

Abstract
G-quadruplexes are thought to play an important role in gene regulation and telomere maintenance, but developing probes for their presence and location is challenging due to their transitory and highly dynamic nature. The majority of probes for G-quadruplexes have relied on antibody or small molecule binding agents, many of which can also alter the dynamics and relative populations of G-quadruplexes. Recently, it was discovered that UVB irradiation of human telomeric DNA and various G-quadruplex forming sequences found in human promoters, as well as reverse Hoogsteen hairpins, produces a unique class of non-adjacent anti cyclobutane pyrimidine dimers (CPDs). Therefore, one can envision using a pulse of UVB light to irreversibly trap these non-B DNA structures via anti CPD formation without perturbing their dynamics, after which the anti CPDs can be identified and mapped. As a first step towards this goal, we report radioactive post- and pre-labeling assays for the detection of non-adjacent CPDs and illustrate their use in detecting trans,anti T=(T) CPD formation in a human telomeric DNA sequence. Both assays make use of snake venom phosphodiesterase (SVP) to degrade the trans,anti T=(T) CPD-containing DNA to the tetranucleotide pTT=(pTT) corresponding to CPD formation between the underlined T’s of two separate dinucleotides while degrading the adjacent syn TT CPDs to the trinucleotide pGT=T. In the post-labeling assay, calf intestinal phosphodiesterase is used to dephosphorylate the tetranucleotides which are then rephosphorylated with kinase and [32P]-ATP to produce radiolabeled mono and diphosphorylated tetranucleotides. The tetranucleotides are confirmed to be non-adjacent CPDs by 254 nm photoreversion to the dinucleotide p*TT. In the pre-labeling assay, radiolabeled phosphates are introduced into non-adjacent CPD forming sites by ligation prior to irradiation thereby eliminating the dephosphorylation and rephosphorylation steps. The assays are also demonstrated to detect the stereoisomeric cis,anti T=(T) CPD.
Keywords: Post-labeling assay, G-quadruplex, Human telomeric DNA, Reverse Hoogsteen hairpin, Non-adjacent DNA photoproduct, Photocrosslink, Cyclobutane pyrimidine dimer
Graphical Abstract

INTRODUCTION
Non-B DNA secondary structures play many roles in vivo, such as Holliday junctions in recombination and slipped structures in mutagenesis.1–4 Most recently, G-quadruplexes (G-quadruplexes) were proposed to play an important role in telomere maintenance and modulating transcription and translation, as well as causing genome instability.5–7 Unfortunately, most non-B DNA structures exist transiently in vivo and are difficult to detect and locate. Small molecule and antibody probes for detecting G-quadruplexes in vivo have been reported,8–17 some of which drive the formation of the structures that they seek to probe and/or require fixation of the cells which chemically alters the DNA.18 Many of the probes also bind to multiple G-quadruplex conformations with varying selectivity.
Recently we discovered that UVB irradiation of G-quad-forming sequences from human telomeres and various promoters results in the formation of a unique class of non-adjacent cyclobutane pyrimidine dimers (CPDs). These CPDs are formed between distant pyrimidines in a sequence with an anti or head-to-tail orientation with both cis and trans stereochemistries between the C5-substituents (Figure 1).19–24 The anti regiochemistry is opposite to that of adjacent syn CPDs that form between sequential pyrimidines with a head to head orientation such as the cis,syn CPDs that are the major photoproduct of B form DNA and trans,syn CPDs that can also form in single strand DNA (Figure 1A). Anti CPD’s cannot form from sequential pyrimidines in B DNA or single stranded DNA owing to conformational constraints25 and can only arise from folded structures in which non-adjacent pyrimidines can be brought together in close proximity. Non-adjacent CPDs with the syn regiochemistry can form in bulge loop DNA26 but have not yet been identified in irradiated G-quadruplexes.
Figure 1.

Idea of using structure-specific photoproducts to detect and map folded DNA structures such as basket G-quadruplexes. A) Structure of adjacent cis,syn and non-adjacent cis and trans,anti T=(T) CPDs where the parentheses denotes non-adjacent nucleotides. B) Whereas UVB irradiation of B DNA produces adjacent cis,syn T=T CPDs, UVB irradiation of non-B DNA folded structures such as the basket G-quadruplex form of human telomeric DNA produces unique non-adjacent anti T=(T) CPDs.
Non-adjacent CPDs from non-B DNA structures can be readily distinguished from adjacent CPDs formed in single strand and B DNA by digestion with Nuclease P1 (NP1), which hydrolyzes the O3’-phosphodiester bond following a canonical nucleotide.27, 28 As a result of its specificity, NP1 digestion of a non-adjacent T=(T) CPD where the parentheses indicate that the second T is not adjacent to the first T, produces the tetranucleotide pTN=(pTN)23, 24 which corresponds to the CPD photoproduct between the underlined T’s of the dinucleotides pTN and pTN. On the other hand, NP1 digestion of adjacent T=T CPDs produces the trinucleotide pNT=T within a single trinucleotide,29 which can be easily distinguished from the tetranucleotide products by HPLC, electrophoresis and mass spectrometry. The stereochemistry of the anti CPDs was established by correlation of the thymine dimers released by acid-catalyzed glycosidic bond hydrolysis with authentic compounds.23, 24 Non-adjacent CPDs have also been detected in UVC-irradiated ethanolic and desiccated DNA by an enzyme-coupled LC-MS/MS assay that involves degrading the DNA to pyrimidine dimers using nuclease P1, snake venom phosphodiesterase, and alkaline phosphatase.30
The anti T=(T) CPDs that form in UVB irradiated human telomeric DNA in vitro were proposed to arise from antiparallel basket type G-quadruplexes in which a pyrimidine from one lateral loop photocyclizes with a non-adjacent pyrimidine on the opposing lateral loop that is in close proximity, resulting in an interloop crosslink (Figure 1b).22, 24 Anti CPDs cannot arise from the parallel or hybrid type G-quadruplex conformations because they lack opposing lateral loops. It was later found that anti T=(T) CPDs can also form between T’s of interior loops of reverse Hoogsteen hairpin conformers of human telomeric DNA21 and in various G-quadruplex forming sequences found in promoters.20 As such, non-adjacent anti CPDs could serve as intrinsic photoprobes for these types of non-B DNA structures. One could, therefore, envision irradiating cells with a pulse of UVB laser light to covalently trap these otherwise unstable non-B DNA structures by anti CPD formation for subsequent analysis (Figure 1B). Because the duration of the laser pulse is sub microsecond and the CPD formation is on the picosecond time scale31 one would effectively be taking a non-perturbing snapshot of the distribution of photoreactive non-B DNA secondary structures present within a living cell at a specific instance in time. Owing to the dynamic and transient nature of the non-B DNA structures, however, anti CPDs are expected to form in small quantities requiring highly sensitive techniques to detect and map their location.
Before embarking on sequencing strategies for mapping anti CPDs, we require evidence that they are indeed being formed in vivo upon UVB irradiation. One highly sensitive method for detecting DNA damage is the radioactive post-labeling assay that has been estimated to be capable of detecting one DNA adduct per human genome.32–35 In this assay, DNA is enzymatically degraded to short oligodeoxynucleotides or mononucleotides containing the DNA adduct. The nucleotides are rephosphorylated with radioactive 32P and identified chromatographically or electrophoretically by comparison to authentic adducts. It was previously shown that degradation of UV irradiated B DNA with a combination of snake venom phosphodiesterase (SVP) and DNAse I produces a trinucleotide of the form pNY=Y (Figure 2A).36 The canonical nucleotide at the 5’-end makes it a good substrate for dephosphorylation with calf intestinal phosphatase and rephosphorylation with [32P]-ATP and kinase36 unlike the product from NP1 (Figure 1A). We previously found that SVP degrades the cis,anti T2=(T7) CPD product of d(GTATCATGAGGTGC) to the tetranucleotide GT=(pAT) corresponding to the photodimer of GpT and pAT,23 which also has canonical nucleotides at the 5’-ends and is, therefore, expected to undergo phosphorylation.
Figure 2.

Post-labeling assay schemes for CPDs. A) DNA containing an adjacent cis,syn T=T CPD is degraded to a trinucleotide with snake venom phosphodiesterase (SVP) and then dephosphorylated with calf intestinal phosphatase (CIP) followed by radiolabeling with [γ-32P]-ATP and kinase to give a phosphorylated trinucleotide product. Irradiation with 254 nm light reverts the CPD to the canonical bases. B) DNA containing a non-adjacent anti T=(T) CPD is degraded to a tetranucleotide with snake venom phosphodiesterase (SVP) and after dephosphorylation with calf intestinal phosphatase (CIP) and radiolabeling with [γ-32P]-ATP and kinase would give mono and diphosphorylated tetranucleotide products. Irradiation with 254 nm light would revert the tetranucleotide products to two dinucleotides. Treatment of the tetra- or dinucleotide products with NP1 would release the 5’-terminal radiolabeled nucleotide. All radiolabeled products are distinguishable by high resolution polyacrylamide gel electrophoresis.
Herein, we report a post-labeling assay for trans,anti T=(T) CPDs formed in folded conformations of human telomeric DNA by using SVP and calf intestinal phosphatase to degrade the irradiated DNA to TT=(TT) that can then be rephosphorylated with [γ-32P]-ATP and kinase to p*TT=(p*TT) (Figure 2B). Furthermore, we show that these tetramer products can be distinguished by high resolution gel electrophoresis from the trinucleotide p*NT=T produced from cis,syn T=T CPDs, and partial degradation products of DNA by subsequent photoreversal to p*TT dinucleotides by 254 nm light. We also show that a cis,anti T=(T) CPD can be similarly detected, and that anti CPD formation can also be easily assayed in vitro by using pre-labeled DNA substrates.
EXPERIMENTAL SECTION
Materials and reagents.
Oligodeoxynucleotides (ODNs) were purchased from Integrated DNA Technologies, Inc. (IDT, Coralville, IA, USA). Snake venom phosphodiesterase (SVP) from Crotalus adamanteus was obtained from Worthington Biochemical (Lakewood, NJ, USA). DNase I (RNase- free) was obtained from New England Biolabs (Ipswich, MA, USA). Calf intestinal phosphatase and T4 DNA ligase were obtained from Promega (Madison, WI, USA). T4 Polynucleotide Kinase (T4 PNK) and HPLC solvents were obtained from ThermoFisher Scientific (Waltham, MA, USA) and 6000 Ci/mmol [γ-32P]-ATP was from Perkin Elmer (Waltham, MA, USA).
Large Scale Preparation of pTT=(pTT).
Tel26 (50 μM) was incubated in 100 μL of 150 mM KCl at 95 °C for 10 min and then rapidly cooled in ice to preferentially form intramolecular folded structures. Samples were left on ice for at least 1 h before irradiation with broadband UVB/UVA light (275–400 nm, centered at 330 nm). Irradiation was carried out in polyethylene microfuge tubes on a bed of ice for 2.5 h at 1 cm distance from two Spectroline XX-15B UV 15W tubes with an approximate intensity of 2.2 mW/cm2 (22 J/m2-s) after filtration through a plate of Pyrex glass. The solution was evaporated to dryness in vacuo and then incubated with 20 units of DNase I in10 mM Tris-HCl (pH 7.6), 2.5 mM MgCl2 and 0.5 mM CaCl2 at 37 °C overnight followed by inactivation at 75 °C for 20 min. The digested samples were dried in vacuo and then dissolved in 100 μL of 10 mM ammonium citrate, 100 μM MgCl2, pH 9.4, to which 10 units of SVP were added and incubated at 37 °C overnight. SVP was deactivated in a boiling water bath for 20 min and removed by phenol extraction after which residual phenol were removed by multiple diethyl ether extractions. The tetramer photoproduct pTT=(pTT) was then isolated and purified by HPLC and analyzed by MS/MS as described below.
Large Scale Dephosphorylation of pTT=(pTT) with Calf Intestinal Phosphatase.
The HPLC fractions containing pTT=(pTT) were evaporated in vacuo and incubated for 3 h at 37 °C with 6 units of calf intestinal phosphatase (CIP) in in 40 μL of 50 mM Tris-HCl (pH 9.3), 1 mM MgCl2, 100 μM ZnCl2, and 1 mM spermidine. After incubation, the enzyme was inactivated by heating to 80 °C for 20 min and removed by phenol extraction after which residual phenol was removed by multiple diethyl ether extractions. The dephosphorylated product TT=(TT) was purified by HPLC and analyzed by MS/MS as described below.
Large Scale Phosphorylation of TT=(TT) with T4 PNK.
HPLC fractions containing TT=(TT) were evaporated to dryness in vacuo and then incubated with 50 units of T4-PNK in 40 μL of 1 mM adenosine triphosphate (ATP), 50 mM Tris-HCl (pH 7.6), 10 mM MgCl2, 5 mM DTT, 100 μM spermidine for 3 h at 37°C. After incubation, the enzyme was inactivated by incubation in a boiling water bath for 20 min and removed by phenol extraction after which traces of phenol removed by multiple diethyl ether extractions. The rephosphorylated product pTT=(pTT) was analyzed by HPLC as described below.
HPLC Analysis and Purification of the Post-labeling Assay Intermediates.
Reversed-phase HPLC was carried out with an X-Bridge BEH column (C18, 4.6 × 75 mm, 3.5 μm, 130 Å) on a System Gold HPLC system with a binary gradient Model 125 pump and a Model 166 UV detector (Beckman Coulter, Inc., Fullerton, CA). Photoproducts were eluted with 1 mL/min of 100% solvent A (50 mM triethylammonium acetate, pH 7.5) for 3 min followed by a linear gradient of 0–20% B (50% acetonitrile in 50 mM triethylammonium acetate, pH 7.5) in solvent A for 3–53 min and detected by absorbance at 260 nm. HPLC fractions corresponding to the enzymatically degraded photoproducts were evaporated in vacuo.
Mass Spectrometry of the Post-labeling Assay Intermediates.
The CPD samples were analyzed in the negative-ion mode on a Waters Synapt-G2 mass spectrometer (Waters, Milford, MA). The source and desolvation temperatures were 120 °C and 150 °C, respectively. A solution of 50/50 (vol/vol) acetonitrile/water was used as the spray solvent. The capillary voltage was 2.8 kV. The sampling cone and extraction cone voltage were 20 V and 4 V, respectively. MS/MS experiments were carried out by collisionally activated dissociation (CAD). The mass width for precursor selection was set at m/z 3, and the collision gas was Argon at a pressure of 1×10−4 mbar. The trap collision energy was tuned to 20 V where both the precursor ions and fragmented ions were observed on the same spectrum.
Post-labeling Assay of UVB Irradiated DNA.
Tel26 (500 pmol) was incubated at 95 °C for 10 min in 20 μL of 150 mM KCl and plunged into an ice water bath for 1 h, after which it was irradiated with UVB light for 2.5 h on ice. The solution was then incubated with 20 units of DNaseI in 20 μL of 10 mM Tris-HCl (pH 7.6), 2.5 mM MgCl2, and 0.5 mM CaCl2 at 37 °C overnight, followed by inactivation of the DNAse I at 75 °C for 20 min. After drying the sample in vacuo, it was incubated with 10 units of SVP in 30 μL of 10 mM ammonium citrate, 100 μM MgCl2 pH 9.4 at 37 °C overnight. The SVP was inactivated in a boiling water bath for 20 min and then treated with 6 units of CIP in 40 μL of 50 mM Tris-HCl (pH 9.3), 1 mM MgCl2, 100 μM ZnCl2, and 1 mM spermidine at 37 °C for 3 h. The enzymes were removed by phenol extraction, and residual phenol was removed by multiple diethyl ether extractions. After evaporating the sample in vacuo, it was treated with 10 units of T4-PNK and 0.5 pmol [γ-32P]-ATP in 40 μL of 50 mM Tris-HCL (pH 7.6), 10 mM MgCl2, 5 mM DTT, 0.1 mM spermidine, 0.1 mM EDTA at 37 °C for 3 h. An aliquot of the sample (4 μL) was irradiated for 30 min with 254 nm light to reverse any CPDs. Unirradiated and irradiated aliquots (4 μL) were then mixed with an equal volume of 2X loading buffer (98% formamide with xylene cyanol dye), heated in a boiling water bath for 5 min, and cooled on ice. The radiolabeled samples were then analyzed on a 20% denaturing polyacrylamide gel (7 M urea, 19:1 acrylamide:bisacrylamide, 0.8 mm thick) at 1500 V until the dye reached 8 cm. The gel was then was scanned with an Amersham Typhoon biomolecular imager.
Post-labeling Assay with Competitor Plasmid DNA.
Competition experiments were carried out at a fixed base pair concentration (650 μM bp) in 100 μL of 150 mM KCl with varying Tel26: plasmid ratios from 1:1 to 1:1000. The solutions were heated to 95 °C and then plunged into an ice bath for 1 h to induce G-quadruplex formation after which they were irradiated with UVB light for 2.5 h on ice. After incubation with DNaseI and SVP following the conditions described above, an aliquot of 10 μL (6.5 nmol bp) from each dilution was then treated with CIP in a total volume of 40 μL as described above. Subsequent radiolabeling and gel electrophoresis was also performed exactly as described above except that the amount of [γ-32P]-ATP was increased to 5 pmol.
Preparation of Internally Labelled Tel26 Sequences.
Tel26, AAA(GGGTTA)3GGGAA, was separately internally radiolabeled at the second T of the 1st and 2nd TTA loop. To introduce the radioactive phosphate in the first TTA loop, the 5’-end of the 19-mer sequence TA(GGGTTA)2GGGAA (500 pmol) was first radiolabeled with [32P]-ATP and PNK and then ligated to the 3’-end of the 7-mer sequence AAAGGGT (1500 pmol) in the presence of the 30-mer scaffold TTCCC(TAACCC)3TTTTTTT (1500 pmol). The ligation reaction was performed with 4 units of T4 DNA ligase in 30 μL of 30 mM Tris-HCl (pH 7.8), 10 mM MgCl2, 10 mM DTT, 1 mM ATP at 4 °C overnight. After incubation, the ligase was deactivated at 75 °C for 20 min and dried in vacuo. The same general procedure was used to introduce the radioactive phosphate into the second TTA loop by ligating 13A-mer AAAGGGTTAGGGT to 5’-end labeled 13B-mer TAGGGTTAGGGAA. The ligation products were purified by denaturing 15% denaturing PAGE (19:1 acrylamide: bisacrylamide) being careful to separate the 26-mer band from the 30-mer scaffold band. The radiolabeling of the 13-mer and 19-mer sequences was carried out with 10 units of T4-PNK and 2.5 pmol [γ-32P]-ATP in 30 μL of 50 mM Tris-HCL (pH 7.6), 10 mM MgCl2, 5 mM DTT, 0.1 mM spermidine, 0.1 mM EDTA at 37°C for 1 h. This reaction was chased with 750 pmol of cold ATP and 5 units of T4-PNK for 1 h at 37 °C. After incubation, the kinase was deactivated for 20 min in a boiling water bath, and the solution dried in vacuo.
SVP degradation of internally labeled UVB irradiated Tel26 sequences.
Purified ligation products of Tel26 radiolabeled in the 1st and 2nd TTA loop were irradiated with UVB light after G-quadruplex induction in 150 mM LiCl, which produces predominantly the trans,anti T=T CPD. The sequences were then digested with DNaseI and SVP following the conditions described above. Parts of the digested products from both sequences were treated with 0.01 units of CIP in 30 μL of 50 mM Tris-HCl (pH 9.3), 1 mM MgCl2, 100 μM ZnCl2, and 1 mM spermidine for 2.5 min at room temperature and immediately quenched in dry ice. CIP was deactivated in 80 °C for 20 min and dried in vacuo. A portion of the digested products and the dephosphorylated products was irradiated for 30 min with 254 nm light to reverse any CPDs present.
SVP degradation and labeling studies with UVB irradiated 14-mer.
The 14-mer GTATCATGAGGTGC (100 μM bp, 100 μL) was irradiated on ice in 10 mM sodium acetate, pH 4.8 either before or after 5’-endlabeling with 0.5 pmol of [γ-32P]-ATP and kinase. Degradation and post-labeling experiments were carried out as generally described above for Tel26.
RESULTS AND DISCUSSION
To validate the proposed enzymatic sequence for post-labeling the trans,anti T=T CPD, the human telomere containing sequence Tel26, AAAGGGTTAGGGTTAGGGTTAGGGAA, was UVB irradiated in large scale to produce the trans,anti T=(T) CPD as the major product21, 22, 24 so that the intermediates could be characterized by MS/MS. The irradiated sample was then digested with SVP to produce a major product, which eluted at 34.2 min from reversed-phase HPLC (Figure 3A). ESI-MS/MS of the product (Figure 4A) showed an [M – 2H]2- ion of m/z 625, which fragmented by an electrocyclic elimination reaction to dehydrothymidinephosphate [pT – H2O – H]− at m/z 321 and [M – (pT – H2O) – H]− at m/z 947 (Figure 5) confirming it to be pTT=(pTT). A fragment ion of m/z 643 was also observed that results from the loss of two dehydrothymidinephosphate groups [M – 2(pT – H2O) – H]−, which corresponds to the monoanion of the CPD of thymidine monophosphate [pT=pT - H]−. Treatment of pTT=(pTT) with calf intestinal phosphatase (CIP) resulted in a slower eluting product with a retention time of 49 min (Figure 3B). ESI-MS/MS of this product confirmed the removal of the two 5’-phosphates to give TT=(TT) by observing the molecular ion [M - 2H]2- at m/z 545, which fragmented to [M - (T - H2O)-H]− at m/z 867 and to [M - 2(T - H2O) - H]− corresponding to [pT=(pT)-H]− at m/z 643 (Figure 4B, 5). Treatment of TT=(TT) with kinase and ATP converted it back to pTT=(pTT) as confirmed by HPLC (Figure 3C).
Figure 3.

HPLC analysis and purification of the intermediates in the post-labeling assay of Tel26 DNA irradiated with UVB light. A) UVB irradiated Tel26 digested with snake venom phosphodiesterase (SVP) for 24 h to give trans,anti pTT=(pTT). B) Dephosphorylation of trans,anti pTT=(pTT) with calf intestinal phosphodiesterase (CIP) to give TT=(TT). C) Rephosphorylation of TT=(TT) from B with polynucleotidyl kinase (PNK) and ATP.
Figure 4.
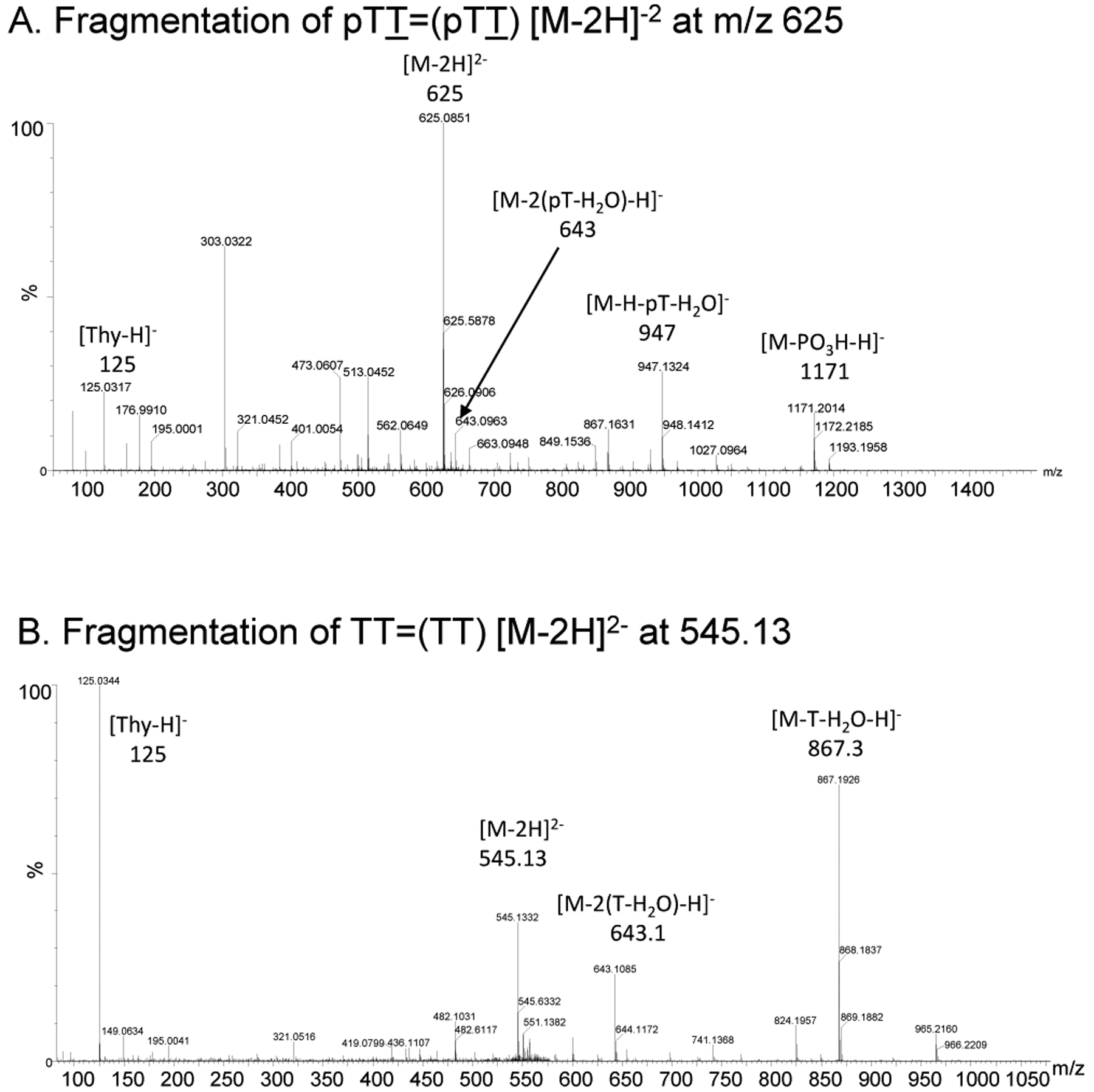
ESI-MS/MS characterization of the intermediates in the post-labeling assay of irradiated Tel26 DNA. A) MS/MS of the [M - 2H]2- ion (m/z 624.11) of trans,anti pTT=(pTT). b) MS/MS of the [M - 2H]2- ion (m/z 545.13) of the dephosphorylated trans,anti TT=(TT).
Figure 5.
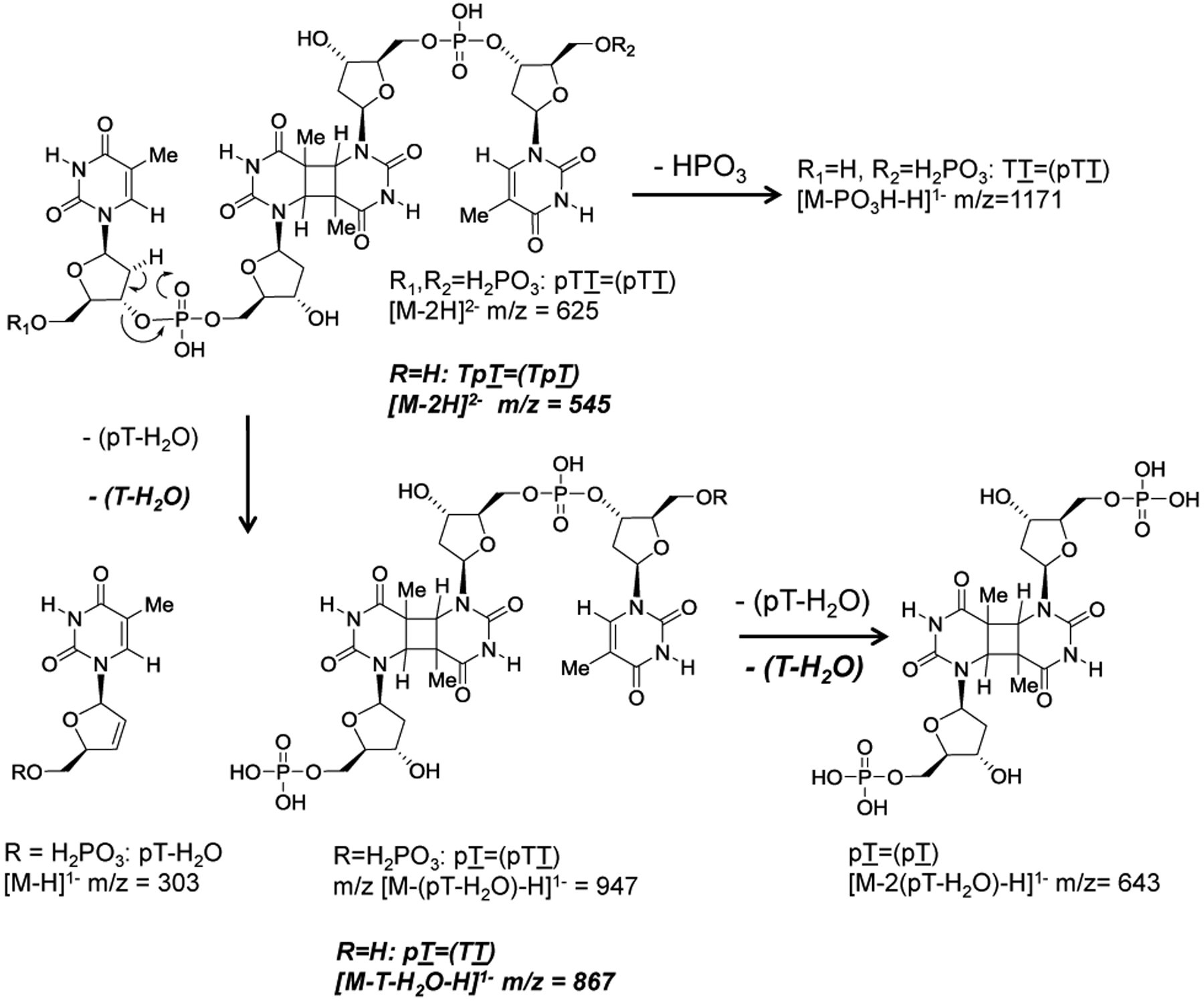
MS/MS fragmentation pathways of the trans,anti T=(T) CPD-containing products of Figure 4. Pathways for pTT=(pTT) are shown in normal font, whereas pathways for TT=(TT) are shown in bold italic font.
Treatment of the dephosphorylated tetramer with [γ-32P]-ATP and polynucleotide kinase, however, resulted in two major radiolabeled bands on high resolution denaturing polyacrylamide gel electrophoresis, a fast moving band and much more slowly moving band (Figure 6 lane 5). A band corresponding to p*GT=T that would have arisen from an adjacent syn T=T CPD was very weak, suggesting that syn CPDs are not produced efficiently in the loops of the G-quadruplex or reverse Hoogsteen structures owing to conformational constraints. Exposure of the products of lane 5 to 254 nm light prior to electrophoresis converted both major products to a single faster moving band (lane 6) corresponding to the dinucleotide p*TT, which was expected to be produced by photoreversal of p*TT=(p*TT) CPD (Figure 2). The formation of two radiolabeled products that photoreverted to the same product suggests that the two initial products were the mono and diphosphorylated products p*TT=(TT) and p*TT=(p*TT), respectively (vide infra). Formation of the two photorevertible radiolabeled products confirms that the trans,anti T=(T) CPD of the human telomere sequence can be detected by a post-labeling assay involving sequential treatment with SVP, CIP and polynucleotidyl kinase and [γ-32P]-ATP.
Figure 6.

Post-labeling assay carried out on UVB irradiated Tel26 in the absence or presence of competing plasmid DNA. Lanes 2, 3 and 5 are authentic standards prepared by radiolabeling TT and HPLC purified GT=T and TT=(TT). 254 nm light was used to photorevert CPD-containing products. Lane headings for the post-labeling reactions refer to the ratio of the base pair concentrations of Tel26 to plasmid DNA. The portion of the gel to the right is shown with a reduced dynamic range to make the photoreversed dinucleotide bands more apparent. An asterisk indicates a radiolabeled phosphate.
To determine whether the post-labeling assay would work in the presence of competitor DNA, we irradiated decreasing amounts of Tel26 in the presence of plasmid DNA at a total concentration of 650 μM bp. The samples were then treated with DNAse I and SVP, followed by CIP and then kinase and [γ-32P]-ATP. DNAse I was added to help digest the plasmid DNA. In this case, only the slower moving radiolabeled product was observed in lanes 7, 9, 11 and 13 (Figure 6). To confirm that the slower moving band was radiolabeled p*TT=(TT), and to distinguish it from radiolabeled partially digested ODNs, each sample was irradiated with 254 nm light to photorevert the tetramer products to dinucleotides. As can be seen in lanes 8,10,12, and 14, the slow-moving band decreases in intensity by the same amount that the fast-moving band corresponding to p*TT increases in intensity. The formation of the trans,anti T=(T) CPD could be visibly detected down to a 40 fmol level in a 5 min exposure of the gel (1:100 lane). In the same gel, a 10-fold lower amount (4 fmol) of the trans,anti T=(T) CPD was detectable by matching a decrease in the amount of the p*TT=(TT) band to an increase in the amount of the p*TT band following photoreversion in the 1:1000 dilution lane, although this was not observed in a second gel. It is likely that this level of sensitivity or better can be achieved, however, if the tetranucleotide products were isolated prior to the post-labeling assay by HPLC, and/or if the p*TT=(TT) product were excised from the electrophoresis gel prior to photoreversion to p*TT and electrophoresis to eliminate interference from background bands.
A lower limit of about 4 fmol is about three times higher than 1.5 fmol of trans,anti T=T CPD that would be produced if one trans,anti T=(T) CPD were to be formed per telomere in 10 million human cells that have 92 telomeres/cell. There would also be much more background interference from partially digested DNA and adjacent cis,syn T=T CPD trinucleotides, but this could be circumvented by isolating the telomeres prior to the post-labeling assay according to published procedures.37 Interestingly, comparatively lower amounts of radiolabeled adjacent CPD products p*NY=Y were observed in comparison to the radiolabeled p*TT=(TT) product at 1:1 dilution even though GT=T was found to be efficiently labeled (lane 3). The lower amount of radiolabeled cis,syn CPDs could either be due to the inefficiency of SVP digestion of DNAseI digestion products, which may be largely duplex, and/or that non-adjacent CPDs form in much higher yield than the adjacent CPDs.
To confirm the identity of the slow and fast migrating radiolabeled bands as either diphosphorylated p*TT=(p*TT) or monophosphorylated p*TT=(TT), internally labeled substrates were used in which the inter-nucleotide phosphate between the two T’s in either the first TTA or the second TTA loop was radiolabeled (Figure 7A). Because this substrate is internally labeled, irradiation followed by SVP would directly produce the radiolabeled diphosphorylated product pT*T=(pTT), without requiring desphosphorylation and rephosphorylation (Figure 7B). The desired substrates were prepared by ligating either the 5’-end-labeled 19-mer (for loop 1) or the 5’-end-labeled 13-mer (for loop 2) to the 7-mer or 13-mer, respectively, using the 30-mer ligation scaffold (Figure 7A). The radiolabeled Tel26 strand was then carefully separated by denaturing PAGE from the 30-mer strand, which would otherwise form a duplex with Tel26 and inhibit folding. As one can see from lane 8 in Figure 8, UVB irradiation of Tel26 internally labeled in loop 1 followed by SVP produces the fast-moving product, which is converted to pT*T (lane 9) following photoreversal with 254 nm light, thereby identifying it as the doubly phosphorylated product pT*T=(pTT). A band corresponding to p*T resulting from degradation of non-photoreacted DNA at that site was also observed, as well as a trimer corresponding to pGT=*T. The same bands were also observed in Tel-26 subjected to the standard post-labeling protocol and using a chase with cold ATP to ensure complete phosphorylation (Figure 8 lane 4). Strangely, a small amount of what appears to be the mono-phosphosphorylated tetramer product was also observed in the pre-labeled products digested with SVP that disappears upon photoreversal, along with slower moving bands only seen following treatment with CIP (vide infra). How the mono-phosphorylated tetramer product and the higher bands would form is not obvious unless there was some phosphatase contaminating the commercial SVP preparation or a buffer. As expected, bands corresponding to pTT=(pTT) were not observable in the SVP degradation products Tel26 pre-labeled in loop 2 (Figure 8 Lane 14) because anti CPD formation can only take place between loops 1 and 3 of a chair or basket G-quadruplex or a reverse Hoogsteen hairpin.
Figure 7.

Preparation and use of internally labeled Tel26 for studies of anti T=(T) CPD formation. A) ODNs (written 5’−3’) used to prepare Tel26 internally labeled in loop 1 and loop 2 where the T’s that undergo trans,anti T=(T) CPD formation are in bold. The 30-mer serves as a ligation scaffold. B) Degradation products the non-adjacent trans,anti T=(T) CPD formed between loop 1 and loop 3 of Tel26 internally labeled at loop 1. The relative mobility of the enzymatic degradation products is compared to the mass to charge ratio assuming that all phosphate groups are fully ionized. An asterisk indicates a radiolabeled phosphate.
Figure 8.

Analysis of the enzymatic degradation products of post- and pre-labeled Tel26 by denaturing polyacrylamide gel electrophoresis. Lanes 1–3: authentic products obtained by end-labeling commercial ODNs. Lanes 4–7: post-labeling assay carried out on UVB irradiated Tel26 followed by dephosphorylation with CIP, with and without CPD photoreversion with 254 nm light as indicated. Lanes 8–13: SVP treatment of UVB irradiated Tel26 that was internally pre-labeled in loop 1 followed by CIP or CIP and NP1, with and without CPD photoreversion with 254 nm light, all as indicated. Lanes 14–18: SVP treatment of UVB irradiated Tel26 that was internally pre-labeled in loop 2 followed by CIP with and without CPD photoreversion with 254 nm light as indicated. Post treatments with CIP (Post CIP) were for either 2.5 min or 30 min as indicated by a or b in the lane headings. Figure prepared from two separate gels carried out under identical conditions as indicated by the vertical line. An asterisk indicates a radiolabeled phosphate.
To confirm the identity of the slow-moving band from the post-labeling assay as the mono-phosphorylated product pTT=(TT), samples containing the fast-moving product from the post-labeling procedure were incubated with CIP for various times to remove the 5’-phosphates. As one can see by comparing lanes 4 & 6 of Figure 8 that CIP converted the fast-moving band to the slow-moving one, both of which disappear in lanes 5 & 7 upon photoreversal with concomitant appearance of the p*TT band. When the SVP products of Tel26 internally labeled in loop 1 (lane 8) were treated with CIP for a short period of time (lane 11), the slower moving band assigned to pTT=(TT) was observed along with more intense bands that migrate even slower that can be assigned to the completely dephosphorylated product T*T=(TT) and the dephosphorylated trimer product GT=*T. There is also one additional band that cannot be assigned. Photoreversal of T*T=(TT) to T*T with 254 nm light resulted in an even slower moving product band, while photoreversal of GT=*T to GT*T caused no observable change in mobility (lane 12). The additional unassigned band in lane 11 disappears upon 254 nm irradiation. Treatment of the Tel26 products internally labeled in loop 2 with CIP only produced GT*T and essentially none of the products expected from an anti CPD’s because they can only form between loops 1 and 3.
The large differences in mobility between the various enzymatic degradation products can be readily understood in terms of their mass-to-charge ratio (Figure 7B). Removal of a single 5’-phosphate group from pTT=(pTT) to produce pTT=(TT) results in the loss of two negative charges but a minor change in molecular weight causing a substantial reduction in electrophoretic mobility. Removal of a second 5’-phosphate to form TT=(TT) results in a loss of an additional two negative charges, resulting in a tetranucleotide with only two negative charges, causing a further reduction in the electrophoretic mobility. Large retardations in electrophoretic mobility upon removal of a 5’-phosphate from oligodeoxynucleotides were observed with 3’-endlabeled substrates.38
To determine whether the cis stereoisomer of the anti T=(T) CPDs can also be detected by the pre- or post-labeling assays, we carried out the assay on the 14-mer sequence, GTATCATGAGGTGC, which we previously showed to form the cis,anti T=(T) CPD between T2 and T7 (underlined) when UVB irradiated under low pH (Figure 9). Because SVP degradation of the cis,anti T2=(T7) CPD includes the 5’-terminal unphosphorylated G, we could either pre-label the sequence with kinase and [γ-32P]-ATP, or post-label the GT=(pAT) product following SVP degradation. When the pre-labeled product was UVB irradiated and then treated with SVP, a major fast migrating band was observed, which was converted to a faster moving band that migrated as p*GT upon 254 nm irradiation (lane 6). Treatment of the SVP degraded product with CIP caused the fast-moving band to disappear and a slow-moving band to appear, both of which disappeared upon 254 nm irradiation with the formation of the p*GT band. This behavior is consistent with the fast-moving SVP degraded band being p*GT=(pAT) and the slow-moving band being p*GT=(AT). The band moving like a nucleotide monophosphate in lanes 5–12 must be p*G resulting from SVP degradation of the undamaged 14-mer.
Figure 9.

Analysis of the enzymatic degradation products of pre- and post-labeled 14-mer containing a cis,anti T=(T) CPD. A) Schematic for the enzymatic reactions carried out on the pre- and post-labeled UVB irradiated 14-mer where the T’s undergoing anti CPD formation are in bold. B) Denaturing PAGE of the enzymatic reaction products. Lanes 5–12: the 5’-pre-labeled UVB irradiated 14-mer was digested with SVP and then dephosphorylated with CIP, where a, b and c refer to the times of 0-, 5- and 10-minutes following addition of CIP, with and without photoreversion by 254 nm light as indicated. Lanes 13–20: The UVB irradiated 14-mer was degraded by SVP and then labeled with PNK and [γ-32P]-ATP, after which it was incubated for increasing time with CIP as described for the pre-labeled experiment, with and without photoreversion with 254 nm light as indicated. An asterisk indicates a radiolabeled phosphate.
When the post-labeling assay was carried out on the 14-mer, the fast-moving band corresponding to p*GT=(pAT) was observed (Figure 9B, lane 13). Upon irradiation with 254 nm light the expected p*GT band appeared along with a faster moving band (lane 14). The only reasonable assignment of this band is that it belongs to p*AT, which resulted from photoreversal of p*GT=(p*AT) that had probably become radiolabeled at A by a known phosphate exchange reaction with [γ-32P]-ATP catalylzed by PNK.39 Further treatment with CIP resulted in the formation of p*GT=(AT) or GT=(p*AT) and eventual loss of all labeling as in the pre-labeled case (lane 15). The apparent efficiency of the exchange labeling reaction suggests that the CIP step could be eliminated from the post-labeling sequence, thereby shortening the procedure. The formation of p*GT and p*AT as separable bands following photoreversal of p*GT=(p*AT) also demonstrates how anti T=T CPDs formed in different sequence contexts can be distinguished and quantified.
4. Conclusions
We describe a radioactive post-labeling assay for the approximately 10 fmol level detection of non-adjacent trans,anti- and cis,anti-T=(T) CPDs indicative of non-B DNA folded structures in the presence of adjacent cis-syn CPDs indicative of native B DNA. Lower levels of detection are expected to be achieved by chromatographic isolation of the tetranucleotide degradation products prior to the postlabeling assay, coupled with isolation of the radiolabeled tetranucleotide band prior to photoreversal with 254 nm light. The ability of 254 nm light to photorevert the anti CPD-containing tetranucleotide products to faster moving dinucleotide products can be used to confirm their identity as non-adjacent CPDs and not partially degraded DNA, or other non-adjacent photoproducts such as (6–4) or Dewar products that cannot be photoreverted. This post-labeling method is also expected to work for non-adjacent anti T=(U), U=(T) and U=(U) CPDs resulting from deaminated C-containing anti CPDs.20, 40 It remains to be established whether this method will also work for regio-isomeric non-adjacent cis,syn and trans,syn CPDs. Because this assay detects non-adjacent CPDs, it will be selective for detecting non-B DNA secondary structures such as chair, basket, and reverse Hoogsteen hairpins containing pyrimidines in interacting loops of sufficient length as found in human telomeres21, 24 and certain promoters.20 It will not be able to detect such structures with short loops or lacking pyrimidines, as well as parallel or hybrid G-quadruplex structures, thereby providing a new type of selectivity for non-B DNA structures that are not specifically detected or identifiable by other methods. The principal advantage of this phototrapping method is that the dynamics of G-quadruplexes are not perturbed in the probe step. Both the post-labeling and pre-labeling assays for anti CPDs should also find application for in vitro studies of non-B structures. Once anti CPDs can be verified to form in vivo, Next-Gen sequencing assays can be developed to map the location of these photoproducts, and hence non-B structures, in vivo. One approach would be to develop antibodies against the anti CPDs and map them by a ChIP-Seq assay.41 Another would be to first photorevert cis,syn CPDs with photolyase and then degrade the DNA with a 3’ to 5’ exonuclease that would be blocked by non-adjacent anti CPD’s and adjacent (6–4) and Dewar products. The ends blocked by anti CPDs, however, could then be specifically restored by photoreversal with 254 nmlight for ligation to sequencing linkers.
ACKNOWLEDGEMENTS
This material is based upon work supported by the National Science Foundation under Grant No. 2003688 (JST) and an NIH/NIGMS Biomedical Mass Spectrometry Resource grant 5R24GM-136766-02 (MLG).
Footnotes
All authors declare no competing financial interest.
REFERENCES
- (1).Sinden RR Slipped strand DNA structures. Front. Biosci 2007, 12, 4788. [DOI] [PubMed] [Google Scholar]
- (2).Saini N, Zhang Y, Usdin K, and Lobachev KS When secondary comes first - the importance of non-canonical DNA structures. Biochimie 2013, 95, 117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Thys RG, Lehman CE, Pierce LC, and Wang YH DNA secondary structure at chromosomal fragile sites in human disease. Curr. Genomics 2015, 16, 60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Sullivan ED, Longley MJ, and Copeland WC Polymerase γ efficiently replicates through many natural template barriers but stalls at the HSP1 quadruplex. J. Biol. Chem 2020, 295, 17802–17815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Bochman ML, Paeschke K, and Zakian VA DNA secondary structures: stability and function of G-quadruplex structures. Nat. Rev. Genet 2012, 13, 770–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Spiegel J, Adhikari S, and Balasubramanian S The Structure and Function of DNA G-Quadruplexes. Trends Chem. 2020, 2, 123–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Linke R, Limmer M, Juranek S, Heine A, and Paeschke K The Relevance of G-Quadruplexes for DNA Repair. Int. J. Mol. Sci 2021, 22, 12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ma DL, Wang M, Lin S, Han QB, and Leung CH Recent Development of G-Quadruplex Probes for Cellular Imaging. Curr. Top. Med. Chem 2015, 15, 1957–1963. [DOI] [PubMed] [Google Scholar]
- (9).Islam MK, Jackson PJ, Rahman KM, and Thurston DE Recent advances in targeting the telomeric G-quadruplex DNA sequence with small molecules as a strategy for anticancer therapies. Future Med. Chem 2016, 8, 1259–1290. [DOI] [PubMed] [Google Scholar]
- (10).Liu HY, Zhao Q, Zhang TP, Wu Y, Xiong YX, Wang SK, Ge YL, He JH, Lv P, Ou TM, Tan JH, Li D, Gu LQ, Ren J, Zhao Y, and Huang ZS Conformation Selective Antibody Enables Genome Profiling and Leads to Discovery of Parallel G-Quadruplex in Human Telomeres. Cell Chem. Biol 2016, 23, 1261–1270. [DOI] [PubMed] [Google Scholar]
- (11).Manna S, and Srivatsan SG Fluorescence-based tools to probe G-quadruplexes in cell-free and cellular environments. RSC Adv. 2018, 8, 25673–25694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Raguseo F, Chowdhury S, Minard A, and Di Antonio M Chemical-biology approaches to probe DNA and RNA G-quadruplex structures in the genome. Chem. Commun 2020, 56, 1317–1324. [DOI] [PubMed] [Google Scholar]
- (13).Yuan JH, Shao W, Chen SB, Huang ZS, and Tan JH Recent advances in fluorescent probes for G-quadruplex nucleic acids. Biochem. Biophys. Res. Commun 2020, 531, 18–24. [DOI] [PubMed] [Google Scholar]
- (14).Di Antonio M, Ponjavic A, Radzevicius A, Ranasinghe RT, Catalano M, Zhang X, Shen J, Needham LM, Lee SF, Klenerman D, and Balasubramanian S Single-molecule visualization of DNA G-quadruplex formation in live cells. Nat. Chem 2020, 12, 832–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jin M, Li J, Chen Y, Zhao J, Zhang J, Zhang Z, Du P, Zhang L, and Lu X Near-Infrared Small Molecule as a Specific Fluorescent Probe for Ultrasensitive Recognition of Antiparallel Human Telomere G-Quadruplexes. ACS Appl. Mater. Interfaces 2021, 13, 32743–32752. [DOI] [PubMed] [Google Scholar]
- (16).Han J-N, Ge M, Chen P, Kuang S, and Nie Z Advances in G-quadruplexes-based fluorescent imaging. Biopolymers 2022, 113, e23528. [DOI] [PubMed] [Google Scholar]
- (17).Zheng B-X, Yu J, Long W, Chan KH, Leung AS-L, and Wong W-L Structurally diverse G-quadruplexes as the noncanonical nucleic acid drug target for live cell imaging and antibacterial study. Chem. Commun 2023. [DOI] [PubMed] [Google Scholar]
- (18).Hoffman EA, Frey BL, Smith LM, and Auble DT Formaldehyde crosslinking: a tool for the study of chromatin complexes. J. Biol. Chem 2015, 290, 26404–26411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Taylor JS Adjacent and Nonadjacent Dipyrimidine Photoproducts as Intrinsic Probes of DNA Secondary and Tertiary Structure(dagger). Photochem. Photobiol 2023, 99, 277–295. [DOI] [PubMed] [Google Scholar]
- (20).Smith-Carpenter JE, and Taylor JS Photocrosslinking of G-Quadruplex-Forming Sequences found in Human Promoters. Photochem. Photobiol 2019, 95, 252–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Lu C, Smith-Carpenter JE, and Taylor JA Evidence for Reverse Hoogsteen Hairpin Intermediates in the Photocrosslinking of Human Telomeric DNA Sequences. Photochem. Photobiol 2018, 94, 685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Smith JE, Lu C, and Taylor JS Effect of sequence and metal ions on UVB-induced anti cyclobutane pyrimidine dimer formation in human telomeric DNA sequences. Nucleic Acids Res. 2014, 42, 5007–5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Su DG, Kao JL, Gross ML, and Taylor JS Structure determination of an interstrand-type cis-anti cyclobutane thymine dimer produced in high yield by UVB light in an oligodeoxynucleotide at acidic pH. J. Am. Chem. Soc 2008, 130, 11328–11337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Su DG, Fang H, Gross ML, and Taylor JS Photocrosslinking of human telomeric G-quadruplex loops by anti cyclobutane thymine dimer formation. Proc. Natl. Acad. Sci. U.S.A 2009, 106, 12861–12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Law YK, Azadi J, Crespo-Hernandez CE, Olmon E, and Kohler B Predicting thymine dimerization yields from molecular dynamics simulations. Biophys. J 2008, 94, 3590–3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Lingbeck JM, and Taylor JS Preparation and characterization of DNA containing a site-specific nonadjacent cyclobutane thymine dimer of the type implicated in UV-induced −1 frameshift mutagenesis. Biochemistry 1999, 38, 13717–13724. [DOI] [PubMed] [Google Scholar]
- (27).Weinfeld M, Soderlind KJ, and Buchko GW Influence of nucleic acid base aromaticity on substrate reactivity with enzymes acting on single-stranded DNA. Nucleic Acids Res. 1993, 21, 621–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Romier C, Dominguez R, Lahm A, Dahl O, and Suck D Recognition of single-stranded DNA by nuclease P1: high resolution crystal structures of complexes with substrate analogs. Proteins 1998, 32, 414–424. [PubMed] [Google Scholar]
- (29).Wang Y, Taylor JS, and Gross ML Nuclease P1 digestion combined with tandem mass spectrometry for the structure determination of DNA photoproducts. Chem. Res. Toxicol 1999, 12, 1077–1082. [DOI] [PubMed] [Google Scholar]
- (30).Douki T, Laporte G, and Cadet J Inter-strand photoproducts are produced in high yield within A-DNA exposed to UVC radiation. Nucleic Acids Res. 2003, 31, 3134–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Schreier WJ, Schrader TE, Koller FO, Gilch P, Crespo-Hernandez CE, Swaminathan VN, Carell T, Zinth W, and Kohler B Thymine dimerization in DNA is an ultrafast photoreaction. Science 2007, 315, 625–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Phillips DH On the origins and development of the (32)P-postlabelling assay for carcinogen-DNA adducts. Cancer Lett. 2013, 334, 5–9. [DOI] [PubMed] [Google Scholar]
- (33).Reddy MV Methods for testing compounds for DNA adduct formation. Regul. Toxicol. Pharmacol 2000, 32, 256–263. [DOI] [PubMed] [Google Scholar]
- (34).Phillips DH Detection of DNA modifications by the 32P-postlabelling assay. Mutat. Res 1997, 378, 1–12. [DOI] [PubMed] [Google Scholar]
- (35).Keith G, and Dirheimer G Postlabeling: a sensitive method for studying DNA adducts and their role in carcinogenesis. Curr Opin Biotechnol 1995, 6, 3–11. [DOI] [PubMed] [Google Scholar]
- (36).Weinfeld M, Liuzzi M, and Paterson MC Enzymatic analysis of isomeric trithymidylates containing ultraviolet light-induced cyclobutane pyrimidine dimers. II. Phosphorylation by phage T4 polynucleotide kinase. J. Biol. Chem 1989, 264, 6364–6370. [PubMed] [Google Scholar]
- (37).Parikh D, Fouquerel E, Murphy CT, Wang H, and Opresko PL Telomeres are partly shielded from ultraviolet-induced damage and proficient for nucleotide excision repair of photoproducts. Nat. Commun 2015, 6, 8214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Hertzberg RP, and Dervan PB Cleavage of DNA with methidiumpropyl-EDTA-iron(II): reaction conditions and product analyses. Biochemistry 1984, 23, 3934–3945. [DOI] [PubMed] [Google Scholar]
- (39).van de Sande JH, Kleppe K, and Khorana HG Reversal of bacteriophage T4 induced polynucleotide kinase action. Biochemistry 1973, 12, 5050–5055. [DOI] [PubMed] [Google Scholar]
- (40).Cannistraro VJ, Pondugula S, Song Q, and Taylor JS Rapid deamination of cyclobutane pyrimidine dimer photoproducts at TCG sites in a translationally and rotationally positioned nucleosome in vivo. J. Biol. Chem 2015, 290, 26597–26609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Nakato R, and Sakata T Methods for ChIP-seq analysis: A practical workflow and advanced applications. Methods 2021, 187, 44–53. [DOI] [PubMed] [Google Scholar]