

Abstract
Mpox (formerly Monkeypox), a zoonotic illness caused by the Mpox virus, belongs to the Orthopoxvirus genus in the family Poxviridae. To design and develop effective antiviral therapeutics against DNA viruses, the DNA‐dependent RNA polymerase (DdRp) of poxviruses has emerged as a promising drug target. In the present study, we modeled the three‐dimensional (3D) structure of DdRp using a template‐based homology approach. After modeling, virtual screening was performed to probe the molecular interactions between 1755 Food and Drug Administration‐approved small molecule drugs (≤500 molecular weight) and the DdRp of Mpox. Based on the binding affinity and molecular interaction patterns, five drugs, lumacaftor (−11.7 kcal/mol), conivaptan (−11.7 kcal/mol), betulinic acid (−11.6 kcal/mol), fluspirilene (−11.3 kcal/mol), and imatinib (−11.2 kcal/mol), have been ranked as the top drug compounds interacting with Mpox DdRp. Complexes of these shortlisted drugs with DdRp were further evaluated using state‐of‐the‐art all‐atoms molecular dynamics (MD) simulations on 200 nanoseconds followed by principal component analysis (PCA). MD simulations and PCA results revealed highly stable interactions of these small drugs with DdRp. After due validation in wet‐lab using available in vitro and in vivo experiments, these repurposed drugs can be further utilized for the treatment of contagious Mpox virus. The outcome of this study may establish a solid foundation to screen repurposed and natural compounds as potential antiviral therapeutics against different highly pathogenic viruses.
Keywords: DNA‐dependent RNA polymerase (DdRp), homology modeling, molecular dynamics simulations, Mpox virus, small molecules, virtual screening
1. INTRODUCTION
Mpox virus, formerly called monkeypox virus, (Poxviridae) is an emerging virus of the Orthopoxvirus genus. Its latest outbreak has led to human‐to‐human (H2H) transmission in non‐endemic areas reinforcing the need for more research and support of countermeasures. Mpox virus is a large (~200 kb), enveloped linear double‐stranded DNA (dsDNA) virus with ~200 genes. 1 Mpox virus can infect a broad range of animal species, suggesting it as a continual threat. 2 Although the virus was first recognized in captive nonhuman primates in 1958, the actual reservoir species is thought to be a rodent species which has yet to be determined. 2 , 3 , 4 Human infections were not identified until 1970 and since then sporadic outbreaks frequently occur in forested regions of Central and West Africa with cases outside this region being travel‐related [WHO Fact Sheets: Monkeypox]. Mpox virus was previously designated in two clades: Clade I (formerly the Congo Basin Clade) and Clade II (formerly the West African Clade), where Clade I was regarded as more virulent. 5 , 6 , 7 The world is currently experiencing the largest recorded Mpox virus outbreak with significant virus transmission in non‐endemic regions. 6 As of December 27, 2022, a total of 83 497 confirmed cases and 72 fatalities in 110 countries with local transmission 8 , 9 have been identified. The WHO recognizes isolates from the 2022 Mpox virus as belonging to Clade II and they are now specifically referred to as Clade IIb. 4 , 5 In response to the current outbreak, public health authorities are recommending the use of approved Orthopoxvirus vaccines such as Jynneos/Imvamune for high‐risk individuals including healthcare workers or those in high‐risk communities. 10 Vaccination against smallpox has been estimated from previous outbreaks to be ~85% effective against preventing Mpox; however, it has yet to be evaluated for Clade IIb. 11
Mpox virus H2H transmission was thought to be infrequent, with the majority of human cases acquiring the virus from contact with infected animals through bites, scratches, or direct contact with an infected animal's blood, body fluid, or lesions. 12 Previous H2H transmission most often occurred from an infected individual to family members within the same household. 3 , 13 , 14 Unlike previous outbreaks, the overwhelming majority of documented Clade IIb cases have been in males and associated with men who have sex with men, with HIV being a common coinfection. 15 , 16 The central conserved region of the Mpox viral genome encodes housekeeping genes which play a major role in the viral life cycle including replication, transcription, and virion assembly. 17 , 18 Terminal regions of the Mpox viral genome encode key proteins that participate in host tropism range and pathogenesis (https://viralzone.expasy.org/). Based on their biological roles, membrane proteins, structural proteins, and DNA‐dependent RNA polymerase (DdRp) have been emerged as potential drug targets of the Mpox virus. 19 , 20 To date, there is no one specific treatment available for Mpox; however, different smallpox antiviral drugs including brincidofovir, tecovirimat, and cidofovir have been approved. 19 In 2019, the US Food and Drug Administration (FDA) (https://www.fda.gov/) approved the JYNNEOS vaccine, a third‐generation poxvirus vaccine, for the prevention of smallpox and Mpox in adults. In the absence of a specific treatment, there is an urgent need to discover small molecule drug compounds as potential inhibitors against Mpox virus for those who are not vaccinated or have suboptimal vaccine responses. Drug repurposing approaches may accelerate the discovery of effective antiviral candidates against Mpox virus, significantly minimizing the cost and time that would be involved in developing a new treatment. 21 , 22 , 23
In the present study, we employed an extensive molecular modeling approach to screen FDA‐approved drug libraries to identify effective small molecules against the DdRp of the Mpox virus. Based on their binding affinity scores and molecular interaction patterns, top hits (lumacaftor, conivaptan, betulinic acid, fluspirilene, and imatinib) were evaluated for drug‐likeness followed by molecular dynamics (MD) simulations on 200 ns to investigate the stability of the docking complexes at the atomic level.
2. METHODOLOGY
A plethora of computer‐aided drug design based methods have been applied to screen small chemical molecules as potential inhibitors against the DdRp of the Mpox virus. The methodology is described in detail as follows:
2.1. Homology modeling and structural evaluation
The peptide sequence of DdRp 147 kD (Accession Number A0A0F6N8L8) was obtained from the UniProt (https://www.uniprot.org/) in FASTA format with a length of 1286 amino acids (aa). Crystal structures of many Mpox viral proteins were not available in the protein data bank (PDB); therefore, homology modeling, also known as the comparative modeling approach, was applied to predict the three‐dimensional (3D) structure model of DdRp. To identify the homologous template structure, the position‐specific iterated BLAST (PSI‐BLAST) algorithm 24 was applied against the comprehensive PDB 25 with high alignment score and lower e‐value parameters. After shortlisting the homologous structure, the automated SWISS‐MODEL server 26 , 27 was employed to deduce the 3D model with a template modeling option. Overall quality of the modeled 3D structure was evaluated using Ramachandran plot calculation by analyzing the phi (Φ) and psi (Ψ) torsion angles and covalent bond quality. Two dedicated servers, SAVES (PROCHECK) 28 and PSVS, 29 were utilized to generate the Ramachandran plot of the DdRp model. Further, the 3D structure model of the DdRp was visualized in different shapes. The predicted final 3D model of DdRp was superimposed over the template structure from vaccinia virus with the aid of the UCSF Chimera package. 30
2.2. Preparation of library and receptor
Initially, a comprehensive library of 2716 FDA‐approved molecules was downloaded from DrugBank 31 in structural data file (SDF) format. Some molecules of the library were found to have a molecular weight (mw) ≥500 g/mol; therefore, we applied a filter to obtain small molecules with a mw ≤500 g/mol to achieve a potential drug target according to the Lipinski rule of five. As many as 1755 drug molecules were shortlisted for preparation and docking. 3D conformation of these 1755 molecules was downloaded from the comprehensive PubChem repository 32 in SDF format. Proceeding to the next step, geometry optimizations of the downloaded molecules were performed using the Merck molecular force field 94 (MMFF94) embedded in the Open Babel (command line) tool, followed by converting them into PDBQT format for docking purposes. 33
Before virtual screening, a modeled 3D structure of DdRp was prepared using MGLTools 34 and UCSF Chimera. 30 DdRp was prepared by adding polar hydrogen atoms and Kollman charges assigned on the structure. The ff14SB force field was applied for protein structure optimization and energy minimization.
2.3. Pocket site prediction and virtual screening
As there is not much information available for pocket site residues of DdRp, we employed the CB‐Dock2 server 35 for cavity detection. The DdRp pocket site residues were encased in a grid box with dimensions 78 × 92 × 92 Å centered at the coordinates X = 187.89, Y = 190.31, and Z = 175.38. The prepared 3D structure of DdRp and geometry optimized ligand structures were selected for the virtual screening. Virtual screening was performed using AutoDock Vina (https://vina.scripps.edu/) software with the combination of pyVSvina, a python script. For further investigations, the docked pose with the minimum full fitness score was considered. Discovery studio, PyMOL (https://pymol.org/2/), UCSF Chimera, 30 and PLIP (Protein−ligand interaction profiler) server 36 were utilized to visualize the 2D and 3D interaction plots of the docking complexes received as a result of virtual screening.
2.4. Drug likeness properties
The ADMET profiling of drug molecules including absorption, distribution, metabolism, excretion (ADME), and toxicity properties plays an important role in shortlisting the drug compounds based on Lipinski's rule of five 37 and Veber's rule. 38 ADMET properties of the top hits were evaluated using the SWISSADME server. 39
2.5. All‐atoms MD simulations
We employed state‐of‐the‐art all‐atoms MD simulations to understand the structural dynamics of the top‐ranked screened complexes using OPLS4 force field parameters in the Desmond simulation package of Schrödinger LLC. Each complex was solvated using the TIP3P water model within the orthorhombic periodic boundary conditions at the distances of 10 Å units as previously defined by. 40 The charge of the complexes was electrically neutralized with balancing Na+/Cl− ions, followed by energy minimization by heating and equilibrium processes before the MD simulations. The complexes were subjected to the minimization protocol based on the steepest descent method, then heated at 0−310 K with the annealing steps of 2000 and the time steps of 0.001 ps. In all runs, we applied a temperature of 310 K and a pressure of 1 bar in the NPT ensemble. The simulation length was 200 ns with a relaxation time of 1 ps for the ligands and the protein. The long‐range electrostatic interactions were calculated using the particle mesh Ewald method, where a cut‐off of 9.0 Å was set for the radius in Coulomb interactions. MD simulations as NPT ensembles were performed using GPUs for the 200 ns production phase applying Nose−Hoover chain thermostat and Martyna−Tobias−Klein barostat methods. The trajectories were harvested at 100 ps intervals for analysis. The behavior and interactions between the ligands and protein were gauged using the Simulation Interaction Diagram tool implemented in the Desmond MD package. The dynamic stability of each system was monitored by computing the backbone RMSD, radius of gyration, and flexibility by root mean squared fluctuation analysis.
2.6. Principal component analysis (PCA)
To determine the important conformations during the simulation, PCA was performed for all complexes using the Bio3D package 41 implemented in the R‐Project and ProDy software. 42 To have a better understanding of the dynamics of the DdRp, cross‐correlation analysis (DCCM) was executed to evaluate the correlated motion between residues in different residues' domains. The Cα atom of each residue interacted with each other in the form of a pair of nodes. Additionally, PCA provides useful information regarding the mutation effect on protein dynamics and can be used to investigate how changes in aa sequence affect the protein's conformational space and how these changes alter the protein's function at the atomic level.
3. RESULTS AND DISCUSSION
3.1. Homology modeling and structural evaluation of DdRp
The solved 3D structure of Vaccinia virus DdRp cotranscription capping complex (PDB ID: 6RIE; Chain A) was selected as a template based on the alignment result with a sequence identity of 99.07% to the DdRp of Mpox virus. Based on the shortlisted template structure, the DdRp 3D structure model was predicted using the SWISS‐MODEL server (Figure 1A,B). As evident from Figure 1, the modeled DdRp structure showed less than 1 Å RMSD with respect to the selected template structure (PDB ID: 6RIE; Chain A) upon superposition using the UCSF Chimera package (Figure 1C). The structural stereochemical property of the modeled structure was evaluated using ϕ and ψ torsion angles of the Ramachandran plot. As shown in Figure 1D, residues totals of 89.9%, 9.7%, and 0.4% were found in the most favored regions, additional allowed regions, and generously allowed regions, respectively. The overall G‐scores (all dihedral angles) −0.11 indicated an acceptable range of the DdRp model. Verify3D and ProSA‐web (−ve) calculated the 0.19 and 0.44 scores respectively, which suggested that the modeled structure was of good quality. Overall, the deduced 3D structure model of DdRp showed excellent geometry which was found to be acceptable for further annotations. After evaluating the structural geometry of the modeled DdRp, coordinates have been uploaded to the Protein Model Database 43 and assigned the following id PM0084406. This submitted model is publicly available and can be utilized in the future to screen therapeutics against Mpox virus. Figure 1E depicts the pocket site residues of DdRp.
Figure 1.
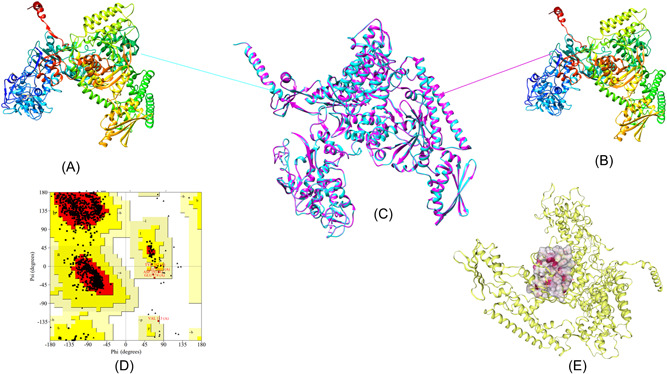
Homology modeling, superimposed structure, structure evaluation, and pocket site annotation: (A) Solved 3D structure of DdRp subunit rpo147 (PDB ID:6RIE) of vaccinia, derived from the RCSB‐PDB. (B) Modeled 3D structure of DdRp of Mpox virus using homology modeling approach utilizing the chain A of PDB ID: 6RIE as template. (C) Superimposed 3D structure of predicted model and template structure less than 1 Å RMSD; cyan color reflects the vaccinia structure while pink color depicts the DdRp of Mpox virus. (D) Calculated Ramachandran plot for the predicted model of DdRp. (E) Surface representation of annotated pocket site residues of DdRp. Protein 3D structure model was visualized and annotated in different shapes using UCSF Chimera package from the resource for biocomputing, visualization, and informatics at the University of California, San Francisco (supported by NIH P41 RR‐01,081). DdRp, DNA‐dependent RNA polymerase; Mpox, monkeypox; 3D, three‐dimensional.
3.2. Virtual screening identified five candidate antivirals
The molecular docking results showed that out of 1755 small drug molecules used in the present study, five drugs, lumacaftor, conivaptan, betulinic acid, fluspirilene, and imatinib, were found to have a higher negative binding energy of −11.7, −11.7, −11.6, −11.3, and −11.2 kcal/mol in comparison with other molecules. These top five molecules were shortlisted for further annotation and interactions analysis based on the calculated binding energies and the docking poses in the pocket site of DdRp. The docking complexes of these shortlisted compounds with DdRp demonstrated a number of molecular interactions including hydrogen bond, hydrophobic, and pi−pi stacking interactions (Figure 2). Table 1 presents detailed interaction patterns of all five top‐ranked compounds with their Pubchem id, mw, binding affinity score, and 3D structures.
Figure 2.
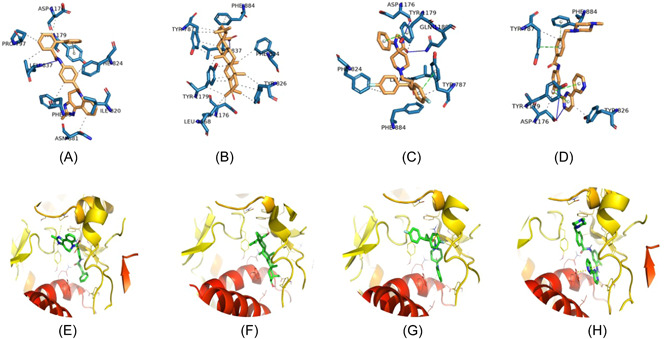
Representation of 2D interaction patterns (A−D) and 3D interaction patterns of top‐ranked compounds with DdRp (E−G). (A) Complex of conivaptan and DdRp. (B) Complex of betulinic acid and DdRp. (C) Complex of fluspirilene and DdRp. (D) Complex of imatinib and DdRp. (E) Close 3D view of DdRp pocket with conivaptan. (F) Pocket view with betulinic acid. (G) Pocket view with fluspirilene; and (H) pocket view with imatinib. 2D and 3D interaction patterns of docking complexes were visualized using PLIP server and PyMOL program, respectively. DdRp, DNA‐dependent RNA polymerase; PLIP, protein−ligand interaction profiler; 3D, three‐dimensional.
Table 1.
List of top ranked compounds and their molecular interaction patterns with DdRp of MPXV.
| Compound name | PubChem id | Binding energy (kcal/mol) | 3D image | Molecular interactions |
|---|---|---|---|---|
| Lumacaftor | 16678941 | −11.7 |
|
Hydrogen bond: LEU837 (2.91 Å), TYR1179 (2.29 Å) Hydrophobic bond: TYR787 (3.43 Å), TYR826 (3.44, 3.88 Å), ILE820 (3.82 Å), PHE824 (3.54, 3.72 Å), PHE884 (3.57, 3.69 Å), ALA833 (3.77 Å) Pi−Pi stacking: TYR826 (4.21 Å), TYR1179 (4.14 Å) |
| Conivaptan | 151171 | −11.7 |
|
Hydrogen bond: LEU837 (3.57 Å) Hydrophobic bond: PRO797 (3.44 Å), ILE820 (3.74, 3.89 Å), PHE824 (3.45 Å), PHE884 (3.83 Å), LEU837 (3.55 Å), ASN881 (3.50 Å), ASP1176 (3.67 Å), TYR1179 (3.76 Å) Pi−Pi stacking: TYR1179 (5.17 Å) |
| Betulinic acid | 64971 | −11.6 |
|
Hydrogen bond: LEU837 (2.18, 3.37 Å) Hydrophobic bond: TYR787 (3.37, 3.60, 3.60, 3.96 Å) PHE824 (3.89 Å), PHE884 (3.41, 3.49 Å), TYR826 (3.60, 3.69, 3.90 Å), TYR1179 (3.32, 3.55, 3.61, 3.76, 3.79 Å), LEU837 (3.60 Å), LEU1168 (3.21 Å), ASP1176 (3.72 Å) |
| Fluspirilene | 3396 | −11.3 |
|
Hydrogen bond: ASP1176 (2.36 Å), GLN1180 (2.34 Å) Hydrophobic bond: TYR787 (3.61, 3.76 Å), TYR1179 (3.73 Å), PHE824 (3.83 Å), PHE884 (3.95 Å), LEU837 (3.70 Å) Pi−Pi stacking: TYR787 (4.25 Å), TYR1179 (3.78 Å) |
| Imatinib | 5291 | −11.2 |
|
Hydrogen bond: ASP1176 (2.45, 3.19 Å) Hydrophobic bond: TYR787 (3.82 Å), TYR826 (3.84 Å), ASP1176 (3.40 Å) Pi−Pi stacking: TYR787(4.05 Å), TYR1179 (4.22, 4.57 Å), PHE884 (4.94 Å) |
Abbreviations: DdRp, DNA‐dependent RNA polymerase; Mpox virus, monkeypox virus.
3.3. Intermolecular contact analysis
3.3.1. Lumacaftor‐DdRp complex
Lumacaftor is an FDA‐approved pharmaceutical drug used to treat cystic fibrosis, a disease of the lungs. 44 The docking results of lumacaftor‐DdRp complex was stabilized with docking energy of −11.7 kcal/mol. Our modeling predicted lumacaftor to form two hydrogen bonds with Leu837 (2.91 Å) and Tyr1179 (2.29 Å), thereby forming stable conformations (Figure 3). Six residues, namely Tyr787 (3.43 Å), Tyr826 (3.44, 3.88 Å), Ile820 (3.82 Å), Phe824 (3.54, 3.72 Å), Phe884 (3.57, 3.69 Å), and Ala833 (3.77 Å), provided stability to complex with hydrophobic interactions in the model. Two residues, Tyr826 (4.21 Å) and Tyr1179 (4.14 Å), established the Pi−Pi stacking interactions.
Figure 3.
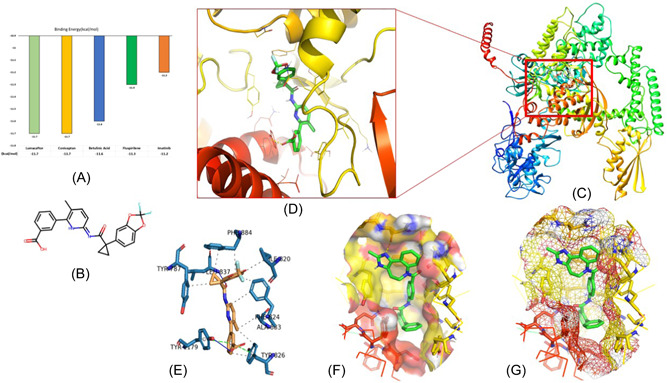
Representation of molecular interaction pattern between lumacaftor and DdRp: (A) Binding energy graph of top‐ranked molecules. (B) 2D structure of lumacaftor molecule. (C) 3D cartoon representation of docking complex of lumacaftor‐DdRp. (D) Close view of binding pattern of lumacaftor (green sticks) to the pocket site of DdRp. Hydrogen bonds are shown as yellow dotted line networks; (E) 3D plot of interaction between lumacaftor and DdRp. The blue, gray, and green lines represent the hydrogen bonds, hydrophobic interactions, and pi‐stacking, respectively. (F) Solid surface transparent representation of pocket site with lumacaftor in green sticks; and (G) mesh surface view of DdRp pocket. Different open‐source programs (UCSF Chimera, PyMOL, and PLIP server) were utilized to visualize and annotate the molecular interactions. DdRp, DNA‐dependent RNA polymerase; PLIP, protein−ligand interaction profiler; 3D, three‐dimensional.
3.3.2. Conivaptan‐DdRp complex
Conivaptan is a drug molecule well known for its role in preventing the binding of the neurohypophysial hormone Arginine vasopressin to V1a and V2 receptors, leading to the reduction of brain edema and the blood‐brain barrier. 45 This drug is approved for the treatment of euvolemic hyponatremia and hypervolemic hyponatremia. 46 The docking complex of conivaptan‐DdRp predicted the formation of a hydrogen bond with Leu837 (3.57 Å), leading to stable conformations. This compound was found to have eight hydrophobic interactions with Pro797(3.44 Å), Ile820 (3.74, 3.89 Å), Phe824 (3.45 Å), Phe884 (3.83 Å), Leu837 (3.55 Å), Asn881 (3.50 Å), Asp1176 (3.67 Å), and Tyr1179 (3.76 Å) (Figure 2A,E). Tyr1179 (5.17 Å) residue showed the Pi−Pi stacking interaction.
3.3.3. Betulinic acid‐DdRp complex
Betulinic acid is exclusively found in the bark of several plant species, principally the white birch (from which it gets its name), eucalyptus, and plane trees. 47 This pentacyclic triterpene possesses a wide range of strong diuretic, antiviral, antimicrobial, antidiabetic, antiparasitic, immunomodulatory, and anticancer activities. 47 , 48 , 49 , 50 , 51 , 52 From the docking analysis, it was modeled that the betulinic acid molecule forms a hydrogen bond with Leu837 (2.18, 3.37 Å). As evident from Figure 2B,F, this compound also manifests hydrophobic interactions with eight residues: Tyr787 (3.37, 3.60, 3.60, 3.96 Å), Phe824 (3.89 Å), Phe884 (3.41, 3.49 Å), Tyr826 (3.60, 3.69, 3.90 Å), Tyr1179 (3.32, 3.55, 3.61, 3.76, 3.79 Å), Leu837 (3.60 Å), Leu1168 (3.21 Å), and Asp1176 (3.72 Å).
3.3.4. Fluspirilene‐DdRp complex
Fluspirilene is one of the FDA‐approved drugs to treat schizophrenia. This traditional antipsychotic drug also showed anti‐glioma stem cell activity based on in vitro and in vivo experiments. 53 The binding affinity of the fluspirilene compound towards the DdRp active site is annotated by two hydrogen bonds with Asp1176 (2.36 Å) and Gln1180 (2.34 Å) residues (Figure 2C,G). Five residues, Tyr787 (3.61, 3.76 Å), Tyr1179 (3.73 Å), Phe824 (3.83 Å), Phe884 (3.95 Å), and Leu837 (3.70 Å), demonstrated hydrogen bonds with DdRp. The fluspirilene molecule was found to have Pi−Pi stacking interactions with Tyr787 (4.25 Å) and Tyr1179 (3.78 Å).
3.3.5. Imatinib‐DdRp complex
Imatinib (also known as “Gleevec” or “Glivec”) is a known tyrosine kinase inhibitor. 54 This oral chemotherapy medication is used to treat a plethora of cancers, blood conditions, and COVID‐19 55 ; [https://go.drugbank.com/drugs/DB00619]. The molecular interaction between the imatinib‐DdRp docked complex showed one hydrogen bond with Asp1176 (2.45, 3.19 Å). Three residues, Tyr787 (3.82 Å), Tyr826 (3.84 Å), and Asp1176 (3.40 Å), formed hydrophobic interactions. As depicted in Figure 2D,H, three residues, Tyr787 (4.05 Å), Tyr1179 (4.22, 4.57 Å), and Phe884 (4.94 Å), interacted through Pi−Pi stacking interactions.
All five drug molecules (lumacaftor, conivaptan, betulinic acid, fluspirilene, and imatinib) screened in this present study were predicted to tightly fit into the binding pocket of the DdRp of Mpox virus in a significant manner. Interestingly, the top three compounds, lumacaftor, conivaptan, and betulinic acid, showed common hydrogen bond interactions with the residue Leu837, while Asp1176 residue commonly formed a hydrogen bond with fluspirilene and imatinib molecules. Sharing common hydrogen bond interactions showed molecules targeting the conserved pocket site for binding with DdRp. Docking results presented in this study may support previous reports on the inhibition mechanism of small chemical molecules against different drug targets of Mpox virus. 21 , 22 , 23
In a recent study, 21 reported fludarabine as a potential inhibitor of the DdRp subunit (A6R) of Mpox virus based on virtual screening. This compound exhibited the binding affinity score of −7.53 kcal/mol. The docking complex of fludarabine compound and A6R predicted the formation of four hydrogen bonds with Gln108, Thr105, Arg94, and Gly93 residues. Based on the extensive molecular docking and interaction pattern investigations, two antivirals, Norov‐29 and bemnifosbuvir, have been proposed as promising inhibitors of DdRp. These compounds exhibited the strong binding affinity and hydrogen bond interactions with pocket site residues of DdRp. Abduljalil, Elfiky (2022). 56 Other key proteins of the Mpox virus including A42R profilin‐like protein, thymidylate kinase, and D9 decapping enzyme, have also emerged as potential drug targets to screen the potential inhibitors using existing cheminformatics methods. 23 , 49 , 57 explored the inhibition potential of mitoxantrone‐related drug compounds against A42R profilin‐like protein of Mpox virus using a computational repurposing approach. Based on the docking and 3D pharmacophore analysis, four PubChem compounds, 153640723, 24848320, 58102019, and 145293737, have been proposed as top‐ranked compounds with the binding energy of −7.2, −6.9, −7.0, and −7.0 kcal/mol, respectively. In another study, Sahoo et al. (2023), 57 presented a computational drug repurposing investigation and reported four US FDA approved compounds (tipranavir, cefiderocol, doxorubicin, and dolutegravir) as potential inhibitors against thymidylate kinase and D9 decapping enzyme of Mpox virus. These compounds showed strong binding patterns and acceptable binding affinity scores with Mpox virus drug targets.
The drug repurposing approach has increasingly become an attractive strategy in the discovery of new uses for approved and investigated drugs to reduce development costs and timelines. 58 In this present study, we have applied this approach to screen promising drug candidates against DdRp of Mpox. Based on extensive molecular modeling investigations, five FDA‐approved drugs (lumacaftor, conivaptan, betulinic acid, fluspirilene, and imatinib) have been proposed as potential inhibitors of DdRp. These screened small drugs possess inhibition potential against a plethora of diseases and antiviral activities are well‐reported. 44 , 45 , 47 , 53 Therefore, these small drugs may be utilized to develop effective antiviral therapy against Mpox after validation in wet lab.
3.4. ADME properties
All five top‐ranked compounds, lumacaftor, conivaptan, betulinic acid, fluspirilene, and imatinib, were selected for the drug‐likeness evaluation based on Lipinski's rule of five 37 and Veber's rule 38 using the SWISSADME server. 39 Calculated ADME properties of these top hits are summarized in Table 2. Different parameters such as number of hydrogen bond acceptors, number of hydrogen bond donors, number of rotatable bonds, consensus logP, molar refractivity, bioavailability score, synthetic accessibility, topological polar surface area (TPSA), and solubility, were described with optimal range of each property. Lumacaftor, conivaptan, betulinic acid, fluspirilene, and imatinib have the following mw, respectively: 452.41, 498.57, 456.70, 475.57, and 493.60 g/mol; these drug molecules have mw ≤500 g/mol. These top‐ranked molecules depicted fewer than 10 hydrogen bond donors and five hydrogen bond donors. All five molecules showed a number of rotatable bonds less than 10 (ranging between 2 and 8). TPSA value of all molecules was found with the range of 35.58−97.75. All five compounds were found to have consensus logP values less than 7, which indicates good permeability across the cell membrane. Lumacaftor, conivaptan, betulinic acid, fluspirilene, and imatinib have molar refractivity values of 113.98, 153.54, 136.91, 145.27, and 154.50, respectively. These molecules present asynthetic accessibility scores of <10, which suggests that compounds can easily be synthesized. 59 , 60 These top‐ranked compounds also validated Lipinski's rule of five and Veber's rule, which state the oral bioavailability of drug molecules. 61 , 62 Taken together, evaluation of ADME properties suggested that these screened molecules harbor favorable drug‐likeness properties and can be utilized for further annotation and the validation.
Table 2.
ADME properties of the screened top ranked compounds against DdRp.
| Molecules | Drug likeliness properties | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MW (g/mol) | Concensus Log Po/w | Number of H‐bond acceptor | Number of H‐bond donors | Molar refractivity | Lipinski | Veber | Bioavailability score | Synthetic accessibility | TPSA (Å2) | Number of rotatable bonds | Solubility (mg/mL) | |
| Lumacaftor | 452.41 | 4.14 | 8 | 2 | 113.98 | Yes | Yes | 0.56 | 3.28 | 97.75 | 6 | 6.97e−06 |
| Conivaptan | 498.57 | 5.01 | 3 | 2 | 153.54 | Yes | Yes | 0.55 | 3.97 | 78.09 | 6 | 2.23e−09 |
| Betulinic acid | 456.70 | 6.13 | 3 | 2 | 136.91 | Yes | Yes | 0.85 | 5.63 | 57.53 | 2 | 9.09e−04 |
| Fluspirilene | 475.57 | 5.19 | 4 | 1 | 145.27 | Yes | Yes | 0.55 | 3.88 | 35.58 | 7 | 1.08e−07 |
| Imatinib | 493.60 | 3.38 | 6 | 2 | 154.50 | Yes | Yes | 0.55 | 3.78 | 86.28 | 8 | 1.05e−07 |
Abbreviations: DdRp, DNA‐dependent RNA polymerase; MW, molecular weight; TPSA, topological polar surface area.
3.5. MD simulation
We employed state‐of‐the‐art all‐atoms MD simulations to understand the structural dynamics of the top‐ranked screened complexes using OPLS4 force field parameters embedded in the Desmond simulation package of Schrödinger LLC. The MD simulation length was 200 ns with a relaxation time of 1 ps for the ligands and the protein. The dynamic stability of each system was monitored by computing the backbone RMSD, hydrogen bonds, and flexibility by root mean squared fluctuation analysis throughout the simulation trajectory. The calculated RMSD plot of the protein backbone depicted the constant range of stability throughout the simulation with a range between 4 and 10 Å. As evident from Figure 4A, almost all ligands showed high‐level fluctuation patterns between 0 and 40 ns. After 40 ns, all five compounds attained stability around 100 ns of a similar trend. However, initial fluctuations did not affect the stability of the docking complexes. Measured ligand RMSD plot represented the constant range of stability with fluctuations throughout the MD simulations. As shown in Figure 4C, the calculated ligand RMSD plot showed the stability of ligand compounds with few small fluctuations in fluspirilene (black), imatinib (red), conivaptan (blue), and lumacaftor (orange). These four molecules demonstrated initial fluctuations between 1 and 40000 ps on average 2.0 Å. After 60 ns, three molecules, fluspirilene (black), imatinib (red), and lumacaftor (orange), showed the second phase of small fluctuations. Betulinic acid (green) demonstrated a straight line without notable fluctuations during MD simulations and indicated minimum conformational changes (Figure 4C). Therefore, betulinic acid (green) significantly contributed to the stability of the complex. Altogether, calculated RMSD plots reflected the minimum conformational changes and all docking complexes were found stable during MD simulations.
Figure 4.
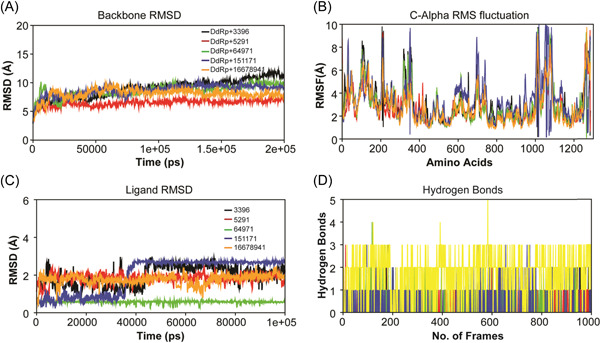
MD simulations on 200 ns: (A) RMSD plot of the backbone; (B) C‐alpha RMS fluctuation plot; (C) ligand RMSD plot; and (D) hydrogen bonds plot (DdRp—fluspirilene complex [black], DdRp—imatinib complex [red], DdRp—betulinic acid complex [green], DdRp—conivaptan complex [blue], and DdRp—lumacaftor [orange]). DdRp, DNA‐dependent RNA polymerase; MD, molecular dynamics.
Root mean square fluctuation (RMSF) plot was calculated to explore the flexibility of the individual residues of the system within the time frame (Figure 4B). The average RMSF values of the lumacaftor, conivaptan, betulinic acid, fluspirilene, and imatinib molecules complex with DdRp were found to be 2.75, 3.32, 2.97, 3.12, 2.79 Å, respectively. We also observed from the RMSF plot that those residues between 1000 and 1100 had higher than predicted fluctuations on 10 Å as compared to other regions of the protein. Few binding site residues belong to this region and were involved in the interaction pattern, which may be a reason for fluctuation in the presence of ligand molecules. The fewer number of fluctuations observed in the docking complexes support the docking results and suggest that the DdRp significantly interacts with lumacaftor, conivaptan, betulinic acid, fluspirilene, and imatinib molecules.
To understand the stability of the molecules' binding pattern to the DdRp, hydrogen bond analysis was conducted for all five docking complexes. Time‐dependent behavior of the hydrogen bonds was investigated and monitored. As shown in Figure 4D, lumacaftor, conivaptan, betulinic acid, fluspirilene, and imatinib compounds depicted sustained hydrogen bonds with DdRp, and all five molecules were predicted to have more than one average hydrogen bond per frame during MD simulations. The investigated hydrogen bond interaction patterns demonstrated that all five molecules maintain a strong interaction profiling with pocket site residues of DdRp and were stable throughout the simulation. Overall, MD simulation analyses were obtained with stable conformation and all five molecules interacted with the pocket site of DdRp with minor conformational changes.
3.6. Interaction fraction analysis
During the MD simulations, all five compounds were also analyzed for the total number of receptor−ligand interactions including hydrogen bonds, hydrophobic interactions, ionic interactions, and water bridges. The analyses were graphically plotted in Figure 5. The interaction fraction of DdRp complex with fluspirilene showed the average binding percentage during MD simulations (Figure 5A). Four residues, Lys835, Thr836, Leu837, and Asp1176, showed hydrogen bond occupancy of less than 20%, while 12 residues, Lys782, Ala784, Tyr787, Ile820, Phe824, Tyr826, Ala833, Lys835, Leu837, Phe884, Phe1154, and Leu1168, represented less than 60% hydrophobic bond occupancy. Only Tyr1179 showed 100% hydrophobic bond occupancy with the fluspirilene molecule. Eight residues, Gly823, Lys835, Thr836, Leu837, Asp1176, Tyr1177, and Gln1180, demonstrated the important role of the water bridge interaction with fluspirilene. Only the Asp1176 residue denoted an ionic interaction. The Asp1176 residue was predicted by MD simulations and docking analysis to establish a hydrogen bond interaction. Interaction fraction analysis of DdRp complex with imatinib represented hydrogen bonds occupancy of less than 90% with Gly823, Thr836, Leu837, Cys1164, Asp1176, Tyr1179, and Gln1180 (Figure 5B). Nine residues, Tyr787, Ile820, Phe824, Tyr826, Ala833, Leu837, Phe884, Leu1168, Tyr1179 demonstrated less than 100% hydrophobic bond occupancy. Tyr787, Lys819, Gln822, Gly823, Val825, Lys834, Lys835, Asn881, Glu1157, Gly1161, Glu1165, Glu1173, Asp1176, Tyr1179 were found to be involved in hydrophobic interaction bonds. From the MD simulations of DdRp complex with betulinic acid, residues Lys835, Asn881, and Asp1176 were observed to be involved in hydrogen bond formation for more than 50% occupancy of the simulation (Figure 5C). Seven residues (Tyr787, Ile820, Phe824, Ala833, Leu837, Phe884, and Tyr1179) were noted to form hydrophobic interaction bonds. Ile449, His450, Lys782, Lys819, Gln822, Gly823, Tyr826, Lys834, Lys835, Thr836, Val879, Asn881, Asp883, Phe884, Asp1176, Tyr1179, and Gln1180 were found to be strongly involved in water bridge interaction formation with imatinib. The interaction fraction analysis of DdRp complex with conivaptan revealed that Tyr787, Ile820, Gly823, Lys835, Thr836, Leu837, Tyr1179, and Gln1180 manifest hydrogen bond interactions with less than 50% occupancy of the simulation time (Figure 5D). Twelve residues, Tyr787, Pro797, Ile820, Phe824, Tyr826, Ala833, Leu837, Phe884, Arg1160, Leu1168, Tyr1177, and Tyr1179 established hydrophobic bond interactions. Thirteen residues (Tyr787, Lys819, Gln822, Thr836, Leu837, Asn881, Phe884, Glu1157, Arg1160, Asp1176, Tyr1179, Gln1180, and Asp1183) were also observed to participate in forming water bridge interactions with conivaptan. In the case of the complex of DdRp and lumacaftor, Gly823, Lys829, Thr836, Leu837, and Glu1165 residues were found to be involved in hydrogen bond interactions formation with more than 100% occupancy simulation time. Eight residues, Tyr787, Phe824, Tyr826, Ala833, Leu837, Phe884, Leu1168, and Tyr1179, were found to be strongly involved in establishing hydrophobic interactions. Thirteen residues (Lys782, Gln822, Gly823, Val825, Ser827, Lys829, Asn881, Gly1161, Glu1165, Asp1176, Leu1178, Tyr1179, and Gln1180) were observed to be involved in water bridge interactions formation (Figure 5E). No ionic interactions were noted in the complexes of imatinib, betulinic acid, conivaptan, and lumacaftor molecules with DdRp. Interaction fraction analysis showed additional interactions during MD simulations when compared to docking results, which significantly contributed to establishing strong interaction patterns with the pocket site of DdRp.
Figure 5.
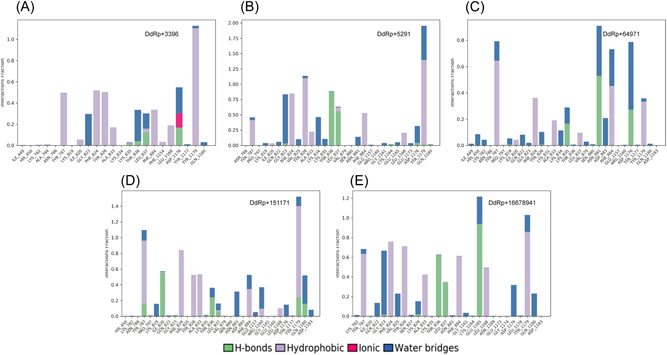
Graphical representation of interaction fraction analysis: (A) Interaction between DdRp and fluspirilene. (B) Interaction between DdRp and imatinib. (C) Interaction between DdRp and betulinic acid. (D) Interaction between DdRp and conivaptan; and (E) interaction between DdRp and lumacaftor. DdRp, DNA‐dependent RNA polymerase.
3.7. PCA
To obtain the conformational state information of the ligand bound protein DdRp, we performed PCA using Cα‐atoms from each trajectory. The first three PCs accounted for 56.63%, 40.05%, and 47.95% of the variance in the motion observed in the trajectories of conivaptan, betulinic acid, and fluspirilene with covariances of 94%, 92%, and 91% respectively, while PCs of imatinib and lumacaftor accounted for 33.51% and 26.92%, sharing the same covariance of 89%.
The 2D diagrams between eigenvectors PC1 and PC2 were utilized to compare the conjoined movements which depict the variance in the conformational distribution. DCCM analysis is shown in Figure 6, with cyan indicating positive cross‐correlation, magenta indicating negative cross‐correlation, and white indicating no cross‐correlation. The lumacaftor shows more cyan, which indicates the strong positive correlation in the region surrounding 200, 500, 700, and 1100 aa. Other complexes, such as conivaptan, have binding site residue in the 200, 400, and 700 aa, showing positive correlation, while both positive and negative cross‐correlation was observed around aa near 800. In the betulinic acid complex, initial 200 aa, there is evidence of both positive and negative cross‐correlation, but the region surrounding 900−1100 aa showed a negative correlation. In addition, imatinib showed positive correlation near 500 and 900 aa with many regions with negative correlation.
Figure 6.
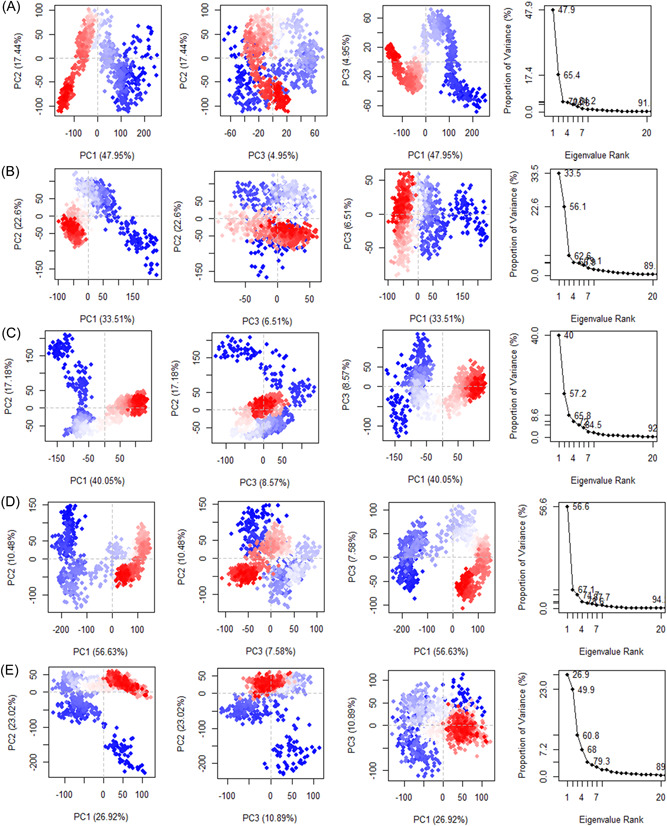
Calculated PCA plots of docking complexes: (A) Fluspirilene; (B) imatinib; (C) betulinic acid; (D) conivaptan; and (E) lumacaftor. PCA, principal component analysis.
4. CONCLUSIONS
The ongoing 2022 Mpox outbreak is a serious public threat across the globe. To date, no effective medicines are available to inhibit the Mpox virus. Due to important roles in the life cycle of the Mpox virus, DdRp has emerged as one of the potential drug targets. In the present study, we report the inhibition potential of FDA‐approved repurposed small drugs against Mpox virus. Based on integrative molecular modeling approaches, we screened the top five drug molecules, namely lumacaftor, conivaptan, betulinic acid, fluspirilene, and imatinib, as potential inhibitors against DdRp. These top‐ranked molecules demonstrated a high range of negative binding affinities and remarkable molecular interaction patterns. MD simulations of these drug molecules in complexes with DdRp exhibited a stable conformation on 200 ns, further confirmed by interaction fraction and PCA analysis. The inhibitory potential of these screened promising drug candidates may also be further validated in the wet lab with the help of cell culture and model organism experiments. In the future, these screened compounds may be utilized for antiviral therapy against emerging strains of poxviruses.
AUTHOR CONTRIBUTIONS
Mansi Dutt, Anuj Kumar, and David J. Kelvin contributed to the conception and design of the study. Mansi Dutt, Anuj Kumar, Madhusmita Rout, Budheswar Dehury, Gustavo Martinez, and Pacifique Ndishimye conducted the experiments. Mansi Dutt and Anuj Kumar drafted the manuscript with the help of Alyson A. Kelvin in the introduction section. David J. Kelvin and Alyson A. Kelvin edited the final manuscript. All authors read and approved the final manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
The authors are thankful to the Nikki Kelvin (The Journal of Infection in Developing Countries) for critical proofreading and editing the final manuscript. This work was supported by grants from the Canadian Institutes of Health Research (OV2170357, SBC171495, and MZ1187236), Genome Canada/Atlantic Genome, Research Nova Scotia, Dalhousie Medical Research Foundation, and the Li‐Ka Shing Foundation. D. J. K. is a Canada Research Chair in Translational Vaccinology and Inflammation.
Dutt M, Kumar A, Rout M, et al. Drug repurposing for Mpox: discovery of small molecules as potential inhibitors against DNA‐dependent RNA polymerase using molecular modeling approach. J Cell Biochem. 2023;124:701‐715. 10.1002/jcb.30397
Mansi Dutt and Anuj Kumar contributed equally to this work.
DATA AVAILABILITY STATEMENT
The data sets presented in this study can be found in public repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
REFERENCES
- 1. Xiang Y, White A. Monkeypox virus emerges from the shadow of its more infamous cousin: family biology matters. Emerg Microbes Infect. 2022;11(1):1768‐1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Silva NIO, de Oliveira JS, Kroon EG, Trindade GS, Drumond BP. Here, there, and everywhere: the wide host range and geographic distribution of zoonotic orthopoxviruses. Viruses. 2020;13(1):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alakunle E, Moens U, Nchinda G, Okeke MI. Monkeypox virus in Nigeria: infection biology, epidemiology, and evolution. Viruses. 2020;12(11):1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alakunle EF, Okeke MI. Monkeypox virus: a neglected zoonotic pathogen spreads globally. Nat Rev Microbiol. 2022;20(9):507‐508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Isidro J, Borges V, Pinto M, et al. Phylogenomic characterization and signs of microevolution in the 2022 multi‐country outbreak of monkeypox virus. Nature Med. 2022;28(8):1569‐1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Likos AM, Sammons SA, Olson VA, et al. A tale of two clades: monkeypox viruses. J Gen Virol. 2005;86(pt 10):2661‐2672. [DOI] [PubMed] [Google Scholar]
- 7.Monkeypox: experts give virus variants new names. 2023. Accessed January 4, 2023. https://www.who.int/news/item/12-08-2022-monkeypox--experts-give-virus-variants-new-names
- 8.2022 Mpox (monkeypox) outbreak: global trends. 2023. Accessed January 4, 2023. https://worldhealthorg.shinyapps.io/mpx_global/
- 9. Vogel L. Making sense of monkeypox death rates. Can Med Assoc J. 2022;194(31):E1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vaccines and immunization for monkeypox: interim guidance , 16 November 2022. Accessed January 4, 2023. https://www.who.int/publications/i/item/WHO-MPX-Immunization
- 11. Monkeypox . Accessed January 4, 2023. https://www.who.int/news-room/fact-sheets/detail/monkeypox
- 12. Pathogen safety data sheets: infectious substances—monkeypox virus—Canada.ca . 2023. Accessed January 4, 2023. https://www.canada.ca/en/public-health/services/laboratory-biosafety-biosecurity/pathogen-safety-data-sheets-risk-assessment/monkeypox-virus.html
- 13. Brown K, Leggat P. Human monkeypox: current state of knowledge and implications for the future. Tropical Med Infec Dis. 2016;1(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hutson CL, Damon IK. Monkeypox virus infections in small animal models for evaluation of anti‐poxvirus agents. Viruses. 2010;2(12):2763‐2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mailhe M, Beaumont AL, Thy M, et al. Clinical characteristics of ambulatory and hospitalized patients with monkeypox virus infection: an observational cohort study. Clin Microbiol Infect. 2023;29:233‐239. 10.1016/J.CMI.2022.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tarín‐Vicente EJ, Alemany A, Agud‐Dios M, et al. Clinical presentation and virological assessment of confirmed human monkeypox virus cases in Spain: a prospective observational cohort study. Lancet. 2022;400(10353):661‐669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shchelkunov SN, Totmenin AV, Safronov PF, et al. Analysis of the monkeypox virus genome. Virology. 2002;297(2):172‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shete AM, Yadav PD, Kumar A, et al. Genome characterization of monkeypox cases detected in India: identification of three sub clusters among A.2 lineage. J Infect. 2023;86(1):66‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang Y, Mu L, Wang W. Monkeypox: epidemiology, pathogenesis, treatment and prevention. Signal Transduct Target Ther. 2022;7(1):373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weaver JR, Isaacs SN. Monkeypox virus and insights into its immunomodulatory proteins. Immunol Rev. 2008;225(1):96‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Altayb HN. Fludarabine, a potential DNA‐dependent RNA polymerase inhibitor, as a prospective drug against monkeypox virus: a computational approach. Pharmaceuticals. 2022;15(9):1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lam HYI, Guan JS, Mu Y. In silico repurposed drugs against monkeypox virus. Molecules. 2022;27(16):5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Preet G, Oluwabusola ET, Milne BF, Ebel R, Jaspars M. Computational repurposing of mitoxantrone‐related structures against monkeypox virus: a molecular docking and 3D pharmacophore study. Int J Mol Sci. 2022;23(22):14287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Altschul S. Gapped BLAST and PSI‐BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389‐3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Berman HM. The protein data bank. Nucleic Acids Res. 2000;28(1):235‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS‐MODEL workspace: a web‐based environment for protein structure homology modelling. Bioinformatics. 2006;22(2):195‐201. [DOI] [PubMed] [Google Scholar]
- 27. Biasini M, Bienert S, Waterhouse A, et al. SWISS‐MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42(Web Server issue):W252‐W258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystal. 1993;26(2):283‐291. [Google Scholar]
- 29. Bhattacharya A, Tejero R, Montelione GT. Evaluating protein structures determined by structural genomics consortia. Proteins: Struct, Funct, Bioinf. 2007;66(4):778‐795. [DOI] [PubMed] [Google Scholar]
- 30. Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605‐1612. [DOI] [PubMed] [Google Scholar]
- 31. Wishart DS, Knox C, Guo AC, et al. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008;36(Database issue):D901‐D906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kim S, Chen J, Cheng T, et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 2020;49(D1):D1388‐D1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open babel: an open chemical toolbox. J Cheminform. 2011;3(1):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785‐2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu Y, Yang X, Gan J, Chen S, Xiao ZX, Cao Y. CB‐Dock2: improved protein‐ligand blind docking by integrating cavity detection, docking and homologous template fitting. Nucleic Acids Res. 2022;50(W1):W159‐W164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Adasme MF, Linnemann KL, Bolz SN, et al. PLIP 2021: expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021;49(W1):W530‐W534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lipinski CA. Lead‐ and drug‐like compounds: the rule‐of‐five revolution. Drug Discovery Today: Technol. 2004;1(4):337‐341. [DOI] [PubMed] [Google Scholar]
- 38. Veber DF, Johnson SR, Cheng H‐Y, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45(12):2615‐2623. [DOI] [PubMed] [Google Scholar]
- 39. Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug‐likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7(1):42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gajula M, Kumar A, Ijaq J. Protocol for molecular dynamics simulations of proteins. Bio‐Protocol. 2016;6(23):e2051. [Google Scholar]
- 41. Grant BJ, Skjærven L, Yao XQ. The Bio3D packages for structural bioinformatics. Prot Sci. 2021;30(1):20‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang S, Krieger JM, Zhang Y, et al. ProDy 2.0: increased scale and scope after 10 years of protein dynamics modelling with python. Bioinformatics. 2021;37(20):3657‐3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Castrignanò T, de Meo PDO, Cozzetto D, Talamo IG, Tramontano A. The PMDB protein model database. Nucleic Acids Res. 2006;34(Database issue):D306‐ D309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Connett GJ. Lumacaftor‐ivacaftor in the treatment of cystic fibrosis: design, development and place in therapy. Drug Des Devel Ther. 2019;13:2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zeynalov E, Jones SM, Seo JW, Snell LD, Elliott JP. Arginine‐vasopressin receptor blocker conivaptan reduces brain edema and blood‐brain barrier disruption after experimental stroke in mice. PLoS One. 2015;10(8):e0136121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li‐Ng M, Verbalis JG. Conivaptan: evidence supporting its therapeutic use in hyponatremia. Core Evid. 2009;4:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lou H, Li H, Zhang S, Lu H, Chen Q. A review on preparation of betulinic acid and its biological activities. Molecules. 2021;26(18):5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hordyjewska A, Ostapiuk A, Horecka A, Kurzepa J. Betulin and betulinic acid: triterpenoids derivatives with a powerful biological potential. Phytochem Rev. 2019;18(3):929‐951. [Google Scholar]
- 49. Jiang W, Li X, Dong S, Zhou W. Betulinic acid in the treatment of tumour diseases: application and research progress. Biomed Pharmacother. 2021;142:111990. [DOI] [PubMed] [Google Scholar]
- 50. Oliveira Costa JF, Barbosa‐Filho JM, de Azevedo Maia GL, et al. Potent anti‐inflammatory activity of betulinic acid treatment in a model of lethal endotoxemia. Int Immunopharmacol. 2014;23(2):469‐474. [DOI] [PubMed] [Google Scholar]
- 51. Oliveira‐Costa JF, Meira CS, Neves MVG, Dos Reis BPZC, Soares MBP. Anti‐Inflammatory activities of betulinic acid: a review. Front Pharmacol. 2022;13:1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. de Sá MS, Costa JFO, Krettli AU, et al. Antimalarial activity of betulinic acid and derivatives in vitro against Plasmodium falciparum and in vivo in P. berghei‐infected mice. Parasitol Res. 2009;105(1):275‐279. [DOI] [PubMed] [Google Scholar]
- 53. Dong Y, Furuta T, Sabit H, et al. Identification of antipsychotic drug fluspirilene as a potential anti‐glioma stem cell drug. Oncotarget. 2017;8(67):111728‐111741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Iqbal N, Iqbal N. Imatinib: a breakthrough of targeted therapy in cancer. Chemother Res Pract. 2014;2014:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Aman J, Duijvelaar E, Botros L, et al. Imatinib in patients with severe COVID‐19: a randomised, double‐blind, placebo‐controlled, clinical trial. Lancet Respiratory Med. 2021;9(9):957‐968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Abduljalil JM, Elfiky AA. Repurposing antiviral drugs against the human monkeypox virus DNA‐dependent RNA polymerase; in silico perspective. J Infect. 2022;85(6):702‐769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sahoo AK, Augusthian PD, Muralitharan I, et al. In silico identification of potential inhibitors of vital monkeypox virus proteins from FDA approved drugs. Mol Divers. 2022. 10.1007/s11030-022-10550-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pushpakom S, Iorio F, Eyers PA, et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discovery. 2018;18(1):41‐58. [DOI] [PubMed] [Google Scholar]
- 59. Ertl P, Schuffenhauer A. Estimation of synthetic accessibility score of drug‐like molecules based on molecular complexity and fragment contributions. J Cheminf. 2009;1(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kumar A, Sharma M, Richardson CD, Kelvin DJ. Potential of natural alkaloids from jadwar (Delphinium denudatum) as inhibitors against main protease of COVID‐19: a molecular modeling approach. Front Mol Biosci. 2022;9:430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kumar A, Mishra DC, Angadi UB, Yadav R, Rai A, Kumar D. Inhibition potencies of phytochemicals derived from sesame against SARS‐CoV‐2 main protease: a molecular docking and simulation study. Front Chem. 2021;9:773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pathak Y, Mishra A, Choudhir G, Kumar A, Tripathi V. Rifampicin and letermovir as potential repurposed drug candidate for COVID‐19 treatment: insights from an in‐silico study. Pharmacological Rep. 2021;73:926‐938. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data sets presented in this study can be found in public repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.