

Abstract
Langerhans cell histiocytosis (LCH) is a rare disease characterized by clonal expansion of CD1a+CD207+ myeloid dendritic cells. The features of LCH are mainly described in children and remain poorly defined in adults; therefore, we conducted a nationwide survey to collect clinical data from 148 adult patients with LCH. The median age at diagnosis was 46.5 (range: 20–87) years with male predominance (60.8%). Among the 86 patients with detailed treatment information, 40 (46.5%) had single system LCH, whereas 46 (53.5%) had multisystem LCH. Moreover, 19 patients (22.1%) had an additional malignancy. BRAF V600E in plasma cell‐free DNA was associated with a low overall survival (OS) rate and the risk of the pituitary gland and central nervous system involvement. At a median follow‐up of 55 months from diagnosis, six patients (7.0%) had died, and the four patients with LCH‐related death did not respond to initial chemotherapy. The OS probability at 5 years post‐diagnosis was 90.6% (95% confidence interval: 79.8–95.8). Multivariate analysis showed that patients aged ≥60 years at diagnosis had a relatively poor prognosis. The probability of event‐free survival at 5 years was 52.1% (95% confidence interval: 36.6–65.5), with 57 patients requiring chemotherapy. In this study, we first revealed the high rate of relapse after chemotherapy and mortality of poor responders in adults as well as children. Therefore, prospective therapeutic studies of adults with LCH using targeted therapies are needed to improve outcomes in adults with LCH.
Keywords: adult, BRAF mutation, Langerhans cell histiocytosis, nationwide survey, prognosis
In this study, we first revealed the high rate of relapse after chemotherapy and mortality of poor responders in adults as well as children. BRAF V600E in plasma cell‐free DNA was associated with a low overall survival rate. Multivariate analysis showed that patients aged ≥60 years at diagnosis had a poor prognosis. Therefore, further exploration of gene mutation and prospective therapeutic studies using targeted therapies could help elucidate the pathogenesis and improve the treatment of adults with LCH.
Abbreviations
- cfDNA
cell‐free DNA
- CNS
central nervous system
- DI
diabetes insipidus
- ltBRAF V600E
BRAF V600E mutation in lesion tissues
- LCH
Langerhans cell histiocytosis
- LCH‐ND
LCH‐associated neurodegeneration
- MS‐LCH
multisystem LCH
- NAD
no active disease
- PG
pituitary gland
- plasma cfBRAF V600E
BRAF V600E mutation in plasma cfDNA
- PLCH
primary pulmonary LCH
- RO
risk organ
- SSm‐LCH
single system with multi‐site LCH
- SSs‐LCH
single system with single‐site LCH
1. INTRODUCTION
Langerhans cell histiocytosis is a rare inflammatory myeloid neoplasm characterized by the infiltration of CD1a+CD207+ myeloid dendritic cells and immune cells 1 and is thus defined as a clonally expanding inflammatory myeloid neoplasm. The incidence of LCH is estimated at three to five cases per million children, 2 and one to two cases per million adults. 3 Badalian‐Very et al. 4 identified the BRAF V600E mutation in more than 50% of LCH cases. In 2016, the revised classification of the Working Group of the Histiocyte Society grouped histiocytic disorders into five groups, 5 with the Langerhans “L” group, including LCH and Erdheim–Chester disease, characterized by clonal mutations in genes involved in the MAPK pathway. Treatment recommendations are based on the site and extension of the disease, including the following disease categories: (MS)‐LCH with or without an RO (hematopoietic system, liver, or spleen), SSm‐LCH, and (SS)‐LCH with a specific site lesion (i.e., vertebral lesions with intraspinal or craniofacial bone lesions exhibiting soft tissue extension).
In pediatric LCH, previous studies have reported that patients younger than 2 years with RO involvement (RO+) and patients with RO+ not responding to initial treatment within 6 weeks had a high mortality rate. 6 , 7 In addition, prospective and large‐scale randomized trials demonstrated that the risk‐adjusted, intensified, and longer treatments have improved the outcomes of pediatric high‐risk patients with MS‐LCH. 7 , 8 , 9 , 10 However, the high recurrence rate of LCH was noted in more than 30% of responders, and 30% of RO+ poor responders died of the disease. 8 , 10 Nevertheless, the specific risk factors of adult patients with LCH have not been determined to date. As only a few retrospective trials, including dozens of cases, have been reported, 11 , 12 , 13 , 14 , 15 there is no standard first‐line therapy. 16
Therefore, we conducted a nationwide survey and retrospectively analyzed the clinical features, treatment, and prognosis of 86 adult Japanese patients with LCH. This survey reveals adult‐specific characteristics that will provide new guidance to improve the ability to plan prospective therapeutic studies that will ultimately improve the outcomes of adult patients with LCH.
2. MATERIALS AND METHODS
2.1. Patients and clinical evaluation
We mailed a questionnaire to an educational hospital certified by the Japanese Society of Hematology and the Japanese Society of Pathology (Appendix S1). We retrospectively analyzed the data of patients referred to hospitals between January 2013 and December 2018. Based on the histopathological findings and immunohistochemical expression of CD1a and/or CD207, all patients diagnosed with LCH were aged ≥20 years. The clinical classification was made according to the management of adult patients with LCH using the Euro‐Histio‐Net criteria. 17 Additional malignancies were classified as diagnosed preceding, concurrent with (within 1 year), or after the LCH diagnosis.
This study was approved by the ethics committee of the Institutional Review Board of the Institute of Medical Science, University of Tokyo (approval numbers: 2019‐35‐1017, 2020‐49‐1119, and 2020–1‐0422) and was conducted according to the principles of the Declaration of Helsinki.
2.2. BRAF V600E analysis
Among the 35 tissue samples subjected to BRAF V600E immunohistochemistry analyses, 30 with available formalin‐fixed paraffin‐embedded blocks were analyzed at our institution. Immunohistochemistry was performed using an anti‐BRAF V600E mouse monoclonal antibody (clone VE1; Spring Bioscience Corp., Pleasanton, CA, USA). At our institution, cfDNA tests were performed. The samples were prepared from peripheral blood plasma and subjected to BRAF V600E genotyping by allele‐specific quantitative polymerase chain reaction, as described previously. 18 The plasma cfDNA and tissue DNA were evaluated by digital‐droplet polymerase chain reaction using a BRAF V600 Screening Kit (Bio‐Rad Laboratories, Hercules, CA, USA) to improve detection accuracy.
Genomic analyses were performed on DNA extracted from LCH tissue biopsies using whole‐exome sequencing. Details are available in the Supplementary Methods.
2.3. Treatment response analysis
At the end of treatment, patient responses were categorized as NAD, defined as the disappearance of the signs or symptoms of disease; partial response (PR), defined as a regression of signs or symptoms of disease without organ dysfunction and new lesions; stable disease (SD), defined as a persistence of signs or symptoms of disease with or without organ dysfunction and the absence of new lesions; or progressive disease (PD), defined as the progression of the signs or symptoms of disease and/or the appearance of new lesions.
2.4. Statistical analysis
All statistical analyses were performed using EZR software (Saitama Medical Center, Jichi Medical University, Saitama, Japan), a graphical user interface for R (R Foundation for Statistical Computing, Vienna, Austria). 19 The probability of event‐free survival (EFS) was calculated from the start of initial chemotherapy until disease progression or death. EFS and overall survival (OS) were estimated using the Kaplan–Meier method and compared using log‐rank analysis. The effects of various parameters on survival were evaluated by univariate and multivariate analyses using the Cox proportional hazards regression model. Factors with a p‐value < 0.05 in the univariate analysis were included in the multivariate analysis of survival. Fisher's exact test was used to compare the distribution of smokers among the patients with LCH and lung involvement and presence of BRAF V600E in cfDNA among the risk of PG and CNS involvement. Statistical significance was set at p < 0.05.
3. RESULTS
3.1. Study population
The questionnaire was sent to 483 hematology and 839 pathology departments, with response rates of 43.9% (n = 212) and 44.0% (n = 369 departments), respectively. We retrospectively studied 148 newly diagnosed cases between January 2013 and December 2018. Patient and clinical characteristics at diagnosis are shown in Table 1. Among the 148 patients, 90 were male (60.8%) and 58 were female (39.2%). The median age at diagnosis was 46.5 years (range: 20–87 years). Moreover, 56 patients (37.8%) were referred for local pain, including 24 (16.2%) for abnormal findings on imaging, 22 (14.9%) for palpable mass, and 21 (14.2%) for rash. In total, 33 (22.3%) patients were referred to the respiratory department, 33 (22.3%) to orthopedics, and 19 (12.8%) to dermatology. The histological diagnoses were made from the bone (n = 57, 38.5%), lung (n = 29, 19.6%), skin (n = 27, 18.2%), and lymph nodes (n = 13, 8.8%) biopsies.
TABLE 1.
Clinical characteristics of patients with Langerhans cell histiocytosis at diagnosis (n = 148).
| Characteristics | Data |
|---|---|
| Sex, n (%) | |
| Male | 90 (60.8) |
| Female | 58 (39.2) |
| Median age at diagnosis, year (range) | 46.5 (20–87) |
| Mean time for diagnosis, months (range) | 15 (0–378) |
| Initial manifestation, n (%) | |
| Local pain | 56 (37.8) |
| Abnormal findings on images | 24 (16.2) |
| Palpable mass | 22 (14.9) |
| Rash | 21 (14.2) |
| Respiratory symptoms | 17 (11.5) |
| Palpable lymph nodes | 10 (6.8) |
| Diabetes insipidus | 6 (4.1) |
| Fever | 6 (4.1) |
| Other | 8 (5.4) |
| Department on first admission, n (%) | |
| Respiratory | 33 (22.3) |
| Orthopedics | 33 (22.3) |
| Dermatology | 19 (12.8) |
| Neurosurgery/Neurology | 14 (9.5) |
| Hematology | 11 (7.4) |
| Endocrinology | 7 (4.7) |
| Oral surgery | 5 (3.4) |
| General internal medicine | 5 (3.4) |
| Gastroenterology | 4 (2.7) |
| Surgery | 4 (2.7) |
| Otorhinolaryngology | 4 (2.7) |
| Other | 9 (6.1) |
| Histological diagnoses, n (%) | |
| Bone | 57 (38.5) |
| Lung | 29 (19.6) |
| Skin | 27 (18.2) |
| Lymph node | 13 (8.8) |
| Soft tissue | 5 (3.4) |
| Liver | 4 (2.7) |
| Pituitary gland/CNS | 3 (2.0) |
| Thyroid gland | 3 (2.0) |
| Other | 7 (4.7) |
Abbreviation: CNS, central nervous system.
3.2. Organ involvement
Among the 86 patients with detailed treatment information, 10 (11.6%) had SSs‐LCH, 22 (25.6%) had SSm‐LCH, 8 (9.3%) had PLCH, and 46 (53.5%) had MS‐LCH (Table 2). SS‐LCH (e.g., SSs‐LCH, SSm‐LCH, and PLCH) occurred predominantly in young adults (Figure 1), with a median of two involved organs per patient (range: 1–8). The most frequently affected organs at diagnosis were the bone (60.5%), lungs (27.9%), PG (25.6%), lymph nodes (24.4%), and skin (23.3%). In total, 17 (19.8%) patients had RO involvement, and 22 patients (25.6%) had special site involvement. At diagnosis, DI, anterior pituitary dysfunction, and LCH‐ND were observed in 19, 11, and three patients, respectively. Last, 29 (29.1%) patients developed DI during their clinical course (Table 2).
TABLE 2.
Organ involvement of Langerhans cell histiocytosis at diagnosis (n = 86).
| Organ involvement | Data |
|---|---|
| Disease classification, n (%) | |
| SSs‐LCH | 10 (11.6) |
| SSm‐LCH | 22 (25.6) |
| PLCH | 8 (9.3) |
| MS‐LCH | 46 (53.5) |
| Median number of involved organs, n (range) | 2 (1–6) |
| Organ involvement, n (%) | |
| Bone | 52 (60.5) |
| Lung | 24 (27.9) |
| Pituitary gland | 22 (25.6) |
| Lymph node | 21 (24.4) |
| Skin | 20 (23.3) |
| Liver | 14 (16.3) |
| Central nervous system | 13 (15.1) |
| Soft tissue | 8 (9.3) |
| Other | 12 (14.0) |
| Risk organ involvement, n (%) | 17 (19.8) |
| Special site involvement, n (%) | 22 (25.6) |
| Permanent consequences, n (%) | |
| Diabetes insipidus | 19 (22.0) |
| Anterior pituitary dysfunction | 11 (12.8) |
| LCH‐associated neurodegeneration | 3 (3.5) |
Note: Risk organs: liver, spleen, or hematopoietic system; Special sites: vertebral lesions with intraspinal or craniofacial bone lesions with soft tissue extensions (orbit, mastoid, sphenoid, or temporal bones).
Abbreviations: MS‐LCH, multisystem LCH; PLCH, primary pulmonary LCH; SSm‐LCH, single system with multi‐site LCH; SSs‐LCH, single system with single‐site LCH.
FIGURE 1.
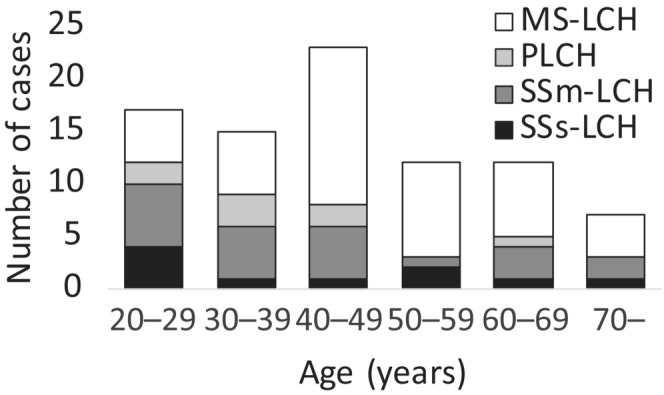
Age and the pattern of disease manifestation at diagnosis in 86 adult patients with Langerhans cell histiocytosis. MS‐LCH, multisystem LCH; PLCH, primary pulmonary LCH; SSm‐LCH, single system with multi‐site LCH; SSs‐LCH, single system with single‐site LCH.
3.3. Additional malignancies
There were 19 non‐LCH malignancies, with nine (47.3%) preceding, four concurrent with (within 1 year; 21.1%), and six (31.6%) occurring after the LCH diagnosis (Table S1). There were 12 (63.2%) solid tumors, three (15.8%) lymphoid malignancies, and four (21.0%) myeloid malignancies (Table S1).
3.4. Initial treatment
The initial therapeutic approaches according to the classification are shown in Table S2. Patients with SSs‐LCH were not treated with chemotherapy for the initial therapy and did not progress to MS‐LCH. Among the 22 patients with SSm‐LCH, eight received chemotherapy. Four patients with SSm‐LCH progressed to MS‐LCH. None of the eight patients with PLCH received chemotherapy at diagnosis, and six patients stopped smoking. PLCH developed into MS‐LCH in two patients. Among the 46 patients with MS, 33 received chemotherapy.
3.5. Initial chemotherapy
Of the 57 patients who required chemotherapy, three had SSs‐LCH, 11 had SSm‐LCH, and 43 had MS‐LCH at the time of chemotherapy administration. Within this cohort, 36 patients (63.1%) received the Japan LCH Study Group (JLSG) Special‐C regimen, 14 comprising nine cycles of 6 mg/m2 (maximum 6 mg) of vinblastine (VBL) on day 1; 2 mg/kg (maximum 60 mg) of oral prednisolone (PSL) on days 1–5; 20 mg/m2 methotrexate on day 15; and 1.5 mg/kg 6‐mercaptopurine on days 1–28. 14 In total, 10 (17.5%) patients received cytarabine (Ara‐C)‐containing regimens of 100 mg/m2 on days 1–5 every 4 weeks. 13 , 20 Three (5.3%) patients received the JLSG‐02 regimen. 10 Three (5.3%) patients received cladribine at 0.12–0.14 mg/kg on days 1–5 every 4 weeks. 11 The median number of lines of chemotherapy was 1 (range 1–4).
At the end of the initial chemotherapy treatment, 49 of the 57 patients had a clinical response (NAD: 33; PR, 16). Among the 53 patients with NAD, PR, and SD, 20 presented with disease recurrence (Table 3). The 57 patients who required systemic chemotherapy were evaluable for EFS at a median follow‐up time of 32 months (range: 1–186 months) after the initial chemotherapy. The probability of EFS at 5 years was 52.1% [95% confidence interval (CI): 36.6–65.5] (Figure 2A). Sex; age ≥60 at diagnosis; >0.3 mg/dL CRP level before therapy; MS‐LCH; regimen; and bone, lung, and CNS, including the pituitary gland, skin, lymph nodes, RO, and special site involvement did not have an effect on EFS (Figure 2B,C).
TABLE 3.
Treatment response and refractory/relapse according to initial chemotherapy regimen (n = 57).
| Regimen | n, (%) | Best response, n | Refractory/relapse, n | |||
|---|---|---|---|---|---|---|
| NAD | PR | SD | PD | |||
| Special‐C 14 | 36 (63.1) | 20 | 12 | 3 | 1 | 14 |
| Cytarabine 13 , 20 | 10 (17.5) | 6 | 3 | 0 | 1 | 5 |
| JLSG‐02 10 | 3 (5.3) | 3 | 0 | 0 | 0 | 0 |
| Cladribine 11 | 3 (5.3) | 1 | 0 | 0 | 2 | 2 |
| Other | 5 (8.8) | 3 | 1 | 1 | 0 | 3 |
| Total | 33 | 16 | 4 | 4 | 24 | |
Abbreviations: Special‐C, the Japan LCH study group Special‐C regimen, comprising vinblastine, prednisolone, methotrexate and 6‐mercaptopurine; JLSG‐02, the Japan LCH study group‐02 regimen; NAD, no active disease; PD, progression disease; PR, partial response; SD, stable disease.
FIGURE 2.
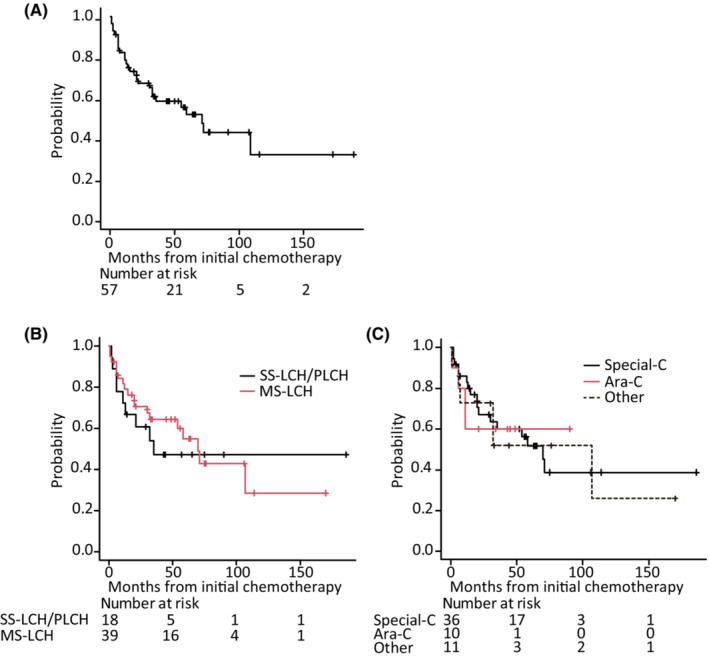
Event‐free survival (EFS) from initial chemotherapy. Kaplan–Meier estimation of EFS from initial chemotherapy of (A) all 57 patients based on (B) disease classification before initial chemotherapy, and (C) initial chemotherapy regimens. MS‐LCH, multisystem LCH; SS‐LCH/PLCH, single system LCH / primary pulmonary LCH; Special‐C, the Japan LCH study group Secial‐C regimen, comprising vinblastine, prednisolone, methotrexate and 6‐mercaptopurine; Ara‐C, cytarabine.
3.6. OS at follow‐up
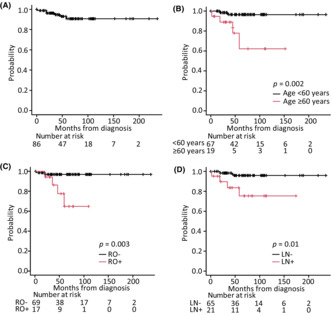
Overall survival was evaluated in 86 patients at a median follow‐up time of 55 months (range: 0–235 months). Six of these patients died during the clinical course of their illness, with the cause of death associated with LCH in four patients (Table 4), whereas the other two patients died of secondary AML or graft‐versus‐host disease. The probability of OS at 5 years was 90.6% (95% CI: 79.8–95.8) (Figure 3A). Kaplan–Meier estimates of 5‐year OS according to disease extension were 100% for SSs‐LCH (n = 10), 95.0% (95% CI: 69.5–99.3) for SSm‐LCH (n = 22), 100% for PLCH (n = 8), and 85.2% (95% CI: 67.0–93.8) for MS‐LCH (n = 46). Patients with MS‐LCH had poorer survival rates than those with SS‐LCH, although this difference was not significant. The log‐rank test revealed that ≥60 years at diagnosis (p = 0.002), RO+ (p = 0.003), and lymph node involvement (p = 0.01) were significant prognostic factors for LCH (Figure 3B–D). Univariate analyses with the Cox proportional hazards regression model revealed that age ≥60 at diagnosis (p = 0.0017), presence of lymph node involvement (p = 0.011), and RO+ (p = 0.0034) were significant poor prognostic factors for OS. The following factors did not affect survival: sex; >0.3 mg/dL CRP level at onset; and bone, lung, pituitary gland, skin, and special site involvement. Multivariate analysis showed that age ≥60 years at diagnosis (hazard ratio: 6.21; 95% CI: 1.12–34.61; p = 0.037) was correlated with shorter survival (Table 5).
TABLE 4.
Cause of death in patients with Langerhans cell histiocytosis included in the study.
| No. | Age | Sex | Disease classification | BRAF V600E | Organ involvement (all) | Initial treatment | Response | Additional malignancy | Cause of death | |
|---|---|---|---|---|---|---|---|---|---|---|
| Tissue | Plasma cfDNA | |||||||||
| IHC | PCR | |||||||||
| 1 | 45 | F | SSm‐LCH | − | + | PG, CNS | Specia‐C | PD | 39y PB | LCH |
| 2 | 61 | F | MS‐LCH | + | + | B, L, PG, LN, LV, ST, SP, DO, CA | 2CdA | PD | − | LCH |
| 3 | 79 | M | MS‐LCH | ND | ND | B, L, SP, BM | Special‐C | PR | 82y AML | Therapy‐related (2nd AML) |
| 4 | 72 | M | MS‐LCH | ND | ND | LN, SP, BM | Ara‐C | PD | − | LCH |
| 5 | 81 | M | MS‐LCH | ND | ND | B, LN | 2CdA | PD | 75y CRC | LCH |
| 6 | 34 | M | MS‐LCH | ND | ND | B, LN, LV, ST, SP, BM | Ara‐C | PR | − | Therapy‐related (GVHD) |
Abbreviations: 2CdA, cladribine; 2nd AML, secondary acute myeloid leukemia; Ara‐C, cytarabine; B, bone; BM, bone marrow; CA, cardiovascular; cfDNA, cell‐free DNA; CNS, central nervous system; CRC, colorectal cancer; DO, digestive organs excluding the liver; GVHD, graft‐versus‐host‐disease; IHC, immunohistochemistry; L, lung; LN, lymph nodes; LV, liver; MS‐LCH, multisystem LCH; ND, no data; PB, phyllodes tumor of the breast; PD, progressive disease; PG, pituitary gland; PR, partial response; SP, spleen; Special‐C, the Japan LCH Study Group Speial‐C regimen, comprising of vinblastine, prednisolone, methotrexate and 6‐mercaptopurine; SSm‐LCH, single system with multi‐site LCH; ST, soft tissue.
FIGURE 3.
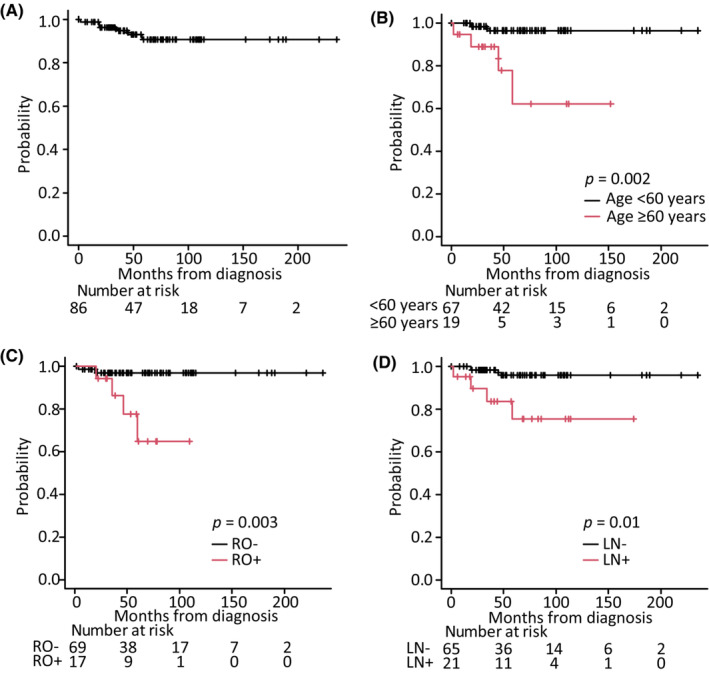
Overall survival (OS) of patients diagnosed with Langerhans cell histiocytosis (LCH). Kaplan–Meier estimation of OS from the LCH diagnosis of (A) all 86 patients based on (B) age, (C) presence of risk organ involvement, and (D) lymph node involvement. LN, lymph node involvement; RO, risk organ involvement.
TABLE 5.
Univariate and multivariate analyses for overall survival of patients with Langerhans cell histiocytosis.
| Number of patients | Univariate analysis | Multivariate analysis | |||
|---|---|---|---|---|---|
| p‐values | Hazard ratio (95% CI) | p‐values | |||
| Age, years | ≥60 | 19 | 0.0017 | 6.21 (1.12–34.61) | 0.037 |
| <60 | 67 | 1 | |||
| Sex | Male | 49 | 0.64 | ||
| Female | 37 | ||||
| Bone involvement | Yes | 52 | 0.92 | ||
| No | 34 | ||||
| Lung involvement | Yes | 24 | 0.78 | ||
| No | 62 | ||||
| Pituitary gland involvement | Yes | 22 | 0.66 | ||
| No | 64 | ||||
| Lymph node involvement | Yes | 21 | 0.011 | 3.87 (0.69–21.84) | 0.125 |
| No | 65 | 1 | |||
| Skin involvement | Yes | 20 | 0.17 | ||
| No | 66 | ||||
| CNS involvement | Yes | 13 | 0.96 | ||
| No | 73 | ||||
| Risk organ a involvement | Yes | 17 | 0.0034 | 4.56 (0.80–26.01) | 0.087 |
| No | 69 | 1 | |||
| Special site b involvement | Yes | 22 | 0.47 | ||
| No | 64 | ||||
Abbreviation: CNS, central nervous system (excluding the pituitary gland).
Risk organs: liver, spleen, or hematopoietic system.
Special sites: vertebral lesions with intraspinal or craniofacial bone lesions with soft tissue extensions (orbit, mastoid, sphenoid, or temporal bones).
3.7. BRAF V600E mutation occurrence in adult patients with LCH
Immunohistochemical analysis of the biopsy specimens of 33 patients revealed that nine (27.3%) were positive for the BRAF V600E mutation. Cell‐free DNA analysis of peripheral blood plasma samples of 23 patients revealed that six (26.1%) were positive for the mutation and were also more likely to have PG/CNS involvement than those without the mutation (p = 0.048) (Figure S1). No difference was found in EFS and OS between BRAF V600E‐mutated (Figure S2A and S2B) or MAPK pathway‐mutated cases and non‐mutated cases. However, a difference was found in OS between plasma cfBRAF V600E‐mutated and non‐mutated cases. The OS probability at 5 years post‐diagnosis of patients with or without plasma cfBRAF V600E was 62.5% (95% CI: 14.2–89.3) versus 100% (95% CI: 75.3–100.0), p = 0.015, respectively (Figure S3A). No difference was found in EFS between positive and negative plasma cfBRAF V600E (Figure S3B). In total, 24 specimens were analyzed using whole‐exome sequencing (WES); however, the analysis of four of them was inadequate due to a low specimen volume, such as that of the PG. Of the remaining 20 specimens, gene mutations were detected in only six cases, while 14 had no detectable gene mutations. The gene mutations harbored a BRAF V600E mutation in two cases, BRAF V600E mutation and MAP2K1 exon2 mutation in one case, KRAS and NRAS mutation in one case each, respectively, and BCR/JAK2 mutation in one case (Table 6).
TABLE 6.
Gene mutations in patients with Langerhans cell histiocytosis.
| No. | Sex | Age | BRAF V600E | WES | Refractory /relapsed disease | Disease classification (overall) | Organ involvement (overall) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Tissue | Plasma cfDNA | ||||||||||
| IHC | PCR | PCR | Organ | PG/CNS | RO | ||||||
| 1 | F | 61 | + | + | + | BCR::JAK2 | PD (Dead) | MS‐LCH | B, L, PG, LN, LV, ST, DO, CV | + | + |
| 2 | F | 55 | + | + | + | BRAF p.V600E | R5 | MS‐LCH | B, PG, S, DO | + | − |
| 3 | M | 36 | + | + | + | BRAF p.V600E | R2 | MS‐LCH | B, PG, LN, CNS | + | − |
| 4 | M | 25 | + | + | N/A | N/A | R1 | MS‐LCH | L, PG, CNS, ST | + | − |
| 5 | M | 59 | + | + | − | None | − | MS‐LCH | B, L | − | − |
| 6 | M | 24 | + | − | − | None | R1 | SSm‐LCH | S | − | − |
| 7 | F | 47 | + | − | − | KRAS p.V14I | R3 | MS‐LCH | B, PG | + | − |
| 8 | F | 66 | + | N/A | N/A | N/A | − | SSm‐LCH | PG, CNS | + | − |
| 9 | F | 54 | + | − | N/A | None | − | MS‐LCH | B, PG, CNS, DO | + | − |
| 10 | F | 45 | − | + | N/A | BRAF p.V600E MAP2K1 p.F53L | − | MS‐LCH | B, LN | − | − |
| 11 | F | 30 | − | + | N/A | None | − | SSm‐LCH | B | − | − |
| 12 | F | 45 | − | N/A | + | N/A | PD (Dead) | SSm‐LCH | PG, CNS | + | − |
| 13 | M | 29 | − | N/A | + | N/A | − | MS‐LCH | L, LV, CNS, DO, T | + | + |
| 14 | F | 40 | NT | NT | + | NT | R1 | SSm‐LCH | B, PG, CNS | + | − |
| 15 | F | 44 | − | − | − | N/A | − | MS‐LCH | PG, S | + | − |
| 16 | M | 47 | − | − | − | NT | R1 | MS‐LCH | B, L, PG | + | − |
| 17 | M | 57 | − | − | − | None | R2 | MS‐LCH | B, PG, ST, TG, DO | + | − |
| 18 | F | 42 | − | − | − | None | − | MS‐LCH | B, L, S | − | − |
| 19 | M | 42 | − | − | − | None | − | MS‐LCH | PG, LN, TG | + | − |
| 20 | M | 62 | − | − | − | None | − | SSm‐LCH | S | − | − |
| 21 | F | 36 | − | − | − | None | SD | SSm‐LCH | PG, CNS | + | − |
| 22 | F | 43 | − | − | − | None | − | MS‐LCH | PG, S, CNS | + | − |
| 23 | M | 53 | − | − | − | None | − | MS‐LCH | B, PG, ST | + | − |
| 24 | F | 66 | − | − | N/A | NRAS p.G12D NRAS p.G13D | R2 | SSm‐LCH | S | − | − |
| 25 | F | 29 | − | − | N/A | None | − | MS‐LCH | S, LN | − | − |
| 26 | F | 41 | − | − | N/A | N/A | − | SSm‐LCH | S | − | − |
| 27 | M | 29 | − | − | N/A | NT | − | SSm‐LCH | B | − | − |
| 28 | M | 27 | − | NT | − | None | R1 | MS‐LCH | B, S, CNS | + | − |
| 29 | F | 40 | − | NT | − | NT | R2 | MS‐LCH | B, LN, S | − | − |
| 30 | M | 25 | − | N/A | − | N/A | R1 | SSm‐LCH | B | − | − |
| 31 | M | 50 | − | N/A | N/A | N/A | − | SSs‐LCH | B | − | − |
| 32 | M | 35 | − | N/A | N/A | N/A | − | MS‐LCH | LN, LV | − | + |
| 33 | M | 25 | − | N/A | N/A | N/A | − | SSs‐LCH | B | − | − |
| 34 | F | 60 | − | N/A | N/A | N/A | − | MS‐LCH | B, PG, CNS, ST, TG | + | − |
| 35 | M | 78 | NT | − | N/A | None | R1 | SSm‐LCH | S | − | − |
| 36 | M | 30 | N/A | N/A | − | N/A | R1 | MS‐LCH | B, LN | − | − |
| 37 | M | 29 | N/A | N/A | − | N/A | − | MS‐LCH | B, L, LV, ST, TG, T | − | + |
Abbreviations: B, bone; cfDNA, cell‐free DNA; CNS, central nervous system; CV, cardiovascular; DO, digestive organs excluding the liver; IHC, immunohistochemistry; L, lung; LN, lymph nodes; LV, liver; MS‐LCH: multisystem LCH; N/A, not available; None, none of the mutant alleles measured by WES; NT, not tested (because of insufficient sample for analysis); PCR, polymerase chain reaction; PD, progression disease; PG, pituitary gland; PG/CNS, pituitary gland and CNS involvement; R No., relapse numbers; RO, risk organ involvement; S, skin; SSm‐LCH, single system with multi‐site LCH; SSs‐LCH, single system with single‐site LCH; ST, soft tissue; T, thymus; TG, thyroid gland; WES, whole‐exome sequencing.
4. DISCUSSION
We found an age onset peak between 20 and 40 years in adult patients with LCH, which is consistent with previous studies. 18 , 21 We similarly noted male predominance (60.8%, 95% CI: 52.5–68.7, p = 0.011). 21 However, in this study, the median age at diagnosis was older, and the duration between onset and diagnosis was longer than those reported previously. 3 , 21
CNS involvement in LCH was divided into two subtypes according to location in the hypothalamic–pituitary region, particularly in the posterior pituitary or in the cerebellum and/or brainstem. The most common manifestations were DI and symptoms of neurodegeneration (ataxia and cognitive dysfunction), with DI reported as the most common disease occurring in up to 30% of patients. 21 LCH‐ND is among the most severe complications of LCH, occurring in 2%–11% of reported cases. 22 In our study, 11 patients developed DI before LCH diagnosis, with a median latency time of 5 years (range: 2–31 years). Three patients with LCH‐ND presented neuropsychiatric or behavioral disorders during LCH diagnosis, suggesting that the diagnosis was delayed. Although LCH is not fatal, a delay in its diagnosis and treatment can lead to irreversible organ impairment. As the clinical presentation of LCH varies, patients were referred to various departments, and histological diagnoses were based on various types of lesions. The diagnosis may also be missed in adult patients with a single LCH lesion detected by a specialist (e.g., respiratory, orthopedic, or dermatologist), who may be unaware that the lesion is a part of a more generalized systemic disorder. Physicians should thus consider prompt diagnosis, systemic evaluation, and appropriate treatment for LCH.
In our study, adult patients with LCH demonstrated an unusually high number of additional malignancies, for which we presumed three causes. First, each malignancy possibly developed from the same cell of origin. This finding was suspected in patients with mixed germ cell tumors and T‐lymphoblastic leukemia / lymphoma (T‐ALL/LBL). Taylor et al. 23 reported that germ cell tumors and hematologic malignancies in the same patient evolved from a common shared precursor. Feldman et al. 24 reported an identical T‐cell receptor γ rearrangement in both T‐ALL/LBL and LCH that confirmed a clonal relationship between the two neoplastic diseases. Second, among the four patients with additional malignancies after LCH diagnosis, three were suspected to have therapy‐related acute myeloid leukemia / myelodysplastic syndromes (AML/MDS). Recently, reports of secondary LCH malignancies associated with chemotherapy have increased. 25 The increase in the levels of cytokines and growth factors associated with LCH may indicate that a systemic biological process precedes both LCH and other neoplastic processes, including potential contributions from random replicative errors. 26 Finally, one patient developed an Langerhans cell sarcoma (LCS), a non‐LCH histiocytosis. Xerri et al. 27 reported that the CDKN2A/B deletion might cause the aggressive transition of LCH to LCS. These results suggest that the aggressiveness of a disease (e.g., from LCH to LCS) is determined by the accumulation of genomic mutations. Thus, genome analysis should be performed to determine whether additional malignancies are associated with mutations in the RAS–BRAF–MEK–ERK pathway, such as hematopoietic cell of origin, tumorigenic agents, or localized inflammatory reactive processes, that are common in LCH.
In our study, the population with lymph node involvement was higher than that reported in previous studies (7%–19%). 3 , 21 , 28 Among the 21 patients with lymph node involvement, 20 presented with MS‐LCH, and one with SSm‐LCH. In total, 19 patients with MS‐LCH and lymph node involvement were treated with chemotherapy according to the MS‐LCH treatment guidelines. 17
Localized pulmonary lesions are strongly associated with smoking and reduced smoking cessation, 29 whereas PLCH occurs predominantly in young smokers. All patients with PLCH were smokers, compared with 35% of those without lung involvement or those prescribed smoking cessation (Figure S4). Two patients with PLCH developed MS‐LCH. In a recent review, smoking cessation was the most important recommendation for patients with PLCH, but treatment of progressive PLCH is based on chemotherapy. 30
In our study, of the 10 patients with SSs‐LCH at diagnosis, bone lesions were detected in nine patients, and skin lesions detected in one patient. The initial treatments were surgery (n = 6) or radiation (n = 3) for the bone lesion and careful observation of the skin lesions. Three patients were administered chemotherapy as secondary treatment, but none developed MS‐LCH. In the cases of SSs‐LCH without CNS risk, local therapy and careful observation are recommended. 17 Of the 22 patients with SSm‐LCH, bone lesions were detected in 10 patients, skin lesions in seven, CNS lesions in four, and lymph nodes in one. Among the four patients who developed MS‐LCH, two were not treated with chemotherapy as initial treatment. Of the 46 patients with MS‐LCH, 13 were not initially treated with chemotherapy. We presume seven patients were not administered chemotherapy, as they were asymptomatic or had developed skin symptoms. In contrast, all six patients, who had bone lesions and some symptoms, repeated inappropriate local therapy, such as operation or radiation, and received chemotherapy after disease progression. (Table S3). Systemic therapy is strongly recommended for patients with SSm‐LCH or MS‐LCH. 17
The probability of EFS at 5 years was 52.1% (95% CI: 36.6–65.5) in all patients who underwent initial chemotherapy. Among the 53 patients with NAD, PR, and SD, 20 had recurrence. Almost all patients responded to frontline chemotherapy, but half of them relapsed within a few years, although we could not determine the prognostic factors for refractory/relapsed disease after initial chemotherapy. The European Consortium for Histiocytosis guideline for adult 17 recommends that systemic therapy be considered for the following disease category: MS‐LCH, SSm‐LCH, and SSs‐LCH with special site lesions. However, there is no standard first‐line therapy in adult LCH. For patients requiring systemic therapy, VBL‐based regimens are mentioned in various chemotherapeutic manuals 17 ; however, they result in poor responses and excessive toxicity. 13 As alternative regimens, the JLSG Special‐C regimen, Ara‐C, 2‐CdA, and MACOP‐B regimens were reported but have never been shown as effective for adults with LCH in a prospective study. For patients with MS‐LCH, 2‐CdA and MACOP‐B regimens were more effective than the Special‐C regimen, but they showed a higher incidence of grade 3–4 adverse events. 11 , 12 Therefore, such intensive therapy may be best reserved for salvage in adult patients. Morimoto et al. 14 reported the outcome of Special‐C regimen for adult patients. All four patients with SS‐LCH achieved NAD, and six of the 10 patients with MS‐LCH showed a good response. Although the Special‐C regimen was not mentioned in the guideline, the regimen is safe and effective and is particularly significant because it is an ambulatory treatment. Cantu et al. 13 reported the outcome of Ara‐C for adult patients with bone lesions, which was superior to VBL/PSL and even to 2‐CdA in terms of response and toxicity. The guideline mentioned that Ara‐C may be the treatment for MS‐LCH with and without RO involvement. 17 However, Cantu et al. did not clarify the disease category; therefore, we could not compare the therapeutic effects of the Special‐C and Ara‐C regimens. In our study, as initial chemotherapy treatments, 36 (63.2%) patients were administered the JLSG Special‐C regimen14 and 10 (17.5%) Ara‐C‐containing regimens. 10 , 13 The same response rate and refractory/relapse rate were observed between the two regimens. However, this study is retrospective; thus, we could not assess the adverse events. Therefore, a randomized trial is required based on risk stratification.
Six of the 86 patients died during the clinical course of their illness, with the cause of death associated with LCH in four patients and therapy‐related death in two. None of the four patients whose deaths were LCH related responded to initial chemotherapy. Minkov et al. 31 reported that response to initial therapy emerged as an important independent prognostic factor for LCH. Of the four refractory patients, two had the BRAF V600E mutation. Prospective trials, including targeted therapies, are required to improve outcomes in patients with chemotherapy‐resistant LCH.
The OS reported in our study was consistent with previous reported results. 21 We found that age, RO+, and lymph node involvement were poor prognostic factors for patients with LCH. Risk organ involvement in adults with LCH was known as a poor prognosis factor. Similarly, RO+ indicated a less favorable prognosis for children. 32 The prognostic impact of lymph nodes was previously reported in a small‐scale study to differ from that of the more aggressive MS‐LCH, in which lymphoreticular organ involvement was defined as a benign localized lymph node infiltration and locally treated in two patients. 3 However, in our study, of the 21 patients with lymph node involvement, 20 had MS‐LCH, suggesting that lymph node involvement was associated with a poor prognosis. Among the four dead patients aged ≥60 years, three who were refractory to initial treatment and one who had secondary AML died of LCH. Therefore, we hypothesized that, in poor prognosis detection, older adults would have more severe biological features (e.g., acquisition of somatic mutations in genes and random replicative errors because of a localized inflammatory reaction) than young adults.
LCH that activates somatic mutations in MAPK pathway genes, most notably the BRAF V600E mutation, has been identified in 56% of LCH cases. 4 In our study, immunohistochemical analysis of biopsy specimens showed a lower positive ratio than that found in previous reports from North America and Europe. 18 Heritier et al. 33 reported that patients with BRAF V600E mutation had an increased risk of frontline treatment and permanent consequences, such as DI and LCH‐ND. Kemps et al. 34 reported the associations of BRAF and MAP2K1 mutations with clinical features and ltBRAF V600E did not correlate with inferior clinical outcome after patient stratification by disease extent. We tried to analyze BRAF and other MAPK pathway mutations. No difference was found in EFS and OS between BRAF V600E‐mutated or MAPK pathway‐mutated cases and non‐mutated cases. However, despite the small sample size, our study demonstrated the feasibility of cfBRAF V600E increasing the risk of PG/CNS involvement and being associated with a low OS rate. Furthermore, no difference was found in OS and EFS between positive ltBRAF V600E detected using IHC or PCR and negative ltBRAF V600E. Wang et al. 35 reported that cfBRAF V600E, rather than ltBRAF V600E, was a prognostic factor and was more closely associated with critical clinical characteristics and treatment outcomes. Cell‐free DNA originates from cell turnover, which involves the natural death of cells through apoptosis or necrosis and is becoming a widely used prognostic and predictive biomarker in oncology. Heitzer et al. 36 reported that cfDNA is associated with the increased proliferation of some cancer cells. Furthermore, CD1a+ cells in patients' circulation may directly lead to the spread of LCH cells and PG/CNS involvement. These reasons suggest that cfBRAF V600E may be associated with poor independent prognostic factors and disease spread in our study.
Patients with BRAF V600E, KRAS, and NRAS mutations tended to relapse repeatedly. We found that a patient who passed away due to refractory LCH had a BRAF V600E mutation combined with a BCR/JAK2 mutation. The reasons mature adult LCH is a poor prognostic factor may be because it is an additional malignancy and harbors genetic mutations in addition to those in the MAPK pathway genes. However, due to the study's retrospective nature, we could not analyze gene mutations in all patients. Further exploration of mutations in MAPK pathway genes and others could help elucidate the pathogenesis and improve the treatment of adults with LCH.
In adults with LCH, we must carefully consider various co‐morbidities and treatment‐related malignancy. If younger adult patients do not respond to initial chemotherapy, then intensified chemotherapy may improve their outcome. However, unlike children and younger adult patients, older adult patients not responding to initial chemotherapy cannot receive intensified treatment. In addition, frequent chemotherapy for refractory/relapsed disease causes therapy‐related malignancy. Therefore, if patients have the MAPK somatic mutation, then a MAPK inhibitor, instead of intensified treatment, may improve their outcome.
In conclusion, LCH is a rare disease in adults, and the biological and standard therapeutic approaches are not well established. However, advances over the past decade have defined LCH as an inflammatory myeloid neoplastic disorder that is evaluated based on the cell of origin where activating MAPK somatic mutations arise. We observed a high recurrence rate of LCH after chemotherapy, a relationship between poor prognosis and chemotherapy resistance, and plasma cfBRAF V600E. Therefore, new therapies, including targeted therapy, and randomized therapeutic trials for adult patients with LCH are needed to improve diagnosis and patient care. Prospective trials and translation of biological discoveries will improve the high recurrence and refractory disease in adults with LCH.
FUNDING INFORMATION
This study was funded by the Japanese National Research Group on Idiopathic Bone Marrow Failure Syndromes (Grant No. 20FC1018).
CONFLICT OF INTEREST STATEMENT
Seiya Imoto receives honoraria (Daiichi Sanko RD Novare, BrightPath Biotherapeutics Co. Ltd. and Liquid Mine corporation). Kazuaki Yokoyama receives research funding (Nippon Shinyaku, Liquid Mine) and belongs to Liquid Mine. The other authors have no conflict of interest.
ETHICAL APPROVAL
Approval of the research protocol by an Institutional Reviewer Board: This study was approved by the Ethics Committee of the Institutional Review Board of the Institute of Medical Science, University of Tokyo (approval numbers 2019‐35‐1017, 2020‐49‐1119 and 2020–1‐0422).
Informed Consent: All informed consent was obtained from the patients.
Registry and the Registration No. of the study.
Trial: N/A.
Animal Studies: N/A.
Supporting information
Appendix S1
ACKNOWLEDGMENTS
The authors would like to thank the patients, their families, and all investigators.
Sato A, Kobayashi M, Yusa N, et al. Clinical and prognostic features of Langerhans cell histiocytosis in adults. Cancer Sci. 2023;114:3687‐3697. doi: 10.1111/cas.15879
REFERENCES
- 1. Collin M, Bigley V, McClain KL, Allen CE. Cell(s) of origin of Langerhans cell histiocytosis. Hematol Oncol Clin North Am. 2015;29(5):825‐838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rigaud C, Barkaoui MA, Thomas C, et al. Langerhans cell histiocytosis: therapeutic strategy and outcome in a 30‐year nationwide cohort of 1478 patients under 18 years of age. Br J Haematol. 2016;174(6):887‐898. [DOI] [PubMed] [Google Scholar]
- 3. Baumgartner I, von Hochstetter A, Baumert B, Luetolf U, Follath F. Langerhans'‐cell histiocytosis in adults. Med Pediatr Oncol. 1997;28(1):9‐14. [DOI] [PubMed] [Google Scholar]
- 4. Badalian‐Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116(11):1919‐1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage‐dendritic cell lineages. Blood. 2016;127(22):2672‐2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gadner H, Grois N, Arico M, et al. A randomized trial of treatment for multisystem Langerhans' cell histiocytosis. J Pediatr. 2001;138(5):728‐734. [DOI] [PubMed] [Google Scholar]
- 7. Gadner H, Grois N, Pötschger U, et al. Improved outcome in multisystem Langerhans cell histiocytosis is associated with therapy intensification. Blood. 2008;111(5):2556‐2562. [DOI] [PubMed] [Google Scholar]
- 8. Gadner H, Minkov M, Grois N, et al. Therapy prolongation improves outcome in multisystem Langerhans cell histiocytosis. Blood. 2013;121(25):5006‐5014. [DOI] [PubMed] [Google Scholar]
- 9. Morimoto A, Ikushima S, Kinugawa N, et al. Improved outcome in the treatment of pediatric multifocal Langerhans cell histiocytosis: results from the Japan Langerhanscell histiocytosis study Group‐96 protocol study. Cancer. 2006;107(3):613‐619. [DOI] [PubMed] [Google Scholar]
- 10. Morimoto A, Shioda Y, Imamura T, et al. Intensified and prolonged therapy comprising cytarabine, vincristine and prednisolone improves outcome in patients with multisystem Langerhans cell histiocytosis: results of the Japan Langerhans cell histiocytosis study Group‐02 protocol study. Int J Hematol. 2016;104(1):99‐109. [DOI] [PubMed] [Google Scholar]
- 11. Saven A, Burian C. Cladribine activity in adult langerhans‐cell histiocytosis. Blood. 1999;93(12):4125‐4130. [PubMed] [Google Scholar]
- 12. Derenzini E, Fina MP, Stefoni V, et al. MACOP‐B regimen in the treatment of adult Langerhans cell histiocytosis: experience on seven patients. Ann Oncol. 2010;21(6):1173‐1178. [DOI] [PubMed] [Google Scholar]
- 13. Cantu MA, Lupo PJ, Bilgi M, Hicks MJ, Allen CE, McClain KL. Optimal therapy for adults with Langerhans cell histiocytosis bone lesions. PLOS One. 2012;7(8):e43257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Morimoto A, Shimazaki C, Takahashi S, et al. Therapeutic outcome of multifocal Langerhans cell histiocytosis in adults treated with the special C regimen formulated by the Japan LCH study group. Int J Hematol. 2013;97(1):103‐108. [DOI] [PubMed] [Google Scholar]
- 15. Duan MH, Han X, Li J, et al. Comparison of vindesine and prednisone and cyclophosphamide, etoposide, vindesine, and prednisone as first‐line treatment for adult Langerhans cell histiocytosis: a single‐center retrospective study. Leuk Res. 2016;42:43‐46. [DOI] [PubMed] [Google Scholar]
- 16. Kobayashi M, Tojo A. Langerhans cell histiocytosis in adults: advances in pathophysiology and treatment. Cancer Sci. 2018;109(12):3707‐3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Girschikofsky M, Arico M, Castillo D, et al. Management of adult patients with Langerhans cell histiocytosis: recommendations from an expert panel on behalf of euro‐Histio‐net. Orphanet J Rare Dis. 2013;8:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kobayashi M, Ando S, Kawamata T, et al. Clinical features and outcomes of adult Langerhans cell histiocytosis: a single‐center experience. Int J Hematol. 2020;112(2):185‐192. [DOI] [PubMed] [Google Scholar]
- 19. Kanda Y. Investigation of the freely available easy‐to‐use software ‘EZR’ for medical statistics. Bone Marrow Transplant. 2013;48(3):452‐458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Allen CE, Flores R, Rauch R, et al. Neurodegenerative central nervous system Langerhans cell histiocytosis and coincident hydrocephalus treated with vincristine/cytosine arabinoside. Pediatr Blood Cancer. 2010;54(3):416‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aricò M, Girschikofsky M, Généreau T, et al. Langerhans cell histiocytosis in adults. Report from the international registry of the histiocyte society. Eur J Cancer. 2003;39(16):2341‐2348. [DOI] [PubMed] [Google Scholar]
- 22. Héritier S, Barkaoui MA, Miron J, et al. Incidence and risk factors for clinical neurodegenerative Langerhans cell histiocytosis: a longitudinal cohort study. Br J Haematol. 2018;183(4):608‐617. [DOI] [PubMed] [Google Scholar]
- 23. Taylor J, Donoghue MT, Ho C, et al. Germ cell tumors and associated hematologic malignancies evolve from a common shared precursor. J Clin Invest. 2020;130(12):6668‐6676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Feldman AL, Berthold F, Arceci RJ, et al. Clonal relationship between precursor T‐lymphoblastic leukaemia/lymphoma and Langerhans‐cell histiocytosis. Lancet Oncol. 2005;6(6):435‐437. [DOI] [PubMed] [Google Scholar]
- 25. Ma J, Laird JH, Chau KW, Chelius MR, Lok BH, Yahalom J. Langerhans cell histiocytosis in adults is associated with a high prevalence of hematologic and solid malignancies. Cancer Med. 2019;8(1):58‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tomasetti C, Li L, Vogelstein B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science. 2017;355(6331):1330‐1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xerri L, Adélaïde J, Popovici C, et al. CDKN2A/B deletion and double‐hit mutations of the MAPK pathway underlie the aggressive behavior of langerhans cell tumors. Am J Surg Pathol. 2018;42(2):150‐159. [DOI] [PubMed] [Google Scholar]
- 28. Grois N, Pötschger U, Prosch H, et al. Risk factors for diabetes insipidus in langerhans cell histiocytosis. Pediatr Blood Cancer. 2006;46(2):228‐233. [DOI] [PubMed] [Google Scholar]
- 29. Vassallo R, Ryu JH, Schroeder DR, Decker PA, Limper AH. Clinical outcomes of pulmonary Langerhans'‐cell histiocytosis in adults. N Engl J Med. 2002;346(7):484‐490. [DOI] [PubMed] [Google Scholar]
- 30. Radzikowska E. Update on pulmonary Langerhans cell histiocytosis. Front Med (Lausanne). 2020;7:582581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Minkov M, Grois N, Heitger A, et al. Response to initial treatment of multisystem Langerhans cell histiocytosis: an important prognostic indicator. Med Pediatr Oncol. 2002;39(6):581‐585. [DOI] [PubMed] [Google Scholar]
- 32. Haupt R, Minkov M, Astigarraga I, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work‐up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60(2):175‐184. doi: 10.1002/pbc.24367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Héritier S, Emile JF, Barkaoui MA, et al. BRAF mutation correlates with hgh‐risk Langerhans cell histiocytosis and increased resistance to first‐line therapy. J Clin Oncol. 2016;34(25):3023‐3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kemps PG, Zondag TCE, Arnardóttir HB, et al. Clinicogenomic associations in childhood Langerhans cell histiocytosis: an international cohort study. Blood Adv. 2023;7(4):664‐679. doi: 10.1182/bloodadvances.2022007947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang CJ, Cui L, Ma HH, et al. Mutation in cell‐free DNA, rather than in lesion tissues, at diagnosis is an independent prognostic factor in children with Langerhans cell histiocytosis. Mol Cancer Ther. 2021;20(7):1316‐1323. [DOI] [PubMed] [Google Scholar]
- 36. Heitzer E, Auinger L, Speicher MR. Cell‐free DNA and apoptosis: how dead cells inform about the living. Trends Mol Med. 2020;26(5):519‐528. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1