

Abstract
Neuromyelitis optica, an autoimmune inflammatory disorder affecting the central nervous system, can occur in a paraneoplastic context, although rare. We report an intriguing case of a 71-year-old woman with a history of triple-negative infiltrating ductal breast carcinoma, manifesting with paraneoplastic neuromyelitis optica that led to significant respiratory failure and required a cervical laminectomy. The patient presented with pain in the left breast, weakness in the lower extremities, and neck pain. The neurological evaluation showed 2/5 muscle strength in all extremities, diffuse hyperreflexia, and loss of multimodal sensation below the shoulder. She developed acute respiratory failure that required mechanical ventilation. Magnetic resonance imaging highlighted a diffuse abnormal increase in T2 signal intensity throughout the posterior and central portion of the cervical and thoracic spinal cord consistent with longitudinally extensive transverse myelitis, and significant cervical cord compression at C3–C4. Magnetic resonance imaging of the brain showed non-enhancing T2/fluid-attenuated inversion recovery (FLAIR) white matter hyperintensities and cerebellar hemispheres. The serum cell-based assay study demonstrated a high anti-aquaporin-4 immunoglobulin G titer (>1:160) confirming the diagnosis of neuromyelitis optica. She was taken for bilateral laminectomy from C3 to C6. Despite intravenous methylprednisolone and plasmapheresis treatment, no significant recovery was achieved, necessitating tracheostomy and percutaneous endoscopic gastrostomy. Subsequent rituximab treatment led to a mild improvement, with no new lesions on repeat magnetic resonance imaging. This case raises suspicion of the potential for neuromyelitis optica to occur as a paraneoplastic phenomenon, strengthening the need for vigilance in patients with malignancies.
Keywords: Paraneoplastic neuromyelitis optica, aquaporin-4, AQP4, NMOSD, triple-negative breast cancer
Introduction
Neuromyelitis optica (NMO) spectrum disorder (NMOSD), characterized by inflammatory lesions of the optic nerves and spinal cord, can present with a wide range of clinical manifestations and disease course, often leading to significant disability. Recent advances in immunology have led to the identification of a specific antibody, anti-aquaporin-4 immunoglobulin G (AQP4-IgG), confirming the diagnosis and differentiating NMO from NMOSD. The pathophysiology of the disease involves the binding of AQP4-IgG to AQP4 water channels, expressed primarily in astrocytes in the central nervous system, leading to a cascade of events culminating in neuroinflammation and neuronal damage. 1
Paraneoplastic syndromes represent another complex facet of neuroimmunology. They are rare disorders triggered by an altered immune system response to a neoplastic process. These syndromes can affect various organ systems and are independent of tumor size or extent of the tumor. 2 Paraneoplastic NMO, although rarely reported, has been associated with various malignancies. 3 Herein, we present a rare case of possible paraneoplastic NMO with respiratory failure in a patient with a history of triple-negative infiltrating ductal carcinoma. The intricacies of this case underscore the complexities of the diagnostic process and the nuances of NMO.
Case presentation
A 71-year-old woman, previously treated for triple-negative infiltrating ductal carcinoma with lumpectomy, chemotherapy, and radiation therapy, along with a history of type 2 diabetes mellitus, obesity (body mass index > 30 kg/m2), and hypertension, presented with a 10-day history of left breast pain, weakness of bilateral lower extremities, and neck pain. The patient was initially diagnosed with breast cancer 1 year before her current presentation based on positron emission tomography-computered tomography (PET/CT) imaging and histopathology (Figure 1). During the recent admission, the physical examination revealed a thickening of the skin in the upper outer left breast, and ultrasound suggested a seroma. Computerized tomography scan of the chest identified an intermediate rim-enhancing collection (2.6 × 4.5 × 2.8 cm) in the deep left breast tissue. Subsequently, her condition worsened with progressive shortness of breath and increased weakness, necessitating emergent intubation and admission to the intensive care unit (ICU). Neurological examination demonstrated significant weakness (2/5 muscle strength) in all four extremities, diffuse hyperreflexia, and loss of multimodal sensation below the shoulders.
Figure 1.
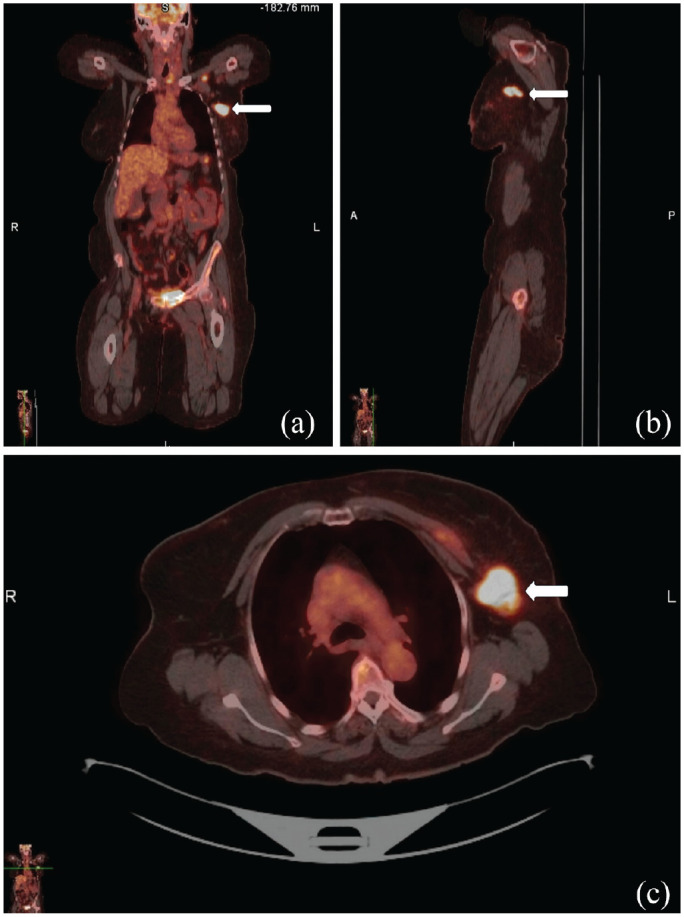
(a) Anterior, (b) lateral, and (c) cross-sectional view of FDG-PET/CT showing FDG-avid left breast mass and axillary lymphadenopathy.
FDG-PET/CT: fluorodeoxyglucose positron emission tomography-computered tomography.
Magnetic resonance imaging (MRI) of the cervical and thoracic spine revealed an abnormal diffuse increase in T2 signal intensity extending from C1 to T7 consistent with longitudinally extended transverse myelitis (LETM), with multi-degenerative changes and moderate spinal canal stenosis at C3–C4 and C4–C5, causing cord compression. The brain MRI showed non-enhancing T2/FLAIR hyperintensities in the cerebral and cerebellar white matter and middle cerebellar peduncles (Figure 2). Serum studies produced a high AQP4-IgG cell-based assay (CBA) titer of >1:160 and AQP4 IgG fluorescence-activated cell sorting titer of >1:10000. Based on clinical, laboratory, and radiographic findings, the diagnosis of NMO was made. Cerebrospinal fluid (CSF) analysis was not completed due to body habitus and acuity. In addition, an abdominal and pelvic CT scan and an ovarian ultrasound were performed to rule out other malignancies or metastatic diseases.
Figure 2.

(a) T2/FLAIR sagittal sequence of the cervical spinal cord, (b) T1 sagittal sequence, and (c) T2 sagittal sequence of the thoracic spinal cord with non-enhancing hyperintense lesion extending from C1 to T6. (e) T2/FLAIR axial view at C1 and (f) T6 depicting a predominantly hyperintense lesion within the central cord. (d) T2/FLAIR sagittal sequence of the brain and (g) axial view showing non-enhancing hyperintensities involving the cerebrum, cerebellum, and cerebellar peduncles.
Due to acute respiratory failure secondary to cervical cord compression and myelopathy, a C3–C6 bilateral laminectomy was performed. The family rejected a breast biopsy to assess for AQP4 antibodies. The patient’s ICU course was complicated by an acute kidney injury. Therefore intravenous immunoglobulin (IVIG) was not considered a suitable option due to worsening renal function. The patient received a 5-day course of intravenous methylprednisolone 1000 mg followed by a 5-day plasmapheresis course due to failure of ventilation weaning. Due to non-improvement, a tracheostomy and percutaneous endoscopic gastrostomy procedures were performed on the 20th day of hospitalization. A decision was made to start rituximab, and she received two infusions 2 weeks apart. Cytokine and antibody levels included CD 19 and CD 20 <1%, IgG 787 mg/dL (600–1540), IgA 60 mg/dL (82–453), and IgM 22 mg/dL (50–300).
During treatment, she was weaned off mechanical ventilation with a slight improvement in weakness and discharged to a rehabilitation facility. Repeat MRI did not show new lesions and stability of previously defined lesions. However, at 3 months of follow-up, the patient showed only minimal improvement in weakness and still experienced significant sensory difficulties with a modified Rankin score of 3. 4
Discussion
NMO is an inflammatory demyelinating disease that predominantly affects the optic nerves, brainstem, and spinal cord leading to optic neuritis and inflammatory myelopathy. The International Panel for NMO Diagnosis (IPND) has provided revised NMOSD diagnostic criteria. 5 They require the presence of at least one core clinical characteristic (optic neuritis, acute myelitis, area postrema syndrome, acute brainstem syndrome, symptomatic narcolepsy or acute diencephalic clinical syndrome with NMOSD-typical diencephalic MRI lesions, symptomatic cerebral syndrome with NMOSD-typical brain lesions), along with positive AQP4-IgG, and exclusion of alternative diagnoses. 5 Our patient presented with the clinical and imaging findings of LETM involving the cervical and thoracic spinal cord (C1–T6). Moreover, the patient demonstrated positive AQP4-IgG serology using the gold standard CBA, thus fulfilling the IPND criteria for NMOSD diagnosis. 5
Acute respiratory failure was likely due to superimposed cervical cord inflammation and edema on prior degenerative disk disease and spondylosis. Paraneoplastic NMOSD is exceptionally rare and only a handful of cases have been reported. 6 Carrillo et al. recently reported the case of a 32-year-old woman with triple-negative breast cancer diagnosed with NMOSD. 7 The patient was adequately treated with good effect with corticosteroids, plasma exchange, and rituximab. As in our patient, several malignancies, including breast cancer, have been associated with paraneoplastic NMOSD. 8 Although not universally accepted, the proposed mechanism suggests molecular mimicry or ectopic expression of neuronal antigens leading to the production of cross-reacting antibodies, leading to autoimmunity. 9 The temporal relationship between cancer treatment and the onset of myelopathy and AQP4 antibodies in serum suggests a possible paraneoplastic origin. NMOSD is typically present in adults in their late thirties to early sixties. However, in cases of paraneoplastic NMOSD, as demonstrated in our patient, the age at diagnosis is typically high, underscoring the possible role of malignancies in triggering this rare condition in older populations. 10
Management of NMOSD is with acute and preventive treatment strategies. Acute treatments aim to stop the ongoing attack and typically accelerate recovery, often achieved with high doses of intravenous steroids, plasma exchange, or IVIGs. 11 Newer biologic therapies aim to prevent further attacks, as seen in our patient who received rituximab. Given the rarity and complexity of paraneoplastic NMOSD, future research must focus on the evaluation of targeted treatment strategies for this distinct subset of patients. 12 Understanding the unique pathophysiological mechanisms and therapeutic responses in paraneoplastic NMOSD will enable clinicians to provide more effective personalized care.
Our case presents several limitations. A definitive link between NMO and malignancy in our patient is complex due to the lack of CSF studies, which makes it difficult to rule out other neuroimmunological processes that could mimic NMO. It is to be noted that serum testing for AQP4 antibodies has a greater sensitivity than CSF. 13 Therefore, the lack of CSF examination does not limit or hinder the diagnosis of NMO. The patient’s moderate response to rituximab treatment may be due to the advanced stage of the disease at the time of initiation. However, more recent therapeutic interventions with different targets as complements were not tested. The lack of direct tissue biopsy and staining to assess for AQP4 is also a limitation. Despite these limitations, this case highlights the importance of considering a paraneoplastic origin in patients with NMOSD and malignancy. Future research should focus on establishing diagnostic criteria for paraneoplastic NMOSD, screening protocols, and evaluating the optimal treatment approach. Longitudinal studies could provide valuable information on disease progression and the impact of early intervention on patient outcomes.
Conclusions
This case of a possible paraneoplastic NMOSD associated with infiltrating ductal carcinoma complicated by acute respiratory failure underlines the clinical complexities and management challenges in such rare presentations. The atypical onset of NMO symptoms correlating with the patient’s cancer journey suggests a possible paraneoplastic etiology. Furthermore, the moderate response to rituximab highlights the necessity of early recognition and intervention in paraneoplastic NMOSD cases. This case emphasizes the importance of heightened clinical vigilance for NMOSD symptoms in patients with malignancies and reinforces the need for future research to enhance our understanding of disease mechanisms, diagnostic criteria, and optimal treatment strategies for paraneoplastic NMOSD.
Footnotes
Author contributions: B.S.S. completed literature review, drafted the initial manuscript, generated illustrations/figures, provided intellectual verification of the topic, and edited the final manuscript. S.S., S.N.C., and V.K. drafted the initial manuscript. M.A.G-D provided intellectual verification on this topic. All authors reviewed the final draft of the manuscript.
Availability of data and materials: All data generated or analyzed during this study are included in this article. Further enquiries can be directed to the corresponding author.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics approval: Our institution does not require ethical approval for reporting individual cases or case series.
Informed consent: Written informed consent was obtained from the patient in the publication of this case report.
ORCID iD: Vincent Kipkorir  https://orcid.org/0000-0002-5301-4102
https://orcid.org/0000-0002-5301-4102
References
- 1. Prasad S, Chen J. What you need to know about AQP4, MOG, and NMOSD. Semin Neurol 2019; 39: 718–731. [DOI] [PubMed] [Google Scholar]
- 2. Dinoto A, Borin GU, Campana G, et al. Investigating paraneoplastic aquaporin-4-IgG-seropositive neuromyelitis optica spectrum disorder through a data-driven approach. Eur J Neurol 2022; 29: 3466–3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ding M, Lang Y, Cui L. AQP4-IgG positive paraneoplastic NMOSD: a case report and review. Brain Behav 2021; 11: e2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Banks JL, Marotta CA. Outcomes validity and reliability of the modified Rankin scale: implications for stroke clinical trials: a literature review and synthesis. Stroke 2007; 38: 1091–1096. [DOI] [PubMed] [Google Scholar]
- 5. Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015; 85: 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shahmohammadi S, Doosti R, Shahmohammadi A, et al. Neuromyelitis optica spectrum disorder (NMOSD) associated with cancer: a systematic review. Mult Scler Relat Disord 2021; 56: 103227. [DOI] [PubMed] [Google Scholar]
- 7. Carrillo P, Gorria T, Santana D, et al. Aquaporin-4-positive triple-negative breast cancer presenting with paraneoplastic neuromyelitis optica spectrum disorder. Biomed Hub 2022; 7: 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sudo A, Chihara N, Takenaka Y, et al. Paraneoplastic NMOSD associated with EG junction adenocarcinoma expressing unprotected AQP4. Neurol Neuroimmunol Neuroinflamm 2018; 5: e482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim W, Lee JE, Kim SH, et al. Cerebral cortex involvement in neuromyelitis optica spectrum disorder. J Clin Neurol 2016; 12: 188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tian DC, Li Z, Yuan M, et al. Incidence of neuromyelitis optica spectrum disorder (NMOSD) in China: a national population-based study. Lancet Reg Health West Pac 2020; 2: 100021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ma X, Kermode AG, Hu X, et al. NMOSD acute attack: understanding, treatment and innovative treatment prospect. J Neuroimmunol 2020; 348: 577387. [DOI] [PubMed] [Google Scholar]
- 12. Asseyer S, Cooper G, Paul F. Pain in NMOSD and MOGAD: a systematic literature review of pathophysiology, symptoms, and current treatment strategies. Front Neurol 2020; 11: 778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Majed M, Fryer JP, McKeon A, et al. Clinical utility of testing AQP4-IgG in CSF: guidance for physicians. Neurol Neuroimmunol Neuroinflamm 2016; 3: e231. [DOI] [PMC free article] [PubMed] [Google Scholar]