

Abstract
The bone morphogenetic protein (BMP) pathway promotes differentiation and induces apoptosis in normal colorectal epithelial cells. However, its role in colorectal cancer (CRC) is controversial, where it can act as context‐dependent tumor promoter or tumor suppressor. Here we have found that CRC cells reside in a BMP‐rich environment based on curation of two publicly available RNA‐sequencing databases. Suppression of BMP using a specific BMP inhibitor, LDN193189, suppresses the growth of select CRC organoids. Colorectal cancer organoids treated with LDN193189 showed a decrease in epidermal growth factor receptor, which was mediated by protein degradation induced by leucine‐rich repeats and immunoglobulin‐like domains protein 1 (LRIG1) expression. Among 18 molecularly characterized CRC organoids, suppression of growth by BMP inhibition correlated with induction of LRIG1 gene expression. Notably, knockdown of LRIG1 in organoids diminished the growth‐suppressive effect of LDN193189. Furthermore, in CRC organoids, which are susceptible to growth suppression by LDN193189, simultaneous treatment with LDN193189 and trametinib, an FDA‐approved MEK inhibitor, resulted in cooperative growth inhibition both in vitro and in vivo. Taken together, the simultaneous inhibition of BMP and MEK could be a novel treatment option in CRC cases, and evaluating in vitro growth suppression and LRIG1 induction by BMP inhibition using patient‐derived organoids could offer functional biomarkers for predicting potential responders to this regimen.
Keywords: BMP, colorectal cancer, EGFR, LRIG1, organoid
In this study, we found that colorectal cancer (CRC) is a bone morphogenetic protein (BMP)‐rich environment and that BMP inhibition suppresses the growth of select populations of CRC organoids through LRIG1‐mediated epidermal growth factor reception downregulation. The effect of BMP inhibition varied among CRC organoids, which could be masked by high levels of environmental growth factors. Accordingly, simultaneous inhibition of BMP and MEK resulted in a combined effect both in vitro and in vivo, especially in CRCs that are dependent on MEK activation.
Abbreviations
- BMP
bone morphogenetic protein
- CRC
colorectal cancer
- CTOS
cancer tissue‐originated spheroid
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- GF
growth factor
- HER
human epidermal growth factor receptor
- HRG
heregulin
- KD
knockdown
- LDN
LDN193189
- LGR5
leucine‐rich repeat containing G protein‐coupled receptor 5
- LRIG1
leucine‐rich repeats and Ig‐like domains protein 1
- TCGA
The Cancer Genome Atlas
- TGF‐β
transforming growth factor‐beta
1. INTRODUCTION
Bone morphogenetic protein ligands are members of the TGF‐β superfamily, which trigger intracellular signaling via specific receptors upstream of common SMAD4 proteins. Transforming growth factor‐β has been extensively studied for its role in cancer pathogenesis, including CRC, especially in the epithelial–mesenchymal transition. 1 , 2 However, the role of BMP ligands in cancer has been studied less extensively. In the normal colorectal epithelium, BMP2, 4, and 7 are secreted from stromal cells, such as myofibroblasts, which promote the differentiation of epithelial cells and suppress the stemness of epithelial stem cells at the crypt base. 3 , 4 In CRC, BMPs are reported both as tumor‐suppressing and tumor‐promoting molecules, depending on the study. Beck et al. reported that BMP signaling is intact and suppresses the growth of human colon cancer cell lines. 5 Lombardo et al. revealed that BMP4 induces differentiation of human CRC stem cells. 6 In contrast, Lorente‐Trigos et al. reported that BMP signaling promotes the growth of xenograft tumors derived from primary human CRC. 7 More recently, Yokoyama et al. reported that autocrine BMP4 signaling is a therapeutic target in CRC cell lines. 8 In addition to the context dependencies of the BMP effect that have been discussed in various cancers, 9 , 10 these controversies might represent intertumor heterogeneity among the CRC population. One possible explanation for the latter is that the mutation status of SMAD4 and TP53 is suggested to determine cell fate by BMP. 11 However, further studies on the heterogeneous effects of BMPs are warranted.
Epidermal growth factor receptor is a receptor tyrosine kinase important for the survival and proliferation of intestinal and colorectal epithelial cells. Therefore, the regulation of EGFR signaling is critical for the pathology of CRC. Indeed, molecular‐targeted drugs for EGFR, such as cetuximab and panitumumab, are clinically used for treating advanced CRC with WT KRAS. 12 We have reported that KRAS mutant CRC can be characterized into two groups: a partially responsive group, in which cetuximab had a substantial growth inhibitory effect, and a resistant group, in which no effect was observed. 13 The CRC cells in the partially responsive group showed a combined effect of cetuximab and trametinib, a MEK inhibitor, both in vitro and in vivo. These observations support targeting EGFR, even in KRAS mutant CRC. However, selecting a sensitive case is essential because cancer is a highly heterogeneous disease.
To address the heterogeneity of cancer, more researchers have been utilizing the 3D organoid culture technique in recent years. Patient‐derived organoids retain the physiological features of parental cancer cells and reflect the heterogeneity of disease subgroups. 14 We have reported and utilized a method for organoid preparation, the CTOS method, which prepares cancer organoids with high purity, yield, and viability by retaining cell–cell interactions throughout the procedure. 14 , 15 In this study, the effect of BMP inhibition was evaluated using a panel of patient‐derived CRC organoids established using the CTOS method. A group of CRC organoids sensitive to BMP inhibition was further investigated for the mechanism of growth suppression, and the possibility of a combination therapy was explored.
2. MATERIALS AND METHODS
2.1. Patient samples and animal studies
This study was approved by the Institutional Ethics Committees at Osaka International Cancer Institute (1,712,225,296, 1,803,125,402) and Kyoto University (R1575, R2444) and was carried out in accordance with the Declaration of Helsinki. The surgical specimens were obtained from Osaka International Cancer Institute and Kyoto University after obtaining written informed consent.
The animal studies were approved by the Institutional Animal Care and Use Committee of Kyoto University and Osaka International Cancer Institute and were carried out according to institutional guidelines.
2.2. Generation and treatment of xenograft tumors
To expand the organoids, they were injected into NOD/SCID mice (CLEA Japan) to generate s.c. xenograft tumors. For the in vivo treatment studies, a mixture of 1000 organoids in Matrigel (Corning Inc.) was transplanted into the flank of BALB/cAJcl‐nu/nu mice (CLEA Japan). LDN193189 (Sigma‐Aldrich) solution was prepared in water and given daily to mice at 3 mg/kg i.p. Trametinib solution was prepared in 0.5% methyl cellulose with 0.2% Tween‐80 and given using oral gavage at 0.3 μg/kg every other day. The s.c. xenograft volume was calculated using the formula: volume = (width)2 × (length)/2, and treatment was started when the volume exceeded 300 mm3. Mice with an excessive tumor volume (>2000 mm3) were killed for ethical reasons.
2.3. Organoid preparation, culture, and cryopreservation
Organoid preparation of CRC patient tissue and xenograft tumors was carried out using the CTOS method as previously described. 13 , 15 Briefly, the tumor specimens were mechanically and enzymatically digested, and the small fragments trapped by 100 μm or 40 μm cell strainers (BD Falcon). They were then collected, cultured, and passaged in suspension in StemPro hESC (Invitrogen) as 3D organoids. Organoids were cryopreserved using CS10 (BioLife Solutions), following the manufacturer's instructions.
2.4. Vector construction and gene transfer
Tet‐pLKO‐puro was a gift from Dmitri Wiederschain (plasmid #21915; Addgene). Oligos for the LRIG1 shRNA coding sequence (shLRIG1‐Fw, CCGGTCCACACGGACCGCCTATAAACTCGAGTTTATAGGCGGTCCGTGTGGATTTTTG; shLRIG1‐Rev, AATTCAAAAATCCACACGGACCGCCTATAAACTCGAGTTTATAGGCGGTCCGTGTGGA) were annealed and cloned into the tet‐pLKO‐puro vector using the AgeI and EcoRI cloning sites. The shRNA constructs were introduced into organoids using lentiviral transduction with the spin infection technique. 16 Organoids were selected with 2.0 μg/mL puromycin (#ant‐pr‐1; InvivoGen), and the transcription of shRNAs was induced with 2.0 μg/mL doxycycline hyclate (#D9891; Sigma‐Aldrich).
2.5. Organoid growth assay
Organoid growth was evaluated in GF‐free medium (Advanced DMEM/F12 [Gibco, Thermo Fisher Scientific] supplemented with 1× GlutaMax [Gibco]) or StemPro medium (StemPro hESC; Invitrogen) containing 2.5% Matrigel GFR (BD Biosciences). Organoids were used for the growth assay 2 days after passage. Approximately 10 organoids (φ70–100 μm) per well in a 96‐well plate were used for each condition. For the assays, LDN, trametinib (Selleck Chemicals), recombinant human EGF (PeproTech), recombinant human HRG (PeproTech), and bafilomycin A1 were added at the indicated doses and time points. To quantify the viability of organoids, an ATP assay was carried out using CellTiter‐Glo (Promega). Chemiluminescence values were obtained using a GloMax Discover Microplate Reader (Promega). Organoids were cultured for 5 days, and the relative ATP levels were adjusted with the initial numbers of organoids. GraphPad Prism 9 software (GraphPad Software) was used to draw sigmoidal dose–response curves.
Additional information for materials and methods are described in Data S1.
3. RESULTS
3.1. Colorectal cancer cells reside in BMP ligand‐rich environment
First, we profiled BMP pathway gene expression in CRC tissues using two publicly available databases. Using RNA‐sequencing data from cancerous and normal tissues registered in TCGA 17 and normal tissues registered in Genotype‐Tissue Expression (GTEx), 18 we found that in normal colorectal tissues, among the major BMPs (2, 4, and 7) of the intestine, BMP4 and BMP7 expressions were increased in CRC tissues. In contrast, BMP2 expression was lower than that in normal tissues (Figure 1A). In addition, single‐cell RNA sequencing data were used to analyze the status of the BMP pathway by cell lineage, and analysis of pooled single‐cell gene expression data from nine CRC patients 19 revealed that BMP4 was mainly expressed in fibroblasts and epithelial tumor cells (Figures 1B,C and S1‐S9.). In contrast, BMP7 was produced by various types of cells, including malignant cells and hematopoietic cells, such as T cells and B cells (Figure 1B,C). The expression of ID1 and 2, major targets of the BMP/SMAD pathway, was upregulated in epithelial cell clusters, suggesting that the BMP pathway is active in cancer cells (Figure 1D). These results suggest that the BMP pathway is involved in the development of the microenvironment in CRC, based on gene expression analysis of clinical tissue‐derived samples.
FIGURE 1.
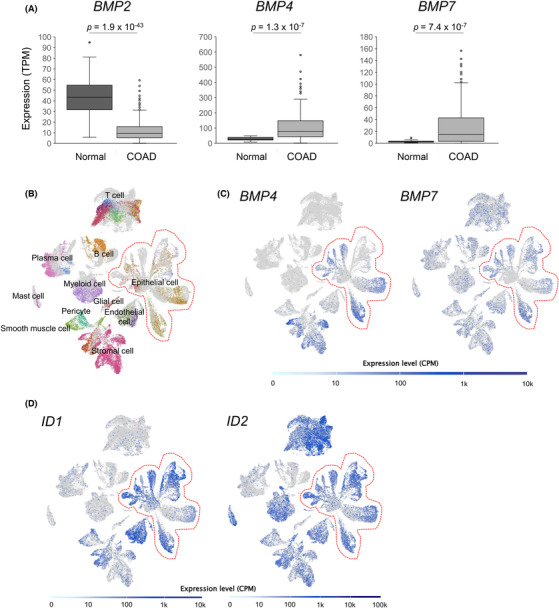
Bone morphogenetic protein (BMP) pathway is active in clinical colorectal cancer (CRC) tumors. (A) Box and whisker plots of BMP2, BMP4, and BMP7 expression (transcripts per million [TPM]) in normal colon (normal n = 41) and colorectal adenocarcinoma (COAD n = 308) tissue specimen. p values calculated by Student's t‐test are indicated. (B–D) Uniform Manifold Approximation and Projection (UMAP) for the single‐cell RNA‐sequencing data of nine CRC patients' specimens. Cell types corresponding to each cluster are designated on the UMAP (B). More detailed cluster labeling is presented in Figures S1‐S9.. (C) BMP4 and BMP7, as well as (D) ID1 and ID2 expression levels, are plotted on the UMAP. Areas enclosed by red dotted lines indicate clusters of epithelial cells. CPM, counts per million.
3.2. Inhibition of BMP pathway suppresses CRC organoid growth with decreased EGFR
To investigate the role of the BMP pathway in CRC cells, the CRC organoid panel (Table S1) was tested for growth inhibition in a defined medium without growth factors using LDN, a BMP receptor inhibitor. Colorectal cancer organoids were confirmed to produce BMP4 protein, which can accumulate during the culture period (Figure S2A), supporting the autocrine production of BMP ligands by CRC cells indicated from omics data (Figure 1C). Significant suppression of ID1 by LDN and recombinant BMP‐inhibitory protein Noggin support that the BMP pathway is active in these CRC organoids and LDN is effective (Figure S2B). We found that the effects of LDN varied among organoids from different cases (Figure 2A,B). This phenomenon was confirmed with Noggin; the growth of C45 and CB3 LDN‐sensitive organoids was significantly inhibited by Noggin, whereas that of C75 and C166 LDN‐resistant organoids was not (Figure 2C). Next, the time course of the activation status of the signaling pathway was evaluated in these LDN‐sensitive and LDN‐resistant organoids. As expected, LDN suppressed phosphorylation of SMAD1/5, which are key signaling molecules of the BMP/SMAD pathways downstream of BMP receptors (Figure 2D). The level of EGFR, a major receptor tyrosine kinase in the pathogenesis of CRC, 20 decreased in these CRC organoids treated with LDN, especially after long‐term treatment (48 h, Figure 2D). Phospho‐ERK1/2 was suppressed by LDN in these organoids (Figure 2D). Another LDN‐sensitive organoid C307 also showed the suppression of EGFR and pERK by LDN, but three other LDN‐resistant organoids, C97, C120, and C324, showed no change by LDN (Figure S3). Note that LDN‐resistant C166 showed downregulation of EGFR and pERK more clearly than CB3, an LDN‐sensitive organoid. These results indicate that suppression of EGFR/MEK/ERK by LDN was predicted to be the mechanism of growth inhibition, which is a required but not sufficient factor for determining the LDN sensitivity. Also, it is worth mentioning that LDN did not directly suppress the EGF‐mediated EGFR activation and downstream ERK phosphorylation (Figure S4A). Therefore, we investigated the effect of MEK/ERK activation on LDN treatment. LDN193189 treatment in the presence of EGF (Figure 2E) attenuated the growth suppression effect of LDN. Addition of exogeneous EGF effectively stimulated ERK phosphorylation in LDN‐treated organoids even with the decreased total EGFR (Figure S4B). Similarly, HRG, a ligand of HER3, also attenuated the effect of LDN (Figure 2F). Furthermore, the effect of LDN on the growth of CRC organoids was examined using a medium containing a cocktail of GFs (insulin‐like GF, hepatocyte GF, and HRG). 15 , 21 Compared to the medium without GFs (Figure 2A), the growth inhibition by LDN was generally much lower (Figure 2G). These results suggest that LDN inhibits CRC organoid proliferation by suppression of MEK/ERK through downregulation of EGFR as an upstream event.
FIGURE 2.
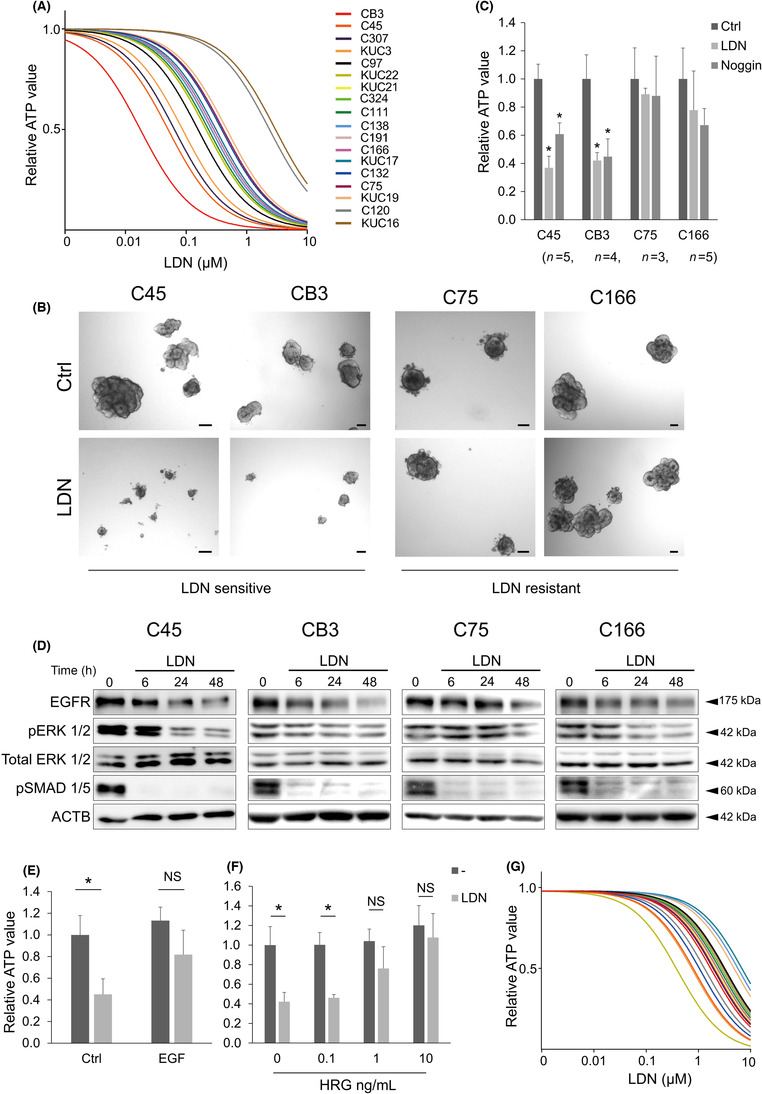
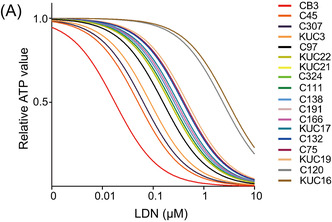
Inhibition of the bone morphogenetic protein (BMP) pathway suppresses colorectal cancer (CRC) organoid growth. (A) Dose–response curves of LDN193189 (LDN) in 18 CRC organoid lines cultured in growth factor‐free medium. (B) Bright‐field images of CRC organoids cultured in the presence (lower panels) or absence (Ctrl, upper panels) of 0.1 μM LDN for 5 days. Scale bar, 100 μm. (C) Cell viability assay results for CRC organoids. Organoids were treated with 0.1 μM LDN or 100 ng/mL Noggin. Number of replicates is indicated below the organoid names. Data are presented as mean + SD. Statistical comparisons were made for each control condition. (D) Immunoblotting analysis of CRC organoids cultured in the presence (LDN) or absence (−) of 0.1 μM LDN for the indicated time (h). Proteins were detected with indicated Ab. (E–G) Cell viability assay results for C45 organoids. Organoids were treated with 0.1 μM LDN with or without 50 ng/mL epidermal growth factor (EGF) (n = 5 for each condition, E) or heregulin (HRG) at the indicated concentration (n = 3 for each condition, F). Data are presented as mean + SD. (G) Dose–response curves of LDN in 16 CRC organoid lines cultured in StemPro hESC medium. *p < 0.01, t‐test with Bonferroni correction. ACTB, β‐actin; EGFR, EGF receptor; NS, not significant.
3.3. Inhibition of BMP induces LRIG1‐mediated degradation of EGFR
Next, we investigated the mechanisms underlying the downregulation of the EGFR/MEK pathway by BMP inhibition. Despite the decreased EGFR protein levels, EGFR mRNA expression was only slightly downregulated in C45 and C75 organoids treated with LDN or Noggin and was not altered in C166 organoids (Figure 3A). In addition, Noggin treatment upregulated the expression of EGFR in CB3 organoids (Figure 3A). These results indicate that the suppression of organoid growth by BMP inhibition was not due to the transcriptional regulation of EGFR. We evaluated the induction of several other negative regulators of the EGFR/MEK pathway by BMP inhibition. Epidermal growth factor receptor is regulated at the post‐translational level, and ligand‐stimulated EGFR proteins are endocytosed either to be degraded or recycled. 22 , 23 Some negative regulators of EGFR, including LRIG1 24 and ERRFI1 (MIG‐6), 25 are known to trigger this degradation machinery. LRIG1 expression increased over time (Figure S5) and was highly upregulated after 48 h of LDN treatment in C45 and CB3 organoids (Figure 3B), and slightly upregulated in C75 organoids (1.47‐fold). However, ERRFI1 was not upregulated in any of the organoids evaluated (Figure S6). Lysosomal degradation, led by ubiquitination, has been reported as a mechanism by which LRIG1 degrades EGFR. 26 , 27 Indeed, EGFR was highly ubiquitinated in the C45 organoid following LDN treatment (Figure 3C). Moreover, bafilomycin A1, a lysosomal inhibitor, inhibited the downregulation of EGFR by LDN (Figure 3D) and growth suppression by LDN (Figure 3E). Noggin also induced LRIG1 expression and EGFR downregulation, confirming this effect is through suppression of the BMP pathway (Figure 3F). LRIG1, a pan‐ErbB negative regulator, did not induce a robust decrease of HER2 or HER3 proteins (Figure 3F). These results indicate that EGFR degradation was involved in the LDN‐induced growth suppression of CRC organoids and further suggest that LRIG1 induction is responsible for the LDN effect.
FIGURE 3.

Bone morphogenetic protein (BMP) inhibition induces LRIG1‐mediated degradation of epidermal growth factor receptor (EGFR). (A,B) Gene expression of EGFR (A) and LRIG1 (B) in colorectal cancer organoids treated with LDN193189 (LDN) or Noggin. Expression level was normalized to control (Ctrl) in each organoid. n = 3 for each condition. Data are presented as mean + SD. Statistical comparisons were made for each control condition. (C) Co‐immunoprecipitation (IP) analysis of lysates from C45 organoids treated with 0.1 μM LDN compared to untreated control (Ctrl). Samples were immunoblotted (IB) with EGFR, ubiquitin, and β‐actin (ACTB) Abs. Input lysates were used as loading controls. (D) Immunoblotting analysis of C45 organoid cultured for 48 h with LDN at the indicated concentration (nM) in the presence (+) or absence (−) of 5 nM bafilomycin A1. Proteins were detected with indicated Abs. (E) Cell viability assay results for C45 organoids. Organoids were treated with 0.1 μM LDN with or without 5 nM bafilomycin A1 (BAF). Data are presented as mean + SD. N = 3. (F) Immunoblotting analysis of C45 organoid cultured with 0.1 μM LDN or 250 ng/mL Noggin for 48 h. Proteins were detected with indicated Abs. *p < 0.05, **p < 0.001, t‐test with Bonferroni correction. HER, human epidermal growth factor receptor; NS, not significant.
3.4. Induction of LRIG1 by BMP inhibition suppresses growth of CRC organoids
In the 18 CRC organoid lines, LRIG1 induction by LDN was inversely correlated with the relative growth of organoids with versus without LDN in the medium (Figure 4A), suggesting that LRIG1 is involved in LDN‐induced growth suppression. However, induction of the ERK negative feedback factor DUSP5, which is reportedly involved in the growth suppression of CRC cell lines treated with LDN, 8 was not correlated with LDN‐induced growth suppression in these 18 CRC organoids (Figure S7). Next, induction of the Wnt target genes was evaluated because the Wnt pathway is crucial in CRC pathogenesis. In addition, LRIG1 is a marker of intestinal and colorectal epithelial stem cells, which have high Wnt activity, 24 , 28 and can be regulated by Wnt through MYC. 29 Induction of LGR5, a Wnt target stem cell marker gene, was correlated with organoid growth under LDN treatment, and the induction of LRIG1 and LGR5 was highly correlated (Figure S8). However, the induction of two major Wnt target genes, AXIN2 and MYC, was not correlated with the growth of CRC organoids treated with LDN (Figure S7). These results suggest that LRIG1 induction is independent of Wnt activation and is regulated by the same mechanism as LGR5.
FIGURE 4.

Induction of LRIG1 by bone morphogenetic protein (BMP) inhibition is involved in the growth suppression of colorectal cancer (CRC) organoids. (A) Scatter plot showing the correlation of growth and LRIG1 induction in the presence of 0.1 μM LDN193189 (LDN) relative to LDN‐free condition. Each dot represents an individual CRC organoid line (n = 18). r, Spearman's rank correlation coefficient; p = 0.000195, Spearman's rank correlation test. (B) Immunoblotting analysis of C45 organoid with inducible shLRIG1 cultured for 48 h with LDN and at the indicated concentration (nM) in the presence (+) or absence (−) of doxycycline (DOX). Proteins were detected with indicated Abs. (C) Cell viability assay results for C45 and CB3 organoids with inducible shLRIG1. Organoids were treated with 0.1 μM LDN in the presence (+) or absence (−) of DOX for 5 days. Data are presented as mean + SD. n = 5. *p < 0.05, t‐test with Bonferroni correction. (D) Representative bright‐field images of C45 organoids with inducible shLRIG1. Organoids were treated with 0.1 μM LDN in the presence (+) or absence (−) of DOX for 5 days. Scale bar, 100 μm. ACTB, β‐actin; Ctrl, control; EGFR, epidermal growth factor receptor; NS, not significant.
To confirm the involvement of LRIG1 in LDN‐induced growth suppression, LRIG1 was knocked down by inducible shRNA in C45 organoids, in which LRIG1 induction by LDN was the most robust (Figure 4A). LRIG1 KD abrogated the downregulation of EGFR by LDN in the C45 organoids (Figure 4B). Accordingly, the inhibition of ERK phosphorylation was slightly reversed. In addition, growth suppression by LDN was inhibited by the LRIG1 KD in CRC organoids (Figure 4C,D). Collectively, the growth inhibition of CRC organoids by the BMP inhibitor was, at least in part, mediated by the induction of the EGFR negative regulator, LRIG1.
3.5. Combined inhibition of BMP and MEK cooperatively suppresses growth of in vitro MEK‐dependent CRC organoids
As mentioned above (Figure 2E–G), the growth‐inhibitory effect of BMP inhibition was attenuated in the GF‐rich microenvironment, possibly due to the strongly stimulated signaling pathways, such as MEK/ERK. The potential of CRC cells to respond to BMP inhibition is attenuated or even masked by the presence of excess GFs. Therefore, we investigated whether BMP inhibition combined with MEK inhibition showed an additional effect in LDN‐sensitive CRC organoids. Trametinib, a specific FDA‐approved MEK inhibitor, was used as a single agent to treat the 15 CRC organoids. Trametinib treatment resulted in variable growth inhibitory effects on these organoids under GF‐free conditions (Figure 5A). The growth inhibitory effects of LDN and trametinib were positively correlated (Figure 5B). The organoids were then treated with trametinib and LDN. As expected, LDN‐sensitive organoid lines showed an additive effect of these two inhibitors (Figure 5C, upper). Western blot analysis confirmed that ERK phosphorylation was suppressed more by the combined treatment than by each single agent (Figure 5D). Of note, organoids resistant to LDN failed to show a clear combination effect (Figure 5C, lower). The combined effect was confirmed using LDN treatment with an additional MEK inhibitor, PD 0325901 (Figure 5E). Furthermore, given that C166 is an in vitro MEK‐independent organoid (Figure 5A,E), it is not surprising that C166 is resistant to growth inhibition by LDN, despite robust suppression of EGFR and pERK (Figure 2D). These results indicate the heterogeneity of the dependency on MEK/ERK signaling among CRC organoid lines and support the association of the LDN effect with in vitro MEK dependency.
FIGURE 5.
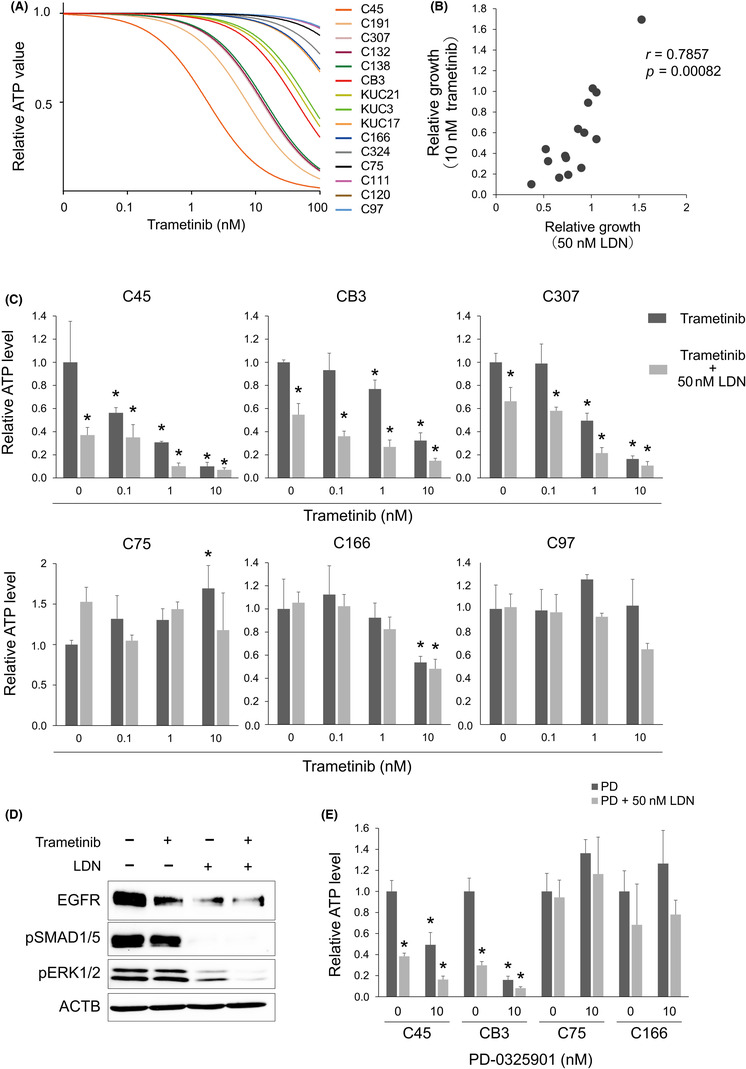
Combined inhibition of bone morphogenetic protein (BMP) and MEK cooperatively suppresses the growth of the in vitro MEK‐dependent colorectal cancer (CRC) organoids. (A) Dose–response curves of trametinib in 15 CRC organoid lines cultured in growth factor‐free medium. (B) Scatter plot showing the correlation of the organoid growth treated with 50 nM LDN193189 (LDN) and with 10 nM trametinib relative to the drug‐free condition. Each dot represents an individual CRC organoid line (n = 15). r, Spearman's rank correlation coefficient; p = 0.00082, Spearman's rank correlation test. (C) Cell viability assay results for the in vitro MEK‐dependent (upper) and ‐independent (lower) organoids. Organoids were treated with trametinib at indicated doses in the absence (dark gray) or presence (light gray) of LDN for 5 days. Data are presented as mean + SD. n = 3. *p < 0.05 compared with drug‐free control, one‐way ANOVA with post‐hoc Tukey test. (D) Immunoblotting analysis of C45 organoid cultured for 48 h with LDN and trametinib. Proteins were detected with indicated Abs. (E) Cell viability assay results for in vitro MEK‐dependent (C45 and CB3) and ‐independent (C75 and C166) organoids. Organoids were treated with 10 nM PD‐0325901 (PD) in the absence (dark gray) or presence (light gray) of LDN for 5 days. Data are presented as mean + SD. n = 3. *p < 0.05 compared with drug‐free control in each organoid line, one‐way ANOVA with post‐hoc Tukey test.
3.6. LDN193189 enhanced growth‐inhibitory effect of trametinib in xenograft tumors of LDN‐sensitive CRC organoids
To evaluate the ability of LDN to suppress the growth of CRC cells in vivo, mice with C45 xenograft tumors were treated with LDN. LDN193189 had no inhibitory effect on tumor growth (Figure 6, upper left), which corresponds to the in vitro results obtained in GF‐rich environments. The combined effects of LDN and trametinib were evaluated in vivo. Trametinib showed an enhanced growth‐inhibitory effect on C45 xenograft tumors when combined with LDN. Similarly, the growth of CB3 xenograft tumors was suppressed more efficiently by trametinib combined with LDN than by trametinib alone (Figure 6, lower left). However, LDN alone had no effect. Notably, the combined effect of LDN and trametinib was not observed in C75 and C166 organoids, reflecting the results of the in vitro organoid growth assay (Figure 6, upper and lower right). Therefore, in CRC cells that are more dependent on MEK/ERK pathway activation, the simultaneous inhibition of the BMP and MEK pathways can be a potent therapeutic target.
FIGURE 6.
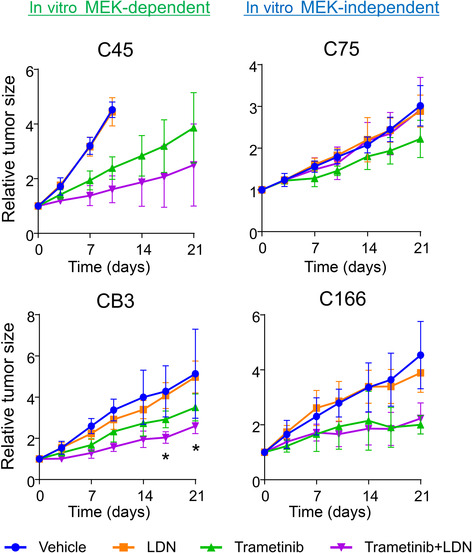
LDN193189 (LDN) enhanced the growth‐inhibitory effect of trametinib in xenograft tumors of LDN‐sensitive colorectal cancer (CRC) organoids. Growth curves of subcutaneous tumors generated by four CRC organoid lines (in vitro MEK‐dependent group: C45 and CB3; in vitro MEK‐independent group: C75 and C48). Blue, treated with vehicle; orange, LND (3 mg/kg) alone; green, trametinib (0.3 mg/kg) alone; purple, combination of LDN (3 mg/kg) and trametinib (0.3 mg/kg). Mean ± SD is shown. n = 4–6 in each treated group. *p < 0.05, trametinib monotherapy versus combination; one‐way ANOVA with post‐hoc Tukey test.
4. DISCUSSION
In this study, we found that CRC is a BMP‐rich environment and that BMP inhibition suppresses the growth of select populations of CRC organoids through LRIG1‐mediated EGFR downregulation. The effect of BMP inhibition varied among CRC organoids, which could be masked by high levels of environmental GFs. Accordingly, simultaneous inhibition of BMP and MEK resulted in a combined effect both in vitro and in vivo, especially in CRCs that are dependent on MEK activation.
Several studies have shown that the BMP/SMAD pathway regulates EGFR signaling; BMP2 suppresses EGFR signaling during chondrocyte maturation, 30 and BMP signaling downregulates EGFR in astrocytes 31 and gastric cancer cells. 32 All these previous reports have shown that the active BMP pathway suppresses EGFR, although the underlying mechanisms are unknown. In the present study, we found that, contrary to these previous findings, BMP inhibition downregulated the total EGFR level in some CRC organoids. We determined that one possible mechanism of this phenomenon is the posttranscriptional downregulation—not transcriptional regulation—of the EGFR protein by LRIG1. In the intestine and colon, LRIG1 is a marker for epithelial stem cells 28 and a subpopulation of the interstitial cells of Cajal. 33 Functionally, LRIG1 induces Cbl‐dependent ubiquitination of the ErbB receptor family, which induces lysosomal degradation. 24 Therefore, LRIG1 can serve as a tumor suppressor in many cancer types. The expression of LRIG1 is correlated with a better prognosis in several cancers, including non‐small‐cell lung cancer, 34 , 35 breast cancer, 24 , 36 and hepatocellular carcinoma. 37 Although its expression is significantly lower in CRC than in normal colorectal tissue, 24 , 38 LRIG1 expression is not a prognostic biomarker in CRC at the protein and mRNA levels. 38 , 39 This low LRIG1 expression might be due to the suppression of LRIG1 by BMP, which is known to suppress the expression of murine intestinal epithelial stemness genes, including Lrig1 and Lgr5, through SMAD‐mediated repression. 40 This is supported by our finding that BMP inhibition upregulates the expression of LRIG1 and LGR5 in CRC organoids. In addition, the degree of LRIG1 induction by BMP inhibition was correlated with sensitivity to LDN and combination therapy with LDN and trametinib in vitro and in vivo. Note that knockdown of LRIG1 with shRNA did not provide complete rescue from LDN‐induced suppression of pERK and growth inhibition. This suggests the existence of additional mechanisms, which should be addressed in future studies. Whether BMP is a tumor suppressor or promoter in CRC is controversial. 5 , 6 , 7 , 8 Such complexities in BMP signaling have been described in various cancers. 9 , 10 As CRC organoids showed varying responses to BMP inhibition in our study, one possible explanation for the controversy regarding the role of BMP in CRC is the heterogeneous nature of the disease. Bone morphogenetic protein is likely a tumor promoter in a certain population of CRC and can be a therapeutic target in such CRC cases.
Although BMP inhibition reduced the growth of CRC organoids in vitro, the addition of a MEK inhibitor was required to see in vivo growth inhibition. Both exogenous EGF and HRG could attenuate the growth‐inhibitory effects of BMP inhibition, suggesting that endogenous ERBB ligands and/or alternative growth signaling pathways might be activated upon BMP blockade. Meanwhile, targeting the EGFR/RAS/RAF/MEK pathway is a major strategy for the treatment of advanced CRC. Recently, combination therapies targeting more than one molecule in the same pathway have been used in clinical trials, such as targeting EGFR and MEK in KRAS WT patients 41 and targeting EGFR and BRAF in BRAF‐mutant patients. 42 Inhibition of BMP could be an option for indirectly suppressing EGFR in combination with MEK inhibitors. Inhibition of BMP is a possible candidate for clinical combination therapy, but it is essential to select potential responders carefully because of the heterogeneous response. The mutation status of frequently mutated genes in CRC, including APC and KRAS, did not show a clear correlation with the in vitro response to LDN (Figure S9). The LDN‐sensitive organoid lines, C45 and CB3, were both APC and KRAS mutants. In line with our previous report that these organoid lines are partially sensitive to cetuximab, 13 these organoids were dependent on EGFR signaling despite constitutively active KRAS mutations. SMAD4 mutations, which are detected in 10% of CRC specimens according to the TCGA database, 17 inhibit SMAD‐mediated signaling by TGF‐β and BMP. 43 Colorectal cancer organoids with SMAD4 mutations tended to show less sensitivity to LDN and LRIG1 induction (Figure S9), suggesting that LRIG1 induction by LDN is a SMAD4‐dependent event. Based on the result from the present study, the SMAD4 wild CRC, which increases LRIG1 expression responding to BMP inhibition, is the potential responder to BMP/MEK inhibition treatment, irrespective of the KRAS mutant status. Further accumulation of CRC cases is needed in the future study to evaluate the correlation between mutation status and sensitivity to LDN/trametinib combination treatment.
Taken together, these findings lead to the establishment of the BMP/SMAD pathway as a therapeutic target, especially in combination with a MEK inhibitor. Organoids prepared from patient tumor tissues are an optimal model to dissect intertumor heterogeneity. Notably, the organoid model allows the detection of biochemical responses to external stimuli, which cannot be assessed using single timepoint snapshot data. An “organoid assay”, such as in vitro LDN sensitivity or LRIG1 induction by LDN, could be useful in predicting prognosis and therapeutic efficacy. Nevertheless, a larger cohort study is necessary to clarify the heterogeneity of the CRC response to BMP.
AUTHOR CONTRIBUTIONS
S. Shimizu: investigation, methodology, writing – review and editing. J. Kondo: conceptualization, investigation, methodology, validation, visualization, writing – original draft preparation, funding acquisition. K. Onuma: investigation, writing – review and editing. R. Coppo: investigation, writing – review and editing. K. Ota, M. Kamada, Y. Harada, Y. Tanaka, M.A. Nakazawa, and Y. Tamada: formal analysis. Y. Okuno: formal analysis, funding acquisition. R.J. Coffey: writing – review and editing. Y. Fujiwara: writing – review and editing. M. Inoue: supervision, validation, writing – review and editing, funding acquisition.
FUNDING INFORMATION
This study was supported by the Japan Society for the Promotion of Science Grant‐in‐Aid for Scientific Research (18H02648 (K.O., M.I.) and 21 K07942 (J.K., M.I.)), a Grant‐in‐Aid from P‐CREATE, Japan Agency for Medical Research and Development 19cm0106203h0004 (J.K., K.O., M.I.), 21am0401004h0003 (J.K., K.O., J.H., M.M., M.I.), a Grant‐in‐Aid from Science and Technology Platform Program for Advanced Biological Medicine, and by a Grant‐in‐Aid from Takeda Science Foundation (M.I.), by Cabinet Office, Government of Japan, Public/Private R&D Investment Strategic Expansion Program (PRISM) (Y.O.), and by National Cancer Institute P50CA236733 and R35197570 (R.J.C.).
CONFLICT OF INTEREST STATEMENT
J.K., K.O., R.C., and M.I. are members of the Department of Clinical Bio‐resource Research and Development at Kyoto University, which is sponsored by KBBM, Inc. Y.F. has received incentive endowments from Taiho Pharmaceutical Company and Chugai Pharmaceutical Company. The other authors declare no conflicts of interest.
ETHICS STATEMENT
Approval of the research protocol by an institutional review board: The Institutional Ethics Committees at Osaka International Cancer Institute and Kyoto University approved this study.
Informed consent: Patients’ samples and information were collected under written informed consent.
Animal studies: The Institutional Animal Care and Use Committee of Kyoto University, and Osaka International Cancer Institute approved the animal studies, which were performed according to institutional guidelines.
Registry and the registration no. of the study/trial: N/A.
Supporting information
Data S1.
Figure S1‐S9.
Table S1.
ACKNOWLEDGMENTS
The authors thank M. Izutsu for secretarial assistance, and A. Manabe and C. Saito for technical assistance. We would like to thank Editage for English language editing. R.J.C. acknowledges the generous support of the Nicholas Tierney GI Cancer Memorial Fund.
Shimizu S, Kondo J, Onuma K, et al. Inhibition of the bone morphogenetic protein pathway suppresses tumor growth through downregulation of epidermal growth factor receptor in MEK/ERK‐dependent colorectal cancer. Cancer Sci. 2023;114:3636‐3648. doi: 10.1111/cas.15882
REFERENCES
- 1. Xu J, Lamouille S, Derynck R. TGF‐beta‐induced epithelial to mesenchymal transition. Cell Res. 2009;19(2):156‐172. doi: 10.1038/cr.2009.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kalluri R, Weinberg RA. The basics of epithelial‐mesenchymal transition. J Clin Invest. 2009;119(6):1420‐1428. doi: 10.1172/JCI39104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. He XC, Zhang J, Tong WG, et al. BMP signaling inhibits intestinal stem cell self‐renewal through suppression of Wnt–β‐catenin signaling. Nat Genet. 2004;36(10):1117‐1121. doi: 10.1038/ng1430 [DOI] [PubMed] [Google Scholar]
- 4. Kosinski C, Li VSW, Chan ASY, et al. Gene expression patterns of human colon tops and basal crypts and BMP antagonists as intestinal stem cell niche factors. Proc Natl Acad Sci. 2007;104(39):15418‐15423. doi: 10.1073/pnas.0707210104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beck SE, Jung BH, Fiorino A, et al. Bone morphogenetic protein signaling and growth suppression in colon cancer. Am J Physiol ‐ Gastrointest Liver Physiol. 2006;291(1):G135‐G145. doi: 10.1152/ajpgi.00482.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lombardo Y, Scopelliti A, Cammareri P, et al. Bone morphogenetic protein 4 induces differentiation of colorectal cancer stem cells and increases their response to chemotherapy in mice. Gastroenterology. 2011;140(1):297‐309.e6. doi: 10.1053/j.gastro.2010.10.005 [DOI] [PubMed] [Google Scholar]
- 7. Lorente‐Trigos A, Varnat F, Melotti A, Altaba AR i. BMP signaling promotes the growth of primary human colon carcinomas in vivo. J Mol Cell Biol. 2010;2(6):318‐332. doi: 10.1093/jmcb/mjq035 [DOI] [PubMed] [Google Scholar]
- 8. Yokoyama Y, Watanabe T, Tamura Y, Hashizume Y, Miyazono K, Ehata S. Autocrine BMP‐4 signaling is a therapeutic target in colorectal cancer. Cancer Res. 2017;77(15):4026‐4038. doi: 10.1158/0008-5472.CAN-17-0112 [DOI] [PubMed] [Google Scholar]
- 9. Jiramongkolchai P, Owens P, Hong CC. Emerging roles of the bone morphogenetic protein pathway in cancer: potential therapeutic target for kinase inhibition. Biochem Soc Trans. 2016;44(4):1117‐1134. doi: 10.1042/BST20160069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ehata S, Miyazono K. Bone morphogenetic protein signaling in cancer; some topics in the recent 10 years. Front Cell Dev Biol. 2022;10:883523. doi: 10.3389/fcell.2022.883523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Voorneveld PW, Kodach LL, Jacobs RJ, et al. The BMP pathway either enhances or inhibits the Wnt pathway depending on the SMAD4 and p53 status in CRC. Br J Cancer. 2015;112(1):122‐130. doi: 10.1038/bjc.2014.560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. García‐Foncillas J, Sunakawa Y, Aderka D, et al. Distinguishing features of cetuximab and panitumumab in colorectal cancer and other solid tumors. Front Oncologia. 2019;9:00849. doi: 10.3389/fonc.2019.00849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tashiro T, Okuyama H, Endo H, et al. In vivo and ex vivo cetuximab sensitivity assay using three‐dimensional primary culture system to stratify KRAS mutant colorectal cancer. PLOS One. 2017;12(3):e0174151. doi: 10.1371/journal.pone.0174151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kondo J, Inoue M. Application of cancer organoid model for drug screening and personalized therapy. Cell. 2019;8(5):470. doi: 10.3390/cells8050470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kondo J, Endo H, Okuyama H, et al. Retaining cell–cell contact enables preparation and culture of spheroids composed of pure primary cancer cells from colorectal cancer. Proc Natl Acad Sci. 2011;108(15):6235‐6240. doi: 10.1073/pnas.1015938108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. de Jeude JFVL, Vermeulen JLM, Montenegro‐Miranda PS, Van den Brink GR, Heijmans J. A protocol for Lentiviral transduction and downstream analysis of intestinal organoids. J Vis Exp JoVE. 2015;98:52531. doi: 10.3791/52531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Muzny DM, Bainbridge MN, Chang K, et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330‐337. doi: 10.1038/nature11252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lonsdale J, Thomas J, Salvatore M, et al. The genotype‐tissue expression (GTEx) project. Nat Genet. 2013;45(6):580‐585. doi: 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee HO, Hong Y, Etlioglu HE, et al. Lineage‐dependent gene expression programs influence the immune landscape of colorectal cancer. Nat Genet. 2020;52(6):594‐603. doi: 10.1038/s41588-020-0636-z [DOI] [PubMed] [Google Scholar]
- 20. Yarom N, Jonker DJ. The role of the epidermal growth factor receptor in the mechanism and treatment of colorectal cancer. Discov Med. 2011;11(57):95‐105. [PubMed] [Google Scholar]
- 21. Wang L, Schulz TC, Sherrer ES, et al. Self‐renewal of human embryonic stem cells requires insulin‐like growth factor‐1 receptor and ERBB2 receptor signaling. Blood. 2007;110(12):4111‐4119. doi: 10.1182/blood-2007-03-082586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tomas A, Futter CE, Eden ER. EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol. 2014;24(1):26‐34. doi: 10.1016/j.tcb.2013.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bakker J, Spits M, Neefjes J, Berlin I. The EGFR odyssey–from activation to destruction in space and time. J Cell Sci. 2017;130(24):4087‐4096. doi: 10.1242/jcs.209197 [DOI] [PubMed] [Google Scholar]
- 24. Wang Y, Poulin EJ, Coffey RJ. LRIG1 is a triple threat: ERBB negative regulator, intestinal stem cell marker and tumour suppressor. Br J Cancer. 2013;108(9):1765‐1770. doi: 10.1038/bjc.2013.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Frosi Y, Anastasi S, Ballarò C, et al. A two‐tiered mechanism of EGFR inhibition by RALT/MIG6 via kinase suppression and receptor degradation. J Cell Biol. 2010;189(3):557‐571. doi: 10.1083/jcb.201002032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Laederich MB, Funes‐Duran M, Yen L, et al. The leucine‐rich repeat protein LRIG1 is a negative regulator of ErbB family receptor tyrosine kinases. J Biol Chem. 2004;279(45):47050‐47056. doi: 10.1074/jbc.M409703200 [DOI] [PubMed] [Google Scholar]
- 27. Gur G, Rubin C, Katz M, et al. LRIG1 restricts growth factor signaling by enhancing receptor ubiquitylation and degradation. EMBO J. 2004;23(16):3270‐3281. doi: 10.1038/sj.emboj.7600342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Powell AE, Wang Y, Li Y, et al. The pan‐ErbB negative regulator Lrig1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell. 2012;149(1):146‐158. doi: 10.1016/j.cell.2012.02.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ordonez‐Moran P, Huelsken J. Lrig1: a new master regulator of epithelial stem cells. EMBO J. 2012;31(9):2064‐2066. doi: 10.1038/emboj.2012.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lees‐Shepard JB, Flint K, Fisher M, et al. Cross‐talk between EGFR and BMP signals regulates chondrocyte maturation during endochondral ossification. Dev Dyn off Publ Am Assoc Anat. 2022;251(1):75‐94. doi: 10.1002/dvdy.438 [DOI] [PubMed] [Google Scholar]
- 31. Scholze AR, Foo LC, Mulinyawe S, Barres BA. BMP signaling in astrocytes downregulates EGFR to modulate survival and maturation. PLOS One. 2014;9(10):e110668. doi: 10.1371/journal.pone.0110668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sun Z, Gao X, Zabkiewicz C, et al. Noggin is associated with a poor prognosis of gastric cancer by promoting the proliferation of gastric cancer cells via the upregulation of EGFR. Int J Oncol. 2020;57(3):813‐824. doi: 10.3892/ijo.2020.5081 [DOI] [PubMed] [Google Scholar]
- 33. Kondo J, Powell AE, Wang Y, et al. LRIG1 regulates ontogeny of smooth muscle−derived subsets of interstitial cells of Cajal in mice. Gastroenterology. 2015;149(2):407‐419.e8. doi: 10.1053/j.gastro.2015.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kvarnbrink S, Karlsson T, Edlund K, et al. LRIG1 is a prognostic biomarker in non‐small cell lung cancer. Acta Oncol. 2015;54(8):1113‐1119. doi: 10.3109/0284186X.2015.1021427 [DOI] [PubMed] [Google Scholar]
- 35. An Y, Zhao Z, Ou P, Wang G. Expression of LRIG1 is associated with good prognosis for human non‐small cell lung cancer. Medicine (Baltimore). 2015;94(47):e2081. doi: 10.1097/MD.0000000000002081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krig SR, Frietze S, Simion C, et al. Lrig1 is an estrogen regulated growth suppressor and correlates with longer relapse free survival in ERα‐positive breast cancer. Mol Cancer Res MCR. 2011;9(10):1406‐1417. doi: 10.1158/1541-7786.MCR-11-0227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang B, Dai C, Tan R, et al. Lrig1 is a positive prognostic marker in hepatocellular carcinoma. OncoTargets Ther. 2016;9:7071‐7079. doi: 10.2147/OTT.S112534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bakherad M, Salimi M, Angaji SA, Mahjoubi F, Majidizadeh T. LRIG1 expression and colorectal cancer prognosis. BMC Med Genomics. 2021;14(1):20. doi: 10.1186/s12920-020-00846-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang Q, Shi W, Wang Q, et al. Clinicopathological and prognostic significance of leucine‐rich repeats and immunoglobulin‐like domains protein 1 (LRIG1) in malignant tumors: a meta‐analysis. J Cancer. 2018;9(16):2895‐2909. doi: 10.7150/jca.24749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Qi Z, Li Y, Zhao B, et al. BMP restricts stemness of intestinal Lgr5+ stem cells by directly suppressing their signature genes. Nat Commun. 2017;8:13824. doi: 10.1038/ncomms13824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Alshammari K, Aung KL, Zhang T, et al. Phase II trial of Trametinib and Panitumumab in RAS/RAF wild type metastatic colorectal cancer. Clin Colorectal Cancer. 2021;20(4):334‐341. doi: 10.1016/j.clcc.2021.07.004 [DOI] [PubMed] [Google Scholar]
- 42. van Geel RMJM, Iersel LBJV. Combined targeted therapy for BRAF mutant metastatic colorectal cancer: are we there yet? Dig Med Res. 2022;5:5. doi: 10.21037/dmr-22-15 [DOI] [Google Scholar]
- 43. Wan R, Feng J, Tang L. Consequences of mutations and abnormal expression of SMAD4 in tumors and T cells. OncoTargets Ther. 2021;14:2531‐2540. doi: 10.2147/OTT.S297855 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.
Figure S1‐S9.
Table S1.