

Abstract
Aggregation of the RNA‐binding protein TAR DNA‐binding protein 43 (TDP‐43) is the key neuropathological feature of neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD). In physiological conditions, TDP‐43 is predominantly nuclear, forms oligomers, and is contained in biomolecular condensates assembled by liquid–liquid phase separation (LLPS). In disease, TDP‐43 forms cytoplasmic or intranuclear inclusions. How TDP‐43 transitions from physiological to pathological states remains poorly understood. Using a variety of cellular systems to express structure‐based TDP‐43 variants, including human neurons and cell lines with near‐physiological expression levels, we show that oligomerization and RNA binding govern TDP‐43 stability, splicing functionality, LLPS, and subcellular localization. Importantly, our data reveal that TDP‐43 oligomerization is modulated by RNA binding. By mimicking the impaired proteasomal activity observed in ALS/FTLD patients, we found that monomeric TDP‐43 forms inclusions in the cytoplasm, whereas its RNA binding‐deficient counterpart aggregated in the nucleus. These differentially localized aggregates emerged via distinct pathways: LLPS‐driven aggregation in the nucleus and aggresome‐dependent inclusion formation in the cytoplasm. Therefore, our work unravels the origins of heterogeneous pathological species reminiscent of those occurring in TDP‐43 proteinopathy patients.
Keywords: aggregation, LLPS, oligomerization, RNA, TDP‐43
Subject Categories: Neuroscience, RNA Biology
Loss of TDP‐43 oligomerization and RNA binding promotes diverse TDP‐43 pathologies providing insights into the origins of heterogeneous pathological species occurring in human disease.
Introduction
Amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD) are two seemingly different, devastating adult‐onset neurodegenerative diseases that exhibit a significant genetic, clinical, and pathological overlap (Ling et al, 2013). The vast majority of ALS patients and up to half of FTLD cases are characterized by the accumulation of aggregated TAR DNA‐binding protein 43 (TDP‐43) in affected neurons (Arai et al, 2006; Neumann et al, 2006; Ling et al, 2013). Importantly, TDP‐43 pathology is not an exclusive hallmark of ALS and FTLD, but is also the main pathological feature in limbic‐predominant age‐related TDP‐43 encephalopathy (LATE; Nelson et al, 2019) and a concomitant pathology in a subset of patients with other neurodegenerative diseases including Alzheimer's, Parkinson's, and Huntington's disease (Chen‐Plotkin et al, 2010). However, TDP‐43 aggregates associated with different clinical subtypes present distinct subcellular localization, namely cytoplasmic, intranuclear, or axonal (Lee et al, 2017; De Rossi et al, 2021), as well as morphological and biochemical properties (Tsuji et al, 2012; Laferriere et al, 2019; Neumann et al, 2020), indicating a distinct molecular origin of these diverse pathological species.
TDP‐43 is a ubiquitously expressed (Ou et al, 1995) nucleic acid‐binding protein (Ou et al, 1995; Buratti & Baralle, 2001) composed of an N‐terminal domain (NTD, amino acids 1–80) involved in self‐oligomerization (Chang et al, 2012; Afroz et al, 2017), two tandem RNA‐recognition motifs (RRMs, amino acids 106‐259) (Buratti & Baralle, 2001; Lukavsky et al, 2013), and an unstructured low complexity region (LCR, amino acids 260–414). The latter contains a transient α‐helix (amino acids 321–340; Conicella et al, 2016) that associates with interaction partners (D'Ambrogio et al, 2009) and was recently shown to coincide with the aggregation core of pathological cytoplasmic TDP‐43 in FTLD brains (Arseni et al, 2022). Under physiological conditions TDP‐43 predominantly localizes to the nucleus (Winton et al, 2008), where it mainly binds UG‐rich intronic sites on pre‐mRNA to regulate alternative splicing (Polymenidou et al, 2011; Tollervey et al, 2011) and undergoes liquid–liquid phase separation (LLPS; Zbinden et al, 2020) to form dynamic nuclear condensates (Wang et al, 2012, 2020; Gasset‐Rosa et al, 2019; Hallegger et al, 2021), which were suggested to localize to specific subnuclear membraneless compartments (Tsuiji et al, 2013; Wang et al, 2020). Despite its predominantly nuclear localization (Winton et al, 2008), TDP‐43 shuttles between the nucleus and the cytoplasm (Ayala et al, 2008), where it plays roles in mRNA stability, transport and translation, miRNA processing, mitochondrial and synaptic function and stress responses (Ling et al, 2013). In particular, TDP‐43 was shown to incorporate into and modulate the dynamics of stress granules (SGs) upon exposure to different temperature, osmotic, oxidative, and chemical stressors (Aulas et al, 2012; Dewey et al, 2012).
RNA binding is essential for TDP‐43 to perform its physiological functions in RNA metabolism (Afroz et al, 2019). In addition, RNA binding precludes TDP‐43 passive leakage out of the nucleus (Ayala et al, 2008; Ederle et al, 2018) and modulates its LLPS behavior (Mann et al, 2019; Yu et al, 2021). In contrast, little is known about the importance of TDP‐43 NTD‐driven self‐oligomerization in physiology. Previous data have shown that nuclear TDP‐43 oligomerization is required for alternative splicing of at least a subset of its known RNA targets (Zhang et al, 2013; Afroz et al, 2017; Jiang et al, 2017; Mompean et al, 2017; Wang et al, 2018). However, the role of self‐oligomerization in the physiological properties of TDP‐43, including its subcellular localization, stability, LLPS behavior, and cytoplasmic functions, remains poorly understood. Also, whether—and if so, how—TDP‐43 RNA binding and oligomerization impact each other in cells is unknown.
The overexpression of TDP‐43 in cellular and animal models results in its aggregation, a phenomenon that has been extensively explored in recent years to recapitulate the main neuropathological hallmark of ALS/FTLD (Hergesheimer et al, 2019). However, TDP‐43 is a tightly autoregulated protein (Ayala et al, 2011; Polymenidou et al, 2011) and overexpression can distort its subcellular (Ederle et al, 2018) and subnuclear (Tsuiji et al, 2013; Gasset‐Rosa et al, 2019) localization, and potentially its functions. We therefore aimed to study the physiological role of TDP‐43 oligomerization and its interplay with RNA binding at near‐physiological protein levels, and to subsequently compare the pathways triggered by their respective impairment. Using human neural cultures and single‐copy expression systems in human cell lines, we show that NTD‐driven TDP‐43 oligomerization and RNA binding are intertwined and required to maintain the half‐life, functionality, and localization of TDP‐43. Upon failure of the ubiquitin‐proteasome system (UPS), monomerization and impaired RNA binding triggered TDP‐43 aggregation via distinct pathways in the cytoplasm and nucleus. Our results underscore the relevance of loss of oligomerization and RNA binding in the initiation of diverse TDP‐43 pathologies and unravel the origins of heterogeneous pathological species occurring in human disease.
Results
Oligomerization and RNA‐binding cooperatively stabilize the half‐life of TDP‐43
To systematically compare the properties of oligomeric, monomeric, and RNA binding‐deficient TDP‐43, we introduced a single copy of an N‐terminally green fluorescent protein (GFP)‐tagged TDP‐43 coding sequence under the control of a doxycycline‐inducible promoter into HEK293 cells using the Flp‐In T‐REx technology (Fig EV1A) (Ling et al, 2010). This construct harbored either six previously described point mutations that disrupt TDP‐43 oligomerization (termed 6M) (Afroz et al, 2017), five point mutations within its RRMs disrupting RNA binding through the RRMs (all F > A, referred to as RRMm) (Buratti & Baralle, 2001; Lukavsky et al, 2013) or both 6M&RRMm (Fig 1A). The resulting four isogenic cell lines (WT, 6M, RRMm, and 6M&RRMm) expressed equal levels of the exogenous GFP‐TDP‐43 RNA (Fig EV1B) and both transgenic RNA and protein were detectable only upon addition of doxycycline (Figs 1B and EV1C and D). Wild type (WT) GFP‐TDP‐43 protein levels displayed a mere fivefold increase compared to endogenous TDP‐43, as determined by immunoblot analysis (Fig EV1D and E). However, despite equal RNA levels (Fig EV1B), protein levels of the GFP‐TDP‐43 mutants were noticeably lower than their WT counterpart (Figs 1B and C, and EV1E–G), and displayed only 1.5‐, 1‐, and 0.5‐fold levels compared to endogenous TDP‐43 in the case of 6M, RRMm, and 6M&RRMm, respectively (Fig EV1E–G). Protein turnover analysis using the translation inhibitor cycloheximide (CHX) showed that the half‐life of the RNA‐binding TDP‐43 mutant (RRMm) was reduced by > 8 h compared to WT GFP‐TDP‐43 (Fig 1D and E), consistent with previous findings (Flores et al, 2019). Interestingly, oligomerization deficiency (6M) had a similar effect on the half‐life of TDP‐43 as loss of RNA binding (RRMm). Furthermore, the combined GFP‐TDP‐43 variant (6M&RRMm) presented a cumulative effect (Fig 1D and E). Since point mutations can affect protein folding and thereby selectively target proteins for degradation (Jayaraj et al, 2020), we confirmed that the introduced mutations do not interfere with the folding of TDP‐43 using far‐UV circular dichroism (CD) (Figs 1F and EV1H) and two‐dimensional nuclear magnetic resonance (2D‐NMR) spectroscopy (Fig EV1I and J) (Afroz et al, 2017; Wang et al, 2019), which revealed that the mutated domains are properly folded. Therefore, our results indicate that loss of oligomerization or RNA‐binding ability similarly reduces the half‐life of TDP‐43. Since incorporation of proteins into functional multimeric complexes has been reported to correlate with longer half‐lives in yeast (Mallik & Kundu, 2018) and mouse brain cells (Dörrbaum et al, 2018), these observations strengthen the link between TDP‐43 functionality and its half‐life.
Figure EV1. Characterization of the expression levels, localization, and folding of the TDP‐43 variants.
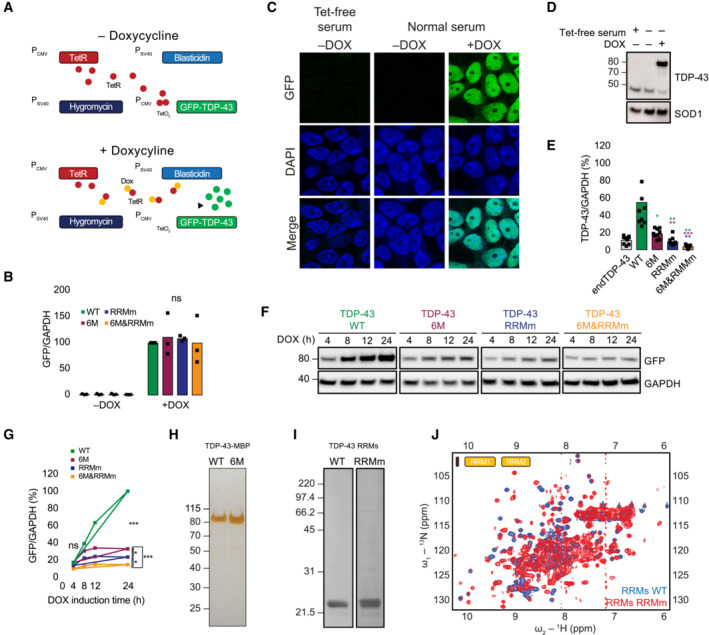
- Schematic representation of the Flp‐In T‐Rex system. Dox: doxycycline, GFP: green fluorescent protein, TetR: tetracycline repressor.
- RNA levels of the GFP‐TDP‐43 variants after 48 h of expression in the isogenic cell lines measured by qPCR with primers specifically targeted to the GFP sequence. N = 3 independent experiments. Two‐way ANOVA with Tukey's multiple comparisons post hoc test.
- Representative image of confocal fluorescence microscopy showing the tight expression regulation of the Flp‐In T‐Rex system, which is unaffected by the residual tetracycline (Tet) present in the regular serum used for the preparation of cell culture medium. Expression of GFP‐TDP‐43 WT is only observable upon addition of doxycycline (DOX) for 48 h. Nuclei are stained with DAPI. Scale bar: 20 μm.
- Western blot analysis of the conditions described in (C).
- Quantification of the TDP‐43 signal from Fig 1B using a total TDP‐43 antibody, including endogenous TDP‐43 (endTDP‐43) and the four GFP‐TDP‐43 variants. N = 3 independent experiments. Repeated measures one‐way ANOVA with Greenhouse–Geisser correction and Tukey's multiple comparisons post hoc test.
- Western blot analysis of a time course of the different GFP‐TDP‐43 variant expression upon induction with DOX.
- Quantification of the GFP signal from (F). N = 3 independent experiments. Two‐way ANOVA with Tukey's multiple comparisons post hoc test.
- Silver‐stained gel showing the purity of 1 μg of the isolated TDP‐43‐MBP variants. (I) Coommasie stained gel showing the purity of the purified TDP‐43 RRM constructs.
- Overlay of 2D 1H‐15N HSQC spectra from purified His‐tagged, 15N‐isotopically labeled TDP‐43 RRMs WT (blue) and RRMm RRMs (red). The presence of dispersed peaks in the spectra indicates that both WT and RRMm RRMs are folded, and are compatible with the formation of α‐helix and β‐strand structures. In case of unfolding, all 1H NMR signals would pool around 8 ppm. ns: not significant, *P < 0.05, **P < 0.01, ***P < 0.001. Graph bars represent mean ± SD.
Figure 1. Oligomerization and RNA‐binding cooperatively stabilize the half‐life of TDP‐43.
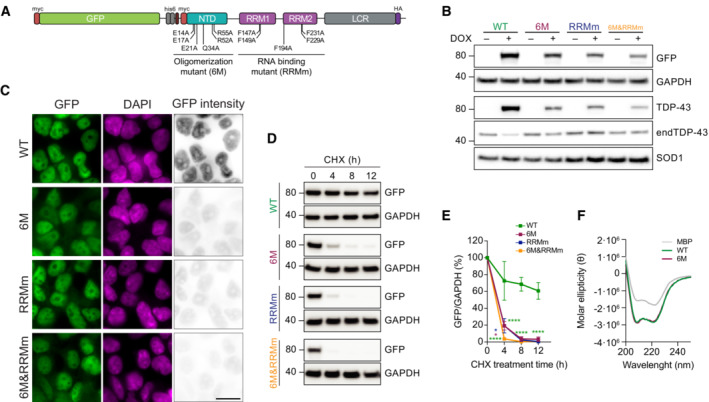
- Schematic representation depicting the mutations disrupting the oligomerization and/or RNA binding property of the GFP‐TDP‐43 variants used to develop the inducible, isogenic HEK293 cell lines. GFP: green fluorescent protein, NTD: N‐terminal domain, RRM: RNA‐recognition motif, LCR: low complexity region.
- Western blot analysis of the generated isogenic cell lines described in (A) after inducing GFP‐TDP‐43 expression for 48 h showing the tightness of the doxycycline (DOX)‐modulated expression system. Note also the different protein levels of the expressed variants. endTDP‐43: endogenous TDP‐43.
- Representative images of widefield fluorescence microscopy of the isogenic cell lines depicted in (B). GFP brightness is adjusted in each condition for optimal visualization of GFP‐TDP‐43 localization. Original intensity values are represented in the right column using grayscale. Cell nuclei are stained with DAPI. Scale bar: 20 μm.
- GFP‐TDP‐43 expression was induced with DOX for 24 h before cycloheximide (CHX) treatment for the indicated times and western blot analysis.
- Quantification of the GFP signal from (D). N = 3 independent experiments. Two‐way ANOVA with Tukey's multiple comparisons post hoc test.
- Average far‐UV CD spectra of purified maltose‐binding protein (MBP) and TDP‐43‐MBP variants from N = 3 independent experiments. ****P < 0.0001. Graph bars represent mean ± SD.
Source data are available online for this figure.
TDP‐43 oligomerization and RNA‐binding preserve its nuclear localization
Due to an active nuclear import and its ability to passively diffuse out of the nucleus, TDP‐43 is a nucleocytoplasmic shuttling protein (Ederle et al, 2018; Pinarbasi et al, 2018). RNA binding retains TDP‐43 in the nucleus by forming bigger macromolecular complexes that slow down its diffusion into the cytoplasm (Ayala et al, 2008; Ederle et al, 2018; Duan et al, 2022). We therefore wondered whether oligomerization also affects the physiological localization of TDP‐43. To address this question, we first measured the mean fluorescence intensity of GFP‐TDP‐43 in the nucleus and the cytoplasm for all four GFP‐TDP‐43 variants. Using DAPI and G3BP as nuclear and cytoplasmic markers, respectively (Fig EV2A), we observed that monomeric GFP‐TDP‐43 (6M) showed a significantly increased cytoplasmic localization compared to WT and RRMm (Figs 1C and 2A). This cytoplasmic mislocalization due to lack of self‐oligomerization was further exacerbated in combination with a loss of RNA binding (6M&RRMm), suggesting an independent, additive contribution of both protein–protein and protein–RNA interactions to the nuclear localization of TDP‐43. Similar observations were obtained in human neurons transduced with HA‐tagged versions of the four TDP‐43 variants (Fig 2B and C).
Figure EV2. Oligomerization and RNA‐binding preserve the nuclear localization of the GFP‐TDP‐43 variants.
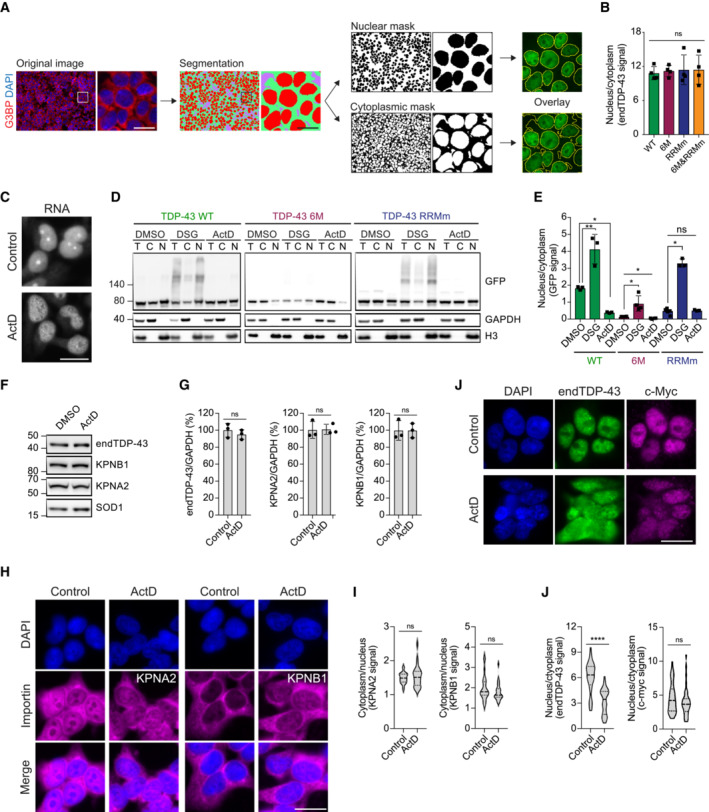
- Schematic of the pipeline for the quantification of nuclear and cytoplasmic GFP‐TDP‐43 fluorescence levels with the Fiji plug‐in Trainable Weka Segmentation. Scale bar: 20 μm.
- Quantification of endogenous TDP‐43 (endTDP‐43) signal from Fig 2D. N = 3 independent experiments. Repeated measures one‐way ANOVA with Greenhouse–Geisser correction and Tukey's multiple comparisons post hoc test. ns: not significant. Graph bars represent mean ± SD.
- Representative images of widefield fluorescence microscopy of HEK293 cells treated with ActD showing the total RNA pattern. Note the absence of rRNA staining in the nucleoli upon ActD treatment indicating that RNA transcription has been halted. Scale bar: 20 μm.
- After expression of GFP‐TDP‐43 (WT, 6M or RBDm) for 48 h, the cell lines were treated with 5 μg/ml ActD for 4 h to inhibit transcription or DSG to cross‐link protein–protein interactions before performing nucleocytoplasmic fractionation and western blot analysis. Note how the fractionation of the RNA‐binding mutant GFP‐TDP‐43 (RRMm) resembles that of ActD‐treated GFP‐TDP‐43 WT, and how the stabilization of TDP‐43 oligomerization through DSG cross‐link does not affect the localization of oligomerization‐deficient GFP‐TDP‐43 as much as it does for the oligomerization‐competent GFP‐TDP‐43 WT.
- Quantification of the GFP signal from (D). N = 3 independent experiments. Repeated measures one‐way ANOVA with Tukey's multiple comparisons post hoc test.
- Expression of the importins involved in the nuclear translocation of TDP‐43 (KPNA2 and KPNB1) was analyzed in HEK293 cells by western blot upon treatment with 5 μg/ml ActD for 4 h.
- Quantification of the endogenous TDP‐43 (endTDP‐43), KPNA2, and KPNB1 signal from (E). Repeated measures one‐way ANOVA with Greenhouse–Geisser correction and Dunnett's multiple comparisons post hoc test. (H) Representative images of widefield fluorescence microscopy of HEK293 cells treated with 5 5 μg/ml ActD for 4 h showing the distribution of KPNA2 and KPNB1. Scale bar: 20 μm.
- Quantification of the nucleocytoplasmic distribution of KPNA2 and KPNB1 in the immunocytochemistry images shown in (H). N = 25 cells. Unpaired two‐tailed t‐test.
- Representative images of widefield fluorescence microscopy of HEK293 cells treated with 5 μg/ml ActD for 4 h showing the distribution of c‐myc, a non‐RNA‐binding cargo of the KPNA2/KPNB1 complex. Scale bar: 20 μm.
- Quantification of the endogenous TDP‐43 (endTDP‐43) and c‐myc levels in the immunocytochemistry images shown in (I). N = 40 cells. Mann–Whitney U‐test. Nuclei are stained with DAPI in (A, H and J). Ns: not significant, *P < 0.05, **P < 0.01, ****P < 0.0001. Graph bars represent mean ± SD. Violin plots show mean and quartiles.
Figure 2. TDP‐43 oligomerization and RNA‐binding preserve its nuclear localization.
- Quantification of nucleocytoplasmic levels of GFP‐TDP‐43 from the immunocytochemistry images shown in Fig 1C. N = 3 independent experiments. One‐way ANOVA with Tukey's multiple comparisons post hoc test.
- Representative maximum intensity Z‐projections from confocal fluorescence imaging of human neurons transduced with TDP‐43‐HA variants and immunolabeled for the HA tag and the neuron‐specific marker MAP2. Scale bar: 10 μm.
- Quantification of nucleocytoplasmic levels of TDP‐43‐HA from the immunocytochemistry images shown in (B). N = 14–20 cells. Kruskal–Wallis test with Dunn's multiple comparisons post hoc test.
- GFP‐TDP‐43 expression was induced with doxycycline (DOX) for 4 h before nucleocytoplasmic fractionation and subsequent analysis of GFP‐TDP‐43 and endogenous TDP‐43 (endTDP‐43) levels by western blot. T: total lysate, C: cytoplasmic fraction, N: nuclear fraction.
- Quantification of the GFP signal from (D). Repeated measures one‐way ANOVA with Greenhouse–Geisser correction and Tukey's multiple comparisons post hoc test. Cyt: cytoplasm, Nuc: nucleus.
- HEK293 cells were treated with ActD to inhibit transcription or subjected to protein–protein cross‐linking with DSG followed by nucleocytoplasmic fractionation and western blot analysis. T: total lysate, C: cytoplasmic fraction, N: nuclear fraction, H3: histone H3.
- Quantification of endTDP‐43 signal from (F). N = 3 independent experiments. Repeated measures one‐way ANOVA with Greenhouse–Geisser correction and Dunnett's multiple comparisons post hoc test.
- Representative images of widefield fluorescence microscopy of HEK293 cells treated with ActD for 4 h and immunolabeled for TDP‐43. Scale bar: 20 μm.
- Quantification of nucleocytoplasmic levels of endTDP‐43 in the immunocytochemistry images shown in (H). N = 3 independent experiments. Unpaired two‐tailed t‐test.
- Representative images of confocal fluorescence microscopy of human neural cultures treated with ActD and ivermectin (IVM) and immunolabeled for TDP‐43 and the neuron‐specific marker MAP2. Scale bar: 20 μm.
- Quantification of nucleocytoplasmic levels of endTDP‐43 in the immunocytochemistry images shown in (J). Kruskal–Wallis test with Dunn's multiple comparisons post hoc test. N = 23–48 fields corresponding to a total of 351–569 neurons per condition. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Graph bars represent mean ± SD. Violin plots show mean and quartiles. Cell nuclei are stained with DAPI in (B, H, J).
Source data are available online for this figure.
We subsequently sought to confirm these results by nucleocytoplasmic fractionation. In line with the immunocytochemistry results (Figs 1C and 2A–C), upon mild lysis and nuclei enrichment by centrifugation, WT GFP‐TDP‐43 was mostly retained in the nuclear fraction (Fig 2D and E), with a small cytoplasmic pool corresponding to the subset of TDP‐43 performing functions in this compartment (Ling et al, 2013). In contrast, oligomerization‐deficient mutations (6M and 6M&RRMm) consistently shifted the majority of GFP‐TDP‐43 to the cytoplasmic fraction (Fig 2D and E), to a larger extent than observed by immunocytochemistry (Fig 2A and C). Importantly, the endogenous protein in the same samples remained predominantly nuclear (Figs 2D and EV2B). This contrast was even more pronounced in the case of GFP‐TDP‐43 lacking RNA‐binding ability (RRMm), which fully shifted its localization to the cytoplasm upon fractionation and subsequent immunoblotting (Fig 2D and E) as opposed to its nuclear localization by immunocytochemistry (Fig 2A and C). We therefore wondered whether monomeric (6M) and RNA binding‐deficient (RRMm) GFP‐TDP‐43 exhibit an increased passive diffusion rate and diffuse out of the nucleus during the fractionation procedure, when active nuclear import is absent. Indeed, stabilization of TDP‐43 oligomers by protein–protein cross‐linking with disuccinimidyl glutarate (DSG) (Afroz et al, 2017) before nucleocytoplasmic fractionation increased the retention of endogenous TDP‐43 in the nucleus (Fig 2F and G). Conversely, when we pretreated the cells with actinomycin D (ActD) to block transcription and decrease the levels of newly synthesized pre‐mRNAs—the main RNA targets bound by TDP‐43 in the nucleus (Polymenidou et al, 2011; Tollervey et al, 2011)—(Appendix Fig S2C; Duan et al, 2022), the localization of endogenous TDP‐43 shifted to the cytoplasm (Figs 2F and G, and EV2D and E), as previously reported (Ayala et al, 2008; Ederle et al, 2018). Importantly, this ActD‐induced translocation of TDP‐43 to the cytoplasm was not due to altered levels (Fig EV2F and G) or distribution (Fig EV2H and I) of the importins involved in active nuclear TDP‐43 import. Moreover, ActD treatment did not affect the localization of c‐myc, which is also transported to the nucleus by the same importin complex as TDP‐43 but does not bind RNA (Fig EV2J and K). Notably and similarly to what we observed for GFP‐TDP‐43 RRMm (Figs 1C, 2A and 2D and E), this efflux of TDP‐43 upon ActD treatment and subsequent sample fractionation (Fig 2F and G) was more pronounced than observed by immunocytochemistry (Figs 2H and I, and EV2J and K). Together, these observations suggest that active nuclear import compensates for the abundant passive TDP‐43 egress from the nucleus in the absence of oligomerization or RNA binding. Indeed, similar results were observed in human neurons, where the combined treatment of ActD and ivermectin, an importin α/β‐mediated nuclear import inhibitor (Wagstaff et al, 2012), increased the cytoplasmic shift of endogenous TDP‐43 as compared to treatment with ActD alone (Fig 2J and K). Collectively, these observations show that RNA binding and protein–protein interactions, especially its self‐oligomerization, involve TDP‐43 in larger macromolecular complexes that are retained in the nucleus.
Oligomerization is required for physiological phase separation of TDP‐43 in the nucleus
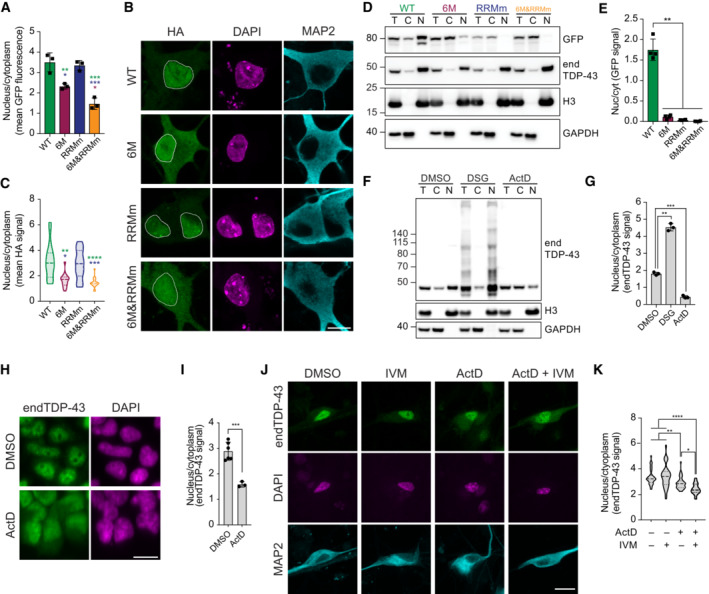
TDP‐43 has been shown to undergo LLPS (Zbinden et al, 2020), a phenomenon visible in the nucleus as small condensates that fuse and split at endogenous protein concentrations (Wang et al, 2012, 2020; Gasset‐Rosa et al, 2019; Hallegger et al, 2021). Also in our model, endogenous TDP‐43 presented this characteristic punctate pattern in the nucleus of HEK293 cells (Fig 3A). Both the number and size of these endogenous TDP‐43 punctate decreased upon treatment with 1,6‐hexanediol (1,6‐HD; Figs 3A and B, and EV3A and B), an alcohol commonly used for its properties to suppress LLPS compartments formed by hydrophobic interactions (Kroschwald et al, 2017; preprint: Perdikari et al, 2022) despite its reported additional effects on nuclear transport (Patel et al, 2007) and kinase and phosphatase activity (Düster et al, 2021). In line with previous data (Gasset‐Rosa et al, 2019; Yu et al, 2021), this indicates that the observed nuclear TDP‐43 punctate are LLPS‐driven condensates.
Figure 3. Oligomerization is required for physiological LLPS of TDP‐43 in the nucleus.
- Representative maximum intensity Z‐projections from confocal fluorescence imaging (thickness of ~10 μm, in steps of 0.21 μm) of HEK293 cells after mock or 1,6‐hexanediol (1,6‐HD) treatment for 15 min stained for endogenous TDP‐43 (endTDP‐43). Scale bar: 5 μm.
- Quantification of three‐dimensional (3D) reconstructions from the images of the conditions shown in (A) depicting the number of nuclear condensates per cell. N = 14–23 cells. Unpaired two‐tailed t‐test.
- Fluorescence microscopy images of 10 μM purified full‐length TDP‐43 and its oligomerization‐deficient counterpart showing different abilities to undergo LLPS and its disruption by 1,6‐HD treatment for 10 min. Scale bar: 10 μm.
- Quantification of the number of condensates in the conditions shown in (C) per 4,000 μm2 field. N = 10 independent experiments. Kruskal–Wallis test with Dunn's multiple comparisons post hoc test.
- Representative maximum intensity Z‐projections (thickness of ~10 μm, in steps of 0.21 μm) from confocal fluorescence microscopy of the isogenic cell lines expressing GFP‐TDP‐43 for 48 h with doxycycline (DOX). Scale bar: 5 μm.
- 3D quantification of the number of nuclear condensates per cell after GFP‐TDP‐43 expression for 48 h. N = 16–22 cells.
- 3D quantification of the volume of the nuclear condensates per cell in the conditions described in (E). N = 51–4,401 condensates. Kruskal–Wallis test with Dunn's multiple comparisons post hoc test.
- GFP‐TDP‐43 expression was induced with DOX for 4 h before crosslinking protein–protein interactions with DSG and subsequent analysis by western blot. *, **, and *** indicate GFP‐TDP‐43 monomers, dimers, and trimers, respectively. °, °°, °°°, and °°°° indicate endTDP‐43 monomers, dimers, trimers, and tetramers.
- Quantification of GFP‐TDP‐43 dimer/monomer ratio based on the GFP signal from (H). N = 3 independent experiments. Repeated measures one‐way ANOVA with Greenhouse–Geisser correction and Tukey's multiple comparisons post hoc test. *P < 0.05, **P < 0.01, ****P < 0.0001. Graph bars represent mean ± SD. Violin plots show mean and quartiles.
Source data are available online for this figure.
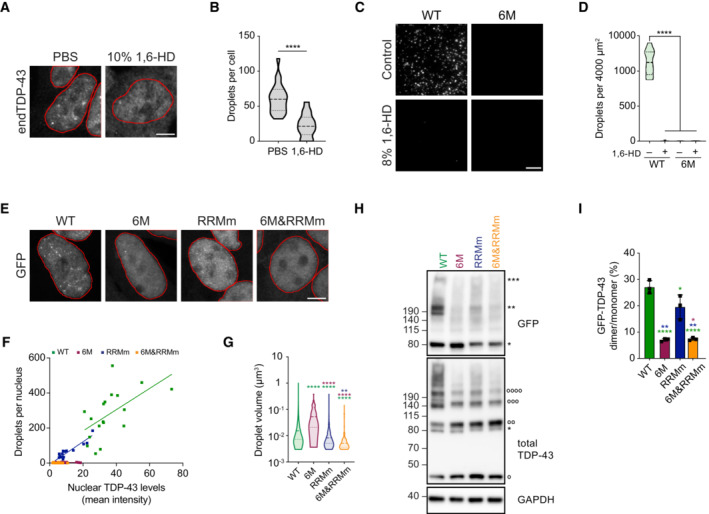
Figure EV3. Oligomerization retains TDP‐43 in the nucleus in both RNA‐bound and ‐unbound states.
- Schematic of the Imaris pipeline for the quantification of the average nuclear TDP‐43 fluorescence as well as the definition of the nuclear TDP‐43 droplets that were subsequently analyzed for total number and volume. Pipeline shown for GFP‐TDP‐43 signal, but the same approach was used with fluorescence derived from the immunostaining of endogenous TDP‐43.
- 3D quantification of the volume of the nuclear droplets per cell in the conditions described in Fig 3A. N = 344–1,679 droplets. Mann–Whitney U‐test.
- Western blot analysis of the cleavage of the maltose‐binding protein (MBP) tag of recombinant full‐length TDP‐43 by TEV protease for the in vitro LLPS assay shown in Fig 2C. Note that the presence of 1,6‐hexanediol (1,6‐HD) does not affect the cleavage of the tag.
- Representative maximum intensity Z‐projections (thickness of ~10 μm, in steps of 0.21 μm) from confocal fluorescence microscopy of the isogenic cell lines expressing GFP‐TDP‐43 for 4 h with doxycycline (DOX). Scale bar: 5 μm.
- 3D quantification of the number of nuclear droplets per cell after GFP‐TDP‐43 expression for 4 h. N = 5 cells.
- Expression of GFP‐TDP‐43 variants was induced for 48 h before DSG cross‐linking of protein–protein interactions and subsequent analysis by western blot. * and ** indicate GFP‐TDP‐43 monomers and dimers, respectively. °, °°, °°°, and °°°° indicate endogenous TDP‐43 (endTDP‐43) monomers, dimers, trimers, and tetramers.
- Expression of GFP‐TDP‐43 variants was induced for 4 h before DSG cross‐linking of protein–protein interactions and subsequent analysis by western blot. * indicates GFP‐TDP‐43 monomers. °, °°, °°°, and °°°° indicate endTDP‐43 monomers, dimers, trimers and tetramers. **P < 0.01, ****P < 0.0001. Violin plots show mean and quartiles.
Although high concentrations of the LCR of TDP‐43 are sufficient for phase separation in vitro (Conicella et al, 2016, 2020), additional interactions must take place for full‐length TDP‐43 to undergo physiological LLPS at far lower concentrations (Wang et al, 2018; Gasset‐Rosa et al, 2019). Recent evidence points towards self‐interaction through the NTD as another driver of TDP‐43 LLPS in vitro (Jiang et al, 2017; Wang et al, 2018) and in a human cell line (Yu et al, 2021). Indeed, at a reported physiological concentration of 10 μM (Maharana et al, 2018; Hallegger et al, 2021), purified full‐length TDP‐43 phase separated into condensates, which also dissolved in the presence of 1,6‐HD (Figs 3C and D, and EV3C). In contrast, oligomerization‐deficient TDP‐43 (6M) did not form condensates under the same conditions, suggesting that NTD interactions are essential for TDP‐43 LLPS (Fig 3C and D).
These findings were reproduced in our isogenic cell lines. Whereas WT GFP‐TDP‐43 formed many nuclear condensates, at comparable protein levels, disruption of oligomerization (6M) virtually suppressed all GFP‐TDP‐43 condensate formation (Fig 3E and F), pointing to an essential role for oligomerization in physiological TDP‐43 LLPS in cells. Interestingly, like the WT protein, RNA binding‐deficient GFP‐TDP‐43 (RRMm) formed nuclear condensates (Figs 3E and F, and EV3D and E), which were smaller in size (Fig 3G). This observation suggests that nuclear TDP‐43 condensates arise independently of specific RNA binding, but does not discard the involvement of RNA in their formation. Quantification of the number of GFP‐TDP‐43 condensates for both WT and RRMm revealed that condensate formation was proportional to their nuclear protein levels (Figs 3F and EV3E). Since local protein concentration modulates (Zbinden et al, 2020) this observation, it further strengthens that indeed the observed condensates in our model are liquid compartments. Disruption of both oligomerization and RNA binding in the combined GFP‐TDP‐43 variant (6M&RRMm) drastically reduced the number of nuclear condensates compared to GFP‐TDP‐43 RRMm (Figs 3E and F, and EV3D and E), indicating that TDP‐43 condensate formation in the absence of RNA binding (RRMm) is also mediated through NTD interactions. This was supported by a biochemical analysis with DSG protein–protein crosslinking, which showed protein complexes at the expected size of GFP‐TDP‐43 dimers for both WT and RRMm, but not for the monomeric variants (6M and 6M&RRMm) at comparable protein levels (Figs 3H and I, and EV3F and G). Similar to its WT counterpart, stabilization of these protein complexes via crosslinking retained GFP‐TDP‐43 RRMm predominantly in the nucleus in our isogenic cell lines despite the lack of specific RNA binding (Fig EV2D and E). Overall, our results indicate that NTD‐driven oligomerization—and not only LCR interactions—are essential for TDP‐43 LLPS in cells, both in the presence and absence of RNA binding.
Loss of RNA binding leads to TDP‐43 oligomers with distinct orientation
The amount of GFP‐TDP‐43 dimers formed by the RNA binding‐deficient variant (RRMm) was significantly reduced compared to the WT protein (Fig 3H and I), suggesting that TDP‐43 oligomerization is modulated by RNA binding. Indeed, treatment with ActD decreased the level of endogenous TDP‐43 dimers detected by protein–protein cross‐linking (Fig 4A and B). Concomitant with this reduction in oligomerization, ActD‐treated cells also displayed a reduced number of TDP‐43 nuclear condensates (Fig 4C and D). To determine whether TDP‐43 oligomerization is exclusively confined to nuclear condensates, we first assessed the exact subnuclear location of the oligomers. For this purpose, we employed proximity ligation assay (PLA) to visualize TDP‐43 dimers with a single monoclonal antibody (mAb) conjugated to two different oligonucleotides. With this approach, only TDP‐43 molecules that come to close proximity (maximum 20 nm apart) allow oligonucleotide hybridization and fluorescent signal amplification. Surprisingly, while abundant PLA signal was detected in the nucleus, only a fraction overlapped with its nuclear condensates, suggesting that TDP‐43 dimerization is not restricted to nuclear condensates (Fig 4E), a result that was confirmed for WT GFP‐TDP‐43 using a mAb against GFP (Figs 4F and EV4A and B).
Figure 4. Loss of RNA binding leads to TDP‐43 oligomers with distinct orientation.
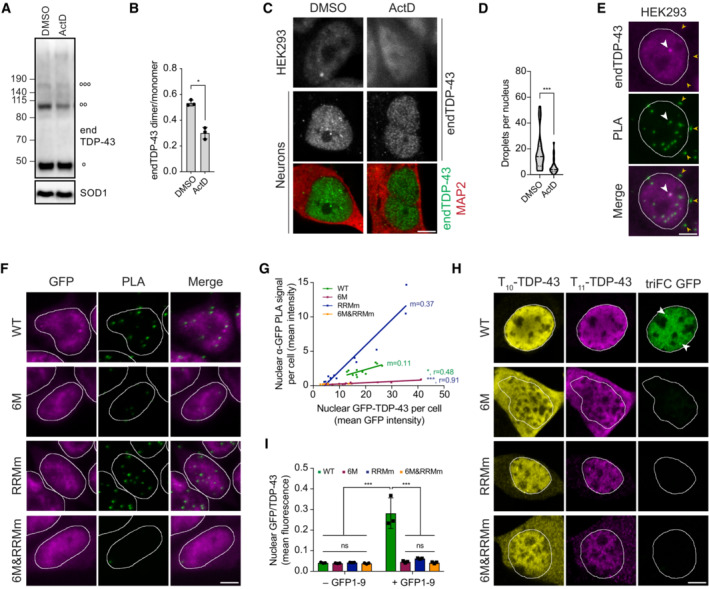
- HEK293 cells were treated with actinomycin D (ActD) for 4 h to inhibit transcription before treatment with the protein–protein cross‐linker DSG and western blot analysis. °, °°, and °°°° indicate endogenous TDP‐43 (endTDP‐43) monomers, dimers and trimers, respectively.
- Quantification of the endTDP‐43 signal from (A). N = 3 independent experiments. Paired two‐tailed t‐test.
- Representative image of confocal fluorescence microscopy of HEK293 cells and neurons treated with 5 μg/ml ActD for 4 h. Scale bar: 5 μm.
- Single‐plane quantification of the number of nuclear condensates per neuron in the conditions described in (C). N = 25–26 cells. Mann–Whitney U‐test.
- Proximity ligation assay (PLA) using a monoclonal anti‐TDP‐43 antibody reveals nuclear and cytoplasmic localization of endTDP‐43 dimers in physiological conditions. Big white arrowheads indicate overlapping GFP‐TDP‐43 condensates and PLA signal. Small yellow arrowheads indicate cytoplasmic PLA signal. Scale bar: 5 μm.
- PLA using a monoclonal anti‐GFP antibody reveals the localization of GFP‐TDP‐43 dimers in the isogenic cell lines upon protein expression with doxycycline (DOX) for 48 h. Note the absence of dimers in the oligomerization‐deficient variants (6M and 6M&RRMm). Scale bar: 5 μm.
- Quantification of the nuclear PLA signal shown in (F) correlated to the protein expression levels of the different TDP‐43 variants, measured as the mean GFP fluorescence. N = 11–13 cells.
- Tripartite GFP complementation assay using a pair of N‐terminally T10‐ and T11‐tagged TDP‐43 constructs co‐transfected with GFP1–9 in motoneuron‐like NSC‐34 cells. Note the cytoplasmic abundance of the monomeric (6M and 6M&RRMm) variants. triFC: trimolecular fluorescence complementation. Scale bar: 5 μm.
- Quantification of the GFP fluorescence levels relative to the T10/T11‐TDP‐43 expression levels as shown in (H). N = 3 biological replicates, with N = 6–35 cells per replicate. Two‐way ANOVA with Tukey's multiple comparisons post hoc test. *P < 0.05, ***P < 0.001. Graph bars represent mean ± SD. Violin plots show mean and quartiles.
Source data are available online for this figure.
Figure EV4. Nuclear TDP‐43 oligomers present different features depending on their RNA binding state.
- Schematic representation of the proximity ligation assay (PLA) to detect homodimers using a single monoclonal antibody conjugated to two different nucleic acid probes, for both tags at the N‐terminus (GFP) or C‐terminus (HA) of the GFP‐TDP‐43 construct.
- PLA using a monoclonal anti‐GFP antibody specifically identifies the presence of GFP‐TDP‐43 WT dimers in the isogenic cell lines upon protein expression with doxycycline (DOX) for 48 h. Note the absence of dimer signal in the absence of GFP‐TDP‐43 expression (–DOX). Nuclei are stained with DAPI. Scale bar: 5 μm.
- Quantification of the nuclear PLA signal obtained using an anti‐HA monoclonal antibody in relation to the protein expression levels of the different TDP‐43 variants, measured as the mean HA signal. N = 9–16 cells.
- Schematic representation of the GFP trimolecular fluorescence complementation (triFC) assay designed to identify TDP‐43 dimers. NLS: nuclear localization signal.
- Quantification of nucleocytoplasmic levels of TDP‐43 in immunocytochemistry images as shown in Fig 4H. N = 6 independent experiments. One‐way ANOVA with Tukey's multiple comparisons post hoc test.
- Quantification of the average nuclear mean T10‐ and T11‐TDP‐43 signals from Fig 4H shows that cells analyzed for reconstituted GFP present comparable protein expression levels of the TDP‐43 variants. N = 30–83. Kruskal–Wallis test with Dunn's multiple comparisons post hoc test.
- Quantification of the nuclear mean GFP fluorescence in relation to the average nuclear mean T10‐ and T11‐TDP‐43 signals from Fig 4H indicated that reconstitution of GFP fluorescence is not due to different protein expression levels of the TDP‐43 variants. N = 30–83. ns: not significant, ***P < 0.001, ****P < 0.0001. Graph bars represent mean ± SD.
In contrast, the oligomerization‐deficient variants (6M and 6M&RRMm) showed a markedly decreased PLA signal, even in cells with comparable protein levels (Fig 4F and G), confirming that the observed PLA signal depends on oligomerization. In line with our DSG cross‐linking results (Fig 3H and I), the RNA‐binding GFP‐TDP‐43 mutant (RRMm) displayed a positive PLA signal (Fig 4F). Interestingly, the mean intensity of PLA‐positive foci was consistently higher than that of its WT counterpart at comparable protein levels (Fig 4F and G). These results were confirmed with the same technique using an alternative mAb against a different tag in the GFP‐TDP‐43 protein (Fig EV4A and C). We sought to further validate the different orientation of the RRMm dimers with an alternative approach using a GFP tripartite fluorescence complementation (triFC) assay that measures physiological nuclear TDP‐43 dimerization (Fig EV4D), as we previously showed (Foglieni et al, 2017; Afroz et al, 2019). We co‐transfected T10‐ and T11‐tagged TDP‐43 variants in mouse motor neuron‐like NSC‐34 cells, which yielded the same difference in nucleocytoplasmic localization (Figs 4H and EV4E) as we previously observed in the isogenic cell lines and in human neurons (Figs 2A–C). As expected, triple transfection of the T10‐ and T11‐tagged TDP‐43 variants with a nuclear‐targeted GFP1‐9 produced positive GFP complementation signal for the WT protein (Fig 4H and I), indicative of dimerization. Interestingly, neither the monomeric TDP‐43 variants (6M and 6M&RRMm) nor RNA binding‐deficient TDP‐43 (RRMm) generated such GFP complementation signal (Fig 4H and I), a finding that did not depend on TDP‐43 variant expression levels (Fig EV4F and G). This suggests that despite our biochemical (Fig 3H and I) and imaging (Figs 4F and G, and EV4C) observations indicating that RNA binding‐deficient TDP‐43 (RRMm) dimerizes, these dimers do not follow the correct orientation to reconstitute GFP fluorescence. This contrast between RRMm and the WT TDP‐43 supports the notion of a distinct orientation of TDP‐43 dimers in the absence of RNA (RRMm). Overall, detection and quantification of dimeric TDP‐43 species by a combination of different imaging and biochemical methodologies supports the view that RNA binding is required for the proper orientation of TDP‐43 dimers.
TDP‐43 partitions in heterogenous nuclear bodies via oligomerization and RNA binding
We next sought to determine whether all nuclear TDP‐43 condensates were of similar structure and composition. For this purpose, we determined the colocalization of endogenous TDP‐43 in HEK293 cells with a broad panel of nuclear membraneless organelles (Fig 5A). This approach revealed that a fraction of TDP‐43 condensates, in particular the largest in size, were labeled with markers of Cajal bodies (Figs 5A and B, and EV5A), the maturation compartments of spliceosomal small nuclear ribonucleoprotein particles. Interestingly, TDP‐43 was present in all Cajal bodies in HEK293 cells and neurons (Figs 5C and D, and EV5A), but Cajal bodies only comprised a small fraction of all TDP‐43 condensates (Fig 5B). Another portion of TDP‐43 was localized within paraspeckles, but, unlike Cajal bodies, not every paraspeckle contained TDP‐43 (Figs 5A–C and EV5B). TDP‐43 was absent from other RNA‐nucleated compartments such as nuclear speckles (Shevtsov & Dundr, 2011) and from protein‐exclusive compartments like promyelocytic leukemia (PML) bodies (Fig 5A and B) (Boisvert et al, 2000). In contrast to WT TDP‐43, albeit RNA binding‐deficient GFP‐TDP‐43 (RRMm) localized to small nuclear condensates (Figs 3E and EV3D), these overlapped with neither Cajal bodies nor paraspeckles (Figs 5E and F, and EV5C and D), in line with previous reports (Tsuiji et al, 2013). Monomeric GFP‐TDP‐43 (6M and 6M&RRMm), which barely forms nuclear condensates (Figs 3E and EV3D), was largely absent from these nuclear compartments (Figs 5E and F, and EV5C and D), highlighting the requirement of TDP‐43 oligomerization for its incorporation within functional nuclear bodies. Again, these results did not stem from differential variant expression levels as similar results were obtained 4 h after GFP‐TDP‐43 expression induction (Fig EV5E and F), when GFP‐TDP‐43 protein levels are comparable (Fig EV1F and G). Collectively, these observations suggest that at least a fraction of TDP‐43 LLPS arises from scaffolding around RNA, as described (Shevtsov & Dundr, 2011; Chujo et al, 2016) an explanation supported by the fact that TDP‐43 is reported to bind small Cajal body‐specific RNAs (Izumikawa et al, 2019) and NEAT1, the architectural RNA of paraspeckles (Polymenidou et al, 2011; Tollervey et al, 2011; Chujo et al, 2016). The remaining, unidentified nuclear TDP‐43 condensates, present in both the WT and RNA binding‐deficient variant (RRMm; Figs 5E and F, and EV5C and D) as well as in endogenous TDP‐43 (Fig 5A–D), might represent a precursor pool for Cajal bodies and paraspeckles, an inert condensate population or even be linked to yet undefined bodies and functions. Whether any of these potentially “RNAless” TDP‐43 nuclear compartments formed by the RNA binding‐deficient variant are identical to the unidentified condensates occurring in physiological conditions and/or have functional roles remains unclear. Overall, the heterogeneous nature of nuclear TDP‐43 condensates suggests that LLPS is required for a wide array of TDP‐43 functions within the nucleus.
Figure 5. Cajal bodies and paraspeckles are the main TDP‐43‐containing nuclear bodies.
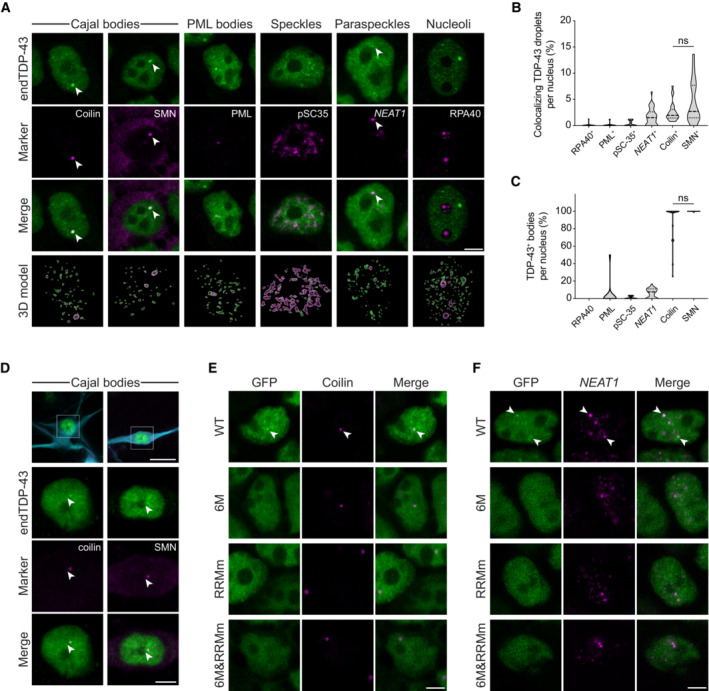
- Representative confocal microscopy images of HEK293 cells probed for endogenous TDP‐43 (endTDP‐43) and different subnuclear compartment markers. Lower panel shows the three‐dimensional (3D) reconstruction of the endTDP‐43 condensates and the indicated nuclear body obtained from confocal Z‐stacks. Scale bar: 5 μm.
- Quantification of the 3D reconstructions shown in (A) depicting the percentage of nuclear TDP‐43 condensates colocalizing with markers of subnuclear compartments. N = 14–38 cells.
- Quantification of the 3D reconstructions shown in (A) depicting the percentage of each of the analyzed subnuclear compartments that colocalize with endTDP‐43. N = 14–38 cells.
- Representative confocal microscopy images of human neurons showing the presence of endTDP‐43 in Cajal bodies. The neuron‐specific marker MAP2 is shown in cyan. Scale bar: 20 μm (inset: 5 μm).
- Representative confocal microscopy images of the isogenic HEK293 lines expressing the different GFP‐TDP‐43 variants for 24 h and stained for the Cajal body marker coilin. Scale bar: 5 μm.
- Representative confocal microscopy images of the isogenic HEK293 lines expressing the different GFP‐TDP‐43 variants for 24 h and hybridized with a fluorescent NEAT1 probe to mark the paraspeckles. Scale bar: 5 μm. ns: not significant. Violin plots show mean and quartiles.
Source data are available online for this figure.
Figure EV5. Nuclear TDP‐43 droplets do not localize to nuclear speckles.
- Representative confocal microscopy images of HEK293 cells stained for the Cajal body marker coilin. Scale bar: 10 μm (5 μm for insets).
- Representative confocal microscopy images of HEK293 cells hybridized with a fluorescent NEAT1 probe to mark the paraspeckles. The field overview is shown as a maximum intensity Z‐projection (thickness of ~10 μm, in steps of 0.21 μm). Scale bar: 10 μm (5 μm for insets).
- Representative confocal microscopy images of the isogenic HEK293 lines expressing the different GFP‐TDP‐43 variants for 24 h and stained for the nuclear speckle marker pSC35.
- Representative confocal microscopy images of the isogenic HEK293 lines expressing the different GFP‐TDP‐43 variants for only 4 h to achieve similar expression levels and stained for the Cajal body marker coilin.
- 3D analysis quantification showing the percentage of Cajal bodies that colocalize with each of the GFP‐TDP‐43 variants as shown in (E). N = 39–54 cells. Kruskal‐Wallis test with Dunn's multiple comparisons post hoc test. ns: not significant, **P < 0.01, ****P < 0.0001. Violin plots show mean and quartiles.
TDP‐43 oligomerization is required for the transcriptome‐wide splicing regulation of its RNA targets
NTD‐mediated oligomerization is required for splicing of at least a subset of the RNA targets of TDP‐43 (Zhang et al, 2013; Afroz et al, 2017; Jiang et al, 2017; Mompean et al, 2017; Wang et al, 2018), but given the broad role of TDP‐43 in regulating splicing events (Polymenidou et al, 2011; Tollervey et al, 2011; Ling et al, 2015; Rot et al, 2017), the question remained whether oligomerization is essential for regulating all or a subset of its splicing targets. RNA‐sequencing (RNA‐seq) of our isogenic cell lines revealed that expression of the WT protein resulted in alternative splicing of > 700 genes when compared to the expression of RNA binding‐deficient GFP‐TDP‐43 (RRMm), which is an established splicing‐deficient TDP‐43 version (D'Ambrogio et al, 2009; Lukavsky et al, 2013). Identified changes included previously reported events of exon inclusion/exclusion, intron retention and alternative polyadenylation site usage depending on TDP‐43 binding (Polymenidou et al, 2011; Tollervey et al, 2011; Rot et al, 2017) (Fig 6A; Appendix Fig S1A). When the same comparison was made between the oligomerization‐deficient (6M) and the RRMm GFP‐TDP‐43 variants, no significant alternatively spliced events were detected (Fig 6A; Appendix Fig S1A). This suggests a lack of splicing functionality of monomeric GFP‐TDP‐43 (6M). In line with this, differentially expressed RNAs or proteins levels were observed in cells expressing monomeric (6M) compared to the RRMm GFP‐TDP‐43 variant (Appendix Fig S1B).
Figure 6. TDP‐43 oligomerization is required for splicing regulation in the nucleus and stress granule incorporation in the cytoplasm.
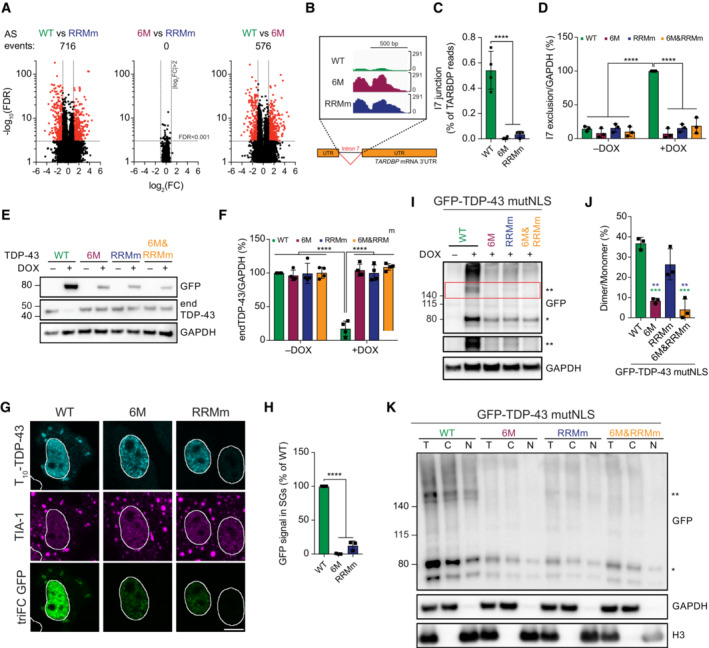
- Volcano plots showing alternative splicing (AS) events upon expression of GFP‐TDP‐43 variants for 48 h.
- RNA sequencing (RNA‐seq) coverage across the intron 7 of the TARDBP gene, showing a strong decrease in the WT, but not the mutant, GFP‐TDP‐43‐expressing cells.
- Quantification of RNA‐seq reads spanning the intron 7 junction. One‐way ANOVA with Tukey's multiple comparisons post hoc test.
- Endogenous TDP‐43 (endTDP‐43) intron 7 exclusion levels after expression of the GFP‐TDP‐43 variants for 48 h in the isogenic cell lines measured by qPCR with primers specifically targeted to the transcripts excluding this region. N = 3 independent experiments. Two‐way ANOVA with Tukey's multiple comparisons post hoc test.
- Western blot analysis of the isogenic HEK293 after GFP‐TDP‐43 expression for 48 h showing that only the WT variant regulates endTDP‐43 levels.
- Quantification of the endTDP‐43 signal from (E). N = 4 independent experiments. Two‐way ANOVA with Tukey's multiple comparisons post hoc test.
- Tripartite GFP complementation assay involving the co‐transfection of a pair of N‐terminally T10‐ and T11‐tagged TDP‐43 constructs in HeLa cells subjected to arsenite stress for 30 min and incubated with recombinant GFP1–9 after fixation to label T10‐ and T11‐TDP‐43 dimers. TriFC: trimolecular fluorescence complementation. Scale bar: 10 μm.
- Quantification of the trimolecular fluorescence complementation (triFC) signal of GFP in the TIA‐1‐marked SGs from the images shown in (G). N = 3 independent experiments. Repeated measures one‐way ANOVA with Greenhouse–Geisser correction and Tukey's multiple comparisons post hoc test.
- Expression of GFP‐TDP‐43 mutNLS variants was induced with doxycycline (DOX) for 4 h before crosslinking protein–protein interactions with DSG and subsequent analysis by western blot. * and ** indicate GFP‐TDP‐43 monomers and dimers, respectively.
- Quantification of the GFP signal from (I). N = 3 independent experiments. Repeated measures one‐way ANOVA with Greenhouse–Geisser correction and Tukey's multiple comparisons post hoc test.
- After expression of GFP‐TDP‐43 mutNLS variants for 48 h, the isogenic lines were treated with DSG to cross‐link protein–protein interactions before performing nucleocytoplasmic fractionation and western blot analysis. * and ** indicate GFP‐TDP‐43 monomers and dimers, respectively. **P < 0.01, ***P < 0.001, ****P < 0.001. Graph bars represent mean ± SD.
Source data are available online for this figure.
A particularly interesting event modulated by TDP‐43 binding is the splicing of an alternative intron (intron 7) in its own 3′ UTR, which results in autoregulation of the TDP‐43 mRNA and protein levels (Ayala et al, 2011; Polymenidou et al, 2011; Avendaño‐Vázquez et al, 2012) (Appendix Fig S1C). Analysis of the 3′ UTR of TDP‐43 by RNA‐seq and qPCR showed that, similar to other alternative splicing events, monomeric GFP‐TDP‐43 (6M) cannot promote the exclusion of intron 7 in the endogenous TARDBP mRNA (Fig 6B–D), resulting in lack of autoregulation at the protein level (Figs 1B and 6E and F). This lack of splicing activity by monomeric TDP‐43 (6M) was not due to its reduced protein concentration, since WT GFP‐TDP‐43 also autoregulated endogenous TDP‐43 at expression levels comparable to that of the 6M variant (Appendix Fig S1D and E). Overall, our data support the requirement of TDP‐43 oligomerization for its broad role in splicing regulation.
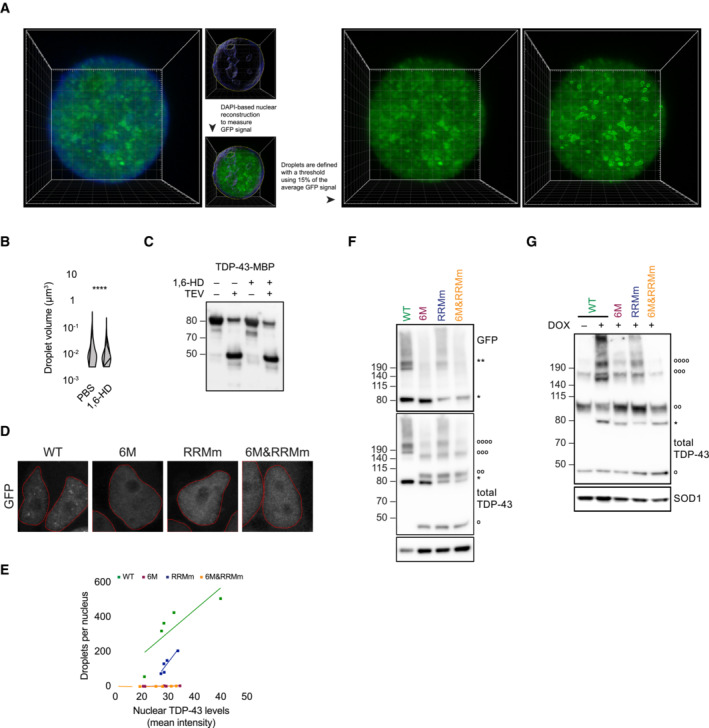
Cytoplasmic TDP‐43 oligomerization is required for its incorporation into stress granules
To understand whether oligomerization is also required for TDP‐43 functions outside of the nucleus, we studied TDP‐43 incorporation into stress granules (SGs) in the cytoplasm (Dewey et al, 2012). We used to the GFP triFC assay (Fig EV4D) to investigate TDP‐43 oligomerization in the well‐established oxidative stress model in HeLa cells (Aulas et al, 2012; Dewey et al, 2012). Expression of T10‐ and T11‐tagged TDP‐43 variants and incubation with recombinant GFP1–9 after fixation revealed that, unlike the WT protein—but similarly to the RNA binding‐deficient mutant (RRMm)—monomeric TDP‐43 (6M) did not incorporate into SGs (Fig 6G and H), suggesting that TDP‐43 oligomerization also takes place in the cytoplasm and is key to TDP‐43 inclusion in SGs. The presence of reconstituted GFP fluorescence in the SGs further indicates that TDP‐43 oligomerization is not only required for its incorporation but also that TDP‐43 exists as a dimer in SGs. To confirm the presence of cytoplasmic TDP‐43 oligomers at near‐endogenous expression levels, we developed isogenic HEK293 Flp‐In T‐REx lines harboring one copy of each of the GFP‐TDP‐43 variants in combination with previously published mutations in the nuclear localization signal that abolish active nuclear import (GFP‐TDP‐43 mutNLS; Elden et al, 2010). Differences between the protein levels of the GFP‐TDP‐43 mutNLS variants were similar to that of their nuclear counterparts (Appendix Fig S1F and H; Fig 1B). Interestingly, the localization of the GFP‐TDP‐43 mutNLS variants differed between the four cell lines. WT and RNA binding‐deficient (RRMm) GFP‐TDP‐43 mutNLS were predominantly present in the cytoplasm, whereas their monomeric counterparts (6M mutNLS and 6M&RRMm mutNLS) significantly shifted their localization to the nucleus (Appendix Fig S1G and H). This suggests that WT and RRMm GFP‐TDP‐43 mutNLS oligomerize in the cytoplasm, which hinders their passive diffusion back into the nucleus. To confirm this, we performed DSG cross‐linking of protein–protein interactions in the GFP‐TDP‐43 mutNLS lines and found that both WT and RRMm GFP‐TDP‐43 mutNLS formed oligomers, albeit their presence was reduced in the RRMm variant (Fig 6I and J). Moreover, DSG cross‐linking followed by nucleocytoplasmic fractionation confirmed the presence of WT and RRM GFP‐TDP‐43 oligomers in the cytoplasmic fraction (Fig 6K). Additionally, exposure of these isogenic lines to oxidative stress further confirmed the requirement of cytoplasmic oligomerization (6M and 6M&RRMm mutNLS) and RNA binding (RRMm mutNLS) for TDP‐43 incorporation into stress granules (Appendix Fig S1I and J). Importantly, albeit less abundant than nuclear oligomerization, cytoplasmic dimerization was also observed at endogenous TDP‐43 levels by PLA (Fig 4E). Altogether, our observations suggest that oligomerization is essential in both the nucleus and the cytoplasm for TDP‐43 to perform its functions in RNA metabolism.
Loss of RNA binding or oligomerization differentially modulate the subcellular localization of TDP‐43 inclusions
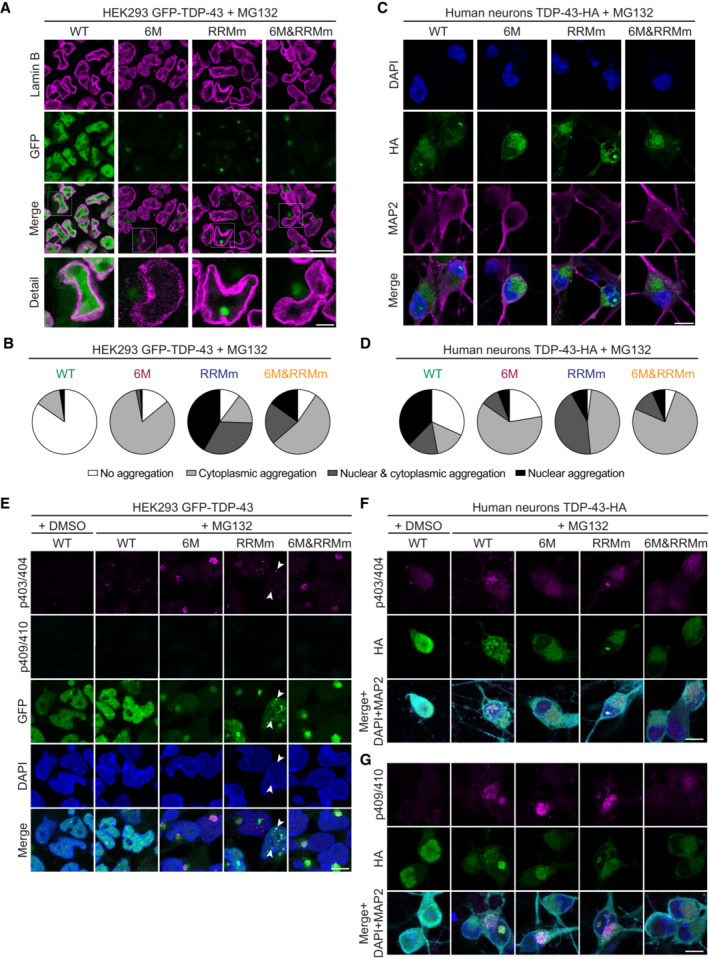
Decline of the cellular proteostasis capacity with age contributes to protein misfolding in neurodegenerative diseases (Jayaraj et al, 2020), often resulting in the accumulation of ubiquitinated inclusions in affected tissues, including TDP‐43 proteinopathies (Arai et al, 2006; Neumann et al, 2006). Since monomeric (6M) and RNA binding‐deficient (RRMm) TDP‐43 showed shorter half‐lives at physiological levels (Fig 1D and E), we sought to determine how failure of the UPS machinery affects the accumulation of these species. By blocking the proteasome with the inhibitor MG132, we observed that both monomeric (6M) and RNA binding‐deficient (RRMm) GFP‐TDP‐43 formed protein inclusions in the isogenic cell lines, in contrast to the WT counterpart, which remained largely diffuse (Fig 7A and B; Appendix Fig S2A). The vast majority of aggregates formed by monomeric GFP‐TDP‐43 (6M and 6M&RRMm) localized to the cytoplasm, in line with previous results showing that high overexpression of oligomerization‐deficient GFP‐TDP‐43 (6M) by transient transfection triggers increased cytoplasmic TDP‐43 aggregation as compared to the WT protein (Afroz et al, 2017). Interestingly, in addition to cytoplasmic inclusions, MG132 treatment resulted in nuclear aggregation of RNA binding‐deficient GFP‐TDP‐43 (RRMm) in > 70% of the cells. The combined loss of oligomerization and RNA binding (6M&RRMm) shifted the aggregation to the cytoplasm, suggesting that the nuclear GFP‐TDP‐43 RRMm aggregation depended on NTD interactions. The observed TDP‐43 aggregation patterns were specific to the inhibition of the UPS degradation pathway, as several classes of proteasome inhibitors, but not disruption of autophagy with bafilomycin A1, yielded similar outcomes in the isogenic cell lines (Appendix Fig S2B and C).
Figure 7. Loss of RNA binding or oligomerization differentially modulate the subcellular localization of TDP‐43 inclusions with pathological signatures.
-
ARepresentative confocal microscopy images of the isogenic GFP‐TDP‐43 lines after 48 h of expression, treated with the proteasome inhibitor MG132 for the last 24 h and stained for lamin B to mark the nuclear envelope. Note the different localization of TDP‐43 inclusions in the oligomerization‐ (6M and 6M&RRMm) versus RNA binding‐deficient (RRMm) variants. Scale bar: 20 μm (5 μm for inset).
-
BQuantification of the localization of GFP‐TDP‐43 inclusions after MG132 treatment for the different variants in the isogenic HEK293 lines as shown in (A). Represented values are averages from N = 3 independent experiments, with N = 189–497 cells quantified per condition and replicate.
-
CRepresentative maximum intensity Z‐projections from confocal fluorescence imaging (thickness of 4 μm, in steps of 1 μm) of human neurons transduced with TDP‐43‐HA variants and treated overnight with the proteasome inhibitor MG132. Scale bar: 10 μm.
-
DQuantification of the differentially localized TDP‐43‐HA inclusions in human neurons as described in (C). Represented values correspond to the quantification of N = 85–97 neurons from two independent experiments.
-
ERepresentative maximum intensity Z‐projections from confocal fluorescence imaging (thickness of 4 μm, in steps of 1 μm) of the same experimental conditions as shown in (A), with the addition of a DMSO control for MG132 treatment. GFP‐TDP‐43‐expressing cells were double immunolabeled for TDP‐43 phosphorylated at the S403/404 and S409/410 epitope. Note the absence of phospho‐S409/410 immunopositivity in all conditions and positivity for phospho‐S403/404 in cytoplasmic inclusions and a subset of nuclear GFP‐TDP‐43 RRMm inclusions (appointed by arrowheads). Scale bar: 10 μm.
-
F, GRepresentative maximum intensity Z‐projections from confocal fluorescence imaging (thickness of 4 μm, in steps of 1 μm) of the same experimental conditions as shown in (C), with the addition of a DMSO control. TDP‐43‐HA expressing human neurons were stained for either phospho‐S403/404 (F) or phospho‐S409/410 (G). Scale bar: 10 μm. Note the re‐localization of the phospho‐S403/404 signal from neuronal nuclei to inclusions (F) and the presence of phospho‐S409/410 signal in only a subset of aggregate‐bearing neurons (G). Nuclei are stained with DAPI in (C, E–G). The neuron‐specific marker MAP2 is shown in magenta or cyan in (C, F, G).
Source data are available online for this figure.
In human neurons treated with the proteasome inhibitor MG132, monomeric TDP‐43‐HA variants (6M and 6M&RRMm) also predominantly aggregated in the cytoplasm, whereas RNA binding‐deficient TDP‐43‐HA (RRMm) additionally presented nuclear inclusions in > 50% of transduced neurons (Fig 7C and D), thus reproducing the distinct TDP‐43 aggregation patterns observed in our isogenic cell lines. Upon MG132 treatment, WT TDP‐43‐HA also formed inclusions in neurons, both in the nucleus and cytoplasm, likely due to higher transgene protein levels in transduced neurons compared to the isogenic GFP‐TDP‐43 lines. Interestingly, and in line with higher protein levels, RRMm TDP‐43‐HA already formed nuclear inclusions in the absence of proteasome inhibition in a subset of transduced human neurons (Appendix Fig S7D). Taken together, our data show that loss of oligomerization shifts TDP‐43 aggregate formation from the nucleus to the cytoplasm.
Subsequently, we probed the different TDP‐43 inclusions formed upon proteasomal inhibition in the isogenic cell lines and human neurons with markers that identify nuclear and cytoplasmic TDP‐43 aggregates in the CNS of ALS/FTLD patients, namely ubiquitin (Arai et al, 2006; Neumann et al, 2006) and TDP‐43 C‐terminally phosphorylated at serines (S) 403/404 or 409/410 (Hasegawa et al, 2008). Both in the isogenic HEK293 lines (Appendix Fig S8A) and in human neurons (Appendix Fig S3B), MG132‐induced cytoplasmic inclusions were starkly immunopositive for ubiquitin. Neuronal nuclear inclusions (NIIs) containing TDP‐43 (WT and RRMm) were also ubiquitin‐positive (Appendix Fig S3B), similar to a subset of the nuclear aggregates arising upon MG132 treatment in HEK293 (Appendix Fig S3A). Likewise, cytoplasmic TDP‐43 inclusions in both the cell lines (Fig 7E) and human neurons (Fig 7F) were overtly phosphorylated at the S403/404 TDP‐43 epitope. Immunopositivity for phospho‐S403/404 was also observed for some NIIs (Fig 7F) and a subset of the smaller nuclear inclusion in the isogenic lines (Fig 7E). Phosphorylation at the more C‐terminal S409/410 TDP‐43 epitope was observed in a fraction of both cytoplasmic and nuclear inclusions in neurons (Fig 7G) and cytoplasmic inclusions formed by GFP‐TDP‐43 mutNLS in the isogenic HEK293 cell line (Appendix Fig S3C). More specifically, the larger and denser inclusions were found immunopositive for phospho‐S409/410. In contrast, none of the inclusions found in the GFP‐TDP‐43 HEK293 lines (WT, 6M, RRMm, and 6M&RRMm) were labeled by the phospho‐S409/410 antibody, despite their immunopositivity for phospho‐S403/404 (Fig 7E; Appendix Fig S3C). This is in line with the previously reported sequential N‐ to C‐terminal phosphorylation pattern in the LCR of TDP‐43 (De Rossi et al, 2021) and indicates a process of TDP‐43 aggregate maturation in our human neurons and isogenic HEK293 cell lines. Taken together, the distinctly localized TDP‐43 inclusions formed in our cellular models harbor the pathological features that characterize TDP‐43 aggregates in the CNS of ALS/FTLD patients.
TDP‐43 aggregates in an LLPS‐ or an aggresome‐dependent manner in the nucleus and cytoplasm, respectively
To understand the origin of cytoplasmic and nuclear TDP‐43 inclusions, we performed live cell imaging of the GFP‐TDP‐43 isogenic lines during treatment with the proteasome inhibitor. While WT GFP‐TDP‐43 condensates merely changed position, fused and split within the nucleus upon MG132 addition, monomeric GFP‐TDP‐43 (6M and 6M&RRMm) formed a single cytoplasmic inclusion whose size increased over time, accompanied by gradual decrease in the diffuse nuclear TDP‐43 signal (Fig 8A; Appendix Fig S4A) resembling the nuclear clearance that has been widely reported in neurons with TDP‐43 pathology in ALS and FTLD patients (Arai et al, 2006; Neumann et al, 2006; Ling et al, 2013). Fluorescence recovery after photobleaching (FRAP) experiments revealed that whereas WT GFP‐TDP‐43 remained diffuse throughout the treatment, cytoplasmic inclusions comprising monomeric TDP‐43 (6M and 6M&RRMm) were immobile structures (Fig 8B; Appendix Fig S4B). A similar aggregation pathway—a single focus expanding in size yielding one solid cytoplasmic inclusion—was also observed for the RNA binding‐deficient GFP‐TDP‐43 (RRMm) in the cytoplasm (Appendix Fig S4C and D), suggesting that the formation of cytoplasmic aggregates upon proteasomal failure requires neither oligomerization nor RNA binding. In contrast, in the nucleus, the elevated protein levels of GFP‐TDP‐43 RRMm caused by proteasome inhibition induced the formation of many, initially dynamic condensates, which eventually fused to form a single solid inclusion (Fig 8B and C), reminiscent of the single intranuclear inclusions found in patients with specific FTLD subtypes (Hiji et al, 2008; Lee et al, 2017). FRAP analysis showed that GFP‐TDP‐43 RRMm in the final inclusion had lost its liquid behavior (Fig 8B). In a subset of cells, GFP‐TDP‐43 RRMm deposited in the nucleoli (Appendix Fig S4E), in line with the protein quality control properties of this phase‐separated compartment (Frottin et al, 2019). Importantly, the formation of nuclear aggregates occurs also in human neurons, in a subset of transduced cells transduced with RRMm TDP‐43‐HA, even in the absence of proteasome inhibition (Appendix Fig S2D). This was accompanied by an increased in nuclear condensates in transduced cells without inclusion (Appendix Fig S4F). Together, these data suggest that TDP‐43 aggregates via LLPS in the nucleus.
Figure 8. TDP‐43 aggregates in an LLPS‐ or an aggresome‐dependent manner in the nucleus and cytoplasm, respectively.
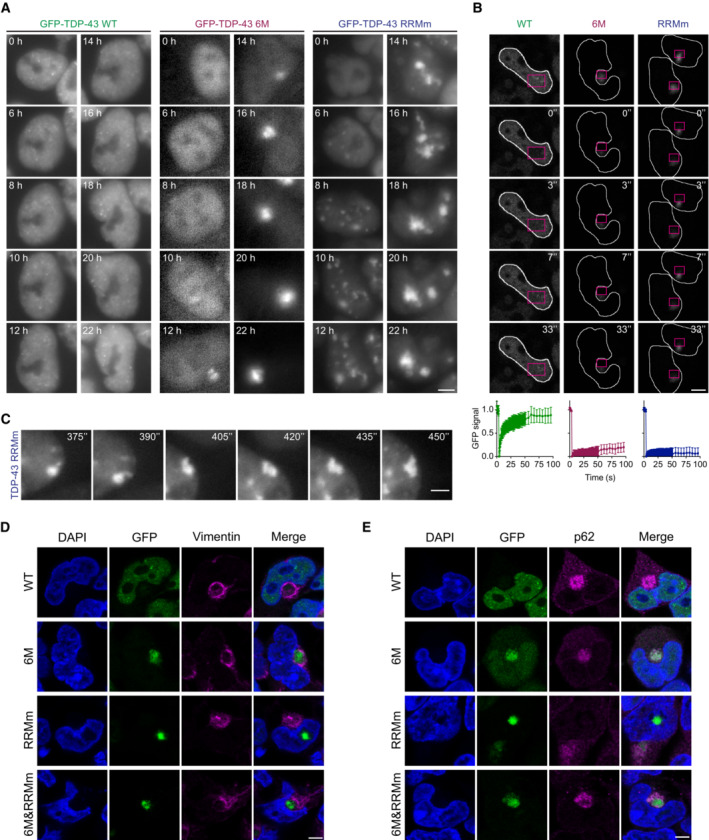
- Representative images of live widefield fluorescence microscopy over the course of the MG132 treatment of the isogenic GFP‐TDP‐43 lines in the conditions described in Fig 7A (24 h of MG132 treatment). Numbers in images indicate the experimental time point in hours (h) of MG132 treatment. Scale bar: 5 μm.
- Representative fluorescent confocal microscopy images of FRAP experiments of the GFP‐TDP‐43 aggregates formed upon MG132 treatment as described in Fig 7A. FRAP was performed in the areas highlighted in magenta. Numbers in images indicate the experimental time point in seconds (") of MG132 treatment. Scale bar: 5 μm. Bottom panel: Measured GFP intensity values are expressed as a fraction of the average pre‐bleach fluorescence levels.
- Live widefield fluorescence microscopy depicting a fusion event and aberrant phase transition of RRMm GFP‐TDP‐43 condensates upon MG132 treatment in the conditions described in Fig 7A. Numbers in images indicate the experimental time point in seconds (") of MG132 treatment. Scale bar: 3 μm.
- Representative confocal microscopy images of the isogenic GFP‐TDP‐43 lines at the endpoint (24 h) of MG132 treatment of the experimental conditions described in Fig 7A and stained for vimentin. Scale bar: 5 μm.
- Representative confocal microscopy images of the isogenic GFP‐TDP‐43 lines at the endpoint (24 h) of MG132 treatment of the experimental conditions described in Fig 7A and stained for p62. Cell nuclei are visualized with DAPI in (D and E). Scale bar: 5 μm.
Source data are available online for this figure.
The MG132‐induced cytoplasmic TDP‐43 inclusions in the human neurons and isogenic lines were consistently found adjacent to the nucleus, in a location occupied by the aggresome (Appendix Fig S9G). The latter is a juxtanuclear accumulation of misfolded proteins resulting from saturation of the chaperone refolding system and/or the UPS degradation pathway (Appendix Fig S4G) which has been linked to the origin of protein aggregates in neurodegenerative diseases (Johnston et al, 1998). Indeed, cytoplasmic inclusions, formed by monomeric and/or RNA binding‐deficient GFP‐TDP‐43 (6M, RRMm, and 6M&RRMm) in the stable lines, were surrounded by a vimentin cage, a characteristic feature of aggresomes (Fig 8D; Appendix Fig S4H) (Johnston et al, 1998). Additionally, cytoplasmic, but not nuclear, TDP‐43 aggregates were positive for p62 (Fig 8E), a critical component of aggresomes (Matsumoto et al, 2018) and a pathological aggregate marker in certain FTLD subtypes (Hiji et al, 2008; Lee et al, 2017). These observations suggest that distinct pathways towards TDP‐43 aggregation are at play in the nucleus and cytoplasm and that monomerization increases TDP‐43 incorporation in cytoplasmic aggresomes upon proteasomal failure, thereby potentially triggering cytoplasmic TDP‐43 aggregation in disease.
Discussion
In this study, we describe the interconnection between NTD‐driven TDP‐43 oligomerization and RNA binding, and show that they cooperatively retain TDP‐43 in the nucleus, instruct its functionality and therefore slow down its turnover. We demonstrate that oligomerization is essential for the broad function of TDP‐43 in splicing regulation and allows its LLPS‐mediated incorporation into nuclear membraneless compartments, including Cajal bodies and paraspeckles. Our work describes for the first time that, under physiological conditions, TDP‐43 oligomers exist in the cytoplasm, where they are required for LLPS‐dependent incorporation into SGs. Moreover, we show that in the absence of RNA binding, TDP‐43 oligomerization is reduced, but the dimers that do form adopt a spatial orientation that is different from the RNA‐bound oligomeric state. Importantly, our results shed light on the molecular mechanisms of two distinct and independent pathways triggering TDP‐43 aggregation, which highlight the importance of TDP‐43 monomerization and/or loss of RNA binding as key early events in the development of TDP‐43 proteinopathies (Fig 9).
Figure 9. Oligomerization and RNA‐binding enable TDP‐43 physiological functions and their disruption drives nuclear and cytoplasmic aggregate formation via distinct pathways.
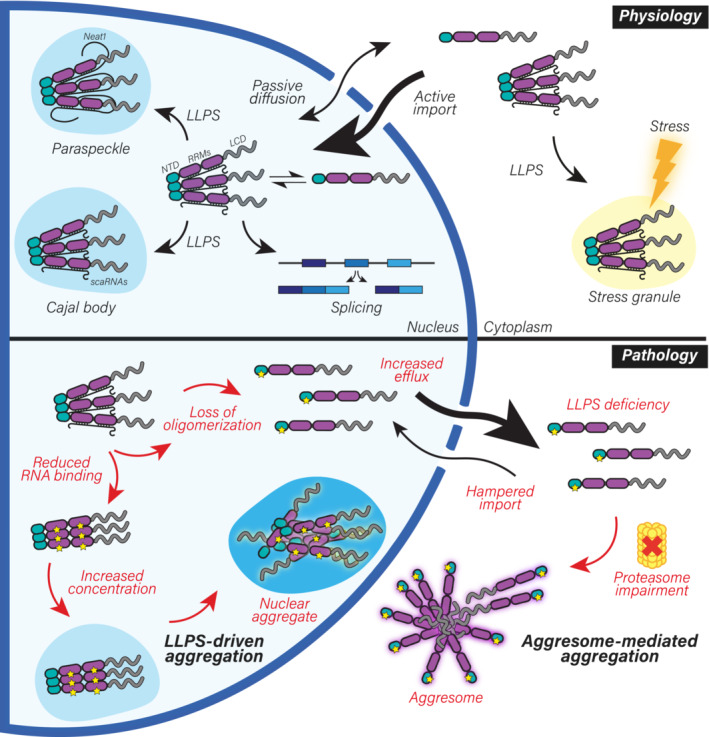
Schematic representation of the role of NTD‐driven oligomerization and RNA binding in TDP‐43 physiology and pathology. Upper panel: In healthy cells, TDP‐43 monomers and oligomers are in a dynamic equilibrium. TDP‐43 is actively imported into the nucleus (Winton et al, 2008), where oligomerization and RNA binding retain it in large macromolecular complexes, limiting passive outflow. In the nucleus, oligomerization and RNA binding are key for the essential roles of TDP‐43 in RNA processing, including alternative splicing. Furthermore, oligomerization enables LLPS of TDP‐43 and in conjunction with binding to specific RNAs—such as small Cajal body‐specific RNAs (scaRNAs) (Izumikawa et al, 2019) and NEAT1 (Polymenidou et al, 2011; Tollervey et al, 2011)—allows its localization to distinct subnuclear compartments, primarily Cajal bodies and paraspeckles. TDP‐43 oligomers are also detected in the cytoplasm, where its LLPS‐mediated incorporation into SGs under stress conditions depends on both oligomerization and RNA binding. Lower panel: In disease, loss of oligomerization or RNA binding disrupts its inclusion into nuclear bodies, increases nuclear efflux, leads to transcriptome‐wide splicing alterations (including TDP‐43 autoregulation) and drives TDP‐43 aggregation via two independent pathways. Upon failure of the ubiquitin‐proteasome degradation machinery observed with aging and in ALS/FTLD patients (Arai et al, 2006; Neumann et al, 2006; Jayaraj et al, 2020), monomeric TDP‐43 aggregates in an aggresome‐dependent manner in the cytoplasm. Notably, the known decline in active nuclear import in disease (Coyne et al, 2021) would further exacerbate TDP‐43 cytoplasmic accumulation. In the nucleus, loss of TDP‐43 RNA‐binding results in enhanced formation of both monomers that escape to the cytoplasm and nuclear TDP‐43 oligomers with distinct orientation. When the concentration of TDP‐43 rises (e.g., due to the aforementioned proteasomal failure), RNA‐binding deficiency modulates TDP‐43 LLPS, culminating in the formation of nuclear aggregates via an LLPS‐mediated pathway. Taken together, RNA binding and oligomerization allow TDP‐43 to maintain its localization and functionality in physiology and their disruption drives LLPS‐dependent and aggresome‐dependent aggregation pathways in the nucleus and cytoplasm, respectively. LCD: low complexity domain, LLPS: liquid–liquid phase separation, NTD: N‐terminal domain, RRMs: RNA recognition motifs.
TDP‐43 oligomerization is a dynamic event that controls the relative amounts of TDP‐43 monomers, dimers, and oligomers in the cell. However, the specialized roles of the individual TDP‐43 species in health and disease remained unknown. Expression of different TDP‐43 variants at near‐physiological levels in human cell lines and in our human neural system revealed that, in comparison to its oligomeric counterpart, monomeric TDP‐43 lacks functionality, becomes more prone to escape the nucleus—likely by passive diffusion (Ederle et al, 2018; Pinarbasi et al, 2018)—and is rapidly degraded. In the event of proteasomal failure, these otherwise short‐lived TDP‐43 monomers are deposited into aggresomes, whose expansion is accompanied by a progressive decrease in nuclear TDP‐43. This cytoplasmic aggregation observed in our human cellular models recapitulates the key pathological TDP‐43 features observed in affected neurons of patients with TDP‐43 proteinopathies, namely its nuclear clearance, loss of function, cytoplasmic aggregation ubiquitination and pathological C‐terminal phosphorylation (Arai et al, 2006; Neumann et al, 2006). Interestingly, the aggresome pathway was previously linked to sporadic ALS/FTLD (Lee et al, 2019) through the disturbance of p62 (Lee et al, 2019; Foster et al, 2021), a key player in aggresome formation (Matsumoto et al, 2018). Furthermore, the aggresome markers HDAC6 and p62 have been reported to colocalize with a subset of cytoplasmic TDP‐43 aggregates in ALS/FTLD patients (Hiji et al, 2008; Lee et al, 2017; Watanabe et al, 2020). When the conditions that favor the monomeric state of TDP‐43 and hamper its proteasomal degradation persist, TDP‐43 monomers contained in the aggresomes may resist clearance by aggrephagy and further mature into compact aggregates due to the high concentration of unfolded LCRs, which in the absence of NTD‐driven organization are not spatially separated (Afroz et al, 2017). Since monomeric TDP‐43 is unable to autoregulate its own levels, the continued production of more TDP‐43 to compensate for its loss of function will only exacerbate this pathological transition. Our data signify that loss of TDP‐43 oligomerization ignites a pathological cascade that culminates in the formation of cytoplasmic TDP‐43 inclusions via the aggresome pathway.
In addition to loss of oligomerization, our data also indicate that the disturbance of the spatial organization of TDP‐43 oligomers due to loss of RNA binding triggers a distinct pathological cascade leading to aggregation in the nucleus. While previous studies have addressed the role of RNA in TDP‐43 pathogenesis (Mann et al, 2019; Wang et al, 2020; Yu et al, 2021), our work provides insights into the molecular mechanism underlying the aberrant phase transition of NTD‐driven RNAless TDP‐43 oligomers into nuclear immobile inclusions. Using a combination of imaging and biochemical assays, we observed that TDP‐43 oligomers present a different orientation in an RNA‐bound or ‐unbound state. RNA‐bound TDP‐43 oligomers enact its physiological functions, maintain its localization and antagonize the formation of pathological aggregates (Afroz et al, 2017 and this study), while RNAless TDP‐43 oligomers, that may or may not have functional roles, undergo aberrant phase separation leading to nuclear aggregation (Yu et al, 2021 and this study). These observations clarify the apparently controversial role of the NTD in aggregate formation, which has been found both synergistic (Zhang et al, 2013; Romano et al, 2015; Sasaguri et al, 2016; French et al, 2019) and antagonistic (Afroz et al, 2017). Based on our observations, we propose that NTD‐mediated TDP‐43 self‐interaction is a double‐edged sword: in the presence of RNA, it is essential for TDP‐43 to perform its functions and undergo physiological LLPS, while in the absence of RNA binding it promotes aberrant LLPS that leads to aggregation.
In addition to the cytoplasmic aggregation of TDP‐43, neuronal intranuclear inclusions have been reported in two of the five recognized FTLD‐TDP subtypes (Arai et al, 2006; Neumann et al, 2006, 2007; Cairns et al, 2007; Josephs et al, 2009; Lee et al, 2017) and are particularly abundant in FTLD cases linked to mutations in valosin‐containing protein (VCP) (Cairns et al, 2007; Neumann et al, 2007), which is involved in nuclear protein quality control degradation (Gallagher et al, 2014). In our human cellular models, we observed LLPS‐driven intranuclear aggregation of RNA binding‐deficient TDP‐43 oligomers upon inhibition of proteasomal degradation. Similarly, nuclear inclusions have also been reported in previous studies employing strong overexpression of TDP‐43 RNA‐binding mutants in cells and neurons (Elden et al, 2010; Wang et al, 2012; Mompean et al, 2017; Flores et al, 2019; Mann et al, 2019). In addition to its LLPS‐mediated nuclear aggregation, RNA binding‐deficient TDP‐43 formed cytoplasmic inclusions via the aggresome pathway. This route is likely favored by the increase of the monomeric TDP‐43 state in the absence of RNA binding, accompanied by nuclear efflux, as previously shown upon proteasome inhibition in cultured neurons (van Eersel et al, 2011). Altogether, our data show that when a cell encounters proteostatic stress, TDP‐43 takes different routes towards inclusion formation, and the selection of the pathway depends on both the protein state (monomeric vs. RNAless) and the subcellular environment (cytoplasm vs. nucleus). While our study provides evidence for the importance of two such routes (nuclear LLPS‐dependent and cytoplasmic aggresome‐mediated), additional aggregation pathways—for example, cytoplasmic LLPS‐mediated (Gasset‐Rosa et al, 2019; Mann et al, 2019; Watanabe et al, 2020)—likely exist and may be triggered under different circumstances and involve other TDP‐43 states. Collectively these distinct pathways may account for the spectrum of cytoplasmic aggregates observed in patients with TDP‐43 proteinopathies.
Why would the majority of TDP‐43 aggregates reside in the cytoplasm in ALS/FTLD patients? Our data indicate that TDP‐43 monomerization and its subsequent nuclear efflux is a more frequent or potent event than decreased RNA binding affinity. Additionally, the high nuclear RNA concentrations that have been shown to prevent LLPS of RNA‐binding proteins (Maharana et al, 2018) may counteract aberrant LLPS and aggregation, even in the absence of specific RRM‐RNA interactions for TDP‐43. This balancing mechanism may be reinforced by the upregulation of some architectural TDP‐43 RNA targets in the nucleus, including NEAT1 (Maharana et al, 2018; Wang et al, 2020). This RNA binding could instruct the proper, RNA‐loaded orientation of TDP‐43 oligomers with a concomitant increase in physiological LLPS in the form of nuclear bodies. In fact, elevated NEAT1 levels and paraspeckle formation have been amply reported in TDP‐43 proteinopathies (Tollervey et al, 2011; Nishimoto et al, 2013; Shelkovnikova et al, 2018; Wang et al, 2020).
The cellular machinery that regulates TDP‐43 oligomerization remains unknown. While protein concentration is a determinant of TDP‐43 oligomerization (Chang et al, 2012; Afroz et al, 2017), the conditions that keep the balance between monomeric and oligomeric TDP‐43 species in healthy cells and, most importantly, increase TDP‐43 monomerization in disease require further investigation. Post‐translational modifications (PTMs) are excellent candidates for such physiological regulation of TDP‐43 oligomerization and a recent study identified a single serine phosphorylation event within the NTD interface that decreases oligomerization in vitro (Wang et al, 2018), albeit its effects within cells or occurrence in disease have not yet been tested. Similarly, specific acetylation events within the RRMs that lower the affinity of TDP‐43 for RNA have been detected in ALS patients (Cohen et al, 2015) and were subsequently reported to trigger nuclear TDP‐43 aggregation via aberrant LLPS (Yu et al, 2021). Supporting the link between reduced RNA affinity and disease, ALS/FTLD‐associated mutations within the RRMs have been shown to disrupt RNA binding and enhance TDP‐43 proteinopathy (Chen et al, 2019). Moreover, since ATP was recently shown to directly bind the NTD of TDP‐43, thereby enhancing its oligomerization (Wang et al, 2019), the decrease in cellular ATP levels with age (Boveris & Navarro, 2008) could also act as a monomerization‐inducing trigger. It is also conceivable that additional, yet unknown, protein interactors of TDP‐43 may act as modifiers of oligomerization. Future work should focus on determining the molecular switches within TDP‐43 and/or protein partners that regulate its oligomerization and RNA binding and on validating the occurrence of diminished TDP‐43 oligomerization and RNA affinity in patient tissues. Such insights will be valuable to inform drug design targeting TDP‐43 oligomerization and RNA binding, including dimer stabilization or recovery of RRM‐RNA interactions, among others.
In conclusion, oligomerization and RNA binding allow TDP‐43 to maintain its localization and function in physiology and their disruption drives distinct aggregation pathways in the nucleus and cytoplasm, indicating that distinct molecular origins may account for the plethora of TDP‐43 aggregation types observed in ALS and FTLD subtypes.
Materials and Methods
Plasmids
The pcDNA5 plasmid containing the GFP‐tagged human TDP‐43 cDNA sequence was a kind gift of Dr. Shuo‐Chien Ling (Ling et al, 2010). The mutations to introduce siRNA resistance in the TDP‐43 coding region without altering the amino acid sequence and the oligomerization‐disrupting mutations in the N‐terminal domain were previously described (Afroz et al, 2017). RNA‐binding disruption mutations were introduced by site‐directed mutagenesis PCR as described before (Afroz et al, 2017), using high‐fidelity Phusion DNA Polymerase (New England Biolabs, M0530) with primers detailed in Appendix Table S1 followed by DpnI (New England Biolabs, R0176) digestion before bacterial transformation and colony selection.
Plasmids encoding T10‐ and T11‐tagged TDP‐43 and TDP‐43 6M have already been reported (Afroz et al, 2017; Foglieni et al, 2017). The sequence encoding TDP‐43 RRMm was amplified by PCR from the pcDNA5 plasmids described above by adding BamHI/XhoI restriction sites using the primers detailed in Appendix Table S1. Amplified products were cloned into the T10 and T11pcDNA3 parental plasmids (Foglieni et al, 2017) between BamHI and XhoI in order to obtain plasmids encoding T10‐HA‐TDP43 RRMm and T11‐β1‐TDP‐43 RRMm. The same amplified sequence was cloned into the plasmids encoding T10‐ and T11‐tagged TDP‐43 6M (Foglieni et al, 2017) between EcoRI and XhoI in order to obtain plasmids encoding for T10‐HA‐TDP43 6M&RRMm and T11‐β1‐TDP‐43 6M&RRMm.
TDP‐43 with a C‐terminal HA tag was directly amplified from the GFP‐TDP‐43 construct described above and inserted into an autoregulatory all‐in‐one TetON cassette previously inserted into pLVX backbone (preprint: Hruska‐Plochan et al, 2021) by Gibson cloning using the NEBuilder HiFi DNA Assembly Cloning Kit (New England Biolabs, E5520S) according to the manufacturer's instructions.
The plasmid encoding the His6‐tagged RRMs of TDP‐43 for bacterial protein expression was previously published (Lukavsky et al, 2013) and mutations disrupting RNA binding were introduced as described above. The plasmid encoding TDP‐43‐MBP‐His6 for bacterial protein expression was a gift from Nicolas Fawzi (Addgene plasmid #104480; http://n2t.net/addgene:104480; RRID:Addgene_104480) (Wang et al, 2018). Point mutations for 6M, S2C, C39S, and C50S were introduced with site‐directed mutagenesis PCR with high fidelity Phusion DNA Polymerase (New England Biolabs, M0530) using primers described in Appendix Table S1. The plasmid encoding the MBP‐His6 was generated using deletion cloning by PCR with high fidelity Phusion DNA Polymerase (New England Biolabs, M0530) using designed primers described in Appendix Table S1.
Recombinant protein expression and purification
Production of TDP‐43 RRMs for NMR studies was performed as previously described (Lukavsky et al, 2013). Production of full‐length TDP‐43 was performed as previously reported (De Rossi et al, 2021). In brief, recombinant protein was expressed in Escherichia coli One Shot BL21 Star (DE3) cells (Thermo Fisher, C601003) in lysogeny broth (Invitrogen, 12780052). When the culture reached OD600 = 0.8 at 37°C, temperature was decreased to 4°C for 45 min and protein expression was subsequently induced with 1 mM isopropyl β‐D‐thiogalactoside (IPTG; Biosolve, 162423) at 16°C for 16 h. Cells were harvested by centrifugation and resuspended in 30 ml of lysis buffer (20 mM Tris pH 8.0 (Biosolve, 0020092391BS), 1 M sodium chloride (NaCl; Sigma Aldrich, 71380), 10 mM imidazole (Sigma Aldrich, 792527), 10 % (v/v) glycerol (Sigma Aldrich, G7757), 1 mM tris(2‐carboxyethyl)phosphine (TCEP; Sigma Aldrich, C4706), 5 protease inhibitor tablets (Roche, 11836170001)) per liter culture. Resuspended pellets were lysed by rod sonication (W‐385 LSL SECFROID Sonicator, Heat System Ultrasonic, 11 cycles, 10 s on, 20 s off) and subsequently centrifuged. The supernatant containing the soluble TDP‐43‐MBP‐His6 was filtered through 0.22 μm syringe filters and applied to a Ni Sepharose® Excel material (Sigma, GE17371201) in a gravity column (Econo glass column Biorad, 7372532). Subsequent washes were performed with lysis buffer and wash buffer (30 mM Tris, pH 8.0, 3 M NaCl, 10% (v/v) glycerol, 1 mM 1,4‐dithiothreitol (DTT; Thermo Fisher, P0861)), the protein was eluted in 30 ml 20 mM Tris pH 8.0, 1 M NaCl, 10 % (v/v) glycerol, 500 mM imidazole, 1 mM TCEP. The eluate was concentrated using Amicon Ultra‐15 concentrators MWCO 10 kDa (Merck Millipore, UFC901024) to a volume of 10 ml and subsequently injected onto a HiLoad 26/60 Superdex 200 pg size‐exclusion column (SEC; GE Healthcare, 17‐1071‐01) in SEC buffer (20 mM Tris, pH 8.0, 300 mM NaCl, 1 mM TCEP, 0.007 % (w/v) tween‐20 (Sigma Aldrich, P1379)) on an Äkta pure system (Cytavia, 25M1). Pure fractions (as analyzed by silver staining) were pulled and concentrated again with Amicon Ultra‐15 concentrators, aliquoted and snap frozen for storage at −80°C. The MBP‐His6 was purified like the full‐length TDP‐43, but purity was achieved after the Ni Sepharose Excel material (Merck, GE17‐3712‐02) and the protein was subsequently dialyzed against a storage buffer (20 mM Tris pH 8.0, 300 mM NaCl, 1 mM TCEP) overnight at 4°C, concentrated the next day using Amicon Ultra‐15 concentrators MWCO 10 kDa (Merck Millipore, UFC901024), flash frozen and kept at −80°C.
Circular dichroism experiments
Pure full‐length TDP‐43‐MBP constructs were thawed on ice and centrifuged for 15 min at 17,000 g and 4°C. The buffer was exchanged to 10 mM sodium phosphate pH 7.4 (0.036 % (w/v) sodium phosphate monobasic (Sigma Aldrich, S3139), 0.2 % (w/v) sodium phosphate dibasic (Sigma Aldrich, S5136)) using 500 μl concentrators with a MWCO of 10 kDa (Merck Millipore, UFC501024) 5 × (11,000 g, 10 min, 4°C). To ensure the solubility of the isolated protein, the concentrate was again centrifuged as described above. Protein concentration was determined by the extinction coefficients and molecular weights of the constructs (ExPASy ProtParam software) and absorbance at 280 nm (NanoDrop, Thermo Scientific). CD spectra of 200 μg/ml recombinant protein were recorded at 20°C from 180 to 250 nm with a bandwidth of 1 nm and a sampling period of 25 s on a Chirascan V100 (Applied Photophysics). Ellipticity (in mdeg) was transformed to molar ellipticity (in deg cm2 dmol−1) with the following formula, where c (in M) is the concentration and L (in cm) represents the pathlength of the cuvette:
Phase separation experiments
Pure concentrated fractions of recombinant protein MBP‐tagged full‐length WT and 6M TDP‐43 were desalted in a buffer of 20 mM sodium phosphate pH 7.2 (0.072 % (w/v) sodium phosphate monobasic, 0.4 %(w/v) sodium phosphate dibasic), 300 mM NaCl, 0.001% (v/v) TWEEN 20, 50 mM l‐arginine (Merck Millipore, A5006), 50 mM l‐glutamic acid (Merck Millipore, G1251) on a HiTrap desalting column (Cytavia, 17140801). An equimolar fluorophore labeling reaction was set up with CF660R maleimide (Biotum Inc., Fremont, CA) previously dissolved in dimethylformamide (DMF) in an N2‐hood and incubated for 16 h at 4°C under constant rotation. Reaction was stopped with 10 mM DTT and subsequently passed through a HiLoad 10/300 Superdex 200 pg SEC (Cytiva, 28990944) in SEC buffer on an Äkta pure system (Cytavia). Labeling position was confirmed by mass spectrometry and labeling ratio was determined by first determining the correct protein concentration:
where A280 is the absorbance of the protein at 280 nm, Amax CF660R is the CF660R dye absorbance at 663 nm (absorbance maximum of CF660R), CF is the correction factor for the amount of absorbance at A280 caused by the dye, and ε is the protein molar extinction coefficient.
The labeling ratio was subsequently calculated with following formula:
where ε′ is the molar extinction coefficient of the CF660R fluorescent dye.
Correctly labeled protein with a labeling efficiency of > 75% was snap frozen and stored at ‐80°C. TDP‐43‐MBP constructs were thawed and centrifuged (17,000 g, 15 min, 4°C) before exchanging the buffer to a to phase separation buffer consisting of 20 mM HEPES pH 7.4 (Biosolve, 0008042359BS), 150 mM NaCl, and 1 mM TCEP with Amicon 10 M WCO concentrators (Merck, UFC501024). The samples were centrifuged 5 × (11,000 g, 10 min, 4°C) and protein concentration was determined as described above. Phase separation of 10 μM TDP‐43‐MBP containing CF660R‐labeled TDP‐43‐MBP at a ratio of 1/200 was induced using a TEV protease (Sigma‐Aldrich, T4455) at a final concentration of 0.0045 mg/ml in phase separation buffer and incubation for 30 min at 22°C in μ‐Slide Angiogenesis glass‐bottom coverslips (ibidi, 81507). Images were acquired on a fluorescence microscope (Nanoimager S, ONI) with an Olympus 100× objective (1.4 NA) using a wavelength of 640 nm and 10% laser power. Reversibility was tested by adding 1,6‐hexanediol (Sigma, 240117) to a final concentration of 8% (w/v) in phase separation buffer from the beginning of the reaction. Image analysis and condensate counts were performed with ImageJ (1.52k).
Cell line culture
All cells were cultured at 37°C with saturated humidity and an atmosphere of 5% CO2, and regularly tested for mycoplasma contamination. HeLa (ATCC, CCL‐2) cells were cultured in DMEM (Gibco, 61965‐059) supplemented with 1% non‐essential amino acids (Gibco, 11140035) and 10% fetal bovine serum (FBS; Gibco, 10270‐106).
Mouse motor neuron‐like hybrid cells NSC‐34 (Bioconcept, CLU140) were proliferated on Matrigel (Corning, 354234)‐coated dishes in Dulbecco's modified Eagle medium (DMEM; Sigma, D5671) supplemented with 10% FBS, 1× GlutaMAX (Gibco, 35050‐061), 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco, 15140‐122). For experiments, differentiation was induced by switching to DMEM/F12 medium (Gibco, 21331‐020) supplemented with 1× GlutaMAX, 1× B27+ supplement (Gibco, 17504‐044), 1× N2 supplement (Gibco, 17502‐048), 100 U/ml penicillin, and 100 μg/ml streptomycin.
HEK293 Flp‐In T‐REx stable lines were cultured in DMEM (Sigma, D5671) supplemented with 10% FBS, 1× GlutaMAX, 15 μg/ml blasticidin S (Gibco, R21001; Invivogen, ant‐bl‐10p), 100 μg/ml hygromycin B (Gibco, 10687010), 100 U/ml penicillin, and 100 μg/ml streptomycin. HEK293 Flp‐In T‐REx stable lines were as previously described (Laferriere et al, 2019). In brief, HEK293 Flp‐In T‐REx (Invitrogen, R78007) cells were cultured in DMEM supplemented with 10% FBS, 1× GlutaMAX, 15 μg/ml blasticidin S, 100 μg/ml zeocin (Gibco, R25001), 100 U/ml penicillin and 100 μg/ml streptomycin. 3 × 105 HEK293 cells/well were plated in a 6‐well plate and transfected after 48 h with 125 ng of the corresponding pcDNA5 plasmid (GFP‐TDP‐43 WT, 6M, RRMm or 6M&RRMm) and 1,125 ng of pOG44 plasmid with 6.25 μl Lipofectamine 2000 (Invitrogen, 11668019) according to the manufacturer's instructions. After recovery for 48 h, cells were passaged to a 10‐cm dish with DMEM supplemented with 10% FBS, 1× GlutaMAX, 15 μg/ml blasticidin S, 100 μg/ml hygromycin B, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were selected for 2–3 weeks, expanded and subsequently frozen, and then cultured in the same medium.
When required, cells were treated with the appropriate chemicals at the following concentrations: 1 μg/ml doxycycline (Clontech, 631311), 2.5 μM MG132 (APExBIO, A2585), 1.25 μM MLN9708 (Selleck Chemicals, S2181), 500 nM bortezomib (BTZ; APExBIO, A2614), 100 nM bafilomycin A1 (Sigma‐Aldrich, SML1661), 100 μg/ml cycloheximide (Sigma‐Aldrich, C4859), 5 μg/ml actinomycin D (Sigma‐Aldrich, A1410), 10% 1,6‐hexanediol, 500 μM or 1 mM sodium arsenite (Alfa Aesar, 41533; Sigma‐Aldrich, 35000‐1L‐R), dimethyl sulfoxide (DMSO; Sigma‐Aldrich, D2650).
Human neural culture
Human neural networks were differentiated from an in‐house‐developed iPSC‐derived self‐renewing human neural stem cell line (iCoMoNSCs) obtained from control human skin fibroblast, as described previously (preprint: Hruska‐Plochan et al, 2021). In brief, 600,000 iCoMoNSCs were plated onto Matrigel‐coated (Corning, 354234) 6‐well plates in DMEM/F12 medium, supplemented with 0.5× B27– supplement (Gibco, 12587‐010), 0.5× N2 supplement; 1× GlutaMAX and 25 ng/ml bFGF (Gibco, PHG0261) and complete medium was changed daily. When confluency reached ~95%, the medium was switched to DMEM/F12 (Gibco, 11330‐032), 0.5× B27+ supplement, 1× N2 supplement, 1× GlutaMAX, 100 U/ml penicillin and 100 μg/ml streptomycin, 5 μM forskolin (Cayman, AG‐CN2‐0089‐M050), 1 μM synthetic retinoid Ec23 (Amsbio, AMS.SRP002‐2), and 500 nM smoothened agonist SAG (Millipore, 5666600) for the first 5 days. The majority of the medium was changed daily. On days 6–10, Ec23 was increased to 2 μM. The majority of the medium was changed daily. On days 11–25, Ec23 was decreased to 10 ng/ml, SAG to 50 nM and 20 ng/ml BDNF (PeproTech, 450‐02), 20 ng/ml GDNF (PeptroTech, 450‐10) and 20 ng/ml CNTF (Alomone labs, C‐240) were added. Two‐thirds of the medium was changed three times per week. From day 26 onwards, media was switched to a neural maturation medium consisting of 1:1 DMEM/F12:neurobasal medium (Gibco, 21103‐049), 1× B27+ supplement, 1× N2 supplement, 1.5× GlutaMAX, 5 μM forskolin, 20 ng/ml BDNF, 20 ng/ml GDNF, 20 ng/ml CNTF, 20 ng/ml NT‐3 (PeproTech, 450‐03), 20 ng/ml IGF‐1 (Stem Cell, 78022), and 10 μM cAMP (Sigma, D0260). Two‐thirds of the medium was changed three times per week. 14–22 weeks old neural cultures were used for the experiments.
Cultures were dissociated into single cell suspension using a Papain Dissociation System (Worthington, LK003150), including passing through 70 μm (Falcon, 08‐771‐2) and 40 μm (Falcon, 08‐771‐1) cell strainers and resuspended in neural maturation medium. Cells were plated onto Matrigel‐coated 96‐well plates (135,000 cells/well) (Greiner Bio‐One, 655090), 8‐well glass‐bottom chambers (between 2,400,000 and 425,000 cells/well) (ibidi, 80,827) or Matrigel‐and‐poly‐D‐lysine‐coated 18‐well glass‐bottom chambers (100,000–120,000 cells/well) (ibidi, 81817). For non‐transduced cultures, medium was regularly refreshed. For expression of TDP‐43‐HA variants, cultures were transduced with 1,000 ng/ml lentivirus in neural maturation medium 2–14 days after subculturing. The following day, the complete medium was refreshed. Two days after transduction, complete medium was exchanged to neural maturation medium containing 1 μg/ml doxycycline to induce transgene expression. When required, doxycycline was included in subsequent medium refreshments. Cultures were fixed at days 2–9 after doxycycline addition and appropriate treatments if required.
When required, neural cultures were treated with the appropriate chemicals at the following concentrations: 1 μg/ml doxycycline, 10 μM MG132, 10 μg/ml actinomycin D, and 25 μM ivermectin (Sigma‐Aldrich, I8898).
Lentiviral vector production
TDP‐43‐HA variants were packaged into lentivirus, harvested and concentrated as described previously (preprint: Hruska‐Plochan et al, 2021). Lentiviral pellets were resuspended in neural maturation medium (containing all supplements but forskolin and cAMP), achieving 10x concentrated preparations of which the lentiviral titer was determined using Lenti‐X GoStix Plus (Takara, 631280). Lentiviral preparations were aliquoted and stored at ‐80°C until use.
Quantitative PCR (qPCR)
HEK293 stable lines were plated at a density of 3 × 105 cells/well in a 6‐well plate (TPP, 92406). Expression of GFP‐TDP‐43 was induced with 1 μg/ml doxycycline after 24 h and cells were harvested 48 h later. Total RNA from cells in a single well of a 6‐well plate was isolated using the RNeasy Plus Mini Kit (Qiagen, 74134) according to the manufacturer's instructions. Complementary DNA (cDNA) corresponding to 1 μg was generated using oligo(dT)20 primers and the SuperScript™ III First‐Strand Synthesis SuperMix (Invitrogen, 18080400) according to the manufacturer's instructions. A qPCR of 50 cycles was performed with 10 ng of cDNA and 6.6 pmol of each of the primers (Appendix Table S2) per reaction using Fast SYBR™ Green Master Mix (Applied Biosystems, 4385612) in an AriaMx Real‐Time PCR System (Agilent, G8830A). Relative fold gene expression was calculated with the 2–∆∆Ct method.
GFP complementation assay
Tripartite GFP complementation experiments were performed as described before (Afroz et al, 2017; Foglieni et al, 2017). Cells grown on poly‐D‐lysine (Sigma‐Aldrich, P6407)‐coated 24‐well plates (ibidi, 82426) or 8‐well glass chambers (ibidi, 80827) to 70% confluency were transfected with Lipofectamine 2000 or jetPRIME (Polyplus‐transfection, 114‐15) according to the manufacturer's instructions. Cells were incubated for at least 1 day before immunocytochemistry analysis.
Protein–protein cross‐linking in cells
Native protein–protein interactions were stabilized by crosslinking with disuccinimidyl glutarate (DSG; Thermo Scientific, 20593 or A35392) as previously reported (Afroz et al, 2017). In brief, cells grown to 80% confluency in 6‐well or 10 cm plates were washed once in cell culture‐grade PBS (Gibco, 10010‐015), scrapped in ice‐cold cell culture‐grade PBS and collected at 300 g and 4°C for 5 min in a 1.5‐ml microfuge tube. Cells were resuspended in 100 or 600 μl ice‐cold cell culture‐grade PBS containing protease inhibitors and a freshly prepared 20 mM or 100 mM DSG solution in DMSO was added to the suspension to a final concentration of 1 mM. After incubation at 25°C and 1,500 rpm for 30 min in a Thermomixer (Eppendorf, 2230000048), the reaction was quenched by addition of Tris base to a final concentration of 20 mM and further incubation for 15 min. Cells were collected by centrifugation at 300 g for 5 min.
Nucleocytoplasmic fractionation of cells
Nucleocytoplasmic fractionation of the GFP‐TDP‐43 (mutNLS) isogenic HEK293 lines was performed following a previously published protocol (Suzuki et al, 2010) with the following changes. Cells in 10‐cm dishes at 80–90% confluency were scrapped in 1 ml cell culture‐grade PBS, collected by centrifugation at 300 g for 5 min and resuspended in 880 μl cell culture‐grade PBS supplemented with 0.1% NP‐40 (Sigma, 74385). The final pellet fraction enriched in nuclei was resuspended in 380 μl of 1× Laemmli buffer and 1× reducing agent (Invitrogen, B0009) in cell culture‐grade PBS supplemented with protease inhibitors, and 10 μl of each three of the final fractions were loaded onto the polyacrylamide gel for western blot analysis.
Immunocytochemistry
For immunocytochemistry experiments, cell lines were plated onto poly‐D‐lysine (Sigma‐Aldrich, P6407)‐coated 96‐ or 24‐well plates (Greiner Bio‐One, 655090; ibidi, 82426) or 8‐well glass chambers (ibidi, 80827). Unless stated otherwise, cell line cultures were fixed in 4% methanol‐free formaldehyde (Thermo Scientific, 28908) in warm medium for 15 min, washed with cell culture‐grade PBS, permeabilized and blocked in 10% donkey serum (Sigma‐Aldrich, S30‐M) and 0.1% Triton X‐100 in cell culture‐grade PBS for 10 min at RT. Neural cultures were plated onto Matrigel‐coated 96‐well plates (135,000 cells/well) or 8‐well glass‐bottom chambers (between 2,400,000 and 425,000 cells/well). Neural cultures were fixed in 4% methanol‐free formaldehyde in cell culture‐grade PBS for 25 min, washed with cell culture‐grade PBS, permeabilized in 0.5% Triton X‐100 in cell culture‐grade PBS for 5 min and blocked in 10% donkey serum and 0.1% Triton X‐100 in cell culture‐grade PBS for 30 min at RT. Primary antibodies (Appendix Table S3) were diluted in the same buffer and incubated with the samples overnight at 4°C. After three washes with cell culture‐grade PBS, Alexa Fluor‐conjugated donkey secondary antibodies (Appendix Table S3) were incubated for 1 h at RT and subsequently further washed with cell culture‐grade PBS. Nuclei were counterstained with 1 μg/ml DAPI (Thermo Scientific, 62248) and samples imaged in cell culture‐grade PBS.
Proximity ligation assay (PLA)
Cell lines plated on 96‐well plates were fixed in pure ice‐cold methanol for 7 min at −20°C, followed by three cell culture‐grade PBS washes. PLA assay was performed using custom labeled antibodies and the Duolink In Situ Detection Reagents Red (Sigma‐Aldrich, DUO92008) as described by the manufacturer but with slight modifications. In brief, antibodies of choice (anti‐TDP‐43, anti‐GFP, and anti‐HA in Appendix Table S3) in a carrier‐free buffer were labeled with MINUS and PLUS probes using the Duolink In Situ Probemaker MINUS (Sigma‐Aldrich, DUO92010) and Duolink In Situ Probemaker PLUS (Sigma‐Aldrich, DUO92009) according to the manufacturer's instructions. After blocking for at least 15 min with blocking solution at room temperature, probe‐conjugate antibodies were incubated overnight at 4°C. After two washes of 5 min each with buffer A (10 mM Tris, 150 mM NaCl 0.05% Tween 20), ligation reaction was performed as indicated by the manufacturer. After an additional two washes of 5 min each with buffer A, amplification reaction was performed as indicated by the manufacturer. The reaction was quenched with buffer B (100 mM Tris, 100 mM NaCl). If required, secondary antibodies were incubated in cell culture‐grade PBS overnight at 4°C, followed by DAPI counterstaining and imaging in cell culture‐grade PBS.
Fluorescence in situ hybridization (FISH)
FISH assay was performed using target probes for NEAT1 (Invitrogen, VX‐01) and the View RNA Cell Plus Assay (Invitrogen, 88‐19000) according to the manufacturer's instructions with slight modifications. HEK293 stable lines were plated at a density of 10 × 104 cells/well onto poly‐D‐lysine‐coated 96‐well plates. After 24 h, expression of GFP‐TDP‐43 was induced and, after further 48 h, cells were fixed in ViewRNA Cell Plus Fixation/Permeabilization Solution for 30 min at room temperature. After five washes with 1× PBS with RNase Inhibitor, ViewRNA Cell Plus Probe Solution containing the target probes was incubated for 2 h at 40°C. Following, the cells were washed with ViewRNA Cell Plus Wash Buffer Solution and Signal amplification was continued as indicated by the manufacturer. Nuclei were counterstained with DAPI and samples were imaged in cell culture‐grade PBS.
RNA sequencing (RNA‐seq)
HEK stable lines were plated at a density of 73 × 105 cells/well in 6‐cm dishes (TPP, 93060). Expression of GFP‐TDP‐43 was induced with 1 μg/ml doxycycline 24 h later and cells were harvested after further 48 h. Total RNA from cells in a single well of a 6‐well plate was isolated using the RNeasy Plus Mini Kit (Qiagen, 74134) according to the manufacturer's instructions with an additional on‐column DNase I (Qiagen, 79254) treatment at room temperature for 15 min to ensure the removal of all traces of genomic DNA. RNA quality was assessed by TapeStation (Agilent, Waldbronn, Germany) before being sent to Fasteris (Plan‐les‐Ouates, Switzerland) for RiboZero library preparation and paired‐end NovaSeq Illumina sequencing. The RNA sequencing data been deposited to the Sequence Read Archive (SRA) with the dataset identifier PRJNA970105. Each sample (four biological replicates per condition) was sequenced twice and the resulting fastq files were merged before the analysis. Trimmomatic (Bolger et al, 2014) was used for quality trimming and adapter clipping of the reads and alignment to the Ensembl GRCh38 reference sequence was performed with STAR v.2.7.6a (Dobin et al, 2013). Gene counts were generated using featureCounts v.1.4.6‐p2 (Liao et al, 2014) with default options based on the Ensembl Homo sapiens GRCh38v.102 gene annotation file. Differentially expressed genes were identified using the DESeq() function from the DESeq2 package v.1.18.1 of the R statistical software (Love et al, 2014), applying an adjusted P‐value cutoff of 0.05. For differential splicing analysis, the aforementioned trimmed reads were aligned to the combined splice junction files from the previous STAR runs for all samples. Counts were calculated for all exons using the dexseq_count.py script from the DEXseq software (Anders et al, 2012) and differential splicing events between conditions were detected using the R software in conjunction with the DEXseq package, applying an adjusted P‐value cutoff of 0.01.
Mass spectrometry and label‐free quantification (LFQ) of whole‐cell lysates
HEK stable lines were plated at a density of 73 × 104 cells/dish in 6‐cm dishes (TPP, 93060). After 24 h, expression of GFP‐TDP‐43 was induced and, after further 48 h, cells were collected by pipetting up and down in cell culture‐grade PBS and centrifugation for 5 min at 300 g. Pellets were snap frozen in liquid nitrogen and stored at −80°C until use. Four biological duplicates per condition were collected with a difference of two passages each, but sample preparation for mass spectrometry (MS) was performed simultaneously for all samples regardless of the replicate.
Samples were prepared by using a commercial iST Kit (PreOmics, P.O.00027) with an updated version of the protocol. Briefly, 50 μg of the samples were solubilized in “Lyse” buffer, boiled at 95°C for 10 min and processed with high intensity focused ultrasound for 30 s at an amplitude of 85%. Samples were then transferred to the cartridge and digested with 50 μl of the “Digest” solution for 60 min at 37°C before stopping the reaction with 100 μl of “Stop” solution. Solutions in the cartridge were removed by centrifugation at 3,800 g, while the peptides were retained by the iST‐filter. Finally, digested peptides were washed, eluted, dried, and re‐solubilized in 20 μl of “LC‐Load” buffer. Samples were further diluted 1:10 prior for MS analysis.
MS analysis of samples was performed on a Q Exactive HF‐X mass spectrometer (Thermo Scientific), equipped with a Digital PicoView source (New Objective) and coupled to an M‐Class UPLC (Waters). Solvent composition at the two channels was 0.1% formic acid for channel A and 0.1% formic acid, 99.9% acetonitrile for channel B. For each sample, 2 μl of peptides were loaded on a commercial MZ Symmetry C18 Trap Column (100 Å, 5 μm, 180 μm × 20 mm, Waters) followed by nanoEase MZ C18 HSS T3 Column (100 Å, 1.8 μm, 75 μm × 250 mm, Waters). The peptides were eluted at a flow rate of 300 nl/min by a gradient from 8 to 27% B in 85 min, 35% B in 5 min, and 80% B in 1 min. Samples were acquired in a randomized order. The mass spectrometers were operated in data‐dependent mode (DDA) acquiring a full‐scan MS spectra (350–1,400 m/z) at a resolution of 120,000 at 200 m/z after accumulation to a target value of 3,000,000, followed by higher‐energy collision dissociation (HCD) fragmentation on the 20 most intense signals per cycle. HCD spectra were acquired at a resolution of 15,000 using a normalized collision energy of 25 and a maximum injection time of 22 ms. The automatic gain control (AGC) was set to 100,000 ions. Only precursors with intensity above 250,000 were selected for MS/MS. Charge state screening was enabled. Singly, unassigned and charge states higher than seven were rejected. Precursor masses previously selected for MS/MS measurement were excluded from further selection for 30 s, and the exclusion window was set at 10 ppm. The samples were acquired using internal lock mass calibration on m/z 371.1012 and 445.1200. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD041795.
The mass spectrometry proteomics data were handled using the local laboratory information management system (LIMS; Türker et al, 2010). The acquired raw MS data were processed by MaxQuant (version 1.6.2.3), followed by protein identification using the integrated Andromeda search engine (Cox & Mann, 2008). Spectra were searched against a Swiss‐Prot Homo sapiens (Human) reference proteome (taxonomy 9606, version from 2019‐11‐02), [PN3] concatenated to its reversed decoyed fasta database and common protein contaminants. Carbamidomethylation of cysteine was set as fixed modification, while methionine oxidation and N‐terminal protein acetylation were set as variable. Enzyme specificity was set to trypsin/P allowing a minimal peptide length of seven amino acids and a maximum of two missed‐cleavages. MaxQuant Orbitrap default search settings were used. The maximum false discovery rate (FDR) was set to 0.01 for peptides and 0.05 for proteins. Label‐free quantification was enabled and a 2‐min window for match between runs was applied. In the MaxQuant experimental design template, each file is kept separate in the experimental design to obtain individual quantitative values.
Protein fold changes were computed based on peptide intensity values reported in the MaxQuant‐generated peptides.txt file, using linear mixed‐effects models (Bates et al, 2015). Peptide intensities reported in the peptides.txt file were processed as follows: all proteins with only one identified and quantified peptide were removed, as well as intensities equal to zero. Non‐zero intensities are log2 transformed and modified using robust z‐score transformation. Afterwards, for each protein, a mixed‐effects model was fitted to the peptide intensities, and fold changes and P‐values were computed based on this model for each contrast (Kuznetsova et al, 2017). For a contrast, all the protein P‐values were adjusted using the Benjamini and Hochberg procedure to obtain the FDR (Benjamini & Hochberg, 1995).
To estimate fold‐changes for proteins for which a mixed‐effects model could not be fit because of an excess in missing measurements, we proceed as follows. First, we compute the mean intensity for all peptides for each condition. For the proteins with no measurements, we impute the peptide intensities using the mean of the 10% smallest average peptide intensities computed in step one. Afterwards, we compute contrasts (differences between conditions) for each peptide and finally, the median of the peptide estimates is used to provide a per protein fold change estimate.
Sodium dodecyl‐sulphate polyacrylamide gel electrophoresis (SDS‐PAGE)
Unless indicated otherwise, cells were lysed in RIPA buffer consisting of 10 mM Tris‐Cl pH 8.0, 1% Triton X‐100 (Sigma‐Aldrich, T9284), 0.1% sodium deoxycholate (Sigma‐Aldrich, D6750), 0.1% SDS (Sigma‐Aldrich, 05030), 140 mM NaCl, supplemented protease inhibitors, 2 mM MgCl2 (Sigma‐Aldrich, M8266), and 1 U/μl Benzonase (Millipore, 71205‐3).
Protein concentration of analyzed samples was measured by bicinchoninic acid assay (Thermo Scientific, 23227) or with Pierce 660 nm Protein Assay Reagent (Thermo Scientific, 22660).
Equal protein amounts from cell lysates or of recombinant proteins were mixed with LDS loading buffer (Invitrogen, B0008) supplemented with reducing agent (Life Technologies, B009) and denatured for 10 min at 90°C or 5 min at 95°C. Samples were loaded on 15‐ or 17‐well BOLT 4–12% Bis‐Tris precast gels (Invitrogen, NW04125BOX or NW04127BOX). SDS–PAGE was performed in MES buffer (Invitrogen, B000202) at 90 V for 15 min and 150 V until the lowest marker band (Thermo Scientific, 26617 or 26619) reached the bottom line.
Coomassie staining
Gels were stained in a staining solution containing 0.25% (w/v) Coomassie Brilliant Blue R‐250 (Bio‐rad, #1610400), 45% (v/v) methanol (Merck Millipore, 67‐56‐1), 10% (v/v) acetic acid (Merck, 100063.1000) for 1 h at room temperature and subsequently double destained for 1 h and overnight at RT in destaining solution (45 % (v/v) methanol, 10% (v/v) acetic acid).
Silver staining
Gels were fixed for 45 min at room temperature in a fixation solution of 30% (v/v) ethanol (VWM Chemicals, 20821.296), 10% (v/v) acetic acid, and subsequently sensitized for 45 min at RT in a sensitizing solution of 7.5% (v/v) ethanol, 6.8% (w/v) sodium acetate (Sigma Aldrich, S2889), 0.2% (w/v) sodium thiosulfate (Sigma Aldrich, S6672), and 0.03125 % glutaraldehyde (v/v) (Sigma Aldrich, G5882). Gels were then washed three times for 5 min with Milli‐Q water at room temperature and incubated for 20 min at room temperature in a silver staining solution of 0.2% w/v silver nitrate (Sigma Aldrich, 31630) and 0.0028% formaldehyde (v/v) (Sigma Aldrich, F8775). The gels were washed for 30 s with Milli‐Q water and immersed in a developing solution of 2.5% (w/v) sodium carbonate (Sigma Aldrich, S2889) and 0.0014 % (v/v) formaldehyde for 5–7 min at room temperature. The reaction was stopped with a stopping solution of 1% (w/v) glycine (Sigma Aldrich, G7126) and washed three times for 5 min with Milli‐Q water at room temperature. All steps were performed using 50 ml of solution.
Western blot
Using an iBlot Gel Transfer Device (IB21001), proteins were blotted onto nitrocellulose membranes (Invitrogen, IB23002 or IB23001), which were subsequently blocked in 5% skimmed milk (Coop) in PBS (PanReac AppliChem, A0965) 0.05% TWEEN 20 (PBS‐T). Primary and horseradish peroxidase‐conjugated secondary antibodies (Appendix Table S3) were incubated in 1% milk in PBS‐T overnight at 4°C or for 1 h at room temperature. Western blots were developed with SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Scientific, 34577) or SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific, 34095) in an ImageQuant LAS 4000 system in semi‐automatic chemiluminescent mode.
Western blot analysis
Analysis of chemiluminescent signals from western blots was performed with Fiji (Schindelin et al, 2012). Raw integrated density (RawIntDent) was calculated for each band by selecting equal‐sized areas including the band corresponding to the protein of interest per condition and one area for background control. Background RawIntDent was subtracted from the signal of interest (GFP, TDP‐43) before normalizing by the values of the loading control (SOD1, GAPDH), previously controlled for background as well. For nucleocytoplasmic fractionation ratios, values were normalized by the respective fraction loading controls before calculating the ratio.
Image acquisition
Confocal fluorescence microscopy images were acquired on Leica SP5 or SP8 Falcon inverted confocal laser microscopes using a 63× objective (NA 1.4) with oil immersion and between 1× and 4 zoom or a 40× air objective (NA 0.9). Widefield microscopy images and live cell imaging were performed using a IN Cell Analyzer 2500HS microscope using a 40X CFI Plan Apo Lambda objective (NA 0.9).
Fluorescence recovery after photobleaching (FRAP)
FRAP experiments and analysis were performed as described before (Scotter et al, 2014) in a Leica SP5 inverted confocal laser microscope using 63X objective (NA 1.4). Cells were plated on 24‐well plates and cultured with phenol red‐free DMEM (Gibco, 31053028).
Image analysis
For quantification of the GFP‐TDP‐43 variants or endogenous TDP‐43 in HEK293 cells, widefield microscopy fields were randomly selected in the DAPI channel and optimal focus obtained using the DAPI and G3BP channels (Fig 2A and I; Appendix Fig S6H). Trainable Weka Segmentation (Arganda‐Carreras et al, 2017) plug‐in from Fiji (Schindelin et al, 2012) was trained to create regions of interest (ROIs) corresponding to the nucleus and cytoplasm of the cells in the field based on the DAPI and G3BP signals, respectively. The ROIs were subsequently used to measure the TDP‐43 signal in the corresponding channel (Appendix Fig S2A), from which the nucleocytoplasmic (N/C) ratio was calculated. The same pipeline was followed to determine the N/C ratio of endogenous TDP‐43 in human neural cultures upon mock, ActD or IVM treatment (Fig 2K), but using DAPI and MAP2 signals to identify the neuron‐specific nuclei and cytoplasm instead.
For analysis of TDP‐43‐HA localization in human neurons (Fig 2C), confocal microscopy fields were randomly selected in the DAPI channel. After transgene expression was verified in the HA channel, optimal focus was obtained using the DAPI and MAP2 channels and single focal planes were acquired. Using Fiji (Schindelin et al, 2012), mean intensity of the HA channel was measured in ROIs manually drawn based on the DAPI and MAP2 signal to identify the nucleus and the cytoplasm and the N/C ratio was subsequently calculated.
Quantification of the number, volume and localization of nuclear droplets in HEK cells was performed using Imaris (RRID:SCR_007370) on confocal microscopy Z‐stacks acquired in steps of 0.21 μm. After three‐dimensional reconstruction of the nucleus, a “Nucleus” surface was created using the DAPI channel with a surface detail of 0.1 μm in absolute intensity mode and thresholding to the nearest ten below. The biggest surface computed based on volume was chosen as the nucleus. Mean intensity signal from the TDP‐43 channel was measured within the “Nucleus” surface to calculate the “nuclear TDP‐43” values. TDP‐43 droplets were modeled by creating a “Droplets” surface using the TD‐43 channel with a surface detail of 0.1 μm and background subtraction mode of 0.1 μm. All bodies bigger than 0.003 μm3 were considered droplets. The “Droplets” surface was used to quantify the number and volume of nuclear TDP‐43 droplets (Fig 3B and F; Appendix Fig S3A and B). For colocalization analysis (Fig 5B and C; Appendix Fig S5B), surfaces of the different nuclear subcompartments were created following the same approach as for the nuclear TDP‐43 droplets. Distance between the center of mass of each droplet to the nearest nuclear body and between each nuclear body to the nearest droplet was calculated in order to determine the percentage of TDP‐43 droplets colocalizing with a known nuclear body and the percentage of TDP‐43+ nuclear bodies of each type, respectively. A distance < 500 nm, the axial resolution limit of confocal microscopy (Fouquet et al, 2015), was considered as colocalization.
Analysis of GFP‐TDP‐43 aggregate localization in mock‐ and MG132‐treated in HEK293 lines (Fig 7B; Appendix Fig S7A–C) was performed in randomly acquired widefield microscopy images with Fiji (Schindelin et al, 2012). Cells were manually divided into four groups using the GFP and DAPI signals: no aggregation, nuclear aggregation, cytoplasmic aggregation and both nuclear and cytoplasmic aggregation. For quantification of TDP‐43‐HA aggregate localization in mock‐ and MG132‐treated human neural cultures (Fig 7D; Appendix Fig S7D), confocal microscopy images were analyzed with Fiji (Schindelin et al, 2012) to identify transduced neurons using the MAP2 and HA channels. Using the HA and DAPI channels, the neurons were manually divided into the four aforementioned groups. Only aggregates were counted, meaning that neurons exhibiting diffuse nuclear extraction of TDP‐43‐HA were classified under “no aggregation.”
Statistical analysis
Depending on the experimental design and whether the dataset followed a normal distribution (as determined by a Shapiro–Wilk normality test), statistical analysis was performed with a unpaired two‐tailed t‐test, a Mann–Whitney U‐test, a repeated measures or unpaired one‐way analysis of variance (ANOVA) with Tukey's or Dunnett's multiple comparisons post hoc test, a two‐way ANOVA with Tukey's multiple comparisons post hoc test or a Kruskal–Wallis test with Dunn's multiple comparisons post hoc test, as specified in the legends of each figure. ns: not significant, *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. Error bars in graphs represent the standard deviation of the dataset (SD). Horizontal lines in violin plots represent the mean and quartiles of the dataset. In cases where the data was represented as a percentage of control, statistical analysis was performed with the original data in order not to skew the variances and the normal distribution of the data (Lew, 2007).
Author contributions
Manuela Pérez‐Berlanga: Conceptualization; data curation; formal analysis; validation; visualization; methodology; writing – original draft; writing – review and editing. Vera I Wiersma: Data curation; formal analysis; validation; visualization; methodology; writing – original draft; writing – review and editing. Aurélie Zbinden: Data curation; software; formal analysis; validation; visualization; methodology; writing – original draft; writing – review and editing. Laura De Vos: Data curation; formal analysis; validation; visualization; methodology. Ulrich Wagner: Data curation; software; formal analysis; methodology. Chiara Foglieni: Formal analysis; validation; investigation; visualization; methodology. Izaskun Mallona: Software; formal analysis. Katharina M Betz: Software; formal analysis. Antoine Cléry: Formal analysis; investigation; methodology. Julien Weber: Resources; formal analysis; methodology. Zhongning Guo: Resources; formal analysis; methodology. Ruben Rigort: Formal analysis. Pierre de Rossi: Formal analysis. Ruchi Manglunia: Formal analysis. Elena Tantardini: Formal analysis. Sonu Sahadevan: Formal analysis. Oliver Stach: Formal analysis. Marian Hruska‐Plochan: Methodology. Frederic H‐T Allain: Supervision; methodology. Paolo Paganetti: Supervision; methodology. Magdalini Polymenidou: Conceptualization; resources; supervision; funding acquisition; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Acknowledgements
We thank Prof. Benjamin Schuler (Department of Biochemistry, University of Zurich) for critical feedback on this work and Dr. Matthias Gstaiger (Institute of Molecular Systems Biology, ETH Zurich, Switzerland) for sharing the Flp‐In T‐REx HEK293 cell line and the pOG44 plasmid. We gratefully acknowledge the support from the Proteomics team at the Functional Genomics Center Zurich (FGCZ) from the University of Zurich (UZH), especially Dr. Paolo Nanni, Dr. Serge Chesnov, and Dr. Witold Wolski. We also like to thank Dr. Jana Doehner, Dr. Joana Delgado Martins, Dr. José María Mateos Melero, and Johannes Riemann from the Center for Microscopy and Image Analysis (ZMB) at the University of Zurich UZH for their kind help with image acquisition and analysis. We are grateful to Dr. Erik Slabber for his technical advice on cloning and PLA and help during manuscript preparation, and to our fellow Polymenidou lab members for the continuous and fruitful discussions on the project.
This work was supported by the Swiss National Science Foundation (grants PP00P3_144862 and PP00P3_176966 to MP), the National Centre for Competence in Research (NCCR) RNA & Disease (51NF40‐182880) and a Sinergia grant (CRSII5_170976 11) and the ALS Association Grant 18‐IIA‐411. MP‐B received a Candoc Grant (Forschungskredit) from the University of Zurich. VIW is supported by the FEBS Long‐Term Fellowship and a Rubicon Research Grant from the Dutch Research Council. PDR received a Career Development Award from the Stiftung Synapsis – Alzheimer Forschung Schweiz. SS was supported by a Swiss Government Excellence Scholarship for Foreign Scholars. Open access funding provided by University of Zurich. Open access funding provided by Universitat Zurich.
The EMBO Journal (2023) 42: e111719
Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the supplementary materials (Extended View and Appendix). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (https://www.ebi.ac.uk/pride/archive/projects/PXD041795) with the dataset identifier PXD041795 and the RNA sequencing data been deposited to the Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/bioproject/970105) with the dataset identifier PRJNA970105.
References
- Afroz T, Hock EM, Ernst P, Foglieni C, Jambeau M, Gilhespy LAB, Laferriere F, Maniecka Z, Pluckthun A, Mittl P et al (2017) Functional and dynamic polymerization of the ALS‐linked protein TDP‐43 antagonizes its pathologic aggregation. Nat Commun 8: 45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afroz T, Pérez‐Berlanga M, Polymenidou M (2019) Structural transition, function and dysfunction of TDP‐43 in neurodegenerative diseases. Chimia (Aarau) 73: 380–390 [DOI] [PubMed] [Google Scholar]
- Anders S, Reyes A, Huber W (2012) Detecting differential usage of exons from RNA‐seq data. Genome Res 22: 2008–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y et al (2006) TDP‐43 is a component of ubiquitin‐positive tau‐negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351: 602–611 [DOI] [PubMed] [Google Scholar]
- Arganda‐Carreras I, Kaynig V, Rueden C, Eliceiri KW, Schindelin J, Cardona A, Sebastian Seung H (2017) Trainable Weka segmentation: a machine learning tool for microscopy pixel classification. Bioinformatics 33: 2424–2426 [DOI] [PubMed] [Google Scholar]
- Arseni D, Hasegawa M, Murzin AG, Kametani F, Arai M, Yoshida M, Ryskeldi‐Falcon B (2022) Structure of pathological TDP‐43 filaments from ALS with FTLD. Nature 601: 139–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aulas A, Stabile S, Vande Velde C (2012) Endogenous TDP‐43, but not FUS, contributes to stress granule assembly via G3BP. Mol Neurodegener 7: 54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avendaño‐Vázquez SE, Dhir A, Bembich S, Buratti E, Proudfoot N, Baralle FE (2012) Autoregulation of TDP‐43 mRNA levels involves interplay between transcription, splicing, and alternative polyA site selection. Genes Dev 26: 1679–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala YM, Zago P, D'Ambrogio A, Xu YF, Petrucelli L, Buratti E, Baralle FE (2008) Structural determinants of the cellular localization and shuttling of TDP‐43. J Cell Sci 121: 3778–3785 [DOI] [PubMed] [Google Scholar]
- Ayala YM, De Conti L, Avendaño‐Vázquez SE, Dhir A, Romano M, D'Ambrogio A, Tollervey J, Ule J, Baralle M, Buratti E et al (2011) TDP‐43 regulates its mRNA levels through a negative feedback loop. EMBO J 30: 277–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates D, Mächler M, Bolker BM, Walker SC (2015) Fitting linear mixed‐effects models using lme4. J Stat Softw 10.18637/jss.v067.i01 [DOI] [Google Scholar]
- Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol 57: 289–300 [Google Scholar]
- Boisvert F‐M, Hendzel MJ, Bazett‐Jones DP (2000) Promyelocytic leukemia (Pml) nuclear bodies are protein structures that do not accumulate RNA. J Cell Biol 148: 283–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveris A, Navarro A (2008) Brain mitochondrial dysfunction in aging. IUBMB Life 60: 308–314 [DOI] [PubMed] [Google Scholar]
- Buratti E, Baralle FE (2001) Characterization and functional implications of the RNA binding properties of nuclear factor TDP‐43, a novel splicing regulator of CFTR exon 9. J Biol Chem 276: 36337–36343 [DOI] [PubMed] [Google Scholar]
- Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ, Foong C, White CL 3rd, Schneider JA, Kretzschmar HA et al (2007) TDP‐43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol 171: 227–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CK, Wu TH, Wu CY, Chiang MH, Toh EK, Hsu YC, Lin KF, Liao YH, Huang TH, Huang JJ (2012) The N‐terminus of TDP‐43 promotes its oligomerization and enhances DNA binding affinity. Biochem Biophys Res Commun 425: 219–224 [DOI] [PubMed] [Google Scholar]
- Chen H‐J, Topp SD, Hui HS, Zacco E, Katarya M, McLoughlin C, King A, Smith BN, Troakes C, Pastore A et al (2019) RRM adjacent TARDBP mutations disrupt RNA binding and enhance TDP‐43 proteinopathy. Brain 142: 3753–3770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen‐Plotkin AS, Lee VM, Trojanowski JQ (2010) TAR DNA‐binding protein 43 in neurodegenerative disease. Nat Rev Neurol 6: 211–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chujo T, Yamazaki T, Hirose T (2016) Architectural RNAs (arcRNAs): a class of long noncoding RNAs that function as the scaffold of nuclear bodies. Biochim Biophys Acta 1859: 139–146 [DOI] [PubMed] [Google Scholar]
- Cohen TJ, Hwang AW, Restrepo CR, Yuan CX, Trojanowski JQ, Lee VM (2015) An acetylation switch controls TDP‐43 function and aggregation propensity. Nat Commun 6: 5845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conicella AE, Zerze GH, Mittal J, Fawzi NL (2016) ALS mutations disrupt phase separation mediated by alpha‐helical structure in the TDP‐43 low‐complexity C‐terminal domain. Structure 24: 1537–1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conicella AE, Dignon GL, Zerze GH, Schmidt HB, D'Ordine AM, Kim YC, Rohatgi R, Ayala YM, Mittal J, Fawzi NL (2020) TDP‐43 α‐helical structure tunes liquid–liquid phase separation and function. Proc Natl Acad Sci U S A 117: 5883–5894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 12: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Coyne AN, Baskerville V, Zaepfel BL, Dickson DW, Rigo F, Bennett F, Lusk CP, Rothstein JD (2021) Nuclear accumulation of CHMP7 initiates nuclear pore complex injury and subsequent TDP‐43 dysfunction in sporadic and familial ALS. Sci Transl Med 13: eabe1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Ambrogio A, Buratti E, Stuani C, Guarnaccia C, Romano M, Ayala YM, Baralle FE (2009) Functional mapping of the interaction between TDP‐43 and hnRNP A2 in vivo. Nucleic Acids Res 37: 4116–4126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rossi P, Lewis AJ, Furrer J, De Vos L, Demeter T, Zbinden A, Zhong W, Wiersma VI, Scialo C, Weber J et al (2021) FTLD‐TDP assemblies seed neoaggregates with subtype‐specific features via a prion‐like cascade. EMBO Rep 22: e53877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewey CM, Cenik B, Sephton CF, Johnson BA, Herz J, Yu G (2012) TDP‐43 aggregation in neurodegeneration: are stress granules the key? Brain Res 1462: 16–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dörrbaum AR, Kochen L, Langer JD, Schuman EM (2018) Local and global influences on protein turnover in neurons and glia. Elife 7: e34202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan L, Zaepfel BL, Aksenova V, Dasso M, Rothstein JD, Kalab P, Hayes LR (2022) Nuclear RNA binding regulates TDP‐43 nuclear localization and passive nuclear export. Cell Rep 40: 111106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Düster R, Kaltheuner IH, Schmitz M, Geyer M (2021) 1,6‐hexanediol, commonly used to dissolve liquid‐liquid phase separated condensates, directly impairs kinase and phosphatase activities. J Biol Chem 296: 100260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ederle H, Funk C, Abou‐Ajram C, Hutten S, Funk EBE, Kehlenbach RH, Bailer SM, Dormann D (2018) Nuclear egress of TDP‐43 and FUS occurs independently of Exportin‐1/CRM1. Sci Rep 8: 7084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eersel J, Ke YD, Gladbach A, Bi M, Götz J, Kril JJ, Ittner LM (2011) Cytoplasmic accumulation and aggregation of TDP‐43 upon proteasome inhibition in cultured neurons. PloS One 6: e22850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elden AC, Kim H‐J, Hart MP, Chen‐Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM et al (2010) Ataxin‐2 intermediate‐length polyglutamine expansions are associated with increased risk for ALS. Nature 466: 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores BN, Li X, Malik AM, Martinez J, Beg AA, Barmada SJ (2019) An intramolecular salt bridge linking TDP43 RNA binding, protein stability, and TDP43‐dependent neurodegeneration. Cell Rep 27: 1133–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foglieni C, Papin S, Salvade A, Afroz T, Pinton S, Pedrioli G, Ulrich G, Polymenidou M, Paganetti P (2017) Split GFP technologies to structurally characterize and quantify functional biomolecular interactions of FTD‐related proteins. Sci Rep 7: 14013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster AD, Flynn LL, Cluning C, Cheng F, Davidson JM, Lee A, Polain N, Mejzini R, Farrawell N, Yerbury JJ et al (2021) p62 overexpression induces TDP‐43 cytoplasmic mislocalisation, aggregation and cleavage and neuronal death. Sci Rep 11: 11474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouquet C, Gilles J‐F, Heck N, Dos Santos M, Schwartzmann R, Cannaya V, Morel M‐P, Davidson RS, Trembleau A, Bolte S (2015) Improving axial resolution in confocal microscopy with new high refractive index mounting media. PloS One 10: e0121096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- French RL, Grese ZR, Aligireddy H, Dhavale DD, Reeb AN, Kedia N, Kotzbauer PT, Bieschke J, Ayala YM (2019) Detection of TAR DNA‐binding protein 43 (TDP‐43) oligomers as initial intermediate species during aggregate formation. J Biol Chem 294: 6696–6709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frottin F, Schueder F, Tiwary S, Gupta R, Körner R, Schlichthaerle T, Cox J, Jungmann R, Hartl FU, Hipp MS (2019) The nucleolus functions as a phase‐separated protein quality control compartment. Science 365: 342–347 [DOI] [PubMed] [Google Scholar]
- Gallagher PS, Clowes Candadai SV, Gardner RG (2014) The requirement for Cdc48/p97 in nuclear protein quality control degradation depends on the substrate and correlates with substrate insolubility. J Cell Sci 127: 1980–1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasset‐Rosa F, Lu S, Yu H, Chen C, Melamed Z, Guo L, Shorter J, Da Cruz S, Cleveland DW (2019) Cytoplasmic TDP‐43 De‐mixing independent of stress granules drives inhibition of nuclear import, loss of nuclear TDP‐43, and cell death. Neuron 102: 339–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallegger M, Chakrabarti AM, Lee FCY, Lee BL, Amalietti AG, Odeh HM, Copley KE, Rubien JD, Portz B, Kuret K et al (2021) TDP‐43 condensation properties specify its RNA‐binding and regulatory repertoire. Cell 184: 4680–4696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y, Beach TG, Buratti E, Baralle F, Morita M et al (2008) Phosphorylated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol 64: 60–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hergesheimer RC, Chami AA, De Assis DR, Vourc'h P, Andres CR, Corcia P, Lanznaster D, Blasco H (2019) The debated toxic role of aggregated TDP‐43 in amyotrophic lateral sclerosis: a resolution in sight? Brain 142: 1176–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiji M, Takahashi T, Fukuba H, Yamashita H, Kohriyama T, Matsumoto M (2008) White matter lesions in the brain with frontotemporal lobar degeneration with motor neuron disease: TDP‐43‐immunopositive inclusions co‐localize with p62, but not ubiquitin. Acta Neuropathol 116: 183–191 [DOI] [PubMed] [Google Scholar]
- Hruska‐Plochan M, Betz KM, Ronchi S, Wiersma VI, Maniecka Z, Hock E‐M, Laferriere F, Sahadevan S, Hoop V, Delvendahl I et al (2021) Human neural networks with sparse TDP‐43 pathology reveal NPTX2 misregulation in ALS/FTLD. bioRxiv 10.1101/2021.12.08.471089 [PREPRINT] [DOI]
- Izumikawa K, Nobe Y, Ishikawa H, Yamauchi Y, Taoka M, Sato K, Nakayama H, Simpson RJ, Isobe T, Takahashi N (2019) TDP‐43 regulates site‐specific 2′‐O‐methylation of U1 and U2 snRNAs via controlling the Cajal body localization of a subset of C/D scaRNAs. Nucleic Acids Res 47: 2487–2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraj GG, Hipp MS, Hartl FU (2020) Functional modules of the Proteostasis network. Cold Spring Harb Perspect Biol 12: a033951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L‐L, Xue W, Hong J‐Y, Zhang J‐T, Li M‐J, Yu S‐N, He J‐H, Hu H‐Y (2017) The N‐terminal dimerization is required for TDP‐43 splicing activity. Sci Rep 7: 6196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JA, Ward CL, Kopito RR (1998) Aggresomes: a cellular response to misfolded proteins. J Cell Biol 143: 1883–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Stroh A, Dugger B, Dickson DW (2009) Evaluation of subcortical pathology and clinical correlations in FTLD‐U subtypes. Acta Neuropathol 118: 349–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroschwald S, Maharana S, Alberti S (2017) Hexanediol: a chemical probe to investigate the material properties of membrane‐less compartments. Matters 3: e201702000010 [Google Scholar]
- Kuznetsova A, Brockhoff PB, Christensen RHB (2017) lmerTest package: tests in linear mixed effects models. J Stat Softw 82 10.18637/jss.v082.i13 [DOI] [Google Scholar]
- Laferriere F, Maniecka Z, Perez‐Berlanga M, Hruska‐Plochan M, Gilhespy L, Hock EM, Wagner U, Afroz T, Boersema PJ, Barmettler G et al (2019) TDP‐43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting disease progression rates. Nat Neurosci 22: 65–77 [DOI] [PubMed] [Google Scholar]
- Lee EB, Porta S, Michael Baer G, Xu Y, Suh ER, Kwong LK, Elman L, Grossman M, Lee VMY, Irwin DJ et al (2017) Expansion of the classification of FTLD‐TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol 134: 65–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SM, Asress S, Hales CM, Gearing M, Vizcarra JC, Fournier CN, Gutman DA, Chin L‐S, Li L, Glass JD (2019) TDP‐43 cytoplasmic inclusion formation is disrupted in C9orf72‐associated amyotrophic lateral sclerosis/frontotemporal lobar degeneration. Brain Commun 1: fcz014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930 [DOI] [PubMed] [Google Scholar]
- Ling SC, Albuquerque CP, Han JS, Lagier‐Tourenne C, Tokunaga S, Zhou H, Cleveland DW (2010) ALS‐associated mutations in TDP‐43 increase its stability and promote TDP‐43 complexes with FUS/TLS. Proc Natl Acad Sci U S A 107: 13318–13323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling SC, Polymenidou M, Cleveland DW (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79: 416–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling JP, Pletnikova O, Troncoso JC, Wong PC (2015) TDP‐43 repression of nonconserved cryptic exons is compromised in ALS‐FTD. Science 349: 650–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukavsky PJ, Daujotyte D, Tollervey JR, Ule J, Stuani C, Buratti E, Baralle FE, Damberger FF, Allain FH (2013) Molecular basis of UG‐rich RNA recognition by the human splicing factor TDP‐43. Nat Struct Mol Biol 20: 1443–1449 [DOI] [PubMed] [Google Scholar]
- Maharana S, Wang J, Papadopoulos DK, Richter D, Pozniakovsky A, Poser I, Bickle M, Rizk S, Guillén‐Boixet J, Franzmann TM et al (2018) RNA buffers the phase separation behavior of prion‐like RNA binding proteins. Science 360: 918–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallik S, Kundu S (2018) Topology and oligomerization of mono‐ and oligomeric proteins regulate their half‐lives in the cell. Structure 26: 869–878 [DOI] [PubMed] [Google Scholar]
- Mann JR, Gleixner AM, Mauna JC, Gomes E, DeChellis‐Marks MR, Needham PG, Copley KE, Hurtle B, Portz B, Pyles NJ et al (2019) RNA binding antagonizes neurotoxic phase transitions of TDP‐43. Neuron 102: 321–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto G, Inobe T, Amano T, Murai K, Nukina N, Mori N (2018) N‐Acyldopamine induces aggresome formation without proteasome inhibition and enhances protein aggregation via p62/SQSTM1 expression. Sci Rep 8: 9585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mompean M, Romano V, Pantoja‐Uceda D, Stuani C, Baralle FE, Buratti E, Laurents DV (2017) Point mutations in the N‐terminal domain of transactive response DNA‐binding protein 43 kDa (TDP‐43) compromise its stability, dimerization, and functions. J Biol Chem 292: 11992–12006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, Rademakers R, Alafuzoff I, Attems J, Brayne C et al (2019) Limbic‐predominant age‐related TDP‐43 encephalopathy (LATE): consensus working group report. Brain 142: 1503–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM et al (2006) Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314: 130–133 [DOI] [PubMed] [Google Scholar]
- Neumann M, Mackenzie IR, Cairns NJ, Boyer PJ, Markesbery WR, Smith CD, Taylor JP, Kretzschmar HA, Kimonis VE, Forman MS (2007) TDP‐43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropathol Exp Neurol 66: 152–157 [DOI] [PubMed] [Google Scholar]
- Neumann M, Frick P, Paron F, Kosten J, Buratti E, Mackenzie IR (2020) Antibody against TDP‐43 phosphorylated at serine 375 suggests conformational differences of TDP‐43 aggregates among FTLD‐TDP subtypes. Acta Neuropathol 140: 645–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimoto Y, Nakagawa S, Hirose T, Okano HJ, Takao M, Shibata S, Suyama S, Kuwako K, Imai T, Murayama S et al (2013) The long non‐coding RNA nuclear‐enriched abundant transcript 1_2 induces paraspeckle formation in the motor neuron during the early phase of amyotrophic lateral sclerosis. Mol Brain 6: 31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou SH, Wu F, Harrich D, Garcia‐Martinez LF, Gaynor RB (1995) Cloning and characterization of a novel cellular protein, TDP‐43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol 69: 3584–3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SS, Belmont BJ, Sante JM, Rexach MF (2007) Natively unfolded nucleoporins gate protein diffusion across the nuclear pore complex. Cell 129: 83–96 [DOI] [PubMed] [Google Scholar]
- Perdikari TM, Murthy AC, Fawzi NL (2022) Molecular insights into the effect of alkanediols on FUS liquid‐liquid phase separation. bioRxiv 10.1101/2022.05.05.490812 [PREPRINT] [DOI]
- Pinarbasi ES, Cağatay T, Fung HYJ, Li YC, Chook YM, Thomas PJ (2018) Active nuclear import and passive nuclear export are the primary determinants of TDP‐43 localization. Sci Rep 8: 7083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M, Lagier‐Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling SC, Sun E, Wancewicz E, Mazur C et al (2011) Long pre‐mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP‐43. Nat Neurosci 14: 459–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano V, Quadri Z, Baralle FE, Buratti E (2015) The structural integrity of TDP‐43 N‐terminus is required for efficient aggregate entrapment and consequent loss of protein function. Prion 9: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rot G, Wang Z, Huppertz I, Modic M, Lenče T, Hallegger M, Haberman N, Curk T, von Mering C, Ule J (2017) High‐resolution RNA maps suggest common principles of splicing and polyadenylation regulation by TDP‐43. Cell Rep 19: 1056–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaguri H, Chew J, Xu YF, Gendron TF, Garrett A, Lee CW, Jansen‐West K, Bauer PO, Perkerson EA, Tong J et al (2016) The extreme N‐terminus of TDP‐43 mediates the cytoplasmic aggregation of TDP‐43 and associated toxicity in vivo. Brain Res 1647: 57–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotter EL, Vance C, Nishimura AL, Lee Y‐B, Chen H‐J, Urwin H, Sardone V, Mitchell JC, Rogelj B, Rubinsztein DC et al (2014) Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP‐43 species. J Cell Sci 127: 1263–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelkovnikova TA, Kukharsky MS, An H, Dimasi P, Alexeeva S, Shabir O, Heath PR, Buchman VL (2018) Protective paraspeckle hyper‐assembly downstream of TDP‐43 loss of function in amyotrophic lateral sclerosis. Mol Neurodegener 13: 30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevtsov SP, Dundr M (2011) Nucleation of nuclear bodies by RNA. Nat Cell Biol 13: 167–173 [DOI] [PubMed] [Google Scholar]
- Suzuki K, Bose P, Leong‐Quong RYY, Fujita DJ, Riabowol K (2010) REAP: a two minute cell fractionation method. BMC Res Notes 3: 294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, Konig J, Hortobagyi T, Nishimura AL, Zupunski V et al (2011) Characterizing the RNA targets and position‐dependent splicing regulation by TDP‐43. Nat Neurosci 14: 452–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuiji H, Iguchi Y, Furuya A, Kataoka A, Hatsuta H, Atsuta N, Tanaka F, Hashizume Y, Akatsu H, Murayama S et al (2013) Spliceosome integrity is defective in the motor neuron diseases ALS and SMA. EMBO Mol Med 5: 221–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji H, Arai T, Kametani F, Nonaka T, Yamashita M, Suzukake M, Hosokawa M, Yoshida M, Hatsuta H, Takao M et al (2012) Molecular analysis and biochemical classification of TDP‐43 proteinopathy. Brain 135: 3380–3391 [DOI] [PubMed] [Google Scholar]
- Türker C, Akal F, Joho D, Panse C, Barkow‐Oesterreicher S, Rehrauer H, Schlapbach R (2010) B‐Fabric: the Swiss army knife for life sciences. In Proceedings of the 13th International Conference on Extending Database Technology, pp 717–720. New York, NY: Association for Computing Machinery; [Google Scholar]
- Wagstaff KM, Sivakumaran H, Heaton SM, Harrich D, Jans DA (2012) Ivermectin is a specific inhibitor of importin α/β‐mediated nuclear import able to inhibit replication of HIV‐1 and dengue virus. Biochem J 443: 851–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IF, Chang HY, Hou SC, Liou GG, Way TD, James Shen CK (2012) The self‐interaction of native TDP‐43 C terminus inhibits its degradation and contributes to early proteinopathies. Nat Commun 3: 766 [DOI] [PubMed] [Google Scholar]
- Wang A, Conicella AE, Schmidt HB, Martin EW, Rhoads SN, Reeb AN, Nourse A, Ramirez Montero D, Ryan VH, Rohatgi R et al (2018) A single N‐terminal phosphomimic disrupts TDP‐43 polymerization, phase separation, and RNA splicing. EMBO J 37: e97452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Lim L, Dang M, Song J (2019) A novel mechanism for ATP to enhance the functional oligomerization of TDP‐43 by specific binding. Biochem Biophys Res Commun 514: 809–814 [DOI] [PubMed] [Google Scholar]
- Wang C, Duan Y, Duan G, Wang Q, Zhang K, Deng X, Qian B, Gu J, Ma Z, Zhang S et al (2020) Stress induces dynamic, cytotoxicity‐antagonizing TDP‐43 nuclear bodies via paraspeckle LncRNA NEAT1‐mediated liquid‐liquid phase separation. Mol Cell 79: 443–458 [DOI] [PubMed] [Google Scholar]
- Watanabe S, Inami H, Oiwa K, Murata Y, Sakai S, Komine O, Sobue A, Iguchi Y, Katsuno M, Yamanaka K (2020) Aggresome formation and liquid–liquid phase separation independently induce cytoplasmic aggregation of TAR DNA‐binding protein 43. Cell Death Dis 11: 909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ, Lee VM (2008) Disturbance of nuclear and cytoplasmic TAR DNA‐binding protein (TDP‐43) induces disease‐like redistribution, sequestration, and aggregate formation. J Biol Chem 283: 13302–13309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Lu S, Gasior K, Singh D, Vazquez‐Sanchez S, Tapia O, Toprani D, Beccari MS, Yates JR, Da Cruz S et al (2021) HSP70 chaperones RNA‐free TDP‐43 into anisotropic intranuclear liquid spherical shells. Science 371: eabb4309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zbinden A, Pérez‐Berlanga M, De Rossi P, Polymenidou M (2020) Phase separation and neurodegenerative diseases: a disturbance in the force. Dev Cell 55: 45–68 [DOI] [PubMed] [Google Scholar]
- Zhang YJ, Caulfield T, Xu YF, Gendron TF, Hubbard J, Stetler C, Sasaguri H, Whitelaw EC, Cai S, Lee WC et al (2013) The dual functions of the extreme N‐terminus of TDP‐43 in regulating its biological activity and inclusion formation. Hum Mol Genet 22: 3112–3122 [DOI] [PMC free article] [PubMed] [Google Scholar]