step sequence (55% overall). The syntheses of (+)-methyl chanofruticosinate (403) and (−)-fruticosine (404) were completed in twenty-four steps and 1.0% overall yield, and twenty-six steps and 0.85% overall yield, respectively.
Scheme 47.
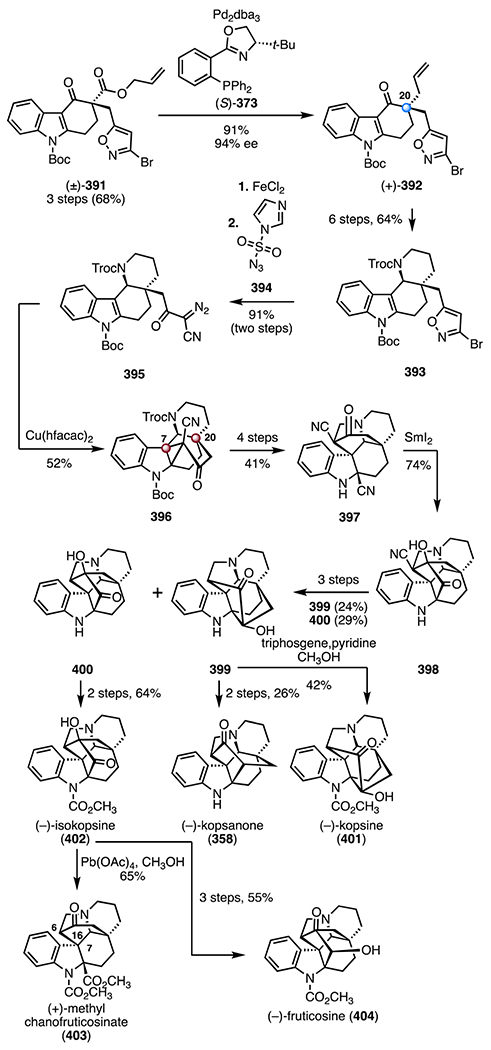
Collective synthesis of kopsia alkaloids by Qin and co-workers.170 Pd2dba3 = tris(dibenzylideneacetone)dipalladium(0), Cu(hfacac)2 = copper(II) hexafluoroacetylacetonate.
(+)-Flavisiamine F (417) was isolated by Morita and co-workers in 2008 from Kopsia flavida. Its structure was elucidated by NMR spectroscopy.188 In 2019, Xia and co-workers reported an enantioselective synthesis of (+)-flavisiamine F (417; Scheme 48).171 The synthetic route employed a photochemical cyclization to form the hexacyclic ring system. The synthesis began with the trifluoromethanesulfonate 406, which was prepared from the known carbazole 405 (lithium bis(trimethylsilyl) amide, bis(trifluoromethanesulfonyl)aniline, 94%). An NHK reaction between the trifluoromethanesulfonate 406 and the chiral acetonide (+)-407 (chromium(II) chloride, nickel(II) chloride) provided a diastereomeric mixture of addition products (1 : 1 dr, not shown), which were oxidized (2-iodoxybenzoic acid) then reduced (lithium triethylborohydride) to provide the allylic alcohol (+)-408 (64% over three steps, >20 : 1 dr). An Overman rearrangement189 was achieved by exposure of the allylic alcohol (+)-408 to sodium hydride and trichloroacetonitrile, to form the allylic acetamide 409 (77%). Opening of the acetonide (trimethylsilyl trifluoromethanesulfonate, N,N-diisopropylethylamine) followed by treatment with potassium carbonate and heating to 130 °C initiated a second [3,3]-sigmatropic rearrangement, to provide the allylic alcohol 411 after acidic work-up (hydrochloric acid, 62%, two steps).190 A five-step sequence was developed to transform the allylic alcohol 411 to the diene 412 (51% overall). ROM of the diene 412 (Grubbs II catalyst), followed by α-iodination (lithium bis(trimethylsilyl) amide, iodine), furnished the iodide 413, which bears the C20 TBCC (70%, two steps). In a key transformation, irradiation of the iodide with blue LEDs (455 nm) in the presence of the iridium catalyst 414 (air, triethylamine) provided the imine 415, which bears the C7 TBCC (77%).191 Exposure of the imine 415 to the masked acyl cyanide 416 and imidazole,192 followed by oxidation and methanolysis (potassium carbonate, hydrogen peroxide; methanol, hydrochloric acid, 38%, three steps) completed the synthesis of (+)-flavisiamine F (417). The synthesis was achieved in eighteen steps and 3.2% overall yield.
Scheme 48.
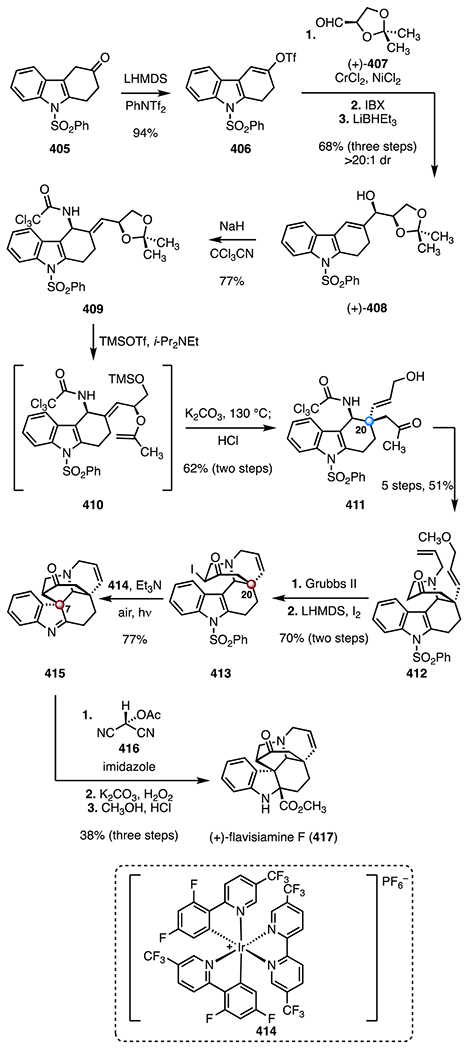
Synthesis of (+)-flavisiamine F (417) by Xia and co-workers.171 LHMDS = lithium bis(trimethylsilyl)amide, PhNTf2 = bis(trifluoromethanesulfonyl)aniline, IBX = 2-iodoxybenzoic acid, TMSOTf = trimethylsilyl trifluoromethanesulfonate, LHMDS = lithium bis(trimethylsilyl)amide.
The Magnus166 and the Macmillan167 syntheses constructed pentacyclic structures that bear the C7 TBCC first (352 and 366), and then generated the C20 TBCC by cycloaddition reactions (353 → 354, 367 → 369). Ma168,169 and Qin170 both installed the C20 quaternary stereogenic center early in the synthetic route by an enantioselective decarboxylative allylic alkylation, or enantioselective Michael addition (372 → 374, 380 → 383, 391 → 392). The C7 TBCC was formed by an oxidative cyclization (377 → 378; 386 → 387 + 388) or cyclopropanation (395 → 396). Xia and co-workers171 formed the C20 center through consecutive sigma-tropic rearrangements (408 → 409 → 411). They installed the C7 TBCC by a novel photochemical cyclization reaction (413 → 415).
2.10. Communesin alkaloids: vicinal TBCCs within a dimeric indole alkaloid
(−)-Communesin F (418) was isolated in 2004 by Hayashi and co-workers from Penisillium expansum.193 Its structure was determined by mass spectrometry and NMR spectroscopy (Scheme 49). (−)-Communesin F (418) possesses a heptacyclic skeleton containing two bicyclic aminals and vicinal TBCCs at C7 and C8. The first total synthesis of (±)-communesin F (418) was reported by Qin and co-workers in 2007,194 followed by a second racemic synthesis by Weinreb and co-workers in 2010.195,196 In the same year, Ma and co-workers disclosed the first enantioselective synthesis of (−)-communesin F (418).197 These syntheses have been previously reviewed.198–202 More recent syntheses by the Funk,203 Yang,204 Chen,205,206 and Movassaghi groups207 are discussed below as they highlight distinct synthetic strategies to prepare TBCCs.
Scheme 49.
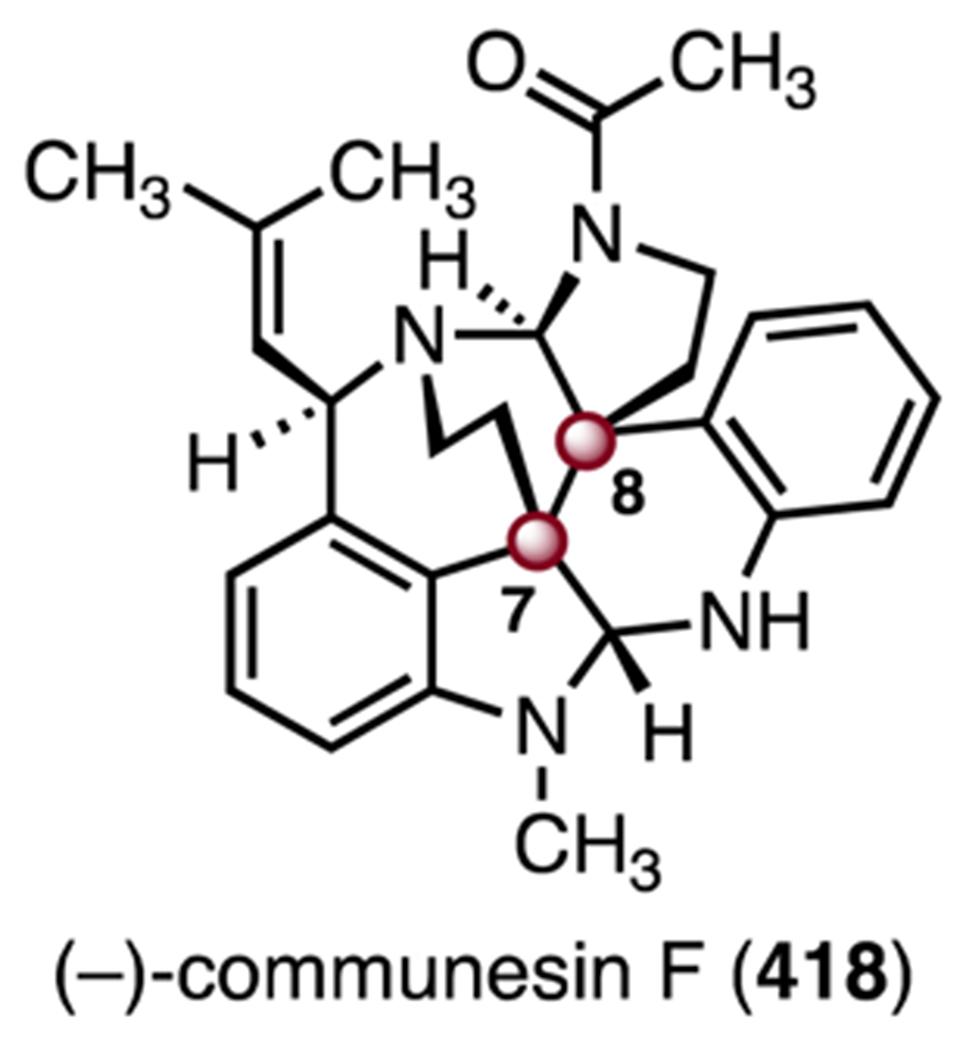
Structure of (−)-communesin F (418).
In 2012, Funk and co-worker disclosed a synthesis of (±)-communesin F (418) that employed a [3 + 2] cycloaddition to form the C7–C8 bond (Scheme 50).203 Treatment of a mixture of the bromoindole 419 and the oxindole 420 with silver(I) carbonate provided the indolenine (±)-422 by an endo-selective [3 + 2] cycloaddition (421) and spontaneous ring-opening (70%).208,209 A four-step sequence was developed to transform the indolenine (±)-422 to the aminal 423 (26% overall). A Heck coupling (palladium(II) acetate, potassium carbonate, 424),210 followed by an S′N substitution (mercury(II) trifluoromethanesulfonate)211 provided the benzazepine 425 (80%, two steps). Carbamate removal (tert-butyldimethylsilyl trifluoromethanesulfonate, 2,6-lutidine; potassium fluoride),212 followed by addition of trimethylaluminum, generated the “twisted-bridge” amide 426 (82%, three steps). The absence of strong conjugation between the nitrogen and the carbonyl increased the reactivity of the lactam residue,213 which was crucial for the eventual formation of the second bicyclic aminal. A stereocontrolled alkylation (potassium bis(trimethylsilyl) amide, iodoacetamide, 77%) followed by reduction (lithium aluminum hydride) provided the hemiacetal 427, which bears the C8 TBCC. A two-step sequence was developed to complete the synthesis (51%, three steps). The synthetic route to (±)-communesin F (418) was completed in fourteen steps and 4.7% overall yield.
Scheme 50.
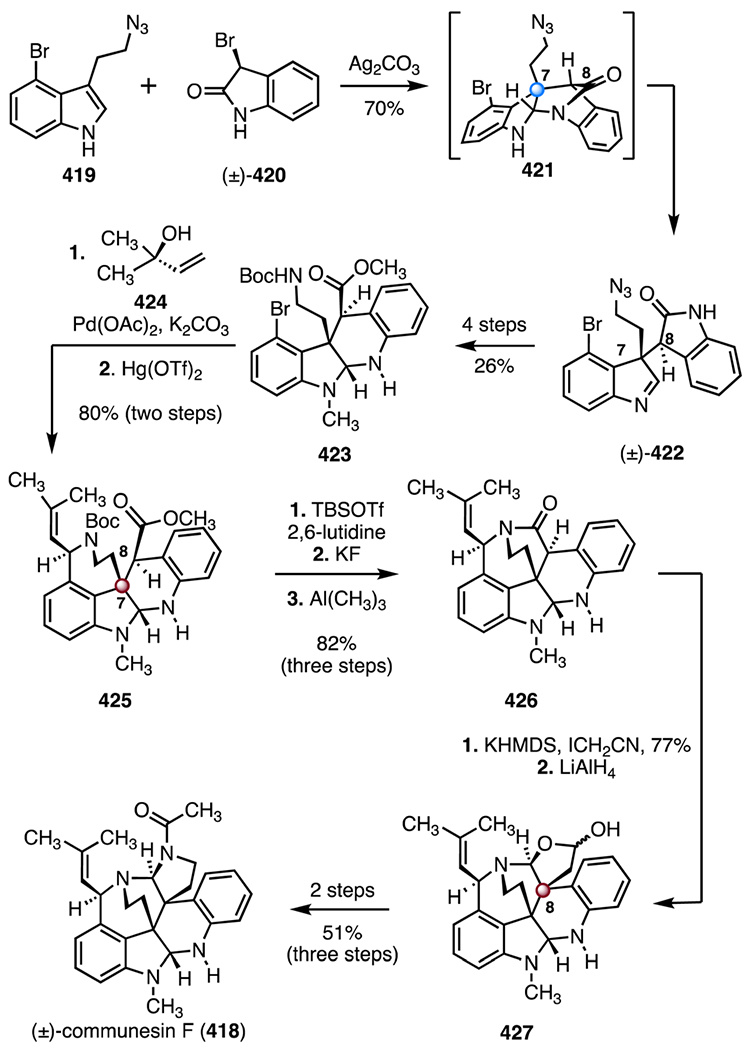
Synthesis of (±)-communesin F (418) by Funk and co-workers.203 Hg(OTf)2 = mercury(II) trifluoromethanesulfonate, TBSOTf = tert-butyldimethylsilyl methanesulfonate, KHMDS = potassium bis(trimethylsilyl)amide.
In 2017, Yang and co-workers reported an enantioselective route to (−)-communesin F (418; Scheme 51).204 The synthesis began with an iridium-catalyzed annulation between the indole 428 and the allylic carbonate (±)-429 (cyclooctadiene iridium chloride dimer, 9-borabicyclo[3.3.1]nonane-n-hexane, (S)-313, potassium tert-butoxide), which provided the aminal (−)-430 (55%, 99% ee). The aminal 430 contains the C7 quaternary center and the C7–C8 bond. This coupling reaction was postulated to proceed by nucleophilic addition of the indole 428 on a π-allyl complex derived from (±)-429 (not shown) followed by subsequent aminal formation. A six-step sequence was developed to transform (−)-430 to the amide 431 (41% overall), which bears the C7 TBCC. Alkylation (potassium tert-butoxide, allyl iodide) and carbamate hydrolysis (potassium hydroxide) provided the benzazepine 432 (75%, two steps). A five-step sequence was developed to transform the benzazepine 432 to the “twisted-bridge” amide 433 (23% overall). Reduction of the amide 433 (lithium aluminum hydride), followed by alcohol activation (methanesulfonyl chloride, triethylamine) provided the iminium intermediate 434, which underwent cyclization to provide, after carbamate cleavage (trifluoroacetic acid) (−)-communesin F (418; 65%, two steps). The synthesis of (−)-communesin F (418) was completed in sixteen steps and 2.5% overall yield.
Scheme 51.
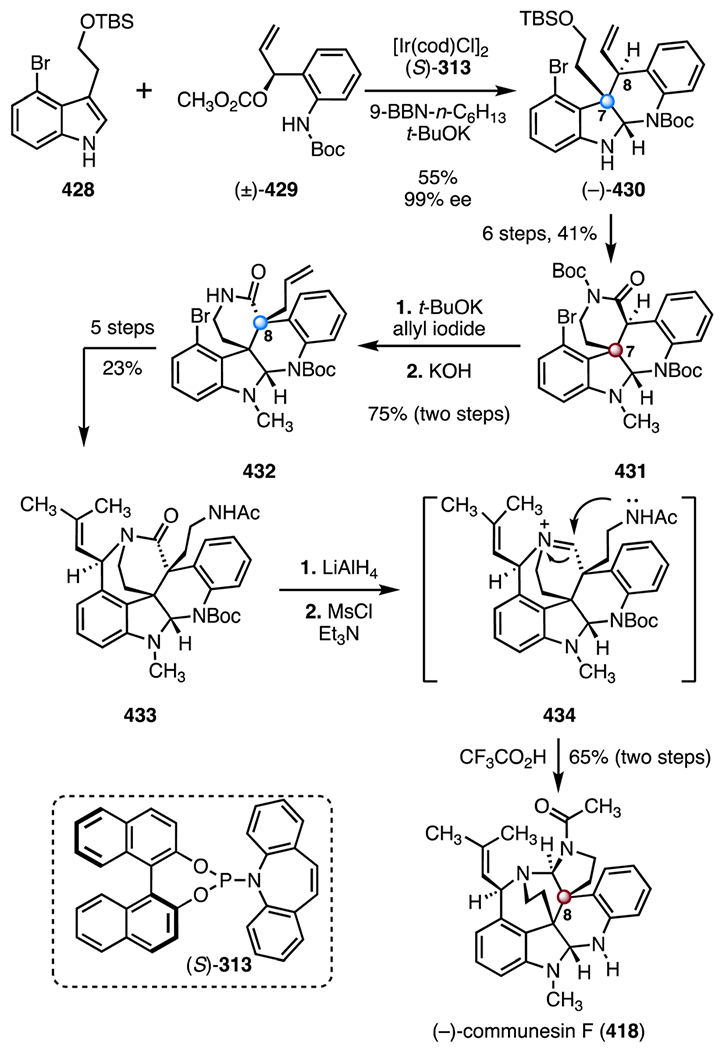
Synthesis of (−)-communesin F (418) by Yang and co-workers.204 [Ir(cod)Cl]2 = cyclooctadiene iridium chloride dimer, 9-BBN-n-C6H13 = 9-borabicyclo[3.3.1]nonane-n-hexane, MsCl = methanesulfonyl chloride.
In 2017, Chen and co-workers disclosed a synthesis of (−)-communesin F (418) that employed an enantioselective silylprolinol-catalyzed fragment coupling to form the C7–C8 linkage (Scheme 52).205,206 Treatment of a mixture of the aldehyde (±)-436 (prepared in three steps and 73% yield from the isatin derivative 435) and the azide (±)-437 with the silylprolinol catalyst (S)-438, followed by in situ reduction (sodium borohydride), provided the alcohol (+)-439 (31%, 86% ee, 1:0.8 dr at C8).214–216 It was proposed that (±)-436 first underwent an elimination of methanol to form an enal (not shown), which effected an enantioselective 1,4-addition by the chiral enamine derived from the condensation of (±)-437 and (S)-438. This step constructed the vicinal C7 and C8 quaternary stereocenters. A seven-step sequence was developed to convert the adduct (+)-439 to the spirolactams 440 (8.4% overall) and 441 (28% overall). A substrate-directed C–H alkenylation (methyl acrylate, palladium(II) acetate, silver(I) carbonate) provided the cinnamate derivatives 442 (94%) and 443 (93%),217 which were converged to the tert-butyloxycarbonyl protected substrate 444 by two-step sequences (71% from 442, 75% from 443). A six-step sequence (4.5% overall) was developed to elaborate 444 to the alcohol 445, which underwent an S′N substitution (pyridinium p-toluenesulfonate) to provide the cyclic amine 446 (52%). Imidate formation (triethyloxonium tetrafluoroborate) and selective carbamate removal (trifluoroacetic acid), followed by exposure to silica gel, provided the imine 447, which bears the C8 TBCC (31%, two steps). A one-flask reduction–acylation (sodium borohydride, acetic acid; lithium aluminum hydride; acetic anhydride, acetic acid) completed the synthesis of (−)-communesin F (418; 43%). The synthetic route was completed in twenty-four steps and 0.018% overall yield.
Scheme 52.
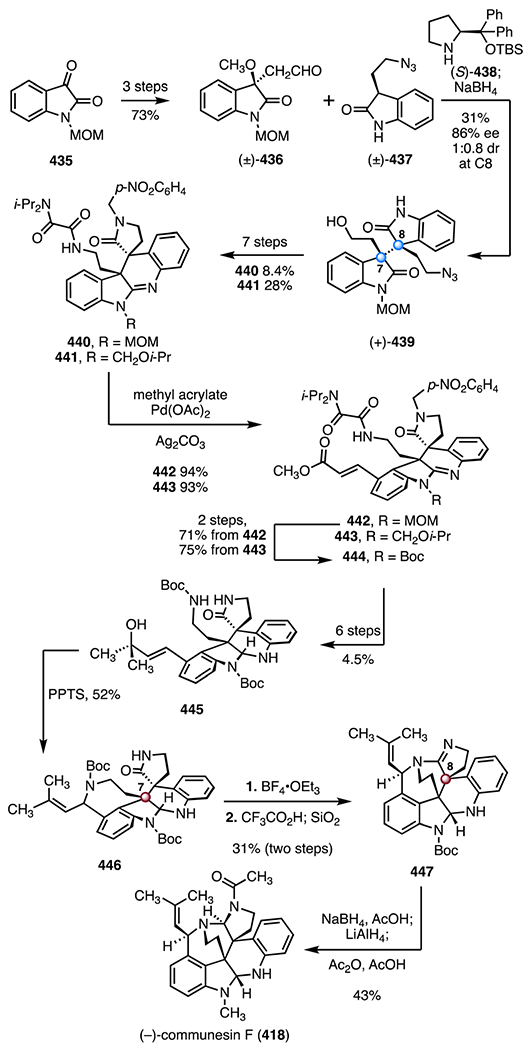
Synthesis of (−)-communesin F (418) by Chen and co-workers,205,206 PPTS = pyridinium p-toluenesulfonate.
In 2016, Movassaghi and co-workers disclosed a biomimetic route218,219 to (−)-communesin F (418; Scheme 53).207 The synthesis employed a diazene-based dimerization as a key step. A four-step sequence was developed to transform the known (−)-448 to the allylic alcohol 449 (44% overall). A palladium-mediated cyclization of the alcohol 449 (bis(acetonitrile)palladium(II) chloride) provided the azapane 450, as a single diastereomer (81%). A two-step sequence then furnished the nitrile 451 (38% overall). The cyclotryptamine 453 was obtained in three steps from the known bromide (+)-452 (26% overall). Coupling with the azapane 451 (4-dimethylaminopyridine) provided the heterodimeric sulfamide 454 (80%). N-Chloro-N-methyl benzamide and 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine were used to oxidize the sulfamide 454 to the diazene 455 (57%). Upon photoexcitation (350 nm), the diazene 455 was converted to the dimeric product 456 by extrusion of dinitrogen and rapid, stereoselective radical recombination (39%).220,221 The stereoselectivity of this reaction was enhanced by the presence of the C6 nitrile. The C7 and C8 TBCCs were established in this single step. Removal of the tert-butyl carbamates (scandium(III) trifluoromethanesulfonate), followed by base-mediated cyclotryptamine opening and cyanide elimination (lithium tert-butoxide), triggered a biomimetic aminal reorganization through the imine–iminium intermediate 457. Addition of pyridinium p-toluenesulfonate and acetic anhydride to the basic reaction mixture provided the heptacycle 458 (55%, two steps). A final deprotection (sodium mercury amalgam) delivered (−)-communesin F (418, 83%). The synthesis of (−)-communesin F (418) was completed in thirteen steps and 1.1% overall yield.
Scheme 53.
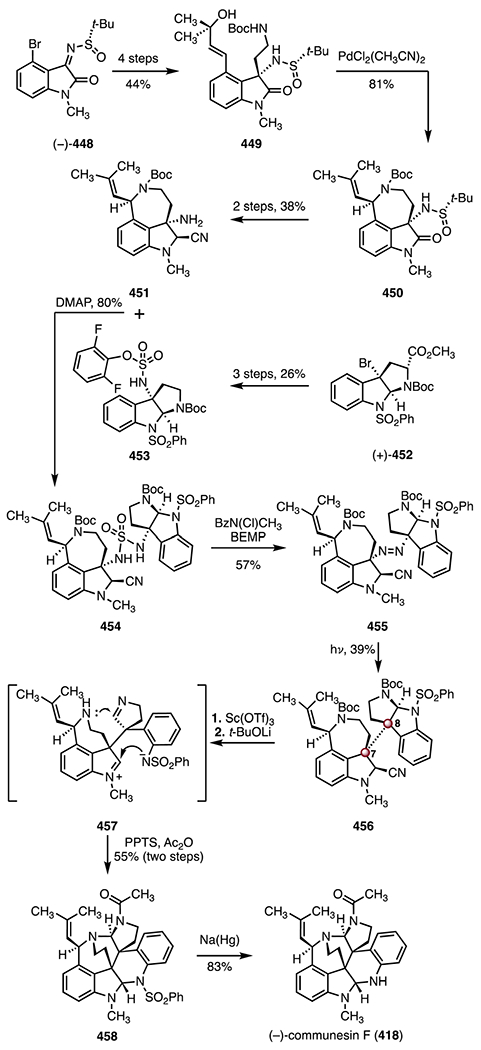
Synthesis of (−)-communesin F (418) by Movassaghi and co-workers.207 DMAP = 4-dimethylaminopyridine, BEMP = 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine, Sc(OTf)3 = scandium(III) trifluoromethanesulfonate, PPTS = pyridinium p-toluenesulfonate.
The C7 TBCC was constructed early (as a quaternary center) in the Funk,203 Yang,204 and Chen205,206 syntheses. Funk203 utilized a [3 + 2] cycloaddition and ring-opening sequence (419 + 420 → 422), while Yang204 employed an iridium-catalyzed annulation (428 + 429 → 430) to form the C7 stereocenter. Chen205,206 established both the C7 and C8 quaternary stereocenters through an organocatalytic transformation (436 + 437 → 439), while Yang204 and Funk203 employed alkylation reactions (426 → 427, 431 → 432) to establish the C8 center. The enhanced reactivity of the twisted amides (426 and 433) was essential to forming the pentacyclic system in the Yang and Funk syntheses. The stereoselective coupling employed by Movassaghi207 (451 + 453 → 456) provided a convergent route to the target while directly furnishing both the C7 and C8 TBCCs in a single step.
3. Discussion
The syntheses discussed above can be arranged around five generalizable approaches to TBCCs. Each transformation employed for TBCC construction is summarized in Table 1. Here we group the strategies as those proceeding by “direct construction” (the TBCC was formed in a single step) or “indirect construction” (the TBCC was constructed by initial formation of a mono or bicyclic quaternary carbon center, followed by a cyclization event). A numerical analysis of TBCC construction by these strategies is arranged as a function of reaction type in Fig. 1. Here we discuss each of these strategies, as well as their functional group compatibility, strategic advantages, and limitations, with the goal of assisting future work toward related targets.
Table 1.
Summary and categorization of TBCC construction transformations
| Method | Scheme | Reaction |
|---|---|---|
| Enolate-, enol-, or enoxysilane-mediated C-C bond formation | ||
| Direct construction of TBCC | ||
| Aldol | 20 | 152 → 153 |
| Oxidative α-alkylation | 37 | 294 → 296 |
| 45 | 377 → 378 | |
| 46 | 386 → 387 + 388 | |
| 48 | 413 → 415 | |
| Indirect construction of TBCC | ||
| 1,2-Additon | 10 | 68 + 69 → 70; 71 → 72, hemiacetal formation |
| Michael addition | 5 | 25 → 27; 29 → 30, intramolecular [2 + 2] |
| 46 | 380 → 383; 385 → 386, Reformatsky | |
| 52 | 436 + 437 → 439; 445 → 446, S’N, 446 → 447, imine formation | |
| α-Alkylation | 2 | 5 → 6; 6 → 7, aldol |
| 3 | 16 → 18; 18 → 20, enolate vinylation | |
| 12 | 96 → 97; 97 → 98, lactonization; 98 → 67, lactonization | |
| 29 | 223 → 225; 226 → 227, intramolecular [4 + 2] | |
| 34 | 269 → 270; 271 → 273, intramolecular [4 + 2] | |
| 38 | 217 → 301; 303 → 304, hemiaminal formation | |
| 50 | 426 → 427; 426 → 427, hemiacetal formation | |
| 51 | 431 → 432; 433 → 418, aminal formation | |
| Aldol | 28 | 219 → 220; 221 → 222, reductive cyclization |
| Robinson annulation | 2 | 2 → 3; 4 → 5, hemiacetal formation |
| Cycloaddition-, sigma-tropic rearrangement-, or ene-based transformation | ||
| Direct construction of TBCC | ||
| Fischer indolization | 40 | 326 → 330 |
| Intramolecular [2 + 2] | 11 | 82 → 84 |
| 18 | 140 → 141 | |
| 21 | 156 → 157 | |
| Intramolecular [3 + 2] | 26 | 199 → 202 |
| Intermolecular [4 + 2] | 31 | 243 + 244 → 245 |
| 44 | 360 + 361 → 366 | |
| Intramolecular [4 + 2] | 24 | 184 → 186 |
| 25 | 195 → 196 | |
| 28 | 214 → 215 | |
| 29 | 226 → 227 | |
| 30 | 238 → 239 | |
| 31 | 248 → 249 | |
| 32 | 256 → 258 | |
| 34 | 271 → 273 | |
| 43 | 353 → 354 | |
| 44 | 367 → 369 | |
| Intermolecular [5 + 2] | 20 | 143 + 148 → 151 |
| Indirect construction of TBCC | ||
| Conia–ene reaction | 15 | 113 → 114; 114 → 115, Pauson–Khand |
| 16 | 122 → 123; 124 → 108, Pauson–Khand | |
| Eschenmoser–Claisen | 23 | 166 → 168; 173 → 174, 6-endo-trig |
| 48 | 409 → 411; 412 → 413, ring-closing metathesis | |
| Intramolecular [2 + 2] | 21 | 156 → 157; 159 → 160, Prins cyclization |
| Intermolecular [3 + 2] | 50 | 419 + 420 → 421; 423 → 425, S’N |
| Intramolecular [3 + 2] | 24 | 181 → 183; 184 → 186, intramolecular [4 + 2] |
| Intermolecular [4 + 2] | 3 | 13 + 14 → 15; 16 → 18, lactonization |
| 7 | 49 → 50; 54 → 55, ring-closing metathesis | |
| 25 | 187 + 188 → 189; 195 → 196, intramolecular [4 + 2] | |
| Intramolecular [4 + 2] | 34 | 271 → 273; 277 → 264, lactonization |
| Intermolecular [5 + 2] | 20 | 143 + 148 → 151; 151 → 152, lactonization |
| Radical mediated C–C bond formation | ||
| Direct construction of TBCC | ||
| 5-exo-trig | 14 | 106 → 107 |
| 35 | 287 → 290 | |
| 6-endo-trig | 26 | 205 → 206 |
| Diazane-mediated radical combination | 14 | 106 → 107 |
| 53 | 455 → 456 | |
| Indirect construction of TBCC | ||
| 5-exo-trig | 12 | 92 → 93; 98 → 67, lactonization |
| 17 | 128 → 129; 124 → 108, Pauson–Khand | |
| Cation-based C–C bond formation | ||
| Direct construction of TBCC | ||
| Pinacol rearrangement | 6 | 42 → 43 |
| 8 | 63 → 64 | |
| Wagner–Meerwein rearrangement | 18 | 134 → 139 |
| 32 | 259 → 262 | |
| Pummerer rearrangement | 43 | 351 → 352 |
| Transition metal-catalyzed C–C bond formation | ||
| Direct construction of TBCC | ||
| Pauson–Khand | 15 | 114 → 115 |
| 16 | 124 → 108 | |
| 17 | 124 → 108 | |
| Allylic alkylation | 39 | 312 → 315 |
| 41 | 340 → 341 | |
| Cyclopropanation | 47 | 395 → 396 |
| Indirect construction of TBCC | ||
| Enolate arylation | 23 | 164 → 165; 169 → 170, Prins cyclization |
| Decarboxylative allylic alkylation | 30 | 234 → 235; 238 → 239, intramolecular [4 + 2] |
| 45 | 372 → 374; 375 → 376, Reformatsky | |
| 47 | 391 → 392; 395 → 396, cyclopropanation | |
| Allylic alkylation | 51 | 428 + 429 → 430; 430 → 431, lactamization |
Fig. 1.
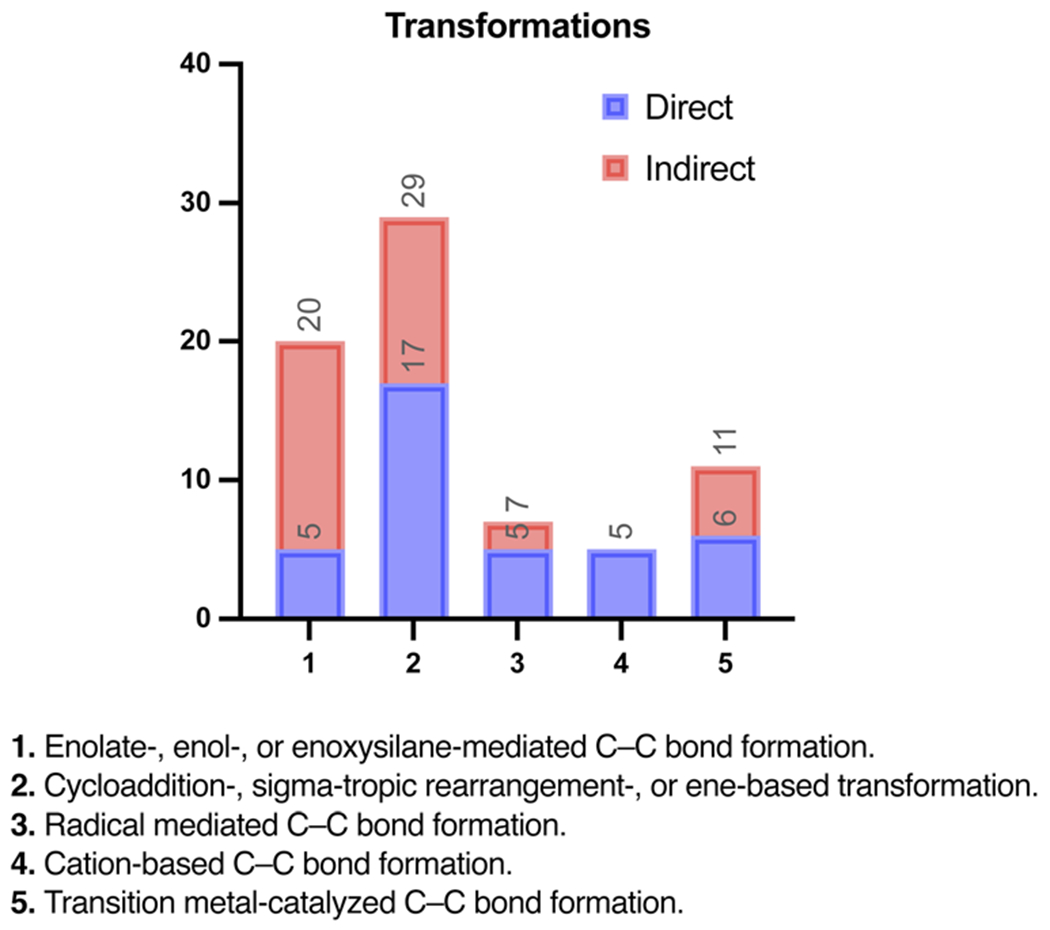
Numerical analysis of TBCC construction transformations, based on Table 1.
3.1. Enolate-, enol-, or enoxysilane-mediated C–C bond formation
The most straightforward methods for the formation of a TBCC employ carbonyl chemistry to facilitate the formation of the last carbon–carbon bond (Scheme 54). Enolate formation by α-deprotonation (e.g., Scheme 3, 16, lithium bis(trimethylsilyl)amide) or 1,4-addition to α,β-unsaturated ketones (e.g., Scheme 5, 25, lithium, ammonia); enol formation under acidic conditions (e.g., Scheme 2, 2, p-toluenesulfonic acid) and enoxysilane formation from carbonyl substrates (e.g., Scheme 28, 216, 217, 218, l-proline; tert-butyl(chloro)diphenylsilane, triethylamine) facilitate C–C bond formation by addition to an appropriate electrophile (e.g., Scheme 3, 16 → 18), Michael acceptors (e.g., Scheme 5, 25 → 27), or carbonyls (e.g., Scheme 28, 219 → 220). While most of the examples shown are substrate-controlled, these bond constructions can be conducted with reagent or catalyst control of absolute stereochemistry, as exemplified by the enantioselective Michael addition shown in Scheme 46 (380 → 383). A limitation of this approach is that it often necessitates using simple nucleophiles (predominantly, bicyclic carbonyl compounds) and electrophiles owing to the steric congestion of the developing quaternary center. Lateral manipulations and a ring-forming reaction are often required if the TBCC was built this way, which translates to the high percentage (80%) of indirect TBCC construction observed under this category (Fig. 1). Direct TBCC construction by carbonyl chemistry can be achieved with specific functional groups (e.g., Scheme 46, 386 → 387 + 388).
Scheme 54.
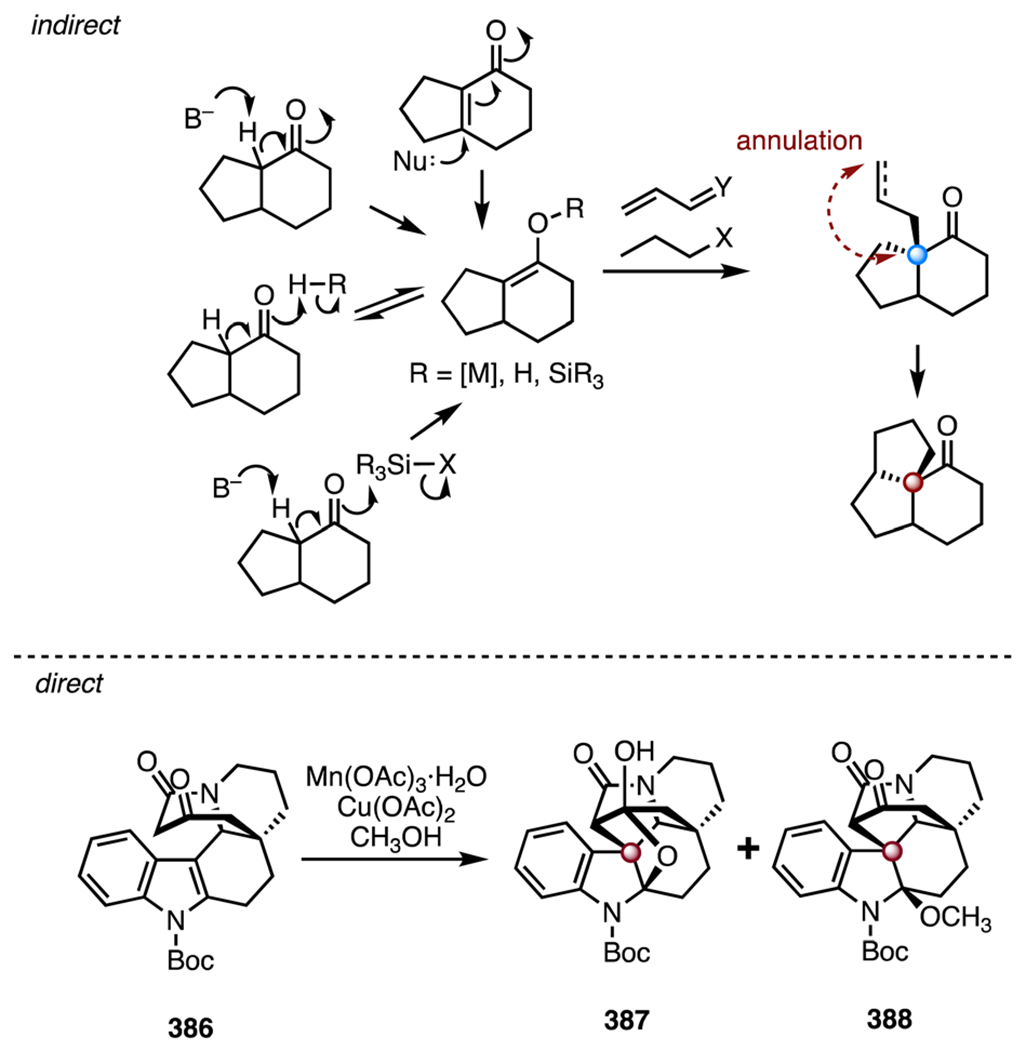
A general scheme for indirect TBCC construction by enolate-, enol-, or enoxysilane-mediated C–C bond formation, and an example of a direct TBCC construction by this strategy.
3.2. Cycloaddition-, sigma-tropic rearrangement-, or ene-based transformation
Pericyclic reactions, including cycloaddition reactions, sigma-tropic rearrangements, and ene reactions, provide a potentially powerful strategy to rapidly generate complex polycyclic scaffolds (Scheme 55). The highest number of examples (29 in total) and the high percentage (59%) of direct TBCC construction (Fig. 1) underscores the compatibility and efficiency of this method in TBCC construction. Trisubstituted sp2-hybridized carbon atoms may be accessed directly from commercially available building blocks or by well-established methods such as olefinations (e.g., Scheme 43, 351 → 352) or cross-coupling reactions (e.g., Scheme 21, 154 + 155 → 156). While [2 + 2] cycloadditions are promoted by irradiation, [3 + 2] and [4 + 2] cycloadditions are often conducted under thermal conditions, creating an opportunity for expanded functional group compatibility, provided the substrate is amenable to heating. Modern variations of classic cycloadditions have been widely utilized as well, which often involve in situ formation of cyclization partners by oxidative dearomatization (e.g., Scheme 31, 247 → 248), enolate generation (e.g., Scheme 32, 256 → 257), or imine formation (e.g., Scheme 24, 184 → 185). These advantages notwithstanding, the implementation of cycloadditions using complex substrates requires a detailed understanding of the electronic, steric, and conformational characteristics of the molecules. In some cases, these properties can be engineered into a substrate. Stereoselectivity in these cycloadditions is often predictable based on ground-state conformational analysis. Although sharing many of the same favorable characteristics, sigmatropic rearrangements and ene reactions are less common, due to the need to install specific functionality, such as aryl hydrazones (e.g., Scheme 40, 327), allylic alcohols (e.g., Scheme 48, 408), or keto-alkynes (e.g., Scheme 15, 113).
Scheme 55.
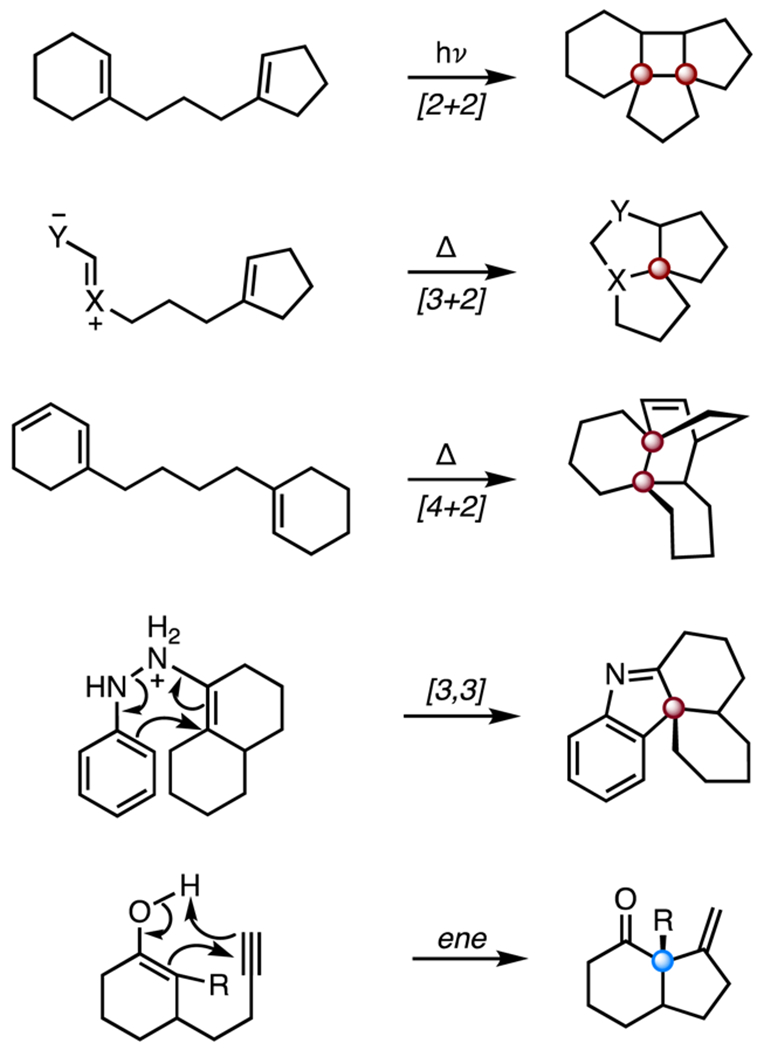
A general scheme for TBCC construction by cycloaddition ([2 + 2], [3 + 2], and [4 + 2]), sigma-tropic rearrangement, or ene reactions.
3.3. Radical mediated C–C bond formation
Radical mediated cyclization or recombination involving trisubstituted carbon atoms is another efficient way to construct TBCCs (Scheme 56). Such bond constructions can be initiated chemically from alkynes (e.g., Scheme 35, 287, azobisisobutyronitrile, tri-n-butyltin hydride) or alkenyl halides (e.g., Scheme 12, 92, azobisisobutyronitrile, tri-n-butyltin hydride), or photochemically from diazenes (e.g., Schemes 53, 455, hv). The orthogonality of radicals to most common heteroatom functional groups often translates to high compatibility and protection of substrates that might be sensitive to two-electron pathways, such as stereocenters that may be epimerized. Radical-mediated transformations are often incorporated into multi-step cascades; this can facilitate complex skeletal reorganizations that are difficult to achieve otherwise (e.g., Scheme 35, 287 → 290). The short lifetimes of open-shell intermediates are a limitation of this strategy, as it is necessary for the substrate to readily adopt a reactive conformation. The relative stability of carbon-centered radicals needs to be carefully considered to achieve multi-step cascade transformations involving multiple carbon–carbon bond-forming steps.
Scheme 56.
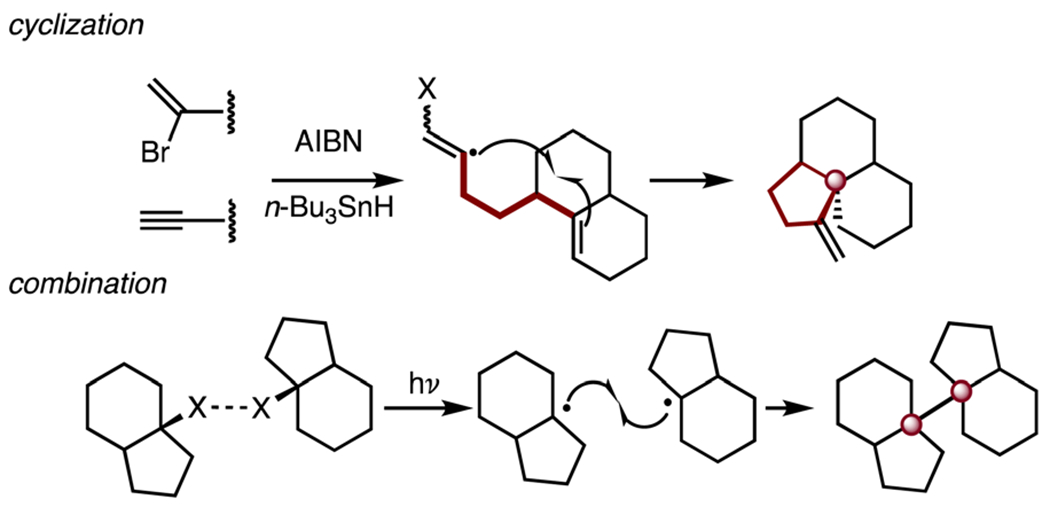
A general scheme for TBCC construction by radical cyclization and recombination.
3.4. Cation-based C–C bond formation
Acid-catalyzed cyclization cascades are a classic approach to constructing polycyclic scaffolds that often mimic the biosynthetic pathway to a given target. The carbocation may be formed by epoxide opening (Scheme 6, 42, trimethylaluminum), ionization of an allylic alcohol (Scheme 8, 63, boron trifluoride diethyl etherate), Nazarov cyclization (Scheme 18, 134, indium(III) hexafluoroantimonate), or Prins cyclization (Scheme 32, 259, tin(IV) chloride). The stereochemical outcomes are governed by well-established stereoelectronic requirements (Scheme 57). Like the radical-mediated transformations, cationic rearrangements also require carefully designed substrates that, in some instances, can be difficult to access. This is illustrated by the low number of cases (5 in total, Fig. 1), although all of these introduce the TBCC directly. Generation of the carbocation generally requires strongly acidic conditions, which excludes the use of acid-sensitive substrates.
Scheme 57.
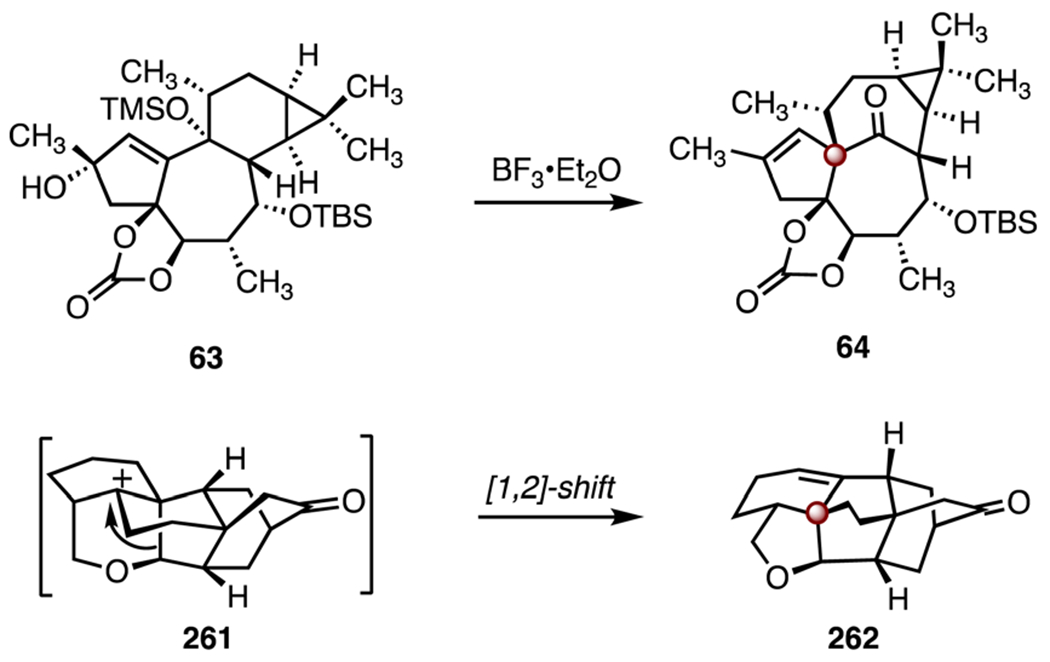
Selected examples of TBCC construction by carbocationic pathways.
3.5. Transition metal-catalyzed C–C bond formation
Transition metal-catalyzed couplings can affect the construction of TBCCs (Scheme 58). Examples presented here include Paulson–Khand reactions (e.g., Scheme 16, 124 → 108), allylic alkylations (e.g., Scheme 39, 312 → 315), and cyclopropanations (e.g., Scheme 47, 395 → 396). Incorporation of chiral ligands (e.g., Scheme 47, 391 → 392) can facilitate enantioselective TBCC formation.
Scheme 58.
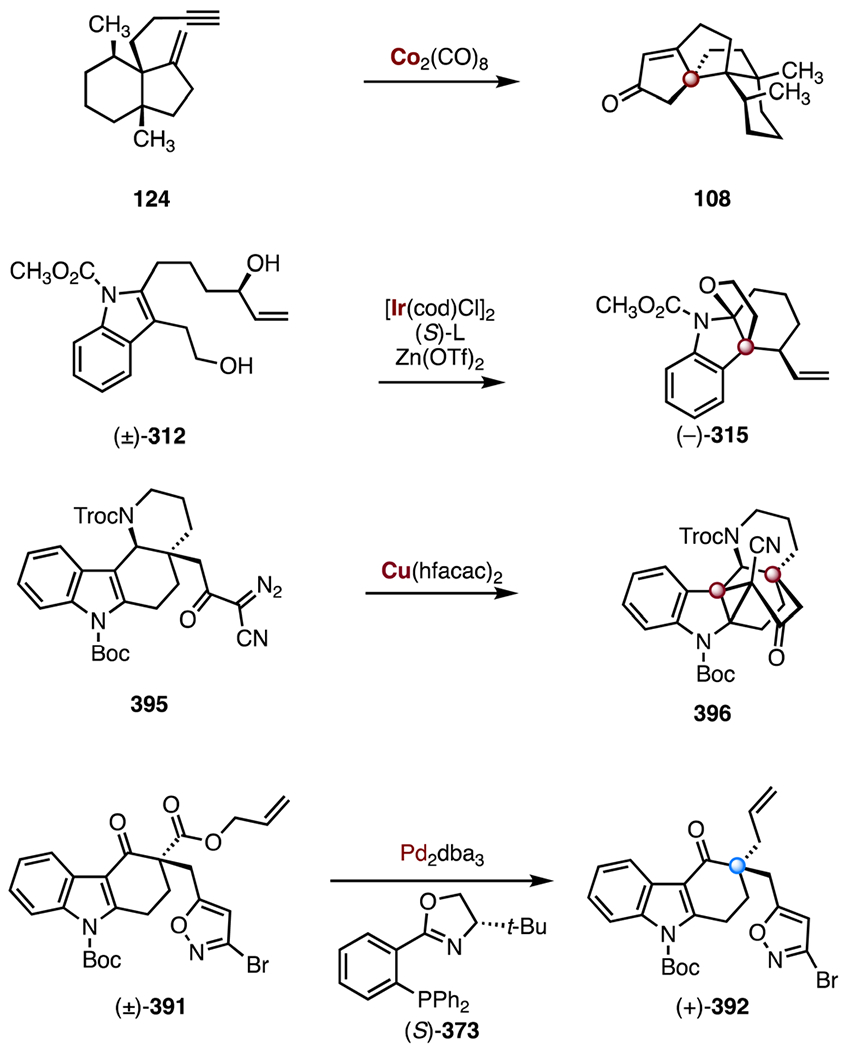
Selected examples of TBCC construction by transition metal-catalyzed C–C bond formation.
6. Acknowledgements
Financial support from the National Institutes of Health (R35-GM131913) is gratefully acknowledged.
Biographies

Zhi Xu was born in Harbin, China in 1997. He obtained his undergraduate degree in chemical biology from Peking University under the supervision of Prof. Xiaoguang Lei. In 2019, Zhi began his graduate studies in the laboratory of Prof. Seth Herzon at Yale University. He is currently a fourth-year graduate student working on total syntheses of diazofluorene and terpenoid natural products.

Xin Li was born in Handan, China in 1992. He obtained his B.S. degree in Chemistry from Renmin University of China (2013). He received his PhD in Organic Chemistry from Oregon State University in 2019 under the supervision of Prof. Rich Carter. He then worked on the synthetic study towards the total synthesis of lomaiviticins with Prof. Seth Herzon at Yale University as a postdoctoral associate. He is currently a Senior Research Investigator at Incyte Cooperation working on small molecular drug discovery.

John A. Rose was born in Fort Wayne IN in 1994. He completed his undergraduate studies at University of Indiana in 2016. He went on to received his PhD at Yale University under the supervision of Seth B. Herzon in 2021 where he worked on synthetic studies towards lomaiviticins. He is currently a Senior Research Scientist at Prelude Therapeutics.

Seth B. Herzon was born in Philadelphia PA in 1979, completed undergraduate studies at Temple University, obtained a PhD from Harvard University, and was a postdoctoral fellow at the University of Illinois, Urbana-Champaign. He is currently the Milton Harris ‘29 PhD professor of Chemistry and Professor of Pharmacology and Therapeutic Radiology at the Yale School of Medicine and a member of the Yale Comprehensive Cancer Center. Herzon’s research focuses on synthetic and translational studies of DNA damaging and microbiome-derived secondary metabolites, and the development of novel therapeutics targeting tumorassociated DNA repair defects.
Footnotes
Conflicts of interest
There are no conflicts to declare.
7 Notes and references
- 1.Ling T and Rivas F, Tetrahedron, 2016, 72, 6729–6777. [Google Scholar]
- 2.Xu P-W, Yu J-S, Chen C, Cao Z-Y, Zhou F and Zhou J, ACS Catal., 2019, 9, 1820–1882. [Google Scholar]
- 3.Zeng X-P, Cao Z-Y, Wang Y-H, Zhou F and Zhou J, Chem. Rev, 2016, 116, 7330–7396. [DOI] [PubMed] [Google Scholar]
- 4.Wang Z, Org. Chem. Front, 2020, 7, 3815–3841. [Google Scholar]
- 5.Prusov EV, Angew. Chem., Int. Ed, 2017, 56, 14356–14358. [DOI] [PubMed] [Google Scholar]
- 6.Li H and Lu Y, Asian J. Org. Chem, 2017, 6, 1130–1145. [Google Scholar]
- 7.Wu G, Wu J-R, Huang Y and Yang Y-W, Chem. - Asian J, 2021, 16, 1864–1877. [DOI] [PubMed] [Google Scholar]
- 8.Pandey G, Mishra A and Khamrai J, Tetrahedron, 2018, 74, 4903–4915. [Google Scholar]
- 9.Xue W, Jia X, Wang X, Tao X, Yin Z and Gong H, Chem. Soc. Rev, 2021, 50, 4162–4184. [DOI] [PubMed] [Google Scholar]
- 10.Büschleb M, Dorich S, Hanessian S, Tao D, Schenthal KB and Overman LE, Angew. Chem., Int. Ed, 2016, 55, 4156–4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Talele TT, J. Med. Chem, 2020, 63, 13291–13315. [DOI] [PubMed] [Google Scholar]
- 12.Lovering F, Bikker J and Humblet C, J. Med. Chem, 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]
- 13.Fujita E, Shibuya M, Nakamura S, Okada Y and Fujita T, J. Chem. Soc., Perkin Trans 1, 1974, 165–177. [Google Scholar]
- 14.Pan S, Chen S and Dong G, Angew. Chem., Int. Ed, 2018, 57, 6333–6336. [DOI] [PubMed] [Google Scholar]
- 15.Iitaka Y and Natsume M, Tetrahedron Lett., 1964, 5,1257–1261. [Google Scholar]
- 16.Kubota T, Matsuura T, Tsutsui T, Uyeo S, Irie H, Numata A, Fujita T and Suzuki T, Tetrahedron, 1966, 22, 1659–1699. [Google Scholar]
- 17.Sun H-D, Huang S-X and Han Q-B, Nat. Prod. Rep, 2006, 23, 673–698. [DOI] [PubMed] [Google Scholar]
- 18.Fujita E, Fujita T, Fuji K and Ito N, Tetrahedron, 1966, 22, 3423–3441. [Google Scholar]
- 19.Fujita E, Shibuya M, Nakamura S, Okada Y and Fujita T, J. Chem. Soc., Chem. Commun, 1972, 1107, DOI: 10.1039/C39720001107. [DOI] [Google Scholar]
- 20.Cornforth JW and Robinson R, J. Chem. Soc, 1949, 1855–1865. [DOI] [PubMed] [Google Scholar]
- 21.Spiegel DA, Njardarson JT and Wood JL, Tetrahedron, 2002, 58, 6545–6554. [Google Scholar]
- 22.Johansson CCC and Colacot TJ, Angew. Chem., Int. Ed, 2010, 49, 676–707. [DOI] [PubMed] [Google Scholar]
- 23.Gao K, Hu J and Ding H, Acc. Chem. Res, 2021, 54, 875–889. [DOI] [PubMed] [Google Scholar]
- 24.Lazarski KE, Moritz BJ and Thomson RJ, Angew. Chem., Int. Ed, 2014, 53, 10588–10599. [DOI] [PubMed] [Google Scholar]
- 25.Luo D, Callari R, Hamberger B, Wubshet SG, Nielsen MT, Andersen-Ranberg J, Hallström BM, Cozzi F, Heider H, Lindberg Møller B, Staerk D and Hamberger B, Proc. Natl. Acad. Sci. U. S. A, 2016, 113, E5082–E5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hecker E, Cancer Res., 1968, 28, 2338–2348. [PubMed] [Google Scholar]
- 27.Zechmeister KFB, Hoppe W, Hecker E, Opferkuch HJ and Adolf W, Tetrahedron Lett., 1970, 11, 4075–4078. [Google Scholar]
- 28.Ogbourne SM, Suhrbier A, Jones B, Cozzi S-J, Boyle GM, Morris M, McAlpine D, Johns J, Scott TM, Sutherland KP, Gardner JM, Le TTT, Lenarczyk A, Aylward JH and Parsons PG, Cancer Res., 2004, 64, 2833–2839. [DOI] [PubMed] [Google Scholar]
- 29.Fujiwara M, Ijichi K, Tokuhisa K, Katsuura K, Shigeta S, Konno K, Wang GY, Uemura D, Yokota T and Baba M, Antimicrob. Agents Chemother, 1996, 40, 271–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vasas A, Rédei D, Csupor D, Molnár J and Hohmann J, Eur. J. Org. Chem, 2012, 2012, 5115–5130. [Google Scholar]
- 31.Alchin DR, Dermatol. Ther, 2014, 4, 157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim S and Winkler JD, Chem. Soc. Rev, 1997, 26, 387–399. [Google Scholar]
- 33.Saunders M, J. Comput. Chem, 1989, 10, 203–208. [Google Scholar]
- 34.Kuwajima I and Tanino K, Chem. Rev, 2005, 105, 4661–4670. [DOI] [PubMed] [Google Scholar]
- 35.Winkler JD, Rouse MB, Greaney MF, Harrison SJ and Jeon YT, J. Am. Chem. Soc, 2002, 124, 9726–9728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanino K, Onuki K, Asano K, Miyashita M, Nakamura T, Takahashi Y and Kuwajima I, J. Am. Chem. Soc, 2003,125, 1498–1500. [DOI] [PubMed] [Google Scholar]
- 37.Nickel A, Maruyama T, Tang H, Murphy PD, Greene B, Yusuff N and Wood JL, J. Am. Chem. Soc, 2004, 126, 16300–16301. [DOI] [PubMed] [Google Scholar]
- 38.Jørgensen L, McKerrall SJ, Kuttruff CA, Ungeheuer F, Felding J and Baran PS, Science, 2013, 341, 878–882. [DOI] [PubMed] [Google Scholar]
- 39.Seebach D and Goliński J, Helv. Chim. Acta, 1981, 64, 1413–1423. [Google Scholar]
- 40.Winkler JD, Harrison ST, Greaney MF and Rouse MB, Synthesis, 2002, 14, 2150–2154. [Google Scholar]
- 41.Nakamura T, Matsui T, Tanino K and Kuwajima I, J. Org. Chem, 1997, 62, 3032–3033. [DOI] [PubMed] [Google Scholar]
- 42.Funk RL, Olmstead TA and Parvez M, J. Am. Chem. Soc, 1988, 110, 3298–3300. [Google Scholar]
- 43.Tang H, Yusuff N and Wood JL, Org. Lett, 2001, 3, 1563–1566. [DOI] [PubMed] [Google Scholar]
- 44.Weatherhead GS, Ford JG, Alexanian EJ, Schrock RR and Hoveyda AH, J. Am. Chem. Soc, 2000,122,1828–1829. [Google Scholar]
- 45.Tadayoshi Konegawa YO, Ikeda H, Sugai T and Ohta H, Synlett, 1997, 1997, 1297–1299. [Google Scholar]
- 46.Brummond KM, Chen H, Fisher KD, Kerekes AD, Rickards B, Sill PC and Geib SJ, Org. Lett, 2002, 4, 1931–1934. [DOI] [PubMed] [Google Scholar]
- 47.Nakamura MNA, Synthesis, 2013, 45, 1421–1451. [Google Scholar]
- 48.Nakanishi K, Habaguchi K, Nakadaira Y, Woods MC, Maruyama M, Major RT, Alauddin M, Patel AR, Weinges K and Baehr W, J. Am. Chem. Soc, 1971, 93, 3544–3546. [Google Scholar]
- 49.Ng CC, Duke RK, Hinton T and Johnston GAR, Eur. J. Pharmacol, 2017, 806, 83–90. [DOI] [PubMed] [Google Scholar]
- 50.Fernandez F, Morishita W, Zuniga E, Nguyen J, Blank M, Malenka RC and Garner CC, Nat. Neurosci, 2007, 10, 411–413. [DOI] [PubMed] [Google Scholar]
- 51.Corey EJ and Su WG, J. Am. Chem. Soc, 1987, 109, 7534–7536. [Google Scholar]
- 52.Crimmins MT, Jung DK and Gray JL, J. Am. Chem. Soc, 1993, 115, 3146–3155. [Google Scholar]
- 53.Baker MA, Demoret RM, Ohtawa M and Shenvi RA, Nature, 2019, 575, 643–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Corey EJ and Su W.-g., Tetrahedron Lett., 1987, 28, 5241–5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wolf C and Moskowitz M, J. Org. Chem, 2011, 76, 6372–6376. [DOI] [PubMed] [Google Scholar]
- 56.Fernández-Ibáñez MÁ, Maciá B, Alonso DA and Pastor IM, Eur. J. Org. Chem, 2013, 2013, 7028–7034. [Google Scholar]
- 57.Obradors C, Martinez RM and Shenvi RA, J. Am. Chem. Soc, 2016, 138, 4962–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fernandez Gonzalez D, Brand JP and Waser J, Chem. - Eur. J, 2010, 16, 9457–9461. [DOI] [PubMed] [Google Scholar]
- 59.Clarke DB, Hinkley SFR and Weavers RT, Tetrahedron Lett., 1997, 38, 4297–4300. [Google Scholar]
- 60.Lee H, Kang T and Lee H-Y, Angew. Chem., Int. Ed, 2017, 56, 8254–8257. [DOI] [PubMed] [Google Scholar]
- 61.Qu Y, Wang Z, Zhang Z, Zhang W, Huang J and Yang Z, J. Am. Chem. Soc, 2020, 142, 6511–6515. [DOI] [PubMed] [Google Scholar]
- 62.Peng C, Arya P, Zhou Z and Snyder SA, Angew. Chem., Int. Ed, 2020, 59, 13521–13525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rosenbaum L-C, Häfner M and Gaich T, Angew. Chem., Int. Ed, 2021, 60, 2939–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Y-P, Fang K, Tu Y-Q, Yin J-J, Zhao Q and Ke T, Nat. Commun, 2022, 13, 2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mori K and Matsui M, Tetrahedron, 1966, 22, 879–884. [Google Scholar]
- 66.Kang T, Song SB, Kim W-Y, Kim BG and Lee H-Y, J. Am. Chem. Soc, 2014, 136, 10274–10276. [DOI] [PubMed] [Google Scholar]
- 67.Bleschke C, Tissot M, Müller D and Alexakis A, Org. Lett, 2013, 15, 2152–2155. [DOI] [PubMed] [Google Scholar]
- 68.Ricker JD, Mohammadrezaei V, Crippen TJ, Zell AM and Geary LM, Organometallics, 2018, 37, 4556–4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Iwasaki K, Wan KK, Oppedisano A, Crossley SWM and Shenvi RA, J. Am. Chem. Soc, 2014, 136, 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brown MK, May TL, Baxter CA and Hoveyda AH, Angew. Chem., Int. Ed, 2007, 46, 1097–1100. [DOI] [PubMed] [Google Scholar]
- 71.Kennedy-Smith JJ, Staben ST and Toste FD, J. Am. Chem. Soc, 2004, 126, 4526–4527. [DOI] [PubMed] [Google Scholar]
- 72.Trost BM, Van Vranken DL and Bingel C, J. Am. Chem. Soc, 1992, 114, 9327–9343. [Google Scholar]
- 73.da Silva Araújo FD, de Lima Fávaro LC, Araújo WL, de Oliveira FL, Aparicio R and Marsaioli AJ, Eur. J. Org. Chem, 2012, 2012, 5225–5230. [Google Scholar]
- 74.Wangun H, Ishida K and Hertweck C, Eur. J. Org. Chem, 2008, 2008, 3781–3784. [Google Scholar]
- 75.Talontsi FM, Dittrich B, Schüffler A, Sun H and Laatsch H, Eur. J. Org. Chem, 2013, 2013, 3174–3180. [Google Scholar]
- 76.Ellerbrock P, Armanino N, Ilg MK, Webster R and Trauner D, Nat. Chem, 2015, 7, 879–882. [DOI] [PubMed] [Google Scholar]
- 77.Kravina AG and Carreira EM, Angew. Chem., Int. Ed, 2018, 57, 13159–13162. [DOI] [PubMed] [Google Scholar]
- 78.Cook SP and Danishefsky SJ, Org. Lett, 2006, 8, 5693–5695. [DOI] [PubMed] [Google Scholar]
- 79.Ellerbrock P, Armanino N and Trauner D, Angew. Chem., Int. Ed, 2014, 53, 13414–13418. [DOI] [PubMed] [Google Scholar]
- 80.Bellina F and Rossi R, Synthesis, 2007, 12, 1887–1889. [Google Scholar]
- 81.Kranz DP, Griesbeck AG, Alle R, Perez-Ruiz R, Neudörfl JM, Meerholz K and Schmalz H-G, Angew. Chem., Int. Ed, 2012, 51, 6000–6004. [DOI] [PubMed] [Google Scholar]
- 82.Ruider SA, Sandmeier T and Carreira EM, Angew. Chem., Int. Ed, 2015, 54, 2378–2382. [DOI] [PubMed] [Google Scholar]
- 83.Park HS, Lee IS, Kwon DW and Kim YH, Chem. Commun, 1998, 2745–2746. [Google Scholar]
- 84.Yin T, Zhang H, Zhang W and Jiang Z, RSC Adv., 2021,11, 36023–36033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Muratake H and Natsume M, Tetrahedron Lett, 2002, 43, 2913–2917. [Google Scholar]
- 86.Muratake H and Natsume M, Angew. Chem., Int. Ed, 2004, 43, 4646–4649. [DOI] [PubMed] [Google Scholar]
- 87.Peese KM and Gin DY, J. Am. Chem. Soc, 2006, 128, 8734–8735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kou KGM, Pflueger JJ, Kiho T, Morrill LC, Fisher EL, Clagg K, Lebold TP, Kisunzu JK and Sarpong R, J. Am. Chem. Soc, 2018, 140, 8105–8109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang Q, Zhang Z, Huang Z, Zhang C, Xi S and Zhang M, Angew. Chem., Int. Ed, 2018, 57, 937–941. [DOI] [PubMed] [Google Scholar]
- 90.Ochiai E, Okamoto T, Sakai S.-i. and Saito A, Yakugaku Zasshi, 1956, 76, 1414–1418. [Google Scholar]
- 91.Sakai S, Yamamoto I, Yamaguchi K, Takayama H, Ito M and Okamoto T, Chem. Pharm. Bull, 1982, 30, 4579–4582. [Google Scholar]
- 92.Wick AE, Felix D, Steen K and Eschenmoser A, Helv. Chim. Acta, 1964, 47, 2425–2429. [Google Scholar]
- 93.Johnson WS and Kinnel RB, J. Am. Chem. Soc, 1966, 88, 3861–3862. [DOI] [PubMed] [Google Scholar]
- 94.Reina M, Gavín JA, Madinaveitia A, Acosta RD and de la Fuente G, J. Nat. Prod, 1996, 59, 145–147. [Google Scholar]
- 95.Marth CJ, Gallego GM, Lee JC, Lebold TP, Kulyk S, Kou KGM, Qin J, Lilien R and Sarpong R, Nature, 2015, 528, 493–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kou KGM, Kulyk S, Marth CJ, Lee JC, Doering NA, Li BX, Gallego GM, Lebold TP and Sarpong R, J. Am. Chem. Soc, 2017, 139, 13882–13896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tambar UK and Stoltz BM, J. Am. Chem. Soc, 2005, 127, 5340–5341. [DOI] [PubMed] [Google Scholar]
- 98.Trost BM and Tang W, J. Am. Chem. Soc, 2002, 124, 14542–14543. [DOI] [PubMed] [Google Scholar]
- 99.Sun F and Yu DQ, Yaoxue Xuebao, 1985, 20, 913–917. [Google Scholar]
- 100.Ambros R, Angerer SV and Wiegrebe W, Arch. Pharm, 1988, 321, 481–486. [DOI] [PubMed] [Google Scholar]
- 101.Tang P, Chen QH and Wang FP, Tetrahedron Lett, 2009, 50, 460–462. [Google Scholar]
- 102.Tashkhodzhaev B, Saidkhodzhaeva SA, Bessonova IA and Antipin MY, Chem. Nat. Compd, 2000, 36, 79–83. [Google Scholar]
- 103.Weber M, Owens K and Sarpong R, Tetrahedron Lett, 2015, 56, 3600–3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gong J, Chen H, Liu X-Y, Wang Z-X, Nie W and Qin Y, Nat. Commun, 2016, 7, 12183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Xie S, Chen G, Yan H, Hou J, He Y, Zhao T and Xu J, J. Am. Chem. Soc, 2019, 141, 3435–3439. [DOI] [PubMed] [Google Scholar]
- 106.Nie W, Gong J, Chen Z, Liu J, Tian D, Song H, Liu X-Y and Qin Y, J. Am. Chem. Soc, 2019, 141, 9712–9718. [DOI] [PubMed] [Google Scholar]
- 107.Owens KR, McCowen SV, Blackford KA, Ueno S, Hirooka Y, Weber M and Sarpong R, J. Am. Chem. Soc, 2019, 141, 13713–13717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhou S, Xia K, Leng X and Li A, J. Am. Chem. Soc, 2019, 141, 13718–13723. [DOI] [PubMed] [Google Scholar]
- 109.Harayama T, Sato T, Nakano Y, Abe H and Takeuchi Y, Heterocycles, 2003, 59, 293–301. [Google Scholar]
- 110.Elamparuthi E, Fellay C, Neuburger M and Gademann K, Angew. Chem., Int. Ed, 2012, 51, 4071–4073. [DOI] [PubMed] [Google Scholar]
- 111.Vuagnoux-d’Augustin M and Alexakis A, Chem. - Eur. J, 2007, 13, 9647–9662. [DOI] [PubMed] [Google Scholar]
- 112.Petrova KV, Mohr JT and Stoltz BM, Org. Lett, 2009, 11, 293–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.White PB, Jaworski JN, Zhu GH and Stahl SS, ACS Catal., 2016, 6, 3340–3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Beierlein JM, Frey KM, Bolstad DB, Pelphrey PM, Joska TM, Smith AE, Priestley ND, Wright DL and Anderson AC, J. Med. Chem, 2008, 51, 7532–7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mukherjee S and Corey EJ, Org. Lett, 2010, 12, 632–635. [DOI] [PubMed] [Google Scholar]
- 116.Attolini M, Bouguir F, Iacazio G, Peiffer G and Maffei M, Tetrahedron, 2001, 57, 537–543. [Google Scholar]
- 117.Zhou S, Guo R, Yang P and Li A, J. Am. Chem. Soc, 2018, 140, 9025–9029. [DOI] [PubMed] [Google Scholar]
- 118.Leeds JP and Kirst HA, Synth. Commun, 1988, 18, 777–782. [Google Scholar]
- 119.Chattopadhyay AK and Hanessian S, Chem. Rev, 2017, 117, 4104–4146. [DOI] [PubMed] [Google Scholar]
- 120.Wu H, Zhang X, Ding L, Chen S, Yang J and Xu X, Planta Med., 2013, 79, 1589–1598. [DOI] [PubMed] [Google Scholar]
- 121.Kang B, Jakubec P and Dixon DJ, Nat. Prod. Rep, 2014, 31, 550–562. [DOI] [PubMed] [Google Scholar]
- 122.Guo L-D, Chen Y and Xu J, Acc. Chem. Res, 2020, 53, 2726–2737. [DOI] [PubMed] [Google Scholar]
- 123.Heathcock CH, Stafford JA and Clark DL, J. Org. Chem, 1992, 57, 2575–2585. [Google Scholar]
- 124.Guo L-D, Hu J, Zhang Y, Tu W, Zhang Y, Pu F and Xu J, J. Am. Chem. Soc, 2019, 141, 13043–13048. [DOI] [PubMed] [Google Scholar]
- 125.Arbain D, Byrne L, Cannon J, Patrick V and White A, Aust. J. Chem, 1990, 43, 185–190. [Google Scholar]
- 126.Panthong A, Kanjanapothi D, Thitiponpunt Y, Taesotikul T and Arbain D, Planta Med., 1998, 64, 530–535. [DOI] [PubMed] [Google Scholar]
- 127.Zhang C-R, Yang S-P and Yue J-M, J. Nat. Prod, 2008, 71, 1663–1668. [DOI] [PubMed] [Google Scholar]
- 128.Trost BM and Metzner PJ, J. Am. Chem. Soc, 1980, 102, 3572–3577. [Google Scholar]
- 129.Ma D, Tang G and Kozikowski AP, Org. Lett, 2002, 4, 2377–2380. [DOI] [PubMed] [Google Scholar]
- 130.Guo L-D, Hou J, Tu W, Zhang Y, Zhang Y, Chen L and Xu J, J. Am. Chem. Soc, 2019, 141, 11713–11720. [DOI] [PubMed] [Google Scholar]
- 131.Molander GA and Etter JB, Tetrahedron Lett., 1984, 25, 3281–3284. [Google Scholar]
- 132.Molander GA and Etter JB, J. Org. Chem, 1986, 51,1778–1786. [Google Scholar]
- 133.Molander GA and McKie JA, J. Org. Chem, 1991, 56, 4112–4120. [Google Scholar]
- 134.Adams GL and Smith AB, The Alkaloids: Chemistry and Biology, ed. Knölker H-J, Academic Press, 2016, vol. 76, pp. 171–257. [DOI] [PubMed] [Google Scholar]
- 135.Ramirez A and Garcia-Rubio S, Curr. Med. Chem, 2003,10, 1891–1915. [DOI] [PubMed] [Google Scholar]
- 136.Smith JM, Moreno J, Boal BW and Garg NK, Angew. Chem., Int. Ed, 2015, 54, 400–412. [DOI] [PubMed] [Google Scholar]
- 137.Teng M, Zi W and Ma D, Angew. Chem., Int. Ed, 2014, 53, 1814–1817. [DOI] [PubMed] [Google Scholar]
- 138.Ren W, Wang Q and Zhu J, Angew. Chem., Int. Ed, 2014, 53, 1818–1821. [DOI] [PubMed] [Google Scholar]
- 139.Jiang S-Z, Zeng X-Y, Liang X, Lei T, Wei K and Yang Y-R, Angew. Chem., Int. Ed, 2016, 55, 4044–4048. [DOI] [PubMed] [Google Scholar]
- 140.Moreno J, Picazo E, Morrill LA, Smith JM and Garg NK, J. Am. Chem. Soc, 2016, 138, 1162–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Picazo E, Morrill LA, Susick RB, Moreno J, Smith JM and Garg NK, J. Am. Chem. Soc, 2018, 140, 6483–6492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Li W, Chen Z, Yu D, Peng X, Wen G, Wang S, Xue F, Liu X-Y and Qin Y, Angew. Chem., Int. Ed, 2019, 58, 6059–6063. [DOI] [PubMed] [Google Scholar]
- 143.Subramaniam G, Hiraku O, Hayashi M, Koyano T, Komiyama K and Kam T-S, J. Nat. Prod, 2007, 70, 1783–1789. [DOI] [PubMed] [Google Scholar]
- 144.Schnoes HK, Biemann K, Mokry J, Kompis I, Chatterjee A and Ganguli G, J. Org. Chem, 1966, 31, 1641–1642. [Google Scholar]
- 145.Ahmad Y, Fatima K, Atta ur R, Occolowitz JL, Solheim BA, Clardy J, Garnick RL and Le Quesne PW, J. Am. Chem. Soc, 1977, 99, 1943–1946. [Google Scholar]
- 146.Zi W, Xie W and Ma D, J. Am. Chem. Soc, 2012, 134, 9126–9129. [DOI] [PubMed] [Google Scholar]
- 147.Sole D, Cancho Y, Llebaria A, Moreto JM and Delgado A, J. Am. Chem. Soc, 1994, 116, 12133–12134. [Google Scholar]
- 148.Solé D, Bosch J and Bonjoch J, Tetrahedron, 1996, 52, 4013–4028. [Google Scholar]
- 149.Li Q, Li G, Ma S, Feng P and Shi Y, Org. Lett, 2013, 15, 2601–2603. [DOI] [PubMed] [Google Scholar]
- 150.Jiao L and Bach T, J. Am. Chem. Soc, 2011, 133, 12990–12993. [DOI] [PubMed] [Google Scholar]
- 151.Jiao L, Herdtweck E and Bach T, J. Am. Chem. Soc, 2012, 134, 14563–14572. [DOI] [PubMed] [Google Scholar]
- 152.Zu L, Boal BW and Garg NK, J. Am. Chem. Soc, 2011, 133, 8877–8879. [DOI] [PubMed] [Google Scholar]
- 153.Trost BM, Zhang T and Sieber JD, Chem. Sci, 2010, 1, 427–440. [Google Scholar]
- 154.Lu Z, Li Y, Deng J and Li A, Nat. Chem, 2013, 5, 679–684. [DOI] [PubMed] [Google Scholar]
- 155.Cardullo F, Donati D, Merlo G, Paio A, Salaris M and Taddei M, Synlett, 2005, 2005, 2996–2998. [Google Scholar]
- 156.Brandau S, Landa A, Franzén J, Marigo M and Jørgensen KA,Angew. Chem., Int. Ed, 2006, 45, 4305–4309. [DOI] [PubMed] [Google Scholar]
- 157.Greshoff M, Ber. Dtsch. Chem. Ges, 1890, 23, 3537–3550. [Google Scholar]
- 158.Kam T-S, Subramaniam G, Sim K-M, Yoganathan K, Koyano T, Toyoshima M, Rho M-C, Hayashi M and Komiyama K, Bioorg. Med. Chem. Lett, 1998, 8, 2769–2772. [DOI] [PubMed] [Google Scholar]
- 159.Lim K-H, Hiraku O, Komiyama K, Koyano T, Hayashi M and Kam T-S, J. Nat. Prod, 2007, 70, 1302–1307. [DOI] [PubMed] [Google Scholar]
- 160.Lim S-H, Sim K-M, Abdullah Z, Hiraku O, Hayashi M, Komiyama K and Kam T-S, J. Nat. Prod, 2007, 70, 1380–1383. [DOI] [PubMed] [Google Scholar]
- 161.Kam T-S, Sim K-M, Koyano T and Komiyama K, Phytochemistry, 1999, 50, 75–79. [Google Scholar]
- 162.Zhou H, He H-P, Kong N-C, Wang Y-H, Liu X-D and Hao X-J, Helv. Chim. Acta, 2006, 89, 515–519. [Google Scholar]
- 163.Tan M-J, Yin C, Tang C-P, Ke C-Q, Lin G and Ye Y, Planta Med, 2011, 77, 939–944. [DOI] [PubMed] [Google Scholar]
- 164.Chen J, Yang M-L, Zeng J and Gao K, Phytochem. Lett, 2014, 7, 156–160. [Google Scholar]
- 165.Qin B, Wang Y, Wang X and Jia Y, Org. Chem. Front, 2021, 8, 369–383. [Google Scholar]
- 166.Gallagher T and Magnus P, J. Am. Chem. Soc, 1983, 105, 2086–2087. [Google Scholar]
- 167.Jones SB, Simmons B, Mastracchio A and MacMillan DWC, Nature, 2011, 475, 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wei Y, Zhao D and Ma D, Angew. Chem., Int. Ed, 2013, 52, 12988–12991. [DOI] [PubMed] [Google Scholar]
- 169.Ni D, Wei Y and Ma D, Angew. Chem., Int. Ed, 2018, 57, 10207–10211. [DOI] [PubMed] [Google Scholar]
- 170.Leng L, Zhou X, Liao Q, Wang F, Song H, Zhang D, Liu X-Y and Qin Y, Angew. Chem., Int. Ed, 2017, 56, 3703–3707. [DOI] [PubMed] [Google Scholar]
- 171.Tong X, Shi B, Liang K, Liu Q and Xia C, Angew. Chem., Int. Ed, 2019, 58, 5443–5446. [DOI] [PubMed] [Google Scholar]
- 172.Achenbach H and Biemann K, J. Am. Chem. Soc, 1965, 87, 4944–4950. [DOI] [PubMed] [Google Scholar]
- 173.Filho JMF, Gilbert B, Kitagawa M, Leme LAP and Durham LJ, J. Chem. Soc. C, 1966, 1260–1266. [Google Scholar]
- 174.Craven BM, Gilbert B and Leme LAP, Chem. Commun, 1968, 955–956. [Google Scholar]
- 175.Crow W and Michael M, Aust. J. Chem, 1955, 8, 129–135. [Google Scholar]
- 176.Jones SB, Simmons B and MacMillan DWC, J. Am. Chem. Soc, 2009, 131, 13606–13607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wei-Shin C, Sao-Han L, Kirfel A, Will G and Breitmaier E, Liebigs Ann. Chem, 1981, 1981, 1886–1892. [Google Scholar]
- 178.Gartshore CJ and Lupton DW, Angew. Chem., Int. Ed, 2013, 52, 4113–4116. [DOI] [PubMed] [Google Scholar]
- 179.Fukuzawa S.-i., Matsuzawa H and Yoshimitsu S.-i., J. Org. Chem, 2000, 65, 1702–1706. [DOI] [PubMed] [Google Scholar]
- 180.Zeng T, Wu X-Y, Yang S-X, Lai W-C, Shi S-D, Zou Q, Liu Y and Li L-M, J. Nat. Prod, 2017, 80, 864–871. [DOI] [PubMed] [Google Scholar]
- 181.Govindachari TR, Pai BR, Rajappa S, Viswanathan N, Kump WG, Nagarajan K and Schmid H, Helv. Chim. Acta, 1962, 45, 1146–1152. [Google Scholar]
- 182.Okino T, Hoashi Y and Takemoto Y, J. Am. Chem. Soc, 2003, 125, 12672–12673. [DOI] [PubMed] [Google Scholar]
- 183.Govindachari TR, Nagarajan K and Schmid H, Helv. Chim. Acta, 1963, 46, 433–444. [Google Scholar]
- 184.Guggisberg A, Hesse M, Von Philipsborn W, Nagarajan K and Schmid H, Helv. Chim. Acta, 1966, 49, 2321–2337. [Google Scholar]
- 185.Bhattacharya A, Chatterjee A and Bose PK, J. Am. Chem. Soc, 1949, 71, 3370–3372. [Google Scholar]
- 186.Battersby AR and Gregory H, J. Chem. Soc, 1963, 22–32. [Google Scholar]
- 187.Trost BM and Xu J, J. Am. Chem. Soc, 2005, 127, 17180–17181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Morita H, Sekiguchi M, Hirasawa Y, Zaima K, Teh C-H and Chan K, Heterocycles, 2008, 76. [Google Scholar]
- 189.Overman LE, Angew. Chem., Int. Ed, 1984, 23, 579–586. [Google Scholar]
- 190.Hoye RC and Rajapakse HA, Synth. Commun, 1997, 27, 663–672. [Google Scholar]
- 191.Chai W, Takeda A, Hara M, Ji S-J and Horiuchi CA, Tetrahedron, 2005, 61, 2453–2463. [Google Scholar]
- 192.Nemoto H, Kawamura T and Miyoshi N,J. Am. Chem. Soc, 2005, 127, 14546–14547. [DOI] [PubMed] [Google Scholar]
- 193.Hayashi H, Matsumoto H and Akiyama K, Biosci., Biotechnol., Biochem, 2004, 68, 753–756. [DOI] [PubMed] [Google Scholar]
- 194.Yang J, Wu H, Shen L and Qin Y, J. Am. Chem. Soc, 2007, 129, 13794–13795. [DOI] [PubMed] [Google Scholar]
- 195.Liu P, Seo JH and Weinreb SM, Angew. Chem., Int. Ed, 2010, 49, 2000–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Seo JH, Liu P and Weinreb SM, J. Org. Chem, 2010, 75, 2667–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Zuo Z, Xie W and Ma D, J. Am. Chem. Soc, 2010, 132, 13226–13228. [DOI] [PubMed] [Google Scholar]
- 198.Trost BM and Osipov M, Chem. - Eur. J, 2015, 21, 16318–16343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Zhang D, Song H and Qin Y, Acc. Chem. Res, 2011, 44, 447–457. [DOI] [PubMed] [Google Scholar]
- 200.Zhang D, Song H and Qin Y, Strategies and Tactics in Organic Synthesis, ed. Harmata M, Academic Press, 2015, vol. 11, pp. 377–408. [Google Scholar]
- 201.Zuo Z and Ma D, Isr. J. Chem, 2011, 51, 434–441. [Google Scholar]
- 202.Morris JC, Nat. Prod. Rep, 2013, 30, 783–805. [DOI] [PubMed] [Google Scholar]
- 203.Belmar J and Funk RL, J. Am. Chem. Soc, 2012, 134, 16941–16943. [DOI] [PubMed] [Google Scholar]
- 204.Liang X, Zhang T-Y, Zeng X-Y, Zheng Y, Wei K and Yang Y-R, J. Am. Chem. Soc, 2017, 139, 3364–3367. [DOI] [PubMed] [Google Scholar]
- 205.Park J, Jean A and Chen DY-K, Angew. Chem., Int. Ed, 2017, 56, 14237–14240. [DOI] [PubMed] [Google Scholar]
- 206.Park J, Jean A and Chen DYK, J. Org. Chem, 2018, 83, 6936–6957. [DOI] [PubMed] [Google Scholar]
- 207.Lathrop SP, Pompeo M, Chang W-TT and Movassaghi M, J. Am. Chem. Soc, 2016, 138, 7763–7769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Fuchs JR and Funk RL, J. Am. Chem. Soc, 2004, 126, 5068–5069. [DOI] [PubMed] [Google Scholar]
- 209.Fuchs JR and Funk RL, Org. Lett, 2005, 7, 677–680. [DOI] [PubMed] [Google Scholar]
- 210.Ku J-M, Jeong B-S, Jew S.-s. and Park H.-g., J. Org. Chem, 2007, 72, 8115–8118. [DOI] [PubMed] [Google Scholar]
- 211.Yamamoto H, Ho E, Namba K, Imagawa H and Nishizawa M, Chem. - Eur. J, 2010, 16, 11271–11274. [DOI] [PubMed] [Google Scholar]
- 212.Sakaitani M and Ohfune Y, Tetrahedron Lett., 1985, 26, 5543–5546. [Google Scholar]
- 213.Wang QP, Bennet AJ, Brown RS and Santarsiero BD, J. Am. Chem. Soc, 1991, 113, 5757–5765. [Google Scholar]
- 214.Liu R and Zhang J, Org. Lett, 2013, 15, 2266–2269. [DOI] [PubMed] [Google Scholar]
- 215.Liu R and Zhang J, Chem. - Eur. J, 2013, 19, 7319–7323. [DOI] [PubMed] [Google Scholar]
- 216.Mukaiyama T, Ogata K, Sato I and Hayashi Y, Chem. - Eur. J, 2014, 20, 13583–13588. [DOI] [PubMed] [Google Scholar]
- 217.Wang Q, Han J, Wang C, Zhang J, Huang Z, Shi D and Zhao Y, Chem. Sci, 2014, 5, 4962–4967. [Google Scholar]
- 218.Lin H-C, Chiou G, Chooi Y-H, McMahon TC, Xu W, Garg NK and Tang Y, Angew. Chem., Int. Ed, 2015, 54, 3004–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Lin H-C, McMahon TC, Patel A, Corsello M, Simon A, Xu W, Zhao M, Houk KN, Garg NK and Tang Y, J. Am. Chem. Soc, 2016, 138, 4002–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Movassaghi M, Ahmad OK and Lathrop SP, J. Am. Chem. Soc, 2011, 133, 13002–13005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Lathrop SP, Kim J and Movassaghi M, Chimia, 2012, 66, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Birch AJ, J. Chem. Soc, 1944, 430–436. [Google Scholar]