

Abstract
Stimulator of interferon genes (STING) pathway is the key innate immune pathway involving in cancer immunity. Emerging new molecules and drug delivery systems have made systemic STING agonist immunotherapy possible and demonstrated efficient tumor eradication in preclinical studies. In this perspective, we will discuss the potential mechanisms of STING agonism as a multifaceted anti-cancer therapy and the pharmacological challenges associated with systemic delivery of STING agonists on the level of organs, tissues, cells, and intracellular compartments. We will present and discuss drug delivery strategies to address these challenges. New advances in the field can unlock the promise of systemic STING agonist as effective and safe cancer immunotherapy.
Keywords: STING agonist, Cancer immunotherapy, Systemic delivery
1. Opportunities of systemic STING agonists for cancer immunotherapy
Innate immunity holds the key to the initiation and maintenance of adaptive immune responses [1]. Antitumor immune response is not an exception, which also depends on the innate immune system to fuel robust and long-lasting immune responses. Over the past decade, accumulating evidence has shown that the cyclic GAM-AMP synthase (cGAS)-stimulator of IFN genes (STING) pathway is a critical innate immune activating pathway in cancer immunity [2,3]. Briefly, tumor-derived DNA is detected by the innate immune cells via intracellular cGAS, which catalyzes the generation of cyclic GAM-AMP (cGAMP). Cytosolic cGAMP activates STING and induces type-I IFNs as well as other proinflammatory cytokines, thereby orchestrating antitumor immunity. Pharmacological activation of STING has been shown to be an effective cancer immunotherapy in various preclinical models [4].
The first generation of STING agonists has been evaluated in clinical trials, including ADU-S100 and MK-1454. Both are cyclic dinucleotide (CDN)-based compounds and are injected directly into tumors. However, these CDN compounds have a very short half-life (< 60 mins) in blood [5]. Even after intratumoral injection, they quickly diffuse away from the tumor, with a half-life of <15 min in humans [6], resulting in poor antitumor efficacy. Moreover, CDN compounds are impermeable to cell membranes, leading to low availability in the cytosol where STING is expressed. Because of these features, the first generation of STING agonists failed to generate significant clinical benefits [5,7]. In 2019, Ramanjulu and co-workers from GSK reported the STING-binding activity of amidobenzimidazole (ABZI) compounds [8]. Further optimizations have led to the development of dimer ABZI (diABZI) as a class of highly potent non-CDN STING agonists, which can eradicate tumors in mice after intravenous injection. Indeed, diABZI is the first example of an effective systemic STING agonist for cancer immunotherapy, which has galvanized the field to develop other systemic STING agonists, including MSA-2. Developed by Pan and co-workers from Merck [9], MSA-2 allows for both subcutaneous and oral administration. Interestingly, MSA-2 forms a pharmacologically active non-covalent dimer in acidic conditions and selectively induces proinflammatory immune activation in the tumor microenvironment (TME). This prodrug-like strategy could potentially increase its therapeutic efficacy while reducing the adverse effect associated with STING activation in normal tissues.
2. Cyclic dinucleotide-manganese particles for systemic STING agonist cancer immunotherapy
Nanomedicine is a promising approach that can enable and improve the systemic delivery of STING agonists for cancer immunotherapy. Different from small-molecule STING agonists, nanomedicine is a versatile platform that can integrate various functions and modulate the bioactivity of STING agonists. In our previous work published in Nature Nanotechnology [10], we reported a prototype nanomedicine for the systemic delivery of STING agonists for cancer immunotherapy. Briefly, we reported that CDNs and metal ions self-assembled into coordination particles and that certain metal ions increased the activity of STING agonists. Specifically, Mn2+ potentiated the IFN-I producing capability of various STING agonists by up to 77 folds. Based on this, we have designed a novel lipid-based particle, termed CDA-Manganese particles (CMP), for the local and systemic delivery of STING agonists. Importantly, our CMP formulation addresses multiple challenges associated with other conventional STING agonists: A) CMP protects CDNs molecules from enzymatic degradation and enables CDN-based systemic immunotherapy with increased pharmacokinetics. We have reported the remarkable therapeutic efficacy of CMP given intravenously in small doses in multiple murine tumor models. B) Intravenous administration of CMP leads to their accumulation among innate immune cells in tumors, including dendritic cells (DCs), macrophages, and myeloid-derived suppressor cells (MDSCs), leading to strong immune activation and conversion of “cold” tumor into “hot” tumor. C) CMP increases the cellular uptake of STING agonists and promotes endosomal escape, thus allowing for efficient targeting of cytosolic STING. D) Mn2+ incorporated within CMP induces STING-independent TBK1 and p65 phosphorylation, elevates STING-dependent IRF3 phosphorylation, and promotes the assembly of IFN-β transcriptional enhanceosome. This allows for robust STING activation and IFN-I response by CMP. E) CMP activates various human STING haplotypes, including those that are insensitive to CDN-mediated STING activation, thus addressing patient heterogeneity and pharmacogenomics.
Moreover, CMP provides unique opportunities for systemic STING agonist immunotherapy. First, CMP is produced by coordination-assisted assembly of lipid nanoparticles in a process that integrates state-of-the-art lipid nanoparticle technology and novel coordination nanotechnology for the simultaneous delivery of STING agonists and immune-activating metal ions. The lipid nanoparticle properties of CMP address the scale-up challenges by utilizing the standard manufacturing procedure for mRNA-LNP production, which is now widely adopted in pharmaceutical manufacturing facilities. The coordination interaction in CMP is an effective way to incorporate STING agonists into a delivery system and offers a new mechanism to formulate other pharmaceutical agents. Second, poly(histidine)11 (H11)-lipid conjugate in CMP is not only used as the coordination ligand for the formation of CMP but also serves as an ionizable component to promote endosomal escape. H11 is hydrophobic and uncharged in the physiological pH but will be protonated once it is internalized into endosomes, which is similar to the role of the ionizable lipid in mRNA-LNP vaccines. This ionizable H11-lipid is endowed with both biocompatibility and effectiveness for the systemic delivery of STING agonists. Third, it is notable that integration of Mn2+ into the delivery system greatly boosts STING activation. Given the extensive roles of metal ions in immune regulation, we believe metal ions could be an important pharmaceutical component in cancer immunotherapeutics. Surprisingly, we have also found that Co2+ exhibited a similar STING agonists potentiation effect as Mn2+. The mechanism of action for Co2+-mediated immune activation is yet to be investigated.
3. Perspectives of systemic STING agonists for cancer immunotherapy
We and others have demonstrated the feasibility and promise of systemic STING agonist cancer immunotherapy [9–11]. Complex mechanisms of action are involved for systemic immunotherapy based on STING agonists. Here, we will discuss the key questions on the impact of systemic STING agonist injection on organs, tissue, cells, and intracellular compartments. We also discuss the potential solutions to the remaining challenges and present pathways for developing effective and safe STING agonists for systemic cancer immunotherapy (Fig. 1).
Fig. 1.
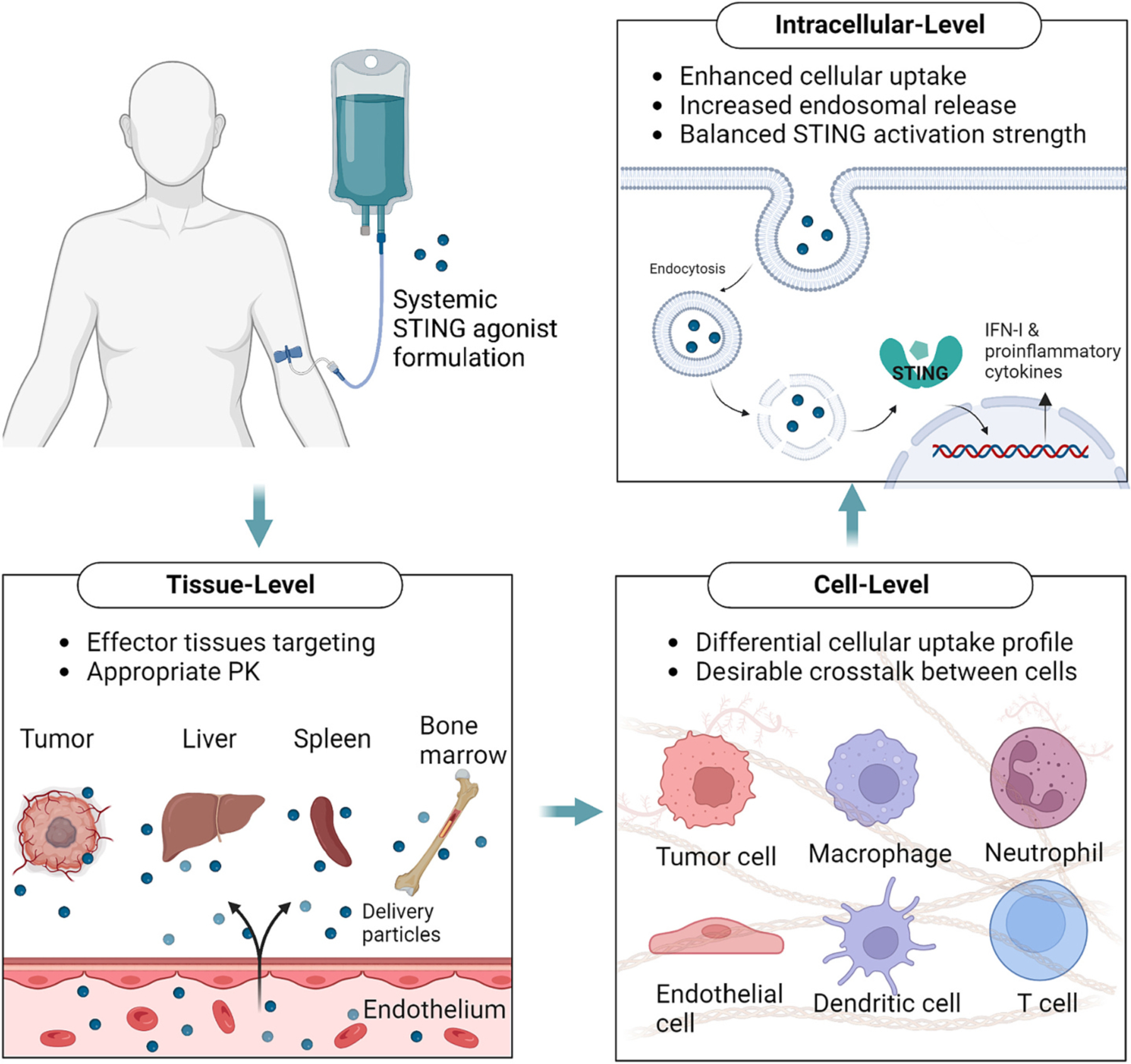
The design framework for systemic delivery of STING agonists. To develop safe and effective systemic STING agonists, we need to address various challenges at tissue-, cell-, and intracellular levels.
3.1. Tissue biasing for systemic STING agonist immunotherapy
The first pharmacological challenge for systemic STING agonist immunotherapy is to identify appropriate target tissues with anti-tumor effects and deliver STING agonists to the right tissues. After systemic administration of STING agonist-loaded nanoparticles, STING activation was observed in the tumor, plasma, as well as other major organs, including the spleen, liver, lung, and kidney [12]. Tumor-intrinsic STING activation plays an indispensable role in turning “cold” tumor “hot” and facilitating downstream T cell responses, which may be partly similar to the effect of intratumoral therapy with STING agonists. Apart from intratumoral STING activation, effective accumulation of STING agonists in lymphatic organs, including bone marrow and spleen, might be supportive in eliciting potent systemic antitumor immunity [13]. However, strong systemic immune activation in the spleen, liver, and other normal tissues may also cause acute inflammation or cytokine storm, which needs to be balanced to achieve a reasonable therapeutic window. Moreover, recent studies have shown that STING activation in endothelial cells plays a key role in supporting subsequent immune activation and T cell infiltration. Specifically, STING activation is involved in remodeling the tumor vasculature and contributes to the antitumor efficacy through type-I IFN signaling by endothelial cells [14,15]. The antitumor effect of a given STING agonist immunotherapy may be attributed to the multiple mechanisms of action discussed above and may be dependent on the animal models.
The rational design of delivery systems could potentially modulate the tissue specificity to enable safe and effective systemic STING agonist therapy. For example, by using an EGFR antibody-STING agonist conjugate, STING agonist could be specifically delivered to the tumor and elicit antitumor immunity [16]. A recent study has reported another coordination nanoparticle, Zn-CDA, that enhances tumor accumulation of STING agonists by disrupting endothelial cells in the tumor vasculature [17]. Disruption of the tumor vasculature further increases the effect of enhanced permeation and retention (EPR) in tumors, thus inducing intratumoral infiltration of immune cells and activation of tumor-associated macrophages and T cells [17]. It is noteworthy that Zn-CDA did not exhibit any significant effect on the hepatic vasculature. However, intravenous administration of most nanoparticles results in their accumulation in the liver, leading to hepato-toxicity and dysfunction as undesirable side effects. In the future, recent developments in drug delivery systems (DDS) for non-hepatic delivery could be applied for non-liver targeted systemic delivery of STING agonists [18,19]. These new DDS may minimize liver accumulation, thus reducing potential side effects while maintaining therapeutic efficacy. In addition, integrating targeting ligands into nanomedicine offers another great opportunity, which may promote the targeted delivery of STING agonists to specific tissues. Lastly, the extended pharmacokinetics and release kinetics of STING agonists delivered via nanoparticles should be considered to adjust the extent of STING activation in different organs and tissues [12,20].
3.2. Selective modulation of the target cell populations
STING agonist is a multifaceted anti-cancer agent which induces complex effects in different cell populations. STING agonists induce IFN-I and proinflammatory cytokine production in antigen-presenting cells (APCs) and exert direct and indirect killing effects in cancer cells, tumor-associated macrophage polarization, DC maturation, activation and/or indirect suppression of natural killer cells. STING agonists can also promote the activation of vascular endothelial cells [21] and disrupt the tumor vasculature [17] while modulating the cytotoxicity and stemness maintenance of T cells [22] and inducing regulatory B cells [23]. Thus, the complex underlying mechanisms of systemic STING agonist immunotherapy need to be further studied.
For cancer cells, the effect of STING agonists may depend on the intrinsic expression level of STING because many types of cancers are deficient in STING activation [24]. The potent anti-tumor effect of STING agonists, even in STING-deficient cancer, may be mediated mainly via STING activation in immune cells and stromal cells within the tumor tissues. However, some cancers were also reported to be directly killed by intrinsic STING activation, such as 4 T1 breast cancer [25], primary B-cell chronic lymphocytic leukemia cells [26], and acute myeloid leukemia [27]. Apart from the direct effect on cancer cells, antigen-specific T cell induction is key for persistent STING-activating immunotherapy [4]. It has been widely reported that STING-induced APC activation and type-I IFN response prime T cell activation for antigen-specific immune responses. However, STING hyperactivation in T cells induces antiproliferative and apoptotic responses [28]. Most recently, a study reported that IV injection of STING agonists expands IL-35+ regulatory B cells in an IRF3-dependent manner. Interestingly, the STING-IL35 axis in B cells reduces the proliferation of NKs and NK-mediated therapeutic response, and these responses can be reversed by IL-35 blockade in B cells [23]. Furthermore, endothelial cells have been demonstrated as a major source of type-I IFN after intratumoral injection of STING agonists, and activated endothelial cells are reported to facilitate the initiation of adaptive immunity [21]. As STING agonists administered intravenously have easy access to vascular endothelial cells [17], endothelial cells within the tumor tissues may play a crucial role in the production of type-I IFN for effective immunotherapy. All of these studies indicate that systemic administration of STING agonists leads to complex crosstalk between various cell populations within the TME, and more studies are needed to dissect the mechanisms of action at the single-cell level.
Novel DDS could potentially promote a preferential uptake of drugs among certain cell populations [29]. Nanodisc-mediated delivery of STING agonists allowed for the delivery of STING agonists to cancer cells and co-localization of STING agonists and tumor antigens in DCs, leading to robust T cell activation [20]. Endothelial cells-targeted DDS might facilitate STING activation in tumor endothelial cells and promote tumor vasculature normalization [30]. Among the immune cells, nanoparticle-based DDS is preferably uptaken by myeloid cells, such as DCs and macrophages. Inflammatory cytokines could be effectively elicited among these cells and further promote the activation status as well as reduce their immunosuppressive functions [31], which contribute to the subsequent T cell responses. In conclusion, rationally designed DDS could optimize the STING agonist uptake profiles by desired cell types and enhance their therapeutic efficacy.
3.3. Intracellular delivery of STING agonists for appropriate STING signaling
The desired target of systemically administered STING agonists is the STING complexes expressed on the endoplasmic reticulum of target cells. Thus, intracellular delivery of STING agonists has been the focus of STING agonist delivery because the anionic CDN STING agonist is not permeable to the cell membrane, and their transporters are inefficient. To solve this problem, various lipid-based, polymer-based, and inorganic nanoparticle-based delivery systems have been developed [29]. A wide range of toolboxes could be applied to improve endocytosis-mediated cellular uptake and endosomal escape of STING agonists for binding to STING [29].
Despite binding to the target is typically the end of the fate of a small molecule drug, nanomedicine is poised to fine-tune the STING signaling and subsequent immune response. There are distinct consequences of different STING signaling strengths. When the STING activation is moderate, it induces a decent amount of type-I IFNs and other immune stimulatory cytokines, which promote the activation of APCs, antigen presentation, antigen-specific T cell elicitation, and activation of other effecter cells, including macrophages and NK cells. However, when STING is overstimulated, it can induce apoptosis, anti-proliferation, and other forms of cell death. Some evidence gathered from animal studies shows that intratumoral injection of CDNs leads to T cell death [28]. However, there are several interpretations of this phenomenon. First, most CD8+ T cells in established tumors are terminally exhausted T cells. Emerging evidence shows that Tim3+PD-1high terminally exhausted CD8+ T cells are immunosuppressive, with a similar functional profile as Tregs [32]. Thus, the initial ablation of this population may be beneficial. Next, nanoparticles, including CMP, cannot be uptaken by T cells effectively, which prevents the potential direct pro-apoptotic effects in subsequently expanded effector T cells. Last but not least, some evidence shows that STING signaling is essential to maintain the stemness of CD8+ T cells for a durable anti-tumor response [22]. Thus, STING agonists formulated into nanoparticles may have unique advantages in reprograming the myeloid cells and potentiating durable effector T cell responses.
3.4. Understanding the sequence of anti-cancer events of STING activation
Besides the potency of STING activation, the timing of STING activation also matters. Many STING agonist therapy induced immediate tumor control and regression after treatment [4,10,17], indicating the initial therapeutic effect is adaptive immunity-independent, which generally takes multiple days to develop [1]. We speculate that the therapeutic effects of systemic STING agonists involve two phases. In Phase I, type-I IFN and other inflammatory cytokine production within the first 24 h of STING agonist administration lead to tumor vascular damage, STING activation-induced apoptosis, innate immune cell activation, and acute inflammation, resulting in tumor regression. In Phase II, activated APCs prime T cell activation and tumor-specific T cells home to tumor, resulting in T cell-mediated anti-cancer immune responses. Thus, the timing of systemic STING agonist therapy should be carefully considered when they are combined with other immunotherapies. For instance, premature dosing of immune checkpoint inhibitors during Phase I after systemic STING agonist therapy may induce higher inflammation and adverse effects. In contrast, delayed dosing of immune checkpoint inhibitors during Phase II after systemic STING agonist therapy may achieve synergistic immunotherapeutic effects. To better control the timing of STING activation, nanomedicine with spatiotemporal control function [33] could also be developed to activate STING agonists on demand.
4. Conclusions
Overall, systemic STING agonist immunotherapy is a promising cancer treatment but a lot of pharmocological challenges remain to be addressed. Further mechanistic study and delivery technology development could potentially help us better understand the pharmacology of systemic STING agonist immunotherapy and catalyze designs of rational systemic delivery strategy of STING agonist. Targeted delivery of STING agonist to the right tissues, right cell populations, and then inducing appropriate intracellular signaling strength at the right time will potentially expand the treatment window and unlock the promise of systemic STING agonists.
Acknowledgment
This work was supported by the NIH (R01AI127070, R01EB022563, R01CA210273, R01DK125087, R01 DE026728, R01DE030691, R01DE026728, and R01DE031951).
Declaration of Competing Interest
J.J.M. declares financial interests for board membership, as a paid consultant, for research funding, and/or as an equity holder in EVOQ Therapeutics, Saros Therapeutics, and Intrinsic Medicine. Y.L.L declares financial interests for board membership and/or as an equity holder in Saros Therapeutics. X.S. is an employee and shareholder in Editas Medicine.
Footnotes
Invited submission
Perspectives in Journal of Controlled Release.
Data availability
Data will be made available on request.
References
- [1].Pulendran B, Ahmed R, Translating innate immunity into immunological memory: implications for vaccine development, Cell 124 (2006) 849–863. [DOI] [PubMed] [Google Scholar]
- [2].Sun L, Wu J, Du F, Chen X, Chen ZJ, Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway, Science 339 (2013) 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Corrales L, McWhirter SM, Dubensky TW, Gajewski TF, The host STING pathway at the interface of cancer and immunity, J. Clin. Invest 126 (2016) 2404–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo S-R, Lemmens E, Banda T, Leong JJ, Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity, Cell Rep. 11 (2015) 1018–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Meric-Bernstam F, Sweis RF, Hodi FS, Messersmith WA, Andtbacka RHI, Ingham M, Lewis N, Chen X, Pelletier M, Chen X, Wu J, McWhirter SM, Muller T, Nair N, Luke JJ, Phase I Dose-Escalation Trial of MIW815 (ADU-S100), an Intratumoral STING Agonist, in Patients with Advanced/Metastatic Solid Tumors or Lymphomas, Clin. Cancer Res 28 (2022) 677–688. [DOI] [PubMed] [Google Scholar]
- [6].Flood BA, Higgs EF, Li S, Luke JJ, Gajewski TF, STING pathway agonism as a cancer therapeutic, Immunol. Rev 290 (2019) 24–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Meric-Bernstam F, Sandhu SK, Hamid O, Spreafico A, Kasper S, Dummer R, Shimizu T, Steeghs N, Lewis N, Talluto CC, Dolan S, Bean A, Brown R, Trujillo D, Nair N, Luke JJ, Phase Ib study of MIW815 (ADU-S100) in combination with spartalizumab (PDR001) in patients (pts) with advanced/metastatic solid tumors or lymphomas, J. Clin. Oncol 37 (2019) 2507. [Google Scholar]
- [8].Ramanjulu JM, Pesiridis GS, Yang J, Concha N, Singhaus R, Zhang S-Y, Tran J-L, Moore P, Lehmann S, Eberl HC, Design of amidobenzimidazole STING receptor agonists with systemic activity, Nature 564 (2018) 439. [DOI] [PubMed] [Google Scholar]
- [9].Pan BS, Perera SA, Piesvaux JA, Presland JP, Schroeder GK, Cumming JN, Trotter BW, Altman MD, Buevich AV, Cash B, Cemerski S, Chang W, Chen Y, Dandliker PJ, Feng G, Haidle A, Henderson T, Jewell J, Kariv I, Knemeyer I, Kopinja J, Lacey BM, Laskey J, Lesburg CA, Liang R, Long BJ, Lu M, Ma Y, Minnihan EC, O’Donnell G, Otte R, Price L, Rakhilina L, Sauvagnat B, Sharma S, Tyagarajan S, Woo H, Wyss DF, Xu S, Bennett DJ, Addona GH, An orally available non-nucleotide STING agonist with antitumor activity, Science 369 (2020). [DOI] [PubMed] [Google Scholar]
- [10].Sun X, Zhang Y, Li J, Park KS, Han K, Zhou X, Xu Y, Nam J, Xu J, Shi X, Wei L, Lei YL, Moon JJ, Amplifying STING activation by cyclic dinucleotide-manganese particles for local and systemic cancer metalloimmunotherapy, Nat. Nanotechnol 16 (2021) 1260–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chin EN, Yu C, Vartabedian VF, Jia Y, Kumar M, Gamo AM, Vernier W, Ali SH, Kissai M, Lazar DC, Nguyen N, Pereira LE, Benish B, Woods AK, Joseph SB, Chu A, Johnson KA, Sander PN, Martinez-Pena F, Hampton EN, Young TS, Wolan DW, Chatterjee AK, Schultz PG, Petrassi HM, Teijaro JR, Lairson LL, Antitumor activity of a systemic STING-activating non-nucleotide cGAMP mimetic, Science 369 (6506) (2020) 993–999. [DOI] [PubMed] [Google Scholar]
- [12].Wehbe M, Wang-Bishop L, Becker KW, Shae D, Baljon JJ, He X, Christov P, Boyd KL, Balko JM, Wilson JT, Nanoparticle delivery improves the pharmacokinetic properties of cyclic dinucleotide STING agonists to open a therapeutic window for intravenous administration, J. Control. Release 330 (2021) 1118–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Curran E, Chen X, Corrales L, Kline Douglas E., Dubensky Thomas W., Duttagupta P, Kortylewski M, Kline J, STING pathway activation stimulates potent immunity against acute myeloid leukemia, Cell Rep. 15 (2016) 2357–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yang H, Lee WS, Kong SJ, Kim CG, Kim JH, Chang SK, Kim S, Kim G, Chon HJ, Kim C, STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade, J. Clin. Invest 129 (2019) 4350–4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jeong S-H, Yang MJ, Choi S, Kim J, Koh GY, Refractoriness of STING therapy is relieved by AKT inhibitor through effective vascular disruption in tumour, Nat. Commun 12 (2021) 4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wu YT, Fang Y, Wei Q, Shi H, Tan H, Deng Y, Zeng Z, Qiu J, Chen C, Sun L, Chen ZJ, Tumor-targeted delivery of a STING agonist improvescancer immunotherapy, Proc. Natl. Acad. Sci. U. S. A 119 (2022), e2214278119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yang K, Han W, Jiang X, Piffko A, Bugno J, Han C, Li S, Liang H, Xu Z, Zheng W, Wang L, Wang J, Huang X, Ting JPY, Fu YX, Lin W, Weichselbaum RR, Zinc cyclic di-AMP nanoparticles target and suppress tumours via endothelial STING activation and tumour-associated macrophage reinvigoration, Nat. Nanotechnol 17 (12) (2022) 1322–1331. [DOI] [PubMed] [Google Scholar]
- [18].Cheng Q, Wei T, Farbiak L, Johnson LT, Dilliard SA, Siegwart DJ, Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–Cas gene editing, Nat. Nanotechnol 15 (2020) 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Loughrey D, Dahlman JE, Non-liver mRNA Delivery, Acc. Chem. Res 55 (2022) 13–23. [DOI] [PubMed] [Google Scholar]
- [20].Dane EL, Belessiotis-Richards A, Backlund C, Wang J, Hidaka K, Milling LE, Bhagchandani S, Melo MB, Wu S, Li N, Donahue N, Ni K, Ma L, Okaniwa M, Stevens MM, Alexander-Katz A, Irvine DJ, STING agonist delivery by tumour-penetrating PEG-lipid nanodiscs primes robust anticancer immunity, Nat. Mater 21 (2022) 710–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, Gaide O, Michielin O, Hwu P, Petrova TV, Martinon F, Modlin RL, Speiser DE, Gilliet M, STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity, Proc. Natl. Acad. Sci 112 (2015) 15408–15413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Li W, Lu L, Lu J, Wang X, Yang C, Jin J, Wu L, Hong X, Li F, Cao D, Yang Y, Wu M, Su B, Cheng J, Yang X, Di W, Deng L, cGAS-STING-mediated DNA sensing maintains CD8(+) T cell stemness and promotes antitumor T cell therapy, Sci. Transl. Med 12 (2020). [DOI] [PubMed] [Google Scholar]
- [23].Li S, Mirlekar B, Johnson BM, Brickey WJ, Wrobel JA, Yang N, Song D, Entwistle S, Tan X, Deng M, Cui Y, Li W, Vincent BG, Gale M Jr., Pylayeva-Gupta Y, Ting JP, STING-induced regulatory B cells compromise NK function in cancer immunity, Nature 610 (2022) 373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sokolowska O, Nowis D, STING signaling in Cancer cells: important or not? Arch. Immunol. Ther. Exp 66 (2018) 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chandra D, Quispe-Tintaya W, Jahangir A, Asafu-Adjei D, Ramos I, Sintim HO, Zhou J, Hayakawa Y, Karaolis DK, Gravekamp C, STING ligand c-di-GMP improves cancer vaccination against metastatic breast cancer, Cancer, Immunol. Res 2 (2014) 901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tang CH, Zundell JA, Ranatunga S, Lin C, Nefedova Y, Del Valle JR, Hu CC, Agonist-mediated activation of STING induces apoptosis in malignant B cells, Cancer Res. 76 (2016) 2137–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Song C, Liu D, Liu S, Li D, Horecny I, Zhang X, Li P, Chen L, Miller M, Chowdhury R, Issa M, Shen R, Yan Y, Zhang F, Zhang L, Zhang L, Bai C, Feng J, Zhuang L, Zhang R, Li J, Wilkinson H, Liu J, Tao W, SHR1032, a novel STING agonist, stimulates anti-tumor immunity and directly induces AML apoptosis, Sci. Rep 12 (2022) 8579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gulen MF, Koch U, Haag SM, Schuler F, Apetoh L, Villunger A, Radtke F, Ablasser A, Signalling strength determines proapoptotic functions of STING, Nat. Commun 8 (2017) 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Garland KM, Sheehy TL, Wilson JT, Chemical and biomolecular strategies for STING pathway activation in Cancer immunotherapy, Chem. Rev 122 (2022) 5977–6039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Shuvaev VV, Brenner JS, Muzykantov VR, Targeted endothelial nanomedicine for common acute pathological conditions, J. Control. Release 219 (2015) 576–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Shae D, Becker KW, Christov P, Yun DS, Lytton-Jean AKR, Sevimli S, Ascano M, Kelley M, Johnson DB, Balko JM, Wilson JT, Endosomolytic polymersomes increase the activity of cyclic dinucleotide STING agonists to enhance cancer immunotherapy, Nat. Nanotechnol 14 (2019) 269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Vignali PDA, DePeaux K, Watson MJ, Ye C, Ford BR, Lontos K, McGaa NK, Scharping NE, Menk AV, Robson SC, Poholek AC, Rivadeneira DB, Delgoffe GM, Hypoxia drives CD39-dependent suppressor function in exhausted T cells to limit antitumor immunity, Nat. Immunol 24 (2022) 267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Rahoui N, Jiang B, Taloub N, Huang YD, Spatio-temporal control strategy of drug delivery systems based nano structures, J. Control. Release 255 (2017) 176–201. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available on request.