

Abstract
The brain primarily relies on glycolysis for mitochondrial respiration but switches to alternative fuels such as ketone bodies (KB) during low glucose availability. Neuronal KB uptake, which does not rely on the glucose transporter 4 (GLUT4) and insulin, has shown promising clinical applications in alleviating the neurological and cognitive effects of disorders with hypometabolic components. However, the specific mechanisms by which such interventions affect neuronal functions are poorly understood. In this study, we pharmacologically blocked GLUT4 to investigate the effects of the exogenous KB D-β-hydroxybutyrate (D-βHb) on mouse brain metabolism during acute insulin resistance (AIR). We found the impact of D-βHb to be distinct across neuronal compartments: AIR decreased synaptic activity and LTP, and impaired axonal conduction, synchronization, and action potential (AP) properties. D-βHb rescued neuronal functions associated with axonal conduction, synchronization and LTP.
Keywords: hippocampus, ketone bodies, beta-hydroxybutyrate, ketogenic diet, insulin resistance, GLUT4, hypometabolism, Schaffer collaterals, pyramidal neurons
2. Introduction
Over the past two decades, research on insulin signaling in the brain has become increasingly important in the contexts of type 2 diabetes mellitus (T2DM) and Alzheimer’s disease (AD) [1,2]. Insulin plays a critical role in memory formation processes in the hippocampus, such that systemic insulin resistance can interfere with hippocampal metabolism and cognitive function, as shown by various studies [3,4]. Additionally, insulin is an essential element in memory processing in the hippocampus and a vital mediator of cognitive impairments in T2DM and AD, due to its function in promoting GLUT4 translocation [5,6]. Although insulin from the blood crosses the blood-brain barrier, research suggests supplementary local insulin synthesis and release in the brain [7] which enhances the pro-cognitive effects of brain glucose [8]. Therefore, impaired glucose uptake in the hippocampus may contribute to cognitive decline associated with aging, T2DM, and AD [9,10,11].
This relationship has led to the hypothesis that brain hypometabolism reflects encroaching neuronal insulin resistance. The hippocampus is highly enriched with the GLUT4 receptor, which is the primary neuronal insulin dependent glucose transporter, particularly in brain areas with high concentrations of insulin receptors and relatively high neuronal activity [8,12,13]. Insulin acts by enhancing GLUT4 translocation across neuronal membranes under regulation of phosphatidylinositol 3-kinase (PI3K), which furthermore promotes memory formation [14,15]. During memory formation, GLUT4 is primarily expressed in the soma rather than in the more metabolically active hippocampal neuropil [16,17], suggesting that insulin effects may be primarily involved in maintenance of neuronal firing. Mouse models of AD or diet-induced obesity (DIO) showed blunting of the pro-cognitive effects of intrahippocampal insulin injections, along with decreased local glucose metabolism [18,19], coupled with neuronal hyperexcitability with epileptiform spikes and impairment of GLUT4 translocation [20].
During periods of lower glucose availability, ketone bodies (KBs) are supplementing metabolism as an alternative fuel [21,22]. Introduction of a ketogenic diet has shown pro-cognitive benefits in patients suffering from T2DM or AD. These benefits may arise from a reduction in neuronal firing rates during ketosis [23,24], increased ATP:ADP ratios [25], increased secretion of GABA due to substantial formation of acetyl-CoA [26], equilibration of the NAD:NADH, and curtailed production of free radicals, observed during ketogenic diet, together may help to temper neuronal hyperexcitability and its metabolic consequences [27,28].
In addition, KBs play a crucial role in protecting mitochondria from acute metabolic stress by preventing mitochondrial permeability transition (mPT) through their effects on intracellular calcium levels [29,30]. Furthermore, KBs can affect neuronal firing by promoting the opening of ATP sensitive K+ channels (K-ATP), thus reducing the cytosolic pool of ATP generated from glycolysis [24,30].
Exogenous KBs, such as D-β-hydroxy-butyrate (DĤb) ester, unlike a ketogenic diet, can raise KB levels in circulation without severe dietary restrictions [31,32]. It is unclear whether administration of exogenous KBs in the presence of normal plasma glucose levels maintains their neuroprotective effects, but recent studies show that DβHb provided to healthy adults increased their overall brain activity and stabilized functional networks [33,34].
Those observations led us to hypothesize that exogenous KB could be utilized to rescue neuronal metabolism during cerebral insulin resistance. To test these hypotheses, we first established a murine hippocampal model of acute insulin resistance (AIR) through the inhibition of GLUT4 by administration of Indinavir [35,36]. We then studied how neuron-specific insulin resistance affected hippocampal neurons in the CA3-CA1 circuit; a model circuit for studying learning and memory. We tested the effects of D-βHb during AIR to establish the therapeutic potential of exogenous KBs in hypoglycemic animals. In expectation of circuit-wide effects on synaptic and axonal function, we obtained field potential recordings in hippocampus of the treated mice and made patch-clamp recordings in hippocampal slices to assess the in vitro effects of AIR and D-βHb on the electrophysiological properties of CA1 pyramidal neurons and CA1 fast-spiking interneurons (FSI). Finally, we used computational modeling to establish a bridge between our electrophysiological results and the dysfunction of Na+/K+ ATPase, as the potential reason for the detrimental changes observed during AIR.
3. Results
Various paradigms of synaptic plasticity associated with learning and memory have been identified in the hippocampus. There is extensive documentation of long-term potentiation (LTP) and long-term depression (LTD), spike-timing-dependent plasticity, and EPSP-spike potentiation in many hippocampal circuits, making the hippocampus a classic system for studying neuroplasticity. Furthermore, the hippocampus’s simple cytoarchitecture makes it an ideal model system. In this study, we utilized Indinavir, a potent GLUT4 receptor blocker, to pharmacologically induce AIR in the hippocampal slices Our investigation focused on the stratum radiatum of the CA1, where numerous synapses are formed between Schaffer collaterals (SCO) and the apical dendrites of the pyramidal neurons.
3.1. Synaptic activity and LTP, but not fiber volleys (FV), are adversely affected by AIR and are not reversed by either 0.1 mM or 1 mM DβHb.
To test synaptic transmission within CA1 under a wide range of energetic demands, we measured the circuit’s response under 3 different stimulation paradigms. First, we applied mild physiological stimulation (not imposing high energetic demands [37,38]), consisting of paired stimulation applied at 25 Hz, repeated every 20 s over 30-60 trials [Fig.1A-F].
Figure 1: Synaptic activity and LTP, but not neuronal firing or PPR, decrease during paired stimulation under AIR, and do not recover during D-βHb administration.

A) Scatter plot of the fEPSP amplitudes evoked by paired-pulse stimulation of Schaffer collaterals. Each pair of circles is the average of 30-40 consecutive responses. Black bars represent mean ± SEM. Control fEPSPs in gray, AIR in red; 0.1 mM D-βHb+AIR in blue; 1 mM D-βHb+AIR in purple. Control: n=26 slices; AIR n=19; 0.1 mM D-βHb+AIR: n=20; 1 mM D-βHb+AIR: n=16. B) Scatter plot of the paired-pulse ratio (PPR) of fEPSP recorded in A. The labels, n, are identical to A). No significant differences (p=0.41, ANOVA). C) On top: Representative recording of a Control experiment where paired stimulation was applied for min. Each circle represents fEPSPs amplitude normalized to the average of the first 30 fEPSPs. The dashed orange line represents the mean amplitude at time −10 to 0 min. The black line represents the mean amplitude at +30 to +40 min. At the bottom: corresponding normalized PPR. D) fEPSPs waveforms recorded in C), showing LTP development. In gray, 30 fEPSP waveforms at t=0 ± 5 min and their average (black). In yellow, 30 fEPSP at time +30 ± 5min and their average (orange). Stimulation artifacts and FVs are removed. E) Scatter plot of LTPs triggered by paired stimulation. Control: n=13; AIR n=11; 0.1 mM D-βHb+AIR: n=12; 1 mM D-βHb+AIR: n=14. F) Scatter plot of the FV amplitudes recorded during A). The labels, n is identical to A). No significant differences (p=0.42, p=0.40; ANOVA). In all plots *p<0.05; **p<0.01; ***p<0.001.
During the AIR condition, we observed a strong decrease in fEPSP amplitudes; −39.23 ± 8.72% (1st stimulus) and −38.79 ± 8.93% (2nd stim.) compared to baseline (Fig. 1A). There was no recovery of amplitude when D-βHb was applied at low (0.1 mM) (+13.10 ± 6.64% and +14.07 ± 6.76%, Fig. 1A, p=0.34, p=0.30) or high (1 mM) concentrations [Fig.1A, −5.98 ± 6.13% and −5.81 ± 6.26%, p=0.88; p=0.56]. There were no effects of either AIR or D-βHb+AIR on PPR [Fig.1B, Control: 1.89; AIR: 1.92; 0.1 mM D-βHb+AIR: 2.00; 1 mM D-βHb+AIR: 1.88; p=0.54], suggesting that the facilitation at the synaptic sites remained largely unchanged.
When applied over sufficient time, our paired stimulation triggered LTP in the CA1, as evident by a gradual increase in the fEPSP amplitudes [Fig.1C, top], but no change in PPR [Fig.1C, bottom]. Therefore, we compared the magnitude of LTP between the groups by calculating a ratio of EPSP amplitudes during the first 10 min with amplitudes at 40 ± 10 min [Fig. 1D-F]. The control condition showed the strongest LTP ratio of 1.53 ± 0.13. LTP induction was almost abolished during AIR [Fig. 1E, 1.06 ± 0.06 ratio; −30.66 ± 9.42% reduction vs Control, p=0.018] and 0.1 mM D-βHb did not reverse this effect [Fig. 1E, 1.08 ± 0.08, +1.25 ± 6.61% AIR vs. D-βHb+AIR 0.1 mM, p > 0.99], 1 mM D-βHb, however, did [Fig. 1E, 1.39 ± 0.10, +21.19 ± 7.64% AIR vs. D-βHb+AIR 1 mM, p=0.044].
Moreover, we found no significant effects of either AIR or D-βHb+AIR on the amplitudes of the fiber volleys (FV) [Fig. 1F, Control: 1.03 ± 0.08 mV; AIR: 0.89 ± 0.07; D-βHb+AIR 0.1 mM: 0.85 ± 0.08; D-βHb+AIR 1 mM: 1.03 ± 0.12; p=0.40]. Next, we tested whether the circuit would behave differently under a stronger stimulation that imposed greater metabolic demands. We stimulated the Schaffer collaterals with 20 pulses applied at 25 Hz, with 20 s breaks between trials. To avoid the onset of LTP from skewing the results, we recorded all train stimulations at least 40 min after the paired stimulation paradigm.
All compared groups showed potentiation of fEPSP amplitudes during the train, reaching peak at stimuli 5-6. Thereafter, the amplitudes experienced a slight decline but remained potentiated [Fig. S1A,B]. Interestingly, throughout the entire stimulation period, the fEPSP amplitudes matched the differences we had seen during paired stimulation [Fig. S1B; at stimulus 5: −34.25 ± 11.80% Control vs. AIR, p=0.031; −40.54 ± 11.48% Control vs. 1 mM D-βHb+AIR, p=0.000044; +3.37 ± 7.80 AIR vs. 0.1 mM D-βHb+AIR, p = 0.99; −25.21 ± 7.36% AIR vs. 1 mM D-βHb+AIR, p=0.0054]. The FV amplitudes followed a different pattern, with potentiation at stimulus 2, followed by a consistent decline to approximately 70% of the initial amplitude [Fig. S1C]. Similar to paired stimulation, no statistically significant differences were observed between groups at any time point during the train [Fig. S1C; p=0.16 to 0.40].
Finally, we examined the hippocampal circuit under conditions of intense non-physiological stimulation. This involved applying long trains of 50 stimuli at 25 Hz, spanning across 60 trials with a 20-second break between each trial. The responses during this specific stimulation varied between trials, making it inappropriate to average them over the entire stimulation period. As a result, we represent the fEPSP and FV amplitudes as heat maps. In these heat maps, each point represents the group’s mean response to a single stimulus at a given time point.
Consistent with our previous results, we observed a period of fEPSP amplitude potentiation followed by a steady decline [Fig. S2A-D; at maximum, trial 4-6, stimuli 7-9: −34.81 ± 10.56% Control vs. AIR, p=0.0084; −48.27 ± 11.37% Control vs. 1 mM D-βHb+AIR, p=0.00065; +10.19 ± 11.39% AIR vs. 0.1mM D-βHb+AIR, p=0.59; −13.66 ± 9.08% AIR vs. 1mM D-βHb+AIR]. Synaptic depression first emerged during the final stimuli of the trains, roughly 3 min into the stimulation and became more prominent with each subsequent train application [Fig. S2A-D]. In general, fEPSP amplitudes of Controls and AIR groups differed substantially through the first 7 min of the recordings [Fig. S2A]. Control and 1 mM D-βHb+AIR groups also differed throughout most of the stimulation trains [Fig. S2B], but the 0.1 mM D-βHb+AIR and AIR alone groups scarcely differed except at the end of the trains [Fig. S2C]. The AIR and 0.1 mM D-βHb group fEPSPs generally remained comparable. FV remained statistically similar between all the groups, with almost the same response pattern [Fig. S2E-G], replicating the results from Fig. 1 and Fig. S1.
Taken together, these results strongly suggest that the effects of AIR and/or D-βHb could differ between cellular compartments (i.e., dendritic versus somatic), have different specificity for various cellular compartments, with synapses being particularly susceptible to metabolic challenges induced by GLUT4 inhibition.
3.2. AIR impairs axonal conduction speeds, but D-βHb treatment restores and enhances axonal conduction in a dose-dependent manner.
In the previous sections, we highlighted how AIR and D-βHb affected fEPSPs and FVs, finding a surprising lack of adverse effects of AIR on FV amplitudes, along with a noticeable reduction in fEPSP amplitudes; all further exacerbated by D-βHb. This difference suggests that AIR and/or D-βHb treatment might have differential effects on cellular compartments. We decided to investigate whether any other axonal properties changed during AIR and whether D-βHb remedied such changes.
We found that inducing AIR resulted in a strong reduction in the conduction velocities (CV) of SCOs [Fig. 2C; −13.07 ± 2.87% Control vs. AIR, p=0.027]. Addition of 0.1 mM D-βHb during AIR recovered CV back to Control levels [Fig. 2C; +12.43 ± 4.42% AIR vs. 0.1 mM D-βHb+AIR, p=0.034; +0.01 ± 0.07% Control vs. 0.1mM D-βHb+AIR, p=0.99] and 1 mM D-βHb significantly improved CV, surpassing the Control [Fig. 2C; +31.79 ± 4.59% for AIR vs. 1 mM D-βHb+AIR, p=4.2E-8; +19.10 ± 4.53% for Control vs. 1 mM D-βHb+AIR, p=0.00057].
Figure 2: Conduction velocity (CV) of Schaffer collaterals decreases under AIR are rescued by 0.1 mM D-βHb, and increase with 1mM D-βHb concentration.

A) Representative hippocampal slice during CV recording. B) Representative averaged FV waveforms, recorded in CA1. The stimulus onset time (t=0) is marked with a dashed line. Stimulus artifacts are removed. C) Conduction velocity scatter plots. Each colored circle represents a CV replicate. Control in gray, AIR in light red, 0.1 mM D-βHb+AIR in blue, and 1 mM D-βHb+AIR in purple. Data are shown as means ± SEM. *p<0.05; ***p<0.001, nested ANOVA, Control: m=62, n=24, N=14; AIR: m=65, n=22, N=13; 0.1 mM D-βHb+AIR: m=64, n=21, N=13; 1 mM D-βHb+AIR: m=50, n=14, N=10. D) Diagram of conduction velocity of an action potential propagating along an axon: distance along the axon (Δx) and the time required for an AP to pass that distance (t2-t1). V represents membrane potential. E) Model of a propagating action potential. Change in V(t) is shown at a longitudinal increment along the axon (x2) and at neighboring increments lying immediately ahead (x1) and behind (x3). Membrane potential at rest (VR) changes over time proportionally to the sum of differences between its value and those of the preceding and superseding increments. Upon reaching the threshold potential (VT), a spike occurs, and the membrane potential reaches peak value (VA). F-G) CV is modulated by VA and VR. Our computational model predicts declining CV due to reductions in the peak of action potentials (F) and/or hyperpolarization of the resting membrane potential (G). CV quantified as percentage, relative to Control.
To enable a better interpretation of these findings, we constructed a computational model of CV by employing an approximation of axonal cable theory (see Methods). Our model examined three processes that determine CV: resting membrane potential (VR), peak membrane potential (VA, the amplitude of AP overshoot), and activation threshold potential (VT, potential for Nav activation). We considered VT to be robust to various physiological conditions, given that it primarily depends on Nav channel type and gating kinetics [39]; therefore, we fixed VT at a constant value of −50 mV in our calculations. We varied VR between −90 and −60 mV and VA between 10 and 30 mV, both of which are considered physiological ranges. We normalized the results to a reference state where VR = −75 mV, VA = 30 mV, and VT = −50 mV. Modeling showed that a decrease in CV required either hyperpolarized resting membrane potential or decreased peak AP amplitude [Fig. 2F-G].
3.3. AIR adversely affects input timing, restored by high D-βHb concentrations.
Changes in the CV of the observed magnitude and resulting differences in input timing would likely desynchronize the hippocampal circuit. Therefore, we investigated whether latency of FVs changes during stimulation with 20 pulse trains.
In all treatment groups, the early FVs showed ~−0.2 ms improvement in latency. During the first 3 min, all experimental groups were comparable and exhibited the same pattern of latency changes. Afterwards, AIR and 0.1 mM D-βHb+AIR groups displayed progressively more prominent delays than Control, while Controls and 1 mM D-βHb+AIR groups remained comparable. During AIR, we observed the largest delays [Fig. 3E-F, Fig. 3I, L; at 15 min, stim. 9-11: +2.69 ± 0.84 ms, Control vs. AIR, p=0.0077; 2.64 ± 0.94 ms, AIR vs.1 mM D-βHb+AIR, p=0.020]. The escalating nature of the delays during the stimulation suggests that the detrimental effects of AIR are further exacerbated over longer periods of time.
Figure 3: Time delays in axonal firing during train stimulation are increased during AIR and are reversed by 1 mM DβHb.

A) Top: Representative example of the FVs recorded at the first stimulus in the control group during stimulation with 20 pulses applied at 25 Hz every 20 s, over 20 min. Bottom: FVs recorded in response to the last stimulus in each train. The FVs recorded at t = 0 are marked in black; the FVs recorded at t = +20 min, are marked in dark gray. B) - D) The same as A) in the AIR, 0.1 mM D-βHb+AIR and 1 mM D-βHb+AIR groups. E) Heat map of the mean, normalized FV peak latencies recorded in the Control group. Each trial is represented by a new row. The columns represent the time points of successive stimuli. Normalized latencies (0 ms) are color-coded in yellow. Delays (>0 ms) are color-coded in green-violet, and latency improvements (<0 ms) are coded in orange-brown. n=24, N=22. F) - H) The same as E) for AIR, n=16, N=15; 0.1 mM D-βb+AIR. n=17, N=15, 1 mM D-βb+AIR. n=14, N=10. I) Statistical comparison of latencies for the Control (E) and AIR (F) groups, performed on means of 3 stimuli x 2 trials blocks. p<0.05 in green, p<0.1 in yellow, p≥0.1 in white. J) The same as I) comparing Control (E) and 1 mM D-βHb+AIR (H) groups. No significant differences, p=0.067 to p=0.99. K) The same as I) comparing AIR (F) and 0.1 mM D-βHb+AIR (G) groups. No significant differences, p=0.41 to p=0.99. L) The same as I) comparing AIR (F) and 1 mM D-βHb+AIR (H) groups.
3.4. AIR increases membrane resistance (Rm) without affecting other intrinsic membrane properties of CA1 pyramidal neurons. D-βHb treatment under AIR does not rescue Rm to normal levels.
Results presented above [Fig. 2] indicate that AIR might negatively impact the membrane properties of hippocampal neurons, therefore we sought to identify the properties most vulnerable to AIR and test DβHb as a recovery agent. Based on dose-response results from the field potential studies, we selected 1 mM D-βHb for use in patch clamp experiments, testing the effects of AIR and D-βHb on intrinsic membrane properties of the CA1 pyramidal neurons (membrane resistance, Rm; membrane capacitance, Cm, and resting membrane potential, Vrest), and their spike thresholds. We found significantly increased Rm of the CA1 pyramidal neurons in the AIR group compared with Controls [Fig. 4B, +43.24 ± 3.73%, p=1.41E-07], which remained increased in the presence of 1 mM D-βHb [Fig. 4B, +27.86 ± 8.93%, p=0.030], while AIR and 1 mM D-βHb+AIR groups were similar (p=0.31). Interestingly, neither Cm [Fig. 4C, 114.25 ± 9.73, 115.52 ± 9.26, 132.20 ± 9.46, Control, AIR, D-βHb+AIR, respectively, p=0.32], spike threshold [Fig. 4E, −46.31 ± 0.76, −44.53 ± 0.80, 44.64 ± 0.73, p=0.17] nor Vrest. [Fig. 4F, −67.26 ± 1.17, −63.57 ± 1.38, −65.11 ± 0.99, p=0.093] were significantly affected by either AIR or D-βHb+AIR treatments.
Figure 4: Membrane resistance (Rm) increases during AIR and is not reversed by D-βHb. Other intrinsic properties did not change under either condition.

A) On the left: A CA1 pyramidal neuron recorded in whole-cell patch clamp mode (outlined in red) with resistances and capacitance within the cell. On the right: A simple electric circuit diagram of the cell with the same resistances. B) Scatter plots of CA1 pyramidal neuron Rm, compared between the experimental groups. Each colored circle represents a value recorded in a single cell. Control in gray, AIR in light red, 1 mM D-βHb+AIR in purple. Vertical black bars represent mean ± SEM. C) Scatter plots of CA1 pyramidal neurons Cm, compared between the experimental groups. The plots and coloring are identical to B). No significant differences (p=0.32). D) Representative example of a 300pA ramp current injection performed in a Control CA1 pyramidal neuron. The dashed line marks the onset (spike threshold) of the first AP. The inset shows the magnified AP with the arrow pointing to the onset. E) Scatter plots of CA1 pyramidal neuron spike thresholds, compared between the experimental groups. The plots are identical to B). No significant differences (p=0.17). F) Scatter plots of CA1 pyramidal neurons RMP (Vrest), compared between the experimental groups. The plots and coloring are identical to B). No significant differences (p=0.09). Control n=28, N=22; AIR n=25, N=17; 1 mM D-βHb+AIR; n=30, N=16. In black, *p<0.05; **p<0.01; ***p<0.001; in gray *p<0.1.3.3 AIR adversely affects input timing, restored by high D-βHb concentrations.
3.5. AIR increases the frequency of spontaneous vesicular release at CA1 synapses (sEPSCs) and mildly increases sEPSC amplitudes. D-βHb does not reverse the increase in frequency but mildly decreases sEPSC amplitudes.
Recovering the membrane back to resting potential during synaptic transmission is the most energy-consuming neuronal process (~% of total ATP produced [40]), therefore, we tested for changes in the frequency and magnitude of quantal EPSCs that might explain the ~35% decrease in fEPSP amplitudes reported above (Fig.1-3).
AIR significantly increased the frequency of sEPSCs, nearly doubling the number of events compared with the Control [Fig. 5A,B; 0.53 vs. 0.96 Hz, +0.42 ± 0.12 Hz, p=0.0031]. Interestingly, D-βHb failed to rectify the increased qEPSC frequency [Fig. 5A,B; D-βHb+AIR 0.93 vs. AIR 0.96 Hz, 0.021 ± 0.15 Hz, p=0.99; D-βHb+AIR vs. Control, +0.40 ± 0.12 Hz, p=0.0091]. However, the amplitude, rise-time, and sEPSCs decay time showed no significant differences between the groups [Fig. 5C-E].
Figure 5: AIR increased sEPSC frequency without altering sEPSC properties and D-βHb (1 mM) does not reverse this effect.

A) Representative examples of the first 2 min of sEPSC recorded in CA1 pyramidal neurons, voltage clamped at Vh = −70 mV (Control in gray; AIR in red; 1.0 mM D-βHb+AIR in purple). Below: averaged sEPSC waveforms from individual cells (light color) and the group mean (full color). B) Scatter plots of the sEPSCs frequency recorded in CA1 pyramidal neurons, compared between the experimental groups. Each colored circle represents frequency recorded in a single cell. Labels as in A) Vertical black bars represent mean ± SEM. *p<0.05. C) Scatter plots of the sEPSC 20-80% rise times, as in B). No significant differences (p=0.51). D) Scatter plots of the sEPSC decay tau times, as in B). No significant differences (p=0.98). E) Scatter plots of the sEPSC amplitudes, as in B). No significant differences (p=0.21). Control n=19, N=19; AIR n=19, N=14; 1 mM D-βHb+AIR, n=23, N=16. **p<0.01.
Given the change in Rm, the lack of significant changes in the amplitudes is surprising. Several studies utilizing different metabolic challenges, such as food deprivation [41] and inhibition of glycolysis [42], showed an increase in mEPSC/sEPSC amplitudes, therefore, we decided to investigate our sEPSCs in more detail. In most central synapses, the distribution of mEPSC/sEPSC amplitudes shows several peaks related to simultaneous release of multiple quanta of neurotransmitter [43], with each subsequent peak linked to an increasing number of concurrent sEPSCs. Capitalizing on the multiplicative nature of this process, we assumed that small changes in amplitudes [Fig. 5E] would scale up. We detected 2-4 distinct peaks in ~85% of the analyzed cells [Fig. S3A-C], with no differences between the groups in the number of cells with multi-peak distributions [Fig. S3C; 89.5% in Control; 84.2% in AIR, 87.5% in D-βHb+AIR]. In line with our initial assumptions, the scaled amplitudes revealed differences between the groups [Fig. S3E]: AIR group had significantly higher amplitudes than D-βHb+AIR [p1: p=0.048; p2: p=0.0054; p3: p=0.0038; p4: p=0.021] and also tested as different from Controls at peak 3 [p=0.007]. Control and D-βHb+AIR groups did not show significant differences at any point [Fig. S3E], despite the same sEPSC frequency as AIR.
We further validated those findings by comparing the normalized amplitude distributions [Fig. S3F-I]. Within the range correlating to peak 2 – peak 4 (sEPSC of ~20pA or more), the AIR group had a significantly higher number of large events (15.19% of total) when compared to Control (7.65%) and D-βHb+AIR (9.48%) [Fig. S3F-I], while Control and D-βHb+AIR groups showed little consistent difference. This result suggests that during AIR, the elevated frequency of events increased the probability of multi-vesicular events occurring, further exacerbated by the ~20% increase in Rm. On the other hand, D-βHb+AIR produced a distribution resembling Control, as if the vesicles had lower glutamate content, as suggested by literature [44,45].
3.6. AIR slightly changes the firing pattern of hippocampal pyramidal neurons and causes increased membrane depolarization with input. Those effects are reversed by D-βHb, which also increases AP amplitudes.
The action potential generation and subsequent recovery of Vm to resting levels entail a large part of the energy budget of pyramidal neurons (~41% of all produced ATP [40]), therefore, we investigated whether pyramidal neurons retain their input-output curves (I-O curve) under AIR and how D-βHb alleviates AIR effects.
First, we focused on the firing frequency [Fig. 6A]. Cells under AIR showed increased firing at lower current injections than D-βHb+AIR [Fig. 6B, 75–150 pA, p=0.0038 to 0.050], but with the same maximal firing rate. These results suggest that the cells activate earlier under AIR than in the Control or D-βHb+AIR conditions. To test this further, we measured the maximum depolarization attained by each neuron during the final 5 ms of each current injection step [Fig. 6D]. AIR neurons had slightly higher depolarization with each injection step compared to Controls [Fig. 6E,F; p=0.0027 to 0.048] or D-βHb+AIR [Fig. 6E,F; p=0.0029 to 0.042].
Figure 6: Neuronal firing is mildly affected by AIR and reversed by DβHb, with DβHb increasing AP overshoot amplitudes.
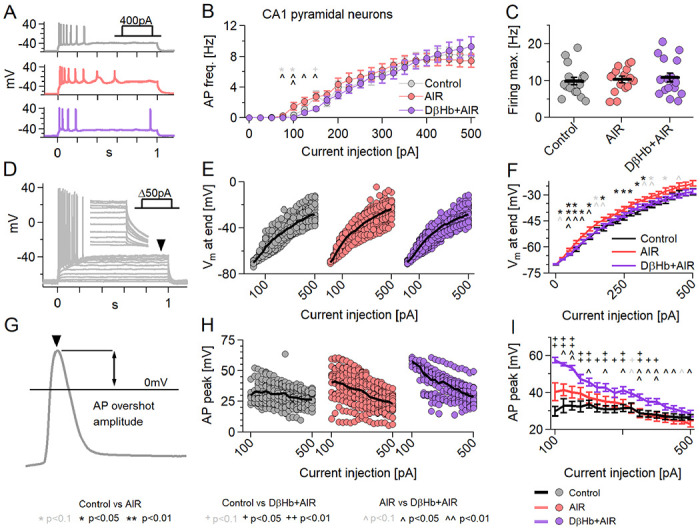
A) Representative examples of responses to 400 pA square current injections into Control (gray), AIR (red), and 1 mM D-βHb+AIR (purple) CA1 pyramidal neurons, held at Vh=−70 mV. B) I-O curves of CA1 pyramidal neurons receiving 21, Δ+25pA injections (max. 0 pA; Vh=−70 mV). Circles represent group mean ± SEM. *Control vs. AIR, + Control vs. 1 mM D-βHb+AIR, ^ AIR vs. 1 mM D-βHb+AIR. *,+,^ in black p<0.05; **,++,^^ in black p<0.01; *,+,^ in gray p<0.1; ANOVAC) Maximum firing rate of the pyramidal neurons in B). Circles represent single neurons; black bars represent group means ± SEM. No significant differences (p=0.778, ANOVA). D) Example of responses to Δ+pA square current injections into a control pyramidal neuron. The arrowhead and inset mark the final membrane depolarization (Vm, last 5 ms of the step). E) Vm at the current injections from B). F) Group means ± SEM of Vm from E). Statistical tests and labels are identical to B). Control n=23, N=20; AIR n=20, N=14; 1 mM D-βb+AIR n=22, N=16. G) Example of AP overshoot measurement. Black line marks Vm= 0 mV. H) Averaged AP overshoot amplitudes from B). I) Group means ± SEM of overshoot amplitudes from H). Statistical tests and labels are identical to B). In all comparisons Control n=23, N=20; AIR n=20, N=14; 1 mM D-βHb+AIR, n=22, N=16.
Next, we investigated the averages of AP peak amplitudes [Fig. 6G], per step [Fig. 6H]. Surprisingly, D-βHb+AIR neurons showed the largest amplitudes, consistently elevated throughout the injections, when compared to Controls or AIR, while AIR neurons remained comparable to Controls [Fig. 6I, p=0.0017 to 0.036, AIR vs. Control; p=0.0093 to 0.044, AIR vs. 1 mM D-βHb+AIR].
3.7. AIR increases AP decay time, while D-βHb accelerates the AP rise without reversing the AIR-induced slower AP decay.
We next proceeded to investigate other properties of the APs: decay time (from the overshoot peak to 0 mV) [Fig. 7A-C] and rise time (from 0 mV to the overshoot peak) [Fig. 7D-F]. We found that Control neurons had significantly lower average AP decay times when compared to AIR [Fig. 7B,C; p=0.047 to 0.0019] and D-βHb+AIR conditions [Fig. 7B,C; p=0.041 to 0.0023], across almost all current injection steps. AIR and D-βHb+AIR cells did not significantly differ at any point [Fig. 7B,C; p=0.32 to 0.99]. D-βHb+AIR cells demonstrated faster rise times compared to Control [Fig. 7E,F; p=0.000094 to 0.0025] and AIR cells [Fig. 6E,F; p=0.047 to 0.0052] in the majority of the injection steps. Interestingly, AIR cells exhibited slightly faster AP rise times exclusively at the lowest current injections [Fig. 4 N,O; p=0.012 to 0.035]. These slightly faster APs were likely due to earlier activation of the cells, as shown in [Fig. 6B-C, E-F]. Fig. 7Fig. 6
Figure 7: AP decay times change during AIR and DβHb + AIR but DβHb + AIR APs have faster rise times.

A) Example of AP decay time measurement. Black line marks Vm= 0mV. B) Mean AP decay times for the current injections. Circles represent averaged amplitudes in single neurons. Black lines represent group means. C) Group means ± SEM of AP decay times from B). Statistical tests and labels are identical to B). D) Example of AP decay time measurement. Black line marks Vm= 0mV. E) Averaged AP rise times for the current injections in B). Circles represent averaged amplitudes in single neurons. Black lines represent group means. F) Group means ± SEM of AP rise times from B). Statistical tests and labels are identical to B). G) An example of the AP IEI measurement. The double arrow marks the latency between AP peaks. H) Averaged IEI recorded during the injections in B). Each point represents the average of IEIs recorded at each step in a single neuron. The black line represents the group mean. I) Group means ± SEM of averaged IEI. *,+,^ p<0.05; ANOVA. In all comparisons Control n=23, N=20; AIR n=20, N=14; 1 mM D-βHb+AIR, n=22, N=16.
Finally, as we did not observe significant differences in firing frequency among the groups [Fig. 6B,C], we examined variations in the timing of AP firing based on the intervals between consecutive APs (AP Inter Event Interval, AP IEI) [Fig. 7G], but we found no clear changes in the AP IEI [Fig. 7H,I].
3.8. AP adaptation is impaired during AIR, with AP amplitudes showing steeper declines, and is not reversed by D-βHb.
The amplitudes of the AP overshoot, decays, and rise times demonstrated steeper slopes in the AIR and D-βHb+AIR groups. The properties of APs during neuronal firing undergo dynamic changes, commonly referred to as spike adaptation or spike frequency adaptation [46] and in our experiments, we see evidence that GLUT4 antagonism intensified these changes.
To test this observation, we compared peak amplitudes of the first APs [Fig. S4A-D], the averaged APs [Fig. 6G-I, Fig. S4E] and the final APs during current injections [Fig. S4F]. The first APs represents neuronal firing after rest (10s between subsequent injections), while the average and final AP – after energy expenditure. As expected, the first APs displayed minimal changes in amplitude during injections [Fig. S4D], with D-βHb+AIR exhibiting consistently larger amplitudes than the Control. AIR, also showing elevated amplitudes, did not differ significantly from either the Control or D-βHb+AIR groups. For the final APs, we observed a sharp decline in amplitude scaling with injected current [Fig. S4F] in both AIR and D-βHb+AIR groups. Control showed lower amplitudes than D-βHb+AIR at 100-300pA injections (p=0.040 to p=6E-05) and remained comparable to AIR up until the very last, large, 400-0pA injections (p=0.032 to p=0.0011), when AIR APs significantly declined [Fig. S4F]. AIR and D-βHb+AIR remained consistently different.
Linear fits to the average AP amplitudes revealed significantly steeper declines [Fig. S4G,H; p=0.00039, AIR vs Control; p=0.0012, D-βHb+AIR vs Control], indicating that prolonged neuronal activity is heavily affected by AIR.
3.9. Changes in AP decay and rise times translate into differences in the widths of FVs.
In our initial analysis of the field potential (FV) data, our focus was solely on the amplitudes of the biological signals. Consistently, we found no significant changes in the FV amplitudes [Fig. 1F, 2C, 3E-G], which supports our findings on AP overshoot amplitudes [Fig. 6G-I] (with the exception of D-βHb+AIR group). Based on these results, we further investigated whether the changes in AP decay and/or rise times also corresponded to changes in FV widths during train stimulation [Fig. S5].
Consistent with our findings on AP decay/rise times [Fig. 7A-F], AIR consistently exhibited the widest FVs throughout the stimulation followed by the Control group, with 1mM D-βHb+AIR FVs as the narrowest [Fig. S5D]. We found differences between AIR and Control (p=3.94E-06 to 0.049), and AIR and 1mM D-βHb+AIR (p=4.04E-05 to 0.046) throughout the whole stimulation period, while Control and 1mM D-βHb+AIR were not different (p=0.079 to 0.99), supporting our AP decay/rise time findings [Fig. 6G-I].
3.10. With abolished GABAergic inhibition, AIR strongly decreased firing rates of CA1 pyramidal neurons and fast-spiking interneurons (FSI), which were not rescued by DβHb.
As the next step, we investigated the properties of CA1 pyramidal neurons and FSIs after pharmacologically abolishing AMPA/kainate excitatory (NBQX) and fast GABAergic inhibitory (gabazine) transmission [Fig. S6].
We first recorded the firing frequency of CA1 pyramidal neurons [Fig. S6A,B] at different activation levels [Fig. S6C,D], finding that AIR prominently decreased their firing rate, when compared with Control [Fig. S6C, p=0.024 to 0.0053]. Surprisingly, the application of 1 mM D-βHb during AIR resulted in a stronger reduction in firing rate, when compared with Control [Fig. S6E, p=0.000027 to 0.035]. AIR and D-βHb+AIR results remained similar throughout the injections [Fig. S6E]. However, all groups differed significantly with respect to the maximum firing rate, with Control having the highest (31.03 ± 3.43) and D-βHb+AIR having the lowest (13.17 ± 2.05) firing rates.
Next, we examined CA1 FSIs using the same drug conditions [Fig. S6E,F]: AIR decreased the firing rate of FSIs compared with Control [Fig. S6G, p=0.012 to 0.048], not reversed by D-βHb [Fig. 5S6H, p=0.00017 to 0.035]. As for pyramidal neurons, the FSIs for AIR and D-βHb+AIR groups remained mostly comparable [Fig. S6G]. None of the groups differed in the maximum firing rate [Fig. S6H, p=0.068].
3.11. Hodgkin-Huxley model predicts that impairments in Na+/K+ ATPase activity result in more depolarized Vrest, lower AP overshoot (Va), and increased neuronal firing.
Here we aim to explain the previous observations of dysregulated firing dynamics in response to AIR. We hypothesize that Indinavir restricts the energy supply of neurons and thus impairs the neuronal Na+/K+ ATPase. To test this hypothesis, we developed a computational model of an isolated CA1 neuron in the absence of inhibition and toggled Na+/K+ ATPase activity through the enzyme kinetic parameter vATPasemax (see supplementary methods, Fig. S7). Figs. S7A,B (at two different temporal scales) shows the predicted changes in neuronal firing dynamics when the Na+/K+ ATPase is impaired. Such an impairment predicts the depolarization of the resting membrane potential (Fig. S7C) and is consistent with trends of our I/O curves (Fig. 4F). Furthermore, this modeled depolarization reduces the magnitude of membrane potential shift required for an action potential. Consequently, the model predicts increased firing frequency (Fig. S7D). Finally, the model predicts that the Na+/K+ ATPase should act more slowly to recover ion gradients after an AP, causing the amplitude of subsequent APs to decline progressively with time (Figs. S7E,F). Such an amplitude decrease is consistent with the trends in measured conduction velocity (Fig. 2).
4. Discussion
Central insulin resistance impairs cognitive performance and promotes hippocampal neurodegeneration and memory loss [47,48]. Our present results highlight the adverse effects of GLUT4 inhibition on synaptic function and LTP, in accord with findings that acute and chronic blockade of GLUT4 are detrimental to performance on hippocampal-mediated memory tasks, thus suggesting that GLUT4 is critical for acquisition and consolidation of memory [8,13]. Memory formation is promoting GLUT4 incorporation into the membrane, resulting in increased glucose flux into neurons [13,49]. This process enables increased metabolic support during high demand. Crucially, most GLUT4s localize in the perikaryon [50,51], likely in close proximity to axo-somatic synapses, which are associated with synaptic plasticity and long-term memory (LTM). Indeed, prolonged blockade of the GLUT4 glucose transporter in the brain results in impaired formation of LTM [13] and decreased levels of hippocampal BDNF [52]. Moreover, hypoglycemia negatively affects memory performance and cognition [53,54, however, see 13]
In this study, we found that AIR increased the frequency of sEPSC events, in line with the previously published data showing similar increases during food deprivation [42], inhibition of glycolysis [41] and ischemia [55], likely due to hypoglycemia-driven Ca2+ accumulation at the presynapse [41,55]. The same studies suggest that presynaptic glycolysis is essential for maintaining synaptic transmission, even at a low frequency, and that mitochondrial respiration cannot substitute it. Normally, glycolysis meets approximately one-third of the energy budget at presynaptic terminals, thus sustaining low-frequency transmission. In addition, impaired glycolysis leads to slower, broader, and smaller AP waveforms, and a depolarization of the resting membrane potential [42]; results which are consistent with ours. Interestingly, D-βHb+AIR treatment, which showed the same increase in the frequency of sEPSCs as AIR, had the lowest sEPSC amplitudes [Fig. S3E]. This result suggests a reduction in the quantal content of synaptic vesicles and is consistent with reports of lowered conversion of glutamate to aspartate under ketosis [44,45].
Although the results of our computational models are consistent with the observed changes in resting state membrane potential and firing frequency during AIR, there were seemingly contradicting results with respect to AP peak amplitude. Specifically, we measured no significant changes to average AP overshoot amplitudes during AIR, our model predicted a decrease due to energy constraints. In the model we did not consider a progressive decline in available energy, which is suggested by the steeper AP adaptation [Fig. S4]. Moreover, neurons may compensate for the energetic limitations by increasing Rm (as reported in 42), while our model assumes that Rm remains unchanged. When those factors are accounted for, the model and experimental data agree: during periods of extensive activity, energy reserves in neurons are progressively depleting and, provided that the neuron fired multiple APs in short succession, the negative effects of hypoglycemia overcome compensatory Rm increases and AP amplitudes decline below Control levels [Fig. S4]. Furthermore, we used the maximal Na+/K+ ATPase activity as a surrogate for ATP availability, and we relied on literature estimates of kinetic and thermodynamic rate constants. A more direct approach might consider ATP concentration as an input variable, were such experimental data available.
Our model aiming to establish the underlying causes of the changes in conduction velocity investigated resting membrane potential (Vrest) and AP peak amplitude as two critical factors. The model explains CV recovery and increases during D-βHb+AIR treatment, since the measured AP peak amplitudes remain consistently elevated and Vrest changes are negligible, but fails for AIR alone, where AP amplitudes remain comparable to Control and Vrest tends to depolarize. This discrepancy might reflect the simplistic nature of our model, which neglects complex ion dynamics, altered membrane permeability or synaptic processes.
The ketogenic diet has served for decades to treat epilepsy and has an emerging wide range of therapeutic applications [30]. Possible mechanisms include modulation of ATP-sensitive K+ channels (KATP), free fatty acid receptor 3 (FFAR3/ GPR41) activation, promotion of GABA synthesis, and epigenetic modifications [56]. The action of D-βHb in modulating potassium flux through KATP channels may be most relevant in the present context [57], since KATP activation results in decreased neuronal firing rates [58,59] and prevented picrotoxin-induced epileptiform activity in the hippocampus [60] and may explain present findings that firing rates and membrane depolarization remained at control levels in the presence of 1 mM D-βHb during AIR (despite elevated Rm).
D-βHb is a precursor of glutamine, which promotes increased GABA levels, as seen in studies of the ketogenic diet in clinical epilepsy [61]. D-βHb also directs glutamate towards GABA production by reducing the conversion of glutamate to aspartate [44,45]. In addition, D-βHb has a direct effect on the vesicular glutamate transporter VGLUT2, by inhibiting Cl− dependent glutamate uptake. VGLUT2 is critical for glutamate excitatory transmission in CA3-CA1 and its conditional VGLUT2 knock-out results in impaired spatial learning and reduced LTP [62]. Together with the increased formation of GABA and/or GABA derivatives, VGLUT2 inhibition could further reduce glutamate excitatory transmission [63].
The present results have important implications for the treatment of conditions such as type 2 diabetes, metabolic syndrome, and seizure disorders that involve insulin hyposensitivity. According to NIH estimates, ~30% of Americans today display symptoms of prediabetes, a percentage expected to increase in the nearby future. Together with the longer average lifespans and demographic changes leading to an expansion in the aging population, obtaining insights into the outcomes of metabolic dysfunction and its potential treatment becomes a more pressing matter each day. Our results provide a unique insight into the potential use of KBs as an inexpensive and relatively risk-free treatment for metabolic disorders and insulin resistance. Medical researchers can use this information to discover new targets for treatment and develop therapies that are more effective. Further research in this area could inform novel treatment approaches that address the complex effects of metabolic disorders on brain health.
5. Materials and Methods
5. 0. Mice
Breeding pairs of transgenic GAD2Cre/GCamp5gTdTomato, Thy1/GCamp6fTdTomato, and C57BL/6J mice were originally obtained from The Jackson Laboratory (stocks 010802, 024477, 024339) or Charles River Laboratories and bred in-house under standard conditions of 12-12 hours light-dark cycle, with food and water available ad libitum. The Institutional Animal Care and Use Committee approved all experiments. Mice of both sexes aged between 30 and 60 days (P30-P60) were used in all experiments.
5.1. Electrophysiology – slice preparation.
Coronal brain slices containing the hippocampus were used in all recordings. Mice were deeply anesthetized with a mixture of isoflurane and air (3% v/v), decapitated, and their brains extracted and cut using a Leica VT1200S vibratome into 300 μm-thick slices in ice-cold ACSF solution containing (in mM): 230 sucrose, 1 KCl, 0.5 CaCl2, 10 MgSO4, NaHCO3, 1.25 NaH2PO4, 0.04 Na-ascorbate, and 10 glucose; 310 ± 5 mOsm, pH adjusted to 7.35 ± 5 with HCl, and, gassed with carbogen (95% O2, 5% CO2) for at least 30 min before use. The slices were then transferred to a Zbicz-type BSC-PC submerged slice chamber (Harvard Apparatus, USA) filled with artificial cerebrospinal fluid (ACSF) containing (in mM): 124 NaCl, 2.5 KCl, 2 CaCl2, 2 MgSO4, 26 NaHCO3, 1.25 NaH2PO4, 0.04 Na-ascorbate, and 10 glucose; 305 ± 5 mOsm/kg, pH adjusted to 7.35 ± 0.05 mOsm/kg, gassed with carbogen and prewarmed to 32 °C. The slices were allowed to recover in the warm ACSF for 15 min, whereupon the heating was switched off, and the chamber gradually cooled down to room temperature (RT; 23 ± 2 °C) over 45 min.
Following the recovery, individual slices were transferred to another BSC-PC chamber filled with gassed ACSF and a mixture of drugs (Indinavir 0.1 mM; Indinavir 0.1 mM and either 0.1- or 1-mM DβHb), where they were incubated for 60 ± 15 min. Next, the slices were transferred to a submerged recording chamber mounted on an upright microscope stage (E600FN, Nikon, Japan) equipped with infrared differential interference contrast (IR-DIC) filters. The slices were kept at RT and superfused continuously with gassed ACSF delivered by a peristaltic pump at a steady rate of ~2 ml/min.
5.2. Field potential recordings.
Evoked field potential responses were triggered with an isolated pulse stimulator (Model 2100, AM Systems, USA) using a CBARC75 concentric bipolar electrode (FHC, USA) placed between CA3 and CA1. The recording electrode was placed 300 ± μm away for the recordings of CA1 FV and fEPSP. During the recordings of axonal conduction velocity (CV), the electrodes were 300 to 900 ± 50 μm apart, and up to 6 different sites were recorded. All stimulation paradigms were applied every 20 s as biphasic rectangular pulses of 240 μs total duration, 50 ± 5 V strength (~60 ± 10% of the maximum response size), and a between-stimuli frequency of 25 Hz (40 ms).
During non-CV recordings, three different types of stimulation were applied: paired, 20 stimuli, and stimuli trains, repeated every 20 s for a total period of 20 min in each case. After each train stimulation, the slice was allowed to recover for 20 min under paired stimulation.
Recording pipettes were pulled from borosilicate glass capillaries (ID 0.86, OD 1.5, BF1-86-10, Sutter Instruments, USA) on a vertical PC-100 puller (Narishige, Japan) and filled with 6% NaCl (in H2O, 1.02 M). Only pipettes with a resistance of 0.2-1.0 MΩ were used. Recording pipettes were allowed to rest at the recording site for 10 min to equilibrate before the start of a recording. All recorded signals were acquired using a MultiClamp 700B amplifier (Molecular Devices, USA), Bessel-filtered at 2 kHz, and digitized at a sampling frequency of 10 kHz using an Axon Digidata 1440A digitizer (Molecular Devices, USA).
5.3. Whole-cell patch-clamp recordings.
Pyramidal neurons of the hippocampal CA1 region were selected for recordings based on their general morphology and position within the pyramidal layer. As an additional verification, recorded cells were screened for GAD2+-driven tdTomato fluorescence. Cells that displayed any level of tdTomato fluorescence were rejected.
Patch pipettes were pulled from borosilicate glass capillaries (BF1-86-10, Sutter Instruments, USA) on a vertical puller (PC100, Narishige, Japan). Pipettes had a resistance of 6–8 MΩ when filled with an internal solution containing (in mM): 136 K-gluconate, 4 Na2ATP, 2 MgCl2, 0.2 EGTA, 10 HEPES, 4 KCl, 7 di-triphospho-creatine, 0.3 Na3GTP; 280–290 ± 5 mOsm/kg, titrated to pH 7.3 ± 0.05 with KOH.
During recordings in voltage clamp (VC), cells were clamped at the holding potential (Vh) = −70 mV with a Multiclamp 700B amplifier (Molecular Devices, USA). Liquid junction potential was not corrected. All cells were compensated for pipette capacitance. During VC experiments, series resistance (Rs) was not compensated. During recordings in current clamp (CC), a holding current was applied to the cells to maintain membrane potential at Vh = −70 ± 3 mV. All cells were bridge-balanced at % of Rs. Cell capacitance was not compensated in either mode. Immediately after establishing the whole-cell configuration, the cell was kept in I=0 mode to monitor the resting membrane potential (Vrest) over a period of ~120 to 180 s. The average value of Vrest over this period is reported as the neuron’s Vrest.
During the recordings, every 5 to 10 min a series of 10 square voltage steps of −10 mV was apllied to monitor changes in the Rs (Vh = −70 mV). All whole-cell currents in response to voltage steps were low-pass filtered at 10 kHz with a Bessel filter and sampled at 20 kHz frequency (Axon Digidata 15B, Molecular Devices, USA). For inclusion in the final dataset, recordings had to meet 3 criteria: 1) Rs after any recorded protocol could not increase by more than 20% compared to the Rs pre-protocol; 2) Rs during any of the recorded protocols could not exceed 30 MΩ; 3) the offset drift by the end of the recordings could not be higher than ± 5 mV.
Spontaneous synaptic currents (sEPSC) were recorded in VC at Vh = −70 mV in a single, continuous sweep for 10 to 15 min, low-pass filtered at 2 kHz with a Bessel filter and sampled at 10 kHz frequency.
To establish the spiking threshold, each neuron was current-clamped at Vh ~ −70 ± 3 mV and subject to a ramp-shaped 200-0 pA current injection with a 1s duration. The spike threshold was determined as the membrane potential at which the first fully developed action potential (AP) was visible. Each ramp injection was repeated 10x, and the averaged threshold value is reported for the neuron.
To establish the firing properties of recorded neurons, each neuron was current-clamped as described previously and subjected to a square-shaped series of current injections with 25pA Δ per step to a maximum of 0 pA, with each step of 1 s duration and inter-step interval (ISI) of 10 s. If necessary, the holding current was corrected during the protocol to maintain Vh at ~ −70 ± 3 mV. During analyses, only fully-formed APs that crossed the 0 mV threshold were included. All data acquisition was performed using pCLAMP software (Molecular Devices, USA).
5.4. Field potential recordings – analyses.
During field potential recordings, the biologically relevant signals are superimposed onto the decaying stimulation artifact. To isolate the stimulation artifacts for subtraction from the signal at the end of each recording, we applied 1 μM TTX to the slice and recorded 30 repetitions of each stimulation paradigm were recorded in the presence of TTX.
Recorded traces were analyzed in IgorPro (Wavemetrics, USA). First, an average of the traces recorded with TTX was subtracted from each trace. The traces were then filtered using a rolling five-point average to remove noise, and the baseline was adjusted to the 1 s period before the stimuli. The amplitudes and latencies of FVs and fEPSPs were measured at their peaks. Amplitudes were measured against 0 mV baselines, while latencies were measured against the onset of the stimulation. During high-frequency stimulation, cells often do not have sufficient time to fully recover their membrane potentials before the next stimulus is applied, leading to a gradual shift in the baseline, therefore, as a correction, we subtracted the median voltage at +35 to +40 ms after each stimulus from each FV or fEPSP amplitude. FV widths were measured at the peaks of positive deflections in the waveform, first after the artifact subtraction and second directly before fEPSP.
All traces were screened for occasional electrical interference and the affected data points were manually removed from the analysis.
To measure axonal conduction velocity (CV), we calculated the time shift (Δt, ms) between the stimulus start and the time of the FV peak. Next, the linear distance between the recording site and the stimulation electrode was divided by the measured Δt. We recorded CV at 3-6 sites over a span of 300-900 μm, with ~1 μm steps between sites. Each CV recording site served as a replicate for statistical analysis (see Statistics). During CV recording, stimulation remained stationary, and the sites were recorded in randomized order.
To calculate changes in FV peak latency during train stimulations the median Δt at stimulus 1-3 of traces 1-2 (ΔtN) was subtracted from each Δt (normalization).
5.5. Field potential recordings – data visualization.
For constructing “heatmap” visualizations of the data, numerical matrices of FV or fEPSP amplitude means were built for each of the four treatment groups. In every matrix each new row represented responses during each subsequent train. Then, two additional columns were added at the end as a color normalization in the heatmap (0, 1 mV), and the matrix was saved as a text file. Afterwards, the text files were imported as text images to ImageJ (NIH, USA), converted from 32- to 16-bit, saved as TIFF images and imported to IgorPro (Wavemetrics, USA), whereupon their look-up table (LUT) was changed from “Grays” to “Spectrum.”
The heat maps for latency changes during train stimulation were generated similarly but using different normalizations: 20 pulse train recordings had normalization boundaries of −1 and +1 ms.
5.6. Whole-cell patch-clamp – sEPSC analyses.
sEPSCs detection was automatized using a convolution-based algorithm in Fbrain 3.03 [64], a customized program running under IgorPro 6 (WaveMetrics, Lake Oswego, USA), kindly provided by the Peter Jonas Lab (IST, Klosterneuburg, Austria). Recorded traces were smoothed by subtracting a 20-term polynomial fitted to the trace and digital Notch (60 ± 0.5 Hz) filtered in FBrain before the analysis. The trace subject to convolution was passed through a digital band-pass filter at 0.001 to 200 Hz. The event detection template had a rise-time time constant τ = 1.5 ms, a decay τ = 6 ms, and an amplitude of −10 pA. The event detection threshold (θ) was set to 4.2 times the standard deviation of a Gaussian function fitted to the all-point histogram of the convolved trace. All events detected by the algorithm were inspected visually. Those that clearly did not show fast rise time and exponential decay kinetics of a typical hippocampal EPSC were removed by hand. Only cells with a rejection ratio lower than 20% were included in the analysis. The analysis was performed using custom-written macros provided by Dr. Maria Kukley (Achucarro Basque Center for Neuroscience, Bilbao, Spain).
Afterwards, individual sEPSC waveforms were extracted from the trace. Each waveform consisted of a 5 ms segment of the trace before the onset of the sEPSC and a 50 ms piece after the onset to capture the full decay of the waveform. Before averaging, all sEPSCs were baseline-adjusted to the pre-onset 5 ms. The peak amplitude, 20-80% onset-to-peak rise time, and decay constant τ were measured for the averaged sEPSC. The decay T was calculated from the monoexponential fit from the sEPSC peak to the final ms of the averaged waveform.
To avoid selection bias during sEPSC extraction, we repeated the Fbrain analysis at least twice in each cell and averaged the results.
5.7. Statistics.
All data acquisitions were made in randomized sequence, with a maximum of 2 slices (field potential recordings) or 2 cells (patch clamp recordings) from a single animal per experimental condition to avoid pseudoreplication. All experimental groups had comparable distributions of ages (close to P50 ± 6) and sex. The order of slice pre-incubation with drugs was randomized for each animal. The numbers of slices or cells and animals used in each experiment are indicated in the figure legends.
Statistical analysis was performed using GraphPad Prism 9.5.1 (GraphPad Software, USA). Significant outliers were removed with the Prism ROUT method at Q = 5%, and the normality of residuals and homoscedasticity were tested for each comparison. If the datasets had normal residuals and equal variances, we used ordinary one-way ANOVA with post hoc Holm-Šídák’s test. If the datasets had normal residuals but unequal variances, we used Brown-Forsythe ANOVA with post hoc Dunnett’s T3 test. If the datasets were not normally distributed but had equal variances, we used Kruskal-Wallis ANOVA with post hoc Dunn’s test. In rare cases where the data was neither normally distributed nor had equal variance, we applied the Brown-Forsythe ANOVA with post hoc Dunnett’s T3 test. If the dataset consisted of multiple replicates, we used nested one-way ANOVA with post hoc Holm-Šídák’s test. The tested groups and p values are indicated in the text or the figure legends. Individual cells or slices are labeled as n, replicates are labeled as m, and the numbers animals used are labeled as N. If the groups tested as being different, we report the p-values of post hoc tests but omit the p-value of the omnibus test. If the groups did not test as different, only the p-value of an omnibus test is reported. For scatter plots, each point represents an individual data point (cell, slice, or replicate) and the horizontal bars represent the group mean ± SEM. In all other graphs, we present mean ± SEM. The heat maps represent the group means.
5.8. Model of conduction velocity
Here we model how AP shape affects the conduction velocity (CV) along an unmyelinated axon. We model an infinitesimally small unit of the axon consisting of three distinct units in a row, separated by a space h. The first and last of these units are kept “fixed” at voltages (the peak membrane potential) and , respectively; this potential difference leads to a voltage flow across the axon which is generally described by the cable equation [65]:
Once the intermediate unit is excited above a threshold voltage , the unit activates and is self-excited up to a voltage . This process then repeats ad infinitum along the entirety of the axon (assuming no backflow of the charge). The conduction velocity (i.e., the rate of this voltage flow) is thus inversely proportional to the reset time (the first time when , with as well). Over the discrete lattice above, the cable equation is given by:
Solving this for and substituting in the boundary conditions, we find:
where K is an integration constant needed to satisfy the initial condition . Substituting in the initial condition and solving for , we find:
Thus:
However, h is an arbitrarily small spacing that we can take to approach zero. Thus:
5.9. Power and sample size calculations.
The sample sizes for all statistical comparisons in this work were determined based on the means and pooled standard deviations from preliminary recordings of 8-10 cells or slices per group, α = 0.05, β = 0.8, corrected for the number of pairwise comparisons (τ), based on the following equations:
Where n is the sample size, σ is standard deviation, ϕ is standard normal distribution function, α is Type I error rate, t is the number of pairwise comparisons, and β is Type II error rate. During the calculations, the normality of residuals and equality of variances were assumed a priori.
All of the calculations were performed in an online calculator available at: http://powerandsamplesize.com/Calculators/Compare-k-Means/1-Way-ANOVA-Pairwise
Supplementary Material
7. Acknowledgments
We thank Dr. Vittorio Gallo (Children’s National Research Institute, USA) for sharing his electrophysiology setup, without which this work would not have been possible; Dr. Kieran Clarke (University of Oxford) for providing D-βHb ester and advice; Dr. Joseph Abbah (Children’s National Research Institute, USA) for help during the preliminary stage of the research; Dr. Richard L. Veech and Dr. M. Todd King (Lab of Metabolic Control, NIH/NIAAA, USA) for providing D-βHb ester and advice; Dr. Ting-Jiun Chen (Mt. Sinai Medical Center, USA) for help and advice with setting up patch clamp recordings; Dr. Peter Jonas (Institute of Science and Technology Austria, Austria) for providing FBrain software; Dr Fernando Fernandez (Boston University, USA) for comments to the first draft of the manuscript; Dr Paul Hruz (Washington University School of Medicine in St. Louis, USA) for help with establishing the GluT4 inhibition model; Dr Nandkishore Prakash and Dave Saxon (Children’s National Research Institute, USA) for advice and discussions; Dr Heidi Matos Galicia (National Institute of Neurological Disorders and Stroke, USA) for advice; Dr. Paul Cumming (Bern University Hospital) for critical reading of the manuscript; and members of the Mujica-Parodi laboratory (LCNeuro, USA) for discussions; C.W. also acknowledges support from the Marie-Josée Kravis Fellowship in Quantitative Biology.
8. Funding
This work was supported by the National Institutes of Health Grant K01NS110981 to N.A.S., the National Science Foundation NSF1926781 to L.M.P. and N.A.S., and the Department of Defense ARO W91NF2010189 to N.A.S.
Footnotes
Competing Interests
Authors declare no competing interest.
11. Data and materials availability
Raw data presented in this work, data tables with analyses and tables with statistics are available upon request. All code is available on GitHub, by following the links provided in the text.
6. References
- 1.de la Monte SM. Insulin Resistance and Neurodegeneration: Progress Towards the Development of New Therapeutics for Alzheimer’s Disease. Drugs. 2017;77(1):47–65. doi: 10.1007/s40265-016-0674-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Felice FG, Ferreira ST. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes. 2014;63(7):2262–2272. doi: 10.2337/db13-1954 [DOI] [PubMed] [Google Scholar]
- 3.McNay EC, Recknagel AK. Brain insulin signaling: a key component of cognitive processes and a potential basis for cognitive impairment in type 2 diabetes. Neurobiol Learn Mem. 2011;96(3):432–442. doi: 10.1016/j.nlm.2011.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao WQ, Chen H, Quon MJ, Alkon DL. Insulin and the insulin receptor in experimental models of learning and memory. Eur J Pharmacol. 2004;490(1-3):71–81. doi: 10.1016/j.ejphar.2004.02.045 [DOI] [PubMed] [Google Scholar]
- 5.McNay EC, Recknagel AK. Brain insulin signaling: a key component of cognitive processes and a potential basis for cognitive impairment in type 2 diabetes. Neurobiol Learn Mem. 2011;96(3):432–442. doi: 10.1016/j.nlm.2011.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghasemi R, Haeri A, Dargahi L, Mohamed Z, Ahmadiani A. Insulin in the brain: sources, localization and functions. Mol Neurobiol. 2013;47(1):145–171. doi: 10.1007/s12035-012-8339-9 [DOI] [PubMed] [Google Scholar]
- 7.Kuwabara T, Kagalwala MN, Onuma Y, et al. Insulin biosynthesis in neuronal progenitors derived from adult hippocampus and the olfactory bulb. EMBO Mol Med. 2011;3(12):742–754. doi: 10.1002/emmm.201100177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McNay EC, Pearson-Leary J. GluT4: A central player in hippocampal memory and brain insulin resistance. Exp Neurol. 2020;323:113076. doi: 10.1016/j.expneurol.2019.113076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Craft S. Insulin resistance syndrome and Alzheimer’s disease: age- and obesity-related effects on memory, amyloid, and inflammation. Neurobiol Aging. 2005;26 Suppl 1:65–69. doi: 10.1016/j.neurobiolaging.2005.08.021 [DOI] [PubMed] [Google Scholar]
- 10.Kerti L, Witte AV, Winkler A, Grittner U, Rujescu D, Flöel A. Higher glucose levels associated with lower memory and reduced hippocampal microstructure. Neurology. 2013;81(20): 1746–1752. doi: 10.1212/01.wnl.0000435561.00234.ee [DOI] [PubMed] [Google Scholar]
- 11.Antal B, McMahon LP, Sultan SF, et al. Type 2 diabetes mellitus accelerates brain aging and cognitive decline: Complementary findings from UK Biobank and meta-analyses. Elife. 2022;11:e73138. Published 2022 May 24. doi: 10.7554/eLife.73138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McEwen BS, Reagan LP. Glucose transporter expression in the central nervous system: relationship to synaptic function. Eur J Pharmacol. 2004;490(1-3): 13–24. doi: 10.1016/j.ejphar.2004.02.041 [DOI] [PubMed] [Google Scholar]
- 13.Pearson-Leary J, McNay EC. Novel Roles for the Insulin-Regulated Glucose Transporter-4 in Hippocampally Dependent Memory. J Neurosci. 2016;36(47):11851–11864. doi: 10.1523/JNEUROSCI.1700-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McNay EC, Fries TM, Gold PE. Decreases in rat extracellular hippocampal glucose concentration associated with cognitive demand during a spatial task. Proc Natl Acad Sci U S A. 2000;97(6):2881–2885. doi: 10.1073/pnas.0583697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grillo CA, Piroli GG, Hendry RM, Reagan LP. Insulin-stimulated translocation of GLUT4 to the plasma membrane in rat hippocampus is PI3-kinase dependent. Brain Res. 2009;1296:35–45. doi: 10.1016/j.brainres.2009.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stranahan AM, Norman ED, Lee K, et al. Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus. 2008;18(11):1085–1088. doi: 10.1002/hipo.20470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ashrafi G, Wu Z, Farrell RJ, Ryan TA. GLUT4 Mobilization Supports Energetic Demands of Active Synapses. Neuron. 2017;93(3):606–615.e3. doi: 10.1016/j.neuron.2016.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Felice FG, Ferreira ST. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes. 2014;63(7):2262–2272. doi: 10.2337/db13-1954 [DOI] [PubMed] [Google Scholar]
- 19.Kothari V, Luo Y, Tornabene T, et al. High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim Biophys Acta Mol Basis Dis. 2017;1863(2):499–8. doi: 10.1016/j.bbadis.2016.10.006 [DOI] [PubMed] [Google Scholar]
- 20.Martínez-François JR, Fernández-Agüera MC, Nathwani N, et al. BAD and KATP channels regulate neuron excitability and epileptiform activity. Elife. 2018;7:e32721. Published 2018 Jan 25. doi: 10.7554/eLife.32721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kashiwaya Y, Takeshima T, Mori N, Nakashima K, Clarke K, Veech RL. D-beta-hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proc Natl Acad Sci U S A. 2000;97(10):5440–5444. doi: 10.1073/pnas.97.10.5440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chikahisa S, Shimizu N, Shiuchi T, Séi H. Ketone body metabolism and sleep homeostasis in mice. Neuropharmacology. 2014;79:399–404. doi: 10.1016/j.neuropharm.2013.12.009 [DOI] [PubMed] [Google Scholar]
- 23.Puchalska P, Crawford PA. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017;25(2):262–284. doi: 10.1016/j.cmet.2016.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma W, Berg J, Yellen G. Ketogenic diet metabolites reduce firing in central neurons by opening K(ATP) channels. J Neurosci. 2007;27(14):3618–3625. doi: 10.1523/JNEUROSCI.0132-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jensen NJ, Wodschow HZ, Nilsson M, Rungby J. Effects of Ketone Bodies on Brain Metabolism and Function in Neurodegenerative Diseases. Int J Mol Sci. 2020;21(22):8767. Published 2020 Nov 20. doi: 10.3390/ijms21228767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hartman AL, Gasior M, Vining EP, Rogawski MA. The neuropharmacology of the ketogenic diet. Pediatr Neurol. 2007;36(5):281–292. doi: 10.1016/j.pediatrneurol.2007.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xin L, Ipek Ö, Beaumont M, et al. Nutritional Ketosis Increases NAD+/NADH Ratio in Healthy Human Brain: An in Vivo Study by 31P-MRS. Front Nutr. 2018;5:62. Published 2018 Jul 12. doi: 10.3389/fnut.2018.00062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maalouf M, Sullivan PG, Davis L, Kim DY, Rho JM. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience. 2007;145(1):256–264. doi: 10.1016/j.neuroscience.2006.11.06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shimazu T, Hirschey MD, Newman J, et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339(6116):211–214. doi: 10.1126/science.1227166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lutas A, Yellen G. The ketogenic diet: metabolic influences on brain excitability and epilepsy. Trends Neurosci. 2013;36(1):32–40. doi: 10.1016/j.tins.2012.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clarke K, Tchabanenko K, Pawlosky R, et al. Kinetics, safety and tolerability of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate in healthy adult subjects. Regul Toxicol Pharmacol. 2012;63(3):401–408. doi: 10.1016/j.yrtph.2012.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kesl SL, Poff AM, Ward NP, et al. Effects of exogenous ketone supplementation on blood ketone, glucose, triglyceride, and lipoprotein levels in Sprague-Dawley rats. Nutr Metab (Lond). 2016;13:9. Published 2016 Feb 4. doi: 10.1186/s12986-016-0069-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weistuch C, Mujica-Parodi LR, Razban RM, et al. Metabolism modulates network synchrony in the aging brain. Proc Natl Acad Sci USA. 2021;118(40):e2025727118. doi: 10.1073/pnas.2025727118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mujica-Parodi LR, Amgalan A, Sultan SF, et al. Diet modulates brain network stability, a biomarker for brain aging, in young adults. Proc Natl Acad Sci U S A. 2020;117(11):6170–6177. doi: 10.1073/pnas.1913042117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hruz PW, Murata H, Qiu H, Mueckler M. Indinavir induces acute and reversible peripheral insulin resistance in rats. Diabetes. 2002;51(4):937–942. doi: 10.2337/diabetes.51.4.937 [DOI] [PubMed] [Google Scholar]
- 36.Hresko RC, Hruz PW. HIV protease inhibitors act as competitive inhibitors of the cytoplasmic glucose binding site of GLUTs with differing affinities for GLUT1 and GLUT4. PLoS One. 2011;6(9):e25237. doi: 10.1371/journal.pone.0025237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Colgin LL, Denninger T, Fyhn M, et al. Frequency of gamma oscillations routes flow of information in the hippocampus. Nature. 2009;462(7271):353–357. doi: 10.1038/nature08573 [DOI] [PubMed] [Google Scholar]
- 38.Buzsáki G, Wang XJ. Mechanisms of gamma oscillations. Annu Rev Neurosci. 2012;35:203–225. doi: 10.1146/annurev-neuro-062111-1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26(1):13–25. doi: 10.1016/s0896-6273(00)81133-2 [DOI] [PubMed] [Google Scholar]
- 40.Howarth C, Gleeson P, Attwell D. Updated energy budgets for neural computation in the neocortex and cerebellum. J Cereb Blood Flow Metab. 2012;32(7):1222–1232. doi: 10.1038/jcbfm.2012.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lujan B, Kushmerick C, Banerjee TD, Dagda RK, Renden R. Glycolysis selectively shapes the presynaptic action potential waveform. J Neurophysiol. 2016;116(6):2523–2540. doi: 10.1152/jn.00629.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Padamsey Z, Katsanevaki D, Dupuy N, Rochefort NL. Neocortex saves energy by reducing coding precision during food scarcity. Neuron. 2022;110(2):280–296.e10. doi: 10.1016/j.neuron.2021.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jensen TP, Zheng K, Cole N, Marvin JS, Looger LL, Rusakov DA. Multiplex imaging relates quantal glutamate release to presynaptic Ca2+ homeostasis at multiple synapses in situ. Nat Commun. 2019;10(1):1414. Published 2019 Mar 29. doi: 10.1038/s41467-019-09216-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Newman JC, Verdin E. β-Hydroxybutyrate: A Signaling Metabolite. Annu Rev Nutr. 2017;37:51–76. doi: 10.1146/annurev-nutr-071816-064916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lund TM, Obel LF, Risa Ø, Sonnewald U. β-Hydroxybutyrate is the preferred substrate for GABA and glutamate synthesis while glucose is indispensable during depolarization in cultured GABAergic neurons. Neurochem Int. 2011;59(2):309–318. doi: 10.1016/j.neuint.2011.06.002 [DOI] [PubMed] [Google Scholar]
- 46.Scott RS, Henneberger C, Padmashri R, Anders S, Jensen TP, Rusakov DA. Neuronal adaptation involves rapid expansion of the action potential initiation site. Nat Commun. 2014;5:3817. Published 2014 May 23. doi: 10.1038/ncomms4817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stranahan AM, Mattson MP. Impact of energy intake and expenditure on neuronal plasticity. Neuromolecular Med. 2008;10(4):209–218. doi: 10.1007/s12017-008-8043-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Piroli GG, Grillo CA, Reznikov LR, et al. Corticosterone impairs insulin-stimulated translocation of GLUT4 in the rat hippocampus. Neuroendocrinology. 2007;85(2):71–80. doi: 10.1159/000101694 [DOI] [PubMed] [Google Scholar]
- 49.Grillo CA, Piroli GG, Hendry RM, Reagan LP. Insulin-stimulated translocation of GLUT4 to the plasma membrane in rat hippocampus is PI3-kinase dependent. Brain Res. 2009;1296:35–45. doi: 10.1016/j.brainres.2009.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leloup C, Arluison M, Kassis N, et al. Discrete brain areas express the insulin-responsive glucose transporter GLUT4. Brain Res Mol Brain Res. 1996;38(1):45–53. doi: 10.1016/0169-328x(95)00306-d [DOI] [PubMed] [Google Scholar]
- 51.Vannucci SJ, Koehler-Stec EM, Li K, Reynolds TH, Clark R, Simpson IA. GLUT4 glucose transporter expression in rodent brain: effect of diabetes. Brain Res. 1998;797(1):1–11. doi: 10.1016/s0006-8993(98)00103-6 [DOI] [PubMed] [Google Scholar]
- 52.42. Miranda M, Morici JF, Zanoni MB, Bekinschtein P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Front Cell Neurosci. 2019;13:363. Published 2019 Aug 7. doi: 10.3389/fncel.2019.00363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Won SJ, Yoo BH, Kauppinen TM, et al. Recurrent/moderate hypoglycemia induces hippocampal dendritic injury, microglial activation, and cognitive impairment in diabetic rats. J Neuroinflammation. 2012;9:182. Published 2012 Jul 25. doi: 10.1186/1742-2094-9-182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.44. Yamada KA, Rensing N, Izumi Y, et al. Repetitive hypoglycemia in young rats impairs hippocampal long-term potentiation. Pediatr Res. 2004;55(3):372–379. doi: 10.1203/01.PDR.0000110523.07240.C [DOI] [PubMed] [Google Scholar]
- 55.Lee SY, Kim JH. Mechanisms underlying presynaptic Ca2+ transient and vesicular glutamate release at a CNS nerve terminal during in vitro ischaemia. J Physiol. 2015;593(13):2793–2806. doi: 10.1113/JP270060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.García-Rodríguez D, Giménez-Cassina A. Ketone Bodies in the Brain Beyond Fuel Metabolism: From Excitability to Gene Expression and Cell Signaling. Front Mol Neurosci. 2021;14:732120. Published 2021 Aug 27. doi: 10.3389/fnmol.2021.732120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Nieuwenhuizen H., Chesebro A. G., Polizu C., Clarke K., Strey H. H., Weistuch C., & Mujica-Parodi L. R. Ketosis regulates K+ ion channels, strengthening brain-wide signaling disrupted by age. bioRxiv, 2023-05. [Google Scholar]
- 58.Ma W, Berg J, Yellen G. Ketogenic diet metabolites reduce firing in central neurons by opening K(ATP) channels. J Neurosci. 2007;27(14):3618–3625. doi: 10.1523/JNEUROSCI.0132-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tanner GR, Lutas A, Martínez-François JR, Yellen G. Single K ATP channel opening in response to action potential firing in mouse dentate granule neurons. J Neurosci. 2011;31(23):8689–8696. doi: 10.1523/JNEUROSCI.5951-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martínez-François JR, Fernández-Agüera MC, Nathwani N, et al. BAD and KATP channels regulate neuron excitability and epileptiform activity. Elife. 2018;7:e32721. Published 2018 Jan 25. doi: 10.7554/eLife.32721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lund TM, Ploug KB, Iversen A, Jensen AA, Jansen-Olesen I. The metabolic impact of β-hydroxybutyrate on neurotransmission: Reduced glycolysis mediates changes in calcium responses and KATP channel receptor sensitivity. J Neurochem. 2015;132(5):520–531. doi: 10.1111/jnc.12975 [DOI] [PubMed] [Google Scholar]
- 62.Fremeau RT Jr, Kam K, Qureshi T, et al. Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science. 2004;304(5678):1815–1819. doi: 10.1126/science.1097468 [DOI] [PubMed] [Google Scholar]
- 63.Juge N, Gray JA, Omote H, et al. Metabolic control of vesicular glutamate transport and release. Neuron. 2010;68(1):99–112. doi: 10.1016/j.neuron.2010.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pernía-Andrade AJ, Goswami SP, Stickler Y, Fröbe U, Schlögl A, Jonas P. A deconvolution-based method with high sensitivity and temporal resolution for detection of spontaneous synaptic currents in vitro and in vivo. Biophys J. 2012;103(7):1429–1439. doi: 10.1016/j.bpj.2012.08.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Koch C, Chapter 6: The Hodgkin and Huxley Model of Action Potential Generation. In: Koch C, Biophysics of Computation: Information Processing in Single Neurons. Oxford University Press. 2004. p. 139–176. Print. [Google Scholar]
- 66.Traub RD, Wong RK, Miles R, Michelson H. A model of a CA3 hippocampal pyramidal neuron incorporating voltage-clamp data on intrinsic conductances. J Neurophysiol. 1991;66(2):635–6. doi: 10.1152/jn.1991.66.2.635 [DOI] [PubMed] [Google Scholar]
- 67.Yu N, Morris CE, Joós B, Longtin A. Spontaneous excitation patterns computed for axons with injury-like impairments of sodium channels and Na/K pumps. PLoS Comput Biol. 2012;8(9):e1002664. doi: 10.1371/journal.pcbi.1002664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Altemus KL, Lavenex P, Ishizuka N, Amaral DG. Morphological characteristics and electrophysiological properties of CA1 pyramidal neurons in macaque monkeys. Neuroscience. 2005;136(3):741–756. doi: 10.1016/j.neuroscience.2005.07.001 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data presented in this work, data tables with analyses and tables with statistics are available upon request. All code is available on GitHub, by following the links provided in the text.