

Abstract
An investigation of a multidimensional proteomics workflow composed of off-gel isoelectric focusing (IEF) and superficially porous liquid chromatography (SPLC) with Fourier transform mass spectrometry (FTMS) was completed in order to assess various figures of merit associated with intact protein measurements. Triplicate analysis performed at both high and low FTMS resolutions on the E. coli proteome resulted in ~900 redundant proteoforms from 3 to 95 kDa. Normalization of the chromatographic axis to identified proteoforms enabled reproducible physicochemical property measurements between proteome replicates with inter-replicate variances of ±3 ppm mass error for proteoforms <30 kDa, ±1.1 Da for proteins >30 kDa, ±12 s retention time error, and ±0.21 pI units. The results for E. coli and standard proteins revealed a correlation between pI precision and proteoform abundance with species detected in multiple IEF fractions exhibiting pI precisions less than the theoretical resolution of the off-gel system (±0.05 vs ±0.17, respectively). Evaluation of differentially modified proteoforms of standard proteins revealed that high sample loads (100s μgrams) change the IEF pH gradient profile, leading to sample broadening that facilitates resolution of charged post-translational modifications (e.g., phosphorylation, sialylation). Despite the impact of sample load on IEF resolution, results on standard proteins measured directly or after being spiked into E. coli demonstrated that the reproducibility of the workflow permitted recombination of the MS signal across IEF fractions in a manner supporting the evaluation of three label-free quantitation metrics for intact protein studies (proteoforms, proteoform ratios, and protein) over 102–103 sample amount with low femtomole detection limits.
Keywords: proteoform, label-free quantitation, chromatography, reversed-phase, isoelectric focusing, mass spectrometry, top-down
Graphical Abstract
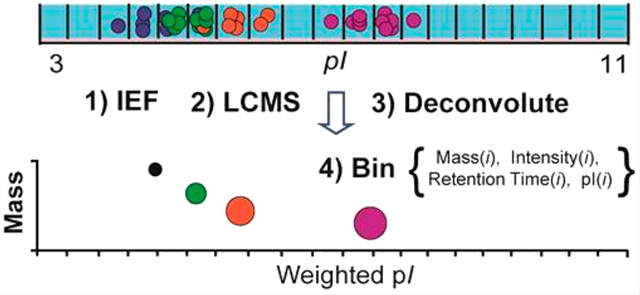
Proteins are versatile macromolecules that support an exceptional array of biological processes (e.g., transport, metabolism, signaling, interactions). Their structural and catalytic flexibility in part stems from chemical variability associated with alternative splicing of RNA, single-nucleotide polymorphisms (SNPs), and post-translational modifications (PTMs). Identifying and quantifying the myriad of functional proteoforms (protein-forms)1 for a given gene remains a modern measurement challenge, particularly in the context of complex mixtures. This is due to analytical challenges associated with processing proteins with diverse physicochemical properties as well as informatics and statistical challenges related to the prediction and identification of proteoforms harboring multiple sequence variations or unknown chemical transformations. Plus, robustness and ruggedness of multidimensional proteomics workflows are further challenged by the sophistication of chromatography and mass spectrometry (MS) methods.2,3 Continued advancement of technologies that help catalog and quantify proteoforms in complex mixtures is crucial for understanding normal physiological mechanisms and changes associated with different disease progressions (e.g., microheterogeneity of epigenetic combinatorial histone forms, glycosylation specific cancer proteoforms, myristoylation of virulence factors).4–8
Top-down mass spectrometry (TDMS) characterizes intact proteins.9 When performed by Fourier transform mass spectrometry (FTMS), which often couples high spectral resolving power and mass accuracy MS1 acquisitions with gasphase fragmentation (i.e., MS/MS), TDMS can identify the expressed gene and discriminate proteoforms of similar chemical composition. The approach contrasts mainstream bottom-up analysis which digests proteins into peptides prior to MS/MS.10 While today’s high-throughput bottom-up MS workflows largely trivialize the identification of thousands of proteins and can characterize numerous classes of target PTMs, TDMS still serves a critical need in biology and clinical investigations by resolving proteoform-microheterogeneity that is often overlooked by bottom-up MS due to the short length of peptides and selectivity of sample protocols (e.g., enrichment of peptides with specific PTMs).11,12 Over the last two decades, TDMS has been applied on simple mixtures to expose the extreme proteoform complexity for target proteins in both denatured and native states (e.g., troponin phosphorylation, Oglycosylated apolipoprotein C−III, myelin basic protein).13–15 In more recent years, with the evolution of mass analyzers, intelligent data acquisition, and biostatistics and bioinformatics resources, top-down has shown promise in proteomics investigations that identify and quantify hundreds to thousands of proteoforms in complex mixtures.16,17 Vital to the successful implementation of TD in proteomics is the advancements of orthogonal liquid chromatography (LC) techniques for intact proteins (e.g., ion exchange chromatography,18 isoelectric focusing (IEF),19 capillary electrophoresis,20 size exclusion,21 in-solution molecular weight separations (e.g., GELFrEE),22 and different LC resin or column configurations23). When combined into multidimensional workflows, these tools serve to greatly expand the observational capacity of TDMS.24–26 For example, Ntai et al. demonstrated the capability to identify and quantify many unique proteoforms from S. cerevisiae using combined GELFrEE and LCMS. Plus, unique to TDMS is it supports label-free quantitation of the expressed genes,27 individual proteoforms,28 or the ratios between related proteoforms5 with recent reports having worked to standardize differential MS for high-throughput quantitative analysis of low mass proteins (<30 kDa).28–33
Superficially porous reversed-phase LC (SPLC) and FTMS also provides high peak capacity, large quantitative dynamic range, and good detection limits for intact proteins studies.34 Zhang et al. previously showed when SPLC-FTMS was integrated after a first dimension separation by solutionbased “offgel” IEF, the workflow effectively separated complex mixtures (e.g., heart tissues and cerebrospinal fluid (CSF)) by their isoelectric point (pI) at increments of ~0.33 ΔpI/cm.19 In comparison to other multidimensional platforms that utilized other pI chromatography strategies, the offgel approach provides advantages in improved pI resolution/separation, liquid phase recovery, and low influence of offgel reagents on downstream orthogonal chromatographic separation.35 The high resolution observed across all three physicochemical property dimensions (pI, hydrophobicity, and mass) not only facilitated protein detection over a broad mass range (>200 kDa), but also improved detection of discrete proteoforms through separation by their unique pI. For example, over 200 differentially sialylated glyco-proteoforms where identified for a single di-N-glycosylated protein in CSF.36 Over the last several years, offgel IEF has been applied in diverse bottom-up proteomics investigations to improve dynamic range for protein identification.37,38 However, implementation of offgel for TD proteomics has been limited because separation of both proteins and proteoforms by pI, hydrophobicity, and mass presents unique complexities not routinely addressed in proteomics workflows that largely emphasize multidimensional LC that maximizes protein identification at the expense of proteoform coverage.39 Factors such as precision and accuracy of the IEF-SPLC-FTMS workflow need to be assessed to enable intelligent data handling that improves throughput of protein identification, as well as, permits effective and reliable access to the three levels of quantifiable information possible with TDMS. To address this need, we examined various figures of merit (FOM) of the 3D workflow. Technical replicates on Escherichia coli (E. coli) benchmarked the precision and accuracy of physicochemical properties in noncalibrated and calibrated environments. Subsequently, standard proteins were analyzed directly and in a mixed-matrix investigation in order to verify many proteome-level results and to help benchmark the quantitative reliability of the IEF-SPLC-FTMS workflow by determination of the quantitative dynamic range and limits of detection (LOD) for the protein, the protein’s individual proteoforms, and the relative ratio between the proteoforms.
MATERIALS AND METHODS
Cell Cultures and Sample Preparation.
A 5 mL Escherichia coli (E. coli) strain NM522 cells starter culture was used to inoculate 1 L of Luria–Bertani (LB) medium incubated at 250 rpm at 37 °C to an OD600 = 1.2. Cells were harvested by centrifugation (3000 rpm, 4 °C, 20 min) and stored at −80 °C. For protein extraction, cells were twice washed in ice chilled 0.1× PBS, followed by centrifugation (3000 rpm, 4 °C, 5 min), and then suspended and lysed in 0.1× PBS (4 °C) by pulse sonication with 10/10 s on/off cycles for 1 min with cell debris removed by centrifugation (4 °C, 16000 rpm, 10 min). The supernatant was concentrated through a 3 kDa MWCO filter (Millipore, Billerica, MA), and proteins were precipitated overnight with acetone at −20 °C. Protein quantification was performed by bicinchoninic acid assay (Pierce, Rockford, IL).
Isoelectric Focusing.
All IEF studies were conducted on a 3100 off-gel fractionator (Agilent, Santa Clara, CA) using pH 3–11 NL, 240 × 3 × 0.5 mm, nonlinear gradient immobilized pH gradient (IPG) strips and buffers from GE Healthcare (Piscataway, NJ). IEF power timetables were from the user manual. For proteome scale analysis studies, 1 mg of E. coli lysate was utilized. For loading studies on target proteins, the standard proteins RNase A, RNase B, α-lactalbumin, bovine serum albumin (BSA), and transferrin (Sigma, St. Louis, MO) were suspended in 750 mM urea, 300 mM thiourea, 200 mM DTT, 1% ampholytes, and 3% glycerol and examined in triplicate at each of four different loading amounts (0.3, 3, 30, and 300 μg). To assist in the electroosmotic flow/mobility for RNase A, RNase B, and α-lactalbumin, an additional 50 μg of BSA was added to each run, while for BSA and transferrin runs, an additional 50 μg of bovine ubiquitin was added. For both spiked protein and E. coli proteome analysis, an IEF buffer condition of 2 M urea, 600 mM thiourea, 200 mM DTT, 0.75% ampholytes, and 2% glycerol were used. For spiked protein analysis in the presence of E. coli, the same proteins and loading amounts were used in the targeted IEF (minus electroosmotic add-ins), but also in the presence of 1 mg of E. coli lysate. All IEF runs were subsequently processed with gel electrophoresis and visualized via silver stain with the ImageLab software (BioRad, Hercules, CA). IEF fractions were reduced with 20 mM DTT at 35 °C for 30 min prior to SPLC-FTMS analysis.
SPLC-FTMS and SPLC-NSD-FTMS.
Optima grade solvents and acids were from Thermo Fisher (Waltham, MA, U.S.A.). Samples were injected via an autosampler (LC Packings), with SPLC performed on an Agilent 1100 Series HPLC with a 0.5 × 75 mm, Poroshell 300SB-C8 column with 300 Å pore size, 5 μm diameter particles (Agilent), and heated to 70 °C at a flow rate of 150 μL/min. Flow split was conducted via a Triversa Nanomate nanoelectrospray robot (Advion Biosystems, Ithaca, NY) with approximately 0.2% of the SPLC eluate directed into a LTQ Orbitrap XL (Thermo Fisher, Waltham, MA). For all studies, solvent conditions of 94.7/5/0.3/0.025% water/ACN/FA/TFA (v/v/v/v) for phase A and 80/20 ACN/IPA with 0.3% FA and 0.025% TFA (v/v/v/v) for phase B were used, and the injected sample was washed to remove ampholytes at 0% B for 5 min prior to SPLC separation. For targeted and spiked standard protein studies, 20 μL of the sample was analyzed with a 9 min linear gradient ranging from 0 to 70% B. For E. coli proteome studies, 25 μL of the sample was analyzed over 50 min with a gradient ranging from 0 to 45% B. Technical replicates were obtained using two distinct LCMS methods utilizing either high or low mass spectral resolving power in order to optimize isotopologue resolution for low mass proteins (<30 kDa) versus sensitivity of larger proteins (>30 kDa), respectively. An automatic gain control (AGC) of 2e5 was used for all runs. For proteins ≤30 kDa, instrument conditions were as follows: a 60k resolving power at m/z 400, positive ion mode, with data acquired from 900–2000 m/z. For proteins >30 kDa, instrument conditions were as follows: 15k resolving power at m/z 400, positive ion mode, with data acquired via selected ion monitoring over a 1200–1800 m/z range.40 Runs for targeted and spiked protein analysis were collected at 1 μscan, while those for the E. coli proteome analysis were collected at 3 μscan. Nozzle skimmer dissociation (NSD) was completed at 30k resolving power at m/z 400, with data acquired from 800 to 2000 m/z.
Data Processing.
Mass Deconvolution.
For high resolving power data sets the modified THRASH algorithm was used for monoisotopic mass determination over time within .raw files.41,42 Peak intensities were integrated by a sliding window algorithm applied across the LCMS elution period. The AutoRespect algorithm in Protein Deconvolution 4.0 (PD4; Thermo Fisher, Pittsburgh, PA) was used for average mass determination over time within low resolving power .raw files. Typical deconvolution settings used were as follows: signal-tonoise (S/N), 1.0; minimum number of detected charges for intact protein, 2; minimum number of detected charges for fragments, 1; isotopologue fit factor, 80%; isotopologue remainder threshold, 80%; monoisotopic mass merge tolerance, 15 ppm; and target average time window, 1.0 or 0.5 min. PD4 outputs were converted into observed mass, retention time, intensity, and estimated grand average hydrophobicity index (GRAVY) values; the was later determined by an internal calibration curve created from known proteins within the sample. Monoisotopic masses are reported for proteoforms ≤30 kDa and proteoforms >30 kDa are reported as average mass. High mass species were considered real if 1/2 of the theoretical number of charge states possible in the spectrum m/z range were detected. Intensity, pI, and SPLC retention time information that derived from the IEF-SPLC-FTMS workflow were compiled for each mass using in-house developed software (Figure 1). Further details on the multidimensional binning procedures are provided in the Supporting Information.
Figure 1.
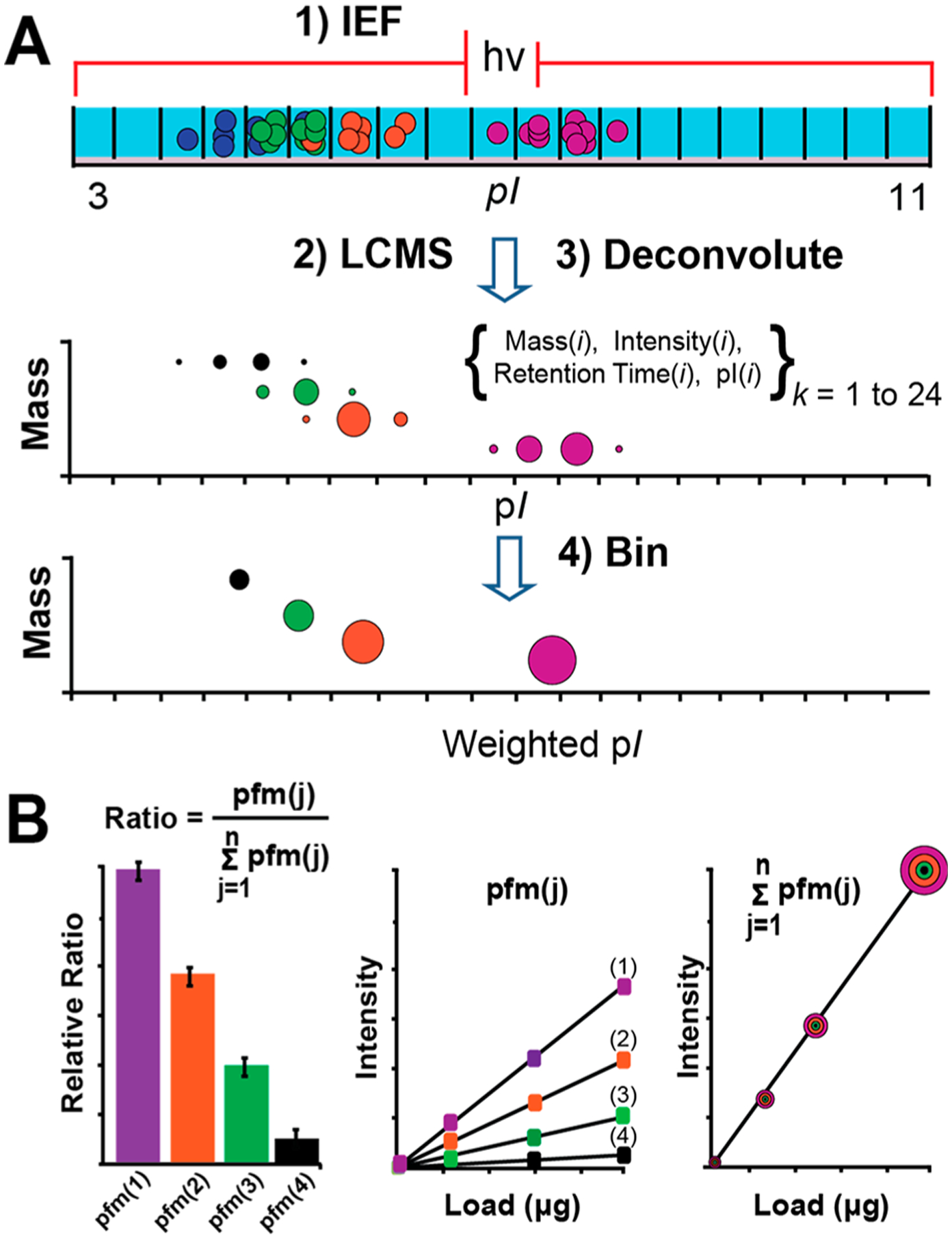
(A) The IEF-SPLC-FTMS workflow. Mass, intensity, RT, and pI information were tabulated for discrete IEF fractions followed by binning of redundant observations and calculation of wpI. (B) Illustration of the three quantitative metrics evaluated (i.e., ratios of proteoforms, individual proteoforms, and proteins).
RESULTS
Benchmark IEF-SPLC-FTMS Figures of Merit for a Complex Mixture.
Proteome Precision.
Initial investigations performed technical replicates on the E. coli proteome in order to benchmark various FOM for the multidimensional workflow. We first tabulated the number of redundant observations between replicates and then determined the precision of the observed mass, retention time (RT), peak intensities, and weighted pI (wpI). On average, 948 and 189 proteoforms were observed in a single replicate performed at high resolving power (<30 kDa) and low resolving power (>30 kDa), respectively, with the majority of proteoforms observed between 5–10 kDa (Tables S1 and S2 and Figure S1). From these, 724 and 163 proteoforms were observed in all replicates (Figure 2). For the redundant observations, ~95% were detected with a mass precision of ±3 ppm for the proteoforms <30 kDa and ±1.1 Da for those >30 kDa (Figure 3A,B), which confirms the expected precision of a mass analyzer employing automatic gain control.43 Inspection of peak intensities of the remaining ~250 nonredundant proteoforms revealed an average relative summed intensity of ≤1.5% or ~18.5-fold lower average intensity compared to that of the redundant proteoforms (Figure 3C). We also noted many of the nonredundant ≤30 kDa species exhibited an 1 Da mass shift relative to proteoforms in other replicates (Figure S2), which was attributed to the miss assignment of 12C100%13C0% isotopes that resulted from baseline interference of the isotopologue fitting.41
Figure 2.
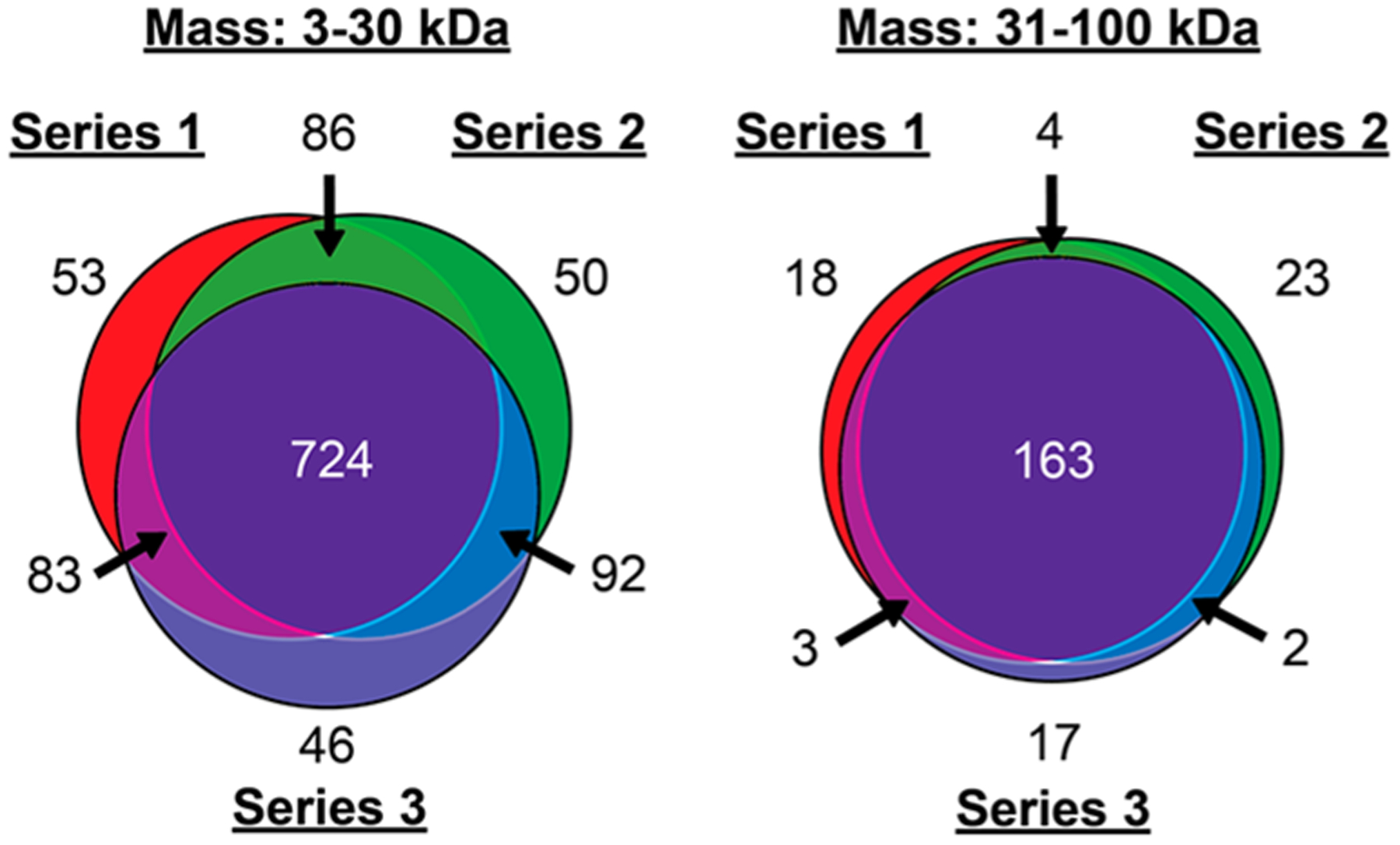
Number of proteoforms observed in triplicate runs on E. coli. The runs were performed at both high and low MS resolving power, corresponding to the 3–30 kDa and >30 kDa ranges, respectively.
Figure 3.
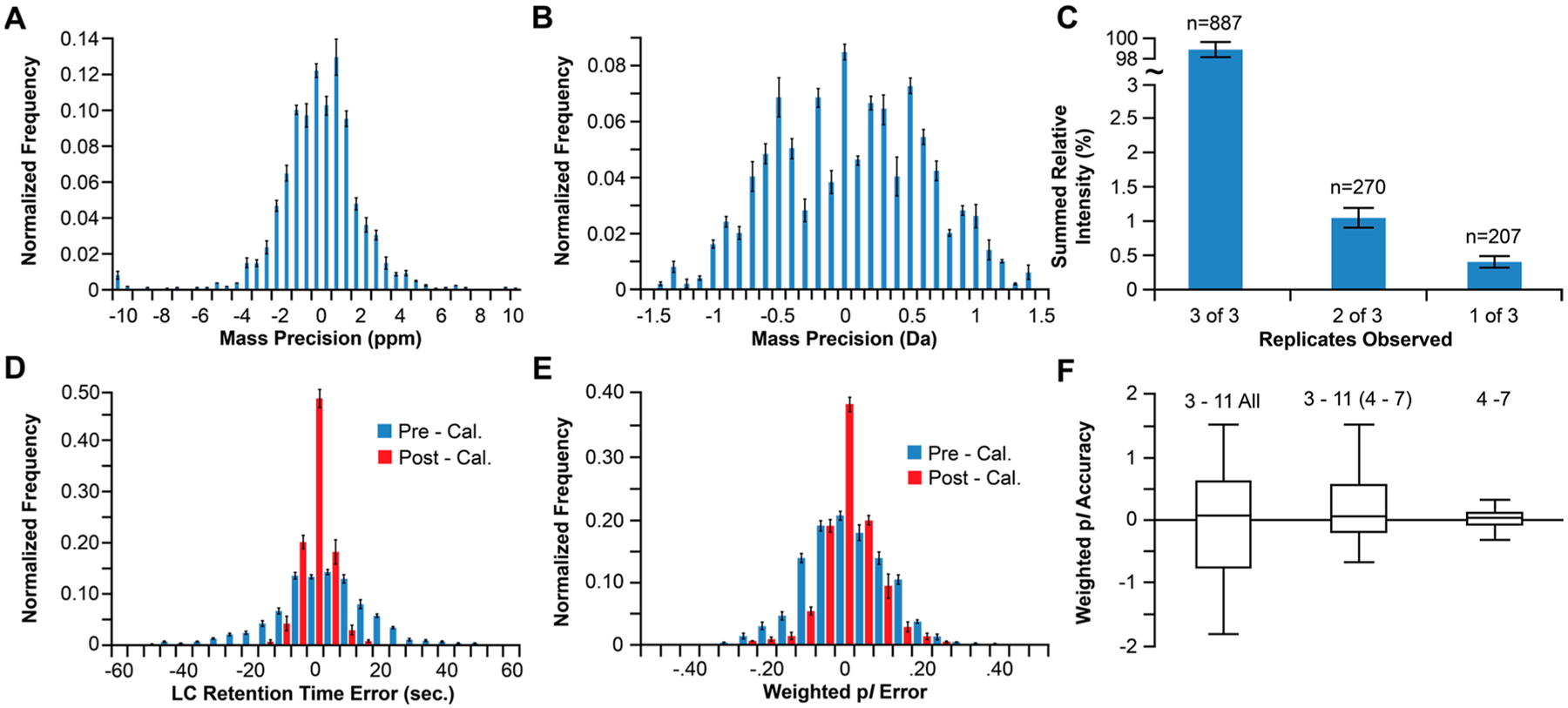
Mass precision histograms for the redundant proteoforms observed in all replicates of the high (A) and low (B) resolving power analysis. (C) The number of proteoforms (n) observed in 1, 2, or all 3 replicates and their relationship to summed relative intensity. (D) LC retention time precision histogram pre- and postaxis normalization. (E) Weighted pI precision histogram pre- and postaxis normalization. (F) Box plot of the average calculated wpI accuracy (i.e., observed wpI − theoretical pI) values.
Next, we compared the observed RT, intensity, and wpI for the ~900 redundant proteoforms in each E. coli replicate both before and after normalization of the two chromatographic axes with proteins identified by NSD. Linear regression analysis of the inter replicate comparison plots showed good agreement between the replicates with average correlation of determination (R2) of 0.99, RT; 0.97, intensity; and 0.99, wpI (Figure S3). Before axis normalization ~95% of observed proteoforms exhibited an inter-replicate RT precision from the mean of ±30 s. This reduced to ±12 s after normalization of the RT axis (Figure 3D), which is consistent with RT reproducibility previously shown for 1D SPLC-FTMS.34 The observed wpI precision from the inter-replicate mean was ±0.33 prior to axis normalization, which reduced to ±0.21 after (Figure 3E). However, further evaluation revealed that the precision estimate was generally independent of species mass but dependent on intensity (Figure S4). For example, proteoforms with relative intensity 10–90% and >90% exhibited precisions of ±0.15 and ±0.05, respectively.
Accuracy of wpI.
For the E. coli proteins identified within the workflow by NSD (Table S3) we performed an assessment of the accuracy of the observed wpIs by comparison to their theoretical values. The assessment was performed for proteins identified across (1) the entire pI 3–11 nonlinear (NL) IPG, (2) only the identified proteins observed in the linear region (pI 4–7) of the pI 3–11 NL data set, and (3) for a separate proteomic analysis of E. coli performed with a pI 4–7, 24 cm, linear IPG. For the pI 3–11 NL IPG, the observed precision was ±0.449 (Figure 3F), although proteins identified near the anode and cathode often deviated from theoretical values over a broad range (−1.78−1.50 ΔpI). For proteins in the pI 4–7 portion of the pI 3–11 NL IPG, the precision reduced to ±0.29 with a range of −0.68−1.489. In the separate analysis on the 24 cm, pI 4–7, linear IPG, the observed precision was ±0.156, with a range of −0.327−0.311. The result for the pI 4–7 linear IPG was consistent with that shown for various reference proteins in human cell lines characterized by 2DGE (±0.13).44 While the improvement over the NL gradient was largely attributed to the increased resolution of the linear IPG, the accuracy of the NL IPG may be improved by accounting for nonlinear IPG when estimating theoretical pI and other factors, such as assessing protein drift near the anode and cathode due to gradient instability that occurs when proteins are focused over a broad pH range over long periods.45 Overall, the results suggest that with increasing pI resolution, the difference between observed and theoretical proteoform wpI values may offer an additional metric for proteoform identification.46
wpI Precision versus Sample Load.
Effect of Sample Load on wpI Precision.
To verify the wpI precision estimates from E. coli proteome, we next examined how the quantity of loaded standard proteins affected the wpI of their individual proteoforms containing various modification classes (Figure 4A and Table S4). The proteins were subjected to IEF from pI 3–11 with three technical replicates performed for four different sample loads (0.3, 3.0, 30, and 300 μg). Silver stain gel electrophoresis on the focused proteins showed that sample loads ≥3 μg broadened between IEF fractions with a maximum of 5–6 fractions observed at the highest levels (Figure 4B).47 The collected fractions were subsequently analyzed by LCMS with mass and intensity data tabulated (Figure S5 and Table S5) and wpI determined for the proteins both independent and relative to the amount loaded (Figure 4C and E, respectively). For each protein the average of the wpIs determined for all replicates at each sample load was 8.70 ± 0.08, 5.59 ± 0.07, 5.41 ± 0.08, and 6.23 ± 0.07 for RNase B, α-Lac, BSA, and transferrin, respectively. Assessment of the wpI versus the quantity loaded showed for BSA and transferrin there was shift in wpI toward the anode with increased quantity (Figure 4E). A similar trend was observed for all but the highest amount for α-Lac. However, RNase B, the most basic protein, the wpI shifted to the cathode for all but the highest load.
Figure 4.
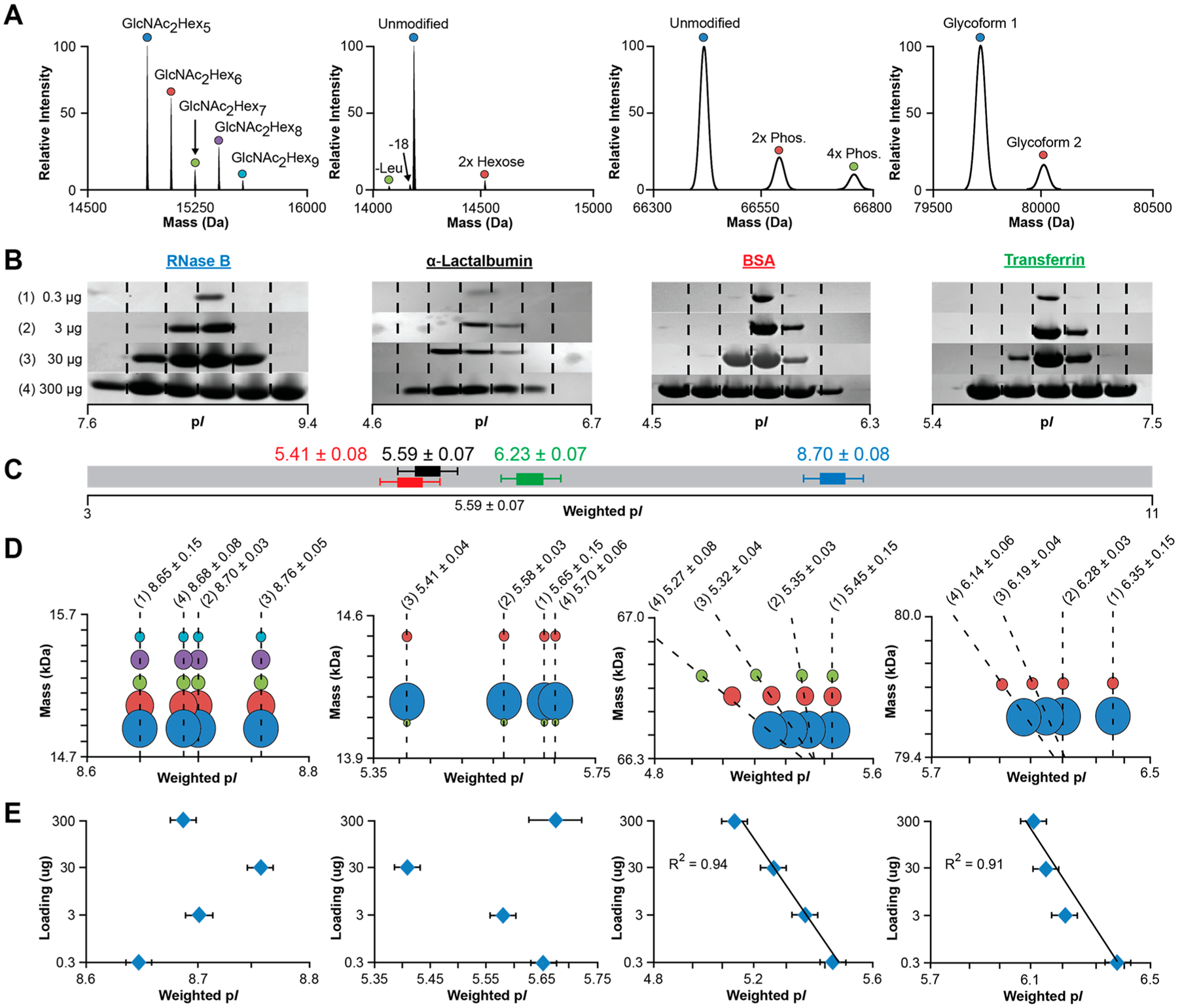
(A) Representative deconvoluted spectra for standard proteins analyzed via 1D SPLC-FTMS. (B) Representative silver stain SDS/PAGE of IEF fractions for RNase B, α-lactalbumin, BSA, and transferrin at 0.3, 3, 30, and 300 μg sample loads. (C) Average wpI across the four sample loadings for the four standard proteins independent of proteoform content. (D) Bubble plots of the averaged wpI (n = 3) for each proteoform at the four sample amounts. The reported pI is for the base (most intense) proteoform for each protein. Dashed lines show related proteoforms concurrently detected at the sample loads highlighted in (B). (E) Averaged wpI (n = 3) for the proteins independent of proteoform content at the different sample loads.
Effect of Modification Class on pI.
For each standard protein we also examined if different proteoforms preferentially separated to distinct isoelectric points. Here the largest wpI difference between the observed proteoforms was determined and then compared across the sample amounts. For RNase B and α-Lac, which consist of charge neutral PTMs, such as nonsialylated N-glycans or amino acid losses, no significant difference in wpI was observed between proteoforms at each quantity loaded (Figure 4D). For BSA and transferrin, which consist of differentially phosphorylated and sialylated proteoforms, the maximum wpI difference between proteoforms at the lowest load (0.3 μg) was negligible (<0.01); however, at the highest amount, a −0.067 ΔwpI per phosphorylation was observed for BSA and −0.09 ΔwpI was observed for the sialylation of transferrin. The data suggests the shallowing pH gradient for BSA and transferrin facilitated the resolution of the modified proteoforms despite the theoretical shifts being less than the resolution of the IPG employed (0.33 ΔwpI/cm; Figure 4D). The ΔwpIs were consistent with the theoretical ΔwpI computed for each PTM class using the IPC program (−0.062 ± 0.006 and −0.105 ± 0.039, respectively), suggesting that theoretical ΔwpI calculations for common PTMs may be utilized in addition to delta mass searches (i.e., acetylation (+42 Da), phosphorylation (+80 Da), sialylation (+291 Da)) and MS/MS analysis to assign unknown proteoforms within complex mixtures. Overall, the results also imply that at low amounts the resolution of the IEF is dictated by the ΔwpI/cm of the IPG and ampholyte concentration, which at 0.33 ΔwpI/cm, the conditions used here, is inadequate to resolve the different modified forms. However, progressively higher loads flatten the local pH gradient leading to broadening, accompanied by destabilization of the pH gradient and drift toward the anode for all but the most basic protein (RNaseB), which shifts toward the cathode.45
Label-Free Quantitation of Proteoforms, Proteoform Ratios, and Proteins.
We next examined if the multidimensional workflow would permit the simultaneous determination of the three quantitative metrics available with top-down workflows (Figure 1B). To evaluate this hypothesis, we determined the limits of detection (LOD) and a linear dynamic range for the standard proteins, which included a new study on the standards spiked into an E. coli lysate to examine how a complex matrix may affect protein quantitation.
Proteoform Spectral Intensity and Proteoform Ratios versus Quantity Loaded.
The results for the nonspiked experiments for RNase B (Figure 5A,B, upper left panels) and α-Lac, BSA, and transferrin (Figure S6) show that all proteoform spectral intensities increased linearly across the 0.3–300 μg protein load. BSA and transferrin spiked into E. coli yielded similar results to the nonspiked data; however, for RNase B and α-Lac, the linear range was limited to 102 range due to interference by other E. coli proteins at the lowest quantity. The LOD for the individual proteoforms ranged from 20–40 ± 6.79 fmol for RNase B, 48–517 ± 27.87 fmol for α-Lac, 18–50 ± 6.35 fmol for BSA, and 36–204 ± 10.75 fmol for transferrin. On average, the LOD for the standards spiked into E. coli increased ~1.7-fold with the observed LOD for the individual proteoforms ranged from 40–150 ± 9.132, 59–1269 ± 10.23, 26–73 ± 10.50, and 65–370 ± 24.28 fmol, respectively. The quantitative response for the individual proteoforms was also reflected in their relative ratios, where a good correlation was observed (Ravg = 0.998; Figure 5A,B and S6, middle panels) for each protein across the linear portions of the calibration curves (Table S5).
Figure 5.
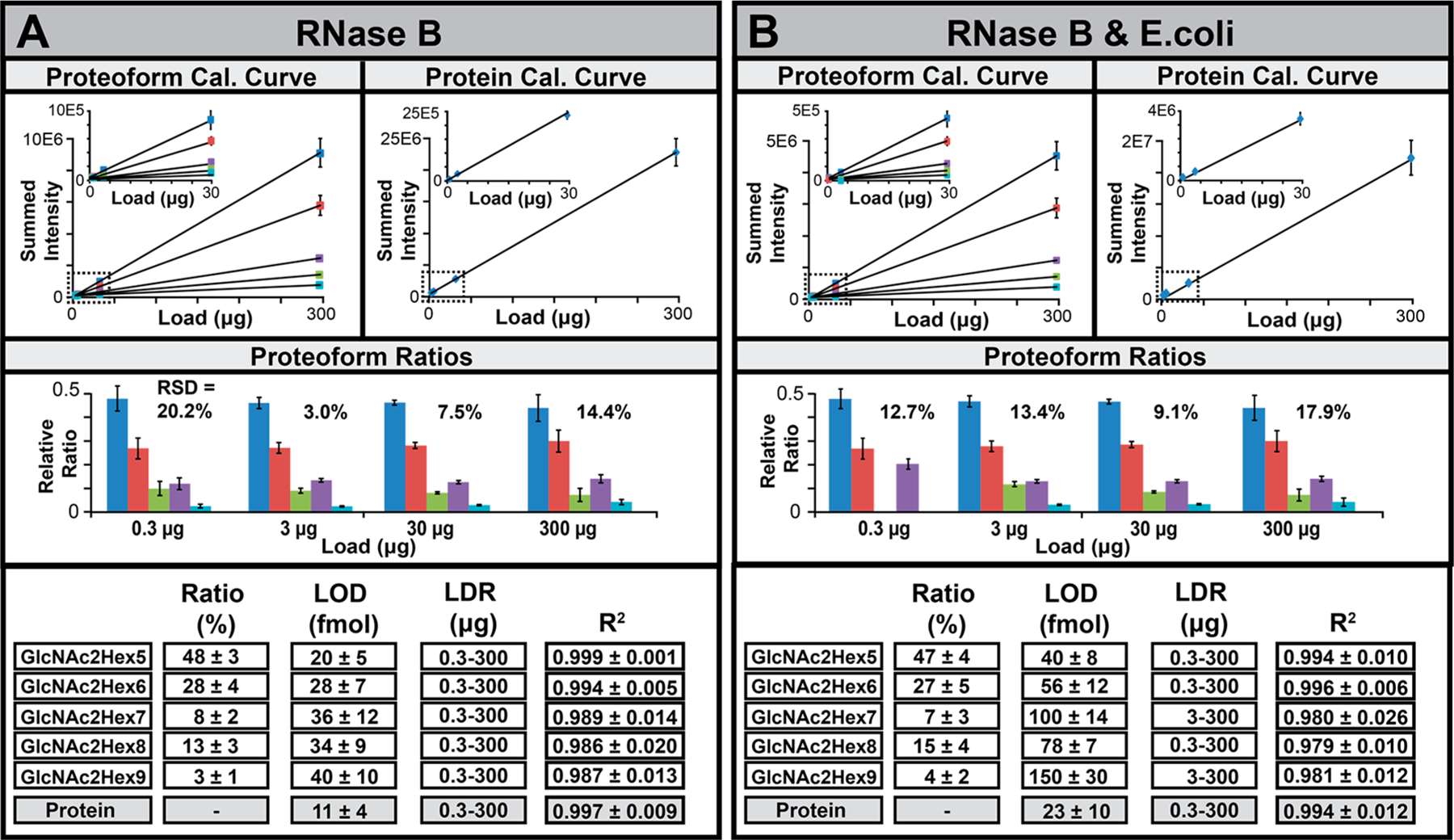
(A, B) Averaged calibration curves (n = 3) for the proteoforms (upper left panels), estimated total protein (upper right panels), proteoform ratios (middle panels), and tabulated FOM (lower panels) and for nonspiked and spiked RNase B (A, B) across the four loading amounts. List of slopes and standard deviations are presented in Table S5.
Protein Spectral Intensity versus Sample Load.
Finally, we summed the signal of all proteoforms for each protein together to generate a theoretical protein-level calibration curve (Figure 5A,B and S5, upper right panels). The estimated theoretical LODs for the nonspiked proteins were 11 ± 3.97 fmol for RNase B, 45 ± 13.04 fmol for α-Lac, 15 ± 4.02 fmol for BSA, and 32 ± 6.96 fmol for transferrin. For the proteins spiked into E. coli, the LODs were 23 ± 10.02, 55 ± 8.97, 22 ± 7.01, and 58 ± 12.99 fmol, respectively. The observed LODs for the nonspiked proteins were consistent with a report on ubiquitin (~7.0 fmol), which manifests in one dominant proteoform.19 The results suggested that summation of proteoform signal across the multidimensional workflow may be a surrogate for quantitative analysis of a protein independent of the number of proteoforms observed. To help verify this, an additional IEF-SPLC-FTMS analysis of nonspiked RNase A (an unmodified form of RNase B) was performed. An LOD of 6.0 ± 2.0 fmol (Table S5) was determined that was ~2-fold improved LOD over RNase B, which suggests that sample loss associated with resolving five individual proteoforms for RNase B versus one for RNase A had a small impact on assay sensitivity.
DISCUSSION
Two-dimensional gel electrophoresis (2DGE), which balances high resolution pI separations in the first dimension through immobilized pH gradients with SDS/PAGE in the second, is a powerful resource for separations of 100 to 1000s of proteins or differentially modified proteoforms that often spread across the pI axis (e.g., glycosylated, phosphorylated, or citrullinated proteoforms).48–50 However, the discontinuity of SDS/PAGE with MS without added steps for protein visualization, spot picking, electroelution, and digestion prior to LCMS adds significant complexity to the experimental design, which in the case of digestion can create a proteoform inference problem due to peptide-level sampling on multiple proteoforms simultaneously. Despite these limitations, the simplicity of the voltage-driven electrophoretic separations through commercial IPG strips promises to be a TDMS-friendly approach by balancing protein and proteoform separations in a single workflow through careful choice of IPG pH range.51,52 In 2DGE, proteins are characterized with visually intuitive mass versus pI Cartesian coordinates; however, off-gel IEF followed by LCMS results in tabulated lists of masses, intensities, and LC retention times (i.e., hydrophobicity) for each IEF fraction. Here, in contrast to the high inter-replicate precision afforded by SPLC and FTMS analysis, the wpI estimated is limited by the resolution (ΔpI/cm) of the liquid-filled compartments across the IPG. Indeed, E. coli proteins typically exhibited the standard deviation from their mean wpI of ±0.15–0.21, which was consistent with our observation that most proteins observed within proteome analysis localize to a single IEF fraction.19 However, the inter-replicate precision improved to ±0.05 for both abundant E. coli proteoforms and individual proteoforms for standard proteins that focused into 2 or more fractions (e.g., Figure 4D). This result indicates that the MS signal intensity for each protein in each IEF fraction was consistent among the replicates and corroborated by the experiments with standard proteins, indicating that, despite sample broadening at higher loads, recombination of MS spectral intensities across multiple IEF fractions permits the generation of calibration curves for individual proteoforms or total protein amounts with a linear response that spans 102–103 orders of magnitude. It is important to note though that the current methodology used to calculate protein amounts is a theoretical value dependent on the detection of each related proteoform and is in itself not directly detected. Additionally, calculated LODs are dependent on the complexity of the background material and may change when different lysates are used. This result is an improvement upon the 101 dynamic range reported by other multidimensional platforms,29,53 although further work on the quantitative reliability of the workflow must be assessed in a biological context on the proteome level. Plus, while the observed linear dynamic range is in general agreement with the reported ~4000 dynamic range of our instrument; it is important to note that, under max injection time conditions, the intensity data is extrapolated from known equations in order to arrive at quantitative trends similar to that of a triple quadrupole mass spectrometer.54,34 Overall, the work highlights baseline figures of merits associated with the IEF-SPLC-FTMS workflow that could be improved upon in diverse ways. For example, future investigations should continue to explore protein pI, size, amount, and PTM pKa impacts on the detection of distinct modification clases (e.g., citrullination which are early indicators of neurodegenerative diseases55–57).
CONCLUSIONS
The work provides the results of an analytical workup of the IEF-SPLC-FTMS platform, emphasizing the characterization of pI precision and quantitative reliability of intact protein measurements observed in technical replicates on the E. coli proteome and standard proteins evaluated at various sample loads. Good reproducibility was observed for ~900 proteoforms redundantly observed in E. coli with procedures for normalizing the physicochemical property axis across the multidimensional data sets, providing improved inter-replicate precision of the physicochemical properties measured. The good reproducibility enabled quantitative assessment of the three different quantitative metrics available within the top-down, where standard proteins and their proteoforms were quantifiable across 102–103 loading amounts with low fmol detection limits.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award No. 1R01GM115739-01A1. Any opinions, findings, conclusions, or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Institutes of Health. This work was also supported by the Multiple Sclerosis Society (PP-1503-04034), The Darrel K. Royal Research Fund for Alzheimer’s Disease (48680-DKR), The Texas Alzheimer’s Research and Care Consortium Investigator Grant Program (354091), and the UT System Neuroscience and Neurotechnology Research Institute (363027). Funding was also provided by the University of Texas at Dallas, the John L. Roach Scholarship in Biomedical Research, and the Friends of Alzheimer’s Disease Research Award.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jasms.0c00355.
Five supporting tables (XLSX)
Additional experimental details and five supporting figures (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jasms.0c00355
The authors declare no competing financial interest.
Contributor Information
John R. Corbett, Department of Pathology, UT Southwestern Medical Center, Dallas, Texas 75390, United States; Department of Bioengineering, UT Dallas, Richardson, Texas 75080, United States
Dana E. Robinson, Department of Pathology, UT Southwestern Medical Center, Dallas, Texas 75390, United States
Steven M. Patrie, Department of Chemistry, Northwestern University, Evanston, Illinois 60208, United States; Department of Pathology, UT Southwestern Medical Center, Dallas, Texas 75390, United States
REFERENCES
- (1).Smith LM; Kelleher NL Proteoform: a single term describing protein complexity. Nat. Methods 2013, 10 (3), 186–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kruve A; Rebane R; Kipper K; Oldekop ML; Evard H; Herodes K; Ravio P; Leito I Tutorial review on validation of liquid chromatography-mass spectrometry methods: part I. Anal. Chim. Acta 2015, 870, 29–44. [DOI] [PubMed] [Google Scholar]
- (3).Kruve A; Rebane R; Kipper K; Oldekop ML; Evard H; Herodes K; Ravio P; Leito I Tutorial review on validation of liquid chromatography-mass spectrometry methods: part II. Anal. Chim. Acta 2015, 870, 8–28. [DOI] [PubMed] [Google Scholar]
- (4).Peng Y; Yu D; Gregorich Z; Chen X; Beyer AM; Gutterman DD; Ge Y In-depth proteomic analysis of human tropomyosin by top-down mass spectrometry. J. Muscle Res. Cell Motil 2013, 34 (3–4), 199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Zhang H; Ge Y Comprehensive analysis of protein modifications by top-down mass spectrometry. Circ.: Cardiovasc. Genet 2011, 4 (6), 711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Wang YC; Peterson SE; Loring JF Protein post-translational modifications and regulation of pluripotency in human stem cells. Cell Res. 2014, 24 (2), 143–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Munshi A; Shafi G; Aliya N; Jyothy A Histone modifications dictate specific biological readouts. J. Genet. Genomics 2009, 36 (2), 75–88. [DOI] [PubMed] [Google Scholar]
- (8).Burnaevskiy N; Fox TG; Plymire DA; Ertelt JM; Weigele BA; Selyunin AS; Way SS; Patrie SM; Alto NM Proteolytic elimination of N-myristoyl modifications by the Shigella virulence factor IpaJ. Nature 2013, 496 (7443), 106–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Patrie SM Top-Down Mass Spectrometry: Proteomics to Proteoforms. Adv. Exp. Med. Biol 2016, 919, 171–200. [DOI] [PubMed] [Google Scholar]
- (10).Zhang Y; Fonslow BR; Shan B; Baek MC; Yates JR Protein Analysis by Shotgun/Bottom-up Proteomics. Chem. Rev 2013, 113 (4), 2343–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Patrie SM; Ferguson JT; Robinson DE; Whipple D; Rother M; Metcalf WW; Kelleher NL Top down mass spectrometry of < 60-kDa proteins from Methanosarcina acetivorans using quadrupole FRMS with automated octopole collisionally activated dissociation. Mol. Cell. Proteomics 2006, 5 (1), 14–25. [DOI] [PubMed] [Google Scholar]
- (12).Tran JC; Zamdborg L; Ahlf DR; Lee JE; Catherman AD; Durbin KR; Tipton JD; Vellaichamy A; Kellie JF; Li M; Wu C; Sweet SM; Early BP; Siuti N; LeDuc RD; Compton PD; Thomas PM; Kelleher NL Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature 2011, 480 (7376), 254–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Mazur MT; Cardasis HL; Spellman DS; Liaw A; Yates NA; Hendrickson RC Quantitative analysis of intact apolipoproteins in human HDL by top-down differential mass spectrometry. Proc. Natl. Acad. Sci. U. S. A 2010, 107 (17), 7728–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wijnker PJ; Murphy AM; Stienen GJ; van der Velden J Troponin I phosphorylation in human myocardium in health and disease. Netherlands heart journal: monthly journal of the Netherlands Society of Cardiology and the Netherlands Heart Foundation 2014, 22 (10), 463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Plymire DA; Wing CE; Robinson DE; Patrie SM Continuous Elution Proteoform Identification of Myelin Basic Protein by Superficially Porous Reversed-Phase Liquid Chromatography and Fourrier Transform Mass Spectrometry. Anal. Chem 2017, 89 (22), 12030–12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Sharma S; Simpson DC; Tolic N; Jaitly N; Mayampurath AM; Smith RD; Pasa-Tolic L Proteomic profiling of intact proteins using WAX-RPLC 2-D separations and FTICR mass spectrometry. J. Proteome Res 2007, 6 (2), 602–10. [DOI] [PubMed] [Google Scholar]
- (17).Roth MJ; Parks BA; Ferguson JT; Boyne MT 2nd; Kelleher NL ″Proteotyping″: population proteomics of human leukocytes using top down mass spectrometry. Anal. Chem 2008, 80 (8), 2857–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Muneeruddin K; Nazzaro M; Kaltashov IA Characterization of intact protein conjugates and biopharmaceuticals using ionexchange chromatography with online detection by native electrospray ionization mass spectrometry and top-down tandem mass spectrometry. Anal. Chem 2015, 87 (19), 10138–45. [DOI] [PubMed] [Google Scholar]
- (19).Zhang J; Roth MJ; Chang AN; Plymire DA; Corbett JR; Greenberg BM; Patrie SM Top-down mass spectrometry on tissue extracts and biofluids with isoelectric focusing and superficially porous silica liquid chromatography. Anal. Chem 2013, 85 (21), 10377–84. [DOI] [PubMed] [Google Scholar]
- (20).McCool EN; Lubeckyj RA; Shen X; Chen D; Kou Q; Liu X; Sun L Deep Top-Down Proteomics Using Capillary Zone Electrophoresis-Tandem Mass Spectrometry: Identification of 5700 Proteoforms from the Escherichia coli Proteome. Anal. Chem 2018, 90 (9), 5529–5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Cai W; Tucholski T; Chen B; Alpert AJ; McIlwain S; Kohmoto T; Jin S; Ge Y Top-Down Proteomics of Large Proteins up to 223 kDa Enabled by Serial Size Exclusion Chromatography Strategy. Anal. Chem 2017, 89 (10), 5467–5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Botelho D; Wall MJ; Vieira DB; Fitzsimmons S; Liu F; Doucette A Top-down and bottom-up proteomics of SDScontaining solutions following mass-based separation. J. Proteome Res 2010, 9 (6), 2863–70. [DOI] [PubMed] [Google Scholar]
- (23).Shen Y; Tolic N; Piehowski PD; Shukla AK; Kim S; Zhao R; Qu Y; Robinson E; Smith RD; Pasa-Tolic L High resolution ultrahigh-pressure long column reversed-phase liquid chromatography for top-down proteomics. Journal of chromatography. A 2017, 1498, 99–110. [DOI] [PubMed] [Google Scholar]
- (24).Zhang X; Fang A; Riley CP; Wang M; Regnier FE; Buck C Multi-dimensional liquid chromatography in proteomics–a review. Anal. Chim. Acta 2010, 664 (2), 101–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wang S; Shi X; Xu G Online Three Dimensional Liquid Chromatography/Mass Spectrometry Method for the Separation of Complex Samples. Anal. Chem 2017, 89 (3), 1433–1438. [DOI] [PubMed] [Google Scholar]
- (26).Valeja SG; Xiu L; Gregorich ZR; Guner H; Jin S; Ge Y Three dimensional liquid chromatography coupling ion exchange chromatography/hydrophobic interaction chromatography/reverse phase chromatography for effective protein separation in top-down proteomics. Anal. Chem 2015, 87 (10), 5363–5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Wiener MC; Sachs JR; Deyanova EG; Yates NA Differential mass spectrometry: a label-free LC-MS method for finding significant differences in complex peptide and protein mixtures. Anal. Chem 2004, 76 (20), 6085–96. [DOI] [PubMed] [Google Scholar]
- (28).Ntai I; LeDuc RD; Fellers RT; Erdmann-Gilmore P; Davies SR; Rumsey J; Early BP; Thomas PM; Li S; Compton PD; Ellis MJ; Ruggles KV; Fenyo D; Boja ES; Rodriguez H; Townsend RR; Kelleher NL Integrated Bottom-Up and Top-Down Proteomics of Patient-Derived Breast Tumor Xenografts. Mol. Cell. Proteomics 2016, 15 (1), 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ntai I; Kim K; Fellers RT; Skinner OS; Smith A. D. t.; Early BP; Savaryn JP; LeDuc RD; Thomas PM; Kelleher NL Applying label-free quantitation to top down proteomics. Anal. Chem 2014, 86 (10), 4961–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Durbin KR; Fornelli L; Fellers RT; Doubleday PF; Narita M; Kelleher NL Quantitation and Identification of Thousands of Human Proteoforms below 30 kDa. J. Proteome Res 2016, 15 (3), 976–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Toby TK; Abecassis M; Kim K; Thomas PM; Fellers RT; LeDuc RD; Kelleher NL; Demetris J; Levitsky J Proteoforms in Peripheral Blood Mononuclear Cells as Novel Rejection Biomarkers in Liver Transplant Recipients. Am. J. Transplant 2017, 17 (9), 2458–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ntai I; Toby TK; LeDuc RD; Kelleher NL A Method for Label-Free, Differential Top-Down Proteomics. Methods Mol. Biol. (N. Y., NY, U. S.) 2016, 1410, 121–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Savaryn JP; Toby TK; Catherman AD; Fellers RT; LeDuc RD; Thomas PM; Friedewald JJ; Salomon DR; Abecassis MM; Kelleher NL Comparative top down proteomics of peripheral blood mononuclear cells from kidney transplant recipients with normal kidney biopsies or acute rejection. Proteomics 2016, 16 (14), 2048–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Roth MJ; Plymire DA; Chang AN; Kim J; Maresh EM; Larson SE; Patrie SM Sensitive and reproducible intact mass analysis of complex protein mixtures with superficially porous capillary reversed-phase liquid chromatography mass spectrometry. Anal. Chem 2011, 83 (24), 9586–92. [DOI] [PubMed] [Google Scholar]
- (35).Moreda-Pineiro A; Garcia-Otero N; Bermejo-Barrera P A review on preparative and semi-preparative offgel electrophoresis for multidimensional protein/peptide assessment. Anal. Chim. Acta 2014, 836, 1–17. [DOI] [PubMed] [Google Scholar]
- (36).Zhang J; Corbett JR; Plymire DA; Greenberg BM; Patrie SM Proteoform analysis of lipocalin-type prostaglandin Dsynthase from human cerebrospinal fluid by isoelectric focusing and superficially porous liquid chromatography with Fourier transform mass spectrometry. Proteomics 2014, 14 (10), 1223–31. [DOI] [PubMed] [Google Scholar]
- (37).Warren CM; Geenen DL; Helseth DL Jr.; Xu H; Solaro RJ Sub-proteomic fractionation, iTRAQ, and OFFGEL-LC-MS/MS approaches to cardiac proteomics. J. Proteomics 2010, 73 (8), 1551–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Jafari M; Primo V; Smejkal GB; Moskovets EV; Kuo WP; Ivanov AR Comparison of in-gel protein separation techniques commonly used for fractionation in mass spectrometry-based proteomic profiling. Electrophoresis 2012, 33 (16), 2516–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Pan S; Chen R; Aebersold R; Brentnall TA Mass Spectrometry Based Glycoproteomics—From a Proteomics Perspective. Mol. Cell. Proteomics 2011, 10 (1), R110.003251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Savaryn JP; Catherman AD; Thomas PM; Abecassis MM; Kelleher NL The emergence of top-down proteomics in clinical research. Genome Med. 2013, 5 (6), 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Horn DM; Zubarev RA; McLafferty FW Automated reduction and interpretation of high resolution electrospray mass spectra of large molecules. J. Am. Soc. Mass Spectrom 2000, 11 (4), 320–32. [DOI] [PubMed] [Google Scholar]
- (42).Barbarini N; Magni P Accurate peak list extraction from proteomic mass spectra for identification and profiling studies. BMC Bioinf. 2010, 11, 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Li H; Wolff JJ; Van Orden SL; Loo JA Native Top-Down ESI-MS of 158 kDa Protein Complex by High Resolution Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Chem 2014, 86 (1), 317–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Bjellqvist B; Basse B; Olsen E; Celis JE Reference points for comparisons of two-dimensional maps of proteins from different human cell types defined in a pH scale where isoelectric points correlate with polypeptide compositions. Electrophoresis 1994, 15 (3–4), 529–39. [DOI] [PubMed] [Google Scholar]
- (45).Cantrell SJ; Babitch JA; Torres S Protein-load effects on the pH gradient of isoelectric focusing in polyacrylamide gel. Anal. Biochem 1981, 116 (1), 168–73. [DOI] [PubMed] [Google Scholar]
- (46).Liu T; Belov ME; Jaitly N; Qian WJ; Smith RD Accurate mass measurements in proteomics. Chem. Rev 2007, 107 (8), 3621–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Hubner NC; Ren S; Mann M Peptide separation with immobilized pI strips is an attractive alternative to in-gel protein digestion for proteome analysis. Proteomics 2008, 8 (23–24), 4862–72. [DOI] [PubMed] [Google Scholar]
- (48).Mayer K; Albrecht S; Schaller A Targeted Analysis of Protein Phosphorylation by 2D Electrophoresis. Methods Mol. Biol. (N. Y., NY, U. S.) 2015, 1306, 167–76. [DOI] [PubMed] [Google Scholar]
- (49).Kleinert P; Kuster T; Arnold D; Jaeken J; Heizmann CW; Troxler H Effect of glycosylation on the protein pattern in 2-Dgel electrophoresis. Proteomics 2007, 7 (1), 15–22. [DOI] [PubMed] [Google Scholar]
- (50).Koshel BM; Wirth MJ Trajectory of isoelectric focusing from gels to capillaries to immobilized gradients in capillaries. Proteomics 2012, 12 (0), 2918–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Barrabes S; Sarrats A; Fort E; De Llorens R; Rudd PM; Peracaula R Effect of sialic acid content on glycoprotein pI analyzed by two-dimensional electrophoresis. Electrophoresis 2010, 31 (17), 2903–12. [DOI] [PubMed] [Google Scholar]
- (52).Zhu K; Zhao J; Lubman DM; Miller FR; Barder TJ Protein pI shifts due to posttranslational modifications in the separation and characterization of proteins. Anal. Chem 2005, 77 (9), 2745–55. [DOI] [PubMed] [Google Scholar]
- (53).Natale M; Caiazzo A; Bucci EM; Ficarra E A Novel Gaussian Extrapolation Approach for 2D Gel Electrophoresis Saturated Protein Spots. Genomics, Proteomics Bioinf 2012, 10 (6), 336–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Page JS; Bogdanov B; Vilkov AN; Prior DC; Buschbach MA; Tang K; Smith RD Automatic gain control in mass spectrometry using a jet disrupter electrode in an electrodynamic ion funnel. J. Am. Soc. Mass Spectrom 2005, 16 (2), 244–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Zabrouskov V; Han X; Welker E; Zhai H; Lin C; van Wijk KJ; Scheraga HA; McLafferty FW Stepwise deamidation of ribonuclease A at five sites determined by top down mass spectrometry. Biochemistry 2006, 45 (3), 987–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Dan A; Takahashi M; Masuda-Suzukake M; Kametani F; Nonaka T; Kondo H; Akiyama H; Arai T; Mann DM; Saito Y; Hatsuta H; Murayama S; Hasegawa M Extensive deamidation at asparagine residue 279 accounts for weak immunoreactivity of tau with RD4 antibody in Alzheimer’s disease brain. Acta neuropathologica communications 2013, 1, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Witalison EE; Thompson PR; Hofseth LJ Protein Arginine Deiminases and Associated Citrullination: Physiological Functions and Diseases Associated with Dysregulation. Curr. Drug Targets 2015, 16 (7), 700–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.