

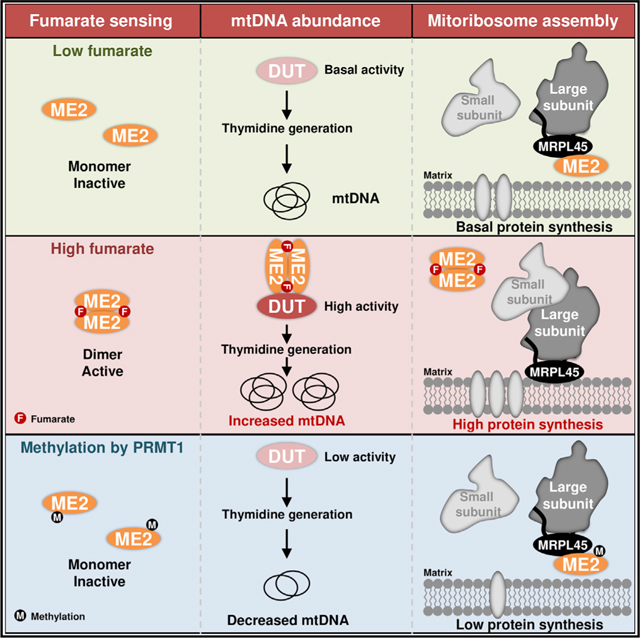
SUMMARY
Mitochondria have an independent genome (mtDNA) and protein synthesis machinery that coordinately activate for mitochondrial generation. Here, we report that the Krebs cycle intermediate fumarate links metabolism to mitobiogenesis through binding to malic enzyme 2 (ME2). Mechanistically, fumarate binds ME2 with two complementary consequences. First, promoting the formation of ME2 dimers, which activate deoxyuridine 5′-triphosphate nucleotidohydrolase (DUT). DUT fosters thymidine generation and an increase of mtDNA. Second, fumarate-induced ME2 dimers abrogate ME2 monomer binding to mitochondrial ribosome protein L45, freeing it for mitoribosome assembly and mtDNA-encoded protein production. Methylation of the ME2-fumarate binding site by protein arginine methyltransferase-1 inhibits fumarate signaling to constrain mitobiogenesis. Notably, acute myeloid leukemia is highly dependent on mitochondrial function and is sensitive to targeting of the fumarate-ME2 axis. Therefore, mitobiogenesis can be manipulated in normal and malignant cells through ME2, an unanticipated governor of mitochondrial biomass production that senses nutrient availability through fumarate.
In brief
Wang et al. report that intramitochondrial ME2 connects mitobiogenesis to metabolism through fumarate. Fumarate promotes ME2 dimerization activating DUT and increasing mtDNA. Fumarate disrupts ME2-MRPL45 interaction, enhancing mitoribosome assembly and mitochondrial protein production. PRMT1 methylates ME2, inhibiting mitobiogenesis. Fumarate-ME2 axis is a regulatable link between nutrient substrate and mitochondrial generation.
Graphical Abstract
INTRODUCTION
Mitochondria are powerhouses of cellular metabolism that are highly integrated into eukaryotic cell bioenergetic requirements (Birsoy et al., 2015; Chandel, 2015; Sullivan et al., 2015). Mitochondrial mass is dynamically regulated by both the nuclear and mitochondrial genomes (nDNA and mtDNA) in response to nutrient availability. Mitochondria maintain a dNTP pool to support the replication of mtDNA, which encodes at least 13 proteins, and harbor unique ribosomal proteins for protein generation (Mansueto et al., 2017; Wallace, 2016; Zong et al., 2016). The mitoribosomal proteins are synthesized in the cytoplasm and assembled in mitochondria (Bogenhagen et al., 2018; Brown et al., 2017; Rackham et al., 2016), where they are required for mtDNA-encoded protein translation. All of the mtDNA-encoded proteins are synthesized within the mitochondria and function in the electron transport chain (ETC), necessitating their residence in the mitochondrial inner membrane (Richter-Dennerlein et al., 2016). Notably, mitochondrial ribosomal protein L45 (MRPL45) directly binds to the inner membrane and is required for mitoribosome assembly (Kehrein et al., 2015; Zeng et al., 2018).
Mammalian cells fuel mitochondria with a variety of nutrients (Chen et al., 2016a; Corbet et al., 2016; DeBerardinis and Chandel, 2016; Faubert et al., 2017). Multiple nutrient-sensing pathways in the cytoplasm and nucleus have been discovered to govern mitobiogenesis-related transcriptional and translational programs. In contrast, how mitochondria sense nutrients and modulate biomass production is unclear.
Dysregulated mitobiogenesis has been implicated in multiple human diseases including cancer and aging (Dorn et al., 2015; Yambire et al., 2019). Notably, hyperactivation of mitobiogenesis has been shown to promote leukemia, liver cancer, and breast cancer (Carew et al., 2004; Jitschin et al., 2014; LeBleu et al., 2014; Martinez-Outschoorn et al., 2011; Skrtić et al., 2011 ; Tohme et al., 2017). Mitobiogenesis regulation is of particular interest in acute myeloid leukemia (AML), a highly lethal hematopoietic neoplasm. AML cells have been reported to have more mitochondria than normal hematopoietic cells (Boultwood et al., 1996) and are dependent upon oxidative metabolism (Baccelli et al., 2019; Jones et al., 2018; Konopleva et al., 2016; Molina et al., 2018). More importantly, chemoresistant AML cells shift to higher oxidative phosphorylation (OXPHOS) and are particularly sensitive to inhibition of cellular respiration (Farge et al., 2017). Agents that inhibit mitochondrial translation (Farge et al., 2017) have shown promising effects in suppressing AML (Jones et al., 2018). Therefore, we set out to identify metabolite-sensing pathways that regulate mitobiogenesis. Unexpectedly, fumarate was defined as a signaling metabolite that acts via malic enzyme 2 to regulate mitochondrial biomass production.
RESULTS
Fumarate upregulates mitochondrial biomass
To define metabolites acting as signaling molecules to regulate mitochondrial biomass, we conducted a cell-based screen. AML cell lines were used due to their sensitivity to OXPHOS (Carew et al., 2004) with normal CD34+ cord blood cells as controls. We exposed cells to different metabolites for 48 h and assayed mtDNA and mitotracker green (MTG) for mitochondrial mass. Glucose mildly increased mitochondrial mass, in agreement with its role as a major carbon source (Figure S1A). Surprisingly, cell-permeable fumarate (dimethyl fumarate, DMF) strongly elevated mtDNA and MTG staining of AML cells, but not of normal cells. Succinate, the mitochondrial precursor of fumarate, only led to a modest mitochondrial mass increase (Figure S1A). Notably, treatment with cell-permeable fumarate, including DMF and diethyl fumarate (DEF), increased mitochondrial mass in a dose- and time-dependent manner (Figures 1A, S1B, and S1C) significantly above control (Figures S1D and S1E). Correspondingly, fumarate levels positively correlate with OXPHOS potential in AML cells (Figures 1B and S1F). Mitochondria number by transmission electron microscopy was 1.98-fold higher in DMF-treated MOLM14 cells (Figures 1C and 1D). Because DMF is an electrophilic compound that modifies proteins on thiols (Kulkarni et al., 2019), we asked whether DMF rather than fumarate elevated mitochondrial mass. Due to the low cell permeability of fumarate, we treated AML cells with a high dose of fumarate (1 mM) compared with fumarate esters, including monomethyl fumarate (MMF) and monoethyl fumarate (MEF). Fumarate and its esters elevated intracellular and mitochondrial fumarate by approximately 2-fold in MOLM14 cells (Figures 1E, S1G, and S1H), with a concurrent increase in mtDNA, MTG intensity (Figures 1F and 1G), and mitochondrial respiration (Figure 1H). Importantly, the mtDNA increase was greater in DMF-treated cells than in cells overexpressing nuclear respiration factor 2 (NRF2), a known nuclear transcription regulator of mitobiogenesis (Guo et al., 2019) (Figures S1I and S1J). [U-13C]-fumarate tracing in MOLM14 cells further demonstrated that the products of mitochondrial fumarate metabolism, including malate and citrate, were efficiently labeled (Figures S1K–S1N). These data indicate that exogenous fumarate is capable of entering into mitochondria and increasing mitochondrial mass.
Figure 1. Fumarate upregulates mitochondrial biomass.
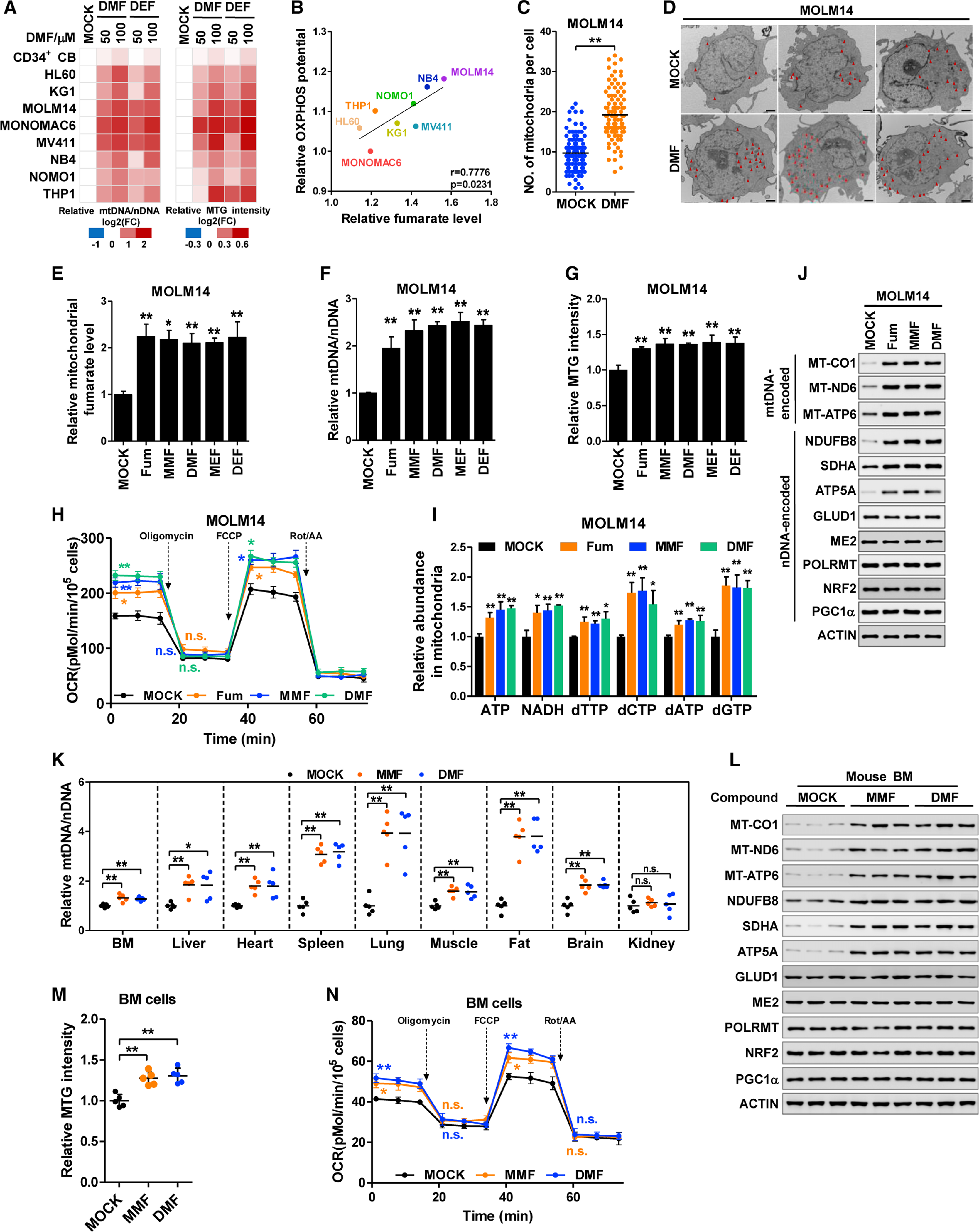
(A) Human cord blood CD34+ cells and AML cell lines were treated with DMF or DEF for 24 h. mtDNA was determined by qPCR and normalized to nDNA (left). Cells were stained with MTG and the fluorescent intensity was normalized to cell number (right). All data were normalized to DMSO-treated group. The fold change (FC) was presented on a log2 scale.
(B) Pearson correlation of fumarate and maximum respiration capacity (OXPHOS potential) of AML cell lines.
(C and D) Control and DMF-treated MOLM14 cells were analyzed with transmission electron microscopy. Mitochondria numbers in 100 cells were counted (C). Representative images were shown in (D) (scale bar, 1 μm).
(E–J) MOLM14 cells were treated with fumarate (Fum) and its esters for 24 h. Mitochondrial fumarate (E), mtDNA copies (F), MTG intensity (G), oxygen consumption rate (H), and mitochondrial ATP, NADH, and dNTPs (I) were quantified. Whole-cell lysates were subjected to western blotting; β-actin (actin) was included as the loading control (J).
(K–N) Mice were injected intraperitoneally with DMSO (MOCK), MMF, or DMF for 7 days. mtDNA copies from multiple tissues were quantified (n = 5) (K). Mitochondrial proteins in the BM cells from three independent mice was determined by western blotting (L). MTG intensity (M) and oxygen consumption rate (N) of BM cells were assayed (n = 5).
All data are shown as mean ± SEM from three independent experiments. *p < 0.05, **p < 0.01; n.s. indicates not significant. See also Figure S1.
We next asked how fumarate modulates mitochondrial mass. As mitochondria have a distinct dNTP pool, we quantified multiple metabolites in nucleotide metabolism. In line with the enhanced respiration, fumarate increased cellular and mitochondrial levels of ATP and NADH, indicating an increase in energy production (Figures 1I, S1G, and S1O). The substrates for DNA synthesis, dTTP, dCTP, dATP, and dGTP, were also increased in fumarate-treated cells (Figures 1I and S1O). These findings suggest that fumarate may promote dNTP anabolism to provide building blocks for mtDNA. This is confirmed by the increased generation of mtDNA after fumarate treatment (Figure S1D).
Mitochondrial proteins are encoded by both mtDNA and nDNA. We tested whether fumarate modulates mitochondrial gene transcription. Notably, fumarate minimally changed mtDNA-encoded mRNAs (Figure S1P) or nDNA-encoded mitochondrial enzymes including mRNA for SDHA (succinate dehydrogenase A) and GLUD1 (glutamate dehydrogenase 1) (Figure S1P). Furthermore, fumarate did not modify mitochondrial RNA polymerase (POLRMT) levels (Figure 1J). mtDNA abundance did not couple with mtDNA transcription, consistent with findings by others (Agaronyan et al., 2015). However, multiple mitochondrial proteins including ETC proteins encoded by either mtDNA (MT-CO1, MT-ND6, and MT-ATP6) or nDNA (NDUFB8, SDHA, and ATP5A) were increased by fumarate treatment (Figure 1J). This corresponded to the enhanced respiration we observed. In contrast, nDNA-encoded mitochondrial proteins such as glutamate dehydrogenase 1 (GLUD1), malic enzyme 2 (ME2), and nuclear transcription factors regulating mitochondrial biogenesis such as NRF2 and peroxisome proliferator-activated receptor-γ coactivator 1α (PGC1α) were not affected by fumarate and its esters (Figure 1J). Therefore, fumarate upregulates mitochondrial dNTPs, mtDNA, and selectively, mtDNA-encoded proteins.
As fumarate esters and high-dose fumarate equivalently elevated mitochondrial mass, we used an esterified form of fumarate in our following experiments. Testing fumarate in vivo, daily intraperitoneal administration of fumarate esters (MMF and DMF) led to a remarkable accumulation of fumarate in multiple organs, but not kidney (Figure S1Q). In agreement, mtDNA abundance was significantly increased in the tested organs except kidney (Figure 1K). mtDNA-encoded proteins were elevated by MMF and DMF (Figures 1L and S1R). Focusing on hematopoietic tissue, MMF and DMF moderately increased MTG staining and mitochondrial respiration of bone marrow (BM) cells (Figures 1M and 1N). Taken together, fumarate upregulates mitochondrial biomass.
Fumarate relies on ME2 to increase mitochondrial biomass
Assessing the mechanism by which fumarate alters mitochondria, we focused on metabolic enzymes that directly bind fumarate including fumarylacetoacetase (FAH), adenylosuccinate lyase (ADSL), argininosuccinate lyase (ASL), SDHA, fumarate hydratase (FH), and malic enzyme 2 (ME2) (Tao et al., 2003) (Figure 2A). We silenced NRF2 (positive control) and fumarate-binding enzymes with short hairpin RNAs (shRNAs) (Figure S2A). Knockdown of NRF2 and ME2, but not the other enzymes, abolished the DMF-mediated increase of mtDNA (Figure 2B). These results demonstrate that ME2 and NRF2 are required for fumarate to elevate mitochondrial mass.
Figure 2. Fumarate relies on ME2 to increase mitochondrial mass.
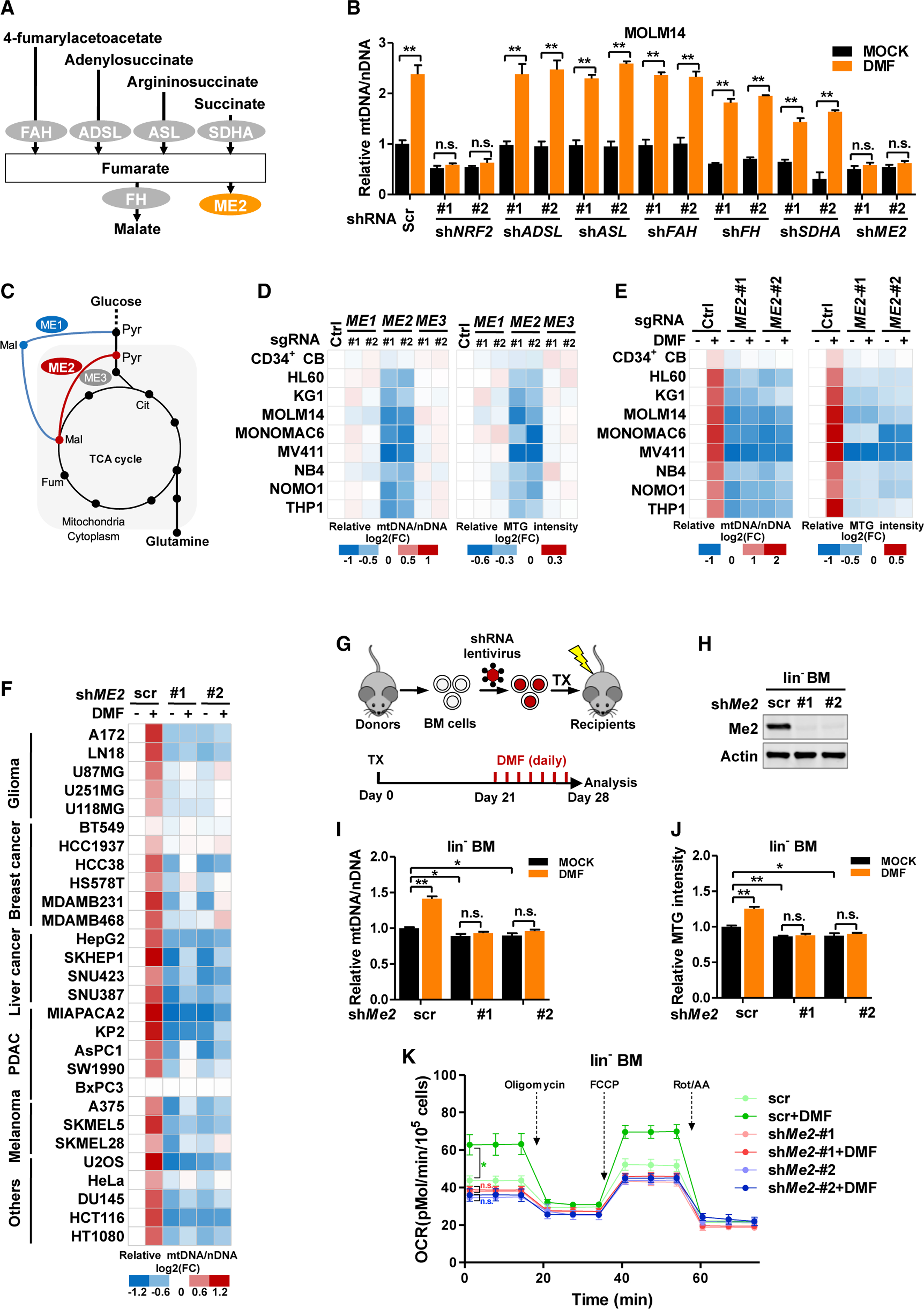
(A) Schematic overview of fumarate-interacting enzymes.
(B) Scrambled control (Scr) or shRNAs targeting fumarate-binding enzymes were stably expressed in MOLM14 cells. mtDNA was determined by qPCR after DMF treatment.
(C) Schematic overview of malic enzymes in central carbon metabolism.
(D and E) Human CD34+ CB cells and AML cells were transduced with control short guide RNA (sgRNA) or sgRNAs targeting malic enzymes. mtDNA abundance (left) and MTG intensity (right) were determined (D). Cells were further treated with DMF. mtDNA abundance (left) and MTG intensity (right) were assayed (E). All data were normalized to the control group.
(F) A panel of solid tumor cell lines was transduced with scrambled control or shRNAs targeting ME2. mtDNA were quantified after DMF treatment. All data were normalized to the scrambled control.
(G–K) Mouse BM cells were transduced with scrambled control or shRNAs targeting Me2. BM cells were transplanted (TX) into lethally irradiated mice. Twentyone days after transplantation, mice were injected intraperitoneally with DMSO (MOCK) or DMF for 7 days (G). Me2 protein (H), mtDNA abundance (I), MTG intensity (J), and oxygen consumption (K) in lin− BM cells were assayed (n = 5).
All data are shown as mean ± SEM from three independent experiments. *p < 0.05, **p < 0.01; n.s. indicates not significant. See also Figure S2 and Table S1.
Mammalian malic enzymes have three paralogues. Notably, ME2 is a mitochondrial enzyme coordinating glucose and glutamine metabolism (Figure 2C) (Jiang et al., 2013). Interestingly, ME2, but not its mitochondrial paralogue ME3, was highly expressed in AML cells (Figure S2B). To evaluate the functions of malic enzymes, we depleted them with short guide RNAs (sgRNAs) and quantified mitochondrial mass (Figure S2C). Knockdown of ME2, but not ME1 or ME3, dramatically lowered mtDNA and MTG intensity in AML cell lines (Figure 2D), accompanied by decreased consumption of glucose and glutamine (Figures S2D and S2E). Importantly, DMF was incapable of increasing mitochondrial mass in ME2-depleted AML cells (Figure 2E). To test the cell specificity of fumarate-induced mitobiogenesis, we collected cancer cell lines with different tissues of origin and stably expressed ME2-targeting shRNAs (Figure S2F). DMF increased mtDNA in 27 of 28 cell lines tested. This increment was abolished in ME2-depleted cells (Figure 2F). Therefore, ME2 is indispensable for fumarate to upregulate mitochondrial mass.
Examining ME2 in vivo, we stably silenced Me2 with shRNAs in lin− BM cells. Three weeks after transplanting control or Me2-knockdown cells, DMF was intraperitoneally injected for 7 days (Figures 2G and 2H). Me2 knockdown led to a moderate decrease of mtDNA, MTG intensity, and oxygen consumption in lin− BM cells (Figures 2I–2K). Importantly, DMF failed to upregulate mitochondria mass or mitochondrial respiration in Me2-knockdown cells (Figures 2I–2K). These observations suggest that Me2 mediates fumarate-induced mitobiogenesis in vivo.
ME2 responds to fumarate by increasing DUT activity and mtDNA
We next investigated how ME2 mediated fumarate signaling. Although ME2 diverts malate from the TCA cycle to produce pyruvate (Figure 2C), citrate, but not pyruvate, was efficiently labeled in a [U-13C]-fumarate tracing assay (Figures S1M and S1N). These data indicate that the catalytic activity of ME2 plays a minor role in mitochondrial carbon flux. Because fumarate is an allosteric activator of ME2 (Tao et al., 2003), we hypothesized that fumarate might conjugate with or physically bind to ME2 to regulate mitochondrial mass.
High-dose fumarate (>1 mM) was previously reported to modify thiols in the form of succination to destabilize or inactivate thiol-containing proteins, such as aconitase 2 (ACO2) (Ternette et al., 2013). We transduced MOLM14 cells with Flag-tagged ME2 and ACO2 and treated them with increasing doses of DMF. Western blotting analysis demonstrated that ACO2 succination was mildly increased by high-dose DMF, while succination of ME2 was undetectable (Figure S3A). Fumarate conjugation would be expected to decrease free thiols on ME2. Interestingly, the biotin-conjugated maleimide assay for free thiols showed no effect on ME2 from high doses of DMF while the pull-down efficacy of ACO2 was decreased (Figure S3B). Besides, ME2 protein was unchanged by MMF and DMF (Figure S3C). Together, these data argue against the possibility that the fumarate ester covalently modifies ME2.
We next evaluated the physical association of fumarate and ME2. The residence of fumarate in the ME2 dimer interface (Figure 3A) indicates that fumarate potentially regulates ME2 oligomerization. As anticipated, fumarate and its esters enhanced ME2 dimerization in glutaraldehyde crosslinking assays (Figures 3B and S3D). We introduced point mutations at the dimer interface to generate fumarate-binding defective mutants (R67F and E59L) (Figure 3A). A catalytic-inactive mutant (CM) was also generated by mutating three key residues (glutamate164, arginine165, and isoleucine166) to alanines in the catalytic center. As anticipated, fumarate-binding defective mutations decreased ME2 activity and abolished fumarate-induced activation, while the CM mutant lost its catalytic activity (Figure S3E). More importantly, R67F and E59L mutants, but not the CM mutant, existed predominantly as monomers (Figure 3C). DMF treatment promoted the dimerization of wild-type ME2 and ME2CM, but not fumarate binding-defective mutants (Figure 3C). Co-immuno-precipitation (coIP) assay further showed that fumarate enhanced the interaction of ME2-HA with wild-type ME2-GFP and its CM mutant, but not R67F and E59L mutants (Figures S3F and S3G). In addition, we silenced ME2 with shRNA and reintroduced wild-type ME2-Flag and its mutants in MOLM14 and KG1 cells (Figures S3H and S3I). ME2 depletion significantly decreased mitochondrial respiration, which was largely restored by the CM mutant, but not the fumarate binding-defective mutants (Figures 3D and S3J). These results suggest that the fumarate-ME2 association is essential to maintain mitochondrial respiration. Importantly, the monomer-to-dimer transition of ME2 then potentially provides a structural basis for fumarate signaling.
Figure 3. ME2 responds to fumarate by increasing DUT activity and mtDNA.
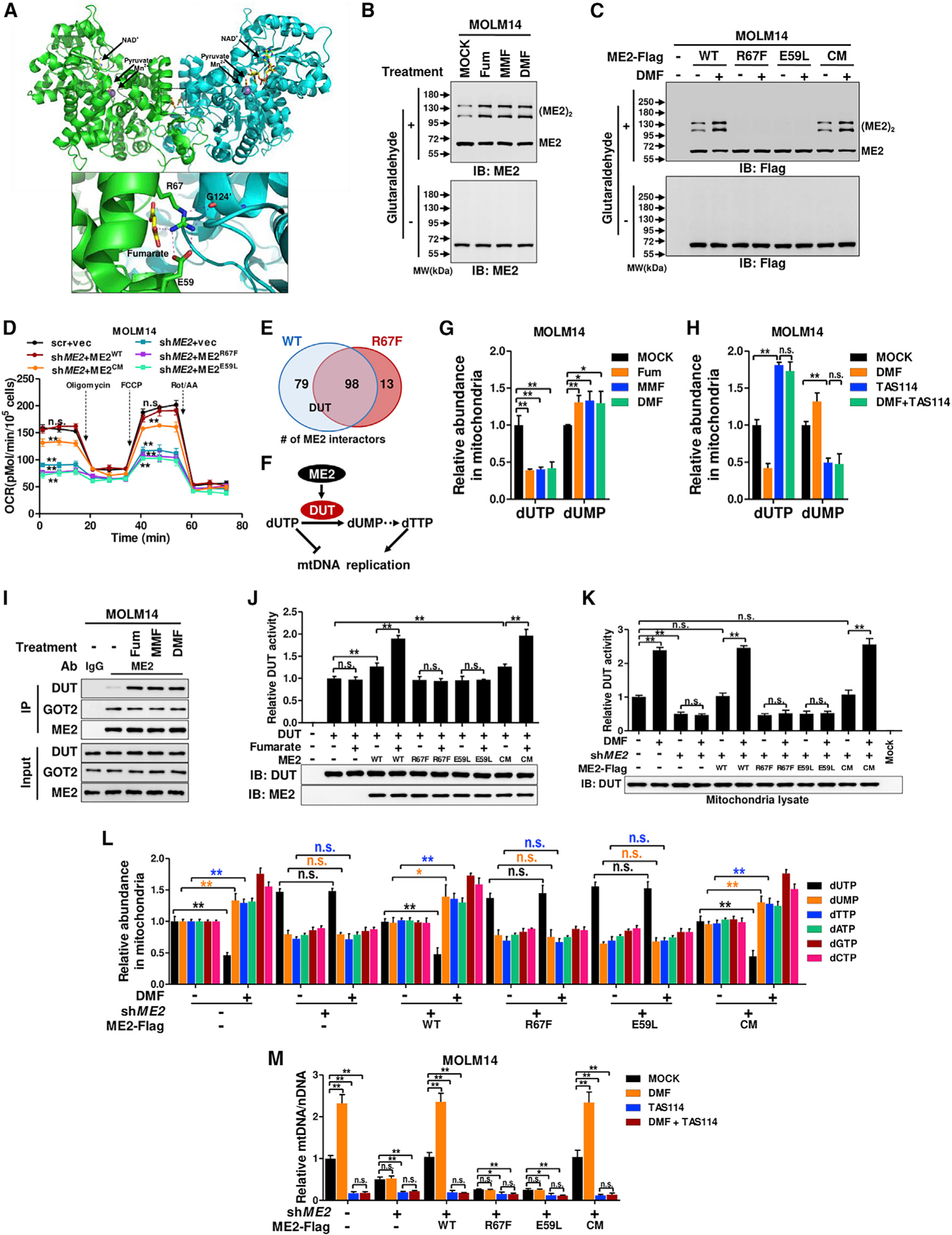
(A) ME2 dimer (PDB: 1PJ4) (Tao et al., 2003) was colored green and cyan on each subunit.
(B) Whole-cell lysate was analyzed by crosslinking after treating MOLM14 cells with fumarate or its esters.
(C) MOLM14 cells expressing ME2-Flag and its mutants were treated with DMF and subjected to crosslinking assay.
(D) The oxygen consumption rate of ME2-knockdown and re-expression MOLM14 cells was determined.
(E) ME2-Flag and its R67F mutant were stably expressed in MOLM14 cells. ME2-interacting proteins were identified by pull-down mass spectrometry. Shown are numbers of wild-type or mutant ME2-interacting mitochondrial proteins.
(F) DUT limits dUTP and enhances dTTP synthesis.
(G) Mitochondrial dUTP and dUMP were determined after treating MOLM14 cells with fumarate or its esters.
(H) Mitochondrial dUTP and dUMP were quantified after treating MOLM14 cells with DMF and TAS114.
(I) Endogenous ME2 was immunoprecipitated to determine its interaction with DUT and GOT2 in fumarate-treated MOLM14 cells.
(J) DUT-Flag was immunopurified from MOLM14 cells and mixed with recombinant ME2 to determine its activity.
(K and L) ME2-knockdown and re-expression MOLM14 cells were treated with DMF for 24 h. Mitochondria lysate was subjected to DUT activity assay (K).
Mitochondrial dUTP, dUMP, and four dNTPs were quantified (L).
(M) mtDNA was determined after treating MOLM14 cells with DMF and TAS114.
All data are presented as mean ± SEM from three independent experiments. *p < 0.05, **p < 0.01; n.s. indicates not significant. See also Figure S3 and Table S2.
We next asked whether ME2 was involved in fumarate-induced upregulation of mtDNA. Because fumarate increased mitochondrial dNTP, we hypothesized that fumarate-ME2 interaction may regulate dNTP metabolism. We immunopurified wild-type ME2-Flag and the R67F mutant from MOLM14 cells to identify ME2 interactors (Figures 3E and S3K). Proteomic profiling revealed that two enzymes in dNTP metabolism, deoxyuridine 5′-triphosphate nucleotidohydrolase (DUT) and dCTP pyrophosphatase 1 (dCTPP1), interacted with ME2 (Figure 3E; Table S2). DUT converts dUTP to dUMP and enables dTTP production (Figure 3F). Interestingly, fumarate decreased dUTP and increased dUMP in MOLM14 cells (Figures 3G and S1G), suggesting an elevation of DUT activity. We therefore focused on DUT because it not only supports thymidine nucleotide production to maintain mtDNA synthesis but also limits dUTP, which can misincorporate into DNA and destabilize mtDNA (Hirmondo et al., 2017) (Figure 3F). As expected, TAS114, a DUT-specific inhibitor, elevated dUTP and decreased dUMP in MOLM14 cells (Figures 3H and S3L). Importantly, TAS114 also abrogated the effect of DMF on mitochondrial dUTP and dUMP (Figure 3H), suggesting DMF dependency on DUT to modulate dUTP and dUMP levels. Since fumarate mediates ME2 dimerization, we asked whether fumarate affected the association of ME2 and DUT. Fumarate treatment increased the binding of ME2 with DUT, but not with another ME2 interactor, glutamic-oxaloacetic transaminase 2 (GOT2) (Figure 3I). CoIP assays further showed that R67F and E59L mutations abolished the interaction of ME2 and DUT (Figure S3M). Notably, fumarate increased the association of DUT with wild-type ME2 and ME2CM, but not the fumarate binding-defective mutants (Figure S3M). These data demonstrate that dimerization of ME2, which is enhanced by fumarate, increases the interaction between ME2 and DUT.
To determine whether ME2 modulates DUT activity, we assayed the catalytic efficiency of immunopurified DUT-Flag in the presence of fumarate or recombinant ME2 (Figure S3N). Fumarate alone was incapable of activating DUT (Figure 3J). Wild-type ME2 and its CM mutant, but not ME2R67F and ME2E59L, mildly increased DUT’s activity. Importantly, fumarate strongly increased the catalytic efficiency of DUT in the presence of ME2 and its CM mutants, but not fumarate binding-defective mutants (Figure 3J). Similar results were observed in DUT activity assays using mitochondria lysates (Figures 3K and S3O). These data indicate that fumarate-induced ME2 dimerization activates DUT.
We next tested whether ME2 modulated dNTP levels. DMF mildly elevated dUMP and dTTP, with a concomitant mild increase of dATP, dGTP, and dCTP (Figure S3P). We further isolated mitochondria from treated cells and found that the mitochondrial dNTPs showed more dramatic changes (Figures 3L and S3O). In contrast, DMF treatment of ME2-knockdown cells rescued by fumarate binding-defective mutants failed to increase dUMP and four dNTPs (Figure 3L). These data demonstrate that fumarate modulates dNTP levels through binding with ME2. Moreover, DMF failed to upregulate mtDNA in ME2-knockdown cells and cells rescued by fumarate binding-defective mutants (Figures 3M and S3Q). Importantly, TAS114 strongly decreased mtDNA copy number, which was no longer elevated by DMF (Figures 3M and S3Q). These results suggest that DUT activity is indispensable for fumarate signaling to increase mtDNA. Together, fumarate signals through ME2 to activate DUT and elevate mitochondrial dNTP levels, resulting in the upregulation of mtDNA.
ME2 responds to fumarate by modulating mitoribosome assembly
In addition to mtDNA, fumarate also upregulated mtDNA-encoded proteins (Figure 1J). Given mitochondrial mRNA levels were modestly affected by fumarate, we asked whether ME2 regulated mitochondrial protein translation. Grouping of ME2 interactors by their annotated functions (https://www.uniprot.org/) revealed a wide distribution across mitochondria biology (Figure 4A). Notably, the fumarate-sensing defective mutant (R67F) showed a dramatic decrease in the number of ME2-interacting mitoribosomal proteins, but not ETC components (Figures 4A and 4B). This observation led us to question whether ME2 regulates the mitoribosome.
Figure 4. ME2 responds to fumarate by modulating mitoribosome assembly.
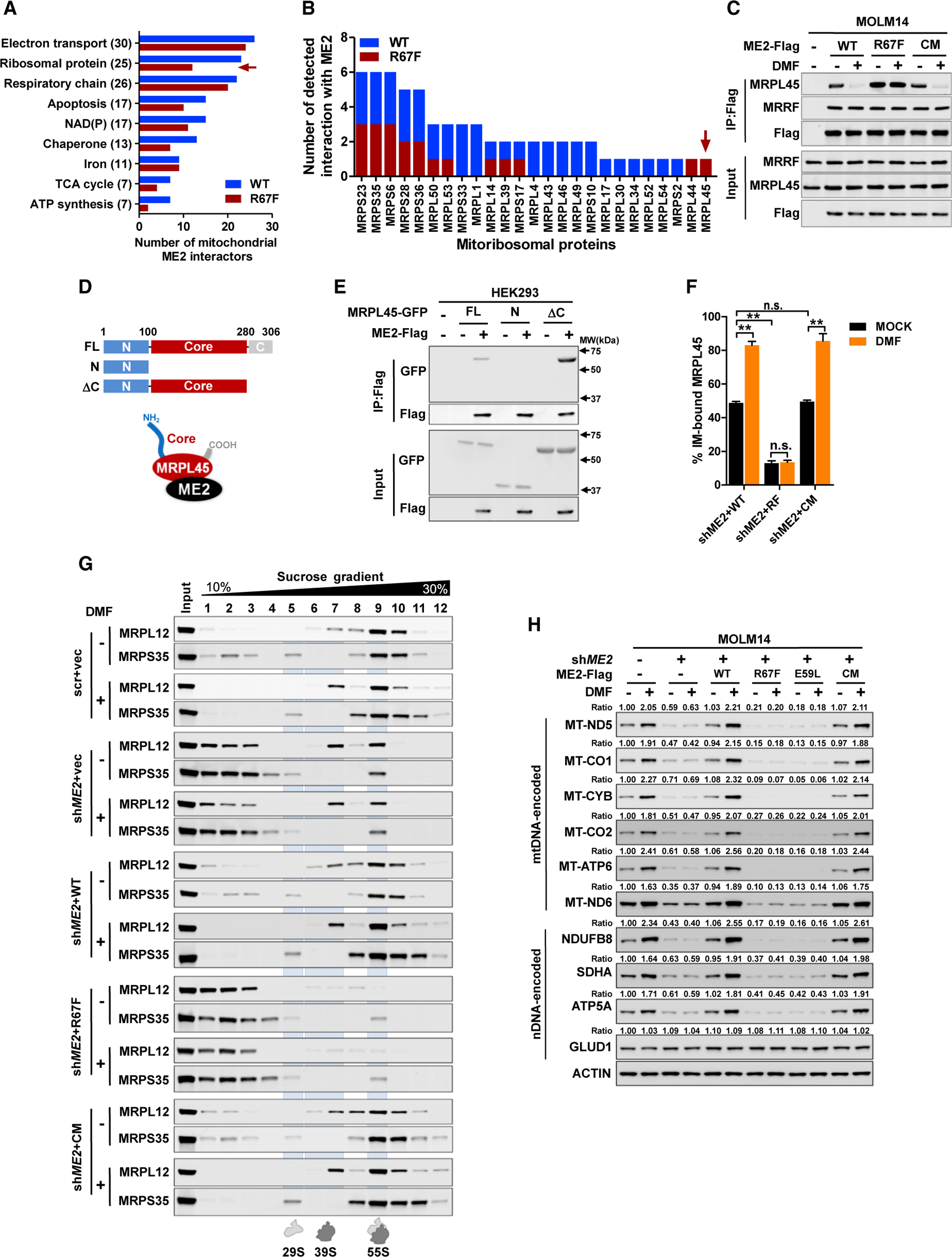
(A) ME2 interactors were functionally grouped; the number on the y axis indicates total number of wild-type or mutant ME2-binding proteins.
(B) ME2-interacting proteins were identified by pull-down mass spectrometry in three independent experiments. The number of detected interactions of ME2 (wild-type and R67F mutant) with mitoribosomal proteins was determined.
(C) MOLM14 cells expressing ME2-Flag and its mutants were treated with DMF. The interaction of ME2 with MRPL45 and MRRF was determined.
(D and E) GFP-tagged full-length MRPL45 (FL) and its mutants (N and ΔC) (D) were co-expressed with ME2-Flag to determine their association (E).
(F) ME2-knockdown and re-expression MOLM14 cells were treated with DMF. Isolated mitochondria were fractionated to determine MRPL45 localization. (G and H) ME2-knockdown and re-expression MOLM14 cells were treated with DMF for 24 h. Isolated mitochondria were loaded on a sucrose gradient to fractionate mitoribosome (G). mtDNA and nDNA-encoded proteins were determined (H).
All data are presented as mean ± SEM from three independent experiments. **p < 0.01; n.s. indicates not significant. See also Figure S4.
We focused on the regulatory proteins of the mitoribosome in the ME2 interactome, including MRPL45 and mitochondrial ribosome recycling factor (MRRF) (Table S2). While MRPL45 anchors the mitoribosome to the mitochondrial inner membrane (Kummer et al., 2018), MRRF dissociates mitoribosomes from mRNA upon translation termination to recycle them (Rorbach et al., 2008). CoIP assays revealed that wild-type ME2 and R67F mutant bound MRRF at similar levels (Figure 4C). However, MRPL45 differentially bound ME2R67F compared with wild-type ME2 and ME2CM. More importantly, DMF disrupted the binding of MRPL45 with wild-type ME2 and ME2CM, but not ME2R67F (Figure 4C). Additionally, semi-endogenous coIP showed that MRPL45 interacted with ME2, but not ME3 (Figure S4A). In vitro pull-down further demonstrated that fumarate, but not malate or succinate, dissociated the ME2-MRPL45 complex (Figure S4B). These results indicate that monomeric ME2 strongly interacts with MRPL45, and that the interaction is disrupted by fumarate.
MRPL45 has tails on both ends to bind to the mitoribosome large subunit and a core domain that directly interacts with the inner membrane (Figure 4D). We truncated MRPL45 to map the ME2-binding region. CoIP assays showed that the full-length MRPL45 and its ΔC mutant, but not the N-mutant, interacted with ME2 (Figures 4D and 4E), indicating that ME2 binds to the core domain of MRPL45 and potentially regulates its inner membrane attachment. To examine this, we isolated mitochondria from ME2-knockdown and re-expression MOLM14 cells and further fractionated mitochondria into mitoplast (MP, without outer membrane), inner membrane (IM), and matrix (Mtx) (Figure S4C). Western blotting demonstrated that in cells rescued by wild-type ME2 or ME2CM, approximately 49% of total MRPL45 located on the inner membrane (Figures 4F and S4C). In contrast, only 13% of MRPL45 resided on the inner membrane in R67F-rescued MOLM14 cells. Exogenous fumarate dramatically increased inner membrane attachment of MRPL45 in control cells, but not in cells rescued by ME2R67F (Figure 4F). Therefore, ME2 monomers dissociate MRPL45 from the inner membrane. Fumarate promotes inner membrane attachment of MRPL45 by abolishing ME2-MRPL45 interaction.
Inner membrane attachment of MRPL45 is a prerequisite for mitoribosome assembly and activity (Kummer et al., 2018). We next evaluated mitoribosome assembly using MRPL12 and MRPS35 as the markers for large and small subunits, respectively. While ME2 knockdown reduced the level of mitoribosome assembly, reintroduction of wild-type ME2 or ME2CM, but not ME2R67F, restored mitoribosome complexing in MOLM14 cells (Figure 4G). Notably, DMF treatment enhanced mitoribosome assembly in control cells and cells that were rescued by wild-type ME2 or ME2CM, but not in ME2-knockdown cells or cells rescued by ME2R67F (Figure 4G). Therefore, fumarate binding is essential for ME2 to regulate mitoribosome assembly.
Mitoribosomes are dedicated to manufacturing proteins in the ETC. Interestingly, mtDNA-encoded ETC genes including MT-ND5, MT-CO1, MT-CYB, MT-CO2, MT-ATP6, and MT-ND6 showed modest changes in their mRNA expression in ME2-knockdown and re-expression cells (Figure S4D). However, ME2 knockdown reduced the protein expression of these mtDNA-encoded genes (Figures 4H and S4E). Re-introducing wild-type ME2 and ME2CM restored the level of mtDNA-encoded proteins. This did not happen with fumarate-sensing defective mutants (Figures 4H and S4E). DMF was unable to enhance mtDNA-encoded protein expression in cells rescued by the R67F and E59L mutants (Figures 4H and S4E). Interestingly, nuclear-encoded ETC proteins (NDUFB8, SDHA, and ATP5A) showed a similar expression pattern (Figures 4H and S4E). Previously, nDNA and mtDNA have been shown to produce mitochondrial proteins in a synchronized manner (Couvillion et al., 2016). To examine whether fumarate coordinates nDNA and mtDNA-encoded protein expression, we silenced MRPL45 and NRF2 and observed a decrease of ETC proteins and MTG intensity (Figures S4F–S4H). Moreover, fumarate-induced mitobiogenesis was blocked in these cells (Figures S4G and S4H). Taken together, ME2 functions as a fumarate-responsive sensor to modulate mitoribosome assembly and mtDNA-encoded protein expression (Figure S4I).
PRMT1 methylates ME2, inhibiting fumarate sensing
Because mitobiogenesis commits a cell to an energetically expensive process, we hypothesized that there may be more than a simple direct relationship of fumarate levels to ME2 initiation of mitobiogenesis. Specifically, we assessed possible post-translational modifications, a well-defined mechanism of modifying enzyme activity and metabolite binding (Xiong and Guan, 2012), that might modulate the fumarate-ME2 interaction. We focused on the fumarate-binding site (Figure S5A) where arginine 67 (R67) is the only residue reported to be methylated (Larsen et al., 2016). We assessed whether methylation of it by arginine methyltransferases (PRMTs) regulates fumarate responsiveness. Notably, treatment with PRMT inhibitors (AMI-1 and AMI-5) revealed that ME2 arginine methylation associated with downregulated activity (Figure S5B). To precisely monitor R67 methylation, we generated a site-specific methylation antibody [α-me-ME2(R67)] (Figures S5C and S5D). We also mutated R67 into lysine (R67K). Notably, wild-type ME2, but not R67K or the R67F mutant, was readily recognized by the site-specific methylation antibody (Figure S5E). AMI-5 treatment resulted in a 4-fold decrease of R67 methylation, with a concomitant increase of ME2 activity. ME2R67F and ME2R67K were barely recognized by the site-specific methylation antibody and were deficient in catalysis (Figure S5E). Notably, AMI-5 reduced R67 methylation of endogenous ME2 in MOLM14 and KG1 cells (Figures 5A and S5F). Further, both R67K and R67F mutations disrupted ME2 dimerization (Figures S5G and S5H). In addition, AMI-5 treatment enhanced the interaction of ME2-HA with wild-type ME2-GFP, but not ME2R67F-GFP (Figure S5I). These data indicate that R67 methylation suppresses ME2 dimerization.
Figure 5. PRMT1 methylates ME2, inhibiting fumarate sensing.
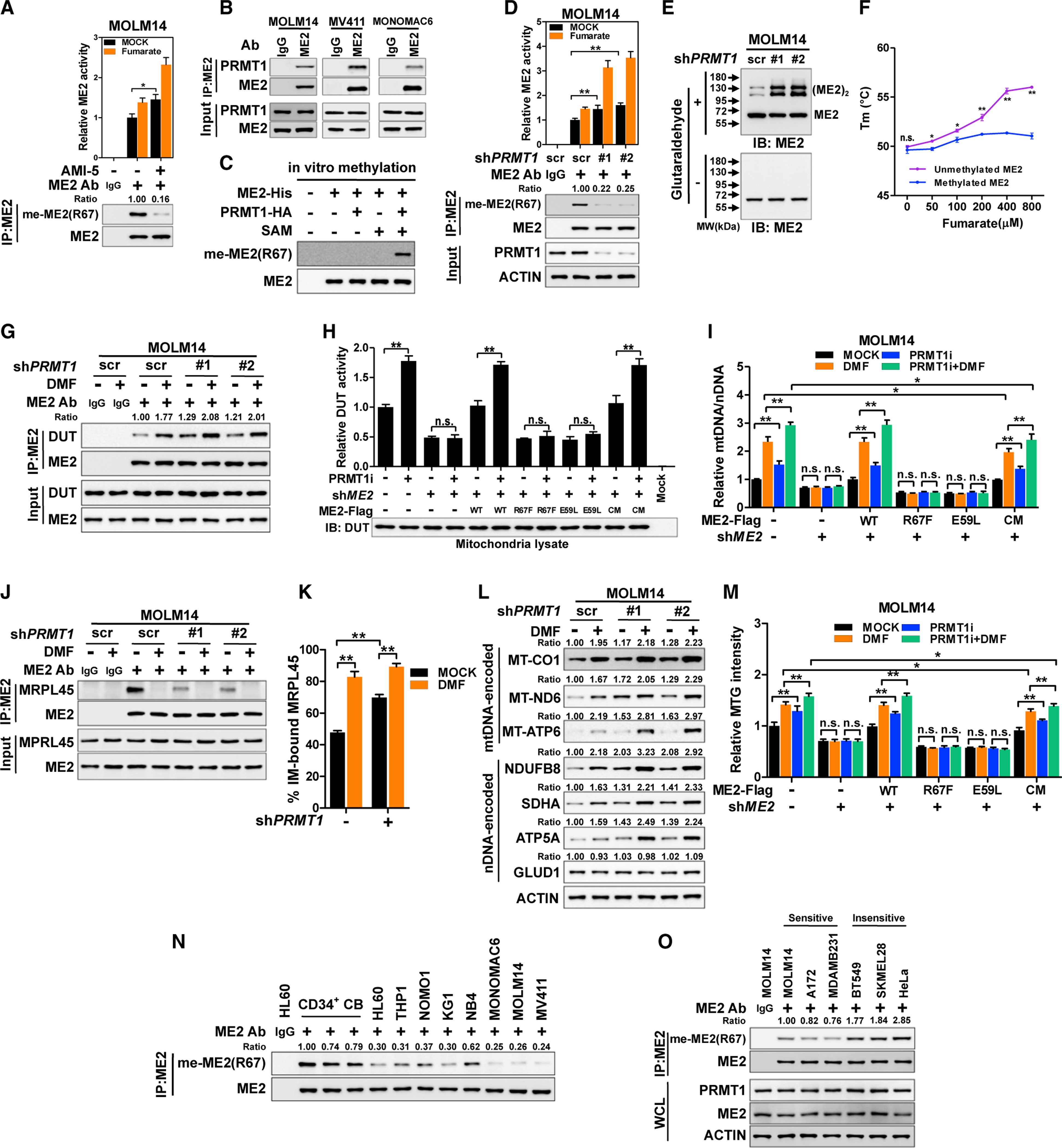
(A) R67 methylation of immunoprecipitated ME2 was determined in MOLM14 cells after AMI-5 treatment for 24 h.
(B) The interaction between ME2 and PRMT1 in AML cells was assayed.
(C) Recombinant ME2-His was incubated with PRMT1-HA in the presence of SAM. R67 methylation was determined.
(D and E) ME2 was immunopurified from control and PRMT1-knockdown MOLM14 cells, and subjected to western blotting and enzymatic activity assay (D).
Whole-cell lysate of MOLM14 cells was subjected to crosslinking assay (E).
(F) The melting temperature (Tm) of unmethylated and methylated ME2 (lanes 4 and 5 in C) was determined.
(G) Control and PRMT1-knockdown MOLM14 cells were treated with DMF. The interaction between ME2 and DUT was determined.
(H and I) ME2-knockdown and re-expression MOLM14 cells were treated with PRMT1i for 24 h. Mitochondrial lysate was subjected to DUT activity assay (H).
Stable cells were treated with PRMT1i and DMF as indicated. mtDNA was quantified (I).
(J–L) Control or PRMT1-knockdown cells were treated with DMF. Interaction of ME2 and MRPL45 was assayed (J). MRPL45 protein in inner-membrane and matrix fractions was quantified (K). The expression of mtDNA and nDNA-encoded proteins was determined (L).
(M) Stable MOLM14 cells were treated with PRMT1i and DMF as indicated. MTG intensity was determined.
(N and O) Endogenous ME2 was immunopurified from CD34+ CB cells, AML cells (N), and representative solid tumor cell lines (O) to determine R67 methylation.
Whole-cell lysate was used to detect PRMT1 and ME2 (O).
All data are presented as mean ± SEM from three independent experiments. *p < 0.05, **p < 0.01; n.s. indicates not significant. See also Figure S5.
To identify the ME2 methylase, we co-expressed ME2-Flag with GFP-tagged PRMTs (PRMT1-PRMT9) (Blanc and Richard, 2017). ME2 selectively interacted with PRMT1 and PRMT4 (also named CARM1) (Figure S5J). PRMT1, but not CARM1, increased R67 methylation and suppressed ME2 activity (Figure S5K). Importantly, endogenous ME2 readily associated with PRMT1 in AML cells (Figure 5B). Overexpression of PRMT1 upregulated the methylation of wild-type ME2, but not ME2R67F (Figure S5L). Furthermore, PRMT1 efficiently methylated recombinant ME2 at R67 in vitro (Figure 5C). Depletion of PRMT1 prominently reduced R67 methylation of endogenous ME2 (Figures 5D and S5M). These results demonstrate that PRMT1 methylates and inhibits ME2.
Next, we tested whether PRMT1 modulates ME2 dimerization. PRMT1 suppressed the binding between ME2-HA and ME2-GFP, which was restored by AMI-5 treatment (Figure S5N). Silencing PRMT1 induced a monomer-to-dimer transition of ME2 in MOLM14 cells (Figure 5E). Fumarate markedly increased the thermal stability of recombinant ME2, but not its methylated form (Figures 5C, 5F, and S5O), consistent with methylation directly suppressing fumarate binding. Together, we conclude that PRMT1 methylates ME2 to block fumarate binding and dimerization.
We next investigated whether PRMT1 regulated mtDNA levels. Notably, the interaction between ME2 and DUT was increased in PRMT1-knockdown cells and was further enhanced by DMF treatment (Figure 5G). These data suggest that PRMT1 potentially inhibits DUT activity through methylating ME2. To test this, we treated ME2-knockdown and re-expression cells with the PRMT1-specific inhibitor TCE5003 (hereafter PRMT1i), and isolated mitochondria to determine endogenous DUT activity. PRMT1i enhanced DUT activity in control cells and cells re-expressing wild-type ME2 or ME2CM, but not in ME2-knockdown cells or cells rescued by fumarate binding-defective mutants (Figures 5H and S5P). Consistently, PRMT1i alone increased mtDNA abundance, which was further upregulated by DMF treatment (Figures 5I and S5Q). mtDNA copy number was not modulated by PRMTi in cells re-expressing fumarate binding-defective mutants (Figures 5I and S5Q), suggesting that PRMT1 decreases DUT activity and mtDNA in a manner dependent on the fumarate-sensing activity of ME2.
We further asked whether PRMT1 regulated mitochondrial protein expression. PRMT1 depletion weakened the binding between ME2 and MRPL45 (Figure 5J). Accordingly, PRMT1 knockdown increased the fraction of inner membrane-bound MRPL45 (Figures 5K and S5R), enhanced mitoribosome assembly (Figure S5S), and increased mtDNA-encoded protein (Figures 5L and S5T). DMF further upregulated these mtDNA-encoded proteins (Figures 5L and S5T). MTG staining assay showed that PRMT1 inhibition increased mitochondria mass in control cells and cells re-expressing wild-type ME2 or ME2CM, but not fumarate sensing-defective mutants (Figures 5M and S5U). Collectively, PRMT1 suppresses fumarate signaling by decreasing inner membrane attachment of mitoribosome and mtDNA-encoded protein expression.
Because the impact of fumarate on mitochondrial mass varies across different cell lines (Figures 2D and 2F), we therefore asked whether ME2 methylation correlates with fumarate-induced mitobiogenesis. While AML cells and their normal counterparts expressed similar levels of PRMT1 protein (Figure S5V), ME2 methylation was generally lower in AML cells (Figure 5N). In addition, cell lines that were sensitive to fumarate-induced mitobiogenesis showed lower R67 methylation (Figures 2F and 5O). Together, PRMT1 negatively regulates fumarate sensing and mitobiogenesis.
ME2-mediated fumarate sensing supports leukemia growth
Oxidative metabolism is a distinctive vulnerability of myeloid malignancies (Pollyea et al., 2018; Skrtić et al., 2011 ; Stevens et al., 2018). In MOLM14 and KG1 cells, knockdown of ME2 led to a proliferative defect, which was rescued by wild-type ME2 and ME2CM, but not the fumarate binding-defective mutants (Figures 6A and S6A). Activating ETC by exogenous pyruvate or SLC1A3, a high-affinity glutamate transporter, partially restored the proliferation of ME2-depleted cells (Figures S6B–S6G). Therefore, ETC activity is only part of the growth defect imposed by ME2 knockdown. Further, DMF moderately upregulated cell proliferation and colony formation in control cells, but not cells re-expressing fumarate binding-defective mutants (Figures 6B, 6C, S6H, and S6I). Investigating the role of fumarate sensing in vivo (NSG mice), ME2 depletion delayed MOLM14 leukemia and improved animal survival (Figure 6D). Wild-type ME2 and ME2CM, but not fumarate binding-defective mutants, restored leukemic aggressiveness (Figure 6D), showing that fumarate-sensing activity of ME2, but not its catalytic activity, plays a major role in regulating AML progression in vivo.
Figure 6. ME2-mediated fumarate sensing supports leukemia growth.
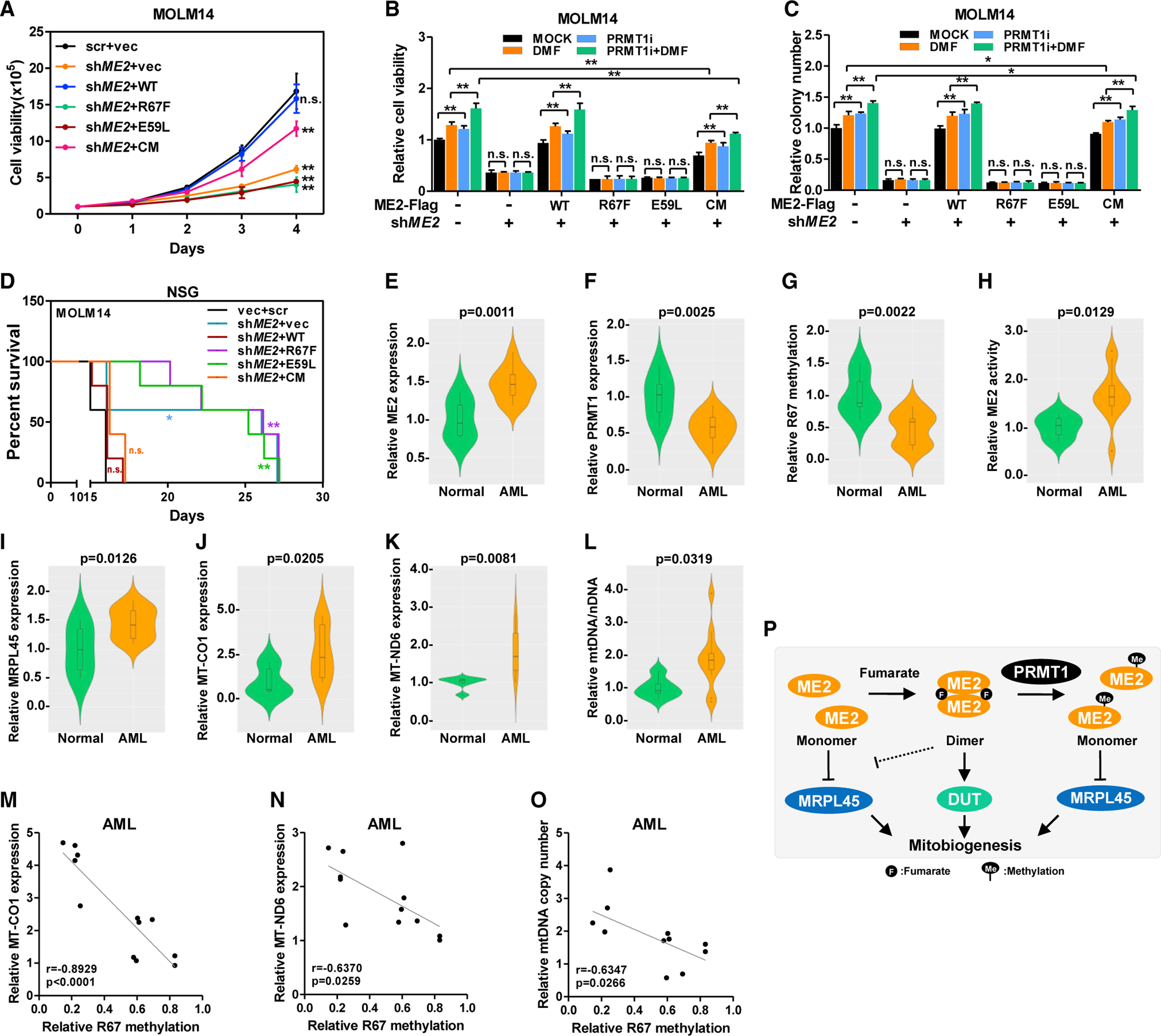
(A–C) Growth curves of stable MOLM14 cells were determined (A). Cells were treated with PRMT1i and DMF. Cell viability was determined by cell counting after 4 days of culture (B). Colonies of MOLM14 cells were counted 7 days after treatment (C).
(D) ME2-knockdown and re-expression MOLM14 cells were transplanted into sublethally irradiated NSG mice to monitor leukemia progression (n = 5).
(E–O) ME2 (E) and PRMT1 (F) protein in normal and leukemic human BM samples were determined. R67 methylation of immunoprecipitated ME2 was determined (G). ME2 activity was assayed in the presence of fumarate (H). MRPL45 (I), MT-CO1 (J), and MT-ND6 (K) were quantified by western blotting. mtDNA was quantified by qPCR (L). Pearson’s correlation of ME2 protein with MT-CO1 (M), MT-ND6 (N), and mtDNA abundance (O) in AML samples was determined.
(P) Working model of ME2-mediated fumarate signaling.
Data are presented as mean ± SEM from three independent experiments. *p < 0.05, **p < 0.01; n.s. indicates not significant. See also Figure S6.
To evaluate the clinical relevance, we collected 12 human leukemic BM (blast percentage > 69%) and six normal human BM samples (Figure S6J). Leukemic BM cells expressed higher ME2 protein than the normal cells, while the PRMT1 protein was decreased in human leukemia (Figures 6E and 6F). Further, ME2 was hypomethylated at R67 and showed higher activity in leukemic samples (Figures 6G and 6H). MRPL45, MT-CO1, and MT-ND6 were also overabundant in AML samples (Figures 6I–6K). Accordingly, leukemic BM showed a 1.84-fold increase of mtDNA (Figure 6L). R67 methylation, but not ME2 protein, negatively correlated with MT-ND6, MT-CO1, and mtDNA copies (Figures 6M–6O and S6K–S6M). Together, ME2 hypomethylation, which promotes fumarate signaling, is significantly linked to mitobiogenesis in human AML.
DISCUSSION
Studies on the control of mitobiogenesis have largely focused on nuclear and cytoplasmic events. However, mtDNA encodes key ETC components critical for mitochondrial function. Mitobiogenesis must, therefore, depend on coordinated generation of both nDNA and mtDNA encoded elements in response to organismal cues. Our results show that ME2 regulates mitochondrial aspects of mitobiogenesis, serving as a fumarate sensor to directly link mitochondrial protein synthesis and mtDNA replication with nutrient supply (Figure 6P). Previously, mitochondrial and nuclear transcription and translation programs have been shown to co-regulate ETC genes (Couvillion et al., 2016). Mitochondria sense the cytosolic translation efficiency and coordinately generate mtDNA-encoded products (Couvillion et al., 2016; Richter-Dennerlein et al., 2016). Here, we found that multiple mtDNA-encoded proteins were decreased in NRF2-knockdown cells (Figure S4G), supporting mitochondrial protein production as tightly coupled to the transcription and translation programs in the nucleus and cytoplasm.
Fumarate has previously been implicated in controlling nDNA participation in mitobiogenesis. DMF suppresses KEAP-mediated clearance of Nrf2, resulting in elevation of mtDNA (Hayashi et al., 2017). In agreement, we found that reducing NRF2 suppressed DMF-induced elevation of mtDNA (Figure 2B). However, we found more marked effects of DMF on both mtDNA and mitochondrial protein translation that were dependent on ME2. Therefore, fumarate may act as a metabolic signal in both mitochondria and the nucleus to promote mitochondrial biomass production.
In addition to DUT and MRPL45, fumarate-ME2 signaling may have other downstream targets. For example, fumarate treatment increased all four dNTPs in mitochondria, implying that fumarate-ME2 axis may modulate enzymes other than DUT in nucleotide metabolism. Two observations further indicate that ME2 may control mitoribosome activity through unknown targets. First, depleting ME2 suppressed mitoribosome assembly (Figure 4G), which could not be explained by the inhibitory effect of ME2 monomers. Second, wild-type ME2 interacted with more mitoribosomal proteins comparing to ME2R67F (Figure 4A). Mitoribosomes are complex, and the function of most of its components is poorly understood. Here we focused on MRPL45 because of its clear role in attaching mitoribosomes to the inner membrane (Kummer et al., 2018). It remains possible that ME2 dimers interact with other regulatory proteins of mitoribosome to fulfill fumarate signaling.
Metabolites have gained increasing recognition as signaling molecules (Frezza, 2017; Haas et al., 2016; Husted et al., 2017). Fumarate is involved in oncogenic signaling at multiple levels. Notably, fumarate is a proto-oncometabolite in FH-mutated renal tumors (Tomlinson et al., 2002). Aberrant accumulation of fumarate (millimolar levels or higher) mediates epigenetic reprogramming and hypoxia signaling (Laukka et al., 2016; Sciacovelli et al., 2016) and reduces apoptosis (Bardella et al., 2012). Despite these close links, the mechanism by which cells sense fumarate in its physiological range remains not well understood. We demonstrate that ME2 serves as a physiological fumarate sensor and regulates mitobiogenesis. While DMF is applied in the treatment of multiple sclerosis (Hayashi et al., 2017), our results suggest that inhibition of ME2 may also be medically useful. ME2-mediated fumarate signaling may be targetable in AML and other mitochondria-related diseases.
Limitations of study
First, the process of mitochondria isolation in our study could perturb metabolite quantification. Rapid mitochondrial isolation would aid in more precise quantification (Chen et al., 2016b). Second, sensors typically have low affinity for the corresponding metabolite. Further studies of binding kinetics are required to validate ME2 as a classic fumarate sensor. Third, we do not know if the ME2 effects of fumarate are sufficient to induce mitobiogenesis because of concurrent fumarate effects on KEAP increasing Nrf2 activity. NRF2 knockdown imperfectly tests whether steady-state Nrf2 is required. Fourth, other nutrient signaling pathways may converge on ME2-modulated mitobiogenesis through PRMT1 activity. Fifth, our conclusions regarding AML must be tempered by the limited samples, cell lines, and in vivo models used. Finally, we expected FH knockdown might recapitulate the exogenous fumarate effect but in contrast, we observed a decrease in MRPL45 and DUT (Figures S6N and S6O), with a modest decrease in mitochondrial mass (Figures 2B and S6P–S6S). Since reduced FH invariably decreases multiple downstream and increases upstream metabolites, it may affect pathways, including ETC function (Tyrakis et al., 2017), that counter the effect of increased fumarate on mitobiogenesis. In support of this notion, Fh1 knockout mouse model displayed decreased mitochondrial respiration of hematopoietic stem cells and defective hematopiesis (Guitart et al., 2017).
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David T. Scadden (david_scadden@harvard.edu).
Materials availability
All reagents are available from the Lead Contact under a material transfer agreement with Harvard University
Data and code availability
All data and code to understand and assess the conclusion of this research are available in the main text, supplementary materials, or MassIVE Database (accession number MSV000086778).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and culture conditions
All cells were cultured at 37°C under 5% CO2 humidified atmosphere. Human embryonic kidney cell line (HEK293), human AML cell lines (HL60, KG1, MOLM14, MONOMAC6, MV411, NB4, NOMO1, THP1), human glioma cell lines (A172, LN18, U87MG, U251MG, U118MG), breast cancer cell lines (BT549, HCC1937, HCC38, HS578T, MDAMB231, MDAMB468), liver cancer cell lines (HepG2, SKHEP1, SNU423, SNU387), pancreatic cancer cell lines (MIAPACA2, KP2, AsPC1, SW1990, BxPC3), melanoma cell lines (A375, SKMEL5, SKMEL28), sarcoma cell lines (U2OS and HT1080), cervical cancer cell line (HeLa), prostate cancer cell line (DU145), colorectal cancer cell line (HCT116) were maintained in RPMI medium 1640 or Dulbecco’s modified Eagle medium (DMEM) (Lonza) supplemented with 10% fetal bovine serum (Invitrogen) in the presence of penicillin, streptomycin, and 2 mM L-glutamine (Corning). Human cord blood CD34+ cells were grown in RPMI 1640 (with serum and glutamine) supplemented with recombinant human growth factors, including 40 ng/mL IL-6 (Peprotech), 50 ng/mL FLT3 ligand (Peprotech), 20 ng/mL stem cell factor (SCF) (Peprotech), and 50 ng/ml TPO (Peprotech).
Normal and leukemic mouse BM cells were maintained in RPMI 1640 (with serum and glutamine) supplemented with recombinant murine growth factors, including 10 ng/mL IL3 (R&D systems), 10 ng/mL SCF (R&D systems), 100 ng/mL IL6 (Peprotech).
Microbe strains
One-shot E. coli BL21 (DE3) (Invitrogen) was grown in LB medium at 37°C and then at 16°C after IPTG induction, for recombinant protein expression.
Mouse models
All animal studies were conducted in compliance with NIH guidelines for the care and use of laboratory animals and were approved by the IACUC of Harvard University. Black 6 (B6) mice (C57BL/6J) and NSG mice (NOD.Prkdcscid.Il2rgnull) were purchased from the Jackson Laboratory. Mouse lineage negative (lin−) bone marrow cells were transduced with control short hairpin and two different short hairpins against Me2. Control and Me2-knockdown leukemic bone marrow cells were transplanted into lethally irradiated animals (9.5 Gy, 6-weeks old, male). In NSG model of human leukemia xenograft, one million human leukemia cells were transplanted into NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (6-weeks old, male) 24 hours after sublethal irradiation (2 Gy).
Human bone marrow samples
Normal and leukemic human bone marrow samples were collected from diagnostic bone marrow aspirations at Southwest Hospital (Chongqing, China). Bone marrow mononuclear cells were isolated by density gradient centrifugation and stored in liquid nitrogen until further use. Written informed consent was obtained from all patients. The procedures related to human subjects were approved by the Ethics Committee of the Institutes of Biomedical Sciences (Fudan University) and Institutional Ethics Review Board of Southwest Hospital.
METHOD DETAILS
Chemicals and treatment of cells
To identify metabolites that regulate mitochondria mass, cells were cultured in RPMI 1640 supplemented with 10% FBS and 2 mM glutamine. The dose of additional nutrient/metabolites was added to physiologically relevant levels. Glucose (10 mM), fructose (5 mM), pyruvate (1 mM), lactate (1 mM), trimethyl citrate (50 μM), dimethyl alpha-ketoglutarate (50 μM), dimethyl succinate (50 μM), dimethyl fumarate (50 μM), dimethyl malate (50 μM), trimethyl oxaloacetate (50 μM), palmitate [bovine serum albumin (BSA)-conjugated, 100 μM], oleate (BSA-conjugated, 100 μM), acetate (5 mM), amino acid mixture (Sigma, #R7131, 1x), glutamine (2 mM), non-essential amino acids (NEAA) (Sigma, #M7145, 1x), and NH4Cl (1 mM) were added to culture media. Cells were cultured for 48 hours before further analysis.
PRMT inhibitor AMI-1 (30 μM), AMI-5 (5 μM), PRMT1-specific inhibitor (PRMT1i, also known as TC-E 5003, 2 μM), and DUT inhibitor (TAS114, 10 μM) were added into culture medium 24 hours before harvesting cells, respectively. Fumarate esters including MMF, DMF, MEF, and DEF (100 μM) were added to medium 24 hours or as indicated before harvest.
Plasmids and transfection
The cDNAs encoding full-length human ME2, ACO2, MRPL45, DUT, NRF2, and SLC1A3 were cloned into Flag, HA, GFP, or His-tagged vectors (pcDNA3.1, pLV-EF1a-IRES, pEGFP-N1, pQCXIH, and pQE-1). Plasmids encoding GFP-PRMTs and NRF2 were generous gifts from Dr. Yanzhong Yang (City of Hope Cancer Center) and Dr. Rong Cai (Shanghai Jiao Tong University School of Medicine), respectively. Point mutations of ME2 were generated by site-directed mutagenesis using the GeneArt Site-Directed Mutagenesis System kit (Invitrogen). Truncated mutants of MRPL45 were cloned into pEGFP-N1. All expression constructs were verified by DNA sequencing. Plasmid transfection was carried out by using FuGENE 6 (Promega).
Immunoprecipitation and western blotting
Cells were lysed in ice-cold NP-40 buffer [50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.3% NP-40] containing protease inhibitor cocktail (Sigma). Immunoprecipitation was carried out either by incubating Flag beads (Sigma) at 4°C with lysate for three hours or by incubating HA, GFP or ME2 antibody with cell lysate for one hour, followed by incubating with Protein-A beads (Millipore) for another two hours at 4°C. After the incubation, beads were washed three times with ice-cold NP-40 buffer. Standard western blotting protocols were adopted.
For proteomic profiling of ME2 interactors, MOLM14 cells expressing vector control, ME2-Flag or the R67F mutant were lysed in NP-40 buffer. ME2 protein was immunoprecipitated with Flag beads and eluted with 0.1 M glycine-HCl (pH 3.0). Mass spectrometric analysis were performed in three independent experiments for vector control (Samples #4553, #4629, and #4630), wildtype ME2 (Samples #4554, #4632, and #4633), and R67F mutant (#4555, #4631, and #4634).
ME2 enzyme activity assay
ME2 enzyme activity was determined as described previously (Tronconi et al., 2010). Flag-tagged ME2 proteins were overexpressed in cells, immunoprecipitated with Flag-beads, eluted by Flag peptides (Sigma), and subjected to activity assay with malate and NAD+ as substrates. Reaction mixture consists of 50 mM HEPES (pH 7.4), 10 mM MnCl2, 4 mM NAD+, 10 mM malate in a total volume of 200 μL. 200 μM fumarate was added to the mixture to determine allosteric activation. Reactions were initiated by adding the enzyme and analyzed at 25°C. Activities were measured by the conversion of NAD+ to NADH, which was monitored by measuring the increase of fluorescence (Ex. 350nm, Em. 470nm) for NADH generation. Endogenous ME2 proteins were immunoprecipitated by using ME2 antibody, on-beads catalytic activity was assayed.
DUT enzyme activity assay
The substrate dUTP (1 mM, final concentration) was added to DUT activity assay buffer [50 mM Tris (pH 7.4), 150 mM NaCl]. Reactions were initiated by adding DUT enzyme into the reaction mixture (100 μL final volume). The reactions were stopped at 0.5 min, 1.0 min, 2.0 min, and 4.0 min by adding 200 μL chloroform and 200 mL methanol, followed by vortexing and centrifugation (5000g, 4°C for 15 minutes). The aqueous phase was dried with nitrogen flow evaporator at 37 °C and subjected to mass spectrometry to quantify the generation of dUMP. Recombinant ME2 was mixed with DUT enzyme at the molar ratio of 1:1 before adding to the reaction mixture to determine the effect of ME2 protein on DUT activity. The rate of dUMP accumulation was calculated and normalized to DUT enzyme to determine its catalytic activity.
Quantification of metabolites
The abundance of intracellular fumarate (Sigma, #MAK060), NADH (Sigma, #MAK037), and ATP (Abcam, #ab113849) were determined by using quantification kits, according to the manufacturer’s instruction. Briefly, 1×106 cells were collected and homogenized on ice in assay buffer provided and centrifuged at 4 °C for 10 min at 13,000g. Supernatants were deproteinized using 10kD spin column (Abcam), analyzed and compared to standard curves. The signals obtained were normalized to cell number.
To quantify intracellular nucleotides, 5×106 cells or isolated mitochondria were washed in 1 mL saline (0.9% NaCl in water) and lysed in ice-cold methanol. Water and chloroform were added to the lysate (methanol:water:chloroform=2:1:2 final volume), followed by vortexing and centrifugation (5000g, 4°C for 15 minutes). The aqueous phase was dried with nitrogen flow evaporator at 37 °C. For [U-13C]-fumarate tracing, 1×106 MOLM14 cells were cultured in RPMI 1640 supplemented with 10% FBS and 2 mM glutamine. Increasing concentrations of [U-13C]-fumarate was added to the culture medium as indicated, metabolites were extracted after incubation for 24 hours. Extracted metabolites were resuspended in 50% acetonitrile and subjected to mass spectrometry analysis. For the quantification of dATP, dTTP, dGTP, dCTP, dUTP, and dUMP, an Ultimate 3000 UHPLC equipped with a refrigerated autosampler (at 8°C) and a column heater (at 30°C) with a Welch Ultimate AQ-C18 column (2.1×250mm i.d., 5 μm) was used for separations. Solvent A was 10 mM ammonium acetate and 0.075% FA in water and solvent B was acetonitrile. The gradient was as follows: 100% A for 1 min at 0.2 mL/min, 95% A at 7 min with 0.2 mL/min, 5% A at 8 min with 0.2 mL/min, 5% A at 12 min with 0.2 mL/min, 100% A at 12.5 min and 100% A at 20 min with 0.2 mL/min. For MS analysis, the UHPLC was coupled to a 6500 Qtrap mass spectrometer (Sciex, USA). The ion transitions at m/z 307.0→195.0, 467.0→369.0, 490.1→392.1, 506.1→408.1, 481.0→383.0, 466.1→367.9 were selected for monitoring dUMP, dUTP, dATP, dGTP, dTTP, and dCTP, respectively. The operating conditions were as follows: spray voltage −4500 V; Orifice temperature 500 °C; GS1 and GS2 50; Curtain Gas 40.
Glucose consumption (Sigma, #GAGO-20) and glutamine consumption (Abcam, #ab197011) was determined by using colorimetric assay kits following the manufacturer’s instructions. Briefly, cells were seeded into six-well plate at 33105 per well. After three hours of cell culture, the supernatant of the medium was collected, deproteinized using 10kD spin column, and subjected to glucose/glutamine detection. The glucose/glutamine uptake was determined by subtracting the final glucose/glutamine concentration from initial glucose/glutamine concentration in the culture medium.
In vitro pulldown assay of ME2 and MRPL45
Recombinant His-tagged MRPL45 and ME2 protein (1 μg each in 0.5 mL NP-40 buffer) were incubated with overhead rotation at 4°C overnight. After the addition of 200 μM fumarate, succinate, or malate, the mixture was further incubated for three hours at 4°C. His-MRPL45 was pulled down with nickel-NTA agarose beads (Invitrogen). Beads were washed three times with 0.3% NP-40 buffer and subjected to western blotting analysis.
Generation of stable cell pools
shRNAs targeting ADSL, ASL, FAH, FH, SDHA, ME2, MRPL45, NRF2, and PRMT1 were used to generate stable knockdown cell pools. Lentivirus was produced by using a two-plasmid packaging system (Δ8.9 and vsvg). Cells were mixed with 8 μg/mL polybrene and spinfected with the lentivirus and selected in 4 μg/mL puromycin for one week.
To generate ME2-knockdown and re-expression stable cell pools, Flag-tagged human wild-type ME2 or its mutants (R67K, R67F, CM) was cloned into the lentiviral pLV-EF1a-IRES-Hygro vector and co-transfected with vectors expressing the Δ8.9 and vsvg genes in HEK293T cells to produce lentiviruses. After transduction, cells were selected in 200 μg/ml hygromycin B for 1 week.
CRISPR editing
Malic enzymes (ME1, ME2, and ME3) were silenced in leukemia cell lines through CRISPR editing using lentiCRISPR v2 with sgRNA sequences targeting ME1, ME2, and ME3 respectively. Oligos were phosphorylated, annealed, and ligated into the lentiCRISPR v2 backbone, which was then transformed into bacteria, isolated, and verified by sequencing. The lentiCRISPR vector expressing sgRNA against GFP was used as control. Lentivirus carrying sgRNA was produced using the two-plasmid packaging system. Leukemia cell lines were transduced and selected in 4 μg/mL puromycin. The depletion effect was verified by western blotting. Targeting sequences for sgRNAs were shown in Table S1.
Quantitative real-time PCR
Total RNA was isolated from cultured cells using the RNeasy Kit (Qiagen) and reverse-transcribed with random primers following the manufacturer’s instructions (RETROscript Kit, Invitrogen). The cDNA was preceded to real-time PCR with gene-specific primers in the presence of SYBR Green PCR Master Mix (Applied Biosystems). PCR reactions were performed in triplicate and the relative amount of cDNA was calculated by the comparative CT method using the β-actin as a control. Primer sequences were listed in Table S1.
Oxygen consumption rate (OCR)
OCR was determined using the XFe96 Extracellular Flux Analyzer (Agilent). Briefly, leukemia cells or mouse BM cells were attached to 96-well plates using Cell-Tak (Corning) at the density of 4×104 or 8×104 cells/well, respectively. Cells were incubated with Seahorse XF RPMI medium buffer (without phenol red, with 10 mM glucose, 2 mM glutamine, and 1 mM pyruvate). Cell Mito Stress Test Kit (Agilent) was used to measure cellular mitochondrial function, 180 μL of Seahorse buffer plus 20 μL each of 2 μM oligomycin, 2 μM FCCP, and 0.5 mM rotenone/antimycin A (AA) was automatically injected to determine the oxygen consumption rate (OCR), according to the manufacturer’s instructions.
In vitro methylation assay
For in vitro methylation assay, HA-tagged PRMT1 protein was overexpressed in HEK293T cells and immunopurified with HA-beads. Recombinant His-tagged ME2 (30 μg) was mixed with PRMT1-HA (on beads) at a molar ratio of approximately 1:1 in methylation reaction buffer (50 mM Tris-HCl, pH 8.0, 20 mM KCl, 5 mM DTT, 4 mM EDTA). The mixture was incubated with or without 200 μM S-adenosyl-L-methionine (Sigma) at 37°C for 1 hour in a final volume of 500 μL. After centrifugation (500g, 4°C for 3 minutes), the supernatant was transferred to an Amicon filter (Millipore, Amicon Ultra-15 Centrifugal Filter Device) for buffer exchange and further analysis.
Protein melting curve analysis
The thermodynamic stability of ME2 was determined using SYPRO-Orange (Invitrogen). Briefly, 45 μL of 1 μM purified ME2 (in 25 mM HEPES, 150 mM NaCl at pH 8.0) was mixed with 15 μL SYPRO-Orange (20X,). 45 μL buffer (25 mM HEPES, 150 mM NaCl, pH 8.0) mixed with 15 μL of 20X SYPRO-Orange was used as control. The mixture was aliquoted in triplicate (20 μL per well) into a 96-well plate. Data of melting curves were collected by using ABI 7500 (Applied Biosystems). Melting curve fluorescent signal was acquired between 20°C and 70°C using a ramping rate of 0.03 °C/s. Melting temperatures (Tm) were determined by fitting the data with Boltzmann model.
Cell proliferation and colony formation assay
To monitor cell proliferation, cells (1×105/mL) were seeded into six-well plates. Viable cells were visualized by methylene blue staining and counted every day for 4 days.
In colony formation assay, AML cells were plated in methylcellulose medium (MethoCult H4434; Stem Cell Technologies) according to the manufacturer’s instructions. Briefly, 4000 cells in 0.5 mL IMDM with 10% FBS were added to 3.5 mL of methylcellulose medium. After thorough vortex mixing, the cell suspension was plated into the six-well plate with 1 mL in each well. Culture plates were incubated at 37°C in a humid atmosphere with 5% CO2. Colonies (>50 μm diameter) were counted after seven days of incubation.
Mitochondria staining and mtDNA quantification
To quantify mitochondria mass, 1×106 cells were stained with 50 nM MitoTracker Green (Invitrogen) for 30 min at 37°C and analyzed with a fluorescence microplate reader (BioTek, Ex. 491nm, Em. 516nm). To determine mtDNA copy number, total DNA was isolated from cell lines or tissues using DNeasy kits (Qiagen). Samples were adjusted to 1 ng/μL final concentration. Nuclear and mitochondrial DNA content was analyzed by qPCR as described previously (Wang et al., 2014). mtDNA content was determined by normalizing mitochondrial DNA abundance [tRNA-Leu(UUR) in human and 16S rRNA in mouse] to nuclear DNA (beta-2-microglobulin, B2M) abundance. Primers for qPCR were listed in Table S1.
Mitochondria isolation and fractionation
Mitochondrial isolation was performed as previously described (Rackham et al., 2016). Briefly, cells were resuspended in mitochondria isolation buffer (MIB) [310 mM sucrose, 10 mM Tris-HCl (pH7.5) and 0.05 % BSA (w/v), with protease inhibitor cocktail (Sigma)], homogenized with Dounce homogenizer, centrifugated at 1000g for 10min at 4°C. The supernatant was further centrifugated at 4500g for 15 min at 4°C, and the pellet was washed once with MIB. Crude mitochondrial pellets were resuspended in MIB with protease inhibitor cocktail.
Mitochondria subfraction was performed as reported previously (Pallotti and Lenaz, 2007). In brief, isolated mitochondria were resuspended in 1 mL of Mitolysis buffer (3 mM HEPES, pH 7.4, 210 mM mannitol, 70 mM sucrose, 0.2 mM EGTA, and protease inhibitor cocktail) with digitonin (0.2 mg/mL). To achieve mitoplast (MP, inner membrane and matrix), mitochondria were lysed with overhead rotation at 4°C for 15 min. 1 mL of Mitolysis buffer was added to stop digitonin extraction. The lysate was centrifugated at 11,000g for 10 min at 4°C to achieve mitoplast pellet. Mitoplast was resuspended in 300 μL Mitolysis buffer and disrupted by a sonicator in ice-cold water bath. Disrupted mitoplast was further centrifugated at 10,000g for 30 min at 4°C, to isolate inner membrane fraction in the pellet and matrix fraction in the supernatant.
Sucrose gradient fractionation
Isolated mitochondria (2 mg) were lysed in 2 mL MitoL buffer [10 mM Tris-HCl (pH 7.4), 260 mM sucrose, 100 mM KCl, 20 mM MgCl2 and 2% digitonin, RNase inhibitor (40 U/ml) and EDTA-free protease inhibitor cocktail (Sigma)] for 20 min at 4°C. The lysate was centrifuged at 9,200g for 45 min at 4°C. The supernatant was loaded on a continuous 10–30% sucrose gradient (in 10 mM TrisHCl, pH 7.5, 100 mM KCl, 20 mM MgCl2 supplemented with RNase and protease inhibitors) and centrifuged at 20,000g for 6 hours at 4°C in an Optima Beckman Coulter ultracentrifuge. Fractions were collected and precipitated with 20% trichloroacetic acid (final concentration). MRPL12 and MPRS35 were used as markers of the mitochondrial ribosomal subunits. 12S rRNA and 16S rRNA were extracted using the RNeasy kit (Qiagen). The RNA was reverse transcribed and detected by qPCR to determine mitoribosome assembly. Primers for qPCR were listed in Table S1.
QUANTIFICATION AND STATISTICAL ANALYSIS
Student’s t tests or one-way ANOVA with Dunnett’s multiple comparisons test were performed to determine statistical significance using GraphPad Prism. All data shown represent the results obtained from three (or as indicated) independent experiments. The p values <0.05 were considered statistically significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Rabbit polyclonal anti-MT-CO1 | Novus biologicals | Cat# NBP2-29949 |
| Rabbit polyclonal anti-MT-ND6 | Novus biologicals | Cat# NBP1-70650; RRID: AB_11006834 |
| Mouse monoclonal anti-MT-ATP6 (clone 1G7-1G2) | Sigma-Aldrich | Cat#: MABS1995 |
| Rabbit monoclonal anti-NDUFB8 (clone JG61-36) | Novus biologicals | Cat# NBP2-75586 |
| Rabbit monoclonal anti-SDHA (clone D6J9M) | Cell Signaling Technology | Cat# 11998; RRID: AB_2750900 |
| Mouse monoclonal anti-ATP5A (clone 15H4) | Abcam | Cat# ab14748; RRID: AB_301447 |
| Rabbit monoclonal anti-GLUD1(clone EPR11369(B)) | Abcam | Cat# ab166618; RRID: AB_2815030 |
| Rabbit monoclonal anti-ME2 (clone EPR7218) | Abcam | Cat#:ab126616; RRID: AB_11131748 |
| Mouse polyclonal anti-POLRMT | Abcam | Cat#:ab167368 |
| Mouse monoclonal anti-NRF2 | Abcam | Cat#: ab89443; RRID: AB_2041334 |
| Rabbit polyclonal anti-PGC1α | Abcam | Cat#: ab54481; RRID: AB_881987 |
| Mouse monoclonal anti-beta-Actin (clone 8H10D10) | Cell Signaling Technology | Cat#:3700; RRID: AB_2242334 |
| Rabbit polyclonal anti-GAPDH | Proteintech | Cat#: 10494-1-AP; RRID: AB_2263076 |
| Mouse monoclonal anti-Histone H3 (clone 96C10) | Cell Signaling Technology | Cat#:3638; RRID: AB_1642229 |
| Mouse monoclonal anti-Flag (clone M2) | Sigma-Aldrich | Cat#:F1804; RRID: AB_262044 |
| Rabbit polyclonal anti-ME1 | Abcam | Cat#:ab97445; RRID: AB_10679994 |
| Rabbit monoclonal anti-ME2 (clone EP7217) | Abcam | Cat#: ab139686 |
| Rabbit monoclonal anti-ME3 (clone EPR10378) | Abcam | Cat#:ab172972 |
| Rabbit polyclonal anti-ACO2 | Proteintech | Cat#: 11134-1-AP |
| Rabbit polyclonal anti-2SC antibody | Discovery Antibodies | Cat#: crb2005017 |
| Rabbit polyclonal anti-DUT | Proteintech | Cat#: 13740-1-AP |
| Rabbit polyclonal anti-GOT2 | Proteintech | Cat#: 14800-1-AP |
| Mouse monoclonal anti-GFP (clone 9F9.F9) | Abcam | Cat#:ab1218; RRID: AB_298911 |
| Mouse monoclonal anti-HA (clone F-7) | Santa Cruz | Cat#:sc-7392; RRID: AB_627809 |
| Rabbit polyclonal anti-MRPL45 | Sigma-Aldrich | Cat# HPA023373; RRID: AB_1854114 |
| Rabbit monoclonal anti-MRRF [clone EPR14140(B)] | Abcam | Cat#: ab181223 |
| Rabbit polyclonal anti-MRPL12 | Abcam | Cat#: ab154961 |
| Rabbit monoclonal anti-MRPS35 [clone EPR11731(2)] | Abcam | Cat#: ab182160 |
| Rabbit polyclonal anti-MT-ND5 | Proteintech | Cat#: 55410-1-AP |
| Rabbit polyclonal anti-MT-CYB | Proteintech | Cat#: 55090-1-AP |
| Rabbit polyclonal anti-MT-CO2 | Proteintech | Cat#: 55070-1-AP |
| Mouse monoclonal anti-His (clone 27E8) | Cell Signaling Technology | Cat# 2366; RRID: AB_2115719 |
| Rabbit monoclonal anti-TOM20 (clone 29) | BD Biosciences | Cat#612278; RRID: AB_399595 |
| Rabbit polyclonal anti-me-ME2(R67) | This paper | N/A |
| Rabbit polyclonal anti-PRMT1 | Abcam | Cat#:2449; RRID: AB_2237696 |
| Rabbit monoclonal anti-Mono-Methyl Arginine (clone Me-R4-100) | Cell Signaling Technology | Cat#:8015; RRID: AB_10891776 |
| Mouse monoclonal anti-CARM1 | Arigo Biolaboratories | Cat#: ARG53996 |
|
| ||
| Bacterial strains | ||
|
| ||
| Escherichia coli BL21 | Invitrogen | C600003 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| MitoTracke Green FM | Invitrogen | M7514 |
| Dimethyl fumarate | Sigma-Aldrich | 242926; CAS: 624-49-7 |
| Diethyl fumarate | Sigma-Aldrich | D95654; CAS: 623-91-6 |
| Sodium fumarate dibasic | Sigma-Aldrich | F1506; CAS: 17013-01-3 |
| Monomethyl fumarate | Sigma-Aldrich | 651419; CAS: 2756-87-8 |
| Monoethyl fumarate | Sigma-Aldrich | 128422; CAS: 2459-05-4 |
| Dimethyl malate | Sigma-Aldrich | 374318; CAS: 617-55-0 |
| Diethyl oxalacetate | Sigma-Aldrich | 171263; CAS: 40876-98-0 |
| MEM Non-essential Amino Acid Solution | Sigma-Aldrich | M7145 |
| MEM Amino Acids solution | Sigma-Aldrich | M5550 |
| [U-13C]-fumarate | Cambridge Isotope Laboratories | CLM-1529; CAS: 201595-62-2 |
| Maleimide-PEG2-Biotin | Thermo Scientific | A39261 |
| Glutaraldehyde | Sigma-Aldrich | 340855; CAS: 111-30-8 |
| TAS114 | MedChemExpress | HY-124062; CAS: 1198221-21-4 |
| Succinate | Sigma-Aldrich | S9637; CAS: 6106-21-4 |
| Malate | Sigma-Aldrich | M1000; CAS: 97-67-6 |
| AMI-1 sodium salt hydrate | Sigma-Aldrich | A9232 |
| AMI-5 | EMD Millipore | 539211; CAS: 17372-87-1 |
| TC-E 5003 | Tocris | 5099 |
| S-adenosylmethionine | Sigma-Aldrich | A9384; CAS: 979-92-0 |
| Unmodified R67 peptide | This paper | N/A |
| Monomethylated R67 peptide | This paper | N/A |
| Recombinant Human ME2 protein | Abcam | ab201360 |
| SYPRO orange protein gel stain | Invitrogen | S6650 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Glucose (GO) Assay Kit | Sigma-Aldrich | GAGO20 |
| Glutamine and Glutamate Determination Kit | Sigma-Aldrich | GLN1 |
| Mitochondria Isolation Kit | Sigma-Aldrich | MITOISO2 |
| Glutamine Detection Assay Kit (Colorimetric) | Abcam | ab197011 |
| Fumarate Assay Kit | Sigma-Aldrich | MAK060 |
| NAD/NADH Quantitation Kit | Sigma-Aldrich | MAK037 |
| Luminescent ATP Detection Assay Kit | Abcam | ab113849 |
| Seahorse XF Cell Mito Stress Test Kit | Agilent | 103015 |
| MagniSort Mouse Hematopoietic Lineage Depletion Kit | Invitrogen | 8804-6829-74 |
|
| ||
| Deposited data | ||
|
| ||
| Proteomic data | This paper | MassIVE:MSV000086778 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Human Cord Blood CD34+ Cells | STEMCELL Technologies | 70008 |
| Human:HL60 | ATCC | CCL-240 |
| Human:THP1 | ATCC | TIB-202 |
| Human:NOMO1 | JCRB Cell Bank | IFO50474 |
| Human:KG1 | ATCC | CCL-246.1 |
| Human:NB4 | DSMZ | ACC-207 |
| Human:MONOMAC6 | DSMZ | ACC-124 |
| Human:MOLM14 | DSMZ | ACC-777 |
| Human:MV411 | ATCC | CRL-9591 |
| Human:HEK293 | ATCC | CRL-1573 |
| Human:A172 | ATCC | CRL-1620 |
| Human:LN18 | ATCC | CRL-2610 |
| Human:U87MG | ATCC | HTB-14 |
| Human:U251MG | Sigma-Aldrich | 09063001 |
| Human:U118MG | ATCC | HTB-15 |
| Human:BT549 | ATCC | HTB-122 |
| Human:HCC1937 | ATCC | CRL-2336 |
| Human: HCC38 | ATCC | CRL-2314 |
| Human: HS578T | ATCC | HTB-126 |
| Human: MDAMB231 | ATCC | HTB-26 |
| Human: MDAMB468 | ATCC | HTB-132 |
| Human: HepG2 | ATCC | HB-8065 |
| Human: SKHEP1 | ATCC | HTB-52 |
| Human: SNU423 | ATCC | CRL-2238 |
| Human: SNU387 | ATCC | CRL-2237 |
| Human: A375 | ATCC | CRL-1619 |
| Human: SKMEL5 | ATCC | HTB-70 |
| Human: SKMEL28 | ATCC | HTB-72 |
| Human: MIAPaCa2 | ATCC | CRM-CRL-1420 |
| Human: KP2 | JCRB Cell Bank | JCRB0181 |
| Human: AsPC1 | ATCC | CRL-1682 |
| Human: SW1990 | ATCC | CRL-2172 |
| Human: BxPC3 | ATCC | CRL-1687 |
| Human: U2OS | ATCC | HTB-96 |
| Human: HeLa | ATCC | CCL-2 |
| Human: DU145 | ATCC | HTB-81 |
| Human: HCT116 | ATCC | CCL-247 |
| Human: HT1080 | ATCC | CCL-121 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6J | The Jackson Laboratory | JAX: 000664 |
| Mouse: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | The Jackson Laboratory | JAX: 005557 |
|
| ||
| Recombinant DNA | ||
|
| ||
| Human ME2 Mission shRNA-#1 | Sigma-Aldrich | TRCN0000064742 |
| Human ME2 Mission shRNA-#2 | Sigma-Aldrich | TRCN0000294007 |
| Human ADSL Mission shRNA-#1 | Sigma-Aldrich | TRCN0000078272 |
| Human ADSL Mission shRNA-#2 | Sigma-Aldrich | TRCN0000078271 |
| Human ASL Mission shRNA-#1 | Sigma-Aldrich | TRCN0000078310 |
| Human ASL Mission shRNA-#2 | Sigma-Aldrich | TRCN0000078312 |
| Human FAH Mission shRNA-#1 | Sigma-Aldrich | TRCN0000296459 |
| Human FAH Mission shRNA-#2 | Sigma-Aldrich | TRCN0000050674 |
| Human SDHA Mission shRNA-#1 | Sigma-Aldrich | TRCN0000028118 |
| Human SDHA Mission shRNA-#2 | Sigma-Aldrich | TRCN0000028093 |
| Human PRMT1 Mission shRNA-#1 | Sigma-Aldrich | TRCN0000310243 |
| Human PRMT1 Mission shRNA-#2 | Sigma-Aldrich | TRCN0000035931 |
| Mouse Me2 Mission shRNA-#1 | Sigma-Aldrich | TRCN0000332396 |
| Mouse Me2 Mission shRNA-#2 | Sigma-Aldrich | TRCN0000332397 |
| pcDNA3.1 | Invitrogen | V80020 |
| pcDNA3.1-ME2-Flag | This study | N/A |
| pcDNA3.1-ME2-HA | This study | N/A |
| lentiCRISPR v2 | Addgene | Cat#52961 |
| pLV-EF1a-IRES-Hygro | Addgene | Cat#85134 |
| pLV-EF1a-IRES-Hygro-ME2-Flag | This study | N/A |
| pLV-EF1a-IRES-Hygro-NRF2-Flag | This study | N/A |
| pLV-EF1a-IRES-Hygro-SLC1A3-Flag | This study | N/A |
| pEGFP-N1-FLAG | Addgene | Cat#60360 |
| pEGFP-N1-FLAG-ME2 | This study | N/A |
| pEGFP-PRMT1-9 | (Herrmann et al., 2009) | N/A |
| pcDNA3.1-MRPL45-HA | This study | N/A |
| pEGFPN1-MRPL45 | This study | N/A |
| pQE1-MRPL45 | This study | N/A |
| pSJ3-ME2-His | This study | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| FlowJo, version 10 | FlowJo | N/A |
| PyMol2.1 | https://pymol.org/2/ | N/A |
| GraphPad Prism 8 | https://www.graphpad.com/ | N/A |
Highlights.
Fumarate promotes monomer-to-dimer transition of ME2 to enhance mitobiogenesis
ME2 dimers activate DUT to foster thymidine generation and an increase of mtDNA
ME2 dimers relieve MRPL45 inhibition to enable mitochondrial protein production
ME2 methylation by PRMT1 inhibits dimerization and delays leukemia progression
ACKNOWLEDGMENTS
We thank the Harvard Center for Mass Spectrometry for the metabolic flux and proteomic analysis, Shanghai Proteomics Professional Technical Platform for Severe Diseases for technical support, and the Core Facility of Basic Medical Sciences, Shanghai Jiao Tong University School of Medicine for support with transmission electron microscopy. We thank Amir Schajnovitz for assistance with animal irradiation and Siang Boon Koh for providing breast cancer cell lines. This work was supported by the National Institutes of Health (U19HL129903 to D.T.S.), Ludwig Cancer Research Center at Harvard, and the Gerald and Darlene Jordan Professorship (to D.T.S.).
Footnotes
DECLARATION OF INTERESTS
D.T.S. declares the following competing interests: Novartis, sponsored research; Agios, director and equity holder; Editas Medicines, director and equity holder; Fate Therapeutics, founder and equity holder; Magenta Therapeutics, director, founder, equity holder, and consultant; Clear Creek Bio, director, equity holder, and consultant; LifeVaultBio, director and equity holder; Fog Pharma, consultant; VCanbio, consultant.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cmet.2021.03.003.
REFERENCES
- Agaronyan K, Morozov YI, Anikin M, and Temiakov D (2015). Mitochondrial biology. Replication-transcription switch in human mitochondria. Science 347, 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccelli I, Gareau Y, Lehnertz B, Gingras S, Spinella JF, Corneau S, Mayotte N, Girard S, Frechette M, Blouin-Chagnon V, et al. (2019). Mubritinib targets the electron transport chain complex I and reveals the landscape of OXPHOS dependency in acute myeloid leukemia. Cancer Cell 36, 84–99.e8. [DOI] [PubMed] [Google Scholar]
- Bardella C, Olivero M, Lorenzato A, Geuna M, Adam J, O’Flaherty L, Rustin P, Tomlinson I, Pollard PJ, and Di Renzo MF (2012). Cells lacking the fumarase tumor suppressor are protected from apoptosis through a hypoxia-inducible factor-independent, AMPK-dependent mechanism. Mol. Cell. Biol. 32, 3081–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, and Sabatini DM (2015). An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 162, 540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc RS, and Richard S (2017). Arginine methylation: the coming of age. Mol. Cell 65, 8–24. [DOI] [PubMed] [Google Scholar]
- Bogenhagen DF, Ostermeyer-Fay AG, Haley JD, and Garcia-Diaz M (2018). Kinetics and mechanism of mammalian mitochondrial ribosome assembly. Cell Rep 22, 1935–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boultwood J, Fidler C, Mills KI, Frodsham PM, Kusec R, Gaiger A, Gale RE, Linch DC, Littlewood TJ, Moss PA, et al. (1996). Amplification of mitochondrial DNA in acute myeloid leukaemia. Br. J. Haematol. 95, 426–431. [DOI] [PubMed] [Google Scholar]
- Brown A, Rathore S, Kimanius D, Aibara S, Bai XC, Rorbach J, Amunts A, and Ramakrishnan V (2017). Structures of the human mitochondrial ribosome in native states of assembly. Nat. Struct. Mol. Biol. 24, 866–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carew JS, Nawrocki ST, Xu RH, Dunner K, McConkey DJ, Wierda WG, Keating MJ, and Huang P (2004). Increased mitochondrial biogenesis in primary leukemia cells: the role of endogenous nitric oxide and impact on sensitivity to fludarabine. Leukemia 18, 1934–1940. [DOI] [PubMed] [Google Scholar]
- Chandel NS (2015). Evolution of mitochondria as signaling organelles. Cell Metab 22, 204–206. [DOI] [PubMed] [Google Scholar]
- Chen WL, Wang YY, Zhao A, Xia L, Xie G, Su M, Zhao L, Liu J, Qu C, Wei R, et al. (2016a). Enhanced fructose utilization mediated by SLC2A5 is a unique metabolic feature of acute myeloid leukemia with therapeutic potential. Cancer Cell 30, 779–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WW, Freinkman E, Wang T, Birsoy K, and Sabatini DM (2016b). Absolute quantification of matrix metabolites reveals the dynamics of mitochondrial metabolism. Cell 166, 1324–1337.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbet C, Pinto A, Martherus R, Santiago de Jesus JP, Polet F, and Feron O (2016). Acidosis drives the reprogramming of fatty acid metabolism in cancer cells through changes in mitochondrial and histone acetylation. Cell Metab 24, 311–323. [DOI] [PubMed] [Google Scholar]
- Couvillion MT, Soto IC, Shipkovenska G, and Churchman LS (2016). Synchronized mitochondrial and cytosolic translation programs. Nature 533, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, and Chandel NS (2016). Fundamentals of cancer metabolism. Sci. Adv. 2, e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn GW 2nd, Vega RB, and Kelly DP (2015). Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev 29, 1981–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R, Bosc C, Sugita M, Stuani L, Fraisse M, et al. (2017). Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov 7, 716–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D, et al. (2017). Lactate metabolism in human lung tumors. Cell 171, 358–371.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C (2017). Mitochondrial metabolites: undercover signalling molecules. Interface Focus 7, 20160100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guitart AV, Panagopoulou TI, Villacreces A, Vukovic M, Sepulveda C, Allen L, Carter RN, van de Lagemaat LN, Morgan M, Giles P, et al. (2017). Fumarate hydratase is a critical metabolic regulator of hematopoietic stem cell functions. J. Exp. Med. 214, 719–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Xu J, Zheng Q, He J, Zhou W, Wang K, Huang X, Fan Q, Ma J, Cheng J, et al. (2019). NRF2 SUMOylation promotes de novo serine synthesis and maintains HCC tumorigenesis. Cancer Lett 466, 39–48. [DOI] [PubMed] [Google Scholar]
- Haas R, Cucchi D, Smith J, Pucino V, Macdougall CE, and Mauro C (2016). Intermediates of metabolism: from bystanders to signalling molecules. Trends Biochem. Sci. 41, 460–471. [DOI] [PubMed] [Google Scholar]
- Hayashi G, Jasoliya M, Sahdeo S, Saccà F, Pane C, Filla A, Marsili A, Puorro G Lanzillo R, Brescia Morra V, et al. (2017). Dimethyl fumarate mediates Nrf2-dependent mitochondrial biogenesis in mice and humans. Hum. Mol. Genet. 26, 2864–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann F, Pably P, Eckerich C, Bedford MT, and Fackelmayer FO (2009). Human protein arginine methyltransferases in vivo–distinct properties of eight canonical members of the PRMT family. J. Cell Sci. 122, 667–677. [DOI] [PubMed] [Google Scholar]
- Hirmondo R, Lopata A, Suranyi EV, Vertessy BG, and Toth J (2017). Differential control of dNTP biosynthesis and genome integrity maintenance by the dUTPase superfamily enzymes. Sci. Rep. 7, 6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husted AS, Trauelsen M, Rudenko O, Hjorth SA, and Schwartz TW (2017). GPCR-mediated signaling of metabolites. Cell Metab 25, 777–796. [DOI] [PubMed] [Google Scholar]
- Jiang P, Du W, Mancuso A, Wellen KE, and Yang X (2013). Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 493, 689–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jitschin R, Hofmann AD, Bruns H, Giessl A, Bricks J, Berger J, Saul D, Eckart MJ, Mackensen A, and Mougiakakos D (2014). Mitochondrial metabolism contributes to oxidative stress and reveals therapeutic targets in chronic lymphocytic leukemia. Blood 123, 2663–2672. [DOI] [PubMed] [Google Scholar]
- Jones CL, Stevens BM, D’Alessandro A, Reisz JA, Culp-Hill R, Nemkov T, Pei S, Khan N, Adane B, Ye H, et al. (2018). Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell 34, 724–740.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehrein K, Schilling R, Möller-Hergt BV, Wurm CA, Jakobs S, Lamkemeyer T, Langer T, and Ott M (2015). Organization of mitochondrial gene expression in two distinct ribosome-containing assemblies. Cell Rep 10, 843–853. [DOI] [PubMed] [Google Scholar]
- Konopleva M, Pollyea DA, Potluri J, Chyla B, Hogdal L, Busman T, McKeegan E, Salem AH, Zhu M, Ricker JL, et al. (2016). Efficacy and biological correlates of response in a phase II study of Venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov 6, 1106–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni RA, Bak DW, Wei D, Bergholtz SE, Briney CA, Shrimp JH, Alpsoy A, Thorpe AL, Bavari AE, Crooks DR, et al. (2019). A chemoproteomic portrait of the oncometabolite fumarate. Nat. Chem. Biol. 15, 391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummer E, Leibundgut M, Rackham O, Lee RG, Boehringer D, Filipovska A, and Ban N (2018). Unique features of mammalian mitochondrial translation initiation revealed by cryo-EM. Nature 560, 263–267. [DOI] [PubMed] [Google Scholar]
- Larsen SC, Sylvestersen KB, Mund A, Lyon D, Mullari M, Madsen MV, Daniel JA, Jensen LJ, and Nielsen ML (2016). Proteome-wide analysis of arginine monomethylation reveals widespread occurrence in human cells. Sci. Signal. 9, rs9. [DOI] [PubMed] [Google Scholar]
- Laukka T, Mariani CJ, Ihantola T, Cao JZ, Hokkanen J, Kaelin WG Jr., Godley LA, and Koivunen P (2016). Fumarate and succinate regulate expression of hypoxia-inducible genes via TET enzymes. J. Biol. Chem. 291, 4256–4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A, Domingos Chinen LT, Rocha RM, et al. (2014). PGC-1a mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 16, 992–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansueto G, Armani A, Viscomi C, D’Orsi L, De Cegli R, Polishchuk EV, Lamperti C, Di Meo I, Romanello V, Marchet S, et al. (2017). Transcription factor EB controls metabolic flexibility during exercise. Cell Metab 25, 182–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Outschoorn UE, Pavlides S, Sotgia F, and Lisanti MP (2011). Mitochondrial biogenesis drives tumor cell proliferation. Am. J. Pathol. 178, 1949–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, McAfoos T, Morlacchi P, Ackroyd J, Agip AA, et al. (2018). An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 24, 1036–1046. [DOI] [PubMed] [Google Scholar]
- Pallotti F, and Lenaz G (2007). Isolation and subfractionation of mitochondria from animal cells and tissue culture lines. Methods Cell Biol 80, 3–44. [DOI] [PubMed] [Google Scholar]
- Pollyea DA, Stevens BM, Jones CL, Winters A, Pei S, Minhajuddin M, D’Alessandro A, Culp-Hill R, Riemondy KA, Gillen AE, et al. (2018). Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 24, 1859–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rackham O, Busch JD, Matic S, Siira SJ, Kuznetsova I, Atanassov I, Ermer JA, Shearwood AM, Richman TR, Stewart JB, et al. (2016). Hierarchical RNA processing is required for mitochondrial ribosome assembly. Cell Rep 16, 1874–1890. [DOI] [PubMed] [Google Scholar]
- Richter-Dennerlein R, Oeljeklaus S, Lorenzi I, Ronsör C, Bareth B, Schendzielorz AB, Wang C, Warscheid B, Rehling P, and Dennerlein S (2016). Mitochondrial protein synthesis adapts to influx of nuclear-encoded protein. Cell 167, 471–483.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorbach J, Richter R, Wessels HJ, Wydro M, Pekalski M, Farhoud M, Ku€hl I, Gaisne M, Bonnefoy N, Smeitink JA, et al. (2008). The human mitochondrial ribosome recycling factor is essential for cell viability. Nucleic Acids Res 36, 5787–5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciacovelli M, Gonç alves E, Johnson TI, Zecchini VR, da Costa AS, Gaude E, Drubbel AV, Theobald SJ, Abbo SR, Tran MG, et al. (2016). Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 537, 544–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skrtić M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, Hurren R, Jitkova Y, Gronda M, Maclean N, et al. (2011). Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 20, 674–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens BM, Khan N, D’Alessandro A, Nemkov T, Winters A, Jones CL, Zhang W, Pollyea DA, and Jordan CT (2018). Characterization and targeting of malignant stem cells in patients with advanced myelodysplastic syndromes. Nat. Commun. 9, 3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, and Vander Heiden MG (2015). Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 162, 552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X, Yang Z, and Tong L (2003). Crystal structures of substrate complexes of malic enzyme and insights into the catalytic mechanism. Structure 11, 1141–1150. [DOI] [PubMed] [Google Scholar]
- Ternette N, Yang M, Laroyia M, Kitagawa M, O’Flaherty L, Wolhulter K, Igarashi K, Saito K, Kato K, Fischer R, et al. (2013). Inhibition of mitochondrial aconitase by succination in fumarate hydratase deficiency. Cell Rep 3, 689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohme S, Yazdani HO, Liu Y, Loughran P, van der Windt DJ, Huang H, Simmons RL, Shiva S, Tai S, and Tsung A (2017). Hypoxia mediates mitochondrial biogenesis in hepatocellular carcinoma to promote tumor growth through HMGB1 and TLR9 interaction. Hepatology 66, 182–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, Leigh I, Gorman P, Lamlum H, Rahman S, et al. (2002). Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 30, 406–410. [DOI] [PubMed] [Google Scholar]
- Tronconi MA, Maurino VG, Andreo CS, and Drincovich MF (2010). Three different and tissue-specific NAD-malic enzymes generated by alternative subunit association in Arabidopsis thaliana. J. Biol. Chem. 285, 11870–11879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrakis PA, Yurkovich ME, Sciacovelli M, Papachristou EK, Bridges HR, Gaude E, Schreiner A, D’Santos C, Hirst J, Hernandez-Fernaud J, et al. (2017). Fumarate hydratase loss causes combined respiratory chain defects. Cell Rep 21, 1036–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC (2016). Genetics: mitochondrial DNA in evolution and disease. Nature 535, 498–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KZ, Zhu J, Dagda RK, Uechi G, Cherra SJ 3rd, Gusdon AM, Balasubramani M, and Chu CT (2014). ERK-mediated phosphorylation of TFAM downregulates mitochondrial transcription: implications for Parkinson’s disease. Mitochondrion 17, 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, and Guan KL (2012). Mechanistic insights into the regulation of metabolic enzymes by acetylation. J. Cell Biol. 198, 155–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yambire KF, Fernandez-Mosquera L, Steinfeld R, Mühle C, Ikonen E, Milosevic I, and Raimundo N (2019). Mitochondrial biogenesis is transcriptionally repressed in lysosomal lipid storage diseases. eLife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng R, Smith E, and Barrientos A (2018). Yeast mitoribosome large subunit assembly proceeds by hierarchical incorporation of protein clusters and modules on the inner membrane. Cell Metab 27, 645–656.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong WX, Rabinowitz JD, and White E (2016). Mitochondria and cancer. Mol. Cell 61, 667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and code to understand and assess the conclusion of this research are available in the main text, supplementary materials, or MassIVE Database (accession number MSV000086778).