

Abstract
As an epitranscriptomic modulation manner, N6‐methyladenosine (m6A) modification plays important roles in various diseases, including hepatocellular carcinoma (HCC). m6A modification affects the fate of RNAs. The potential contributions of m6A to the functions of RNA still need further investigation. In this study, we identified long noncoding RNA FAM111A‐DT as an m6A‐modified RNA and confirmed three m6A sites on FAM111A‐DT. The m6A modification level of FAM111A‐DT was increased in HCC tissues and cell lines, and increased m6A level was correlated with poor survival of HCC patients. m6A modification increased the stability of FAM111A‐DT transcript, whose expression level showed similar clinical relevance to that of the m6A level of FAM111A‐DT. Functional assays found that only m6A‐modified FAM111A‐DT promoted HCC cellular proliferation, DNA replication, and HCC tumor growth. Mutation of m6A sites on FAM111A‐DT abolished the roles of FAM111A‐DT. Mechanistic investigations found that m6A‐modified FAM111A‐DT bound to FAM111A promoter and also interacted with m6A reader YTHDC1, which further bound and recruited histone demethylase KDM3B to FAM111A promoter, leading to the reduction of the repressive histone mark H3K9me2 and transcriptional activation of FAM111A. The expression of FAM111A was positively correlated with the m6A level of FAM111A‐DT, and the expression of methyltransferase complex, YTHDC1, and KDM3B in HCC tissues. Depletion of FAM111A largely attenuated the roles of m6A‐modified FAM111A‐DT in HCC. In summary, the m6A‐modified FAM111A‐DT/YTHDC1/KDM3B/FAM111A regulatory axis promoted HCC growth and represented a candidate therapeutic target for HCC.
Keywords: DNA replication, epigenetic modulation, hepatocellular carcinoma, histone methylation, N6‐methyladenosine
The m6A modification level of FAM111A‐DT was increased in hepatocellular carcinoma (HCC), and increased m6A level was correlated with poor survival of HCC patients. m6A‐modified FAM111A‐DT promoted HCC cellular proliferation, DNA replication, and HCC tumor growth. m6A‐modified FAM111A‐DT epigenetically activated FAM111A via binding and recruiting YTHDC1 and KDM3B to the FAM111A promoter region, leading to the reduction of the repressive histone mark H3K9me2.
Abbreviations
- CCK‐8
Cell Counting Kit‐8
- ChIP
chromatin immunoprecipitation
- ChIRP
chromatin isolation by RNA purification
- EdU
5‐ethynyl‐2′‐deoxyuridine
- HCC
hepatocellular carcinoma
- HNRNP
heterogeneous nuclear ribonucleoprotein
- HR
hazard ratio
- IGF2BP
insulin‐like growth factor 2 mRNA‐binding protein
- IHC
immunohistochemistry
- LIHC
liver hepatocellular carcinoma
- lncRNA
long noncoding RNA
- m6A
N6‐methyladenosine
- MeRIP
methylated RNA immunoprecipitation
- NC
negative control
- PCNA
proliferating cell nuclear antigen
- qPCR
quantitative polymerase chain reaction
- RIP
RNA immunoprecipitation
- RNA‐seq
RNA sequencing
- SELECT
single‐base elongation‐ and ligation‐based qPCR amplification method
- TCGA
The Cancer Genome Atlas
1. INTRODUCTION
Liver cancer is one of the most prevalent malignancies worldwide with a relatively poor prognosis. 1 Hepatocellular carcinoma (HCC) is the major histological subtype of liver cancer. 1 The effects of surgical resection and system treatment are both limited for HCC, leading to a less than 20% 5‐year survival rate of HCC. 2 Therefore, it is urgent to deeply investigate the molecular alterations of HCC to develop a more efficiently targeted therapy.
Whole‐genome and ‐exome sequencings have identified several mutational signatures of HCC and the recurrent mutations of coding and noncoding regions, such as the coding genes TP53, CTNNB1, AXIN1, and ARID1A, and the noncoding genes NEAT1 and MALAT1. 3 , 4 , 5 Apart from genomic alterations, epigenetic alterations of HCC were intensively investigated. 6 , 7 , 8 , 9 Aberrant DNA methylation modification, histone acetylation modification, histone methylation modification, and noncoding RNAs were frequently reported to induce aberrant gene expression in HCC. 10 , 11 , 12 , 13 As a class of noncoding RNAs, long noncoding RNAs (lncRNAs) are defined as longer than 200 nucleotides in length with limited protein‐coding potential. 14 lncRNAs mainly function as gene expression modulators and change the expressions and functions of proteins involved in various pathophysiological processes. 15 , 16 , 17 , 18 , 19 In HCC, many lncRNAs have been revealed to play oncogenic or tumor‐suppressive roles, including PAARH, ADORA2A‐AS1, and HOMER3‐AS1, which we previously reported. 20 , 21 , 22 , 23 , 24
Recently, aberrant epitranscriptomic modifications of RNAs were found in various diseases and presented critical roles in development, homeostasis, and various diseases. 25 Among these epitranscriptomic modifications of RNAs, N6‐methyladenosine (m6A) is one of the most widespread and conserved RNA modifications. 26 m6A modification has been detected in nearly all types of RNAs, including mRNAs, rRNAs, snRNAs, and so on. 27 Current studies have established m6A modification as the critical determinant of RNA fate. 28 m6A modification modulates RNA stability, conformation, folding, translation, pre‐mRNAs splicing, nuclear export, chromatin modification, genome integrity, transcription of target genes, and so on. 29 , 30 , 31 , 32 m6A is installed by m6A methyltransferases. The METTL3‐METTL14‐WTAP methyltransferase complex is the major component responsible for the deposition of m6A in RNAs. 33 , 34 , 35 The methyl group in m6A can be removed by demethylases, mainly including FTO and ALKBH5. 36 The functional consequences of m6A modification are mainly mediated by m6A‐binding proteins, which were also termed as m6A readers. Different readers mediate the different effects of m6A modification on RNA fate. The well‐known m6A readers include YTH domain family proteins (YTHDC1, YTHDC2, YTHDF1, YTHDF2, YTHDF3), insulin‐like growth factor 2 mRNA‐binding proteins (IGF2BPs), and heterogeneous nuclear ribonucleoprotein (HNRNP) family proteins. 37 , 38 Although m6A modification of mRNAs have been intensively investigated, the contributions of m6A modification to the regulatory roles of lncRNAs are still unclear.
Through analyzing The Cancer Genome Atlas (TCGA) liver hepatocellular carcinoma (LIHC) data, several m6A‐related lncRNAs have been identified to be correlated with clinical prognosis of HCC patients. 39 Among these m6A‐related lncRNAs in HCC, we further detected their expressions, associations with prognosis, and their m6A modification levels, and found that not only the expression level but also the m6A modification level of FAM111A‐DT was increased in HCC and correlated with poor survival of HCC patients. We further identified m6A‐modified, but not nonmodified, FAM111A‐DT as oncogenic lncRNA in HCC. The mechanisms underlying the oncogenic roles of m6A‐modified FAM111A‐DT were also investigated.
2. MATERIALS AND METHODS
2.1. Human tissue samples
Eighty‐two pairs of HCC tissues and adjacent noncancerous liver tissues were acquired at the Affiliated Hospital of Youjiang Medical University for Nationalities from HCC patients with written informed consents. The clinicopathological characteristics of these 82 cases are shown in Table S1. This study was undertaken following the Declaration of Helsinki and approved by the Institutional Review Board of the Affiliated Hospital of Youjiang Medical University for Nationalities (approval no. YYFY‐LL‐2022‐103).
2.2. Cell lines and cell culture
Human HCC cell lines SK‐HEP‐1 (cat. no. TCHu109), HuH‐7 (cat. no. SCSP‐526), and Hep3B (cat. no. SCSP‐5045) were purchased from the Chinese Academy of Sciences Cell Bank. Human HCC cell line SNU‐398 (cat. no. CRL2233) and human immortalized liver cell lines THLE‐2 (cat. no. CRL‐2706) and THLE‐3 (cat. no. CRL‐11233) were obtained from the American Type Culture Collection (ATCC). SK‐HEP‐1 and Hep3B cells were cultured in Eagle's Minimum Essential Medium (cat. no. 11095080, Invitrogen) added with 10% fetal bovine serum (cat. no. 10099141, FBS, Invitrogen). HuH‐7 cell was cultured in Dulbecco's modified Eagle's medium (cat. no. 11965092, Invitrogen) added with 10% FBS. SNU‐398 cell was cultured in RPMI 1640 medium (cat. no. 11875093, Invitrogen) added with 10% FBS. THLE‐2 and THLE‐3 cells were cultured using the BEGM Bullet Kit (cat. no. CC‐3170, Lonza) following the provided protocol. All cells were maintained at 37°C containing 5% CO2 and routinely tested as mycoplasma‐free.
2.3. RNA extraction and quantitative polymerase chain reaction (qPCR)
Total RNA was extracted using the RNA isolater Total RNA Extraction Reagent (cat. no. R401, Vazyme). Reverse transcription was performed using RNA and the HiScript III RT SuperMix for qPCR (cat. no. R323, Vazyme) to generate complementary DNA (cDNA). The cDNA was further subjected to qPCR using the ChamQ Universal SYBR qPCR Master Mix (cat. no. Q711, Vazyme) on StepOnePlus Real‐Time PCR System (cat. no. 4376600, Applied Biosystems). The sequences of primers used in qPCR were as follows: 5′‐GCAAAGCCGTTTCTTCCTA‐3′ (sense) and 5′‐CCTGTGGTTCAACTACTTCAAT‐3′ (antisense) for FAM111A‐DT, 5′‐ACAAAACAGCCAGAGACAAT‐3′ (sense) and 5′‐GTGGGTAGAAGCCAAGGA‐3′ (antisense) for AL031985.3, 5′‐TCAGTATGAACGCAAGGG‐3′ (sense) and 5′‐GCAACAAGCACAGCCAGT‐3′ (antisense) for AC145207.5, 5′‐CGGTGTGTATCTTTTGGG‐3′ (sense) and 5′‐ATCATTGACTTGTGTCTGC‐3′ (antisense) for SNHG21, 5′‐ACGGGACAGTCAGAAGAT‐3′ (sense) and 5′‐GAGGCACGGTAAGGGTTA‐3′ (antisense) for AC012467.2, 5′‐GCACAACGGGGATGTAGC‐3′ (sense) and 5′‐AAACTTTGGGCAGCGACT‐3′ (antisense) for SREBF2‐AS1, 5′‐AACCATCCGTTCATCTTCA‐3′ (sense) and 5′‐TGGCTCTTGGGTCTCCTCT‐3′ (antisense) for FAM111A, 5′‐GTCTTTCCATCCACTCACGTCT‐3′ (sense) and 5′‐GGACAACTAGATGCCGAGGTAG‐3′ (antisense) for NEAT1, 5′‐TGGAGAAATAGTAGATGGC‐3′ (sense) and 5′‐GGTGAGGAAGTAAAAACAG‐3′ (antisense) for MALAT1, 5′‐CATCTTGGCTCCTCCGAATGTG‐3′ (sense) and 5′‐TCTTGCCAGGTGTTGTTCTGC‐3′ (antisense) for CASC9, 40 5′‐GTCGGAGTCAACGGATTTG‐3′ (sense) and 5′‐TGGGTGGAATCATATTGGAA‐3′ (antisense) for GAPDH. GAPDH served as an endogenous control. Relative expression was calculated using the comparative Ct method.
2.4. Vectors, siRNAs, and stable cell lines construction
METTL3‐, METTL14‐, and FTO‐overexpressing vectors were purchased from GenePharma. ON‐TARGETplus Human METTL3 siRNA SMART Pool (cat. no. L‐005170‐02‐0010), ON‐TARGETplus Human YTHDC1 siRNA SMART Pool (cat. no. L‐015332‐02‐0010), ON‐TARGETplus Human HNRNPA2B1 siRNA SMART Pool (cat. no. L‐011690‐01‐0010), ON‐TARGETplus Human HNRNPG siRNA SMART Pool (cat. no. L‐011691‐01‐0010), ON‐TARGETplus Human HNRNPC siRNA SMART Pool (cat. no. L‐011869‐03‐0010), and ON‐TARGETplus Human KDM3B siRNA SMART Pool (cat. no. L‐020378‐01‐0010) were purchased from Horizon Discovery. The transfection of vectors and siRNAs were performed using the GP‐transfect‐Mate (cat. no. G04009, GenePharma).
Wild‐type and m6A modification sites mutated FAM111A‐DT overexpressing lentiviruses (LV11 vector) were purchased from GenePharma. Two pairs of cDNA oligonucleotides targeting FAM111A‐DT and one pair of cDNA oligonucleotide targeting FAM111A were synthesized and cloned into the shRNA lentivirus‐expressing vector (LV‐2N vector) (GenePharma) to generate shRNA lentivirus targeting FAM111A‐DT or FAM111A. Scrambled nontargeting shRNA lentivirus were used as negative control (NC). The sequences of shRNA oligonucleotides were as follows: 5′‐GATCCGCACACTGCAATGTCCAAACGTTCAAGAGACGTTTGGACATTGCAGTGTGCTTTTTTG‐3′ (sense) and 5′‐AATTCAAAAAAGCACACTGCAATGTCCAAACGTCTCTTGAACGTTTGGACATTGCAGTGTGCG‐3′ (antisense) for shRNA‐FAM111A‐DT‐1, 5′‐GATCCGCACTGTAAGCCCTTGAATGGTTCAAGAGACCATTCAAGGGCTTACAGTGCTTTTTTG‐3′ (sense) and 5′‐AATTCAAAAAAGCACTGTAAGCCCTTGAATGGTCTCTTGAACCATTCAAGGGCTTACAGTGCG‐3′ (antisense) for shRNA‐FAM111A‐DT‐2, 5′‐GATCCGCGCAAACTCTGTGTTTATGCTTCAAGAGAGCATAAACACAGAGTTTGCGCTTTTTTG‐3′ (sense) and 5′‐AATTCAAAAAAGCGCAAACTCTGTGTTTATGCTCTCTTGAAGCATAAACACAGAGTTTGCGCG‐3′ (antisense) for shRNA‐FAM111A, 5′‐GATCCGTTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAACTTTTTTG‐3′ (sense) and 5′‐AATTCAAAAAAGTTCTCCGAACGTGTCACGTTCTCTTGAAACGTGACACGTTCGGAGAACG‐3′ (antisense) for shRNA‐NC.
To construct FAM111A‐DT stably overexpressed and control HCC cells, SK‐HEP‐1 and SNU‐398 cells were infected with wild‐type or m6A modification sites‐mutated FAM111A‐DT‐overexpressing lentiviruses. Ninety‐six hours after infection, the cells were treated with 800 μg/mL neomycin (cat. no. ant‐gn‐1, InvivoGen) for 4 weeks to select FAM111A‐DT‐overexpressed cells. To construct FAM111A‐DT stably depleted HCC cells, SK‐HEP‐1 and SNU‐398 cells were infected with shRNA lentivirus targeting FAM111A‐DT. Ninety‐six hours after infection, the cells were treated with 2 μg/mL puromycin (cat. no. ant‐pr‐1, InvivoGen) for 4 weeks to select FAM111A‐DT‐silenced cells. To construct HCC cells with concurrent FAM111A‐DT overexpression and FAM111A depletion, FAM111A‐DT stably overexpressed SK‐HEP‐1 cells were infected with shRNA lentivirus targeting FAM111A and selected with 800 μg/mL neomycin and 2 μg/mL puromycin. To construct HCC cells with concurrent FAM111A‐DT knockdown and FAM111A overexpression, FAM111A‐DT stably depleted SNU‐398 cells were infected with FAM111A‐overexpressing lentiviruses and selected with 800 μg/mL neomycin and 2 μg/mL puromycin.
2.5. Site‐specific m6A modification detection
Site‐specific m6A modification detection was performed using the previously reported single‐base elongation‐ and ligation‐based qPCR amplification method (termed “SELECT”). 41 Simultaneously, detections of nonmodified 498 A site, 774 A site, or 876 A site were performed for controlling the initial RNA input amounts and used as input for m6A sites 501, 779, and 881 respectively. The sequences of probes used were as follows: 5′‐tagccagtaccgtagtgcgtgCGTCTCGTCTCGGGAGCTG‐3′ (up) and 5′‐5phos/CCTGAAAGGGGGCTGCCAcagaggctgagtcgctgcat‐3′ (down) for m6A 501, 5′‐tagccagtaccgtagtgcgtgGTCTCGTCTCGGGAGCTGTCC‐3′ (up) and 5′‐5phos/GAAAGGGGGCTGCCACGCcagaggctgagtcgctgcat‐3′ (down) for A 498, 5′‐tagccagtaccgtagtgcgtgCTCTGGCTAATGATTCTGGACAG‐3′ (up) and 5′‐5phos/CCAATCCTGTGGTTCAACTACTTCcagaggctgagtcgctgcat‐3′ (down) for m6A 779, 5′‐tagccagtaccgtagtgcgtgGCTAATGATTCTGGACAGTCCAA‐3′ (up) and 5′‐5phos/CCTGTGGTTCAACTACTTCAATGcagaggctgagtcgctgcat‐3′ (down) for A 774, 5′‐tagccagtaccgtagtgcgtgACAGAACTCATTGGTTTCTGCAG‐3′ (up) and 5′‐5phos/TTTTTGTTTTGTTGTTGCTTTTGGcagaggctgagtcgctgcat‐3′ (down) for m6A 881, 5′‐tagccagtaccgtagtgcgtgGAACTCATTGGTTTCTGCAGTTTTT‐3′ (up) and 5′‐5phos/GTTTTGTTGTTGCTTTTGGTAAAGGcagaggctgagtcgctgcat‐3′ (down) for A 876. The sequences of primers used in qPCR for SELECT were: 5′‐ATGCAGCGACTCAGCCTCTG‐3′ (sense) and 5′‐TAGCCAGTACCGTAGTGCGTG‐3′ (antisense). 41
2.6. RNA immunoprecipitation (RIP) and methylated RNA immunoprecipitation (MeRIP) assays
RNA immunoprecipitation assays were performed in SK‐HEP‐1 cells with wild‐type or mutated FAM111A‐DT overexpression using the EZ‐Magna RIP RNA‐Binding Protein Immunoprecipitation Kit (cat. no. 17‐701, Millipore) and YTHDC1‐specific antibody (cat. no. 77422, Cell Signaling Technology). Enriched wild‐type or mutated FAM111A‐DT was detected by qPCR with the primers: 5′‐GCAAAGCCGTTTCTTCCTA‐3′ (sense) and 5′‐CCTGTGGTTCAACTACTTCAAT‐3′ (antisense) for wild‐type FAM111A‐DT, 5′‐AGTAGTTGAACCACAGGATTGGT‐3′ (sense) and 5′‐CAGAACTCATTGGTTTCTGCAGA‐3′ (antisense) for mutated FAM111A‐DT. MeRIP assays were performed in indicated tissues and cells using the Magna MeRIP m6A Kit (cat. no. 17‐10499, Millipore). Enriched RNA was detected by qPCR.
2.7. Chromatin isolation by RNA purification (ChIRP) assay
Chromatin isolation by RNA purification assays were performed in indicated cells using the EZ‐Magna ChIRP RNA Interactome Kit (cat. no. 17‐10495, Millipore) following the provided protocol. The sequences of FAM111A‐DT antisense DNA probes were as follows: 1, 5′‐agactcaagctgccacagtg‐3′; 2, 5′‐gctgcaaattaaggagcact‐3′; 3, 5′‐gaagaaacggctttgctggg‐3′; 4, 5′‐atcccatagagcacattaga‐3′; 5, 5′‐ggcatgcacaaaaatttcct‐3′; 6, 5′‐ctcaaatgttaccacctctg‐3′; 7, 5′‐agtaagattcatttgccacc‐3′; 8, 5′‐tgtatcactgcttgagctta‐3′; 9, 5′‐tgctacaaccacacacacta‐3′; 10, 5′‐cttacagtgctcatggaagt‐3′. The enriched DNA was detected using qPCR with the primers: 5′‐ATTTACAGGCGGGGACAG‐3′ (sense) and 5′‐TAAAAACTCGGGTGTGGG‐3′ (antisense) for FAM111A promoter; 5′‐GACGCTTTCTTTCCTTTCGC‐3′ (sense) and 5′‐CTGCCCATTCATTTCCTTCC‐3′ (antisense) for GAPDH promoter.
2.8. Chromatin immunoprecipitation (ChIP) assay
Chromatin immunoprecipitation assays were performed in indicated cells using the EZ‐Magna ChIP A/G Chromatin Immunoprecipitation Kit (cat. no. 17‐10086, Millipore) and a KDM3B antibody (cat. no. 5377, Cell Signaling Technology), an H3K9me2 antibody (cat. no. ab1220, Abcam), an H3K4me3 antibody (cat. no. ab8580, Abcam), or an H3K27ac antibody (cat. no. ab4729, Abcam) following the provided protocol. The enriched DNA was detected using qPCR with the primers: 5′‐ATTTACAGGCGGGGACAG‐3′ (sense) and 5′‐TAAAAACTCGGGTGTGGG‐3′ (antisense) for FAM111A promoter.
2.9. Cellular proliferation and DNA replication assays
Cellular proliferation was assessed using Cell Counting Kit‐8 (CCK‐8) assays as we previously described. 22 Briefly, 2000 indicated cells resuspended in 100 μL complete medium were seeded into a 96‐well plate. After culture for the indicated time, 10 μL CCK‐8 reagent (cat. no. CK04, Dojindo) was added to each well. After culture for another 2 h, the absorbance values at 450 nm were detected using the Synergy 2 microplate reader (BioTek) to indicate the viable cell number. DNA replication was assessed using 5‐ethynyl‐2′‐deoxyuridine (EdU) incorporation assays. EdU incorporation assays were performed using the Cell‐Light EdU Apollo567 In Vitro Kit (cat. no. C10310‐1, RiboBio). The percentage of EdU‐positive cells was detected using the Imager.M2 fluorescence microscope (Carl Zeiss) and calculated as the ratio of EdU‐positive cells to total cells.
2.10. In vivo tumor growth assay
Five‐week‐old male BALB/C athymic nude mice were purchased from Shanghai SLAC Laboratory Animal Co. and fed in specific pathogen‐free conditions. The use of mice was reviewed and approved by the Institutional Review Board of the Affiliated Hospital of Youjiang Medical University for Nationalities. Indicated cells were subcutaneously injected into the back flank of mice. Subcutaneous tumor volumes were measured every week and calculated following the formula: volume = 0.5 × length × width2. At the 28th day after inoculation, subcutaneous tumors were resected, weighed, and subjected to immunohistochemistry (IHC) staining with primary antibodies against Ki67 (cat. no. 9027, Cell Signaling Technology) or proliferating cell nuclear antigen (PCNA) (cat. no. 13110, Cell Signaling Technology).
2.11. Statistical analysis
Statistical analyses were performed using the GraphPad Prism 6.0 software. Mann–Whitney test, Wilcoxon matched‐pairs signed‐rank test, log‐rank test, Student's t‐test, one‐way ANOVA followed by Dunnett's multiple comparisons test, Pearson chi‐square test, and Spearman correlation analysis were conducted as indicated in the figure and table legends. p < 0.05 was considered statistically significant.
3. RESULTS
3.1. The expression of FAM111A‐DT was increased in HCC and correlated with poor survival of HCC patients
Although 25 prognostic m6A‐related lncRNAs in HCC were identified in the previous report, whether the m6A modification levels of these lncRNAs were genuinely involved in HCC initiation and progression was still unknown. Thus, we first measured m6A modification level of the six most prognosis‐related lncRNAs in 10 pairs of HCC tissues and adjacent noncancerous liver tissues using MeRIP assays. The results showed that the m6A modification level of FAM111A‐DT had the highest increase in HCC tissues compared with adjacent noncancerous liver tissues (Figure S1). Thus, we focused on FAM111A‐DT.
To assess the clinical significance of FAM111A‐DT in HCC, TCGA‐LIHC RNA sequencing (RNA‐seq) data were analyzed by the online tool Kaplan–Meier Plotter (https://kmplot.com/analysis/index.php?p=service&cancer=liver_rnaseq). 42 The result showed that increased expression of FAM111A‐DT was correlated with poor overall survival of HCC patients (Figure 1A). The TCGA‐LIHC data also showed that the expression of FAM111A‐DT was increased in HCC tissues compared with normal liver tissues (Figure 1B). Further analysis of the TCGA‐LIHC data revealed that the expression of FAM111A‐DT was correlated with poor differentiation and high alpha fetoprotein (AFP) level (Figure 1C,D). To further confirm the clinical significance of FAM111A‐DT in HCC, we measured FAM111A‐DT expression in our HCC cohort, and the results showed that the expression of FAM111A‐DT was also increased in HCC tissues (Figure 1E). In our HCC cohort, increased expression of FAM111A‐DT was also correlated with poor overall survival of HCC patients (Figure 1F). Furthermore, analysis of the correlation between FAM111A‐DT expression and clinicopathological features of HCC showed that high expression of FAM111A‐DT was correlated with high AFP level, poor differentiation, and advanced clinical stage (Table S1). Consistent with the expression of FAM111A‐DT in HCC tissues, the expression of FAM111A‐DT was also increased in human HCC cell lines SK‐HEP‐1, HuH‐7, Hep3B, and SNU‐398 compared with immortalized human liver cell lines THLE‐2 and THLE‐3 (Figure 1G).
FIGURE 1.
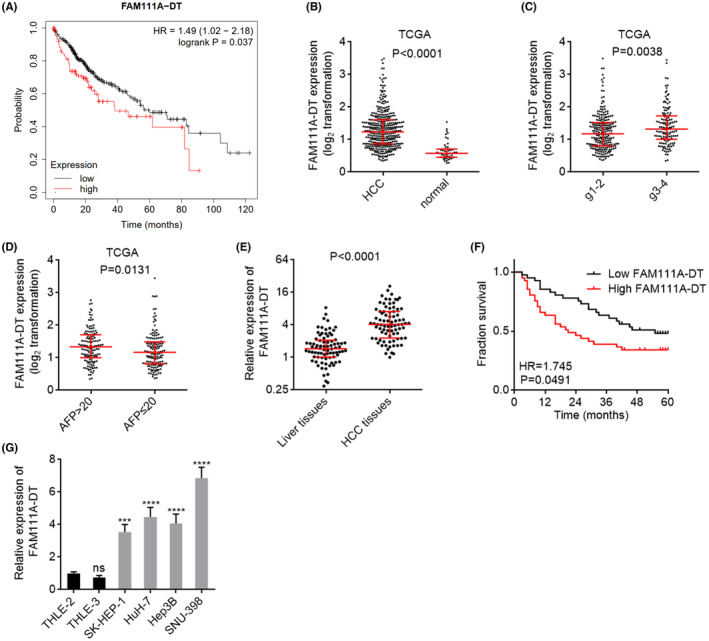
FAM111A‐DT was highly expressed and correlated with poor survival in hepatocellular carcinoma (HCC). (A) The correlation between FAM111A‐DT expression and overall survival according to the TCGA liver hepatocellular carcinoma (LIHC) data, analyzed by the online tool Kaplan–Meier Plotter. p = 0.037, HR = 1.49 by log‐rank test. (B) The expression of FAM111A‐DT in 371 HCC tissues and 50 normal liver tissues according to the TCGA‐LIHC data. Results are shown as median with interquartile range. p < 0.0001 by Mann–Whitney test. (C) The expression of FAM111A‐DT in 232 HCC tissues with grade 1 and 2, and 134 HCC tissues with grade 3 and 4, according to the TCGA‐LIHC data. Results are shown as median with interquartile range. p < 0.0001 by Mann–Whitney test. (D) The expression of FAM111A‐DT in 131 HCC tissues with AFP > 20, and 147 HCC tissues with AFP ≤ 20, according to the TCGA‐LIHC data. Results are shown as median with interquartile range. p < 0.0001 by Mann–Whitney test. (E) The expression of FAM111A‐DT in 82 pairs of HCC tissues and adjacent noncancerous liver tissues was measured by qPCR. Results are shown as median with interquartile range. p < 0.0001 by Wilcoxon matched‐pairs signed‐rank test. (F) Kaplan–Meier survival analysis of the correlation between FAM111A‐DT expression and overall survival in our HCC cohort containing 82 cases. p = 0.0491, hazard ratio (HR) = 1.745 by log‐rank test. (G) The expression of FAM111A‐DT in immortalized liver cell lines THLE‐2 and THLE‐3, and HCC cell lines SK‐HEP‐1, HuH‐7, Hep3B, and SNU‐398 was measured by qPCR. Results are shown as mean ± standard deviation (SD) of n = 3 independent experiments. ***p < 0.001, ****p < 0.0001, ns, not significant, by one‐way ANOVA followed by Dunnett's multiple comparisons test.
3.2. FAM111A‐DT was m6A RNA methylation modified
To identify whether FAM111A‐DT was m6A modified, MeRIP assays were performed in SK‐HEP‐1 and SNU‐398 cells. The results showed that FAM111A‐DT had m6A modification (Figure 2A). Ectopic expression of METTL3 or METTL14 increased the m6A modification level of FAM111A‐DT, while FTO overexpression decreased the m6A modification level of FAM111A‐DT (Figure 2B), which further supports the existence of m6A modification in FAM111A‐DT. m6A modification sites of FAM111A‐DT were predicted using two online tools SRAMP (http://www.cuilab.cn/sramp) and RMBase (https://rna.sysu.edu.cn/rmbase/m6Amod.php). 43 , 44 Three sites (501, 779, and 881) on FAM111A‐DT were predicted to be m6A modified by both tools (Figure 2C and Table S2). The site‐specific detection of m6A modification levels on 501, 779, and 881 sites was conducted using the previously reported SELECT assays. 41 m6A modification hinders the single‐base elongation and nick ligation efficiencies, therefore leading to the dramatic reduction of final ligation products, which were subjected to qPCR (Figure 2D). SELECT assays revealed the existence of m6A modification in 501, 779, and 881 sites, whose m6A modification levels were upregulated by METTL3 and METTL14 and downregulated by FTO (Figure 2E–G).
FIGURE 2.
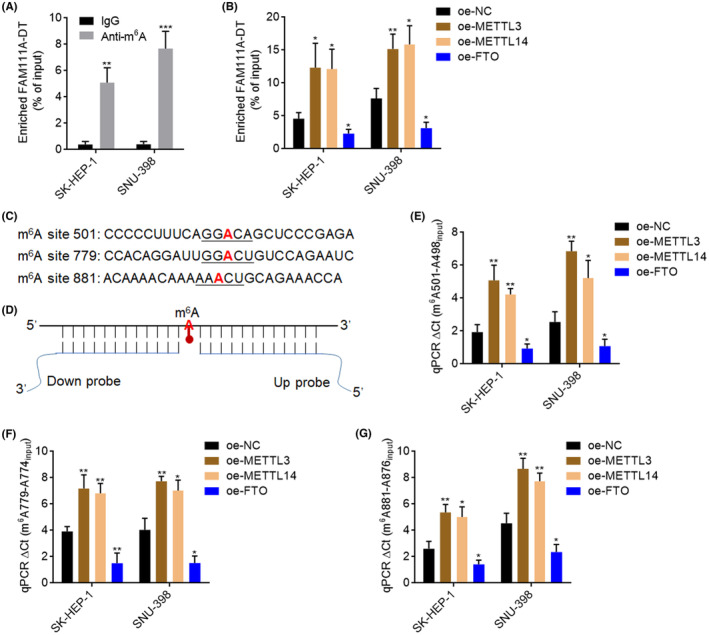
FAM111A‐DT was N6‐methyladenosine (m6A) RNA methylation modified. (A) MeRIP assays were performed in SK‐HEP‐1 and SNU‐398 cells to enrich m6A‐modified RNA, followed by qPCR to assess m6A modification level of FAM111A‐DT. (B) MeRIP assays were performed in SK‐HEP‐1 and SNU‐398 cells with METTL3, METTL14, or FTO overexpression or control to enrich m6A‐modified RNA, followed by qPCR to assess m6A modification level of FAM111A‐DT. (C) The predicted m6A modification sites on FAM111A‐DT by online tools SRAMP and RMBase. (D) Schematic of the SELECT m6A detection method. (E–G) m6A modification levels of 501 site (E), 779 site (F), and 881 site (G) on FAM111A‐DT in SK‐HEP‐1 and SNU‐398 cells with METTL3, METTL14, or FTO overexpression or control were measured by SELECT. Results are shown as mean ± SD of n = 3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 by Student's t‐test.
3.3. m6A modification level of FAM111A‐DT was increased in HCC and correlated with poor prognosis of HCC patients
Next, we measured the m6A modification level of FAM111A‐DT in HCC cells and tissues using SELECT assays. The results showed that the m6A modification levels of 501, 779, and 881 sites on FAM111A‐DT were all increased in HCC cell lines compared with immortalized liver cell lines (Figure 3A–C). Furthermore, the m6A modification levels of 501, 779, and 881 sites on FAM111A‐DT were also increased in HCC tissues compared with paired adjacent noncancerous liver tissues (Figure 3D–F). Kaplan–Meier survival analyses showed that high m6A modification levels of 501, 779, and 881 sites were all significantly correlated with poor overall survival of HCC patients (Figure 3G–I). The hazard ratio (HR) values calculated by m6A modification levels of FAM111A‐DT were higher than those calculated by FAM111A‐DT expression, which suggested that m6A‐modified FAM111A‐DT may have greater significance than general FAM111A‐DT in HCC.
FIGURE 3.
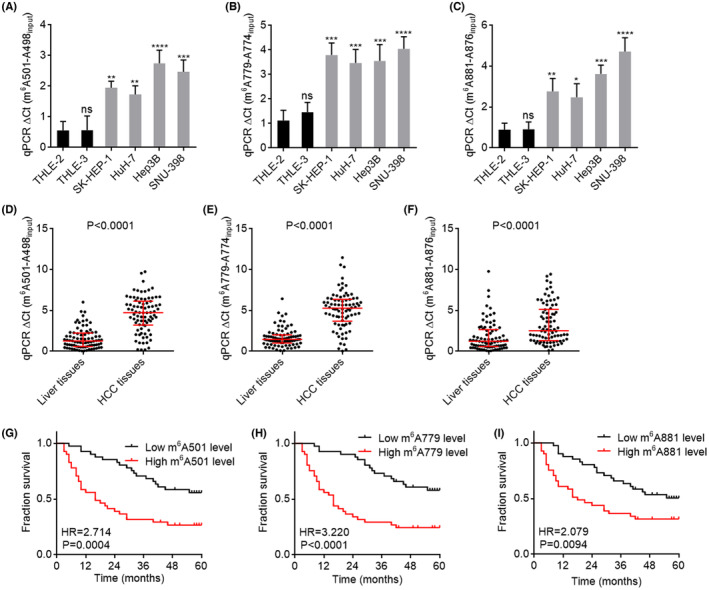
m6A modification level of FAM111A‐DT was increased and correlated with poor survival in hepatocellular carcinoma (HCC). (A–C) N6‐methyladenosine (m6A) modification levels of 501 site (A), 779 site (B), and 881 site (C) on FAM111A‐DT in immortalized liver cell lines THLE‐2 and THLE‐3, and HCC cell lines SK‐HEP‐1, HuH‐7, Hep3B, and SNU‐398 were measured by SELECT. Results are shown as mean ± SD of n = 3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns, not significant, by one‐way ANOVA followed by Dunnett's multiple comparisons test. (D–F) m6A modification levels of 501 site (D), 779 site (E), and 881 site (F) on FAM111A‐DT in 82 pairs of HCC tissues and adjacent noncancerous liver tissues were measured by SELECT. Results are shown as median with interquartile range. p < 0.0001 by Wilcoxon matched‐pairs signed‐rank test. (G–I) Kaplan–Meier survival analysis of the correlation between m6A modification levels of 501 site (G), 779 site (H), or 881 site (I) on FAM111A‐DT and overall survival in our HCC cohort containing 82 cases. p and HR values were calculated by log‐rank test.
3.4. m6A modification increased the stability of FAM111A‐DT transcript
To investigate the potential contributions of m6A modification to FAM111A‐DT, we evaluated the stability of FAM111A‐DT transcript. After the blockage of new RNA synthesis using α‐amanitin, the degradation of FAM111A‐DT transcript was detected. We found that ectopic expression of METTL3 or METTL14 elongated the half‐life of FAM111A‐DT, while ectopic expression of FTO shortened the half‐life of FAM111A‐DT (Figure 4A,B). m6A modification levels of 501, 779, and 881 sites on FAM111A‐DT were all positively correlated with FAM111A‐DT expression in HCC tissues (Figure 4C–E), supporting the positive regulation of FAM111A‐DT by m6A modification. Furthermore, the TCGA‐LIHC data showed that FAM111A‐DT expression level was positively correlated with METTL3, METTL14, and WTAP in HCC tissues (Figure 4F–H), further supporting the positive regulation of FAM111A‐DT by m6A.
FIGURE 4.
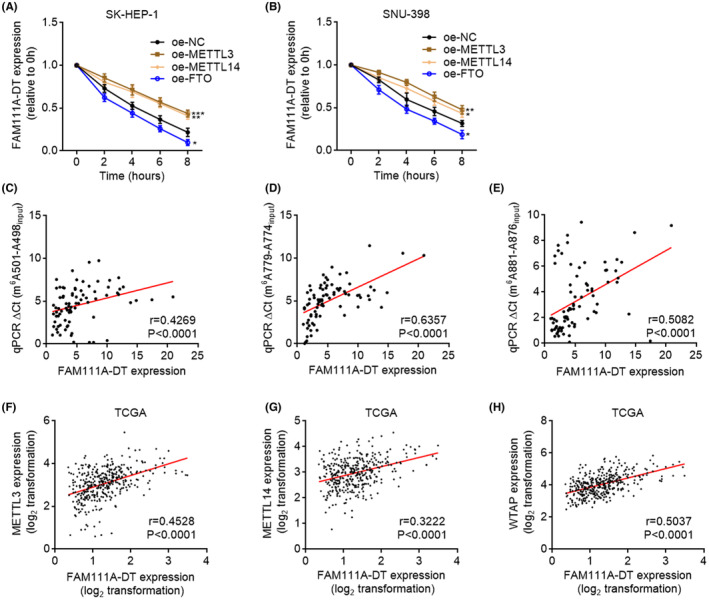
N6‐methyladenosine (m6A) modification increased the transcript stability of FAM111A‐DT. (A, B) The stability of FAM111A‐DT over time was measured after blocking new RNA synthesis with α‐amanitin (50 μM) in SK‐HEP‐1 (A) or SNU‐398 (B) cells with METTL3, METTL14, or FTO overexpression or control. Results are shown as mean ± SD of n = 3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 by Student's t‐test. (C–E) The correlation between m6A modification levels of 501 site (C), 779 site (D), or 881 site (E) on FAM111A‐DT and FAM111A‐DT expression in our cohort containing 82 hepatocellular carcinoma (HCC) tissues. p‐ and r‐values were calculated by Spearman correlation analysis. (F–H) The correlation between METTL3 (F), METTL14 (G), or WTAP (H) and FAM111A‐DT expression in 371 HCC tissues according to the TCGA liver hepatocellular carcinoma (LIHC) data. p‐ and r‐values were calculated by Spearman correlation analysis.
3.5. FAM111A‐DT upregulated FAM111A expression in an m6A‐depedent manner
Considering the important clinical significance of FAM111A‐DT in HCC, we next investigated the downstream molecular targets of FAM111A‐DT. The TCGA‐LIHC data were analyzed to search the genes whose expression was correlated with FAM111A‐DT by R2 Genomics Analysis and Visualization Platform (http://r2.amc.nl). FAM111A was identified as the most significantly correlated gene (Figure 5A). To investigate whether FAM111A‐DT modulated FAM111A expression and whether the regulation was correlated with m6A modification of FAM111A‐DT, we constructed HCC cells with stable overexpression of wild‐type or 501, 779, and 881 sites‐mutated FAM111A‐DT (Figure 5B–D). Ectopic expression of wild‐type FAM111A‐DT, but not mutated FAM111A‐DT, significantly upregulated the expression of FAM111A (Figure 5E,F). Furthermore, we constructed HCC cells with stable knockdown of FAM111A‐DT (Figure 5G,H). Knockdown of FAM111A‐DT significantly downregulated the expression of FAM111A (Figure 5I,J). To investigate whether FAM111A‐DT regulated the generation or degradation of FAM111A mRNA, HCC cells with stable overexpression or knockdown of FAM111A‐DT were treated with α‐amanitin to block new RNA synthesis, and then the degradation of FAM111A mRNA was measured. The results showed that neither overexpression nor knockdown of FAM111A‐DT changes the half‐life of FAM111A mRNA (Figure S2A,B), which suggested that FAM111A‐DT regulated the generation of FAM111A mRNA. Consistent with the TCGA‐LIHC data, the expression of FAM111A was also significantly positively correlated with FAM111A‐DT in our HCC cohort (Figure 5K). Moreover, the expression of FAM111A was significantly positively correlated with m6A modification levels of 501, 779, and 881 sites on FAM111A‐DT in HCC tissues (Figure 5L–N), supporting the positive regulation of FAM111A by m6A‐modified FAM111A‐DT.
FIGURE 5.
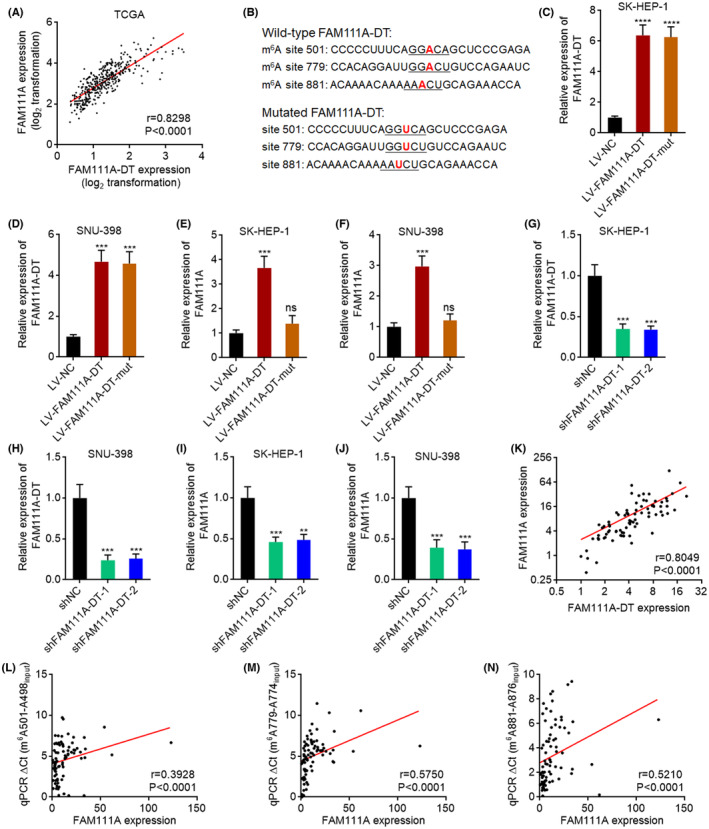
FAM111A‐DT increased FAM111A expression in an N6‐methyladenosine (m6A)‐dependent manner. (A) The correlation between FAM111A and FAM111A‐DT expression in 371 hepatocellular carcinoma (HCC) tissues according to the TCGA liver hepatocellular carcinoma (LIHC) data. p‐ and r‐values were calculated by Spearman correlation analysis. (B) Schematic of the mutation of m6A modification sites 501, 779, and 881 on FAM111A‐DT. (C, D) The expression of FAM111A‐DT in SK‐HEP‐1 (C) and SNU‐398 (D) cells with wild‐type or m6A modification sites‐mutated FAM111A‐DT stable overexpression was measured by qPCR. (E, F) The expression of FAM111A in SK‐HEP‐1 (E) and SNU‐398 (F) cells with wild‐type or mutated FAM111A‐DT stable overexpression was measured by qPCR. (G, H) The expression of FAM111A‐DT in SK‐HEP‐1 (G) and SNU‐398 (H) cells with FAM111A‐DT stable knockdown was measured by qPCR. (I, J) The expression of FAM111A in SK‐HEP‐1 (I) and SNU‐398 (J) cells with FAM111A‐DT stable knockdown was measured by qPCR. (K) The correlation between FAM111A and FAM111A‐DT expression in our cohort containing 82 HCC tissues. p‐ and r‐values were calculated by Spearman correlation analysis. (L–N) The correlation between m6A modification levels of 501 site (L), 779 site (M), or 881 site (N) on FAM111A‐DT and FAM111A expression in our cohort containing 82 HCC tissues. p‐ and r‐values were calculated by Spearman correlation analysis. For (C–J), results are shown as mean ± SD of n = 3 independent experiments. **p < 0.01, ***p < 0.001, ****p < 0.0001, ns, not significant, by one‐way ANOVA followed by Dunnett's multiple comparisons test.
3.6. m6A‐modified FAM111A‐DT directed the demethylation of H3K9me2 at the FAM111A promoter region
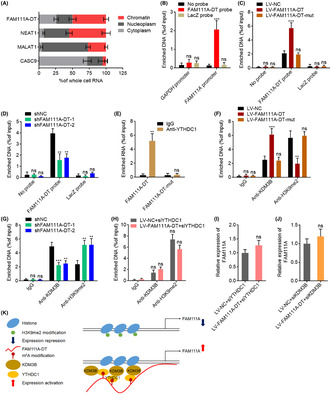
To investigate the mechanisms underlying the upregulation of FAM111A transcription by m6A‐modified FAM111A‐DT, we first detected the subcellular localization of FAM111A‐DT. The results showed that FAM111A‐DT was a chromatin‐associated RNA (Figure 6A). ChIRP assays revealed the specific binding of FAM111A‐DT to FAM111A promoter (Figure 6B). Overexpression of wild‐type, but not mutated, FAM111A‐DT, showed stronger binding to the FAM111A promoter region (Figure 6C), suggesting that m6A‐modified FAM111A‐DT bound to the FAM111A promoter region. Knockdown of FAM111A‐DT reduced the binding of FAM111A‐DT to the FAM111A promoter region (Figure 6D). Reducing the m6A modification level of FAM111A‐DT by METTL3 knockdown reduced the chromatinic localization of FAM111A‐DT and also the binding of FAM111A‐DT to the FAM111A promoter region (Figure S3A,B). m6A‐modified transcript could be bound by m6A reader YTHDC1, which further interacts with and recruits the H3K9me2 demethylase KDM3B to m6A‐associated chromatin regions, inducing H3K9me2 demethylation and gene activation. 45 To investigate whether m6A‐modified FAM111A‐DT modulated FAM111A expression in such a manner, we first detected whether m6A‐modified FAM111A‐DT bound to YTHDC1. RIP assays with YTHDC1‐specific antibody showed that YTHDC1 specifically bound to wild‐type FAM111A‐DT, but not 501, 779, and 881 sites‐mutated FAM111A‐DT (Figure 6E), indicating that the binding between YTHDC1 and FAM111A‐DT was m6A dependent. ChIP assays showed that ectopic expression of wild‐type, but not mutated, FAM111A‐DT significantly increased the binding of KDM3B to the FAM111A promoter region (Figure 6F). Consistently, ectopic expression of wild‐type, but not mutated, FAM111A‐DT significantly decreased H3K9me2 modification level at the FAM111A promoter region (Figure 6F). Conversely, FAM111A‐DT knockdown decreased the binding of KDM3B to the FAM111A promoter region and increased H3K9me2 modification level at the FAM111A promoter region (Figure 6G). Neither overexpression nor knockdown of FAM111A‐DT changed H3K4me3 and H3K27ac modification levels at the FAM111A promoter region (Figure S3C,D). Depletion of YTHDC1 largely abolished the increase in KDM3B binding and the reduction in H3K9me2 level at the FAM111A promoter region caused by FAM111A‐DT overexpression (Figure 6H). Depletion of YTHDC1 or KDM3B largely abolished the increased expression of FAM111A caused by FAM111A‐DT overexpression (Figure 6I,J). In the context of depletion of other nuclear m6A readers HNRNPA2B1, HNRNPG, and HNRNPC, FAM111A‐DT overexpression also increased the expression of FAM111A (Figure S3E–G). These data supported that the positive regulation of FAM111A by FAM111A‐DT was dependent on YTHDC1 and KDM3B. Collectively, these findings showed that m6A‐modified FAM111A‐DT bound to FAM111A promoter, and also bound and recruited YTHDC1 and KDM3B to FAM111A promoter, inducing H3K9me2 demethylation and activating FAM111A expression (Figure 6K). The TCGA‐LIHC data revealed that FAM111A expression was positively correlated with METTL3, METTL14, WTAP, YTHDC1, and KDM3B expression in HCC tissues (Figure S4A–E), further supporting the molecular mechanisms underlying the positive modulation of FAM111A by FAM111A‐DT. Several m6A modification sites were predicted in FAM111A mRNA. Thus, we further investigated whether m6A modification also regulated FAM111A mRNA stability. After the blockage of new RNA synthesis using α‐amanitin, the degradation of FAM111A mRNA was detected. The results revealed that overexpression of METTL3, METTL14, or FTO had no effects on the degradation of FAM111A mRNA (Figure S4F).
FIGURE 6.
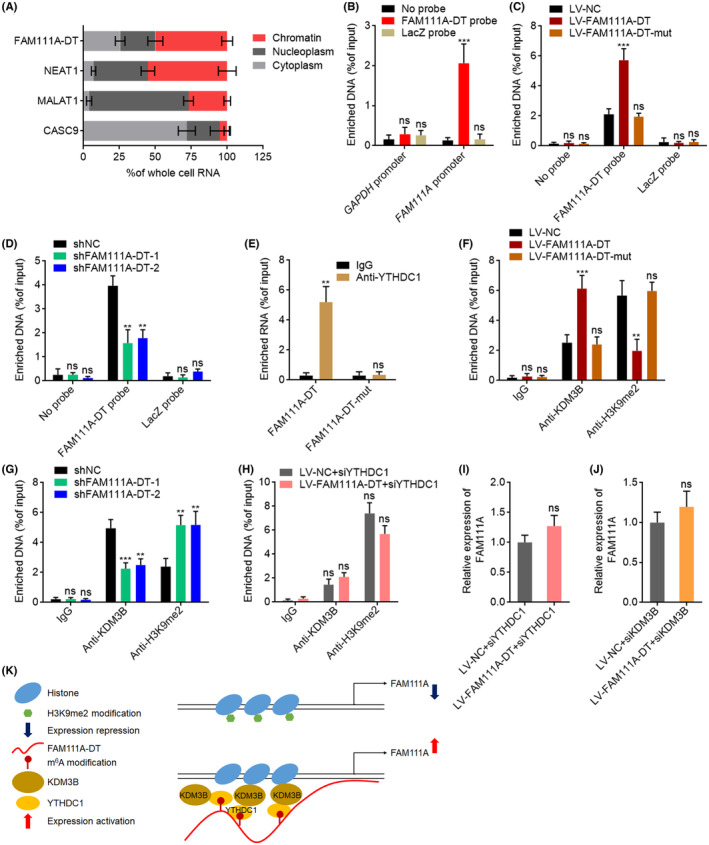
N6‐methyladenosine (m6A)‐modified FAM111A‐DT directed the demethylation of H3K9me2 at the FAM111A promoter region. (A) Subcellular localization of FAM111A‐DT and control genes analyzed with qPCR in biochemically fractionated SK‐HEP‐1 cells. (B) Chromatin isolation by RNA purification (ChIRP) assays with FAM111A‐DT antisense probes or control probes were performed in SK‐HEP‐1 cells to detect the binding of FAM111A‐DT to the FAM111A promoter region. (C) ChIRP assays with FAM111A‐DT antisense probes or control probes were performed in SK‐HEP‐1 cells with wild‐type or mutated FAM111A‐DT overexpression to detect the binding of FAM111A‐DT to the FAM111A promoter region. (D) ChIRP assays with FAM111A‐DT antisense probes or control probes were performed in SNU‐398 cells with FAM111A‐DT knockdown to detect the binding of FAM111A‐DT to the FAM111A promoter region. (E) RIP assays with YTHDC1‐specific antibody were performed in SK‐HEP‐1 cells with wild‐type or mutated FAM111A‐DT overexpression to detect the binding of YTHDC1 to wild‐type or mutated FAM111A‐DT. (F) ChIP assays with KDM3B‐ or H3K9me2‐specific antibodies were performed in SK‐HEP‐1 cells with wild‐type or mutated FAM111A‐DT overexpression to detect the binding of KDM3B to the FAM111A promoter region and the H3K9me2 modification levels at the FAM111A promoter region. (G) ChIP assays with KDM3B‐ or H3K9me2‐specific antibodies were performed in SNU‐398 cells with FAM111A‐DT knockdown to detect the binding of KDM3B to the FAM111A promoter region and the H3K9me2 modification levels at the FAM111A promoter region. (H) ChIP assays with KDM3B‐ or H3K9me2‐specific antibodies were performed in SK‐HEP‐1 cells with FAM111A‐DT overexpression and YTHDC1 depletion to detect the binding of KDM3B to the FAM111A promoter region and the H3K9me2 modification levels at the FAM111A promoter region. (I) The expression of FAM111A in SK‐HEP‐1 cells with FAM111A‐DT overexpression and YTHDC1 depletion was measured by qPCR. (J) The expression of FAM111A in SK‐HEP‐1 cells with FAM111A‐DT overexpression and KDM3B depletion was measured by qPCR. (K) Schematic of the modulatory mechanisms of m6A‐modified FAM111A‐DT on FAM111A expression. Results are shown as mean ± SD of n = 3 independent experiments. **p < 0.01, ***p < 0.001, ns, not significant, by one‐way ANOVA followed by Dunnett's multiple comparisons test (B–D, F, G) or Student's t‐test (E, H–J).
3.7. The expression of FAM111A was increased in HCC and correlated with poor survival of HCC patients
Consistent with the clinical significances of FAM111A‐DT in HCC, the TCGA‐LIHC data showed that the expression of FAM111A was also increased in HCC tissues compared with normal liver tissues (Figure S5A). Analysis of TCGA‐LIHC data by Kaplan–Meier Plotter showed that increased expression of FAM111A was also correlated with poor overall survival of HCC patients (Figure S5B). In our HCC cohort, we also found that the expression of FAM111A was increased in HCC tissues (Figure S5C), and increased expression of FAM111A was also correlated with poor overall survival of HCC patients (Figure S5D).
3.8. FAM111A‐DT promoted HCC cellular proliferation and DNA replication
FAM111A was known to promote S‐phase entry and DNA replication. 46 TCGA‐LIHC patients were classified into FAM111A high‐ and low‐expression groups, or FAM111A‐DT high‐ and low‐expression groups. TCGA‐LIHC data were subjected to Gene set enrichment analysis (GSEA), which showed that the genes in DNA replication pathways were significantly enriched in the FAM111A high‐expression group (Figure 7A). Consistent with FAM111A, the genes in DNA replication pathways were also significantly enriched in the FAM111A‐DT high‐expression group (Figure 7A), which suggested that FAM111A‐DT may also participate in DNA replication. Therefore, we further investigated the roles of FAM111A‐DT in HCC cellular proliferation and DNA replication. CCK‐8 assays showed that HCC cells with overexpression of wild‐type FAM111A‐DT, but not m6A modification sites‐mutated FAM111A‐DT, showed faster cell proliferation compared with control HCC cells (Figure 7B,C). HCC cells with FAM111A‐DT stable knockdown showed slower cell proliferation compared with control HCC cells (Figure 7D,E). EdU incorporation assays were performed to evaluate DNA replication. HCC cells with overexpression of wild‐type FAM111A‐DT, but not mutated FAM111A‐DT, had more EdU incorporation (Figure 7F), which indicated quicker DNA replication. HCC cells with FAM111A‐DT knockdown had less EdU incorporation (Figure 7G), which indicated slower DNA replication.
FIGURE 7.
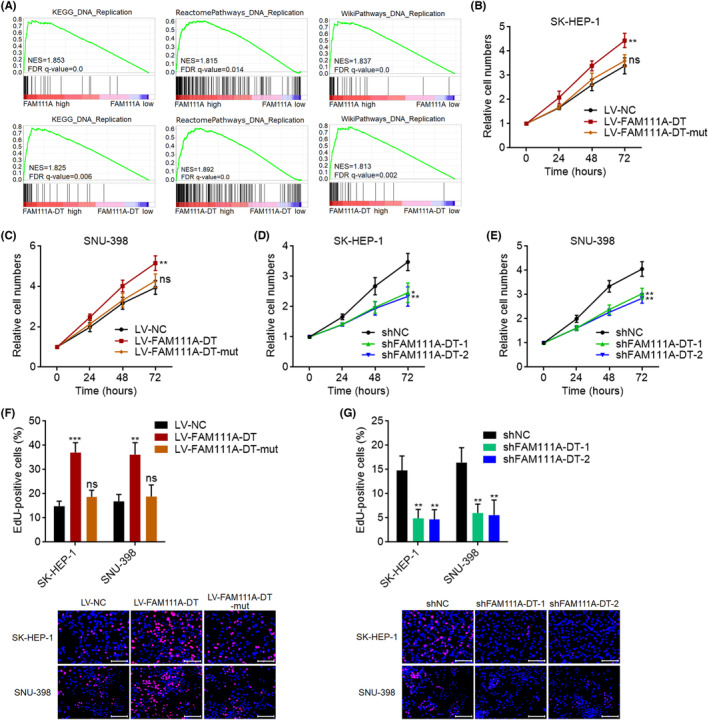
FAM111A‐DT promoted hepatocellular carcinoma (HCC) cellular proliferation and DNA replication. (A) GSEA of DNA replication gene signatures in the FAM111A high‐expression group versus FAM111A low‐expression group, or FAM111A‐DT high‐expression group versus FAM111A‐DT low‐expression group. NES, normalized enrichment score. (B, C) Cellular proliferation of SK‐HEP‐1 (B) or SNU‐398 (C) cells with wild‐type or mutated FAM111A‐DT stable overexpression was detected by CCK‐8 assays. (D, E) Cellular proliferation of SK‐HEP‐1 (D) or SNU‐398 (E) cells with FAM111A‐DT stable knockdown was detected by CCK‐8 assays. (F) DNA replication of SK‐HEP‐1 and SNU‐398 cells with wild‐type or mutated FAM111A‐DT stable overexpression was detected by EdU incorporation assays. Scale bars, 100 μm. (G) DNA replication of SK‐HEP‐1 and SNU‐398 cells with FAM111A‐DT stable knockdown was detected by EdU incorporation assays. Scale bars, 100 μm. Results are shown as mean ± SD of n = 3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant, by one‐way ANOVA followed by Dunnett's multiple comparisons test.
3.9. FAM111A‐DT promoted HCC tumor growth in vivo
We next assessed the roles of FAM111A‐DT in HCC tumor growth in vivo. Wild‐type or mutated FAM111A‐DT stably overexpressed SK‐HEP‐1 cells were subcutaneously injected into the flanks of nude mice. Subcutaneous tumor growth curves showed that ectopic expression of wild‐type, but not mutated, FAM111A‐DT significantly promoted tumor growth (Figure 8A). At the 28th day after injection, SK‐HEP‐1 cells with wild‐type, but not mutated, FAM111A‐DT overexpression formed heavier and larger tumors than control SK‐HEP‐1 cells (Figure 8B,C). Furthermore, SNU‐398 cells with FAM111A‐DT stable knockdown were subcutaneously injected into the flanks of nude mice. FAM111A‐DT knockdown significantly repressed subcutaneous tumor growth (Figure 8D–F). Proliferation marker Ki67 and PCNA IHC staining showed that the subcutaneous tumors formed by wild‐type, but not mutated, FAM111A‐DT‐overexpressed cells had more Ki67‐ and PCNA‐positive cells, while FAM111A‐DT knockdown reduced the number of Ki67‐ and PCNA‐positive cells (Figure 8G–J).
FIGURE 8.
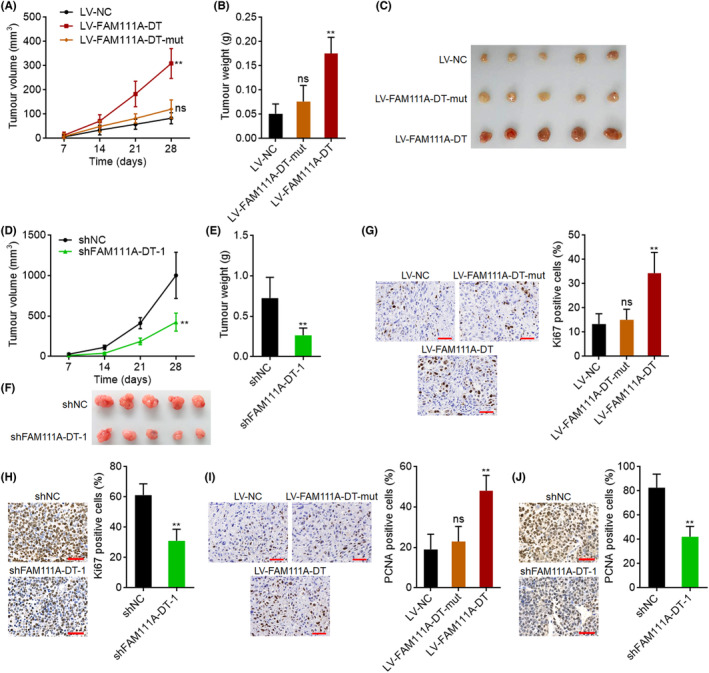
FAM111A‐DT promoted hepatocellular carcinoma (HCC) tumor growth in vivo. (A–C) Tumor volume (A), weight (B), and photograph (C) of subcutaneous tumors formed by SK‐HEP‐1 cells with wild‐type or mutated FAM111A‐DT stable overexpression. (D–F) Tumor volume (D), weight (E), and photograph (F) of subcutaneous tumors formed by SNU‐398 cells with FAM111A‐DT stable knockdown. (G) Ki67 immunohistochemistry (IHC) staining of subcutaneous tumors derived from (C). Scale bars, 50 μm. (H) Ki67 IHC staining of subcutaneous tumors derived from (F). Scale bars, 50 μm. (I) Proliferating cell nuclear antigen (PCNA) IHC staining of subcutaneous tumors derived from (C). Scale bars, 50 μm. (J) PCNA IHC staining of subcutaneous tumors derived from (F). Scale bars, 50 μm. Results are shown as mean ± SD of n = 5 mice in each group. **p < 0.01, ns, not significant, by Kruskal–Wallis test followed by Dunn's multiple comparisons test (A, B, G, I) or Mann–Whitney test (D, E, H, J).
3.10. FAM111A was essential for the roles of FAM111A‐DT in HCC
To assess whether the roles of FAM111A‐DT in HCC were dependent on FAM111A, we depleted FAM111A in FAM111A‐DT‐overexpressed SK‐HEP‐1 cells. CCK‐8 assays showed that depletion of FAM111A reversed the quicker cell proliferation caused by FAM111A‐DT overexpression (Figure S6A). EdU incorporation assays showed that depletion of FAM111A reversed the increased EdU incorporation caused by FAM111A‐DT overexpression (Figure S6B), suggesting that depletion of FAM111A reversed the quicker DNA replication caused by FAM111A‐DT overexpression. Furthermore, we overexpressed FAM111A in SNU‐398 cells with FAM111A‐DT knockdown. CCK‐8 assays showed that FAM111A overexpression rescued the cell proliferation repressed by FAM111A‐DT knockdown (Figure S6C). EdU incorporation assays showed that FAM111A overexpression rescued the decreased EdU incorporation caused by FAM111A‐DT knockdown (Figure S6D), suggesting that FAM111A overexpression reversed the slower DNA replication caused by FAM111A‐DT knockdown.
4. DISCUSSION
Here, we identified a novel aberrant m6A modification event in HCC and confirmed the specific m6A modification sites on FAM111A‐DT. We found that m6A modification level of FAM111A‐DT was increased in HCC tissues and cell lines. Increased m6A modification level of FAM111A‐DT was correlated with poor clinical outcome of HCC patients. Our study suggested that m6A modification of RNAs may be potential prognostic biomarkers for HCC.
The major contribution of m6A on RNAs is the regulation of stability or degradation of modified RNAs. 47 Here, we also found that m6A modification increased the stability and decreased the degradation of FAM111A‐DT transcript. The expression of FAM111A‐DT was positively correlated with the m6A modification level of FAM111A‐DT and also positively correlated with the expression of the METTL3‐METTL14‐WTAP methyltransferase complex in HCC tissues, supporting the positive regulation of FAM111A‐DT expression by m6A. Consistent with the clinical relevance of the m6A modification level of FAM111A‐DT in HCC, the expression of FAM111A‐DT was also increased in HCC tissues and cell lines and correlated with poor clinical outcome of HCC patients. Although FAM111A‐DT has been reported to be upregulated and associated with poor prognosis in thyroid carcinoma, 48 further investigations are needed to elucidate whether FAM111A‐DT is a general cancer‐related lncRNA or an HCC‐specific lncRNA.
Functional investigations found that only m6A‐modified FAM111A‐DT promoted HCC cellular proliferation, DNA replication, and HCC tumor growth. Mutation of the m6A modification sites abolished the roles of FAM111A‐DT, indicating that m6A also influences the function of modified RNAs. Mechanistic explorations identified FAM111A as the downstream target of m6A‐modified FAM111A‐DT. FAM111A‐DT was found to directly bound to the FAM111A promoter region. As an m6A reader, YTHDC1 bound to m6A‐modified FAM111A‐DT. YTHDC1 further bound and recruited KDM3B to the FAM111A promoter region. KDM3B is a lysine‐specific demethylase, which demethylates Lys‐9 of histone H3 (H3K9). 49 H3K9me2 is a repressive histone mark. 50 The recruitment of YTHDC1 and KDM3B by m6A‐modified FAM111A‐DT to the FAM111A promoter region induced demethylation and the reduction of H3K9me2 level at the FAM111A promoter region, leading to the transcriptional activation of FAM111A. Thus, our study provides a link between epitranscriptomic m6A modification and epigenetic regulation of gene transcription. The major effects of m6A modification were changing pre‐mRNA splicing, RNA stability, degradation, and translation. 51 Only a few studies found that some m6A modifications regulate transcription through histone modification and DNA demethylation, relying on different m6A readers. 45 , 52 The expression of FAM111A was positively correlated with the m6A modification level of FAM111A‐DT and also positively correlated with the expression of the METTL3‐METTL14‐WTAP methyltransferase complex, YTHDC1, and KDM3B in HCC tissues, supporting the m6A‐modified FAM111A‐DT/YTHDC1/KDM3B/FAM111A regulatory axis. Although several m6A modification sites were also predicted in FAM111A transcript, m6A modification did not post‐transcriptionally regulate FAM111A transcript stability. FAM111A is a single‐stranded DNA‐binding serine protease, which promotes DNA synthesis. 53 FAM111A was also reported as a replication factor needed for PCNA loading during DNA replication. 46 Through protecting replication forks, FAM111A was revealed to promote cell survival after drug treatment. 54 FAM111A was also reported to be associated with poor prognosis of diffuse lower‐grade glioma, the risk of aggressive prostate cancer, and distal metastases of cervical cancer. 55 , 56 , 57 Here, we also found that FAM111A was increased in HCC and correlated with poor survival of HCC patients. FAM111A showed oncogenic roles in HCC, which was also essential for the roles of FAM111A‐DT in HCC.
Taken together, this study identified a new aberrant m6A modification event‐increased m6A modification of FAM111A‐DT in HCC, which was correlated with prognosis of HCC patients. m6A‐modified FAM111A‐DT promoted HCC cell growth and DNA replication through epigenetically activating FAM111A, highlighting the m6A‐modified FAM111A‐DT/YTHDC1/KDM3B/FAM111A regulatory axis as a potential therapeutic target for HCC.
AUTHOR CONTRIBUTIONS
HW, JP, and XW designed the study. XW, ZX, JP, HW, YH, JN, MY, QF, ZH, and GL performed the experiments. JP, HW, XW, ZX, QW, and JW analyzed the data. HW, JP, ZX, and XW are the major contributors in writing the manuscript. All authors read and approved the final manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
ETHICS STATEMENTS
Approval of the research protocol by an Institutional Review Board: Affiliated Hospital of Youjiang Medical University for Nationalities Institutional Review Board (approval no. YYFY‐LL‐2022‐103).
Informed Consent: Written informed consent was obtained from each patient.
Registry and the Registration No. of the study/trial: N/A.
Animal Studies: The animal research in our study was approved by the Institutional Review Board of the Affiliated Hospital of Youjiang Medical University for Nationalities.
Supporting information
Figures S1–S6
Table S1
Table S2
ACKNOWLEDGMENTS
This work was supported by the Guangxi Natural Science Foundation Project (2020GXNSFAA259019, 2019GXNSFBA245023) and Guangxi Science and Technology Project—Science and Technology Base and Talents Special Project (2021AC20006).
Pu J, Xu Z, Huang Y, et al. N6 ‐methyladenosine‐modified FAM111A‐DT promotes hepatocellular carcinoma growth via epigenetically activating FAM111A . Cancer Sci. 2023;114:3649‐3665. doi: 10.1111/cas.15886
Jian Pu and Zuoming Xu contributed equally to this study.
Contributor Information
Xianjian Wu, Email: xianjian_wu@sina.com.
Huamei Wei, Email: weihuamei@yeah.net.
DATA AVAILABILITY STATEMENT
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- 1. Villanueva A. Hepatocellular carcinoma. N Engl J Med. 2019;380:1450‐1462. [DOI] [PubMed] [Google Scholar]
- 2. Jemal A, Ward EM, Johnson CJ, et al. Annual report to the nation on the status of cancer, 1975‐2014, featuring survival. J Natl Cancer Inst. 2017;109:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schulze K, Imbeaud S, Letouze E, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47:505‐511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fujimoto A, Totoki Y, Abe T, et al. Whole‐genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44:760‐764. [DOI] [PubMed] [Google Scholar]
- 5. Fujimoto A, Furuta M, Totoki Y, et al. Whole‐genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat Genet. 2016;48:500‐509. [DOI] [PubMed] [Google Scholar]
- 6. Nomoto S, Kinoshita T, Kato K, et al. Hypermethylation of multiple genes as clonal markers in multicentric hepatocellular carcinoma. Br J Cancer. 2007;97:1260‐1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sengupta I, Mondal P, Sengupta A, et al. Epigenetic regulation of Fructose‐1,6‐bisphosphatase 1 by host transcription factor speckled 110 kDa during hepatitis B virus infection. FEBS J. 2022;289:6694‐6713. [DOI] [PubMed] [Google Scholar]
- 8. Wen J, Huang Z, Wei Y, et al. Hsa‐microRNA‐27b‐3p inhibits hepatocellular carcinoma progression by inactivating transforming growth factor‐activated kinase‐binding protein 3/nuclear factor kappa B signalling. Cell Mol Biol Lett. 2022;27:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yan W, Han Q, Gong L, et al. MBD3 promotes hepatocellular carcinoma progression and metastasis through negative regulation of tumour suppressor TFPI2. Br J Cancer. 2022;127:612‐623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mudbhary R, Hoshida Y, Chernyavskaya Y, et al. UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell. 2014;25:196‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yuan JH, Yang F, Chen BF, et al. The histone deacetylase 4/SP1/microrna‐200a regulatory network contributes to aberrant histone acetylation in hepatocellular carcinoma. Hepatology. 2011;54:2025‐2035. [DOI] [PubMed] [Google Scholar]
- 12. Li J, Li MH, Wang TT, et al. SLC38A4 functions as a tumour suppressor in hepatocellular carcinoma through modulating Wnt/beta‐catenin/MYC/HMGCS2 axis. Br J Cancer. 2021;125:865‐876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shao YY, Chen PS, Lin LI, et al. Low miR‐10b‐3p associated with sorafenib resistance in hepatocellular carcinoma. Br J Cancer. 2022;126:1806‐1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang X, Jiang Q, Li J, et al. KCNQ1OT1 promotes genome‐wide transposon repression by guiding RNA‐DNA triplexes and HP1 binding. Nat Cell Biol. 2022;24:1617‐1629. [DOI] [PubMed] [Google Scholar]
- 15. Yuan JH, Yang F, Wang F, et al. A long noncoding RNA activated by TGF‐beta promotes the invasion‐metastasis cascade in hepatocellular carcinoma. Cancer Cell. 2014;25:666‐681. [DOI] [PubMed] [Google Scholar]
- 16. Li G, Kryczek I, Nam J, et al. LIMIT is an immunogenic lncRNA in cancer immunity and immunotherapy. Nat Cell Biol. 2021;23:526‐537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li JK, Chen C, Liu JY, et al. Long noncoding RNA MRCCAT1 promotes metastasis of clear cell renal cell carcinoma via inhibiting NPR3 and activating p38‐MAPK signaling. Mol Cancer. 2017;16:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lu L, Huang J, Mo J, et al. Exosomal lncRNA TUG1 from cancer‐associated fibroblasts promotes liver cancer cell migration, invasion, and glycolysis by regulating the miR‐524‐5p/SIX1 axis. Cell Mol Biol Lett. 2022;27:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li Y, Ding T, Hu H, et al. LncRNA‐ATB participates in the regulation of calcium oxalate crystal‐induced renal injury by sponging the miR‐200 family. Mol Med. 2021;27:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wei H, Xu Z, Chen L, et al. Long non‐coding RNA PAARH promotes hepatocellular carcinoma progression and angiogenesis via upregulating HOTTIP and activating HIF‐1alpha/VEGF signaling. Cell Death Dis. 2022;13:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pu J, Zhang Y, Wang A, et al. ADORA2A‐AS1 restricts hepatocellular carcinoma progression via binding HuR and repressing FSCN1/AKT axis. Front Oncol. 2021;11:754835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pu J, Li W, Wang A, et al. Long non‐coding RNA HOMER3‐AS1 drives hepatocellular carcinoma progression via modulating the behaviors of both tumor cells and macrophages. Cell Death Dis. 2021;12:1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhu XT, Yuan JH, Zhu TT, Li YY, Cheng XY. Long noncoding RNA glypican 3 (GPC3) antisense transcript 1 promotes hepatocellular carcinoma progression via epigenetically activating GPC3. FEBS J. 2016;283:3739‐3754. [DOI] [PubMed] [Google Scholar]
- 24. Yuan JH, Liu XN, Wang TT, et al. The MBNL3 splicing factor promotes hepatocellular carcinoma by increasing PXN expression through the alternative splicing of lncRNA‐PXN‐AS1. Nat Cell Biol. 2017;19:820‐832. [DOI] [PubMed] [Google Scholar]
- 25. Liu Y, Zhou J, Li X, et al. tRNA‐m1A modification promotes T cell expansion via efficient MYC protein synthesis. Nat Immunol. 2022;23:1433‐1444. [DOI] [PubMed] [Google Scholar]
- 26. Wu Y, Xu X, Qi M, et al. N6‐methyladenosine regulates maternal RNA maintenance in oocytes and timely RNA decay during mouse maternal‐to‐zygotic transition. Nat Cell Biol. 2022;24:917‐927. [DOI] [PubMed] [Google Scholar]
- 27. Paramasivam A, Priyadharsini JV. RNA N6‐methyladenosine: a new player in autophagy‐mediated anti‐cancer drug resistance. Br J Cancer. 2021;124:1621‐1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cheng Y, Xie W, Pickering BF, et al. N6‐Methyladenosine on mRNA facilitates a phase‐separated nuclear body that suppresses myeloid leukemic differentiation. Cancer Cell. 2021;39:958‐972.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Boulias K, Greer EL. Biological roles of adenine methylation in RNA. Nat Rev Genet. 2022;24:143‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang L, Yi X, Xiao X, Zheng Q, Ma L, Li B. Exosomal miR‐628‐5p from M1 polarized macrophages hinders m6A modification of circFUT8 to suppress hepatocellular carcinoma progression. Cell Mol Biol Lett. 2022;27:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dai YZ, Liu YD, Li J, et al. METTL16 promotes hepatocellular carcinoma progression through downregulating RAB11B‐AS1 in an m6A‐dependent manner. Cell Mol Biol Lett. 2022;27:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xu W, He C, Kaye EG, et al. Dynamic control of chromatin‐associated m6A methylation regulates nascent RNA synthesis. Mol Cell. 2022;82:1156‐1168.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ito‐Kureha T, Leoni C, Borland K, et al. The function of Wtap in N6‐adenosine methylation of mRNAs controls T cell receptor signaling and survival of T cells. Nat Immunol. 2022;23:1208‐1221. [DOI] [PubMed] [Google Scholar]
- 34. Dong L, Chen C, Zhang Y, et al. The loss of RNA N6‐adenosine methyltransferase Mettl14 in tumor‐associated macrophages promotes CD8+ T cell dysfunction and tumor growth. Cancer Cell. 2021;39:945‐957.e10. [DOI] [PubMed] [Google Scholar]
- 35. Wei X, Huo Y, Pi J, et al. METTL3 preferentially enhances non‐m6A translation of epigenetic factors and promotes tumourigenesis. Nat Cell Biol. 2022;24:1278‐1290. [DOI] [PubMed] [Google Scholar]
- 36. Sun W, Li Y, Ma D, et al. ALKBH5 promotes lung fibroblast activation and silica‐induced pulmonary fibrosis through miR‐320a‐3p and FOXM1. Cell Mol Biol Lett. 2022;27:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Weng H, Huang F, Yu Z, et al. The m6A reader IGF2BP2 regulates glutamine metabolism and represents a therapeutic target in acute myeloid leukemia. Cancer Cell. 2022;40:1566‐1582.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ma S, Sun B, Duan S, et al. YTHDF2 orchestrates tumor‐associated macrophage reprogramming and controls antitumor immunity through CD8+ T cells. Nat Immunol. 2023;24:255‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen B, Yang Z, Lang Z, et al. M6A‐related lncRNAs predict clinical outcome and regulate the tumor immune microenvironment in hepatocellular carcinoma. BMC Cancer. 2022;22:867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Klingenberg M, Gross M, Goyal A, et al. The long noncoding RNA cancer susceptibility 9 and RNA binding protein heterogeneous nuclear ribonucleoprotein L form a complex and coregulate genes linked to AKT signaling. Hepatology. 2018;68:1817‐1832. [DOI] [PubMed] [Google Scholar]
- 41. Xiao Y, Wang Y, Tang Q, Wei L, Zhang X, Jia G. An elongation‐ and ligation‐based qPCR amplification method for the radiolabeling‐free detection of locus‐specific N6‐methyladenosine modification. Angew Chem Int Ed Engl. 2018;57:15995‐16000. [DOI] [PubMed] [Google Scholar]
- 42. Menyhart O, Nagy A, Gyorffy B. Determining consistent prognostic biomarkers of overall survival and vascular invasion in hepatocellular carcinoma. R Soc Open Sci. 2018;5:181006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhou Y, Zeng P, Li YH, Zhang Z, Cui Q. SRAMP: prediction of mammalian N6‐methyladenosine (m6A) sites based on sequence‐derived features. Nucleic Acids Res. 2016;44:e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xuan JJ, Sun WJ, Lin PH, et al. RMBase v2.0: deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res. 2018;46:D327‐D334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li Y, Xia L, Tan K, et al. N6‐Methyladenosine co‐transcriptionally directs the demethylation of histone H3K9me2. Nat Genet. 2020;52:870‐877. [DOI] [PubMed] [Google Scholar]
- 46. Alabert C, Bukowski‐Wills JC, Lee SB, et al. Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat Cell Biol. 2014;16:281‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Murakami S, Jaffrey SR. Hidden codes in mRNA: control of gene expression by m6A. Mol Cell. 2022;82:2236‐2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang Y, Jin T, Shen H, Yan J, Guan M, Jin X. Identification of long non‐coding RNA expression profiles and co‐expression genes in thyroid carcinoma based on the cancer genome atlas (TCGA) database. Med Sci Monit. 2019;25:9752‐9769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhu Z, Wu X, Li Q, et al. Histone demethylase complexes KDM3A and KDM3B cooperate with OCT4/SOX2 to define a pluripotency gene regulatory network. FASEB J. 2021;35:e21664. [DOI] [PubMed] [Google Scholar]
- 50. Filion GJ, van Steensel B. Reassessing the abundance of H3K9me2 chromatin domains in embryonic stem cells. Nat Genet. 2010;42:4; author reply 5–6. [DOI] [PubMed] [Google Scholar]
- 51. Niu X, Yang Y, Ren Y, Zhou S, Mao Q, Wang Y. Crosstalk between m6A regulators and mRNA during cancer progression. Oncogene. 2022;41:4407‐4419. [DOI] [PubMed] [Google Scholar]
- 52. Deng S, Zhang J, Su J, et al. RNA m6A regulates transcription via DNA demethylation and chromatin accessibility. Nat Genet. 2022;54:1427‐1437. [DOI] [PubMed] [Google Scholar]
- 53. Welter AL, Machida YJ. Functions and evolution of FAM111 serine proteases. Front Mol Biosci. 2022;9:1081166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kojima Y, Machida Y, Palani S, et al. FAM111A protects replication forks from protein obstacles via its trypsin‐like domain. Nat Commun. 2020;11:1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ji X, Ding F, Gao J, et al. Molecular and clinical characterization of a novel prognostic and immunologic biomarker FAM111A in diffuse lower‐grade glioma. Front Oncol. 2020;10:573800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schaid DJ, McDonnell SK, FitzGerald LM, et al. Two‐stage study of familial prostate cancer by whole‐exome sequencing and custom capture identifies 10 novel genes associated with the risk of prostate cancer. Eur Urol. 2021;79:353‐361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fernandez‐Retana J, Zamudio‐Meza H, Rodriguez‐Morales M, et al. Gene signature based on degradome‐related genes can predict distal metastasis in cervical cancer patients. Tumour Biol. 2017;39:1010428317711895. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S6
Table S1
Table S2
Data Availability Statement
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.