

Summary
In this work, we propose an approach to generate whole-slide image (WSI) tiles by using deep generative models infused with matched gene expression profiles. First, we train a variational autoencoder (VAE) that learns a latent, lower-dimensional representation of multi-tissue gene expression profiles. Then, we use this representation to infuse generative adversarial networks (GANs) that generate lung and brain cortex tissue tiles, resulting in a new model that we call RNA-GAN. Tiles generated by RNA-GAN were preferred by expert pathologists compared with tiles generated using traditional GANs, and in addition, RNA-GAN needs fewer training epochs to generate high-quality tiles. Finally, RNA-GAN was able to generalize to gene expression profiles outside of the training set, showing imputation capabilities. A web-based quiz is available for users to play a game distinguishing real and synthetic tiles: https://rna-gan.stanford.edu/, and the code for RNA-GAN is available here: https://github.com/gevaertlab/RNA-GAN.
Keywords: deep learning, generative adversarial network, synthetic biomedical data, variational autoencoder, artificial intelligence, generative model
Graphical abstract
Highlights
-
•
The RNA-GAN model is proposed as a solution for the RNA-to-image synthesis problem
-
•
A VAE reduces RNA-seq data’s dimensionality, preserving tissue differences
-
•
RNA-GAN achieves superior image quality compared with a traditional GAN
-
•
Experts rate RNA-GAN synthetic tiles higher than GAN synthetic tiles
Motivation
The acquisition of multi-modal biological data for the same sample, such as RNA sequencing and whole-slide imaging (WSI), has increased in recent years, enabling studying human biology from multiple angles. However, despite these emerging multi-modal efforts, for the majority of studies, only one modality is typically available, mostly due to financial or logistical constraints. Given these difficulties, cross-modal data imputation and cross-modal synthetic data generation are appealing as solutions for the multi-modal data scarcity problem. Currently, most studies focus on generating a single modality (e.g., WSI), without leveraging the information provided by additional data modalities (e.g., gene expression profiles).
Carrillo-Perez et al. report RNA-GAN, a method that enables multi-modal integration of RNA expression and imaging data to generate synthetic tissue tiles. The model, trained on expression data, produces high-quality lung and brain tissue images preferred by experts and demonstrates imputation capabilities.
Introduction
Biomedical data have become increasingly multi-modal, which has allowed us to better capture the complexity of biological processes. In the multi-modal setting, several technologies are used to obtain data from the same patient, providing a richer representation of their biological status and disease state. In current clinical practice, often demographic, clinical, molecular, and imaging data are collected on patients. Making these data modalities available helps advance the goals of precision medicine.1,2 For example, DNA and RNA sequencing are now widely used for the characterization of patients with cancer.3,4 Somatic mutation and gene expression profiles can be used to improve diagnosis, define disease subtypes, and determine the treatment regimen for patients with cancer.5,6 Similarly, in pathology, tissue slides are the cornerstone for a variety of tasks. This includes primary diagnosis based on visual examinations by pathologists as well as treatment recommendations based on insights revealed by, e.g., immunohistochemistry stains.7 Specifically for oncology, tissue slides are a valuable resource to observe morphological and texture changes, which reflect the tumor and its microenvironment.7,8,9 Since the digitization of tissue slides to whole-slide image (WSI) data (specifically hematoxylin and eosin [H&E]-stained images), they have become a key data source for training deep learning models in a wide range of clinically relevant endpoints.10
The association of molecular and morphologic patterns is also gaining interest in the research community, especially in the area of computational pathology.11 In particular, the relationship between genomic features and WSI features has recently been demonstrated, with several studies showing that these two modalities are complementary. For example, morphological features from WSI data have been shown to associate with genomic mutations, gene expression profiles, and methylation patterns.5,12,13 The effect of these variations are visually examined and can be spotted in the tissue both by pathologists and deep learning models.5,14 However, some genomic variations are utterly rare,15 limiting the available data. This fact slows downs the creation of machine learning models for their detection using a routinely obtained data source, such as H&E images. Furthermore, the variety of morphologic patterns that can be found is limited to the small pool of patients, losing a global perspective of the disease. Moreover, in terms of machine learning models, studies have shown that the integration of both modalities leads to an improvement in the performance of diagnostic and prognostic tasks in cancer when sufficient data are available.5,16,17,18,19
However, both modalities are not always available due to financial or logistical constraints. For example, the Genome Express Omnibus (GEO) database20 has numerous RNA sequencing (RNA-seq) datasets available, but few datasets have the corresponding WSIs. Similarly, most medical centers have large archives of tissue slides but not yet the means to generate matched gene expression data. New multi-modal datasets are being created to deal with these issues,21 yet the problem still occurs for most clinical datasets. Thus, opportunities for training models that require multi-modal data are missed, slowing down progress in advancing precision medicine.22,23
Data scarcity is a concerning problem in the machine learning community, especially in the context of recent successes for non-medical applications where huge amounts of data are available.24,25 Specifically in biomedical problems, large and diverse cohorts are necessary to develop accurate clinical decision support systems that depend on machine learning algorithms.26 To overcome the scarcity of heterogeneous annotated data in real-world biomedical settings, synthetic data are increasingly being considered.27 Generative models, which impute synthetic data that are indistinguishable from real data, potentially offer a solution to deal with this issue. Within generative models, generative adversarial networks (GANs) and variational autoencoders (VAEs) have been widely used for multiple data generation tasks and have obtained exceptional performances in previous studies.28,29 In both cases, the models learn a latent space to draw samples that cannot be discerned from real data. VAEs learn a latent space by solving the task of accurately reconstructing the original data using an encoder and a decoder,30 and GANs are unsupervised generative models based on a game theoretic scenario where two different networks compete against each other.31 The synthetic data generated by these models can expand the diversity of samples that a model is trained on, potentially increasing the predictive performance and also improving the model generalization capabilities. It is important to emphasize that this comes at almost zero cost once the model is trained, contrary to generating new data. In addition, synthetic data have the advantage that, when they can serve as a faithful representation of real patient data, they can be easily shared without any regulatory hurdles for protected health information.
Several studies have focused on the generation of single-modality synthetic data for both RNA gene expression and WSI data. For example, the generation of gene expression data has been mainly in the context of data imputation and has been researched by leveraging the latent space of VAEs. Qiu et al. showed that β-VAEs, a special case of VAEs, can impute RNA-seq data.32 Similarly, Way et al. proposed a VAE trained on pan-cancer TCGA data that is able to encode tissue characteristics in the latent space and also leverages biological signals.33 Recently, Vinas et al. presented an adversarial methodology for the generation of synthetic gene expression profiles that closely resemble real profiles and capture biological information.34 The generation of high-quality WSI tiles has also been researched in recent years given the success of GANs in generating natural images.35,36 For example, Quiros et al. showed that GANs are able to capture morphological characteristics of cancer tissues, placing similar tissue tiles closer in the latent space, while generating high-quality tiles.37,38
However, current research focuses on generating or imputing single modalities without leveraging the information provided from other data types. For non-medical applications, cross-modal data generation has made enormous progress thanks to the availability of large paired data, e.g., paired text and image data. Unsupervised learning methods such as GANs, transformers,39 and diffusion models40 have been developed to leverage the relationship between these two modalities, enabling the generation of images based on their textual description41,42,43 or generating textual descriptions of given images.44
While cross-modal generation has proven successful for natural images in non-medical applications,41,42 the relation between WSIs and gene expression needs yet to be explored for cross-modal synthetic data generation. For this use case, we were inspired by the observation that the relation between textual descriptions and their corresponding images is similar to the relation between WSIs and genomic data, as they are describing the same phenomenon from two different perspectives.
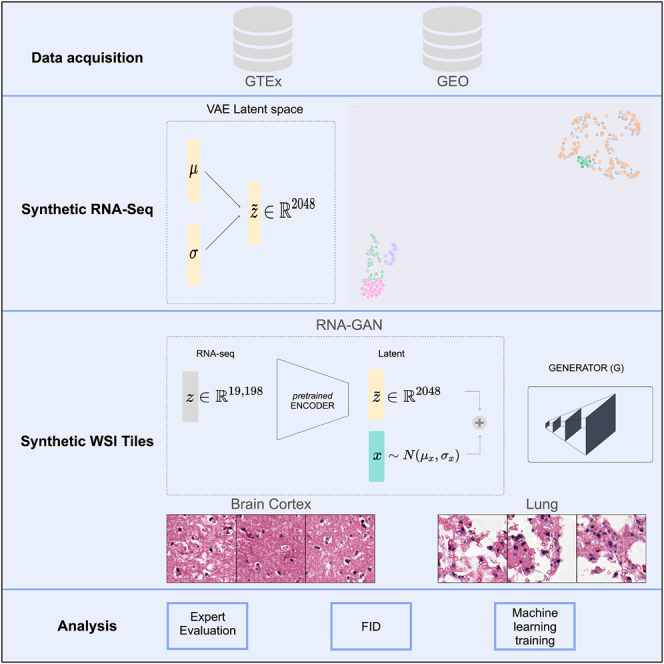
Therefore, in this work, we develop an architecture for cross-modal synthetic data generation using WSIs and genomic information for heterogeneous healthy tissues as a use case. Specifically, we explore the generation of WSI tiles using gene expression profiles of healthy lung and brain cortex tissue (Figure 1). First, we train a VAE that reduces the dimensionality of the RNA-seq data. Then, using the latent representation of the gene expression as input, we present a GAN-based architecture (named RNA-GAN) that generates image tiles for healthy lung and brain cortex tissues. Based on evaluations of blinded pathologists, we show that the quality of generated WSIs can be significantly improved when the GAN is infused with gene expression data.
Figure 1.

Model architecture for gene expression, WSIs, and combined data using VAE and GANs
(A) β-VAE architecture for the generation of synthetic gene expression data. The model uses as input the expression of 19,198 genes. Both the encoder and the decoder are formed by two linear layers of 6,000 and 4,096, respectively. The latent μ and σ vectors have a feature size of 2,048.
(B) GAN architecture for generating tiles by sampling from a random normal distribution. The architecture chosen was a deep convolutional GAN (DCGAN),45 using as input a feature vector of size 2,048. The final size of the tiles generated is 256 × 256, the same as the size of the real tiles.
(C) RNA-GAN architecture where the latent representation of the gene expression is used for generating tiles. The gene expression profile of the patient is used in the β-VAE architecture to obtain the latent representation. Then, a feature vector is sampled from a scaled random normal distribution (values ranging between [−0.3,0.3]) and added to the latent representation. A DCGAN is trained to use this vector as input and generate a 256 × 256 sample. The discriminator receives synthetic and real samples of that size.
Results
A β-VAE model can build a representative latent space that discriminates between healthy tissues
As a first step, we aimed to create an accurate, distinguishable latent representation of healthy multi-tissue gene expression using a β-VAE architecture (Figure 1A). The goal was to reduce the dimensionality of the gene expression profile while maintaining the differences among the tissues. To do so, we use the traditional approach for training a β-VAE, i.e., reconstructing the input from the latent space (see STAR Methods). The β-VAE model was able to accurately reconstruct the gene expression by forwarding the latent representation through the decoder and obtaining a mean absolute error percentage of 39% (root-mean-square error [RMSE] of 0.631) on the test set for multiple tissues.
To verify that the latent representation learnt by the β-VAE accurately maps to the different tissues, the uniform manifold approximation and projection (UMAP) algorithm45 was used to visualize the real gene expression data as well as reconstructions of latent representations on the test set. For lung and brain samples, two separated clusters can be distinguished, showing how the model is characterizing the two tissues in the latent space (Figure 2A, “real” versus “reconstruction”).
Figure 2.

UMAP visualization of β-VAE embedding of multi-tissue expression profiles
(A) UMAP visualization of the real and reconstructed gene expression profiles of lung and brain cortex healthy tissues. Generated gene expression profiles, by sampling from the latent space and interpolating to the respective tissue, are also plotted.
(B) Shifting real gene expression profiles between the two tissues. The latent representation of all the available samples is obtained, and the difference vectors between the cluster centroids are computed.
(C) UMAP visualization of real gene expression profiles of multiple tissues and generated one from brain cortex tissue.
To further validate the learned latent space, we tested what happens when interpolating data in it. By interpolating in the latent space, we should be able to “transform” a randomly drawn sample to a gene expression profile that looks like it originated from one of the tissues (i.e., synthetic gene expression generation). To do so, we need to calculate the cluster centroid vector over the real data latent representations of the desired tissue and add this centroid vector to randomly drawn samples from the β-VAE latent distribution. This procedure allows us to generate synthetic gene expression data that look like real brain or lung gene expression data. When projecting these synthetic samples in the UMAP space, they indeed fall in the same clusters as the original data (Figure 2A, “generated” versus “real”).
We can also perform other operations in the latent space. For example, we should be able to “shift” the gene expression from one tissue into what it would look like if it originated from another tissue. In this case, we need to add the difference vectors between the cluster centroids of the respective tissues to the latent representation of a given sample gene expression. For example, we can shift a real brain gene expression profile to a lung gene expression profile, and vice versa. Visualizing these new samples in the UMAP space verifies that these operations can indeed be successfully performed (Figure 2B). Next, the representation capabilities of the β-VAE can also be extended to multiple tissues, showing a diverse representation with well-differentiated clusters and maintaining the generative capabilities across the multiple tissues (Figure 2C). The RMSE value distribution of the tissues is presented in Figure S3.
GANs generate quality synthetic WSI tiles preserving real data distribution differences
Next, we developed a traditional GAN model to generate synthetic WSI tiles for brain cortex and lung tissue. The model was able to generate good-quality images, preserving the morphological structures and showing few artifacts (Figures 3A and S1A). In some tiles, checkerboard artifacts are noticeable, which is a known problem in GANs.46
Figure 3.

A GAN generates realistic lung and brain cortex tiles maintaining the distribution of the real tiles
(A) Tiles generated by the GAN model for brain tissue on the top and for lung tissue on the bottom.
(B) UMAP representation of the real patients in the lung and brain cortex dataset.
(C) UMAP representation of generated tiles using the GAN model. 600 tiles are generated per patient and then used to compute the feature vectors and the UMAP visualization.
Despite the artifacts, the main cell types can be observed in the tiles, such as epithelial, connective, and muscle tissues. In addition, there is a clear distinction between the tiles generated for the brain cortex and for the lung, preserving the characteristics of the corresponding real tiles. Specifically, the brain cortex tissue is grouped in a set of layers that form a homogeneous and continuous layer (the outer plexiform layer, outer granular layer, outer pyramidal cell, inner granular layer, inner pyramidal layer, and polymorphous layer).47 These characteristics can be observed in the synthetic brain tiles, i.e., they appear more homogeneous and contain less white spaces compared with the synthetic lung tissue tiles. The synthetic lung tissue tiles also present the characteristics of real tiles, showing the terminal bronchioles, respiratory bronchioles, alveolar ducts, and alveolar sacs in some cases.
To test if the generated tiles have the same distribution as the real ones, the feature vectors outputted from one of the last convolutional layers of an Inception V3 network pretrained on ImageNet were obtained for the 600 generated tiles. Then, these feature vectors were projected and visualized using the UMAP algorithm, showing a similar distribution between the tissues for both real (Figure 3B) and synthetic samples (Figure 3C).
Using latent gene expression profiles as input on GANs improves synthetic H&E tile quality and reduces training time
Next, we used latent gene expression profiles as input instead of a random normal distribution for a GAN model generating WSI tiles. The gene expression was first forwarded through the pretrained β-VAE to reduce the dimensionality and to encode it in the latent space. Then, that representation plus noise sampled from a narrowed random normal distribution (values between [−0.3,0.3]) was used as input to the generator, which outputs the synthetic tile (Figure 1C). This model generates synthetic tiles with fewer artifacts and better quality of the morphological structures (Figures 4A and S1B). To demonstrate that the gene expression latent representation provides actual information to generate the tiles and that the model does not mainly focus on the random signal (as with conventional GANs), we also created a GAN that samples only from a scaled random normal distribution (values between [−0.3,0.3]). This model was not able to produce quality samples of any tissue (Figure 4C), even when sufficient training time was used. Hence, the RNA-seq data in the RNA-GAN is the main signal that guides its generation process and shows that the latent RNA-seq distribution is enough to generate synthetic tiles. The scaled normal distribution only adds sufficient noise to vary the generated tiles.
Figure 4.

A gene expression-infused GAN improves tile quality
(A) Tiles generated using the RNA-GAN model for lung and brain cortex healthy tissues.
(B) UMAP visualization of the patients by generating tiles using their gene expression. The model preserves the distribution differences between the two tissues.
(C) Generated tiles of model trained using only random Gaussian data on a small range ([−0.3,0.3]) does not generate high-quality tiles, showing that the gene expression distribution is essential for synthetic tile generation.
(D) Brain cortex and lung tissue tiles generated using an external dataset (GEO: GSE120795), showing the generalization capabilities of the model.
We also obtained the feature vectors from one of the last convolutional layers of the Inception V3 architecture pretrained on ImageNet to observe if the distribution of the synthetic tiles was similar to that from real patients. The differences between the tissues were preserved, as well the tissue inner-cluster distribution (Figure 4B).
To test the generalization capabilities of the trained models, we also used as input external brain cortex and lung tissue RNA-seq data (GEO: 120795). The model was able to successfully generate tissue samples with characteristics similar to those obtained with the training data (Figure 4D). We then tested whether a model trained on real data can distinguish the synthetic generated tiles from this GEO cohort. This model reached an accuracy of 80.5%, an F1 score of 79.7%, and an area under the curve (AUC) of 0.805, showing that a model trained on real tiles can accurately classify the synthetic tiles. Finally, we observed that the RNA expression-infused GAN model needed fewer training epochs compared with the regular GAN model (Figure 5).
Figure 5.

A gene expression profile-infused GAN converges faster: Brain cortex and lung tissue tiles generated at the same epoch during training for the model with and without gene expression profiles
The visualized epoch is the last epoch of training for the models using RNA-seq data.
(A) Brain cortex generation at training epoch 24 for GAN and RNA-GAN models, with similar performance and quality between the generated tiles; however, less diversity is obtained when not using gene expression profiles.
(B) Lung tissue generation at training epoch 11 for both the GAN and RNA-GAN models. A comparison of both models shows noticeable differences in the quality of the generated tiles. The model using gene expression profiles outputs better morphological features and less artifacts and has a higher overall quality.
To show the usefulness of these synthetic tiles in a clinical application, we decided to use them paired with self-supervised learning to show the improvements that can be obtained in a classification task between glioblastoma (GBM) and lung adenocarcinoma (LUAD) tiles. We firstly generated 10,000 tiles (5,000 per class) and trained a ResNet-50 architecture in a self-supervised learning (SSL) manner using the SimCLR framework.48 SimCLR is a contrastive learning method that maximizes the agreement between two different augmented versions of the same image, learning a relevant feature representation of the image. Once the pretraining has been performed, the learned weights are used as the initialization weights for the downstream task and compared with training the model from scratch. Then, we took 1,000 real tiles from patients with GBM and LUAD from The Cancer Genome Atlas (TCGA) project, respectively, and performed a 5-fold cross-validation in two settings: training from scratch, and using the SSL weights as initialization. The details of the SSL training are presented in the STAR Methods section. The model using SSL weights outperformed the one training from scratch in both terms of accuracy and F1 score. The model using SSL weights obtained a mean accuracy of 85.50% ± 1.54% and an F1 score of 85.49% ± 1.54%, while the model trained from scratch obtained a mean accuracy of 76.71% ± 4.51% and an F1 score of 76.67% ± 4.52%. Both the accuracy and F1 score obtained across the splits and the confusion matrices are presented in Figure S4.
Expert evaluation of synthetic tiles
Next, we asked a panel of board-certified anatomic pathologists with different subspecialty expertise (M.G.O., H.V., R.B.W., C.S.K., Matt Van den Rijn, J.S.) to rate the quality of brain cortex and lung cortex tiles (N = 18) on a scale from 1 (worst) to 5 (best). These pathologists were not informed about the presence of synthetic data in the examples to prevent any bias when examining the tiles. Instead, they were told that the tiles presented were going to be used to train machine learning models and that we wanted to score their quality of that task. The set of questions asked and the procedure are further described in the STAR Methods section.
The pathologists’ evaluations of the morphological structures resulted in a mean score of 3.55 ± 0.95 for real brain, 2.88 ± 0.62 for GAN brain, and 2.94 ± 0.64 for RNA-GAN brain. For the lung tissue, the mean score for the real samples was 2.26 ± 1.14, 1 ± 0.55 for the GAN lung, and 1.73 ± 0.79 for the RNA-GAN lung. Hence, the pathologists rated the real samples as best quality, with second-best ratings for the samples from the RNA-GAN and the worst ratings for those from the conventional GAN. In the case of the RNA-GAN lung generated tiles, one of the pathologists scored the generated tiles higher than the real tiles in terms of quality (mean value of 1.2 compared with the score of real tiles, 0.8). Nevertheless, the rest of the pathologists scored the synthetic tiles lower than the real ones, but this difference was never greater than one point (real lung scores: [4,1.4,2,0.8,2.4,3]; RNA-GAN lung scores: [3,1,1.2,1.2,1.6,2.4]). The difference was greater in the GAN lung generated tiles, where the scores obtained were (1,0.6,1.2,0.6,0.6,2). This was not the case for the brain tissue, where pathologists scored similarly GAN and RNA-GAN tiles, even though they scored slightly better the RNA-GAN-generated tiles (real brain tile scores: [2.3,3.3,3,4.3,3.3,5]; RNA-GAN brain tiles scores: [2,3,3,3,2.7,4]; GAN brain tiles scores: [2.3,2.7,3,3,2.3,4]).
The preference in the pathologists’ evaluations for the RNA-GAN lung over GAN synthetic tiles is statistically significant (p = 0.025), while there was no statistically significant difference in ratings between the real and RNA-GAN lung tiles scores (p = 0.052). On the contrary, generated brain tissue tiles were significantly different from real tiles both for GAN (p = 0.043) and RNA-GAN (p = 0.038), and no significant difference in ratings was found between RNA-GAN and GAN (p = 0.305). However, a bigger mean evaluation score difference was found between real and GAN tiles than between real and RNA-GAN tiles, again confirming that the quality of RNA-GAN synthetic tiles is closer to the quality of real tiles (Figure 6A). In addition, the mean difference in the evaluation between GAN and RNA-GAN tiles was bigger than zero, showing the preference of pathologists for the RNA-GAN tiles over GAN tiles (Figure 6B).
Figure 6.

Expert evaluation of synthetic slides
(A) Difference in morphological structure quality of synthetic (generated by GAN and RNA-GAN) and real tissues based on the pathologists’ evaluations. The difference between real tiles and generated tiles was bigger for GAN than for RNA-GAN.
(B) Difference in morphological structure quality between the synthetic generated tiles by the GAN and RNA-GAN based on the pathologists’ evaluations. Pathologists evaluated the tiles generated using RNA-GAN better compared with only GAN.
Finally, pathologists detected the tissue of origin of the tiles with a 100% ± 0% accuracy for both real and RNA-GAN tiles, while this drops to 74.98% ± 20.40% accuracy for the GAN tiles.
Discussion
Previously, in biomedical problems, imputed samples and synthetic data were generated in an isolated way, not using the information provided by other modalities. In contrast, in this work, we studied the generation of WSI tiles of lung and brain cortex tissues by leveraging the corresponding gene expression profiles. This idea was inspired by recent advances in cross-modal data generation for non-medical images, including the generation or modification of images based on text prompts (e.g., DALL-E 2,42 Imagen,48 Parti,49 and related models41,42,44). Here, we extrapolated this idea to biomedical data by treating RNA-seq data as prior text to contextualize image generation.50,51 This differs from other approaches presented in literature, where cost- and time-expensive transcriptomic profile levels were predicted from an easy- and cheap-to-obtain paired H&E sample.12 However, our approach can be situated in the digital biomarker study area and serve as a general purpose framework for other modalities. In addition, there is a growing archive of RNA-seq samples for which the H&E tissue samples have not been digitized missing that modality, and there are existing databases with publicly available gene expression data. Our proposed methodology can serve as an imputation method for generating the paired tissue slide from the RNA-seq profile of the patient.
Several studies have been reported using GTEx data where gene expression variations have been studied across tissues and characterized several genetic variations.52,53,54,55 These studies investigate how genetic variants affect gene regulation and complex traits in humans at the transcriptomic level, as well as the genetic bases of diseases. Thus, our proposed model can provide a solution to study those variations on tissues where only RNA-seq is available and model the effect that they might have on its morphology.
Moreover, even though we are using healthy tiles as a use case to show our methodology capabilities, it can be further applied to other applications. The use of our method is potentially useful for rare diseases. In this context, synthetic data generation can be used as a tool to increase the availability of samples in scarce settings and can be used to study the variations between healthy and diseased H&E tiles. It is widely known that scarce data are one of the bottlenecks in the development of deep learning techniques56 in medicine, and our proposed methodology can serve as a solution in this context. By pretraining models using recently developed SSL techniques,57 global features can be learned with the synthetically generated data and then fine-tuned on the real samples. In addition, data quality has been presented in the literature as essential to obtain powerful machine learning models,58 and in this work, we have shown how RNA-GAN-generated tiles have higher quality both in terms of pathologists’ evaluations and image quality metrics (e.g., Fréchet inception distance [FID]) than those generated by a traditional GAN. In addition, the proposed generative framework could be further adapted to other imaging modalities including gene expression and computed tomography scans.
The use of deep learning techniques is revolutionizing medicine, allowing the creation of systems that can accurately detect genetic variants or perform prognosis prediction. Nevertheless, for training these models, enough data are required, which can be especially challenging for rare mutation variants or rare cancer types (e.g., pediatric cancers).56 Synthetic data generation can be used as a tool to increase the availability of samples in scarce settings, augmenting the number of samples. Using the generated data and SSL techniques, models can learn global features that can be further fine-tuned in downstream tasks with only a few samples.57 However, with traditional generative approaches, it is not possible to regulate the effect of certain factors in tissue generation, apart from performing label guidance. This limits its potential in cases where we want to study the effect of an up- or downregulated gene in tissue morphology, expanding the diversity of samples. In this work, we proposed a methodology, RNA-GAN, that can be used under this assumption when trained on the required data, creating in silico simulations of specific genomic variations and studying their effect on the tissue. An example would be the generation of prostate cancer tiles where the TP53 mutation can be found, helping to increase the performance of detection models.14 Moreover, the effect of specific mutations in a specific gene on morphologic characteristics of tissue has been studied but not from the expression of multiple genes.59,60 Given that the latent representation of the gene expression is being used, the effect of thousands of genes can be reflected in the generated tissue tile, not limited to specific expression values.
From a machine learning perspective, since biomedical data are becoming increasingly multi-modal, there is a growing interest in developing multi-modal predictive models for advancing the goals of precision medicine. However, obtaining multi-modal biological data is a slow and costly process. Multimodal data are sometimes available for widely studied diseases, but this is not common and definitely not the case for pediatric and rare diseases. In addition, not all medical facilities have the required expertise or instruments to collect each data modality for a patient. Therefore, cross-modal data imputation32,61,62 and generation of cross-modal synthetic data is a promising approach to complete datasets by leveraging the potential of deep learning models.27
As a first step in our work, we developed a β-VAE model that reduces the dimensionality of gene expression data. We have shown that the β-VAE model is able to capture the latent representation of multiple human tissues and that it obtains a representative latent feature vector that accurately distinguishes between the tissues (Figure 2). To test the accuracy, practicality, and capabilities of the learned latent space, we performed certain sanity checks. For example, we tested the ability to generate synthetic gene expression profiles and to interpolate samples between classes. Our analysis shows that the β-VAE model indeed allows us to obtain a compact, accurate representation of tissue gene expression profiles. We later use the encoder part of the β-VAE to reduce the dimensionality of gene expression profiles, which in turn will guide the generation of the WSI tiles. Note that this compact representation could also be used for downstream tasks including prognosis or treatment outcome prediction, but this analysis was out of the scope of this work.
Next, we trained a traditional GAN model on brain cortex and lung tissue data to generate WSI tiles. We explored the possibility of using the same architecture to generate both tissues, but the model collapsed and only generated brain cortex tiles. We hypothesize that this could be due to the homogeneity of brain tissue compared with lung tissue, making it easier for the model to generate brain cortex tiles and reduce the loss. This can also be observed in the number of necessary epochs to generate quality tiles for each tissue. While the model only needs 34 epochs for training convergence for brain cortex tissue, it requires 91 epochs for lung tissue. Clearly, it is more difficult to generate the lung tiles, probably because lung tissues are more heterogeneous compared with the brain cortex. Importantly, the synthetic tiles preserved the distribution of real tiles, showing two well-differentiated clusters using a UMAP projection of the feature vectors (Figure 3C). Pathologists were asked if they could observe any kind of artifact on the synthetic tissue (e.g., image aberrations). From the presented GAN tiles, in 70% of cases, pathologists detected certain artifacts (mean percentage across pathologists), while this was only the case for 17% of the real tiles.
Finally, we trained the gene expression profile-infused GAN model, both on brain cortex and lung tissue data. The quality of the generated tiles improved significantly compared with tiles from a regular GAN, based on the evaluation of expert pathologists. In addition, pathologists reported significantly fewer artifacts in RNA-GAN images (56% compared with 70% for GAN images), even though pathologists scored RNA-GAN tiles for brain and tissue lower than real tiles. Thus, this could affect their use in a downstream task. However, the score was better still than GAN-generated tiles. While a decreased quality compared with real tiles is expected, our model can provide data in a situation that is unfeasible to obtain (if the histology sample has been destroyed or is not available anymore) and therefore serves as a solution for data imputation and augmentation purposes.
Another advantage of the RNA-GAN model over the GAN model is that it needed less training epochs for reaching high-quality results (using the same amount of training tiles). While the GAN lung model was trained during 94 epochs for obtaining tiles without major artifacts and good tissue structure, the RNA-GAN lung model only needed 11 epochs. This reduces the number of training epochs by 88%. We computed the FID 60k for both models, where the GAN model obtained a value of 87.85 and the RNA-GAN a value of 83.89. While the RNA-GAN model obtains a lower FID value than the traditional GAN model, these values are still high compared with other generative models,38 yet future work is warranted to further improve these results.
In both cases, checkerboard artifacts were present in the generated tiles. This effect is usually produced by the deconvolution operation, which is used to go from a lower-resolution image to a higher-resolution one.63 The problem relies on an uneven overlap that is produced with the operation, which is even more extreme in a two-dimensional space. This produces a checkerboard-like pattern of varying magnitudes since the uneven overlap of the two axes (taking into account the kernel size and stride of the convolution operation) are multiplied together. We refer to the reader to this work for a visual example of the phenomenon causing the problem.64 Other factors can also produce such artifacts, like the image compression algorithm used to save the images. We tried different alternatives to mitigate this effect, such as firstly resizing the image using interpolation and then applying a traditional convolution operation. However, while the artifacts were reduced, they were still present in our case. We hypothesize that since H&E lung images contain a higher quantity of white space, the model tries to “fill” it with color even when the uneven overlap is avoided, producing the observable checkerboard effect. Newer architectures and models might reduce this effect, and having a model that is trained with a variety of tissues. The panel of expert pathologists was not aware of this typical effect in synthetic image generation, and, being unbiased to this issue and thinking that all images were real, they just gave a lower quality score if artifacts were present. However, this effect can have an effect on the downstream task due to the model associating these features to specific classes. This would not be a problem if the images were being used for pretraining purposes, where more general features are learned. However, more work is needed to get rid of the checkerboard effect in future models.
Once we have a trained generative model, generating or imputing missing values with synthetic data comes at very low cost. Adding these low-cost synthetic samples to real data can help to train more complex deep learning models, which are data hungry. While the synthetic generation of WSIs and imputation of RNA-seq data has been studied in the literature,32,33,37 to our knowledge, generation of synthetic WSI tiles using gene expression profiles has not yet been explored. Here, we showed that infusing GANs with gene expression data for WSI tile generation not only increases the quality of the resulting tiles but additionally requires less compute time compared with the regular, single-modality approach. We further study the implication of these healthy tiles for pretraining machine learning models. We used RNA-GAN synthetically generated tiles for pretraining a ResNet-50 architecture using SSL, specifically the SimCLR framework. We showed how pretraining on the synthetically generated tiles improves the performance over training from scratch for the classification task of distinguishing between GBM and LUAD on a stratified 5-fold CV while also reducing the standard deviation. When the confusion matrices obtained across the five test set splits are compared (see Figure S4), we can observe how the correctly classified samples are improved for the two classes, and the misclassifications are reduced when training using the SSL weights. These results show the promise of using synthetic data in scarce data settings, where there is not sufficient data to learn. In addition, it has been shown in previous studies that fine-grained data might be more suitable for medical tasks compared with using natural images for pretraining purposes.65
We further showed how our model was able to generalize to gene expression profiles outside of the training dataset, still generating realistic synthetic tiles. While a vast number of gene expression datasets are publicly available, most of these datasets do not provide matching WSIs. This limits their use in multi-modal classification models.16,18,23,57,66 With our model, WSI tiles can be imputed from the gene expression, which opens new possibilities in publicly available datasets.
Even though we have used healthy samples to show the capabilities of the proposed methodology, it can be further expanded to cancer tissue. In this work, we aimed to test the advances in cross-modality synthetic generation of natural images and show that they can be applied in a gene expression profile to H&E tile setting. We focused on two well-differentiated tissues, such as lung and brain, to facilitate the task of observing tissue characteristics and limiting the non-detection factor based on the lack of expertise in a given tissue. Detecting the tissue of origin based on a single tile can be an arduous task, and therefore the tile quality and tissue characteristics can be occluded by the diversity of tissues when presented against a pathologist panel for visual quality examination. In addition, given that different architectures needed to be trained per tissue, we decided to focus on two of the most prominent ones in order to reduce computational requirements.
In summary, our work shows the promise of cross-modal synthetic biological data generation to obtain better-quality multi-modal data for training complex, data-hungry deep learning models. This is especially useful for multi-modal problems16,18 and datasets with missing modalities but also for problems with small datasets restricted by expensive data collection or rare diseases. In future work, we intend to extend this approach to more heterogeneous tissues and complex diseases and also explore cross-modal synthetic data generation with different architectures. We will also explore other GAN architectures35,36,67 and diffusion models.40,68
Limitations of the study
RNA-GAN needs to be independently trained per tissue, which increases the number of models that are required if multiple tissues are generated. The VAE is dependent on the protein-coding genes selected based on those sequenced in the GTEx project. If other genes have been sequenced, it could give a worse performance when the latent representation is obtained.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited Data | ||
| GTEx RNA-Seq and H&E Data | https://gtexportal.org/home/ | GTEx Brain - Cortex; GTEx Lung; GTEx Pancreas; GTEx Stomach; GTEx Liver |
| GEO RNA-Seq data | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE120795 | GSE120795 |
| Software and Algorithms | ||
| RNA-GAN code | This paper |
https://github.com/gevaertlab/RNA-GAN https://doi.org/10.5281/zenodo.8041872 |
Resource availability
Lead contact
Further information and requests should be directed to lead contact Olivier Gevaert (ogevaert@stanford.edu).
Materials availability
This study did not generate new unique reagents.
Methods details
Data
Data was obtained from The Genotype-Tissue Expression (GTEx) project.69 The GTEx project aims to build a public resource of healthy tissue-spefic characteristics, providing gene expression and WSI among other data types. The data was collected from 54 non-disease tissue and across almost 1,000 individuals. We collected the RNA-Seq and WSIs from brain cortex, lung, pancreas, stomach and liver tissues. There were a total of 246 samples of brain cortex tissue, 562 samples of lung tissue, 328 samples of pancreas tissue, 356 of stomach tissue, and 226 samples of liver tissue. To validate the generalization capabilities in generating tiles from the gene expression of other cohorts, the GEO series 120795 was used.70
RNA-seq data preprocessing
Gene expression data from the GTEx project contains a total of 56,201 genes. This number would require huge computational capabilities, and it difficulties the training of the machine learning models. Therefore, we reduced the feature dimension and obtained the expression of 19,198 protein coding genes for further experiments. The data was log transformed, and the Z score normalization was applied to the gene expression using the training set data, in order to not include the validation or the test set information on the normalization process.
In the generalization experiments, the gene expression from lung and brain cortex tissue of the GEO series 120795 was used. However, not all the previously selected protein coding genes where among those sequenced in this dataset. Therefore, for those missing in this external cohort, we initialized them as zero for the generation of the tiles. Data was normalized using the mean and standard deviation from the training set of the GTEx data and log transformed.
WSI data preprocessing
GTEx WSIs were acquired in SVS format and downsampled to 20× magnification (0.5μm px−1). The size of WSIs is usually over 10k × 10k pixels, and therefore, cannot be directly used for training machine learning models to generate the data. Instead, tiles of a certain dimension are taken from the tissue, and they are used to train the models, which is consistent with related work in state-of-the-art WSI processing.5,71,72 In our work we took nonoverlapping tiles of 256 × 256 pixels. Firstly, a mask of the tissue in the higher resolution of the SVS file was obtained using the Otsu threshold method.73 Tiles containing more then 60% of the background and with low-contrast were discarded. A maximum of 4,000 tiles were taken from each slide. For the preprocessing of the images we relied on the python package openslide,74 that allows us to efficiently work with WSI images. The tiles were saved in an LMDB database using as index the number of the tile. This approach enables to reduce the number of generated files, and structure the tiles in an organized way for a faster reading while training.
βVAE architecture for synthetic gene expression generation and experiments
We chose the βVAE model for the generation of synthetic gene expression data.75 The βVAE model is an extension of the VAE where a β parameter is introduced in the loss function. The original auto-encoder is formed by two networks, the encoder and the decoder. The encoder encodes the input into a lower dimensionality representation, and then it is used to reconstruct the input using the decoder, by learning the function hθ(x) ≈ x being θ the parameters of the neural network. To learn this function, we want to minimize the reconstruction error between the input and the output. The most common loss function is the root mean squared error (RMSE). However, for the VAE we want to learn a probability distribution of the latent space, which allows us to later sample from it to generate new samples. The assumption of the VAE is that the distribution of the data x, P(x) is related to the distribution of the latent variable z, P(z). The loss function of the VAE, which is the negative log likelihood with a regularizer is as follows:
| (Equation 1) |
where the first term is the reconstruction loss and the second term is the Kullback-Leibler (KL) divergence between the encoder’s distribution qθ(z|x) and p(z) which is defined as the standard normal distribution p(z) = N(0,1).
For the βVAE we introduce the parameter β, which controls the effect of the KL divergence for the total loss:
| (Equation 2) |
If β = 1, we have the standard loss of the VAE. If β = 0, we would only focus on the reconstruction loss, approximating the model to a normal autoencoder. For the rest of the values, we are regularizing the effect of the KL divergence on the training of the model, making the latent space smoother and more disentangled.75
For the final architecture, we empirically determined to use two hidden layers of 6,000 and 4,096 neurons each for both the encoder and the decoder, and a size of 2,048 for the latent dimension. Given that we were going to use the latent representation for the generation of the tiles, we followed the same dimensionality as the output of the convolutional layers of state-of-the-art convolutional neural networks.76 We used batch norm between the layers and the LeakyReLU as the activation function. A β = 0.005 was used in the loss function. We used the Adam optimizer for the training with learning rate equal to 5 × 10−5, along with a warm-up and a cosine learning rate scheduler. We trained the model for 250 epochs with early stopping based on the validation set loss, and a batch size of 128. A schema of the architecture is presented in Figure 1A.
We divided the dataset in 60-20-20% training, validation and test stratified splits. We trained two different models, one for brain cortex and lung tissue data, and the other with all the tissues described in previous subsections (lung, brain cortex, stomach, pancreas, and liver).
GAN architecture for synthetic WSI tiles generation and experiments
GANs have been succesfully used for generating high-fidelity images. In this work we use the Deep Convolutional GAN architecture presented by Radford et al.77 On early experiments we used the minmax loss function described on the original Godfellow et al.31 work. However, this loss function led to a lack of diversity in the generation of the samples, and a diminished quality. Therefore, we decided to use the Wasserstein loss introduced by Arjovsky et al.,78 also adding the gradient penalty proposed by Gulrajani et al.79 In this case the discriminator (or critic, as called in the paper) does not classify between real and synthetic samples, but for each sample it outputs a number. The discriminator training just tries to make the output bigger for real samples and smaller for synthetic samples. This simplifies the loss function of both networks, where the discriminator tries to maximize the difference between its output on real instances and its output on synthetic instances as follows:
| (Equation 3) |
and the generator tries to maximize the discriminator’s output for its synthetic instances as follows:
| (Equation 4) |
We trained two DCGANs, one per tissue, by sampling from a normal random distribution (scheme depicted in Figure 1B). We samples different number of tiles per image for the training of the network, finally selecting 600 tiles per image because the quality of the image and the artifacts were highly improved by augmenting the number of tiles. We used the Adam optimizer for both the generator and the discriminator, with a learning rate equal to 1 × 10−3 for the generator, a learning rate equal to 4 × 10−3 for the discriminator and betas values (0.5,0.999) in both cases. Data augmentations such as color jitter and random vertical and horizontal flips were used during training. The brain tissue GAN was trained during 39 epochs while the lung tissue GAN was trained during 91 epochs. For the training of the GANs, the Python package Torchgan was used.80
RNA-GAN architecture for synthetic WSI tiles generation and experiments
After the successful generation of the tiles using a traditional GAN approach, we explored the generation of synthetic tiles by using the gene expression profile of the patient. We combined the pretrained βVAE with the DCGAN architecture, using the encoding in the latent space as the input for training the generator. To generate different tiles from the same gene expression profile, we sample a noise vector from a narrowed random normal distribution (values ranging between [−0.3,0.3]) and add it to the latent encoding. Therefore, the input to the generator would be:
| (Equation 5) |
We trained two DCGANs, one per tissue, and the pipeline is depicted in Figure 1 C). We finally selected 600 tiles per image to train the generator. We used the Adam optimizer for both the generator and the discriminator, with a learning rate equal to 1 × 10−3 for the generator, a learning rate equal to 4 × 10−3 for the discriminator and betas values (0.5,0.999) in both cases. Data augmentations such as color jitter and random vertical and horizontal flips were used during training. The brain tissue GAN was trained during 24 epochs while the lung tissue GAN was trained during 11 epochs. For the training of the GANs, the Python package Torchgan was used.80
To validate the generalization capabilities of the trained model, the GEO series 120795 was used. It contains gene expression profiles from healthy tissues, where we took the expression of lung and brain cortex tissues. For obtaining machine learning performance metrics, one hundred images were generated per tissue. Then, a Resnet-18 was trained from scratch using 10 epochs and early stopping based on a 20% of data as validation set. A learning rate value of 3e−5 and AdamW optimizer were used. Finally, the model was tested on the GEO synthetically generated data, and accuracy, F1-Score and AUC was computed.
To test the use of the synthetic tiles for pretraining machine learning models, we used SimCLR to train a Resnet-50 backbone on 10000 synthetically generated tiles (5000 per class). We trained the model for 100 epochs, using the SGD optimizer with a learning rate equal to 6 × 10−2, a momemtum value of 0.9 and a weight decay equal to 5 × 10−4, and we used a cosine annealing learning rate scheduler. For both training from scratch and fine-tuning the SSL weights, we used AdamW as the optimizer, with a weight decay of 0.01 and a learning rate value of 3 × 10−5. Both models were trained for 40 epochs using a batch size of 4, and a stratified 5--Fold CV was used.
We release an online quiz where users can try to distinguish between real and synthetic samples, obtaining a score on how well they performed. The quiz is available in the following URL: https://rna-gan.stanford.edu/. The code and checkpoints for the proposed models is available in this Github repository: https://github.com/gevaertlab/RNA-GAN.
Expert pathologist evaluation form
To evaluate the quality of the synthetic tiles, we presented a form to expert pathologists (Figure S2). The pathologists were not informed that some presented tiles were synthetic, to omit any kind of biases in the evaluation. Instead, we informed the pathologists that these tiles were going to be used to create machine learning classifiers, and we wanted to evaluate their quality for this task. Three questions were asked to the experts.
-
1.
Is the tile from brain cortex or lung tissue?
-
2.
Quality of the morphological structures: Being 1 very bad and 5 very good, how would you rate the morphological features present in the tile for an assessment of the tissue?
-
3.
Do you find artifacts in the image? (e.g. image aberrations) (Yes/No)
Ten tiles were randomly selected from 600 synthetically generated tiles per architecture. Then, tiles were visually inspected and the three with the highest quality were selected, leaving with a total of 18 tiles (between real, GAN generated, and RNA-GAN generated). This number of tiles was selected to enable clinicians to complete the form in less than 10 min. We used the same selection criteria for both cases (traditional GAN and RNA-GAN), and we manually selected them so both synthetic tiles could be reviewed in the best condition possible. Since synthetic tiles generated by the networks could present checkerboard artifacts (in less extend in RNA-GAN generated tiles), it was something that we wanted to omit therefore we used the following criteria.
-
•
No checkerboard artifacts were present in the tile.
-
•
Tissue characteristics were clearly visible in the tile.
Quantification and statistical analysis
Model performance was primarily assessed using expert pathologist’s evaluations, FID scores, Accuracy, and F1-Score. Further details can be found in the methods details and Figure Legends.
Acknowledgments
We would like to thank Matt Van den Rijn for his collaboration evaluating the synthetic images. F.C.-P. and L.J.H. were supported by grants PID2021-128317OB-I00, funded by MCIN/AEI/10.13039/501100011033, and Project P20-00163, funded by Consejería de Universidad, Investigación e Innovación, both also funded by “ERDF A way of making Europe.” F.C.-P. was also supported by a predoctoral scholarship from the Fulbright Spanish Commission. Research reported here was further supported by the National Cancer Institute (NCI) under award R01 CA260271. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. M.P. was supported by a fellowship from the Belgian American Educational Foundation. The results shown here are in whole or in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Author contributions
Conceptualization, F.C.-P. and O.G.; methodology, F.C.-P., M.P., and O.G.; investigation, F.C.-P., M.P., M.G.O., H.V., R.B.W., C.S.K., L.J.H., J.S., and O.G.; writing – original draft, F.C.-P, M.P., and O.G.; writing – review & editing, M.G.O., H.V., R.B.W., C.S.K., L.J.H., and J.S.; funding acquisition, O.G. and L.J.H.; resources, O.G.; supervision, O.G.
Declaration of interests
The authors declare no competing interests.
Published: July 19, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2023.100534.
Supplemental information
Data and code availability
-
•
This paper analyzes existing, publicly available data. These accession numbers for the dataset are listed in the key resources table.
-
•
All original code has been deposited at GitHub and is publicly available as of the date of publication. The URL and an archival DOI are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Hodson R. Precision medicine. Nature. 2016;537:49. doi: 10.1038/537S49a. [DOI] [PubMed] [Google Scholar]
- 2.König I.R., Fuchs O., Hansen G., von Mutius E., Kopp M.V. What is precision medicine? Eur. Respir. J. 2017;50 doi: 10.1183/13993003.00391-2017. [DOI] [PubMed] [Google Scholar]
- 3.Hadjadj D., Deshmukh S., Jabado N. Entering the era of precision medicine in pediatric oncology. Nat. Med. 2020;26:1684–1685. doi: 10.1038/s41591-020-1119-6. [DOI] [PubMed] [Google Scholar]
- 4.Nakagawa H., Fujita M. Whole genome sequencing analysis for cancer genomics and precision medicine. Cancer Sci. 2018;109:513–522. doi: 10.1111/cas.13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coudray N., Ocampo P.S., Sakellaropoulos T., Narula N., Snuderl M., Fenyö D., Moreira A.L., Razavian N., Tsirigos A. Classification and mutation prediction from non–small cell lung cancer histopathology images using deep learning. Nat. Med. 2018;24:1559–1567. doi: 10.1038/s41591-018-0177-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bignell G.R., Greenman C.D., Davies H., Butler A.P., Edkins S., Andrews J.M., Buck G., Chen L., Beare D., Latimer C., et al. Signatures of mutation and selection in the cancer genome. Nature. 2010;463:893–898. doi: 10.1038/nature08768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Williams B.J., Bottoms D., Treanor D. Future-proofing pathology: The case for clinical adoption of digital pathology. J. Clin. Pathol. 2017;70:1010–1018. doi: 10.1136/jclinpath-2017-204644. [DOI] [PubMed] [Google Scholar]
- 8.Heindl A., Nawaz S., Yuan Y. Mapping spatial heterogeneity in the tumor microenvironment: A new era for digital pathology. Lab. Invest. 2015;95:377–384. doi: 10.1038/labinvest.2014.155. [DOI] [PubMed] [Google Scholar]
- 9.Cheng J., Mo X., Wang X., Parwani A., Feng Q., Huang K. Identification of topological features in renal tumor microenvironment associated with patient survival. Bioinformatics. 2018;34:1024–1030. doi: 10.1093/bioinformatics/btx723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van der Laak J., Litjens G., Ciompi F. Deep learning in histopathology: The path to the clinic. Nat. Med. 2021;27:775–784. doi: 10.1038/s41591-021-01343-4. [DOI] [PubMed] [Google Scholar]
- 11.Lehrer M., Powell R.T., Barua S., Kim D., Narang S., Rao A. Advances in Biology and Treatment of Glioblastoma. Springer; 2017. Radiogenomics and histomics in glioblastoma: The promise of linking image-derived phenotype with genomic information; pp. 143–159. [DOI] [Google Scholar]
- 12.Schmauch B., Romagnoni A., Pronier E., Saillard C., Maillé P., Calderaro J., Kamoun A., Sefta M., Toldo S., Zaslavskiy M., et al. A deep learning model to predict RNA-seq expression of tumours from whole slide images. Nat. Commun. 2020;11:3877. doi: 10.1038/s41467-020-17678-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng H., Momeni A., Cedoz P.-L., Vogel H., Gevaert O. Whole slide images reflect DNA methylation patterns of human tumors. NPJ Genom. Med. 2020;5 doi: 10.1038/s41525-020-0120-9. 11–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen M., Zhang B., Topatana W., Cao J., Zhu H., Juengpanich S., Mao Q., Yu H., Cai X. Classification and mutation prediction based on histopathology H&E images in liver cancer using deep learning. NPJ Precis. Oncol. 2020;4:14. doi: 10.1038/s41698-020-0120-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rowlands C., Thomas H.B., Lord J., Wai H.A., Arno G., Beaman G., Sergouniotis P., Gomes-Silva B., Campbell C., Gossan N., et al. Comparison of in silico strategies to prioritize rare genomic variants impacting RNA splicing for the diagnosis of genomic disorders. Sci. Rep. 2021;11 doi: 10.1038/s41598-021-99747-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carrillo-Perez F., Morales J.C., Castillo-Secilla D., Gevaert O., Rojas I., Herrera L.J. Machine-learning-based late fusion on multi-omics and multi-scale data for non-small-cell lung cancer diagnosis. J. Pers. Med. 2022;12:601. doi: 10.3390/jpm12040601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carrillo-Perez F., Morales J.C., Castillo-Secilla D., Molina-Castro Y., Guillén A., Rojas I., Herrera L.J. Non-small-cell lung cancer classification via RNA-seq and histology imaging probability fusion. BMC Bioinf. 2021;22:454. doi: 10.1186/s12859-021-04376-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheerla A., Gevaert O. Deep learning with multimodal representation for pancancer prognosis prediction. Bioinformatics. 2019;35:446–454. doi: 10.1093/bioinformatics/btz342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schneider L., Laiouar-Pedari S., Kuntz S., Krieghoff-Henning E., Hekler A., Kather J.N., Gaiser T., Fröhling S., Brinker T.J. Integration of deep learning-based image analysis and genomic data in cancer pathology: A systematic review. Eur. J. Cancer. 2022;160:80–91. doi: 10.1016/j.ejca.2021.10.007. [DOI] [PubMed] [Google Scholar]
- 20.Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M., et al. NCBI Geo: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013;41:991–995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jennings C.N., Humphries M.P., Wood S., Jadhav M., Chabra R., Brown C., Chan G., Kaye D., Bansal D., Colquhoun C., et al. Bridging the gap with the UK Genomics Pathology Imaging Collection. Nat. Med. 2022;28:1107–1108. doi: 10.1038/s41591-022-01798-z. [DOI] [PubMed] [Google Scholar]
- 22.Zhang D., Kabuka M. Multimodal deep representation learning for protein interaction identification and protein family classification. BMC Bioinf. 2019;20:531. doi: 10.1186/s12859-019-3084-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen R.J., Lu M.Y., Wang J., Williamson D.F.K., Rodig S.J., Lindeman N.I., Mahmood F. Pathomic fusion: an integrated framework for fusing histopathology and genomic features for cancer diagnosis and prognosis. IEEE Trans. Med. Imaging. 2022;41:757–770. doi: 10.1109/TMI.2020.3021387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Silver D., Huang A., Maddison C.J., Guez A., Sifre L., Van Den Driessche G., Schrittwieser J., Antonoglou I., Panneershelvam V., Lanctot M., et al. Mastering the game of go with deep neural networks and tree search. Nature. 2016;529:484–489. doi: 10.1038/nature16961. [DOI] [PubMed] [Google Scholar]
- 25.Brown T., Mann B., Ryder N., Subbiah M., Kaplan J.D., Dhariwal P., Neelakantan A., Shyam P., Sastry G., Askell A., et al. Language models are few-shot learners. Adv. Neural Inf. Process. Syst. 2020;33:1877–1901. [Google Scholar]
- 26.Trister A.D. The tipping point for deep learning in oncology. JAMA Oncol. 2019;5:1429–1430. doi: 10.1001/jamaoncol.2019.1799. [DOI] [PubMed] [Google Scholar]
- 27.Chen R.J., Lu M.Y., Chen T.Y., Williamson D.F.K., Mahmood F. Synthetic data in machine learning for medicine and healthcare. Nat. Biomed. Eng. 2021;5:493–497. doi: 10.1038/s41551-021-00751-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Creswell A., White T., Dumoulin V., Arulkumaran K., Sengupta B., Bharath A.A. Generative adversarial networks: An overview. IEEE Signal Process. Mag. 2018;35:53–65. doi: 10.1109/MSP.2017.2765202. [DOI] [Google Scholar]
- 29.Wei R., Mahmood A. Vol. 9. IEEE Access; 2020. Recent Advances in Variational Autoencoders with Representation Learning for Biomedical Informatics: A Survey; pp. 4939–4956. [DOI] [Google Scholar]
- 30.Kingma D.P., Welling M. Auto-encoding variational bayes. arXiv. 2013 doi: 10.48550/arXiv.1312.6114. Preprint at. [DOI] [Google Scholar]
- 31.Goodfellow I., Pouget-Abadie J., Mirza M., Xu B., Warde-Farley D., Ozair S., Courville A., Bengio Y. Generative adversarial nets. Adv. Neural Inf. Process. Syst. 2014;27 doi: 10.48550/arXiv.1406.2661. [DOI] [Google Scholar]
- 32.Qiu Y.L., Zheng H., Gevaert O. Genomic data imputation with variational auto-encoders. GigaScience. 2020;9:082. doi: 10.1093/gigascience/giaa082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Way G.P., Greene C.S. PACIFIC SYMPOSIUM ON BIOCOMPUTING 2018: Proceedings of the Pacific Symposium. World Scientific; 2018. Extracting a biologically relevant latent space from cancer transcriptomes with variational autoencoders; pp. 80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Viñas R., Andrés-Terré H., Liò P., Bryson K. Adversarial generation of gene expression data. Bioinformatics. 2022;38:730–737. doi: 10.1093/bioinformatics/btab035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brock A., Donahue J., Simonyan K. Large scale GAN training for high fidelity natural image synthesis. arXiv. 2018 doi: 10.48550/arXiv.1809.11096. Preprint at. [DOI] [Google Scholar]
- 36.Karras T., Aittala M., Laine S., Hörkönen E., Hellsten J., Lehtinen J., Aila T. Alias-free generative adversarial networks. Adv. Neural Inf. Process. Syst. 2021;34 [Google Scholar]
- 37.Claudio Quiros A., Coudray N., Yeaton A., Sunhem W., Murray-Smith R., Tsirigos A., Yuan K. International Conference on Medical Image Computing and Computer-Assisted Intervention. Springer; 2021. Adversarial learning of cancer tissue representations; pp. 602–612. [Google Scholar]
- 38.Quiros A.C., Murray-Smith R., Yuan K. Pathologygan: Learning deep representations of cancer tissue. arXiv. 2019 doi: 10.48550/arXiv.1907.02644. Preprint at. [DOI] [Google Scholar]
- 39.Vaswani A., Shazeer N., Parmar N., Uszkoreit J., Jones L., Gomez A.N., Kaiser L., Polosukhin I. Attention is all you need. Adv. Neural Inf. Process. Syst. 2017;30 doi: 10.48550/arXiv.1706.03762. [DOI] [Google Scholar]
- 40.Ho J., Jain A., Abbeel P. Denoising diffusion probabilistic models. Adv. Neural Inf. Process. Syst. 2020;33:6840–6851. [Google Scholar]
- 41.Ramesh A., Pavlov M., Goh G., Gray S., Voss C., Radford A., Chen M., Sutskever I. International Conference on Machine Learning. PMLR; 2021. Zero-shot text-to-image generation; pp. 8821–8831. [Google Scholar]
- 42.Ramesh A., Dhariwal P., Nichol A., Chu C., Chen M. 2022. Hierarchical text-conditional image generation with clip latents. Preprint at. [DOI] [Google Scholar]
- 43.Tao M., Tang H., Wu F., Jing X.-Y., Bao B.-K., Xu C. Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition. 2022. DF-GAN: A simple and effective baseline for text-to-image synthesis; pp. 16515–16525. [Google Scholar]
- 44.Alayrac J.-B., Donahue J., Luc P., Miech A., Barr I., Hasson Y., Lenc K., Mensch A., Millican K., Reynolds M., et al. Flamingo: A visual language model for few-shot learning. arXiv. 2022 doi: 10.48550/arXiv.2204.14198. Preprint at. [DOI] [Google Scholar]
- 45.McInnes L., Healy J., Melville J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv. 2018 doi: 10.48550/arXiv.1802.03426. Preprint at. [DOI] [Google Scholar]
- 46.Schwarz K., Liao Y., Geiger A. On the frequency bias of generative models. Adv. Neural Inf. Process. Syst. 2021;34:18126–18136. [Google Scholar]
- 47.Peinado M.A. Histology and histochemistry of the aging cerebral cortex: An overview. Microsc. Res. Tech. 1998;43:1–7. doi: 10.1002/(SICI)1097-0029(19981001)43:1%3C1::AID-JEMT1%3E3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 48.Saharia C., Chan W., Saxena S., Li L., Whang J., Denton E., Ghasemipour S.K.S., Ayan B.K., Mahdavi S.S., Lopes R.G., et al. 2022. Photorealistic text-to-image diffusion models with deep language understanding. Preprint at. [DOI] [Google Scholar]
- 49.Yu J., Xu Y., Koh J.Y., Luong T., Baid G., Wang Z., Vasudevan V., Ku A., Yang Y., Ayan B.K., et al. Scaling autoregressive models for content-rich text-to-image generation. arXiv. 2022 doi: 10.48550/arXiv.2206.10789. Preprint at. [DOI] [Google Scholar]
- 50.Fu Y., Jung A.W., Torne R.V., Gonzalez S., Vöhringer H., Shmatko A., Yates L.R., Jimenez-Linan M., Moore L., Gerstung M. Pan-cancer computational histopathology reveals mutations, tumor composition and prognosis. Nat. Cancer. 2020;1:800–810. doi: 10.1038/s43018-020-0085-8. [DOI] [PubMed] [Google Scholar]
- 51.Kather J.N., Heij L.R., Grabsch H.I., Loeffler C., Echle A., Muti H.S., Krause J., Niehues J.M., Sommer K.A.J., Bankhead P., et al. Pan-cancer image-based detection of clinically actionable genetic alterations. Nat. Cancer. 2020;1:789–799. doi: 10.1038/s43018-020-0087-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.GTEx Consortium, Laboratory, Data Analysis &Coordinating Center LDACC—Analysis Working Group, Statistical Methods groups—Analysis Working Group, Enhancing GTEx eGTEx groups, NIH Common Fund, NIH/NCI, NIH/NHGRI, NIH/NIMH, NIH/NIDA, Biospecimen Collection Source Site—NDRI, et al., (2017). Genetic effects on gene expression across human tissues. Nature 550, 204–213 10.1038/nature24277 [DOI]
- 53.GTEx Consortium The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–1330. doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.The GTEx Consortium Cell type-specific genetic regulation of gene expression across human tissues. Science. 2020;369 6509:8528. doi: 10.1126/science.aaz8528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ferraro N.M., Strober B.J., Einson J., Abell N.S., Aguet F., Barbeira A.N., Brandt M., Bucan M., Castel S.E., Davis J.R., et al. Transcriptomic signatures across human tissues identify functional rare genetic variation. Science. 2020;369:5900. doi: 10.1126/science.aaz5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Varoquaux G., Cheplygina V. Machine learning for medical imaging: Methodological failures and recommendations for the future. NPJ Digit. Med. 2022;5:48. doi: 10.1038/s41746-022-00592-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lipkova J., Chen R.J., Chen B., Lu M.Y., Barbieri M., Shao D., Vaidya A.J., Chen C., Zhuang L., Williamson D.F.K., et al. Artificial intelligence for multimodal data integration in oncology. Cancer Cell. 2022;40:1095–1110. doi: 10.1016/j.ccell.2022.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Esteva A., Chou K., Yeung S., Naik N., Madani A., Mottaghi A., Liu Y., Topol E., Dean J., Socher R. Deep learning-enabled medical computer vision. NPJ Digit. Med. 2021;4:5. doi: 10.1038/s41746-020-00376-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tang X., Shigematsu H., Bekele B.N., Roth J.A., Minna J.D., Hong W.K., Gazdar A.F., Wistuba I.I. EGFR tyrosine kinase domain mutations are detected in histologically normal respiratory epithelium in lung cancer patients. Cancer Res. 2005;65:7568–7572. doi: 10.1158/0008-5472.CAN-05-1705. [DOI] [PubMed] [Google Scholar]
- 60.Lee M.V., Katabathina V.S., Bowerson M.L., Mityul M.I., Shetty A.S., Elsayes K.M., Balachandran A., Bhosale P.R., McCullough A.E., Menias C.O. BRCA-associated cancers: role of imaging in screening, diagnosis, and management. Radiographics. 2017;37:1005–1023. doi: 10.1148/rg.2017160144. [DOI] [PubMed] [Google Scholar]
- 61.Ortuño F.M., Loucera C., Casimiro-Soriguer C.S., Lepe J.A., Camacho Martinez P., Merino Diaz L., de Salazar A., Chueca N., García F., Perez-Florido J., et al. Highly accurate whole-genome imputation of SARS-CoV-2 from partial or low-quality sequences. GigaScience. 2021;10 12:078. doi: 10.1093/gigascience/giab078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aghili M., Tabarestani S., Adjouadi M. Addressing the missing data challenge in multi-modal datasets for the diagnosis of alzheimer’s disease. J. Neurosci. Methods. 2022;375 doi: 10.1016/j.jneumeth.2022.109582. [DOI] [PubMed] [Google Scholar]
- 63.Dumoulin V., Visin F. A guide to convolution arithmetic for deep learning. arXiv. 2016 doi: 10.48550/arXiv.1603.07285. Preprint at. [DOI] [Google Scholar]
- 64.Odena A., Dumoulin V., Olah C. Deconvolution and checkerboard artifacts. Distill. 2016 doi: 10.23915/distill.00003. [DOI] [Google Scholar]
- 65.Hosseinzadeh Taher, M.R., Haghighi, F., Feng, R., Gotway, M.B., Liang, J. (2021). A systematic benchmarking analysis of transfer learning for medical image analysis. In: Domain Adaptation and Representation Transfer, and Affordable Healthcare and AI for Resource Diverse Global Health: Third MICCAI Workshop, DART 2021, and First MICCAI Workshop, FAIR 2021, Held in Conjunction with MICCAI 2021, Strasbourg, France, September 27 and October 1, 2021, Proceedings 3, pp. 3–13. Springer [DOI] [PMC free article] [PubMed]
- 66.Vanguri R.S., Luo J., Aukerman A.T., Egger J.V., Fong C.J., Horvat N., Pagano A., Araujo-Filho J.d.A.B., Geneslaw L., Rizvi H., et al. Multimodal integration of radiology, pathology and genomics for prediction of response to PD-(L) 1 blockade in patients with non-small cell lung cancer. Nat. Cancer. 2022;3:1151–1164. doi: 10.1038/s43018-022-00416-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Viazovetskyi Y., Ivashkin V., Kashin E. European Conference on Computer Vision. Springer; 2020. StyleGAN2 distillation for feedforward image manipulation; pp. 170–186. [Google Scholar]
- 68.Ho J., Saharia C., Chan W., Fleet D.J., Norouzi M., Salimans T. Cascaded diffusion models for high fidelity image generation. J. Mach. Learn. Res. 2022;23 47:1–33. https://jmlr.org/papers/v23/21-0635.html [Google Scholar]
- 69.GTEx Consortium. Thomas J., Salvatore M., Phillips R., Lo E., Shad S., Hasz R., Walters G., Garcia F., Young N., et al. The genotype-tissue expression (GTEx) project. Nat. Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Suntsova M., Gaifullin N., Allina D., Reshetun A., Li X., Mendeleeva L., Surin V., Sergeeva A., Spirin P., Prassolov V., et al. Atlas of RNA sequencing profiles for normal human tissues. Sci. Data. 2019;6:36–39. doi: 10.1038/s41597-019-0043-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lu M.Y., Chen T.Y., Williamson D.F.K., Zhao M., Shady M., Lipkova J., Mahmood F. AI-based pathology predicts origins for cancers of unknown primary. Nature. 2021;594:106–110. doi: 10.1038/s41586-021-03512-4. [DOI] [PubMed] [Google Scholar]
- 72.Lu M.Y., Williamson D.F.K., Chen T.Y., Chen R.J., Barbieri M., Mahmood F. Data-efficient and weakly supervised computational pathology on whole-slide images. Nat. Biomed. Eng. 2021;5:555–570. doi: 10.1038/s41551-020-00682-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Otsu N. A threshold selection method from gray-level histograms. IEEE Trans. Syst. Man Cybern. 1979;9:62–66. doi: 10.1109/TSMC.1979.4310076. [DOI] [Google Scholar]
- 74.Goode A., Gilbert B., Harkes J., Jukic D., Satyanarayanan M. Openslide: A vendor-neutral software foundation for digital pathology. J. Pathol. Inform. 2013;4:27. doi: 10.4103/2153-3539.119005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Higgins I., Matthey L., Pal A., Burgess C.P., Glorot X., Botvinick M.M., Mohamed S., Lerchner A. ICLR. 2017. beta-VAE: Learning basic visual concepts with a constrained variational framework. [Google Scholar]
- 76.He K., Zhang X., Ren S., Sun J. Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition. 2016. Deep residual learning for image recognition; pp. 770–778. [Google Scholar]
- 77.Radford A., Metz L., Chintala S. Unsupervised representation learning with deep convolutional generative adversarial networks. arXiv. 2015 doi: 10.48550/arXiv.1511.06434. Preprint at. [DOI] [Google Scholar]
- 78.Arjovsky M., Chintala S., Bottou L. International Conference on Machine Learning. PMLR; 2017. Wasserstein generative adversarial networks; pp. 214–223. [Google Scholar]
- 79.Gulrajani I., Ahmed F., Arjovsky M., Dumoulin V., Courville A.C. Improved training of Wasserstein GANS. Adv. Neural Inf. Process. Syst. 2017;30 [Google Scholar]
- 80.Pal A., Das A. Torchgan: A flexible framework for GAN training and evaluation. J. Open Source Softw. 2021;6:2606. doi: 10.21105/joss.02606. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This paper analyzes existing, publicly available data. These accession numbers for the dataset are listed in the key resources table.
-
•
All original code has been deposited at GitHub and is publicly available as of the date of publication. The URL and an archival DOI are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.