

Abstract

The electronic quenching of NO(A2Σ+) with molecular partners occurs through complex non-adiabatic dynamics that occurs on multiple coupled potential energy surfaces. Moreover, the propensity for NO(A2Σ+) electronic quenching depends heavily on the strength and nature of the intermolecular interactions between NO(A2Σ+) and the molecular partner. In this paper, we explore the electronic quenching mechanisms of three systems: NO(A2Σ+) + CH4, NO(A2Σ+) + CH3OH, and NO(A2Σ+) + CO2. Using EOM-EA-CCSD calculations, we rationalize the very low electronic quenching cross-section of NO(A2Σ+) + CH4 as well as the outcomes observed in previous NO + CH4 photodissociation studies. Our analysis of NO(A2Σ+) + CH3OH suggests that it will undergo facile electronic quenching mediated by reducing the intermolecular distance and significantly stretching the O–H bond of CH3OH. For NO(A2Σ+) + CO2, intermolecular attractions lead to a series of low-energy ON–OCO conformations in which the CO2 is significantly bent. For both the NO(A2Σ+) + CH3OH and NO(A2Σ+) + CO2 systems, we see evidence of the harpoon mechanism and low-energy conical intersections between NO(A2Σ+) + M and NO(X2Π) + M. Overall, this work provides the first detailed theoretical study on the NO(A2Σ+) + M potential energy surface of each of these systems and will inform future velocity map imaging experiments.
Introduction
Nitric oxide (NO) is an atmospherically important free radical generated in the combustion of fossil fuels and biomass. NO is often quantified using laser-induced fluorescence (LIF) on the A2Σ+ ← X2Π transition band.1−4 However, electronic quenching can interfere with the ability of LIF to provide an accurate quantification of NO. Electronic quenching occurs when a collision between NO(A2Σ+) and an atomic or molecular partner causes nonradiative relaxation back to the electronic ground state, NO(X2Π). Such pathways compete with fluorescence and cause the concentration of NO quantified through LIF to appear lower than the true concentration.5−7
Electronic quenching pathways can be separated into two general classes: reactive and nonreactive. In nonreactive electronic quenching, which is illustrated in eq 1, a collision between NO(A2Σ+) and a molecular partner induces nonradiative relaxation down to NO(X2Π). The energy released by this nonradiative NO(A2Σ+) → NO(X2Π) electronic transition is divided between the translational, rotational, and vibrational degrees of freedom of the two molecules. As a result, electronic quenching can cause a change in the vibrational and rotational states of both molecules. Equations 2a–2c illustrate different possible reactive electronic quenching pathways for NO(A2Σ+) and CO2 proposed in the literature.8,9 In all these, the collision between NO(A2Σ+) and CO2 causes a chemical reaction.
| 1 |
| 2a |
| 2b |
| 2c |
To correct inaccuracies in the quantification of NO through LIF, the electronic quenching cross-sections of NO(A2Σ+) with various molecular partners have been experimentally determined.6,7,9−11Table 1 summarizes the electronic quenching cross-section of NO(A2Σ+) with four representative molecular partners. As seen in the table, H2O has the largest electronic quenching cross section, indicating that it is the most effective of these molecules at quenching NO(A2Σ+). The electronic quenching cross section for NO(A2Σ+) + CO2 is also quite large at 68.3 Å2 at 294 K. In contrast, NO(A2Σ+) + CO has an electronic quenching cross section around 5% that of NO(A2Σ+) + H2O while NO(A2Σ+) + CH4 does not undergo appreciable electronic quenching at 296 K. At lower temperatures, NO(A2Σ+) + H2O, NO(A2Σ+) + CO2, and NO(A2Σ+) + CO have larger electronic quenching cross sections, consistent with collision complexes playing a role in facilitating the quenching. Though these experimental electronic quenching cross sections are helpful for correcting LIF measurements, they do not reveal the photochemical pathways responsible for electronic quenching.
Table 1. Experimentally Determined Electronic Quenching Cross Sections of NO(A2Σ+) with Molecular Partners CH4, CO, CO2, and H2O near Room Temperature9,12.
| molecular partner | electronic quenching cross section (Å2) | temperature (K) |
|---|---|---|
| CH4 | <0.001 | 296 |
| CO | 6.55 | 294 |
| CO2 | 68.3 | 294 |
| H2O | 121 | 294 |
While the photochemical mechanisms responsible for the electronic quenching of NO(A2Σ+) with atomic partners have been well studied, the non-adiabatic dynamics associated with the electronic quenching of NO(A2Σ+) with molecular partners remain relatively unexplored.13−17 Three recent studies of the NO(A2Σ+) + O2 system provide a notable exception to this.18−20 In the first of these, Few and co-workers used time-resolved Fourier-transform infrared emission spectroscopy to probe the pathways for nonreactive electronic quenching.18 Their analysis revealed a bimodal distribution of NO(X2Π, νNO′ = 2–22) products, suggesting the presence of two nonreactive electronic quenching channels. They attributed the formation of NO(X2Π) with high vibrational excitation to a channel with an O2(X3Σg) or O2(a1Δg) co-product. Few and co-workers speculated that the low νNO′ NO(X2Π) were generated in a second nonreactive electronic quenching channel which involves either the formation of O2(c1Σu) via a harpoon mechanism or the generation of O2(X3Σg–) through an inefficient process. Recent work by Blackshaw and co-workers used velocity map imaging (VMI) to probe, with quantum state resolution, the formation of NO(X2Π, νNO = 0) through NO(A2Σ+) + O2 electronic quenching.19 By analyzing the total kinetic energy release (TKER), they concluded that O2 receives a significant fraction of the available energy. Phase space theory simulations of the TKER distributions strongly suggested that the co-product of the nonreactive electronic quenching is O2(c1Σu–), consistent with most of the available energy inducing electronic excitation of the O2. Recent theoretical work by Soulié and Paterson, performed using the multireference methods SA-CASSCF and XMS-CASSCF, developed cuts of the NO + O2 potential energy surfaces (PESs) to rationalize the experimental observations on this system.20,21 They argued that their PESs are consistent with two nonradiative relaxation channels for NO(A2Σ+) + O2. The first proceeds through a transient ion-pair generated by electron transfer from NO(A2Σ+) to O2. This pathway exhibited a strong dependence on the intermolecular orientation and likely results in significant vibrational excitation in the products due to the large molecular geometry changes caused by the transient electron transfer. The second channel proceeds via conical intersections accessed when the O2 bond length becomes significantly elongated. Collectively, the previous studies on NO(A2Σ+) + O2 illustrate the complex chemical physics that can be associated with NO(A2Σ+) electronic quenching.
As summarized in Figure 1, our two recent computational studies provide insights into NO(A2Σ+) + H2O and NO(A2Σ+) + CO electronic quenching mechanisms.22,23 Here, we introduce the notation D2 to represent the NO(A2Σ+) + M electronic state, while D0 and D1 are associated with NO(X2Π) + M. Note that NO(X2Π) + M is doubly-degenerate at very large intermolecular distances but splits into two low-lying electronic states due to intermolecular interactions with the molecular partner. Figure 1a shows that the NO + H2O and NO + CO intermolecular interactions are attractive for the D2 state and strongly repulsive for D1. In our previous work, we demonstrated that the attractive intermolecular interactions on D2 can be rationalized using the harpoon mechanism. Specifically, electron density shifts from the NO(A2Σ+) 3sσ Rydberg orbital to the intermolecular partner’s lowest unoccupied molecular orbital. This creates a transient ion-pair that experiences attractive Coulombic intermolecular interactions. Figure 1 further shows that both NO + H2O and NO + CO possess D1–D2 conical intersections that are energetically downhill in energy from the asymptotic limit. These D2–D1 conical intersections facilitate the NO(A2Σ+) + H2O and NO(A2Σ+) + CO electronic quenching.
Figure 1.

Panel (a) shows the relative energies of the D1 (dashed lines) and D2 (solid lines) states of the NO + H2O (orange) and NO + CO (blue) systems as a function of the intermolecular distance. Panel (b) shows the relative energies of the D1 and D2 states of NO + H2O as a function of one of the OH bond lengths of water, rOHA, at a fixed intermolecular distance of RON = 1.787 Å. All energies are reported relative to an optimized geometry on D2 with an intermolecular distance of 10 Å. Adapted with permission from the American Chemical Society, ref (22), and the Royal Society of Chemistry, ref (23).
Figure 1 also highlights significant differences between NO + H2O and NO + CO, which allow for a mechanistic rationalization of the large difference between the electronic quenching cross-sections of NO(A2Σ+) + H2O and NO(A2Σ+) + CO. NO + H2O has significantly stronger attractive intermolecular interactions on D2 than NO + CO. Moreover, the D2 PES of NO + H2O funnels a wide variety of initial intermolecular orientations to the same low-energy conformation shown in the inset of Figure 1a. In contrast, the NO + CO D2 PES is much more anisotropic, with only a narrow range of initial intermolecular orientations producing attractive intermolecular interactions that lead to a D2–D1 conical intersection. Figure 1b shows that the pathway to the D2–D1 conical intersection for NO + H2O involves the significant stretching of one of the O–H bonds of H2O. Nonreactive electronic quenching will produce H2O with substantial vibrational excitation in an O–H local mode. Alternatively, a reactive electronic quenching channel is available, which will produce HONO and a H-atom, consistent with previous experimental observations made by Umemoto and co-workers.24 Only nonreactive electronic quenching is observed at low collision energies for NO(A2Σ+) + CO.
In this computational study, three different systems are considered: NO(A2Σ+) + CH4, NO(A2Σ+)+CH3OH, and NO(A2Σ+)+CO2. In choosing these systems, we sought to explore how the electronic structure of the molecular partner affects the photochemistry of NO(A2Σ+). CH4 is a larger polyatomic molecule than H2O with no low-lying π molecular orbitals (MOs). CO2 represents a polyatomic extension of our previously studied CO, with multiple heavy atoms and frontier MOs with π-symmetry. CH3OH represents a hybrid between methane, which has a very small electronic quenching cross section, and H2O, which has a very large electronic quenching cross section. Before outlining the goals of our work, we summarize previous experimental and theoretical work on the NO + CH4 and NO + CO2 systems; to the best of our knowledge, the NO + CH3OH system has not been previously studied.
Several previous studies have computationally and spectroscopically explored the NO(X2Π) + CH4 complex.25−28 Crespo-Otero et al. characterized the NO(X2Π) + CH4 PESs at the RCCSD(T)/aug-cc-pVTZ level of theory.25 This study identified multiple complexes stabilized by intermolecular interactions between NO and a C–H bond or NO and a CH3 face. The NO(X2Π) + CH4 complexes were found to be significantly impacted by the Jahn–Teller effect, with the interaction potential driving geometric distortions away from more symmetric structures. As such, Cs geometries were generally more stable than higher-symmetry C3v geometries. The lowest-energy NO(X2Π) + CH4 complex identified in this study has the NO interacting with a CH3 face and oriented nearly perpendicular to the intermolecular bond. Experimentally, the earliest work used REMPI on the A ← X band to probe the structure of NO(X2Π) + CH4. Such spectra were interpreted by Musgrave et al. to reveal a vibrational progression in the NO bending mode that originally appeared to suggest effective C3v structures for both NO(X2Π) + CH4 and NO(A2Σ+) + CH4.26 The direct observation of NO(X2Π) + CH4 rovibrational transitions was accomplished by Wen and Meyer using near IR-REMPI double resonance spectroscopy.27 Careful analysis of their spectra revealed that NO(X2Π) + CH4 is best characterized as a Jahn–Teller distorted system with the NO preferentially oriented perpendicular to the intermolecular bond and undergoing large-amplitude vibrational motions. Recently, Kidwell and co-workers used VMI to probe the product state distributions associated with the infrared photodissociation of the NO(X2Π) + CH4 complex.28 They rationalized the observed product state distributions through a careful analysis of symmetry-restrictions and a pair of Jahn–Teller NO(X2Π) + CH4 PESs (D0 and D1).
The dissociation dynamics of the NO(A2Σ+) + CH4 complex have been experimentally interrogated in a series of studies by Lawrance and co-workers.29,30 These experiments begin by electronically exciting to above the dissociation threshold of NO(A2Σ+) + CH4. The NO(A2Σ+) generated by the fragmentation of the complex was then probed using REMPI. Changing the photon energy used in the REMPI scheme allows one to selectively analyze different ro-vibrational states of NO(A2Σ+). Using VMI detection and energy conservation, Lawrance and co-workers obtained correlated product state distributions for the NO(A2Σ+) and CH4 products. The NO(A2Σ+) is produced in a broad distribution of rotational states which span the entire energetically accessible range. In contrast, the dissociation of NO(A2Σ+) + CH4 strongly favors small changes to the rotational angular momentum of CH4, with the dominant product channels having ΔJ = 0 or ΔJ = 1 for CH4. Consistent with several previous studies, Lawrance and co-workers showed that NO + CH4 is more strongly bound in the excited state than in the ground state. Specifically, the most recently measured NO(X2Π) + CH4 and NO(A2Σ+) + CH4 binding energies are 108 ± 2 and 203 ± 2 cm–1, respectively.30 Finally, note that while the NO(A2Σ+) + CH4 complex has been studied experimentally, it has not, to the best of our knowledge, been characterized computationally.
Turning to the NO(A2Σ+) + CO2 system, several experimental studies attempted to characterize the products of the reactive electronic quenching channel(s).7,8,31,32 Early work by Cohen and Heicklen used gas chromatography to identify CO as the major product of the reactive electronic quenching, which they attributed to eq 2b.31 More recent work by Azcárate et al. utilized infrared spectroscopy to determine that 26% of NO(A2Σ+) + CO2 electronic quenching produces CO and NO2.32 Settersten and co-workers used LIF to monitor the kinetics associated with the regeneration of NO(X2Π, νNO′ = 0) through NO(A2Σ+) + CO2 electronic quenching.7 They found that approximately 60% of the electronic quenching results in the formation of NO(X2Π) in its vibrational ground state. Note that this population represents a portion of that following the nonreactive electronic quenching channel (eq 1) along with all the population following the reactive quenching channel given by eq 2a; the endothermicity of eq 2a ensures that NO(X2Π) will only be formed in its ground vibrational state. Interestingly, the formation of NO(X2Π, νNO = 0) accounts for only approximately 30% of the electronic quenching population for NO + CO, NO + O2, and NO + H2O.7
The most complete experimental characterization of the NO(A2Σ+) + CO2 electronic quenching pathways was performed by Hancock and co-workers.8 Here, the reactive and nonreactive electronic quenching product distributions were measured using Fourier transform infrared emission spectroscopy. Focusing first on nonreactive electronic quenching, they found that CO2 is produced in a wide range of vibrational states, with approximately 62% of the available energy ending up in the vibrational degrees of freedom of CO2. While NO(X2Π) is also observed in a broad distribution of vibrational states, approximately 80% of the electronic quenching results in the formation of NO(X2Π) with νNO′ = 0 or νNO = 1, with the major product being NO(X2Π) in its vibrational ground state. Hancock and co-workers attribute approximately 6% of this to nonreactive quenching (eq 1) based on an extrapolation of the distribution of NO(X2Π, νNO′ ≥ 2) population determined using infrared emission. The remaining 74% is ascribed to reactive quenching via eq 2a, along with additional nonreactive electronic quenching that exceeds that predicted by their extrapolation.
Hancock and co-workers further argue that the other reactive quenching pathways (eqs 2b and 2c) are inconsistent with the observed infrared emission spectra. Specifically, they rule out the second reactive electronic quenching pathway (eq 2b) due to the absence of vibrationally hot NO2 and CO in the emission spectra. Because this reaction is highly exothermic, it should produce vibrationally excited NO2 and CO. The formation of NO2 observed in previous studies is instead ascribed to the reaction between O atoms generated via eq 2a and NO. Turning to eq 2c, the generated NCO is known to undergo reactions with NO. The infrared emission spectra associated with these reactions have previously been measured and are distinct from that measured by Hancock and co-workers.
In this study, we apply similar computational methodologies to those employed previously in the studies summarized in Figure 1. We identify the most important regions of the D2 PES for NO + CH4, NO + CH3OH, and NO + CO2 and develop a mechanistic analysis of the photochemical pathways responsible for electronic quenching in each of these systems. In doing so, we explore the viability of both nonreactive and reactive electronic quenching pathways. Throughout, we attempt to develop rationalizations for the experimentally known electronic quenching cross-sections and product state distributions. We additionally explore the extent to which the harpoon mechanism explains the long-range intermolecular attractive interactions in these systems. Finally, our work makes clear predictions about the NO(A2Σ+) + M photochemistry of all three systems which will inform future experimental studies.
Methods
As in our previous studies, we employ the equation-of-motion electron attachment coupled-cluster singles and doubles (EOM-EA-CCSD) methodology in this work.22,23 This allows us to use the closed-shell NO+ + M reference to avoid the challenges associated with using an open-shell reference. EOM-EA-CCASD will accurately describe all target doublet states of NO + M whose dominant electron configurations can be generated by adding an electron to a virtual orbital of the NO+ + M reference. This is true regardless of whether a NO + M electronic excited state has valence, Rydberg, or charge-transfer character.33−35 The EOM-EA-CCSD methodology provides dynamic electron correlation to the target states by incorporating electronic configurations that are doubly excited (and higher) from the NO+ + M reference. EOM-EA-CCSD will have much lower accuracy for target states with significant double excitation character; such states would have dominant electronic configurations consistent with exciting an electron in the NO+ + M reference and adding an electron to a virtual orbital.33,34 All states analyzed in this study were verified to consistently have dominant single excitation character; singly excited determinants accounted for over 93% of the D0, D1, and D2 states for each geometry analyzed in this study.
Throughout this study, we employed a protocol designed to effectively balance accuracy and computational cost. The basis sets used for the geometry optimizations ranged from aug-cc-pVDZ to d-aug-cc-pVTZ and are clearly identified in the text. Larger basis sets were required for the geometry optimizations when the intermolecular interactions were especially weak and while mapping out pathways near D2–D1 conical intersections. For NO–CO2, we performed the single-point calculations using the d-aug-cc-pVQZ basis set for the N and O atoms of NO and the aug-cc-pVQZ basis set for the C and O atoms of CO2; we refer to this basis set below as AVQZ. Because NO(A2Σ+) + M has significant Rydberg character, using a doubly augmented basis set is necessary to achieve accurate energetics. As described in the text, we did assess the impact of using the smaller d-aug-cc-pVTZ basis set for the single-point calculations and verified that doing so does not significantly affect the overall mechanistic picture; see Figures S24–S34 in the Supporting Information. Similar results were observed in our previous study of NO(A2Σ+) + CO.22 As a result, we used EOM-EA-CCSD/d-aug-cc-pVTZ to describe the energetics of NO(A2Σ+) + CH3OH. Löwdin spin densities and partial charges were investigated to determine the electronic properties of the D2 and D1 states. This analysis was performed at the EOM-EA-CCSD/aug-cc-pVTZ level of theory. All calculations were performed using Q-Chem 6.0 and analyzed using IQmol 3.0.1.36 For NO + CH3OH, we also analyzed natural transition orbitals using wxMacMolPlt.37,38
The weak intermolecular interactions associated with NO(A2Σ+) + CH4 necessitated a more careful treatment of basis set superposition and basis set incompleteness errors. As such, we performed a three-point extrapolation to the complete basis set (CBS) limit using the formula
| 3 |
where ECBS is the extrapolated energy in the CBS limit and α is a fitting parameter.39 Our three-point extrapolations were based on N = 2, 3, and 4 with AVNZ taken to be d-aug-cc-pVNZ for the N and O atoms and aug-cc-pVNZ for C and H atoms. Figure S1 shows an example of our three-point extrapolation to the CBS limit. For all geometries analyzed, the R2 of the linear regression was always greater than or equal to 0.999.
Results and Discussion
NO + CH4
For the NO(A2Σ+) + CH4 system, we constructed a wide variety of initial intermolecular orientations and allowed each to optimize on D2 using EOM-EA-CCSD/aug-cc-pVTZ without any constraints. Each initial conformation relaxed into one of four molecular geometries. Two of these, which are depicted in the inset of Figure 2, have the NO interacting with one of the CH3 faces of CH4, while the other two have the NO interacting with a C–H bond. All four stationary points exhibited C3v symmetry despite the geometry optimizations beginning from C1 initial geometries.
Figure 2.

Energy of the D2 state of the ON-H3CH (blue data) and NO-H3CH (red data) conformations plotted against the intermolecular distance of the two interacting atoms. The geometry optimizations were calculated using EOM-EA-CCSD/aug-cc-pVTZ and a three-point extrapolation to the CBS limit was performed for the electronic energies. All energies are reported relative to a D2-optimized geometry with an intermolecular distance of 20 Å.
In order to obtain accurate molecular geometries and interaction energies for the four stationary points, we re-optimized them using EOM-EA-CCSD with the d-aug-cc-pVTZ basis set for the N and O atoms and the aug-cc-pVTZ basis set for the C and H atoms. Harmonic vibrational frequency analysis demonstrated that the ON–H3CH and NO–H3CH complexes are true minima, while the ON–HCH3 and NO–HCH3 complexes are saddle points. For the saddle points, the vibrational modes with imaginary frequencies correspond to motions toward the ON–H3CH and NO–H3CH geometries. The minimum energy geometry of the ON–H3CH complex has an intermolecular distance of RNC = 3.17 Å and an electronic binding energy of 295.7 cm–1 in the CBS limit. This is in reasonable agreement with the NO(A2Σ+) + CH4 binding energy determined by recent velocity map imaging experiments, 203 ± 2 cm–1.30 Better agreement with experiment would require the incorporation of anharmonic zero-point energy. Turning to the other minimum energy geometry, the NO–H3CH complex has an intermolecular distance of ROC = 3.25 Å and an electronic binding energy of 82.1 cm–1.
Figure 2 shows the results of relaxed scans on D2 for the ON–H3CH and NO–H3CH complexes along the intermolecular distance, RNC or ROC. We performed a three-point extrapolation to the CBS limit at every geometry in these scans and the energies are reported relative to the energy of the D2 state when the molecules are 20 Å apart. The relative energies are negative for both potential energy curves at larger values of R, indicating attractive intermolecular interactions. Consistent with the analysis described above, Figure 2 shows that the ON–H3CH complex has a significantly deeper well than the NO–H3CH complex. Both potential energy curves become strongly repulsive at closer intermolecular distances. The molecular geometries remained in the C3v point group throughout the attractive region of the PES and only dropped to lower symmetry in the repulsive region.
We now compare the D2 PES with those of D0 and D1. Previous computational work on NO(X2Π) + CH4 revealed eight different local minima, encompassing ON–H3CH, NO–H3CH, ON–HCH3, and NO–HCH3 complexes with both C3v and Cs symmetry.25 In contrast, NO(A2Σ+) preferentially interacts with a CH3 face of CH4; we identified no minimum-energy geometries where NO(A2Σ+) interacts with a C–H bond. Both theory and experiment agree that NO(X2Π) + CH4 undergoes Jahn–Teller distortion away from C3v structures, with the lowest energy conformations having Cs symmetry with the NO oriented perpendicular to the intermolecular bond.25,27 In contrast, we find no evidence of Jahn–Teller distortions in the attractive region of the D2 PES; all geometries in the attractive region of Figure 2 belonged to the C3v point group. Repeating representative constrained geometry optimizations from molecular geometries distorted into the C1 point group resulted in the same C3v structures. Moreover, Figure S2 in the Supporting Information shows that the D2 energy increases significantly as the O–N–C angle of ON–H3CH is decreased from 180° (C3v) to 90° (Cs). Figure S3 further shows the same behavior for the NO–H3CH complex. We posit that the preference for higher-symmetry structures on D2 originates from the unpaired electron of NO residing in the more symmetric 3sσ MO instead of one of the 2pπ* MOs.
Our observation that the NO(A2Σ+) + CH4 PES supports only C3v minimum energy structures is consistent with the recent experimental work reported by Lawrance and co-workers in which velocity map imaging was used to obtain correlated product state distributions for the photodissociation of NO(A2Σ+) + CH4.30 Upon NO(X2Π) + CH4 → NO(A2Σ+) + CH4 electronic excitation, the NO will re-orient itself from being perpendicular with the intermolecular bond (Cs symmetry) to being oriented head-on with the CH3 face (C3v symmetry). As such, the vibrational relaxation on NO(A2Σ+) + CH4 will include hindered rotation of the NO resulting from a torque imposed on the NO by the D2 PES. This supports the experimental observation that the photodissociation of NO(A2Σ+) + CH4 produces NO(A2Σ+) in a broad range of final rotational states, including the highest rotational state that was energetically allowed under the experimental conditions. In contrast, the NO(A2Σ+) + CH4 photodissociation imparts little rotational energy to the CH4.
Figure 3 shows how the energies of the other electronic states vary with the intermolecular distance, RNC, for the ON–H3CH complex. The energy gap between the D2 and D1 states remain above 3.94 eV across all intermolecular distances despite the D2 potential energy curve ranging from weakly attractive to strongly repulsive. As such, there is no pathway for electronic quenching at low-collision energies for NO(A2Σ+) + CH4 in the ON–H3CH conformation. Figure S4 in the Supporting Information shows that the same behavior is observed with the NO–H3CH conformation. This is consistent with the experimental observation that the electronic quenching cross section of NO(A2Σ+) + CH4 at T = 296 K is less than 0.001 Å2.12
Figure 3.

Potential energy curves of the ON–H3CH complex showing how the relative energy depends on the intermolecular distance, RNC. The calculations were performed at the EOM-EA-CCSD/AVQZ//EOM-EA-CCSD/aug-cc-pVTZ level of theory, and all energies are reported relative to a D2-optimized geometry with an intermolecular distance of 20 Å. The AVQZ basis set uses d-aug-cc-pVQZ for the N and O atoms and aug-cc-pVQZ for the C and H atoms.
NO + CH3OH
We begin our analysis of this system by considering intermolecular orientations in which the NO(A2Σ+) is interacting with the CH3 face of CH3OH. Figure 4a demonstrates that the D2 PES supports weakly bound ON–H3COH and NO–H3COH complexes. For the ON–H3COH orientation, the strongest intermolecular attractions on D2 occur at an intermolecular distance of 3.1 Å, where the energy is −0.018 eV relative to an optimized geometry with an intermolecular distance of 20 Å. The most stable NO–H3COH geometry occurs at a significantly larger intermolecular distance, 4.6 Å, but has a comparable energy, −0.017 eV. Both minimum-energy geometries exhibit nearly C3v symmetry and have large D2–D1 energy gaps of over 4.8 eV, suggesting that these pathways do not facilitate electronic quenching. Finally, as discussed in more detail in Figure S6 in the Supporting Information, the weak nature of these intermolecular attractions necessitated using a larger basis set for the geometry optimizations consisting of d-aug-cc-pVTZ for the C, N, and O atoms and aug-cc-pVDZ for the H atoms.
Figure 4.

Panel (a) shows the energy of the D2 state as a function of the intermolecular distance when the NO is interacting with the CH3 group of CH3OH. The blue data are for ON–H3COH geometries, while the red data are for NO–H3COH geometries. Panel (b) shows the energy of the D2 state as a function of the intermolecular N–C–O angle (θNCO) at fixed intermolecular distances RNC = 3.0 Å (red data), RNC = 3.2 Å (orange data), RNC = 3.4 Å (blue data), and RNC = 3.6 Å (purple data).The geometry optimizations were performed using EOM-EA-CCSD with a d-aug-cc-pVTZ basis set for the C, N, and O atoms and an aug-cc-pVDZ basis set for the H atoms. The single-point energies were evaluated at the EOM-EA-CCSD/d-aug-cc-pVTZ level of theory and all energies are reported relative to a D2-optimized geometry with an intermolecular distance of 20 Å.
In order to ascertain the overall importance of the ON-H3COH complexes on the NO(A2Σ+) + CH3OH photochemistry, we show in Figure 4b how the D2 energy depends on the intermolecular N–C–O bond angle (θNCO) at fixed intermolecular distances, RNC, between the nitrogen atom of NO and the carbon atom of CH3OH. This varies the relative intermolecular orientation from the NO being head-on with the CH3 face (θNCO = 180°) to the NO approaching the methyl group from the side (θNCO = 90°). At all four intermolecular distances considered in Figure 4b, the energy of the D2 state is lower when the NO interacts with the methyl group from the side rather than head-on. Figure 4b shows that when RNC ≥ 3.6 Å, the D2 potential supports a nearly barrierless transition from the local minimum at θNCO = 180° to the lower-energy conformations with θNCO ≤ 90°. As RNC decreases, a barrier grows between the local minimum at θNCO = 180° and the conformations with θNCO ≤ 90°. Overall, the flatness of the D2 potential at RNC ≥ 3.6 Å and θNCO > 130° and the development of a barrier at smaller RNC suggest that collisions that begin with the NO approaching the methyl group can become trapped in the local minimum at θNCO = 180°. For those collisions that reach conformations with θNCO ≤ 90°, unconstrained geometry optimizations on D2 show that the system will ultimately reach conformations in which the nitrogen of NO is interacting with the oxygen of CH3OH. Finally, Figure S7 in the Supporting Information shows that NO–H3COH conformations with θOCO = 180° are separated from lower-energy conformations with θOCO = 90° by a very small barrier of 0.002–0.003 eV. Unconstrained geometry optimizations from conformations with θOCO = 90° lead to geometries in which the oxygen of NO is interacting with the oxygen of CH3OH.
Figure 5a shows
how the electronic energies of the D0, D1, and
D2 states varies with the intermolecular distance, RNO, for conformations in which the nitrogen
of NO interacts with the oxygen of CH3OH. At large intermolecular
distances, RNO > 3.75 Å, the intermolecular
interactions are very weakly attractive on D2. As the molecules
move closer together, the strength of the intermolecular attractions
increases significantly on D2, growing from  eV when RNO = 3.95 Å to
eV when RNO = 3.95 Å to  eV when RNO = 1.78 Å. In contrast, the D0 and D1 states
become strongly repulsive as the intermolecular distance is reduced,
with
eV when RNO = 1.78 Å. In contrast, the D0 and D1 states
become strongly repulsive as the intermolecular distance is reduced,
with 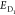 increasing by over 2.75 eV when RNO decreases from 3.95 to 1.78 Å. Figure
S8 in the Supporting Information shows
that the D3, D4, and D5 states remain
well separated from the D2 throughout this range of intermolecular
distances. Finally, note that the abrupt end to this figure reflects
our difficulty obtaining converged optimized geometries at smaller
intermolecular distances due to the presence of a D2–D1 conical intersection; we will return to this point below.
increasing by over 2.75 eV when RNO decreases from 3.95 to 1.78 Å. Figure
S8 in the Supporting Information shows
that the D3, D4, and D5 states remain
well separated from the D2 throughout this range of intermolecular
distances. Finally, note that the abrupt end to this figure reflects
our difficulty obtaining converged optimized geometries at smaller
intermolecular distances due to the presence of a D2–D1 conical intersection; we will return to this point below.
Figure 5.

Panel (a) shows the energy of the D0 (light orange), D1 (orange), and D2 (green) states as a function of the intermolecular distance, RNO, when the N of NO is interacting with O of CH3OH. The inset shows the optimized geometry with RNO = 1.78 Å. Panel (b) shows the energy of the D1 and D2 states as a function of the OH-bond length (rOH) at a fixed intermolecular distance of RNO = 1.78 Å. The geometry optimizations were performed using EOM-EA-CCSD with a d-aug-cc-pVTZ basis set for the C, N, and O atoms and an aug-cc-pVDZ basis set for the H atoms. The single-point energies were evaluated at the EOM-EA-CCSD/d-aug-cc-pVTZ level of theory and all energies are reported relative to a D2-optimized geometry with an intermolecular distance of 20 Å.
In order to understand the physical origin of the intermolecular attractions on D2, we analyze in Figure S9 in the Supporting Information the total Löwdin spin densities and partial charges on NO and CH3OH as a function of RNO. At large intermolecular distances, both molecules are neutral and the spin density is localized on the NO. As the molecules move closer together, the spin density begins to delocalize onto CH3OH, eventually becoming primarily localized onto CH3OH for RNO ≤ 2.55 Å. As this occurs, NO develops a partial positive charge and CH3OH a partial negative charge, consistent with intermolecular electron transfer. Similar behavior was observed in our previous studies of the NO(A2Σ+) + CO and NO(A2Σ+) + H2O systems and is consistent with the harpoon mechanism, wherein the formation of a transient ion-pair increases the strength of the intermolecular attractions.22,23,40−43
In Figures S10–S12, we analyze the natural transition orbitals associated with the D0 → D1 and D0 → D2 transitions at representative intermolecular distances. Figure S10 shows that at large RNO, the D1 and D2 states exhibit clear 2pπ* and 3sσ electronic character, respectively. As the molecules move closer together, the singly occupied molecular orbital (SOMO) of the D2 state becomes increasingly delocalized between the two molecules, with the region of space around the OH group of methanol especially gaining electron density. At RNO = 1.78 Å, the SOMO for D2 has its greatest amplitude around the OH group of CH3OH; see Figure S12 in the Supporting Information. This further demonstrates that the D0 → D2 transition develops significant charge-transfer character as RNO is reduced, consistent with the harpoon mechanism.
Returning to Figure 5, panel (b) shows
clear evidence for a D2–D1 conical intersection
at RNO =
1.78 Å and an extended O–H bond length of rOH ≈ 1.33 Å. On D1, stretching
the O–H bond is associated with a significant increase in energy.
In contrast, the D2 potential is nearly flat along this
coordinate, with the energy decreasing from −0.61 to −0.68
eV along the path shown in Figure 5b. Figure S13 in the Supporting Information shows a similar D2–D1 conical intersection at an alternative conformation in which the
NO lies above the methyl group of CH3OH and RNO = 1.78 Å. Figure S14 in the Supporting Information shows that at larger intermolecular
distances, increasing rOH is not as energetically
favorable on D2 and does not as readily decrease 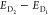 . For example, at RNO = 1.90 Å,
. For example, at RNO = 1.90 Å,  increases from −0.56 eV at rOH = 1.10 Å to −0.46 eV at rOH = 1.33 Å, while
increases from −0.56 eV at rOH = 1.10 Å to −0.46 eV at rOH = 1.33 Å, while 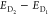 remains larger than 1.03 eV.
remains larger than 1.03 eV.
As with NO(A2Σ+) + H2O, we surmise that the pathways shown in Figure 5 support both nonreactive and reactive electronic quenching. Focusing first on nonreactive electronic quenching, Figure 5 shows an energetically downhill pathway toward a D2–D1 conical intersection that will facilitate rapid internal conversion. Because reaching the D2–D1 conical intersection requires the significant stretching of an O–H bond of CH3OH, we predict that nonreactive NO(A2Σ+) + CH3OH electronic quenching will produce CH3OH in a range of product states with vibrational excitation in the O–H stretch. The similarities between Figures 1 and 5b suggest a competing reactive electronic quenching pathway with products H + CH3ONO. Our calculations show that the reaction
| 4 |
is thermodynamically feasible. Specifically, the electronic ΔE = −52.8 kcal/mol when both products are generated in their ground electronic states and CH3ONO is in its trans-conformation.
We additionally considered potential electronic quenching pathways associated with stretching the O–C bond of methanol, rOC. From a thermodynamic perspective, the reaction
| 5 |
is feasible, with an electronic ΔE = −77.8 kcal/mol when both products are generated
in their ground electronic states and HONO is in its trans-conformation.
In Figure S15 in the Supporting Information, we evaluate how the energies of the D2 and D1 states change as rOC is increased at
a fixed intermolecular distance of RNO = 1.78 Å. These potential energy curves differ markedly from
those shown in Figure 5b. The energy of the D2 state increases significantly
as the O–C bond is stretched, ranging from −0.61 eV
when rOC = 1.44 Å to −0.30
eV when rOC = 1.68 Å. At the same
time, the D2 and D1 states remain energetically
well separated, with 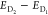 ranging from 1.53 to 1.36 eV. The absence
of a D2–D1 conical intersection in Figure S15 along with the significant energetic
cost of increasing rOC suggest that this
pathway will not play an important role in NO(A2Σ+) + CH3OH photochemistry.
ranging from 1.53 to 1.36 eV. The absence
of a D2–D1 conical intersection in Figure S15 along with the significant energetic
cost of increasing rOC suggest that this
pathway will not play an important role in NO(A2Σ+) + CH3OH photochemistry.
NO + CO2
We first considered the possibility that NO(A2Σ+) favorably interacts with the carbon atom of CO2. We analyzed both ON–CO2 and NO–CO2 complexes and varied the intermolecular distance and the angle NO makes with the CO2 (θONC for ON–CO2 or θNOC for NO–CO2). Throughout these constrained geometry optimizations, the atom of NO interacting with CO2 was constrained to be at a 90° angle from one of the C=O bonds. As shown in Figure S16 in the Supporting Information, the intermolecular interactions in these conformations become increasingly repulsive as the intermolecular distance is reduced from 4.0 to 3.0 Å. This is true for all intermolecular orientations that we considered. As a result, we did not further consider conformations in which the NO(A2Σ+) directly interacts with the carbon atom of CO2.
We next considered pathways in which the nitrogen atom of NO interacts with one of the oxygen atoms of CO2. Figure 6 shows how the energy of the D2 state varies with the intermolecular bond angle θNOC for the larger intermolecular distances, RNO, of 3.5, 3.3, 3.1, and 2.9 Å. These data were obtained by performing constrained geometry optimizations on D2 at fixed values of θNOC and RNO using the EOM-EA-CCSD/AVQZ//EOM-EA-CCSD/aug-cc-pVDZ level of theory; the AVQZ basis consists of d-aug-cc-pVQZ for the N and O atoms of NO and aug-cc-pVQZ for the C and O atoms of CO2. The conformations analyzed in Figure 6 are all nearly planar; non-planar geometries were consistently found to be higher in energy. For each of the intermolecular distances, the energy increases as θNOC decreases, suggesting an energetic preference for the nitrogen atom of NO and an oxygen atom of CO2 to approach each other nearly head-on. Additionally, the intermolecular attractions between the two molecules increase as RNO decreases, causing the two molecules to move closer together.
Figure 6.

Energy of the D2 state of ON + OCO as a function of the intermolecular angle θNOC at the intermolecular distances RNO = 3.5 Å (blue), RNO = 3.3 Å (orange), RNO = 3.1 Å (green), and RNO = 2.9 Å (yellow). These data were calculated at the EOM-EA-CCSD/AVQZ//EOM-EA-CCSD/aug-cc-pVDZ level of theory. All energies are reported relative to a D2-optimized geometry with an intermolecular distance of 20 Å.
In Figures S17–S19 in the Supporting Information, we compare the data summarized in Figure 6 to conformations in which the oxygen atom of NO is interacting with one of the oxygen atoms of CO2. The qualitative picture for these NO + OCO conformations is the same as that shown in Figure 6. However, the energies of the NO + OCO conformations are consistently less attractive than the corresponding ON + OCO conformations by as much as 0.023 eV. This indicates that while there are attractive intermolecular interactions for NO + OCO conformations, they are not as favorable as the orientations shown in Figure 6.
As shown in Figure 7, the cuts of the D2 PES along
θNOC are
dramatically different for intermolecular distances RON ≤ 2.8 Å then what is seen in Figure 6. For RNO = 2.8 Å and RNO = 2.7 Å,
there is a general increase in energy as θNOC decreases
until a barrier is reached around θNOC = 97°
and  eV for RNO =
2.8 Å and θNOC = 107° and
eV for RNO =
2.8 Å and θNOC = 107° and 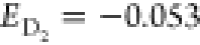 eV for RNO =
2.7 Å. At smaller θNOC after the barrier, the
energy sharply decreases as CO2 adopts a bent conformation
with a O–C–O bond angle of around 140°. In addition,
the linear to bent isomerization of CO2 is accompanied
by an elongation of both C–O bond lengths, rOC, with the largest elongation occurring at the C–O
bond that is interacting with the NO. For example, at RON = 2.7 Å, the rOC are
1.167 and 1.172 Å when θNOC = 180° and
1.218 and 1.258 Å when θNOC = 80°. The
barrier preceding the linear to bent isomerization of CO2 further decreases at RNO = 2.6 Å
and disappears for RNO ≤ 2.5 Å.
As such, the D2 PES drives ON–OCO into conformations
in which the CO2 is significantly bent. Finally, Figure 7 shows that the strength
of the attractive intermolecular interactions between the two molecules
increases as the molecules move closer together. For example, at RNO = 2.8 Å, the minimum energy is −0.12
eV, while at RNO = 2.4 Å, the minimum
energy is −0.50 eV. At the minimum energy geometry with RNO = 2.4 Å, the O–C–O angle
is 138.4° and the rOC are 1.210 and
1.268 Å.
eV for RNO =
2.7 Å. At smaller θNOC after the barrier, the
energy sharply decreases as CO2 adopts a bent conformation
with a O–C–O bond angle of around 140°. In addition,
the linear to bent isomerization of CO2 is accompanied
by an elongation of both C–O bond lengths, rOC, with the largest elongation occurring at the C–O
bond that is interacting with the NO. For example, at RON = 2.7 Å, the rOC are
1.167 and 1.172 Å when θNOC = 180° and
1.218 and 1.258 Å when θNOC = 80°. The
barrier preceding the linear to bent isomerization of CO2 further decreases at RNO = 2.6 Å
and disappears for RNO ≤ 2.5 Å.
As such, the D2 PES drives ON–OCO into conformations
in which the CO2 is significantly bent. Finally, Figure 7 shows that the strength
of the attractive intermolecular interactions between the two molecules
increases as the molecules move closer together. For example, at RNO = 2.8 Å, the minimum energy is −0.12
eV, while at RNO = 2.4 Å, the minimum
energy is −0.50 eV. At the minimum energy geometry with RNO = 2.4 Å, the O–C–O angle
is 138.4° and the rOC are 1.210 and
1.268 Å.
Figure 7.

Energy of the D2 state of ON + OCO as a function of the intermolecular angle θNOC at the intermolecular distances RNO = 2.8 Å (blue), RNO = 2.7 Å (orange), RNO = 2.6 Å (green), RNO = 2.5 Å (yellow), and RNO = 2.4 Å (light blue). These data were calculated at the EOM-EA-CCSD/AVQZ//EOM-EA-CCSD/aug-cc-pVDZ level of theory. All energies are reported relative to a D2-optimized geometry with an intermolecular distance of 20 Å. As indicated by the insets, the low-energy conformations at smaller θNOC are associated with the linear-to-bent isomerization of CO2.
Figure S20 shows the
energies of the
D1 and D2 states as a function of θNOC for the representative intermolecular distances RNO = 2.8 Å, RNO = 2.6 Å, and RNO = 2.4 Å.
The transition from linear to bent CO2, which causes a
drop in the energy of the D2 state, results in a significant
increase in the energy of the D1 state. As a result, this
isomerization causes the two states to become significantly closer
together in energy. For example, at RNO = 2.4 Å, the D2–D1 energy gap,  , is 4.80 eV when θNOC =
180° and 2.68 eV when θNOC = 160°, where
CO2 first isomerizes into a bent geometry. Collectively,
our analysis suggests that decreasing RNO and the associated linear-to-bent isomerization of CO2 play a significant role in pushing ON + CO2 toward a
D2–D1 conical intersection that can facilitate
electronic quenching.
, is 4.80 eV when θNOC =
180° and 2.68 eV when θNOC = 160°, where
CO2 first isomerizes into a bent geometry. Collectively,
our analysis suggests that decreasing RNO and the associated linear-to-bent isomerization of CO2 play a significant role in pushing ON + CO2 toward a
D2–D1 conical intersection that can facilitate
electronic quenching.
Figure S21 in the Supporting Information extends the analysis shown in Figure 7 to NO + OCO conformations. Similar to the RNO = 2.8 Å and RNO = 2.7 Å data in Figure 7, the energy of D2 initially increases as θOOC decreases, reaching a barrier that precedes a drop in energy associated with CO2 adopting a bent geometry. The energies in Figure S21 are significantly less negative than those shown in Figure 7, indicative of weaker intermolecular attractions in NO + OCO conformations than ON + OCO conformations. The intermolecular attractions in NO + OCO conformations become even weaker as the molecules move closer together. Moreover, the barrier preceding the linear-to-bent isomerization of CO2 in the NO + OCO conformations fall above the asymptotic limit. This is quite different from Figure 7, where the barrier to bent CO2 for ON + OCO conformations consistently lies below the asymptotic limit and eventually disappears at closer intermolecular distances. Figure S21 therefore provides additional support for NO + OCO conformations being significantly less important for electronic quenching than ON–OCO conformations.
In order to better understand the origin of the attractive intermolecular interactions between NO(A2Σ+) and CO2 shown in Figures 6 and 7, we analyze in Figures 8 and 9 the Löwdin partial charges and spin densities for ON + OCO conformations at RON= 2.9 and RON= 2.5 Å. These figures additionally show the singly occupied molecule orbital (SOMO) of the D2 state at representative θNOC. The SOMOs shown in Figure 8 demonstrate that at RON= 2.9 Å, an intermolecular distance where the linear-to-bent isomerization of CO2 does not occur, the electron in the 3sσ Rydberg orbital on NO becomes somewhat delocalized onto CO2 as θNOC is reduced. As a result, the Löwdin analysis reveals evidence of charge-transfer, with CO2 gaining a partial charge ranging from −0.19 to −0.37 and a spin density ranging from 0.20 to 0.38. Figure 9 shows that at RON= 2.5, a distance where CO2 does become bent for θNOC < 140°, there is a dramatic change in the distribution of electron density in the complex. Specifically, CO2 develops a partial charge of approximately −0.77 and nearly all of the spin density becomes localized on CO2, both of which clearly indicate electron transfer. At the same time, the SOMO changes from having 3sσ character on NO to 2pπ* character on the CO2. As such, the un-paired electron has transferred from a diffuse Rydberg orbital centered on NO to the LUMO of CO2.
Figure 8.

Panel (a) shows the Löwdin population analysis of the total spin densities and partial charges of NO and CO2 as a function of θNOC at RNO = 2.9 Å. The total spin density of a molecule is obtained by summing together the spin densities of all atoms belonging to that molecule. The total partial charges are obtained analogously. Panel (b) shows the SOMOs for the D2 state of ON + OCO at representative θNOC and RNO = 2.9 Å.
Figure 9.

Panel (a) shows the Löwdin population analysis of the total spin densities and partial charges of NO and CO2 as a function of θNOC at RNO = 2.5 Å. The total spin density of a molecule is obtained by summing together the spin densities of all atoms belonging to that molecule. The total partial charges are obtained analogously. Panel (b) shows the SOMOs for the D2 state of ON + OCO at representative θNOC and RNO = 2.5 Å. Note that the electronic character of the SOMO changes from 3sσ on NO at θNOC = 180° to 2pπ* on CO2 for θNOC < 140°. This, along with the increased charge and spin density on CO2 for θNOC < 140°, is consistent with electron transfer occurring between the two molecules, i.e., the harpoon mechanism.
Collectively, the analysis presented in Figure 9 allows us to rationalize several aspects of the D2 PES shown in Figure 7. Specifically, the strong intermolecular attractions on the D2 state of ON + OCO originate from the harpoon mechanism in which electron transfer from NO to CO2 creates a transient ion-pair which coulombically attract one another. Moreover, the linear-to-bent isomerization of CO2 reflects electron transfer from NO(A2Σ+) to CO2 as the CO2 radical anion is experimentally known to exist in a significantly bent geometry with an O–C–O angle of approximately 127° ± 8°.44
An overview of
the photochemical pathway responsible for NO(A2Σ+) + CO2 electronic
quenching is provided in Figure 10. Note that we employed the larger aug-cc-pVTZ basis
set for these geometry optimizations to obtain an improved description
of the region in the vicinity of the D2–D1 conical intersection. Consistent with Figure 6, the two molecules initially approach each
other head-on in a linear arrangement and experience relatively weak
intermolecular attractions ranging from −0.03 to −0.11
eV as RNO is reduced. As in Figure 7, the sudden drop in  at RNO = 2.5
Å is associated with CO2 adopting a bent conformation.
As shown by the inset in Figure 10, the lowest energy complex at this intermolecular
distance is non-planar. The D2 potential becomes significantly
more attractive after the linear-to-bent isomerization of the CO2. Figure 10 further suggests that the D2 PES is effective at funneling
population to a D2–D1 conical intersection
which occurs at approximately RNO = 1.93
Å and
at RNO = 2.5
Å is associated with CO2 adopting a bent conformation.
As shown by the inset in Figure 10, the lowest energy complex at this intermolecular
distance is non-planar. The D2 potential becomes significantly
more attractive after the linear-to-bent isomerization of the CO2. Figure 10 further suggests that the D2 PES is effective at funneling
population to a D2–D1 conical intersection
which occurs at approximately RNO = 1.93
Å and  eV. At the approximate D2–D1 conical intersection, the CO2 molecule has an
O–C–O bond angle of 133.1° and O–C bond
lengths of 1.30 and 1.19 Å, while the N–O bond length
of NO is 1.09 Å. Comparing this to the geometry at RNO = 20 Å, where the O–C–O bond angle
is 180.0°, the O–C bond lengths are 1.16 Å, and the
N–O bond length is 1.06 Å, we see that the pathway shown
in Figure 10 causes
a significant distortion to the geometry of CO2 and a smaller
change to the bond length NO.
eV. At the approximate D2–D1 conical intersection, the CO2 molecule has an
O–C–O bond angle of 133.1° and O–C bond
lengths of 1.30 and 1.19 Å, while the N–O bond length
of NO is 1.09 Å. Comparing this to the geometry at RNO = 20 Å, where the O–C–O bond angle
is 180.0°, the O–C bond lengths are 1.16 Å, and the
N–O bond length is 1.06 Å, we see that the pathway shown
in Figure 10 causes
a significant distortion to the geometry of CO2 and a smaller
change to the bond length NO.
Figure 10.

Energy of the D0 (light orange), D1 (orange), and D2 (green) states as a function of the intermolecular distance, RNO, when the N of NO is interacting with an O of CO2. The insets show representative molecular geometries along this pathway. These calculations were performed at the EOM-EA-CCSD/AVQZ//EOM-EA-CCSD/aug-cc-pVTZ level of theory and all energies are reported relative to a D2-optimized geometry with an intermolecular distance of 20 Å.
Figure S23 in the Supporting Information shows an alternative pathway to a D2–D1 conical intersection for ON + OCO in which
the O atom of NO remains
oriented away from the C atom of CO2 throughout. Beginning
at RNO = 2.6 Å, the system adopts
a planar conformation with a bent CO2. As in Figure 10, the linear-to-bent
isomerization of CO2 is associated with a significant increase
in the attractive intermolecular interactions on D2. The
D2 potential funnels the two molecules closer together
until a D2–D1 conical intersection is
reached. This D2–D1 conical intersection
occurs at a nonplanar geometry at approximately RNO = 1.925 Å and 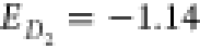 eV. At this approximate D2–D1 conical intersection, the O–C–O bond angle
is 134.3°, the O–C bond lengths are 1.30 and 1.18 Å,
and the N–O bond length is 1.09 Å.
eV. At this approximate D2–D1 conical intersection, the O–C–O bond angle
is 134.3°, the O–C bond lengths are 1.30 and 1.18 Å,
and the N–O bond length is 1.09 Å.
We now turn to the connection between our computational analysis of NO(A2Σ+) + CO2 photochemistry and existing experimental data. The large room temperature electronic quenching cross section of NO(A2Σ+) + CO2 reported in Table 1 is consistent with the presence of multiple pathways to D2–D1 conical intersections that are downhill in energy from the asymptotic limit. Moreover, our analysis suggests that the D2 PES can effectively funnel a wide range of initial intermolecular orientations to D2–D1 conical intersections, particularly those in which the N atom of NO is oriented toward an O atom of CO2. The significant distortions to the geometry of CO2 observed at our approximate D2–D1 conical intersections are consistent with the experimental observation that NO(A2Σ+) + CO2 electronic quenching releases a large fraction of the available energy, approximately 62%, into the vibrational degrees of freedom of CO2.8 In particular, the pathways shown in Figures 10 and S23 are consistent with the formation of CO2 with significant vibrational excitation in its bending and asymmetric stretching modes. Additionally, experiments show a clear preference for NO(X2Π) being produced with νNO′ = 0 or νNO = 1.7 This is consistent with the fact that the NO geometry is not nearly as distorted at the D2–D1 conical intersection as CO2; the N–O bond length is 1.09 Å at the D2–D1 conical intersection and 1.16 Å at the equilibrium geometry of NO(X2Π).
Turning to reactive quenching, we do not believe that the pathways shown in Figures 10 and S23 support the direct production of CO + O(3P) atoms via eq 2a for several reasons. First, these pathways only involve a modest increase in the O–C bond length of around 0.14 Å. In contrast, the pathways to a D2–D1 conical intersection for NO + H2O and NO + CH3OH shown in Figures 1 and 5 require an O–H bond to stretch by a significantly greater amount, 0.36–0.41 Å. Second, because the most elongated O–C bond in Figures 10 and S23 contains the O atom that is interacting with the NO, these molecular geometries appear more consistent with the formation of NO2 + CO via eq 2b than NO(X2Π) + CO + O(3P). This contrasts with the pathways shown in Figures 1 and 5 where the H atom of the stretched O–H bond is not directly interacting with the NO and hence can freely dissociate from the complex. Finally, the geometries of the D2–D1 conical intersections shown in Figures 10 and S23 more closely resemble NO + CO2 than the products of either eq 2a or 2b, consistent with nonreactive electronic quenching. Moreover, upon internal conversion from D2 to D1, the D1 PES will rapidly drive the NO(X2Π) and CO2 molecules apart.
Based on our computational analysis and the existing experimental data, we hypothesize that the experimentally observed production of CO originates from the reaction between highly vibrationally excited CO2 produced through eq 1 and NO(X2Π)
| 6 |
Using standard thermodynamic values, eq 6 becomes exothermic if greater than 42.96% of the available energy from the NO(A2Σ+) + CO2 electronic quenching is partitioned into the vibrational degrees of freedom of the CO2. Hancock and co-workers experimentally demonstrated that NO(A2Σ+) + CO2 electronic quenching produces highly vibrationally excited CO2, with approximately 62% of the available energy partitioned into the vibrational degrees of freedom of CO2. As such, NO(A2Σ+) + CO2 electronic quenching readily produces CO2 with enough internal energy to make eq 6 thermodynamically favorable. Note that eq 6 also accounts for the formation of NO2 which has also been observed experimentally.
Conclusions
Our exploration of the electronic quenching of NO(A2Σ+) with molecular partners provides new insights into the photochemistry of open-shell molecular systems. In the case of NO(A2Σ+) + CH4, the only attractive complexes are of C3v symmetry, which quickly become repulsive at smaller intermolecular distances. The absence of a low-energy D2–D1 conical intersection is consistent with the near-zero electronic quenching cross sections of NO(A2Σ+) + CH4 observed experimentally. Our work supports the experimental observation that the photodissociation of NO(A2Σ+) + CH4 produces NO(A2Σ+) in a broad range of final rotational states, while imparting little rotational energy to CH4. Specifically, upon NO(X2Π) + CH4→NO(A2Σ+) + CH4 electronic excitation, the NO re-orients itself from being perpendicular with the intermolecular bond (Cs symmetry) to being oriented head-on with the CH3 face (C3v symmetry). As a result, vibrational relaxation on the D2 PES away from the Franck–Condon region imposes a torque on the NO, consistent with NO(A2Σ+) receiving much more rotational energy than CH4 when the complex dissociates.
NO(A2Σ+) + CH3OH presents a very different story. Although there are weakly bound complexes where NO interacts with the CH3 face, the strongest attractive intermolecular interactions occur in conformations where the N atom of NO interacts with the O atom of CH3OH. We attribute the strong attractive intermolecular interactions on D2 to the harpoon mechanism, with transient electron transfer occurring from NO(A2Σ+) to CH3OH. In addition, similar to NO(A2Σ+) + H2O, our results suggest that NO(A2Σ+) + CH3OH has the potential to undergo both reactive and nonreactive electronic quenching. This is because the downhill pathway to a D2–D1 conical intersection involves both decreasing the intermolecular distance and significantly stretching the O–H bond of CH3OH. The reactive pathway will produce H and CH3ONO, while the nonreactive pathway will result in CH3OH being formed in a range of product states with a vibrational progression in the O–H stretch. Future experimental and ab initio dynamics studies on this system are warranted to test the mechanistic predictions made in this study as well as determine the relative branching between reactive and non-reactive electronic quenching.
Finally, we discuss the conclusions of our mechanistic study of the NO(A2Σ+) + CO2 system. We showed that the most energetically favorable conformations on the D2 PES have the N atom of NO interacting with an O atom of CO2. The attractive intermolecular interactions increase as the molecules grow closer together and eventually drive the isomerization of CO2 into a bent conformation. The linear-to-bent isomerization of CO2 results in significantly increased intermolecular attractions and reflects the formation of a transient NO+ + CO2– ion pair through the harpoon mechanism. We further showed that the D2 PES provides multiple, energetically downhill pathways to D2–D1 conical intersections which facilitate electronic quenching. These pathways induce significant geometric distortions to CO2, consistent with the experimental observation that NO(A2Σ+) + CO2 electronic quenching releases a large fraction of the available energy into the vibrational degrees of freedom of CO2. Finally, we hypothesize that the highly vibrationally excited CO2 produced through NO(A2Σ+) + CO2 electronic quenching will subsequently react with ground-state NO to produce the experimentally observed CO and NO2. Future velocity map imaging experiments, in conjunction with ab initio dynamics simulations, on this system are needed to shed light on the electronic quenching pathways responsible for producing NO(X2Π, νNO ≤ 1) as well as test whether the CO and NO2 products are produced directly (eqs 2a–2c) or through the subsequent reaction of vibrationally hot CO2 (eq 6).
Acknowledgments
Computational resources were provided through the Center for Computational and Applied Mathematics at California State University, Fullerton. J.A.U. acknowledges funding from Project RAISE, U.S. Department of Education HSI-STEM award number P031C160152. We acknowledge helpful conversations with Dr. Nathanael M. Kidwell.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpca.3c03981.
PESs of the NO + CH4 system along with tabulated data to construct all PES plots; PESs of the NO + CH3OH system and tabulated data to construct all PES plots: Löwdin spin and partial charge plots for NO + CH3OH at various RNO; SOMOs for NO + CH3OH over various intermolecular distances; comparison of O–H and O–C stretching pathways in the NO + CH3OH system with tabulated values; comparison of pathways for the NO + CO2 system in which the C of CO2 is interacting with the NO; comparison between the D2 states of the NO + CO2 system with either the O or N of NO approaching the O of CO2 at various intermolecular distances; PESs of both long and short-range intermolecular distances based on the θNOC or θOOC of both NO + OCO and ON + OCO complexes along with tabulated data; PESs of conical intersection pathways for the NO + CO2 system with the tabulated data; and results of assessing the basis set dependence for NO + CH4 and NO + CO2 (PDF)
The authors declare no competing financial interest.
Special Issue
Published as part of The Journal of Physical Chemistry virtual special issue “Marsha I. Lester Festschrift”.
Supplementary Material
References
- Eckbreth A. C.Laser Diagnostics for Combustion Temperature and Species; CRC Press, 1996. [Google Scholar]
- Kohse-Hoinghaus K.; Jeffries J. B.. Applied Combustion Diagnostics; CRC Press, 2002. [Google Scholar]
- Finlayson-pitts B. J.; Pitts J. N.. Chemistry of the Upper and Lower Atmosphere; Elsevier, 2000. [Google Scholar]
- Barker J. R.Progress and Problems in Atmospheric Chemistry; World Scientific, 1995; Vol. 3. [Google Scholar]
- Sánchez-González R.; Eveland W. D.; West N. A.; Mai C. L. N.; Bowersox R. D. W.; North S. W. Low-temperature collisional quenching of NO A2Σ+ (v′ = 0) by NO(X2Π) and O2 between 34 and 109 K. J. Chem. Phys. 2014, 141, 074313. 10.1063/1.4892980. [DOI] [PubMed] [Google Scholar]
- Drake M. C.; Ratcliffe J. W. High temperature quenching cross sections for nitric oxide laser-induced fluorescence measurements. J. Chem. Phys. 1993, 98, 3850–3865. 10.1063/1.465047. [DOI] [Google Scholar]
- Settersten T. B.; Patterson B. D.; Kronemayer H.; Sick V.; Schulz C.; Daily J. W. Branching ratios for quenching of nitric oxide A2Σ+ (ν′ = 0) to X2Π (ν″ = 0). Phys. Chem. Chem. Phys. 2006, 8, 5328–5338. 10.1039/b608619e. [DOI] [PubMed] [Google Scholar]
- Burgos Paci M. A.; Few J.; Gowrie S.; Hancock G. Products of the quenching of NO A2Σ+ (v = 0) by N2O and CO2. Phys. Chem. Chem. Phys. 2013, 15, 2554. 10.1039/c2cp43878j. [DOI] [PubMed] [Google Scholar]
- Settersten T. B.; Patterson B. D.; Gray J. A. Temperature- and species-dependent quenching of NO A2Σ+ (v′ =0) probed by two-photon laser-induced fluorescence using a picosecond laser. J. Chem. Phys. 2006, 124, 234308. 10.1063/1.2206783. [DOI] [PubMed] [Google Scholar]
- Paul P. H.; Gray J. A.; Durant J. L.; Thoman J. W. Collisional quenching corrections for laser-induced fluorescence measurements of NO A2Σ+. AIAA J. 1994, 32, 1670–1675. 10.2514/3.12158. [DOI] [Google Scholar]
- Settersten T. B.; Patterson B. D.; Carter C. D. Collisional Quenching of NO A2Σ+ (v’=0) Between 125 and 294 K. J. Chem. Phys. 2009, 130, 204302. 10.1063/1.3138178. [DOI] [PubMed] [Google Scholar]
- Furlanetto M. R.; Thoman J. W.; Gray J. A.; Paul P. H.; Durant J. L. Near-resonant electronic energy transfer in the electronic quenching of NO A2Σ+ by hydrocarbons and ammonia. J. Chem. Phys. 1994, 101, 10452–10457. 10.1063/1.468441. [DOI] [Google Scholar]
- Kay J. J.; Coy S. L.; Wong B. M.; Jungen C.; Field R. W. A quantum defect model for the s, p, d, and f Rydberg series of CaF. J. Chem. Phys. 2011, 134, 114313. 10.1063/1.3565967. [DOI] [PubMed] [Google Scholar]
- Sharples T. R.; Luxford T. F. M.; Townsend D.; McKendrick K. G.; Costen M. L. Rotationally Inelastic Scattering of NO(A2Σ+) + Ar: Differential Cross Sections and Rotational Angular Momentum Polarization. J. Chem. Phys. 2015, 143, 204301. 10.1063/1.4935962. [DOI] [PubMed] [Google Scholar]
- Luxford T. F. M.; Sharples T. R.; Townsend D.; McKendrick K. G.; Costen M. L. Comparative stereodynamics in molecule-atom and molecule-molecule rotational energy transfer: NO(A2Σ+) + He and D2. J. Chem. Phys. 2016, 145, 084312. 10.1063/1.4961258. [DOI] [PubMed] [Google Scholar]
- Steill J. D.; Kay J. J.; Paterson G.; Sharples T. R.; Kłos J.; Costen M. L.; Strecker K. E.; McKendrick K. G.; Alexander M. H.; Chandler D. W. Rotational Alignment of NO (A2Σ+) from Collisions with Ne. J. Phys. Chem. A 2013, 117, 8163–8174. 10.1021/jp402019s. [DOI] [PubMed] [Google Scholar]
- Luxford T. F. M.; Sharples T. R.; McKendrick K. G.; Costen M. L. Experimental Testing of ab initio Potential Energy Surfaces: Stereodynamics of NO(A2Σ+) + Ne Inelastic Scattering at Multiple Collision Energies. J. Chem. Phys. 2016, 145, 174304. 10.1063/1.4966688. [DOI] [PubMed] [Google Scholar]
- Few J.; Fletcher J. D.; Hancock G.; Redmond J. L.; Ritchie G. A. D. An FTIR Emission Study of the Products of NO A2Σ+ (v=0, 1) + O2 Collisions. Phys. Chem. Chem. Phys. 2017, 19, 11289–11298. 10.1039/c7cp00904f. [DOI] [PubMed] [Google Scholar]
- Blackshaw K. J.; Quartey N. K.; Korb R. T.; Hood D. J.; Hettwer C. D.; Kidwell N. M. Imaging the nonreactive collisional quenching dynamics of NO (A2Σ+) radicals with O2 (X3Σg–). J. Chem. Phys. 2019, 151, 104304. 10.1063/1.5109112. [DOI] [PubMed] [Google Scholar]
- Soulié C.; Paterson M. J. Molecular properties and excited state van der Waals potentials in the NO A2Σ+ + O2 X3Σg– collision complex. Phys. Chem. Chem. Phys. 2022, 24, 7983–7993. 10.1039/d1cp05286a. [DOI] [PubMed] [Google Scholar]
- Soulié C.; Paterson M. J. Multi-State Electronic Quenching: Non-Adiabatic Pathways in NO A2Σ+ + O2 X3Σg– Scattering. J. Chem. Phys. 2022, 157, 164304. 10.1063/5.0112556. [DOI] [PubMed] [Google Scholar]
- Guardado J. L.; Hood D. J.; Luong K.; Kidwell N. M.; Petit A. S. Stereodynamic Control of Collision-Induced Nonadiabatic Dynamics of NO (A2Σ+) with H2, N2, and CO: Intermolecular Interactions Drive Collision Outcomes. J. Phys. Chem. A 2021, 125, 8803–8815. 10.1021/acs.jpca.1c05653. [DOI] [PubMed] [Google Scholar]
- Guardado J. L.; Urquilla J. A.; Kidwell N. M.; Petit A. S. Reactive quenching of NO (A2Σ+) with H2O leads to HONO: a theoretical analysis of the reactive and nonreactive electronic quenching mechanisms. Phys. Chem. Chem. Phys. 2022, 24, 26717–26730. 10.1039/d2cp04214b. [DOI] [PubMed] [Google Scholar]
- Umemoto H.; Terada N.; Tanaka K.; Takayanagi T.; Kurosaki Y.; Yokoyama K. Production processes of H(D) atoms in the reactions of NO(A2Σ+) with C2H2, C2H4, H2O, and their isotopic variants. Chem. Phys. 2000, 259, 39–47. 10.1016/s0301-0104(00)00184-1. [DOI] [Google Scholar]
- Crespo-Otero R.; Suardiaz R.; Montero L. A.; García de la Vega J. M. Potential energy surfaces and Jahn-Teller effect on CH4···NO complexes. J. Chem. Phys. 2007, 127, 104305. 10.1063/1.2752805. [DOI] [PubMed] [Google Scholar]
- Musgrave A.; Bergeron D. E.; Wheatley R. J.; Wright T. G. Electronic spectroscopy of the deuterated isotopomers of the NO·methane molecular complex. J. Chem. Phys. 2005, 123, 204305. 10.1063/1.2125748. [DOI] [PubMed] [Google Scholar]
- Wen B.; Meyer H. The near IR spectrum of the NO(X2Π)–CH4 complex. J. Chem. Phys. 2009, 131, 034304. 10.1063/1.3175556. [DOI] [PubMed] [Google Scholar]
- Davis J. P.; Neisser R. W.; Kidwell N. M. Infrared Activated Signatures and Jahn–Teller Dynamics of NO–CH4 Collision Complexes. J. Phys. Chem. A 2023, 127, 5171–5182. 10.1021/acs.jpca.3c01410. [DOI] [PubMed] [Google Scholar]
- Wright D. S.; Holmes-Ross H. L.; Lawrance W. D. Dissociation of the NO–CH4 van der Waals complex: Binding energy and correlated motion of the molecular fragments. Chem. Phys. Lett. 2007, 435, 19–23. 10.1016/j.cplett.2006.12.060. [DOI] [Google Scholar]
- Holmes-Ross H. L.; Gascooke J. R.; Lawrance W. D. Correlated Product Distributions in the Photodissociation of A ~ State NO–CH4 and NO–N2 van der Waals Complexes. J. Phys. Chem. A 2022, 126, 7981–7996. 10.1021/acs.jpca.2c06312. [DOI] [PubMed] [Google Scholar]
- Cohen N.; Heicklen J. Reaction of NO(A 2Σ+) with carbon dioxide. J. Phys. Chem. 1967, 71, 558–563. 10.1021/j100862a015. [DOI] [Google Scholar]
- Codnia J.; Manzano F. A.; Azcárate M. L. Efficiency determination of the laser-induced reaction of NO(A2Σ+) with CO2. J. Photochem. Photobiol., A 2011, 223, 65–69. 10.1016/j.jphotochem.2011.08.002. [DOI] [Google Scholar]
- Krylov A. I. Equation-of-motion coupled-cluster methods for open-shell and electronically excited species: The Hitchhiker’s guide to fock space. Annu. Rev. Phys. Chem. 2008, 59, 433–462. 10.1146/annurev.physchem.59.032607.093602. [DOI] [PubMed] [Google Scholar]
- Krylov A. I.The Quantum Chemistry of Open-Shell Species, 2017; Vol. 30, pp 151–224. [Google Scholar]
- Faraji S.; Matsika S.; Krylov A. I. Calculations of non-adiabatic couplings within equation-of-motion coupled-cluster framework: Theory, implementation, and validation against multi-reference methods. J. Chem. Phys. 2018, 148, 044103. 10.1063/1.5009433. [DOI] [PubMed] [Google Scholar]
- Epifanovsky E.; Gilbert A. T. B.; Feng X.; Lee J.; Mao Y.; Mardirossian N.; Pokhilko P.; White A. F.; Coons M. P.; Dempwolff A. L.; et al. Software for the frontiers of quantum chemistry: An overview of developments in the Q-Chem 5 package. J. Chem. Phys. 2021, 155, 084801. 10.1063/5.0055522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode B. M.; Gordon M. S. MacMolPlt: A graphical user interface for GAMESS. J. Mol. Graph. Model. 1998, 16, 133–138. 10.1016/s1093-3263(99)00002-9. [DOI] [PubMed] [Google Scholar]
- Plasser F.; Wormit M.; Dreuw A. New tools for the systematic analysis and visualization of electronic excitations. I. Formalism. J. Chem. Phys. 2014, 141, 024106. 10.1063/1.4885819. [DOI] [PubMed] [Google Scholar]
- Giner E.; Scemama A.; Toulouse J.; Loos P. F. Chemically accurate excitation energies with small basis sets. J. Chem. Phys. 2019, 151, 144118. 10.1063/1.5122976. [DOI] [PubMed] [Google Scholar]
- Li F.; Dong C.; Chen J.; Liu J.; Wang F.; Xu X. The harpooning mechanism as evidenced in the oxidation reaction of the Al atom. Chem. Sci. 2018, 9, 488–494. 10.1039/c7sc03314a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winner J. D.; West N. A.; McIlvoy M. H.; Buen Z. D.; Bowersox R. D. W.; North S. W. The role of near resonance electronic energy transfer on the collisional quenching of NO (A2Σ+) by C6H6 and C6F6 at low temperature. Chem. Phys. 2018, 501, 86–92. 10.1016/j.chemphys.2017.12.001. [DOI] [Google Scholar]
- Paul P. H.; Gray J. A.; Durant J. L.; Thoman J. W. Collisional quenching corrections for laser-induced fluorescence measurements of NO A2Σ+. Am. Inst. Aeronaut. Astronaut. 1994, 32, 1670–1675. 10.2514/3.12158. [DOI] [Google Scholar]
- Paul P. H.; Gray J. A.; Durant J. L.; Thoman J. W. Collisional Electronic Quenching Rates for NO A2Σ+ (ν’=0). Chem. Phys. Lett. 1996, 259, 508–514. 10.1016/0009-2614(96)00763-4. [DOI] [Google Scholar]
- Hartman K. O.; Hisatsune I. C. Infrared Spectrum of Carbon Dioxide Anion Radical. J. Chem. Phys. 1966, 44, 1913–1918. 10.1063/1.1726961. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.