

Abstract
The current rapid extinction of species leads not only to their loss but also the disappearance of the unique features they harbour, which have evolved along the branches of the underlying evolutionary tree. One proxy for estimating the feature diversity (FD) of a set S of species at the tips of a tree is ‘phylogenetic diversity’ (PD): the sum of the branch lengths of the subtree connecting the species in S. For a phylogenetic tree that evolves under a standard birth–death process, and which is then subject to a sudden extinction event at the present (the simple ‘field of bullets’ model with a survival probability of s per species) the proportion of the original PD that is retained after extinction at the present is known to converge quickly to a particular concave function as t grows. To investigate how the loss of FD mirrors the loss of PD for a birth–death tree, we model FD by assuming that distinct discrete features arise randomly and independently along the branches of the tree at rate r and are lost at a constant rate . We derive an exact mathematical expression for the ratio of the two expected feature diversities (prior to and following an extinction event at the present) as t becomes large. We find that although has a similar behaviour to (and coincides with it for ), when , is described by a function that is different from . We also derive an exact expression for the expected number of features that are present in precisely one extant species. Our paper begins by establishing some generic properties of FD in a more general (non-phylogenetic) setting and applies this to fixed trees, before considering the setting of random (birth–death) trees.
Keywords: Feature diversity, Phylogenetic diversity, Birth–death processes, Extinction
Introduction
Phylogenetic trees provide a way to quantify biodiversity and the extent to which it might be lost in the face of the current mass extinction event. One such biodiversity measure is phylogenetic diversity (PD), which associates to each subset S of extant species, the sum of the branch lengths of the underlying evolutionary tree that connects (just) these species to the root of the tree. This measure, pioneered by Faith (1992), provides a more complete measure of biodiversity than merely counting the number of species in S (see e.g. Miller et al. 2018). Moreover, if new features evolve along the branches of a tree and are never lost, then the resulting features present amongst the species in the subset S (the feature diversity (FD) of S) are directly correlated with the phylogenetic diversity of S (Wicke et al. 2021). However, when features are lost, it has recently been shown mathematically that under simple (deterministic or stochastic) models of feature gain and loss, FD necessarily deviates from PD except for very trivial types of phylogenetic trees (Theorems 2 and 3 of Rosindell et al. (2022)). More generally, the question of the extent to which PD captures feature or functional diversity has been the subject of considerable debate in the biological literature (see Devictor et al. 2010; Mazel et al. 2017, 2018, 2019; Owen et al. 2019; Tucker et al. 2018, 2019).
In this paper, we investigate a related question: under a standard phylogenetic diversification model and a simple stochastic process of feature gain and loss, what proportion of feature diversity is expected to be lost in a mass extinction event at the present? And how does this ratio compare with the expected phylogenetic diversity that will be lost? Although the latter ratio (for PD) has been determined in earlier work, here we provide a corresponding result for FD, and show how it differs from PD when the rate of feature loss is non-zero. Our results suggest that the relative loss of FD under a mass extinction event at the present is greater than the relative loss of PD. We also investigate the number of features that are expected to be found in just one species at the present.
We begin by considering the properties of FD in a more general setting based purely on the species themselves (i.e. not involving any underlying phylogenetic tree or network) and then consider FD on fixed phylogenetic trees before presenting the results for (random) birth–death trees. Some of the results of these earlier sections are applied in the later sections.
General properties of feature diversity without reference to phylogenies
This section considers the generic properties of expected FD for sets of species, and thus, no underlying phylogenetic tree or stochastic process that generates a tree, or model of feature evolution is assumed. We mostly follow the notation from Wicke et al. (2021).
Definitions
Let X be a labelled set of species, and for each , let be a non-empty set of discrete features (e.g. genes, genomic inserts, traits) that are associated with species x.
The collection of ordered pairs is called a feature assignment on X.
For any subset A of X, let and let be a function assigning some richness or novelty to a feature .
- The feature diversity of some subset A of a set of species X is defined as,
2.1
A default option for is to set , which simply counts the number of features present. Notice that for any subset A of X we have with equality if and only if no features are shared by any two species.
Feature diversity loss under a ‘Field of Bullets’ model of extinction at the present
Definition 1.1
Consider a sudden extinction event taking place across a set of species (Raup 1993). In the generalised field of bullets (g-FOB) model, each species either survives the extinction event (with probability ) or disappears (with probability ), and these survival events are assumed to be independent among the species. We write (or, more briefly, ) to denote the vector and we let denote the (random) subset of X corresponding to the species that survive the extinction event. If for all , then we have the simpler field of bullets (FOB) model.
We define the following quantity:
which is the proportion of feature diversity that survives the extinction event. In the case of the FOB model, we denote this ratio by .
Proposition 1.1
For the g-FOB model,
where are the normalised values (which sum to 1).
For the FOB model, the equation in Proposition 2.1 simplifies to
| 2.2 |
where , the number of species in X that possess feature f.
Proof
We have where is the Bernoulli variable that takes the value 1 if at least one species in X with feature f survives the g-FOB extinction event, and 0 otherwise. Applying linearity of expectation and noting that gives the result.
Notice that at . The behaviour between these two extreme values of s is described next.
Proposition 1.2
Under the FOB model, the following hold:
-
(i)
for all .
-
(ii)
for a value of if and only if the sets in are pairwise disjoint, in which case for all .
-
(iii)
If the sets are not pairwise disjoint, then is a strictly concave increasing function of s.
Proof
Part (i): Since in Eq. (2.2) and for all , the claimed inequality is immediate. Part (ii): If then for all , and if then for every . Part (iii): By Eq. (2.2),
and
for all , from which the claimed results follow.
Approximating by its expected value
For each , let be a labelled set of n species with feature assignment . Let denote the (random) set of species after a g-FOB extinction event with survival probability vector , which assigns each a corresponding survival probability . Note that we make no assumption regarding how the species in and are related (e.g. they may be disjoint, overlapping or nested), or any apriori relationship between and .
In the following result, we provide a sufficient condition under which is likely to be close to its expected value (readily computed via Eq. (2.1)) when the number of species n is large. This condition allows some species to contribute proportionately more FD than other species do on average, and this proportion can grow with n, provided that it does not grow too quickly.
Proposition 1.3
- For ,
where . If as then .
- Let (the average contribution of each species to the total FD), and suppose that for each , , where and (e.g. . We then have the following convergence in probability as :
Proof
Let be a sequence of Bernoulli random variables where each takes the value of 1 if species survives and 0 otherwise. For the g-FOB model, the random variables are independent. We can write where is the ratio Observe that for any particular value of , if we change (from 0 to 1 or visa versa) to give then:
We now apply McDiarmid’s inequality McDiarmid (1989) to obtain (for each ):
| 2.3 |
This establishes Part (a). Part (b) now follows immediately, and Part (c) follows from Part (b), since the condition in Part (c) implies that
as .
Remark
Note that Proposition 2.3(c) can fail when the condition stated in Part (c) does not hold, even for the simpler FOB model. We provide a simple example to demonstrate this. Let and let for and where f and g are distinct features with . In this case:
Therefore, does not converge in probability to 0 (or to any constant) as .
Consequences for phylogenetic diversity
Proposition 2.2 and Proposition 2.3 provide a simple way to derive certain results concerning phylogenetic diversity - both on rooted trees and also for rooted phylogenetic networks (specifically for the subNet diversity measure described in Wicke and Fischer (2018)). To each edge e of a rooted phylogenetic tree (or network), associate some unique feature and give it the value , where is the length of edge e in the tree (or network). For any subset Y of X (the leaf set of T) we then have:
It follows that for the simple field of bullets models, PD satisfies the concavity properties described in Proposition 2.2, where the condition that the sets are pairwise disjoint corresponds to the tree being a star tree. These results were established for rooted trees by specific tree-based arguments (see e.g. Sect. 5 of Lambert and Steel (2013)), but they directly follow from the more general framework above, and extend beyond trees.
Using this same link between PD and FD, Proposition 2.3 provides a further application to any sequence of rooted phylogenetic trees with n leaves and ultrametric edge lengths. For example, the ratio of surviving PD to original PD under the FOB model converges in probability to the expected value of this ratio for a sequence of trees if the total PD of grows at least as fast as , where L is the height of the tree and . This condition holds, for example, for Yule trees (Stadler and Steel 2012).
The feature diversity ratio for a model of feature evolution on a phylogenetic tree
Consider a rooted binary phylogenetic tree , in which each edge has a positive length that corresponds to a temporal duration, with the root of being placed at the top of a stem edge at time 0, and with each leaf in the leaf set of being placed at time t (as in Fig. 1). For convenience, we will assume in this section that for all features; however, this assumption can be relaxed (e.g. by allowing to take values in a fixed interval [a, b] where according to some fixed distribution, and independently between features) without altering the results significantly.
Fig. 1.

An example of feature gain and loss on a tree and the impact of extinction at the present. Left: New features arise (indicated by ) and disappear (indicated by −) along the branches of the tree. To simplify this example, no features are present at time 0; however, our results do not require this restriction. In total, there are five features present amongst the leaves of the tree at time t, namely . Right: An extinction event at the present (denoted by ) results in the loss of three of the extant species, leaving just three features being present among the leaves of the resulting pruned tree, namely . Thus, the ratio of surviving features to total features is
We let denote the (possibly empty) set of features present at time 0 (i.e. at the top of the stem edge), and we assume throughout this section that is bounded by some fixed constant B, independent of n.
On , we apply a stochastic process in which (discrete) features arise independently along the branches of this tree at rate r, and each feature that arises is novel (i.e. it has not appeared earlier elsewhere in the tree). Once a feature arises, it is then carried forward in time along the branches of (and is passed on to the two lineages arising at any speciation event). In addition, any feature can be lost from a lineage at any point according to a continuous-time pure-death process that operates at rate . This model was investigated in a different setting in Huson and Steel (2004) and studied more recently in Rosindell et al. (2022). Under this process, each leaf x of will have a (possibly empty) set of features (). Fig. 1 illustrates the processes described. Note that (for any ) and are now random variables.
Let denote the number of features at the end of any path P in that starts at time and ends at time . Then is described by a continuous-time Markov process that has a constant birth rate and a linear death rate. It is then a classical result Feller (1950) that has expected value , where (the number of features present at time 0), and converges to a Poisson distribution with mean as grows. Moreover, if then has a Poisson distribution for any value of (Feller (1950) p. 461), and so, regardless of the value of , the random variable has a Poisson distribution with mean where is the length of the path from the root to leaf x. The (random) number of features at any leaf x of (i.e. ) has the same distribution as . Note that for distinct leaves x and y of , the random variables and are not independent.
Notational convention: Henceforth we will write in place of and we will also write in place of when is the subset of the set of leaves of that survive under a FOB model with a survival probability s. We will let denote the set of features present at the root vertex at the top of the stem edge.
Lemma 1.1
Set . Then for any value of the following hold:
-
(i)
The random variable has a Poisson distribution.
-
(ii)The expected value of satisfies the following bound:
Proof
Part (i): We use induction on n. Since , the result for base case () holds by the results mentioned in the previous paragraph. Thus, suppose that , and let be a binary tree with n leaves and with . Then:
where the trees (with ) are subtrees of obtained by deleting the stem edge and its endpoints (and setting the set of features at the top of the stem edge of and equal to the empty set), and G is the number of features that arise on the stem edge of and are present in at least one leaf of .
Notice that , and G are independent random variables, and, by induction, and each have a Poisson distribution. Conditional on the number X of features that are present at the end of the stem edge, G has a binomial distribution with parameters X and p where p is the probability that a single feature present at the end of the stem edge is present in at least one leaf of . Since X has a Poisson distribution, and a Poisson number of Bernoulli random variables is Poisson, it follows that , being the sum of three independent Poisson variables, also has a Poisson distribution. This establishes the induction step and thus Part (i).
Part (ii): We have:
Now, for each , we have where denotes the length of the path from the top of the stem edge of to the leaf x. Thus
Example () Consider the process described on with . Let denote the length of the stem edge, and the length of each of the two pendant edges. Then has a Poisson distribution with expected value
and has expected value
In particular,
When , the right-hand side of this equation equals s, but for all other values it is strictly greater than s. Moreover, by differentiating it can be verified that this ratio is monotone decreasing as increases for all positive values of and ; in particular, for , and , this ratio is always less than the expected proportion of PD that survives in .
A limit result for sequences of trees
The main result of this section is Theorem 3.1, and its proof relies on establishing a sequence of preliminary lemmas.
Lemma 1.2
Let be a fixed constant. Given a sequence of trees with leaf set , let be the event that for every . Then .
Proof
We combine the Bonferroni inequality with a standard right-tail probability bound for a Poisson variable. Firstly, observe that:
| 3.1 |
Now, can be written as the sum of two independent random variables where has a Poisson distribution with mean , and with probability 1 ( counts the features at if , counts the features in that are remain present at , and B is the global bound on described near the start of Sect. 3). Thus, the Chernoff bound on the right hand tail of a Poisson variable (Mitzenmacher and Upfal 2005, p. 97) gives and so as n grows. Applying this to the inequality in (3.1) establishes the result.
Lemma 1.3
Let be a sequence of rooted binary trees, with having leaf set , and suppose that for some constant . Then converges in probability to 1 as .
Proof
First observe that we can write as the sum of two independent random variables, namely , where is the FD value when , and K is the number of features at the time at the root that are also present in at least one leaf. In particular, which is assumed to be bounded by a constant B (independent of n). Thus
By Lemma 3.1, , so , and thus:
| 3.2 |
as . We now apply Chebyshev’s inequality to obtain:
| 3.3 |
and since
we have:
and so the term on the right of Eq. (3.3) is bounded above by , which converges to 0 as by Eq. (3.2).
Lemma 1.4
Let be a sequence of rooted binary trees, with having leaf set , and suppose that for some constant . Suppose that for each , , where . Then as .
Proof
Let , where is the sequence of Bernoulli random variables with if species survives the FOB extinction event and otherwise, and is the ratio , where the numerator is as defined in Eq. (2.1) with for all f. Observe that for any particular value of , if we change (from 0 to 1 or visa versa) to give then:
We now apply McDiarmid’s inequality to obtain (for each ):
| 3.4 |
Now, by Proposition 2.2(i) (taking for all f) and, by assumption, . Thus we obtain:
Therefore, as . Since this holds for all , we obtain the claimed result.
We can now state the main result of this section. Recall that is the number of features present among leaves of that survive the FOB extinction event.
Theorem 1.1
Let be a sequence of binary trees and let features evolve on according to the stochastic feature evolution process described. If for some constant , and if converges to a constant as n grows we have:
as .
Before we proceed to the proof, we provide the following comments.
Remarks
Theorem 3.1 can fail without the condition . Fig. 2 provides a simple example of a sequence of trees for which does not converge in probability to any constant value as n grows. In this example, and the tree has height with one leaf having an incident edge of length and the remaining leaves having incident edges of length 1/n.
A sufficient condition for the inequality in Theorem 3.1 to hold is that for some and the proportion of pendant edges of of length is at least for all . Briefly, the reason for this is that the expected number of features arising on each of these pendant edges and surviving to the end of this edge is bounded away from 0 and the features associated with distinct pendant edges are always different from each other, and different from any other features arising in the tree.
Fig. 2.
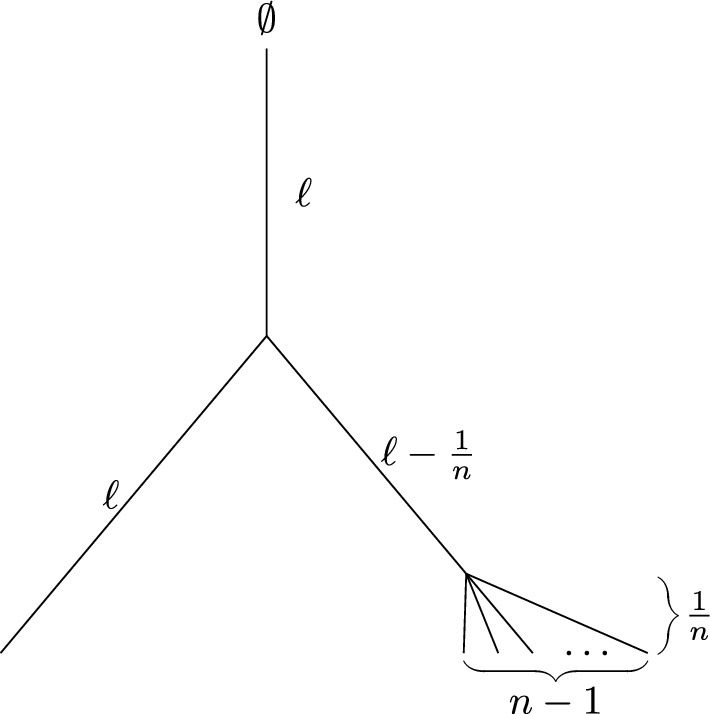
For the tree shown (with fixed and ) the sequence of trees has the property that does not converge in probability to any constant value as n grows
Proof of Theorem 3.1
Let be the event that , and let be the event described in Lemma 3.2 with . Then, , by Lemma 3.4. Now,
and since as (by Lemma 3.2) we have . Since this holds for all , it follows that as n grows.
Moreover, from Lemma 3.3, we have Now we can write as follows:
The first two terms on the right of this equation each converge in probability to 1 as , whereas the third (deterministic) term converges to . This completes the proof.
We will apply Theorem 3.1 in the next section to establish a result for FD loss on birth–death trees.
Feature diversity ratios in birth–death trees
In this section, we continue to investigate the stochastic model of feature gain and loss, but rather than considering fixed trees, we will now allow the trees themselves to be stochastically generated, following the simple birth–death processes that are common in phylogenetics. Thus, there will now be three stochastic processes in play: the linear-birth/linear-death process that generates the tree, the constant-birth/linear-death process of feature gain and loss operating along the branches of the tree, and the simple FOB extinction event at the present.
Definitions
Let denote a birth–death tree grown from a single lineage for time t with birth and death parameters and , respectively. We will assume throughout that (since otherwise the tree is guaranteed to die out as t becomes large).
On , we impose the model of feature gain and loss from the previous section with parameters r and . We now apply the FOB model in which each extant species (i.e. leaves of that are present at time t) has a probability of surviving and of becoming extinct (independently across the extant species), and we let denote the tree obtained from by removing the species at the present that did not survive this process. We refer to as the pruned tree, and the leaves of and that are present at time t as the extant species (or leaves) of these trees (to contrast them from leaves of that lie at the endpoints of any extinct lineages). If , there may now be fewer features present among the (probably reduced number of) extant species in than there were in .
Let be the discrete random variable that counts the number of features present in at least one of the extant leaves of , and let denote the number of these features that are also present in at least one extant leaf in the pruned tree .
Expected feature diversity
Next, we consider the expected values of and , where this expectation is across all three processes (the birth–death process that generates , feature gain and loss on this tree, and the species that survive the extinction event at the present under the FOB model). Of particular interest is the ratio of these expectations, and their limit as t becomes large. Specifically, let:
Note that is a function of five parameters (); however, we will show that it is just a function of s and two other parameters. Notice also that once these parameters are fixed, and are monotone increasing functions of s taking the value 0 at and 1 at . Moreover, and are both independent of r (the rate at which features arise along any lineage) as we formally show shortly.
Relationship to phylogenetic diversity (PD)
Recall that for a rooted phylogenetic tree T with branch lengths, the PD value of a subset S of leaves (PD(S, T)) is the sum of the lengths of the edges of the subtree of T that connect S and the root of the tree.
In the special case where , and where no features are present at time 0, (conditioned on ), has a Poisson distribution with a mean of r times , where is the (random) set of leaves at time t that survive the extinction event at the present. Consequently, . Similarly, , where is the set of extant leaves of . Thus, in this special case we have , where:
The function was explicitly determined in Lambert and Steel (2013), Mooers et al. (2012) as follows:
| 4.1 |
Calculating
We first recall a standard result from birth–death theory. Consider a linear birth–death process (starting with a single individual at time 0), with a birth rate , a death rate . For the individuals present at time t, sample each individual independently with sampling probability . Let () denote the number of these sampled individuals and let be the probability that . Then
| 5.1 |
In particular, converges to 0 if and converges to a strictly positive value if (Kendall 1948), (Yang and Rannala 1997).
The number of species at time t in that have a copy of particular feature f that arose at some fixed time in is described exactly by the birth–death process with parameters and and survival probability s at the present; it follows that as t becomes large, it becomes increasingly certain that none of the species at time t in the pruned tree will contain feature f if , whereas if , there is a positive limiting probability that f will be present in the extant leaves of the pruned tree.
Since (as given by Eqn. (4.1)) for all values of s when , in this section we will assume that (in addition to our universal assumption that ). Our main result provides an explicit formula for in Part (a), and describes some of its key properties in Parts (b) and (c).
Theorem 1.2
Given and , let , . Then:
where
andConditional on the non-extinction of , converges in probability to as .
is an increasing concave function that satisfies for all s.
Remarks
-
(i)
Notice that although depends on five parameters (), Theorem 5.1(a) reveals that just three derived parameters suffice to determine , namely s and the ratios and (these determine and , since and ). Notice also that and .
-
(ii)Our proof relies on establishing the following exact expression for :
where is the number of features present at time .5.2 In particular, this also provides an exact expression for . Notice that the ratio of the expected number of features present in the pruned tree, divided by the expected number of species in the pruned tree is , and this ratio converges to as when (via a further analysis of Eq. (5.2) in the Appendix).
-
(ii)
We saw in Sect. 4.3 that when , . At the other extreme, if and are fixed, and we let , then converges to s (since and in Theorem 5.1(a)). Informally, when is large compared to , most of the features present among the extant leaves of will have arisen near the end of the pendant edges incident with these extant leaves (a formalisation of this claim appears in Rosindell et al. (2022)); if we now apply the FOB model then the expected proportion of these features that survive will be close to the expected proportion of leaves that survive, namely s.
Illustrative examples
First, consider a Yule tree (i.e. ) grown for time t. Figure 3 (left) plots for values of . When , describes the proportional loss of expected PD in the pruned tree, and as increases, converges towards s (the expected proportion of leaves that survive extinction at the present). Figure 3 (right) plots for birth–death trees with , showing a similar trend, however with ranging higher above the curve .
Fig. 3.

Left: The function as a function of s for pure-birth Yule trees (). Right: The function for birth–death trees with . The curves within each graph show the effect of increasing , for values of between 0 (top) and 10 (bottom). The top curve also corresponds to the phylogenetic diversity ratio
Simulations
We ran simulations to test the expected relationship between and s and to get estimates of standard deviation. All simulations were run in R version 4.2.1 Team (2021). We first simulated 500 Yule trees (age = 100, , repeated and filtered to keep 500 250-tip trees) and 500 birth–death trees (age = 100, , repeated and filtered to keep 500 trees with 250-300 tips) using the sim.bd.age function in the package TreeSim (Stadler 2011). Features were then evolved on each tree, followed by separate extinction events.
Keeping the rate of feature gain fixed (), we modelled five different rates of feature loss (). We estimated feature gain and loss on each edge using the Gillespie algorithm (Gillespie 1976), where time until the next event (either feature gain or loss) was drawn from an exponential distribution with rate ), where k is the number of currently existing features at the start of the edge. The type of event was then determined with a Bernoulli draw with probability of a gain equal to . At each split on the tree, all existing features were copied to descendent edges. Each gain event created a new unique feature, and each loss event randomly selected an existing feature to eliminate from the current edge. At the end of the simulation the presence of features on each tip of the tree was recorded.
Extinction events were simulated by randomly selecting a proportion s of tips to delete, with s ranging from 0.05 to 0.95 by intervals of 0.05. The proportion of unique features remaining after extinction events was recorded, and the results are shown in Fig. 4.
Fig. 4.

Simulation results for remaining feature diversity as a function of s for pure-birth Yule trees (, left) and birth–death trees (, right). Results shown for two of the five feature evolution parameter scenarios. Each point represents one simulation on one simulated tree ( trees). The solid curves are the theoretical relationships between the expected proportion of remaining feature diversity and s for both of the shown feature evolution parameter scenarios
Resulting ratios of remaining features generally tracked expectations (the bias for birth–death trees when is likely due to our theoretical results conditioning on t rather than n). The standard deviation (SD), calculated for each on both Yule and birth–death trees, were fairly consistent for all except , where it was noticeably higher, as shown in Table 1. Because the number of total events along an edge (gains and losses) is described by a Poisson distribution, its variance increases with the mean, and this may explain the higher standard deviation at the highest loss rate.
Table 1.
Mean standard deviations of proportions of remaining feature diversity (each value is averaged over the 19 values of s between 0.05 and 0.95)
| 0 | 0.5 | 1 | 2 | 10 | |
|---|---|---|---|---|---|
| Yule | |||||
| Birth–death |
Features that appear in only one extant species
Let denote the number of features that are present in precisely one species in , and let . The following result describes a simple relationship between and .
Proposition 1.4
| 5.3 |
Proof
Let denote the number of features present at time t among the leaves of . Consider evolving for an additional (short) period into the future. Then, conditional on (the number of leaves of present at time t) and :
Applying the expectation operator (and using ) and letting leads to the equation stated.
Concluding comments
In this paper, we have considered, in order, three types of data to quantify the expected loss of feature diversity: sets of features across species (without any model of feature evolution or phylogeny), sets of features at the tips of a given phylogenetic tree, and sets of features at the tips of a random (birth–death) tree. The results of the earlier sections also proved helpful in establishing certain results in later sections.
In terms of wider significance to biodiversity conservation, our results and graphs in Sect. 5 suggest that the extent of relative feature diversity loss following extinction at the present is likely to be greater than that predicted by relative phylogenetic diversity loss for any given extinction rate .
Of course, our results are based on simple models (of feature gain and loss, and extinction at the present) and so exploring how these results might extend to more complex and realistic biological models would be a worthwhile topic for future work.
Acknowledgements
We thank François Bienvenu and James Rosindell for helpful suggestions on an earlier draft of this manuscript, and Ailene MacPherson for technical advice regarding the simulations. We also thank the two anonymous reviewers for further helpful comments and the New Zealand Marsden Fund (MFP-UOC2005) for supporting this research.
Appendix: Proof of Theorem 5.1
Part (a): Consider a birth–death tree with parameters , a feature evolution model with parameters , and a survival probability s for leaves at the present. Let , and let be the random variable that has the same distribution as with the initial condition (i.e. no features at the root of the tree). Let be the number of features that are present at the root of and also in the pruned tree . Then
Let and . The two random variables and are not independent (they are linked by both the underlying tree and the pruning event at the present) however, applying the expectation operator gives:
| 7.1 |
where is the number of features present at the root of .
Now, consider , and the events that can occur in the interval .
A new feature arises (with probability );
A speciation event occurs (with probability ;
The lineage (and hence the tree) dies (with probability .
None of the above occur (with probability
Let X be the random variable taking values in which denotes which of these four events occurs. In Case (a), the new feature is also present in with probability and so
For Case (b), , where is an independent copy of . Thus,
For Cases (c) and (d), we have: Thus, by the law of total expectation,
Consequently, the function satisfies the first-order linear differential equation:
| 7.2 |
subject to the initial condition . Solving Eq. (7.2) gives:
| 7.3 |
By making a change of variable we can rewrite Eq. (7.3) as:
| 7.4 |
Combining this equation and Eq. (7.1) provides the explicit expression for described earlier (Eq. (5.2)).
We now substitute in the expression for from Eq. (5.1) (with and ). For , we have:
| 7.5 |
Multiplying the numerator and denominator of the integrand by gives , and then by making the substitution , we obtain:
| 7.6 |
for the value of described in the theorem. Combining Eqs. (7.1) and (7.6) gives:
| 7.7 |
Thus, for ,
where the two terms of order o(1) (which converge to zero as t grows) refer to the last term on the right of Eq. (7.7) which is bounded above by the constant and so is asymptotically negligible in comparison to the term in Eq. (7.6). This gives the limit for as stated in Part (a) for fixed .
In the case where (which implies ), we use the corresponding expression for from Eq. (5.1) with and to obtain:
By a similar approach to the above we are led to the second equation in Part (a).
Part (b): Let be a birth–death tree with rates where , let denote the number of leaves of present at time t, and let be the event that (i.e. the non-extinction of ).
Conditional on the event , the number of leaves in tends to infinity (with probability 1) as (Jagers 1992), and so we can define a sequence of trees by letting denote the tree at the first time when has k extant leaves (we ignore leaves of that have already become extinct by time ).
Next, we establish that for a constant (in order to apply Theorem 3.1). The tree has n extant pendant edges, and the length of a randomly selected pendant edge in has a strictly positive probability p of having length at least (dependent on and ), by Theorem 3.1 of Stadler and Steel (2012). Now, is bounded below by the total number of features that arises on the n pendant edges and survive to the end of the edge (since all these features will necessarily be distinct from each other, and from other features that arise in the tree). Moreover, for each edge having length at least the expected number of features that arise on this edge and survive to the end of the edge is at least . Thus the expected number of features contributed by the pendant edges to is at least .
Thus, we can now apply Theorem 3.1, since exists, and equals , so (and thus ) converges to as n (respectively t) grows.
Part (c): We apply Proposition 2.2. By Part (i) of that result, and conditioning on we obtain , and so, taking expectation again (over the distribution of ) gives: , and thus
The inequality is clear since, for any choice of , we have with probability 1.
For concavity, Proposition 2.2 implies that the conditional expectation is concave as a function of s, and (by taking expectation over the distribution of ), is also concave as a function of s. Finally, by Part (a) of the current theorem, converges (deterministically) to and so this function is also concave as a function of s.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Devictor V, Mouillot D, Meynard C, Jiguet F, Thuiller W, Mouquet N. Spatial mismatch and congruence between taxonomic, phylogenetic and functional diversity: the need for integrative conservation strategies in a changing world. Ecol Lett. 2010;13:1030–1040. doi: 10.1111/j.1461-0248.2010.01493.x. [DOI] [PubMed] [Google Scholar]
- Faith DP. Conservation evaluation and phylogenetic diversity. Biol Cons. 1992;61(1):1–10. doi: 10.1016/0006-3207(92)91201-3. [DOI] [Google Scholar]
- Feller W. An introduction to probability theory and its applications. 3. London: Wiley; 1950. [Google Scholar]
- Gillespie D. A general method for numerically simulating the stochastic time evolution of coupled chemical reactions. J Comput Phys. 1976;22:403–434. doi: 10.1016/0021-9991(76)90041-3. [DOI] [Google Scholar]
- Huson D, Steel M. Phylogenetic trees based on gene content. Bioinformatics. 2004;20:2044–2049. doi: 10.1093/bioinformatics/bth198. [DOI] [PubMed] [Google Scholar]
- Jagers P. Stabilities and instabilities in population dynamics. J Appl Probab. 1992;29:770–780. doi: 10.2307/3214711. [DOI] [Google Scholar]
- Kendall DG. On the generalized birth-and-death process. Ann Math Stat. 1948;19:1–15. doi: 10.1214/aoms/1177730285. [DOI] [Google Scholar]
- Lambert A, Steel M. Predicting the loss of phylogenetic diversity under non-stationary diversification models. J Theor Biol. 2013;337:111–124. doi: 10.1016/j.jtbi.2013.08.009. [DOI] [PubMed] [Google Scholar]
- Mazel F, Mooers AO, Riva GVD, Pennell MW. Conserving phylogenetic diversity can be a poor strategy for conserving functional diversity. Syst Biol. 2017;66(6):1019–1027. doi: 10.1093/sysbio/syx054. [DOI] [PubMed] [Google Scholar]
- Mazel F, Pennell MW, Cadotte MW, Diaz S, Riva GVD, Grenyer R, Leprieur F, Mooers AO, Mouillot D, Tucker CM, Pearse WD. Prioritizing phylogenetic diversity captures functional diversity unreliably. Nat Commun. 2018;9:2888. doi: 10.1038/s41467-018-05126-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazel F, Pennell MW, Cadotte MW, Diaz S, Riva GVD, Grenyer R, Leprieur F, Mooers AO, Mouillot D, Tucker CM, Pearse WD. Reply to: “Global conservation of phylogenetic diversity captures more than just functional diversity”. Nat Commun. 2019;10:858. doi: 10.1038/s41467-019-08603-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDiarmid C. Surveys in combinatorics, London mathematical society lecture notes series 141. Cambridge: Cambridge University Press; 1989. On the method of bounded differences; pp. 148–188. [Google Scholar]
- Miller JT, Jolley-Rogers G, Mishler BD, Thornhill AH. Phylogenetic diversity is a better measure of biodiversity than taxon counting. J Syst Evol. 2018;56(6):663–667. doi: 10.1111/jse.12436. [DOI] [Google Scholar]
- Mitzenmacher M, Upfal E. Probability and computing: Randomized algorithms and probabilistic analysis. Cambridge: Cambridge University Press; 2005. [Google Scholar]
- Mooers A, Gascuel O, Stadler T, Li H, Steel M. Branch lengths on birth–death trees and the expected loss of phylogenetic diversity. Syst Biol. 2012;61(2):195–203. doi: 10.1093/sysbio/syr090. [DOI] [PubMed] [Google Scholar]
- Owen NR, Gumbs R, Gray CL, Faith DP. Global conservation of phylogenetic diversity captures more than just functional diversity. Nat Commun. 2019;10:859. doi: 10.1038/s41467-019-08600-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raup DM. Extinction: bad genes or bad luck? Oxford: Oxford University Press; 1993. [Google Scholar]
- Rosindell J, Manson K, Gumbs R, Pearse W, Steel M (2022) Phylogenetic biodiversity metrics should account for both accumulation and attrition of evolutionary heritage. Technical Report 2022.07.16.499419, BioRxiv [DOI] [PMC free article] [PubMed]
- Stadler T. Simulating trees with a fixed number of extant species. Syst Biol. 2011;60:676–684. doi: 10.1093/sysbio/syr029. [DOI] [PubMed] [Google Scholar]
- Stadler T, Steel M. Distribution of branch lengths and phylogenetic diversity under homogeneous speciation models. J Theor Biol. 2012;297(2):33–40. doi: 10.1016/j.jtbi.2011.11.019. [DOI] [PubMed] [Google Scholar]
- Team RC (2021) A language and environment for statistical computing (accessed on 7 August 2021)
- Tucker CM, Aze T, Cadotte MW, Cantalapiedra JL, Chisholm C, Díaz S. Assessing the utility of conserving evolutionary history. Biol Rev. 2019;94:1740–1760. doi: 10.1111/brv.12526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker CM, Davies TJ, Cadotte MW, Pearse WD. On the relationship between phylogenetic diversity and trait diversity. Ecology. 2018;99(6):1473–1479. doi: 10.1002/ecy.2349. [DOI] [PubMed] [Google Scholar]
- Wicke K, Fischer M. Phylogenetic diversity and biodiversity indices on phylogenetic networks. Math Biosci. 2018;298:80–90. doi: 10.1016/j.mbs.2018.02.005. [DOI] [PubMed] [Google Scholar]
- Wicke K, Mooers A, Steel M. Formal links between feature diversity and phylogenetic diversity. Syst Biol. 2021;70:480–490. doi: 10.1093/sysbio/syaa062. [DOI] [PubMed] [Google Scholar]
- Yang Z, Rannala B. Bayesian phylogenetic inference using DNA sequences: a Markov chain Monte Carlo method. Mol Biol Evol. 1997;14(7):717–724. doi: 10.1093/oxfordjournals.molbev.a025811. [DOI] [PubMed] [Google Scholar]