

Summary
Genetically encoded fluorescent indicators are powerful tools for tracking cellular dynamic processes. Engineering these indicators requires balancing screening dimensions with screening throughput. Herein, we present a functional imaging-guided photoactivatable cell selection platform, Faculae (functional imaging-activated molecular evolution), for linking microscopic phenotype with the underlying genotype in a pooled mutant library. Faculae is capable of assessing tens of thousands of variants in mammalian cells simultaneously while achieving photoactivation with single-cell resolution in seconds. To demonstrate the feasibility of this approach, we applied Faculae to perform multidimensional directed evolution for far-red genetically encoded calcium indicators (FR-GECIs) with improved brightness (Nier1b) and signal-to-baseline ratio (Nier1s). We anticipate that this image-based pooled screening method will facilitate the development of a wide variety of biomolecular tools.
Keywords: genetically encoded fluorescent indicator, directed evolution, photoactivatable cell tagging, fluorescence imaging, far-red calcium indicator, pooled screening
Graphical abstract
Highlights
-
•
Development of a microscopy imaging-guided cell selection strategy, Faculae
-
•
Recombinase-assisted mutant library preparation
-
•
Multidimensional directed evolution of far-red fluorescent calcium indicators
-
•
Nier1 indicators exhibit improved brightness and sensitivity in neurons
Motivation
Genetically encoded fluorescent indicators enable the non-invasive optical recording of cellular activities. Engineering these indicators often requires the simultaneous assessment of multiple key parameters such as brightness, sensitivity, and response kinetics. Herein, we report the design and implementation of a microscopic imaging-guided cell selection technology that can evaluate multiple parameters in a mammalian cell population and screen 104-105 variants at a time.
Lin et al. develop an imaging-guided pooled library screening platform that evaluates tens of thousands of variants in cultured mammalian cells in a single run. Cells of interest are phototagged and recovered via flow cytometry for genotyping. This screening platform supports the directed evolution of genetically encoded fluorescent indicators, such as far-red calcium indicators.
Introduction
Genetically encoded fluorescent indicators are powerful tools for monitoring the dynamics of cellular physiology. Among these, fluorescent protein (FP)-based intensiometric biosensors are particularly significant due to their large dynamic ranges and good spectra compatibility for multiplexed imaging. Therefore, they have been widely used in recording neural activities, including neurotransmitter release,1 calcium ion dynamics,2 and membrane potential fluctuations.3
Assessment of fluorescent indicators typically includes key parameters such as brightness, signal-to-baseline ratio (ΔF/F0), dynamic range (Fmax/Fmin), photostability, protein stability, etc. Improving indicator performance on these fronts often requires labor-intensive protein engineering and screening efforts. Notably, there is a general trade-off between screening throughput and the number of parameters being evaluated in single-trial measurements. For example, fluorescence-activated cell sorting (FACS)-based screens can evaluate brightness of FP libraries larger than 106 in a single run4 but are incapable of measuring dynamic parameters such as dynamic range of biosensors in individual cells. Array-based screens, such as those performed in multiwell plates, enable both static and dynamic signal detection but are usually limited to the screening of thousands of mutants at a time and require laborious procedures to prepare the library.5,6,7 Recently, microscope-based pooled screens have been applied for linking the visually examined phenotypes to the genotypes of pooled variants,8,9,10,11,12,13 which are well suited for high-throughput biosensor screening owing to their abilities to measure dynamic parameters with high spatiotemporal resolution, while simultaneously acquiring genetic information on promising candidate mutants.
Several microscopy-based pooled screening platforms have been recently developed to engineer genetically encoded fluorescent indicators.14,15 In these methods, cultured cells expressing a library of biosensor mutants are initially evaluated under a fluorescence microscope. Thereafter, target cells with desired properties (e.g., improved brightness, SBR, photostability, etc.) are identified and retrieved for genotyping, either by fluorescent tagging and subsequent cell sorting or by direct aspiration from the plate. For example, Photopick utilized patterned illumination to phototag target cells expressing a green-to-red photoconvertible FP, which were subsequently sorted with flow cytometry. This method has been successfully employed to screen far-red genetically encoded voltage indicators in engineered HEK 293T cells.14 One limitation of Photopick is its requirement of using a green-to-red photoconvertible FP marker, which impedes its application to GFP- or RFP-based indicators. In another approach, termed Opto-MASS, genetically encoded neurotransmitter indicators in mammalian cell libraries are screened in microwell array formats, with identified high-ranking cells aspirated by micropipettes.15 This aspiration-based retrieval method is time consuming and low throughput. Therefore, a generalizable and automated microscopy-based cell selection method is still needed.
Far-red genetically encoded calcium indicators (FR-GECIs) have become promising tools for deep-tissue imaging and multiplexed imaging due to their FR spectral property.16,17,18 Recently, a series of intensiometric FR-GECIs (termed near-infrared genetically encoded Ca2+ indicator for optical imaging, abbreviated as NIR-GECOs) have been developed that consist of a calcium-binding domain, calmodulin (CaM)-RS20, inserted into a monomeric infrared FP (mIFP).17,18 However, NIR-GECOs still suffer from low molecular brightness and limited fluorescence response and require further improvement in both dimensions.
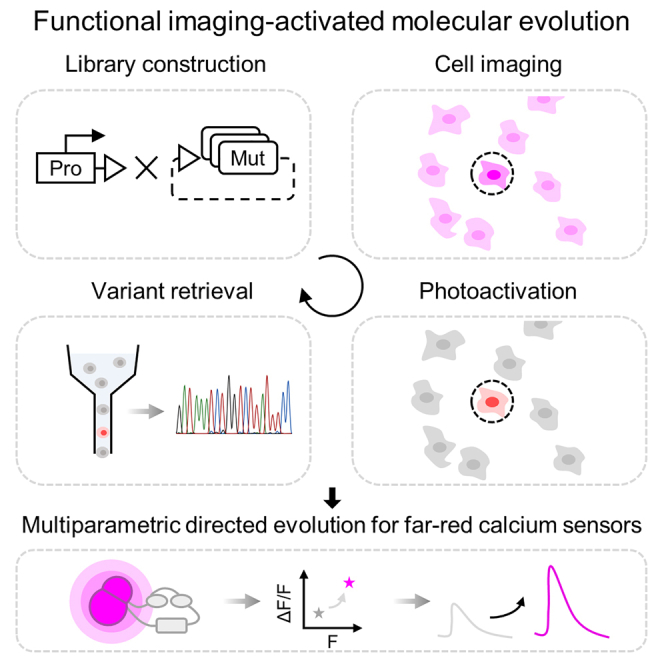
Herein, we report a microscopic imaging-guided pooled screening strategy, termed functional imaging-activated molecular evolution (Faculae), that can, in principle, be used for the evolution of a wide variety of fluorescent indicators. First, we construct biosensor libraries in mammalian cells efficiently through recombinase-based locus-specific integration. Then, we screen the pooled library under the microscope, followed by photoactivating the target cell population via focused illumination with single-cell resolution and on a timescale of seconds per cell. Thereafter, phototagged candidate cells are retrieved by FACS for further genotyping. To demonstrate its capability, we applied Faculae to perform multidimensional screening of FR-GECIs and generated Nier1s and Nier1b with improved performance.
Results
Characterization of imaging-guided cell selection platform
To enable cell selection under microscope, we achieved fast and precise optical labeling of target cells by photoactivating FPs through partially focused laser beam. Phototaggable (photoconvertible, activatable, or switchable) FPs have been applied as fluorescent markers for visualizable phenotypes in many studies.19,20,21 Although the digital micromirror device (DMD) has been widely used for spatially selective activation, it is generally time consuming to activate cells within each field of view due to low power density.11 For the optical tagging of hundreds and thousands of cells, we reasoned that a focal illumination approach, which could restrict the photoactivating light directly on one cell, would be more efficient.22 We filled the back aperture of the objective (20×, 0.75 NA) with a partially collimated 405 nm laser to generate a disc-like illumination pattern with a diameter of ∼15 μm at the focal plane, with an estimated power density of 80 W/cm2 at its center (Figures 1A, S1A, and S1B). To test the photoactivation kinetics, we expressed photoactivatable mCherry23 (PAmCherry) in human embryonic kidney 293T (HEK 293T) cells. Under focal illumination, the fluorescence of PAmCherry in targeted cells increased by more than 100 times after 1.5 s activation, while the ratio of fluorescence intensity of targeted cells to neighboring cells reached a maximum of 50 ± 10 (mean ± SEM) at approximately 2.5 s (Figures 1B and 1C). We repeated the above experiment with PA-GFP,24 whose intensity increased by 49-fold following 1 s activation with a focused 405 nm laser (Figure S1C). Taken together, the above characterization demonstrated the capability of activating either red or green fluorescence with single-cell spatial resolution and second-scale temporal resolution. We decided to use 1–1.5 s photoactivation for subsequent experiments to shorten the activation time and to reduce the off-target activation background.
Figure 1.

Faculae enabled precise photoactivation and retrieval of target cells
(A) Optical setup of focal illumination (violet dashed line) and wide-field imaging (black line).
(B) Target HEK 293T cells could be precisely photoactivated in seconds. In snapshots (first line), the red dashed line represents the target cell, and the gray dashed line represents the neighbor cell. Scale bar: 10 μm. ntarget cell = 15; nneighbor cell = 15. Error bars represent SEM.
(C) Fluorescence ratio (Ftarget/Fneighbor) peaked after around 2.5 s photoactivation. ntarget cell = 15; nneighbor cell = 15. Error bars represent SEM.
(D) HEK 293T cells transfected with GFP-P2A-PAmCherry were mixed with 20-fold more cells transfected with miRFP680-P2A-PAmCherry. GFP+ cells were selected for photoactivation (PA) followed by FACS analysis.
(E) Tiled images and zoomed-in views of co-cultured cells expressing GFP or miRFP680. Gray arrows represent cells for activation.
(F) Top: 90 GFP+ cells were optically activated while 44 cells were detected in the PAmCherry+ gate during flow cytometry analysis. Bottom: 40 (∼91%) were true positives (GFP+ cells), while 4 were false positives (3 negative cells and 1 miRFP680+ cell).
We next tested the efficiency and specificity of retrieving photoactivated cells with FACS. We transiently expressed either GFP-P2A-PAmCherry (GFP+/PAmCherry+) or miRFP680-P2A-PAmCherry (miRFP680+/PAmCherry+) in two separate pools of HEK 293T cells. We randomly doped a small portion of GFP+/PAmCherry+ cells (5%) into miRFP680+/PAmCherry+ cells (95%) and co-cultured the mixed population in the glass-bottom dish. Under the microscopy, GFP+ cells were readily distinguishable from miRFP680+ ones both by visual examination and by automated image analysis (Figures 1D and 1E). We photoactivated 50–100 GFP+/PAmCherry+ cells with pulsed 405 nm laser spot illumination within a total of 4 min. Cells were lifted off the glass-bottom dish with trypsinization and sorted for mCherry+ cells by flow cytometry. Overall, we recovered 44% ± 8% (mean ± SD) of photoactivated cells with a precision of 96% ± 6% (defined as the ratio of true positives over the sum of true positives and false positives) (Figure 1F; Table S1), thus confirming that we can accurately activate and recover cells with the desired phenotype.
To achieve imaging-based pooled screening, we next tried to use a recombinase-based method to generate large-scale mutant libraries in mammalian cells.25 Recombinase-based integration ensures the irreversible single-copy insertion of sequences at a pre-defined “landing-pad” locus within the mammalian cell genome (Figure 2A). To generate the lentiviral landing pad (LLP) cell line, we transduced LLP virus encoding either a CAG-attP or CMV-attP construct into HEK 293T cells at a low multiplicity of infection (MOI) and screened 16 clones (10 from the CAG construct and 6 from the CMV construct) with satisfactory expression level and high integration efficiency. Each LLP cell line was transfected with a mixture of attB-EGFP and attB-mCherry plasmids. All of these cell lines did not produce EGFP+/mCherry+ cells, confirming the single-locus integration (Figures S2A and S2B). By comparing the fluorescence intensity and the fraction of cells exhibiting single-integration (i.e., EGFP+/mCherry− and EGFP+/mCherry−) cells, we identified one of the CAG clones with the highest expression level and the highest recombination efficiency (Figures S2B and S2C). We termed this cell line CAG-LLP and used it for constructing mutant libraries.
Figure 2.

Construction and screening of miRFP680 library
(A) Scheme of gene integration in CAG-LLP cell line. ORF, open reading frame.
(B) Construction of miRFP680 library.
(C) Pipeline of screening of fluorescent protein by Faculae.
(D) The stitching image and segmented regions of interest (ROIs) of the cell library expressing miRFP680 mutants. The regions of nuclear localized EGFP in each cell were clearly identified.
(E) Cells with relatively high miRFP brightness were photoactivated under microscope (red scatters in black gate).
(F) PAmCherry+ cells were recovered by FACS (black gate).
(G) The proportion of sequencing results of retrieved PAmCherry+ cells. The double letters represented residues 202 and 203, e.g., “TV” represents T202, V203.
We evaluated the feasibility and throughput of Faculae by screening against an FR FP library. miRFP680 is an engineered monomeric FR FP derived from truncation of the Rhodopseudomonas palustris bacterial phytochrome photoreceptor RpBphP2, which consists of a Per-ARNT-Sim (PAS) domain and a cGMP phosphodiesterase/adenylate cyclase/FhlA (GAF) domain with a pocket incorporating linear tetrapyrrole biliverdin IXa (BV) as a chromophore.26 Two mutations, D202T and I203V, have been introduced into the conserved PXSDIP motif to increase the fluorescence quantum yield by stabilizing the chromophore.26 We randomly mutated these two key residues and screened the mutant library for brightness with Faculae. A nuclear-localized EGFP was co-expressed and served both as a reference for expression level and a marker for facilitating image segmentation (Figures 2B and 2C). We screened over 4 × 104 cells under the microscope and photoactivated 117 cells (0.3%) exhibiting high brightness (Figures 2D–2F and S2D; Table S2). The screening-photoactivation process was completed within 1 h. Following FACS, 48 retrieved single cells were lysed, and 17 of them (35%) were successfully PCR amplified for Sanger sequencing. 12 out of 17 (71%) of the sequencing results converged to miRFP680 (202T/203V) (Figure 2G; Table S2). We further confirmed that miRFP680 is the brightest candidate in the recovered mutants (Figure S2E). Together, the above experiments demonstrated that Faculae could enrich mutants exhibiting desired phenotypes in the image-based screening from a pooled library.
Multidimensional imaging-guided screening of improved FR calcium sensor
We next applied Faculae to improve the performance of NIR-GECO (Figure 3A). First, to evaluate both the baseline brightness (F0) and the SBR (ΔF/F0) of the mutants within each cell, we sought to manipulate the intracellular calcium concentration through drug treatment (Figure 3B). Our initial attempt at using a combination of ionomycin and CaCl2 in HEK 293T cells failed, as the calcium concentration oscillates rather than reaching a steady state (Figure S3A). To inhibit the fluctuations, we added 2-APB to the imaging buffer to block gap junction and store-operated Ca2+ entry,27,28 which resulted in steady calcium levels (Figure S3B). We applied this ionomycin/CaCl2/2-APB treatment protocol to compare the SBR of GCaMP6s with GCaMP5g, which were readily resolved (Figures S3C–S3E). We then utilized this strategy to compare the SBRs of NIR-GECO2G and NIR-GECO1. Time-lapse imaging showed that NIR-GECO2G had higher SBR (ΔF/F0 = −28% ± 2%) than NIR-GECO1 (ΔF/F0 = −15% ± 1%) (Figures 3C and 3D), which was consistent with the previous report,17 thus demonstrating the general applicability of our approach.
Figure 3.

A multiparametric strategy for NIR-GECO evaluation
(A) Schematic representation of NIR-GECO.
(B) Workflow of NIR-GECO screening by Faculae.
(C) Example images of HEK 293T cells expressing the indicated nuclear targeted NIR-GECOs. The fluorescence response in the presence of 2 μM ionomycin and 1 mM CaCl2 is shown.
(D) The response traces of NIR-GECOs in the presence of 2 μM ionomycin and 1 mM CaCl2. Shadow areas represent SEM. ncell = 9 for NIR-GECO2G; ncell = 9 for NIR-GECO1.
(E) Cells with relatively high brightness and large SBR were photoactivated under microscope (red scatters in black gate). Red dashed line represents −ΔF/F0 = 0.28, the average SBR of NIR-GECO2G.
(F) The proportion of sequencing results of retrieved PAmCherry+ cells. 50 μM 2-APB was present throughout imaging.
See also Figure S3.
Next, we performed a model screen by mixing cells expressing either NIR-GECO2G or NIR-GECO1 with a ratio of 1–20. To improve the precision of measurement and to minimize the effect of cell movement, we localized NIR-GECOs and superfolder GFP29 (sfGFP) to the nucleus. The calcium-insensitive sfGFP could serve as an expression reference and a marker for motion correction30 (Figure 3B). We reduced cell migration by optimizing the coating material in the culture dish prior to seeding cells and starving cells from serum for 16–20 h prior to the measurement (Figure S3F). We screened over 5 × 104 cells and photoactivated 85 cells (0.2%) with relatively high brightness and large fluorescence change for FACS (Figure 3E; Table S2). Notably, despite taking up merely 5% in the initial pool, NIR-GECO2G constitutes 93% of sequencing results (Figure 3F), indicating that Faculae had the potential to enrich high-performance candidates in a pooled biosensor library.
We created several combinational mutagenesis libraries starting from NIR-GECO2G.17 First, we constructed two libraries targeting the mIFP domain and the CaM-RS20 domain, respectively (Figures 4A and 4B). In each library, 17 sites based on previously reported mutations in GECIs were targeted for random codon mutagenesis.30,31,32,33,34,35 Plasmid libraries with an average of ∼2 targeted codon mutations per gene were generated by previously reported methods36 (Figure S4A). To increase the copy number of high-performance mutants, we enriched cells with high basal fluorescence by FACS and cultured them until screening (Figures S4B and S4C). Based on the screening results of these two libraries, we picked V133 and S466 from the mIFP library and N302 from the CaM library as high-mutation-rate residues for further saturated combinational mutagenesis (Figures 4A, 4B, and S4D–S4F). In the previous research, these three sites were mutated in the evolution for NIR-GECO1.18 We analyzed ∼1 × 105 cells carrying ∼8,000 possible combinations. Compared with the first two libraries, the combined library had clearly shifted to higher F0 and ΔF/F0 (Figures 4C and 4D). Meanwhile, different from previous libraries, the sequencing results from the combined library revealed completely no template, suggesting that these selected mutants were promising targets with better performance (Figure 4E). Since no dominant mutants were observed (Figure 4F), we evaluated all of them by measuring their brightness and response to ionomycin/CaCl2/2-APB treatment in HEK 293T cells (Figures 4G and S4G). The fold change in basal brightness of these mutants ranged from 1.3 ± 0.1 (mean ± SEM, same below; NIR-GECO2G-133S/302T/466M, abbreviated as STM) to 3.4 ± 0.1 (NIR-GECO2G-133D/302Q/466V, abbreviated as DQV), and the fold change in SBR ranged from 0.73 ± 0.02 (NIR-GECO2G-133R/302Q/466P, abbreviated as RQP) to 1.5 ± 0.1 (STM), compared with the template. All of the mutants showed increased brightness. Among these, five mutants exhibited significantly improved SBR (Table S3). We also confirmed that the photostability of most variants was not compromised (Figure S4H; Table S3).
Figure 4.

Evolving NIR-GECO2G by Faculae
(A) Predicted structure of NIR-GECO2G (predicted on Uni-Fold37). The chromophore and mutation sites are highlighted.
(B) Design of the mutagenesis sites in indicated libraries.
(C) The screening contour of three libraries. In each library, the contour lines from center to periphery represent relative estimated density from 83% to 17%.
(D) In combined library, cells with relatively high brightness and SBR were photoactivated under microscope (red scatters in black gate).
(E) The proportion of template (NIR-GECO2G) and mutants in sequencing results.
(F) Sequence logo of the screening result in the combined library.
(G) Characterization of SBR and brightness for each mutant in HEK 293T. For SBR tests, at least eight cells of one mutant were calculated. For brightness tests, at least three replicates were analyzed by flow cytometry. Error bars represent SEM.
We selected several candidates with improved properties and evaluated their response to physiological signals in rat cultured hippocampal neurons (Table S4). We performed whole-cell patch clamp and stimulated action potential firing via injecting current pulses of 300 pA for 10 ms. We measured the fluorescent response to 1–10 action potentials (50 Hz pulses) and quantified kinetic parameters including half-rise time and half-decay time. We identified two NIR-GECO2G mutants with better performance than the template: 133Q/302K/466M (named Nier1s) and 133E/302T/466C (named Nier1b). Both Nier1s and Nier1b are uniformly distributed in the neuronal cytoplasm (Figure 5A). Compared to the template, Nier1s exhibited similar brightness (1.2- ± 0.2-fold) and enhanced action potential response (−5.8% ± 0.9% versus 2.9% ± 0.5% for single action potential), while Nier1b showed improved brightness (1.9- ± 0.2-fold) and similar fluorescence change (−3% ± 0.6% for single action potential) (Figures 5B–5E, S5A, and S5B; Table S4). Both indicators showed better signal-to-noise ratio (SNR) than the template with similar response kinetics (Figures 5F–5H). We next characterized FR-GECIs in vitro. Nier1s and Nier1b had similar absorption and emission peaks, pKa values, and Hill coefficients (Figures S5C–S5J; Table S5). Compared to NIR-GECO2G, Nier1s has similar molecular brightness (defined as the product of extinction coefficient and quantum yield) and 2-fold higher dynamic range (Fmax/Fmin = 24 versus 12), while Nier1b has 1.6-fold higher molecular brightness but lower dynamic range (Fmax/Fmin = 4.5 versus 12). The Kd values of Nier1s (146 nM) and Nier1b (308 nM) are comparable to that of NIR-GECO2G (194 nM).
Figure 5.

Nier performance in cultured neurons
(A) Representative images of Nier1s and Nier1b. Scale bar: 20 μm.
(B) Brightness of Nier1s (64 cells) and Nier1b (79 cells) normalized to NIR-GECO2G (105 cells). Cells were imaged at days in vitro (DIV) 16–18 with no supply of exogenous BV. ∗∗∗p < 0.001 (Mann-Whitney test).
(C) Averaged responses to 1 action potential (AP) for NIR-GECO2G, Nier1s and Nier1b. Shaded areas represent SEM.
(D) Averaged responses to 10 APs for NIR-GECO2G, Nier1s, and Nier1b. Shaded areas represent SEM.
(E–H) Measured fluorescence change (E), SNR at 48 Hz imaging rate (F), half-rise time (G), and half-decay time (H) as functions of AP numbers. Error bars represent SEM.
(C–H) ncell = 10 for NIR-GECO2G; ncell = 10 for Nier1s; ncell = 10 for Nier1b.
We performed another round of evolution from Nier1s to demonstrate the efficiency and convenience of Faculae. We constructed a randomly mutated library of 14 sites based on previously reported mutations in iBB-GECO1, an FR calcium indicator engineered from NIR-GECO2.38 In the first round of enrichment, we photoselected 1,986 cells within 50 min and retrieved 800 cells from FACS, which were amplified for the next round of iterative library construction (Figure 6A). In the second round of enrichment, we observed a substantial increase in the average brightness (2.1 × 102 versus 4.4 × 102), with few changes in the average fluorescence response (−26.2% versus −26.0%) of the cell population (Figure 6B). We sequenced individual cells following the same procedure as in the evolution of Nier1s and found that 8 out of 21 clones were Nier1s, while 7 clones contained 323ΔGG deletion (Figure S6). We evaluated Nier1s-323ΔGG and other mutants in HEK 293T cells (Figure 6C; Table S6). Nier1s-323ΔGG has lower brightness (0.82 ± 0.04) despite its higher SBR (1.06 ± 0.05) compared with Nier1s. Unexpectedly, Nier1s-S373Y and Nier1s-Y271F/I378C showed increased baseline brightness (1.54 ± 0.07; 1.24 ± 0.09) and ΔF/F0 (1.15 ± 0.02; 1.10 ± 0.03). These results demonstrate that Faculae is fast and convenient for iterative evolution.
Figure 6.

Iterative evolution by Faculae
(A) Pipeline for Faculae-based iterative evolution starting from Nier1s.
(B) The screening contour of the libraries in two rounds. In each library, the contour lines from center to periphery represent relative estimated density from 83% to 17%.
(C) Characterization of the SBR (n ≥ 10 cells per mutant) and brightness (n ≥ 2 replicates) for each mutant in HEK 293T cells. Error bars represent SEM.
Discussion
We developed Faculae, a functional imaging-guided cell selection strategy for engineering genetically encoded fluorescent indicators. We achieved multiparametric screen of 0.4–1 × 105 cells within 1.5 h and photoactivation of ∼2 × 103 cells within 50 min. Faculae has several advantages. In terms of screening throughput per round, it is on par with Photopick (0.6–1 × 105) and outperforms Opto-MASS (0.1–0.2 × 105) by several fold. Faculae also enables fast photoselection of target cells, with 1–1.5 s spot illumination per cell, which compares favorably against the aspiration-based approach in Opto-MASS (typically requiring 1–2 min per cell).
Faculae and Photopick share a conceptually similar phototagging mechanism but differ in one key aspect: while Photopick uses DMD-based wide-field illumination to photoconvert multiple cells in parallel at a low power density (0.04 W/cm2), Faculae applies spot illumination to photoconvert individual cells one at a time with a high power density (80 W/cm2). Faculae is thus more efficient than Photopick when only a few cells (<2 × 103) need to be retrieved. If one expects to collect a large population of cells from the pool, the Photopick approach would be more suitable.
Faculae has enabled the measurement of tens of thousands of mutants, allowing us to rapidly improve the brightness and SBR of FR-GECIs. We envision that Faculae has extensive applications for sensor engineering. It is compatible with biosensors of various spectra, including green, red, FR, and near-infrared, because both PA-GFP and PAmCherry can provide a high photoactivation contrast ratio for FACS. The CAG-LLP HEK 293T line ensures strong expression of target proteins, making Faculae suitable for unoptimized biosensors with low fluorescent intensity.
In addition to brightness and SBR, Faculae can in principle be leveraged to assess many other properties of biosensors. For example, the photostability of fluorescent indicators can be tested by photobleaching illumination, while the substrate affinity can be assayed by dosing treatment, as previously reported.11,39 Furthermore, subcellular protein distribution and its dynamic changes could also be readily monitored due to the high spatiotemporal resolution of fluorescence imaging.9,13 Since Faculae is performed in mammalian cells, it would be particularly useful for the directed evolution of membrane proteins, such as indicators based on G protein-coupled receptors and rhodopsins,1,3 which has proven difficult for bacterial screens. Similarly, Faculae would be valuable for engineering tools that are designed to perturb the mammalian systems, such as chemogenetic tools and optogenetic tools,40,41 or those that rely on complex mammalian pathways, such as biosensors for kinase activity.42
The FR-GECIs, namely Nier1s and Nier1b, showed improved SBR and brightness compared to template, emphasizing the importance of large-scale multiparameter screens in mammalian cells. During directed evolution for NIR-GECO1, the three key mutations D133V, I302N, and F466S were introduced to the sensor scaffold.18 However, our screening experiments identified better combinations (e.g., NIR-GECO2G-133Q/302K/466M for Nier1s and NIR-GECO2G-133E/302T/466C for Nier1b) on these sites, resulting in better FR-GECIs for recording neural activity. Although array-based screening of FR-GECI mutants in bacterial lysate in the original studies achieved multidimensional characterization, it was still limited by screening throughput and did not match the intended mammalian environment, which might lead to suboptimal mutations.43
The gene recovery efficiency of Faculae depends on the FACS retrieval yield and the success of single-cell PCR, both of which are imperfect. The average FACS retrieval yield is 44% ± 8% (mean ± SD, 2 trials), which is close to a previously reported yield of 54%.11 The PCR recovery rate varies from 24% to 51%. We speculate that the single copy of the gene in the LLP cell line causes the low recovery rate, making it difficult to amplify the mutant gene. We can compensate for the gene information loss by increasing the redundancy of mutants in the initial cell library.
As an image-based directed evolution methodology in mammalian system, Faculae can be further optimized on the following aspects: library construction and phenotyping.44,45 First, the library scale can be increased by incorporating newly developed recombinase into the LLP technique.46 Second, the phenotyping throughput can be further improved by applying large field-of-view setups and advanced cell detection approaches47,48 so that we can derive complex phenotypic information from a greater number of cells. Overall, we anticipate that Faculae will become a generalizable method for biomolecular engineering.
Limitations of the study
Faculae is a pooled screening platform based on imaging. While it can measure both static and dynamic parameters such as brightness and SBR across multiple fields of view, it is not suited for measuring kinetic parameters such as fluorescence response time constants. Faculae also relies on FACS for sorting tagged cells, which requires that drug treatment during the imaging session should not damage cells.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Dulbecco’s Modified Eagle’s medium (DMEM) | Gibco | C11995500BT |
| Fetal Bovine Serum (FBS) | Gibco | 100099044 |
| Trypsin-EDTA (0.25%) | Gibco | 25200056 |
| Neurobasal™ Medium | Gibco | 21103049 |
| B-27™ Supplement | Gibco | 17504044 |
| GlutaMAX™ Supplement | Gibco | 35050061 |
| Penicillin-streptomycin | Beyotime | C0222 |
| HBSS | Gibco | 14025092 |
| Blasticidin | Selleck | S7419 |
| Matrigel® Matrix | Corning | 356234 |
| poly-D-lysine | Sigma | P7280-5X5M |
| Laminin Mouse Protein | Gibco | 23017015 |
| Opti-MEM® Medium | Gibco | 31985062 |
| Ionomycin | Cayman | 11932 |
| Protease K | Thermo | EO0491 |
| HEPES | Amresco | 0511 |
| EGTA | Sigma | 03777-10G |
| 2-APB | Abcam | ab120124 |
| Gabazine | Abcam | ab120042 |
| NBQX | Abcam | ab120045 |
| D-AP5 (APV) | Abcam | ab120003 |
| Critical commercial assays | ||
| Phanta® Max Super-Fidelity DNA Polymerase | Vazyme | P505-d2 |
| ClonExpress II One Step Cloning Kit | Vazyme | C112-02 |
| DNA extraction kit | TIANGEN | DP118-02 |
| E.Z.N.A.® Gel Extraction kit | Omega | D2500 |
| Ultra-Competent Cell Preps Kit | Sangon | B529303-0200 |
| Lipofectamine® 2000 Reagent | Gibco | 11668019 |
| Lipofectamine® 3000 Reagent | Gibco | L3000008 |
| Experimental models: Cell lines | ||
| HEK 293T | ATCC | Cat#CRL-3216 |
| Oligonucleotides | ||
| Primers for mutation and single cell PCR, see Table S7 | This paper | N/A |
| Recombinant DNA | ||
| Plasmids for corresponding experiments, see Table S8 | This paper | N/A |
| Software and algorithms | ||
| FIJI/ImageJ | National Institutes of Health | Version 1.53t (https://imagej.net/Fiji) |
| MATLAB | The Mathworks, Inc., Natick, MA | R2018b (or later) |
| Micro-Manager | Arthur et al.49 | 2.0.0-gamma1 |
| Custom code | This paper | See Method S1 |
| LabVIEW | National Instruments Corp. | Version 2015 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Peng Zou (zoupeng@pku.edu.cn).
Materials availability
Reagents in this study are available by request to lead contact, Dr. Peng Zou (zoupeng@pku.edu.cn).
Experimental model and subject details
Cultured cell models
Human embryonic kidney 293T (HEK 293T) cells were incubated in Dulbecco’s modified eagle medium (DMEM, Gibco) medium with 10% v/v fetal bovine serum (FBS, Gibco) at 37°C with 5% CO2.
All experiments were done in accordance with the Experimental Animal Management Ordinance of Beijing, P. R. China. The protocol was approved by The Institutional Animal Care and Use Committee in Peking University. We did not select animal sex, and animals were housed in standard conditions. Primary rat hippocampal neurons were cultured as previous described.50 Briefly, glass coverslips were incubated with 20 μg/mL poly-D-lysine (Sigma) solution and 10 μg/mL Laminin Mouse Protein (Gibco) solution for 24–48 h, respectively. Neurons were isolated from brains of neonatal Sprague-Dawley rat pups (postnatal day 0), and collected and diluted by neuronal culture medium (Neurobasal medium, B-27 supplement, GlutaMAX supplement and penicillin-streptomycin) to a final cell density of 6×104 cells/mL. 1 mL cell suspension was added to each 24-well containing pre-coated glass coverslip. Half of the neuron culture medium was replaced with fresh medium once every 4 days.
Method details
Molecular cloning
Plasmids were constructed using seamless cloning technique. The inserts and the vectors were PCR amplified (Vazyme, P505), gel purified (Omega, D2500) and then mixed with enzymes following manufacturer’s instructions (Vazyme, C112). Successful clones were verified by Sanger sequencing. Site-directed mutagenesis and linker insertion/deletion were achieved by PCR overlap extension. The NIR-GECO1 gene and Bxb1 gene were synthesized by company. The descriptions of primers and plasmids used in this study are in Table S7 and Table S8. The plasmid maps are available from the corresponding authors upon request.
Cell transfection
HEK 293T cells were seeded in a 24-well plate and grown to 90% confluent for transfection. For each well, 500 ng plasmid and 1 μL Lipofectamine 2000 Reagent were mixed in Opti-MEM Medium and incubated for 5–10 min at room temperature. The mixture was added to the cell culture medium in the absence of serum for 4–8 h. Thereafter, cells were digested by Trypsin-EDTA (0.25%, Gibco), reseeded on a sterile 14-mm glass coverslip or a glass dish pre-treated with Matrigel Matrix, and incubated in the complete medium (DMEM+10%FBS) for 24–48 h before imaging.
Neurons were transfected on DIV7-9 (7–9 days in vitro). For each well, 500 ng plasmid DNA was mixed with 1.5 μL of Lipofectamine 3000 Reagent in the Neurobasal medium, before incubating with neurons for 45 min. The transfected neurons were imaged on DIV16-18.
Library construction
The NIR-GECO plasmid libraries (mIFP lib and CaM lib) were constructed via combinatorial codon mutagenesis36 and MEGAWHOP method.51 Mutations were introduced by designing oligonucleotide contain NNK triplet at the target sites. The upstream and downstream of primers contained 16 nucleotides for annealing (full length: 35 bp). The 17 forward-mutagenesis primers were mixed in equimolar quantities to generate the forward primer pool. The reverse primer pool was created by equally combined reverse complement of each of forward-mutagenesis primers (Table S7). The forward fragment PCR reaction contained 10 ng template, 1 μL forward primer pool (10 μM totally), 1 μL tail primer (10 μM), 12.5 μL 2×Phanta Max Buffer, 0.5 μL dNTP (10 mM each), 0.5 μL Phanta Max Super-Fidelity DNA Polymerase and 9 μL ddH2O. The primer pair in reverse fragment PCR reaction was replaced by reverse primer pool and head primer. The forward and reverse reaction were used as templates for joining PCR without purification (1 μL each). The PCR program was set as below: 1) 95°C for 3 min; 2) 95°C for 15 s; 3) 50°C for 30 s, cooling to 50 °C at 0.5 °C/s; 4) 72°C for 60 s; 5) Repeat steps 2 through 4 for 8 additional cycles if in fragment PCR, or 19 additional cycles if in joining PCR; 6) 72°C for 3 min; 7) Hold at 4°C. The PCR product was purified by gel extraction and used as the template for a second round of fragment-joining PCR step. The second-round PCR product was purified for MEGAWHOP. Mutations in combined lib were introduced by overlap extension PCR. The PCR product was purified for MEGAWHOP.
To create plasmid library from PCR products, 50 ng template attB plasmid (template) and 500–750 ng fragment (megaprimer) were mixed in 25 μL MEGAWHOP PCR reaction consisting of 12.5 μL 2×Phanta Max Buffer, 0.5 μL dNTP (10 mM each), 0.5 μL Phanta Max Super-Fidelity DNA Polymerase and ddH2O. The PCR program was set as below: 1) 95°C for 3 min; 2) 95°C for 15 s; 3) 60°C for 15 s; 4) 72°C for 4 min; 5) Repeat steps 2 through 4 for 24 additional cycles; 6) 72°C for 3 min; 7) Hold at 4°C. The reaction was monitored by agarose gel electrophoresis as previously described.51 The MEGAWHOP product was transferred into ultra-competent DH5α after DpnI treatment. Each 2 μL product was used to transform 1 tube of competent E. coli cells (100 μL). Transformation produced ∼1×105 constructs for 25 μL MEGAWHOP reaction. The competent cells were prepared using commercial kit based on rubidium chloride protocol (Sangon, B529303-0200). Plasmids were extracted from construct mixture (TIANGEN, DP118-02).
For cell library construction, CAG-LLP cells were seeded on a 6-cm dish to reach 90% confluence. Cells were transfected following Lipofectamine 3000 protocol (5.5 μg DNA, 11 μL P3000, 16 μL Lipofectamine 3000). After 6–8 h, cells were digested by trypsin and reseeded on a 10-cm dish. After 2 days, GFP+/mIFP+ cells were collected by FACS (∼200k) and expanded for at least 4 days. Then cells with strong GFP/mIFP signals were FACS enriched (∼100k) and seeded on a 35-mm glass dish. After 1 day, medium was changed to Opti-MEM Medium for reduced-serum cell culture, which could reduce cell movement during imaging. Cells were screened 1 day later. For miRFP680 screening and NIR-GECO screening test, cell libraries were produced and collected as described above but without expansion and FACS enrichment step.
Stable cell line construction
Lentivirus was produced in HEK293T cells. First, cells were seeded and grow to reach 90% confluence. Next, a DNA mix was prepared by combining expression plasmid, delta8.9 and VSVG at a mass ratio of 1:1:0.7 in Opti-MEM. The DNA was mixed with P3000 and then incubated with Lipofectamine 3000 at a ratio of 1:2:3 (w:v/v). Next, the above mix was added to cells and incubated at 37°C for 6–8 h. The old medium was aspirated and new complete medium was replenished. Cells were incubated for 40–48 h. The supernatant was collected and filtered through a 0.45-μm syringe. This lentivirus-contained medium was aliquoted and stored at −80°C.
For LLP cell line construction, HEK293T cells were infected with ∼0.1MOI when cell confluence reached 60%. After 48 h, cells were collected and sorted. BFP+ single cells were seeded into a 96-well plate. Each well contain one clone in 150 μL complete medium. Cell clones grew at least 2 weeks before further validation. The desired clone was kept and expanded for following experiments. Cell culture medium was supplemented with 2.5 μg/mL Blasticidin (Selleck, S7419).
Imaging apparatus and confocal microscopy
All of the fluorescence imaging experiments were conducted on an inverted fluorescence microscope (Nikon-TiE) equipped with a 20 ×0.75 NA dry immersion objective lens (except for calcium imaging in neuron and photobleaching test, in which a 40× NA 1.3 oil immersion objective lens was used), four laser lines (Coherent OBIS 405 nm, 488 nm, 561 nm, and 637 nm), a spinning disk confocal unit (Yokogawa CSU-X1), and two scientific CMOS cameras (Hamamatsu ORCA-Flash 4.0 v2). The microscope, lasers, and cameras were controlled with either custom-built software written in LabVIEW (National Instruments, 15.0 version) or Micro-Manager 2.0 (MM2.0) and could switch between confocal and wide-field imaging modes. The spectra properties of filters and dichroic mirrors for various fluorescent indicators used in this study are summarized in Table S9. Image analysis was performed in ImageJ/Fiji (version 1.53t). ImageJ/Fiji and MM2.0 were run within MATLAB.
Time-lapse imaging
For ionomycin/CaCl2/2-APB test, the transfected/recombined HEK293T cells were seeded on a 35-mm glass dish to reach 60–80% confluence. Cells were maintained in an extracellular (EC) Ca2+-free medium containing 5 mM KCl, 135 mM NaCl, 1 mM MgCl2, 0.4 mM KH2PO4, 10 mM glucose, and 20 mM HEPES (pH 7.4) at room temperature. Cells were incubated in EC with 50 μM 2-APB for at least 10 min before imaging. Time-lapse images were acquired with 20 s interval by LabVIEW. After 10 time points (∼200 s), the medium was replaced with EC containing 50 μM 2-APB, 2 μM Ionomycin and 1 mM CaCl2 by peristaltic pump.
Imaging-based screening
For NIR-GECO screening, cells were incubated in EC with 50 μM 2-APB for at least 10 min before imaging. The first step was to acquire ∼500 2-channel images (330 μm × 330 μm each, defined as “pre-library” and “pre-marker”) on a glass dish. Second, the buffer was replaced with EC containing 50 μM 2-APB, 2 μM Ionomycin and 1 mM CaCl2. After 12 min, the second round of image acquisition of the same positions was performed (“post-library” and “post-marker”). All these images were processed through flat-filed correcting, including background subtraction. The post-marker images were segmented by StarDist.52 ROIs were used to calculate nuclear sizes, intensities and xy coordinates. Brightness was defined as the maximum fluorescence (Fpre-library) of each cell. SBR was calculated as (Fpost-library/Fpost-marker - Fpre-library/Fpre-marker)/(Fpre-library/Fpre-marker). For each library, 1.0–1.2×105 cells were screened in 1 or 2 dishes. Cells with both high brightness and large SBR (greater than the averaged values for the template) were manually gated. The positions of these cells were retrieved under microscope by moving the stage automatically. Cells were photoactivated one by one for 1 s using focal illumination of 405 nm laser with 10% power (Coherent OBIS 405 nm, 50 mW). After photoactivation, the cells were maintained in EC containing 1 μM Ionomycin and 1 mM EGTA for 10 min. This step aimed to reduce intracellular concentration of calcium ion. Cells were digested, centrifuged and resuspended in PBS with 2% FBS. The cell suspension was kept on ice until FACS.
For miRFP680 screening, cells were incubated in HBSS for at least 10 min before imaging. the first step was to acquire 500–1000 2-channel images (defined as “library” and “marker”) on a glass dish. All these images were processed through flat-filed correcting, including background subtraction. The marker images were segmented by StarDist. ROIs were used to calculate nuclear sizes, intensities and xy coordinates. Brightness was defined as the fluorescence (Flibrary and Fmarker) of each cell. Cells with high brightness were gated and photoactivated. After photoactivation, cells were digested, centrifuged and resuspended in HBSS with 2% FBS. The cell suspension was kept on ice until FACS.
For single cell nested PCR amplification, 96-well PCR plates were used to receive PAmCherry+ single cells. Each well contained 5 μL PBS with 125 μg/mL Protease K. The plates were quickly frozen in liquid nitrogen after FACS. The lysates were treated in 56°C for 15 min and 95°C for 5 min. Next, 1 μL forward primer-1 (10 μM), 1 μL reverse primer-1 (10 μM), 12.5 μL 2×Phanta Max Buffer, 0.5 μL dNTP (10 mM each), 0.5 μL Phanta Max Super-Fidelity DNA Polymerase and 4.5 μL ddH2O were added into the lysate in each well. After the first round of PCR, 3 μL reaction was used as template to perform the second round of PCR, while the primer pair was forward primer-2 and reverse primer-2. The PCR program was set as below: 1) 95°C for 3 min; 2) 95°C for 15 s; 3) 61°C for 15 s; 4) 72°C for 90 s; 5) Repeat steps 2 through 4 for 24 additional cycles if in first round PCR, or 34 additional cycles if in second round PCR; 6) 72°C for 3 min; 7) Hold 4°C. The PCR products were gel purified and verified by Sanger sequencing.
Flow cytometry
Cells were disassociated by trypsin and centrifuged at 300 g for 3 min. Cells were re-suspended in Hank’s Balanced Salt Solution (HBSS, Gibco) containing 2% FBS and kept on ice until flow cytometry. Astrios EQ (BeckMan Coulter) was used for FACS sorting. The sorted cells were either lysed for PCR (see “Imaging-based screening” section) or cultured in DMEM with 10% FBS (see “Stable cell line construction” section). LSRFortessa was used for flow cytometry analysis (Becton Dickinson). Optical configuration of LSRFortessa: BFP (Ex: 405 nm, Em: 450/50), GFP (Ex: 488 nm, Em: 530/30), mCherry (Ex: 561 nm, Em: 610/20), miRFP680/mIFP (Ex: 640 nm, Em: 730/45).
Electrophysiology
For single-cell electrophysiology recording, cultured neurons were incubated in Tyrode’s buffer containing 20 μM Gabazine, 10 μM NBQX and 25 μM APV. The electrophysiology experiments were performed at room temperature. Borosilicate glass electrodes (Sutter) were pulled to a tip resistance of 2.5–5 MΩ. The glass electrode was filled with internal solution containing 145 mM potassium gluconate, 5 mM MgCl2, 10 mM HEPES, and 4 mM Na2-ATP (pH 7.2, with KOH). The glass electrode’s position was adjusted by a Sutter MP285 micro-manipulator. Membrane potentials were recorded under whole-cell current clamp under I = 0 mode (Axopatch 200B, Axon Instruments). Recorded membrane potential data were filtered with a 5 kHz internal Bessel filter in the amplified and digitized with a National Instruments PCIe-6353 data acquisition 1067 (DAQ) board. The microscope (Nikon Ti-E), the camera (Hamamatsu ORCA-Flash 4.0 v2) and electrophysiology recording system were controlled with a customized software written in LabVIEW 2015 (National Instruments), and the data were extracted and analyzed with a home-made script written in MATLAB R2018b (MathWorks).
Brightness and photobleaching test
HEK293T cells were transfected with pcDNA3.1-NIR-GECO-mut-3NLS-P2A-EGFP-3NLS using Lipofectamine 2000 Reagent as described in “Cell Culture and Transfection”. For brightness test, cells were analyzed using flow cytometry. The brightness of each mutant was calculated as the median intensity of mIFP+ cells. For photobleaching test, cells were continuously illuminated under microscope with 40× NA 1.3 objective. The power density of 637 nm laser was 9.0 W/cm2.
Protein purification and in vitro characterization
The gene encoding Nier1s, Nier1b and NIR-GECO2G, with a polyhistidine tag on the N-terminus, was expressed from the pJC vector in E. coli strain BL21 (DE3). The bacteria were lysed with an ultrasonic cell disruptor and then centrifuged at 20000 rpm for 30 min. Extracted protein was purified by Ni-NTA agarose (QIAGEN). The eluted sample was dialyzed in PBS containing 1 mM DTT overnight. Proteins were further concentrated with an Amicon Ultra-15 Centrifugal Filter Device (Merck). The buffer was exchanged to 10 mM MOPS, 100 mM KCl (pH 7.2). We prepared a CaEGTA buffer (30 mM MOPS, 100 mM KCl, 10 mM EGTA, 10 mM CaCl2) and an EGTA buffer (30 mM MOPS, 100 mM KCl, 10 mM EGTA).
Absorbance spectra of FR-GECIs were measured with NanoDrop 2000c (Thermo). Excitation and emission of FR-GECIs were measured with the Fluorescence Spectrophotometer F-7000 (HITACHI). For determination of the extinction coefficient, a ratio of maximum absorbance values of Q and Soret bands was calculated, assuming the latter to have the extinction coefficient of 39900 M−1 cm−1. We used integrating sphere (FLS 980) to measure the absolute quantum yields of the FR-GECI solutions.
We performed pH titrations by diluting protein into buffers (pH from 2 to 11) containing 30 mM trisodium citrate, 30 mM sodium borate, 30 mM MOPS, 100 mM KCl, 10 mM EGTA, and either no CaCl2 or 10 mM CaCl2. Fluorescence intensities as a function of pH were then fitted by a sigmoidal binding function to determine the apparent pKa. Ca2+ titrations were carried out using Calcium calibration buffer (Calcium Calibration Buffer Kit, Biotium, 59100). Fluorescence intensities against Ca2+ concentrations were fitted by a sigmoidal binding function to determine the Hill coefficient and Kd.
Wide-field calcium imaging in rat hippocampal neuron culture
The pAAV-hSyn-NIR-GECO-mut-IRES-EGFP-3NLS plasmid was transfected into cultured rat hippocampal neurons as described above. To analyze sensor brightness in neurons, more than 50 cells were snapshotted in mIFP/GFP channel for each mutant. 3–10 cells for each mutant were stimulated to fire 1–10 AP by patch clamp and the calcium ion flux was recorded simultaneously. To stimulate firing of 1–10 individual action potential (AP), 1–10 300-pA current pulses were injected to cultured neurons for 10 ms duration at a repetition rate of 50 Hz under I-CLAMP NORMAL mode (at 21159.48 Hz). The interval of each step from 1 to 10 AP was around 12 s. Fluorescence images were acquired at 48 Hz camera frame rate with 2-by-2 binning. neurons were illuminated with 637 nm laser at 3.3 W/cm2.
Quantification and statistical analysis
Both the electrical data and the fluorescence images were analyzed with home-built scripts written in MATLAB R2018b (MathWorks). MATLAB code is available from Method S2. For neural calcium imaging, fluorescence intensities of cells were extracted from the mean pixel values of a manually drawn ROI around the soma. Following camera bias subtraction (100 and 400 for 1-by-1 and 2-by-2 binning, respectively), the fluorescence signal was corrected for photobleaching and smoothed by Savitzky-Golay filter. Statistical analysis was performed with Excel (Microsoft Excel 2019) and Origin (version 2022b).
Acknowledgments
We thank Dr. Jun Chu (Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences) for providing the miRFP680 plasmid, Dr. Pingyong Xu (Institute for Biophysics, Chinese Academy of Sciences) for providing the PAmCherry plasmid, and Dr. Kenneth A. Matreyek for helpful discussion on the LLP method. We thank Shaoran Zhang and Zirun Zhao for advice on writing and Dr. Yuxin Fang, Yu Chen, and Jun Yang for advice on experimental design. We thank the flow cytometry core at the National Center for Protein Sciences at Peking University, particularly Dr. Hongxia Lyu, Dr. Jia Luo, Xuefang Zhang, and Huan Yang for technical assistance. We thank Mingxing Chen and Wenjun Yan at the Analytical Instrumentation Center of Peking University for the help with spectrum test. This work was supported by the Ministry of Science and Technology (2022YFA1304700 and 2018YFA0507600) and the National Natural Science Foundation of China (32088101). P.Z. is sponsored by the Bayer Investigator Award.
Author contributions
Conceptualization, P.Z. and C.L.; methodology, P.Z. and C.L.; software, C.L.; validation, P.Z., C.L., and L.L.; investigation, C.L. and L.L.; writing – original draft, P.Z. and C.L.; writing – review & editing, P.Z., C.L., and L.L.; funding acquisition, P.Z.; supervision, P.Z.
Declaration of interests
The authors declare no competing interests.
Published: July 27, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2023.100544.
Supplemental information
Data and code availability
-
•
All the data published in this paper will be available from the lead contact upon request.
-
•
The MATLAB code is available as supporting information in this manuscript.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Sabatini B.L., Tian L. Imaging neurotransmitter and neuromodulator dynamics in vivo with genetically encoded indicators. Neuron. 2020;108:17–32. doi: 10.1016/j.neuron.2020.09.036. [DOI] [PubMed] [Google Scholar]
- 2.Inoue M. Genetically encoded calcium indicators to probe complex brain circuit dynamics in vivo. Neurosci. Res. 2021;169:2–8. doi: 10.1016/j.neures.2020.05.013. [DOI] [PubMed] [Google Scholar]
- 3.Xu Y., Zou P., Cohen A.E. Voltage imaging with genetically encoded indicators. Curr. Opin. Chem. Biol. 2017;39:1–10. doi: 10.1016/j.cbpa.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Babakhanova S., Jung E.E., Namikawa K., Zhang H., Wang Y., Subach O.M., Korzhenevskiy D.A., Rakitina T.V., Xiao X., Wang W., et al. Rapid directed molecular evolution of fluorescent proteins in mammalian cells. Protein Sci. 2022;31:728–751. doi: 10.1002/pro.4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Villette V., Chavarha M., Dimov I.K., Bradley J., Pradhan L., Mathieu B., Evans S.W., Chamberland S., Shi D., Yang R., et al. Ultrafast two-photon imaging of a high-gain voltage indicator in awake behaving mice. Cell. 2019;179:1590–1608.e23. doi: 10.1016/j.cell.2019.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dong A., He K., Dudok B., Farrell J.S., Guan W., Liput D.J., Puhl H.L., Cai R., Wang H., Duan J., et al. A fluorescent sensor for spatiotemporally resolved imaging of endocannabinoid dynamics in vivo. Nat. Biotechnol. 2022;40:787–798. doi: 10.1038/s41587-021-01074-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wardill T.J., Chen T.W., Schreiter E.R., Hasseman J.P., Tsegaye G., Fosque B.F., Behnam R., Shields B.C., Ramirez M., Kimmel B.E., et al. A neuron-based screening platform for optimizing genetically-encoded calcium indicators. PLoS One. 2013;8 doi: 10.1371/journal.pone.0077728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kanfer G., Sarraf S.A., Maman Y., Baldwin H., Dominguez-Martin E., Johnson K.R., Ward M.E., Kampmann M., Lippincott-Schwartz J., Youle R.J. Image-based pooled whole-genome CRISPRi screening for subcellular phenotypes. J. Cell Biol. 2021;220 doi: 10.1083/jcb.202006180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan X., Stuurman N., Ribeiro S.A., Tanenbaum M.E., Horlbeck M.A., Liem C.R., Jost M., Weissman J.S., Vale R.D. High-content imaging-based pooled CRISPR screens in mammalian cells. J. Cell Biol. 2021;220 doi: 10.1083/jcb.202008158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hasle N., Cooke A., Srivatsan S., Huang H., Stephany J.J., Krieger Z., Jackson D., Tang W., Pendyala S., Monnat R.J., Jr., et al. High-throughput, microscope-based sorting to dissect cellular heterogeneity. Mol. Syst. Biol. 2020;16 doi: 10.15252/msb.20209442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee J., Liu Z., Suzuki P.H., Ahrens J.F., Lai S., Lu X., Guan S., St-Pierre F. Versatile phenotype-activated cell sorting. Sci. Adv. 2020;6 doi: 10.1126/sciadv.abb7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chien M.-P., Brinks D., Testa-Silva G., Tian H., Phil Brooks F., Adam Y., Bloxham B., Gmeiner B., Kheifets S., Cohen A.E. Photoactivated voltage imaging in tissue with an archaerhodopsin-derived reporter. Sci. Adv. 2021;7:eabe3216. doi: 10.1126/sciadv.abe3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kudo T., Lane K., Covert M.W. A multiplexed epitope barcoding strategy that enables dynamic cellular phenotypic screens. Cell Syst. 2022;13:376–387.e8. doi: 10.1016/j.cels.2022.02.006. [DOI] [PubMed] [Google Scholar]
- 14.Tian H., Davis H.C., Wong-Campos J.D., Park P., Fan L.Z., Gmeiner B., Begum S., Werley C.A., Borja G.B., Upadhyay H., et al. Video-based pooled screening yields improved far-red genetically encoded voltage indicators. Nat. Methods. 2023 doi: 10.1038/s41592-022-01743-5. 2023/01/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rappleye M., Gordon-Fennel A., Castro D.C., Matarasso A.K., Zamorano C.A., Stine C., Wait S.J., Lee J.D., Siebart J.D., Suko A., et al. Opto-MASS: a high-throughput engineering platform for genetically encoded fluorescent sensors enabling all optical in vivo detection of monoamines and neuropeptides. bioRxiv. 2022 doi: 10.1101/2022.06.01.494241. [DOI] [Google Scholar]
- 16.Shemetov A.A., Monakhov M.V., Zhang Q., Canton-Josh J.E., Kumar M., Chen M., Matlashov M.E., Li X., Yang W., Nie L., et al. A near-infrared genetically encoded calcium indicator for in vivo imaging. Nat. Biotechnol. 2021;39:368–377. doi: 10.1038/s41587-020-0710-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qian Y., Cosio D.M.O., Piatkevich K.D., Aufmkolk S., Su W.C., Celiker O.T., Schohl A., Murdock M.H., Aggarwal A., Chang Y.F., et al. Improved genetically encoded near-infrared fluorescent calcium ion indicators for in vivo imaging. PLoS Biol. 2020;18 doi: 10.1371/journal.pbio.3000965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qian Y., Piatkevich K.D., Mc Larney B., Abdelfattah A.S., Mehta S., Murdock M.H., Gottschalk S., Molina R.S., Zhang W., Chen Y., et al. A genetically encoded near-infrared fluorescent calcium ion indicator. Nat. Methods. 2019;16:171–174. doi: 10.1038/s41592-018-0294-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Victora G.D., Schwickert T.A., Fooksman D.R., Kamphorst A.O., Meyer-Hermann M., Dustin M.L., Nussenzweig M.C. Germinal Center Dynamics Revealed by Multiphoton Microscopy with a Photoactivatable Fluorescent Reporter. Cell. 2010;143:592–605. doi: 10.1016/j.cell.2010.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tomura M., Yoshida N., Tanaka J., Karasawa S., Miwa Y., Miyawaki A., Kanagawa O. Monitoring cellular movement in vivo with photoconvertible fluorescence protein “Kaede” transgenic mice. Proc. Natl. Acad. Sci. USA. 2008;105:10871–10876. doi: 10.1073/pnas.0802278105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Medaglia C., Giladi A., Stoler-Barak L., De Giovanni M., Salame T.M., Biram A., David E., Li H., Iannacone M., Shulman Z., Amit I. Spatial reconstruction of immune niches by combining photoactivatable reporters and scRNA-seq. Science. 2017;358:1622–1626. doi: 10.1126/science.aao4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Callaway E.M., Katz L.C. Photostimulation using caged glutamate reveals functional circuitry in living brain slices. Proc. Natl. Acad. Sci. USA. 1993;90:7661–7665. doi: 10.1073/pnas.90.16.7661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Subach F.V., Patterson G.H., Manley S., Gillette J.M., Lippincott-Schwartz J., Verkhusha V.V. Photoactivatable mCherry for high-resolution two-color fluorescence microscopy. Nat. Methods. 2009;6:153–159. doi: 10.1038/nmeth.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patterson G.H., Lippincott-Schwartz J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science. 2002;297:1873–1877. doi: 10.1126/science.1074952. [DOI] [PubMed] [Google Scholar]
- 25.Matreyek K.A., Stephany J.J., Chiasson M.A., Hasle N., Fowler D.M. An improved platform for functional assessment of large protein libraries in mammalian cells. Nucleic Acids Res. 2020;48:e1. doi: 10.1093/nar/gkz910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matlashov M.E., Shcherbakova D.M., Alvelid J., Baloban M., Pennacchietti F., Shemetov A.A., Testa I., Verkhusha V.V. A set of monomeric near-infrared fluorescent proteins for multicolor imaging across scales. Nat. Commun. 2020;11:239. doi: 10.1038/s41467-019-13897-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bai D., del Corsso C., Srinivas M., Spray D.C. Block of specific gap junction channel subtypes by 2-aminoethoxydiphenyl borate (2-APB) J. Pharmacol. Exp. Therapeut. 2006;319:1452–1458. doi: 10.1124/jpet.106.112045. [DOI] [PubMed] [Google Scholar]
- 28.Bootman M.D., Collins T.J., Mackenzie L., Roderick H.L., Berridge M.J., Peppiatt C.M. 2-aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. Faseb. J. 2002;16:1145–1150. doi: 10.1096/fj.02-0037rev. [DOI] [PubMed] [Google Scholar]
- 29.Pédelacq J.D., Cabantous S., Tran T., Terwilliger T.C., Waldo G.S. Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 2006;24:79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- 30.Subach O.M., Barykina N.V., Anokhin K.V., Piatkevich K.D., Subach F.V. Near-infrared genetically encoded positive calcium indicator based on GAF-FP bacterial phytochrome. Int. J. Mol. Sci. 2019;20 doi: 10.3390/ijms20143488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Subach O.M., Subach F.V. GAF-CaMP3-sfGFP, An Enhanced Version of the Near-Infrared Genetically Encoded Positive Phytochrome-Based Calcium Indicator for the Visualization of Neuronal Activity. Int. J. Mol. Sci. 2020;21 doi: 10.3390/ijms21186883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao Y., Araki S., Wu J., Teramoto T., Chang Y.-F., Nakano M., Abdelfattah A.S., Fujiwara M., Ishihara T., Nagai T., Campbell R.E. An expanded palette of genetically encoded Ca2+ indicators. Science. 2011;333:1888–1891. doi: 10.1126/science.1208592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen T.W., Wardill T.J., Sun Y., Pulver S.R., Renninger S.L., Baohan A., Schreiter E.R., Kerr R.A., Orger M.B., Jayaraman V., et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dana H., Sun Y., Mohar B., Hulse B.K., Kerlin A.M., Hasseman J.P., Tsegaye G., Tsang A., Wong A., Patel R., et al. High-performance calcium sensors for imaging activity in neuronal populations and microcompartments. Nat. Methods. 2019;16:649–657. doi: 10.1038/s41592-019-0435-6. [DOI] [PubMed] [Google Scholar]
- 35.Zarowny L., Aggarwal A., Rutten V.M.S., Kolb I., GENIE Project. Patel R., Huang H.Y., Chang Y.F., Phan T., Kanyo R., et al. Bright and High-Performance Genetically Encoded Ca2+ Indicator Based on mNeonGreen Fluorescent Protein. ACS Sens. 2020;5:1959–1968. doi: 10.1021/acssensors.0c00279. [DOI] [PubMed] [Google Scholar]
- 36.Belsare K.D., Andorfer M.C., Cardenas F.S., Chael J.R., Park H.J., Lewis J.C. A simple combinatorial codon mutagenesis method for targeted protein engineering. ACS Synth. Biol. 2017;6:416–420. doi: 10.1021/acssynbio.6b00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Z., Liu X., Chen W., Shen F., Bi H., Ke G., Zhang L. Uni-Fold: an open-source platform for developing protein folding models beyond AlphaFold. bioRxiv. 2022 doi: 10.1101/2022.08.04.502811. Preprint at. 2022.2008.2004.502811. [DOI] [Google Scholar]
- 38.Hashizume R., Fujii H., Mehta S., Ota K., Qian Y., Zhu W., Drobizhev M., Nasu Y., Zhang J., Bito H., Campbell R.E. A genetically encoded far-red fluorescent calcium ion biosensor derived from a biliverdin-binding protein. Protein Sci. 2022;31:e4440. doi: 10.1002/pro.4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koveal D., Rosen P.C., Meyer D.J., Díaz-García C.M., Wang Y., Cai L.-H., Chou P.J., Weitz D.A., Yellen G. A high-throughput multiparameter screen for accelerated development and optimization of soluble genetically encoded fluorescent biosensors. Nat. Commun. 2022;13:2919. doi: 10.1038/s41467-022-30685-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sternson S.M., Roth B.L. Chemogenetic tools to interrogate brain functions. Annu. Rev. Neurosci. 2014;37:387–407. doi: 10.1146/annurev-neuro-071013-014048. [DOI] [PubMed] [Google Scholar]
- 41.Tischer D., Weiner O.D. Illuminating cell signalling with optogenetic tools. Nat. Rev. Mol. Cell Biol. 2014;15:551–558. doi: 10.1038/nrm3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mehta S., Zhang Y., Roth R.H., Zhang J.F., Mo A., Tenner B., Huganir R.L., Zhang J. Single-fluorophore biosensors for sensitive and multiplexed detection of signalling activities. Nat. Cell Biol. 2018;20:1215–1225. doi: 10.1038/s41556-018-0200-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tian L., Hires S.A., Mao T., Huber D., Chiappe M.E., Chalasani S.H., Petreanu L., Akerboom J., McKinney S.A., Schreiter E.R., et al. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat. Methods. 2009;6:875–881. doi: 10.1038/nmeth.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lawson M., Elf J. Imaging-based screens of pool-synthesized cell libraries. Nat. Methods. 2021;18:358–365. doi: 10.1038/s41592-020-01053-8. [DOI] [PubMed] [Google Scholar]
- 45.Hendel S.J., Shoulders M.D. Directed evolution in mammalian cells. Nat. Methods. 2021;18:346–357. doi: 10.1038/s41592-021-01090-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Durrant M.G., Fanton A., Tycko J., Hinks M., Chandrasekaran S.S., Perry N.T., Schaepe J., Du P.P., Lotfy P., Bassik M.C., et al. Systematic discovery of recombinases for efficient integration of large DNA sequences into the human genome. Nat. Biotechnol. 2023;41:488–499. doi: 10.1038/s41587-022-01494-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Werley C.A., Chien M.-P., Cohen A.E. Ultrawidefield microscope for high-speed fluorescence imaging and targeted optogenetic stimulation. Biomed. Opt Express. 2017;8:5794–5813. doi: 10.1364/BOE.8.005794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pachitariu M., Stringer C. Cellpose 2.0: how to train your own model. Nat. Methods. 2022;19:1634–1641. doi: 10.1038/s41592-022-01663-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Edelstein A.D., Tsuchida M.A., Amodaj N., Pinkard H., Vale R.D., Stuurman N. Advanced methods of microscope control using μManager software. J. Biol. Methods. 2014;1 doi: 10.14440/jbm.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu S., Lin C., Xu Y., Luo H., Peng L., Zeng X., Zheng H., Chen P.R., Zou P. A far-red hybrid voltage indicator enabled by bioorthogonal engineering of rhodopsin on live neurons. Nat. Chem. 2021;13:472–479. doi: 10.1038/s41557-021-00641-1. [DOI] [PubMed] [Google Scholar]
- 51.Miyazaki K. In: Directed Evolution Library Creation: Methods and Protocols. Arnold F.H., Georgiou G., editors. Humana Press; 2003. Creating random mutagenesis libraries by megaprimer PCR of whole plasmid (MEGAWHOP) pp. 22–28. [DOI] [PubMed] [Google Scholar]
- 52.Schmidt U., Weigert M., Broaddus C., Myers G. In: Cell detection with star-convex polygons. Cham A.F.F., Schnabel J.A., Davatzikos C., Alberola-López C., Fichtinger G., editors. Springer International Publishing; 2018. pp. 265–273. held in. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All the data published in this paper will be available from the lead contact upon request.
-
•
The MATLAB code is available as supporting information in this manuscript.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.