

Summary
Colorectal cancer (CRC) develops in part through the deregulation of different signaling pathways, including activation of the WNT/β-catenin and PI3K/AKT pathways. Additionally, the lysine methyltransferase enhancer of zeste homologue 2 (EZH2) is commonly overexpressed in CRC. EZH2 canonically represses gene transcription by trimethylating lysine 27 of histone H3, but also has non-histone substrates. Here, we demonstrated that in CRC, active AKT phosphorylated EZH2 on serine 21. Phosphorylation of EZH2 by AKT induced EZH2 to interact with and methylate β-catenin at lysine 49, which increased β-catenin’s binding to the chromatin. Additionally, EZH2-mediated β-catenin trimethylation induced β-catenin to interact with TCF1 and RNA polymerase II and resulted in dramatic gains in genomic regions with β-catenin occupancy. EZH2 catalytic inhibition decreased stemness but increased migratory phenotypes of CRC cells with active AKT. Overall, we demonstrated that EZH2 modulates AKT-induced changes in gene expression through the AKT/EZH2/β-catenin axis in CRC.
Subject areas: Cancer, Epigenetics, Molecular biology
Graphical abstract
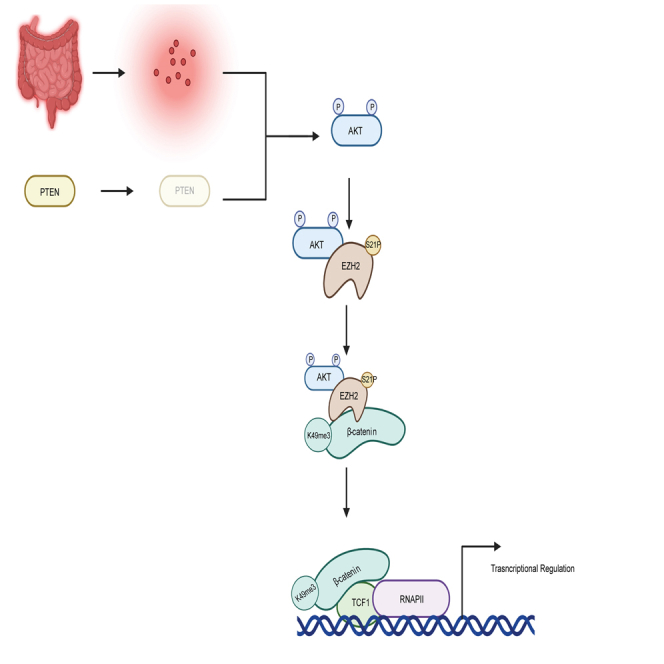
Highlights
-
•
Activation of AKT induces pS21-EZH2 in colorectal cancer
-
•
pS21-EZH2 trimethylates β-catenin at K49
-
•
β-catenin K49me3 interacts with RNAPII and TCF1
-
•
AKT-dependent gene expression is modulated by EZH2 through methylation of β-catenin
Cancer; Epigenetics; Molecular biology
Introduction
Colorectal cancer (CRC) is the third leading cause of cancer-related mortalities in the US.1 CRC develops in part through the deregulation of different signaling pathways, including activation of the WNT/β-catenin and PI3K/AKT pathways.2,3 Activating mutations in the WNT/β-catenin pathway occur in almost 90% of CRCs and are key initiating and promoting events in CRC development.4 Activation of the WNT pathway results in β-catenin accumulation in the nucleus where it binds to T cell factor (TCF) transcription factors to activate gene expression.5 Mutations in the PI3K/AKT pathway are critical for invasive properties and malignant transformation in CRC.6 The two most common genetic PI3K/AKT pathway alterations in CRC are activating mutations in the PI3K catalytic subunit gene PIK3CA or loss of the pathway suppressor PTEN.7 PTEN loss occurs in approximately 30% of CRCs and has been functionally implicated in CRC proliferation and progression.8 Additionally, chronic inflammation is a major risk factor for CRC development and the PI3K/AKT pathway can also be activated by reactive oxygen species (ROS) generated by activated immune cells at inflammatory sites.9,10
Polycomb repressive complex 2 (PRC2) is a chromatin regulator that is involved in human development, tissue homeostasis, and cancer.11,12 Core components of PRC2 include EZH2 (enhancer of zeste homolog 2), Suz12 (suppressor of zeste 12), and EED (embryonic ectoderm development).11 EZH2 functions as a lysine methyltransferase and EZH2-containing PRC2 canonically catalyzes trimethylation of histone 3 at lysine 27 (H3K27me3), which leads to chromatin compaction and transcriptional repression.13 H3K27me3 is involved in establishing chromatin domains that maintain transcriptional repression of hundreds of lineage-specific genes, reinforcing the maintenance of the current gene expression program and upholding cell identity.14,15 Initial studies showed that EZH2 acts as an oncogene by promoting cancer proliferation and progression.16 However, other studies revealed tumor suppressive functions of EZH2, highlighting unanticipated complexity of the role of PRC2 in cancer.17 Additionally, whereas EZH2 was consistently reported to be overexpressed in colon cancers, EZH2 expression levels have been found to be correlated positively or negatively with the survival of CRC patients depending on the study.18,19 These conflicting results suggest that there are context-dependent functions of EZH2 in cancer.
Emerging studies suggest that EZH2 can act noncanonically to regulate gene expression. For example, EZH2 interacts with and methylates various transcription factors, including androgen receptor (AR), GATA Binding Protein 4 (GATA4), related orphan receptor A (RORa), signal transducer and activator of transcription 3 (STAT3) and β-catenin, which either positively or negatively regulates their transcriptional activity.20,21,22,23,24 For example, EZH2-mediated methylation of STAT3 and GATA4 enhances and represses, respectively, the transcriptional activity of these transcription factors.20,23 Additionally, phosphorylation of EZH2 at Serine 21 (pS21-EZH2) by AKT has been shown to mediate the noncanonical roles of EZH2 in several of these studies. For example, AKT-mediated pS21-EZH2 induces EZH2 to interact with AR and STAT3 to induce the transcriptional activity of these transcription factors in prostate cancer and glioblastoma stem cells, respectively.21,23
Here, we investigated the non-canonical role of EZH2 in CRC. We show that, following activation of the PI3K/AKT pathway, AKT phosphorylates EZH2. We further demonstrate that phosphorylation of EZH2 by active AKT is required for EZH2 to interact with and methylate β-catenin, a key factor in CRC. Methylation of β-catenin regulates its transcriptional activity such that it has increased genome-wide chromatin binding and increased interaction with TCF1 and RNA polymerase II (RNAPII). Our work suggests that following phosphorylation by AKT, EZH2 fine-tunes β-catenin’s transcriptional activity resulting in modulation of the expression of genes involved in cell migration and metabolic processes.
Results
AKT activation mediates EZH2 phosphorylation at serine 21
Because AKT has previously been demonstrated to regulate EZH2 in other cancer types21,23 and the PI3K/AKT pathway is commonly activated in CRC,8 we explored connections between AKT activity and EZH2 in CRC. To experimentally determine if AKT regulates EZH2 in CRC, we activated AKT by H2O2 treatment, which resulted in increased interaction between AKT and EZH2 in SW480 and RKO CRC cell lines and CRC organoids (Figure 1A - whole-cell extract and input in S1A, S1B, S1C). As a negative control, EZH2 was not detected in AKT immunoprecipitations (IPs) from H2O2-treated EZH2 knockdown (KD) cells and EZH2 KD did not alter the H2O2-induced increase in whole-cell levels of AKT phosphorylated on serine 473 (pS473-AKT) (Figures 1A and S1A). AKT phosphorylates EZH2 at Serine 21 (S21) to regulate EZH2’s activity in prostate cancer and glioblastoma stem cells.21,23 To test if EZH2 is phosphorylated by AKT in CRC, we utilized an anti-pAKT substrate antibody, which recognizes the phosphorylated consensus sequence in AKT substrates (RXXS∗/T∗). Anti-pAKT substrate signal at EZH2’s expected size was increased in EZH2 IPs from H2O2 -treated compared to untreated SW480 cells and CRC organoids (Figures 1B and S1D). The H2O2-induced increase in the EZH2 anti-pAKT substrate signal was blocked when cells were co-treated with AKT inhibitor (GSK690693) and H2O2, suggesting that the increase in phosphorylated EZH2 following H2O2 is caused by AKT activity (Figure 1B). AKT inhibitor treatment did not alter EZH2 levels in the nuclear lysates used for the IPs (input; Figure 1B). To determine if EZH2 phosphorylation is occurring at S21, we performed anti-HA IPs in SW480 cells transfected with HA-tagged EZH2 wild type (WT) and HA-tagged EZH2 S21A phospho-null (PN) plasmids. The EZH2 pAKT substrate band was detected in HA-EZH2 WT but not in HA-EZH2 S21A IPs from cells treated with H2O2 (Figure 1C).
Figure 1.

AKT activation mediates EZH2 phosphorylation at Serine 21
(A) Western blots of AKT immunoprecipitations (IP) performed using nuclear lysates prepared from EZH2 knockdown (KD) and empty vector (EV) SW480 cells untreated or treated with 250 μM H2O2 for 30 min. See Figure S1A for western blots of nuclear lysates used for IP (input) and total protein prior to cellular fractionation (whole-cell extract-WCE).
(B) EZH2 IP performed using nuclear lysates prepared from SW480 cells untreated or treated with 10 μM AKT inhibitor (AKTi, GSK-690693) for 48 h followed by no additional treatment or co-treatment with 250 μM H2O2 for 30 min.
(C) HA IP performed using nuclear lysates prepared from HA-EZH2 wildtype (WT) and HA-EZH2 phospho-null (PN) S21A expressing SW480 cells treated as in A.
(D) AKT IP performed using nuclear lysates prepared from empty vector (EV) and PTEN knockdown (KD) Caco2 cells.
(E) AKT IP performed using nuclear lysates prepared from EV and PTEN KD RKO cells. See also Figure S1.
PTEN loss, which activates the PI3K/AKT pathway, is also common in CRC.8 Interestingly, we identified a significant negative correlation between PTEN and EZH2 expression in TCGA CRC samples (Figure S1E). As expected, PTEN KD in SW480 cells induced AKT phosphorylation (Figure S1F). Therefore, we performed additional experiments in PTEN KD cells to complement our findings using H2O2 treatment. Like H2O2 treatment, PTEN KD in SW480 cells increased the interaction between EZH2 and AKT compared to empty vector (EV) KD cells (Figure S1G). In addition, activation of AKT via PTEN KD increased AKT interaction with EZH2 in additional CRC cell lines, Caco2, RKO and Lovo (Figures 1D, 1E, and S1H). AKT activation by H2O2 treatment or PTEN KD did not alter global H3K27me3 levels in any cell line tested (Figures 1C–1E). Altogether, these results suggest that AKT activation in CRC induces AKT to interact with and phosphorylate EZH2 at S21.
AKT-mediated EZH2 phosphorylation at S21 induces EZH2 to interact with RNAPII
EZH2 phosphorylation at S21 induces EZH2 to act as a transcriptional coactivator in other cancer types.21,23 To preliminarily determine if AKT activation switches EZH2’s function to become non-canonically involved in transcriptional activation, we tested if EZH2 interacts with RPB1, the largest subunit of the RNAPII complex. Treating SW480 cells with H2O2 induced EZH2 to interact with RNAPII without altering RNAPII or EZH2 protein levels in the whole-cell extracts (WCE) or nuclear input fractions (Figures 2A, 2B, and S2A). Interestingly, inhibiting AKT activity abolished the H2O2-induced increase in interaction between EZH2 and RNAPII (Figure 2B). As a control for the effectiveness of AKT inhibition, AKT inhibitor-treated samples had decreased levels of phosphorylated GSK3β, a known substrate for AKT, in input and whole-cell fractions as compared to non-inhibitor-treated samples (Figure S2A). For the IPs in Figures 2A and 2B, RNaseA was added to the nuclear lysates to exclude the possibility that EZH2 was interacting with RNAPII through RNA bridging as has been shown previously.25,26 PTEN KD also increased the interaction of EZH2 with RNAPII in SW480 cells (Figure S2B). Additionally, H2O2 increased the interaction of AKT with RNAPII (Figure 2C). However, EZH2 KD decreased the H2O2-induced AKT-RNAPII interaction without altering total RNAPII or AKT levels in the nuclear input fractions (Figure 2C). Finally, to determine if EZH2 phosphorylation at S21 drives EZH2 to interact with RNAPII, HA IPs were performed in SW480 cells expressing similar levels of HA-EZH2 WT or HA-EZH2 S21A. Intriguingly, RNAPII was detected in HA-EZH2 WT but not in HA-EZH2 S21A IPs from cells treated with H2O2 (Figure 2D). Altogether, these data suggest that phosphorylation of EZH2 at S21 via AKT induces EZH2 to interact with RNAPII.
Figure 2.

AKT-mediated EZH2 phosphorylation at S21 induces EZH2 to interact with RNAPII
(A) Western blots of EZH2 immunoprecipitations (IP) performed using nuclear lysates prepared from SW480 cells untreated or treated with 250 μM H2O2 for 30 min. IP with IgG serves as a negative control. Input is nuclear lysates used for IP.
(B) EZH2 IP performed using nuclear lysates prepared from SW480 cells treated with 10 μM AKT inhibitor (AKTi, GSK-690693) or DMSO for 48 h followed by no additional treatment or co-treatment with 250 μM H2O2 for 30 min. IP with beads only serves as a negative control. See Figure S2A for western blots of whole-cell extract (WCE) and input.
(C) AKT IP performed using nuclear lysates prepared from EZH2 knockdown (KD) and empty vector (EV) SW480 cells treated as in A.
(D) HA IP performed using nuclear lysates prepared from HA-EZH2 wildtype (WT) and HA-EZH2 phospho-null (PN) S21A expressing SW480 cells treated as in A. IP with IgG serves as a negative control. See also Figure S2.
AKT-mediated EZH2 phosphorylation induces EZH2’s interaction with β-catenin
In prostate cancer, EZH2 phosphorylation at S21 promotes its interaction with androgen receptor (AR) to induce AR’s transcriptional activity.21 Because the WNT/β-catenin pathway is hyperactive in almost 90% of CRCs,4 we tested whether AKT-induced phosphorylation of EZH2 induced EZH2 to interact with β-catenin and if EZH2 mediated β-catenin’s interaction with RNAPII in CRC. AKT activation by either H2O2 treatment or PTEN KD increased the interaction between β-catenin and EZH2 in SW480 cells, CRC organoids, RKO, Caco2, HEK293T, and Lovo cells (Figures 3A–3D, S3A, and S3B). H2O2 and PTEN KD did not alter β-catenin levels in the nuclear input fractions in any of the cell lines tested (Figures 3A, 3C, 3D, S3A, and S3B). However, β-catenin levels were increased in the nuclear input fraction prepared from CRC organoid cells following H2O2 treatment (Figure 3B).
Figure 3.

AKT-mediated EZH2 phosphorylation induces EZH2’s interaction with β-catenin
(A) Western blots of EZH2 immunoprecipitations (IP) performed using nuclear lysates prepared from SW480 cells untreated or treated with 250 μM H2O2 for 30 min. IgG IP serves as a negative control. Input is nuclear lysates used for IP.
(B) EZH2 IP performed using nuclear lysates prepared from human colorectal cancer organoids treated as in A.
(C and D) β-catenin IP performed using nuclear lysates prepared from empty vector (EV) and PTEN knockdown (KD) RKO (C) and Caco2 (D) cells.
(E) β-catenin IP performed using nuclear lysates prepared from EZH2 KD and EV SW480 cells treated as in A.
(F) β-catenin IP performed using nuclear lysates prepared from PTEN KD and EV SW480 cells treated with 2 μM EZH2 inhibitor (EZH2i, GSK-503) or DMSO for 72 h.
(G) HA IP performed using nuclear lysates prepared from HA-EZH2 wildtype (WT) and HA-EZH2 phospho-null (PN) S21A expressing SW480 cells treated as in A. See also Figure S3.
Based on our findings so far, we hypothesized that EZH2 regulates β-catenin’s interaction with the transcriptional machinery. H2O2 and PTEN KD induced an increase in interaction between β-catenin and RNAPII compared to untreated cells (Figure S3C). To determine if EZH2 played a role in this interaction, we treated EV and EZH2 KD SW480 cells with H2O2, which increased β-catenin’s interaction with RNAPII and EZH2 in EV cells (Figure 3E). EZH2 KD reduced the H2O2-induced interaction between β-catenin and RNAPII without altering RNAPII levels in the nuclear input fraction (Figure 3E). As a control, EZH2 was not detected in β-catenin IPs in EZH2 KD cells (Figure 3E). Furthermore, treating SW480 cells with an EZH2 inhibitor (GSK503, EZH2i) blocked the PTEN KD-mediated β-catenin-RNAPII interaction (Figure 3F). We hypothesized that AKT activation is required for EZH2 to interact with β-catenin and RNAPII. Consistent with this hypothesis, inhibiting AKT activity reduced the interaction of β-catenin with RNAPII and EZH2 after H2O2 treatment without affecting the levels of EZH2, RNAPII, or β-catenin in the whole-cell extract fraction (Figure S3D). To test whether EZH2 phosphorylation at S21, which is mediated by AKT, drives EZH2 to interact with β-catenin, we transfected SW480 cells with HA-tagged WT and phosphorylation null EZH2 constructs and performed anti-HA IPs. Interestingly, H2O2 treatment and PTEN KD induced HA-EZH2 WT but not HA-EZH2 S21A to interact with β-catenin (Figures 3G and S3E). These data suggest that phosphorylation of EZH2 at S21 via active AKT induces EZH2 to interact with RNAPII and β-catenin. In addition, β-catenin’s interaction with RNAPII likely requires EZH2 enzymatic activity because inhibiting EZH2 activity blocked the AKT activity-dependent interaction between β-catenin and RNAPII.
EZH2 mediates β-catenin trimethylation at K49
EZH2 has been reported to methylate lysines of non-histone proteins, including Lysine 49 (K49) of β-catenin,20,22,23,24 and we have determined that AKT activation induced EZH2 to interact with β-catenin. To test if H2O2 or/and PTEN KD induces β-catenin tri-methylation at K49, we utilized FLAG-tagged constructs of WT β-catenin or a β-catenin mutant that is methylation null at K49. IPs using an anti-trimethyl lysine antibody showed FLAG signal at the expected size of β-catenin being increased in IPs from H2O2 treated and PTEN KD cells expressing FLAG-β-catenin WT but not FLAG-β-catenin K49R relative to their respective controls (Figures 4A and 4B). These results suggested that the trimethylation of β-catenin was occurring at K49 in response to AKT activation. Our finding that AKT activation induced EZH2 to interact with β-catenin (Figure 2) led us to hypothesize that β-catenin trimethylation is mediated by EZH2. FLAG IPs in SW480 cells transfected with FLAG-β-catenin WT and β-catenin IPs in EV and PTEN KD cells confirmed that H2O2 and PTEN KD increased β-catenin trimethylation (Figures S4 and 4C). Consistent with our hypothesis, EZH2 inhibition reduced H2O2 and PTEN KD-mediated β-catenin trimethylation without altering total levels of FLAG-β-catenin or β-catenin (Figures S4 and 4C). These results suggest that activation of AKT induces EZH2 to interact with and trimethylate β-catenin at K49.
Figure 4.

EZH2 mediates β-catenin trimethylation at K49
(A) Trimethyl lysine (Me3-K) immunoprecipitation (IP) performed using nuclear lysates prepared from Flag β-catenin WT and Flag β-catenin K49R expressing SW480 cells treated with 250 μM H2O2 for 30 min and Flag β-catenin WT expressing untreated cells. Input is nuclear lysates used for IP. IP with IgG serves as a negative control.
(B) Trimethyl lysine (Me3-K) IP performed using nuclear lysates prepared from Flag β-catenin WT and Flag β-catenin K49R expressing PTEN knockdown (KD) and empty vector (EV) SW480 cells. IP with beads only serves as a negative control.
(C) β-catenin IP preformed using nuclear lysates prepared from EV or PTEN KD SW480 cells treated with 2 μM EZH2 inhibitor (EZH2i, GSK-503) or DMSO for 72 h. See also Figure S4.
EZH2-mediated trimethylation of β-catenin induces β-catenin to interact with TCF1 and RNAPII
During WNT pathway activation, β-catenin interacts with TCF proteins to induce WNT target gene expression.27 TCF IPs showed that PTEN KD induced TCF1 to interact with β-catenin (Figure 5A). β-catenin and FLAG IPs in SW480 cells also demonstrated that PTEN KD and H2O2 treatment induced TCF1 interaction with β-catenin (Figures 5B and 5C). EZH2 inhibition reduced the PTEN KD and H2O2–induced β-catenin interaction with TCF1 (Figures 5A–5C). Because EZH2 inhibition reduced the interaction of β-catenin and TCF1, we hypothesized that this interaction is promoted by EZH2 trimethylating β-catenin. To test this hypothesis, FLAG IPs were performed in SW480 cells expressing similar levels of FLAG-β-catenin WT and FLAG-β-catenin K49R. H2O2 treatment increased the interaction of FLAG-β-catenin WT but not FLAG-β-catenin K49R with TCF1 (Figure 5D). Similarly, PTEN KD slightly increased the interaction of FLAG-β-catenin WT with TCF1 relative to EV whereas the interaction between FLAG-β-catenin K49R and TCF1 was reduced relative to FLAG-β-catenin WT in EV and PTEN KD cells (Figure S5A). Additionally, FLAG-β-catenin K49R had reduced interaction with RNAPII as compared to FLAG-β-catenin WT in PTEN KD SW480 cells (Figure 5E). Altogether, these findings suggest that EZH2-mediated trimethylation of β-catenin induces β-catenin’s interaction with RNAPII and TCF1 after AKT activation.
Figure 5.

EZH2-mediated trimethylation of β-catenin induces β-catenin to interact with TCF1 and RNAPII
(A) Western blots of TCF1 immunoprecipitation (IP) performed using nuclear lysates prepared from PTEN knockdown (KD) and empty vector (EV) SW480 cells treated with 1 μM EZH2 inhibitor (EZH2i, GSK-503) or DMSO for 72 h. IgG IP serves as a negative control. Input is nuclear lysates used for IP.
(B) β-catenin IP performed using nuclear lysate prepared from PTEN KD and EV SW480 cells.
(C) Flag IP performed using nuclear lysates prepared from Flag-tagged β-catenin WT expressing SW480 cells treated with 1 μM EZH2 inhibitor (EZH2i, GSK-503) or DMSO for 72 h with or without co-treatment with 250 μM H2O2 for 30 min. Cells not expressing Flag plasmid were used as a negative control for the IP.
(D) Flag IPs were performed using nuclear lysate prepared from FLAG-β-catenin WT and FLAG-β-catenin K49R expressing SW480 cells. IP with beads only serves as a negative control.
(E) Flag IP performed using nuclear lysates prepared from FLAG-β-catenin WT and FLAG-β-catenin K49R expressing PTEN KD or EV SW480 cells. See also Figure S5.
To confirm our hypothesis that AKT activation induces AKT to interact with and phosphorylate EZH2, we induced AKT activation by performing base editing of the PIK3CA genomic loci. Glutamic acid was replaced by lysine at 453 or 545 (E453K or E545K) (Figure S5B) producing common gain of function mutations of PIK3CA that resulted in increased pAKT levels (Figure S5C). SW480 cells harboring the PIK3CA E453K or E545K mutation had increased EZH2 interaction with and phosphorylation by AKT relative to control EV SW480 cells with WT PIK3CA (Figure S5D). PIK3CA E453K and E545K cells also had increased interaction of β-catenin with RNAPII, EZH2, and TCF1 relative to EV control cells (Figure S5E).
EZH2 phosphorylation increases its binding to chromatin
Next, we wanted to determine how the AKT-EZH2-β-catenin axis altered the binding of these proteins to chromatin. Because we demonstrated that phosphorylation of EZH2 at S21 by AKT caused EZH2 to interact with RNAPII and β-catenin (Figures 2D and 3G), we tested whether S21 phosphorylation of EZH2 affected EZH2’s binding to chromatin. EZH2 KD SW480 cells were rescued with HA-EZH2 WT, HA-EZH2 S21A, or EV followed by H2O2 treatment. H2O2 treatment increased EZH2 levels relative to mock treatment in the chromatin fraction isolated from HA-EZH2 WT-expressing cells (Figure 6A). Chromatin-bound EZH2 was not detected in cells expressing either HA-EZH2 S21A or EV even though levels of EZH2 in the whole-cell fraction were similar between HA-EZH2 WT and HA-EZH2 S21A expressing cells (Figure 6A). Additionally, EZH2 levels were higher in the chromatin fraction of cells rescued with an EZH2 phospho-mimetic mutant construct, HA-EZH2 S21D, relative to cells rescued with HA-EZH2 WT or EV (Figure 6B). EZH2 KD SW480 cells expressing HA-EZH2 WT also had increased levels of chromatin-bound TCF1 after H2O2 treatment than mock-treated or HA-EZH2 S21A expressing cells (Figure 6A). We next tested if the binding of RNAPII and/or TCF1 to chromatin is dependent on AKT and/or EZH2 catalytic activity as we have shown the activity of these enzymes is required for β-catenin to interact with RNAPII and TCF1. Treating cells with AKT or EZH2 inhibitors blocked the H2O2-induced increase of RNAPII and TCF1 levels in the chromatin fraction but had minimal effect on the chromatin levels of these proteins in untreated cells (Figure 6C). While AKT and EZH2 inhibitors did not alter the levels of RNAPII in whole-cell fractions, they reduced the total level of TCF1 (Figure 6C). Because we have shown that the interaction of β-catenin with TCF1 and RNAPII is dependent on β-catenin’s methylation by EZH2 (Figures 3E, 3F, and 5), we tested if trimethylation of β-catenin alters β-catenin’s chromatin binding. Interestingly, PTEN KD induced an increase in FLAG-β-catenin WT but not FLAG-β-catenin K49R levels in the chromatin fraction (Figure 6D). Altogether, these results suggest that phosphorylation of EZH2 by AKT regulates EZH2’s binding to chromatin. Additionally, EZH2-mediated β-catenin trimethylation regulates β-catenin’s binding to chromatin.
Figure 6.

Phosphorylation of EZH2 increases its binding to chromatin
(A) Western blots of chromatin lysate prepared from HA-EZH2 wildtype (WT), HA-EZH2 phospho-null (PN) S21A and HA-empty vector (EV) expressing SW480 cells treated with 250 μM H2O2 for 30 min. Whole-cell extract (WCE) serves as a control for the chromatin extract.
(B) Western blots of chromatin lysate prepared from HA-EZH2 WT, HA-EZH2 S21D and HA-EV expressing SW480 cells.
(C) Western blots of chromatin lysate prepared from SW480 cells untreated and treated with 1 μM EZH2 inhibitor (EZH2i, GSK-503) for 72 h or with 10 μM AKT inhibitor (AKTi, GSK-690693) for 48 h and followed by no additional treatment or co-treatment with 250 μM H2O2 for 30 min.
(D) Western blots of chromatin lysate prepared from FLAG-β-catenin WT and FLAG-β-catenin K49R expressing PTEN knockdown (KD) and EV expressing SW480 cells. WCE serves as a control for chromatin extract.
EZH2 inhibition blocks the PTEN knockdown-induced increase in β-catenin enrichment over the genome
We previously demonstrated that phosphorylation of EZH2 at S21 increased EZH2’s binding to chromatin (Figure 6A). To determine how phosphorylation alters the genome-wide binding pattern of EZH2, CUT&RUN for HA-tagged EZH2 in PTEN KD and EV SW480 cells transfected with HA-EZH2 WT or HA-EZH2 S21A was performed. A portion of the called HA-EZH2 WT peaks in PTEN KD overlapped with the called peaks in EV cells (84 peaks; Figure 7A, top). A larger portion of the called HA-EZH2 WT peaks were specific for PTEN KD SW480 cells (942 peaks; Figure 7A, bottom). There were only 64 HA-EZH2 WT peaks uniquely called in EV cells. Cells transfected with HA-EZH2 S21A had a very low number of called peaks in EV (40 peaks) and PTEN KD (41 peaks) samples, the majority of which were also present in the HA-EZH2 WT samples (Figure 7A). These data confirmed our hypothesis that phosphorylation of EZH2 by AKT increases EZH2 binding to chromatin. Our chromatin data suggested that EZH2-mediated β-catenin trimethylation increased β-catenin binding to the chromatin (Figure 6D). To systematically define the genome-wide enrichment pattern of β-catenin, ChIP-seq for FLAG-tagged β-catenin was performed in EV and PTEN KD SW480 cells. PTEN KD increased the enrichment of FLAG-β-catenin over the chromatin in an EZH2 activity-dependent manner (Figures 7C and S6A). Similar to HA-tagged EZH2, a portion of the called FLAG-β-catenin peaks in PTEN KD cells overlapped with FLAG-tagged β-catenin peaks in EV cells (2525 peaks; Figure 7B, middle). A larger portion of the called FLAG-tagged β-catenin peaks were specific only for PTEN KD cells (8453 peaks; Figure 7B, bottom). There were an additional 2323 FLAG-tagged β-catenin peaks uniquely called in EV cells (Figure 7B, top). Inhibiting EZH2 drastically reduced the number of called FLAG-β-catenin peaks in EV and PTEN KD cells to 58 and 288 peaks, respectively, all of which were also present in the non-inhibitor treated samples (Figure 7B). Profile plots of the FLAG-tagged β-catenin peaks demonstrated increased enrichment in PTEN KD cells relative to EV cells, which was further decreased in EZH2 inhibitor-treated EV and PTEN KD cells (Figure 7C). This finding was consistent in a second ChIP-seq experiment (Figure S6A). Altogether, these data suggest that EZH2 phosphorylation and subsequent β-catenin trimethylation increases the enrichment of EZH2 and β-catenin at numerous loci across the genome.
Figure 7.

EZH2 inhibition blocks the PTEN knockdown-induced increase in β-catenin enrichment over the genome
(A) Metagenomic heatmap for HA CUT&RUN prepared from HA-EZH2 wildtype (WT) and HA-EZH2 S21A expressing PTEN knockdown (KD) and empty vector (EV) SW480 cells.
(B) Metagenomic heatmap for FLAG ChIP-seq prepared from FLAG-β-catenin WT and FLAG-β-catenin K49R expressing PTEN KD and EV expressing SW480 cells.
(C) Average ChIP-seq read intensity for all peaks shown in B.
(D) Heatmap for differentially expressed genes (DEGs) in PTEN KD versus EV clustered manually based on the effect of EZH2 inhibitor (GSK503, 1 μM, 72 h).
(E) ALDEFLUOR assay showing the percentage of ALDH+ cells in cells treated as in D. Red lines indicate mean +/− SEM.
(F) Brightfield images of spheroids following treatment as in D. Scale bar = 2000 microns.
(G) Quantification of spheroids normalized to spheroids counts for untreated EV cells. Results are represented as mean of 3 biological replicates +/− SEM.
(H) Barplot for the hallmark analysis of DEGs in cluster 2.
(I) Gene expression of the indicated genes by RT-qPCR in SW480, LS174T and RKO cells treated as in D. Expression of all the genes was normalized to the house keeping gene RHOA and then to untreated EV cells. Results are represented as mean of 3 biological replicates +/− SEM.
(J) EV and PTEN KD cells were treated with DMSO or 2 μM EZH2 inhibitor (EZH2i, GSK-503) for 72 h followed by plating cells in the upper chamber of a transwell insert. Brightfield images of crystal violet–stained migrated cells were taken after 48 h. Scale bar = 200 microns.
(K) Quantification of migration normalized to migration counts for untreated EV cells. Results are represented as mean of 3 biological replicates +SEM. Significance was determined by one-way ANOVA with the Tukey multiple comparisons test. All significant comparisons are shown. ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001. See also Figures S6 and S7.
Increased enrichment of β-catenin across the genome regulates gene expression
To determine the impact of the AKT-EZH2-β-catenin axis on transcription, we performed RNA-seq with and without EZH2 inhibition in EV and PTEN KD SW480 cells. PTEN KD resulted in 265 upregulated and 141 downregulated genes compared to EV (|log 2FC|>0.5; p < 0.05; Figure S6B). As expected, PTEN KD upregulated the expression of genes involved in the PI3K-AKT pathway whereas PTEN KD downregulated genes were involved in metabolic processes such as fatty acid metabolism (Figure S6C). Inhibiting EZH2 activity altered the expression of PTEN KD-regulated genes (Figure 7D). Therefore, we manually clustered PTEN KD-regulated genes into four clusters based on the effect of the EZH2 inhibitor on their expression. In cluster one, EZH2 inhibition reduced the expression of PTEN KD-upregulated genes (Figure 7D). GO analysis of genes in this cluster showed enrichment of pathways related to cell signaling and transcription initiation (Figure S6D). In cluster two, we detected a further increase in the expression of PTEN KD-upregulated genes in response to the EZH2 inhibitor (Figure 7D). GO analysis of this cluster indicated enrichment of pathways related to tissue morphogenesis, cell migration, and epithelial-to-mesenchymal transition (EMT) (Figure S6D). While the EZH2 inhibitor increased the expression of PTEN KD downregulated genes in cluster three, it further reduced gene expression of PTEN KD downregulated genes in cluster four (Figure 7D). GO analysis of these two clusters showed enrichment of pathways associated with different metabolic processes (Figure S6D).
Through the integration of genomic binding and RNA-seq profiles, we determined 25, 51, 14, and 11 genes from clusters 1, 2, 3, and 4, respectively, that exhibited increased binding of FLAG-β-catenin in PTEN KD cells (Figure S6E), as exemplified by MYBPC2 from cluster 1, FGF3 from cluster 2, STEAP3 from cluster 3, and ZNF438 from cluster 4 (Figure S7A). We also determined that 263 peaks had increased binding of both β-catenin and EZH2 in PTEN KD relative to EV cells such as NTM (Figure S7B). Additionally, a small number of EZH2 peaks overlapped with differentially expressed genes in response to PTEN KD as exemplified by P2RX5 and CLSTN2 (Figure S7C). However, the majority of EZH2 peaks gained in PTEN KD cells did not overlap with FLAG-β-catenin peaks and were not associated with differentially expressed genes in PTEN KD cells. Altogether these results suggest that EZH2-mediated β-catenin trimethylation fine-tunes the transcriptional activity of β-catenin to regulate gene expression in PTEN KD SW480 cells.
PTEN KD enhances cell stemness and migration in an EZH2 dependent manner
Activation of AKT is known to increase the proportion of CRC stem cells.28 To determine the effect of PTEN KD on cancer cell stemness, we measured aldehyde dehydrogenase (ALDH) activity, a commonly used robust marker of cancer stem cells.29 PTEN KD significantly increased the percentage of ALDH+ cells compared to EV and parental SW480 cells (Figure 7E). EZH2 inhibition blocked the PTEN KD-induced enrichment of ALDH+ cells (Figure 7E) suggesting that the PTEN KD-mediated increase in stemness is dependent on EZH2 catalytic activity. To confirm these results, we performed a spheroid assay in which only cells that exhibit stem cell characteristics form spheres. PTEN KD significantly increased the number of spheres in comparison to EV, while EZH2 inhibition reduced spheroid formation in PTEN KD SW480 cells (Figures 7F and 7G).
GO analysis of cluster 2 genes that were defined as being increased in expression with PTEN KD and further increased with EZH2 inhibition demonstrated enrichment for genes involved in migration, morphogenesis, and epithelial to mesenchymal transition (EMT) (Figure S6D). Therefore, we decided to narrow our focus to cluster 2 genes and performed hallmark analysis using the Human MSigDB Collections.30 Consistent with our previous analysis, cluster 2 genes were significantly associated with apical junctions and EMT hallmarks (Figures 7H and S7D). β-catenin appeared to directly regulate some EMT-related genes as PTEN KD increased the binding of FLAG-β-catenin to their promoter regions, as exemplified by ANO1 and LAMA3 (Figure S7E). Because the cluster 2 genes were defined as being further increased by EZH2 inhibition, we explored the effect of EZH2 inhibition alone on gene expression pathways using our RNA-seq data. Hallmark analysis of the 255 genes (log2FC > 0.5; p < 0.05) upregulated in response to EZH2 inhibitor in EV cells showed that the most significantly enriched hallmark was the EMT hallmark (Figure S7F). To confirm these findings, we performed RT-qPCR in additional CRC cell lines following PTEN KD and/or EZH2 inhibition. PTEN KD increased expression of EMT marker genes, SNAI2, and FGF3, in SW480 and LS174T cells. EZH2 inhibition in PTEN KD cells increased expression of SNAI1 and further upregulated SNAI2 and FGF3 as compared to PTEN KD alone (Figure 7I). Similar to SW480 and LS174T cells, inhibiting EZH2 in PTEN KD RKO cells increased expression of SNAI1 and SNAI2 (Figure 7I). These results were consistent with our RNA-seq data where EZH2 inhibition upregulated EMT marker genes in PTEN KD cells. To determine the outcome of EZH2 inhibition on EV and PTEN KD CRC cell migration, we performed transwell migration assays. PTEN KD increased SW480 cell migration compared to EV as expected (Figures 7J and 7K). EZH2 inhibitor treatment trended toward inducing increased cell migration of EV cells and significantly increased PTEN KD cell migration (Figures 7J and 7K). Our results suggest that even though EZH2 inhibition blocks PTEN KD-induced cancer stemness, EZH2 inhibition enhances the PTEN KD-mediated increases in expression of EMT marker genes and cell migration.
Discussion
In this study, we reveal the mechanism by which EZH2 regulates β-catenin in CRC. We determined that activation of PI3K/AKT, a pathway activated in a significant portion of CRCs, results in EZH2 phosphorylation at S21 (pS21-EZH2) in CRC, similar to what has been found in other cancer types.21,23 Our data further demonstrate that phosphorylation of EZH2 via AKT is the molecular switch that drives EZH2 to interact with and methylate β-catenin. Inhibition of AKT signaling blocks EZH2’s interaction with and methylation of β-catenin. Additionally, a phospho-null EZH2 mutant (HA-EZH2 S21A) did not interact with β-catenin following AKT activation confirming that phosphorylation at S21 drives EZH2’s interaction with β-catenin. These findings agree with other studies in which phosphorylation of EZH2 by AKT induces EZH2’s interaction with other transcription factors such as AR and STAT3 in prostate cancer and glioblastoma, respectively.21,23
Previously, EZH2 has been reported to modulate β-catenin’s transcriptional activity in other cell types. For example, in liver cancer stem cells, EZH2-mediated methylation of β-catenin at K49 increased β-catenin’s transcriptional activity and promoted WNT signaling, while in embryonic stem cells β-catenin methylation at the same site by EZH2 led to gene repression during development.24,31 Here, we demonstrated that EZH2 trimethylates β-catenin, which fine-tunes β-catenin’s transcriptional activity in CRC. We determined that EZH2’s catalytic activity is essential for modulating β-catenin regulated gene expression. Treating cells with an EZH2 inhibitor (GSK503) blocked the H2O2 or PTEN KD-mediated interaction of β-catenin with RNAPII and TCF1. Additionally, a methylation-null mutant construct, FLAG-β-catenin K49R, had less interaction with RNAPII than wildtype β-catenin in response to H2O2 or PTEN KD. While we did not detect any differences in the nuclear level of wild type or mutant β-catenin following AKT activation, FLAG-β-catenin K49R had reduced binding to the chromatin in response to PTEN KD. These findings suggest that trimethylation of β-catenin alters its chromatin binding not its nuclear localization. Our findings are unique from previous studies because we demonstrated that AKT-mediated phosphorylation of EZH2 at S21 (pS21-EZH2) drives EZH2 to interact with and methylate β-catenin, which enhances β-catenin’s and RNAPII’s binding to the chromatin. Based on our findings, we propose that the AKT-mediated phosphorylation of EZH2 results in EZH2’s regulation of β-catenin being dependent on the activity of EZH2, in contrast to other studies that have found that EZH2 regulates β-catenin independently of its catalytic activity.32 It should be noted that, like the majority of CRC, most of the cell lines used in our study have either APC or CTNNB1 mutations resulting in constitutive activation of the WNT/β-catenin pathway.27,33,34,35 However, we found similar results in RKO cells, which have wildtype APC and CTNNB1 and have normal canonical Wnt signaling. We suggest that aberrant activation of WNT/β-catenin pathway is not required for but may enhance EZH2’s regulation of β-catenin in CRC.
We also demonstrated that the AKT-EZH2-β-catenin axis regulates EZH2’s and β-catenin’s binding to chromatin and enrichment across the genome. While a phospho-mimetic EZH2 (EZH2 S21D) showed increased binding to chromatin, phospho-null EZH2 had reduced binding to chromatin relative to WT EZH2 following AKT activation. Our genome-wide binding profiles for HA-EZH2 and FLAG-β-catenin confirmed our hypothesis that EZH2 and β-catenin binding to the chromatin is dependent on EZH2 phosphorylation at S21 and EZH2 catalytic activity, respectively. Phospho-null EZH2 had a reduced number of called HA-EZH2 peaks in both EV and PTEN KD cells. Treating with an EZH2 inhibitor also drastically reduced the number of called FLAG-β-catenin peaks in both EV and PTEN KD cells. These findings are consistent with other findings in which EZH2 induced the binding of AR, cMyc, and P300 to the chromatin to mediate gene expression.36,37 While previous studies suggest that EZH2 methylation of other non-histone proteins leads exclusively to gene activation or repression,20,21 our findings propose a model in which EZH2-mediated β-catenin methylation increases β-catenin’s binding to the chromatin and fine-tunes β-catenin activity to both repress and activate gene expression. The exact mechanism of how methylation of β-catenin leads to gene activation and repression is unclear. It is possible that methylation of β-catenin represses gene expression by blocking β-catenin acetylation, which is associated with active gene expression, as has been reported previously.38 It is also possible that EZH2-mediated β-catenin methylation activates gene expression through increased recruitment of TCF1 and RNAPII to chromatin. In breast cancer, phosphorylation of EZH2 by AKT was shown to attenuate EZH2’s catalytic activity by reducing its binding to histones.39 Thus, AKT activation resulted in reduced H3K27me3 levels.39 In our study, neither activation nor inhibition of AKT affected global levels of H3K27me3 in CRC cells, suggesting that the AKT pathway does not regulate global levels of H3K27me3 in CRC. A potential explanation for the discrepancy in findings between the two studies is that, in CRC cells following EZH2 phosphorylation, EZH1 may take over for EZH2 in the PRC2 complex to maintain H3K27me3 levels as recently reported.40 While we see an increase in the binding of phosphorylated EZH2 to chromatin, we have not specifically examined how phosphorylation alters EZH2’s binding to and/or methylation of histones.
Our data demonstrate that AKT activation increases cancer cell stemness in an EZH2-dependent manner as indicated by EZH2 inhibition blocking the PTEN KD associated increases in percent ALDH+ cells and spheroid formation. These results are in agreement with findings from other groups in which inhibiting the repressive function of EZH2 reduced the cancer stem cell population in ovarian cancer41 and promoted human stem cell differentiation.42 Our data indicate that AKT activation also promotes β-catenin-mediated upregulation of EMT-related genes in an EZH2-dependent manner. We show that increased β-catenin enrichment over some EMT-related genes is associated with their increased gene expression in PTEN KD compared to EV cells. Treating cells with an EZH2 inhibitor resulted in reduced β-catenin enrichment across the genome. However, this finding was unexpectedly accompanied by further upregulation of EMT-related genes. Related to this result, EZH2 inhibition also increased expression of EMT-related genes in EV cells, which is similar to previous findings by other groups where EZH2 repressed genes to maintain cell identity.43,44,45,46 It is unexpected that EZH2 inhibition reduces cancer stemness but increases EMT-related changes following AKT activation as cancer stemness and EMT are generally linked. It is possible that AKT-mediated EZH2 phosphorylation induces EZH2 to promote cancer stemness and EMT in epithelial colon cells by interacting with and methylating β-catenin. However, inhibiting EZH2 activity enhances colon epithelial cell migration by increasing the expression of EMT-related genes, which are usually repressed by PRC2. We speculate that pS21-EZH2 induces the expression of EMT genes through methylating β-catenin while inhibiting EZH2 attenuates PRC2 repression of EMT genes, which cumulatively results in increased EMT gene expression and migration.
Based on our EZH2 CUT&RUN data, phosphorylation of EZH2 following AKT activation in PTEN KD cells greatly increased the number of genomic loci bound by EZH2. However, very few of these peaks were associated with changes in gene expression as they did not overlap with PTEN KD-regulated genes in our RNA-seq data. Most of the EZH2 peaks also did not overlap with FLAG-β-catenin peaks in PTEN KD cells in our ChIP-seq data. Because EZH2 inhibition blocked the PTEN KD-induced increase in β-catenin binding sites and altered expression of the PTEN-KD regulated genes, we conclude that EZH2-induced trimethylation of β-catenin modulates gene expression in PTEN KD cells. However, we suggest that most of the increased binding of EZH2 to chromatin following AKT-mediated phosphorylation on S21 is not regulated by EZH2’s interaction with β-catenin and does not have a direct effect on gene expression. Future work will explore the mechanism and relevance of the increased binding of phosphorylated EZH2 to chromatin.
Overall, we propose a model where activation of AKT induces EZH2 phosphorylation, which mediates β-catenin methylation. β-catenin methylation increases β-catenin’s interaction with TCF1 and RNAPII and increases β-catenin’s binding to the chromatin to regulate gene expression. Extrapolation of the findings in this study suggests a need for the development of a context-specific strategy for the therapeutic targeting of EZH2 in CRC. The oncogenic role of EZH2 in cancers led to the development of many EZH2 inhibitors that are currently in clinical trials.47 However, our findings suggest that treatment with catalytic EZH2 inhibitors will likely lead to different clinical outcomes depending on the mutations present in the specific CRC being treated. Our findings suggest that, in AKT-activated CRC, therapeutic approaches based on perturbation of the interaction between EZH2 and β-catenin or EZH2 and AKT will be more effective than targeting EZH2 activity as EZH2 catalytic inhibitors will block all activity dependent roles of EZH2, including EZH2’s methylation of histone and non-histone substrates.
Limitations of the study
In the present study, we demonstrated that AKT induces EZH2-mediated β-catenin tri-methylation at K49 via EZH2 phosphorylation at S21. Methylation of β-catenin induces β-catenin’s interaction with RNAPII. However, the precise function of the β-catenin-RNAPII complex is not clear. Additional studies are required to investigate the effect of the β-catenin-RNAPII complex on RNAPII transcription initiation and transcription elongation. Although we used several CRC cell lines and organoids to study the AKT/EZH2/β-catenin axis, we did not perform in vivo experiments. Future research should investigate the role of the AKT/EZH2/β-catenin axis on tumor progression and metastasis using animal models.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-RBP1 | Cell Signaling Technology | Cat#2629, RRID: AB_2167468 |
| Rabbit monoclonal anti-H3K27me3 | Cell Signaling Technology | Cat#9733, RRID: AB_2616029 |
| Rabbit monoclonal anti-H3K4me3 | Cell Signaling Technology | Cat#9751,RRID: AB_261602 |
| Rabbit polyclonal anti-Tri-Methyl-lysine | PTM BIO | Cat#PTM-601 |
| Rabbit monoclonal anti-β-catenin | Cell Signaling Technology | Cat#8480, RRID:AB_11127855 |
| Rabbit monoclonal anti-Vimentin | Cell Signaling Technology | Cat#5741, RRID:AB_10695459 |
| Rabbit monoclonal anti-total AKT | Cell Signaling Technology | Cat#4691, RRID:AB_91578 |
| Rabbit monoclonal anti-Phospho-AKT(Ser473) | Cell Signaling Technology | Cat#4060, RRID:AB_2315049 |
| Mouse monoclonal anti-TCF1 | Santa-Cruz Biotechnology | Cat#Sc-271453, RRID:AB_10649799 |
| Rabbit monoclonal anti-H3 | Cell Signaling Technology | Cat#4499, RRID:AB_10544537 |
| Rabbit monoclonal anti-GAPDH | Cell Signaling Technology | Cat#5174, RRID:AB_10622025 |
| Mouse Monoclonal anti-FLAG | Sigma-Aldrich | Cat#F3165, RRID:AB_259529 |
| Rabbit monoclonal anti-HA | Cell Signaling Technology | Cat#3724, RRID:AB_1549585 |
| Rabbit polyclonal anti-HA | EpiCypher | Cat#13-2010 |
| Rabbit monoclonal anti-TCF1 | Cell Signaling Technology | Cat#2203, RRID:AB_2199302 |
| Rabbit monoclonal anti- Phospho-Akt Substrate (RXXS∗/T∗) | Cell Signaling Technology | Cat#9614, RRID:AB_331810 |
| Rabbit monoclonal anti-EZH2 | Cell Signaling Technology | Cat# 5246, RRID:AB_10694683 |
| Bacterial and virus strains | ||
| MISSION shRNA Bacterial Stock, EZH2 | Sigma-Aldrich | Cat# SHCLNG-NM, TRCN0000010474 |
| MISSION shRNA Bacterial Stock, PTEN | Sigma-Aldrich | Cat# SHCLNG-NM, TRCN0000002748 |
| MISSION shRNA Bacterial Stock, TRC2 pLKO.5-puro Empty Vector | Sigma-Aldrich | Cat# SHC201 |
| Chemicals, peptides, and recombinant proteins | ||
| GSK690693 | Sigma-Aldrich | Cat#SML0428 |
| GSK503 | Sigma-Aldrich | Cat#SML2718 |
| Hydrogen Peroxide (30%) | Fisher-chemical | Cat#H325-500 |
| McCoy’s 5A | Corning | Cat#10-050-CV |
| DMEM | Corning | Cat#10-013-CV |
| RPMI 1640 | Corning | Cat#10-040-CV |
| Advanced DMEM/F12 | Gibco | Cat#12634-010 |
| N2 supplement | Fisher | Cat#17502048 |
| B27 supplement | Fisher | Cat#17504044 |
| EGF | R&D Systems | Cat#236-EG |
| Noggin | R&D Systems | Cat#6057-NG |
| Protease Inhibitor Tablets | Thermo Scientific | Cat#88266 |
| Phosphatase Inhibitor Cocktail 2 | Sigma-Aldrich | Cat#P5726 |
| N-ethylmaleimide | Acros Organics | Cat#128-53-0 |
| pAG-MNase | EpiCypher | Cat#15-1116 |
| Matrigel (Growth Factor Reduced) | Corning | Cat#354230 |
| Human FGF-basic (bFGF) | Invitrogen | Cat#13256-029 |
| Insulin | Sigma-Aldrich | Cat#19278 |
| 16% Formaldehyde solution (w/v), methanol free | Thermo Scientific | Cat#28908 |
| Fetal Bovine Serum | Corning | Cat#35-015-CV |
| Puromycin | Sigma-Aldrich | Cat# P8833 |
| Lipofectamine 3000 | Invitrogen | Cat#L3000015 |
| DMSO | VWR | Cat#97063-136 |
| Critical commercial assays | ||
| Diagenode iDeal ChIP-seq Kit for transcription factor | Diagenode | Cat#C01010055 |
| RNeasy Mini Kit | Qiagen | Cat#74104 |
| ALDEFLUOR assay | Stem Cell Technologies | Cat#01700 |
| CUTANA™ DNA Purification Kit | EpiCypher | Cat#14-0050 |
| NEBNext Ultra II DNA Library Prep kit | New England BioLabs | Cat#E7645 |
| Maxima first strand cDNA synthesis kit | Thermo Fisher Scientific | Cat#K1642 |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO: GSE226589 |
| Experimental models: Cell lines | ||
| Human: SW480 | ATCC | Cat#CCL-228, RRID:CVCL_0546 |
| Human: RKO | ATCC | Cat#CRL-2577, RRID:CVCL_0504 |
| Human: Lovo | ATCC | Cat#CCL-229, RRID:CVCL_0399 |
| Human: LS174T | ATCC | Cat#CL-188, RRID:CVCL_1384 |
| Human: HEK293T | ATCC | Cat#CRL-3216, RRID:CVCL_0063 |
| Human: Caco2 | ATCC | Cat#HTB-37, RRID:CVCL_0025 |
| 519 human colorectal cancer organoids | Miller et al.48 | N/A |
| SW480 PIK3CA E453K and E453K mutant cells | This paper | N/A |
| Oligonucleotides | ||
| Primers and oligos for RT-qPCR, plasmid construction and base editing, see Table S1 | This paper | N/A |
| Recombinant DNA | ||
| pCMV-HA-EZH2 | Addgene | RRID:Addgene_24230 |
| FLAG- β-catenin WT | Addgene | RRID:Addgene_16828 |
| FLAG- β-catenin K49R | Addgene | RRID:Addgene_44750 |
| pLenti-U6-tdTomato-P2A-BlasR (LRT2B) | Addgene | RRID:Addgene_110854 |
| pLenti-FNLS-P2A-Puro | Addgene | RRID:Addgene_110841 |
| pCMV-HA-EZH2 S21A | This paper | N/A |
| pCMV-HA-EZH2 S21D | This paper | N/A |
| Software and algorithms | ||
| FastQC (v0.11.9) | Simon Andrews | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| STAR (v2.7.10a) | Alex Dobin, dobin@cshl.eu | https://github.com/alexdobin/STAR |
| clusterProfiler (v1.38.3) | Yu et al.49 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| EnhancedVolcano | Blighe, Rana, and Lewis50 | https://bioconductor.org/packages/devel/bioc/vignettes/EnhancedVolcano/inst/doc/EnhancedVolcano.html |
| Bowtie2 | Langmead et al.51 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| MACS (v2.2.7.1) | Zhang et al.52 | https://github.com/macs3-project/MACS |
| Bedtools (v2.26.0) | Quinlan & Hall53 | https://bedtools.readthedocs.io/en/latest/ |
| deeptools | Ramírez et al.54 | https://deeptools.readthedocs.io/en/latest/ |
| GVIS (v3.5.1) | Hahne & Ivanek55 | https://bioconductor.org/packages/release/bioc/html/Gviz.html |
| Deseq2 | Love et al.56 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Metascape pathway analysis | Zhou et al.57 | https://metascape.org/gp/index.html#/main/step1 |
| UCSC xenabrowser | Goldman et al.58 | https://xenabrowser.net/ |
| RStudio (V 4.2.2) | R | https://posit.co/download/rstudio-desktop/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Heather O’Hagan (hmohagan@indiana.edu).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact upon request.
Experimental model and study participant details
Cell lines
All cell lines were maintained in a humidified atmosphere at 37°C with 5% CO2. Our study included SW480, RKO, Lovo, LS174T, Caco2 and HEK293T cell lines and human CRC organoids. SW480, Caco2 and Lovo cells were cultured in McCoy 5A media (Corning, #10-050-CV), and RKO cells were cultured in RPMI media (Corning, #10-040-CV) supplemented with 10% FBS (Corning, 35-015-CV). LS174T and HEK293T cells were cultured in DMEM 1X (Corning, #10-013-CV) with 10% FBS (Corning, #35-015-CV) without antibiotics. CRC human organoids were cultured in organoid media (advanced DMEM/F12 (Gibco, #12634-010) supplemented with EGF (R&D Systems, #236-EG), Noggin (R&D systems, #6057-NG), N2 supplement (17502048, Fisher), B27 supplement (Fisher, #17504044), HEPES, and Penn/Strep) as in.48 All cell lines were purchased from the ATCC and authenticated and tested for Mycoplasma using the Universal mycoplasma detection kit (ATCC, 30-1012K) on January 30, 2023. All cell lines were derived from male patients except CRC organoids and HEK293T cells were derived from female patients. All cells used in experiments were passaged fewer than 15 times with most being passaged fewer than 10 times. For hydrogen peroxide (H2O2) treatments, 30% H2O2 (Fisher, #H325-500) was diluted in PBS immediately prior to treatment at 250 mmol/L for 30 minutes at 37°C. Cells were starved in media lacking serum for 24 hours prior to treatment. GSK503 (Sigma, #SML2718) and GSK690693 (Sigma, #SML0428) were solubilized in DMSO (VWR, #97063-136) prior to treatment. Treatment dosages and durations are defined in the figure legends.
Method details
Generation of stable knockdown cell lines
For knockdown of EZH2 (Sigma, SHCLNG-NM_004456, #TRCN0000010474), PTEN (Sigma, SHCLNGNM_000314, #TRCN0000002748), and empty vector (EV) TRC2 (Sigma, #SHC201), the lentiviral shRNA knockdown protocol from The RNAi Consortium Broad Institute was used. Briefly, 4 x 105 HEK293T cells were plated on day 1 in DMEM 1X containing 10% FBS. On day 2, cells were transfected with shRNA of interest, EV control, and packaging plasmids. On day 3, the media was replaced with fresh DMEM containing 10% FBS. Approximately 24 hours later, media containing lentiviral particles was collected, and fresh DMEM + 10% FBS was added. The added media were collected 24 hours later and pooled with media harvested on day 4. The pooled media was then filtered using a 0.45 mm filter and concentrated using a Spin-X concentrator (Corning, #431490). To perform the knockdown, concentrated virus plus polybrene was added to experimental cells 24 hours after plating. Cells were treated with puromycin (2 μg/mL) (Sigma-Aldrich, #P8833) after 24 hours to select for knockdown cells.
Plasmids and transient transfections
HA-EZH2 plasmid mutant constructs were generated using site directed mutagenesis of pCMV-HA-EZH2 a gift from Kristian Helin (Addgene plasmid # 24230; http://n2t.net/addgene:24230; RRID:Addgene_2423059). Wildtype and mutant constructs were then PCR amplified using primers to encompass HA-EZH2 + add NotI and PacI for subcloning into pQXCIH (Clontech). See Table S1 for the primer sequences. FLAG-β-catenin plasmid constructs were purchased from Addgene (FLAG- β-catenin WT (Addgene plasmid # 16828; http://n2t.net/addgene:16828; RRID: Addgene_16828) and FLAG- β-catenin K49R (Addgene plasmid # 44750; http://n2t.net/addgene:44750; RRID:Addgene_44750). All FLAG- β-catenin constructs were a gift from Eric Fearon.60,61 Transient transfection was performed with Lipofectamine 3000 (Invitrogen) per the manufacturer's protocol.
Nuclear Immunoprecipitations (IPs)
3.5 x 106 or 1.2 x 106 cells were cultured in 150 mm plates for approximately 72 hours or 96 hours, respectively. Cell pellets were used to perform nuclear extraction using CEBN [10 mmol/L HEPES, pH 7.8, 10 mmol/L KCl, 1.5 mmol/L MgCl2, 0.34 mol/L sucrose, 10% glycerol, 0.2% NP-40, 1X protease inhibitor cocktail (Sigma, #P5726), 1X phosphatase inhibitor (Thermo, #88266), and N-ethylmaleimide (Acros organics, #128-53-0)] and then washed with CEB buffer (CEBN buffer without NP-40) containing all the inhibitors. To extract the nuclear fraction, after washing the cell pellets with CEBN buffer, they were resuspended in modified RIPA (50 mM Tris pH7.5, 150 mM NaCl, 5 mM EDTA, 50 mM NaF, all inhibitors) and sonicated using Bioruptor® Pico (Diagenode). To exclude the possibility of DNA-dependent protein interactions, the nuclear fractions were incubated with spermine (50 mM) and spermidine (15 mM) for 60 minutes on a rotator at 4°C. Positively charged spermines compete proteins off the DNA. The nuclear extract was rotated with an antibody and protein A/G magnetic beads overnight. The next day, beads were washed, and proteins were eluted and analyzed by Western blot. For some IPs, RNAseA (25 μg/ml) was added during the overnight incubation.
Chromatin extraction
3.5 x 106 or 1.2 x 106 cells were cultured in 150 mm plates for approximately 72 hours or 96 hours, respectively. Cell pellets were used to perform nuclear extraction using CEBN and then washed with CEB buffer containing all the inhibitors. To extract the soluble nuclear fraction, after washing the cell pellets with CEB buffer, they were resuspended in soluble nuclear buffer (2 mmol/L EDTA, 2 mmol/L EGTA, all inhibitors) and rotated at 4°C for 30 minutes. The remaining cell pellet, i.e., the total chromatin fraction, was lysed using 4% SDS and a qiashredder (Qiagen) and analyzed by Western blot.
Chromatin immunoprecipitation-sequencing
Chromatin immunoprecipitation (ChIP) was performed using anti-FLAG antibody (Sigma, #F3165) and the Diagenode iDeal ChIP-seq for transcription factors Kit (Diagenode, #C01010055) as per the manufacturer’s protocol. SW480 cells were fixed, and chromatin DNA-protein was cross-linked with 1% formaldehyde (Thermo-Scientific, Cat#28908) for 10 min, then quenched with 125 mM glycine for 5 min, and finally washed with phosphate-buffered saline. Fixed cells were sonicated with a Bioruptor Pico (Diagenode) and lysed in a lysis buffer containing protease inhibitors. DNA-protein complexes were immunoprecipitated with FLAG antibody and washed. Cross-links were reversed at 65C overnight in Elution buffer. DNA was extracted by PCR purification kit (QIAquick PCR Purification Kit, #28104). Libraries were generated for sequencing using the NEBNext Ultra II DNA Library Prep kit for Illumina (NEB, E7645) as per the manufacturer’s protocol, followed by sequencing.
CUT&RUN
Cleavage Under Targets & Release Using Nuclease (CUT&RUN) was performed according to the EpiCypher CUTANA CUT&RUN protocol. 5 x 105 of live cells were harvested, washed and re-suspended in wash buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, 0.5 mM Spermidine and 1x Protease Inhibitor), followed by incubation at RT for 10 min with activated ConA magnetic beads (CUTANA™ Concanavalin A, #21-1401) which were washed and re-suspended in bead activation buffer (20 mM HEPES, pH 7.9, 10 mM KCl, 1 mM CaCl2, 1 mM MnCl2). After binding to activated beads, cells were permeabilized in antibody buffer (wash buffer + 0.01% digitonin + 2 mM EDTA) and incubated with HA antibody (HA Tag CUTANA, #13-2010) on nutator overnight at 4°C. On the next day, the cell-bead slurry was washed twice with cold digitonin buffer (wash buffer + 0.01% digitonin) and then incubated with pAG-MNase (EpiCypher, #15-1116) for 10 min at RT, followed by two washes with cold digitonin buffer. MNase was then activated by addition of CaCl2 to cleave targeted chromatin for 2 hours at 4°C. After chromatin digestion, MNase activity was stopped and chromatin fragments released into supernatant by adding stop buffer (340 mM NaCl, 20 mM EDTA, 4 mM EGTA, 50 μg/ml RNase A, and 50 μg/ml Glycogen) and incubating at 37°C for 10 min. The DNA was purified from the collected supernatant using the CUTANA™ DNA Purification Kit (EpiCypher, #14-0050) per manufacturer’s instruction. Finally, 5 ng of purified CUT&RUN enriched DNA was used to prepare Illumina library using the NEBNext Ultra II DNA Library Prep kit (NEB, #E7645) as per the manufacturer’s protocol, followed by sequencing.
RNA sequencing
RNA extraction was performed using the RNAeasy mini kit (Qiagen, #74104) as per manufacture’s protocol. Libraries were generated for sequencing using the NEBNext Ultra II DNA Library Prep kit for Illumina (NEB, E7645) as per the manufacturer’s protocol, followed by sequencing.
Migration assay
5 x 104 SW480 cells in serum-free media were plated into transwell in 24-well plates (Corning, #40578) for 48 hours with media containing 10% FBS at the bottom. Transwell inserts were stained using Hema 3 Stat Pack (Thermo Fisher Scientific, #123-869). Migration inserts were randomized prior to manual quantification and the outer 5% of the inserts were not included during quantification to reduce edge-effect bias. All images were taken on an EVOS FL Auto microscope.
ALDEFLUOR assay
SW480 cell (8.0 × 105) were cultured in 100 mm dishes. PTEN KD, EV and parental cells were treated with EZH2 inhibitor (GSK503) and incubated at 37°C, 5% CO2 for 72 hours. After the incubation time (30 minutes), ALDEFLUOR assay (Stem Cell Technologies, #01700) was performed as described previously.29 Briefly, to measure ALDH activity, the ALDEFLUOR assay (Stem Cell Technologies, 01700) was used consisting of 1 million cells/1 mL ALDEFLUOR assay buffer and bodipyaminoacetaldehyde (BAAA) substrate +/- ALDH inhibitor diethylamino benzaldehyde (DEAB; 5 μL, 1.5 mM). Cells were incubated for 30 minutes at 37°C, centrifuged and resuspended in ALDEFLUOR assay buffer. Flow cytometry analysis was performed on a LSRII flow cytometer (BD Biosciences) at IU Flow Cytometry Core Facility. ALDH activity was measured using 488 nm excitation and the signal was detected using the 530/30 filter. For each experiment, 10,000 events were analyzed. ALDH+ percentage gate was determined by sample specific negative control (DEAB) ALDH+ gate. Further data analysis was done in FlowJo software (Becton, Dickinson & Company, RRID:SCR_008520).
Spheroid formation assay
5 × 103 cells were mixed with Matrigel (Corning, Growth Factor Reduced, # 354230) and then platted in 24-well plates (Costar, #3738). The plate was incubated in a tissue culture incubator for 10 minutes to allow Matrigel to solidify followed by the addition of stem cell media [DMEM-F12 (Corning, #10-017-CV) with 100 U Penicillin–Streptomycin, 0.4% BSA, 10 ng/ml bFGF (Invitrogen, #13256-029), 20 ng/ml EGF (Biolegend, #585,506), 5 μg Insulin (Sigma, #19278)]. The plates were incubated for 20 days at 37°C, 5% CO2. Spheroids were manually quantified and the periphery of Matrigel were not included during quantification to reduce edge-effect bias. Images were taken using an EVOS FL Auto microscope (Life Technologies).
RNA isolation and RT-qPCR
Isolation of total RNA from cell pellets was performed using the RNAeasy mini kit (Qiagen, #74104) as per the manufacturer’s protocol. Maxima first strand cDNA synthesis kit (Thermo Fisher, #K1642) for quantitative reverse transcription PCR was used to synthesize cDNA. cDNA was amplified using gene-specific primers and FastStart Essential DNA Green Master (Roche, #06402712001). Cq values of genes of interest were normalized to housekeeping gene ROHA expression. qPCR primer sequences are listed in Table S1.
Base editing of PIK3CA
To generate sgRNA expressing plasmids, E453K or E453K specific oligos were annealed and cloned into pLenti-U6-tdTomato-P2A-BlasR (LRT2B), a gift from Lukas Dow (Addgene plasmid #110854; http://n2t.net/addgene:110854; RRID:Addgene_11085462). SW480 cells were then transfected with E453K, E453K, or empty vector LRT2B plasmids and pLenti-FNLS-P2A-Puro, a gift from Lukas Dow (Addgene plasmid #110841; http://n2t.net/addgene:110841; RRID:Addgene_110841) at a 1:3 ratio of sgRNA:pLenti-FNLS using lipofectamine 3000. The next day media was changed to fresh media containing puromycin. Following puromycin selection, cells were grown in MEK inhibitor (25 nM Trametinib) for 7 days to select for cells containing mutant PIK3CA. To check for successful base editing, genomic DNA was isolated using the DNeasy kit (Qiagen), PCR amplified using E545K or E453K AP PCR primers, and purified PCR products (Qiagen PCR purification kit) were sequenced by Sanger sequencing using the respective sequencing primers. See Table S1 for the oligo and primer sequences.
Sequencing analysis
Sequencing read quality control for all samples was assessed with FastQC (v0.11.9). For RNA-seq analysis, read alignment was performed using STAR63 (v2.7.10a) against the hg38 reference genome, raw read counts for genes were obtained with STAR (v2.7.10a), and Deseq256(v3.24.0) was used for differential expression analysis. Gene Set Enrichment Analysis using the Molecular Signatures Database collections and database were run with clusterProfiler49(v1.38.3) and volcanoplot was visualized with EnhancedVolcano.50 For ChIP-seq and CUT&RUN analysis, read alignment was performed using Bowtie251(v2.4.2) against the hg38 reference genome, peaks were called with MACS52(v2.2.7.1), and intersecting peak regions between replicates and different samples were uncovered with Bedtools53(v2.26.0). Peaks located within 1 kb window between ChIP-seq and CUT&RUN were considered to be the intersecting peak region between ChIP-seq and CUT&RUN. Heatmaps and Counts Per Million normalized bigWigs were created using deepTools54(v3.5.1) bamCoverage, computeMatrix, and plotHeatmap. Gviz55(1.34.1) was utilized for creating gene tracks. Metascape pathway analysis was performed for pathway analysis.57 The correlation between EZH2 and PTEN expression in CRC was performed using UCSC xenabrowser58
Quantification and statistical analysis
Spheroids and migration insert were manually quantified, and the outer part was not included during quantification to avoid edge-bias effect.
Results are represented as mean +/- SEM unless otherwise stated. Significance was determined by one-way ANOVA with the Tukey multiple comparisons test using RStudio. For all figures, ∗ P ≤ 0.05, ∗∗ P ≤ 0.01, ∗∗∗ P ≤ 0.001, ∗∗∗∗ P ≤ 0.0001. All significant comparisons are shown. Statistical details for each experiment are included in the figures and figure legends.
Acknowledgments
We would like to thank the Indiana University Center for Genomics and Bioinformatics for their assistance with library preparation and sequencing. We also thank Christiane Hassel and the Indiana University Flow Cytometry facility for their assistance.This work was funded, in part, with a Core Pilot Grant [to H.M.O.H.] from the Indiana Clinical and Translational Sciences Institute funded, in part by Grant NumberULITR002529 from the National Institutes of Health (NIH), National Center for Advancing Translational Science, Clinical and Translational Sciences Award. This work was also supported by a Research Enhancement Grant and Elwert Award [to H.M.O.H.] from the Indiana University School of Medicine (IUSM) and pilot funding [to H.M.O.H.] from the IU Simon Comprehensive Cancer Center (IUSCCC) Tumor Microenvironment & Metastasis Program and the IUSCCC P30 Support Grant (P30CA082709). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or IUSM. Additional pilot funding was provided by the Van Andel Institute through the Van Andel Institute - Stand Up to Cancer Epigenetics Dream Team and the NCI SPORE Project, Epigenetic Therapies – New Approaches Developmental Research Program [to H.M.O.H.]. Stand Up to Cancer is a division of the Entertainment Industry Foundation, administered by AACR. A. Ghobashi was supported by the Doane and Eunice Dahl Wright Fellowship generously provided by Ms. Imogen Dahl.
Author contributions
A.H.G.: Conceptualization, formal analysis, computational analysis, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. T.T.V.: Validation, investigation. J.W.K.: Investigation. C.A.L.: Investigation. P.C.H.: Resources. H.M.O.H.: Conceptualization, resources, supervision, funding acquisition, writing–review and editing. All authors reviewed and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: August 16, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107630.
Supplemental information
Data and code availability
-
•
Sequencing data have been deposited at GEO and are publicly available as of publication. The accession number is listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Siegel R.L., Miller K.D., Wagle N.S., Jemal A. Cancer statistics, 2023. CA. Cancer J. Clin. 2023;73:17–48. doi: 10.3322/caac.21763. [DOI] [PubMed] [Google Scholar]
- 2.Koveitypour Z., Panahi F., Vakilian M., Peymani M., Seyed Forootan F., Nasr Esfahani M.H., Ghaedi K. Signaling pathways involved in colorectal cancer progression. Cell Biosci. 2019;9:97. doi: 10.1186/s13578-019-0361-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmad R., Singh J.K., Wunnava A., Al-Obeed O., Abdulla M., Srivastava S.K. Emerging trends in colorectal cancer: Dysregulated signaling pathways. Int. J. Mol. Med. 2021;47:14–25. doi: 10.3892/ijmm.2021.4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ji Y., Lv J., Sun D., Huang Y. Therapeutic strategies targeting Wnt/β-catenin signaling for colorectal cancer. Int. J. Mol. Med. 2022;49:1–17. doi: 10.3892/IJMM.2021.5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nusse R., Clevers H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell. 2017;169:985–999. doi: 10.1016/j.cell.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 6.Vogelstein B., Papadopoulos N., Velculescu V.E., Zhou S., Diaz L.A., Jr., Kinzler K.W. Cancer Genome Landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cancer Genome Atlas Network. Bainbridge M.N., Chang K., Dinh H.H., Drummond J.A., Fowler G., Kovar C.L., Lewis L.R., Morgan M.B., Newsham I.F., et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Molinari F., Frattini M. Functions and regulation of the PTEN gene in colorectal cancer. Front. Oncol. 2013;3:326–328. doi: 10.3389/fonc.2013.00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mariani F., Sena P., Roncucci L. Inflammatory pathways in the early steps of colorectal cancer development. World J. Gastroenterol. 2014;20:9716–9731. doi: 10.3748/wjg.v20.i29.9716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kennel K.B., Greten F.R. Immune cell - produced ROS and their impact on tumor growth and metastasis. Redox Biol. 2021;42 doi: 10.1016/j.redox.2021.101891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laugesen A., Højfeldt J.W., Helin K. Role of the Polycomb Repressive Complex 2 (PRC2) in Transcriptional Regulation and Cancer. Cold Spring Harb. Perspect. Med. 2016;6:a026575. doi: 10.1101/cshperspect.a026575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu X., Liu X. PRC2, Chromatin Regulation, and Human Disease: Insights From Molecular Structure and Function. Front. Oncol. 2022;12:894585–894618. doi: 10.3389/fonc.2022.894585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan J.Z., Yan Y., Wang X.X., Jiang Y., Xu H.E. EZH2: Biology, disease, and structure-based drug discovery. Acta Pharmacol. Sin. 2014;35:161–174. doi: 10.1038/aps.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schuettengruber B., Bourbon H.M., Di Croce L., Cavalli G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell. 2017;171:34–57. doi: 10.1016/j.cell.2017.08.002. [DOI] [PubMed] [Google Scholar]
- 15.Piunti A., Shilatifard A. The roles of Polycomb repressive complexes in mammalian development and cancer. Nat. Rev. Mol. Cell Biol. 2021;22:326–345. doi: 10.1038/s41580-021-00341-1. [DOI] [PubMed] [Google Scholar]
- 16.Bracken A.P., Kleine-Kohlbrecher D., Dietrich N., Pasini D., Gargiulo G., Beekman C., Theilgaard-Mönch K., Minucci S., Porse B.T., Marine J.C., et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007;21:525–530. doi: 10.1101/gad.415507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koppens M., Van Lohuizen M. Context-dependent actions of Polycomb repressors in cancer. Oncogene. 2016;35:1341–1352. doi: 10.1038/onc.2015.195. [DOI] [PubMed] [Google Scholar]
- 18.Vilorio-Marqués L., Martín V., Diez-Tascón C., González-Sevilla M.F., Fernández-Villa T., Honrado E., Davila-Batista V., Molina A.J. The role of EZH2 in overall survival of colorectal cancer: A meta-Analysis. Sci. Rep. 2017;7:1–8. doi: 10.1038/s41598-017-13670-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohuchi M., Sakamoto Y., Tokunaga R., Kiyozumi Y., Nakamura K., Izumi D., Kosumi K., Harada K., Kurashige J., Iwatsuki M., et al. Increased EZH2 expression during the adenoma-carcinoma sequence in colorectal cancer. Oncol. Lett. 2018;16:5275–5281. doi: 10.3892/ol.2018.9240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He A., Shen X., Ma Q., Cao J., von Gise A., Zhou P., Wang G., Marquez V.E., Orkin S.H., Pu W.T. PRC2 directly methylates GATA4 and represses its transcriptional activity. Genes Dev. 2012;26:37–42. doi: 10.1101/gad.173930.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu K., Wu Z.J., Groner A.C., He H.H., Cai C., Lis R.T., Wu X., Stack E.C., Loda M., Liu T., et al. EZH2 Oncogenic Activity in Castration-Resistant Prostate Cancer Cells Is Polycomb-Independent. Science. 2012;338:1465–1469. doi: 10.1126/science.1227604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee J.M., Lee J.S., Kim H., Kim K., Park H., Kim J.Y., Lee S.H., Kim I.S., Kim J., Lee M., et al. EZH2 Generates a Methyl Degron that Is Recognized by the DCAF1/DDB1/CUL4 E3 Ubiquitin Ligase Complex. Mol. Cell. 2012;48:572–586. doi: 10.1016/j.molcel.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 23.Kim E., Kim M., Woo D.H., Shin Y., Shin J., Chang N., Oh Y.T., Kim H., Rheey J., Nakano I., et al. Phosphorylation of EZH2 Activates STAT3 Signaling via STAT3 Methylation and Promotes Tumorigenicity of Glioblastoma Stem-like Cells. Cancer Cell. 2013;23:839–852. doi: 10.1016/j.ccr.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoffmeyer K., Junghans D., Kanzler B., Kemler R. Trimethylation and Acetylation of β-Catenin at Lysine 49 Represent Key Elements in ESC Pluripotency. Cell Rep. 2017;18:2815–2824. doi: 10.1016/j.celrep.2017.02.076. [DOI] [PubMed] [Google Scholar]
- 25.Zovoilis A., Cifuentes-Rojas C., Chu H.P., Hernandez A.J., Lee J.T. Destabilization of B2 RNA by EZH2 Activates the Stress Response. Cell. 2016;167:1788–1802.e13. doi: 10.1016/j.cell.2016.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X., Goodrich K.J., Gooding A.R., Naeem H., Archer S., Paucek R.D., Youmans D.T., Cech T.R., Davidovich C. Targeting of Polycomb Repressive Complex 2 to RNA by Short Repeats of Consecutive Guanines. Mol. Cell. 2017;65:1056–1067.e5. doi: 10.1016/j.molcel.2017.02.003. [DOI] [PubMed] [Google Scholar]
- 27.Macdonald B.T., Tamai K., He X. Developmental Cell Review Wnt/b-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y.K., Zhu Y.L., Qiu F.M., Zhang T., Chen Z.G., Zheng S., Huang J. Activation of Akt and MAPK pathways enhances the tumorigenicity of CD133+ primary colon cancer cells. Carcinogenesis. 2010;31:1376–1380. doi: 10.1093/carcin/bgq120. [DOI] [PubMed] [Google Scholar]
- 29.Sriramkumar S., Sood R., Huntington T.D., Ghobashi A.H., Vuong T.T., Metcalfe T.X., Wang W., Nephew K.P., O’Hagan H.M. Platinum-induced mitochondrial OXPHOS contributes to cancer stem cell enrichment in ovarian cancer. J. Transl. Med. 2022;20:246–314. doi: 10.1186/s12967-022-03447-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu P., Wang Y., Huang G., Ye B., Liu B., Wu J., Du Y., He L., Fan Z. Lnc-β-Catm elicits EZH2-dependent β-catenin stabilization and sustains liver CSC self-renewal. Nat. Struct. Mol. Biol. 2016;23:631–639. doi: 10.1038/nsmb.3235. [DOI] [PubMed] [Google Scholar]
- 32.Jung H.Y., Jun S., Lee M., Kim H.C., Wang X., Ji H., McCrea P.D., Park J.I. PAF and EZH2 induce wnt/β-catenin signaling hyperactivation. Mol. Cell. 2013;52:193–205. doi: 10.1016/j.molcel.2013.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brattain M.G., Levine A.E., Chakrabarty S., Yeoman L.C., Willson J.K.V., Long B. Heterogeneity of human colon carcinoma. Cancer Metast. Rev. 1984;3:177–191. doi: 10.1007/BF00048384. [DOI] [PubMed] [Google Scholar]
- 34.Sparks A.B., Morin P.J., Vogelstein B., Kinzler K.W. Mutational analysis of the APC/β-catenin/Tcf pathway in colorectal cancer. Cancer Res. 1998;58:1130–1134. [PubMed] [Google Scholar]
- 35.Da Costa L.T., He T.C., Yu J., Sparks A.B., Morin P.J., Polyak K., Laken S., Vogelstein B., Kinzler K.W. CDX2 is mutated in a colorectal cancer with normal APC/β-catenin signaling. Oncogene. 1999;18:5010–5014. doi: 10.1038/sj.onc.1202872. [DOI] [PubMed] [Google Scholar]
- 36.Wang J., Yu X., Gong W., Liu X., Park K.S., Ma A., Tsai Y.H., Shen Y., Onikubo T., Pi W.C., et al. EZH2 noncanonically binds cMyc and p300 through a cryptic transactivation domain to mediate gene activation and promote oncogenesis. Nat. Cell Biol. 2022;24:384–399. doi: 10.1038/s41556-022-00850-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang J., Park K.-S., Yu X., Gong W., Earp H.S., Wang G.G., Jin J., Cai L. A cryptic transactivation domain of EZH2 binds AR and AR’s splice variant, promoting oncogene activation and tumorous transformation. Nucleic Acids Res. 2022;50:10929–10946. doi: 10.1093/nar/gkac861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen X., Wang C., Jiang Y., Wang Q., Tao Y., Zhang H., Zhao Y., Hu Y., Li C., Ye D., et al. Bcl-3 promotes Wnt signaling by maintaining the acetylation of β-catenin at lysine 49 in colorectal cancer. Signal Transduct. Target. Ther. 2020;5:52. doi: 10.1038/s41392-020-0138-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cha T.L., Zhou B.P., Xia W., Wu Y., Yang C.C., Chen C.T., Ping B., Otte A.P., Hung M.C. Molecular biology: Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science. 2005;310:306–310. doi: 10.1126/science.1118947. [DOI] [PubMed] [Google Scholar]
- 40.Le H.Q., Hill M.A., Kollak I., Keck M., Schroeder V., Wirth J., Skronska-Wasek W., Schruf E., Strobel B., Stahl H., et al. An EZH2-dependent transcriptional complex promotes aberrant epithelial remodelling after injury. EMBO Rep. 2021;22 doi: 10.15252/embr.202152785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zong X., Wang W., Ozes A., Fang F., Sandusky G.E., Nephew K.P. EZH2-mediated downregulation of the tumor suppressor DAB2IP maintains ovarian cancer stem cells. Cancer Res. 2020;80:4371–4385. doi: 10.1158/0008-5472.CAN-20-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu Y., Deng P., Yu B., Szymanski J.M., Aghaloo T., Hong C., Wang C.Y. Inhibition of EZH2 Promotes Human Embryonic Stem Cell Differentiation into Mesoderm by Reducing H3K27me3. Stem Cell Rep. 2017;9:752–761. doi: 10.1016/j.stemcr.2017.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Comet I., Riising E.M., Leblanc B., Helin K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat. Rev. Cancer. 2016;16:803–810. doi: 10.1038/nrc.2016.83. [DOI] [PubMed] [Google Scholar]
- 44.Cardenas H., Zhao J., Vieth E., Nephew K.P., Matei D. EZH2 inhibition promotes epithelial-to-mesenchymal transition in ovarian cancer cells. Oncotarget. 2016;7:84453–84467. doi: 10.18632/oncotarget.11497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Akizu N., Martìnez-Balbàs M.A. EZH2 orchestrates apicobasal polarity and neuroepithelial cell renewal. Neurogenesis. 2016;3:1–7. doi: 10.1080/23262133.2016.1250034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gallardo A., Molina A., Asenjo H.G., Lopez-Onieva L., Martorell-Marugán J., Espinosa-Martinez M., Griñan-Lison C., Alvarez-Perez J.C., Cara F.E., Navarro-Marchal S.A., et al. EZH2 endorses cell plasticity to non-small cell lung cancer cells facilitating mesenchymal to epithelial transition and tumour colonization. Oncogene. 2022;41:3611–3624. doi: 10.1038/s41388-022-02375-x. [DOI] [PubMed] [Google Scholar]
- 47.Kim K.H., Roberts C.W.M. Targeting EZH2 in cancer. Nat. Med. 2016;22:128–134. doi: 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miller S.A., Policastro R.A., Sriramkumar S., Lai T., Huntington T.D., Ladaika C.A., Kim D., Hao C., Zentner G.E., O’Hagan H.M. LSD1 and aberrant DNA methylation mediate persistence of enteroendocrine progenitors that support BRAF-mutant colorectal cancer. Cancer Res. 2021;81:3791–3805. doi: 10.1158/0008-5472.CAN-20-3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu G., Wang L.G., Han Y., He Q.Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. Omi. A J. Integr. Biol. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blighe, K., Rana, S., and Lewis, M. EnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labeling. R package version 1.18.0
- 51.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Quinlan A.R., Hall I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–W165. doi: 10.1093/NAR/GKW257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hahne F., Ivanek R. Visualizing Genomic Data Using Gviz and Bioconductor. Methods Mol. Biol. 2016;1418:335–351. doi: 10.1007/978-1-4939-3578-9_16. [DOI] [PubMed] [Google Scholar]
- 56.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550–621. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A.H., Tanaseichuk O., Benner C., Chanda S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019;10:1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goldman M.J., Craft B., Hastie M., Repečka K., McDade F., Kamath A., Banerjee A., Luo Y., Rogers D., Brooks A.N., et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020;38:675–678. doi: 10.1038/s41587-020-0546-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bracken A.P., Pasini D., Capra M., Prosperini E., Colli E., Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22:5323–5335. doi: 10.1093/emboj/cdg542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kolligs F.T., Hu G., Dang C.V., Fearon E.R. Neoplastic Transformation of RK3E by Mutant β-Catenin Requires Deregulation of Tcf/Lef Transcription but Not Activation of c- myc Expression. Mol. Cell Biol. 1999;19:5696–5706. doi: 10.1128/mcb.19.8.5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Winer I.S., Bommer G.T., Gonik N., Fearon E.R. Lysine residues Lys-19 and Lys-49 of β-catenin regulate its levels and function in T cell factor transcriptional activation and neoplastic transformation. J. Biol. Chem. 2006;281:26181–26187. doi: 10.1074/jbc.M604217200. [DOI] [PubMed] [Google Scholar]
- 62.Zafra M.P., Schatoff E.M., Katti A., Foronda M., Breinig M., Schweitzer A.Y., Simon A., Han T., Goswami S., Montgomery E., et al. Optimized base editors enable efficient editing in cells, organoids and mice. Nat. Biotechnol. 2018;36:888–893. doi: 10.1038/nbt.4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Sequencing data have been deposited at GEO and are publicly available as of publication. The accession number is listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.