

Abstract
Summary
Structure determination is a key step in the functional characterization of many non-coding RNA molecules. High-resolution RNA 3D structure determination efforts, however, are not keeping up with the pace of discovery of new non-coding RNA sequences. This increases the importance of computational approaches and low-resolution experimental data, such as from the small-angle X-ray scattering experiments. We present RNA Masonry, a computer program and a web service for a fully automated modeling of RNA 3D structures. It assemblies RNA fragments into geometrically plausible models that meet user-provided secondary structure constraints, restraints on tertiary contacts, and small-angle X-ray scattering data. We illustrate the method description with detailed benchmarks and its application to structural studies of viral RNAs with SAXS restraints.
Availability and implementation
The program web server is available at http://iimcb.genesilico.pl/rnamasonry. The source code is available at https://gitlab.com/gchojnowski/rnamasonry.
1 Introduction
Non-coding RNAs (ncRNAs) are involved in the regulation of many cellular processes. New families of ncRNAs are being continuously discovered (Kalvari et al. 2021). The function of newly characterized ncRNAs can be deciphered from their structure in an approach that proved very successful for proteins (Miao et al. 2020). The experimental determination of high-resolution 3D structures of ncRNAs (e.g. by X-ray crystallography, nuclear magnetic resonance, or cryo-electron microscopy) is however difficult, and only a very small fraction of ncRNA families have high-resolution structures available for at least one member (134 out of 4108 according to Rfam 14.9 (Kalvari et al. 2021)). Computational structure prediction methods offer an alternative to experimental structure determination, but the purely theoretical models are often of limited accuracy. One promising approach is the computational modeling with the use of restraints derived from low-resolution experimental data (Bernetti et al. 2021, Mazzanti et al. 2021, Fang et al. 2022). To address this issue, we developed RNA Masonry, a computer program for fully automated modeling of RNA molecules by assembly of recurrent 3D motifs, with the use of low-resolution restraints. The 3D motifs (further referred to as fragments) are retrieved from the RNA Bricks database (Chojnowski et al. 2014), which catalogues recurrent substructures observed in experimentally determined, high-resolution RNA structural models available in the Protein Data Bank (wwPDB Consortium 2018).
RNA Masonry provides two basic functionalities: (i) de novo fragment assembly and (ii) RNA model refinement. The input is either RNA sequence with the secondary structure or a preliminary RNA 3D structural model. The input atomic model is analyzed by the ClaRNA program (Walen et al. 2014) to produce the secondary structure, which is then used as input to RNA Masonry in the de novo modeling mode. Pseudo-knots in the secondary structure are removed and used as additional contact restraints.
RNA Masonry is based on hierarchical organization of RNA molecules, which are composed of 3D motifs defined at a secondary structure level; double-stranded helices, single-stranded fragments, and various types of loops. During the model assembly, the motifs are used as a whole, which strictly preserves the input secondary structure. The program accepts restraints for long-range tertiary interactions. Additionally, the model building can be restrained with a goodness-of-fit to the experimental small-angle X-ray scattering (SAXS) data, which is calculated using FoXS (Schneidman-Duhovny et al. 2016) or CRYSOL (Franke et al. 2017) depending on user preferences.
There are other methods available that can assemble RNA 3D structures from fragments of experimentally determined structures like Assemble (Jossinet et al. 2010) and RNAComposer (Popenda et al. 2012). There are also approaches designed specifically for the purpose of modeling with SAXS restraints (Gajda et al. 2013), or filtering large sets of tentative models (Yang et al. 2010). To the best of our knowledge, however, RNA Masonry is the only tool that combines statistical potential with experimental restraints and uses a regularly updated database of fragments (RNA Bricks is updated weekly, http://genesilico.pl/rnabricks2).
2 Materials and methods
2.1 Input processing
If the input is an RNA atomic model (mmCIF or PDB format), the program annotates its secondary structure with ClaRNA which is further processed analogously to a secondary structure given directly on input for de novo modeling.
Pseudo-knots are removed from the input secondary structure using the K2N library (Smit et al. 2008) to maximize the number of nested base pairs (Ponty 2006), and stored as additional restraints. The pseudo-knot free secondary structure is subsequently decomposed into structural motifs and encoded as an RNA motif-graph introduced in the RNA Bricks database as described previously (Chojnowski et al. 2014). Briefly, the graph nodes correspond to RNA structural motifs (double-stranded helices, loops, and single-stranded fragments). The graph edges denote nucleotides shared by two neighboring motifs. Owing to the absence of pseudo-knots in the basic data structure, the motif graphs are trees, albeit not necessarily rooted. Pseudo-knots are nonetheless enforced at the level of structural restraints.
2.2 Selecting RNA 3D fragments
After building the motif-graph, which is a basic data structure used in RNA Masonry, 3D fragments from RNA Bricks database are assigned to each of its nodes. A fragment assigned to a node must match its secondary structure (stem or a loop with specific topology), but no sequence information is considered. If the number of fragments assigned to a node is fewer than a user-defined threshold (10 by default) additional fragments are generated by circular permutations (e.g. by changing ends of symmetrical, internal loops). If needed, new fragments are also automatically built from smaller ones by introducing insertions using ModeRNA (Rother et al. 2011). Finally, each fragment is mutated to the target sequence. Any steric conflicts, unsatisfied pseudo-knot restraints, or implausible geometries introduced at this stage are ignored, but penalized later during fragment assembly steps by the SimRNA (Boniecki et al. 2016) scoring function.
2.3 Starting RNA model assembly
If RNA 3D structure is given as an input, it is used as a starting model without any further modifications. Otherwise, the program assembles a starting model with a possibly small number of severe clashes, which increases the convergence of the subsequent refinement step.
In principle, the aim is to find a configuration of fragments, assigned to each of the motif-graph nodes, which minimizes the number of clashes in a complete model. A clash occurs when two glycosidic nitrogens (N1 for pyrimidines, N9 for purines) are closer than 6 Å apart. To reduce the combinatorial complexity of the task, the assembly process is initiated from the terminal elements of the structure (the motif graph ‘leaves’) and proceeds iteratively by adding a single node at a time. At each step, from all possible partial models only statistically relevant representatives are selected as described by Hajdin et al. (2010).
2.4 Model optimization
The starting model is optimized in a replica exchange Monte-Carlo simulation with a geometric distribution of temperature levels. We use a scoring function based on the SimRNA statistical potential (Boniecki et al. 2016) that enables the use of the long-range tertiary contact restraints (e.g. to indicate pseudo-knots). If SAXS curve is given on input the score is additionally multiplied by a correction factor penalizing poor agreement of a full-atom model representation with experimental data. The correction factor quantifies deviations of the χ2 value from 1 with a mixture of two Gaussians with variances 1 and 10, where a perfect fit (χ2 of 1) corresponds to a value 1.5.
In a single step, a random motif-graph node is selected. Next, from a set of fragments assigned to the node a random one is selected and inserted into corresponding position in the model. For all motifs except terminal loops, this operation changes conformation of a whole domain in the RNA model. The user-provided secondary structure is preserved at each step. The best-scored (lowest pseudo-energy) model constructed during the entire simulation is returned to the user.
3 Structural studies of viral RNAs with SAXS restraints
We used an early version RNA Masonry to model the adenovirus virus-associated RNA (VA-I) structure with SAXS restraints (Dzananovic et al. 2017). Subsequently, a crystal structure of VA-I has been determined and turned out to be inconsistent with the results of scattering experiments with goodness-of-fit parameter (χ2) estimated using CRYSOL of 2.8 (Hood et al. 2019) (Fig. 1A). We used RNA Masonry to optimize the fit of the structure to the SAXS curve, while preserving its secondary structure, which could have been very accurately determined using a crystal structure model. The resulting model fits better not only to the experimental data (χ2 = 1.5), but also to the ab initio reconstruction obtained independently using DAMMIF (Franke and Svergun 2009) (Fig. 1B). This result supports a hypothesis that in solution and in the absence of crystal-lattice constraints VA-I may be flexible and sample additional conformations, possibly at the expense of the pseudo-knot disruption (Hood et al. 2019).
Figure 1.
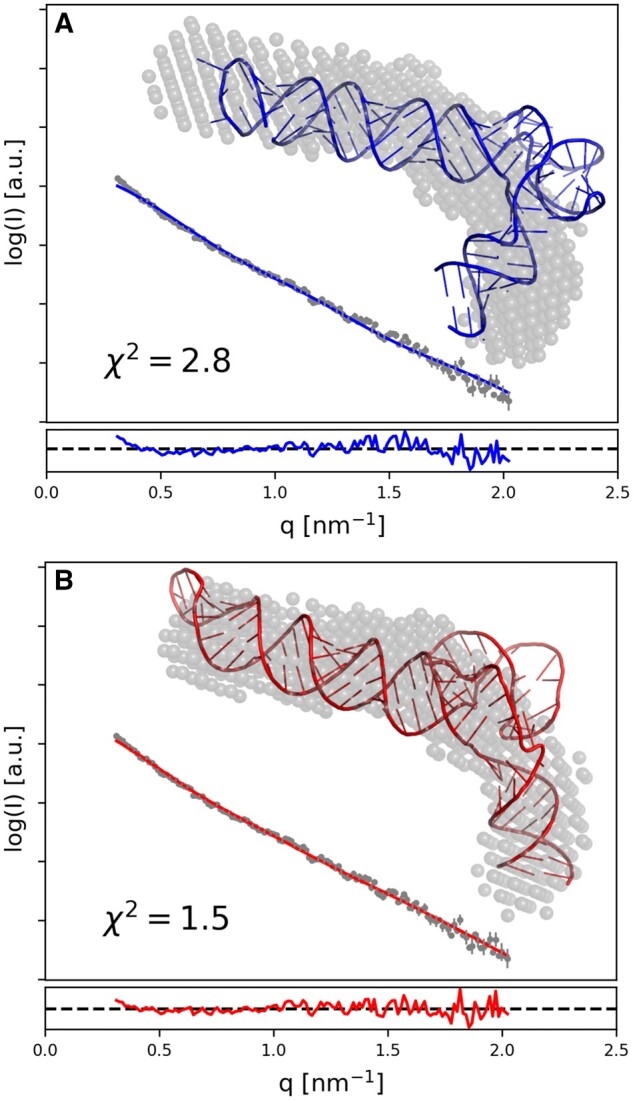
Crystal structure of adenovirus virus-associated RNA (A) was observed to have a conformation inconsistent with results of earlier SAXS experiments. RNA Masonry was used to optimize the fit to experimental SAXS data preserving the crystal structure secondary contacts (B). An ab initio model obtained with DAMMIF isshown with grey spheres. Residuals of model-calculated and experimental SAXS curve fit are shown in the bottom panels.
4 Implementation
RNA Masonry and all utility programs were implemented in Python 3 and C++ with an extensive use of routines from the Computational Crystallography Toolbox (cctbx) (Grosse-Kunstleve et al. 2002), Numpy, Scipy, NetworkX, Biopython (Cock et al. 2009), ClaRNA, ModeRNA, and K2N libraries. The web server engine used in this work is a part of rna-tools toolbox (Magnus et al. 2020) and can be freely reused for new applications.
5 Conclusions
RNA Masonry is a computer program for fully automated modeling of RNA 3D structures by fragment assembly. The RNA structure representation used here significantly reduces the number of degrees of freedom, which in principle equals the number of joints (internal loops) in the target structure. The small number of degrees of freedom significantly reduces the modeling time (e.g. compared to SimRNA simulations). In the case of modeling with SAXS data it also reduces the risk of overfitting. The SAXS experiments provide a number of unique observations much smaller than the already limited number of experimental data points (Konarev and Svergun 2015). This increases the impact of experimental errors on models with more parameters. However, in cases where SAXS curves represent an average over multiple model states, a small number of RNA Masonry model parameters may be not enough to explain the complexity of experimental data. Overall, we show using simulated and experimental SAXS data that RNA Masonry can provide accurate models in most of the cases (see Supplementary data).
It must be stressed, that the presented approach may be unable to model fine details of RNA structures (e.g. non-canonical interactions) that are not encoded within the 3D motifs used for assembly. These, however, can be modeled using complementary high-resolution approaches (e.g. using QRNAS (Stasiewicz et al. 2019), SimRNA, or ROSETTA/FARFAR (Das et al. 2010)). This issue does not affect alternative approach where SAXS data are used for filtering large decoy sets generated using methods that can explicitly model non-canonical interactions, for example MC-Sym and MC-Fold pipeline (Yang et al. 2010).
The current RNA Masonry version selects RNA fragments for model building based exclusively on canonical secondary structure constraints. It was observed, however, that the presence of certain motif types and coaxial stacking between adjacent helices can be reliably predicted from sequence (Tyagi and Mathews 2007, Cruz and Westhof 2011). Both these a priori information could be in principle used during model building in RNA Masonry to increase reliability of resulting models. This can be achieved by a more restrictive selection of fragments for model assembly and penalizing the absence of expected stacking interactions by the SimRNA scoring function. We plan such an extension in further releases of the software.
Supplementary Material
Acknowledgements
Computational resources for SimRNA simulations used in the benchmark study were provided by the Poznań Supercomputing and Networking Center at the Institute of Bioorganic Chemistry, Polish Academy of Sciences through the Polish Grid Infrastructure.
Contributor Information
Grzegorz Chojnowski, International Institute of Molecular and Cell Biology, Warsaw 02-109, Poland; European Molecular Biology Laboratory, Hamburg Unit, Hamburg 22607, Germany.
Rafał Zaborowski, International Institute of Molecular and Cell Biology, Warsaw 02-109, Poland.
Marcin Magnus, ReMedy International Research Agenda Unit, IMol Polish Academy of Sciences, Warsaw, Poland.
Sunandan Mukherjee, International Institute of Molecular and Cell Biology, Warsaw 02-109, Poland.
Janusz M Bujnicki, International Institute of Molecular and Cell Biology, Warsaw 02-109, Poland.
Supplementary data
Supplementary data are available at Bioinformatics online.
Funding
G.C. was supported by the European Research Council (ERC, 261351, grant to J.M.B.). S.M. was supported by the National Science Center, Poland (NCN, 2021/43/D/NZ1/03360). J.M.B. was supported by the Polish National Science Center (NCN, 2017/26/A/NZ1/01083). M.M. was supported by the ‘Regenerative Mechanisms for Health-ReMedy’ (MAB/2017/2), carried out within the International Research Agendas Program of the Foundation for Polish Science co-financed by the European Union under the European Regional Development Fund. Funding for open access charge: European Molecular Biology Laboratory.
Conflict of interest
None declared.
Data availability
The data underlying this article are available in the online supplementary material.
References
- Bernetti M, Hall KB, Bussi G.. Reweighting of molecular simulations with explicit-solvent SAXS restraints elucidates ion-dependent RNA ensembles. Nucleic Acids Res 2021;49:e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boniecki MJ, Lach G, Dawson WK. et al. SimRNA: a coarse-grained method for RNA folding simulations and 3D structure prediction. Nucleic Acids Res 2016;44:e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chojnowski G, Walen T, Bujnicki JM.. RNA bricks–a database of RNA 3D motifs and their interactions. Nucleic Acids Res 2014;42:D123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cock PJA, Antao T, Chang JT. et al. Biopython: freely available python tools for computational molecular biology and bioinformatics. Bioinformatics 2009;25:1422–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz JA, Westhof E.. Sequence-based identification of 3D structural modules in RNA with RMDetect. Nat Methods 2011;8:513–21. [DOI] [PubMed] [Google Scholar]
- Das R, Karanicolas J, Baker D.. Atomic accuracy in predicting and designing noncanonical RNA structure. Nat Methods 2010;7:291–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzananovic E, Chojnowski G, Deo S. et al. Impact of the structural integrity of the three-way junction of adenovirus VAI RNA on PKR inhibition. PLoS One 2017;12:e0186849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Gallego J, Wang Y-X.. Deriving RNA topological structure from SAXS. In: Tainer, J.A. (ed.), Methods in Enzymology. Elsevier, 2022, 479–529. [DOI] [PubMed] [Google Scholar]
- Franke D, Petoukhov MV, Konarev PV. et al. ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J Appl Crystallogr 2017;50:1212–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke D, Svergun DI.. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J Appl Crystallogr 2009;42:342–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajda MJ, Martinez Zapien D, Uchikawa E. et al. Modeling the structure of RNA molecules with small-angle X-ray scattering data. PLoS One 2013;8:e78007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse-Kunstleve RW, Sauter NK, Moriarty NW. et al. The computational crystallography toolbox: crystallographic algorithms in a reusable software framework. J Appl Crystallogr 2002;35:126–36. [Google Scholar]
- Hajdin CE, Ding F, Dokholyan NV. et al. On the significance of an RNA tertiary structure prediction. RNA 2010;16:1340–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood IV, Gordon JM, Bou-Nader C. et al. Crystal structure of an adenovirus virus-associated RNA. Nat Commun 2019;10:2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jossinet F, Ludwig TE, Westhof E.. Assemble: an interactive graphical tool to analyze and build RNA architectures at the 2D and 3D levels. Bioinformatics 2010;26:2057–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalvari I, Nawrocki EP, Ontiveros-Palacios N. et al. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res 2021;49:D192–D200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarev PV, Svergun DI.. A posteriori determination of the useful data range for small-angle scattering experiments on dilute monodisperse systems. IUCrJ 2015;2:352–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnus M, Antczak M, Zok T. et al. RNA-Puzzles toolkit: a computational resource of RNA 3D structure benchmark datasets, structure manipulation, and evaluation tools. Nucleic Acids Res 2020;48:576–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzanti L, Alferkh L, Frezza E. et al. Biasing RNA coarse-grained folding simulations with small-angle X-ray scattering data. J Chem Theory Comput 2021;17:6509–21. [DOI] [PubMed] [Google Scholar]
- Miao Z, Adamiak RW, Antczak M. et al. RNA-Puzzles round IV: 3D structure predictions of four ribozymes and two aptamers. RNA 2020;26:982–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponty Y. Modélisation de séquences génomiques structurées, génération aléatoire et applications. Université Paris Sud-Paris XI, 2006.
- Popenda M, Szachniuk M, Antczak M. et al. Automated 3D structure composition for large RNAs. Nucleic Acids Res 2012;40:e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rother M, Rother K, Puton T. et al. ModeRNA: a tool for comparative modeling of RNA 3D structure. Nucleic Acids Res 2011;39:4007–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Hammel M, Tainer JA. et al. FoXS, FoXSDock and MultiFoXS: single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles. Nucleic Acids Res 2016;44:W424–W429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit S, Rother K, Heringa J. et al. From knotted to nested RNA structures: a variety of computational methods for pseudoknot removal. RNA 2008;14:410–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasiewicz J, Mukherjee S, Nithin C. et al. QRNAS: software tool for refinement of nucleic acid structures. BMC Struct Biol 2019;19:5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi R, Mathews DH.. Predicting helical coaxial stacking in RNA multibranch loops. RNA 2007;13:939–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walen T et al. ClaRNA: a classifier of contacts in RNA 3D structures based on a comparative analysis of various classification schemes. Nucleic Acids Res 2014;42:e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- wwPDB Consortium. Protein Data Bank: the single global archive for 3D macromolecular structure data. Nucleic Acids Res 2018;47:D520–D528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Parisien M, Major F. et al. RNA structure determination using SAXS data. J Phys Chem B 2010;114:10039–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the online supplementary material.